
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporation of U(IV) in monazite–cheralite ceramics under oxidizing and inert atmospheres†
Alison
El Monjid
 a,
Stéphanie
Szenknect
*a,
Adel
Mesbah
b,
Myrtille O. J. Y.
Hunault
c,
Denis
Menut
c,
Nicolas
Clavier
a and
Nicolas
Dacheux
a
a,
Stéphanie
Szenknect
*a,
Adel
Mesbah
b,
Myrtille O. J. Y.
Hunault
c,
Denis
Menut
c,
Nicolas
Clavier
a and
Nicolas
Dacheux
a
aICSM, Univ. Montpellier, CNRS, CEA, ENSCM, Site de Marcoule, Bagnols/Cèze, France. E-mail: stephanie.szenknect@cea.fr
bUniv. Lyon, Université Claude Bernard Lyon 1, CNRS, IRCELYON, F-69626 Villeurbanne, France
cSynchrotron SOLEIL, L'Orme des Merisiers, Départementale 128, 91190 Saint-Aubin, France
First published on 9th January 2024
Abstract
This work is the first attempt to prepare Nd1−xCaxUxPO4 monazite–cheralite with 0 < x ≤ 0.1 by a wet chemistry method. This method relies on the precipitation under hydrothermal conditions (T = 110 °C for four days) of the Nd1−xCaxUxPO4·nH2O rhabdophane precursor, followed by its thermal conversion for 6 h at 1100 °C in air or Ar atmosphere. The optimized synthesis protocol led to the incorporation of U and Ca in the rhabdophane structure. After heating at 1100 °C for 6 h in air, single-phase monazite–cheralite samples were obtained. However, α-UP2O7 was identified as a secondary minor phase in the samples heated under Ar atmosphere. The U speciation in the samples converted in an oxidising atmosphere was carefully characterized using synchrotron radiation by combining HERFD-XANES and XRD. These results showed the presence of a minor secondary phase containing hexavalent uranium and phosphate with a stoichiometry of U![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P = 0.78. This highly labile uranyl phosphate phase incorporated 21 mol% of the uranium initially precipitated with the rhabdophane precursor. This phase was completely removed by a washing protocol. Thus, single-phase monazite–cheralite was obtained through the wet chemistry route described in this work with a maximum U loading of x = 0.08.
:P = 0.78. This highly labile uranyl phosphate phase incorporated 21 mol% of the uranium initially precipitated with the rhabdophane precursor. This phase was completely removed by a washing protocol. Thus, single-phase monazite–cheralite was obtained through the wet chemistry route described in this work with a maximum U loading of x = 0.08.
Introduction
Monazite is a natural phosphate-based mineral that contains light rare earth elements uranium and thorium with amounts up to 16 wt% UO2 and 29 wt% ThO2, respectively.1–3 In spite of the presence of mobile elements incorporated in the monazite structure and high doses of self-irradiation, the chemical durability of natural monazite under weathering conditions is remarkable.4–8 These properties meet the requirements of ceramic matrices for the long-term storage of high level nuclear waste (HLW) including actinides or long life fission products. Therefore, LnPO4 monazite and its derivative have been studied for several decades for the specific conditioning of minor actinides (Am, Cm, and Np) and Pu.8–11 These studies have been conducted in several countries (i.e. France, Netherlands, Germany, Russia, USA, UK, Japan) where the separation of long-life radionuclides, especially minor actinides and plutonium derived from dismantled nuclear weapons, was being considered. Indeed, the chemical flexibility of the monazite-type structure allows the incorporation of actinide elements in the trivalent and tetravalent oxidation states either by the direct substitution of Ln3+ or by coupled substitution of 2 Ln3+ by Ca2+ and An4+.12 The coupled substitution leads to the formation of Ln1−2xCaxAnxPO4 monazite–cheralite solid solutions. This mechanism is the most documented in the literature and allows complete Ca substitution resulting in the formation of Ca0.5An0.5PO4 (An = Th, U, Np).13–17Thus, the monazite nine-coordinate site is able to incorporate large amounts of tetravalent cations through appropriate charge compensation with a divalent element like Ca. Podor and Cuney18 gave two steric criteria for the existence of such a Ln1−2xCaxAnxPO4 monazite–cheralite solid solution,
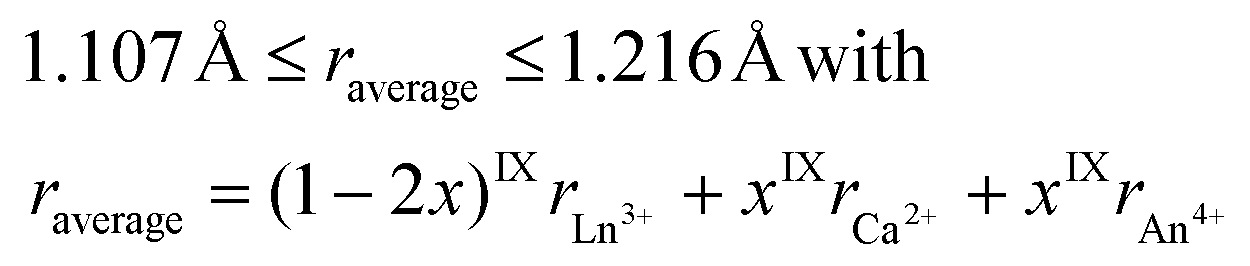 | (1) |
 | (2) |
Using these rules with IXrNd3+ = 1.163 Å, IXrCa2+ = 1.18 Å, and IXrAn4+ = 1.09 Å, 1.05 Å, 1.03 Å, and 1.01 Å15 for Th, U, Np and Pu, respectively, allows the calculation of their maximum incorporation in NdPO4. This calculation shows that the coupled substitution can be complete for Th and U (i.e. x = 0.50) whereas it is limited to xmax = 0.48 and xmax = 0.41 for Np and Pu, respectively.
These steric considerations indicate that high incorporation rates of tetravalent An are achievable in the monazite/cheralite solid solution. Nevertheless, such high doping rates are generally obtained using dry chemistry routes, which could present difficulties for the multivalent elements (U, Np and Pu in particular)17,20 due to the relative instability of tetravalent actinide ions. Compared to Np(IV), the stability domains of U(IV) and Pu(IV) are less extended, as U(IV) is oxidised to U(VI) at lower potential and Pu(IV) is reduced to Pu(III) at higher potential.
Terra et al.21 synthesized La1−2xCaxThx−yUyPO4 solid solutions (with 0.1 ≤ x ≤ 0.4 and y = x/5) as well as end-members by a dry chemistry route. For that purpose, AnO2 was mixed with La2O3, Ca(HPO4)·2H2O and NH4(H2PO4) and the mixture underwent several cycles of milling and calcination at 1200 °C under Ar atmosphere. At least two cycles were required to obtain homogeneous single-phase samples. Bregiroux et al.15 also investigated the formation of Ca0.5U0.5PO4 prepared by a similar solid–solid reaction. They performed analyses of samples fired at several temperatures from 400 to 1400 °C under Ar atmosphere. Below 1100 °C, α-UP2O7 was found to co-exist with the cheralite phase, whereas the phosphates decomposed at temperatures higher than 1300 °C, leading to the formation of CaO and UO2 and the volatilization of P4O10(g). Thus, the range of temperature that allows the preparation of single-phase cheralite incorporating U was found to be rather constrained. It is worth noting that this synthesis route did not allow the preparation of Ca0.5Ce0.5PO4 cheralite. The authors showed that the reduction of tetravalent Ce occurred during the thermal treatment, leading to the formation of a mixture of CePO4 monazite and Ca2P2O7. Similar results were obtained by Bregiroux et al.20 using the same protocol to prepare Ca0.5Pu0.5PO4. However, they also observed the partial incorporation of Pu(IV) in the cheralite when they fired the mixtures under air.
On the other hand, dustless wet chemistry methods have also been widely used to easily prepare LnPO422 and AnPO4 monazite (with An = Pu, Am–Bk).23–27 Thus, the precipitation of LnPO4·nH2O or AnPO4·nH2O rhabdophane precursor followed by its thermal conversion leads to the preparation of single-phase monazite incorporating trivalent actinides. In contrast, the incorporation of high loadings of tetravalent actinides in monazite–cheralite appeared difficult using wet chemistry routes. Qin et al.28 developed a wet chemistry method for the preparation of Ln1−2xCaxThxPO4·nH2O (Ln = Pr and Nd) Th-rhabdophane solid solutions. Using this hydrothermal protocol, under acidic conditions, the limit of incorporation of Th in the solid solution was around 14 mol%. Beyond this limit, thorium phosphate–hydrogen phosphate hydrate (TPHPH), Th2(HPO4)(PO4)2·H2O, was formed as a secondary phase. The thermal conversion of Nd1−2xCaxThxPO4·nH2O (with 0 ≤ x ≤ 0.15) to monazite/cheralite occurred between 750 and 800 °C under air.29 Single phase monazite–cheralite solid solutions were obtained only for x ≤ 0.1. For higher incorporation rates, α-ThP2O7 was found as a secondary phase. Additionally, Qin et al.30 demonstrated the feasibility of the direct sintering of Nd0.8Ca0.1Th0.1PO4·nH2O rhabdophane and succeeded in obtaining dense monazite pellets at a temperature (i.e. 1400 °C) significantly lower than those reported in the literature. Finally, the chemical durability of the Nd0.8Ca0.1Th0.1PO4 monazite was investigated from a thermodynamic point of view.31 Solubility experiments demonstrated the exceptional chemical stability of this phase in environmental conditions. The results obtained guarantee the long-term efficient immobilization of Th in the monazite–cheralite matrix.
Thus, this type of wet process makes it possible to limit the risks of contamination of the facilities by the dissemination of radioactive dust associated with dry route processes. It also guarantees the preparation of dense and homogeneous pellets of monazite–cheralite incorporating up to 10 mol% of thorium. However, a similar protocol has not been tested for actinide elements prone to oxidation. Therefore, this work is a first attempt to prepare monazite–cheralite incorporating U(IV) by adapting the protocol developed by Qin et al.29 Using uranium allows anticipating potential difficulties in the preparation of Np-doped monazite–cheralite by a wet chemistry route, without the difficulties linked to the handling of these highly radioactive radioelements. As the solubility of U(VI) is much higher than that of U(IV) in solution, particular attention was paid to characterizing the oxidation state of uranium in order to guarantee the long-term stability of uranium incorporated in the monazite–cheralite structure.
Results and discussion
Synthesis of U-rhabdophane
The chemical composition of the synthesized precursors was determined by elemental analyses of the solution after complete dissolution of an aliquot of each sample (Table 1). Regardless of the uranium content, the analyses indicated a slight deviation from the target composition. The P/(Nd + Ca + U) ratio was constant regardless of the value of x (0.025 ≤ x ≤ 0.1) and was systematically slightly higher than 1. The incorporation of uranium in the rhabdophane structure implies the coupled substitution of two Nd3+ by one Ca2+ and one U4+. The results presented in Table 1 show a slight deviation from this mechanism. Whereas the neodymium and uranium mole fractions were close to the expected values, the Ca content was found to be lower than the target value despite the large excess of Ca introduced in the initial cation mixture. One assumption to explain these results would be the partial oxidation of uranium to the hexavalent state during the hydrothermal treatment. In that case, uranium introduced in the system that oxidised could also have partly precipitated with phosphate ions as a secondary phase. Another possibility might be the direct substitution of four Nd3+ by three U4+ and a vacancy. This type of direct substitution has been already reported in natural samples32 but never evidenced in synthetic ones.| Target value |
|
|
|
|
|---|---|---|---|---|
| x | 1 − 2x | x | x | 1.00 |
| 0.025 | 0.96 ± 0.05 | 0.017 ± 0.001 | 0.026 ± 0.001 | 1.11 ± 0.06 |
| 0.05 | 0.90 ± 0.05 | 0.042 ± 0.002 | 0.056 ± 0.003 | 1.12 ± 0.06 |
| 0.075 | 0.85 ± 0.04 | 0.064 ± 0.003 | 0.082 ± 0.004 | 1.13 ± 0.06 |
| 0.1 | 0.82 ± 0.04 | 0.085 ± 0.004 | 0.099 ± 0.005 | 1.12 ± 0.06 |



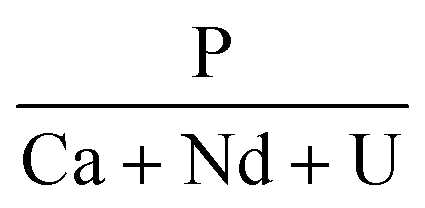
The PXRD patterns of the prepared samples are plotted in Fig. 1 and compared to the rhabdophane reference.33 They confirmed that the prepared samples were single-phase and only composed of the rhabdophane structure under its monoclinic form or that the amount of secondary phase is below the detection limit of this technique.
 | ||
| Fig. 1 PXRD patterns of the prepared Nd1−2xCaxUxPO4·nH2O samples. XRD lines associated with SmPO4·0.667H2O rhabdophane are included as reference33 (black vertical bars). | ||
Rietveld refinements of the PXRD diagrams were carried out in order to show the impact of uranium incorporation on the unit cell parameters. The results displayed in Table 2 show a contraction of the unit cell volume with the increase of the uranium mole content. By considering the effective ionic radii of Nd3+, Ca2+ and U4+ in the 9-fold coordination (1.163, 1.18 and 1.05 Å, respectively),19 this observation indicated that uranium was incorporated in the rhabdophane structure.
| x | a (Å) | b (Å) | c (Å) | β (°) | V (Å3) | |
|---|---|---|---|---|---|---|
| Rhabdophanes | 0.025 | 28.3216(1) | 7.0062(1) | 12.1320(1) | 115.47(1) | 2173.36(1) |
| 0.05 | 28.3231(1) | 7.0039(1) | 12.1251(1) | 115.46(1) | 2171.62(1) | |
| 0.075 | 28.3174(1) | 7.0012(1) | 12.1222(1) | 115.40(1) | 2170.94(1) | |
| 0.1 | 28.3192(1) | 6.9979(1) | 12.1210(1) | 115.40(1) | 2169.87(1) | |
| ① | 0.025 | 6.7383(1) | 6.9523(1) | 6.4083(1) | 103.69(1) | 291.68(1) |
| 0.05 | 6.7359(1) | 6.9490(1) | 6.4087(1) | 103.69(1) | 291.45(1) | |
| 0.075 | 6.7319(1) | 6.9437(1) | 6.4048(1) | 103.71(1) | 291.01(1) | |
| 0.1 | 6.7293(1) | 6.9409(1) | 6.4064(1) | 103.70(1) | 290.71(1) | |
| 0.1 | 6.7241(1) | 6.9365(1) | 6.3994(1) | 103.70(1) | 289.97(1) | |
| ② | 0.025 | 6.7380(1) | 6.9520(1) | 6.4078(1) | 103.69(1) | 291.63(1) |
| 0.05 | 6.7356(1) | 6.9489(1) | 6.4084(1) | 103.69(1) | 291.42(1) | |
| 0.075 | 6.7327(1) | 6.9453(1) | 6.4079(1) | 103.71(1) | 291.10(1) | |
| 0.1 | 6.7287(1) | 6.9410(1) | 6.4053(1) | 103.72(1) | 290.61(1) | |
Finally, the prepared rhabdophanes were all characterized by SEM. A micrograph of the rhabdophane sample with x = 0.1 is presented in Fig. 2a. The needle-like morphology of the grains was similar to the one observed by Qin et al.28 for Th-rhabdophane. Grains exhibiting a different morphology were not evidenced. These observations show the absence of any significant amount of secondary phases. Similar results were obtained for the other samples, regardless of the uranium content.
 | ||
| Fig. 2 SEM images recorded in SE mode for Nd0.8Ca0.1U0.1PO4·nH2O sample (a) and the corresponding monazite obtained after conversion in air (b) or in Ar atmosphere (c). | ||
Thermal conversion of U-rhabdophane to monazite–cheralite
As the PXRD and SEM analyses of the rhabdophane precursors indicated the incorporation of uranium in single-phase rhabdophane, TGA/DTA experiments were performed under both atmospheres to follow the dehydration process leading from U-rhabdophane to U-monazite–cheralite. Fig. 3 shows representative examples of TGA/DTA curves obtained for Nd0.8Ca0.1U0.1PO4·nH2O samples heated in air and Ar atmosphere. The complete data set is supplied in Fig. S1 of the ESI.†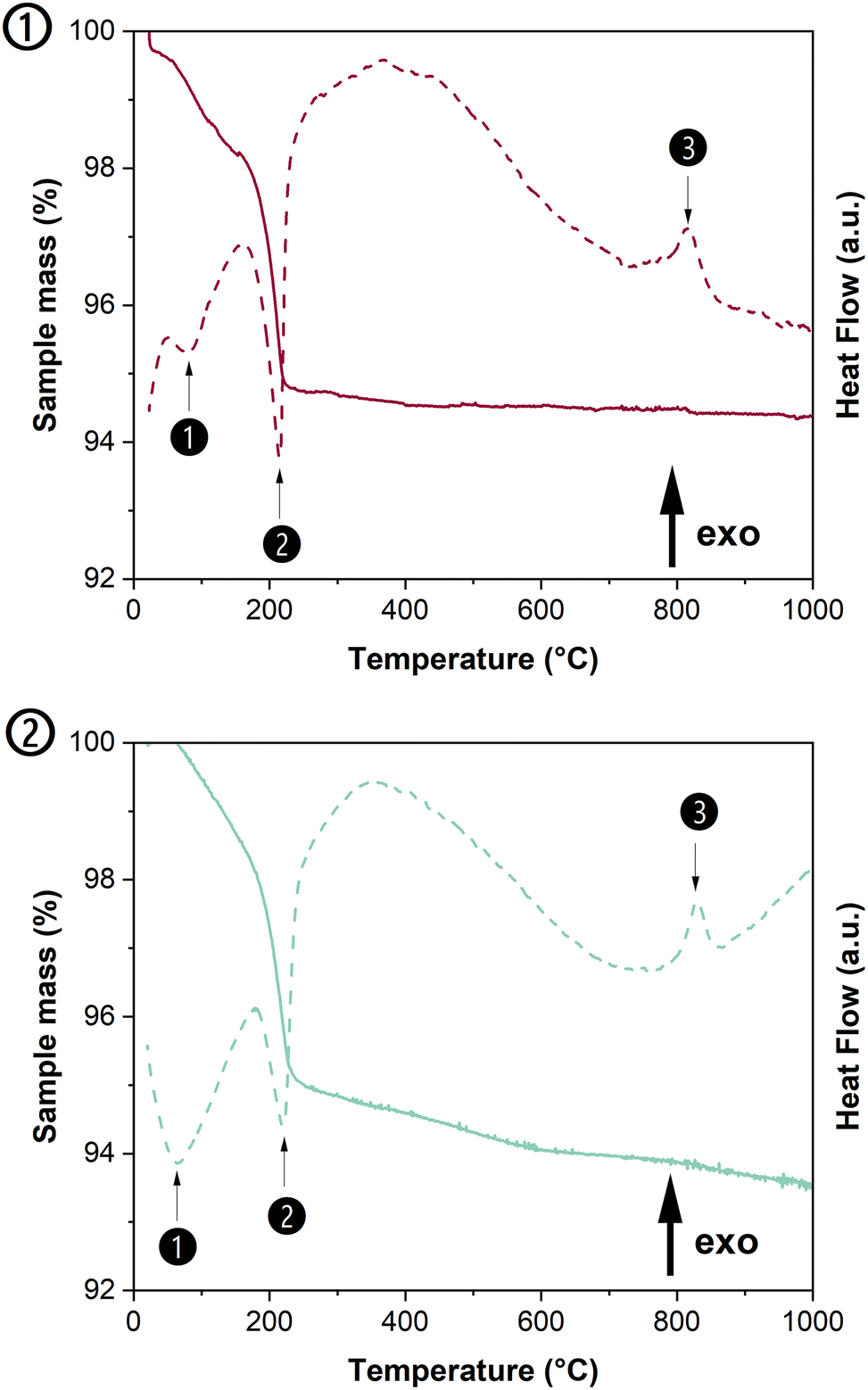 | ||
| Fig. 3 TGA and DTA scans obtained when heating Nd0.8Ca0.1U0.1PO4·nH2O sample in air (①) or inert conditions (Ar, ②). | ||
According to Fig. 3, the dehydration process was similar in both atmospheres. All the samples showed two-step dehydration leading to anhydrous rhabdophane. The first weak endothermic peak occurred at a very low temperature (i.e. below 100 °C) and was attributed to the formation of the hemihydrate form Nd0.8Ca0.1U0.1PO4·0.5H2O,34 while the second deep endothermic peak (around 200 °C) corresponds to the complete dehydration of the rhabdophane structure leading to the hexagonal form.35 The measured water content was found in the range of 0.64 to 0.79 H2O molecule per formula unit in the samples prepared (Table 3). It was found to be close to the amount of structural water proposed by Mesbah et al.33 (i.e. 0.667 H2O per formula unit) and in the range usually observed for Th-rhabdophane or LnPO4·nH2O rhabdophanes due to the adsorption of variable amounts of water molecules onto the surface of the samples or to their incorporation in the zeolitic channels.29,31,36 The exothermic peak observed between 690 and 780 °C corresponded to the irreversible phase transition from U-rhabdophane to U-monazite–cheralite. The temperature of full dehydration of rhabdophane was determined from the derivative of the TGA curve, whereas the temperature of phase transition was that of the maximum of the DTA exothermic peak. The temperatures of dehydration and phase transition obtained for all the prepared samples are listed in Table 3 and compared to the data reported in the literature. From these results, it appeared that the incorporation of uranium and calcium into the structure had low impact on the dehydration temperature of rhabdophanes.
| X | Dehydration temperature (°C) | Conversion temperature (°C) | H2O per formula unit (total amount) | Ref. | |
|---|---|---|---|---|---|
| ① | 0 | 244 | 784 | 0.8–1.1 | 35 |
| 230 ± 5 | 700 ± 50 | 37 | |||
| 210 ± 3 | 693 ± 1 | 0.68 ± 0.05 | 29 | ||
| 0.025 | 223 ± 5 | 826 ± 5 | 0.79 ± 0.05 | This work | |
| 0.050 | 221 ± 5 | 824 ± 5 | 0.64 ± 0.05 | This work | |
| 0.075 | 217 ± 5 | 833 ± 5 | 0.67 ± 0.05 | This work | |
| 0.100 | 214 ± 3 | 762 ± 1 | 0.79 ± 0.05 | 29 | |
| 213 ± 5 | 817 ± 5 | 0.72 ± 0.05 | This work | ||
| ② | 0 | 230 ± 5 | 700 ± 50 | 37 | |
| 0.025 | 214 ± 5 | 805 ± 5 | 0.77 ± 0.05 | This work | |
| 0.050 | 217 ± 5 | 837 ± 5 | 0.75 ± 0.05 | This work | |
| 0.075 | 216 ± 5 | 832 ± 5 | 0.65 ± 0.05 | This work | |
| 0.100 | 221 ± 5 | 828 ± 5 | 0.65 ± 0.05 | This work | |
To investigate the influence of uranium incorporation on the conversion temperature of the phase transition from U-rhabdophane to U-monazite–cheralite, the data obtained by DTA were plotted as a function of the average ionic radius in Fig. S2 of the ESI.† The effective ionic radii used to calculate the average ionic radius are reported in Table S1 of the ESI.† The temperature of phase transition from U-rhabdophane to U-monazite–cheralite solid solution was found to be higher than the temperature of conversion of Th-rhabdophane of equivalent average ionic radius obtained by Qin et al.29 and closer to the data obtained by Kijkowska.35 for LnPO4·nH2O (with Ln = La to Gd). This observation led to the conclusion that the thermal field of existence of U-rhabdophanes was larger than that obtained for Th-rhabdophanes. This result suggested that the respective thermodynamic properties of the U- and Th-rhabdophanes slightly differ. Kijkowska.35 prepared rhabdophane samples by crystallisation from boiling 2 mol L−1 H3PO4 solution containing 0.02 mol L−1 of lanthanide elements. The data obtained by Kijkowska.35 were also systematically higher than those measured by Jonasson and Vance37 for the conversion of rhabdophane to monazite. Not only was the protocol of synthesis of the hydrated rhabdophanes different (refluxing Ln4(P2O7)3 in 0.1 mol L−1 HNO3 solution), but also Jonasson and Vance37 did not specify the nature of the atmosphere used in their DTA experiments. This comparison showed that several factors could affect the temperature of transition, although there is a general decrease of the conversion temperature with the increase of the average ionic radius of the cations.
In order to validate the final objective of this study, Nd1−2xCaxUxPO4·nH2O rhabdophanes were heated for 6 hours under oxidizing (air) or inert (Ar) atmosphere at 1100 °C. This temperature was chosen to ensure the complete conversion of U-rhabdophane into U-monazite–cheralite (based on the results of TG analyses), allow the complete decomposition of the α-UP2O7 reaction intermediate15 and avoid the decomposition of phosphates and the formation of CaO, UO2 and P4O10(g) that occurred at 1300 °C.15
The thermally fired samples were first observed by SEM (Fig. 2b and c). After thermal conversion in air, the morphology of the crystals evolved markedly, whereas the changes were less pronounced under Ar atmosphere. The width of the needle-like crystals increased (about 6 times wider) and their progressive rounding was observed. The observation of both samples did not reveal the presence of secondary phases exhibiting a different morphology.
The powders obtained after conversion in air or under Ar atmosphere were then analyzed by PXRD (Fig. 4). According to the PXRD diagrams of the samples converted in air, the obtained powders were all single-phase monazite–cheralite, whereas the presence of two additional peaks (at 17.9° and 20.7°) was detected for x ≥ 0.05 in the samples converted under Ar. These two peaks could be attributed to small amounts of α-UP2O7 phase, which was observed as a reaction intermediate during the irreversible phase transition from rhabdophane to monazite–cheralite by Bregiroux et al.15 This phase was also frequently formed as an intermediate in the synthesis of uranium phosphates such as U(UO2)(PO4)2 or U2O(PO4)2.38,39 However, under Ar atmosphere, Bregiroux et al.15 observed the complete disappearance of α-UP2O7 between 900 and 1000 °C and prepared monophasic Ca0.5U0.5PO4 cheralite above 1100 °C. Several explanations can be suggested to explain the presence of α-UP2O7 in our prepared samples. First, part of the uranium could segregate into α-UP2O7 phase during the thermal treatment of the rhabdophane precursor due to the lack of Ca to fully compensate the charge in the monazite–cheralite structure. Second, part of the uranium might have precipitated in a phase other than rhabdophane during the hydrothermal synthesis. This secondary phase would be converted into α-UP2O7 during the thermal treatment under Ar and not completely decompose after the heat treatment at 1100 °C. However, the presence of α-UP2O7 phase was not evidenced after conversion in air, which indicates different mechanisms of phase transition, probably involving different reaction intermediates or different behaviour of α-UP2O7 in air due to U(IV) oxidation.39
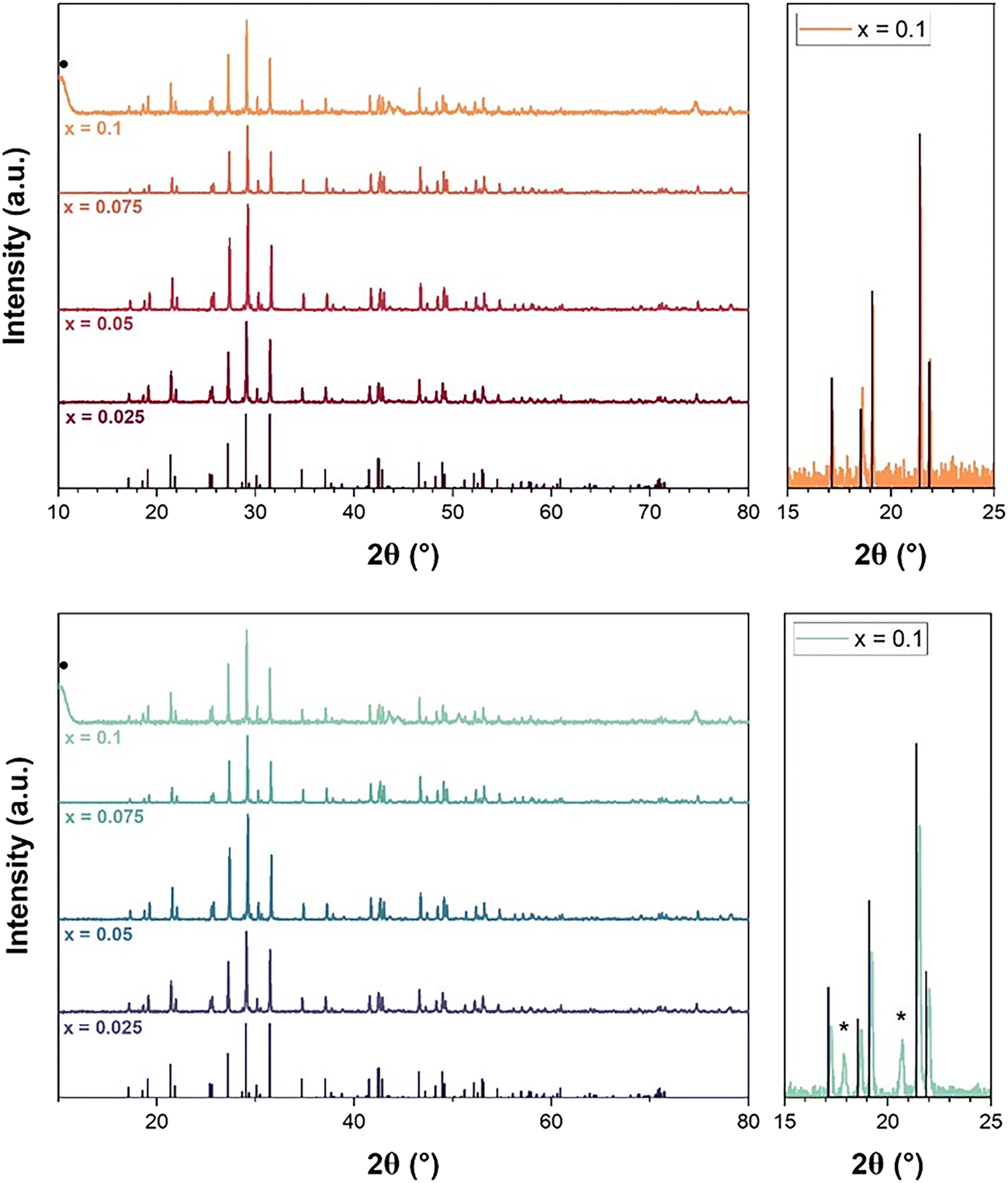 | ||
| Fig. 4 PXRD patterns obtained after thermal conversion of U-rhabdophanes to U-monazite–cheralites at 1100 °C for 6 h in air (a) or under argon atmosphere (b). XRD lines associated with monazite NdPO4 are indicated by black vertical bars,40 while black dot and stars indicate sample holder and additional peaks attributed to α-UP2O7 secondary phase, respectively. | ||
The PXRD diagrams of the obtained monazite–cheralite were refined using the Rietveld method (Fig. S3 of the ESI†). The unit cell parameters are listed in Table 2. Fig. 5 shows the variation of the unit cell volume of the U-monazite–cheralite solid solutions versus the target uranium mole fraction x. (Fig. S4 of the ESI† shows the difference between the experimental cell volume obtained by Rietveld refinement of the diffractograms and that calculated from the proportional combination of the end-members.) The unit cell volumes of the U-monazite–cheralite samples converted either under air or Ar atmosphere decreased linearly with the incorporation rate. When comparing the unit cell volumes of the prepared monazite–cheralite with x ≤ 0.1 with the data reported in the literature for the end-members, a slight deviation from Retger's law was evidenced. Nevertheless, considering the low incorporation rates of uranium in the prepared samples, it is difficult to draw conclusions on the ideality of the solid solution. Moreover, considering the presence of the additional α-UP2O7 phase, the real uranium mole fraction in the monazite–cheralite phase converted under Ar is expected to be lower than the target x value and so does the deviation from Retger's law.
 | ||
| Fig. 5 Refined unit cell volumes of U-monazite–cheralite solid solutions Nd1−2xCaxUxPO4 prepared in air ① or under argon atmosphere ②. Black dots correspond to the unit cell volumes of the endmembers reported in ref. 15, 21, 36, 41–46. Continuous line corresponds to the fitted Retger's law, whereas green area corresponds to the 95% confidence level band. | ||
The cell volumes obtained by refinement of the PXRD diagrams obtained using a laboratory X-ray source revealed that uranium was well incorporated into the structure, since the unit cell volume decreased when the incorporation rate increased. Nevertheless, a difference can be observed between the diffractogram obtained in the laboratory and that recorded on the MARS beamline of the SOLEIL synchrotron for the Nd0.8Ca0.1U0.1PO4 sample after heat treatment in air. This difference could be due to sample preparation (plate vs. capillary), the two Kapton confinements or the possible evolution of the sample under the beam.
Raman and FTIR spectroscopies
The monazite–cheralite samples obtained by thermal conversion in air and Ar atmosphere were also characterized by Raman and FTIR spectroscopies. The main objective was to check that U(VI) (in the uranyl form) was not present in the prepared samples.47 The Raman and FTIR spectra of monazite–cheralites converted in air or under Ar-atmosphere are presented in Fig. 6.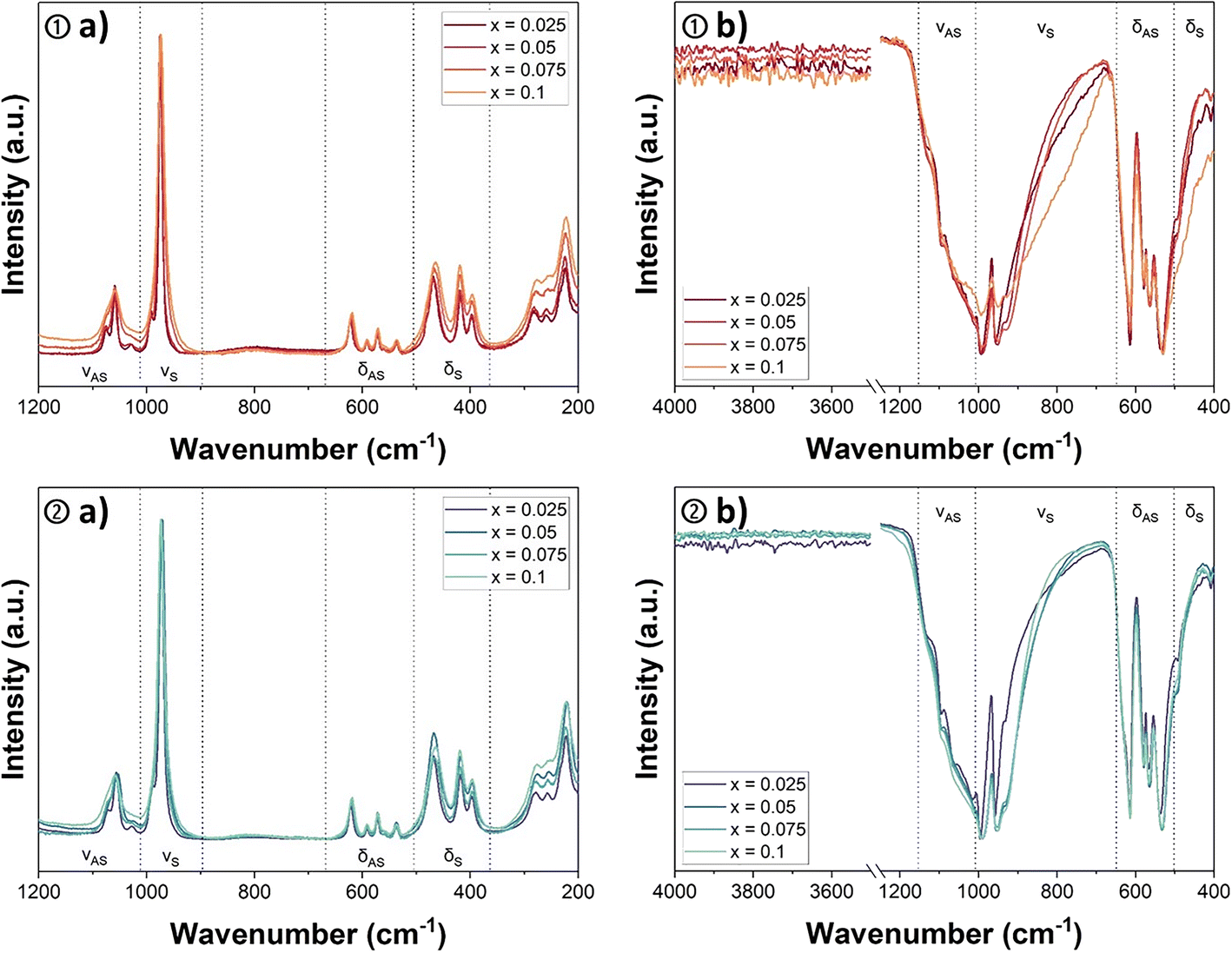 | ||
| Fig. 6 Raman spectra recorded of U-monazite–cheralite solid solutions Nd1−2xCaxUxPO4 prepared by conversion in air ① or under argon atmosphere ② in the 200–1200 cm−1 range (a) and associated FTIR spectra in the 400–4000 cm−1 range (b). | ||
The Raman spectra (Fig. 6a) of the Nd1−2xCaxUxPO4 samples (with x ≤ 0.1) presented the deformation modes of the PO4 tetrahedron in the range between 300 and 800 cm−1 and the characteristic bands of the stretching modes in the 900–1200 cm−1 range. The positions of the different bands and their assignments, based on the work of Clavier et al.,48 are listed in Table 4. The comparison of the Raman spectra of the monazite–cheralites obtained by thermal treatment under air or Ar atmosphere did not reveal any particular differences. The positions of the vibration bands were found to be very close. For instance, the strongest band, assigned to the symmetric stretching vibration of the P–O bonds in phosphate groups νS(P–O), was located in the narrow wavenumber range between 972 and 976 cm−1 irrespective of the atmosphere of conversion. The absence of an intense and narrow vibration band in the domain of 820–880 cm−1, characteristic of the symmetric stretching vibration νS (frequently labelled ν1 in the literature) of the uranyl bond (U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O),49,50 suggested the absence of UO22+ entities and the stabilization of tetravalent uranium during the thermal conversion, even in air.
O),49,50 suggested the absence of UO22+ entities and the stabilization of tetravalent uranium during the thermal conversion, even in air.
| Raman spectroscopy (cm−1) | FTIR spectroscopy (cm−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| δ S(P–O) | δ AS(P–O) | ν S(P–O) | ν AS(P–O) | δ S(P–O) | δ AS(P–O) | ν S(P–O) | ν AS(P–O) | ||
| Ref. 48 | 404, 428, 476 | 539 (567), 577, 598, 632 | 987 | 1004, 1042, 1071, 1092 | 492 | 538, 570, 585, 623 (642) | (933), 960, 995 | 1022 (1070), 1103, 1137 | |
| ① | x = 0.025 | 398, 419, 469 | 538, 572, 592, 622 | 974 | 992, 1029, 1059, 1075 | 493 | 532, 564, 579, 614 | 928, 957, 992 | 1010, 1061, 1092, 1131 |
| x = 0.05 | 398, 419, 467 | 537, 573, 592, 622 | 975 | 1030, 1060, 1076 | 493 | 532, 565, 579, 615 | 930, 951, 990 | 1092 | |
| x = 0.075 | 398, 419, 467 | 536, 572, 591, 621 | 975 | 1059 | 493 | 532, 564, 579, 614 | 928, 950, 991 | 1092 | |
| x = 0.100 | 396, 419, 464 | 536, 571, 591, 619 | 974 | 1060 | 492 | 530, 566, 579, 615 | 928, 951, 993 | 1012, 1061 | |
| ② | x = 0.025 | 397, 419, 469 | 537, 572, 592, 622 | 973 | 990, 1028, 1057, 1072 | 491 | 537, 567, 579, 615 | 930, 958, 994 | 1014, 1061, 1094, 1127 |
| x = 0.050 | 396, 418, 468 | 537, 572, 592, 621 | 972 | 1055 | 493 | 532, 565, 579, 615 | 928, 952, 990 | 1010, 1092 | |
| x = 0.075 | 397, 418, 466 | 537, 573, 590, 621 | 973 | 1058 | 493 | 533, 565, 580, 615 | 930, 952, 990 | 1096, 1125 | |
| x = 0.100 | 394, 419, 467 | 536, 572, 591, 622 | 976 | 1064 | 491 | 532, 567, 580, 614 | 930, 951, 990 | 1094 | |
The FTIR spectra of the monazite–cheralite samples converted under air or Ar did not differ strongly (Fig. 6b). The sharp band observed between 950 and 957 cm−1 was assigned to the νS(P–O) symmetrical stretching mode. The position of this band was not significantly influenced by the uranium content or the atmosphere of conversion. The infrared active antisymmetric stretching vibration of UO22+, νAS (also called ν3), is usually observed in the range of 910 to 962 cm−1.49–51 The potential overlap of the νS(P–O) and νAS(UO22+) characteristic bands in infrared spectroscopy could lead to misinterpretation in the case of uranyl phosphates. However, the absence of a strong active νS (often referred to as ν1) band in the Raman spectra and the similarity between the spectra of samples converted under oxidizing or inert atmosphere led to the conclusion that the uranyl moiety was not present in the monazite–cheralite samples prepared or that the quantity was below the detection limit of this technique. Infrared spectroscopy also shows the absence of vibrations associated with symmetric and anti-symmetric elongation of the O–H bond of water molecules (normally located between 3650 and 3750 cm−1). This confirms that the solids characterized are totally dehydrated. Additionally, the symmetric and anti-symmetric stretching vibrations of the (P–O–P) bond observable at 737 and 955 cm−1, respectively, in the FTIR spectra of α-UP2O7 were not detected in the samples calcined under argon,52 although this phase was identified by XRD. The absence of these bands in the FTIR spectra presented in Fig. 6b ② shows that the amount of α-UP2O7 was below the detection limit of this technique.
Synchrotron analyses
HERFD-XANES analysis of the Nd0.8Ca0.1U0.1PO4 sample prepared by thermal conversion of U-rhabdophane at 1100 °C for 6 hours in air was performed at the M4 edge of uranium. Thanks to its high spectral resolution, this technique allowed the detection of the presence of several oxidation states of uranium in the monazite–cheralite sample. Fig. 7 shows the measured spectra of the two references as well as that of Nd0.8Ca0.1U0.1PO4. Metatorbernite, Cu(UO2)2(PO4)2·nH2O, was chosen as a pure UO22+ bearing phosphate reference whose associated HERFD-XANES spectrum exhibited three distinct spectral features characteristic of UO22+ compounds. The three main fluorescence peaks were assigned to the transitions of 3d3/2 electrons to the 5fδ/ϕ, 5fπ* and 5fσ* valence orbitals.53,54 These peaks appeared at 3728.3, 3730.4, and 3734.3 eV, respectively, in the metatorbernite spectrum. The second reference was a pure U(IV) bearing phosphate phase, diuranium oxophosphate, (U2O)(PO4)2. This spectrum presented only one large peak at 3726.9 eV assigned to U(IV). Due to different local environments, the position of the U(IV) peak in the (U2O)(PO4)2 reference was shifted towards higher energy compared to that of UO2 (reported at 3723 eV by Massonnet et al.).55 In order to determine the relative proportion of U(IV) and U(VI) oxidation states within the monazite–cheralite converted in air, the HERFD-XANES spectrum of the Nd0.8Ca0.1U0.1PO4 sample was fitted by a linear combination of the two reference spectra. Fig. 7 shows the calculated spectrum and the difference from the measured spectrum. Even if the quality of the fit was disputable due to variations of the local environment of uranium in the monazite–cheralite compared to the two references, it gave a semi-quantitative indication of the proportion of U(IV) and U(VI) in the monazite–cheralite sample, with obtained contents of 84 ± 2% and 16 ± 2%, respectively.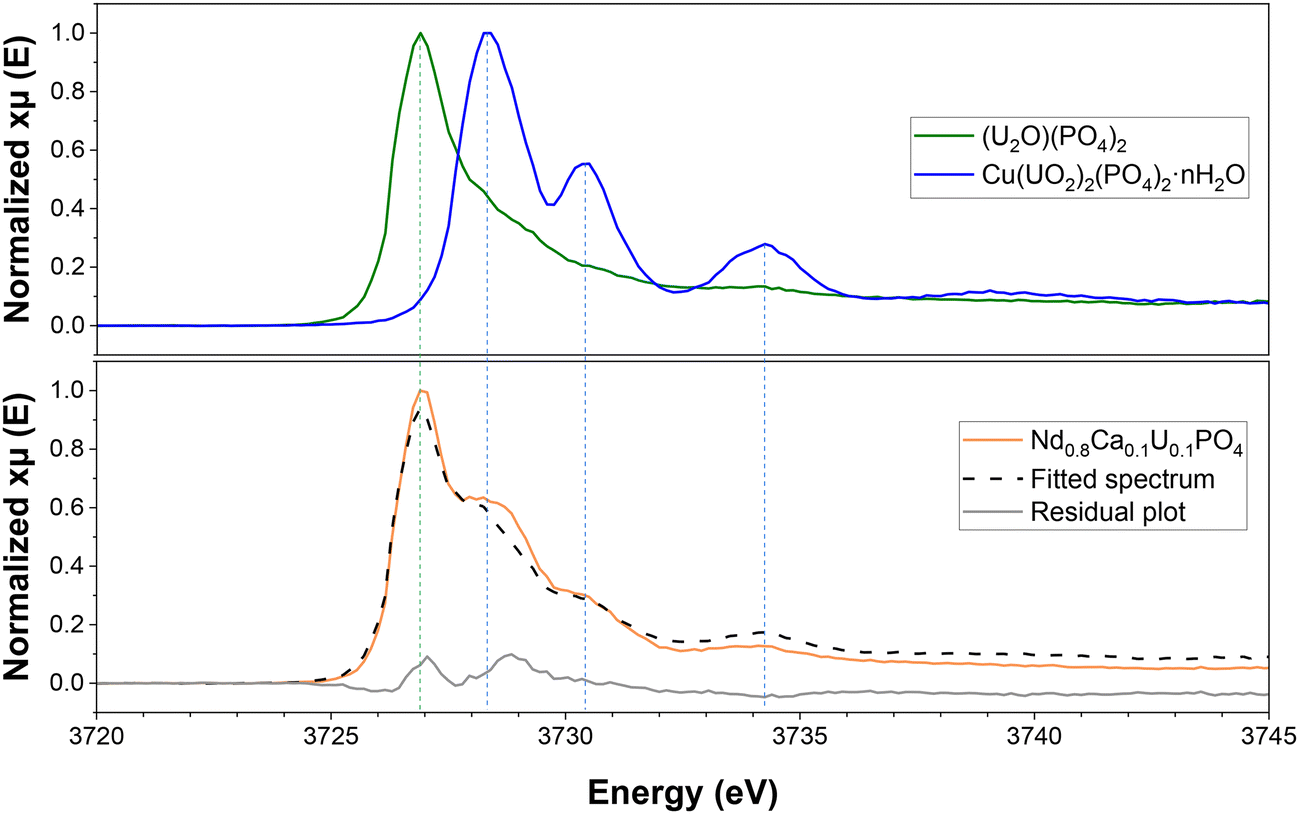 | ||
| Fig. 7 U M4-edge HERFD-XANES spectra of Nd0.8Ca0.1U0.1PO4 converted under air. Reference spectra of U(IV) and U(VI) phosphates were obtained using (U2O)(PO4)2 and Cu(UO2)2(PO4)2·nH2O, respectively. Dashed black line corresponds to the spectrum of Nd0.8Ca0.1U0.1PO4 fitted by linear combination of the two references. | ||
Since the uranium content of the prepared sample was equal to 10 wt%, the mass fraction of U(VI) in the monazite–cheralite was less than 2 wt%. This low amount might explain the absence of the bands characteristic of the vibrations of UO22+ entities in the FTIR and Raman spectra of this sample (a fortiori, in the other samples with lower uranium contents). It is noteworthy that the incorporation of uranyl in the monazite–cheralite structure is very unlikely. Thus, the presence of a minor secondary phase was considered. As the PXRD pattern obtained using a laboratory X-ray source did not evidence the presence of a secondary phase, synchrotron X-ray diffraction analysis of the Nd0.8Ca0.1U0.1PO4 sample was performed after its thermal conversion in air. Fig. 8 presents the SPXRD diagram of this sample obtained with the CX3 station of the MARS beamline at the SOLEIL synchrotron.
 | ||
| Fig. 8 SPXRD pattern obtained for Nd0.8Ca0.1U0.1PO4 monazite–cheralite converted in air at 1100 °C for 6 hours. PXRD patterns reported for monazite NdPO4 (black vertical bars);40 *indicates additional peaks observed in the experimental pattern. | ||
The SPXRD pattern recorded at the MARS beamline was also refined by the Rietveld method (Fig. S3†). The obtained unit cell parameters of the monazite–cheralite are reported in Table 2 and Fig. 5. The unit cell volume was found to be lower than the one obtained by refinement of the laboratory PXRD pattern. However, thanks to the high sensitivity of the experimental device, several small additional peaks were detected between 8° and 17° 2θ (zoom in Fig. 8). The nature of the U(VI) secondary phase was not identified, although the HERFD-XANES spectrum confirmed that it likely incorporated the uranyl moiety rather than uranate.56 This phase could have been formed by precipitation during the hydrothermal synthesis of the U-rhabdophane precursor or during the thermal treatment of conversion under air.
In order to evaluate the composition of this secondary phase, 100 mg of the Nd0.8Ca0.1U0.1PO4 sample was washed for 5 days in 100 mL of a 0.1 mol L−1 HNO3 solution at 40 °C. The washed sample was also characterized at the MARS beamline. The resulting XRD diagram showed the disappearance of the additional peaks attributed to the presence of a secondary phase (Fig. S5 of the ESI†). Additionally, the U M4-edge HERFD-XANES spectrum of the washed Nd0.8Ca0.1U0.1PO4 sample was also recorded at the MARS beamline (Fig. S6 of the ESI†). It showed the complete disappearance of the U(VI) component. The Ca, U, Nd and P elemental concentrations in the washing solution were determined by ICP-AES analysis. The results indicated that 21.3 mol% of the uranium initially present in the solid phase was dissolved in 5 days, in addition to 2.4 mol% of phosphorus and 4.8 mol% of Ca. The mole fraction of Nd released in the solution was negligible, indicating that the monazite–cheralite phase was virtually not dissolved during the washing step. Assuming that the released elements came only from a single additional phase, its stoichiometry would be approximately Ca:P = 0.15 and U:P = 0.78. Alternatively, Ca and U phosphates could have precipitated in two separate phases of unknown stoichiometry. Nevertheless, these results demonstrated that the U(VI) contained in the sample before washing was completely incorporated as a secondary phase. This more soluble uranyl phosphate phase was removed by the washing protocol. 21 mol% of the uranium precipitated initially with the rhabdophane precursor was incorporated in the secondary phase, which was evidenced by synchrotron measurements only. Thus, the limit of incorporation of tetravalent uranium in the monazite–cheralite structure through the wet chemistry route described in this work reached x = 0.08 rather than 0.1, as initially stated. Additionally, using the real value of x in the washed monazite–cheralite brings the cell volume calculated by Rietveld refinement closer to the value estimated using Retger's law (Fig. 5). This observation indicates that the main reason for deviation from Retger's law is an overestimation of the uranium mole fraction.
Experimental
Synthesis
To avoid the oxidation of tetravalent U by the introduction of nitrate ions in the system, analytical grade metal chloride salts and H3PO4 (ACS Reagent, ≥85 wt%, Sigma Aldrich) and HCl (ACS Reagent, 37%, Carlo Erba) solutions were used for the preparation of the monazite–cheralite hydrated precursor of the rhabdophane structure Nd1−2xCaxUxPO4·nH2O (with x ≤ 0.1). NdCl3·6H2O and CaCl2·nH2O (Sigma Aldrich) were first dissolved in 0.1 mol L−1 hydrochloric acid to obtain a cation concentration ranging from 1 to 1.5 mol L−1. The uranium solution was obtained by dissolving uranium metal chips (kindly provided by CETAMA) in hydrochloric acid.57 In this very acidic hydrochloric medium, U(IV) is stabilized against oxidation by the formation of UCl62− in the starting mixture and then by complexation with phosphates during the further mixture with phosphoric acid. The metal chips were washed with dichloromethane, acetone and ethanol to remove the traces of oil required for their long-term storage. Then, the dried uranium metal chips were weighed and dissolved in 6 mol L−1 HCl solution cooled in an ice bath to obtain a final elemental concentration of 0.6 mol L−1. The accurate elemental concentrations of the solutions containing each cation were then measured using inductively coupled plasma optical emission spectroscopy (ICP-OES, Thermo Fisher Scientific, iCAP 7000 series).The hydrothermal synthesis protocol of the Th-rhabdophane developed by Qin et al.28,29 was adapted to allow the incorporation of uranium. Specifically, Qin et al.28 showed that a Ca:Th molar ratio of 10 was mandatory to obtain single-phase Nd1−2xCaxThxPO4·nH2O samples. Thus, the synthesis method consists of mixing adequate volumes of each solution in a beaker under magnetic stirring in order to obtain a mixture with the stoichiometrically expected proportions of Nd and U in the solid phase. The Ca:U stoichiometric ratio varied with the U mole fraction of the sample, x, so that Ca/U = 100x. Once the solutions were mixed, 5 mol L−1 H3PO4 solution was added dropwise to the previous batch with a 3 mol% excess with regard to Nd, Ca and U. The mixture was stirred for 15 minutes before being introduced into the autoclave (Parr 4744). In order to guarantee an identical autogenous pressure for all the syntheses, deionized water was added to the mixture to obtain a final volume of 25 mL and a constant dead volume of 20 mL. The autoclave was then sealed and placed in an oven at 110 °C for four days. The precipitate obtained was washed twice with water and once with ethanol through cycles of centrifugation at 14500 rpm for 5 minutes. The powder was then dried in an oven at 90 °C overnight.
Finally, the monazite–cheralite hydrated precursor underwent thermal treatment under air or under Ar at 1100 °C for 6 hours in a muffle furnace or a tubular furnace, respectively.
ICP-OES
The chemical composition of the synthesized rhabdophanes was determined by ICP-OES (Thermo Fisher Scientific, iCAP 7000 series) measurements after their complete dissolution. For this purpose, about 20 mg of powder was weighed, then dissolved in fresh aqua regia. After complete dissolution, the solutions were diluted in 0.2 mol L−1 HNO3 solution (ACS Reagent, 70%, Sigma-Aldrich) and analyzed by ICP-OES. The ICP-OES was initially calibrated using several standard solutions prepared by the dilution of Th, U, Ca, Nd and P elemental solutions certified at 1000 ppm (SCP Science) in 0.2 mol L−1 HNO3 solution. 3 to 4 of the recommended emission wavelengths were selected for each element. Three replicates for each measurement were performed. The stoichiometry of each compound was then calculated using the average elemental concentrations.PXRD/SPXRD
Powder X-ray diffraction patterns (PXRD) of the synthetized compounds were obtained using a Bruker D8 Advance diffractometer equipped with a Cu Kα1,2 X-ray source (λ = 1.5418 Å), a 60 mm sealed Göbel mirror and a LynxEye linear position sensitive detector in θ/θ step scan mode. The patterns were acquired at room temperature in the range of 5 to 100° (2θ) with a step size of 0.02°. The counting time for one sample was about 3 hours.Structural analyses of selected samples were also performed using the SPXRD (synchrotron PXRD) set-up of the CX3 end-station of the MARS beamline at the SOLEIL synchrotron (Saint-Aubin, France).58 About 20 mg of each sample was placed in 0.6 mm Kapton capillary tubes (3 cm long). The capillary was inserted in a second Kapton capillary for the confinement of the radioactive powder. Then, the powder was sealed in the tube using epoxy resin. XRD patterns were recorded at 17 keV in the transmission Debye–Scherrer geometry. The monochromatic beam was sized at 300 × 150 μm2 (H × V). The 2D hybrid pixel detector PILATUS3 2M CdTe (Dectris AG, Switzerland) was located at 680 mm from the sample on a 2θ motorized rotation axis. 11 images were recorded using a 5° 2θ step between 0 and 50°. 60 s were needed to record one image and the total signal was thus obtained in about 6 min. The 11 images were merged and integrated using the multi-geometry class of the pyFAI library59 to reduce and reconstruct 1D powder diffraction patterns. The setup's geometry correction and instrumental resolution function were done using a NIST SRM LaB6 660b powder (lattice parameter: 4.15689 Å). During the pattern recording, the samples were continuously spun around the capillary axis after mounting and alignment on a goniometer head.
For both the synchrotron and laboratory X-ray sources, PXRD patterns were refined by the Rietveld method using the Fullprof_Suite package.60 The rhabdophane precursors as well as the monazite–cheralites were refined in the monoclinic C233 and P21/n61 space groups, respectively. In both cases, the instrumental function was extracted using Si and LaB6, respectively. During refinement, the profile parameters, such the zero-point shift, unit cell parameters, and asymmetry, were refined and a size model was introduced in order to evaluate the microstructural effect. When needed, the atomic positions and thermal displacement parameters were also refined.
SEM
The morphology of the prepared compounds was investigated by scanning electron microscopy using an FEI QUANTA 200 ESEM FEG. For this purpose, a secondary electron detector (SE) and/or a backscattered electron detector (BSE) were used at room temperature with an accelerating voltage of 2 kV in order to obtain high-resolution images. In terms of sample preparation, the powder was deposited on a polished SEM pad, then dispersed with ethanol in order to deagglomerate the powder as much as possible. No further preparation, such as metallization, was carried out before analysis.TGA/DTA
Thermogravimetric (TGA) and thermodifferential (TDA) analyses were carried out using a SETARAM Setsys Evolution apparatus equipped with a Pt thermocouple. For this purpose, 10 to 15 mg of powder was weighed, then introduced into an alumina crucible. During the analysis, the sample was heated to 1000 °C at a heating rate of 5 °C min−1 and a 20 mL min−1 flow rate of air or Ar. The TGA curves allowed the determination of the water content of the samples, whereas the DTA curves showed the temperature of transition from rhabdophane to monazite–cheralite.Vibrational spectroscopies (Raman/FTIR)
Fourier transform infrared spectroscopy in attenuated total reflection mode (ATR-FTIR) was performed using a PerkinElmer Spectrum 100 spectrometer operating with a Globar MIR source in combination with a cesium iodide (CsI) beamsplitter and a deuterated triglycine sulphate DTGS detector through CsI transmission windows. To carry out these analyses, the powdered samples were deposited at the surface of a 2 mm diameter top-plate diamond crystal stacked on a KRS-5 (thallium bromide-iodide) bottom-plate microfocus condenser without preparation. High pressure was exerted on the sample and adjusted until stabilisation of the signal. Spectra were recorded from 380 to 4000 cm−1 adding 4 scans and operating with background correction. The nominal resolution of the spectra was 4 cm−1.The Raman spectra were recorded with a Horiba-Jobin Yvon Labram Aramis equipped with an edge filter and a Nd:YAG laser (532 nm). Before starting the analyses, the instrument was calibrated with silicon, using the first order Si line at 520.7 cm−1. Once the calibration was completed and the sample was deposited on a glass lamella, the laser was focused on the sample using a confocal microscope with, in optimal conditions, an axial resolution lower than 2 μm and a spot diameter smaller than 1 μm laterally. For the acquisition of spectra, this microscope was equipped with a 532 nm excitation laser (i.e. about 60 mW), a ×100 objective and no filter. An accumulation of 5 spectra per sample was carried out for a total analysis time of approximately 5 minutes. Data processing, such as spectral normalization, was carried out using ORIGIN software.
HERFD-XANES
High-energy resolution fluorescence detected XANES (HERFD-XANES) data were measured at the MARS beamline of the SOLEIL synchrotron (Saint-Aubin, France).58 The storage ring was operated in the top-up mode at an electron current of 500 mA, 2.5 GeV. The beam size at the sample surface was 300 μm × 150 μm (H × V). Spectra were measured at the U M4-edge (3728 eV) using the double-crystal monochromator (DCM) equipped with a pair of Si(111) crystals. Higher harmonics rejection and vertical focusing were achieved using the Si strip of each mirror inserted before and after the DCM with a 4 mrad incidence angle. The incident energy was calibrated using the absorption K-edge of potassium from a KBr pellet (3608.4 eV). HERFD-XANES was performed using the crystal-analyzer emission spectrometer in the Rowland geometry and a KETEK single-element silicon solid-state detector. The Mβ emission line of U (3339 eV) was analyzed using the 220 reflection of a Si(220) bent, stripped crystal analyzer with a curvature radius of 1 m. The energy resolution of the emission spectrometer was 0.8 eV at 6.676 keV. The samples were oriented at 45° with respect to the incident beam. A He-filled balloon was used to reduce the scattering of the incident and emitted X-rays by the air between the sample, the crystal analyzer and the detector. Samples were prepared as pellets for each compound. All the spectra collected were normalized at the maximum of absorption using Athena software,62 then fitted as a linear combination of different references to quantify the contributions of U(IV), U(V), and U(VI). Reference spectra were obtained using (U2O)(PO4)263–66 as the U(IV) standard and Cu(UO2)2(PO4)2·nH2O67 as the U(VI) standard in a phosphate environment.
Conclusion
In this work, Nd1−2xCaxUxPO4·nH2O (with x ≤ 0.1) rhabdophane-type precursors were prepared by a wet chemistry route followed by hydrothermal treatment at 110 °C for four days. A large excess of Ca in the initial mixture of cations was necessary to obtain single-phased rhabdophanes using this protocol. Nd1−2xCaxUxPO4 monazite–cheralite solid solutions were then obtained by thermal conversion of the rhabdophane precursors at 1100 °C for 6 hours either under oxidising (air) or inert (Ar) atmosphere. This temperature was chosen based on TG analyses to guarantee complete phase transition and to avoid the decomposition of the phosphate group and the formation of CaO, UO2 and P4O10(g). The obtained monazite–cheralite samples were characterized using a wide set of analytical techniques. When heated in Ar atmosphere, PXRD revealed the presence of an α-UP2O7 secondary phase in the monazite–cheralite samples, whereas the samples converted under air were single-phase. Special attention was paid to the oxidation state of uranium in the monazite–cheralite samples obtained by conversion of the rhabdophane precursors under air. FTIR and Raman spectroscopies did not reveal the presence of uranyl moieties in these samples. However, the U M4-edge HERFD-XANES spectrum of the Nd0.8Ca0.1U0.1PO4 compound showed the coexistence of about 80 mol% U(IV) and 20 mol% U(VI) present as uranyl in the sample. U(VI) was associated with the presence of a minor secondary phase in the Nd0.8Ca0.1U0.1PO4 sample detected only by SPXRD. This soluble phase was easily removed by washing the sample in a 0.1 mol L−1 HNO3 solution. Finally, the limit of incorporation of uranium in the Nd1−2xCaxUxPO4 monazite–cheralite prepared by this wet chemistry route reached around x = 0.08.The conversion mechanism of U-rhabdophane to monazite–cheralite under Ar atmosphere was very similar to that studied by Qin et al.29 for Th-monazite–cheralites. It is therefore likely that the presence of α-UP2O7 results from the presence of an excess of uranium (relative to Ca) associated with the presence of vacancies in the U-rhabdophane structure. In this case, the excess U segregates upon heat treatment to form α-UP2O7. However, this phenomenon was not observed when heating in air, due to the lower stability of α-UP2O7 under these oxidizing conditions.39 The α-UP2O7 would then be converted into a uranyl phosphate phase with U:P = 0.78 stoichiometry, which coexisted as a minor phase with the monazite–cheralite. This phase, whose presence could affect the long-term behavior of the ceramic in the field of immobilization of actinides, was easily removed by washing in dilute acid to obtain a pure monazite–cheralite phase containing tetravalent uranium only. Using this washing step will allow the study of the chemical durability of Nd1−2xCaxUxPO4 monazite–cheralite (with x = 0.08) from kinetic and thermodynamic points of view in a forthcoming dedicated study.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to ICSM staff for their technical support, especially J. Lautru for his meticulous work with ESEM and EDS analyses, B. Baus-Lagarde for ICP-AES analyses and A. Jonchère for his help in using FTIR and Raman spectroscopies. The authors would also like to thank the SOLEIL synchrotron for providing beamtime, as well as the University of Montpellier for funding Alison El Monjid's PhD work.References
- L. A. Boatner, in Phosphates: Geochemical, Geobiological, and Materials Importance, ed. M. J. Kohn, J. Rakovan and J. M. Hughes, 2002, vol. 48, pp. 87–121 Search PubMed.
- C. M. Gramaccioli and T. V. Segalstad, Am. Mineral., 1978, 63, 757–761 CAS.
- G. R. Lumpkin, Can. Mineral., 1998, 36, 339–353 CAS.
- G. R. Lumpkin, Y. Gao, R. Giere, C. T. Williams, A. N. Mariano and T. Geisler, Mineral. Mag., 2014, 78, 1071–1095 CrossRef.
- L. A. Boatner and B. C. Sales, Monazite, Radioactive waste forms for the future, North Holland, Amsterdam (Netherlands), 1988 Search PubMed.
- A. M. Seydoux-Guillaume, X. Deschanels, C. Baumier, S. Neumeier, W. J. Weber and S. Peuget, Am. Mineral., 2018, 103, 824–827 CrossRef.
- J. M. Montel, Kornprobst and Vielzeuf, J. Metamorph. Geol., 2000, 18, 335–342 CrossRef CAS.
- N. Dacheux, N. Clavier and R. Podor, Am. Mineral., 2013, 98, 833–847 CrossRef CAS.
- L. A. Boatner, G. W. Beall, M. M. Abraham, C. B. Finch, P. G. Huray and M. Rappaz, Sci. Basis Nucl. Waste Manage., 1980, 2, 289–296 CAS.
- R. C. Ewing and L. Wang, Rev. Mineral. Geochem., 2002, 48, 673–699 CrossRef CAS.
- B. Omel'Yanenko, T. Livshits, S. Yudintsev and B. Nikonov, Geol. Ore Deposits, 2007, 49, 173–193 CrossRef.
- N. Clavier, R. Podor and N. Dacheux, J. Eur. Ceram. Soc., 2011, 31, 941–976 CrossRef CAS.
- N. Dacheux, N. Clavier, A.-C. Robisson, O. Terra, F. Audubert, J.-É. Lartigue and C. Guy, C. R. Chim., 2004, 7, 1141–1152 CrossRef CAS.
- X. Deschanels, V. Picot, B. Glorieux, F. Jorion, S. Peuget, D. Roudil, C. Jégou, V. Broudic, J. Cachia and T. Advocat, J. Nucl. Mater., 2006, 352, 233–240 CrossRef CAS.
- D. Bregiroux, O. Terra, F. Audubert, N. Dacheux, V. Serin, R. Podor and D. Bernache-Assollant, Inorg. Chem., 2007, 46, 10372–10382 CrossRef CAS PubMed.
- P. Raison, R. Jardin, D. Bouexiere, R. Konings, T. Geisler, C. Pavel, J. Rebizant and K. Popa, Phys. Chem. Miner., 2008, 35, 603–609 CrossRef CAS.
- A. Tabuteau, M. Pagès, J. Livet and C. Musikas, J. Mater. Sci. Lett., 1988, 7, 1315–1317 CrossRef CAS.
- R. Podor and M. Cuney, Am. Mineral., 1997, 82, 765–771 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- D. Bregiroux, R. Belin, P. Valenza, F. Audubert and D. Bernache-Assollant, J. Nucl. Mater., 2007, 366, 52–57 CrossRef CAS.
- O. Terra, N. Dacheux, N. Clavier, R. Podor and F. Audubert, J. Am. Ceram. Soc., 2008, 91, 3673–3682 CrossRef CAS.
- O. Terra, N. Clavier, N. Dacheux and R. Podor, New J. Chem., 2003, 27, 957–967 RSC.
- J. M. Cleveland, The Chemistry of Plutonium, Gordon & Breach Science Publishers, New York, 1970 Search PubMed.
- C. E. Bamberger, R. G. Haire, H. E. Hellwege and G. M. Begun, J. Less-Common Met., 1984, 97, 349–356 CrossRef CAS.
- C. W. Bjorklund, J. Am. Chem. Soc., 1957, 79, 6347–6350 CrossRef CAS.
- C. Keller and K. H. Walter, J. Inorg. Nucl. Chem., 1965, 27, 1253–1260 CrossRef CAS.
- D. E. Hobart, G. M. Begun, R. G. Haire and H. E. Hellwege, J. Raman Spectrosc., 1983, 14, 59–62 CrossRef CAS.
- D. Qin, A. Mesbah, C. Gausse, S. Szenknect, N. Dacheux and N. Clavier, J. Nucl. Mater., 2017, 492, 88–96 CrossRef CAS.
- D. W. Qin, A. Mesbah, N. Clavier, S. Szenknect and N. Dacheux, Cryst. Growth Des., 2019, 19, 2794–2801 CrossRef CAS.
- D. W. Qin, A. Mesbah, J. Lautru, S. Szenknect, N. Dacheux and N. Clavier, J. Eur. Ceram. Soc., 2020, 40, 911–922 CrossRef CAS.
- D. Qin, A. Shelyug, S. Szenknect, A. Mesbah, N. Clavier, N. Dacheux and A. Navrotsky, Appl. Geochem., 2023, 148, 105504 CrossRef CAS.
- R. Podor, PhD thesis, University of Lorraine, 1994.
- A. Mesbah, N. Clavier, E. Elkaim, C. Gausse, I. B. Kacem, S. Szenknect and N. Dacheux, Cryst. Growth Des., 2014, 14, 5090–5098 CrossRef CAS.
- A. Mesbah, N. Clavier, E. Elkaim, S. Szenknect and N. Dacheux, J. Solid State Chem., 2017, 249, 221–227 CrossRef CAS.
- R. Kijkowska, Thermochim. Acta, 2003, 404, 81–88 CrossRef CAS.
- A. Shelyug, A. Mesbah, S. Szenknect, N. Clavier, N. Dacheux and A. Navrotsky, Front. Chem., 2018, 6, 604 CrossRef CAS PubMed.
- R. Jonasson and E. Vance, Thermochim. Acta, 1986, 108, 65–72 CrossRef CAS.
- V. Brandel and N. Dacheux, J. Solid State Chem., 2004, 177, 4743–4754 CrossRef CAS.
- V. Brandel, N. Dacheux and M. Genet, J. Solid State Chem., 1996, 121, 467–472 CrossRef CAS.
- K. M. Kurbanov, V. A. Efremov and V. P. Orlovsky, Kristallografiya, 1986, 31, 800–802 CAS.
- D. F. Mullica, D. A. Grossie and L. A. Boatner, J. Solid State Chem., 1985, 58, 71–77 CrossRef CAS.
- R. Jardin, C. C. Pavel, P. E. Raison, D. Bouëxière, H. Santa-Cruz, R. J. M. Konings and K. Popa, J. Nucl. Mater., 2008, 378, 167–171 CrossRef CAS.
- Y. Ni, J. M. Hughes and A. N. Mariano, Am. Mineral., 1995, 80, 21–26 CAS.
- S. V. Ushakov, K. B. Helean, A. Navrotsky and L. A. Boatner, J. Mater. Res., 2001, 16, 2623–2633 CrossRef CAS.
- E. Du Fou de Kerdaniel, PhD thesis, University of Paris Sud-Paris XI, 2007.
- J. M. Montel, J. L. Devidal and D. Avignant, Chem. Geol., 2002, 191, 89–104 CrossRef CAS.
- G. Lu, A. J. Haes and T. Z. Forbes, Coord. Chem. Rev., 2018, 374, 314–344 CrossRef CAS PubMed.
- N. Clavier, A. Mesbah, S. Szenknect and N. Dacheux, Spectrochim. Acta, Part A, 2018, 205, 85–94 CrossRef CAS PubMed.
- K. W. Bagnall and M. W. Wakerley, J. Inorg. Nucl. Chem., 1975, 37, 329–330 CrossRef CAS.
- R. L. Frost, Spectrochim. Acta, Part A, 2004, 60, 1469–1480 CrossRef PubMed.
- J. Čejka, J. Čejka and A. Muck, Thermochim. Acta, 1985, 86, 387–390 CrossRef.
- N. Dacheux, V. Brandel and M. Genet, New J. Chem., 1995, 19, 15–25 CAS.
- T. Vitova, I. Pidchenko, S. Biswas, G. Beridze, P. W. Dunne, D. Schild, Z. Wang, P. M. Kowalski and R. J. Baker, Inorg. Chem., 2018, 57, 1735–1743 CrossRef CAS PubMed.
- Y. Podkovyrina, I. Pidchenko, T. Prüßmann, S. Bahl, J. Göttlicher, A. Soldatov and T. Vitova, J. Phys.: Conf. Ser., 2016, 712, 012092 CrossRef.
- M. Massonnet, L. Claparede, J. Martinez, P. M. Martin, M. O. J. Y. Hunault, D. Prieur, A. Mesbah, N. Dacheux and N. Clavier, Inorg. Chem., 2023, 62, 7173–7185 CrossRef CAS PubMed.
- L. Amidani, M. Retegan, A. Volkova, K. Popa, P. M. Martin and K. O. Kvashnina, Inorg. Chem., 2021, 60, 16286–16293 CrossRef CAS PubMed.
- N. Dacheux, V. Brandel and M. Genet, New J. Chem., 1995, 19, 1029–1036 CAS.
- B. Sitaud, P. L. Solari, S. Schlutig, I. Llorens and H. Hermange, J. Nucl. Mater., 2012, 425, 238–243 CrossRef CAS.
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS PubMed.
- C. Frontera and J. Rodriguez-Carvajal, Phys. B, 2003, 335, 219–222 CrossRef CAS.
- M. Keskar, G. P. Shelke, M. Shafeeq, R. A. Phatak, S. K. Sali and S. Kannan, J. Solid State Chem., 2019, 278, 120850 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- N. Dacheux, PhD thesis, University of Paris Sud-Paris XI, 1995.
- J. H. Albering and W. Jeitschko, Z. Kristallogr., 1995, 210, 878 CAS.
- N. Dacheux, N. Clavier, G. Wallez and M. Quarton, Solid State Sci., 2007, 9, 619–627 CrossRef CAS.
- P. Bénard, D. Louer, N. Dacheux, V. Brandel and M. Genet, An. Quim., 1996, 92, 79–87 Search PubMed.
- N. Clavier, F. Cretaz, S. Szenknect, A. Mesbah, C. Poinssot, M. Descostes and N. Dacheux, Spectrochim. Acta, Part A, 2016, 156, 143–150 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt03251e |
| This journal is © The Royal Society of Chemistry 2024 |