
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An automated electrochemistry platform for studying pH-dependent molecular electrocatalysis†
Michael A.
Pence
 ab,
Gavin
Hazen
ab and
Joaquín
Rodríguez-López
*ab
ab,
Gavin
Hazen
ab and
Joaquín
Rodríguez-López
*ab
aBeckman Institute for Advanced Science and Technology, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: joaquinr@illinois.edu
bDepartment of Chemistry, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 5th August 2024
Abstract
Comprehensive studies of molecular electrocatalysis require tedious titration-type experiments that slow down manual experimentation. We present eLab as an automated electrochemical platform designed for molecular electrochemistry that uses opensource software to modularly interconnect various commercial instruments, enabling users to chain together multiple instruments for complex electrochemical operations. We benchmarked the solution handling performance of our platform through gravimetric calibration, acid–base titrations, and voltammetric diffusion coefficient measurements. We then used the platform to explore the TEMPO-catalyzed electrooxidation of alcohols, demonstrating our platforms capabilities for pH-dependent molecular electrocatalysis. We performed combined acid–base titrations and cyclic voltammetry on six different alcohol substrates, collecting 684 voltammograms with 171 different solution conditions over the course of 16 hours, demonstrating high throughput in an unsupervised experiment. The high versatility, transferability, and ease of implementation of eLab promises the rapid discovery and characterization of pH-dependent processes, including mediated electrocatalysis for energy conversion, fuel valorization, and bioelectrochemical sensing, among many applications.
Introduction
Electrochemical methods offer a sustainable alternative to traditional chemical reactions, using electricity to perform molecular transformations that would otherwise require excessive heat or environmentally unfriendly reagents.1 However, many electrochemical reactions require an electrocatalyst to occur as they otherwise display large overpotentials at a given electrode. The use of freely diffusing redox-active molecular electrocatalysts, i.e., mediated electrocatalysis, helps with lowering the overpotential for electrochemical reactions while providing precise control over the nature of the catalyst.2 In addition, certain molecular electrocatalysts enable transformations beyond electron transfer, for example by facilitating proton-coupled electron transfer (PCET).Studies of molecular electrocatalysis are often long and tedious, requiring screening of many catalysts and substrate combinations, different scan rates, and manifold solution conditions and reagent concentrations.3,4 Mechanistic studies of mediated electrocatalytic systems often are conducted by titrating in increasing amounts of reactants into the solution and observing the resulting changes of the current–potential curve using cyclic voltammetry (CV).5–10 Further, investigations of PCET reactions additionally require screening the behavior of the system by performing CV at different concentrations of acid or base in solution, for which knowledge of pH is essential for mechanistic insight.11 As the complexity of large experimental campaigns increases, manual experimentation becomes too slow to explore the massive chemical and parameter space needed for comprehensive understanding of molecular electrocatalytic systems. We can turn to laboratory automation to speed up the exploration process, as automation offers a solution to lengthy exploration campaigns that can hinder the discovery of breakthrough electrochemical systems.12
Automated platforms can increase experimental throughput by improving operational efficiency and enabling passive experimentation over long time frames without interruption. Automation additionally improves the repeatability and reproducibility of experiments, by removing human error and allowing the exact same experimental workflow to be run across replicate experimental platforms. Automated and self-driving laboratories have found use in a broad range of chemistry and materials science problems13–20 and are now becoming increasingly mainstream in electrochemistry as well. For example, automated and high throughput platforms for electrochemical experimentation have been used for applications ranging from energy storage,21–27 electrocatalysis,28–31 electrosynthesis,32–36 and even single entity electrochemistry.37 However, these platforms are not designed for the titration-type electrochemical experiments that are crucial in molecular electrocatalysis studies, highlighting the need for a specialized platform for this type of study.
In this work, we present a new platform that incorporates solution handling, electrochemistry, and pH measurement to study pH-dependent molecular electrocatalysis. We achieve this by building the eLab application programming interface (API) which works in tandem with our recently reported HardPotato API. The modular and open-source nature of eLab enabled us to combine our solution handling platform with various off-the-shelf commercial instruments to perform a range of analytical experiments such as gravimetric calibration, acid–base titrations, and multi-variate electrochemical screening campaigns. Here, we highlight this new platform to screen the behavior of alcohol electrooxidation using (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) as a freely diffusing electrocatalyst.38 We demonstrate the strength of our platform for titration measurements by screening activity of TEMPO electrocatalysis with various substrates, over a range of different solution pH values, and showing the pH dependence of homogeneous oxidation rate constants for selected systems.
Experimental
Chemicals and materials
All chemicals used in this work were used as purchased without further purification. Isopropyl alcohol, glycerol, and ethylene glycol (ACS grade) were purchased from Macron. TEMPO (98%) and acetaldehyde (99.5%) were purchased from Sigma-Aldrich. Ethanol (USP grade) was purchased from Decon Labs. Trifluoroethanol (99%) was purchased from Acros Organics. Sodium hydroxide (Reagent grade) was purchased from Ward Science. 85 w/w% phosphoric acid (ACS grade) was purchased from Fischer Chemical. Sodium bicarbonate (ACS grade) was purchased from Avantor. Anhydrous sodium carbonate (ACS grade) and standard pH reference buffers (4.00, 7.00, 10.00) were purchased from VWR. DI water was obtained from a Milli-Q purification system.Electrochemical measurements
All electrochemical measurements were performed using a CH Instruments 760E bipotentiostat. Electrochemical experiments were carried out in a three-electrode setup, with a 3 mm diameter glassy carbon working electrode, a graphite rod counter electrode, and an Ag/AgCl (3 M KCl) reference electrode with a salt bridge. Electrochemical experiments were carried out at room temperature without gas purging. All cyclic voltammograms are reported in IUPAC convention.Software and hardware
All experiments in this work were carried out automatically using Python (3.12.4). We used our previously developed API, Hardpotato, to control the bipotentiostat.39 We additionally used the numpy (1.24.4), pandas (1.4.3), pyserial (3.5), and scikit-learn (1.1.1) libraries in our code.40–43 We developed an API to control a variety of instrumentation, including a fluidic selection valve (RUNZE SV-07), a syringe pump (RUNZE SY-08), an Arduino based pH meter (Gravity pH Meter v2.0) and pH probe (Mettler Toledo Micro pH Electrode S7), a hot-plate with stirring and thermocouple probe (IKA C-MAG HS-7), an analytical balance (Ohaus E0RR80), and a bipotentiostat (CH Instruments 760e). All instruments were connected to the computer with USB and controlled through Python. A bill of materials and guide to assembling the solution handling platform is available in ESI Notes S1, Table S1, and Fig. S1.† A bill of materials and guide to assembling the Arduino-based pH meter is available in ESI Notes S3, Table S2, and Fig. S2.†Results and discussion
Hardware and software development
A variety of open-source platforms for laboratory automation have been developed that come in a range of architectures and costs, with many platforms built around well-plate robots or flow-based systems.44–48 Despite their popularity, well-plate robot-based platforms do not easily perform titration-like experiments needed for molecular electrochemistry, while flow-based systems lack compatibility with standard electroanalytical approaches. To this end, eLab was developed to suit the needs of electrochemical experimenters, particularly in the field of molecular electrocatalysis. We custom built our platform with core objectives in mind including the ability to perform multi-substrate investigations, precise dispensing for a wide range of reagent concentrations, compatibility with the standard 3-electrode electrochemical setup, a compact footprint that allows deployment inside a glovebox, and a platform that can be quickly and easily implemented while still being compatible with other open-source tools and libraries.Our previous automated electrochemistry platform, the Electrolab, utilized a gantry-based system with integrated hardware controlled by a single Arduino-based board.26 While the Electrolab offers flexibility, such as a multi-nozzle system for dispensing, sparging, and drying, its versatility is not always warranted for all types of problems. In our current work, we aimed to maintain the Electrolab's advantages while simplifying the overall system. This effort yielded a platform with rapid installation and intuitive operation, achieved by focusing on modular connectivity for existing instruments rather than building each component from scratch. Here we employed the traditional 3-electrode electrochemical setup for simplified implementation by the end user, but we believe that for systems where the history or quality of the electrode is critical to the experiment, commercially available disposable electrodes or electrode arrays could be used instead.
Fig. 1a provides an overview schematic of the platform, illustrating the data and fluidic connections between the hardware components. The core of the automated platform is the solution handling hardware, which includes a 16-port fluidic selection valve and a 5 mL bidirectional syringe pump. The use of a syringe pump allows for more precise dispensing and the fluidic selection valve enables many combinations of catalysts and substrates to be explored in one single experiment. Our platform can be configured for various experiments by interconnecting instruments as needed. For example, the setup for pH-dependent electrochemical measurements (Fig. 1b) involves connecting a pH meter and potentiostat to the computer, along with the solution handling hardware. The experimental cell consists of a scintillation vial containing a fluid line, three electrochemical electrodes, and a pH probe. Throughout this work, we employed a range of experimental setups that integrated the base solution handling system with a pH meter, a potentiostat, a thermocouple probe, and an analytical balance. All experiments in this work were performed automatically, with all instruments being controlled via either our eLab or Hard Potato APIs.
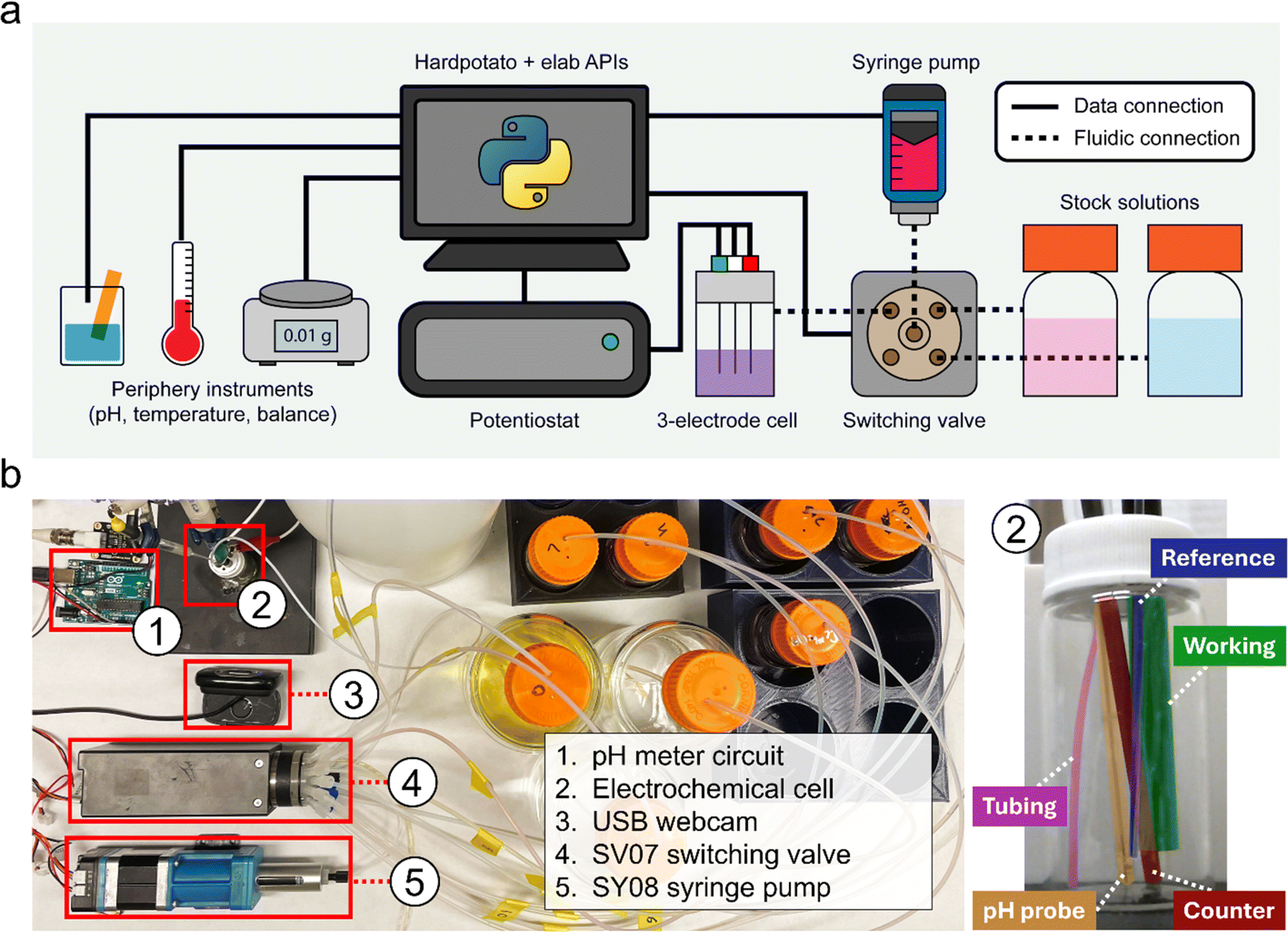 | ||
| Fig. 1 Experimental setup. (a) Generalized schematic of the hardware setup, showing the data and fluidic connections between different components. (b) Labelled image of the hardware setup configured for combined pH and electrochemistry measurements. | ||
We developed the eLab API in Python to control the various hardware components of our platform through high level commands. The new API enables complex multi-instrument operations to be executed with relatively simple commands. Instructions for installing and using the API are found in ESI Note S2, Fig. 2a shows an example script that dispenses a mixture of two solutions and performs a cyclic voltammogram. The script requires a total of 10 lines of code to execute a series of multi-instrument operations, with 7 lines being dedicated to initializing libraries and instruments (Fig. 2b). One important aspect of our API is ‘bundling’ of instruments together, as seen in line 6 of Fig. 2a, which allows a single command to control multiple instruments simultaneously. For example, calling the calibrate_pH() method coordinates the pump, valve, and pH meter to dispense various standard pH buffers, measure voltages in the known buffers, and create a calibration curve.
 | ||
| Fig. 2 Example of a typical measurement and corresponding script. (a) Python script that combines both fluid handling and electrochemistry in 10 lines, and the resulting data. The dispensed and mixed solution is 1.2 mM TEMPO in a 0.1 M solution of pH 9.6 bicarbonate–carbonate buffer. (b) Images at different points of the experimental operations performed in the last three lines of the script, as well as the resulting voltammogram. TEMPO solution is dyed blue for visual effect. | ||
Aside from ease of use, using eLab helps enable reproducibility and transferability. The script serves as a comprehensive record of the experiment, detailing the exact parameters sent to the instruments. This script can be shared from lab to lab, and the implemented instruments can be easily modified to fit that lab's instrumentation. To integrate new instruments into the eLab API, the user can create functions that translate commands and parameters into lower-level serial commands provided in the instruments' data sheets. ESI Note S4 and Table S3† provide a detailed guide on adding instruments to the eLab API, empowering users to leverage the platform's modular nature and adapt it to the instruments available to them.
Benchmarking hardware performance
Before using our automated platform to study pH dependent electrocatalysis, we first performed benchmarking experiments to better understand the limitations of the solution handling hardware. We performed three benchmarking experiments that were designed to identify a lower limit of volume that could reliably be dispensed. Initially, we performed a gravimetric calibration experiment, where we used the automated solution handling equipment to dispense a target amount of water onto a balance, where the corresponding mass of the dispensed water was measured. Next, an acid–base titration of 0.1 M H3PO4 with 1 M NaOH as the titrant was performed. Lastly, diffusion coefficient measurements of TEMPO at varied concentrations and scan rates demonstrated the limits of our automated electrochemical platform for CV measurements.The gravimetric calibration experiment was performed by connecting a switching valve, syringe pump, temperature probe, and analytical balance. The switching valve and syringe pump were used to dispense a target volume of water, and the mass of the dispensed water and external temperature were recorded before taring the balance and performing the next measurement. The actual volume was calculated using the density of water at the average recorded temperature of 22.1 °C ± 0.2 °C. Fig. 3a shows the relative errors of all recorded volume points. Notably, target volumes above 50 μL had average relative errors below 2.0%, an improvement from our previous peristaltic pump-based solution handling platform which had 2.0% relative error at target volumes of 100 μL.26
 | ||
| Fig. 3 Benchmarking of solution handling hardware. (a) Results of gravimetric calibration of the platform with water, showing the relative error of the actual dispensed volume for a given target volume. The black line is the mean relative error (N = 5), and the red error bar represents one standard deviation from the mean. Inset is zoomed in to show dispensing behavior below 200 μL. (b) Automated titration of 4 mL of 0.1 M H3PO4 with 1 M NaOH. The black line is the mean relative error (N = 4), and the red error bar represents one standard deviation from the mean. (c) Distribution of TEMPO diffusion coefficients measured, with the inset showing the explored parameter space of scan rates and analyte concentration (N = 400). 0.1 M of a bicarbonate-carbonate buffer with solution pH of 9.6 was used as a supporting electrolyte. | ||
The acid–base titration experiment was performed by connecting a switching valve, syringe pump, temperature probe, and pH meter. The pH meter was calibrated automatically using standard pH 4.00, 7.00, and 10.00 buffers. 4 mL of 0.1 M H3PO4 was dispensed, followed by 100 aliquots of 100 μL of 1 M NaOH as a titrant. The solution pH was measured after each dispensing operation, and the titration experiment was repeated 4 times in total. Fig. 3b shows the resulting titration curve. The pKa values for the first two deprotonations were measured to be 2.227 ± 0.007 and 6.91 ± 0.03. The measured pKa values differ slightly from the reported values of 2.16 and 7.21, but differences in temperature over the course of the experiment may explain the deviations from reported values (Fig. S4†).49 This experiment showcases our platform's capability for titration experiments, highlighting its versatility beyond automated electrochemical experiments previously demonstrated.
Measuring the diffusion coefficient measurement of TEMPO was performed by connecting a switching valve, syringe pump, temperature probe, and potentiostat. 400 random combinations of scan rates ranging from 10 mV s−1 to 1 V s−1 and analyte concentrations ranging from 0.1 mM to 6 mM were generated, as seen in the inset of Fig. 3c. A solution of 6 mM TEMPO in 0.1 M bicarbonate buffer was mixed with a solution of 0.1 M bicarbonate buffer to achieve the desired concentration. The different concentrations were dispensed in random orders, and CVs were performed at all corresponding scan rates for each concentration. The cell was flushed with water and primed before changing concentrations, which was determined to be appropriate upon evaluating three different cleaning protocols to assess sample carryover between experiments (ESI Note S5 and Table S4†). We measured a reference-adjusted E1/2 of 0.78 V ± 0.01 V vs. SHE, in line with the reported literature value of 0.75 V vs. SHE (Fig. S5†).38,50
Diffusion coefficients were calculated using the Randles–Ševčík equation with baseline-subtracted peak currents (ESI Note S6 and Fig. S6 and S7†), and the resulting distribution is shown in Fig. 3c. The mean diffusion coefficient was determined to be 6 × 10−6 cm2 s−1, which is in good agreement with previously reported values, but the resulting distribution had a large standard deviation of ±2 × 10−6 cm2 s−1.51 We have seen in our previous work that large distributions of diffusion coefficient measurements are often skewed by inaccurate concentration volumes that result from dispensing errors, as may be the case here. Beyond dispensing inaccuracies, distortions in the current–potential curve due to uncompensated solution resistance or electron transfer kinetic issues can add variance to calculated diffusion coefficients. The peak-to-peak separation of all CVs, plotted against scan rate and concentration (Fig. S8†) indicates the presence of uncompensated solution resistance in our data. We see that the largest outliers in the distribution are from experiments where less than 200 μL of analyte were dispensed (Fig. S9†). When we remove concentration points that correspond to sub-200 μL dispensed volume of analyte, our standard deviation improves from ±2 × 10−6 cm2 s−1 to ± 1 × 10−6 cm2 s−1. Based on this observation, we use 200 μL as the minimum dispensing volume for the following experiment, where we perform titration experiments with 200 μL aliquots.
Cyclic voltametric acid–base titrations
As a case study, we used our platform to perform acid–base titrations alongside cyclic voltammetry to study the electrochemical oxidation of alcohols and aldehydes catalyzed by 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO). TEMPO-catalyzed oxidation proceeds by the catalytic cycle shown in Scheme 1. TEMPO is oxidized at the electrode surface to form TEMPO+, which then reacts irreversibly with the deprotonated form of the alcohol or aldehyde (as a geminal diol), transferring two electrons and a proton from the substrate to TEMPO+, and forming TEMPOH and the respective product molecule. The deprotonation of the substrate molecule prior to the rate limiting irreversible chemical step indicates that the kinetics of the reaction should be affected by the concentration of OH− in solution. TEMPOH may be oxidized at the electrode to TEMPO or engage in comproportionating with TEMPO+ to complete the catalytic cycle.38,52–54 | ||
| Scheme 1 The catalytic cycle of TEMPO-mediated oxidation of alcohols and aldehydes and structures of substrates used. | ||
The use of the 16-port fluidic switching valve enabled us to screen 6 substrates molecules in one experimental run with no human intervention. We examined a substrate scope consisting of isopropanol, ethanol, acetaldehyde, trifluoroethanol, ethylene glycol, and glycerol (Scheme 1). We performed acid–base titration experiments using 1 M NaOH, performing pH measurements and CV after dispensing each 200 μL aliquot of NaOH. TEMPO and substrate were dispensed along with NaOH to keep the catalyst and substrate concentrations constant throughout the experiment at 2 mM and 100 mM, respectively. The solution was stirred by bubbling air when each aliquot of the mixture was dispensed, and the solution pH was measured after allowing the solution to settle. CV was then performed by sweeping from 0 V to 1 V, at scan rates of 1000 mV s−1, 500 mV s−1, 100 mV s−1, and 50 mV s−1. The electrode underwent pretreatment by anodization in 1 M NaOH for 30 s prior to each substrate's voltammetric titration experiment to promote efficient oxidation of the TEMPOH to TEMPO.50
Fig. 4 shows 684 voltammograms obtained during titrations of 6 different substrates. The voltammograms demonstrate the pH-dependent kinetics of TEMPO-catalyzed alcohol and aldehyde oxidations. Voltammograms taken with both substrate and catalyst in solution show increased current from the CVs taken with no substrate, showing that TEMPO behaves electrocatalytically in the presence of alcohols and aldehydes. Even over a range of pH values spanning less than 1 unit, the catalytic current of the voltammograms with substrate notably increased in more basic conditions, consistent with previously reported behavior of TEMPO-catalyzed alcohol oxidations. All experiments in Fig. 4 took place over the course of 16 hours, which is less than 3 hours per substrate. The experiments are highly complex routines consisting of dispensing aliquots of multiple solutions, stirring solutions, measuring the solution pH, and performing CV measurements. Considering the complexity of the experiment workflow, we estimate it would take a researcher at least 5 hours to perform the entirety of a titration experiment for one substrate, which is twice the time our automated platform takes. However, our measurements were fully unsupervised, allowing these experiments to be performed passively, potentially freeing the experimenter to focus on other aspects of laboratory work. It is important to note that there is a trade-off between the time of the experiment and experimental precautions such as cleaning the cell, priming the cell, and syringe pressure equilibration. More rigorous experiments will require even more stringent protocol for solution handling, leading to longer experimental times. The impact of such experimental details can be easily evaluated by rerunning the script with minor changes. For example, rerunning the multi-substrate campaign without the anodic pretreatment step dramatically distorts the resulting current–potential curves (Fig. S10†) indicating that pretreatment significantly improves electron-transfer kinetics at the electrode surface.
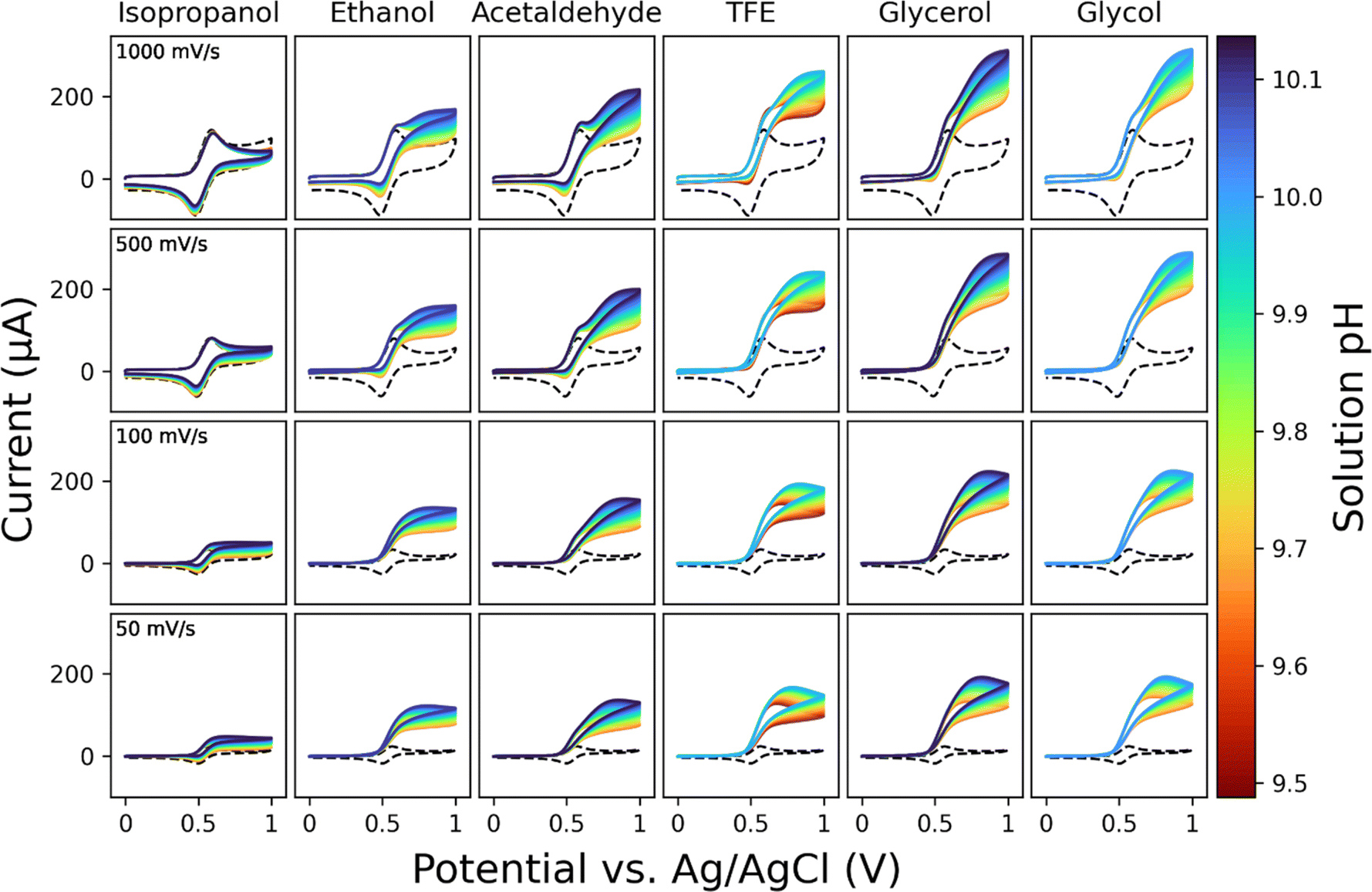 | ||
| Fig. 4 Results of pH dependent voltammetric screening of TEMPO catalyzed alcohol oxidation. Each column represents a different substrate molecule, and each row is a different scan rate. Dashed lines are voltammograms in the absence of substrate. | ||
Having a pH meter inside the electrochemical cell allows the solution pH to be recorded before running a CV. This enables us to examine CVs recorded in solutions with pH values closest to a specified target value, facilitating comparisons between different substrates without the influence of pH variations on the system's kinetics. Fig. 5a shows CVs of 100 mM substrate and 2 mM TEMPO, collected at scan rates of 50 mV s−1 in a pH-adjusted supporting electrolyte with an average pH of 9.63 ± 0.01. The CV responses for all substrates exhibit clear catalytic enhancement compared to substrate-less conditions, with many CVs demonstrating mechanistically informative non-idealities such as curve crossover at the foot of the wave (Fig. S11†) indicating that the reaction follows the ECE' mechanism shown in Scheme 1.55,56
 | ||
| Fig. 5 Comparison of all substrate CVs at a single pH and scan-rate. (a) Overlaid voltammetric responses of all six substrates at pH 9.6. Solution contained 2 mM TEMPO and 100 mM of substrate in 0.1 M bicarbonate–carbonate buffer. Voltammograms were performed at 50 mV s−1. (b) Bar graph showing the ratio of plateau currents in the presence of substrate to peak currents in the absence of substrate. | ||
To easily compare the relative activities of TEMPO catalysis toward various substrates, we calculated the ratio of peak currents in the presence of substrate to those in the absence of substrate. The differences in current ratios serve as an indicator for the difference in catalytic activity of TEMPO towards the various substrates are shown in Fig. 5b. Isopropanol, a secondary alcohol, reacts much more slowly than the other substrates with primary alcohols. On the other hand, oxidation of acetaldehyde is kinetically facile, showing a current that is only slightly lower than that of ethanol, where oxidation of the alcohol is kinetically limiting despite the latter having the possibility of undergoing two catalytic cycles. Electronic effects are evident when comparing ethanol and trifluoroethanol; the highly electron-withdrawing trifluoro group should facilitate the removal of the alpha proton of the alcohol, which is reflected by a higher current ratio. Additionally, the number of primary alcohol groups on the substrate has a significant impact: glycol and glycerol, both having two primary alcohols, exhibit similarly enhanced activity (∼twofold) compared to ethanol. There is little difference between glycerol and glycol, showing that the secondary alcohol on glycerol makes little difference in the measured current response. The ability to quickly perform multi-substrate investigations provides us with a wealth of data on substrate activity that can be compared for similar solution compositions.
Finally, we proceeded to calculate observed homogeneous oxidation rate constants, kobs, for all solution conditions to better understand how TEMPO catalysis depends on the concentration of base in solution and the substrate identity. This was accomplished through eqn (1) by using the ratio between the peak current, ipeak, of CVs recorded in the absence of substrate and the plateau current, iplateau, in the presence of substrate:
 | (1) |
We chose to measure the rate of isopropanol electrooxidation to ensure the calculated rate constants were reasonable, as our group has previously measured the turnover frequency (TOF) of this system to be 0.51 s−1 at pH 10, which was validated using simulations.51Fig. 6a shows CVs performed at 50 mV s−1 in pH 10 solution that were used to calculate a kobs value of 0.59 s−1. Since our iplateau value was calculated almost 500 mV more positive than the E1/2 of our TEMPO catalyst, we can assume that the TOF of the system is equal to our kobs.58 Thus, the TOF for electrooxidation of isopropanol measured with our platform matches well with what has been previously reported. Making use of our automated setup, it is also possible to carry out a systematic variation of pH in our experiment to compare the reaction kinetics vs. OH− concentration to better understand the pH dependance of TEMPO catalysis. Fig. 6b shows the log–log plot of the observed rate constants of isopropanol electrooxidation as a function of the OH− concentration, calculated from CVs performed at 50 mV s−1. The slope of this log–log plot gives us a reaction order of 1.3 with respect to OH−. We might expect the reaction to be first-order with respect to OH− due to a pre-equilibrium approximation of the deprotonation of the alcohol prior to rate-limiting step in Scheme 1, but the complex nature of reactions occurring near an electrode surface may cause deviations from such approximations. Regardless, the average reaction order of OH− across all substrates was calculated to be 0.9 ± 0.2, using the slopes of the linear fits with the highest R2 for each substrate as the reaction order (Fig. S12†). The ability to carry out informationally-rich kinetic analyses in an unsupervised fashion, like that shown in Fig. 6b, and to determine important kinetic parameters such as TOF and reaction orders demonstrates the capabilities of our automated platform for mechanistic analysis.
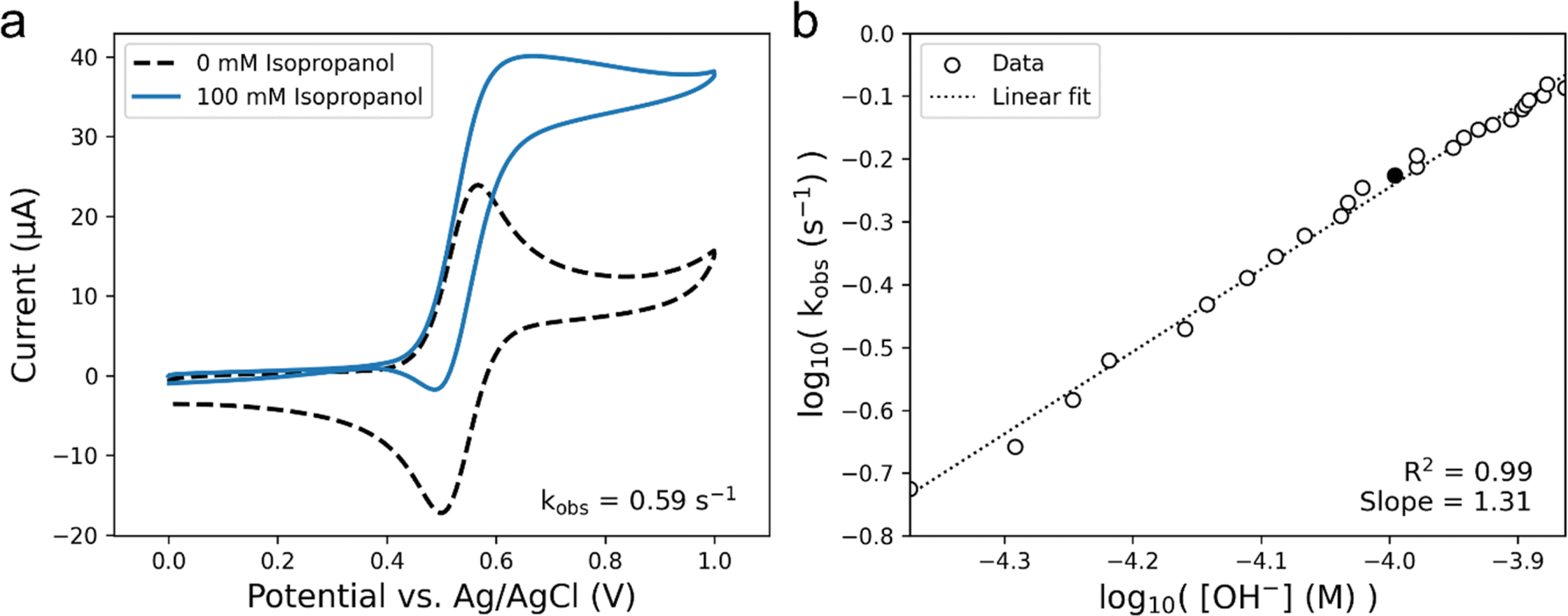 | ||
| Fig. 6 Calculating kinetic information from voltammetric data. (a) CVs of 2 mM TEMPO with and without 100 mM of isopropanol in pH 10 bicarbonate-carbonate buffer solution. Voltammograms were performed at 50 mV s−1. An observed rate constant, kobs, was calculated to be 0.59 s−1. (b) log–log plot of kobsversus OH− concentration, and the linear fit of the data. The shaded data point corresponds to the pH 10 CV in panel (a). | ||
Conclusions
In this work we introduced eLab as a new automated electrochemical platform specifically designed for performing titration-type experiments aimed at studying molecular electrocatalysis. We developed our platform to be modular, allowing easy integration with existing instruments in the user's laboratory. We benchmarked our platform through various experiments including gravimetric calibration, acid–base titration, and the voltametric estimation of diffusion coefficients to identify the limitations of our solution handling system. Our platform executed a complex multi-substrate experimental workflow that resulted in almost 700 CVs over the course of 16 hours. Programmatic analysis of the acquired data allowed for rapid determination of important mechanistic parameters such as TOFs and reaction orders.Our platform is built on hardware and software that is easy to implement and operate. In fact, a replica of this platform was built at a completely different laboratory over the course of a day and running automated electrochemistry experiments before the day was over. This, however, was facilitated by the fact that the same hardware was used, and no changes had to be made to the API. For platforms built upon different hardware and instrumentation, some time will need to be spent adding the hardware commands to the API. To this end, we made sure that our API is open-source and available for anyone to edit as needed, empowering users to modify the API to best suit the platform that they are developing.
One major limitation of our platform in its current state is the lack of hardware for controlling gas flow to, for example, deoxygenate a solution by sparging with inert gas. While we have demonstrated the ability to sparge with inert gas in our previous platform, the ElectroLab, we have not yet added this capability to our new automated platform.26 Accordingly, we focused on using eLab to automate experiments that were not affected by the presence of oxygen. However, the highly modular nature of our platform and the small footprint of the core solution handling system enables it to be installed inside of a glovebox for performing air-free electrochemistry experiments.
The next direction for our platform is incorporating it with machine learning, enabling fully autonomous electrochemical experiments. Electrochemistry is beginning to integrate machine learning to aid in determining reaction mechanisms.59–61 Recent work has shown the combination of deep learning and automated experimentation allows for fully closed-loop mechanistic studies of molecular electrochemistry, however this type of workflow has yet to be applied to electrocatalytic systems.28 We are excited to explore such a closed-loop system for the study of molecular electrocatalysis using our new automated platform. Additionally, the modular nature of our platform allows for the use of more advanced electroanalytical methods such as temperature-controlled voltammetry, automated electrolysis, and downstream analysis of electrolysis products, enabling users to gain a comprehensive mechanistic understanding of complex systems. We believe that such a system will enable rapid discovery of new effective electrocatalysts, and accelerate molecular electrocatalysis for energy conversion, fuel valorization, bioelectrochemical sensing, and other practical applications.
Data availability
The code for the eLab API can be found at https://github.com/jrlLAB/elabAPI. All experimental scripts and resulting data can be found in the ESI† file ‘Data.zip’.Author contributions
M. A. P.: conceptualization, data curation, formal analysis, investigation, software, methodology, visualization, writing – original draft, writing – review & editing. G. H.: investigation, methodology, software, writing – review & editing. J. R. L.: conceptualization, project administration, resources, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Beckman Institute for Advanced Science and Technology Graduate Fellows Program with support from the Arnold and Mabel Beckman Foundation. We acknowledge Seth Putnam for helpful discussion.Notes and references
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- E. C. R. McKenzie, S. Hosseini, A. G. C. Petro, K. K. Rudman, B. H. R. Gerroll, M. S. Mubarak, L. A. Baker and R. D. Little, Chem. Rev., 2022, 122, 3292–3335 CrossRef CAS.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- D. G. Boucher, A. D. Pendergast, X. Wu, Z. A. Nguyen, R. G. Jadhav, S. Lin, H. S. White and S. D. Minteer, J. Am. Chem. Soc., 2023, 145, 17665–17677 CrossRef CAS PubMed.
- S. Hosseini, J. N. Janusz, M. Tanwar, A. D. Pendergast, M. Neurock and H. S. White, J. Am. Chem. Soc., 2022, 144, 21103–21115 CrossRef CAS PubMed.
- E. C. R. McKenzie, S. Hosseini, M. Tanwar, M. Neurock, S. D. Minteer and S. C. Jacobson, J. Phys. Chem. C, 2023, 127, 17335–17344 CrossRef CAS.
- C. Sandford, L. R. Fries, T. E. Ball, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2019, 141, 18877–18889 CrossRef CAS PubMed.
- M. K. Homer, D.-Y. Kuo, F. Y. Dou and B. M. Cossairt, J. Am. Chem. Soc., 2022, 144, 14226–14234 CrossRef CAS PubMed.
- M. E. O'Reilly, R. S. Kim, S. Oh and Y. Surendranath, ACS Cent. Sci., 2017, 3, 1174–1179 CrossRef PubMed.
- D. J. Martin, B. D. McCarthy, E. S. Rountree and J. L. Dempsey, Dalton Trans., 2016, 45, 9970–9976 RSC.
- G. Crabtree, Joule, 2020, 4, 2538–2541 CrossRef CAS.
- N. L. Bell, F. Boser, A. Bubliauskas, D. R. Willcox, V. S. Luna and L. Cronin, Nat. Chem. Eng., 2024, 1, 180–189 CrossRef.
- S. Li, E. R. Jira, N. H. Angello, J. Li, H. Yu, J. S. Moore, Y. Diao, M. D. Burke and C. M. Schroeder, Nat. Commun., 2022, 13, 2102 CrossRef CAS PubMed.
- M. Christensen, L. P. E. Yunker, F. Adedeji, F. Häse, L. M. Roch, T. Gensch, G. dos Passos Gomes, T. Zepel, M. S. Sigman, A. Aspuru-Guzik and J. E. Hein, Commun. Chem., 2021, 4, 1–12 CrossRef PubMed.
- B. P. MacLeod, F. G. L. Parlane, A. K. Brown, J. E. Hein and C. P. Berlinguette, Nat. Mater., 2022, 21, 722–726 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, H. Hao, V. Rathore, J. Derasp, T. Gaudin, N. H. Angello, M. Seifrid, E. Trushina, M. Guy, J. Liu, X. Tang, M. Mamada, W. Wang, T. Tsagaantsooj, C. Lavigne, R. Pollice, T. C. Wu, K. Hotta, L. Bodo, S. Li, M. Haddadnia, A. Wołos, R. Roszak, C. T. Ser, C. Bozal-Ginesta, R. J. Hickman, J. Vestfrid, A. Aguilar-Granda, E. L. Klimareva, R. C. Sigerson, W. Hou, D. Gahler, S. Lach, A. Warzybok, O. Borodin, S. Rohrbach, B. Sanchez-Lengeling, C. Adachi, B. A. Grzybowski, L. Cronin, J. E. Hein, M. D. Burke and A. Aspuru-Guzik, Science, 2024, 384, eadk9227 CrossRef CAS PubMed.
- N. J. Szymanski, B. Rendy, Y. Fei, R. E. Kumar, T. He, D. Milsted, M. J. McDermott, M. Gallant, E. D. Cubuk, A. Merchant, H. Kim, A. Jain, C. J. Bartel, K. Persson, Y. Zeng and G. Ceder, Nature, 2023, 624, 86–91 CrossRef CAS PubMed.
- R. H. J. Xu, L. P. Keating, A. Vikram, M. Shim and P. J. A. Kenis, Chem. Mater., 2024, 36, 1513–1525 CrossRef CAS.
- S. V. Kalinin, D. Mukherjee, K. Roccapriore, B. J. Blaiszik, A. Ghosh, M. A. Ziatdinov, A. Al-Najjar, C. Doty, S. Akers, N. S. Rao, J. C. Agar and S. R. Spurgeon, npj Comput. Mater., 2023, 9, 1–16 CrossRef.
- K. Darvish, M. Skreta, Y. Zhao, N. Yoshikawa, S. Som, M. Bogdanovic, Y. Cao, H. Hao, H. Xu, A. Aspuru-Guzik, A. Garg and F. Shkurti, arXiv, 2024, preprint, arXiv:2401.06949, DOI:10.48550/arXiv.2401.06949, https://arxiv.org/abs/2401.06949.
- A. Dave, J. Mitchell, S. Burke, H. Lin, J. Whitacre and V. Viswanathan, Nat. Commun., 2022, 13, 5454 CrossRef CAS PubMed.
- R. Duke, S. Mahmoudi, A. Preet Kaur, V. Bhat, I. C. Dingle, N. C. Stumme, S. K. Shaw, D. Eaton, A. Vego and C. Risko, Digital Discovery, 2024, 3, 163–172 RSC.
- L. Su, M. Ferrandon, J. A. Kowalski, J. T. Vaughey and F. R. Brushett, J. Electrochem. Soc., 2014, 161, A1905 CrossRef.
- J. Noh, H. A. Doan, H. Job, L. A. Robertson, L. Zhang, R. S. Assary, K. Mueller, V. Murugesan and Y. Liang, Nat. Commun., 2024, 15, 2757 CrossRef CAS PubMed.
- I. Oh, M. A. Pence, N. G. Lukhanin, O. Rodríguez, C. M. Schroeder and J. Rodríguez-López, Device, 2023, 1, 100103 CrossRef.
- M. A. Pence, O. Rodríguez, N. G. Lukhanin, C. M. Schroeder and J. Rodríguez-López, ACS Meas. Sci. Au, 2023, 3, 62–72 CrossRef CAS PubMed.
- H. Sheng, J. Sun, O. Rodríguez, B. B. Hoar, W. Zhang, D. Xiang, T. Tang, A. Hazra, D. S. Min, A. G. Doyle, M. S. Sigman, C. Costentin, Q. Gu, J. Rodríguez-López and C. Liu, Nat. Commun., 2024, 15, 2781 CrossRef CAS PubMed.
- T. Erichsen, S. Reiter, V. Ryabova, E. M. Bonsen, W. Schuhmann, W. Märkle, C. Tittel, G. Jung and B. Speiser, Rev. Sci. Instrum., 2005, 76, 062204 CrossRef.
- E. Lindner, Z.-L. Lu, H. A. Mayer, B. Speiser, C. Tittel and I. Warad, Electrochem. Commun., 2005, 7, 1013–1020 CrossRef CAS.
- V. Ryabova, A. Schulte, T. Erichsen and W. Schuhmann, Analyst, 2005, 130, 1245–1252 RSC.
- A. F. Zahrt, Y. Mo, K. Y. Nandiwale, R. Shprints, E. Heid and K. F. Jensen, J. Am. Chem. Soc., 2022, 144, 22599–22610 CrossRef CAS.
- Y. Mo, G. Rughoobur, A. M. K. Nambiar, K. Zhang and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 20890–20894 CrossRef CAS PubMed.
- K. M. Kulesa, E. A. Hirtzel, V. T. Nguyen, D. P. Freitas, M. E. Edwards, X. Yan and L. A. Baker, Anal. Chem., 2024, 96, 8249–8253 CrossRef CAS PubMed.
- A. Juneau, M. Abdolhosseini, C. Rocq, H. D. M. Pham, M. Pascall, R. Z. Khaliullin, S. Canesi, E. McCalla and J. Mauzeroll, ChemElectroChem, 2024, 11(13), e202400193 CrossRef CAS.
- B. H. R. Gerroll, K. M. Kulesa, C. A. Ault and L. A. Baker, ACS Meas. Sci. Au, 2023, 3, 371–379 CrossRef CAS PubMed.
- S. E. Alden, L. Zhang, Y. Wang, N. V. Lavrik, S. N. Thorgaard and L. A. Baker, Anal. Chem., 2024, 96, 9177–9184 CrossRef CAS PubMed.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- O. Rodríguez, M. A. Pence and J. Rodríguez-López, Anal. Chem., 2023, 95, 4840–4845 CrossRef PubMed.
- W. McKinney, Proceedings of the 9th Python in Science Conference, 2010, pp. 56–61 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. F. del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke and T. E. Oliphant, Nature, 2020, 585, 357–362 CrossRef CAS PubMed.
- N. F. Delaney, J. I. Rojas Echenique and C. J. Marx, J. Lab. Autom., 2013, 18, 171–177 CrossRef PubMed.
- A. Yamada, S. Mohri, M. Nakamura and K. Naruse, Sensor. Actuator. B Chem., 2010, 143, 464–469 CrossRef CAS.
- S. Baas and V. Saggiomo, HardwareX, 2021, 10, e00219 CrossRef PubMed.
- L. Saar, H. Liang, A. Wang, A. McDannald, E. Rodriguez, I. Takeuchi and A. G. Kusne, MRS Bull., 2022, 47, 881–885 CrossRef.
- M. O'Brien, A. Hall, J. Schrauwen and J. van der Made, Tetrahedron, 2018, 74, 3152–3157 CrossRef.
- CRC handbook of chemistry and physics a ready-reference book of chemical and physical data, CRC Press, Taylor & Francis Group, Boca Raton, 101st edn, 2020, pp. 2020–2021 Search PubMed.
- J. B. Gerken, Y. Q. Pang, M. B. Lauber and S. S. Stahl, J. Org. Chem., 2018, 83, 7323–7330 CrossRef CAS PubMed.
- A. Mishra, J. Kim, M. Zorigt, A. I. B. Romo, R. Gaddam, J. E. Braun, D. Ziviani and J. Rodríguez-López, ACS Sustain. Chem. Eng., 2023, 11, 6241–6249 CrossRef CAS.
- E. Carroll, S. L. Parker, A. Fukushima, S. Downey, D. Miller, Z. A. Nguyen, D. G. Boucher and S. D. Minteer, Chem. Bio. Eng., 2024, 1(5), 427–437 CrossRef CAS PubMed.
- M. Rafiee, B. Karimi and S. Alizadeh, ChemElectroChem, 2014, 1, 455–462 CrossRef.
- D. P. Hickey, D. A. Schiedler, I. Matanovic, P. V. Doan, P. Atanassov, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 16179–16186 CrossRef CAS PubMed.
- K. J. Lee, B. D. McCarthy and J. L. Dempsey, Chem. Soc. Rev., 2019, 48, 2927–2945 RSC.
- C. Amatore, J. Pinson, J. M. Savéant and A. Thiebault, J. Electroanal. Chem. Interfacial Electrochem., 1980, 107, 59–74 CrossRef CAS.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem, 2017, 1, 1–14 CrossRef.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- G. F. Kennedy, J. Zhang and A. M. Bond, Anal. Chem., 2019, 91, 12220–12227 CrossRef CAS PubMed.
- H. Chen, E. Kätelhön and R. G. Compton, Curr. Opin. Electrochem., 2023, 38, 101214 CrossRef.
- B. B. Hoar, W. Zhang, S. Xu, R. Deeba, C. Costentin, Q. Gu and C. Liu, ACS Meas. Sci. Au, 2022, 2, 595–604 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00186a |
| This journal is © The Royal Society of Chemistry 2024 |