
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComposite machine learning strategy for natural products taxonomical classification and structural insights†
Qisong
Xu
 a,
Alan K. X.
Tan
a,
Liangfeng
Guo
a,
Yee Hwee
Lim
ab,
Dillon W. P.
Tay
*a and
Shi Jun
Ang
*acd
a,
Alan K. X.
Tan
a,
Liangfeng
Guo
a,
Yee Hwee
Lim
ab,
Dillon W. P.
Tay
*a and
Shi Jun
Ang
*acd
aInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 8 Biomedical Grove, #07-01 Neuros Building, Singapore 138665, Republic of Singapore. E-mail: dillon_tay@isce2.a-star.edu.sg
bSynthetic Biology Translational Research Program, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Republic of Singapore
cInstitute of High Performance Computing (IHPC), Agency for Science, Technology and Research (A*STAR), 1 Fusionopolis Way, #16-16 Connexis, Singapore 138632, Republic of Singapore. E-mail: ang_shi_jun@ihpc.a-star.edu.sg
dDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Republic of Singapore
First published on 23rd September 2024
Abstract
Taxonomical classification of natural products (NPs) can assist in genomic and phylogenetic analysis of source organisms and facilitate streamlining of bioprospecting efforts. Here, a composite machine learning strategy marrying graph convolutional neural networks (GCNNs) and eXteme Gradient boosting (XGB) is proposed and validated for taxonomical classification of NPs in five kingdoms (Animalia, Bacteria, Chromista, Fungi, and Plantae). Our composite model, trained on 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 092 NPs from the LOTUS database, achieved five-fold cross-validated classification accuracy of 97.4%. When employed to classify out-of-sample NPs from the NP Atlas database, accuracies of 82.8% for bacteria and 86.6% for fungi were obtained. Dimensionality-reduced representations of the molecular embeddings from our composite model revealed distinct clusters of NPs that suggest a basis for enhanced classification performance. The top critical substructures from the NPs of each kingdom were also identified and compared to provide insights on structure–taxonomy relationships. Overall, this study showcases the potential of composite machine learning models for robust taxonomical classification of NPs, which can streamline discovery of NPs.
092 NPs from the LOTUS database, achieved five-fold cross-validated classification accuracy of 97.4%. When employed to classify out-of-sample NPs from the NP Atlas database, accuracies of 82.8% for bacteria and 86.6% for fungi were obtained. Dimensionality-reduced representations of the molecular embeddings from our composite model revealed distinct clusters of NPs that suggest a basis for enhanced classification performance. The top critical substructures from the NPs of each kingdom were also identified and compared to provide insights on structure–taxonomy relationships. Overall, this study showcases the potential of composite machine learning models for robust taxonomical classification of NPs, which can streamline discovery of NPs.
1 Introduction
Natural products (NPs) are synthesized by biological organisms1 in response to environmental stimuli for their adaptation, interaction, and use in chemical warfare throughout nature.2–4 Due to their rich bioactivity, NPs have been employed as agrochemicals,5 food preservatives,6,7 cosmetics,8 and most notably, as pharmaceuticals9 where approximately 80% of antibiotics are NP-derived.10 Recent developments in artificial intelligence (AI) offer unprecedented opportunities to investigate, classify, and characterize NPs.11 Some examples of these include: (1) differentiating between NP and non-NP compounds,12 (2) classifying terrestrial and marine NPs,13 (3) visualizing NP chemical space via generative topographic maps,14 and (4) taxonomically classifying NPs.15,16 In particular, taxonomical classification of NPs can facilitate bioprospecting efforts by providing insights on taxonomic groups producing natural products with interesting bioactivity, thus narrowing investigations to those organisms with shared evolutionary histories.17 Some examples include: identifying taxonomic groups that produce drug-like molecular scaffolds18 to guide search efforts for new therapeutic compounds or facilitating the search for natural pesticides19 and herbicides20 by focusing on plant or microbial species with similar defensive mechanisms. However, current capabilities21 only cover three kingdoms (Plantae, Bacteria, and Fungi) which limits the utility of such classifications. Expanding to include more kingdoms would enable more precise and efficient bioprospecting with greater coverage. The convergence of two factors – new advancements in machine learning algorithms22 capable of more effectively tackling this challenge, and the availability of larger curated datasets,23 makes it an opportune time to undertake this work. Here, we demonstrate a composite machine learning strategy to expand taxonomical classification of NPs to encompass the five kingdoms of Plantae, Bacteria, Fungi, Animalia, and Chromista24 (Fig. 1). | ||
| Fig. 1 Previous studies12,13,21,25,26 have focused on substructure and atom-pair information to predict up to three natural product (NP) kingdoms. In this work, composite machine learning models are developed taxonomical classification of NPs in up to five different kingdoms. | ||
Graph convolutional neural networks (GCNNs)22,27,28 are first employed to effectively extract key structural features of NPs as molecular fingerprints that are then used as the input for traditional machine learning algorithms29 like Support Vector Machines (SVMs)30 or eXtreme Gradient Boosting (XGB).31 The combination of molecular fingerprints generated by GCNNs and XGB yielded the most robust classification models (97.4% balanced accuracy), providing improvements of ∼15% in balanced accuracy over incumbent model architectures.21 Our composite models could also be used to characterize complex molecular targets32 or molecules crafted through generative chemistry.33
2 Materials and methods
2.1 Dataset preparation
An open-source and well-annotated database of natural products from the LOTUS initiative was utilized.23 This involved an original dataset (LOTUS version from February 2021) consisting of 276518 NPs retrieved from the official LOTUS website (https://lotus.naturalproducts.net/download). The SMILES (Simplified Molecular Input Line Entry System)34 of NPs were first canonicalized, giving rise to 276499 unique isomeric SMILES that illustrate the diversity of NPs with varying stereochemistry. Seven kingdoms were identified from the annotations of the original LOTUS dataset: Animalia, Archaea, Bacteria, Chromista, Fungi, Plantae, and Protozoa. 266663 isomeric SMILES (96.44% of the original 276499 isomeric SMILES) only held a single kingdom label (Fig. 2A). A detailed breakdown of the 266663 single kingdom isomeric SMILES showed that the largest population of characterized NPs originated from Plantae, followed by Fungi, Bacteria, and Animalia kingdoms (Fig. 2B).
 | ||
| Fig. 2 Distribution of isomeric SMILES in the LOTUS database. (A) Distribution of isomeric SMILES by number of kingdoms, and (B) kingdom distribution of isomeric SMILES belonging to only a single kingdom. | ||
Removing isomeric information from the 266663 single kingdom isomeric SMILES reduced them to 133876 unique non-isomeric SMILES (Fig. 3A) of which 133233 (99.52% out of 133876) hold single-kingdom labels. This suggests that despite the presence of stereochemistry, different stereoisomers of the same non-isomeric SMILES originate mostly from the same kingdom. Similarly, the kingdom distribution for the 133233 non-isomeric SMILES is also dominated by NPs from the Plantae kingdom (Fig. 3B). The final curated dataset for multiclass classification and structural analysis comprised of SMILES from the top five kingdoms (Animalia, Bacteria, Chromista, Fungi, and Plantae), totalling 133092 unique non-isomeric entries, as the kingdoms of Protozoa and Archaea each contributed less than 1% to the dataset.
 | ||
| Fig. 3 Distribution of non-isomeric SMILES in the LOTUS database. (A) Distribution of non-isomeric SMILES by number of kingdoms and (B) kingdom distribution of non-isomeric SMILES belonging to only a single kingdom. | ||
Subsequent machine learning models were trained on these 133092 unique, single kingdom label, non-isomeric SMILES for multiclass classification to five kingdoms (Animalia, Bacteria, Chromista, Fungi, and Plantae). It is important to note the limitation that models trained on this non-isomeric SMILES dataset do not consider chirality information when performing taxonomical classification. Addressing this limitation and incorporating chirality and multiple kingdom labels into future models represents a possible avenue for further research, potentially further enhancing the accuracy of NP taxonomical classification.
2.2 Multiclass classification
A two-step composite method of machine learning algorithms was considered for multiclass classification. First, a GCNN approach employing directed message-passing neural networks (D-MPNN) and feed-forward neural network (FFNN) where non-isomeric NP SMILES were utilized as inputs (Fig. 4).28 The D-MPNN consists of a message passing phase that transmits atomic and bond features to construct the molecular embedding of an NP, followed by a readout phase of the molecular embedding via FFNN to predict its kingdom category.22 Additionally, the use of seven different machine learning (ML) classifiers were also investigated using three types of fingerprints, namely, MAP4 fingerprints,25 MPN fingerprints (from D-MPNN) and last_FFN fingerprints (from the last layer of the FFNN) to develop composite ML models. MPN and last_FFN fingerprints were derived from a GCNN model trained on the entire curated dataset (133092 non-isomeric SMILES). The dimension of the MAP4 fingerprints was 1,024, while both MPN and last_FFN fingerprints have dimensions of 1,100, which is the default in Chemprop. The seven ML classifier algorithms explored include Gaussian Naive Bayes (NB), K-Nearest Neighbors (KNN), Quadratic Discriminant Analysis (QDA), Random Forest (RF), Light Gradient Boosting Machine (LGBM), eXtreme Gradient Boosting (XGB), and Support Vector Machine (SVM) with linear kernel. The hyperparameters of the GCNN models were optimized and the molecular fingerprints calculated using the respective chemprop.hyperparameter_optimization and chemprop.fingerprint objects from the Chemprop package.28 During the optimization of the GCNN models, a total of 200 epochs with batch size of 50 were found to be sufficient. On the other hand, the seven ML models were trained using the scikit-learn package.29 For all learning algorithms, a five-fold cross validation via stratified sampling of the five kingdoms was implemented to evaluate the multiclass classification performance of the training and validation set. The performance of all multiclass classification models were evaluated by balanced accuracy, Matthews correlation coefficient (MCC), and F1 score. The mean and standard errors of the metrics from five-fold cross validation were also used to compare the classification performance. The classification metrics were calculated based on the counts of true positive (TP), false positive (FP), true negative (TN), false negative (FN), as follows: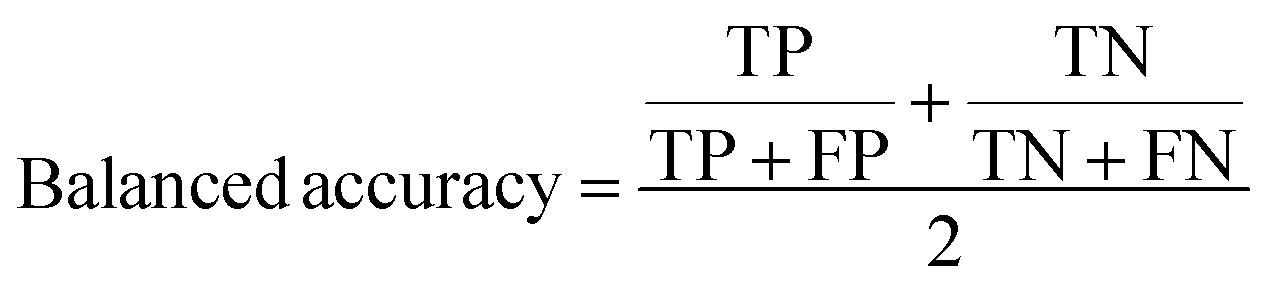

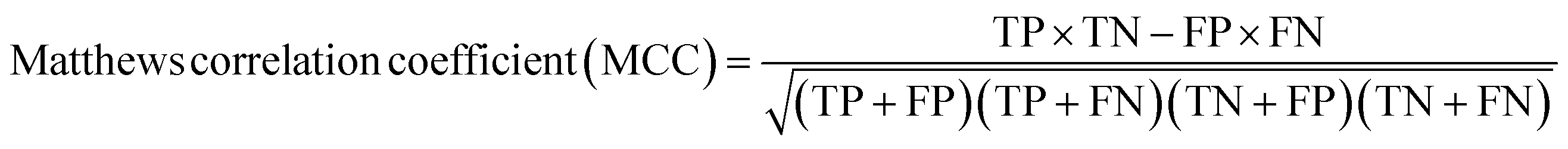
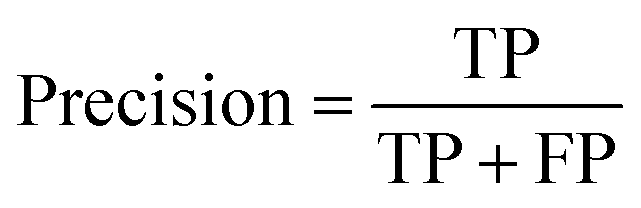


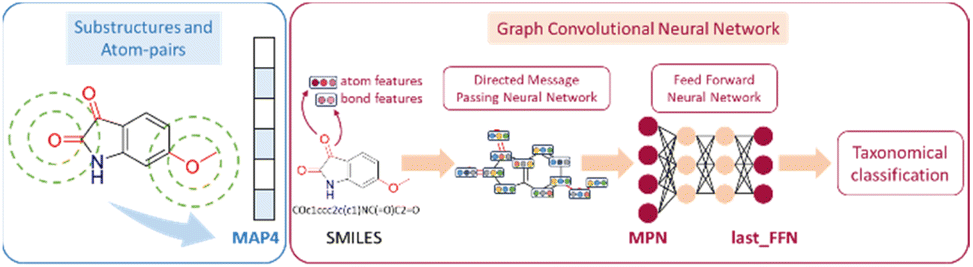 | ||
| Fig. 4 Using the molecular structure of 6-methoxyisatin as an example, the generated MAP4, MPN and last_FFN fingerprints are illustrated.22,25 | ||
2.3 Structural analysis
In addition to optimizing multiclass classification models, the molecular embeddings were also analyzed to verify the validity of the trained models. After five-fold cross validation of all models, a final GCNN model was trained using the full dataset of 133092 non-isomeric SMILES. Through t-distributed stochastic neighbor embedding (t-SNE), the high-dimensional MAP4 fingerprints as well as MPN and last_FFN fingerprints from the final GCNN model were dimensionally reduced and elucidated. A series of four different perplexity parameters [102, 103, 104 and 105] were explored to ensure impartiality when visualizing the two-dimensional (2D) projection clustering of NPs from different kingdoms. To evaluate the performance of t-SNE for different perplexity values, the Kullback–Leibler (KL) divergence between the original and fitted distribution of molecular fingerprints was assessed while the separation among clusters was quantified by Davies–Bouldin (DB) score. Molecular embeddings and their evaluation were performed using the t-SNE and DB_score objects in scikit-learn package.29
Finally, we analyzed the substructures of the NP molecules to identify critical structural fragments and their combinations that are characteristic of their kingdom source. The Monte Carlo Tree Search was employed to determine critical substructures using the chemprop.interpret object in Chemprop package.28 By analyzing the Bemis-Murcko scaffolds of NPs,35 the top critical scaffolds from each kingdom were identified.36
3 Results and discussion
A GCNN model was trained on the curated 133092 non-isomeric SMILES dataset to perform taxonomical classification of NPs from their structures. Subsequently, different ML models and molecular fingerprints were investigated to evaluate their influence on multiclass classification performance. To assess the transferability of the developed classification models, the best models were applied to NP taxonomical classification beyond the training set. Finally, we structurally analyzed NPs through dimensionally reduced molecular embeddings to identify and compare critical substructures of NPs in each kingdom.
3.1 Multiclass classification
092 non-isomeric SMILES dataset gave a slightly poorer balanced accuracy of 82.2% and MCC of 87.0% for the validation set.21 Pursuing further improvement, we explored additional ML models to supplement the prediction capabilities of simple GCNN models for more accurate taxonomical classification.
| Algorithm | MAP4 | GCNN-MPN | GCNN-last_FFN | |||
|---|---|---|---|---|---|---|
| BA | MCC | BA | MCC | BA | MCC | |
| a BA = Balanced Accuracy. MCC = Matthews Correlation Coefficient. GCNN = Graph Convolutional Neural Network. NB = Gaussian Naïve Bayesian. QDA = Quadratic Discriminant Analysis. RF = Random Forest. SVM = Support Vector Machine. LGBM = Light Gradient-Boosting Machine. XGB = eXtreme Gradient Boosting. Error of 1 standard deviation shown. | ||||||
| GCNN | — | — | — | — | 85.6 ± 0.8 | 87.3 ± 0.5 |
| NB | 20.0 ± 0.0 | 0.0 ± 0.0 | 30.8 ± 0.4 | 39.0 ± 0.7 | 67.5 ± 0.8 | 83.9 ± 0.5 |
| QDA | 29.4 ± 0.1 | 12.8 ± 0.4 | 63.1 ± 0.3 | 79.6 ± 0.4 | 96.5 ± 0.4 | 96.5 ± 0.1 |
| KNN | 54.5 ± 4.5 | 59.8 ± 4.4 | 82.5 ± 0.9 | 87.0 ± 0.6 | 93.9 ± 0.6 | 96.8 ± 0.1 |
| RF | 58.1 ± 0.9 | 67.6 ± 0.2 | 84.8 ± 0.9 | 91.5 ± 0.3 | 96.8 ± 0.5 | 97.8 ± 0.1 |
| LGBM | 68.3 ± 0.8 | 72.5 ± 0.3 | 90.2 ± 0.8 | 93.5 ± 0.2 | 97.2 ± 0.3 | 97.9 ± 0.1 |
| XGB | 72.1 ± 0.9 | 76.7 ± 0.3 | 91.1 ± 0.6 | 93.8 ± 0.1 | 97.4 ± 0.2 | 97.9 ± 0.1 |
| SVM | 82.2 ± 0.5 | 87.0 ± 0.3 | 90.9 ± 0.5 | 92.4 ± 0.2 | 97.1 ± 0.3 | 97.9 ± 0.1 |
Seven ML algorithms (NB, QDA, KNN, RF, LGBM, XGB, and SVM) based on three different types of fingerprints (MAP4, MPN, and last_FFN) were further explored as a composite strategy to supplement GCNN classification capability (Tables 1 and S1†). Classification models developed from MPN and last_FFN fingerprints provided better classification performance than those constructed from MAP4 fingerprints. This is evident from the high balanced accuracy and MCC in training and validation for all algorithms using MPN and last_FFN fingerprints. Unlike MAP4 fingerprints that comprise of circular substructure and atom-pair information,25 both MPN and last_FFN fingerprints are based on more detailed, information-rich graph representations of NPs suitable for accurate classification (Fig. 4). Furthermore, ML models based on last_FFN fingerprints offer the best classification performance that could potentially be attributed to its feed-forward neural network that facilitates additional learning drawn from MPN molecular embeddings.22
The classification performance of NPs also depended on the nature of the ML algorithm. For all three types of fingerprints, classification models based on NB and QDA generally performed poorly, as observed from their low accuracies and MCC scores (Tables S1† and 1). This may be due to the probabilistic nature of NB and QDA, which is sensitive to the kingdom populations and skew higher probabilities toward the major class.37,38 For KNN, the balanced accuracy and MCC improved for all three fingerprints, together with comparable classification performance for both training and validation sets. RF and SVM are two high-performing models that provided significant improvements in both classification accuracy and precision. This is because RF combines results from multiple trees to describe complex decision boundaries,39 while SVM is resilient to outliers by identifying optimal hyperplanes that maximize class separation.30 Finally, ensemble learning strategies involving tree-based models such as LGBM40 and XGB31 demonstrated good performance, in-line with their ability to handle imbalanced classes well and prevent overfitting with regularization. To this end, the prediction performance of the GCNN-XGB composite model developed based on last_FFN fingerprints significantly outperformed those from simple GCNN models and the MAP4-SVM model from previous studies (Fig. 5).21
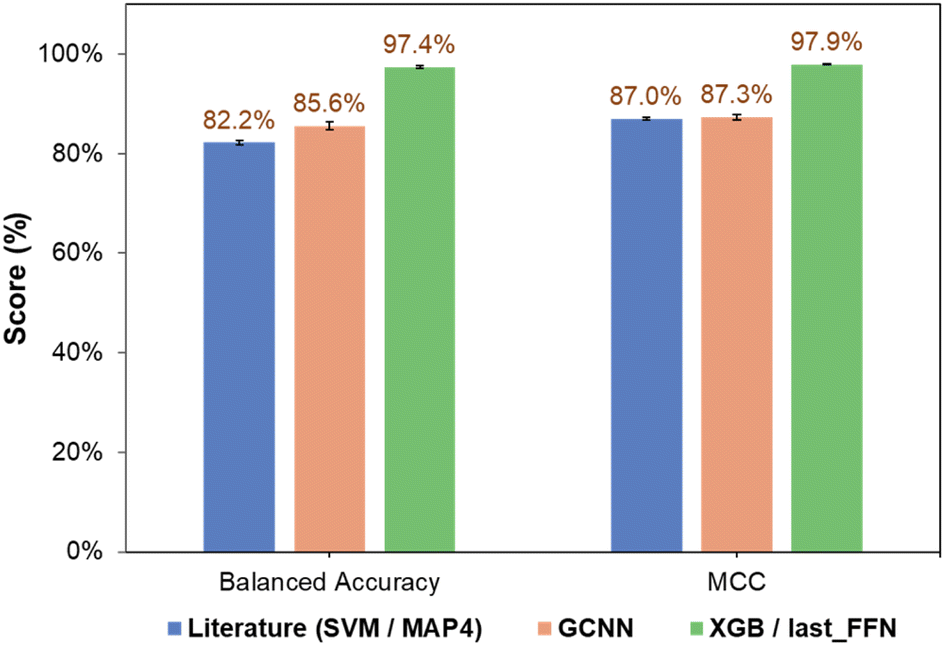 | ||
| Fig. 5 Comparison of overall performance of SVM classification model using MAP4 fingerprints from Capecchi and Reymond21 and current work (GCNN and GCNN-SVM developed using last_FFN fingerprints). | ||
In addition, the influence of traditional ML algorithms and molecular fingerprints on individual classification performance for each kingdom is reported in detail (Fig. S5–S10†). High accuracies and F1 scores were observed for each kingdom when ML models were constructed with MPN and last_FFN fingerprints, demonstrating the advantages of MPN and last_FFN fingerprints over MAP4 fingerprints. In terms of ML algorithms, NB and QDA models performed poorly in classification (low accuracy and F1 score) for most kingdoms. The classification accuracies and F1 scores decrease from Plantae to Chromista, again mirroring their population sizes in the dataset. On the other hand, SVM30 classified accurately for each kingdom despite the differences in kingdom populations. This is because SVMs provide multiple class separation despite the difference in occurrences. RF39 demonstrated excellent training classification performance across different kingdoms due to its ability to handle complex, high-dimensional data. Finally, ensemble learning strategies involving tree-based models such as LGBM40 and XGB31 also performed well due to their leaf-wise growth strategy focusing on the most significant splits and in-built regularization respectively. Overall, the composite strategy of layering XGB on top of last_FFN fingerprints provided the best classification model for the accurate taxonomical classification of NPs.
372 NPs present, 13136 NPs (7446 from Bacteria and 5690 from Fungi) are not found in the LOTUS database used for training our models. GCNN-SVM, GCNN-LGBM, and GCNN-XGB composite models with comparable performance were evaluated on the NP Atlas test set.
The composite GCNN-XGB model performed markedly better at classification compared to simple GCNN and composite GCNN-MPN models (Table 2). However, it trades bacterial NP classification accuracy for fungal NP accuracy when compared to the literature benchmark SVM model using MAP4 molecular fingerprints.
136 NP Atlas test set
| Model | Molecular fingerprint | Bacterial NP accuracy (%) | Fungal NP accuracy (%) |
|---|---|---|---|
| SVM | MAP4 | 89.9 | 81.6 |
| GCNN | last_FFN | 81.1 | 86.0 |
| GCNN-SVM | MPN | 80.2 | 82.9 |
| GCNN-SVM | last_FFN | 83.2 | 86.5 |
| GCNN-LGBM | MPN | 82.7 | 82.9 |
| GCNN-LGBM | last_FFN | 82.7 | 86.3 |
| GCNN-XGB | MPN | 84.0 | 82.6 |
| GCNN-XGB | last_FFN | 82.8 | 86.6 |
3.2 Structural analysis of NPs
To verify the remarkable classification performance of the developed GCNN and composite ML models, structural analyses of MAP4, MPN and last_FFN fingerprints were performed through the t-SNE dimensionality reduction algorithm. Kullback–Leibler (KL) divergence values for each of the molecular fingerprints decrease with perplexity value (Fig. S11a†). For all explored perplexity values however, last_FFN fingerprints possessed the lowest Davies–Bouldin (DB) score amongst all other fingerprints (Fig. S11b†) and provided the most distinct kingdom clusters. Using the perplexity value of 104 as a comparison, MAP4 fingerprints displayed NPs with highly overlapping structural features (Fig. 6A), which led to the poor classification performance observed in the previous section. On the other hand, MPN fingerprints exhibited substantial separation in the molecular features of NPs from different kingdoms (Fig. 6B), as the MPN fingerprints describe the structural similarity of NPs based on its chemical graph representation. Employing FFNs to extract additional learning from MPN fingerprints yielded last_FFN fingerprints that facilitated the best NP taxonomical classification with well-separated clusters of NPs belonging to their respective kingdoms (Fig. 6C). | ||
| Fig. 6 Visualization of the 2D projection using t-SNE (perplexity value of 104) using (A) MAP4, (B) MPN and (C) last_FFN fingerprints from the final trained GCNN model. | ||
Next, the critical substructures in NPs contributing to the classification of kingdom origins were determined. A Monte Carlo Tree Search (MCTS) was used to identify critical chemical fragments in the molecular structures of NPs. The top ten critical substructures deemed by the trained GCNN model as the most informative for NP taxonomical classification are listed for each kingdom (Fig. 7). Critical chemical substructures were identified as possessing a rationale score of more than 0.8, calculated from the chemprop.interpret object in Chemprop package.28
 | ||
| Fig. 7 Combinations of critical substructures identified in the five kingdoms of (A) Plantae, (B) Fungi (C) Chromista, (D) Bacteria, and (E) Animalia. | ||
Interestingly, the identified NP scaffolds also share structural similarities with essential starting fragments for drug discovery.42 The critical substructures for NPs in Plantae, Fungi and Chromista mainly consist of oxygen-based heterocycles (Fig. 7A–C). For plant NPs, the critical substructures tend to be simpler in nature, including furan-like43 (A5, A9), pyran-like44 (A3, A4, A8), and lactones45 (A6). In fungal NPs, molecular systems of fused rings (B5, B8) and linked rings (B10) were found to be critical. For chromists, the critical substructures identified in their NPs tended toward more complex fused ring systems (C3, C7, C8) and macrocycles (C2, C9). On the other hand, bacterial NPs typically consist of nitrogen-based heterocycles,46 including pyridine (D6), thiazole (D5), phenazine (D2) and phenoxazine-like moieties (D10) (Fig. 7D), with a few critical lactone fragments also identified (D4, D7, D9). Nitrogen-fused heterocycles such as pyrrole (E5) and indole (E3) were found to be important for critical substructures in animal NPs (Fig. 7E),47 on top of three- (E9) and four-fused (E1, E7, E9) ring systems resembling steroids.48 The benzene ring is a highly common and critical substructure across all five kingdoms (A1, B2, C1, D1 and E2). Owing to the high structural stability conferred by resonance, the planar aromatic rings offer stable building blocks that are ubiquitously found in nature. As fragments such as benzene and furan (A9, E8) are shared between kingdoms, individual fragments cannot inform taxonomical classification. Instead, it is the unique combination and connectivity of these fragments that drive differentiation between kingdoms. This underscores the importance of analyzing the broader structural context of NP structures via the right molecular fingerprinting technique rather than relying solely on the presence of individual substructures. All of the substructures described above are critical to the synthesis of stable NPs with differing levels of structural complexity.2,49
Structural analyses such as these provide valuable insights into the key fragments and potential fragment combinations characteristic of each kingdom, supporting in silico bioprospecting efforts to systematically identify the biochemical origins of novel NPs.17 Furthermore, the identified relationships between critical fragments and the corresponding kingdoms from which the NPs originate can prompt future genomic and phylogenetic analyses of different organisms to reveal the fundamental biosynthesis pathways of NPs occurring in nature.50 Overall, by leveraging on GCNNs, the structural features of NPs are effectively captured through molecular graphs, facilitating the formation of well-separated clusters corresponding to the five kingdoms. Identifying these critical substructures also enhances the explainability and interpretability of our composite machine learning models, offering a clearer understanding of how they utilize structural information for taxonomical classification.
4 Conclusion
Using a composite machine learning strategy, we optimized a multiclass classification model for taxonomical classification of NPs from their structures. By analyzing the LOTUS database, we determined the kingdom-specific critical substructures of NPs for five kingdoms (Animalia, Bacteria, Chromista, Fungi, and Plantae). GCNN models trained on 133092 non-isomeric SMILES across these five kingdoms were found to classify with a slightly superior performance to those of previous studies. Notably, the classification performance within each kingdom were found to increase with NP populations (i.e. data quantity). Three types of molecular fingerprints (MAP4, MPN, and last_FFN) were explored using seven different ML algorithms (NB, KNN, QDA, RF, LGBM, XGB, and SVM). The composite GCNN-XGB model merging last_FFN fingerprints with XGB yielded the best classification performance of 97.4% balanced accuracy on the validation set. When extended to classifying NPs outside of the training set from the NP Atlas database, the composite GCNN-XGB model achieved accuracies of 82.8% for Bacteria and 86.6% for Fungi. t-SNE embeddings of the three different molecular fingerprints revealed that last_FFN fingerprints gave the most well-separated clusters of NPs that resulted in remarkable classification performance. Finally, the top critical substructures characteristic for NPs in each kingdom were identified and compared to provide insights to structure–taxonomy relationships. Overall, this study demonstrates the potential of a composite machine learning strategy for taxonomically classifying NPs and to provide structural insights. Adopting this approach not only accelerates the classification of NP origins to screen for novel bioactive candidates but can also highlight kingdom-unique structural features of NPs to guide future efforts in virtual screening for bioprospecting as well as genomic and phylogenetic analyses of different organisms. Future avenues to enhance taxonomy classification include adopting advanced strategies such as hybrid data-based learning,51 multi-level learning,52 or meta-learning53 to further extend the generalizability of trained models across various dimensions, such as molecular size, functional groups, and structural complexity.
Code availability
The code used to train and evaluate composite models for taxonomical classification of natural products is available from GitHub at https://github.com/SIBERanalytics/NPTaxonomy.Data availability
The dataset used in this work to develop taxonomical classification of NPs was acquired from the LOTUS initiative (https://lotus.naturalproducts.net/download).23 Processed LOTUS SMILES dataset,54 MPN and last_FFN fingerprints55 are available in (.csv) format on figshare. The screening dataset was obtained from the NP Atlas database (NP Atlas v2023_06 from https://www.npatlas.org/download).41 Processed NP Atlas MPN fingerprints, and last_FFN fingerprints56 available in (.csv) format on figshare. Deep learning of NPs and Monte Carlo Tree Search were performed using the Chemprop package in python.28 The machine learning of fingerprints, calculation of t-distributed stochastic neighbor embedding, Kullback–Leibler divergence and Davies–Bouldin score were achieved using the scikit-learn package in python.29 Pre-trained composite GCNN-SVM (MPN fingerprints)57 and GCNN-SVM (last_FFN fingerprints)58 classification models for NP taxonomical classification can be downloaded from figshare. Pre-trained composite GCNN-LGBM and GCNN-XGB classification models using both MPN and last_FFN fingerprints are available from GitHub at https://github.com/SIBERanalytics/NPTaxonomy.Author contributions
S. J. A. conceptualized, designed, and supervised the study. Y. H. L. acquired funding and conceptualized the study. D. W. P. T. supervised the study. The following authors conducted the investigation and performed data analysis: Q. X., A. K. X. T., L. G., and D. W. P. T. Data curation was done by Q. X. Data visualization was performed by Q. X. and D. W. P. T. Q. X. wrote the manuscript with inputs from all authors. All authors participated in reviewing and editing the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge financial support from the Agency for Science, Technology and Research (A*STAR), Singapore (C233017006) for this work. Q. Xu thanks National Research Foundation Singapore for the funding through SGUnited Jobs Initiative (P20J3d1014). This work was supported by A*STAR Computational Resource Centre and the National Supercomputing Centre, Singapore (https://www.nscc.sg) through the use of their high performance computing facilities.Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- M. Grigalunas, S. Brakmann and H. Waldmann, J. Am. Chem. Soc., 2022, 144, 3314–3329 CrossRef CAS PubMed.
- Y.-M. Shi and H. B. Bode, Nat. Prod. Rep., 2018, 35, 309–335 RSC.
- S. Wöll, S. H. Kim, H. J. Greten and T. Efferth, Nat. Prod. Bioprospect., 2013, 3, 1–7 CrossRef.
- T. C. Sparks, D. R. Hahn and N. V. Garizi, Pest Manag. Sci., 2017, 73, 700–715 CrossRef CAS PubMed.
- S. González-Manzano and M. Dueñas, Foods, 2021, 10(2), 300 CrossRef PubMed.
- S. C. Lourenço, M. Moldão-Martins and V. D. J. M. Alves, Molecules, 2019, 24, 4132 CrossRef PubMed.
- J. B. Sharmeen, F. M. Mahomoodally, G. Zengin and F. Maggi, Molecules, 2021, 26(3), 666 CrossRef CAS PubMed.
- F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206–220 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- M. W. Mullowney, K. R. Duncan, S. S. Elsayed, N. Garg, J. J. J. van der Hooft, N. I. Martin, D. Meijer, B. R. Terlouw, F. Biermann, K. Blin, J. Durairaj, M. Gorostiola González, E. J. N. Helfrich, F. Huber, S. Leopold-Messer, K. Rajan, T. de Rond, J. A. van Santen, M. Sorokina, M. J. Balunas, M. A. Beniddir, D. A. van Bergeijk, L. M. Carroll, C. M. Clark, D.-A. Clevert, C. A. Dejong, C. Du, S. Ferrinho, F. Grisoni, A. Hofstetter, W. Jespers, O. V. Kalinina, S. A. Kautsar, H. Kim, T. F. Leao, J. Masschelein, E. R. Rees, R. Reher, D. Reker, P. Schwaller, M. Segler, M. A. Skinnider, A. S. Walker, E. L. Willighagen, B. Zdrazil, N. Ziemert, R. J. M. Goss, P. Guyomard, A. Volkamer, W. H. Gerwick, H. U. Kim, R. Müller, G. P. van Wezel, G. J. P. van Westen, A. K. H. Hirsch, R. G. Linington, S. L. Robinson and M. H. Medema, Nat. Rev. Drug Discovery, 2023, 22, 895–916 CrossRef CAS PubMed.
- P. Ertl, S. Roggo and A. Schuffenhauer, J. Chem. Inf. Model., 2008, 48, 68–74 CrossRef CAS PubMed.
- F. Pereira, Mol. Inform., 2021, 40, 2060034 CrossRef CAS PubMed.
- Y. Zabolotna, P. Ertl, D. Horvath, F. Bonachera, G. Marcou and A. Varnek, Mol. Inform., 2021, 40, 2100068 CrossRef CAS PubMed.
- Y. Djoumbou Feunang, R. Eisner, C. Knox, L. Chepelev, J. Hastings, G. Owen, E. Fahy, C. Steinbeck, S. Subramanian, E. Bolton, R. Greiner and D. S. Wishart, J. Cheminf., 2016, 8, 61 Search PubMed.
- H. W. Kim, M. Wang, C. A. Leber, L.-F. Nothias, R. Reher, K. B. Kang, J. J. J. van der Hooft, P. C. Dorrestein, W. H. Gerwick and G. W. Cottrell, J. Nat. Prod., 2021, 84, 2795–2807 CrossRef CAS PubMed.
- K. Santana, L. D. do Nascimento, A. Lima e Lima, V. Damasceno, C. Nahum, R. C. Braga and J. Lameira, Front. Chem., 2021, 9, 662688 CrossRef CAS PubMed.
- F. Zhu, C. Qin, L. Tao, X. Liu, Z. Shi, X. Ma, J. Jia, Y. Tan, C. Cui, J. Lin, C. Tan, Y. Jiang and Y. Chen, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 12943–12948 CrossRef CAS PubMed.
- A. L. Hans and S. Saxena, in Bioprospecting of Plant Biodiversity for Industrial Molecules, 2021, pp. 335–344, DOI:10.1002/9781119718017.ch16.
- D. Schein, M. S. N. Santos, S. Schmaltz, L. E. P. Nicola, C. F. Bianchin, R. G. Ninaus, B. B. d. Menezes, R. C. d. Santos, G. L. Zabot, M. V. Tres and M. A. Mazutti, Processes, 2022, 10, 2001 CrossRef CAS.
- A. Capecchi and J.-L. Reymond, J. Cheminf., 2021, 13, 82 Search PubMed.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, J. Chem. Inf. Model., 2024, 64, 9–17 CrossRef CAS PubMed.
- A. Rutz, M. Sorokina, J. Galgonek, D. Mietchen, E. Willighagen, A. Gaudry, J. G. Graham, R. Stephan, R. Page, J. Vondrášek, C. Steinbeck, G. F. Pauli, J.-L. Wolfender, J. Bisson and P.-M. Allard, eLife, 2022, 11, e70780 CrossRef CAS PubMed.
- T. Cavalier-Smith, Biol. Lett., 2009, 6, 342–345 CrossRef PubMed.
- A. Capecchi, D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 43 CAS.
- A. Capecchi and J.-L. Reymond, Biomolecules, 2020, 10 Search PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, ICML, 2017, pp. 1263–1272 Search PubMed.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss and V. Dubourg, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- C. Cortes and V. Vapnik, Mach. Learn., 1995, 20, 273–297 Search PubMed.
- T. Chen and C. Guestrin, presented in part at the Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, California, USA, 2016 Search PubMed.
- F. Hemmerling and J. Piel, Nat. Rev. Drug Discovery, 2022, 21, 359–378 CrossRef CAS PubMed.
- D. W. P. Tay, N. Z. X. Yeo, K. Adaikkappan, Y. H. Lim and S. J. Ang, Sci. Data, 2023, 10, 296 CrossRef PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed.
- Y. Chen, C. Rosenkranz, S. Hirte and J. Kirchmair, Nat. Prod. Rep., 2022, 39, 1544–1556 RSC.
- H. Zhang, Proc. Seventeenth Int. Florida Artif. Intell. Res. Soc. Conf., 2004, 1, 562–567 Search PubMed.
- T. Hastie, R. Tibshirani and J. Friedman, The elements of statistical learning: data mining, inference, and prediction, Springer Science & Business Media, 2009 Search PubMed.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- G. Ke, Q. Meng, T. Finley, T. Wang, W. Chen, W. Ma, Q. Ye and T.-Y. Liu, presented in part at the Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, California, USA, 2017 Search PubMed.
- J. A. van Santen, E. F. Poynton, D. Iskakova, E. McMann, T. A. Alsup, T. N. Clark, C. H. Fergusson, D. P. Fewer, A. H. Hughes, C. A. McCadden, J. Parra, S. Soldatou, J. D. Rudolf, E. M. L. Janssen, K. R. Duncan and R. G. Linington, Nucleic Acids Res., 2022, 50, D1317–D1323 CrossRef CAS PubMed.
- S. F. Martin, J. Org. Chem., 2017, 82, 10757–10794 CrossRef CAS PubMed.
- T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001–1011 CrossRef CAS PubMed.
- D. Kumar, P. Sharma, H. Singh, K. Nepali, G. K. Gupta, S. K. Jain and F. Ntie-Kang, RSC Adv., 2017, 7, 36977–36999 RSC.
- B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502–10566 CrossRef CAS PubMed.
- J. A. Joule, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2016, vol. 119, pp. 81–106 Search PubMed.
- F. Liu, L. Anand and M. Szostak, Chem.–Eur. J., 2023, 29, e202300096 CrossRef CAS PubMed.
- J. A. R. Salvador, J. F. S. Carvalho, M. A. C. Neves, S. M. Silvestre, A. J. Leitão, M. M. C. Silva and M. L. Sá e Melo, Nat. Prod. Rep., 2013, 30, 324–374 RSC.
- S. Wang, G. Dong and C. Sheng, Chem. Rev., 2019, 119, 4180–4220 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- T. Liu, J. Huang, T. Liao, R. Pu, S. Liu and Y. Peng, IRBM, 2022, 43, 62–74 CrossRef.
- Z. Liu, L. Lin, Q. Jia, Z. Cheng, Y. Jiang, Y. Guo and J. Ma, J. Chem. Inf. Model., 2021, 61, 1066–1082 CrossRef CAS PubMed.
- X. Qian, B. Ju, P. Shen, K. Yang, L. Li and Q. Liu, ACS Omega, 2024, 9, 23940–23948 CrossRef CAS PubMed.
- Q. Xu, A. K. X. Tan, L. Guo, Y. H. Lim, D. W. P. Tay and S. J. Ang, Curated LOTUS database, figshare, 2024, DOI:10.6084/m9.figshare.25745325.v1.
- Q. Xu, A. K. X. Tan, L. Guo, Y. H. Lim, D. W. P. Tay and S. J. Ang, LOTUS fingerprints, figshare, 2024, DOI:10.6084/m9.figshare.25745448.
- Q. Xu, A. K. X. Tan, L. Guo, Y. H. Lim, D. W. P. Tay and S. J. Ang, NP Atlas fingerprints, figshare, 2024, DOI:10.6084/m9.figshare.25745421.
- Q. Xu, A. K. X. Tan, L. Guo, Y. H. Lim, D. W. P. Tay and S. J. Ang, NP Taxonomy Classification Model (MPN/SVM), figshare, 2024, DOI:10.6084/m9.figshare.25745634.
- Q. Xu, A. K. X. Tan, L. Guo, Y. H. Lim, D. W. P. Tay and S. J. Ang, NP Taxonomy Classification Model (Last_FFN/SVM), figshare, 2024, DOI:10.6084/m9.figshare.25745598.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00155a |
| This journal is © The Royal Society of Chemistry 2024 |