
Mechanistic insights into an enantioselective synthetic strategy for 1,3-disubstituted planar chiral ferrocenes†
Feiyun
Jia
 *a,
Chenghua
Zhang
a,
Yongsheng
Yang
a,
Xueting
Zheng
b and
Mingsong
Shi
*b
*a,
Chenghua
Zhang
a,
Yongsheng
Yang
a,
Xueting
Zheng
b and
Mingsong
Shi
*b
aSchool of Pharmacy, North Sichuan Medical College, Nanchong, Sichuan 637100, P. R. China. E-mail: jiaFY@nsmc.edu.cn
bNHC Key Laboratory of Nuclear Technology Medical Transformation, Mianyang Central Hospital, School of Medicine, University of Electronic Science and Technology of China, Mianyang, Sichuan 621099, China. E-mail: therotyonth@163.com
First published on 26th November 2024
Abstract
Direct construction of 1,3-disubstituted planar chiral ferrocenes (PCFs) is a challenging task. Herein, we have computationally investigated an enantioselective synthetic strategy for 1,3-disubstituted PCFs using density functional theory (DFT) methods to explore its principal characteristics and find plausible solutions to mechanistic issues. A cooperative palladium/norbornene (NBE)-catalyzed enantioselective relay remote C–H bond activation mechanism is established. The obtained results indicate that the total free energy barrier of the conversion process is 31.8 kcal mol−1, which is reasonable under the studied reaction conditions. The rate-determining step consists of a combination of β-carbon elimination and protodepalladation. Calculations of multiple ortho-C–H activation pathways indicate that the acetate-assisted direct C–H activation is the most kinetically favorable route. Meanwhile, computations of a competitive side reaction pathway confirm that the faster the extrusion of the NBE group, the lower the formation probability of the ortho-substituted by-product. Furthermore, in the protodepalladation step, the acidic ligand (s)-Boc-L-Val-OH (L) most likely acts as a proton donor. Enantioselectivity calculations reveal that the high NBE olefin insertion barrier prevents the formation of the Rp isomer and is most likely responsible for the enantioselectivity of this transformation. The findings of the present work can deepen the understanding of PCF construction strategies and pave the way for the synthesis of 1,3-disubstituted PCFs.
Introduction
Planar chiral ferrocenes (PCFs) are extensively studied by organic chemists because of their utilization in asymmetric catalysis, materials science, and biomedical applications.1–5 The introduction of more than two different substituents into metallocycles is the main method for breaking the plane of symmetry of planar rings and creating chirality. Owing to the recent advances in ortho-C–H activation strategies, the synthesis of 1,2-disubstituted PCFs has made significant progress.6–8 The unique value of 1,3-disubstituted PCFs in remote chiral induction makes their synthesis an important task.2,9,10 However, the direct construction of 1,3-disubstituted PCFs is challenging because the meta-C–H bond in the cyclopentadienyl ring is distant from the functional group.Recently, a successful strategy for the synthesis of 1,3-disubstituted ferrocenes has been reported.11 As shown in Scheme 1, Zhou et al. synthesized 1,3-disubstituted PCFs via the enantioselective remote C–H bond activation with high functional group tolerance. The originality of this method lies in the successful introduction of a transient directing norbornene (NBE) group to relay the remote C–H bond activation during the reaction. NBE has been effectively used in relaying remote C–H activation as a transient group attached to the aryl ring.12–14 However, the applicability of this approach to the cyclopentadienyl ring of ferrocene has not been confirmed because of the existence of certain structural differences between the ferrocene substrate and aromatic ring. Zhou's group constructed desired 1,3-PCFs by screening NBE and mono-N-protected amino acid (MPAA) ligand derivatives. In a previous computational study, we reported a mechanism for NBE relaying the remote enantioselective C–H bond activation in aromatic rings.15 Therefore, this strategy can be potentially applied for the synthesis of PCFs.
 | ||
| Scheme 1 Enantioselective remote C–H activation for the synthesis of 1,3-disubstituted PCFs. | ||
Scheme 2 shows a plausible catalytic mechanism proposed by Zhou et al. According to this scheme, the reaction is initiated by a divalent active palladium catalyst, and ortho-C–H activation is accomplished with the assistance of the MPAA ligand. The subsequent crucial palladation relay step consists of NBE insertion and meta-C–H activation. Thus, the Pd catalyst is placed at the meta-position. After the oxidative addition and reductive elimination reactions with another initiating reactant, an electrophilic reagent and important meta-C–C bonds are formed. Here, intermediate V can undergo β-carbon elimination, NBE extrusion, and protodepalladation to release the final main product 3 and regenerate the active palladium catalyst or directly participate in protodepalladation to produce by-product 4.
 | ||
| Scheme 2 Proposed mechanism for synthesizing 1,3-disubstituted PCFs. | ||
Despite the significant efforts of Zhou et al. aimed to explore this catalytic strategy, many mechanistic details remain ambiguous. First, although the role of the NBE group as a transient mediator relay was demonstrated for common aromatic rings, the mechanism of its insertion and extrusion into ferrocene has not been confirmed computationally. It was hypothesized that the NBE extrusion rate was negatively correlated with the by-product yield. In other words, the slower the NBE extrusion rate, the more favorable the formation of by-products. Therefore, the NBE dependence of the products and competition mechanism between the by-products and main products must be investigated in detail. Second, experiments have shown that during the protodepalladation step, the proton source is not the solvent but another active proton donor in the reaction system. Which species are most likely to act as proton donors? Third, the strategy of chiral induction using MPAA ligands for constructing chiral carbon centers has been utilized in previous C–H activation studies.16 According to the literature, the mechanism of C–H activation for the aryl ring in the reaction system where MPAA coexists with palladium acetate remains unclear. This situation becomes even more confusing after the substrate is changed from a general aromatic ring to the cyclopentadienyl ring of ferrocene. Therefore, the rate-determining step of this process must be clarified.
Theoretical computational methods are powerful and effective tools for elucidating organic reaction mechanisms.16–19 In this study, density functional theory (DFT) calculations were performed to investigate the principal characteristics of this transformation and find plausible solutions to mechanistic issues in the synthesis strategy of PCFs. We hope that our efforts will deepen the understanding of PCF construction strategies and pave the way for the synthesis of 1,3-disubstituted PCFs.
Computational details
Full geometry optimizations were performed using the PBE0-D3 functional20–23 in the gas phase with the def2-SVP24,25 basis set. The PBE0 functional has been demonstrated to be particularly suitable for describing systems containing transition metals, including iron.26–30 Vibrational frequencies were calculated to confirm the minima with all positive frequencies and transition states with single imaginary frequencies. Thermodynamic quantities such as thermal corrections to enthalpy and Gibbs free energy were computed at the same level of theory. Intrinsic reaction coordinates31,32 were applied to connect the stationary states. To obtain more accurate solvation-corrected relative Gibbs energies, the solvation model density (solvent: = dimethylformamide (DMF)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethyl sulfoxide (DMSO) (4:1))33 continuum method and def2-TZVP34 basis set were applied for single-point energy calculations.
:dimethyl sulfoxide (DMSO) (4:1))33 continuum method and def2-TZVP34 basis set were applied for single-point energy calculations.
In addition, we compared the energies of the active catalyst and several key iron-containing intermediates in different spin states. As shown in Table S1 of the ESI,† calculations indicate that all iron-containing species have the lowest free energy at low spin states (spin = 1). The natural population analysis (NPA)35 charge was obtained at the same computational level as the single-point energies, and non-covalent interaction (NCI) analysis was performed using the Multiwfn and VMD programs.36,37 The computed structures were visualized using the CYLView software,38 and all calculations were performed utilizing the Gaussian 09 software package.39
Results and discussion
Enantioselective synthesis of 1,3-disubstituted PCFs
 | ||
| Scheme 3 Computational model reaction. | ||
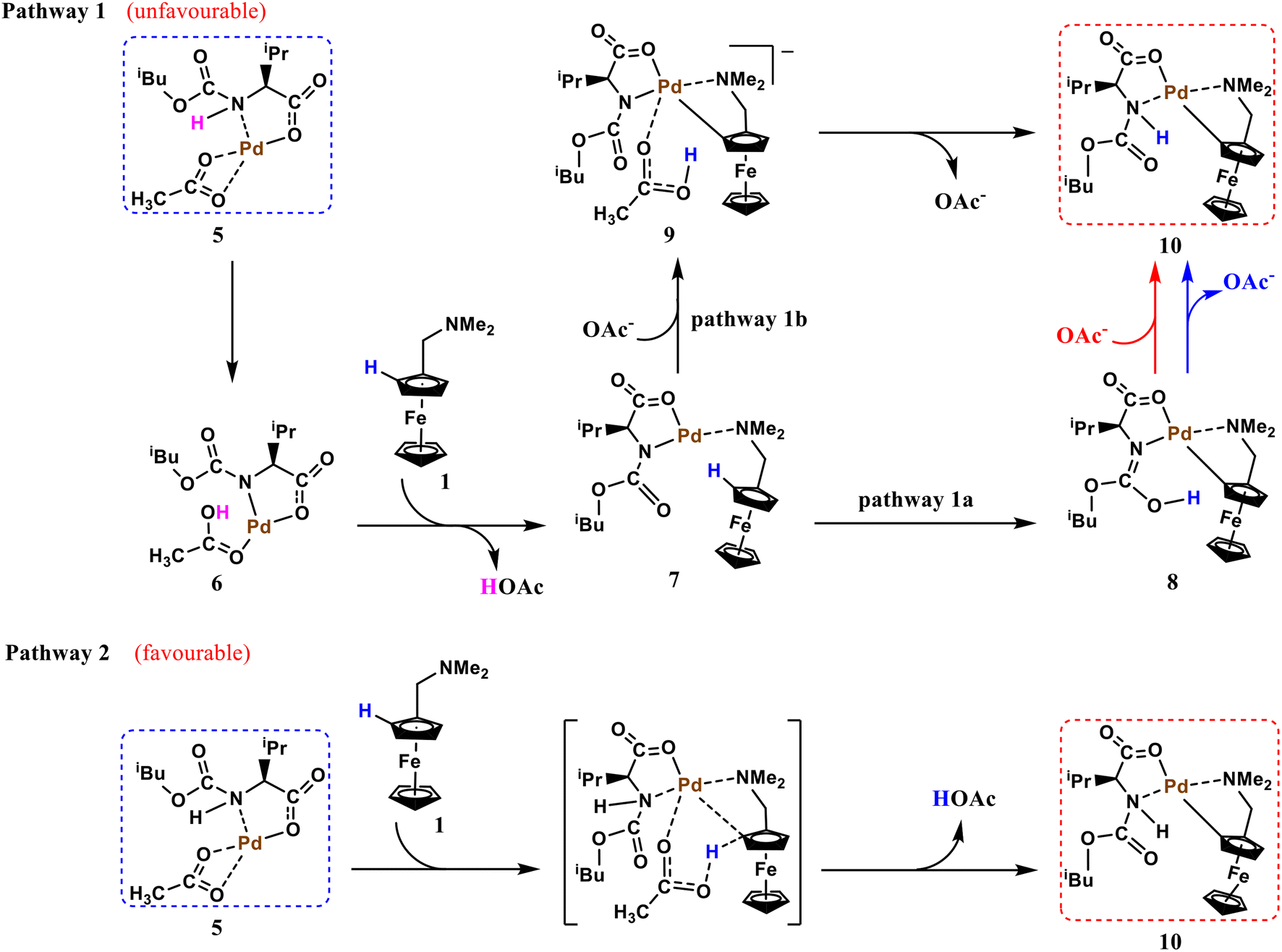
Before starting calculations, the form of the active Pd catalyst had to be clarified for this transformation. For a similar reaction system, Yu et al. investigated the form of the active catalyst by conducting nuclear magnetic resonance studies and ligand synthesis. The active catalyst was found to be an MPAA ligand coordinated to Pd(II) with high affinity via a bidentate binding mode.40 This conclusion was confirmed in a subsequent work.41 As shown in Scheme 4 for species 5, it is also used as the starting active palladium catalyst in the present study.
 | ||
| Scheme 4 Possible ortho-C–H activation mechanisms. | ||
According to Scheme 2, the transformation is initiated by ortho-C–H activation, which represents is the first key step in the proposed strategy. In similar reaction systems involving aryl substrates, ortho-C–H activation generally proceeds via two different mechanisms.42 The first reaction occurs with the assistance of the external acetate base, and the other reaction is directly driven by the internal base of the ligand. Inspired by these mechanisms, we propose two possible ortho-C–H activation pathways using ferrocene as a substrate (Scheme 4). In pathway 1, the N–H bond of the ligand is first activated by the internal acetate base, opening a coordination site at the palladium center. After the acetate molecule is released, palladium is coordinated to the nitrogen atom of the directing group of the ferrocene substrate 1 to form intermediate 7. With the assistance of the ligand internal base or external acetate base, concerted metalation–deprotonation (CMD)43 occurs to activate the neighboring –C–H bond and form a palladium ring (pathway 1a or 1b). Subsequently, intermediate 8 or 9 undergoes intramolecular hydrogen migration to reform the N–H bond. Unlike pathway 1, pathway 2 does not require a prior activation of the N–H bond. Ortho-C–H activation proceeds directly with the assistance of the external acetate base to form the activation intermediate 10.
Fig. 1 shows the mechanism of ortho-C–H activation along pathway 1. The pathway (pathway 1b, ts3) assisted by the external acetate base represented by the blue dashed line has been ruled out first. The ts3 energy barrier is 7.9 kcal mol−1 (18.3 vs. 10.4 kcal mol−1) higher than the internal base-assisted transition state ts2 (pathway 1a). Intermediate 8 belongs to two possible intramolecular proton migration mechanisms. The unassisted direct proton migration pathway (red dashed line, ts5) with a calculated barrier of 38.4 kcal mol−1 has been excluded. Hydrogen migration assisted by the external acetate base proceeds in two steps. The calculated free energy barrier for the first hydrogen migration step ts4 is 12.5 kcal mol−1. These results indicate that activating the N–H bond in pathway 1 requires a free energy barrier of 18.3 kcal mol−1. Clearly, pathway 1 is kinetically unfavorable relative to pathway 2 (the subsequent calculations reveal that the total free energy barrier for pathway 2 is only 10.7 kcal mol−1) and thus has been excluded. At this point, the second step of hydrogen migration was not calculated. By comparing ts2 and ts3 structures, the external acetate base-assisted CMD process appears to be more crowded at the metal-palladium center, which may be responsible for the high energy barrier.
 | ||
| Fig. 1 Energy barrier profiles calculated for ortho-C–H activation along pathway 1 and computed transition state structures with selected bond distances expressed in angstroms. | ||
Next, we calculated the reaction energy barrier for pathway 2. The ortho-C–H activation along pathway 2 is assisted by the internal acetate base. As shown in Fig. 2, the palladium atom of the active catalyst 5 is weakly coordinated to the N atom of the directing group on substrate 1 to form complex 12. This is a kinetically favorable process with a free energy reduction of 4.2 kcal mol−1. Compound 12 has two distinct conformations, 12a and 12b, with a relatively large free energy difference of 15.7 kcal mol−1. This phenomenon can be attributed to structural differences, i.e., the different coordination forms of acetate to metallic palladium: one resembling a bidentate and the other resembling a monodentate. In these calculations, the more stable conformation 12a was used. The subsequent ortho-C–H activation was accomplished using the CMD process. The CMD transition state ts6 consists of a six-membered palladium ring. In ts6, the N atom in the ligand moves away from the palladium center to reduce the steric hindrance of the transition state. Calculation results reveal that pathway 2 with a free energy barrier of 10.7 kcal mol−1 is more kinetically favorable than pathway 1.
 | ||
| Fig. 2 Energy barrier profiles computed for ortho-C–H activation along pathway 2. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond is used to coordinate with palladium to form complex 14. This is a kinetically unfavorable process with an elevated free energy barrier of 15.1 kcal mol−1. Therefore, during the formation of intermediates 10–14, the coordination mode of palladium to the carboxyl group of the ligand changes from bidentate to monodentate to reduce steric hindrance, which decreases coordination and stability. As shown in Fig. 3, a four-membered ring transition state ts7 is formed with the weakening and elongation of the Pd–C bond. Subsequently, the Pd–C bond is broken, and a new C–C bond is generated to release intermediate 15. The calculated free-energy barrier for this process is 17.5 kcal mol−1. Herein, we also propose an alternative reaction pathway, in which a ligand exchange occurs and the acetate replace ligand L, thus reducing the steric hindrance of the palladium center. However, as indicated by the red dashed line in Fig. 3, the free energy barrier for the acetate-assisted NBE olefin insertion is even higher (19.2 kcal mol−1).
C double bond is used to coordinate with palladium to form complex 14. This is a kinetically unfavorable process with an elevated free energy barrier of 15.1 kcal mol−1. Therefore, during the formation of intermediates 10–14, the coordination mode of palladium to the carboxyl group of the ligand changes from bidentate to monodentate to reduce steric hindrance, which decreases coordination and stability. As shown in Fig. 3, a four-membered ring transition state ts7 is formed with the weakening and elongation of the Pd–C bond. Subsequently, the Pd–C bond is broken, and a new C–C bond is generated to release intermediate 15. The calculated free-energy barrier for this process is 17.5 kcal mol−1. Herein, we also propose an alternative reaction pathway, in which a ligand exchange occurs and the acetate replace ligand L, thus reducing the steric hindrance of the palladium center. However, as indicated by the red dashed line in Fig. 3, the free energy barrier for the acetate-assisted NBE olefin insertion is even higher (19.2 kcal mol−1).
 | ||
| Fig. 3 Energy barrier profile obtained for the NBE olefin insertion and computed structures of the transition states with selected bond distances expressed in angstroms. | ||
A new CMD process was performed using the bridging relay role of NBE to activate the meta-C–H bond. The CMD process typically requires the assistance of a base. In the studied reaction system, the carboxyl group in the MPAA anionic ligand and acetate counter ion are most likely to serve as bases. Thus, we calculated the meta-C–H activation reactions involving these two bases separately. In Fig. 4, the black line represents ligand assistance, and the red dashed line denotes acetate assistance. Both pathways form six-membered ring transition states (ts9 and ts10) and exhibit free energy barriers of 22.3 and 22.6 kcal mol−1, respectively, differing by only 0.3 kcal mol−1. This suggests that both competing pathways for meta-C–H activation may be realized. After the transition state, meta-Pd–C3 bonds are formed with the release of neutral ligands and acetate molecules. At this point, metallation–deprotonation at the meta-position of the ferrocene substrate is completed through the relaying action of NBE.
 | ||
| Fig. 4 Energy barrier profiles computed for meta-C–H activation. | ||
 | ||
| Fig. 5 Energy barrier profiles constructed for the oxidative addition and reductive elimination processes. | ||
 | ||
| Fig. 6 Energy barrier profiles obtained for protodepalladation to access by-product 4 and β-carbon elimination. | ||
Before elucidating the mechanism of protodepalladation for the by-product formation, it is necessary to identify possible proton sources. The active H+ donors in the studied reaction system include acidic ligands such as MPAA and acetic acid molecules. The obtained calculation results are shown in Fig. 6. The blue dashed line represents the acidic ligand as a proton donor, and the red line indicates acetic acid. The calculated protodepalladation barrier for the acidic ligand serving as a proton donor is 24.9 kcal mol−1 (ts14), whereas the pathway barrier for acetic acid providing active H+ species is higher and amounts to 27.0 kcal mol−1 (ts15). Hence, acidic ligand is a more efficient active H+ donor in the studied reaction system. Furthermore, regardless of the proton donor, the protodepalladation pathway of intermediate 25 has a higher free energy barrier relative to that of β-C elimination (24.9 and 27.0 vs. 16.9 kcal mol−1). Therefore, under the standard reaction conditions, the NBE extrusion pathway is more kinetically favorable and represents the main reaction channel for this transformation. This conclusion is in good agreement with the experimental findings stating that the faster the NBE extrusion rate, the greater the probability of avoiding by-product 4 formation.11
 | ||
| Fig. 7 Energy barrier profiles constructed for protodepalladation to obtain the final main product 3. | ||
Total barrier profile and rate-determining step for the transformation process
To better understand the entire transformation mechanism, the total free energy barrier profile is presented in Fig. 8. The calculated total free energy barrier for this transformation is 31.8 kcal mol−1, which is a reasonable value under the studied reaction conditions. The rate-determining step consists of the β-carbon elimination and protodepalladation processes. Moreover, the entire reaction is thermodynamically favorable with a total free energy reduction of 35.6 kcal mol−1. | ||
| Fig. 8 Total barrier profiles obtained for the conversion process. | ||
Origin of enantioselectivity
Next, we discuss the origin of the high enantioselectivity of the transformation process. In previous calculations, we explored the mechanism of Sp isomer formation. In this section, we investigate the Rp isomer formation mechanism. According to Scheme 2, the first step, ortho-C–H activation, is the turning point in the formation of different planar chiralities. Therefore, we first calculated the ortho-C–H activation step toward the Rp isomer. The relevant free-energy barrier profiles are shown in Fig. 9. For a better comparison, both ortho-C–H activation mechanisms are provided. The gray line represents a pathway toward the formation of the main Sp isomer, and the red line denotes a pathway toward the Rp isomer formation. Unexpectedly, the free energy barrier of the transition state ts6R toward the Rp isomer by-product in the ortho-C–H activation step is 3.6 kcal mol−1 lower than that of ts6 (7.1 vs. 10.7 kcal mol−1), which clearly contradicts the experimental findings. Therefore, we compared the second step in the formation of the two different isomers—NBE olefin insertion. For intermediate 10R, it is kinetically more favorable to return to the reactants rather than to move forward (14.6 vs. 24.7 kcal mol−1). Therefore, we believe that the higher NBE olefin insertion energy barrier prevents the formation of the Rp isomer and is most likely responsible for the enantioselectivity of this transformation. Next, we also preliminarily investigated what led to ts7R's higher free energy barrier. The NPA charges of several key atoms in ts7 and ts7R are listed as shown in Fig. S1 of the ESI.† However, no significant differences were found. We then proceeded to perform NCI analysis for both the transition state structures. As shown in Fig. S2,† a larger steric repulsion around the palladium in ts7R is found. We speculate that it may be responsible for its larger free energy.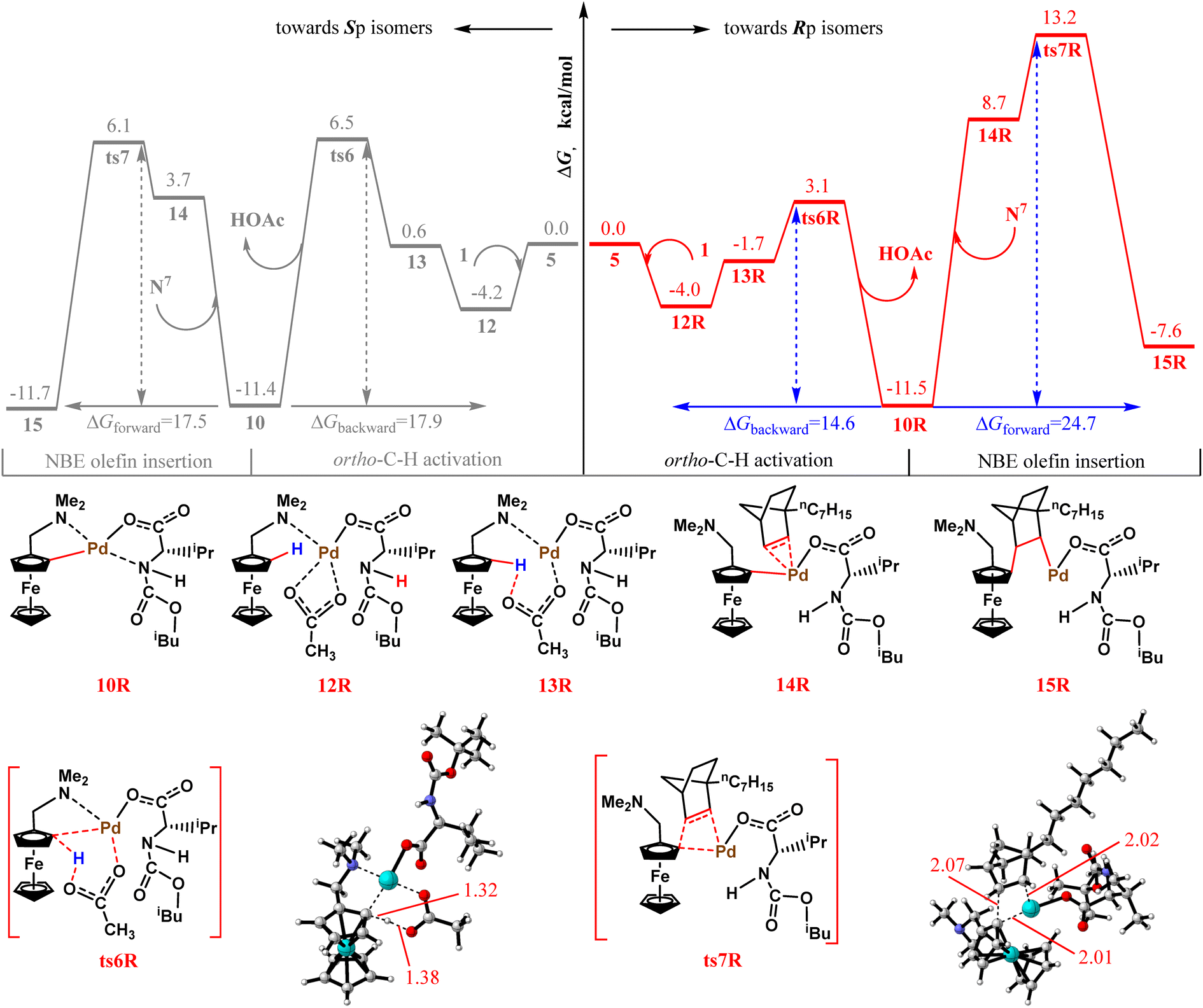 | ||
| Fig. 9 Energy barrier profiles obtained for ortho-C–H activation and NBE olefin insertion toward the Rp or Sp isomers. | ||
It is important to note that due to the extreme complexity of chemical reactions, we cannot computationally rule out all possible pathways.
Conclusions
In this study, DFT calculations were performed to investigate the enantioselective synthetic mechanism of the 1,3-disubstituted PCFs. The obtained results indicate that the transformation possesses a reasonable total free energy barrier of 31.8 kcal mol−1 under the standard reaction conditions. β-Carbon elimination and protodepalladation constitute the rate-determining step of the conversion process. The acetate-assisted pathway is kinetically most favorable for the critical ortho-C–H activation step. Screening suitable transient media to promote the extrusion of NBE is conducive to avoiding the generation of ortho-substitution by-products. In addition, the acidic ligand (L) in the studied system is most likely to provide protons during the protodepalladation step. Finally, the large energy barrier difference for the NBE olefin insertion step is most likely responsible for enantioselectivity of the studied transformation. This study can help pave the way for the synthesis of 1,3-disubstituted PCFs. Next, we plan to further focus on the factors influencing the enantioselectivity of this strategy through electronic and steric analyses.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22203056), NHC Key Laboratory of Nuclear Technology Medical Transformation (Mianyang Central Hospital) (grant no. 2022HYX001), Bureau of Science & Technology Nanchong City of China (grant no. 19SXHZ0251), and Incubation Project of Mianyang Central Hospital (grant no. 2020FH09).Notes and references
- B. Ye and N. Cramer, Chiral cyclopentadienyl ligands as stereocontrolling element in asymmetric C-H functionalization, Science, 2012, 338(6106), 504–506 CrossRef CAS.
- U. Caniparoli, I. Escofet and A. M. Echavarren, Planar Chiral 1,3-Disubstituted Ferrocenyl Phosphine Gold(I) Catalysts, ACS Catal., 2022, 12(6), 3317–3322 CrossRef CAS PubMed.
- T. Muraoka, K. Kinbara and T. Aida, Mechanical twisting of a guest by a photoresponsive host, Nature, 2006, 440(7083), 512–515 CrossRef CAS PubMed.
- J. Brettar, T. Bürgi, B. Donnio, D. Guillon, R. Klappert, T. Scharf and R. Deschenaux, Ferrocene-Containing Optically Active Liquid-Crystalline Side-Chain Polysiloxanes with Planar Chirality, Adv. Funct. Mater., 2005, 16(2), 260–267 CrossRef.
- M. Patra and G. Gasser, The medicinal chemistry of ferrocene and its derivatives, Nat. Rev. Chem., 2017, 1(9), 0066 CrossRef CAS.
- M. Ogasawara, S. Arae, S. Watanabe, K. Nakajima and T. Takahashi, Kinetic Resolution of Planar-Chiral Ferrocenylphosphine Derivatives by Molybdenum-Catalyzed Asymmetric Ring-Closing Metathesis and Their Application in Asymmetric Catalysis, ACS Catal., 2016, 6(2), 1308–1315 CrossRef CAS.
- D. Y. Zhu, P. Chen and J. B. Xia, Synthesis of Planar Chiral Ferrocenes by Transition-Metal-Catalyzed Enantioselective C−H Activation, ChemCatChem, 2015, 8(1), 68–73 CrossRef.
- D. W. Gao, Q. Gu, C. Zheng and S. L. You, Synthesis of Planar Chiral Ferrocenes via Transition-Metal-Catalyzed Direct C-H Bond Functionalization, Acc. Chem. Res., 2017, 50(2), 351–365 CrossRef CAS PubMed.
- T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, Light-Driven Open−Close Motion of Chiral Molecular Scissors, J. Am. Chem. Soc., 2003, 125(19), 5612–5613 CrossRef CAS.
- T. Chuard, S. J. Cowling, M. Fernandez-Ciurleo, I. Jauslin, J. W. Goodby and R. Deschenaux, Planar chirality: a fascinating symmetry breaking which leads to ferroelectricity in ferrocenyl liquid crystals, Chem. Commun., 2000,(21), 2109–2110 RSC.
- L. Zhou, H. G. Cheng, L. Li, K. Wu, J. Hou, C. Jiao, S. Deng, Z. Liu, J. Q. Yu and Q. Zhou, Synthesis of planar chiral ferrocenes via enantioselective remote C-H activation, Nat. Chem., 2023, 15(6), 815–823 CrossRef CAS PubMed.
- J. Wang and G. Dong, Palladium/Norbornene Cooperative Catalysis, Chem. Rev., 2019, 119(12), 7478–7528 CrossRef CAS PubMed.
- H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator, Nature, 2018, 558(7711), 581–585 CrossRef CAS PubMed.
- D. E. Hill, J.-Q. Yu and D. G. Blackmond, Insights into the Role of Transient Chiral Mediators and Pyridone Ligands in Asymmetric Pd-Catalyzed C–H Functionalization, J. Org. Chem., 2020, 85(21), 13674–13679 CrossRef CAS.
- F. Jia, J. Luo and B. Zhang, Mechanistic Insight into Palladium-Catalyzed Enantioselective Remote meta-C−H Arylation and Alkylation by Using Density Functional Theory (DFT) Calculations, Adv. Synth. Catal., 2020, 362(8), 1686–1695 CrossRef CAS.
- G. J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y. D. Wu, Computational organic chemistry: bridging theory and experiment in establishing the mechanisms of chemical reactions, J. Am. Chem. Soc., 2015, 137(5), 1706–1725 CrossRef CAS.
- Y. Li, H. Chen, L.-B. Qu, K. N. Houk and Y. Lan, Origin of Regiochemical Control in Rh(III)/Rh(V)-Catalyzed Reactions of Unsaturated Oximes and Alkenes to Form Pyrdines, ACS Catal., 2019, 9(8), 7154–7165 CrossRef CAS.
- B. Zhang, X. Xu, L. Tao, Z. Lin and W. Zhao, Rhodium-Catalyzed Regiodivergent Synthesis of Alkylboronates via Deoxygenative Hydroboration of Aryl Ketones: Mechanism and Origin of Selectivities, ACS Catal., 2021, 11(15), 9495–9505 CrossRef CAS.
- F. Jia, Y. Yang, J. Luo and B. Zhang, A stereoselective nontraditional Heck reaction mechanism: Origins of enantioselectivity and diastereoselectivity, J. Catal., 2023, 428, 115158 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110(11), 5029–5036 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110(13), 6158–6170 CrossRef CAS.
- L. Goerigk and S. Grimme, A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions, Phys. Chem. Chem. Phys., 2011, 13(14), 6670–6688 RSC.
- S. Grimme, Density functional theory with London dispersion corrections, WIREs Comput. Mol. Sci., 2011, 1(2), 211–228 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- A. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97(4), 2571–2577 CrossRef.
- S. J. Campbell, C. La, Q. Zhou, J. Le, J. Galvez-Reyes, C. Banach, K. N. Houk, J. R. Chen and S. E. Paulson, Characterizing Hydroxyl Radical Formation from the Light-Driven Fe(II)–Peracetic Acid Reaction, a Key Process for Aerosol-Cloud Chemistry, Environ. Sci. Technol., 2024, 58(17), 7505–7515 CrossRef CAS.
- M. Zhao, W. Xu, Y. D. Wu, X. Yang, J. Wang and J. S. Zhou, Cobalt-Catalyzed Enantioselective Reductive Arylation, Heteroarylation, and Alkenylation of Michael Acceptors via an Elementary Mechanism of 1,4-Addition, J. Am. Chem. Soc., 2024, 146(29), 20477–20493 CrossRef CAS PubMed.
- M. Borgini, Q.-N. Huang, P.-P. Chen, S. J. Geib, K. N. Houk and P. Wipf, Rhodium(I)-Catalyzed Annulation of Bicyclo[1.1.0]butyl-Substituted Dihydroquinolines and Dihydropyridines, J. Am. Chem. Soc., 2024, 146(22), 14927–14934 CrossRef CAS.
- L. Cheng, M.-M. Li, M.-L. Li, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, Nickel-Catalyzed Regio- and Enantioselective Hydroarylation of 1,3-Dienes with Indoles, CCS Chem., 2022, 4(8), 2612–2619 CrossRef CAS.
- F. Jia, Y. Yang and B. Zhang, Mechanism insight into palladium-catalyzed site-selective C–H functionalization of carbazoles, Catal. Sci. Technol., 2023, 13(23), 6812–6822 RSC.
- K. Fukui, A formulation of the reaction coordinate, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- K. Fukui, The path of chemical reactions-the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100(8), 5829–5835 CrossRef.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83(2), 735–746 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- C. Y. Legault, CYLview20, Université de Sherbrooke, 2020, (http://www.cylview.org) Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, , Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- K. M. Engle and J.-Q. Yu, Developing Ligands for Palladium(II)-Catalyzed C–H Functionalization: Intimate Dialogue between Ligand and Substrate, J. Org. Chem., 2013, 78(18), 8927–8955 CrossRef CAS PubMed.
- B. E. Haines, J.-Q. Yu and D. G. Musaev, Enantioselectivity Model for Pd-Catalyzed C–H Functionalization Mediated by the Mono-N-protected Amino Acid (MPAA) Family of Ligands, ACS Catal., 2017, 7(7), 4344–4354 CrossRef CAS.
- D. G. Musaev, A. Kaledin, B.-F. Shi and J.-Q. Yu, Key Mechanistic Features of Enantioselective C–H Bond Activation Reactions Catalyzed by [(Chiral Mono-N-Protected Amino Acid)–Pd(II)] Complexes, J. Am. Chem. Soc., 2012, 134(3), 1690–1698 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway, Chem. Lett., 2010, 39(11), 1118–1126 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Coordinates of all the optimized structures (XYZ). See DOI: https://doi.org/10.1039/d4cy01130a |
| This journal is © The Royal Society of Chemistry 2025 |