
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst speciation and deactivation in the ruthenium-mediated transformation of ethynyl-β-ionol to α,β-unsaturated esters for vitamin A synthesis†
Asad
Saib
 ab,
Roman
Goy
c,
Jonathan
Medlock
c,
Bettina
Wüstenberg
c,
Gabriele
Kociok-Köhn
d,
Catherine L.
Lyall
abd,
John P.
Lowe
abd and
Ulrich
Hintermair
*abe
ab,
Roman
Goy
c,
Jonathan
Medlock
c,
Bettina
Wüstenberg
c,
Gabriele
Kociok-Köhn
d,
Catherine L.
Lyall
abd,
John P.
Lowe
abd and
Ulrich
Hintermair
*abe
aDepartment of Chemistry, University of Bath, Claverton Down, BA2 7AY Bath, UK. E-mail: u.hintermair@bath.ac.uk
bDynamic Reaction Monitoring Facility, University of Bath, Claverton Down, BA2 7AY Bath, UK
cDSM-Firmenich, Wurmisweg 576, 4303 Kaiseraugst, Switzerland
dMaterials and Chemical Characterisation Facility, University of Bath, Bath BA2 7AY, UK
eInstitute for Sustainability, University of Bath, Bath BA2 7AY, UK
First published on 21st November 2024
Abstract
The catalytic anti-Markovnikov addition of carboxylic acids to propargylic alcohols to furnish unsaturated esters is an appealing transformation due to its mild conditions, good selectivity and high atom economy. Treatment of the γ-hydroxy α,β-unsaturated esters with Brønsted acids gives access to enals which serve as important building blocks for the production of vitamins and aroma compounds from biogenic terpenes. Unfortunately, current turnover numbers (TON) for this ruthenium-catalysed reaction are too low for industrial application (<100). Here we present a detailed investigation into the speciation and deactivation of the most active [(dppe)RuII(MA)2] catalyst in the anti-Markovnikov addition of carboxylic acids to ethynyl-β-ionol. Multi-nuclear high resolution FlowNMR spectroscopy gave insight into a range of kinetically relevant carboxylate complexes, allowed quantifying catalyst deactivation kinetics, and showed a pronounced influence of the carboxylic acid on catalyst stability. Systematic optimisation of reaction parameters resulted in significant improvements in catalyst productivity to reach TONs of >450 for ethynyl-β-ionol and >2000 for phenylacetylene.
1 Introduction
Vitamin A (a group of retinoids) is an important micronutrient that can be ingested from animal and plant sources as well as from fortified supplements.1 As an essential contributor to the maintenance of many biological functions in the human body,2 a deficiency of vitamin A can lead to blindness as well as child mortality.3,4 As of 2021, the global vitamin A market reached over $500 million.5 Increasing demands in developing countries compounded with the rising cost of raw materials mandates the development of more sustainable and more efficient processes for the preparation of vitamin A.6,7The first industrially viable synthesis of vitamin A was developed by Otto Isler8,9 in 1947 through a modification of the Arens and Van Dorp syntheses starting from the biogenic terpene β-ionone (Scheme 1).10,11 A Darzens reaction generates a β-C14-aldehyde that is reacted with a Grignard reagent to elongate the carbon chain, after which catalytic hydrogenation and elimination steps lead to the vitamin A retinol.8 Numerous variations of this synthesis have been developed in industry since, with the predominant routes employing the use of a C15 + C5 Julia Olefination (Rhône-Poulenc/Adisseo), a C15 + C5 Wittig–Horner reaction (BASF and most Chinese producers) and a C14 + C6 Grignard reaction based on Isler's original synthesis (DSM; Scheme 1).12–17 The evolution of these methods and their impact on the field are comprehensively documented in a recent review article which chronicles the advancements and challenges in vitamin A production over the decades.18
 | ||
| Scheme 1 The main industrial approaches to vitamin A acetate used (1.1–1.3) and newly proposed synthetic route to retinol (1.4). | ||
Although the two main routes to vitamin A acetate (DSM and BASF) have been refined to a high level, there is still room for improvement through the use of catalytic methods that improve the economic and environmental sustainability of the process further.19 A promising alternative synthetic route to retinol utilises ethynyl-β-ionol obtained from the treatment of β-ionone with acetylene/NH3 in a single step.20 With a rearrangement of ethynyl-β-ionol followed by a simple aldol condensation with acetone and repeated treatment of acetylene/NH3, a second rearrangement would yield retinal that may be reduced to retinol (Scheme 1, 1.4).21–23 Established and scalable protocols for the acetylide addition, aldol condensation and reduction steps are readily available, which renders the efficiency of the two Ru-mediated Meyer–Schuster (MS)-like transformations in the new proposed route to vitamin A a key aspect for the economic viability of the process. Nevertheless, the relatively high cost contribution of the catalyst means that detailed investigation of the rearrangement of ethynyl-β-ionol is of high interest, as only limited TONs of ∼90 have been reported so far without any effective catalyst recycling methods.24
The Brønsted acid-catalysed rearrangement of secondary and tertiary α-acetylenic propargyl alcohols to α,β-unsaturated carbonyls was first reported by Meyer and Schuster in 1922.25,26 Carbinols were treated with a selection of solid acid catalysts under relatively harsh conditions to give ketones with 100% atom efficiency.26 Ensuing research by Rupe et al. demonstrated that tertiary alcohols containing an α-acetylenic group did not always result in the anticipated aldehydes, but may yield α,β-unsaturated methyl ketones through an enyne intermediate (the Rupe rearrangement).27 Nevertheless, the requisite for high temperatures and strong acids generally curtailed the interest in the Meyer–Schuster rearrangement (MSR) until the early 1990s. More recent literature has shown the use of milder reaction conditions (RT – 50 °C) by employing Lewis acidic transition metal catalysts28–32 based on gold, silver, vanadium, rhenium and titanium.33,34 Ruthenium, displaying a rich alkyne chemistry,35–43 was soon discovered as one of the most efficient homogeneous catalysts for the MS-like anti-Markovnikov addition of carboxylic acids to terminal alkynes44–46 which after an acid-catalysed rearrangement yield enals as formal MSR products under mild conditions (Scheme 2).
 | ||
| Scheme 2 Ru-catalysed isomerising condensation of propargylic alcohols with benzoic acid. | ||
In particular, Dixneuf showed [{κ2-1,2-bis(diphenylphosphino)ethane}RuII{η3-CH2C(Me)CH2}2] ([(dppe)Ru(MA)2]) to be an effective catalyst for the MS-like transformation of a variety of tertiary alkynols when used with benzoic acid to give the corresponding benzoate esters in yields of 51–90% (Scheme 2).30 Mixtures of Z and E stereoisomers were typically obtained within a range of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 8:1 in favour of the Z isomer. A stepwise binding–isomerisation–addition–elimination mechanism was proposed on the basis of stoichiometric test experiments and in situ NMR spectroscopy, with formal anti-Markovnikov addition of benzoic acid to the alkynol inducing the formation of a 3-hydroxy-1-propen-1-yl benzoate intermediate via a reactive η1-vinylidene ruthenium complex (III, Scheme 3).29,47
:2 to 8:1 in favour of the Z isomer. A stepwise binding–isomerisation–addition–elimination mechanism was proposed on the basis of stoichiometric test experiments and in situ NMR spectroscopy, with formal anti-Markovnikov addition of benzoic acid to the alkynol inducing the formation of a 3-hydroxy-1-propen-1-yl benzoate intermediate via a reactive η1-vinylidene ruthenium complex (III, Scheme 3).29,47
 | ||
| Scheme 3 Proposed mechanism for the Ru-mediated anti-Markovnikov addition of carboxylic acids to propargylic alcohols.47 | ||
Homogeneous catalysis has shown significant potential for efficient and sustainable fine chemical production.48 However, due to the economic importance of separations on scale,49 imperfect selectivity and catalyst deactivation can pose significant problems for process development. Mechanistic studies, including kinetic analyses and new operando reaction monitoring techniques for investigating transition metal catalysis50,51 as well as biocatalysis52 can allow for deeper insights into these limitations and enable rational improvement of catalyst performance as opposed to empirical optimisation approaches.53–57 In particular, online multinuclear high-resolution FlowNMR spectroscopy has become a powerful tool for interrogating complex and dynamic catalytic systems in solution.58–62 Contrary to offline and in situ approaches,63 this operando technique allows reaction systems to be studied under realistic conditions,64 and allows for straightforward reaction progress kinetic analysis (RPKA)65 by way of variable time normalisation (VTNA)66 of the concentration profiles. Here we have applied these methods to better understand the mechanism of the Ru-mediated transformation of ethynyl-β-ionol in the context of vitamin A production, leading to the identification of optimal process conditions that significantly improved the productivity of the catalytic system.
2 Results and discussion
2.1 Optimisation of reaction conditions
|
|
||||
|---|---|---|---|---|
| Solvent | Conversion of 2a (%) | Yield of 3aa (%) | Stereoselectivity (Z/E) | Yield of 4a (%) |
| Ethyl acetate | 98 | 96 | 83:17 |
1.4 |
| Acetone | 95 | 93 | 84:16 |
1.2 |
| Toluene | 98 | 88 | 84:16 |
6.6 |
| Benzene | 98 | 86 | 82:18 |
6.2 |
| Cyclohexane | 92 | 77 | 82:18 |
4.8 |
| Chloroform | 96 | 76 | 88:12 |
6.0 |
| Dichloromethane | 84 | 43 | 90:10 |
18.0 |
| Methanol | 73 | 42 | 80:20 |
13.0 |
 :12 (Table S2†). As the pKa values of these acids are all within the same order of magnitude, it appears that sterically bulkier acids lead to higher product yields in this reaction (Table 2).
:12 (Table S2†). As the pKa values of these acids are all within the same order of magnitude, it appears that sterically bulkier acids lead to higher product yields in this reaction (Table 2).
| Benzoic acid pKa = 4.20
|
Pivalic acid pKa = 4.78
|
Acetic acid pKa = 4.76
|
Cyclohexane carboxylic acid pKa = 4.82
|
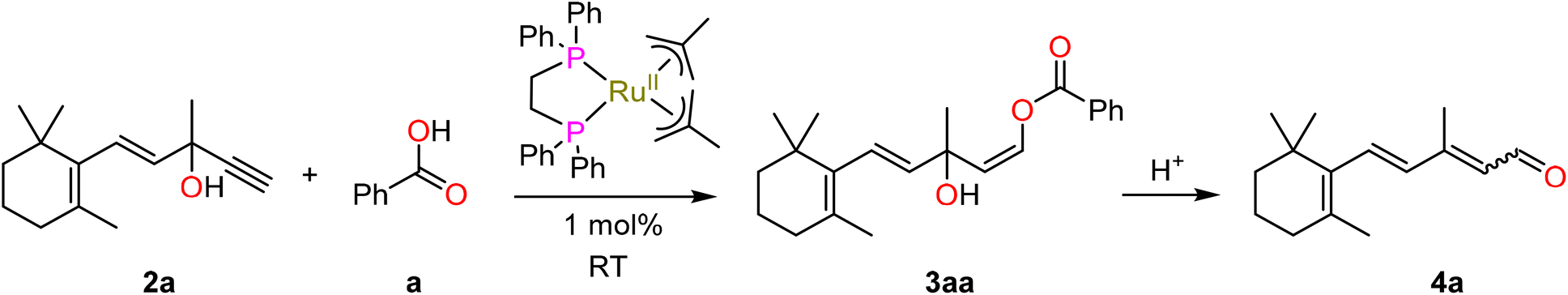
1-Adamantane carboxylic acid pKa = 4.90
|
|
|---|---|---|---|---|---|
| a Sample mixture not homogeneous for first 2 hours. | |||||
| Conversion of 2a | 86% | 97% | 27% | 96% | 100% |
| Product yield | 85% | 97% | 12% | 95% | 98%a |
| Z/E | 84:16 |
88:12 |
84:16 |
84:16 |
88:12 |




Reactions with adamantane carboxylic acid and benzoic acid showed the formation of ∼2% of aldehyde product 4a in solution from in situ ester hydrolysis, which is generally undesirable due the instability of the free aldehyde (Fig. S35†). While adamantane carboxylic acid was only partially soluble under the reaction conditions employed, the significantly cheaper pivalic acid gave a homogeneous reaction mixture and produced a high amount of the desired adduct (up to 97%) with no formation of aldehyde observed, increasing the selectivity to >99% and was therefore selected for further studies.
| Ligand | Bite angle (°)67,72 | Cone angle (°)71 | ν(CO)Pd(L2)(CO) (cm−1)70 | Yield of 3ab [%] | |
|---|---|---|---|---|---|

|
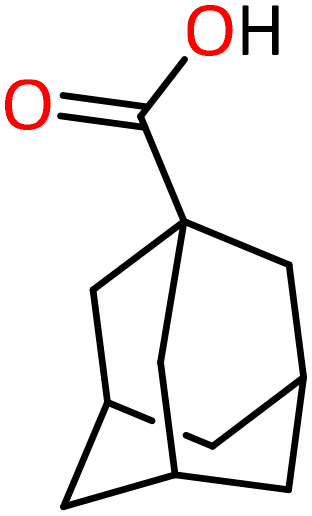
dppe (L1) | 85 | 178 | 2070.1 | 95 |

|
dmpe (L2) | 78 | 156 | 2068.8 | 83 |

|
(R,R)-Dipamp (L3) | 86 | N/A | N/A | 78 |

|
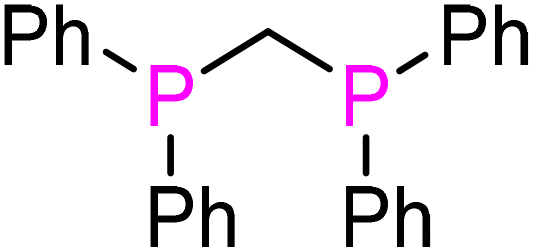
(S,S)-Norphos (L4) | 97 | N/A | 2069.2 | 57 |
|
dppm (L5) | 72 | 168 | 2072.6 | 55 |

|
dppb (L6) | 98 | 188 | 2064.2 | 33 |
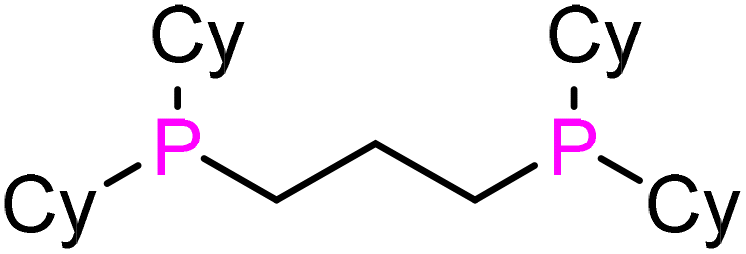
|
dcypp (L7) | 84 | 191 | 2061.1 | 16 |

|
dppp (L8) | 91 | 183 | 2066.2 | 12 |

|
(R,R)-DIOP (L9) | 98 | 196 | 2066.8 | 10 |

|
DPE-Phos (L10) | 102 | 210 | 2064.4 | 7 |

|
DiPrF (L11) | 96 | N/A | 2066.6 | 4 |
The purity of [(dppe)Ru(MA)2] was investigated following the observation of a fine, black precipitate in NMR samples containing carboxylic acids and [(dppe)Ru(MA)2] in anhydrous acetone and ethyl acetate. Initially, the [(dppe)Ru(MA)2] complex was synthesised by refluxing [(COD)Ru(MA)2] with dppe in hexane according to literature.75 After filtration of an acetone solution of the so-obtained crude [(dppe)Ru(MA)2] through a 0.2 μm PTFE membrane and recrystallisation by addition of n-hexane, clean batches of [(dppe)Ru(MA)2] were obtained that did not contain any insoluble material (presumed to be small amounts of ruthenium black from the RuCl3 starting material) and this additional purification procedure was applied in all following experiments (for further details see the ESI† 2.1).
2.2 Catalyst productivity and stability
 | ||
| Fig. 1 Maximum catalyst TONs at varying loadings of [(dppe)Ru(MA)2] for the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with pivalic acid (1 M) in anhydrous acetone (15 mL) at 20 °C (theoretical isoyield curve included to illustrate full conversion). Reactions were performed to their maximum yield (as indicated in brackets) obtained by quantitative ex situ1H NMR spectroscopy against 1,3,5-trimethoxybenzene (167 mM) as internal standard after 50 hours. | ||
Investigations into the stability and productivity of this Ru-catalysed reaction have not been reported in the literature. Many transition metal catalysts used in similar contexts, such as [ReOCl3(OPPh3)(SMe2)], [VO(OR)3], [CpRuCl(PMe3)2] and [Ru(η3-2-C3H4Me)(CO)(dppf)] [SbF6], required catalyst loadings greater than 2 mol%.31 While a TON of 100 has been reported for the Sc(OTf)3 catalysed transformation of ethoxyacetylene,77 the highest reported TON for the addition of carboxylic acids to terminal propargyl alcohols using a ruthenium catalyst is 91 (for 2-phenyl-3-butyn-2-ol with 1 mol% of [(dppe)Ru(MA)2] at 50 °C in toluene).30
|
|
||||
|---|---|---|---|---|

|

|

|

|
|
| 2a | 2b | 2c | 2d | |
| 18 hours | 84% | 78% | 87% | 87% |
| 40 hours | 97% | 93% | 97% | 96% |
| Z/E ratio | 88:12 | 96:4 | 90:10 | 92:8 |

|

|

|
|
|
| 2e | 2a + 1% 1 | 2f | 2g | |
| 18 hours | 83% | 85% | 80% | 80% |
| 40 hours | 97% | 96% | 95% | 94% |
| Z/E ratio | 90:10 |
90:10 |
92:8 |
96:4 |

The results showed yields ranging from 78–87% after 18 hours and 93–97% after 40 hours (Table 4 and Fig. S39† for an example). As the overall yield difference after 40 hours across all substrates tested was only 4% and Z/E ratios in the products all fell within a 10% margin, it may be concluded that neither the functional groups in ethynyl-β-ionol nor the presence of possible derivatives were contributing significantly to the observed deactivation of the catalyst formed from [(dppe)Ru(MA)2] under the conditions applied.
2.3 Operando 1H FlowNMR analysis
Over the course of 40 h, comparing conversion as derived from the FlowNMR experiment with data from offline sampling showed near identical values. Both types of experiments reached close to 97% conversion and exhibited the same Z/E ratios (88:12) at 1 mol% catalyst with no formation of 4a observed. FlowNMR reactions were repeated under the same conditions over the timespan of several months to determine the consistency of the method, and the reaction profiles showed a high level of reproducibility (Fig. 2).
 | ||
| Fig. 2 Conversion profiles of the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with pivalic acid (1.1 equivalents) and pivalic anhydride (0.2 equivalents) using [(dppe)Ru(MA)2] at 1 mol% to form 3ab in anhydrous acetone (15 mL) at 20 °C. Conversion profiles obtained from quantitative 1H FlowNMR spectroscopy at 4 mL min−1 and 1H NMR spectroscopy for the batch time points, both against trimethoxybenzene (167 mM) as internal standard. | ||
:12 at 1 mol% catalyst loading (Fig. 3). In ethyl acetate, adding the same amount of water decreased conversion by up to 30% compared to anhydrous conditions, in this case with a diminished product selectivity as evidenced by the appearance of a multitude of minor 1H NMR signals from unidentified side products. Nevertheless, the Z/E ratios of 88:12 in 3ab were unchanged in both cases and the amount of 4a consistently below 0.3%. Since no significant moisture sensitivity was observed in the same reaction with phenylacetylene, we ascribe the detrimental effect of water on the reaction with ethynyl-β-ionol due to this specific substrate rather than an intrinsic catalyst sensitivity to water. The fact that ethyl acetate was found to be even more sensitive to moisture than acetone may be due to trans-esterification with pivalic acid liberating acetic acid in situ, which has previously been shown to lead to decreased catalyst performance (Table 2).
 | ||
| Fig. 3 Conversion profiles of the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with pivalic acid (1 M) using [(dppe)Ru(MA)2] at a catalyst loading of 1 mol% in anhydrous acetone or ethyl acetate (15 mL) with and without the addition of degassed water (33 mM) to form 3ab at 20 °C. Data from quantitative 1H FlowNMR spectroscopy at 4 mL min−1. | ||
In order to reduce the sensitivity of the Ru-mediated transformation of ethynyl-β-ionol for application on larger scale, where rigorous exclusion of moisture may be difficult (i.e. costly) to implement due to the use of hygroscopic carboxylic acids, we investigated the effectiveness of using a mixture of carboxylic acid and their corresponding anhydride. The use of 0.13 M pivalic anhydride (substituting 0.26 M of the pivalic acid loading) in anhydrous acetone showed an 11% increase in conversion after 18 hours compared to the use of pivalic acid only (Fig. 4). The effect seemed to be purely kinetic, as despite a 37% faster initial rate in the presence of anhydride the reaction eventually converged towards similar yields and maintained the same stereoselectivity after 45 hours to when using reagent grade acid alone. Thus, in order to ensure consistency across different reagent batches and solvents, all following Ru-mediated transformations of ethynyl-β-ionol used a combination of 1.1 equivalents of acid plus 0.2 equivalents of anhydride relative to substrate to eliminate possible influences of moisture.
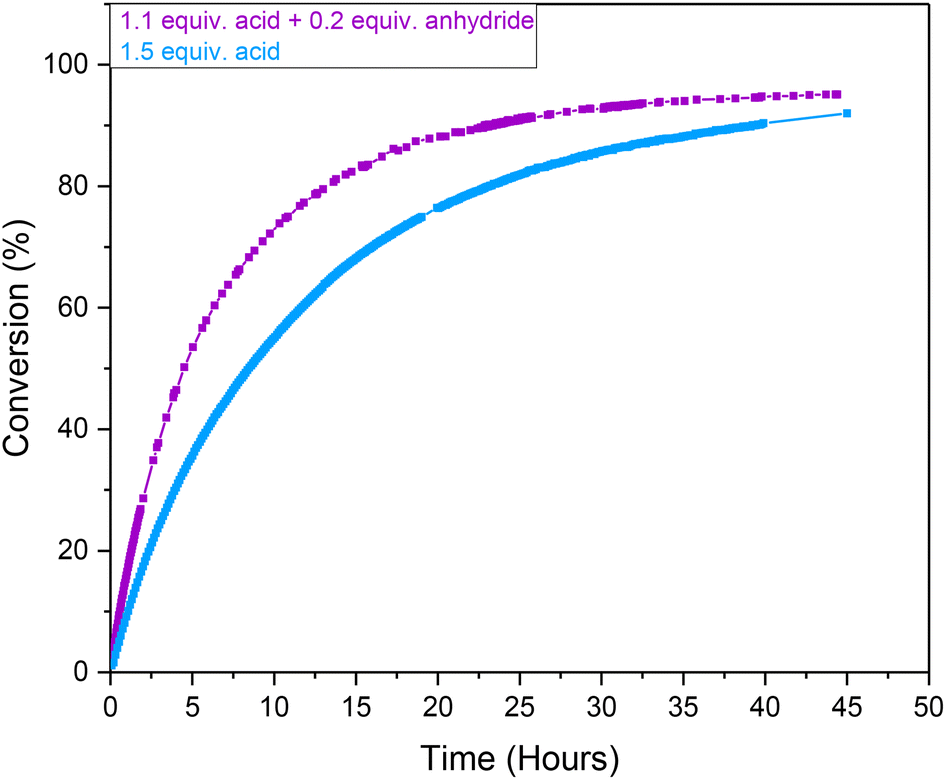 | ||
| Fig. 4 Conversion profiles of the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) catalysed by [(dppe)Ru(MA)2] at 1 mol% to form 3ab in anhydrous acetone (15 mL) at 20 °C from quantitative 1H FlowNMR spectroscopy at 4 mL min−1 with pivalic acid (1.1 equiv.) plus pivalic anhydride (0.2 equiv.) and pivalic acid (1.5 equiv.) only. | ||
 | ||
| Fig. 5 Concentration profiles of the product (sum of Z and E3ab) for the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by various amounts of [(dppe)Ru(MA)2] in anhydrous acetone (15 mL) at 20 °C measured by quantitative 1H FlowNMR spectroscopy at 4 mL min−1. | ||
The time-adjusted plot (Fig. 6) showed the initial rates of the reaction to follow a clean first-order dependence on [Ru], but progressive deactivation occurred from ∼1/3 of the final conversion at the respective catalyst loading, leading to a limitation in TON as evidenced by the linear relationship between [Ru] concentration and TONmax (Fig. S41†).
 | ||
| Fig. 6 Time-adjusted product formation profiles (data from Fig. 5) for a reaction order in [Ru] = 1. | ||
Varying the amount of pivalic acid‡ at 1 mol% [Ru] showed that the product yield was independent of the amount of pivalic acid added to the system, with all reaction profiles giving a perfect overlay without any time-adjustment (Fig. S42 and S43†). This zero order of the reaction in the concentration of acid [A] implies that the carboxylic acid is not involved in the turnover-limiting step (TLS) of the catalytic cycle (Scheme 3). Varying the amount of ethynyl-β-ionol [S] showed the rate of product formation to be first-order in substrate concentration (Fig. S44 and S45†), consistent with global first-order kinetics of the productive cycle:
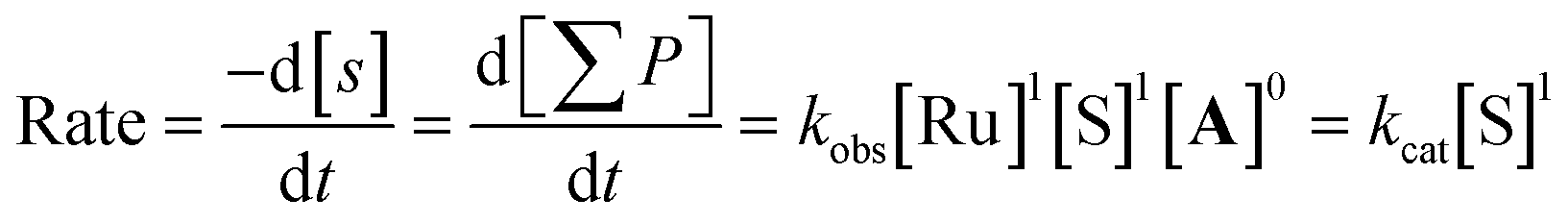
To probe the nature of the observed TON limitation (Fig. 1, 6 and S41†), “same excess” experiments were performed with different concentrations of substrate.57,65,80 Employing a 0.25 mol% catalyst loading, where deactivation/inhibition became apparent from ∼30% conversion with 0.66 M ethynyl-β-ionol, a mixture of 0.46 M substrate + 0.2 M product (mimicking 30% conversion) was used. Comparison of the time-adjusted concentration profiles showed that irreversible catalyst deactivation rather than product inhibition was responsible for the TON limitation in the Ru-mediated transformation of ethynyl-β-ionol (Fig. 7). Further confirmation for the absence of significant product inhibition came from a recycling experiment with a reaction that had afforded 98% conversion after 24 hours using 1 mol% [(dppe)Ru(MA)2] to which a second portion of substrate plus one equivalent of acid was added. After another 24 hours the reaction had reached a total conversion of 96.5% with a TON of 193, whereas starting with a catalyst loading of 0.5 mol% a conversion of only 57% was achieved (TON = 114, see Fig. 1). As further discussed in 2.6 below, these observations suggest substrate and catalyst concentration to be part of the deactivation term of the rate law (rather than product concentration).
 | ||
| Fig. 7 Concentration profiles of the product (sum of Z and E forms of 3ab) for the Ru-mediated transformation of ethynyl-β-ionol (pure substrate vs. time-adjusted substrate/product mixture) with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by [(dppe)Ru(MA)2] (0.25 mol%) in anhydrous acetone (15 mL) at 20 °C measured by quantitative 1H FlowNMR spectroscopy at 4 mL min−1. | ||
A similar analysis was carried out with the minimally functionalised substrate phenylacetylene to investigate how substrate-dependent the observed catalyst deactivation may be, as was the case for the moisture sensitivity of the reaction (Fig. 3). Varying the loading of [(dppe)Ru(MA)2] for the slower reaction with phenylacetylene showed a behaviour similar to ethynyl-β-ionol as the substrate (Fig. S46†), with a good initial overlay of the time-adjusted reaction progress profiles for first-order in [Ru] up until ∼40% conversion (Fig. 8). Although deactivation was not as pronounced as with ethynyl-β-ionol (Fig. 6) it was still noticeable with phenylacetylene, suggesting a general catalyst deactivation mechanism in the MSR catalysed by [(dppe)Ru(MA)2].
 | ||
| Fig. 8 Time-adjusted reaction progress profiles (order in [Ru] of 1.0) of the Ru-mediated transformation of phenylacetylene (0.66 M) with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by various amounts of [(dppe)Ru(MA)2] in anhydrous acetone (15 mL) to form 3bb at 20 °C from quantitative 1H FlowNMR spectroscopy at 4 mL min−1. | ||
2.4 Identification of catalytic intermediates
Quantitative 31P{1H} FlowNMR spectra were acquired in an interleaved manner with the 1H FlowNMR acquisitions that yielded the reaction progress data discussed above to further investigate and understand the observed catalyst deactivation. Being specific to the dppe ligand in the ruthenium catalyst, operando31P{1H} FlowNMR data (flip angle = 90°, dummy scans = 0, acquisition time = 0.4 s, relaxation delay = 0.5 s, number of scans = 60) processed with flow correction factors to ensure that the data was quantitative provides unique insight into the formation of reaction intermediates during and after turnover, important mechanistic information not accessible through conventional ex situ analyses and model reactions.81 | ||
| Fig. 9 Exemplary 31P{1H} FlowNMR spectra acquired during the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with benzoic acid (1 M) catalysed by [(dppe)Ru(MA)2] at 1 mol% in 15 mL of anhydrous acetone at 20 °C. | ||
31P{1H} DOSY analysis showed Ab and Bb to be of similar size in solution based on their identical diffusion coefficients (both 1.27 × 10−9 m2 s−1 compared to 2.09 × 10−9 m2 s−1 for [(dppe)Ru(MA)2]; Fig. S51†). Variable temperature NMR experiments on a mixture of Ab and Bb in the absence of substrate showed coalescence of the signals of Bb to a singlet above 40 °C and increased peak separation of the two doublets at lower temperatures (Fig. 10). The sharp singlet of Ab started to broaden below −5 °C but could not be resolved at lower temperatures. Whereas at room temperature the ratio of Ab to Bb was 1:1 under the conditions applied, higher temperatures favoured Ab (90:10 at 70 °C) and lower temperatures yielded more Bb (35:65 at −35 °C), suggesting a dynamic equilibrium between the two complexes in solution.
 | ||
| Fig. 10 31P{1H} variable temperature NMR spectra of a mixture of Ab and Bb generated from the addition of benzoic acid (0.2 mmol) to [(dppe)Ru(MA)2] (0.05 mmol) in anhydrous acetone. | ||
In acetone at room temperature, the ratio of Ab to Bb generated from [(dppe)Ru(MA)2] was ∼1:1 regardless of the excess of benzoic acid used. Repeating the reaction in different solvents showed the formation of Ab and Bb from [(dppe)Ru(MA)2] to proceed equally well in a range of organic media (except for acetonitrile where [(dppe)Ru(MA)2] remained untouched§), with different populations of AbversusBb (Table 5). The addition of small amounts of water to these samples shifted the distribution in favour of Bb in all cases, suggesting the apparent solvent effect to be a reflection of their residual moisture contents (with less polar solvents generally being drier; for more details see Table S7 and Fig. S52†). Consistent with this notion, generating Ab and Bb from [(dppe)Ru(MA)2] in acetone using a benzoic acid/anhydride mixture saw 10% more Ab being formed compared to using benzoic acid only (Fig. S53 and S54†).
| [Ru(MA)2(dppe)] | +2 equiv. of benzoic acid | +20 equiv. of degassed H2O | ||
|---|---|---|---|---|
| Ab (%) | Bb (%) | Ab (%) | Bb (%) | |
| Ethyl acetate | 30 | 70 | 12 | 88 |
| Acetone | 50 | 50 | 5 | 95 |
| Toluene | 98 | 2 | 2 | 98 |
| Dichloromethane | 91 | 9 | 33 | 66 |
| Acetonitrile | 0 | 0 | 0 | 0 |
Based on these observations, we propose complex Bb to be a water adduct of the symmetrical bis-carboxylate complex Ab (Scheme 4). This assignment was further corroborated by the identification of a 31P cross peak of Bb to a 1H NMR singlet at 15.5 ppm, indicative of a strong hydrogen bonding resonance (Fig. S50†).82,83
 | ||
| Scheme 4 Formation of complexes Ab and Bb from [(dppe)Ru(MA)2] with benzoic acid and residual water. | ||
Reacting [(dppe)Ru(MA)2] with acetic, pivalic, and adamantane carboxylic acid in acetone also gave full conversion of the complex within 30 minutes at room temperature (see Fig. S55 and S56†), but only formed complexes with NMR signatures consistent with the symmetrical bis-carboxylate complexes A according to 1H and 31P{1H} NMR spectroscopy, with integration indicating a 1:2 dppe to carboxylate ratio (Fig. S57 and S58†). Without the addition of substrate, A were found to be stable in solution for at least 2 weeks at room temperature, except for when acetic acid was used where Aa decomposed within a few hours. The addition of water to these samples resulted in the formation of the corresponding B complexes observed in the form of broad 31P{1H} NMR signals around 75–79 ppm, depending on the carboxylic acid used (Fig. S59†). Single crystals obtained from solutions of Ap and Aad (using pivalic and adamantane carboxylic acid, respectively) showed the corresponding carboxylic acid adducts of A [(dppe)RuII(η2-OOCR)(η1-OOCR)(RCOOH)] (A′) (Fig. 11). When redissolving single crystals of A′ in acetone, they quantitatively reformed complexes with NMR signatures consistent with symmetrical bis-carboxylate complexes A (Fig. S10–S14†), showing the coordination of the third carboxylic acid to be reversible and entropically disfavoured in solution at room temperature.
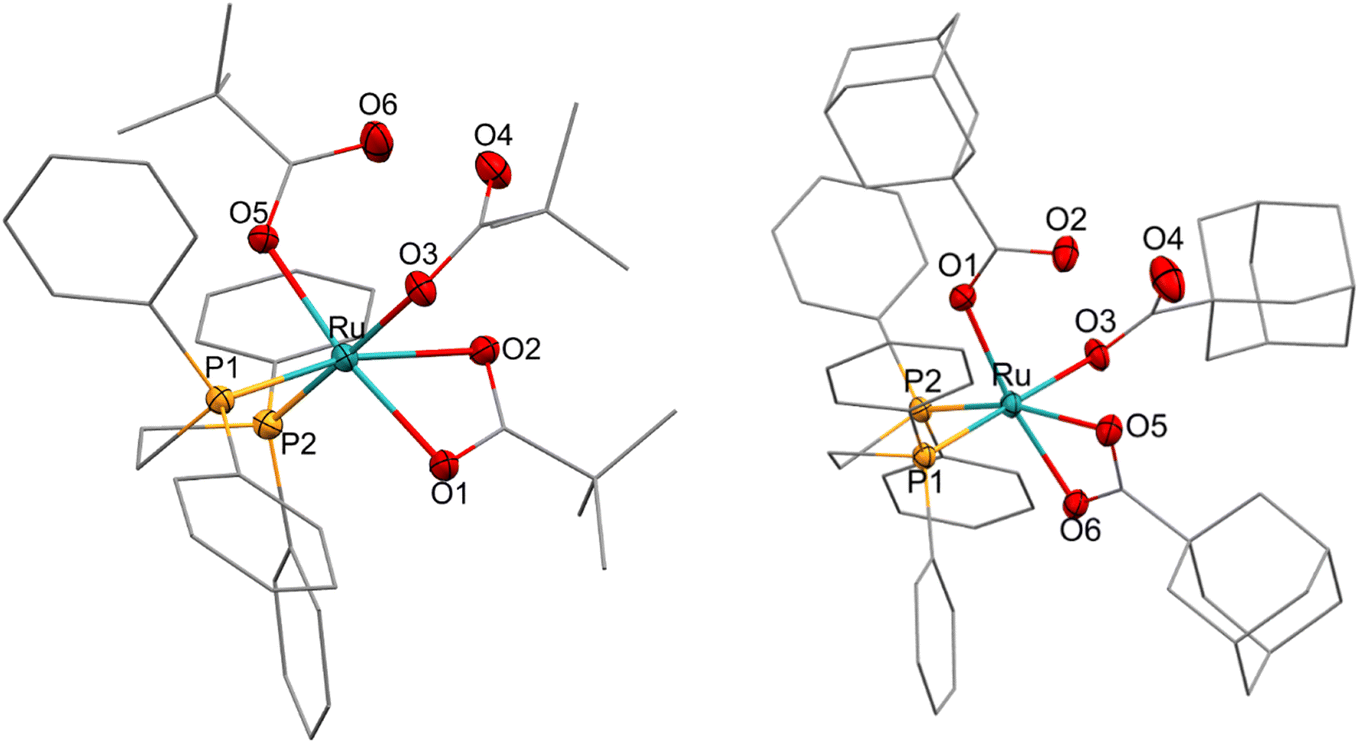 | ||
Fig. 11 X-ray crystal structures of complexes 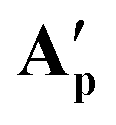 [(dppe)RuII(η2-OOCCtBu)(η1-OOCCtBu)(tBuCCOOH)] and [(dppe)RuII(η2-OOCCtBu)(η1-OOCCtBu)(tBuCCOOH)] and 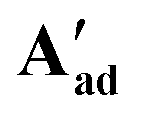 [(dppe)RuII(η2-OOCC(CH2)6(CH)3)(η1-OOCC(CH2)6(CH)3)((CH2)6(CH)3CCOOH)] with selected atoms labelled. Thermal ellipsoids shown at 50% probability level, hydrogen atoms and solvent molecules omitted for clarity (for full details see section 3.0 of the ESI†). [(dppe)RuII(η2-OOCC(CH2)6(CH)3)(η1-OOCC(CH2)6(CH)3)((CH2)6(CH)3CCOOH)] with selected atoms labelled. Thermal ellipsoids shown at 50% probability level, hydrogen atoms and solvent molecules omitted for clarity (for full details see section 3.0 of the ESI†). | ||
These A′ structures, which have not yet been reported in the literature, resemble the unsymmetrical water adduct complexes B observed in solution (Scheme 5), with an additional carboxylic acid bound to the ruthenium centre instead of a water molecule (Fig. 11). Both structures of A′ were slightly distorted octahedral 18-electron RuII complexes with no unusual structural features84,85 apart from the observation that the Ru–O distances trans to phosphorus were longer (2.17–2.20 Å) than those trans to oxygen (2.10 Å; see Table S8†). The slight deviations in the P–Ru–P angles of 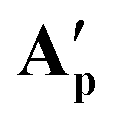 (84.8°) and
(84.8°) and 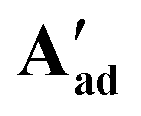 (85.3°) compared to [(dppe)Ru(MA)2] (85.7°) were likely induced by the steric demand of the pivalate and adamantane groups. In the crystal structure of
(85.3°) compared to [(dppe)Ru(MA)2] (85.7°) were likely induced by the steric demand of the pivalate and adamantane groups. In the crystal structure of  , hydrogen-bonding was observed between the proton on O4 and the pendant carboxylate oxygen O6 (bond lengths can be found in Table S8†), likely stabilising the association of the third carboxylic acid that was not bound to the metal centre in solution. Although this hydrogen atom could not be refined in the XRD structure of
, hydrogen-bonding was observed between the proton on O4 and the pendant carboxylate oxygen O6 (bond lengths can be found in Table S8†), likely stabilising the association of the third carboxylic acid that was not bound to the metal centre in solution. Although this hydrogen atom could not be refined in the XRD structure of  it likely is present in the same manner (and required for charge balance in a neutral RuII complex). To the best of our knowledge,
it likely is present in the same manner (and required for charge balance in a neutral RuII complex). To the best of our knowledge, 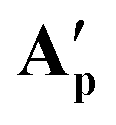 and
and  represent the first examples of a mononuclear transition metal complex featuring the same carboxylic acid in three different binding modes.
represent the first examples of a mononuclear transition metal complex featuring the same carboxylic acid in three different binding modes.
 | ||
| Scheme 5 Interconversion of carboxylate complexes observed to form from [(dppe)Ru(MA)2] and carboxylic acids in solution and the solid state. | ||
When solutions containing Aad, Ab and Ap were crystallised in the presence of moisture, dimeric forms of the corresponding bis-carboxylate complexes were obtained that featured two bridging carboxylates and an aqua ligand bridging across the two Ru centres, denoted B′ (Fig. 12).
 | ||
Fig. 12 X-ray crystal structures of complexes for 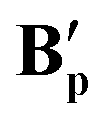 [{(dppe)RuII(η1-OOCCtBu)}2(μ-OOCCtBu)2(μ-OH2)], [{(dppe)RuII(η1-OOCCtBu)}2(μ-OOCCtBu)2(μ-OH2)],  [{(dppe)RuII(η1-OOCC(CH2)6(CH)3)}2(μ-OOCC(CH2)6(CH)3)2(μ-O)], and [{(dppe)RuII(η1-OOCC(CH2)6(CH)3)}2(μ-OOCC(CH2)6(CH)3)2(μ-O)], and  [{(dppe)RuII(η1-OOCCPh)}2(μ-(OOCCPh))2(μ-O)] with selected atoms labelled. Thermal ellipsoids shown at 50% probability level, hydrogen atoms and solvent molecules omitted for clarity (full details see in section 3.0 in the ESI†). [{(dppe)RuII(η1-OOCCPh)}2(μ-(OOCCPh))2(μ-O)] with selected atoms labelled. Thermal ellipsoids shown at 50% probability level, hydrogen atoms and solvent molecules omitted for clarity (full details see in section 3.0 in the ESI†). | ||
All three structures of B′ were slightly distorted octahedral 18-electron RuII complexes where each metal featured two κ2 carboxylates and one η1 carboxylate in addition to a chelating dppe ligand (see also Table S8†).86,87 All three dimers were bridged by a water molecule as the sixth ligand to each metal as shown for  in Fig. 12, although for
in Fig. 12, although for 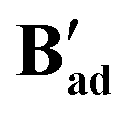 and
and  the corresponding water protons could not be located in the XRD refinement (but as with
the corresponding water protons could not be located in the XRD refinement (but as with 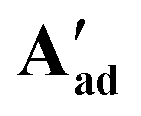 are likely present for reasons of charge balance). No major deviations in the Ru–O–Ru angles were observed (119.5–122.9°), and these values are concurrent with related water-bridged RuII dimers.86,88 Furthermore, inter-ligand hydrogen bonding between the bridging water molecule and pendant carboxylates was observed akin to the solid-state structures of A′ and the solution structures of B (see above). As with the corresponding A′ crystals, redissolving crystals of B′ gave solution phase NMR spectra of their symmetrical, mono-nuclear bis-carboxylate complexes A in all cases (Fig. S15–S21†). These various carboxylate complexes observed to form from [(dppe)Ru(MA)2] thus all relate to the symmetrical species A as an entry point to the catalytic cycle of the Ru-mediated MS-like transformation (Scheme 5).
are likely present for reasons of charge balance). No major deviations in the Ru–O–Ru angles were observed (119.5–122.9°), and these values are concurrent with related water-bridged RuII dimers.86,88 Furthermore, inter-ligand hydrogen bonding between the bridging water molecule and pendant carboxylates was observed akin to the solid-state structures of A′ and the solution structures of B (see above). As with the corresponding A′ crystals, redissolving crystals of B′ gave solution phase NMR spectra of their symmetrical, mono-nuclear bis-carboxylate complexes A in all cases (Fig. S15–S21†). These various carboxylate complexes observed to form from [(dppe)Ru(MA)2] thus all relate to the symmetrical species A as an entry point to the catalytic cycle of the Ru-mediated MS-like transformation (Scheme 5).
2.5 Operando 31P{1H} FlowNMR analysis
Monitoring the Ru-mediated transformation of ethynyl-β-ionol with the pivalic acid/anhydride mixture catalysed by 1 mol% [(dppe)Ru(MA)2] by 31P{1H} FlowNMR spectroscopy saw the precursor transform quantitatively into Ap over the first 30 minutes (Fig. S60†), with no other significant 31P{1H} signals detected under the conditions applied. As the reaction progressed over the following 44 hours, the concentration of Ap steadily fell to zero with three new peaks building up instead: a sharp singlet at 32.8 ppm which was confirmed to be the bis-oxide of dppe (Fig. S61†), a broad resonance centred around 63 ppm (FWHM = 540 kHz, denoted X1p) (Fig. 13) and a transient singlet (Xp-transient) at 79.1 ppm. At the end of the reaction when Ap had completely vanished from the spectra X1p held ca. 65% of the initial Ru concentration alongside about 20% dppe-oxide (Fig. S60†).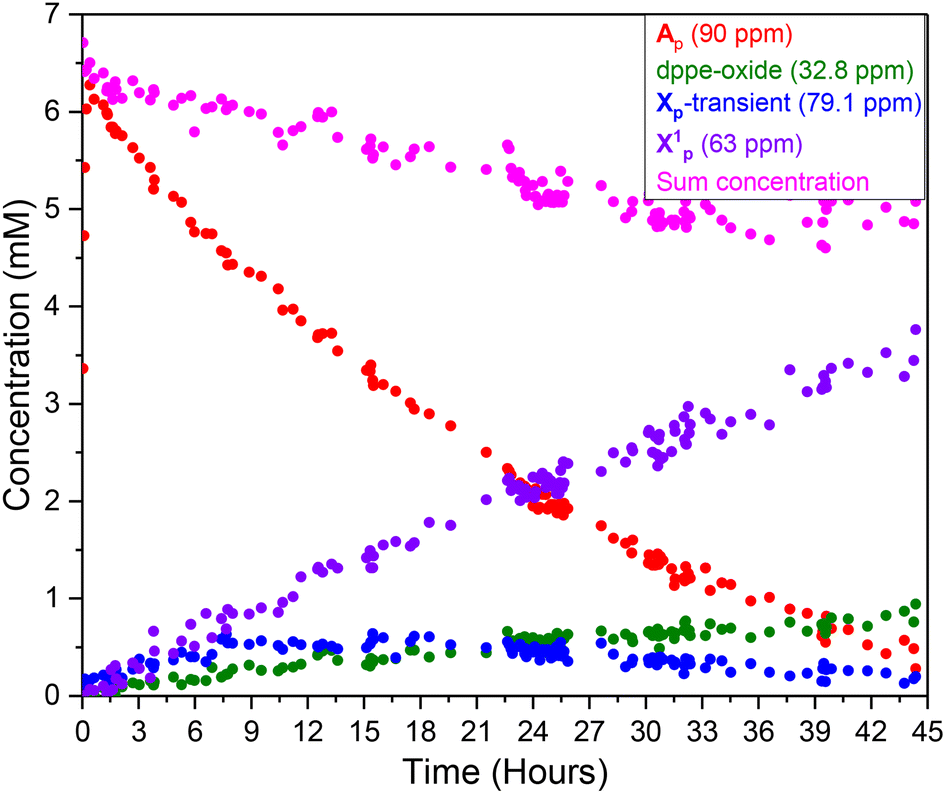 | ||
| Fig. 13 31P{1H} NMR concentration profile of the major quantifiable reaction species observed throughout the Ru-mediated transformation of ethynyl-β-ionol with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by 1 mol% [(dppe)Ru(MA)2] (0.66 mM) in anhydrous acetone (15 mL) at 20 °C from quantitative 31P{1H} FlowNMR spectroscopy at 4 mL min−1. | ||
Qualitatively, a similar behaviour of the consumption of A and formation of deactivation species was observed when the reaction was carried out in different solvents, at different catalyst loadings, when omitting the anhydride, or with phenylacetylene as the substrate (Fig. S62†). With other carboxylic acids, similar trends were also observed, with X1 as the dominant species at the end of the reaction. The 31P NMR chemical shifts of the deactivation species were slightly different, for instance, with adamantane carboxylic acid X1ad was observed around 72 ppm, while a more distinct deactivation species X2ad was found at 53.3 ppm (Fig. S63†). Irrespective of the reaction conditions, the trends in the operando31P{1H} FlowNMR spectra were always the same: the [(dppe)Ru(MA)2] precursor quickly activated to complex A (with no detectable amounts of B under anhydrous turnover conditions) which then steadily decayed over the course of the reaction, with about 20% of dppe-oxide forming and most of the remainder of the Ru/P material ending up in the deactivation species X1 and X2.
While initial tests showed [(dppe)Ru(MA)2] not to be particularly air-sensitive, solutions of A were found to be susceptible to oxidation when exposed to air (Fig. S64†). Given that air-free conditions were employed in our experiments, the consistent detection of small amounts of dppe-oxide suggests its formation to be either due to trace amounts of residual O2 or oxygen exchange reactions involving either the solvent or, more likely, the carboxylic acids used in the reaction. Although we have not been able to find similar precedence for ruthenium, palladium(II) complexes are well known to oxygenate phosphines with carboxylates.89–91 Adding dppe-oxide to a reaction with [(dppe)Ru(MA)2] from the start showed no change to the initial rate of the catalysis or the overall product yield (Fig. S65†). When dppe was added to a reaction with [(dppe)Ru(MA)2] from the start no product formation was observed at all, instead leading to the formation of a new, catalytically inactive species with two mutually coupling triplets at 57.2 and 54.0 ppm (2JPP = 18.2 Hz) in the 31P{1H} NMR spectra (Fig. S66†) indicative of the formation of cationic [RuII(η2-OOCR)(dppe)2]+ complexes.92 This observation likely stems from the strongly coordinating nature of dppe which preferentially binds to the ruthenium centre blocking substrate coordination.
In an attempt to isolate and characterise some Ru-containing deactivation species from the catalysis, a post-reaction mixture predominantly showing X2b at 53.3 ppm in the 31P{1H} NMR spectrum (Fig. S25†) was concentrated and layered with hexane resulting in crystallisation of a mono-carbonyl aqua complex (X2b·H2O, Fig. 14 left).
 | ||
| Fig. 14 X-ray crystal structures of [(dppe)RuII(η1-OOCPh)2(CO)(H2O)] and [(dppe)RuII(η1-OOCPh)2(CO)2] with selected atoms labelled. Thermal ellipsoids shown at 50% probability level, hydrogen atoms and solvent molecules omitted for clarity (full details see Table S9†). | ||
Independent synthesis of X2b was attempted by bubbling carbon monoxide through an acetone solution of a mixture of Ab and Bb at room temperature. Once the colour of the solution had changed from yellow to colourless over the course of 18 hours, 31P{1H} NMR spectroscopic analysis showed exclusive formation of the singlet at 53.3 ppm observed in the post-catalytic reaction mixtures (for full characterisation see Fig. S26–S29†). However, the NMR data was inconsistent with the solid-state structure of X2b·H2O, where two inequivalent 31P resonances would be expected. Therefore, crystals were grown from the synthesised sample which revealed a symmetrical bis-carbonyl structure (Fig. 14 right) consistent with the single 31P resonance observed in solution. The solution structure of [(dppe)Ru(η1-OOCPh)2(CO)2] was further confirmed by the use of 13CO (Fig. S67 and S68†) with coupling simulations (Fig. S69 and S70†) as well as IR spectroscopy showing two characteristic carbonyl bands at 2043 and 1996 cm−1 (Fig. S30†).93 These data unambiguously assigned the bis-carbonyl complex to the 31P{1H} singlet of X2b at 53.3 ppm in acetone, and the mono-carbonyl aqua complex X2b·H2O isolated from a post-reaction mixture must thus have been a minor side product present below the NMR detection limit. When tested for catalytic activity, X2b gave no product formation in the Ru-mediated transformation of ethynyl-β-ionol with benzoic acid (0.5 mol%) in acetone at room temperature after one week (Fig. S71 and S72†). Thus, the formation of such RuII carbonyl complexes, possibly arising from small amounts of aldehydes generated in situ (Fig. S73†), contributes to catalyst deactivation in this Ru-mediated transformation. The formation of [(PPh3)3Ru(H)(Cl)(CO)] from [(PPh3)3RuCl2] refluxed in methanol with formaldehyde is known,94 and carbonyl complexes are often formed during alcohol dehydrogenation reactions.95,96
2.6 Mechanistic relevance of [(dppe)RuII(η2-OOCR)2] (A)
When the steady decrease of A observed in the 31P{1H} FlowNMR data was compared to the reaction progress from the 1H FlowNMR data it became clear that the end of catalytic activity coincided with the complete decay of A (Fig. 15 and S74†). At the beginning of the reaction product formation began immediately as soon as any amount of A had formed from the [(dppe)Ru(MA)2] precursor, suggesting A to be either an in-cycle intermediate or an on-cycle reservoir of catalytically active material. | ||
| Fig. 15 Product yield profile (sum of Z and E forms of 3ab) versus the amount of complex A during the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with pivalic acid (0.73 M) and pivalic anhydride (0.13 M), catalysed by [(dppe)Ru(MA)2] at 0.5 mol% in anhydrous acetone (15 mL) at 20 °C from quantitative 1H and 31P{1H} FlowNMR spectroscopy at 4 mL min−1. Gap between 18–21 hours due to acquiring quantitative calibration spectra. | ||
Correlating the rates of decay of A with their initial concentrations from experiments at different catalyst loadings (Fig. 5 and S75†) showed their decline during the catalysis in steady-state to be ruthenium-dependent (Fig. 16). Applying VTNA to the normalised data revealed catalyst deactivation (as the rate of [A] decaying) to be half order in [A] (Fig. 16 and S76†).¶
 | ||
| Fig. 16 Decay profiles of Ap during the experiments shown in Fig. 5 (left) and time-adjusted data for a decay order in [Ap] = 0.5 (right). | ||
Given that all evidence suggests A to be monomeric in solution, the half order in its rate of decay implies a mechanism that involves its rapid, reversible fragmentation into two components, such as liberation of a carboxylate or de-coordination of the dppe ligand in small amounts. Correlating the decline of A with the concentration of ethynyl-β-ionol by VTNA showed a first-order dependence in the concentration of substrate (Fig. 17 and S77†) as also indicated by the same excess experiments (Fig. 7). Correlating the decline of A with the concentration of carboxylic acid showed a 0th dependence in concentration of carboxylic acid (Fig. S78 and S79†).
 | ||
| Fig. 17 Left: concentration profiles of Ap during the Ru-mediated transformation of ethynyl-β-ionol at different loadings (0.33 M, 0.66 M, 1 M) with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by [(dppe)Ru(MA)2] (6.6 6mM) in anhydrous acetone (15 mL) at 20 °C from quantitative 31P{1H} FlowNMR spectroscopy at 4 mL min−1. Right: Time-adjusted decay profiles for a reaction order of [S] = 1. | ||
The global kinetics of the reaction including catalyst deactivation thus are:
| Rate = kcat[S]1 − kd[S]1 |
| with kcat = [A]1, kd = [A]0.5 |
| where [A]initial = [Ru]initial |
 | ||
| Fig. 18 VTNA plot for the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by various amounts of [(dppe)Ru(MA)2] in anhydrous acetone (15 mL) to form 3ab, adjusted for experimentally observed catalyst deactivation (Fig. 6). | ||
To analyse whether the nature of the substrate had an influence on kd as it had on kcat (see above) we correlated both rates for the two substrates ethynyl-β-ionol and phenylacetylene (Fig. 19). The fact that each were linear with identical slopes but distinct intercepts suggested the same mechanism to be operational for the two substrates,97,98 with more pronounced deactivation in the case of ethynyl-β-ionol that was not simply due to faster turnover.
 | ||
| Fig. 19 Initial rates of product formation from 1H FlowNMR data plotted against initial rates of catalyst deactivation from 31P{1H} FlowNMR data for the Ru-mediated transformation of phenylacetylene and ethynyl-β-ionol. Reactions were carried out with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by 1 mol% [(dppe)Ru(MA)2] using a substrate concentration of 0.66 M in anhydrous acetone (15 mL) at 20 °C. | ||
Comparing the stability of A across different carboxylic acids under otherwise identical conditions, it was observed that a more sterically demanding acid resulted in slower decays of A during the catalysis (Fig. 20). The influence was significant, with adamantane carboxylic acid slowing the rate of catalyst deactivation by a factor of 15 compared to acetic acid, but without reducing the rate of product formation.
 | ||
| Fig. 20 Rate decay of A obtained from 31P{1H} FlowNMR data of the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with different carboxylic acids (1 M) catalysed by 1 mol% [(dppe)Ru(MA)2] in anhydrous acetone (15 mL) at room temperature. | ||
With A being involved in the productive cycle that is first order in substrate but zero order in acid, the binding of substrate to A can be identified as the TLS (Scheme 6). This must coincide with either the dissociation of a carboxylate, leading to a cationic ruthenium species, or the rearrangement of a carboxylate from η2 to η1 coordination, maintaining a neutral complex. Given large excess of acid present in solution we consider the neutral pathway more likely. The resulting alkyne complex II then quickly rearranges the bound alkynol and couples it with one of the η1 carboxylates (presumably via a vinylidene intermediate III)30,47,99 to eliminate the product after association of another carboxylic acid to IV to reform I (i.e.A).
 | ||
| Scheme 6 Proposed catalytic cycle with reversible substrate binding to intermediate I (= complex A) identified as the turnover-limiting step (TLS) consistent with the experimentally observed reaction kinetics from multi-nuclear FlowNMR spectroscopy. | ||
Based on the kinetic data obtained, A appears to be the bifurcation point that also leads into catalyst deactivation by substrate-induced carboxylate loss which generates coordinatively unsaturated species that engage in irreversible and unproductive side reactions that ultimately lead to inactive carbonyl complexes and dppe-oxide. This mechanistic picture aligns with the observed influences of the substrate and the acid on deactivation, where more strongly binding substrates lead to faster turnover but also more pronounced deactivation, and sterically more demanding acids decrease deactivation by protecting temporarily formed low-coordination species without slowing down turnover.
2.7 Maximising catalyst productivity
With ligand and substrate effects tuned to a reasonable level, we investigated whether higher reaction temperatures may allow for increased TONs by outpacing catalyst deactivation. Reaction progress and catalyst deactivation profiles were compared at 20, 40 and 60 °C using 0.5 mol% catalyst under optimised reaction conditions (Fig. 21). Increased reaction temperatures brought forth higher rates of product formation but sped up catalyst deactivation even more to yield lower TONs at higher temperatures, again suggesting a dissociative process being responsible for catalyst deactivation. | ||
| Fig. 21 Product formation from 1H FlowNMR data overlayed with decay of Ap from 31P{1H} FlowNMR data of the Ru-mediated transformation of ethynyl-β-ionol (0.66 M) with pivalic acid (0.73 M) and pivalic anhydride (0.13 M) catalysed by 0.5 mol% [(dppe)Ru(MA)2] in anhydrous acetone (15 mL) at different temperatures. | ||
Given the unfavourable temperature-dependency of the reaction we tried adjusting concentrations to maximise productivity. Based on the kinetics a combination of relatively high catalyst concentration, high substrate concentration, bulky carboxylic acids and low reaction temperature should provide the highest TON for the Ru-mediated transformation of ethynyl-β-ionol. This was explored in a series of reactions for pivalic (Table 6) and adamantane carboxylic acid (Table 7) due to the former being significantly cheaper and more soluble than the latter.
| Entry | Substrate (M) | Catalyst (mM) | Catalyst loading (%) | Acid (M) | Temperature (°C) | Yield of 3ab (%) | TON |
|---|---|---|---|---|---|---|---|
| 1 | 0.67 | 6.7 | 1 | 1.0 | 20 | 96 | 96 |
| 2 | 1.33 | 6.7 | 0.5 | 1.8 | 20 | 95 | 190 |
| 3 | 0.67 | 3.3 | 0.5 | 1.0 | 20 | 62 | 124 |
| 4 | 2.66 | 6.7 | 0.25 | 3.6 | 20 | 61 | 244 |
| 5 | 5.23 | 6.7 | 0.125 | 5.7 | 20 | 34 | 272 |
| 6 | 10.4 | 6.7 | 0.0625 | 11 | 20 | 18 | 280 |
| 7 | 5.23 | 6.7 | 0.125 | 5.23 | 40 | 17 | 136 |
| 8 | 5.23 | 6.7 | 0.125 | 5.23 | 0 | 38 | 300 |
| Entry | Substrate (M) | Catalyst (mM) | Catalyst loading (%) | Acid (M) | Temperature (°C) | Yield of 3ae (%) | TON |
|---|---|---|---|---|---|---|---|
| 1 | 0.667 | 6.67 | 1 | 1 | 20 | 99 | 99 |
| 2 | 1.333 | 6.67 | 0.5 | 1.8 | 20 | 99 | 198 |
| 3 | 2 | 6.67 | 0.375 | 2.5 | 20 | 99 | 297 |
| 4 | 1.333 | 6.67 | 0.1 | 1.8 | 20 | 25 | 250 |
| 5 | 2.666 | 3.34 | 0.25 | 3.6 | 20 | 71 | 284 |
| 6 | 5.23 | 6.67 | 0.125 | 5.7 | 20 | 58 | 462 |
| 7 | 5.23 | 3.34 | 0.0625 | 11 | 20 | 24 | 384 |
| 8 | 5.23 | 6.67 | 0.125 | 5.23 | 40 | 42 | 336 |
| 9 | 5.23 | 3.34 | 0.0625 | 5.5 | 40 | 13 | 214 |
| 10 | 5.23 | 3.34 | 0.125 | 5.5 | 0 | 15 | 245 |
Doubling the substrate concentration at unchanged catalyst concentration indeed increased the TON by a factor of 2 (entries 1 and 2). This is 1.5 times greater than what was observed when the substrate concentration was maintained and the catalyst concentration halved (entry 3), consistent with the finding that kcat is first order in [Ru] and kd is half order in [Ru]. Increasing the substrate concentration further resulted in even larger TONs of 280 (entries 4–6), albeit at lower conversion levels. Increasing the reaction temperature by 20 °C (entries 5 and 7) halved conversion and TON due to the pronounced influence of temperature on catalyst deactivation but lowering it by 20 °C only increased productivity slightly (entries 5 and 8). Nevertheless, these mechanism-guided modifications provided a more than three-fold improved catalyst productivity in the Ru-mediated transformation of ethynyl-β-ionol over literature procedures (entries 1 and 8).
Using the bulkier adamantane carboxylic acid instead of pivalic acid (Table 7), the catalyst reached TONs of almost 300 at full conversion simply by increasing the substrate concentration up to 2 M (entries 1–3) as expected from the observed influence of acid (Fig. 20). Increasing the substrate concentration further to 5.23 M resulted in a TON of 462 (entry 6), with lower conversion and noticeably more side products observed in the 1H NMR spectra (Fig. S83†). This was at least partially due to the system exceeding the solubility limit of adamantane carboxylic acid (∼2.5 M in dry acetone at room temperature) leading to a heterogeneous reaction mixture. Reducing the catalyst concentration at these high substrate and acid loadings provided slightly higher TONs (entry 7), again with lower conversions and side products due to over-saturation. Increasing the reaction temperature by 20 °C (which also increases the solubility of the acid) gave lower conversions and TONs (entries 8 and 9) due to accelerated catalyst deactivation as previously seen with pivalic acid. Lowering the reaction temperature by 20 °C (entry 10) provided worse performance as well, presumably due to even lower acid solubility.
Applying the best conditions identified to the minimally functionalised substrate phenylacetylene (where catalyst deactivation was observed to be slower) gave rise to TONs of >2000 (Table S10†), representing a 20-fold increase in catalyst productivity compared to literature. Based on the mechanistic insights derived from this study further improvements are likely possible with more extensive reaction engineering.
3 Conclusions
Our investigation of the ruthenium-mediated anti-Markovnikov addition of carboxylic acids to ethynyl-β-ionol using the so-far best catalyst [(dppe)Ru(MA)2] has unveiled mechanistic insights and optimisation strategies for enhancing catalyst productivity for industrial applications relevant to vitamin A synthesis. The use of high-resolution operando FlowNMR spectroscopy was instrumental in discerning the kinetic relevance of key reaction intermediates, some of which could be synthesised independently and analysed by single crystal XRD. A key finding is the identification of [(dppe)RuII(η2-RCOO)2] complexes (A) as crucial intermediates in the catalysis. These complexes coordinate the substrate as the turnover-limiting step of the cycle, and all subsequent steps leading to product formation must be rapid as no other reaction intermediates could be resolved by 31P{1H} Flow NMR spectroscopy at 500 MHz. Pronounced catalyst deactivation characterised by a half-order decay of A suggested a dissociative mechanism involving ligand loss that ultimately leads to inactive carbonyl complexes and phosphine oxide. The kinetics of the productive cycle show that higher substrate concentrations, the use of sterically demanding carboxylic acids (such as pivalic and adamantane carboxylic acid), and lower reaction temperatures improve catalytic turnover numbers (TONs). Optimisation of these parameters led to a significant increase in catalyst productivity, achieving TONs of 462 with ethynyl-β-ionol and >2000 with phenylacetylene. The insights obtained from this study likely extend to other substrates and related catalysts to enhance the utility of this atom-economic reaction in sustainable, large-scale production of not only vitamin A but also other fine chemicals for health care applications.Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Crystallographic details can be retrieved from the Cambridge Crystallographic Data Centre under deposition numbers 2296242–2296250.Conflicts of interest
RG, JM and BW are employees of DSM-Firmenich who are an industrial producer of vitamins. The other authors declare no conflicts of interest.Acknowledgements
This work was supported by the Royal Society (UF160458; fellowship to UH), the EPSRC (Dynamic Reaction Monitoring Facility at the University of Bath EP/P001475/1), and DSM-Firmenich (CASE studentship to A. S.). The authors would like to thank colleagues at DSM-Firmenich (Werner Bonrath, Claude Furer, Marcel Joray, Estel Canet-Martinez, Charalampos Panagos, Ralph Waechter and Helen Yeman) for their contributions to this project, as well as Andrew Weller (University of York) and Jordi Burés (University of Manchester) for helpful discussions.References
- D. N. D'Ambrosio, R. D. Clugston and W. S. Blaner, Nutrients, 2011, 3, 63–103 CrossRef.
- P. Trumbo, A. A. Yates, S. Schlicker and M. Poos, in Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc, The National Academies Press, Washington, DC, USA, 2001, p. 161 Search PubMed.
- E. Mayo-Wilson, A. Imdad, K. Herzer, M. Y. Yakoob and Z. A. Bhutta, BMJ, 2011, 343, 1–19 CrossRef.
- S. Akhtar, A. Ahmed, M. A. Randhawa, S. Atukorala, N. Arlappa, T. Ismail and Z. Ali, J. Health Popul. Nutr., 2014, 31, 413–423 Search PubMed.
- Maximize Market Research, Vitamin A Market – Global Industry Analysis and Trends (2023-2029), https://www.maximizemarketresearch.com/market-report/global-vitamin-a-market/121081/, (accessed 22 July 2023).
- PR Newswire, Vitamins - Global Strategic Business Report 2015-2020: DSM and BASF - Market Leaders in the Global Vitamins Market, https://www.prnewswire.com/news-releases/vitamins---global-strategic-business-report-2015-2020-dsm-and-basf---market-leaders-in-the-global-vitamins-market-300054205.html, (accessed 5 September 2021).
- G. L. Parker, L. K. Smith and I. R. Baxendale, Tetrahedron, 2016, 72, 1645–1652 CrossRef CAS.
- O. Isler, W. Huber, A. Ronco and M. Kofler, Helv. Chim. Acta, 1947, 30, 1911–1927 CrossRef CAS.
- O. Isler and R. Ruegg, US2798095A, 1957.
- J. F. Arens and D. A. Van Dorp, Nature, 1946, 157, 190–191 CrossRef CAS.
- D. A. Van Dorp and J. F. Arens, Nature, 1947, 160, 189 CrossRef CAS.
- R. H. Lindlar and R. Dubuis, US3715404A, 1973.
- O. Isler, H. Lindlar, M. Montavon, R. Rüegg and P. Zeller, Helv. Chim. Acta, 1956, 39, 249–259 CrossRef CAS.
- O. Isler, Pure Appl. Chem., 1979, 51, 447–462 CAS.
- M. Julia, US3781313A, 1973.
- W. Sarnecki and H. Pommer, DE1060386B, 1960.
- B. Wüstenberg, M.-A. Müller, J. Schütz, A. Wyss, G. Schiefer, G. Litta, M. John and W. Hähnlein, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2020, pp. 1–26 Search PubMed.
- W. Bonrath, B. Gao, P. Houston, T. McClymont, M.-A. Müller, C. Schäfer, C. Schweiggert, J. Schütz and J. A. Medlock, Org. Process Res. Dev., 2023, 27, 1557–1584 CrossRef CAS.
- M. Eggersdorfer, D. Laudert, U. Létinois, T. McClymont, J. Medlock, T. Netscher and W. Bonrath, Angew. Chem., Int. Ed., 2012, 51, 12960–12990 CrossRef CAS PubMed.
- F. Aquino, W. Bonrath, F. Pace, P. Ruckstuhl and K. Witzgall, WO2020025512A1, 2020.
- W. Bonrath, J. Schütz, T. Netscher and B. Wüstenberg, CH WO16059151, 2016.
- W. Bonrath, T. Netscher, J. Schütz and B. Wüstenberg, CH WO16059152, 2016.
- W. Bonrath, J. A. Medlock, B. Wüstenberg and J. Schütz, CH WO15004117, 2015.
- S. D. Barnicki, J. S. Kanel and K. W. Hampton, US8466328B2, 2013.
- S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429–438 CrossRef CAS.
- K. H. Meyer and K. Schuster, Ber. Dtsch. Chem. Ges., 1922, 55, 815–819 CrossRef.
- H. Rupe and H. Werdenberg, Helv. Chim. Acta, 1935, 18, 542–546 CrossRef CAS.
- D. A. Engel and G. B. Dudley, Org. Biomol. Chem., 2009, 7, 4149–4158 RSC.
- H. Doucet, B. Martin-Vaca, C. Bruneau and P. H. Dixneuf, J. Org. Chem., 1995, 60, 7247–7255 CrossRef CAS.
- M. Picquet, C. Bruneau and P. H. Dixneuf, Chem. Commun., 1997, 1201–1202 RSC.
- V. Cadierno, P. Crochet, S. E. García-Garrido and J. Gimeno, Dalton Trans., 2010, 39, 4015–4031 RSC.
- Y. Sugawara, W. Yamada, S. Yoshida, T. Ikeno and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902–12903 CrossRef CAS.
- S. Banerjee, S. B. Ambegave, R. D. Mule, B. Senthilkumar and N. T. Patil, Org. Lett., 2020, 22, 4792–4796 CrossRef CAS PubMed.
- B. M. Trost and C. K. Chung, J. Am. Chem. Soc., 2006, 128, 10358–10359 CrossRef CAS PubMed.
- C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311–323 CrossRef CAS.
- A. Odedra and R. S. Liu, in Metal Vinylidenes and Allenylidenes in Catalysis: From Reactivity to Applications in Synthesis, ed. C. Bruneau and P. H. Dixneuf, Wiley-VCH, Weinheim, 2008, pp. 193–216 Search PubMed.
- C. Bruneau and P. H. Dixneuf, in C-H Bond Activation and Catalytic Functionalisation I, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2015, pp. 137–188 Search PubMed.
- B. Li and P. H. Dixneuf, Organomet. Chem., 2014, 48, 119–194 CrossRef.
- Y. Yamamoto, T. Arakawa, R. Ogawa and K. Itoh, J. Am. Chem. Soc., 2003, 125, 12143–12160 CrossRef CAS.
- J. R. Johansson, T. Beke-Somfai, A. S. Stålsmeden and N. Kann, Chem. Rev., 2016, 116, 14726–14768 CrossRef CAS PubMed.
- S. Costin, N. P. Rath and E. B. Bauer, Adv. Synth. Catal., 2008, 350, 2414–2424 CrossRef CAS.
- J. Jeschke, M. Korb, T. Rüffer, C. Gäbler and H. Lang, Adv. Synth. Catal., 2015, 357, 4069–4081 CrossRef CAS.
- S. W. Roh, K. Choi and C. Lee, Chem. Rev., 2019, 119, 4293–4356 CrossRef CAS.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS.
- V. Cadierno, S. E. García-Garrido and J. Gimeno, Adv. Synth. Catal., 2006, 348, 101–110 CrossRef CAS.
- I. Emme, C. Bruneau, A. de Meijere and P. H. Dixneuf, Synlett, 2000, 2000, 1315–1317 CrossRef.
- M. Picquet, A. Fernández, C. Bruneau and P. H. Dixneuf, Eur. J. Org. Chem., 2000, 2000, 2361–2366 CrossRef.
- J. G. de Vries and S. D. Jackson, Catal. Sci. Technol., 2012, 2, 2009 RSC.
- A. B. de Haan, H. B. Eral and B. Schuur, Industrial Separation Processes: Fundamentals, 2nd edn, Walter de Gruyter GmbH, Berlin/Boston, 2020 Search PubMed.
- R. H. Crabtree, in The Organometallic Chemistry of the Transition Metals, 4th edn, John Wiley & Sons, Inc., Hoboken, NJ, 2005, vol. 1, pp. 235–273 Search PubMed.
- Q. L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352–5353 CrossRef CAS PubMed.
- D. A. Bruce, J. Am. Chem. Soc., 2009, 131, 4174–4175 CrossRef.
- K. Grabow and U. Bentrup, ACS Catal., 2014, 4, 2153–2164 CrossRef CAS.
- D. A. Foley, C. W. Doecke, J. Y. Buser, J. M. Merritt, L. Murphy, M. Kissane, S. G. Collins, A. R. Maguire and A. Kaerner, J. Org. Chem., 2011, 76, 9630–9640 CrossRef CAS PubMed.
- A. Ray, T. Bristow, C. Whitmore and J. Mosely, Mass Spectrom. Rev., 2018, 37, 565–579 CrossRef CAS PubMed.
- D. A. Foley, J. Wang, B. Maranzano, M. T. Zell, B. L. Marquez, Y. Xiang and G. L. Reid, Anal. Chem., 2013, 85, 8928–8932 CrossRef CAS PubMed.
- D. A. Foley, E. Bez, A. Codina, K. L. Colson, M. Fey, R. Krull, D. Piroli, M. T. Zell and B. L. Marquez, Anal. Chem., 2014, 86, 12008–12013 CrossRef CAS PubMed.
- A. M. R. Hall, J. C. Chouler, A. Codina, P. T. Gierth, J. P. Lowe and U. Hintermair, Catal. Sci. Technol., 2016, 6, 8406–8417 RSC.
- A. Bara-Estaún, C. L. Lyall, J. P. Lowe, P. G. Pringle, P. C. J. Kamer, R. Franke and U. Hintermair, Faraday Discuss., 2021, 229, 422–442 RSC.
- A. M. R. Hall, R. Broomfield-Tagg, M. Camilleri, D. R. Carbery, A. Codina, D. T. E. Whittaker, S. Coombes, J. P. Lowe and U. Hintermair, Chem. Commun., 2018, 54, 30–33 RSC.
- J. H. Vrijsen, I. A. Thomlinson, M. E. Levere, C. L. Lyall, M. G. Davidson and U. Hintermair, Polym. Chem., 2020, 11, 3546–3550 RSC.
- D. B. G. Berry, A. Codina, I. Clegg, C. L. Lyall, J. P. Lowe and U. Hintermair, Faraday Discuss., 2019, 220, 45–57 RSC.
- D. A. Foley, A. L. Dunn and M. T. Zell, Magn. Reson. Chem., 2016, 54, 451–456 CrossRef CAS PubMed.
- A. Saib, A. Bara-Estaún, O. J. Harper, D. B. G. Berry, I. A. Thomlinson, R. Broomfield-Tagg, J. P. Lowe, C. L. Lyall and U. Hintermair, React. Chem. Eng., 2021, 6, 1548–1573 RSC.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef.
- P. W. van Leeuwen, P. C. Kamer, J. N. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS PubMed.
- J. W. Raebiger, A. Miedaner, C. J. Curtis, S. M. Miller, O. P. Anderson and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 5502–5514 CrossRef CAS.
- J. A. Bilbrey, A. H. Kazez, J. Locklin and W. D. Allen, J. Comput. Chem., 2013, 34, 1189–1197 CrossRef CAS PubMed.
- T. Kégl, N. Pálinkás, L. Kollár and T. Kégl, Molecules, 2018, 23, 1–11 CrossRef PubMed.
- J. A. Bilbrey, A. H. Kazez, J. Locklin and W. D. Allen, J. Chem. Theory Comput., 2013, 9, 5734–5744 CrossRef CAS PubMed.
- P. Dierkes and P. W. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1530 RSC.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- N. Mathew, B. R. Jagirdar, R. S. Gopalan and G. U. Kulkarni, Organometallics, 2000, 19, 4506–4517 CrossRef CAS.
- Z. Zhiqing, X. Huiming, S. Chunshui, Q. Jiqiang, W. Zhongping and W. Jiyuan, CN104292087A, 2015.
- R. H. Crabtree, Chem. Rev., 2015, 115, 127–150 CrossRef CAS.
- F. Justaud, A. Hachem and R. Grée, Eur. J. Org. Chem., 2021, 514–542 CrossRef CAS.
- A. M. R. Hall, P. Dong, A. Codina, J. P. Lowe and U. Hintermair, ACS Catal., 2019, 9, 2079–2090 CrossRef CAS.
- T. Gensch, M. Teders and F. Glorius, J. Org. Chem., 2017, 82, 9154–9159 CrossRef CAS PubMed.
- D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 10852–10866 CrossRef CAS.
- Catalysis as it goes, Nat. Catal., 2018, 1, 165–166 Search PubMed.
- P. E. Hansen and J. Spanget-Larsen, Molecules, 2017, 22, 550–571 CrossRef.
- M. N. C. Zarycz and C. Fonseca Guerra, J. Phys. Chem. Lett., 2018, 9, 3720–3724 CrossRef CAS.
- T. M. P. Pagoto, L. L. G. Sobrinho, A. E. Graminha, A. P. M. Guedes, M. C. Carroccia, P. F. de Oliveira, E. P. Silveira-Lacerda, V. M. Deflon, D. C. Tavares, M. Pivatto, A. A. Batista and G. Von Poelhsitz, C. R. Chim., 2015, 18, 1313–1319 CrossRef CAS.
- X. L. Lu, S. Y. Ng, J. J. Vittal, G. K. Tan, L. Y. Goh and T. S. A. Hor, J. Organomet. Chem., 2003, 688, 100–111 CrossRef CAS.
- C. Grünwald, M. Laubender, J. Wolf and H. Werner, J. Chem. Soc., Dalton Trans., 1998, 833–840 RSC.
- R. W. Mitchell, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 846–854 RSC.
- B. Heiser, E. A. Broger and Y. Crameri, Tetrahedron: Asymmetry, 1991, 2, 51–62 CrossRef CAS.
- C. Amatore, A. Jutand, F. Khalil, M. A. M'Barki and L. Mottier, Organometallics, 1993, 12, 3168–3178 CrossRef CAS.
- C. Amatore, G. Le Duc and A. Jutand, Eur. J. Chem., 2013, 19, 10082–10093 CrossRef CAS.
- C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS.
- I. W. Wyman, T. J. Burchell, K. N. Robertson, T. S. Cameron and M. A. S. Aquino, Organometallics, 2004, 23, 5353–5364 CrossRef CAS.
- M. S. Quinby and R. D. Feltham, Inorg. Chem., 1972, 11, 2468–2476 CrossRef CAS.
- N. Ahmad, J. J. Levision, S. D. Robinson, M. F. Uttley, E. R. Wonchoba and G. W. Parshall, Inorg. Synth., 1974, 15, 45–64 CrossRef CAS.
- D. G. Gusev, Organometallics, 2020, 39, 258–270 CrossRef CAS.
- D. H. Nguyen, X. Trivelli, F. Capet, Y. Swesi, A. Favre-Réguillon, L. Vanoye, F. Dumeignil and R. M. Gauvin, ACS Catal., 2018, 8, 4719–4734 CrossRef CAS.
- C. Alamillo-Ferrer, G. Hutchinson and J. Burés, Nat. Rev. Chem., 2022, 7, 26–34 CrossRef.
- A. Martínez-Carrión, M. G. Howlett, C. Alamillo-Ferrer, A. D. Clayton, R. A. Bourne, A. Codina, A. Vidal-Ferran, R. W. Adams and J. Burés, Angew. Chem., Int. Ed., 2019, 58, 10189–10193 CrossRef.
- J. A. Varela, C. González-Rodríguez and C. Saá, in Ruthenium in Catalysis, ed. P. H. Dixneuf and C. Bruneau, Springer, Heidelberg, New York, Dordrecht, London, 2014, pp. 237–287 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2296242–2296250. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cy01032a |
| ‡ Systematic variation of multiple reaction parameters at once, one of the key appeals of VTNA, is unfortunately not possible in systems with pronounced inhibition or deactivation.58 |
| § Consistently, no activity of [(dppe)Ru(MA)2] in the catalytic transformation of alkynols was observed when using acetonitrile as the solvent. |
| ¶ The slight deviation of the 2 mol% data towards the end of the reaction is caused by a non-zero amount of A left at full conversion (i.e. incomplete catalyst deactivation). |
| This journal is © The Royal Society of Chemistry 2025 |