
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the impact of abnormal coordination in macrocyclic N-heterocyclic carbene ligands on bio-inspired iron epoxidation catalysis†
Greta G.
Zámbó
 a,
Carla A.
Esslinger
a,
Michael J.
Sauer
a,
Isabelle
Rüter
b,
Robert M.
Reich
a,
Serhiy
Demeshko
b,
Franc
Meyer
b and
Fritz E.
Kühn
*a
a,
Carla A.
Esslinger
a,
Michael J.
Sauer
a,
Isabelle
Rüter
b,
Robert M.
Reich
a,
Serhiy
Demeshko
b,
Franc
Meyer
b and
Fritz E.
Kühn
*a
aSchool of Natural Sciences, Department of Chemistry and Catalysis Research Centre, Molecular Catalysis, Technical University of Munich, Lichtenbergstraße 4, 85748 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de
bGeorg-August-Universität Göttingen, Institut für Anorganische Chemie, Tammannstraße 4, 37077 Göttingen, Germany
First published on 12th September 2024
Abstract
The first macrocyclic abnormal N-heterocyclic carbene (aNHC) FeIII complex, featuring a calix[4]3-methyl-1,2,3-triazole-5-ylidene ligand system is synthesised and characterised inter alia via EA, SC-XRD, NMR and UV/vis spectroscopy. Including Mössbauer spectroscopy, SQUID and DFT calculations, the impact of the aNHC on the FeIII complex and its corresponding FeII derivative is investigated. A comprehensive study of the aNHC Fe complexes in their performance in homogenous epoxidation reactions is reported and compared to the established benchmark catalysts. The complexes demonstrate efficient and selective catalytic activity in the epoxidation of cis-cyclooctene with H2O2, with TOFs up to almost 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. Additionally, the epoxidation of more challenging olefinic substrates is possible. The reactivity under oxidative conditions of both complexes is investigated. NMR measurements reveal the formation of a mono-oxidised triazole ligand as degradation product. HR-ESI-MS measurements, supported by DFT calculations indicate the formation of an oxoiron species.
000 h−1. Additionally, the epoxidation of more challenging olefinic substrates is possible. The reactivity under oxidative conditions of both complexes is investigated. NMR measurements reveal the formation of a mono-oxidised triazole ligand as degradation product. HR-ESI-MS measurements, supported by DFT calculations indicate the formation of an oxoiron species.
Introduction
Inspired by nature, organometallic iron N-heterocyclic carbene (NHC) systems have gained attention as attractive alternatives to established catalyst systems due to their activity in oxidation reactions.1–4 Following the model of cytochrome P450, various non-heme iron complexes have been designed to mimic its ability to undergo fundamental chemical transformations such as the oxidation of hydrocarbons and oxygen transport.1 This includes the epoxidation of olefins, which represents an essential approach for the modification of organic intermediates in the production of various fine and bulk chemicals.5 In this context, the investigation of the activity of macrocyclic tetradentate iron complexes in the epoxidation of alkenes has evoked interest.6 Previously, focus was laid primarily on examining the mechanisms of corresponding porphyrin-like systems featuring exclusively N-donor ligands. High valent iron intermediates of oxidation state IV or V are considered to be the active species in these reactions.1,7–12 However, most catalytic reactions are starting from iron(II) pre-catalysts, which requires a pre-oxidation to form an active iron(III) hydroperoxo species, usually involving an undesired Fenton-type radical step.2,13–16 The radical formation can affect catalyst activity, represented by the turnover frequency (TOF) and stability, represented by the turnover number (TON).17 Even if the results on mechanistic studies of iron NHC complexes in epoxidation catalysis are limited,2 NHC ligands are expected to be superior due to their favourable electronic properties as strong σ-donors possessing high kinetic stability.4,18,19 The current benchmark catalyst for homogenous olefin epoxidation is an iron(II) tetracarbene complex with a ligand system consisting of four imidazole-2-ylidene units connected via methylene bridges and two labile acetonitrile ligands in trans positions to each other (Fig. 1, A).20,21 Complex A reaches a TOF of ca. 50000 h−1 with a TON of 480. The unfavourable Fenton-type reactivity in the oxidation reaction could be suppressed applying its iron(III) homologue B (Fig. 1) as catalyst by increasing the TOF to > 180000 h−1.22 Adding Lewis acids like Sc(OTf)3 to the catalytic reaction increases activity and stability of the catalyst significantly (TOF > 400000 h−1, TON ≈ 1200).20 These results outperform previous established epoxidation catalysts based on rhenium (TOFs up to ca. 40000 h−1)5 or molybdenum (TOFs up to ca. 50000 h−1).23 However, for application of iron NHC complexes in industrial catalysis, the stability has to be considerably enhanced without compromising the remarkable activity. The electronic properties of A and B can be adjusted by introducing substituents into the NHC backbone, impacting both their respective TON and TOF, with a lag in activity compared to A and B.24
 | ||
| Fig. 1 Chemical structure of the precursor of the most active catalyst FeII[cCCCC]imA in the epoxidation of cis-cyclooctene with H2O2 and its FeIII[cCCCC]im derivative B and the structure of the abnormal coordinating iron NHC complexes FeII[cCCCC]trz1 and FeIII[cCCCC]trz2 (this work). | ||
Abnormal NHCs (aNHCs) are considered to be stronger σ-donors than normal coordinating imidazole-2-ylidene units as in complexes A and B.25,26 Introducing aNHCs as ligands in transition metal catalysis has proven to be effective in adjusting the electronic characteristics of the central metal and enhancing its catalytic performance. Among these, 1,2,3-triazol-5-ylidenes turned out as a promising aNHC subclass due to their straightforward synthesis via click chemistry.26–29 In order to deepen the understanding of the influence of the NHC σ-donor strength on the iron catalyst performance in oxidation reactions, our group recently designed a ligand which is geometrically similar to the calix[4]imidazol in complex A and B. However, the designed ligand differs in its NHC units and mode of coordination with the metal, changing from a normal NHC to an aNHC.30 To date only one macrocyclic abnormal coordinating iron NHC complex is reported in literature (Fig. 1, 1).30 Complex 1 represents a promising candidate for olefin epoxidation30 due the high donor abilities of its tetra NHC ligand consisting of 1,2,3-triazole-5-ylidene moieties.26–29 First epoxidation reactions of cis-cyclooctene with H2O2 applying 1 as pre-catalyst reach conversions up to 97% with a TOF of 41000 h−1 and a TON of 200.30
In this work, on the one hand, the synthesis and characterisation of the iron(III) homologue 2 (Fig. 1) of the first macrocyclic aNHC iron complex 1 is reported. The new compound 2 and if applicable, 1, are characterised using NMR spectroscopy, high-resolution electrospray ionisation mass spectroscopy (HR-ESI-MS), single crystal X-ray diffraction (SC-XRD), UV/VIS spectroscopy, Mössbauer spectroscopy, elemental analysis (EA) and multiple DFT calculations. On the other hand, catalytic in-depth studies using complex 1 and 2 in the epoxidation of olefins and the comparison to the so far most active catalyst allow to gain valuable information for effective ligand design. Additional DFT calculations regarding the modification of the triazole ligand backbone provide further information about the theoretical influence of the aNHC electrons on the iron centre.
Results and discussion
Synthesis and characterisation of aNHC FeII and FeIII complexes 1 and 2
Thianthrenyl hexafluorophosphate proved to be an efficient one-electron oxidant to oxidise iron(II) complexes to their iron(III) derivatives selectively and in high yields.22,24,31 Complex 2 is synthesised by an outer-sphere one-electron oxidation of 1.00 eq. of the previously reported aNHC iron complex 1 with 1.00 eq. thianthrenyl hexafluorophosphate (Scheme 1). 2 can be isolated in 96% yield as yellow solid. | ||
| Scheme 1 Synthesis of iron(III) complex 2via the reaction of iron(II) complex 1 with thianthrenyl hexafluorophosphate as one-electron oxidant. | ||
The paramagnetism of complex 2 poses challenges for the NMR spectroscopy analysis and complicates the comparison with 1. The 1H-NMR spectrum shows a significant low field shift of the methylene bridge protons from 6.12 ppm for 1 to 44.33 ppm for 2, confirming its paramagnetism (see ESI,† Fig. S1). Whereas the signal for the methyl substituent protons with 4.31 ppm is barley shifted (vs. 4.21 ppm for 1).
Single crystals of 2 suitable for SC-XRD were obtained by the slow diffusion of diethyl ether into a solution of 2 in acetonitrile under an argon atmosphere. In general, the crystal structure of 2 differs only marginally from that of complex 1.30 Similar to its iron(II) counterpart, complex 2 displays a distorted octahedral coordination sphere around the iron centre (Fig. 2). Axial positions are occupied by two acetonitrile ligands, while the tetradentate ligand is coordinating equatorially. The bond angles around the iron centre in 1 and 2 are very close to the ideal octahedral angle with a mean deviation of 0.50° for 1 and 0.29° for 2. Varying from the geometry of previous characterised macrocyclic tetracarbene iron complexes with imidazole moieties in a saddle-distorted conformation,3 the aNHC ligand exhibits an ideal square-planar geometry. Selected structural parameters of complexes A, B, 1 and 2 are summarised in Table 1. The Fe–CNHC distances of 2 with 1.950(2) and 1.943(3) Å are slightly longer than measured for 1 with 1.925(2) and 1.931(2) Å.30 In contrast to the comparison of FeII[cCCCC]im complex A (ref. 21) with FeII[cCCCC]trz complex 1,302 and B (ref. 22) show similar bond Fe–CNHC length. The geometric parameters are in conformity with DFT calculated values in gas phase and solution of both structures 1 and 2 (see ESI,† Table S7). Removing one molecule of acetonitrile as labile ligand does not affect the geometry of the structure to a greater extent.
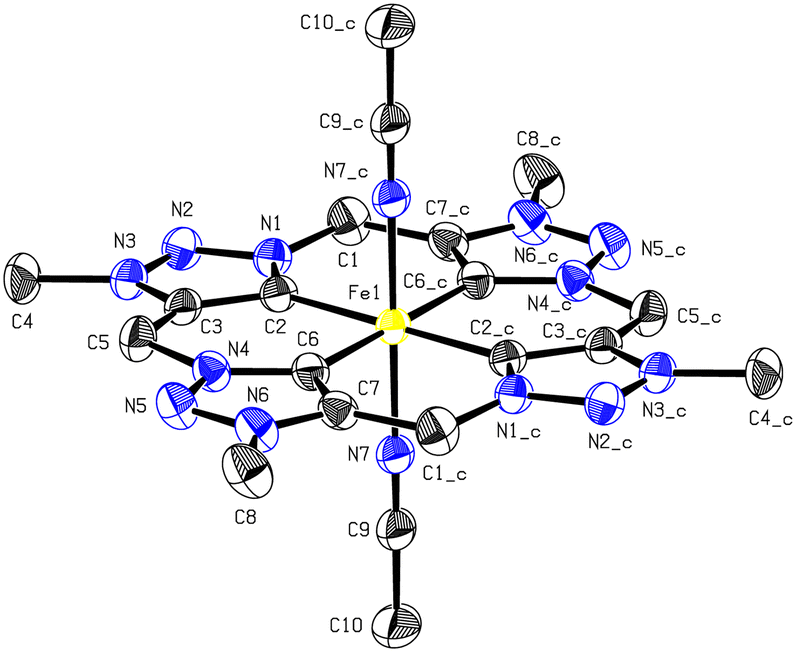 | ||
| Fig. 2 ORTEP-style representation of the cationic fragment of iron(III) calix[4]3-methyl-1,2,3-triazole-5-ylidene hexafluorophosphate 2. Hydrogen atoms and PF6− anions are omitted for clarity and thermal ellipsoids are shown at 30% probability level. | ||
| Bond lengths [Å] | Bond angles [°] | ||||
|---|---|---|---|---|---|
| Fe–CNHCa | Fe–NCMeb | MeCN–Fe–NCMe | CNHC–Fe–CNHCc | CNHC–Fe–NCMed | |
| a Average distance between iron centre and carbene carbon atom. b Average distance between iron centre and axial ligands. c Average angle between carbene and the carbene in trans position. d Average angle between carbene, iron centre and axial ligand. | |||||
| A | 1.907(3) | 1.932(2) | 177.09(10) | 178.93(67) | 90.00(66) |
| B | 1.941(2) | 1.922(8) | 179.04(9) | 179.09(8) | 90.00(23) |
| 1 | 1.928(2) | 1.9247(17) | 180.00 | 180.00 | 90.00(8) |
| 2 | 1.947(3) | 1.926(2) | 180.00 | 180.00 | 90.00(10) |
The electronic influence of the ligand in 1 and 2 is investigated via DFT by calculation of the electronic charge. Löwdin population analysis32 was applied for that reason. Although theoretically calculated charge values are not realistic charge distribution representations, relative trends can be examined for the comparison of similar molecules.33 Löwdin population analysis revealed a more negative value for the FeII centre 1 due to the lower oxidation state, however with only a difference of 0.32 atomic units compared to 2 (Table 2). Hence the additional positive charge of 2 is not solely located at the iron atom but compensated by the ligand system. This is also corroborated by the electrostatic potential (ESP) mapped total density plots (see ESI,† Fig. S26). As expected, the iron(II) complex features overall a more negative charge. The removal of one MeCN molecule increases the Löwdin charge to 0.33 and 0.39 less negative values for iron(II) and iron(III), respectively. This shows that not only the equatorial ligand scaffold influences the electronic characteristics, but also the axial labile ligands. Their substitution was reported to influence the catalytic activity of olefin epoxidation not only statistically by vacating and blocking a catalytic site but also from an electron density point of view.34
| Complex | 1 | 2 | 1-MeCN | 2-MeCN |
|---|---|---|---|---|
| Values are given in atomic units and were rounded to the third decimal digit. | ||||
| Löwdin | −1.793 | −1.475 | −1.399 | −1.142 |
To gain further information about the electronic properties of 1 and 2, solid material of the complexes was studied using 57Fe Mössbauer spectroscopy at 80 K. The respective spectra show a quadrupole doublet with a small isomer shift and a large quadrupole splitting (Fig. 3). The integer iron(II) spin system feature a sharp line in its quadrupole doublet. In contrast, a broadened asymmetric doublet was observed for iron(III) complex 2, which is in accordance with Mössbauer spectra of half-spin systems and can be ascribed to slow relaxation processes.35 The isomer shifts of both compounds with δ = 0.13 mm s−1 for 1 and δ = −0.01 mm s−1 for 2 are in the range of octahedral iron(II) (δ = −0.18–0.50 mm s−1, S = 0) respectively iron(III) low spin species (δ = −0.18–0.25 mm s−1, S = 1/2).3,36–38 Nevertheless, the isomer shift of 1 is slightly more positive than for A (ref. 39) (Table 3).
 | ||
| Fig. 3 Zero-field 57Fe Mössbauer spectrum of solid 1 and 2 at 80 K. The red line represents a simulation with δ = 0.13 mm s−1 and ΔEQ = 3.34 mm s−1 (1, top) and δ = −0.01 mm s−1 and ΔEQ = 2.12 mm s−1 (2, bottom). | ||
The Fe–ligand bond lengths can have a considerable influence on the isomer shift through compression of the s-orbitals resulting in a higher electron density.37 In both cases, oxidising the iron(II) complex to iron(III) moves the isomer shift to lower values, although the length of the Fe–carbene bond increases. This can be ascribed to the decreased number of d-electrons, so that less shielding of the core electron density is obeserved for the iron(III) species. The quadrupole splitting with |ΔEQ| = 3.34 mm s−1 for 1 and |ΔEQ| = 2.12 mm s−1 for 2 are rather large for low-spin complexes, but in line with iron tetra NHC complexes3,36,37 with a heteroleptic ligand coordination sphere (different equatorial donor atoms and axial ligands) and an axial elongated octahedral ligand field.40
The magnetic susceptibility of complexes 1 and 2 were analysed using a SQUID magnetometer (see ESI,† Fig. S14 and S15), confirming the diamagnetic (S = 0) ground state of 1 and the absence of any SCO in the temperature range from 2–400 K. This is in line with a strong ligand field induced by the aNHC ligand and with observations for all related six-coordinate tetra(NHC) ligated complexes including A.3 The SQUID measurement for 2 validate its S = 1/2 low-spin state with the absence of any SCO in the measured temperature area.
In view of occupancy, the molecular frontier orbitals of DFT calculated structures 1 and 2 were analysed (see ESI,† Fig. S27 and S28). The vacating of one catalytic site by MeCN dissociation does not notably affect the occupied orbitals of 1 (HOMO − 1 and HOMO, see ESI,† S29). Regarding the six-coordinate complex 1, the LUMO does not exhibit iron(II) participation, while the LUMO + 1 is of eg symmetry. This order of the unoccupied orbitals is reversed for the five-coordinate iron(II) species. A strong electronic influence also from the axial ligands is inferred.
The generation of an electrophilic oxygen species for catalytic epoxidation is expected to proceed via overlap of the HOMO of the incoming oxo species with the five-coordinate iron complex LUMO. The comparison of 1-MeCN and 2-MeCN show a fairly similar LUMO, asymmetrically distorted along the z-axis and with slightly more carbene participation in case of the iron(III) centre, attributed to the stabilisation of the additional positive charge. A direct comparison of the α-HOMOs of 2 with B show similar symmetry, taking into account the inherent asymmetry of the triazole rings in comparison to imidazole rings.
Reactivity of aNHC FeII and FeIII complexes 1 and 2 with H2O2
UV/Vis spectroscopy of complex 1 (ref. 30) and 2 was performed in dry and degassed acetonitrile at 20 °C under an argon atmosphere to maintain anhydrous and oxygen-free conditions. Handling the measurements under these inert conditions is necessary due to the sensitivity of both the catalyst precursor and the active catalyst to atmospheric moisture and oxygen. Two absorption bands and an absorption shoulder around 337 nm are visible for complex 2 (Fig. 4). The major band seems to be consisting of two bands in close proximity, appearing at a wavelength of 441 nm. The second band is observed at 302 nm. In order to gain information about the formation of the high valent active catalytic species in the oxidative catalysis with FeIII[cCCCC]trz, 1.50 eq. of the oxidising agent H2O2 was added to the solution of 2. The UV/vis spectrum shows the disappearance of the two absorption bands by generating a shoulder around 378 nm and a second one around 450 nm.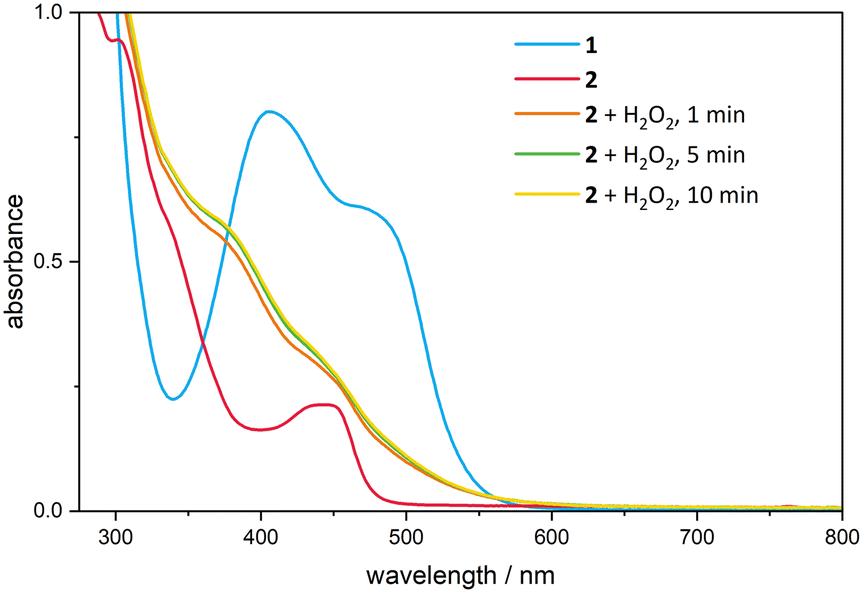 | ||
| Fig. 4 UV/Vis spectrum of 1 (blue) and 2 (red); c = 0.20 mM, T = 20 °C in MeCN with the addition of 1.50 eq. H2O2 to 2 with a reaction time of 1 min (orange), 5 min (green) and 10 min (yellow) in the absence of substrate. | ||
Decomposition studies of the iron epoxidation catalyst A, respectively B identify the Fe-NHC bond as weak spot. Direct oxidation of one of the carbenes results in metal de-coordination, which induces protonation of the remaining NHC moieties.41 The addition of 10.0 eq. H2O2 (50% aq.) to a solution of iron(III) complex 2 in dry and degassed acetonitrile under argon is investigated further with 1H-NMR spectroscopy. After 21 h, selective formation of another species, is detectable. In addition to the paramagnetic complex broad singlet at 4.34 ppm, three singlets occur at 8.85, 8.69 and 8.42 ppm, each with an integral of 1, which are assigned to protons in carbene positions of the diamagnetic ligand (see ESI,† Fig. S2). Additionally, four singlets occur at 6.12, 6.09, 5.75 and 5.49 ppm, each with an integral of 2, which are attributed to the protons of the ligand bridges. The singlet of the methyl substituents splits into four singlets at 4.31, 4.28, 4.25 and 3.92 ppm caused by asymmetric effects of the resulting species. This is in accordance with the disappearance of the two absorption bands in the UV/Vis experiment, indicating a direct oxidation of the complex ligand by H2O2, resulting in the formation of a mono-oxidised and three-fold protonated ligand, which is consistent with the observations regarding the degradation of B under oxidative conditions.41
Decomposition studies of catalyst 1via HR-ESI-MS reveal the formation of a mono-deprotonated FeIII complex (m/z = 217.56) and an oxidised iron compound (m/z = 225.56) after adding 10 eq. H2O2 to a solution of complex 1 in dry, degassed acetonitrile (see ESI,† Fig. S16). Even under ambient conditions without adding an oxidant, an oxidised iron compound (m/z = 226.06) was detected, in addition to the main signal for unreacted complex 1 (m/z = 218.06; see ESI,† Fig. S17). According to mechanistic studies regarding iron epoxidation catalysts with trans labile coordinating sites by Que et al.,42 the reaction between the Fe complex and H2O2 initiates with the formation of a FeIII–OOH intermediate by a simple ligand exchange process after a crucial single electron transfer (Fenton reaction). Mechanistic studies indicate that this can lead to either a high-valent iron(V) oxo compound or an iron(IV) oxo species with an oxyl radical, depending on whether the cleavage pathway is homolytic or heterolytic. The heterolytic pathway is less favourable, as electron-deficient oxidants such as H2O2 cannot stabilise the resulting radical so that it cannot attack the oxygen source and the catalyst.11,42 The corresponding oxoiron species with the aNHC scaffold (aNHC–FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and aNHC–FeVO) are validated via DFT calculations (see ESI†). The HOMOs of both proposed species include the FeO bond of π* symmetry, making them capable of π-donation into the alkene π*–LUMO in order to break the double bond for the epoxide formation. In contrast, the LUMO of the oxoiron(IV) compound is entirely ligand centered while the iron(V) species exhibits a metal centered LUMO, rotated by 90° in comparison to the HOMO (see ESI,† S31 and S32).
O and aNHC–FeVO) are validated via DFT calculations (see ESI†). The HOMOs of both proposed species include the FeO bond of π* symmetry, making them capable of π-donation into the alkene π*–LUMO in order to break the double bond for the epoxide formation. In contrast, the LUMO of the oxoiron(IV) compound is entirely ligand centered while the iron(V) species exhibits a metal centered LUMO, rotated by 90° in comparison to the HOMO (see ESI,† S31 and S32).
Catalytic olefin epoxidation reactions
Complexes 1 and 2 were investigated with regard to their suitability as catalysts for the epoxidation of olefins, with a comparison to the most active non-heme iron complexes A, respectively B reported in literature.20 Established standard conditions for oxidation reactions with iron NHC catalysts comprise 20 °C, the use of H2O2 (101 μmol mL−1, 1.50 eq.) as oxidising agent, MeCN as solvent and cis-cyclooctene (67.3 μmol mL−1, 1.00 eq.), widely applied as model substrate.2 Hydrogen peroxide was chosen as the oxidising agent due to its superiority over other common peroxides such as tert-butyl hydroperoxide (TBHP) or the urea-hydrogen peroxide adduct in iron-NHC epoxidation catalysis.22,43 Previous optimising studies revealed that the best results were achieved with an excess of 1.50 eq. of H2O2.22,43 Product quantification was performed using GC-FID for the model substrate (cis-cyclooctene) and 1H NMR spectroscopy for the screening of various other olefinic substrates.Initial evaluation employing the abnormally coordinated FeII macrocycle 1 demonstrate the remarkable difference in activity and stability of this complex class in comparison to the iron catalyst A with normal NHC coordination.22,30 At 20 °C different catalyst concentrations of FeII pre-catalyst 1 are screened (Fig. 5a). A nearly linear relationship between catalyst loading and conversion is expected if no degradation products22,41,44 prevent catalytic activity. Nevertheless, this trend remains imperceptible in this case (Table 4, entry 1–4). At 0.1 mol% a maximal conversion of 6% (TON = 60) and at 0.5 mol% of 37% (TON = 74) has been detected. However, if the catalyst loading is doubled to 1 mol%, the maximal conversion only increases to 49% (TON = 49). At a concentration of 2 mol% of 1 a conversion of 84% (TON = 42) is reached after 5 min. These results indicate a fast degeneration, possibly promoted by higher catalyst concentrations. A common method to increase the catalyst lifetime is to reduce the reaction temperature.22,30,45 Hence, at a catalyst loading of 1 mol% of 1 the conversion and catalyst stability increases from 84% (TON = 80) for 10 °C over 92% (TON = 92) for 0 °C to full conversion (TON = 100) for −10 °C after 10 min with raising the stability respectively (Fig. 5b and Table 4, entry 2, 5–7). Remarkably, unlike the constantly increasing TON, the TOF does not necessarily decrease with lower temperature and can be pushed up to 32100 h−1 at 0 °C (Table 4, entry 9). As stability is one of the biggest challenges of iron catalysts,22,34,43,46 including complexes with Fe–carbene bonds4,47,48 and many iron complexes decompose rather quickly after the addition of aqueous H2O2, influenced by the unavoidable presence of water,17,22,49 TBHP (101 μmol mL−1, 1.50 eq.) in n-decane solution is used in a single reaction with 1 (Fig. 5c). However, at standard conditions and a catalyst loading of 1 mol% a significantly smaller amount of epoxide is formed after 10 min with a TON of 22 (Table 4, entry 12), which is less than half as much when TBHP is substituted with aqueous H2O2. All reactions are carried out at atmospheric conditions by starting the reaction with the addition of the catalyst from a preformed stock solution in dry and degassed acetonitrile, which was handled under argon. All experiments achieve a selectivity of >99% towards the product epoxide.
 | ||
| Fig. 5 Time-dependent epoxidation of cis-cyclooctene (67.3 μmol mL−1, 1.00 eq.) in MeCN using 1 as catalyst and H2O2 or TBHP (101 μmol mL−1, 1.50 eq.) as oxidant, (a) at different catalyst loadings (20 °C, H2O2, air), (b) at various reaction temperatures (1 mol% of 1, H2O2, air), (c) with different oxidising agents (1 mol% of 1, 20 °C, air) and (d) at different ambient conditions (1 mol% of 1, 20 °C, H2O2). Conversions are determined by GC-FID. | ||
| Entry | Catalyst | T [°C] | Loading [mol%] | Additive | X [%] | S [%] | TOF [h−1] | TON | Oxidant | Atmosphere |
|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions: acetonitrile as solvent, cis-cyclooctene (67.3 μmol mL−1, 1.00 eq.), H2O2 or TBHP (101 μmol mL−1, 1.50 eq.), if stated Sc(OTf)3 (6.73 μmol mL−1, 0.10 eq.). Selectivity is related to the epoxide. Conversions are determined by GC-FID. TOFs are determined after 10 s. TONs are determined when maximal conversion is reached. T = temperature. X = conversion. S = selectivity, selectivity = yield(epoxide)/conversion(substrate). | ||||||||||
| 1 | 1 | 20 | 2 | — | 84 | >99 | 13000 |
42 | H2O2 | Air |
| 2 | 1 | 20 | 1 | — | 49 | >99 | 16600 |
49 | H2O2 | Air |
| 3 (ref. 30) | 1 | 20 | 0.5 | — | 37 | >99 | 24500 |
74 | H2O2 | Air |
| 4 | 1 | 20 | 0.1 | — | 6 | >99 | 20100 |
60 | H2O2 | Air |
| 5 | 1 | 10 | 1 | — | 84 | >99 | 26500 |
80 | H2O2 | Air |
| 6 | 1 | 0 | 1 | — | 92 | >99 | 22700 |
92 | H2O2 | Air |
| 7 | 1 | −10 | 1 | — | 100 | >99 | 25000 |
100 | H2O2 | Air |
| 8 (ref. 30) | 1 | 10 | 0.5 | — | 49 | >99 | 29900 |
99 | H2O2 | Air |
| 9 (ref. 30) | 1 | 0 | 0.5 | — | 72 | >99 | 32100 |
145 | H2O2 | Air |
| 10 (ref. 30) | 1 | −10 | 0.5 | — | 89 | >99 | 30200 |
177 | H2O2 | Air |
| 11 | 1 | −10 | 0.1 | — | 20 | >99 | 40800 |
202 | H2O2 | Air |
| 12 | 1 | 20 | 1 | — | 22 | >99 | 6800 | 22 | TBHP | Air |
| 13 | 1 | 20 | 1 | — | 46 | >99 | 14000 |
46 | H2O2 | Argon |
| 14 (ref. 30) | 1 | 20 | 0.5 | Sc(OTf)3 | 97 | 96 | 40900 |
194 | H2O2 | Air |
| 15 | 1 | 20 | 0.1 | Sc(OTf)3 | 25 | 97 |
59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 200 200
|
248 | H2O2 | Air |
| 16 | 2 | 20 | 0.5 | — | 70 | >99 | 44000 |
142 | H2O2 | Air |
| 17 | 2 | 20 | 0.5 | Sc(OTf)3 | 99 | 97 | 51800 |
197 | H2O2 | Air |
Regarding mechanistic studies towards non-heme iron epoxidation catalysts with trans labile coordinating sites (vide supra) the impact of atmospheric conditions on the catalysis can significantly affect the reaction outcome. The presence of oxygen under aerobic conditions enhances the formation of epoxide products via radical pathways, by suppressing diol product formation, as oxygen traps the radical intermediates. Under argon, the reaction is slower and yields less epoxides, demonstrating the catalysts sensitivity to oxygen.11,42 To investigate the influence of atmospheric conditions on 1 a catalytic reaction was handled completely under an argon atmosphere with a catalyst loading of 1 mol% (Fig. 5d). The TON with 46 vs. 49 and the maximal conversion is comparable to the reaction under atmospheric conditions with a high selectivity (S > 99%) for both reactions (Table 4, entry 2 and 12). The comparable performance under both conditions suggests a mechanism less dependent on radical intermediates and a higher iron stabilisation due to the macrocyclic aNHC ligand system.
The formation of an iron(III) hydroperoxo intermediate with oxidising agents such as hydrogen peroxide after the one-electron oxidation of the iron(II) catalyst precursor appears to be essential for the formation of the active species.2,11,42,50 Applying the abnormally coordinated FeIII macrocycle 2 (0.34 μmol mL−1, 0.005 eq.) as catalyst precursor in the epoxidation of cis-cyclooctene with H2O2 under standard conditions achieves a significant higher conversion within 10 min compared to 1 (Fig. 6a). The maximal conversion of 71% is reached after 5 min. An initial TOF of 44000 h−1 and TON of 142 is achieved (Table 4, entry 16).
 | ||
| Fig. 6 Time-dependent epoxidation of cis-cyclooctene (67.3 μmol mL−1, 1.00 eq.) in MeCN at 20 °C using 1 or 2 (0.34 μmol mL−1, 0.005 eq.) as catalyst and H2O2 (101 μmol mL−1, 1.50 eq.) as oxidant, (a) without additive and (b) with the addition of Sc(OTf)3 (6.73 μmol mL−1, 0.10 eq.). Conversions are determined by GC-FID. | ||
Previous catalytic studies have employed strong Lewis acids such as Sc(OTf)3 in the oxidation process, utilising the normally coordinated FeII and FeIII NHC macrocycle A and B as catalysts, which provides notably enhanced performance.20 Although an epoxidation mechanism involving iron carbenes as catalyst precursors remains uncertain, it can be inferred, that Sc3+ triggers the in situ oxidation of the FeII complex to the active FeIII catalyst, based on observations of related complexes.20 Furthermore, it might be assumed that heterolytic cleavage of the O–O bond in a FeIII–OOH species, initially formed upon reaction of the FeIII catalyst with H2O2, is facilitated by Sc(OTf)3, resulting in the generation of an active oxoiron(IV) or (V) species.6–10 Additionally, the application of Lewis acids in the catalytic reaction demonstrated to reactivate μ2-oxodiiron(III) FeIII–O–FeIII species, which has been identified as a crucial deactivation product.20,41,44 The addition of 0.10 eq. Sc(OTf)3 to the reaction with 0.05 mol% of FeII complex 1 under standard conditions results in an extended lifetime of the catalyst (TON = 194 vs. 74). The reaction is completed after 30 min with an enhanced conversion of 97% (vs. 37%) with a TOF of 40900 h−1 (Table 4, entry 14).30 However, applying FeIII complex 2 as catalyst under the same conditions the reaction is completed after 10 min with a maximum conversion of 99% (vs. 70% without additive) and a TON of 197 (vs. 142), which is comparable to the results with 1 (Fig. 6b). In contrast to the respective experiments with A and B (ref. 20), the TOF is pushed to 51800 h−1 with an increased conversion in the first minutes of the reaction (Table 4, entry 17), which indicates a slower in situ single oxidation of 1 to 2versus the instantaneous in situ single oxidation of A to B. The observed enhancement in activity and stability upon the introduction of Lewis acids to the reaction involving compounds 1 and 2 suggests the potential regeneration of degraded species, analogous to the mechanism observed in systems A and B.20,44 Otherwise, the time-dependent conversion of cis-cyclooctene is anticipated to be nearly identical for complex 2 with and without the additive. Unfortunately, the addition of Sc(OTf)3 to the catalytic reaction with 1 or 2 decreases the product selectivity slightly to 96 and 97%, respectively. No byproduct formation other than 1,2-cyclooctanediol was observed.
Epoxides not only serve as final products in industrial applications but also function as crucial building blocks in synthetic organic chemistry.51–54 In this context, a screening of a variety of olefin substrates is conducted using complex 1 for epoxidation catalysis, as the resulting epoxides are of great value as intermediates in various industrial processes. Additionally, the catalyst tolerance of different functional groups is assessed. The resulting epoxides find wide application as monomers in polymerisation processes,55 stabilisers for halogenated hydrocarbons or oil-soluble bases in cosmetic formulations,51–54 as well as constituents in epoxy resins and as reactive diluents for such resins.5256
All experiments concerning the epoxidation of various olefin substrates are performed using 0.5 mol% of FeII catalyst 1 and 0.10 eq. Sc(OTf)3 at standard conditions and are analysed via1H-NMR spectroscopy. Samples were taken after 10 s and 1 h (Table 5). Catalyst 1 demonstrates the ability to transform all employed substrates. Highest conversion is obtained for the benchmark substrate cis-cyclooctene. The smaller ring cis-cyclohexene is converted to 33% after 10 s (S > 99%), with a TOF of 23800 h−1 and to 39% after 1 h with a significant decrease in selectivity (38%). The electron density of the double bond plays a decisive role in the reactivity of the respective substrate.34 After 1 h, lowest conversion of 6% is achieved for allyl chloride, but with a high selectivity of >99%. Replacing the chloride substituent, which possesses an –I effect towards the double bond, by an electron donating hydroxyl group, a conversion of 34% is obtained, with a TOF of 15100 h−1. However, the increased electrophilic character of the double bond likely leads to a reduced selectivity of 11%. Styrene, as aromatic system is transformed to 27% after 1 h with no selectivity towards the epoxide due to overoxidation. Catalyst 1 shows rather low reactivity to the more challenging olefinic systems trans-chalcone, which bears additionally an electron-withdrawing carbonyl group in proximity to the double bond, provoking a reduced electron density. Acyclic 1-hexene with its terminal double bond is converted to 29% after 1 h. Complex 1 exhibits relatively high activity and stability towards cis- and trans-oct-2-ene (conv. = 57 and 51%, 1 h) attributed to the enhanced reactivity of the internal double bond influenced by neighbouring alkyl groups. Notably the TOFs differ significantly with 28600 h−1 for cis-oct-2-ene and 4500 h−1 for trans-oct-2-ene. This preference for cis conversion over trans is also a commonly observed for other iron epoxidation catalysts.22,49,56 In order to determine whether the respective substrates are also converted without additives, the experiments were carried out under same conditions without the addition of Sc(OTf)3 with a reaction time of 5 min (Table 5, right column). With the exception of allyl chloride and allyl alcohol, all substrates were successfully transformed with high selectivity. In general, an increased nucleophilic nature of the alkene, induced by inductive effects, correlates with enhanced reactivity towards the epoxide, consistent with an electrophilic active species.22,57 A highly substituted alkene has a higher reactivity than its terminal counterpart due to the +I effect of the substituents. In addition, the -M effect of the carbonyl in chalcone reduces the reactivity of the alkene.56
| X 10 s [%] (S) | X 1 h [%] (S) | TOF [h−1] | TON | X 5 min [%] (S)b | |
|---|---|---|---|---|---|
| Reaction conditions: acetonitrile as solvent, substrate, (67.3 μmol mL−1, 1.00 eq.), FeII complex 1 (1.35 μmol mL−1, 0.005 eq.), and H2O2 (50% aq., 101 μmol mL−1, 1.50 eq.), using Sc(OTf)3 (6.73 μmol mL−1, 0.10 eq.) as additive, 20 °C. X = conversion; S = selectivity, related to the epoxide. TOFs are determined after 10 s. TONs are determined after 1 h. Conversions are determined by 1H-NMR spectroscopy, applying benzene as external standard.a Conversions are determined by GC-FID.b Without the addition of Sc(OTf). | |||||

|
57 (>99) | 97 (96) | 40900 |
194 | 37 (>99) |

|
33 (>99) | 39 (38) | 23800 |
78 | 25 (>99) |

|
0 (−) | 6 (>99) | — | 13 | 0 (−) |

|
21 (0) | 34 (11) | 15100 |
67 | 9 (8) |

|
19 (24) | 27 (0) | 13300 |
54 | 24 (89) |

|
4 (92) | 13 (5) | 3000 | 26 | 2 (83) |

|
6 (> 99) | 29 (49) | 4100 | 58 | 9 (>99) |

|
40 (64) | 51 (45) | 28600 |
91 | 27 (96) |
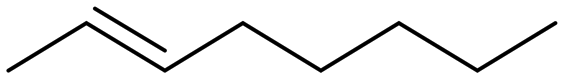
|
6 (>99) | 57 (74) | 4500 | 114 | 11 (>99) |
Conclusions
In summary, the first macrocyclic aNHC iron(III) complex 2 was successfully synthesised. 1H-NMR spectroscopy confirms the paramagnetism of 2. Characterisation via SC-XRD analysis reveals an octahedral geometry with the tetradentate ligand arranged in an ideal square-planar coordination, as in its iron(II) counterpart 1. Mössbauer measurements as well as SQUID data allow to identify both complexes as low spin species (1: S = 0 and 2: S = ½). Epoxidation reactions applying aNHC iron(II) and (III) complexes 1 and 2 and screening of optimal conditions show efficient substrate conversion. However, the performance of the so far most active macrocyclic iron catalyst with classical NHC coordination could not be exceeded or even reached. These findings suggest that a more electron rich iron centre is not necessarily associated with higher activity, due to reduced catalyst stability and vice versa. Decomposition studies via1H-NMR spectroscopy identify the Fe-aNHC bond as weak spot by detecting the mono-oxidised and three-fold protonated ligand after the addition of H2O2. HR-ESI-MS measurements supported by DFT studies imply the possible formation of an oxoiron(IV) or (V) species under oxidative conditions. While these results provide insights into the correlation between ligand design and activity in oxidation catalysis of iron NHC complexes, the exact nature of the active species remains unclear, and further investigation is necessary. Importantly, this study contributes to the limited analytical and experimental data available on iron aNHC complexes, offering valuable information for future research in this area.Experimental
General considerations
Unless otherwise stated, all manipulations were performed under argon atmosphere using standard Schlenk and glovebox techniques. Dry and degassed solvents were obtained from an MBraun solvent purification system, degassed via freeze–pump–thaw (3 cycles) and stored over molecular sieve (3 or 4 Å) prior to use. Fe(btsa)2(THF),56,58 thianthrenyl hexafluorophosphate59 and the iron(II) calix[4]3-methyl-1,2,3-triazole-5-ylidene hexafluorophosphate complex301 were synthesised according to literature procedures. All other reagents were purchased from commercial suppliers and used without further purification. NMR spectra were recorded on a Bruker Advanced Ultrashield AV400 (400 MHz) at a temperature of 297 K. Chemical shifts (δ) are reported in ppm and referenced to the residual signal of the deuterated solvent.60 Elemental analyses (C/H/N/S) were performed by the microanalytical laboratory at Technische Universität München. High resolution electrospray ionization mass spectrometry (HR-ESI-MS) data were acquired on a Thermo Fisher Exactive Plus Orbitrap. UV/vis spectra were recorded on an Agilent Technologies Cary 60 UV/vis spectrophotometer with a concentration of 0.20 mM complex in dry and degassed acetonitrile at 20 °C. Solid material of Fe complexes 1 and 2 was studied using 57Fe Mössbauer spectroscopy at 80 K. 57Fe Mössbauer spectra were measured using a 57Co source in a Rh matrix using an alternating constant acceleration Wissel Mössbauer spectrometer equipped with a Janis closed-cycle helium cryostat. Transmission data were collected, and isomer shifts are reported relative to iron metal at ambient temperature. Experimental data were simulated with mf2.SL software.61 GC analysis was performed with an Agilent Technologies 7890B GC-FID system with a 7693A Automatic Liquid Sampler for 150 samples with G4513A Autoinjector using a HP-5 column (30 m × 320 μm × 0.25 μm).Synthesis of iron(III) calix[4]3-methyl-1,2,3-triazole-5-ylidene hexafluorophosphate (2)
Under Schlenk-conditions 44.7 mg thianthrenyl hexafluoro-phosphate (124 μmol, 1.00 eq.) are dissolved in 2 mL dry and degassed acetonitrile resulting in a dark blue solution. 100 mg of Fe(II)[cCCCC]trz complex 1 (124 μmol, 1.00 eq.) are dissolved in 3 mL dry and degassed acetonitrile resulting in an orange solution and added to the thianthrenyl hexafluorophosphate solution. The dark purple mixture is stirred at r.t. for 1 h. Subsequently, the addition of 5 mL dry and degassed diethyl ether while stirring leads to the precipitation of a yellow solid and turns the solution from dark purple via purple into a colourless supernatant. The yellow solid is separated from the solution via Whatman-filtration and washed with dry and degassed diethyl ether (3 × 2 mL). After drying under vacuum, 113 mg of the product (120 μmol, 96%) are obtained as yellow solid. 1H-NMR (400 MHz, CD3CN): δ [ppm] = 44.49 (s, 8 H, CH2), 4.38 (s, 12 H, CH3). Analytical calculation (%) for C20H26F18FeN14P3: C 25.20; H 2.75; N 20.57. Found: C 24.91 H 2.63; N 19.52.Catalytic procedures
All catalytic reactions were conducted in a cryostat (JulaboFP-50) under atmospheric conditions, unless otherwise stated. Acetonitrile (HPLC-grade) as solvent was applied for all experiments, which are screened via GC (substrate: cis-cyclooctene). The screening of other substrates was performed using 1H-NMR spectroscopy and deuteride acetonitrile as solvent. The catalyst was added from a preformed stock solution in dry and degassed acetonitrile, which was handled under argon corresponding to the appropriate stoichiometry to a solution of the respective substrate (67.3 μmol mL−1, 1.00 eq.). Hydrogen peroxide (50% aq., 101 μmol mL−1, 1.50 eq.) was used as oxidising agent and, if required, Sc(OTf)3 as additive 8.41 μmol mL−1, (0.10 eq.). The reaction was started upon addition of the catalyst stock solution, by adding the catalyst solution all at once. The reaction was terminated by adding electrolytically precipitated activated MnO2 in order to decompose the excess of H2O2 in the reaction solution. After filtration over activated neutral alumina (separation of the catalyst), GC samples were prepared for each experiment using 200 μL filtrate, diluted with 1300 μL MeCN and p-xylene (0.90 μL mL−1) as external standard for each chosen time point. For the screening via1H-NMR spectroscopy, 500 μL filtrate was added to 1 μL benzene as external standard. Control experiments without catalyst were performed for all reactions. An additional blank experiment with a simple iron salt, iron(II) chloride in the presence of H2O2 was conducted to highlight the importance of iron complexes associated with NHCs due to minimal product and unselective side-product formation. All reactions were conducted at least twice. Analogous, the additive Sc(OTf)3 itself shows minimal unselective catalytic activity.20Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors thank Patrick Mollik for HR-ESI-MS measurements. G. G. Z., C. A. E. and M. J. S. gratefully acknowledge support from TUM Graduate School. Part of this work has been supported by the Deutsche Forschungsgemeinschaft (DFG) in the framework of the Research Unit 5215 (FOR 5215 “Bioinspired Oxidation Catalysis with Iron Complexes”; project Me1313/18-1/445916766 to F.M.).References
- L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed.
- G. G. Zámbó, J. F. Schlagintweit, R. M. Reich and F. E. Kühn, Catal. Sci. Technol., 2022, 12, 4949–4961 RSC.
- T. P. Schlachta and F. E. Kühn, Chem. Soc. Rev., 2023, 52, 2238–2277 RSC.
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552–561 RSC.
- A. J. Jasniewski and L. Que Jr, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- M. Borrell, E. Andris, R. Navrátil, J. Roithová and M. Costas, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- X. Lu, X.-X. Li, Y.-M. Lee, Y. Jang, M. S. Seo, S. Hong, K.-B. Cho, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2020, 142, 3891–3904 CrossRef CAS PubMed.
- S. Kal and L. Que Jr, Angew. Chem., Int. Ed., 2019, 58, 8484–8488 CrossRef CAS PubMed.
- K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que, J. Am. Chem. Soc., 2002, 124, 3026–3035 CrossRef CAS PubMed.
- S. M. Hölzl, P. J. Altmann, J. W. Kück and F. E. Kühn, Coord. Chem. Rev., 2017, 352, 517–536 CrossRef.
- W. N. Oloo and L. Que Jr, Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS PubMed.
- D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251–259 CrossRef CAS.
- R. Mas-Ballesté and L. Que, J. Am. Chem. Soc., 2007, 129, 15964–15972 CrossRef PubMed.
- E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS.
- H. J. H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
- J. W. Kück, R. M. Reich and F. E. Kühn, Chem. Rec., 2016, 16, 349–364 CrossRef PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- N. Demirel, J. Haber, S. I. Ivlev and E. Meggers, Organometallics, 2022, 41, 3852–3860 CrossRef CAS PubMed.
- F. Dyckhoff, J. F. Schlagintweit, R. M. Reich and F. E. Kühn, Catal. Sci. Technol., 2020, 10, 3532–3536 RSC.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797–3804 CrossRef CAS PubMed.
- J. W. Kück, M. R. Anneser, B. Hofmann, A. Pöthig, M. Cokoja and F. E. Kühn, ChemSusChem, 2015, 8, 4056–4063 CrossRef PubMed.
- A. Schmidt, N. Grover, T. K. Zimmermann, L. Graser, M. Cokoja, A. Pöthig and F. E. Kühn, J. Catal., 2014, 319, 119–126 CrossRef CAS.
- M. A. Bernd, F. Dyckhoff, B. J. Hofmann, A. D. Böth, J. F. Schlagintweit, J. Oberkofler, R. M. Reich and F. E. Kühn, J. Catal., 2020, 391, 548–561 CrossRef CAS.
- A. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- R. Maity and B. Sarkar, JACS Au, 2021, 2, 22–57 CrossRef PubMed.
- G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed.
- K. O. Marichev, S. A. Patil and A. Bugarin, Tetrahedron, 2018, 74, 2523–2546 CrossRef CAS.
- G. G. Zámbó, J. Mayr, M. J. Sauer, T. P. Schlachta, R. M. Reich and F. E. Kühn, Dalton Trans., 2022, 51, 13591–13595 RSC.
- J. M. Smith and J. R. Long, Inorg. Chem., 2010, 49, 11223–11230 CrossRef CAS PubMed.
- P. O. Löwdin, J. Chem. Phys., 1950, 18, 365–375 CrossRef.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504–1518 CrossRef CAS.
- E.-M. H. J. Esslinger, J. F. Schlagintweit, G. G. Zámbó, A. M. Imhof, R. M. Reich and F. E. Kühn, Asian J. Org. Chem., 2021, 10, 2654–2662 CrossRef CAS.
- M. Blume, Phys. Rev. Lett., 1965, 14, 96 CrossRef CAS.
- P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer spectroscopy and transition metal chemistry: fundamentals and applications, Springer Science & Business Media, 2010 Search PubMed.
- E. Bill, in Practical approaches to biological inorganic chemistry, Elsevier, 2020, pp. 201–228 Search PubMed.
- C. Schremmer, C. Cordes, I. Klawitter, M. Bergner, C. E. Schiewer, S. Dechert, S. Demeshko, M. John and F. Meyer, Chem. – Eur. J., 2019, 25, 3918–3929 CrossRef CAS PubMed.
- J. Donat, P. Dubourdeaux, M. Clémancey, J. Rendon, C. Gervasoni, M. Barbier, J. Barilone, J. Pécaut, S. Gambarelli and P. Maldivi, Chem. – Eur. J., 2022, 28, e202201875 CrossRef CAS PubMed.
- S. C. Chan, P. Gupta, X. Engelmann, Z. Z. Ang, R. Ganguly, E. Bill, K. Ray, S. Ye and J. England, Angew. Chem., Int. Ed., 2018, 57, 15717–15722 CrossRef CAS PubMed.
- F. Dyckhoff, J. F. Schlagintweit, M. A. Bernd, C. H. Jakob, T. P. Schlachta, B. J. Hofmann, R. M. Reich and F. E. Kühn, Catal. Sci. Technol., 2021, 11, 795–799 RSC.
- M. R. Bukowski, P. Comba, A. Lienke, C. Limberg, C. Lopez de Laorden, R. Mas-Ballesté, M. Merz and L. Que Jr, Angew. Chem., Int. Ed., 2006, 45, 3446–3449 CrossRef CAS PubMed.
- J. W. Kück, A. Raba, I. I. Markovits, M. Cokoja and F. E. Kühn, ChemCatChem, 2014, 6, 1882–1886 CrossRef.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, V. D'Elia, M. P. Högerl, J.-M. Basset and F. E. Kühn, Dalton Trans., 2016, 45, 6449–6455 RSC.
- J. F. Schlagintweit, F. Dyckhoff, L. Nguyen, C. H. Jakob, R. M. Reich and F. E. Kühn, J. Catal., 2020, 383, 144–152 CrossRef CAS.
- W. N. Oloo, A. J. Fielding and L. Que Jr, J. Am. Chem. Soc., 2013, 135, 6438–6441 CrossRef CAS PubMed.
- M. J. Ingleson and R. A. Layfield, Chem. Commun., 2012, 48, 3579–3589 RSC.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- R. Mas-Ballesté, M. Costas, T. Van Den Berg and L. Que Jr, Chem. – Eur. J., 2006, 12, 7489–7500 CrossRef PubMed.
- A. R. McDonald and L. Que Jr, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- S. T. Oyama, Mechanisms in homogeneous and heterogeneous epoxidation catalysis, Elsevier, 2011 Search PubMed.
- H. H. Szmant, Organic building blocks of the chemical industry, John Wiley & Sons, 1989 Search PubMed.
- C. A. de Parrodi and E. Juaristi, Synlett, 2006, 2006, 2699–2715 CrossRef.
- I. Kim, S. M. Kim, C.-S. Ha and D.-W. Park, Macromol. Rapid Commun., 2004, 25, 888–893 CrossRef CAS.
- T. P. Schlachta, G. G. Zámbó, M. J. Sauer, I. Rüter, C. A. Hoefer, S. Demeshko, F. Meyer and F. E. Kühn, J. Catal., 2023, 426, 234–246 CrossRef CAS.
- E. A. Mikhalyova, O. V. Makhlynets, T. D. Palluccio, A. S. Filatov and E. V. Rybak-Akimova, Chem. Commun., 2012, 48, 687–689 RSC.
- M. M. Olmstead, P. P. Power and S. C. Shoner, Inorg. Chem., 1991, 30, 2547–2551 CrossRef CAS.
- H. J. Shine, B.-J. Zhao, J. N. Marx, T. Ould-Ely and K. H. Whitmire, J. Org. Chem., 2004, 69, 9255–9261 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- mf2.SL, Max-Planck Institute for Chemical Energy Conversion, Mülheim/Ruhr, Germany, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR-spectra, crystallographic data, analytical methods, buried volume and topographic steric map calculations, thermochromism, magnetic susceptibility, HR-ESI-MS spectra, DFT calculations. CCDC 2362784. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cy00992d |
| This journal is © The Royal Society of Chemistry 2024 |