
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of redox treatments on the low-temperature water gas shift reaction over Pt/CeO2 catalysts†
Clément
Molinet-Chinaglia
 ,
Elizabeth
Vera
,
Philippe
Vernoux
,
Laurent
Piccolo
and
Stéphane
Loridant
*
,
Elizabeth
Vera
,
Philippe
Vernoux
,
Laurent
Piccolo
and
Stéphane
Loridant
*
CNRS, IRCELYON, UMR 5256, Université Claude Bernard-Lyon 1, Villeurbanne, F-69100, France. E-mail: stephane.loridant@ircelyon.univ-lyon1.fr
First published on 9th September 2024
Abstract
Pt/CeO2 catalysts are promising for the low-temperature water gas shift (LT-WGS) reaction, which is an important step to produce H2 from syngas. When prepared by impregnation of platinum salt and calcination at 500 °C, they contain Pt2+ single atoms (SAs) and/or PtOx clusters which need to be converted into Pt0 nanoparticles (NPs) to obtain higher activity for the LT-WGS reaction. In this work, it was shown that reducing pretreatments at 250 °C under H2 promote the molar activity of catalysts containing from 0.10 to 1.06 wt% Pt by increasing the number of Pt0 NPs formed during reaction at 230 °C. An improvement was also obtained via pretreatment at 500 °C but only for low-Pt-content catalysts, underlying the importance of the pretreatment temperature. Furthermore, it was shown that all prepared Pt/CeO2 catalysts which slowly deactivate over reaction time can be regenerated by oxidative post-treatment at only 230 °C, which is industrially interesting. Even more original, a strong improvement in activity of the low-Pt-content catalysts was observed after a 12 h oxidative post-treatment at 500 °C. This treatment was shown to redisperse and reoxidize Pt atoms into PtOx species different from the initial ones. Such species are highly reducible on the surface of CeO2 and easily transformed into active Pt0 NPs.
1. Introduction
The water gas shift reaction (CO + H2O → CO2 + H2) is an important step to produce H2, by removing CO from syngas in different processes such as the Haber–Bosch process, metal oxide reduction and methanol synthesis.1 A major effort has been made to develop catalysts for the production of pure H2 for fuel cell applications because of CO poisoning of Pt-based electrodes.1 Among all the catalysts, the Pt/CeO2 system is promising for the low-temperature water gas shift (LT-WGS)1–5 reaction compared to Cu–ZnO–Al2O3 because of its high activity,6,7 selectivity and stability.8–11 The high cost of Pt can be mitigated by using very low noble metal loading, which is facilitated by stabilization of Pt single atoms (cations) on ceria.12,13 Agglomeration of Pt atoms was previously observed under WGS reaction conditions: the Pt/CeO2 catalyst composed of mostly isolated atoms rapidly forms metallic Pt nanoparticles (Pt0 NPs) under WGS conditions above 180 °C.14 However, there is still a major debate on the nature of the active sites. Recently, a study demonstrated by using environmental transmission electron microscopy (ETEM) that the dynamic nature of the perimeter Pt0–Ovacancy–Ce3+ sites is a key mechanistic step for the WGS reaction.14 Furthermore, the necessity to form Pt0 NPs to activate the catalyst was evidenced.15–17 In particular, Pt NPs of size close to 4 nm were found to be more active for low-temperature WGS reaction than isolated atoms and subnanometer clusters.16Redox treatments have often been used to activate Pt/CeO2 catalysts before CO oxidation18–20 or to regenerate them afterwards.21,22 For the WGS reaction, Lee et al. observed the formation of a ceria nanolayer around Pt particles after reduction under H2 at 250 °C and under N2 at 800 °C.23,24 These treatments slow down the dissociation of H2 and thus limit its competitive adsorption in favour of CO. Pastor-Pérez et al. showed that the combination of calcination, plasma and H2 treatments increases the activity of Pt/CeO2 catalysts by the formation of electron-enriched Pt NPs.25 Furthermore, the size and morphology of Pt nanoparticles and hence the activity of Pt/CeO2 catalysts can be tuned by low-temperature redox treatments below 500 °C.26,27 In particular, an oxidizing post-treatment can redisperse Pt NPs28 and desorb poisoning carbonates from the surface of CeO2.29
Our previous study has highlighted a significant influence of Pt content on the molar activity of Pt/CeO2 catalysts for the LT-WGS reaction, increasing by a factor of 2.5 from 0.1 to 0.6 wt% and stabilizing above. Below 0.6 wt%, PtOx species initially present are ultradispersed and in strong interaction with CeO2. Their low reducibility limits their activation under reaction mixture and, consequently, their catalytic activity.17 A suitable reductive treatment can allow their transformation into Pt0 NP active species.
To gain further insights, this work describes the impact of different pre- and post-treatments on the LT-WGS activity of Pt/CeO2 catalysts with various Pt contents: pre-reduction with and without consecutive oxidation was explored at two different temperatures (250 °C and 500 °C) to favour the formation of Pt0 NP active species. An oxidative post-treatment at 500 °C for 12 h to maximize redispersion and another one at the reaction temperature (230 °C) for only 10 min to approach viable conditions for an industrial process were also attempted. The Pt/CeO2 catalysts were characterized by microRaman spectroscopy, annular dark-field scanning transmission electron microscopy (ADF-STEM), temperature-programmed reduction by H2 and CO (H2-TPR and CO-TPR, respectively) and operando diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) in connection with their catalytic activities in order to understand the influence of the different treatments on the structural modifications of Pt species.
2. Experimental
2.1 Preparation
Nine Pt/CeO2 catalysts with various loadings were prepared by wet impregnation of 2 g of CeO2 (Solvay Special Chem Company, specific surface area: 150 m2 g−1, pore diameter: 7 nm, pore volume: 0.28 cm3 g−1) with 24 mL of deionized water containing between 4 and 80 mg (to obtain Pt loadings in the range 0.1–1.72 wt%) of [Pt(NH3)4](NO3)2 (Sigma-Aldrich, >99%) at 60 °C for 4 h under stirring at 400 rpm. Water was then removed with a rotary evaporator at 70 °C and 200 mbar. The obtained powder was finally calcined under 100 mL min−1 of synthetic air at 500 °C for 4 h with a heating rate of 10 °C min−1. The catalysts are labelled xPt in the following, where x corresponds to the weight percent of Pt determined by XRF.172.2 Catalytic testing and pretreatments
A catalyst mass of 200 mg was placed in a straight Pyrex reactor with an internal diameter of 1 cm. Before any catalytic test, an initial oxidizing pretreatment named OX500 was performed under 100 mL min−1 of a 20% O2/Ar mixture at 500 °C for 1 h (Fig. S1a†). To study the influence of reductive pretreatment, a subsequent treatment under 20% H2 for 1 h was performed at either 250 °C (RED250, Fig. S1b†) or 500 °C (RED500, Fig. S1c†). Furthermore, REDOX250 and REDOX500 define a catalyst that has been treated before reaction by the initial oxidizing pretreatment at 500 °C followed by a reducing one under 20% H2 for 1 h and then an oxidizing one under 20% O2 flow for 1 h either at 250 °C or at 500 °C, respectively. These protocols are described in Fig. S1d and e,† respectively. Note that a fresh catalyst was used for each pretreatment.Catalytic testing for the WGS reaction was detailed in a previous study.17 After pretreatment, measurements were performed at 230 °C under 40 NmL min−1 of H2O, 10 NmL min−1 of CO, 65 NmL min−1 of Ar and 5 NmL min−1 of He (feed composition H2O/CO/Ar/He: 33.3%/8.3%/54.2%/4.2%). Helium was used as an internal standard for analysis using an INFICON Transpector CPM 3 mass spectrometer. The GHSV was set to 600 mL min−1 g−1. Preliminary measurements varying the mass of catalyst and its granulometry demonstrated the absence of external and internal diffusion limitations for a large range of conversion. The granulometry was then fixed to 106–180 μm.
The influence of oxidative post-treatments on the activity was also studied. For each catalyst after OX500 pretreatment and WGS reaction, a first short treatment of 10 min under 20% O2 at 230 °C (ReOx230_1) was performed followed by 1 h of reaction at 230 °C (Fig. S1f†). Then, a long oxidizing treatment of 12 h at 500 °C under 20% O2 (ReOx500) was added and also followed by 1 h of reaction at 230 °C. Finally, a last short treatment of 10 min at 230 °C under oxidizing conditions was performed (ReOx230_2) followed by 1 h of reaction at 230 °C (see the complete sequence in Fig. S1f†).
2.3 Characterization
Diffuse reflectance infrared Fourier-transform (DRIFT) spectra with a spectral resolution of 4 cm−1 were obtained using a Thermo Scientific Nicolet IR-TF 6700 spectrometer equipped with a ceramic source. Operando DRIFTS analyses were performed in a Harrick-HVC-DRP cell similar to a fixed-bed reactor with a gas feed from the top to the bottom and a powdered solid (around 50 mg) retained by a stainless-steel grid. A calibration of the powder bed temperature as a function of the cell temperature was carried out using an IR pyrometer. The DRIFT cell was equipped with ZnSe windows absorbing weakly in the IR. The IR beam scattered at the surface of the sample was analysed by a high-sensitivity MCT detector cooled by liquid nitrogen. During operando analysis, the feed composition was H2O/CO/He (2%/0.5%/97.5) with a GHSV value of 1000 mL min−1 g−1. Water vapor was added using a saturator at 18 °C. Analyses were performed every minute during the reaction with background subtraction performed at 230 °C after OX500 pretreatment. An INFICON Transpector CPM 3 mass spectrometer was placed at the vent to analyse the inlet and outlet gases and determine the catalytic activity, similarly to the method used during catalytic tests. The protocol of oxidative pretreatment and post-treatments performed during the catalytic tests (Fig. S1†) were applied to the catalysts in the DRIFTS cell.MicroRaman spectra were recorded with a LabRAM HR (Horiba) spectrometer equipped with an Open Electrode CCD detector cooled at −75 °C. An Ar ion laser at 514 nm (power limited to 0.3 mW) was focused with a ×50 objective, leading to a spatial resolution of ca. 2 μm. The backscattered light was recollected, sent to an edge filter to remove the Rayleigh light, and spatially dispersed using a 300 lines per mm diffraction grating, leading to a spectral resolution of 4 cm−1.
A Titan ETEM G2 80–300 kV (FEI) microscope equipped with a spherical aberration corrector was operated at 300 kV for the analyses, which were mainly carried out in annular dark-field scanning transmission electron microscopy (ADF-STEM) mode with a resolution of 0.136 nm. For each preparation, a few milligrams of sample were ground and ultrasonically suspended in ethanol. Two to three drops of the suspension were deposited on the surface of a holey carbon film covering a 300 mesh copper grid.
Temperature-programmed reduction (TPR) curves were obtained using a 9 Omnistar GSD 301 O2 (Pfeiffer Vacuum) mass spectrometer. The sample (50 mg) was placed in a U-shaped reactor (internal diameter of 4 mm) on a quartz fiber pad and heated by a tubular furnace. The sample was pretreated under 40 mL min−1 of 20% O2/N2 at 500 °C for 1 h. Then, it was reduced under 40 mL min−1 of either 1% H2/He or 1% CO/He by heating from 25 °C to 500 °C at a rate of 10 °C min−1. When the temperature reached 500 °C, the feed was replaced by pure H2 or CO and maintained for 1 h before cooling.
3. Results and discussion
3.1 Influence of reductive and redox pretreatments on the WGS activity
Only peaks of cubic ceria were observed by X-ray diffraction up to 1.06 wt% Pt after oxidizing pretreatment at 500 °C (OX500) indicating high dispersion of Pt species. This is consistent with the low Pt surface density (below 0.22Pt nm−2) due to the high surface area of the CeO2 support. Additionally, X-ray photoelectron spectroscopy (XPS) showed that Pt species mostly contain Pt2+ cations.26 Therefore, one can conclude that Pt/CeO2 catalysts contain ultra-dispersed oxidized Pt species (i.e. Pt2+ SAs and PtOx subnanometer clusters).The WGS molar activity at 230 °C strongly increases up to 0.6 wt% Pt and stabilizes above (Fig. 1).17
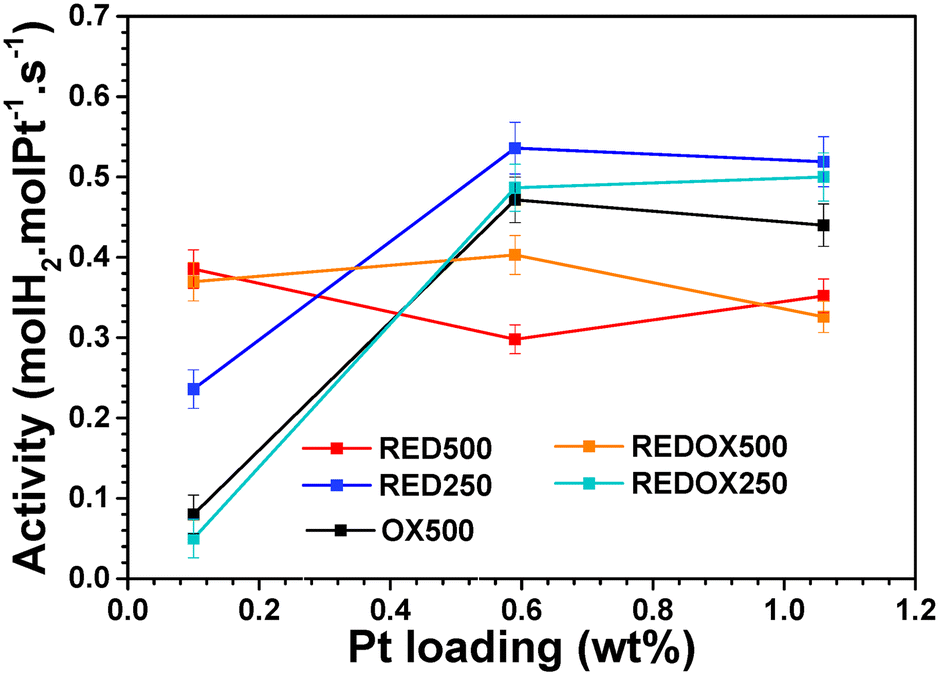 | ||
| Fig. 1 Evolution of molar activity with Pt loading after oxidizing OX500, reducing RED250 and RED500, reducing-oxidizing REDOX250 and REDOX500 pretreatments. Reaction conditions: 230 °C, H2O/CO/Ar/He: 33.3/8.3/54.2/4.2 vol% feed, GHSV = 600 mL min−1 gcat−1, time on stream: 45 min. | ||
Thus, different reductive and redox pretreatments (see the protocols in Fig. S1a–e†) were attempted for three representative catalysts: 0.10Pt, 0.59Pt and 1.06Pt. For each catalyst, the activity rose to a maximal value after a few minutes (time topti) and then slowly decreased over time (Fig. S2†). Therefore, data are compared both at topti and after 45 min in the following.
The evolutions over time in molar activity and CO2/H2 ratio are provided for 0.10Pt, 0.59Pt and 1.06Pt catalysts in Fig. S3, S4 and S5,† respectively. The latter parameter was significantly higher than 1 during the first 5 min of reaction after the OX500 pretreatment due to the reduction of both PtOx and ceria induced by CO.17 Interestingly, the reductive pretreatment at 500 °C, named RED500, and the redox one, REDOX500, strongly increase the molar activity of the 0.10Pt catalyst by a factor of 4 (Fig. 1). Conversely, the RED500 pretreatment significantly decreases the activity of 0.59Pt and 1.06Pt catalysts (by about 25–50%). For the reductive pretreatment at 250 °C, named RED250, the molar activity of the three catalysts slightly increased especially for 0.10Pt. The WGS molar activity of the three catalysts reduced at 250 °C and reoxidized at 250 °C before reaction (protocol REDOX250 in Fig. S1d†) is close to the reference values measured after OX500 pretreatment. However, this is not the case after REDOX500 pretreatment, suggesting that the structural modification of Pt species is reversible at 250 °C but not at 500 °C.
The activation of catalysts after reducing treatments under H2 could be due to an increase in the amount of Pt0 NPs active for the LT-WGS reaction by decreasing the amount of Pt2+ cations strongly anchored in the subsurface layers of ceria.30,31 To address this question, the influence of pretreatments on the nature of Pt species and the redox properties of Pt/CeO2 catalysts was investigated by H2-TPR analysis and operando DRIFT spectroscopy.
3.2 Impact of the reductive and redox pretreatments on the nature of Pt species and redox properties of Pt/CeO2 catalysts
For the nine Pt/CeO2 catalysts and the CeO2 support, a first H2-TPR analysis (named TPR1) was performed after the initial OX500 pretreatment. Structural modifications during TPR1 are assumed to be similar to that during RED500 since a temperature plateau at 500 °C under H2 was applied in both cases. TPR1 was followed by an oxidative treatment at 500 °C for 1 h, leading to a catalyst state equivalent to that after REDOX500 pretreatment. The second H2-TPR analysis (named TPR2) subsequently carried out allows comparison of reducing properties after OX500 and REDOX500 pretreatments. The curves plotted in Fig. S6a and b† respectively contain one main non-symmetrical reduction peak. For TPR 1 achieved after OX500 pretreatment, the peak temperature is above 250 °C for Pt loading below 0.59 wt% (Fig. 2a). According to Resasco et al., this high-temperature reducibility arises from the presence of Pt SAs at the surface of CeO2.32 For Pt content from 0.59 to 1.06 wt%, the reduction temperature, which ranges between 211 and 222 °C, could be related to the presence of PtOx clusters.32 | ||
| Fig. 2 Evolution of (a) Tmax peak temperature and (b) total H2 consumption with Pt loading for TPR1 and TPR2 analyses from RT up to 500 °C under a 1% H2/He flow. | ||
Finally, the reduction temperature is much lower for 1.43Pt and 1.72Pt catalysts (125 and 93 °C, respectively). Such low-temperatures reveal that either Pt0 NPs are present at the beginning of the TPR analysis or are easily formed under H2. For the second TPR analysis (TPR2) used to probe reducing properties after REDOX500 pretreatment, the peak temperature is shifted below 150 °C for all the prepared Pt/CeO2 catalysts (Fig. 2a). This indicates the presence, even for low-Pt-content (LPC) catalysts, of Pt0 clusters or NPs which are already present after REDOX500 pretreatment or easily formed from highly reducible PtOx species. Indeed, metallic Pt species are able to dissociate H2, leading to the surface reduction of CeO2 by H spillover.33,34 Hence, the reducibility improvement of the 0.1Pt catalyst reflects a major structural change of Pt species after REDOX500 pretreatment compared to OX500. Pt0 NPs can then be easily formed under reaction mixtures, leading to a strong enhancement of molar activity of the 0.10 catalyst (Fig. 1). Interestingly, REDOX500 pretreatment did not reoxidize and redisperse Pt atoms as at the initial state, in agreement with the changes of activity observed after REDOX500 pretreatment compared to OX500 (Fig. 1). Note that irreversible structural changes after such a redox cycle were previously proposed from the decrease in intensity of the PtOx Raman band.26,35
The reducibility of Pt/CeO2 catalysts was reported to be a key parameter determining the activity.6,11,36–38 In this work, improvements of reducibility and activity seem to be related for LPC catalysts, typically 0.10Pt. However, REDOX500 pretreatment leads to a decrease in activity for 0.59Pt and 1.06Pt in spite of a significant improvement of reducibility. It shows that for these Pt contents, other parameters determine the catalytic activity. The decrease in activity after RED500 and REDOX500 pretreatments could arise from Pt aggregation and poisoning by carbonates.29,39–41 However, ADF-STEM images of the 1.06Pt catalyst recorded after RED500 pretreatment (Fig. S7†) did not reveal the presence of Pt NPs significantly larger than 1.4 nm, the typical size determined after the WGS reaction at 230 °C.17 Therefore, if Pt aggregation occurs during RED500, it should be limited to the subnanometer species and should have no impact on catalytic activity since they are poorly active.17
The total H2 consumption rates measured between RT and 500 °C during TPR1 are much higher than the quantity required to reduce PtO to Pt0 (Table S1†). This reveals a high reduction rate of CeO2 and hence the presence of a high amount of oxygen vacancies and Ce3+ cations at the surface of CeO2. Furthermore, the rate increases with Pt loading (Fig. 2b), which can be explained by a higher number of Pt0 NPs, leading to a higher extent of H spillover.42
The H2 consumption rates during TPR2 are significantly higher for 0.10Pt and 0.25Pt catalysts compared to TPR1 (Fig. 2b and Table S1†). For LPC catalysts, reduction at 500 °C under H2 could favour extraction of Pt2+ cations anchored in the subsurface layers of ceria, increasing the number of Pt atoms available to form Pt0 NPs at the surface.17,43 A higher number of Pt0 NPs favours both reduction of ceria and catalytic activity.17 However, no significant change is observed for high-Pt-content (HPC) catalysts (Fig. 2b), showing that in this case, reduction at 500 °C has no impact on the number of Pt0 particles and the H spillover over the CeO2 surface.
A strong increase in activity of the 0.10Pt catalyst was also observed during operando DRIFT analysis after RED500 pretreatment (Fig. S8†). Furthermore, the spectrum recorded after this pretreatment contains ν(CO) bands, in contrast to that after OX500 (Fig. 3a). Indeed, the absence of ν(CO) bands after the oxidative pretreatment arises from the presence of Pt2+ SAs which weakly adsorb CO and are poorly active for the reaction.17,32,44 After reduction, Pt NPs are formed on CeO2 with characteristic ν(CO) bands at 2063, 2046 and 2009–1969 cm−1 corresponding to CO molecules adsorbed on the terrace, edge and periphery sites, respectively (see ref. 17 and references therein), leading to observation of a broad band for the 0.1Pt catalyst.
 | ||
| Fig. 3 Operando DRIFT spectra of the 0.10Pt catalyst after OX500 and RED500 pretreatments: (a) 1900–2200 cm−1 region, (b) 3100–3800 cm−1 region, (c) 2650–3000 cm−1 region and (d) 900–1800 cm−1 region. Cm: monodentate carbonate, Cbi: bidentate carbonate, F1 and F2: bridged formates. Reaction conditions: T = 230 °C, PCO = 5 mbar, PH2O = 20 mbar, GHSV = 1000 mL min−1 g−1. The background corresponds to the spectrum of KBr recorded at 230 °C and the ν(CO) gas signal is subtracted. | ||
Furthermore, the formation of Pt NPs is accompanied by the intensification of the band at 2120 cm−1 (Fig. 3a) due to electronic transition of Ce3+ cations17,32,45 and of the ν(O–H) band at 3630 cm−1 (Fig. 3b) attributed to hydroxyl species near oxygen vacancies (OH type II-B).46,47 Note that a broad band around 3400 cm−1 typical of H bonding is superimposed.
These features confirm that a treatment at 500 °C under H2 favours the formation of Pt0 NPs and the reduction of CeO2. Moreover, OH groups of type II-B are often considered as the active species for the water gas shift reaction.48,49 Hence, the increase in their concentration could arise from the activation of the catalyst. Finally, the formation of Pt0 NPs is accompanied by an increase in the amount of different formates and carbonates (Fig. 3c and d) with DRIFT bands similar to those observed on the spectra of the HPC catalysts after OX500 pretreatment (see ref. 17 and references therein).
In summary, a reductive pretreatment under H2 at 500 °C allows the activation of an LPC catalyst, essentially composed of Pt2+ SAs, by the formation of Pt0 NPs more active than oxidic Pt2+ SAs and PtOx clusters.16,17,44,48 Inversely, the activity of a HPC catalyst is decreased by this same reductive pretreatment. It does not appear to result from Pt aggregation but could be due to poisoning by carbonates.29,39–41 Note that carbonate formation was observed by DRIFT spectroscopy for both LPC and HPC catalysts, but those species are absorbed on the overall surface of CeO2, while carbonate species poisoning Pt0 NPs should be located around them and hence correspond only to a small fraction of the observed carbonates.
3.3 Influence of oxidative post-treatments on the activity of Pt/CeO2 catalysts
A slight deactivation was evidenced during the catalytic testing whatever the pretreatment (see Fig. S2–S5† and relative deviations reported in Table S2†). Therefore, two oxidative post-treatments were applied to regenerate the catalysts. The first one, called ReOx500, was performed at high temperature (500 °C) for 12 h to boost redispersion of Pt0 NPs26,27 and removal of carbonates.29 It was limited at 500 °C to avoid sintering of CeO2. The second reoxidation, called ReOx230, took place at the reaction temperature (230 °C) for only 10 min to approach viable conditions for an industrial process. Consequently, four catalytic tests were successively performed: the first one after OX500 pretreatment, a second one after ReOx230 post-treatment (ReOx230_1), a third one after ReOx500, and finally the last one after a second ReOx230 post-treatment (ReOx230_2). The overall protocol is described in Fig. S1f.† As this study was achieved with a new series of catalysts, some differences in activity after OX500 can be found mainly for the 0.10Pt catalyst.After the first post-treatment ReOx230_1, the molar activity re-increased during the first few minutes toward the maximal values measured after OX500 pretreatment at the optimal time topti (Fig. S9† and 4a) and was significantly higher than the value measured after OX500 pretreatment and 45 min on stream (see the relative deviations reported in Table S3† for the different catalysts). Even if the molar activity slowly decreased again after topti, it remained higher than this value for a similar time (Fig. S9†). Hence, catalyst regeneration is obtained after a reoxidation step at 230 °C for only 10 min. To our knowledge, it was reported that a regeneration of Pt/CeO2 catalysts can occur by a treatment in air at temperatures higher than 400 °C29 but not at 230 °C.
 | ||
| Fig. 4 Evolution with Pt loading of molar activity (a) at topti (maximal value after approx. 1 min on stream), at 45 min of reaction after OX500 pretreatment and at topti after ReOx230_1 first post-treatment, and (b) at topti after OX500 pretreatment and after ReOx500 post-treatment, at 45 min of reaction after ReOx500 and at topti after ReOx230_2 second post-treatment. | ||
After ReOx500, the molar activity increases compared to the OX500 state for a Pt content below 1.06 wt% (Fig. 4b and S10†) with an increase of between 18% and 41% for the maximal values at topti and between 8% and 27% after 45 min on stream (Table S3†). To our knowledge, such improvement in catalytic activity has never been reported. However, for a Pt content higher than 1.06 wt%, a loss of activity is observed after ReOx500, underlying the impact of Pt content on the evolution of activity. Nevertheless, whatever the Pt loading, the molar activity again gradually decreases over time (Fig. S10 and S11 and Table S3†).17 Therefore, an oxidative post-treatment at 500 °C for 12 h can generate more active sites for LPC catalysts but not suppress the slow deactivation process during WGS reaction at 230 °C. However, the second post-treatment ReOx230_2 carried out after ReOx500 (Fig. S1f†) gave rise to activities close to those obtained after ReOx500 both for the maximal values at topti and after 45 min on stream (Fig. 4b and S11†), confirming the regenerative effect of an oxidative post-treatment at 230 °C.
To understand the activation after an oxidative post-treatment at 500 °C for 12 h (ReOx500), catalytic tests were performed on the 0.52Pt catalyst after a 12 h oxidative pretreatment instead of the usual 1 h (OX500). The extension of pretreatment time from 1 to 12 h led to an overall decrease in activity of about 7% (Fig. S12†), which could arise from dispersion of PtOx clusters and anchoring of Pt atoms in the subsurface layers of CeO2. Note that the evolutions over time were similar with a maximal value after around 4 min on stream and a slow deactivation afterwards. Again, the 0.52Pt catalyst was activated by ReOx500 post-treatment with the same magnitude. Hence, a post-treatment at 500 °C is required to obtain activation of LPC catalysts. The origin of activation is explained in the following, crossing different techniques.
To determine the nanostructure of Pt species on the surface of CeO2, ADF-STEM images were recorded after the different oxidative post-treatments at 230 °C and 500 °C. After WGS reaction at 230 °C, an average diameter of 1.4 nm was determined from statistical analysis for catalysts with Pt content above 0.42 wt%.17 After the ReOx230_1 first post-treatment (Fig. 5a and b), some Pt NPs with similar size were still present at the surface of the support. It reveals that if redispersion occurred, it would involve a limited number of Pt atoms. Hence, regeneration by oxidative post-treatment at 230 °C probably arises from another phenomenon which could be decarbonation of poisoning species upon reoxidation.
 | ||
| Fig. 5 ADF-STEM images of the 0.25Pt catalyst after (a and b) ReOx230_1, (c and d) ReOx500, and (e and f) ReOx_2 post-treatment and WGS reaction. The red circles indicate Pt NPs. | ||
Nevertheless, no Pt NPs are observed after the ReOx500 post-treatment (Fig. 5c and d), suggesting a redispersion of Pt NPs as Pt SAs and/or clusters not observed in the ADF-STEM images. This redispersion does not lead to the initial OX500 state. Otherwise, no activation would be observed. However, Pt NPs with diameters ranging between 1 and 2 nm were again observed on 0.25Pt (Fig. 5e and f) after ReOx230_2 post treatment and the last WGS reaction of the post-treatment protocol (Fig. S1f†), showing that such species are stabilized under reaction conditions.
Raman spectra were also recorded to determine the structural changes after the different post-treatments (Fig. 6). The main band at 464 cm−1 arises from the F2g vibrational mode of cubic CeO2 and the less intense features at 264 and 402 cm−1 are attributed to surface modes of the clean CeO2 (111) surface.50 The band at 550–600 cm−1, also called D band, is attributed to the LO mode, which is activated by the presence of defects such as oxygen vacancies.50 The D band is superimposed to a band at 560 cm−1 corresponding to ν(Pt–O–Ce) vibrations.51,52 Another band at 650 cm−1 is attributed to either νs(Pt–O–Ce)52 or ν(Pt–O)51 stretching vibrations of PtOx species anchored on the surface of CeO2. For the sake of simplicity, it is labelled ν(PtOx) in the following. Finally, the band at 826 cm−1 arises from ν(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibrations of peroxo species adsorbed on oxygen vacancies.
O) vibrations of peroxo species adsorbed on oxygen vacancies.
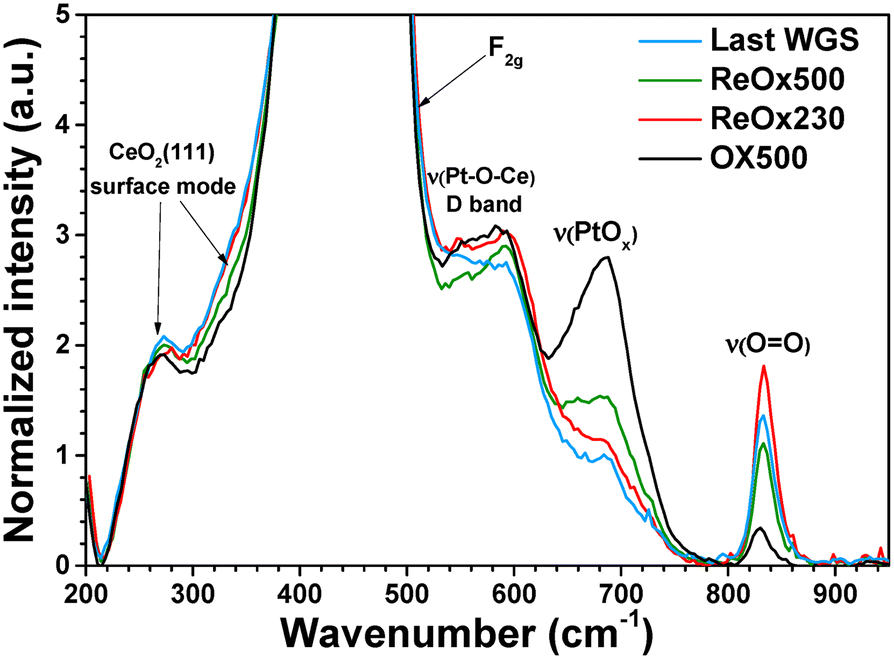 | ||
| Fig. 6 Raman spectra of 0.25Pt catalyst recorded in ambient air after OX500 pretreatment, after ReOx230_1 and ReOx500 post-treatments and after ReOx230_2 and WGS reaction (called 'Last WGS'). The spectra correspond to the average of spectra recorded from a mapping of 20 analysed areas and are presented after baseline subtraction and normalization to the amplitude of the F2g band. | ||
A reduction of PtOx species during WGS reaction is evidenced by the damping of the ν(PtOx) band.17 This band was still quite low after ReOx230 (Fig. 6), showing that if Pt reoxidation occurs it is limited. Note that the small ν(PtOx) feature may result either from the reoxidation of Pt0 atoms or the presence of Pt2+ cations that were not reduced during the reaction. The Raman spectrum recorded after ReOx500 shows a slight increase in the intensity of the ν(Pt–Ox) band at 680 cm−1, reflecting an oxidation of Pt atoms on the surface during the oxidative post-treatment at 500 °C. It is consistent with the redispersion of Pt NPs observed by microscopy. However, the intensity of the ν(PtOx) band does not reach that of the initial one, showing irreversible structural evolution after reduction under reaction conditions. Finally, the ν(PtOx) band in the spectrum recorded after ReOx230_2 post-treatment and the last WGS reaction of the sequence was quite low, showing again reduction of PtOx species under reaction conditions. Furthermore, the ν(OO) band at 826 cm−1 reveals the formation of peroxo species on the surface of catalysts after WGS reaction and exposure to ambient air. Those species arise from the reduction of O2 molecules chemisorbed on oxygen vacancies formed during the reaction. Such reduction induces the reoxidation of Ce3+ cations and/or Pt0 atoms. However, the process stops at this step since the dissociation of peroxo species leading to the formation of O− does not take place.53 Hence, reoxidation is only partial. Either a slight increase or a slight decrease in the ν(OO) band is observed after ReOx230 and ReOx500, respectively. However, its intensity remains much higher than after OX500, revealing the presence of a higher amount of oxygen vacancies than at the initial state.
From these results, it can be concluded that both reoxidation and partial redispersion of Pt0 atoms take place during an oxidative post-treatment at 500 °C for 12 h, while only limited reoxidation occurs during an oxidative post-treatment at 230 °C for 10 min.
As CO is more reducing than H227 and is the main reducing molecule under WGS reaction conditions, CO-TPR analysis was preferred to H2-TPR to probe the reducibility of Pt/CeO2 catalysts after oxidative post-treatments. Fig. 7 shows the production of CO2 and H2 and the consumption of CO during CO-TPR analyses between 20 °C and 500 °C for the three chosen Pt/CeO2 catalysts. They were carried out after 120 min of WGS reaction at 230 °C and ReOx500 post-treatment without air exposure. Very few differences are observed in the CO2 production and CO consumption profiles for the three Pt contents.
 | ||
| Fig. 7 CO consumption, CO2 production and MS signal intensity at mass 2 (H2) during TPR analysis under 1% CO/He for (a) 0.10Pt, (b) 0.52Pt and (c) 1.43Pt catalysts after 120 min of WGS reaction at 230 °C and ReOx500 post-treatment. | ||
During CO-TPR analyses, CO is adsorbed and rapidly reduces PtO into Pt0 and CeO2 into CeO(2-x) according to eqn (1) or (2), respectively:54
| PtO + CO → Pt0 + CO2 | (1) |
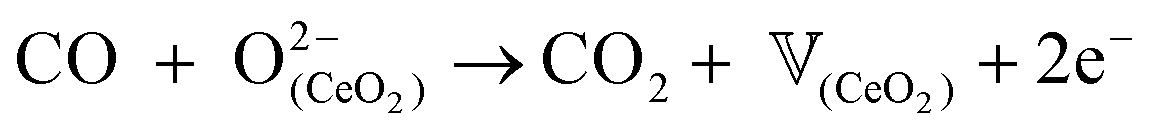 | (2) |
![[Doublestruck V]](https://www.rsc.org/images/entities/char_e179.gif) (CeO2) corresponds to an oxygen vacancy at the surface of the CeO2 support.
(CeO2) corresponds to an oxygen vacancy at the surface of the CeO2 support.
At higher temperature, a CO2 production peak is accompanied by H2 production (see the H2 signal intensity in Fig. 7) and can be attributed to the WGS reaction with hydroxyl species according to (eqn (3)):55
 | (3) |
On LPC catalysts, both CO2 production and CO consumption take place at much lower temperatures after ReOx500 post-treatment than after OX500 pretreatment (Fig. S13†). In particular, the peak temperatures of CO2 production after ReOx500 are much lower (Fig. 8), with values below 150 °C, which were obtained only for HPC catalysts after OX500.17
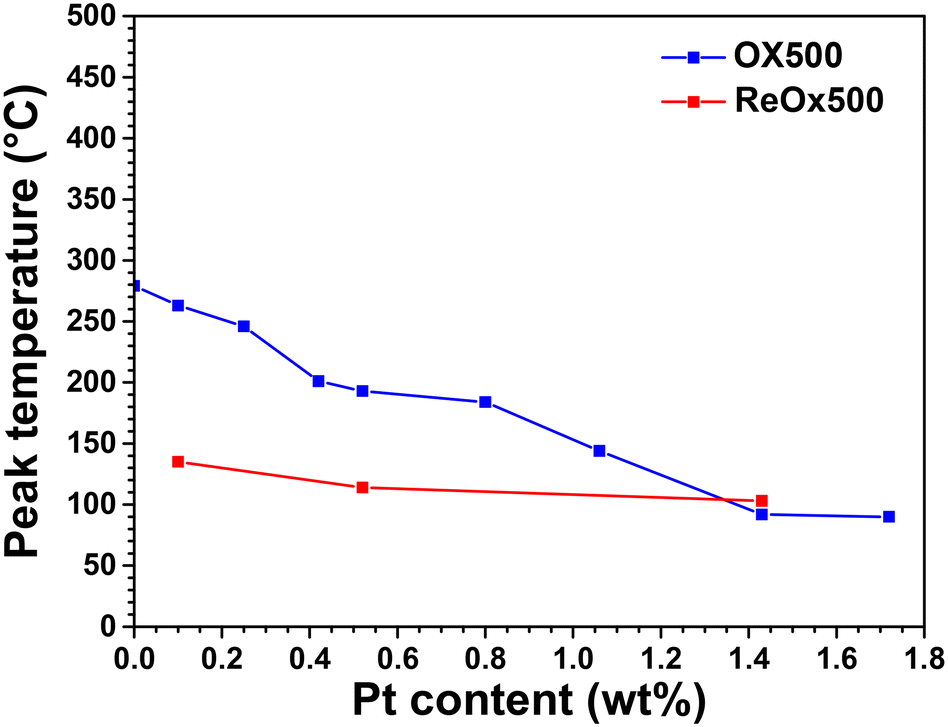 | ||
| Fig. 8 Evolution of peak temperature of CO2 production after OX500 pretreatment and after 120 min of WGS reaction at 230 °C followed by ReOx500 post-treatment. | ||
Thus, combining WGS reaction conditions and ReOx500 post-treatment strongly increases the reducibility of LPC catalysts, revealing an easy formation of Pt0 NPs under CO, which in turn leads to reduction of CeO2.17 Note that the same trends were observed for H2-TPR analysis (see Fig. S14† and associated comments) which is quite sensitive to the presence of Pt0 NPs because it allows reduction of CeO2 by dissociative adsorption of H2 and H spillover.33,34
Operando DRIFTS studies were carried out in order to better understand the origin of the activation of LPC catalysts after ReOx500. It was important to check that the same activity trends were obtained in the DRIFTS cell as in a conventional fixed-bed reactor. The activities of the different Pt/CeO2 catalysts during operando DRIFTS experiments are compared in Fig. S15.† Again, an activation is observed after ReOx500 for the LPC catalysts (0.10Pt, 0.25Pt and 0.52Pt). It is even more pronounced, which may be due to differences in feed composition.
The temporal evolutions of operando DRIFTS spectra between 1800 and 2200 cm−1 of the 0.25Pt, 0.52Pt and 1.06Pt catalysts are plotted for the first 10 min of reaction in Fig. 9a, b and c, respectively. For the three catalysts, the first spectrum recorded under reaction conditions after OX500 contains a ν(CO) band at 2090 cm−1 indicating the presence of PtOx clusters (Fig. 9a–c).32,54 This observation may seem to contradict the H2-TPR analysis, which indicates the presence of Pt2+ SAs for Pt loading below 0.59 wt% (Fig. 2a). However, PtOx clusters are probably rapidly formed at 230 °C under reaction mixture by the cleavage of Pt–O–Ce bonds and Pt diffusion, and are therefore immediately observed during operando DRIFT experiments. Indeed, reactive adsorption of CO was previously shown even at RT.1
 | ||
| Fig. 9 Operando DRIFT spectra recorded between 1800 and 2200 cm−1 during the first 10 min and at apparent steady state after 1 h of WGS reaction for the (a and d) 0.25Pt, (b and e) 0.52Pt and (c and f) 1.06Pt catalysts after OX500 pretreatment and ReOx230_1, ReOx500 and ReOx230_2 post-treatments. For the temporal evolution, the spectra are plotted from purple to dark red, each spectrum corresponding to approximately 1 min under reaction conditions. Reaction conditions: T = 230 °C, PCO = 5 mbar, PH2O = 20 mbar, GHSV = 1000 mL min−1 g−1. The background corresponds to the spectrum of KBr powder recorded at 230 °C and the ν(CO)gas signal is subtracted. | ||
Afterwards, PtOx species are rapidly reduced to form metallic NPs giving rise to characteristic ν(CO) bands at about 2065, 2045, and 2005 cm−1 attributed to CO chemisorption on terrace, edge and periphery sites of Pt0 NPs.17 The typical band at 2090 cm−1 was not observed after ReOx230_1 post-treatment, indicating that such treatment cannot form stable PtOx clusters from Pt0 NPs or very few. However, after ReOx500, the first spectrum of the three catalysts contains a shoulder at 2100 cm−1 showing the formation of PtOx clusters during this post-treatment (Fig. 9a–c). Furthermore, the spectra exhibit an intense band at 2010 cm−1 during the first minutes of reaction, which is then damped. For instance, it vanishes after 2 min of reaction for the 1.06Pt catalyst (Fig. 9c). This feature reflects the decrease in the amount of Pt atoms at the periphery of Pt NPs, and thus their agglomeration.
Moreover, the formation rate of Pt0 NPs is higher after ReOx500 than after OX500 for LPC catalysts: Indeed, for 0.25Pt and 0.52Pt catalysts after ReOx500, the DRIFT spectra rapidly evolve like the 1.06Pt one after OX500 pretreatment, leading to spectra typical of Pt0 NPs. Thus, the redispersion of Pt0 NPs during the oxidative post-treatment at 500 °C results in a higher number of reducible PtOx clusters than after OX500.
After 1 h on stream, no signal was detected on 0.10Pt even after ReOx500 (Fig. S16†). However, for 0.25Pt, a higher absorbance of the ν(CO) bands at 2076–2065 cm−1 is observed after ReOx500 than after OX500 together with a lower absorbance of the band at 2010 cm−1 (Fig. 9d). For 0.52Pt, the ν(CO) absorbance decreases sharply after ReOx230_1 and the band at 2050 cm−1 is slightly blue-shifted after ReOx500 (Fig. 9e), which is attributed to the presence of less defects in Pt0 NPs.56 As expected from the absence of impact of the oxidative post-treatments on the molar activities of 1.06Pt and 1.43Pt catalysts, no significant modification of the relative intensity of the bands at 2067, 2046 and 2012 cm−1 is observed at the steady state (Fig. 9f and S16b†).
Regarding the other spectral ranges, very few differences are observed between the three Pt contents. Between 3300 and 3800 cm−1 (Fig. S17†), the bands at 3680 and 3630 cm−1 are attributed to OH species of type II-A and II-B, respectively.46,47,57 Furthermore, two types of formates, denoted F1 and F2, and characterized by bands at 2935, 2842 and 2723 cm−1 and at 2944, 2831 and 2713 cm−1 were identified (Fig. S18†). As it was proposed from DFT calculations that bidentate formates are not stable on CeO2 in contrast to bridged ones,58 these bands could be attributed to two types of bridged formates. Finally, the 900–1800 cm−1 spectral range contains bands due to monodentate and bidentate carbonates as well as the two types of bridged formates (Fig. S19†). Hence, even if some species could be desorbed during the ReOx230 and ReOx500 post-treatments, the adsorbed species observed at the steady state of WGS reaction remain similar.
Thus, the oxidative post-treatment at 500 °C for 12 h triggers the formation of PtOx clusters on LPC catalysts which can rapidly evolve to Pt0 NP active species under WGS conditions at 230 °C, as schematized in Fig. 10, and then promotes the molar activity of ca. 50%, reaching the values obtained for HPC catalysts. However, they remain lower than the maximal value obtained for Pt content around 0.6 wt%, which was ca. 0.6 molH2 molPt−1 s−1 (Fig. 4).
 | ||
| Fig. 10 Scheme of the influence of oxidative post-treatment at 500 °C for 12 h on Pt species formation during the water gas shift reaction for 0.25Pt and 1.06Pt catalysts. | ||
4. Conclusions
The low reducibility under WGS conditions of Pt2+ SAs and/or PtOx clusters limits the operando formation of Pt0 NPs active for the WGS reaction at the surface of LPC catalysts. This study has shown that reducing pretreatments at 250 and 500 °C under H2 as well as an oxidative post-treatment under O2 at 500 °C can promote their reducibility. The gain in activity was attributed to an increase in the number of Pt0 NPs formed under LT-WGS conditions.For the reductive pretreatment, it was concluded that the choice of temperature is critical to activate the catalyst. Indeed, a reduction temperature of 250 °C increases the activity whatever the Pt content. After a reducing pretreatment at 500 °C, a 4-fold increase in activity compared to the initial state was measured for the 0.1Pt catalyst which contains mostly SAs. Conversely, catalysts with higher Pt content containing PtOx clusters and Pt0 clusters are deactivated by the same reductive treatment. The results support that a reductive pretreatment and the reaction conditions irreversibly modify the structure of Pt species, even if an oxidizing treatment is carried out afterwards.
Despite a slight deactivation of Pt/CeO2 catalysts, a recovery of activity was demonstrated after oxidative post-treatment at 230 °C for only 10 min, which is industrially interesting. Such a regeneration does not arise from redispersion of Pt NPs.
Finally, an original highlight of this work is the strong increase in activity observed after a 12 h oxidative post-treatment at 500 °C for LPC catalysts. Even if this treatment allows for redispersion and oxidation of Pt atoms, the initial Pt2+ SAs and/or PtOx clusters are not recovered. In fact, such treatment leads to the formation of highly reducible PtOx clusters on the surface of CeO2, which are easily transformed into Pt0 NP active species.
Data availability
The data supporting this article are available in the published article and its ESI.†Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The Lyon Doctoral School of Chemistry (ED206) is acknowledged for the scholarship of C. Molinet-Chinaglia. This work was supported by the French National Research agency (Agence Nationale de la Recherche (ANR)) in the framework of the DYCAT project (ANR-19-CE05-0038).References
- D. B. Pal, R. Chand, S. N. Upadhyay and P. K. Mishra, Renewable Sustainable Energy Rev., 2018, 93, 549–565 CrossRef CAS.
- P. Panagiotopoulou and D. I. Kondarides, Catal. Today, 2007, 127, 319–329 CrossRef CAS.
- H.-S. Roh, D.-W. Jeong, K.-S. Kim, I.-H. Eum, K. Y. Koo and W. L. Yoon, Catal. Lett., 2011, 141, 95–99 CrossRef CAS.
- C. G. Maciel, T. de F. Silva, E. M. Assaf and J. M. Assaf, Appl. Energy, 2013, 112, 52–59 CrossRef CAS.
- I. D. González, R. M. Navarro, W. Wen, N. Marinkovic, J. A. Rodriguéz, F. Rosa and J. L. G. Fierro, Catal. Today, 2010, 149, 372–379 CrossRef.
- G. Jacobs, Appl. Catal., A, 2004, 258, 203–214 CrossRef CAS.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- C. Ratnasamy and J. P. Wagner, Catal. Rev.: Sci. Eng., 2009, 51, 325–440 CrossRef CAS.
- O. Thinon, F. Diehl, P. Avenier and Y. Schuurman, Catal. Today, 2008, 137, 29–35 CrossRef CAS.
- B. Liu, A. Goldbach and H. Xu, Catal. Today, 2011, 171, 304–311 CrossRef CAS.
- T. Bunluesin, R. J. Gorte and G. W. Graham, Appl. Catal., B, 1998, 15, 107–114 CrossRef CAS.
- L. Piccolo, S. Loridant and P. Christopher, in Supported Metal Single Atom Catalysis, John Wiley & Sons, Ltd, 2022, pp. 377–423 Search PubMed.
- H. Yan, N. Zhang and D. Wang, Chem Catal., 2022, 2, 1594–1623 CrossRef CAS.
- Y. Li, M. Kottwitz, J. L. Vincent, M. J. Enright, Z. Liu, L. Zhang, J. Huang, S. D. Senanayake, W.-C. D. Yang, P. A. Crozier, R. G. Nuzzo and A. I. Frenkel, Nat. Commun., 2021, 12, 914 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- K. Yuan, Y. Guo, Q.-L. Lin, L. Huang, J.-T. Ren, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, J. Catal., 2021, 394, 121–130 CrossRef CAS.
- C. Molinet-Chinaglia, L. Cardenas, P. Vernoux, L. Piccolo and S. Loridant, Materials Today Catalysis, 2024, 4, 100046 CrossRef.
- A. Holmgren, F. Azarnoush and E. Fridell, Appl. Catal., B, 1999, 22, 49–61 CrossRef CAS.
- K.-M. Lee, M. Brito, J. DeCoster, K. Linskens, K. Mehdi, W.-I. Lee, E. Kim, H. Kim, G. Kwon, C.-Y. Nam and T. Kim, Mol. Catal., 2022, 528, 112465 CrossRef CAS.
- E. M. Slavinskaya, A. I. Stadnichenko, J. E. Quinlivan Domínguez, O. A. Stonkus, M. Vorokhta, B. Šmíd, P. Castro-Latorre, A. Bruix, K. M. Neyman and A. I. Boronin, J. Catal., 2023, 421, 285–299 CrossRef CAS.
- D. J. Fullerton, A. V. K. Westwood, R. Brydson, M. V. Twigg and J. M. Jones, Catal. Today, 2003, 81, 659–671 CrossRef CAS.
- D. Eisenbeil, P. Demel, M. Haas, H. Hamel, B. Betz, A. Dreizler, C. Beidl and M. Votsmeier, Top. Catal., 2023, 66, 943–953 CrossRef CAS.
- J. Lee, D. Shin, E. Lee, C. Li, J. M. Kim, J. W. Han and D. H. Kim, Appl. Catal., B, 2022, 305, 121038 CrossRef CAS.
- J. Lee, C. Li, S. Kang, J. Park, J. M. Kim and D. H. Kim, J. Catal., 2021, 395, 246–257 CrossRef CAS.
- L. Pastor-Pérez, V. Belda-Alcázar, C. Marini, M. M. Pastor-Blas, A. Sepúlveda-Escribano and E. V. Ramos-Fernandez, Appl. Catal., B, 2018, 225, 121–127 CrossRef.
- G. Ferré, M. Aouine, F. Bosselet, L. Burel, F. J. C. S. Aires, C. Geantet, S. Ntais, F. Maurer, M. Casapu, J.-D. Grunwaldt, T. Epicier, S. Loridant and P. Vernoux, Catal. Sci. Technol., 2020, 10, 3904–3917 RSC.
- A. M. Gänzler, M. Casapu, P. Vernoux, S. Loridant, F. J. Cadete Santos Aires, T. Epicier, B. Betz, R. Hoyer and J.-D. Grunwaldt, Angew. Chem., Int. Ed., 2017, 56, 13078–13082 CrossRef PubMed.
- K. Morgan, A. Goguet and C. Hardacre, ACS Catal., 2015, 5, 3430–3445 CrossRef CAS.
- X. Liu, W. Ruettinger, X. Xu and R. Farrauto, Appl. Catal., B, 2005, 56, 69–75 CrossRef CAS.
- M. Farnesi Camellone, F. Dvořák, M. Vorokhta, A. Tovt, I. Khalakhan, V. Johánek, T. Skála, I. Matolínová, S. Fabris and J. Mysliveček, ACS Catal., 2022, 12, 4859–4871 CrossRef CAS.
- A. Bruix, Y. Lykhach, I. Matolínová, A. Neitzel, T. Skála, N. Tsud, M. Vorokhta, V. Stetsovych, K. Ševčíková, J. Mysliveček, R. Fiala, M. Václavů, K. C. Prince, S. Bruyère, V. Potin, F. Illas, V. Matolín, J. Libuda and K. M. Neyman, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS PubMed.
- J. Resasco, L. DeRita, S. Dai, J. P. Chada, M. Xu, X. Yan, J. Finzel, S. Hanukovich, A. S. Hoffman, G. W. Graham, S. R. Bare, X. Pan and P. Christopher, J. Am. Chem. Soc., 2020, 142, 169–184 CrossRef CAS PubMed.
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2016, 120, 25870–25879 CrossRef CAS.
- A. Beck, D. Kazazis, Y. Ekinci, X. Li, E. A. Müller Gubler, A. Kleibert, M.-G. Willinger, L. Artiglia and J. A. van Bokhoven, ACS Nano, 2023, 17, 1091–1099 CrossRef CAS PubMed.
- C. Molinet-Chinaglia, L. Piccolo and S. Loridant, ChemCatChem, 2023, 15, e202300627 CrossRef CAS.
- V. Palma, A. Pietrosanto, M. Martino, E. Reverchon and I. De Marco, Chem. Eng. Trans., 2017, 57, 967–972 Search PubMed.
- L. Pastor-Pérez, E. V. Ramos-Fernández and A. Sepúlveda-Escribano, Int. J. Hydrogen Energy, 2019, 44, 21837–21846 CrossRef.
- J. Ashok, M. H. Wai and S. Kawi, ChemCatChem, 2018, 10, 3927–3942 CrossRef CAS.
- X. Wang, R. J. Gorte and J. P. Wagner, J. Catal., 2002, 212, 225–230 CrossRef CAS.
- W. Ruettinger, X. Liu and R. J. Farrauto, Appl. Catal., B, 2006, 65, 135–141 CrossRef CAS.
- S. Hilaire, X. Wang, T. Luo, R. J. Gorte and J. Wagner, Appl. Catal., A, 2004, 258, 271–276 CrossRef CAS.
- J. Lee, P. Tieu, J. Finzel, W. Zang, X. Yan, G. Graham, X. Pan and P. Christopher, JACS Au, 2023, 3, 2299–2313 CrossRef CAS PubMed.
- F. Maurer, J. Jelic, J. Wang, A. Gänzler, P. Dolcet, C. Wöll, Y. Wang, F. Studt, M. Casapu and J.-D. Grunwaldt, Nat. Catal., 2020, 1–10 Search PubMed.
- X. Li, Doctoral Thesis, ETH Zurich, 2023 Search PubMed.
- C. Binet, A. Badri and J.-C. Lavalley, J. Phys. Chem., 1994, 98, 6392–6398 CrossRef CAS.
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. Knop-Gericke, Z. Paál and R. Schlögl, J. Catal., 2006, 237, 1–16 CrossRef CAS.
- C. Binet, M. Daturi and J.-C. Lavalley, Catal. Today, 1999, 50, 207–225 CrossRef CAS.
- Y. Li, M. Kottwitz, J. L. Vincent, M. J. Enright, Z. Liu, L. Zhang, J. Huang, S. D. Senanayake, W.-C. D. Yang, P. A. Crozier, R. G. Nuzzo and A. I. Frenkel, Nat. Commun., 2021, 12, 914 CrossRef CAS PubMed.
- C. M. Kalamaras, S. Americanou and A. M. Efstathiou, J. Catal., 2011, 279, 287–300 CrossRef CAS.
- S. Loridant, Catal. Today, 2021, 373, 98–111 CrossRef CAS.
- W. Lin, A. A. Herzing, C. J. Kiely and I. E. Wachs, J. Phys. Chem. C, 2008, 112, 5942–5951 CrossRef CAS.
- M. S. Brogan, T. J. Dines and J. A. Cairns, J. Chem. Soc., Faraday Trans., 1994, 90, 1461–1466 RSC.
- M. Daniel and S. Loridant, J. Raman Spectrosc., 2012, 43, 1312–1319 CrossRef CAS.
- A. Holmgren, B. Andersson and D. Duprez, Appl. Catal., B, 1999, 22, 215–230 CrossRef CAS.
- H. Zhu, Z. Qin, W. Shan, W. Shen and J. Wang, J. Catal., 2004, 225, 267–277 CrossRef CAS.
- P. Bazin, O. Saur, J. C. Lavalley, M. Daturi and G. Blanchard, Phys. Chem. Chem. Phys., 2005, 7, 187–194 RSC.
- C. Li, Y. Sakata, T. Arai, K. Domen, K. Maruya and T. Onishi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 1451–1461 RSC.
- G. N. Vayssilov, M. Mihaylov, P. St. Petkov, K. I. Hadjiivanov and K. M. Neyman, J. Phys. Chem. C, 2011, 115, 23435–23454 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Protocols for pre-treatments and post-treatments, temporal evolutions of catalytic data, evolutions of molar activity with Pt loading, relative deviations of molar activity, ADF-STEM images, operando DRIFTS spectra, H2-TPR curves and data, CO-TPR curves and data. See DOI: https://doi.org/10.1039/d4cy00741g |
| This journal is © The Royal Society of Chemistry 2024 |