
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural dynamics of PtSn/SiO2 for propane dehydrogenation†
Kewei
Yu
 ab,
Matthew
Scarpelli
a,
Sagar
Sourav
ab,
Alfred
Worrad
ab,
J. Anibal
Boscoboinik
c,
Lu
Ma
d,
Steven N.
Ehrlich
d,
Nebojsa
Marinkovic
d,
Weiqing
Zheng
*ab and
Dionisios G.
Vlachos
*ab
ab,
Matthew
Scarpelli
a,
Sagar
Sourav
ab,
Alfred
Worrad
ab,
J. Anibal
Boscoboinik
c,
Lu
Ma
d,
Steven N.
Ehrlich
d,
Nebojsa
Marinkovic
d,
Weiqing
Zheng
*ab and
Dionisios G.
Vlachos
*ab
aDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy St., Newark, DE, USA. E-mail: weiqing@udel.edu; vlachos@udel.edu
bDelaware Energy Institute, University of Delaware, 221 Academy St., Newark, DE, USA
cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
dNational Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 24th July 2024
Abstract
PtSn bimetallic catalysts are among the best-performing propane dehydrogenation (PDH) catalysts. However, understanding these catalysts remains limited due to the intricate nature of bimetallic systems and their dynamic structural evolution under reaction conditions. To address this challenge, we employ various in situ/operando techniques, including UV-vis, CO diffuse reflective infrared Fourier transform spectroscopy (CO-DRIFTS), near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), and operando X-ray absorption spectroscopy (XAS), to elucidate the structural dynamics of PtSn/SiO2 catalysts under reduction and working conditions. Our investigation reveals that the interactions between Pt, Sn, and SiO2 support are strongly influenced by the synthesis procedures and the initial catalyst structure. Exposure to H2 causes a reversible Sn–OH formation observed by modulation excitation spectroscopy (MES). A sequentially impregnated catalyst with a nominal Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn ratio of 1:3 and a co-impregnated catalyst with a ratio of 1:2 exhibit optimal performance for PDH. Despite distinct synthesis procedures and bulk structures, these two catalysts exhibit comparable surface properties and PDH performance, attributed to the dynamic migration of Sn species and formation of a Pt-rich metal surface under reductive atmospheres.
:Sn ratio of 1:3 and a co-impregnated catalyst with a ratio of 1:2 exhibit optimal performance for PDH. Despite distinct synthesis procedures and bulk structures, these two catalysts exhibit comparable surface properties and PDH performance, attributed to the dynamic migration of Sn species and formation of a Pt-rich metal surface under reductive atmospheres.
1 Introduction
Propylene is a crucial petrochemical product with high demand globally.1–4 Propane dehydrogenation (PDH) has become economically viable with the shale gas revolution. Considerable research efforts have been devoted to developing and enhancing PDH catalysts.1,3,5 Among these, Pt-based catalysts are top-performing despite their relatively high cost.6 PtSn-based catalysts, in particular, have been commercialized due to their remarkable activity and selectivity.1,7,8 The synergistic effect between Pt and Sn, attributed to geometric and electronic factors, is pivotal to their superior performance.1,9 Specifically, Sn atoms break large Pt ensembles, suppressing structure-sensitive side reactions like coke formation. Additionally, they facilitate electron transfer to the Pt 5d band, weakening the adsorption of coke precursors, such as ethylene, leading to more facile olefin desorption.1 However, the exact mechanisms behind Sn′s promotional effects and the most active phase remain unclear due to the structural complexity of supported bimetallics.According to the Pt–Sn phase diagram, five Pt–Sn intermetallic phases exist alongside individual Pt and Sn species.10–12 Realistic catalysts usually comprise a mixture of these species with different sizes, depending on the synthesis, manifesting the system complexity. Numerous catalyst structures have been proposed. For example, Liu et al. suggested partially reduced Sn species interacting with Pt clusters at the metal/support interface on MFI zeolite,13 and found that smaller PtSn clusters perform better than nanoparticles.14 Ye et al. suggested that Sn–Pt sequential impregnation leads to Pt nanoparticles with a hexagonal Pt1Sn1 intermetallic shell,12 whereases Deng et al. found that Pt–Sn sequential impregnation renders a Pt–Sn alloy core and Sn-rich surface coexisting with SnO2 species.15 Xu et al., on the other hand, demonstrated that the supported PtSn nanoparticles possess a significant compositional variation.16 All these structural variances give rise to distinct catalytic properties of these catalysts. Further adding to the complexity is that the distribution of Pt and Sn, and consequently the prevalence of these species, can change dynamically with the reaction conditions. For example, Wang et al. used an acid to etch away the surface Sn species, revealing that the nanoparticle surface can self-adjust to a Pt3Sn1 configuration regardless of the bulk nanoparticle composition after reduction.17 Xing et al. found a Sn1Pt single-atom alloy phase forming after reduction, which evolves into a Pt3Sn1 phase during PDH.18 This dynamic behavior renders traditional ex situ characterizations inadequate for building structure–performance relations. Therefore, it is crucial to monitor the catalyst structure in situ/operando to capture the catalyst structure dynamics, and to identify the true active phase.
Recently, Motagamwala et al. reported a highly stable and selective PtSn/SiO2 PDH catalyst capable of operating at the thermodynamic limit with minimal deactivation.19 The superior performance was attributed to the weak interaction of PtSn nanoparticles with SiO2. Our study aims to delve into this catalyst and understand the Pt–Sn–SiO2 interactions by comparing catalysts synthesized with different methods and Pt:Sn ratios using in situ/operando techniques. We monitor the structure dynamics of the catalysts via techniques including UV-vis, CO diffuse reflective infrared Fourier transform spectroscopy (CO-DRIFTS), near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), and operando X-ray absorption spectroscopy (XAS). Alloyed PtSn is observed in the co-impregnated catalyst, while separated phases dominate the sequentially impregnated ones. Sn dynamics is revealed during the reduction process, engaging in stronger interactions with SiO2, elucidating key factors underlying the catalyst's superior performance and the optimal surface compositional range.
2 Materials and methods
2.1 Catalyst synthesis
PtSn/SiO2 with various Pt and Sn ratios were synthesized through incipient wetness impregnation (IWI). SiO2 (Fuji Silysia G6) was calcined at 700 °C for 1 h prior to impregnation. For sequentially impregnated samples, SnCl2 aqueous solution with a SnCl2:HCl molar ratio of 1:3 (made with concentrated HCl, SnCl2·2H2O, and DI water) was first impregnated into 1 g of SiO2. The resulting powder was subsequently dried under atmospheric conditions for 24 h before further drying at 120 °C in an air oven for 4 h and calcination at 500 °C for 3 h using a muffle furnace. The resulting powder is denoted as Sn/SiO2. Pt was introduced by impregnating an H2PtCl6 aqueous solution on Sn/SiO2 with the same subsequent drying process described above. The dried catalyst was directly reduced at 600 °C in 10% H2 for 2 h to obtain the final catalyst PtSn/SiO2-seq. For co-impregnated samples, the SnCl2 and H2PtCl6 aqueous solutions were premixed and then impregnated into 1 g of SiO2. The resulting powder was subsequently dried under atmospheric conditions for 24 h before further drying at 120 °C in an air oven for 4 h. The dried powder was directly reduced in 10% H2/He atmosphere at 600 °C for 2 h to obtain the final catalyst PtSn/SiO2-co. The nominal loading of Pt in all catalysts was 2 wt%. Nominal Sn loadings were varied with the atomic ratio of Pt and Sn indicated as xPtySn. Some catalysts were also calcined at 500 °C and then reduced in 10% H2 at 600 °C for comparison.
2.2 Characterizations
X-ray photoelectron spectrometry (XPS) data was obtained using a Thermo Fisher K-Alpha+ XPS system equipped with an Al-Kα X-ray monochromatic source and a hemispherical analyzer. Near ambient pressure (NAP) XPS was measured in a lab-based XPS system from Specs, capable of measuring at gas pressures up to ∼2 mbar and sample temperatures from −180 °C to 1000 °C, equipped with monochromated Al (1486.6 eV) X-ray source.
X-ray absorption spectroscopy (XAS) was performed at NSLS-II 7-BM QAS at Brookhaven National Laboratory. Samples were pelletized into discs of 11 mm diameter and heated in a Linkam HFS600E-PB4 stage (−195–600 °C). A portable gas handling system was used to supply certain gases to sample reactors. Data was collected in fluorescence mode.
In situ CO DRIFTS was conducted in a Praying Mantis high-temperature reaction chamber and measured in situ with a Nicolet iS50 FTIR spectrometer. The samples were loaded in the cell and reduced in 10% H2 at 600 °C for 30 min and ramped to 400 °C. The gas was then switched to 10% H2, 1% CO balanced by Ar. Spectra were measured during ramp down every 50 °C.
Diffuse-reflectance UV-visible (DR UV-vis) spectra were recorded with an AVANTES AvaSpec-ULS2048CLEVO-RS spectrometer equipped with an AvaLight-XE light source.
Modulation excitation spectroscopy (MES) was conducted in a custom-made low-dead volume spectroscopic cell.20 The gas flow was regulated using 8 thermal mass flow controllers (Brooks Instrument). Gas switching was realized by two 4-port/2-position switching valves (VICI Valco). Time-series data, including UV-vis and DRIFTS spectra, were Fourier-transformed, and only the fundamental frequency was chosen to obtain the phase domain spectra after the inverse-Fourier transform. The gases are switched between 10% H2 in Ar or 10% O2 in Ar, 50 sccm total, with a switching period of 120 s (60 s per switch). Typically, an integration time of 100 ms and an averaging number of 10 are used for UV-vis measurement. An integration time of ∼9 ms is used for DRIFTS measurement. The MES spectroscopic data were analyzed with software developed by our group, available on GitHub at https://github.com/worradal/wavey.
Bright-field high-resolution transmission electron microscopy (BF-HRTEM) was conducted on a JEOL 2010F transmission electron microscope with a field emission gun emitter at 200 kV. Samples were diluted in acetone and deposited on a lacey carbon film on copper grids provided by the Electron Microscopy Sciences. In situ heating experiments were conducted with a vacuum heating holder (Protochips) with a thermal E-chip (Protochips, Fusion) in JEOL 2010F.
2.3 Catalytic performance evaluation
Non-oxidative PDH reactions were conducted in an atmospheric pressure continuous flow tubular quartz reactor of 7 mm inner diameter. Typically, 50 mg pelletized catalyst (40–60 mesh) were mixed with quartz pellets to form a catalyst bed 15 mm in length. The bed was fixed by quartz wool and quartz pellets above and below it. The C3H8, He, and H2 flows were regulated by calibrated thermal mass flow controllers from MKS Instruments. Typical flow was 5 sccm C3H8 balanced by He, totaling 54.2 sccm.Conversion and selectivity were calculated based on the differences in inlet and outlet gas compositions. As shown in eqn (1) and (2), [C3H8]0 represents the inlet propane concentration; terms without the subscript represent outlet concentrations. The change in volumetric flow rate is neglected due to the high dilution. The carbon balance was 100 ± 5% in all cases.
 | (1) |
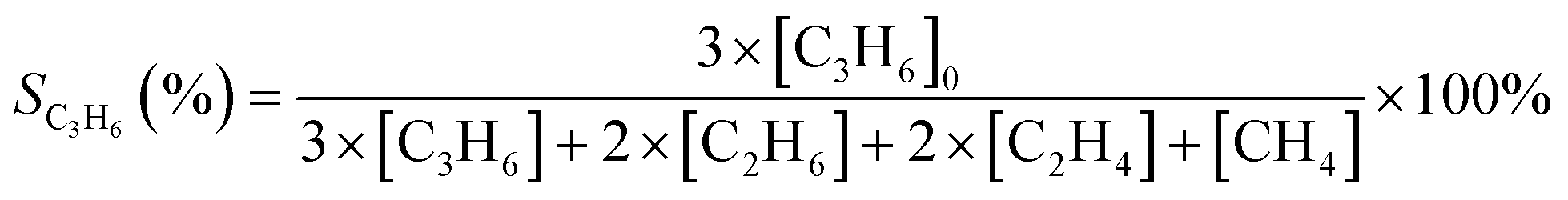 | (2) |
3 Results and discussion
3.1 Sn–SiO2 interactions in Sn/SiO2 and 1Pt3Sn/SiO2-seq
It has been hypothesized that the weak interaction between Pt, Sn, and the support is essential for the stability of Pt–Sn bimetallic catalysts.19,21–23 To vary the Pt–Sn interactions, we synthesize PtSn/SiO2 catalysts with different Pt:Sn ratios by co-impregnation and sequential impregnation as described in section 2.1.
Structural characterizations of Sn/SiO2 were conducted to elucidate the nature of Sn species on the SiO2 support and their interactions. Sn/SiO2 has been extensively investigated for various acid-catalyzed reactions, typically containing Sn4+ species as the Lewis acid site.24–28 The sample underwent calcination in air at 500 °C (denoted as Sn/SiO2-500C) and then was impregnated with Pt precursor when the sequentially impregnated PtSn/SiO2 catalyst was made. In Fig. 1a, the XRD data of fresh Sn/SiO2 post-calcination exhibits a pattern like that of the bare SiO2 support, indicating the highly dispersed Sn species, most likely in the form of Sn4+, or small clusters below 3 nm. Minor diffraction patterns corresponding to tetragonal rutile SnO2 exist (black triangles), suggesting the presence of small-sized crystalline SnO2 clusters. EDS mapping analysis of Sn/SiO2 in Fig. S2† shows the uniform dispersion of Sn over the support, while the HRTEM images confirm the existence of SnO2 clusters (Fig. S1a†).
 | ||
| Fig. 1 Characterizations of Sn/SiO2. (a) XRD of Sn/SiO2; (b) FTIR of pyridine adsorbed on Sn/SiO2-500C, Sn/SiO2-500C-600R, and 1Pt3Sn/SiO2-seq. Band intensities are normalized to the feature bands of SiO2 (2104–1730 cm−1). (c) MES DR UV-vis of Sn/SiO2 at 500 °C, original spectra during alternating O2–H2 feed; (d) phase domain UV-vis spectra after phase-sensitive detection (PSD) analysis. Gas feed: 10% O2/Ar or 10% H2/Ar, total 50 sccm gas flow. Modulation frequency = 1/120 Hz (period = 120 s). | ||
However, after the reduction of Sn/SiO2 at 600 °C in 10% H2 (denoted as Sn/SiO2-500C-600R), the SnO2 clusters vanish in XRD (Fig. 1a) and TEM (Fig. S1b†). Due to the facile reduction of SnO2,29,30 it is likely that SnO2 clusters got reduced to SnO and subsequently diffused into the porous SiO2 support.31In situ reduction of Sn/SiO2 was conducted with a TEM holder capable of heating in vacuum, using the electron beam as the reducing agent.32 As shown in Fig. S3,† at 600 °C, the SnO2 clusters undergo gradual reduction and disappear after 40 min of heating. Sn species may be prone to evaporation in vacuum under electron beam irradiation, affirming the high mobility of reduced Sn species at high temperatures.
Pyridine FTIR shed light on the surface chemical properties of Sn/SiO2. In Fig. 1b, adsorbed pyridine exhibits bands at 1613 cm−1 (ν8a), 1491 cm−1 (PyH+ and coordinatively bound pyridine), and 1454 cm−1 (ν19b), characteristic of Lewis acid sites, likely originating from the Lewis acidic Sn4+ species.24,33,34 Reduction of Sn/SiO2 does not alter the intensity of the band but leads to a red shift of the 1613 cm−1 band to 1607 cm−1, attributed to the reduction of Sn4+ to Sn2+, resulting in a decrease in the Lewis acid strength of the Sn sites.26,35 This observation aligns with previous studies indicating the reduction of isolated Sn4+ to Sn2+ on Sn/SiO2 at 500 °C.26 Therefore, SnO2 clusters are unlikely to be present under PDH reaction conditions due to their instability in a reductive atmosphere.
DR UV-vis spectra further confirmed the structure of Sn/SiO2.24,36–38 The spectra were collected at 500 °C in alternating H2 and O2 atmospheres (Fig. 1c). A main absorption band at 236 nm can be assigned to tetrahedral Sn sites.36–38 A weak shoulder around 306 nm, extending beyond 350 nm, is attributed to SnO2, which has a Rutile structure consisting of a mixture of corner and edge-sharing SnO6 octahedra.38,39
Interestingly, alternating the gas feed between H2 and O2 at 500 °C induces reversible changes in the UV-vis spectra (Fig. 1c, top). Phase domain spectra (Fig. 1d) reveal changes in characteristic bands at 213 nm, 288 nm, and 377 nm. The bands at 213 nm and 288 nm exhibit the same phase (Fig. S4†) and can be related to the redox behavior of the tetrahedral Sn4+ and the SnO2 clusters, respectively, as per previous peak assignments. Upon switching from O2 to H2, the intensities of the 213 nm and 288 nm bands decrease while the 377 nm band increases, suggesting the reduction of Sn4+ species into forms (discussed below) represented by the 377 nm band.40
The UV-vis band at 377 nm, previously unreported in the literature, likely corresponds to the formation of Sn2+–OH through the reduction of Sn4+. This inference is supported by its intensification upon switching from O2 to H2. To further investigate this, we conducted MES-DRIFTS, a technique more sensitive to –OH species. In Fig. 2a and S5a,† under alternating H2 and O2 feeds, the SiO2 support shows minor changes, with an almost flat profile between 4000 and 1500 cm−1. The perturbations observed in the –OH vibration region (3745 and 3735 cm−1) may arise from the reversible formation of hydrogen bonds upon introducing hydrogen.41,42 Gas switching on Pt/SiO2 induces band variation at 2042 cm−1 in Fig. S5b† due to Pt-hydride formation.43 Little change is observed in the O–H region (Fig. 2b), suggesting that H2 prefers to interact with Pt rather than –OH when Pt nanoparticles are present.
 | ||
| Fig. 2 Phase domain MES-DRIFTS-FTIR in O–H vibration region for: (a) SiO2; (b) Pt/SiO2; (c) Sn/SiO2; and (d) 1Pt3Sn/SiO2-seq. Gas feed: 10% O2/Ar or 10% H2/Ar, total 50 sccm gas flow. Modulation frequency = 1/120 Hz (period = 120 s). Temperature: 400 °C. All figures have the same y-scale. | ||
On the other hand, significant changes are observed in Sn-containing samples. In Fig. S5c,† a broad and intensive background shift between 3636 cm−1 and 2000 cm−1 is present for Sn/SiO2, absent in SiO2 (Fig. S5a†). This background shift likely originates from Sn species, indicating a strong interaction between Sn and the SiO2 support, resulting in a wide-range background alteration. A similar background shift, albeit with weaker intensity, is observed for 1Pt3Sn/SiO2-seq (Fig. S5d†), extending only to 3636 cm−1, suggesting less Sn species in direct contact with SiO2 compared to Sn/SiO2. In the O–H vibration region of Sn/SiO2 (Fig. 2c), besides changes in isolated silanol groups at 3744 cm−1, perturbations are also observed at slightly lower wavenumbers between 3739 and 3636 cm−1. These changes also exist in 1Pt3Sn/SiO2-seq (Fig. 2d), but not in SiO2 (Fig. 2a) and Pt/SiO2 (Fig. 2b), indicating their association with Sn species. This is likely due to the formation and removal of Sn–OH in H2 and O2 atmospheres, previously reported around 3665 cm−1.24,44 This agrees with the UV-vis band changes at 377 nm in Fig. 1d.
Based on the spectroscopic results, Sn in 1Pt3Sn/SiO2-seq is partially alloyed with Pt, whereas Sn alone exhibits a strong interaction with the SiO2 support, forming Sn2+–OH species under a reducing environment. This dynamic behavior of the PtSn catalyst under alternating gas environments provides valuable insights into the relative location of Pt and Sn species in the catalyst.
3.2 Pt–Sn interactions in PtSn/SiO2-seq and PtSn/SiO2-co
The impregnation of Pt on Sn/SiO2 followed by subsequent reduction at 600 °C leads to a significantly lower pyridine band area (Fig. 1b) indicating interactions between the Pt precursor with Sn sites during impregnation and the persistence of isolated Sn sites on SiO2. This is likely due to the preferential adsorption of [PtCl6]2− ions on the Lewis acidic Sn4+ sites, driven by electrostatic interactions.45 Upon reduction, part of the surface Sn4+ is incorporated into the Pt nanoparticles, forming a PtSn alloy. Pt loadings were kept constant across all samples, while Sn loadings were varied. In Fig. 3a, XRD of Pt/SiO2 shows fcc Pt(111) diffraction pattern at 39.801°, consistent with standard Pt structure (PDF 03-065-2868). XRD of the sequentially impregnated PtSn/SiO2 with increasing Sn:Pt ratio shows a continuous shift of the Pt(111) pattern toward lower angles, suggesting the incorporation of Sn into Pt and the resulting expansion of Pt lattice.10,15 The broadening of the Pt(111) pattern with increasing Sn:Pt ratio suggests random incorporation of Sn, yielding Pt–Sn nanoparticles with a distribution of Sn:Pt ratios that systematically shifts with increasing Sn:Pt ratio. Despite increasing the Sn:Pt ratio to 4:1, no Pt1Sn1 intermetallic phase was detected in XRD. HAADF-STEM images show particles with an average size of 2.10 nm (Fig. 3c and S8†). HRTEM images show particles with a lattice spacing of 2.33–2.34 Å, consistent with a Pt3Sn1 intermetallic phase and the XRD results. This suggests Sn species on the SiO2 support not alloyed with Pt, consistent with the spectroscopic findings in the previous section. EDS mappings (Fig. 3e–h) show an almost overlapping distribution of Pt and Sn elements on SiO2 support, confirming alloy formation.
 | ||
| Fig. 3 Structural characterizations of PtSn/SiO2. (a) XRD of PtSn/SiO2-seq with varying Pt:Sn ratios; (b) XRD of PtSn/SiO2-co with varying Pt:Sn ratios; (c and d) HAADF-STEM of 1Pt3Sn/SiO2-seq; (e–h) EDS mapping of 1Pt3Sn/SiO2-seq. | ||
For comparison, PtSn/SiO2 with comparable Pt and Sn loadings were also synthesized by co-impregnation. Sn and Pt precursors were pre-mixed before one-step impregnation. Upon mixing the Pt and Sn precursor solutions, the Pt precursor changed color from a bright yellow to dark brown, suggesting the formation of Pt–Sn complexes.19,21,46–48 This ensures the molecular-level mixing between Pt and Sn. In Fig. 3b, Pt1Sn1 intermetallic phase starts forming in the 1Pt1Sn/SiO2-co sample. Further increase in the Sn amount led to the disappearance of Pt and Pt3Sn1 patterns, leaving only the Pt1Sn1 phase. This contrasts with PtSn/SiO2-seq, where no Pt1Sn1 phase was observed, indicating a higher degree of alloying for co-impregnated samples. Additionally, the particle sizes of co-impregnated samples are much less uniform than sequentially impregnated ones, especially for high Sn-loading samples (Fig. S6†). This suggests that the metal complex–support interaction during the impregnation phase is weakened in co-impregnation, likely due to the formation of Pt–Sn complexes.46,48 After calcination and reduction of co-impregnated samples, the resulting catalysts have much broader particle size distributions and complicated phase compositions (Fig. S7†). Overall, PtSn/SiO2-seq samples show the most uniform particle size distribution across the whole composition range. Therefore, direct reduction was used for all catalyst synthesis.
In situ CO-DRIFTS provides valuable insights into the surface properties of PtSn catalysts. Fig. 4 shows the CO adsorption on three samples: 1Pt3Sn/SiO2-seq, 1Pt2Sn/SiO2-co, and Pt/SiO2. Sn-containing samples exhibit a lower CO stretching frequency than Pt/SiO2 at all temperatures. This phenomenon can be attributed to the electronic effect of Sn on Pt, weakening CO adsorption on the Sn-modified Pt surface.49–51 Interestingly, the 1Pt3Sn/SiO2-seq and 1Pt2Sn/SiO2-co show almost the same CO stretching frequencies despite different Sn loadings and synthesis methods, suggesting similar catalytic surface structures. Beyond band position, peak shapes also differ: in Fig. 4c, Pt/SiO2 displays a main peak with a shoulder at higher wavenumbers, assigned to the high-Miller-index step or corner and terrace sites on Pt nanoparticles, respectively.52,53 In contrast, Sn-containing samples show a single band with no shoulder at higher wavenumbers. This hints at Sn blocking the low coordination step sites, and partially covering Pt particles, resulting in smaller Pt ensembles, as reported in the literature.1,50,54Fig. 4d depicts increasing CO coverages as the temperature decreases. The relative coverage increases substantially more for Sn-containing samples than Pt/SiO2, indicating stronger CO adsorption on pure Pt surface than Sn-containing Pt surface. As the temperature drops from 100 °C to 50 °C, the coverage on 1Pt3Sn/SiO2-seq continues to increase, while it decreases slightly for 1Pt2Sn/SiO2-co. This suggests that the Sn-rich 1Pt2Sn/SiO2-co nanoparticles may undergo surface segregation of Sn below 100 °C.50 Consequently, the catalyst surface structure may markedly differ at low temperatures compared to working conditions, underscoring the importance of examining the catalyst under working conditions.
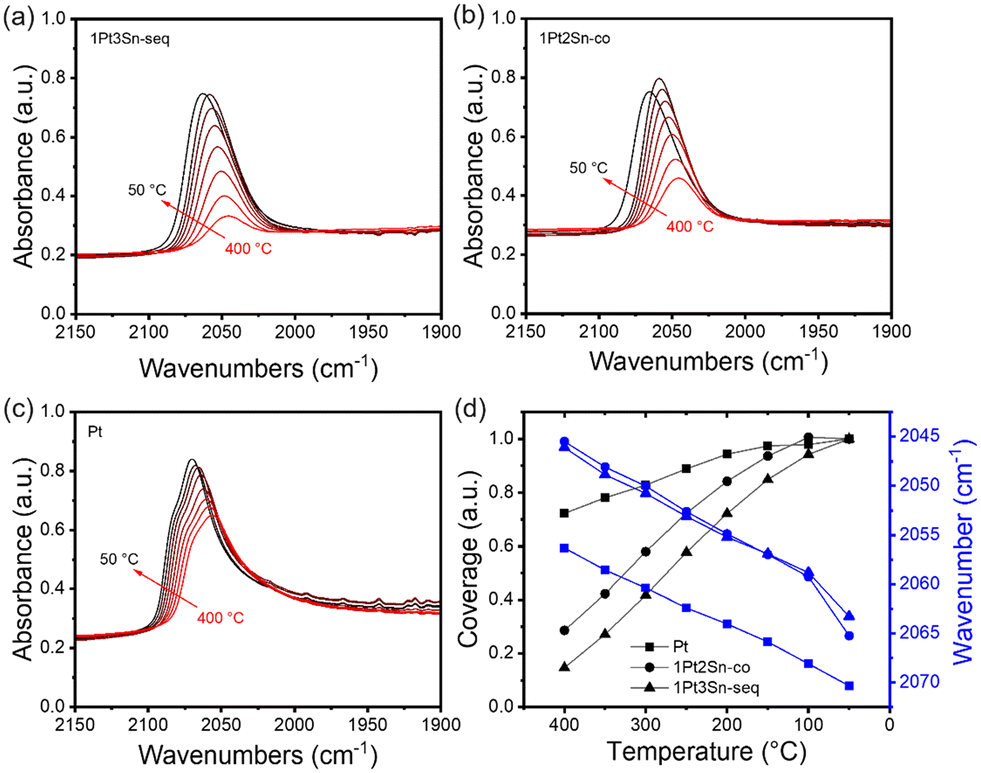 | ||
| Fig. 4 Temperature-programmed in situ CO-DRIFTS. (a) 1Pt3Sn/SiO2-seq; (b) 1Pt2Sn/SiO2-co; (c) Pt/SiO2; (d) comparison of temperature versus peak area and peak center location. Band area obtained by integration of 2120–1960 cm−1. Samples are reduced in 10% H2 at 600 °C for 30 min, then ramped to 400 °C, and switched to 10% H2, 1% CO cofeed. Spectra collected during further ramp-down every 50 °C. | ||
3.3 Structural dynamics of PtSn catalyst under working conditions using in situ XAS and NAP-XPS
In situ XAS reveals the Pt–Sn interaction of the bulk catalyst during reduction. Fig. 5a displays the temperature-dependent Pt LIII-edge XANES spectra of 1Pt3Sn/SiO2-seq in 5% H2. As the sample is heated from 100 °C to 200 °C, contrary to the typical downward shift in edge energy and white line energy during reduction,55,56 both edge energy and white line energy shift upward by about 1 eV. This energy shift has been associated with Pt interaction with Sn, caused by electron donation from Sn to Pt,56,57 suggesting an increase in Pt–Sn alloying during reduction. The white line intensity reflects the vacancy population of Pt 5d orbital.56 With increasing temperature, the Pt LIII white line intensity slightly decreases, as shown in the inset of Fig. 5a. This is likely due to further alloying and a stronger electron donation from Sn. In the Fourier-transformed R space EXAFS spectra in Fig. 5b, no peak is present at ∼1.7 Å corresponding to Pt–O, suggesting a reduced Pt state at as low as 100 °C.15,58 The peak shapes are similar to those reported in the literature, suggesting the coexistence of Pt and PtSn phases.56,58,59 | ||
| Fig. 5 In situ XAS of 1Pt3Sn/SiO2-seq. (a) Pt LIII-edge XANES spectra, inset: zoom in view of white line region; (b) Fourier transform of the Pt LIII-edge EXAFS spectra; (c) Sn K-edge XANES spectra, isosbestic points indicated by the arrows; (d) Fourier transforms of the Sn K-edge EXAFS spectra, inset: zoom in view of 0–6 Å region, standard samples' intensities are multiplied by 0.5. Reduction condition: 5% H2/N2, 20 sccm total flow. | ||
Fig. 5c shows the Sn K-edge XANES spectra during the initial ramp-up of the reduction. The series of spectra exhibit 5 isosbestic points marked by the arrows (see Fig. S9†), indicating that the spectra are a linear combination of two principal components.60 The SnO2 standard (red dotted line) shares all 5 isosbestic points with the sample spectra, suggesting that a SnO2-like phase is one of the two components across all Pt–Sn catalysts. Conversely, Sn0 fails to capture the second isosbestic point, suggesting that the second component may have a slightly different structure than pure Sn0, likely influenced by Pt alloying. The white line intensity in Fig. 5c decreases rapidly as the sample is ramped from room temperature to 200 °C. At the same time, a minor change occurs at higher temperatures, suggesting that Sn species are predominantly reduced below 200 °C.
Fig. 5d shows Sn K-edge EXAFS spectra. The fresh 1Pt3Sn/SiO2-seq sample measured at room temperature (blue line) shows strong Sn–O scattering at 1.52 Å, indicating that Sn in the air-exposed fresh catalyst is mostly oxidized. The radial distance is slightly lower than 1.60 Å of SnO2 (red dotted line in Fig. 5d), suggesting that Sn is tetrahedrally rather than octahedrally coordinated with O as in bulk SnO2.61,62 Despite exposure to air, 1Pt3Sn/SiO2-seq lacks Sn–Sn scattering between 2.5 Å and 4 Å, which is prominent in bulk SnO2, indicating that the Sn species, although oxidized, are highly dispersed, consistent with our XRD and TEM characterizations in previous sections. As the reduction temperature rises, the intensity of Sn–O scattering between 1.5 Å and 2.0 Å decreases and shifts towards higher radial distances, as shown in Fig. 5d inset. These shifts are clear indications of the continuous Sn reduction.
XPS provides surface properties of the fresh catalysts (Fig. 6 and S10†). The calcined Sn/SiO2 has an initial Sn:Si atomic ratio of 0.0080, close to 0.0078 of 1Pt3Sn/SiO2-seq, indicating that the impregnation of Pt and subsequent reduction in H2 did not significantly affect the Sn dispersion on the SiO2 support. This suggests that most Sn remains close to the SiO2 support, while a small amount may get alloyed with Pt. XRD results in Fig. 3a also suggest a low degree of alloying in the sequentially impregnated samples, confirming this observation.
 | ||
| Fig. 6 NAP-XPS surface Sn oxidation states composition evolution with temperature. (a) 1Pt3Sn/SiO2-seq; (b) 1Pt2Sn/SiO2-co. Measurement conducted 1 mbar H2 or C3H8 at indicated temperatures. Samples are heated up to 600 °C in H2 then switched to C3H8 atmosphere at 600 °C. Original spectra in Fig. S8.† | ||
In contrast, 1Pt2Sn/SiO2-co has a Sn:Si ratio of 0.0032, suggesting a lower Sn dispersion. This aligns with the TEM results in Fig. S6,† as well as the low Pt:Si ratio of 0.0025, compared to 0.0051 of 1Pt3Sn/SiO2-seq, confirming larger metal particles in co-impregnated samples. The data suggests that Sn species tend to alloy with Pt rather than disperse on SiO2, consistent with the XRD results in Fig. 3b.
Apart from surface atomic ratios, the Sn oxidation states of the fresh catalysts also provide insight into the degree of alloying between Sn and Pt. Sn 3d5/2 core level peak of Sn/SiO2 centers around 488.6 eV, indicative of Sn4+ species, while 1Pt3Sn/SiO2-seq centers around 486.6 eV, and 1Pt2Sn/SiO2-co around 486.0 eV. The shift toward lower binding energy after Pt impregnation suggests that the Sn species in 1Pt3Sn/SiO2-seq and 1Pt2Sn/SiO2-co are more reduced, likely due to the partial alloying with Pt.
Next, samples were heated to 600 °C in 1 mbar H2 to assess the surface Sn oxidation state evolution. In Fig. S11,† Sn/SiO2 contains mostly Sn4+ species (81%). As the temperature rises, the fraction of Sn4+ in Sn/SiO2 decreases rapidly while Sn0 and Sn2+ increase. In the literature, bulk SnO2 is typically reduced above 500 °C.26,28,30,63 It is noteworthy that, in our case, surface reduction happens at a much lower temperature. The initial Sn4+ species are likely highly dispersed oligomers and are reduced more easily than bulk SnO2, forming Sn–OH species mentioned in the UV-vis and FTIR results. Additionally, the copper plate used to hold the sample may facilitate the dissociation of H2 and, thereby, the reduction of Sn species due to the small sample thickness on the copper plate.
The Pt-containing samples in Fig. 6 show a lower Sn4+ percentage before reduction, suggesting a smaller amount of Sn4+ species on the SiO2 support. 1Pt2Sn/SiO2-co contains 2% Sn4+ and 77% Sn0 prior to reduction, indicating considerable alloying between Sn and Pt. Furthermore, the Sn4+ percentages remain relatively constant during reduction for both PtSn samples, suggesting that these Sn4+ species are most likely isolated and have strong interaction with the support, making them difficult to reduce with H2.
With the rising temperature, 1Pt3Sn/SiO2-seq shows a decreasing Sn2+ percentage and an increasing Sn0 percentage, which follows the reduction process. However, 1Pt2Sn/SiO2-co shows the opposite trend: while the percentages of Sn2+ and Sn0 remain relatively constant, a slight increase in Sn2+ and a decrease in Sn0 are observed. This hints at a distinct structure of these samples despite a similar Pt and Sn content. It is likely that as the temperature increases, the Sn species in the Sn-rich 1Pt2Sn/SiO2-co nanoparticles becomes mobile and interacts with SiO2 at the periphery of the alloy nanoparticles, increasing the apparent oxidation states measured at the sample surface. Meanwhile, on 1Pt3Sn/SiO2-seq, Pt-rich nanoparticles facilitate H2 dissociation and assist the reduction and incorporation of the neighboring Sn species.
3.4 Propane dehydrogenation over PtSn/SiO2
It is known that adding Sn promotes the stability of PDH Pt catalysts. Fig. 7a illustrates the time-on-stream conversion of PtSn/SiO2-seq catalysts with various Sn loadings. Pt/SiO2 shows high initial activity but quickly deactivates in the first 10 min, with selectivity toward C3H6 also dropping significantly, likely due to the increasing relative contribution from gas phase cracking as the catalyst deactivates. Increased Sn loading maximizes the activity at a 1:3 Pt:Sn ratio for sequentially impregnated samples and a 1:2 ratio for co-impregnated samples.
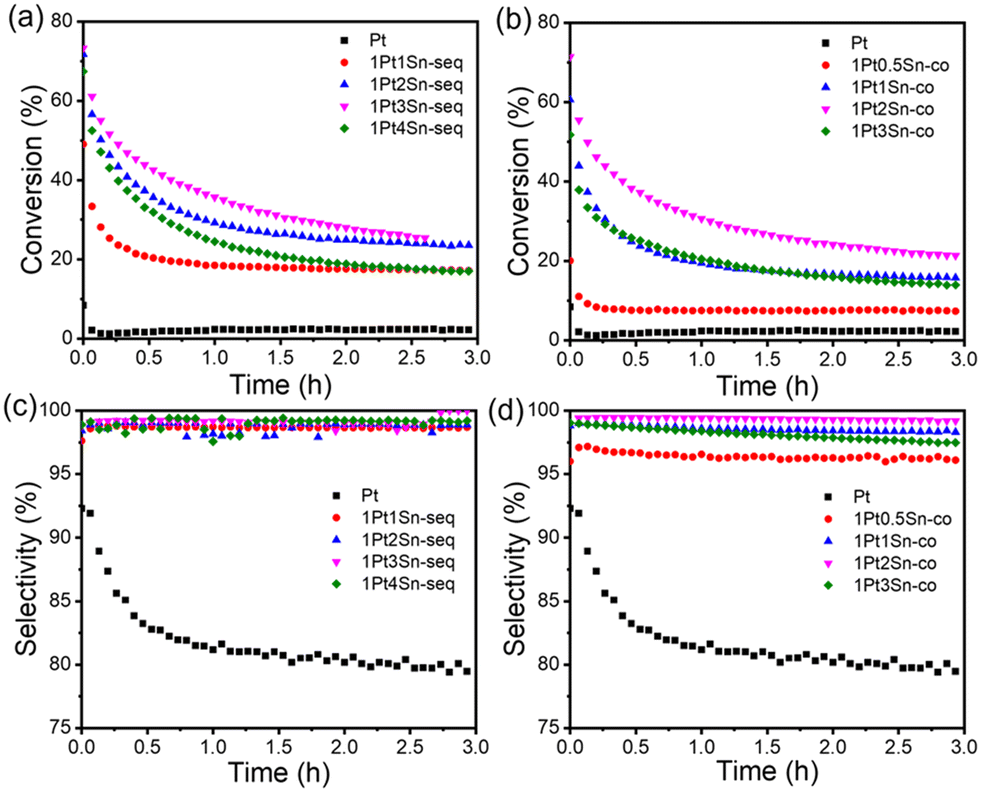 | ||
| Fig. 7 PDH activity of PtSn/SiO2 catalysts. (a) Conversion vs. time of PtSn/SiO2-seq; (b) conversion vs. time of PtSn/SiO2-co; (c) selectivity vs. time of PtSn/SiO2-seq; (d) selectivity vs. time of PtSn/SiO2-co. Reaction condition: 600 °C, 50 mg catalyst, 5% C3H8/He, 54.2 sccm total flow. Equilibrium conversion at 600 °C: 88.5%. | ||
To better understand the optimal surface configuration, we estimate the surface site densities and Pt:Sn atomic ratios in Table 1. For Pt/SiO2, the Pt site density (from pulse CO chemisorption) is 38.61 μmol g−1, and the Pt loading (from ICP analysis) is 1.63 wt%, resulting in a dispersion of 46.18% and a mean particle diameter of 2.39 nm, consistent with the 2.47 nm average size from HRTEM (Fig. S12†). CO chemisorption does not work for the Sn-containing samples due to the surface segregation of Sn at low temperatures.15,64 Instead, we estimate their Pt site densities from the CO band areas in DRIFTS. The Pt site densities and TOF for 1Pt3Sn/SiO2-seq and 1Pt2Sn/SiO2-co are summarized in Table 1. Despite the distinct synthesis procedures, bulk compositions (Fig. 3a and b), and particle sizes, these samples have comparable surface site density, TOF, and deactivation profile. Upon reduction, the nanoparticle surfaces likely self-adjust to a specific surface Pt:Sn ratio with similar active sites. The comparable DRIFTS CO frequencies of both Sn-containing samples at all temperatures in Fig. 4d also corroborate this point, as the CO frequency typically changes with the surface Pt:Sn ratio due to the electron donation from Sn to Pt.1,49
| Sample | CO chemisorption (μmol g−1) | Normalized DRIFTS CO area (a.u.) | Pt site density (μmol g−1) | TOF (s−1) | XPS Pt:Sn ratio |
XPS Pt:Si ratio |
XPS Sn:Si ratio |
|---|---|---|---|---|---|---|---|
| Pt/SiO2 | 38.61 | 0.3859 | 38.61 | — | — | 0.0038 | — |
| 1Pt3Sn/SiO2-seq | — | 0.3054 | 30.55 | 0.89 | 0.79 | 0.0056 | 0.0071 |
| 1Pt2Sn/SiO2-co | — | 0.3177 | 31.78 | 0.83 | 1.34 | 0.0063 | 0.0028 |
| Sn/SiO2 | — | — | — | — | — | — | 0.0093 |
Based on the XRD result in Fig. 3b, 1Pt2Sn/SiO2-co contains Pt1Sn1 phase in the bulk. Further increase of Sn amount results in a similar XRD pattern, suggesting that a Sn:Pt ratio higher than one does not alter the Pt1Sn1 pattern of the diffraction result. Therefore, the bulk of the nanoparticles 1Pt2Sn/SiO2-co have a composition of Pt1Snx (x ≥ 1). However, its XPS surface Pt:Sn ratio is above 1, suggesting surface Pt enrichment compared with the bulk. Additionally, during NAP-XPS in H2, the initial Pt:Sn ratio of the sample before reduction is 1.17, slightly lower than that measured during reduction, suggesting that a reductive environment could pull Pt to the surface driven by the chemical potential of H2.
On the other hand, the Pt(111) diffraction pattern of 1Pt3Sn/SiO2-seq is between that of Pt and Pt3Sn1 (Fig. 3a), indicating a bulk composition of Pt3Snx (x < 1). However, its XPS surface Pt:Sn ratio is below 1. This is understandable because much of the Sn species in 1Pt3Sn/SiO2-seq is on the support rather than in the metal particles, and XPS only gives the total surface composition, including the surface of the support and surface of the nanoparticles. Indeed, the XPS Sn:Si ratio of 1Pt3Sn/SiO2-seq is just slightly lower than Sn/SiO2, suggesting that most of the Sn species remain on the support after Pt addition, in direct contrast with 1Pt2Sn/SiO2-co, whose XPS Sn:Si ratio is significantly lower than Sn/SiO2. Therefore, despite the low XPS Pt:Sn ratio in 1Pt3Sn/SiO2-seq, the nanoparticle surface can still be Pt-rich and likely similar to that of 1Pt2Sn/SiO2-co due to their similar reactivity and CO DRIFTS results.
The discrepancy in compositions from different techniques highlights the concentration gradient from the nanoparticle bulk to the surface. While the final surface compositions for both samples are similar, 1Pt2Sn/SiO2-co features a Sn-rich core and Pt-rich surface, driven by the chemical potential of reductive atmospheres; whereas 1Pt3Sn/SiO2-seq features a Pt-rich core and Sn-rich surface, driven by the high Sn concentration at the support surface and the metal–support interface.
Based on the above analysis, the optimal surface Pt–Sn composition for PDH can be estimated. Co-impregnated samples show an increasing trend of TOF to a synthesis Sn:Pt ratio of 2.0 (Fig. S13†). Despite being a surface-sensitive technique, XPS still provides information from deeper than a few atomic layers near the surface. Therefore, the optimal surface Pt:Sn ratio can be slightly higher than 1.34 of 1Pt2Sn/SiO2-co. On the other hand, it cannot be higher than the ratio for the bulk of 1Pt3Sn/SiO2-seq, estimated to be 6.2 from XRD result based on Vegard's law.65,66 Therefore, a surface with 1.34 < Pt:Sn < 6.2 is optimal, indicating that a Pt-rich surface is essential for high PDH performance.
4 Conclusions
The PtSn/SiO2 catalyst is complex due to the various species present and its dynamic structure upon exposure to different temperatures and atmospheres. In this work, we synthesized the PtSn/SiO2 catalyst using different procedures and Pt:Sn ratios. Sequential impregnation generates separated Pt and Sn, while co-impregnation generates highly alloyed PtSn nanoparticles. The PDH activity is maximum at a 1:3 Pt:Sn ratio for the sequentially impregnated sample and a 1:2 ratio for the co-impregnated sample. Through in situ UV-vis and DRIFTS, we discovered that in addition to the reduction of Sn in alloy nanoparticles, Sn4+ species on SiO2 also get reduced, forming Sn2+–OH species in H2 at 500 °C. By in situ surface-sensitive techniques, such as XPS and CO DRIFTS, the surfaces of 1Pt3Sn/SiO2-seq and 1Pt2Sn/SiO2-co are alike and possess similar catalytic properties despite the distinct synthesis and bulk structure. In 1Pt2Sn/SiO2-co, Sn atoms migrate from the Sn-rich nanoparticles to the PtSn–support interface. In contrast, in 1Pt3Sn/SiO2-seq, Sn species close to the Pt-rich nanoparticles tend to get reduced and incorporated into them. The opposite Pt–Sn concentration gradient in co-impregnated and sequentially-impregnated samples highlights the effect of synthesis procedures on the surface structure and the need for a Pt-rich surface for optimal PDH activity and stability. This study focused on the dynamics during catalyst reduction. To obtain a full picture, the catalyst dynamics during regeneration is also worth investigating in future work.
Data availability
The data supporting this article have been included as part of the ESI.† The MES spectroscopic data were analyzed with software developed by our group, available on GitHub at https://github.com/worradal/wavey.Author contributions
Kewei Yu: methodology, investigation, data collection, writing – original draft; Matthew Scarpelli: data collection, investigation; Sagar Sourav: methodology, investigation, formal analysis, editing; Alfred Worrad: methodology, formal analysis, software; J. Anibal Boscoboinik: resources, methodology; Lu Ma, Steven N. Ehrlich, Nebojsa Marinkovic: resources, methodology; Weiqing Zheng: conceptualization, supervision, writing – review & editing; Dionisios G. Vlachos: conceptualization, resources, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation's “Designing Materials to Revolutionize and Engineer our Future” (DMREF) program under Award Number 2323700. This work was partly carried out at the Singh Center for Nanotechnology, supported by the NSF National Nanotechnology Coordinated Infrastructure Program under grant NNCI-2025608. Ex situ X-ray photoelectron spectroscopy conducted at the University of Delaware was sponsored by the National Science Foundation, Major Research Instrumentation, Award Number: CHE-1428149. This research used the Near ambient X-ray photoelectron spectroscopy of the Center for Functional Nanomaterials (CFN), a U.S. Department of Energy (DOE) Office of Science User Facility at Brookhaven National Laboratory under Contract No. DE-SC0012704. This research also used the 7-BM QAS beamline X-ray absorption spectroscopy of the National Synchrotron Light Source II, a U.S. DOE Office of Science User Facility operated by the Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- Z.-P. Hu, D. Yang, Z. Wang and Z.-Y. Yuan, Chin. J. Catal., 2019, 40, 1233–1254 CrossRef CAS.
- C. Li and G. Wang, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- N. S. Zadeh and S. Talebi, Shale Gas - New Aspects and Technologies, 2018, DOI:10.5772/intechopen.76542.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- M. Martino, E. Meloni, G. Festa and V. Palma, Catalysts, 2021, 11, 1070 CrossRef CAS.
- M. Farsi, A. Jahanmiri and M. R. Rahimpour, Asia-Pac. J. Chem. Eng., 2013, 8, 862–869 Search PubMed.
- C. DiGiulio, Overview of Honeywell UOP Oleflex™ in 2018 Spring AICHE Meeting, 2018 Search PubMed.
- Y. Liu, X. Zong, A. Patra, S. Caratzoulas and D. G. Vlachos, ACS Catal., 2023, 13, 2802–2812 CrossRef CAS.
- D. Y. DeSario and F. J. DiSalvo, Chem. Mater., 2014, 26, 2750–2757 Search PubMed.
- V. Grolier and R. Schmid-Fetzer, J. Alloys Compd., 2008, 450, 264–271 Search PubMed.
- C. Ye, M. Peng, Y. Wang, N. Zhang, D. Wang, M. Jiao and J. T. Miller, ACS Appl. Mater. Interfaces, 2020, 12, 25903–25909 CrossRef CAS PubMed.
- L. Liu, M. Lopez-Haro, C. W. Lopes, S. Rojas-Buzo, P. Concepcion, R. Manzorro, L. Simonelli, A. Sattler, P. Serna, J. J. Calvino and A. Corma, Nat. Catal., 2020, 3, 628–638 CrossRef CAS.
- H. Wan, L. Qian, N. Gong, H. Hou, X. Dou, L. Zheng, L. Zhang and L. Liu, ACS Catal., 2023, 13, 7383–7394 CrossRef CAS.
- L. Deng, H. Miura, T. Shishido, S. Hosokawa, K. Teramura and T. Tanaka, ChemCatChem, 2014, 6, 2680–2691 CrossRef CAS.
- C. Xu, S. Tan, Y. Tang, S. Xi, B. Yao, A. Wade, B. Zhao, S. Lu, Y. Du, M. Tian, C. He, L. Ma, X. Fu, J. Shi, J. Lu, A. G. R. Howe, S. Dai, G. Luo and Q. He, Appl. Catal., B, 2024, 341, 123285 CrossRef CAS.
- J. Wang, X. Chang, S. Chen, G. Sun, X. Zhou, E. Vovk, Y. Yang, W. Deng, Z.-J. Zhao, R. Mu, C. Pei and J. Gong, ACS Catal., 2021, 11, 4401–4410 CrossRef CAS.
- Y. Xing, L. Kang, J. Ma, Q. Jiang, Y. Su, S. Zhang, X. Xu, L. Li, A. Wang, Z.-P. Liu, S. Ma, X. Y. Liu and T. Zhang, Chin. J. Catal., 2023, 48, 164–174 CrossRef CAS.
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed.
- B. S. Patil, P. D. Srinivasan, E. Atchison, H. Zhu and J. J. Bravo-Suárez, React. Chem. Eng., 2019, 4, 667–678 RSC.
- N. Kaylor and R. J. Davis, J. Catal., 2018, 367, 181–193 CrossRef CAS.
- L. Deng, H. Miura, T. Shishido, Z. Wang, S. Hosokawa, K. Teramura and T. Tanaka, J. Catal., 2018, 365, 277–291 CrossRef CAS.
- H. Zhu, D. H. Anjum, Q. Wang, E. Abou-Hamad, L. Emsley, H. Dong, P. Laveille, L. Li, A. K. Samal and J.-M. Basset, J. Catal., 2014, 320, 52–62 CrossRef CAS.
- P. Ferrini, J. Dijkmans, R. De Clercq, S. Van de Vyver, M. Dusselier, P. A. Jacobs and B. F. Sels, Coord. Chem. Rev., 2017, 343, 220–255 CrossRef CAS.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- H. Wang, H. Huang, K. Bashir and C. Li, Appl. Catal., A, 2020, 590, 117291 CrossRef CAS.
- Y. N. Palai, A. Shrotri, M. Asakawa and A. Fukuoka, Catal. Today, 2021, 365, 241–248 CrossRef CAS.
- Y. Yue, J. Fu, C. Wang, P. Yuan, X. Bao, Z. Xie, J.-M. Basset and H. Zhu, J. Catal., 2021, 395, 155–167 CrossRef CAS.
- B.-S. Kim, J. Lee, H.-S. Yoon and S.-K. Kim, Mater. Trans., 2011, 52, 1814–1817 CrossRef CAS.
- F. Lan, X. Wang, X. Xu, R. Zhang and N. Zhang, React. Kinet., Mech. Catal., 2012, 106, 113–125 CrossRef CAS.
- M. L. Colaianni, P. J. Chen, J. T. Yates and M. Arbab, Appl. Surf. Sci., 1993, 68, 467–475 CrossRef CAS.
- T. Ma, S. Wang, M. Chen, R. V. Maligal-Ganesh, L.-L. Wang, D. D. Johnson, M. J. Kramer, W. Huang and L. Zhou, Chem, 2019, 5, 1235–1247 CAS.
- M. Tamura, K. Shimizu and A. Satsuma, Appl. Catal., A, 2012, 433–434, 135–145 CrossRef CAS.
- M. E. Z. Velthoen, S. Nab and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2018, 20, 21647–21659 RSC.
- G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723–736 RSC.
- S. Roy, K. Bakhmutsky, E. Mahmoud, R. F. Lobo and R. J. Gorte, ACS Catal., 2013, 3, 573–580 CrossRef CAS.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS.
- W. Dai, Q. Lei, G. Wu, N. Guan, M. Hunger and L. Li, ACS Catal., 2020, 10, 14135–14146 CrossRef CAS.
- J. A. Toledo-Antonio, R. Gutiérrez-Baez, P. J. Sebastian and A. Vázquez, J. Solid State Chem., 2003, 174, 241–248 CrossRef CAS.
- C.-M. Fan, Y. Peng, Q. Zhu, L. Lin, R.-X. Wang and A.-W. Xu, J. Phys. Chem. C, 2013, 117, 24157–24166 CrossRef CAS.
- B. A. Morrow and A. J. McFarlan, J. Phys. Chem., 1992, 96, 1395–1400 Search PubMed.
- R. S. McDonald, J. Phys. Chem., 1958, 62, 11 Search PubMed.
- A. I. Serykh, O. P. Tkachenko, V. Y. Borovkov, V. B. Kazansky, M. Beneke, N. I. Jaeger and G. Schulz-Ekloff, Phys. Chem. Chem. Phys., 2000, 2, 5647–5652 RSC.
- P. Fink, B. Camara, E. Welz and P. D. Ty, Z. Chem., 1971, 11, 473–474 Search PubMed.
- K. P. de Jong, Synthesis of Solid Catalysts, John Wiley & Sons, 2009 Search PubMed.
- G. T. Baronetti, S. R. de Miguel, O. A. Scelza, M. A. Fritzler and A. A. Castro, Appl. Catal., 1985, 19, 77–85 Search PubMed.
- N. Prakash, M.-H. Lee, S. Yoon and K.-D. Jung, Catal. Today, 2017, 293–294, 33–41 Search PubMed.
- J. F. Young, R. D. Gillard and G. Wilkinson, J. Chem. Soc., 1964, 5176–5189 Search PubMed.
- Q. Wang, D. Tichit, F. Meunier and H. Guesmi, J. Phys. Chem. C, 2020, 124, 9979–9989 Search PubMed.
- A. Moscu, Y. Schuurman, L. Veyre, C. Thieuleux and F. Meunier, Chem. Commun., 2014, 50, 8590–8592 Search PubMed.
- K. Balakrishnan and J. Schwank, J. Catal., 1992, 138, 491–499 Search PubMed.
- M. J. Lundwall, S. M. McClure and D. W. Goodman, J. Phys. Chem. C, 2010, 114, 7904–7912 CrossRef CAS.
- D. M. Haaland, Surf. Sci., 1987, 185, 1–14 CrossRef CAS.
- R. D. Cortright and J. A. Dumesic, J. Catal., 1994, 148, 771–778 Search PubMed.
- D. Eggart, X. Huang, A. Zimina, J. Yang, Y. Pan, X. Pan and J.-D. Grunwaldt, ACS Catal., 2022, 12, 3897–3908 CrossRef CAS.
- Y. Uemura, Y. Inada, K. K. Bando, T. Sasaki, N. Kamiuchi, K. Eguchi, A. Yagishita, M. Nomura, M. Tada and Y. Iwasawa, Phys. Chem. Chem. Phys., 2011, 13, 15833–15844 RSC.
- J. H. Kim, S. M. Choi, S. H. Nam, M. H. Seo, S. H. Choi and W. B. Kim, Appl. Catal., B, 2008, 82, 89–102 CrossRef CAS.
- G. J. Siri, J. M. Ramallo-López, M. L. Casella, J. L. G. Fierro, F. G. Requejo and O. A. Ferretti, Appl. Catal., A, 2005, 278, 239–249 Search PubMed.
- A. Borgna, S. M. Stagg and D. E. Resasco, J. Phys. Chem. B, 1998, 102, 5077–5081 CrossRef CAS.
- S. Calvin, XAFS for Everyone, CRC Press, 2013 Search PubMed.
- S. R. Bare, S. D. Kelly, W. Sinkler, J. J. Low, F. S. Modica, S. Valencia, A. Corma and L. T. Nemeth, J. Am. Chem. Soc., 2005, 127, 12924–12932 CrossRef CAS PubMed.
- C. Hammond, D. Padovan, A. Al-Nayili, P. P. Wells, E. K. Gibson and N. Dimitratos, ChemCatChem, 2015, 7, 3322–3331 Search PubMed.
- M. Haneda, Y. Ota, Y. Doi and M. Hattori, J. Mater. Sci., 2016, 51, 10949–10959 CrossRef CAS.
- L. Deng, X. Liu, Z. Wu, J. Xu, Z. Zhou and M. Xu, Catal. Lett., 2023, 153, 3665–3677 Search PubMed.
- A. R. Denton and N. W. Ashcroft, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 3161–3164 CrossRef CAS PubMed.
- E. Antolini, F. Colmati and E. R. Gonzalez, J. Power Sources, 2009, 193, 555–561 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00725e |
| This journal is © The Royal Society of Chemistry 2024 |