
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct dehydrogenation of methanol to formaldehyde over ZnO–SiO2-based catalysts†
Ankur Ghosh
Chowdhury
a,
Ulrich
Arnold
 *a,
Krassimir
Garbev
b,
Michael
Bender
c and
Jörg
Sauer
a
*a,
Krassimir
Garbev
b,
Michael
Bender
c and
Jörg
Sauer
a
aInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: ulrich.arnold@kit.edu
bInstitute for Technical Chemistry (ITC), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cBASF SE, Carl-Bosch-Straße 38, 67056 Ludwigshafen am Rhein, Germany
First published on 11th July 2024
Abstract
Direct dehydrogenation of methanol to formaldehyde and hydrogen is a “dream reaction” requiring catalysts, which are not only active in this highly endothermic reaction but also stable under harsh reaction conditions. Previous reports showed that materials with Zn2SiO4 exhibit a relatively high activity along with considerable long-time stability. However, neither detailed information on the physicochemical properties of such zinc silicates nor information on deactivation mechanisms was provided and discussed. In this study, the Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Si ratio has been varied to obtain different phases of zinc silicate and to investigate their specific activities in the methanol dehydrogenation reaction. Amorphous ZnO and SiO2, as well as crystalline phases of zinc oxide and zinc silicate, viz. α-Zn2SiO4 and β-Zn2SiO4, were present in almost all materials in different concentrations. The β-Zn2SiO4 phase was found to be relatively unstable in methanol dehydrogenation similar to ZnO, which is readily reduced to metallic Zn. Since detailed material characterization was not reported in studies before, the catalytic role of different phases present in zinc silicate materials for the target reaction remained unclear. Some aspects of this role are addressed within this work with a focus on α-Zn2SiO4 and its potential as a catalyst for direct methanol dehydrogenation.
:Si ratio has been varied to obtain different phases of zinc silicate and to investigate their specific activities in the methanol dehydrogenation reaction. Amorphous ZnO and SiO2, as well as crystalline phases of zinc oxide and zinc silicate, viz. α-Zn2SiO4 and β-Zn2SiO4, were present in almost all materials in different concentrations. The β-Zn2SiO4 phase was found to be relatively unstable in methanol dehydrogenation similar to ZnO, which is readily reduced to metallic Zn. Since detailed material characterization was not reported in studies before, the catalytic role of different phases present in zinc silicate materials for the target reaction remained unclear. Some aspects of this role are addressed within this work with a focus on α-Zn2SiO4 and its potential as a catalyst for direct methanol dehydrogenation.
Introduction
Although formaldehyde production has been carried out for decades on a world scale, it still bears considerable potential for optimization. A highly attractive option is the direct dehydrogenation of methanol, which yields hydrogen as a valuable by-product instead of water, which is inherently formed in conventional oxidative processes, as described in the following.Methanol is industrially produced from fossil raw materials using mixtures of CO, H2 and small amounts of CO2, at relatively high pressures from 50 to 100 bar and at temperatures between 200 and 300 °C using a Cu/ZnO/Al2O3 catalyst.1 Approximately 30% of the produced methanol is used to synthesize formaldehyde, which is employed in a variety of branches of the chemical industry.2,3 Currently, the water ballast process (with silver catalysts) and the Formox process (with iron oxide–molybdenum oxide catalysts) are primarily used in the industry to produce formaldehyde.4 In the Formox process, formaldehyde is produced through partial oxidation of methanol (reaction (2)), whereas in the water ballast process dehydrogenation (1) and partial oxidation (2) take place in parallel.4 Water formation can also occur through direct oxidation of formaldehyde and hydrogen.5
| CH3OH → CH2O + H2 | (1) |
| CH3OH + 1/2O2 → CH2O + H2O | (2) |
Monomeric formaldehyde is a very reactive chemical, which is stable in gas phase only from 110 to 300 °C depending on its gas phase concentration.6,7 Therefore, the product gas is cooled from reaction temperature to temperatures below 300 °C. The reactor is followed with a gas scrubber, where formaldehyde is condensed and removed from the effluent gas stream by using water as the primary solvent for absorption.8 Formalin, as a commercially available formaldehyde source, is usually a 40 vol% solution of formaldehyde in water, containing 10 to 12 vol% methanol as a stabilizer.9 For the energy efficient manufacturing of oxymethylene ethers (OMEs), highly concentrated formaldehyde or dry formaldehyde is required as feed.10–13 In order to do that, the commercially available formalin is concentrated via evaporation or vacuum distillation in many stages. In the final step, formaldehyde concentrations in the range of 80 to 90 vol% can be obtained, which is utilized as feed for manufacturing a series of formaldehyde derivatives. On the other hand, direct dehydrogenation of methanol to formaldehyde offers the possibility to produce anhydrous formaldehyde directly, and hydrogen as a lucrative by-product can be obtained.
Numerous studies have been published already, which disclose various catalyst materials such as metal oxides, metal alloys, zeolites and material combinations regarding the direct dehydrogenation of methanol.14,15 Catalyst deactivation is a crucial issue, and it seems that every system exhibits its very own deactivation mechanisms, viz. coking, volatilization of the active component, sintering or catalyst poisoning. Recent studies show that Na, Zn and Ga are promising elements for catalyst materials, whose salts and oxides exhibit remarkable catalytic activity.14–19 Regarding the work presented here, the focus is on ZnO materials, which gained some attention in recent years in the field of alkane dehydrogenation like the direct dehydrogenation of propane.20–27 A very recent study shows that bulk gallium oxide (Ga2O3) is also active in the non-oxidative methanol dehydrogenation, exhibiting an initial methanol conversion of 72% and a formaldehyde selectivity of 77% at 550 °C.16 The catalyst suffers deactivation mainly due to coking, whereas the reduction of Ga to a lower oxidation state is proposed to occur to some extent at and above 650 °C. ZnO, on the other hand, is readily reduced to Zn, which rapidly enters the gas phase because of its relatively high vapor pressure under reaction conditions.17 Metallic zinc can afterwards be observed at the reactor outlet, where it condenses on the outer wall.28 Apart from only a handful of fixed bed experimental studies, numerous adsorption–desorption experiments have been performed for methanol decomposition with ZnO systems.29–35
Secco pioneered the research to understand ZnO volatility, where it was peculiar to see that in the absence of any oxygen scavenging species, the ZnO material was still susceptible to oxygen loss at temperatures around 1000 °C,36 which is a relatively low temperature for thermal decomposition of metal oxides. In the 1980s, temperature programmed desorption (TPD) measurements combined with X-ray photoelectron spectroscopy (XPS) methods were used to comprehend the various adsorption sites and their specific adsorbates during methanol decomposition on the ZnO surface.37,38 Desorption of various oxidation products CO2 and H2O in the range from 350 to 800 °C was concluded to be the main reason for ZnO reduction, which is proposed to be active towards CO and H2 oxidation. Akhter found that on a specific defected surface of ZnO, the decomposition of methanol proceeded via oxidation, thereby forming methoxy species, which in turn form formate species at lower temperatures. The formate species eventually decompose to CO, CO2 and H2 above 400 °C.29 It was also concluded that oxidation reactions like conversion of CO to CO2 or H2 to H2O can be the reason for ZnO reduction.34
Throwing light on the ZnO material, Sagou and coworkers prepared ZnO from various precursors and found that ZnO prepared from zinc nitrate enabled a slightly higher formaldehyde yield of 22.4%, compared to ZnO obtained from zinc acetate. Further investigations were performed on various support materials, such as silica, γ-alumina, a mixture of silica and alumina, magnesium oxide and amorphous titanium oxide. It was revealed that supporting ZnO on silica doubled the formaldehyde yield to 44% and reduced the coking of ZnO.28
Mild oxidizing agents like H2O and CO2 in the feed counteract the deactivation problem by competing directly with CH3OH for the same active sites.39,40 This is quite different in alkane dehydrogenation chemistry, where the CO2 reacts with coke to release CO, thereby revealing a rather positive effect on the dehydrogenation reaction.23,41 The effect of CO2 has been investigated recently in our own work and it could be demonstrated that CO2 competes for the active sites.19 Thus, methanol conversion decreases with increasing CO2 content in the feed. However, at particular concentrations and temperatures, catalyst deactivation can be reduced to some extent.
Zinc oxide is an amphoteric solid displaying some manner of acidity and basicity depending on its reaction partner.42,43 Here, some characteristics are illustrated. At high temperatures, reactions with various transition metal oxides and typical catalyst support materials like SiO2, Al2O3 or TiO2 result in spinel type structures with the general formula AxByOz, where A and B represent the two metals.21,44–46 For example, industrially produced zinc titanate has been used to carry out H2S absorption from flue gases of coal gasifiers.47,48 The catalytic process sometimes requires temperatures from 650 to 750 °C, thereby also requiring the ZnO material to be chemically and thermally stable to enable an oxidation reaction. Usually, various phases of zinc titanate exist (ZnTiO3, Zn2TiO4, Zn2Ti3O8), which show different activities and stabilities in a specific reaction. Here, the formation of various phases can be attributed to two main factors. Firstly, the quantities of ZnO and TiO2, i.e. the specific Zn:Ti ratios are important for specific phase formation and secondly, the synthesis method followed by temperature treatment or calcination, which is crucial to obtain the desired phases of the material. Referring to previous investigations, it can be stated that several reports are available regarding the various types of stable and unstable zinc silicate phases in the ZnO–SiO2 system.49–51
Sagou and co-workers also described a zinc silicate catalyst, where ZnO was chemically bonded to the SiO2 support.28 The new material led to an initial decrease in methanol conversion. However, a constant catalyst stability for more than 500 h on stream was reached at relatively high CH3OH concentration in N2 carrier gas feed. The material was described to be a zinc–silica spinel catalyst with the chemical formula Zn2SiO4. Elemental analysis of the synthesized material revealed a content of 43.1 wt% Zn and 21.2 wt% Si and the rest is oxygen, which corresponds to the chemical formula Zn1Si1.02O3.3.28 It can be suspected that the material was not pure Zn2SiO4, but a mixture of various zinc silicates.
Within this study, the preparation of a bulk zinc silicate catalyst is addressed, by combining various amounts of Zn with Si at different calcination temperatures. By performing these iterations, various materials were obtained, which contained different phases of the zinc silicate material. These phases were identified and quantified using XRD and Raman spectroscopy. After a catalyst screening, one of the materials was chosen for deeper investigation. The tests revealed more information on the specific stable and active phases inside the zinc silicate material. Overall, the objective here was to obtain a high anhydrous formaldehyde yield alongside an indication of a stable zinc-based catalyst.
Experimental section
Materials and catalyst synthesis
The following chemicals were used as purchased: zinc nitrate hexahydrate (Zn(NO3)2·6H2O, Sigma-Aldrich, reagent grade 98%) as the zinc precursor and tetraethyl orthosilicate (TEOS, Sigma-Aldrich, for synthesis) as the precursor for silica. The molar ratio of Zn:Si was varied and four different ratios, namely 0.75, 1, 1.5 and 2, were chosen. The zinc salt was dissolved in an excess of ethanol, which served as a solvent. To this, the required amount of TEOS was fed in, while constantly stirring the solution at room temperature. After 30 minutes, nitric acid was added to the solution and the pH was adjusted to 4 thereby activating the sol.52
The solution was then allowed to stand until complete evaporation of the solvent took place through air convection.53 Within two days, the gel-like material turned into a white amorphous powder, which is known as the gel-desolvation effect.54 Afterwards, the powder was dried for 12 h at 120 °C. Calcination was performed in an oven under airflow at two different temperatures, viz. 600 and 900 °C. The heating rate was adjusted to 5 °C min−1 with about 30 mL min−1 of synthetic airflow and a dwell time of 5 h for the specified temperatures. This resulted into dumbbell-shaped particles.
The nomenclature of the samples was performed in a way that the number of the zinc silicate (ZS) represents the increasing Zn:Si ratio, where ZS1, ZS2, ZS3 and ZS4 represent a molar ratio of 0.75, 1, 1.5 and 2, respectively. This designation was combined with C1 and C2, which represent the calcination temperatures of 600 and 900 °C, respectively.
Material characterization
Phase compositions of the different zinc silicate catalysts were determined by means of XRD with Cu-Kα (40 mA, 45 kV) radiation. For the measurements, a Bruker D8 Advance diffractometer with an in situ reaction cell (XRK 900) from the company Anton Parr was used. The measured data were then analyzed quantitatively for various phases of zinc silicate using the software Topas6 from Bruker AXS. The direct Rietveld refinement method was used to quantify the weight percentages of various phases and amorphous structures. Meanwhile, due to the lack of structural data of β-Zn2SiO4, the reflections of this phase were fitted by introducing peaks in the position of the observed reflection corresponding to the ICDD card 014-0653. For α-Zn2SiO4 and ZnO, the structures from the ICSD entries 257027 and 34474 were used respectively. Detailed explanation of the quantification method can be found within the ESI.†N2 physisorption measurements were performed using a Quantachrome Novawin analyzer. Prior to the measurements, the samples were degassed for removal of moisture and absorbed gases at 130 °C with 20 h holding time. During the measurement, adsorption and desorption isotherms were recorded and the BJH fitting was applied to determine the surface areas of the catalysts. Not just BET measurements, but also using the weight of the sample and the relative pressure, multi-point surface area measurements were performed. The maximum deviation within the surface area measured by the two different methods was less than 1.5%, showing the accuracy of a single measurement.
For the characterization of the chemical composition of the catalyst bulk, ICP-OES measurements were undertaken. An Anton Paar Multiwave 3000 instrument was used for sample preparation, where a maximum amount of 500 mg of sample was completely dissolved in concentrated hydrogen fluoride solution (40%). An Agilent 725 ICP-OES spectrometer was then used to detect the ions, which were generated by ionizing the solution with the help of argon plasma. Here, the flow rate of Ar was maintained at 15 L min−1.
Confocal Raman spectroscopic analysis was performed on some zinc samples dusted on glass slides. A WITec alpha300 R equipped with a UHTS300 spectrometer (300 mm focal length) and a Zeiss microscope was employed for single spot measurements. A 488 nm laser operated at 50 mW (measured on the sample) was used as an excitation source. The measurements were performed with a 100× objective with a numerical aperture of 0.9 using a 1800 lines per mm holographic grating (space resolution better than 1 cm−1). A high performance back illuminated CCD camera with 96% quantum efficiency was used for detection. Typical acquisition times were between 5 and 20 s for 5–10 scans. 2D Raman mappings of typical areas of 15 × 15 μm were performed in 1 μm steps. The phase identification was aided with the RRUFF mineral database and our own database integrated in the WITec True Match program. The data processing and preparation of the Raman images were performed with the Project 5.3+ software from WITec. The single spectra were corrected with a cosmic ray removal algorithm and consequent subtraction of the background calculated by a shape algorithm. High quality single spectra of different zinc silicate polymorphs and zinc oxide were used for imaging of the 2D phase distribution using the module True Component Analyst of the Project 5.3+ software.
Material testing
The catalytic experiments were carried out in a laboratory setup, which is specially designed and built for the high temperature methanol dehydrogenation reaction. The plant comprises a dosing unit, a fixed bed reactor made from quartz glass, a separate section for analytical instruments and a section consisting of an absorption column for the safe disposal of formaldehyde gas.The dosing unit consists of a mass flow controller (MFC) and a mass flow meter (MFM) for dosing liquid methanol. The instruments were purchased from Bronkhorst. The MFM is coupled with a controlled evaporator mixer (CEM) to evaporate methanol. The MFC is also connected with the CEM, where N2 is used as the carrier gas and as a dilutant for the evaporated methanol.
The reactor unit contains a setup, where the quartz glass reactor (inside diameter: 9 mm, outside diameter: 12 mm) is heated with the help of an electric oven (length of heating zone: 450 mm) purchased from Horst GmbH, which in turn is controlled with an external temperature controller. The temperature of the catalyst bed is measured with the help of a 1 mm thin thermocouple, which is protected by a quartz glass tube from the outside (inside diameter: 3 mm, outside diameter: 5 mm).
During the feeding stage, liquid methanol flows under overpressure from He gas (purity: 6.0) firstly into the CEM. Coming from the CEM, the nitrogen-diluted methanol feed directly enters the top of the reactor. The catalyst fixed bed is placed precisely in between two quartz glass plugs. Quartz glass beads are positioned before the fixed bed, to ensure a homogeneous mixing of the feed before it reaches the catalyst bed.
Regarding the design of experiments, three different types of fixed bed experiments were performed within the scope of this study. Initially, there was a small and quick screening test performed. Here, the various synthesized zinc silicates were tested on the fixed bed. To do this, the particles were pressed, ground and sieved. The sieve fraction in between 350 and 500 μm was chosen for carrying out catalytic testing. All reactions were carried out at atmospheric pressure. For this, three reaction temperatures were tried, viz. 500, 550 and 600 °C. The amount of methanol in feed along with the amount of catalyst in the bed was kept constant at 4.2% CH3OH in N2 carrier gas and 200 mg of catalyst, respectively. Methanol conversion and formaldehyde selectivity were determined by online gas chromatography once after 20 min and also after 3.5 h from the start of the test. The relative error during a consecutive measurement of methanol conversion, in a stable and steady-state operation of the plant, was less than 2%. Therefore, fluctuations in methanol conversion more than this value were considered in the error calculation, are reflected by the corresponding error bars, and are understood to be a direct indication of catalyst deactivation. The screening test was performed to compare primarily two things: firstly, it was used to calculate the standard deviation for the methanol conversion from the start of the experiment until 3.5 hours in TOS. Secondly, the standard deviation values were used to characterize the C2 catalysts before and after usage as catalysts.
The screening tests were followed by a reaction-temperature study to determine catalyst performances at varying temperatures. Here, the concentration of methanol was 4.2% in N2 carrier gas. The amount of catalyst was fixed at 250 mg and the calculated WHSV at standard temperature and pressure was 212 h−1. The investigated temperature range was from 400 to 700 °C. The temperature was increased with a rate of 10 °C min−1. The target temperature was kept constant for 1 h in order to reach steady state. Two measurements were performed in succession. Here as well, the fluctuations in methanol conversion arising from the two consecutive measurements are indicated in terms of error bars calculated from standard deviation values.
For the experimentation, a starting time of 20 min was fixed, since all these four catalysts reached steady state after 20 minutes. On the other hand, after 3.5 hours, ZS1-C2 and ZS2-C2 were inactive especially at reaction temperatures >575 °C. That is why we have chosen a maximum of 3.5 h to display the activity of all the catalysts.
Lastly, long-term experiments were performed to test the longevity of some specific zinc silicate materials. During all trials, the amount of methanol in the feed along with the catalyst loading was kept constant, viz. 4.2% CH3OH in N2 carrier gas and 250 mg of catalyst was used, respectively. The reaction temperature for all the trials was kept constant at 550 °C. The choice of feed composition, catalyst loading and reaction conditions was driven by a good balance between catalyst stability and activity. Increasing the methanol concentration in the feed led to faster catalyst deactivation, which hindered the study of the catalysts at longer reaction times. Therefore, a methanol concentration of up to 5% was found to be suitable for the experiments.
The evaluation of the catalytic performance included methanol conversion and selectivity to products. The corresponding calculations were carried out as follows:
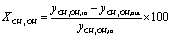 | (3) |
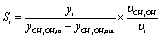 | (4) |
 | (5) |
| SH2O = 100 − SH2 | (6) |
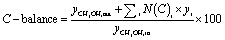 | (7) |
Results and discussion
Initially, surface properties have been determined and the dependency of the BET surface area of various zinc silicate materials on the Zn:Si ratio and calcination temperature has been studied (Fig. 1). For C1 substances, increasing the Zn:Si ratio led to a decrease in surface area. This can be explained by a decreasing amount of highly porous silica in the material caused by an increase in the relative amounts of zinc.55 For C2 materials, in contrast, the surface area goes through a minimum at a Zn:Si molar ratio of 1 and increases steadily as the zinc loading increases. The highest BET surface area was observed for ZS4-C2, which was 32 m2 g−1 followed by ZS3-C2 with 26 m2 g−1. Comparing the C1 and C2 materials, it can be stated that irrespective of the Zn:Si ratio, significant differences in the surface area can occur by increasing the calcination temperature. In previous work, it was concluded that at higher calcination temperatures, silica reacts to a larger extent with ZnO and leads to the formation of new structures, thereby also reducing the surface area of the material.53 The results during this study fit quite well with the fact that the bonding of ZnO with SiO2 requires higher temperatures than 875 °C.56,57 Here, it can be proposed that the increasing trend of the surface area of the C2 materials for Zn:Si > 1 is an indication of either unreacted ZnO and/or SiO2 within the material.
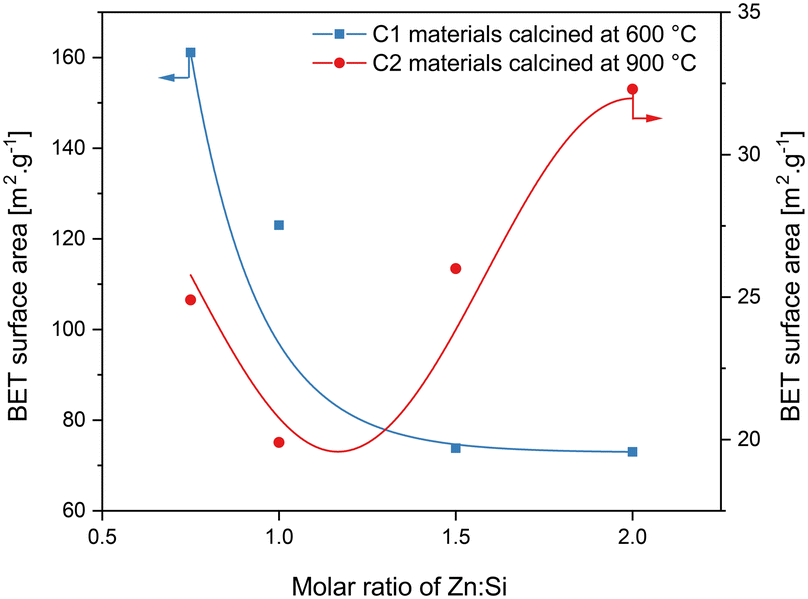 | ||
| Fig. 1 Molar Zn:Si ratio versus BET surface area of materials calcined at 600 °C (C1 materials) and 900 °C (C2 materials). | ||
The XRD patterns of all C1 and C2 materials are shown in Fig. 2. The thinner lines indicate the C1 materials, with a decreasing order of the Zn:Si ratio depicted from top to bottom. The first noticeable difference is the overall lower crystallinity of the C1 materials at lower zinc contents, viz. in ZS1-C1 and in ZS2-C1. As the zinc concentration increases, ZnO crystals can be detected, as in the case of ZS3-C1 and ZS4-C1. Below this Zn:Si ratio, the material is mostly amorphous, probably comprising only amorphous ZnO and SiO2.56
 | ||
| Fig. 2 XRD diffractograms of zinc silicate C1 and C2 materials (thinner lines: C1 materials and thicker lines: C2 materials). | ||
Therefore, it can be concluded that a certain calcination temperature is necessary for the formation of zinc silicates, which promotes the incorporation of ZnO into SiO2. The further discussion primarily focuses on the C2 materials. This is because the C1 materials contain less zinc silicate, which is because of the lower calcination temperature. The materials rather contain unreacted and sometimes amorphous ZnO, which is known to be an unstable material under the harsh conditions of methanol dehydrogenation. Fig. 2 and 3 show relevant information on the C2 materials. Chiefly, two main zinc silicate phases could be identified by XRD and Raman spectroscopy, viz. α-Zn2SiO4 and β-Zn2SiO4. The primary particles of the mixture can be described as Zn–O–Si phases, which are responsible for the formation of various zinc silicate phases and polymorphs.58
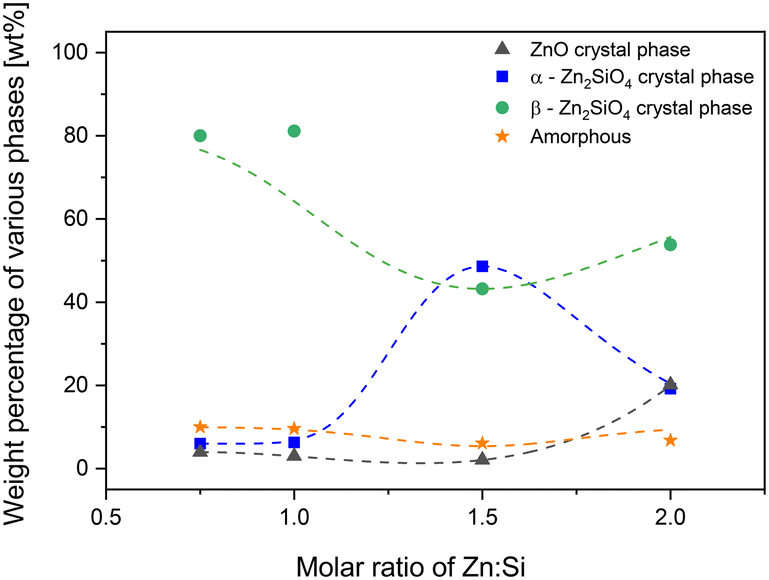 | ||
| Fig. 3 Percentage of zinc silicate and zinc oxide phases in the C2 materials as a function of Zn:Si ratio. | ||
Other zinc silicate phases, e.g. zinc metasilicate (ZnSiO3), hemimorphite (Zn4Si2O7(OH)2·H2O) or sauconite (Zn3Si4O10(OH)2·4H2O), were not observed. The reason for this could be that these phases are not stable at 900 °C and atmospheric pressure. In the previous literature on zinc silicates, a phase with a composition of Zn1.7SiO4 was reported, which was said to be an unstable phase under environmental conditions.50,58 Consequently, such a phase was not observed. As shown in Fig. 3, the two materials ZS1-C2 and ZS2-C2 with Zn:Si ratios of 0.75 and 1, respectively, exhibited relatively lower crystallinity. Here, the dominating phase is the β-Zn2SiO4 phase (ICDD reference code: 00-014-0653) with a content of approximately 80 wt% in both materials. In the ZS3-C2 material, the α-Zn2SiO4 phase (ICDD reference code: 00-037-1485) prevails. Interestingly, proceeding to ZS4-C2, one can see an increment in the ZnO and β-Zn2SiO4 phases (ICDD reference code: 00-036-1451), alongside a decrease in α-Zn2SiO4. The reason for the increase in ZnO could be an incomplete reaction of ZnO with SiO2, either due to insufficient temperature or due to short calcination time. The ZS3-C2 material exhibited a slightly higher crystallinity amongst all the C2 materials.
The choice of the C2 materials for a more detailed investigation is due to a two-fold reasoning, viz. because of the higher material crystallinity of the C2 materials and a lower ZnO content within the materials. It was assumed that the unreacted ZnO would automatically lead to a lower material stability, since it is known to be readily reduced to metallic zinc during the methanol decomposition reaction.59,60
Catalyst testing
Fig. 4 depicts the results from the screening of the zinc silicates, where only the C2 materials were tested. Firstly, it is worth mentioning that in general, the standard deviation of methanol conversion increased with rising reaction temperature for all the materials irrespective of the Zn:Si ratio. This indicates a catalyst deactivation occurring in all materials, however with different intensities. In general, the higher the Zn:Si ratio, the higher the methanol conversion for a specific temperature, which can be understood by looking at the amount of the active component, i.e. the absolute amount of Zn inside the materials. By analyzing the results obtained with the different C2 materials, it can be assumed that the β-Zn2SiO4 phase is relatively inactive towards methanol dehydrogenation compared to ZnO and the α-Zn2SiO4 phase. As can be seen from Fig. 3 and 4, ZS1-C2 and ZS2-C2 are rich in β-Zn2SiO4 phases and the methanol conversion is low. However, at this point, it was assumed to be a relatively stable phase since it was observed by ICP-OES measurements that ZS1-C2 and ZS2-C2 had lost only 0.1 and 1.2 wt% zinc from their matrix after 3.5 h TOS.
 | ||
| Fig. 4 Methanol conversion (XCH3OH, shown as black squares) and formaldehyde selectivity (SCH2O, shown as red dots) of the C2 catalysts at different temperatures. | ||
A high fluctuation in the methanol activity was observed above 550 °C compared to lower temperatures for all the materials. This can be attributed to the loss of unreacted ZnO from the calcination step, which is susceptible to reduction when subjected to reductive atmospheres and increasing temperatures.36 Another prominent deactivation mechanism was the formation of carbonaceous deposits. This was later proved with the help of Raman spectroscopy and is a subject of discussion in the latter part of this section. Overall, the ZS3-C2 material was slightly more stable than the ZS4-C2 catalyst. Fig. 3 reveals that ZS4-C2 contains more ZnO than ZS3-C2; this could be a possible reason for the higher fluctuation in methanol conversion for the ZS4-C2 catalyst. This also explains the higher zinc loss of ZS4-C2 (16 wt%) compared to ZS3-C2 (2.9 wt%) at the end of these experiments. Moreover, XRD of the spent ZS3-C2 and ZS4-C2 revealed that the ZnO present in both materials was reduced, especially in the case of the ZS4-C2 catalyst after the reaction, thereby complementing the results of ICP-OES measurements.
Hereafter, the ZS3-C2 material was chosen for a reaction temperature study, whose results are portrayed in Fig. 5. It becomes clear from this test that the zinc silicate material exhibits a stable behavior even at higher temperature, unlike the previously tested ZnO catalysts.19 The selectivity to formaldehyde increased at temperatures above 550 °C. At these temperatures, the second largest by-product was CO followed by CO2. Methyl formate (depicted in reaction (8)) and small quantities of methane and dimethyl ether were formed throughout the entire temperature range. Besides direct methanol dehydrogenation, the following side reactions can occur:
2CH2O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) HCOOCH3 HCOOCH3 | (8) |
| CH3OH + CH2O HCOOCH3 + H2 | (9) |
| 2CH3OH HCOOCH3 + 2H2 | (10) |
| 2CH3OH CH3OCH3 + H2O | (11) |
| CH3OH + H2O CO2 + 3H2 | (12) |
| CH3OH CO + 2H2 | (13) |
| ZnO + CO Zn + CO2 | (14) |
 | ||
| Fig. 5 Methanol conversion and product selectivity of ZS3-C2 as a function of temperature. | ||
A long-term experiment was carried out and a reaction temperature of 550 °C was chosen, due to the relatively high activity of the ZS3-C2 catalyst at this temperature. The experiment was run for more than 24 h and it can be seen from Fig. 6 that the methanol conversion decreased exponentially within the first 12 h. During the first 4 h, the CH2O selectivity increased, whereas the CO, CO2 and HCOOCH3 selectivities decreased. CO2 was observed in the product stream within the first 12 h, whose concentration decreased steadily over the course of the reaction. As described in reaction (14), formation of CO2 could be due to the reduction of ZnO with initially formed CO, which could explain the initial deactivation of the ZS3-C2 catalyst. Dimethyl ether formation remained unchanged during the entire experiment, which suggests that a stable active site on the zinc silicate material catalyzes its formation. However, the formation of CH3OCH3 is coupled with the formation of H2O, through reaction (11). In addition, since water was not observed during the entire experiment, reaction (12) is quite likely as well. It is important to mention here that the methanol steam reforming yields not only CO2 and H2, but also syngas as a reaction product. Therefore, the high selectivity to CO can be well addressed to this reaction. After 24 h, the methanol conversion settled at approximately 20% alongside a constant formaldehyde selectivity of around 80%, until the next 12 h.
 | ||
| Fig. 6 Long-term experiment with the ZS3-C2 catalyst at 550 °C. | ||
Raman spectroscopy was used to study the spent catalysts. After the long-term experiment, the ZS3-C2 catalyst mainly consisted of the α-Zn2SiO4 phase (Fig. 7). In contrast to the fresh catalyst (Fig. 3), almost no β-Zn2SiO4 was detected in the spent material indicating the instability of this phase in a harsh reductive environment.67 Possibly, the following reactions and their rates compete and determine the material stability and activity: ZnO reduction to metallic Zn, formation and decomposition of various zinc silicate phases from unreacted ZnO and SiO2 from the calcination step and lastly, coke formation on the freshly formed zinc silicate material. Increasing temperature is associated with an increase in the kinetics of all the above-mentioned reactions.4,28,34 The band at 435 cm−1 represents zincite or ZnO, the signals in the range of 840–990 cm−1 correspond to α-Zn2SiO4 and the bands within 1200–1650 cm−1 can be attributed to deposited carbon.68 These intense bands of carbon show its disorderly nature, as similar carbon depositions were found in various other dehydrogenation studies.16 The crystalline α-Zn2SiO4 phase dominates the sample after the reaction, complementing the XRD results. Between the large α-Zn2SiO4 crystals, some organic carbon was also observed, which confirms the deactivation through carbon species formed during the reaction. Additional information on the results of the characterization methods of the fresh and spent catalysts is given in the ESI.†
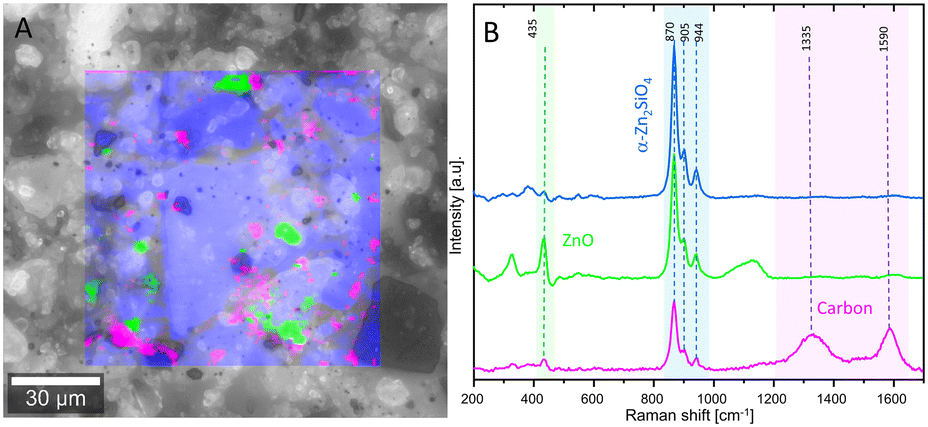 | ||
| Fig. 7 A: Raman image revealing the phases in ZS3-C2 after the reaction overlaid on an optical image. The bandwidths for the individual phases were used to generate this map; B: basic Raman spectra of the α-Zn2SiO4, ZnO and carbon phases. | ||
Conclusions
By chemically combining ZnO and SiO2, the resulting materials exhibit a remarkable performance as catalysts in the direct dehydrogenation of methanol to formaldehyde. The main findings within this study are as follows:1. A molar ratio of Zn:Si from 1.5 to 2 accompanied with a calcination temperature of 900 °C leads to a material with an enriched α-Zn2SiO4 phase. Although ZnO is more active, this material also shows relatively good activity and higher stability regarding ZnO reduction.
2. The generalized proposition made in the past, in which by combining ZnO and SiO2 in a molar ratio of 2:1, a very stable Zn2SiO4 material can be achieved, was disproved.
3. In general, increasing the Zn content leads to the formation of loose and unreacted ZnO, which results in higher activity but lower stability of the catalysts.
4. Apart from α-Zn2SiO4, there was also a β-Zn2SiO4 structure detected within the materials. This phase was identified and characterized by XRD, Raman spectroscopy and by referring to a handful of literature catering to this particular zinc silicate structure. However, this phase turned out to be neither stable nor significantly active towards methanol dehydrogenation.
The resulting product mixtures can be used advantageously in processes, in which large quantities of water have a disruptive effect. As an example, the synthesis of oxymethylene ethers (OMEs) was already mentioned. OME synthesis from methanol/formaldehyde mixtures has been investigated intensely in recent years and is considered as a preferred pathway compared to various other routes. By employing zinc silicates as catalysts, appropriate feeds could be generated, given that the current system can be further optimized in terms of stability, methanol conversion and formaldehyde selectivity. An absorption column after the fixed bed reactor can help to achieve methanol/formaldehyde mixtures, which can then be fed to a different reactor to synthesize OMEs. Moreover, the several by-products of the reaction pose no challenge, since gases like H2, CO and CO2, which are produced in higher amounts, are all non-condensable.
Since the α-Zn2SiO4 structure exhibits stable catalyst characteristics and leads to good results, it is desirable to find an alternative method to manufacture zinc silicates with a significantly enriched or even pure α-Zn2SiO4 phase. The sol–gel procedure employed within this study yields materials that contain other phases as impurities, making them unstable and unsuitable for long-term catalytic applications. In future work, the preparation procedure should be varied and the influence of different parameters on the final material properties should be studied. Typical examples are the ratio of components in the educt mixtures, the type of precursors and temperature-related parameters, such as the employed temperature program and calcination temperature. Monitoring of solidification and crystallization by thermal analysis, especially by differential scanning calorimetry, could be instructive to identify possible phase transformations. The use of structure-directing agents could also be an option to improve the uniformity and catalytic performance. Another possibility is the use of suitable supports and depositing catalytically active zinc silicate layers in a defined way on their surfaces. Other catalyst synthesis techniques like hydrothermal or solvothermal methods should also be taken into account.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would kindly like to thank the following colleagues, Armin Lautenbach for support with ICP-OES analysis, Doreen Neumann-Walter and Thomas Nicola Otto for the BET measurements, Lucas Warmuth for his inputs in regards to the catalyst synthesis and Diana Deutsch for synthesizing the catalysts. Furthermore, special appreciation is extended from the side of the authors to the Federal Ministry of Education and Research within the research project NAMOSYN - Nachhaltige Mobilität durch synthetische Kraftstoffe (Funding indicator of Project NAMOSYN: FKZ 03SF0566K0).Notes and references
- U. Mondal and G. D. Yadav, Green Chem., 2021, 23, 8361–8405 RSC.
- M. I. Malik, N. Abatzoglou and I. E. Achouri, Catalysts, 2021, 11, 893 CrossRef CAS.
- M. Drexler, P. Haltenort, T. A. Zevaco, U. Arnold and J. Sauer, Sustainable Energy Fuels, 2021, 5, 4311–4326 RSC.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, Weinheim, 1997 Search PubMed.
- F. Eichner, E. Turan, J. Sauer, M. Bender and S. Behrens, Catal. Sci. Technol., 2023, 13, 2349–2359 RSC.
- R. Maurer and A. Renken, Chem. Eng. Res. Des., 2003, 81, 730–734 CrossRef CAS.
- United Nations Environment Programme and World Health Organization and International Labour Organisation, Formaldehyde - Environmental Health Criteria 89, GENEVA, 1991 Search PubMed.
- G. J. Millar and M. Collins, Ind. Eng. Chem. Res., 2017, 56, 9247–9265 CrossRef CAS.
- G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, Formaldehyde: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 Search PubMed.
- M. Drexler, P. Haltenort, U. Arnold, J. Sauer, S. A. Karakoulia and K. S. Triantafyllidis, Catal. Today, 2022, 424, 113847 CrossRef.
- M. Drexler, P. Haltenort, U. Arnold and J. Sauer, Chem. Ing. Tech., 2022, 94, 256–266 CrossRef CAS.
- T. Grützner, H. Hasse, N. Lang, M. Siegert and E. Ströfer, Chem. Eng. Sci., 2007, 62, 5613–5620 CrossRef.
- S. Schemme, S. Meschede, M. Köller, R. C. Samsun, R. Peters and D. Stolten, Energies, 2020, 13, 3401 CrossRef CAS.
- S. Su, P. Zaza and A. Renken, Chem. Eng. Technol., 1994, 17, 34–40 CrossRef CAS.
- N. Y. Usachev, I. M. Krukovskii and S. A. Kanaev, Pet. Chem., 2004, 44, 379–394 Search PubMed.
- M. Merko, G. W. Busser and M. Muhler, ChemCatChem, 2022, 14, e202200258 CrossRef CAS.
- A. Mušič, J. Batista and J. Levec, Appl. Catal., A, 1997, 165, 115–131 CrossRef.
- S. Ruf, A. May and G. Emig, Appl. Catal., A, 2001, 213, 203–215 CrossRef CAS.
- A. Ghosh Chowdhury, D. Deutsch, U. Arnold and J. Sauer, Proceedings of the DGMK-Conference The Role of Catalysis for the Energy Transition, Report 2022-3, 2022, pp. 186–198 Search PubMed.
- D. Zhao, K. Guo, S. Han, D. E. Doronkin, H. Lund, J. Li, J.-D. Grunwaldt, Z. Zhao, C. Xu, G. Jiang and E. V. Kondratenko, ACS Catal., 2022, 12, 4608–4617 CrossRef CAS.
- M. Nadjafi, A. M. Kierzkowska, A. Armutlulu, R. Verel, A. Fedorov, P. M. Abdala and C. R. Müller, J. Phys. Chem. C, 2021, 125, 14065–14074 CrossRef CAS.
- Y. Luo, C. Wei, C. Miao, Y. Yue, W. Hua and Z. Gao, Chin. J. Chem., 2020, 38, 703–708 CrossRef CAS.
- Y. Luo, C. Miao, Y. Yue, W. Hua and Z. Gao, Microporous Mesoporous Mater., 2020, 294, 109864 CrossRef CAS.
- C. Chen, Z.-P. Hu, J.-T. Ren, S. Zhang, Z. Wang and Z.-Y. Yuan, Mol. Catal., 2019, 476, 110508 CrossRef CAS.
- D. Zhao, Y. Li, S. Han, Y. Zhang, G. Jiang, Y. Wang, K. Guo, Z. Zhao, C. Xu, R. Li, C. Yu, J. Zhang, B. Ge and E. V. Kondratenko, iScience, 2019, 13, 269–276 CrossRef CAS PubMed.
- C. Chen, Z. Hu, J. Ren, S. Zhang, Z. Wang and Z.-Y. Yuan, ChemCatChem, 2019, 11, 868–877 CrossRef CAS.
- G. Liu, L. Zeng, Z.-J. Zhao, H. Tian, T. Wu and J. Gong, ACS Catal., 2016, 6, 2158–2162 CrossRef CAS.
- M. Sagou, T. Deguchi and S. Nakamura, Proceedings of the Worldwide Catalysis Seminars, July, 1988, on the Occasion of the 30th Anniversary of the Catalysis Society of Japan, Elsevier, 1989, p. 139 Search PubMed.
- S. Akhter, W. H. Cheng, K. Lui and H. H. Kung, J. Catal., 1984, 85, 437–456 CrossRef CAS.
- L. Chan and G. L. Griffin, Surf. Sci., 1985, 155, 400–412 CrossRef CAS.
- W. H. Cheng, S. Akhter and H. H. Kung, J. Catal., 1983, 82, 341–350 CrossRef CAS.
- J. Kiss, D. Langenberg, D. Silber, F. Traeger, L. Jin, H. Qiu, Y. Wang, B. Meyer and C. Wöll, J. Phys. Chem. A, 2011, 115, 7180–7188 CrossRef CAS PubMed.
- S. Ruan, Z. Li, H. Shi, W. Wang, X. Ren and X. Shao, J. Phys. Chem. C, 2019, 123, 9105–9111 CrossRef CAS.
- K. M. Tawarah and R. S. Hansen, J. Catal., 1984, 87, 305–318 CrossRef CAS.
- G. Zwicker, K. Jacobil and J. Cunningham, Int. J. Mass Spectrom. Ion Processes, 1984, 60, 213–223 CrossRef CAS.
- E. A. Secco, Can. J. Chem., 1960, 38, 596–601 CrossRef CAS.
- K. Lui, M. Vest, P. Berlowitz, S. Akhter and H. H. Kung, J. Phys. Chem., 1986, 90, 3183–3187 CrossRef CAS.
- J. M. Vohs and M. A. Barteau, Surf. Sci., 1986, 176, 91–114 CrossRef CAS.
- D. Chadwick and K. Zheng, Catal. Lett., 1993, 20, 231–242 CrossRef CAS.
- K.-D. Jung, O.-S. Joo, S.-H. Han, S.-J. Uhm and I.-J. Chung, Catal. Lett., 1995, 35, 303–311 CrossRef CAS.
- Y. Ren, F. Zhang, W. Hua, Y. Yue and Z. Gao, Catal. Today, 2009, 148, 316–322 CrossRef CAS.
- J. M. Rubio-Caballero, J. Santamaría-González, J. Mérida-Robles, R. Moreno-Tost, A. Jiménez-López and P. Maireles-Torres, Appl. Catal., B, 2009, 91, 339–346 CrossRef CAS.
- A. R. Yacob and K. S. Kabo, Adv. Mater. Res., 2015, 1107, 326–332 Search PubMed.
- M. Fabián, P. Bottke, V. Girman, A. Düvel, K. L. Da Silva, M. Wilkening, H. Hahn, P. Heitjans and V. Šepelák, RSC Adv., 2015, 5, 54321–54328 RSC.
- B. R. Strohmeier, Surf. Sci. Spectra, 1994, 3, 128–134 CrossRef CAS.
- L. Su, L. Miao, J. Miao, Z. Zheng, B. Yang, R. Xia, P. Chen and J. Qian, J. Asian Ceram. Soc., 2016, 4, 185–190 CrossRef.
- K. Jothimurugesan and S. K. Gangwal, Ind. Eng. Chem. Res., 1998, 37, 1929–1933 CrossRef CAS.
- M. C. Woods, S. K. Gangwal, K. Jothimurugesan and D. P. Harrison, Ind. Eng. Chem. Res., 1990, 29, 1160–1167 CrossRef CAS.
- S. Zh. Karazhanov, P. Ravindran, P. Vajeeston, A. G. Ulyashin, H. Fjellvåg and B. G. Svensson, J. Phys.: Condens. Matter, 2009, 21, 485801 CrossRef PubMed.
- M. Kanzaki, J. Mineral. Petrol. Sci., 2018, 113, 263–267 CrossRef CAS.
- D. Ehrt and S. Flügel, J. Mater. Sci. Eng. A, 2011, 1, 312 CAS.
- R. K. Iler, The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica, Wiley, New York, Chichester, 1979 Search PubMed.
- B. C. Babu and S. Buddhudu, Phys. Procedia, 2013, 49, 128–136 CrossRef CAS.
- G. Ertl, Handbook of Heterogeneous Catalysis, Wiley-VCH, Chichester, Weinheim, 2008 Search PubMed.
- P. Krasucka, W. Stefaniak, A. Kierys and J. Goworek, Microporous Mesoporous Mater., 2016, 221, 14–22 CrossRef CAS.
- M. Takesue, A. Suino, K. Shimoyama, Y. Hakuta, H. Hayashi and R. L. Smith Jr., J. Cryst. Growth, 2008, 310, 4185–4189 CrossRef CAS.
- R. F. Samigullina and T. I. Krasnenko, Mater. Res. Bull., 2020, 129, 110890 CrossRef CAS.
- A. Roy, S. Polarz, S. Rabe, B. Rellinghaus, H. Zähres, F. E. Kruis and M. Driess, Chemistry, 2004, 10, 1565–1575 CrossRef CAS PubMed.
- K. M. Tawarah, Kinetics and mechanism of methanol decomposition over zinc oxide, Iowa State University, 1982 Search PubMed.
- T. Imoto, Y. Harano and Y. Nishi, Bull. Chem. Soc. Jpn., 1964, 37, 1181–1186 CrossRef CAS.
- D. Kaiser, L. Beckmann, J. Walter and M. Bertau, Catalysts, 2021, 11, 869 CrossRef CAS.
- C. J. Baranowski, A. M. Bahmanpour and O. Kröcher, Appl. Catal., B, 2017, 217, 407–420 CrossRef CAS.
- A. M. Bahmanpour, F. Héroguel, C. J. Baranowski, J. S. Luterbacher and O. Kröcher, Appl. Catal., A, 2018, 560, 165–170 CrossRef CAS.
- E. A. G. Engku Ali, K. A. Matori, E. Saion, S. A. H. Aziz, M. H. M. Zaid and I. M. Alibe, ASM Sci. J., 2018, 1, 75–85 Search PubMed.
- D. B. Pal, R. Chand, S. N. Upadhyay and P. K. Mishra, Renewable Sustainable Energy Rev., 2018, 93, 549–565 CrossRef CAS.
- D. S. Newsome, Catal. Rev.: Sci. Eng., 1980, 21, 275–318 CrossRef CAS.
- K. Breuer, J. H. Teles, D. Demuth, H. Hibst, A. Schäfer, S. Brode and H. Domgörgen, Angew. Chem., Int. Ed., 1999, 38, 1401–1405 CrossRef CAS PubMed.
- L. Bokobza, J.-L. Bruneel and M. Couzi, Journal of Carbon Research, 2015, 1, 77–94 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: BET surface areas and Zn contents of the zinc silicates (Table S1), XRD patterns of the zinc silicates (Fig. S1), parameters for the quantification of phases within the zinc silicates (Table S2), optical and Raman images of ZS3-C2 before the reaction (Fig. S2), content of crystalline and amorphous phases in the ZS3-C2 and ZS4-C2 materials before and after the reaction (Fig. S3), optical and Raman images of ZS3-C2 after the reaction (Fig. S4), zinc loss of the C2 catalysts after the reaction (Table S3), and time on stream experiment to determine catalyst coking and deactivation (Fig. S5). See DOI: https://doi.org/10.1039/d4cy00541d |
| This journal is © The Royal Society of Chemistry 2024 |