
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling palladium particle size and dispersion as a function of loading by chemical vapour impregnation: an investigation using propane total oxidation as a model reaction†
Liam A.
Bailey
 *,
Mark
Douthwaite
,
Thomas E.
Davies
,
David J.
Morgan
and
Stuart H.
Taylor
*
*,
Mark
Douthwaite
,
Thomas E.
Davies
,
David J.
Morgan
and
Stuart H.
Taylor
*
Max Planck-Cardiff Centre on the Fundamentals of Heterogeneous Catalysis FUNCAT, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Translational Research Hub, Maindy Road, Cardiff CF24 4HQ, UK. E-mail: baileyla2@cardiff.ac.uk; taylorsh@cardiff.ac.uk
First published on 24th July 2024
Abstract
A series of Pd/Al2O3 catalysts with metal weight loadings of 1.0 wt%, 2.5 wt%, and 5.0 wt% were synthesised by chemical vapour impregnation (CVI) and used for the total oxidation of propane. All the catalysts were highly active for propane total oxidation. Extensive characterisation showed essentially identical catalyst structural and chemical characteristics, with consistent nanoparticle size, dispersion, and metal oxidation state regardless of metal loading. The major difference between catalysts was the number of surface palladium sites which scaled directly with metal loading. Turnover frequency calculations showed that the intrinsic activity of each catalyst is the same, with conversion scaling with the number of active sites. The number of active sites was normalised experimentally with catalyst performance proving to be identical regardless of weight loading. This study shows that CVI is a technique that can produce active catalysts with high levels of control and consistency of active metal nanoparticles as a function of loading. The same level of control over dispersion and activity was not achieved when catalysts were prepared by conventional aqueous impregnation. The fundamental understanding of CVI is important for the design of highly active catalysts, which is exemplified for propane total oxidation, but has wider significance for other applications of supported metal catalysts.
Introduction
The method of catalyst synthesis can lead to significant differences in the properties and activity. It can influence many factors including nanoparticle size, distribution, shape and oxidation state.1 Traditional impregnation and precipitation techniques are popular due to ease of synthesis but can demonstrate poor control over nanoparticle size, especially when metal loading is varied.2,3 Previous studies have suggested that the size distribution broadens, while dispersion decreases when metal weight loading increases. The precursor used can also introduce surface poisons, for example, chloride ions from metal chloride precursors, which remain on the surface after synthesis and have been found to be detrimental to catalytic activity. Colloidal methods like sol-immobilisation have been designed with the intention of greater particle size control through use of protecting groups.4,5Chemical vapour impregnation (CVI) is a preparation method that can negate some of the drawbacks of the more traditional impregnation techniques. CVI is a simple technique to use experimentally, metals are deposited onto a support using volatile metal precursors.6 Typically an acetylacetonate precursor is sublimed to deposit the complex onto the surface and then the ligands are removed via thermal decomposition to form metal nanoparticles. This makes it a much cleaner technique than traditional impregnation methods as there are less opportunities for impurities and poisons to be introduced. CVI is also a solventless technique allowing better compliance with the 12 principles of green chemistry through the reduction of waste production.7
Initial research into CVI has predominately focused on catalysts used for liquid phase reactions, demonstrating it can produce highly active catalysts for oxidation reactions due to a high level of control over nanoparticle size and distribution.8–10 Recently there has been an increase in study into CVI catalysts used for gas phase applications, with high performance catalysts being synthesised for CO2 hydrogenation,11–18 ammonia decomposition,19 and NH3-SCR reactions.20 However, there has not been a more in-depth study into CVI as a synthesis technique investigating how factors like metal loading can influence activity.
Supported metal nanoparticle catalysts are particularly important for environmental pollution control. Volatile organic compounds (VOCs) are a major cause of air pollution, especially in urban areas,21 and have a significant negative impact on both the environment and human health.22,23 They are emitted from a wide range of anthropogenic sources, however, in urban environments they typically are emitted from automobiles due to the incomplete combustion of fuel.24,25 As air pollution is the leading cause of preventable death worldwide,26 it is imperative that the emissions of VOCs are reduced, with wide ranging legislation enacted to achieve this aim.27–29 While there are several methods for removing VOCs, catalytic oxidation has proved effective due to lower operating cost, higher selectivity, and the ability to treat low concentration streams.30,31
The total oxidation of propane has been extensively researched because it is a good model for VOC total oxidation.32 Many recent studies has focussed on metal oxide catalysts due to cheaper cost of production, with Co, Mn, Ce, and Fe based metal oxides all being exceptionally active.33–45 However, noble metal supported catalysts a favoured industrially due to greater activity and high durability.46 Supported palladium on alumina catalysts have also been extensively studied for propane total oxidation.47 Despite the depth of study there is still debate in the literature as to the most active oxidation state and palladium nanoparticle size. Although there is a consensus that PdO is the active form for propane oxidation.48–52
This work aims to investigate in more detail CVI as a synthesis technique for supported palladium catalysts, using propane total oxidation as a model reaction. Catalyst activity is studied as a function of active metal loading and performance correlated with the structural properties of the catalysts to provide insight into the scope of CVI as an alternative preparation technique.
Experimental
Catalyst preparation
Catalyst characterisation
Catalyst activity evaluation
All reactions were performed at atmospheric pressure using a fixed bed continuous flow microreactor. 0.3 g of the catalyst was packed between plugs of quartz wool in a stainless-steel tube (1 cm i.d.). Catalysts were pre-treated under a flow of N2 (250 mL min−1) at 200 °C for 1 hour. A pre-made gas mix of 1000 ppm propane/10% O2/N2 (BOC specialty gas) was used at a flow rate of 250 ml min−1 to give a GHSV of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. Reactions were performed between 200–500 °C and were monitored using an in-line Fourier transform infrared (FTIR) process gas analyser (Gasmet) and gas concentrations calculated using Calcmet software. Concentration readings were taken at 25 °C intervals once the temperature stabilised with readings repeated until it was certain steady state had been reached.
000 h−1. Reactions were performed between 200–500 °C and were monitored using an in-line Fourier transform infrared (FTIR) process gas analyser (Gasmet) and gas concentrations calculated using Calcmet software. Concentration readings were taken at 25 °C intervals once the temperature stabilised with readings repeated until it was certain steady state had been reached.
Results and discussion
Catalytic activity of Pd/Al2O3 catalysts synthesised by CVI
To begin the investigation, blank reactions with no catalyst and the alumina support material were conducted. No propane conversion was observed with an empty reactor tube over the temperature range studied, and only 22% propane conversion was exhibited over the alumina support at 500 °C. This shows that homogeneous gas phase reactions in the reactor tube were negligible for propane conversion and that the contribution of alumina alone was relatively low compared to the Pd-containing catalysts. The performance of the 1.0 wt%, 2.5 wt% and 5.0 wt% Pd/Al2O3 catalysts synthesised by CVI for propane oxidation are shown in Fig. 1. All the CVI catalysts were active, with full conversion achieved at 450 °C for the 1.0 wt% catalyst, and 400 °C for the 2.5 wt% and 5.0 wt% catalysts. Catalyst performance increased with Pd loading, with the 5.0 wt% catalyst the most active, followed by the 2.5 wt%, then the 1 wt% Pd/Al2O3 catalyst. This is demonstrated by the T20 and T50 values (Table 1). However, when the activity of the catalyst is normalised to the mass of palladium present in each reaction, very similar rates of propane conversion are seen, suggesting similar intrinsic activity of Pd sites. All catalysts showed high selectivity for carbon dioxide; in all cases over 99% conversion to CO2 was observed, indicating that these catalysts are very selective for the deep oxidation of propane. The catalysts synthesised here were also found to be more active than analogous materials synthesised by the more traditional method of wet impregnation (Fig. S1 and Table S1†) suggesting that CVI is a superior method of making catalysts for this application.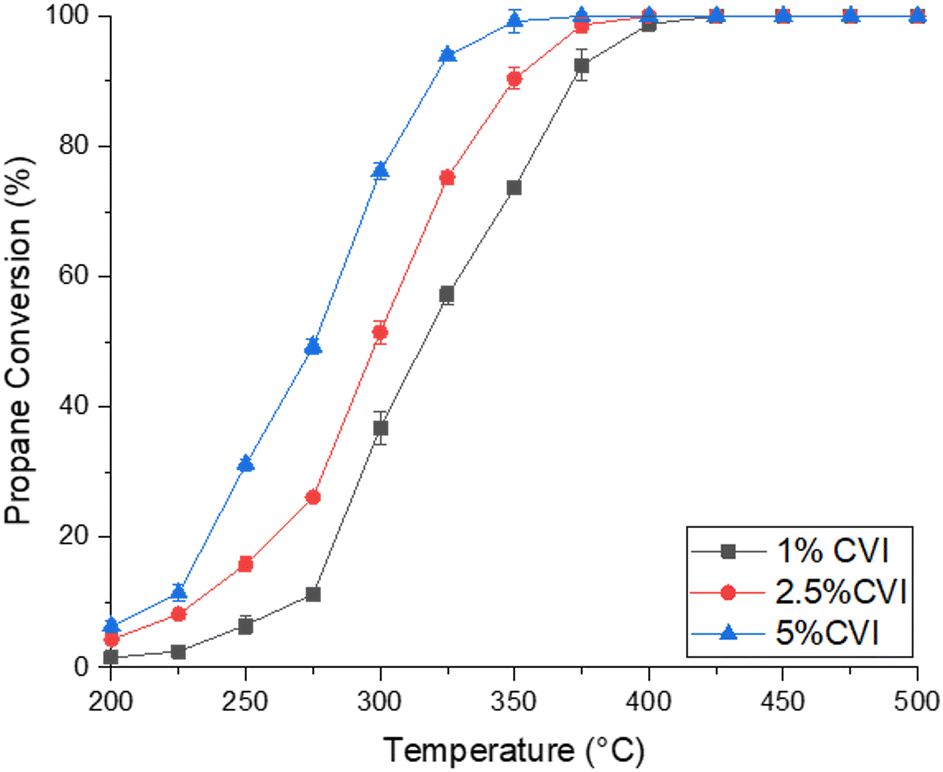 | ||
| Fig. 1 Propane conversion as a function of temperature for the Pd/Al2O3 catalysts synthesised by chemical vapour impregnation. (Black square) 1.0 wt% Pd/Al2O3, (red circle) 2.5 wt% Pd/Al2O3, (blue triangle) 5.0 wt% Pd/Al2O3. Reaction conditions: 1000 ppm propane/10% O2/N2, gas hourly space velocity (GHSV) 50000 h−1, temperature range 200–500 °C. | ||
000 h−1, temperature range 200–500 °C
| Catalyst | T 20 (°C) | T 50 (°C) | Activity at 250 °C (molC6H8 gPd−1 s−1) |
|---|---|---|---|
| 1.0 wt% Pd/Al2O3 | 283 | 316 | 4.03 × 10−6 (±4.08 × 10−8) |
| 2.5 wt% Pd/Al2O3 | 260 | 298 | 3.92 × 10−6 (±4.32 × 10−8) |
| 5.0 wt% Pd/Al2O3 | 236 | 276 | 3.93 × 10−6 (±3.00 × 10−8) |
Catalyst stability was also probed with 5 cycles of catalyst testing performed on each sample, along with 50 hours time on line at 275 °C (Fig. S2 and S3†). All cycles line up suggesting stability over multiple tests, while less than a 5% drop off in activity was noted over the 50 hours of testing. These tests show limited change in catalyst activity for all materials demonstrating high stability for catalysts synthesised by CVI.
Catalyst characterisation
MP-AES was performed on the samples and can be seen in Table 2. This demonstrated that the target loading palladium loading and actual palladium loading is very similar, identifying CVI as accurate technique for producing supported palladium catalysts.| Sample | Particle size distributiona (nm) | Average nanoparticle sizea (nm) | Pd weight loadingb (%) | BET (m2 g−1) |
|---|---|---|---|---|
| a Calculated from TEM imaging. Average of 200 nanoparticles. b Calculated using MP-AES. | ||||
| 1.0 wt% Pd/Al2O3 | 2.0–8.0 | 4.0 | 0.98 | 128 |
| 2.5 wt% Pd/Al2O3 | 2.0–8.0 | 4.5 | 2.51 | 126 |
| 5.0 wt% Pd/Al2O3 | 2.0–8.0 | 4.5 | 4.95 | 124 |
Transmission electron microscopy was used to gain insight on the size and distribution of the supported Pd particles in the materials (Fig. 2a–c). All the catalysts had distinct palladium nanoparticles of a consistent size with an average nanoparticle size of 4.0 nm, 4.5 nm, and 4.5 nm measured for the 1.0 wt% CVI, 2.5 wt% CVI, and 5.0 wt% CVI catalysts, respectively. Consistent control over the nanoparticle size distribution was also noted with a diameter range of 2–8 nm for all CVI catalysts. Size distribution plots (Fig. S4†) shows a normal distribution of nanoparticles ranging 3–5 nm with few outliers closer to 2 nm and 8 nm. High resolution AC-STEM imaging was also performed for all three of the catalysts prepared by CVI (Fig. 2d–f). Analysis showed the alumina support was decorated with palladium nanoparticles, and also clusters under 1 nm in diameter. These clusters formed on all CVI catalysts irrespective of loading, but were more prevalent on the 1% CVI catalyst. This imaging suggests that the size and distribution of palladium nanoparticles are largely independent of weight loading and the particles formed are very consistent.
 | ||
| Fig. 2 Electron microscopy micrographs of the catalysts prepared by CVI. a–c: TEM images (50k× magnification), d–f: AC-STEM images (80k× magnification). a and d: 1.0 wt% CVI, b and e: 2.5 wt% CVI, c and f: 5.0 wt% CVI. | ||
The physicochemical properties of the samples were probed by XRD and BET analysis of N2 physisorption. All catalysts exhibited major peaks at 2θ = 37.1°, 39.8°, 46.0° and 67.0° corresponding to (311), (222), (400), and (440) planes of γ-alumina, respectively (Fig. 3 and Table 2).56 Additionally, a peak at 2θ = 34° was observed, corresponding to PdO (101)/(002) planes for all of the catalysts. This peak increases in intensity as the weight loading increases, suggesting that more PdO was present in the 5.0 wt% Pd catalyst. This observation can be important for rationalising the increase in performance with weight loading for the CVI catalysts, as PdO is considered to be the active species for propane total oxidation.48 None of the samples show scattering for metallic palladium, suggesting that the metal was solely in its oxide form which is expected given heat treatment under oxidising conditions. Overall, there was minimal difference in the scattering pattern besides changes in intensity between weight loadings suggesting that differences in the large-scale catalyst structure was negligible. Analysis of the surface area showed very little difference between catalysts, suggesting little impact on the wider catalyst structure with loading.
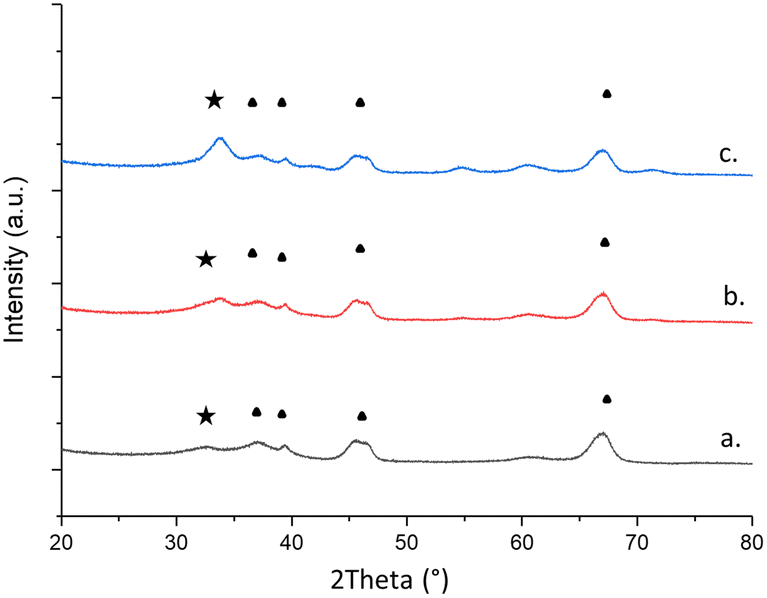 | ||
| Fig. 3 XRD patterns of (a.) 1.0 wt% Pd/Al2O3, (b.) 2.5 wt% Pd/Al2O3 and (c.) 5.0 wt% Pd/Al2O3 catalysts. (Black star) PdO, JCPDS no. 00-006-0515. (Black triangle) γ-Al2O3, JCPDS no. 10-0425. | ||
X-Ray photoelectron spectroscopy (XPS) analysis of the catalysts (Pd 3d, Fig. 4) revealed that Pd was predominantly in the 2+ oxidation state (Table S2†), which correlates with the XRD analysis. It should be noted that with the 1.0 wt% CVI sample some metallic Pd was observed, but is likely to be attributed to the photoreduction of PdO to Pd(0) or smaller particles during the analysis. These findings align with the literature, where it has been reported that PdO is more active than metallic Pd for propane oxidation, it implies that differences in the chemical state of the metal is not the reason for differences in activity. The binding energy of the 1% CVI catalyst is interesting as it increased compared to the other catalysts, with a value of 337.3 eV. This is a shift of 0.5 eV from the 336.8 eV expected from bulk PdO. Jürgensen et al. suggested an increase of 0.6 eV in the palladium binding energy is indicative of the presence of very small nanoparticles.57 Thus, the XPS for the 1% catalyst indicates that it could have smaller palladium particles than the other catalysts, which corresponds to the increased number of small clusters identified in the AC-STEM imaging, an observation which is supported by the change in FWHM of the Pd 3d peaks with increasing metal loading.58 XPS was also used to probe the surface concentration of the palladium (Table S2†). Unsurprisingly, surface concentration of palladium increases with weight loading which can explain the differences in conversion identified. The surface concentration scales in a linear fashion suggesting that the size distribution of the metal nanoparticles remains consistent, generally agreeing with the microscopy obtained (Fig. S5†).
 | ||
| Fig. 4 Pd 3d core-level XPS spectra for (a.) 1.0 wt% Pd/Al2O3, (b.) 2.5 wt% Pd/Al2O3 and (c.) 5.0 wt% Pd/Al2O3 catalysts. | ||
CO DRIFTs was performed on the samples to probe the palladium sites present. The spectra recorded for the CVI catalysts are shown in Fig. 5. For all three samples there are three peaks at 1926 cm−1, 1975 cm−1, and 2088 cm−1, corresponding to linear CO bonding of Pd+, Pd0, and bridged CO bonding on Pd0 respectively,59 with the intensity of each peak increasing with weight loading. The peak for the linear Pd0–CO stretch is largely obscured by the linear Pd+–CO peak and appears as a shoulder. There are no peaks in the 2180–2160 cm−1 region where a PdO–CO stretching vibration is typically expected, showing that the surface palladium is easily reducible. This has been identified for other palladium-based catalysts in the literature as evidence of easy cycling between Pd oxidation states helping to explain improved performance.60,61 This is further evidence that the structure of the nanoparticles formed by CVI are very similar in nature irrespective of the metal loading across the range 1.0–5.0 wt%.
 | ||
| Fig. 5 DRIFT spectra of CO adsorbed at room temperature for (a.) 1.0 wt% Pd/Al2O3, (b.) 2.5 wt% Pd/Al2O3 and (c.) 5.0 wt% Pd/Al2O3 catalysts. | ||
CO chemisorption was performed on the catalysts with results shown in Table 3. The concentration of CO adsorbed increases in a linear relationship with the palladium weight loading. Accordingly, the number of palladium sites present on the catalyst surface can be measured, the number increasing linearly with weight loading. These measurements agree with the TEM analysis and help explain the increase in activity with increased Pd loading. As palladium is the active site for this reaction, having more surface sites expectedly enhances conversion. CO chemisorption was also used to calculate the dispersion of the palladium for each catalyst. The dispersion for all CVI catalysts was similar, at ca. 30%. This is again consistent with other complimentary characterisation data for these catalysts, suggesting that catalysts with consistent nanoparticle structure are formed via CVI regardless of weight loading.
| Sample | CO adsorbed (mmol g−1) | Palladium surface sitesa (g−1) | Dispersiona (%) | TOFb (10−3)(s−1) |
|---|---|---|---|---|
| a Calculated from CO chemisorption assuming a stoichiometry of 2. b Calculated at 250 °C. | ||||
| 1.0 wt% Pd/Al2O3 | 1.73 × 10−2 | 2.08 × 1019 | 30 | 1.43 |
| 2.5 wt% Pd/Al2O3 | 3.28 × 10−2 | 3.95 × 1019 | 28 | 1.49 |
| 5.0 wt% Pd/Al2O3 | 6.68 × 10−2 | 8.05 × 1019 | 28 | 1.48 |
Calculation of the catalytic turnover frequency (TOF) provides a measure of the intrinsic activity per active site (Table 3). The measured TOFs for the CVI catalysts showed consistent values regardless of weight loading with values of 1.43 × 10−3 s−1, 1.49 × 10−3 s−1 and 1.48 × 10−3 s−1 for the 1.0 wt%, 2.5 wt%, and 5.0 wt% CVI catalysts respectively. This agrees with the similar rates calculated (Table 1) where activity was normalised to grams of palladium present in the reaction, and shows that the intrinsic activity per site is the same regardless of Pd loading when the CVI method is employed.
Post reaction XRD and CO chemisorption (Fig. S6 and Table S3†) were performed on all samples after 50 hours time on line with little change noted from the pre-reaction samples. No change was seen on the x-ray diffraction patterns while a very small decrease in nanoparticle dispersion was noted for the 5 wt% Pd/Al2O3 sample which can be explained by error of the technique. This links well with the stability testing performed which demonstrated no difference in catalyst activity with extended use suggesting that these catalysts are stable over prolonged use.
Assessing intrinsic palladium site activity
To probe this effect further, the mass, and hence volume, of catalyst used in a reaction for total propane oxidation was varied to ensure the same number of palladium sites were present in the catalyst bed, based on the number of sites measured by CO chemisorption. This was performed for the CVI catalysts and also for comparison with analogous catalysts synthesised by wet impregnation (WI) (Fig. 6). For the CVI catalysts, propane conversion was identical at each temperature point and activity curves were superimposable within experimental error. This shows that activity for these catalysts are dominated by the number of surface palladium active sites with the intrinsic activity for each site being the same. It further identifies CVI as a technique for producing highly uniform materials regardless of metal loading, allowing a scalable method to prepare catalysts with varying numbers of surface sites without changing the characteristics of the nanoparticles. This trend was not identified for the WI catalysts, where large differences in propane total oxidation activity occurs as a function of metal loading. Activity decreases with weight loading suggesting that several factors are influencing performance. Previous studies have suggested that impregnation synthesis methods have poor control over factors like dispersion and nanoparticle size distribution, with these decreasing and increasing with increased weight loading respectively.2,3 Characterisation performed on the wet impregnation catalysts (Tables S4 and S5, and Fig. S7–S10†) show large differences in nanoparticle structure and characteristics suggesting that the high uniformity of the CVI prepared samples is characteristic of the technique.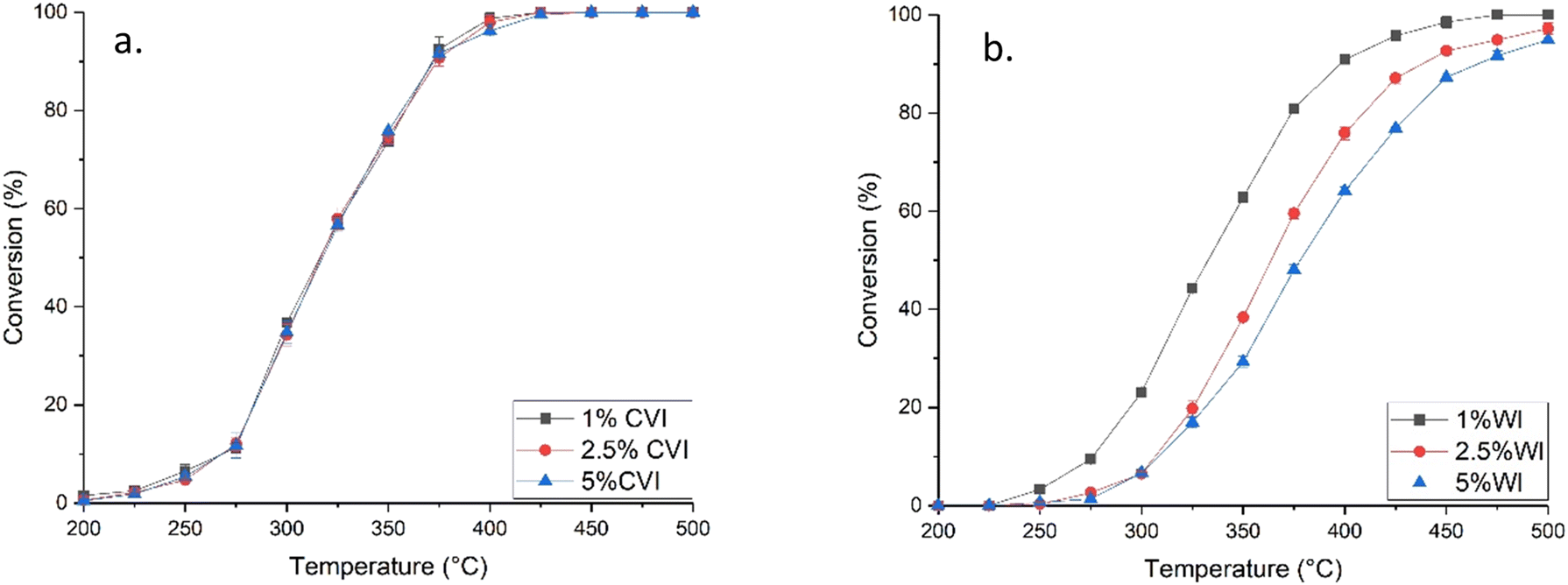 | ||
| Fig. 6 Propane conversion as a function of temperature for Pd/Al2O3 catalysts synthesised by a. CVI and b. WI when the volume of sample was varied to normalise activity to Pd sites. (Black square) 1.0 wt% Pd/Al2O3, (red circle) 2.5 wt% Pd/Al2O3, (blue triangle) 5.0 wt% Pd/Al2O3. Reaction conditions: 1000 ppm propane/10% O2/N2, temperature range 200–500 °C. | ||
Conclusions
Chemical vapour impregnation is a relatively unstudied catalyst synthesis technique, but one that can be readily employed in the laboratory with simple and inexpensive equipment. Pd/Al2O3 catalysts with varying weight loading were prepared by this method and studied for the total oxidation of propane. In all cases the CVI catalysts were highly active with activity ascribed to high nanoparticle dispersion and the formation of many active sites. Characterisation showed control over uniformity in nanoparticle structure and composition with activity per active site equivalent, and activity scaling linearly with metal loading which scaled with the number of palladium sites. This was studied experimentally by normalising the sample volume to ensure the same number of palladium sites were present. Near identical performance was noted, that was not seen for analogous samples prepared via wet impregnation exploitation of these controllable properties that are accessible by CVI provides an important tool in the process of catalyst design. The high level of control over particle size and distribution is very desirable, though additional study is needed to see whether the consistent average nanoparticle size of 4 nm is characteristic of this technique, or if the method can be optimised further to give a range of average nanoparticle size.Data availability
All relevant data is contained within the manuscript and ESI.†Author contributions
The manuscript was written through contributions of all authors. L. A. B. synthesised catalysts, conducted experiments and data analysis. L. A. B., M. D., T. E. D., and D. J. M. conducted material characterisation and corresponding data processing. L. A. B. and S. H. T. contributed to the design of study and provided technical advice and result interpretation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
XPS data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195. The authors would like to thank the CCI-Electron Microscopy Facility which has been partially funded by the European Regional Development Fund through the Welsh Government and The Wolfson Foundation. All authors gratefully acknowledge Cardiff University and the Max Planck Centre for Fundamental Heterogeneous Catalysis (FUNCAT) for financial support.Notes and references
- J. A. Schwarz, C. Contescu and A. Contescu, Chem. Rev., 1995, 95, 477–510 CrossRef CAS.
- Y. J. Huang and J. A. Schwarz, Appl. Catal., 1987, 30, 239–253 CrossRef CAS.
- B. A. T. Mehrabadi, S. Eskandari, U. Khan, R. D. White and J. R. Regalbuto, in Advances in Catalysis, ed. C. Song, Academic Press, 2017, vol. 61, pp. 1–35 Search PubMed.
- A. Villa, D. Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036–3041 RSC.
- S. Campisi, D. Ferri, A. Villa, W. Wang, D. Wang, O. Kröcher and L. Prati, J. Phys. Chem. C, 2016, 120, 14027–14033 CrossRef CAS.
- M. M. Forde, R. D. Armstrong, C. Hammond, Q. He, R. L. Jenkins, S. A. Kondrat, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. Willock, C. J. Kiely and G. J. Hutchings, J. Am. Chem. Soc., 2013, 135, 11087–11099 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- M. M. Forde, R. D. Armstrong, R. McVicker, P. P. Wells, N. Dimitratos, Q. He, L. Lu, R. L. Jenkins, C. Hammond, J. A. Lopez-Sanchez, C. J. Kiely and G. J. Hutchings, Chem. Sci., 2014, 5, 3603–3616 RSC.
- M. M. Forde, L. Kesavan, M. I. bin Saiman, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, S. H. Taylor, C. J. Kiely and G. J. Hutchings, ACS Nano, 2014, 8, 957–969 CrossRef CAS PubMed.
- R. Su, M. M. Forde, Q. He, Y. Shen, X. Wang, N. Dimitratos, S. Wendt, Y. Huang, B. B. Iversen, C. J. Kiely, F. Besenbacher and G. J. Hutchings, Dalton Trans., 2014, 43, 14976–14982 RSC.
- H. Bahruji, M. Bowker, W. Jones, J. Hayward, J. R. Esquius, D. J. Morgan and G. J. Hutchings, Faraday Discuss., 2017, 197, 309–324 RSC.
- M. Bowker, N. Lawes, I. Gow, J. Hayward, J. R. Esquius, N. Richards, L. R. Smith, T. J. A. Slater, T. E. Davies, N. F. Dummer, L. Kabalan, A. Logsdail, R. C. Catlow, S. Taylor and G. J. Hutchings, ACS Catal., 2022, 12, 5371–5379 CrossRef CAS PubMed.
- N. Lawes, K. J. Aggett, L. R. Smith, T. J. A. Slater, M. Dearg, D. J. Morgan, N. F. Dummer, S. H. Taylor, G. J. Hutchings and M. Bowker, Catal. Lett., 2024, 154, 1603–1610 CrossRef CAS.
- N. Lawes, N. F. Dummer, S. Fagan, O. Wielgosz, I. E. Gow, L. R. Smith, T. J. A. Slater, T. E. Davies, K. J. Aggett, D. J. Morgan, S. H. Taylor, G. J. Hutchings and M. Bowker, Appl. Catal., A, 2024, 679, 119735 CrossRef CAS.
- H. Bahruji, J. R. Esquius, M. Bowker, G. Hutchings, R. D. Armstrong and W. Jones, Top. Catal., 2018, 61, 144–153 CrossRef PubMed.
- H. Bahruji, R. D. Armstrong, J. Ruiz Esquius, W. Jones, M. Bowker and G. J. Hutchings, Ind. Eng. Chem. Res., 2018, 57, 6821–6829 CrossRef CAS.
- J. Ruiz Esquius, H. Bahruji, S. H. Taylor, M. Bowker and G. J. Hutchings, ChemCatChem, 2020, 12, 6024–6032 CrossRef CAS.
- N. Lawes, I. E. Gow, L. R. Smith, K. J. Aggett, J. S. Hayward, L. Kabalan, A. J. Logsdail, T. J. A. Slater, M. Dearg, D. J. Morgan, N. F. Dummer, S. H. Taylor, M. Bowker, C. R. A. Catlow and G. J. Hutchings, Faraday Discuss., 2023, 242, 193–211 RSC.
- L. A. Parker, N. Richards, L. Bailey, J. H. Carter, E. Nowicka, S. Pattisson, N. F. Dummer, Q. He, L. Lu, C. J. Kiely, S. E. Golunski, A. Roldan and G. J. Hutchings, Catal. Lett., 2024, 154, 1958–1969 CrossRef CAS.
- A. Cooper, T. E. Davies, D. J. Morgan, S. Golunski and S. H. Taylor, Catalysts, 2020, 10, 294 CrossRef CAS.
- C. He, J. Cheng, X. Zhang, M. Douthwaite, S. Pattisson and Z. Hao, Chem. Rev., 2019, 119, 4471–4568 CrossRef CAS PubMed.
- Y. Guo, M. Wen, G. Li and T. An, Appl. Catal., B, 2021, 281, 119447 CrossRef CAS.
- A. Evuti, Int. J. Eng. Sci., 2013, 2, 145–153 Search PubMed.
- B. Barletta, S. Meinardi, F. Sherwood Rowland, C.-Y. Chan, X. Wang, S. Zou, L. Yin Chan and D. R. Blake, Atmos. Environ., 2005, 39, 5979–5990 CrossRef CAS.
- H. Huo, Q. Zhang, K. He, Z. Yao, X. Wang, B. Zheng, D. G. Streets, Q. Wang and Y. Ding, Environ. Pollut., 2011, 159, 2954–2960 CrossRef CAS PubMed.
- P. J. Landrigan, R. Fuller, N. J. R. Acosta, O. Adeyi, R. Arnold, N. Nil Basu, A. B. Baldé, R. Bertollini, S. Bose-O'Reilly, J. I. Boufford, P. N. Breysse, T. Chiles, C. Mahidol, A. M. Coll-Seck, M. L. Cropper, J. Fobil, V. Fuster, M. Greenstone, A. Haines, D. Hanrahan, D. Hunter, M. Khare, A. Krupnick, B. Lanphear, B. Lohani, K. Martin, K. V. Mathiasen, M. A. McTeer, C. J. L. Murray, J. D. Ndahimananjara, F. Perera, J. Potočnik, A. S. Preker, J. Ramesh, J. Rockström, C. Salinas, L. D. Samson, K. Sandilya, P. D. Sly, K. R. Smith, A. Steiner, R. B. Stewart, W. A. Suk, O. C. P. van Schayck, G. N. Yadama, K. Yumkella and M. Zhong, Lancet, 2018, 391, 462–512 CrossRef PubMed.
- S. Scirè and L. F. Liotta, Appl. Catal., B, 2012, 125, 222–246 CrossRef.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- M. S. Kamal, S. A. Razzak and M. M. Hossain, Atmos. Environ., 2016, 140, 117–134 CrossRef CAS.
- A. Krishnamurthy, B. Adebayo, T. Gelles, A. Rownaghi and F. Rezaei, Catal. Today, 2020, 350, 100–119 CrossRef CAS.
- M. N. Taylor, W. Zhou, T. Garcia, B. Solsona, A. F. Carley, C. J. Kiely and S. H. Taylor, J. Catal., 2012, 285, 103–114 CrossRef CAS.
- B. A. Tichenor and M. A. Palazzolo, Environ. Prog., 1987, 6, 172–176 CrossRef CAS.
- P. M. Shah, A. N. Day, T. E. Davies, D. J. Morgan and S. H. Taylor, Appl. Catal., B, 2019, 253, 331–340 CrossRef CAS.
- B. Solsona, T. E. Davies, T. Garcia, I. Vázquez, A. Dejoz and S. H. Taylor, Appl. Catal., B, 2008, 84, 176–184 CrossRef CAS.
- B. Solsona, T. García, G. J. Hutchings, S. H. Taylor and M. Makkee, Appl. Catal., A, 2009, 365, 222–230 CrossRef CAS.
- B. Solsona, T. García, R. Sanchis, M. D. Soriano, M. Moreno, E. Rodríguez-Castellón, S. Agouram, A. Dejoz and J. M. López Nieto, Chem. Eng. J., 2016, 290, 273–281 CrossRef CAS.
- B. Solsona, R. Sanchis, A. M. Dejoz, T. García, L. Ruiz-Rodríguez, J. M. López Nieto, J. A. Cecilia and E. Rodríguez-Castellón, Catalysts, 2017, 7, 96 CrossRef.
- B. Solsona, T. Garcia, E. Aylón, A. M. Dejoz, I. Vázquez, S. Agouram, T. E. Davies and S. H. Taylor, Chem. Eng. J., 2011, 175, 271–278 CrossRef CAS.
- P. M. Shah, J. W. H. Burnett, D. J. Morgan, T. E. Davies and S. H. Taylor, Catalysts, 2019, 9, 475 CrossRef CAS.
- P. M. Shah, L. A. Bailey and S. H. Taylor, Catalysts, 2023, 13, 114 CrossRef CAS.
- P. M. Shah, L. A. Bailey, D. J. Morgan and S. H. Taylor, Catalysts, 2023, 13, 794 CrossRef CAS.
- G. Salek, P. Alphonse, P. Dufour, S. Guillemet-Fritsch and C. Tenailleau, Appl. Catal., B, 2014, 147, 1–7 CrossRef CAS.
- Z. Zhu, G. Lu, Z. Zhang, Y. Guo, Y. Guo and Y. Wang, ACS Catal., 2013, 3, 1154–1164 CrossRef CAS.
- Y. Xie, Y. Yu, X. Gong, Y. Guo, Y. Guo, Y. Wang and G. Lu, CrystEngComm, 2015, 17, 3005–3014 RSC.
- K. Aggett, T. E. Davies, D. J. Morgan, D. Hewes and S. H. Taylor, Catalysts, 2021, 11, 1461 CrossRef CAS.
- M. Taylor, E. N. Ndifor, T. Garcia, B. Solsona, A. F. Carley and S. H. Taylor, Appl. Catal., A, 2008, 350, 63–70 CrossRef CAS.
- M. V. Twigg, Catal. Today, 2011, 163, 33–41 CrossRef CAS.
- A. K. Khudorozhkov, I. A. Chetyrin, A. V. Bukhtiyarov, I. P. Prosvirin and V. I. Bukhtiyarov, Top. Catal., 2017, 60, 190–197 CrossRef CAS.
- Y. Yazawa, H. Yoshida, N. Takagi, S. Komai, A. Satsuma and T. Hattori, Appl. Catal., B, 1998, 19, 261–266 CrossRef CAS.
- R. J. Farrauto, M. C. Hobson, T. Kennelly and E. M. Waterman, Appl. Catal., A, 1992, 81, 227–237 CrossRef CAS.
- F. H. Ribeiro, M. Chow and R. A. Dallabetta, J. Catal., 1994, 146, 537–544 CrossRef CAS.
- R. F. Hicks, H. Qi, M. L. Young and R. G. Lee, J. Catal., 1990, 122, 295–306 CrossRef CAS.
- M. Macino, A. J. Barnes, S. M. Althahban, R. Qu, E. K. Gibson, D. J. Morgan, S. J. Freakley, N. Dimitratos, C. J. Kiely, X. Gao, A. M. Beale, D. Bethell, Q. He, M. Sankar and G. J. Hutchings, Nat. Catal., 2019, 2, 873–881 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- P. Canton, G. Fagherazzi, M. Battagliarin, F. Menegazzo, F. Pinna and N. Pernicone, Langmuir, 2002, 18, 6530–6535 CrossRef CAS.
- N. Richards, J. H. Carter, E. Nowicka, L. A. Parker, S. Pattisson, Q. He, N. F. Dummer, S. Golunski and G. J. Hutchings, Appl. Catal., B, 2020, 264, 118501 CrossRef.
- A. Jürgensen, N. Heutz, H. Raschke, K. Merz and R. Hergenröder, Anal. Chem., 2015, 87, 7848–7856 CrossRef PubMed.
- I. Aruna, B. R. Mehta, L. K. Malhotra and S. M. Shivaprasad, J. Appl. Phys., 2008, 104, 064308 CrossRef.
- Y. Zhang, Y. Cai, Y. Guo, H. Wang, L. Wang, Y. Lou, Y. Guo, G. Lu and Y. Wang, Catal. Sci. Technol., 2014, 4, 3973–3980 RSC.
- E. J. Jang, J. Lee, D. G. Oh and J. H. Kwak, ACS Catal., 2021, 11, 5894–5905 CrossRef CAS.
- L. Meng, A.-P. Jia, J.-Q. Lu, L.-F. Luo, W.-X. Huang and M.-F. Luo, J. Phys. Chem. C, 2011, 115, 19789–19796 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00665h |
| This journal is © The Royal Society of Chemistry 2024 |