
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHematin supported on Colour Catcher®: a biodegradable heterogeneous catalyst for halogen-free CO2 cycloadditions†
Caterina
Damiano
 a,
Alessia
Fata
a,
Matteo
Cavalleri
a,
Gabriele
Manca
*b and
Emma
Gallo
*a
a,
Alessia
Fata
a,
Matteo
Cavalleri
a,
Gabriele
Manca
*b and
Emma
Gallo
*a
aDepartment of Chemistry, Università degli Studi di Milano, Via Golgi 19, 20136 Milan, Italy. E-mail: emma.gallo@unimi.it
bICCOM-CNR, Istituto di Chimica dei Composti OrganoMetallici, Via Madonna del Piano 10, I-50019 Sesto Fiorentino, Italy
First published on 30th May 2024
Abstract
This work describes the activity of biocompatible hemin derivatives in catalyzing the cycloaddition of CO2 to epoxides and aziridines. The process sustainability was enhanced through the use of a catalytic material obtained by heterogenizing hematin onto Colour Catcher® sheets, a cellulose-based support with complete biodegradability. The material exhibited remarkable efficiency and stability, enabling consecutive use without adding additional co-catalysts, thereby enhancing the eco-tolerance of the system. The generality of the reaction was demonstrated by testing various substrates under both homogeneous and heterogeneous conditions and the reaction mechanism was suggested through DFT calculations.
Introduction
Carbon dioxide is the primary greenhouse gas responsible for global warming and the resulting climate change, which is provoking a significant increase in extreme weather events that impact our health, environment, and economy.In order to achieve an economy with net-zero greenhouse gas (GHG) emissions by 2050, both governments and the scientific community are doing many efforts for mitigating the environmental impact of CO2 produced by human activities.
Nowadays, the most important tools in ‘CO2 management’ are carbon capture and storage (CCS), which focuses on capturing and storing CO2, and carbon capture and utilization (CCU), which implies that the capture of carbon dioxide is followed by its conversion into valuable products.1–4 This strategy not only prevents the release of CO2 into the atmosphere as a greenhouse gas but also allows it to be utilized as a C1 source5,6 in the synthesis of a wide range of useful products, in alignment with circular economy principles.
Among various synthetic methodologies that use CO2 as a starting material, the synthesis of cyclic carbonates and oxazolidin-2-ones by the respective CO2 cycloaddition to epoxides7,8 and aziridines9–11 offers the added benefit of being 100% atom-efficient procedures (Scheme 1).12
 | ||
| Scheme 1 General scheme of CO2 cycloaddition to epoxides and aziridines forming cyclic carbonates and oxazolidin-2-ones, respectively. | ||
Both cyclic carbonates and oxazolidin-2-ones are valuable chemicals employed in various fields. Whilst cyclic carbonates find applications as electrolytic solvents in lithium batteries, reagents for polycarbonate synthesis and in the production of drugs and cosmetics,13–15 oxazolidin-2-ones are chiral auxiliaries, pharmaceutical compounds and also used as useful intermediates in organic synthesis.10,16–19
The formation of the five-membered ring is facilitated by the synergistic action of electrophilic and nucleophilic species. It is generally assumed that the electrophile coordinates the heteroatom of the three-membered ring to promote the ring-opening reaction by the nucleophilic species. Subsequently, the reaction can proceed as illustrated in Scheme 2.8,20–24
 | ||
| Scheme 2 General mechanism of CO2 cycloaddition to epoxides and aziridines. | ||
Porphyrin-based catalysts have been shown to be efficient catalysts for CO2 cycloaddition to epoxides and aziridines whether under homogeneous or heterogeneous conditions.25 Considering that the contemporary presence of an electrophile and nucleophile is required, several binary and bifunctional porphyrin-based systems have been developed.20,21,26–30 The binary systems typically consist of an electrophilic porphyrin species, called the catalyst, and a source of the nucleophilic agent (i.e., an ammonium salt), which is commonly defined as the co-catalyst.
On the other hand, in a bifunctional porphyrin system, the tetrapyrrolic core serves as the electrophilic center responsible for interacting with the heteroatom of the three-membered cycle, while the porphyrin periphery carries the nucleophile responsible for the ring-opening reaction.31–37 The use of bifunctional catalysts, rather than binary systems, improves the sustainability and cost-effectiveness of the process by reducing the number of species involved and minimizing the necessary purification steps. The environmental friendliness of the procedure can be further enhanced by employing biocompatible catalysts and/or by immobilizing the catalyst onto a solid support.31,32,34
Porphyrin-like complexes inherently exhibit biocompatibility due to the tetrapyrrolic scaffold, which is present in essential biological molecules, including hemin and hematin. These latter molecules play pivotal roles in biological functions and present different pharmaceutical and catalytic activities.38 Hemin, the oxidized form of heme, is an iron(III) protoporphyrin IX species that shows a chloride ligand in the axial position. Its cost-effective extraction and purification make it an ideal choice for a wide range of applications. Conversely, in the case of hematin, also known as hydroxyhemin, the iron(III) center bears a hydroxyl axial group in place of the chloride atom (Fig. 1).39
 | ||
| Fig. 1 Molecular structures of hemin (1), Fe(OMe)protoporphyrin IX (2) and the sodium salt of hematin (3). | ||
As mentioned earlier, the sustainability of the process can be improved by immobilizing the active molecule onto a surface. A variety of supports has been employed and the utilization of materials with a cellulose-like structure is especially attractive for obtaining low-toxic and biocompatible heterogeneous materials.40,41
We recently reported that porphyrins can be readily anchored onto Colour Catcher®(CC) sheets, a cellulose-based material commonly used in everyday life to capture colors in laundry water.42,43 This absorbent material, with a remarkable ability to bind porphyrins, stands as a highly innovative choice owing to its full biodegradability and exceptional mechanical, thermal and chemical stability that prevents the leaching of active molecules. The intrinsic coloration of porphyrins enabled them to be effectively immobilized onto CC sheets, resulting in a material that exhibited strong performance in sensing42,44–46 and catalysis.43
Given our expertise in utilizing metal porphyrins and free porphyrins in combination with ammonium salts or chloride sources both in homogeneous27,47–52 and heterogeneous53 CO2 cycloadditions, we have studied the catalytic activity of environmentally-friendly porphyrins derived from natural sources, such as hemin and hematin.
In this paper, we present a catalytic protocol involving the utilization of hemin (1), Fe(OMe)protoporphyrin IX (2), and sodium salt of hematin (3) under homogeneous conditions to mediate the CO2 cycloaddition to three-membered rings. In addition, the immobilization of hematin onto Colour Catcher® sheets yielded a highly effective and no-toxic heterogeneous 3@CC catalyst that promoted the synthesis of oxazolidin-2-ones. Notably, when 3@CC was used, the presence of a nucleophilic co-catalyst was not required resulting in a convenient and halogen-free procedure.
Results and discussion
Cycloaddition of CO2 to epoxide under homogeneous conditions
The model reaction between styrene oxide and CO2, forming 4-phenyl-1,3-dioxolan-2-one (4), was run in the presence of the three different catalysts, hemin (1), Fe(OMe)protoporphyrin IX (2) and the sodium salt of hematin (3) (Fig. 1), in order to evaluate how the nature of the axial ligand influences the catalytic activity.It is important to note that both the nature of the axial ligand on iron and the peripheral substituents on the pyrrole rings significantly influence the solubility of the complex in both organic solvents and water. Complexes 1 and 3 exhibited poor solubility in common organic solvents, except for DMSO. On the other hand, complex 2, which was synthesized by refluxing commercially available hemin (1) in methanol, displayed good solubility not only in DMSO but also in dichloroethane (DCE), tetrahydrofuran (THF) and methanol.
It's worth noting that hemin (1) tends to aggregate in water54 forming various non-monomeric species with a resulting poor solubility in aqueous environments. To achieve complete solubilization in water, hemin can be converted into the sodium salt of hematin (3) by treating compound 1 with aqueous NaOH.
The synthesis of compound 4 was run in the presence of 1, 2, or 3 iron(III) catalyst and tetrabutyl ammonium chloride (TBACl) as the co-catalyst, by applying different experimental conditions (Table 1). The reaction was initially conducted at 100 °C and 0.6 MPa of CO2 for four hours by using styrene oxide as the solvent. The desired compound was obtained in varying yields depending on the different solubility of the three catalysts in the reaction medium. When catalyst 3 was used, the reaction productivity was hindered by the limited solubility of the complex in styrene oxide (Table 1, run 3). On the other hand, thanks to the higher solubility of complexes 1 and 2 in styrene oxide, compound 4 was formed in almost a quantitative yield in the presence of these two catalysts (Table 1, runs 1 and 2). Given these findings and considering the higher solubility of complex 2 in styrene oxide, the reaction conditions were optimized by using catalyst 2. When the synthesis of 4 was performed without the co-catalyst, only traces of the product were detected (Table 1, run 4), even when stronger reaction conditions (125 °C, 1.2 MPa of CO2 for 16 hours) were applied and using DCE as the reaction solvent (Table 1, run 5). These results indicated that, under these experimental conditions, the axial ligand of the catalyst remained tightly bound to the iron center and was not available to act as a nucleophilic species for opening the epoxide ring and consequently, the presence of TBACl was necessary (Scheme 2). Nevertheless, it is worth highlighting that the catalyst/TBACl ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 used in this study was more convenient than that we have previously reported by using a ruthenium-based porphyrin catalyst (catalyst/TBACl = 1:10).51
:2 used in this study was more convenient than that we have previously reported by using a ruthenium-based porphyrin catalyst (catalyst/TBACl = 1:10).51
|
|
|||||
|---|---|---|---|---|---|
| Run | Cat. (mol%) | TBACl (mol%) | CO2 (MPa) | T (°C) | Yield (%) |
| a The reaction was performed in styrene oxide for 4 hours. b The reaction was performed in DCE for 16 hours. | |||||
| 1 | 1 (0.7) | 1.4 | 0.6 | 100 | 96 |
| 2 | 2 (0.7) | 1.4 | 0.6 | 100 | 94 |
| 3 | 3 (0.7) | 1.4 | 0.6 | 100 | 40 |
| 4 | 2 (0.7) | — | 0.6 | 100 | Traces |
| 5b | 2 (0.7) | — | 1.2 | 125 | 10 |
| 6 | 2 (0.35) | 0.7 | 0.6 | 100 | 71 |
| 7 | 2 (0.7) | 1.4 | 0.6 | 75 | 20 |
| 8 | 2 (0.7) | 1.4 | 0.6 | 125 | 100 |
| 9 | 2 (0.7) | 1.4 | 0.3 | 100 | 73 |
| 10 | 2 (0.7) | 1.4 | 1.2 | 100 | 99 |

Then, the optimization of reaction parameters was performed by varying catalytic loading, temperature and CO2 pressure. As reported in Table 1, a reduction of the product yield was observed by halving the catalyst/co-catalyst amount to 0.35 and 0.7 mol%, respectively (Table 1, run 6) and maintaining all the other parameters as reported before.
Also a decrease of the temperature to 75 °C (Table 1, run 7) or CO2 pressure to 0.3 MPa (Table 1, run 9) had a negative effect on the reaction productivity. Conversely, quantitative yields of 4 were achieved by increasing the reaction temperature to 125 °C (Table 1, run 8) or the CO2 pressure to 1.2 MPa (Table 1, run 10).
Considering data reported in Table 1, the reactivity of ten different epoxides with diverse electronic and steric features was investigated by running reactions for 4 hours at 0.6 MPa of CO2 and 100 °C (Fig. 2). The steric hindrance of the substituent on the epoxide ring had a negative impact on the reaction efficiency and reaction yields decreased as the steric bulk of the reagent increased (Fig. 2, compounds 9 and 12). When a similar steric hindrance was present on the epoxide ring, the catalytic activity was further inhibited by the presence of an electron-donor substituent (compare yields between compounds 5 and 6 as well as compounds 11 and 12). Thus, the reaction performed well when utilizing electron-poor epoxides with linear and short alkyl chain substituents on the ring.
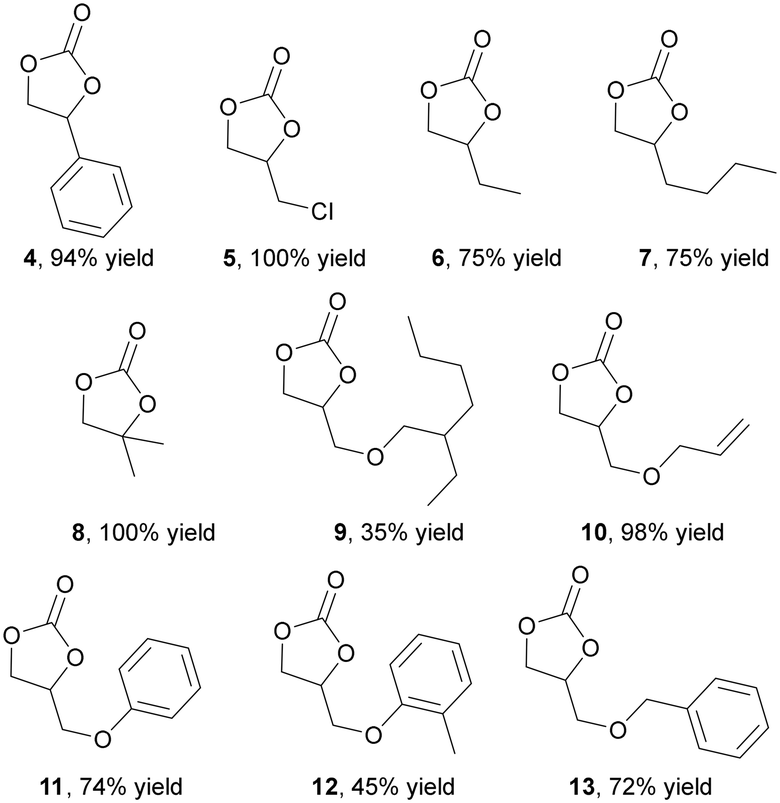 | ||
| Fig. 2 Synthesis of cyclic carbonates 4–13 mediated by the 2 (0.7 mol%)/TBACl (1.4 %mol) binary system run at 100 °C, 0.6 MPa CO2 in epoxide as the reaction solvent for four hours. | ||
Cycloaddition of CO2 to aziridines under homogeneous and heterogeneous conditions
Based on the data obtained up to now, we evaluated the activity of 1, 2, and 3 catalysts in CO2 cycloaddition to N-butyl-2-phenyl aziridine forming the corresponding N-butyl-5-phenyloxazolidin-2-one (14a) as the major isomer. According to our previous optimizations of the CO2 cycloaddition to N-alkyl aziridines,47 the reaction was run for 12 hours using dichloroethane (DCE) as the reaction solvent, under 1.2 MPa of CO2 pressure and at 125 °C (Scheme 3). It is important to emphasize that only catalyst 2 exhibited complete solubility in DCE, whereas catalysts 1 and 3 remained active despite being only sparingly soluble in the reaction medium.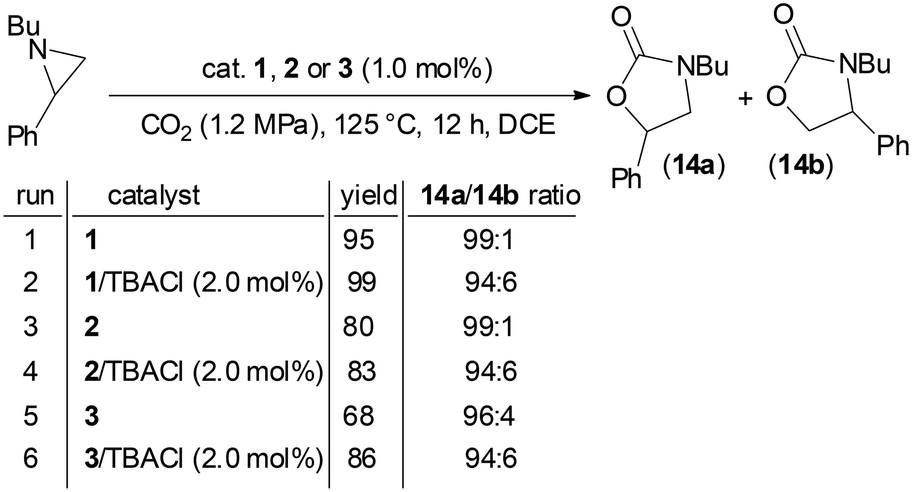 | ||
| Scheme 3 Synthesis of N-alkyl oxazolidin-2-one 14a/14b promoted by 1, 2 or 3 in the presence and in the absence of TBACl. | ||
As reported in Scheme 3, the model reaction was conducted both in the presence and absence of TBACl and to our delight, the reaction proceeded effectively even without any co-catalyst.
The efficiency of the TBACl-free catalytic reactions proved that all the catalysts have a bifunctional nature and consequently, the eco-compatibility of the procedure increased due to both the reduction of the reaction components and the absence of noxious halogen species.
Considering the activity exhibited by complex 3 (Scheme 3, run 5) and the presence of anionic COO− functionalities in the ligand periphery, this complex was grafted onto Colour Catcher® sheets through electrostatic interactions between the COO− group of the porphyrin and the positively charged groups (cationic polymer) of the Colour Catcher® surface.55 Complex 1 was dissolved in an aqueous NaOH solution to be converted into 3 and then the CC sheet was immersed in the 3-containing solution to form the 3@CC material.
The quantity of porphyrin loaded onto the CC sheet was directly influenced by both the porphyrin concentration and the immersion duration, and the maximum loading of porphyrin 3 onto the CC surface was reached by dipping the CC sheet in a 1.0 × 10−3 M solution of 3 for 7 hours (see the ESI† for further details). Thus, to test the reproducibility in the anchoring process, we always left a 25 cm2 Colour Catcher® sheet immersed in 100 mL of 1.0 × 10−3 M solution for 8.0 hours (Fig. 3). Then, the so-obtained 3@CC was dried and no porphyrin leaching was observed by washing the sheets with NaOH 0.1 M and distilled water. The quantity of porphyrin loaded onto the CC sheet was determined through UV-vis spectroscopy by measuring the porphyrin concentration before and after the immersion of the CC into the porphyrin solution (see the ESI† for further details). The amount of porphyrin grafted onto the CC sheet was consistent across several preparations of 3@CC demonstrating the reproducibility of the anchorage procedure.
 | ||
| Fig. 3 Preparation of the 3@CC heterogeneous catalyst. | ||
The resulting 3@CC material was analyzed by UV-vis spectrophotometry, scanning electron microscopy (SEM), energy dispersive X-ray analysis (EDX), inductively coupled plasma-optical emission spectroscopy (ICP-OES), X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), Fourier-transform infrared spectroscopy (FTIR) and nuclear magnetic resonance (NMR) (see the ESI† for all analytic details).
The presence of porphyrin 3 on the CC surface was firstly confirmed by comparing the UV-vis spectrum of 3 in water with that of 3@CC obtained in solid state. As depicted in Fig. 4, even if the profile of the solid-supported 3@CC exhibits broader bands in comparison to those of the sodium salt of hematin (3), the typical absorption profile of porphyrin 3 was observed in the spectrum of 3@CC and the distinct absorptions of the Soret and Q bands remain discernible.
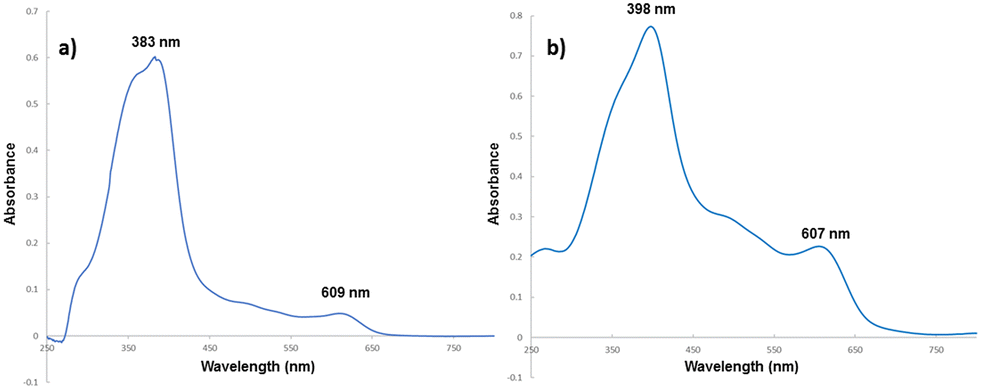 | ||
| Fig. 4 UV-vis spectra of (a) 3 and (b) 3@CC. | ||
In accordance to the UV-vis features of other anionic porphyrins anchored to Colour Catcher® sheets,42,43 the Soret band of 3@CC was red-shifted compared to its free counterparts. Both 3 and 3@CC spectra showed a splitting of the Soret band indicating that the aggregation phenomenon, usually present in homogeneous aqueous solution (Fig. 4a),54 was maintained on the solid surface (Fig. 4b). A sample of 3@CC, presenting a catalytic loading of 0.054 mg cm−2, was analyzed by SEM and EDX techniques (Fig. 5). The SEM analysis revealed a homogeneous texture of 3@CC and the EDX spectra disclosed the presence of iron (not present in the unfunctionalized Colour Catcher® sheet, see the EDX spectrum in the ESI†) on the solid surface confirming the efficiency of the grafting procedure. The presence of iron in the CC fibers was also proved by XPS analysis and ICP-OES analysis showed 0.11% w/w of iron in the 3@CC material. Finally, TGA analysis demonstrated that 3@CC was stable up to 200 °C to prove the excellent thermal stability of the material. Unfortunately, FT-IR and 13C NMR spectroscopy were inadequate for characterizing 3@CC due to the predominance of the cellulose-based matrix that hindered the detection of the expected hematin signals in both acquired spectra. All data collected up to now (see the ESI† for details) underlined the suitability of Colour Catcher® as a solid support for the efficient and reproducible immobilization of negatively charged porphyrin species.42,43 The stability and biocompatibility nature of the 3@CC material validated its employment as an eco-friendly catalyst in CO2 cycloaddition to aziridines.
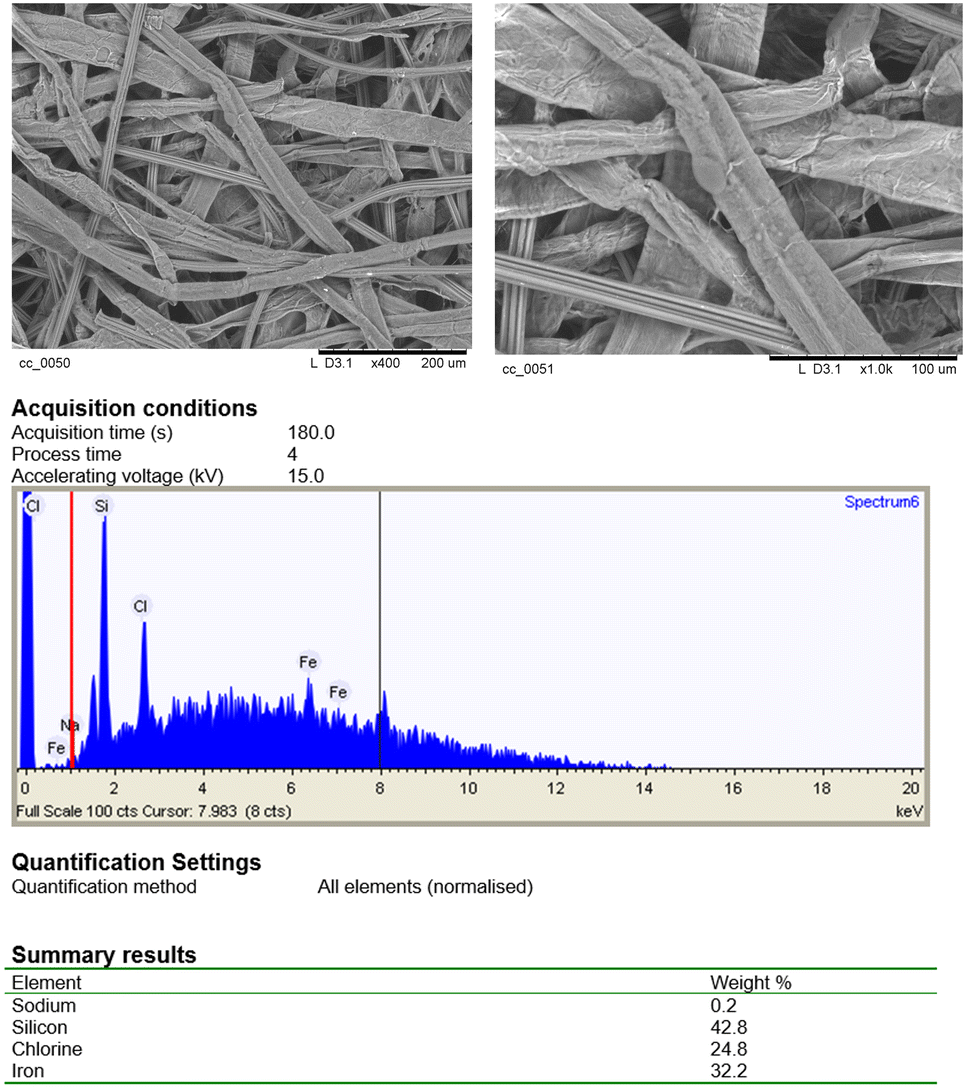 | ||
| Fig. 5 SEM and EDX analyses of 3@CC (0.054 mg cm−2). | ||
After characterizing the 3@CC material, its chemical and physical stability under operating CO2 pressures and temperatures were tested by heating DCE, DMSO (dimethyl sulfoxide) or DMF (N,N-dimethylformamide) suspensions of 3@CC at 150 °C under 1.5 MPa of CO2 for 12 hours. The obtained data revealed a very high stability of 3@CC in DCE in which no traces of leached porphyrin were detected by UV-vis spectroscopy. On the contrary, a slight porphyrin leaching was observed by employing DMSO and DMF, whose UV-vis analysis revealed the release of 3% and 10% of 3, respectively. Despite the porphyrin leaching, SEM-EDX analyses of all the heterogeneous materials showed no changes in the structure and/or composition of 3@CC after the treatment under harsh conditions.
In view of the experimental conditions already used for the homogeneous CO2 cycloaddition to N-butyl-2-phenyl aziridine (1.2 MPa of CO2 at 125 °C for 12 hours) and the high stability of 3@CC in DCE, we employed this heterogeneous material to investigate the scope of the CO2 cycloaddition to N-alkyl aziridines by applying the experimental conditions reported in Scheme 3.
As illustrated in Fig. 6, the reaction was productive with various aziridine substrates even at the low 3@CC loading of 0.1 mol% and in the absence of any nucleophilic source. It is worth noting that when the reaction between N-butyl-2-phenyl aziridine and CO2 was carried out in the presence of the porphyrin-free Colour Catcher® sheet and applying experimental conditions outlined in Fig. 6, the formation of the desired 14a/14b mixture was not observed.
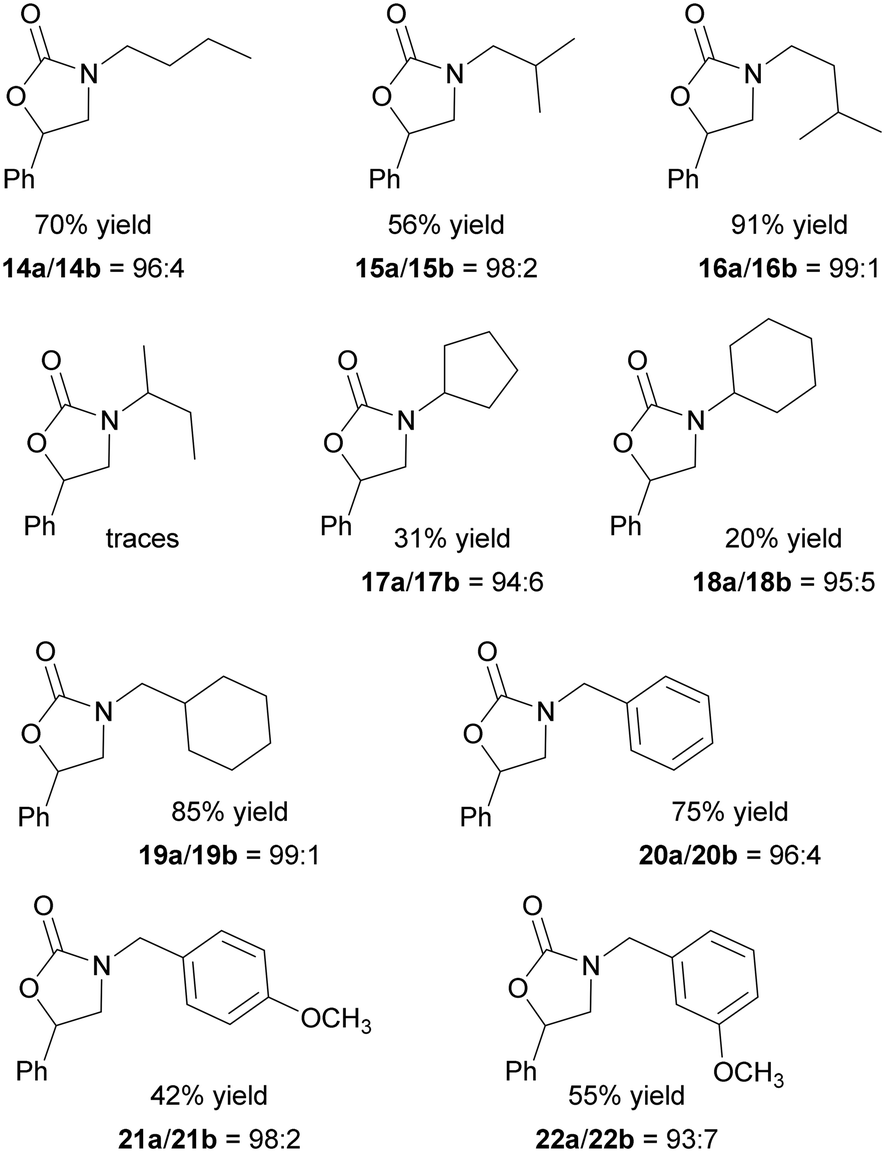 | ||
| Fig. 6 Synthesis of N-alkyl oxazolidin-2-ones 14–22 mediated by 3@CC (0.1 mol%) run at 125 °C, 1.2 MPa CO2 in DCE as the reaction solvent for 12 hours. | ||
The reaction efficiency was influenced by the steric hindrance of the N-alkyl substituent and the presence of a –CH2– spacer between the nitrogen atom and the bulky group was necessary to achieve satisfactory yields (Fig. 6, compounds 14–16, 19–22). When the cyclic substituent was directly linked to the nitrogen atom of the aziridine, its steric bulk hampered an efficient coordination to the iron center, resulting in the reduced activation of the molecule towards the ring-opening reaction. Consequently, the corresponding N-alkyl oxazolidin-2-ones were obtained in low yields (Fig. 6, compounds 17–18). Even if the electronic effects of substituents on reaction efficiency were less pronounced, the presence of electron-donating groups had an adverse impact on reactivity. As a result, lower productivities were observed in the synthesis of 21 and 22 compared to that of 20. The data obtained indicated a good regioselectivity in all the reactions mediated by 3@CC, and the 5-substituted regioisomer was always formed as the major product.
The very good activity and versatility of 3@CC prompted us to investigate the chemo-physical characteristics of the heterogeneous material at the end of a catalytic reaction to establish possible modifications of the porphyrin structure that could hamper its fruitful recycle. Thus, 3@CC was recovered after the complete conversion of N-butyl aziridine into 14, washed with DCE and analyzed by UV-vis, ICP-MS and SEM-EDX spectroscopy. The post-catalysis UV-vis spectrum of 3@CC was comparable to that of the starting material (Fig. 4b) confirming that 3 was still grafted on the CC surface. In addition, ICP-MS analysis showed the presence of 0.11% w/w of iron to validate the absence of catalyst leaching and SEM-EDX analysis confirmed the preservation of the homogenous structure of 3@CC.
The stability of the heterogeneous catalyst was further proven by its excellent activity for four consecutive catalytic reactions. After the synthesis of 14, 3@CC was recovered, washed with DCE and subsequently reused three additional times. The desired compound was formed with a 70% overall yield and a 14a/14b regioselectivity of 94:6.
The catalytic performances shown by 3@CC in the synthesis of N-alkyl oxazolidin-2-ones make this heterogeneous material a competitive alternative to other heterogeneous catalysts already reported in the literature.56–62 Among these, Cu, In and Co-based metal–organic frameworks (MOFs) have been used to synthesize N-alkyl oxazolidin-2-ones and they showed very good activity and recyclability. Even if these catalysts worked under experimental conditions milder than those used in the presence of 3@CC (e. g. 30 °C, 1.0 MPa CO2 (ref. 57 and 58) or 60 °C, 0.5 MPa CO2 (ref. 56)), the presence of a co-catalyst was always required. Thus, the bifunctional nature, biodegradability and low-toxicity of 3@CC (both hematin and cellulose-based support are bio-derived species) as well as the application of a halogen-free procedure compensate for the drawback of the slightly harsher experimental conditions required.
To broaden the scope of the reaction, we explored CO2 cycloaddition to 1-(3,5-bis(trifluoromethyl)phenyl)-2-phenylaziridine to form the corresponding N-aryl-oxazolidin-2-one (23) under homogeneous conditions by using the 1 (1.0 mol%)/TBACl (2.0 %mol) binary system at 125 °C, 1.2 MPa CO2 for 12 hours. The product was obtained in a yield of 95% and with a 23a/23b regioisomer ratio of 95:5 (Scheme 4). Unfortunately, the reaction did not occur in the absence of TBACl as a co-catalyst. This data aligns with our expectations and indicates a lower reactivity of N-aryl aziridine compared to N-alkyl analogues due to both electronic and steric factors, which hinders the reaction to proceed in the absence of a chloride source.
 | ||
| Scheme 4 Synthesis of N-aryl oxazolidin-2-ones 23 and 24 mediated by the 3@CC (0.1 mol%)/TBACl (2.0%) binary system. | ||
When the synthesis of 23 was conducted under heterogeneous conditions by employing 3@CC as the catalyst, a slight decrease in both yield and regioselectivity was noted (Scheme 4). Interestingly, the catalytic activity was not influenced by the electronic characteristics of the aromatic substituent on the nitrogen atom. In fact, N-aryl oxazolidin-2-one 24 was obtained in a yield comparable to that obtained in the synthesis of 23 (Scheme 4). In both cases, the presence of TBACl was required.
Study of the reaction mechanism of oxazolidin-2-one formation
Given that the CO2 cycloaddition to N-alkyl aziridines occurred in the absence of any nucleophilic species, which contrasts with the requirement for a halogen source in the analogous reaction involving epoxides, two significant questions arise for suggesting mechanistic hypotheses. First, what additional role does the three-membered ring play in the mechanism, beyond serving as the reaction substrate? Second, in the absence of TBACl, who acts as the nucleophile responsible for the ring-opening reaction?To address these questions, we explored the ability of the three-membered ring (aziridine or epoxide) to coordinate the iron metal of the porphyrin complex and promote the cleavage of the Fe–X bond to release a X− nucleophilic agent essential for the ring-opening step. Thus, Fe(OMe)protoporphyrin IX (2) was dissolved in DCE and the UV-vis spectrum was recorded both in the absence and in the presence of either N-butyl aziridine or styrene oxide. Note that the test was conducted by using 2 to achieve a homogeneous solution in which it is easier to estimate weak interactions. As detailed in the ESI,† while a slight modification of the UV-vis spectrum was observed in the reaction between 2 and N-butyl aziridine, no interaction was registered by treating 2 with styrene epoxide. These results indicated that aziridine, but not epoxide, can weakly coordinate iron during the reaction and facilitates the departure of the trans axial group (Cl in 1, OMe in 2, and OH in 3), which can act as a nucleophile for opening the three-membered ring (Scheme 5). Thus, the presence of TBACl becomes unnecessary. Subsequently, as the three-membered ring opens, the reaction with CO2 can occur and the mechanism proceeds as previously described for analogous porphyrin-mediated catalytic reactions.20,27,47–49
 | ||
| Scheme 5 Suggested mechanism of CO2 cycloaddition to N-alkyl aziridines. | ||
This assumption was also supported by the lack of reactivity observed when N-aryl aziridines were tested in the absence of TBACl. These less basic and more sterically encumbered substrates exhibit reduced capacity to coordinate the iron metal, making them less effective in promoting the cleavage of the axial ligand bond. To better support the mechanism outlined in Scheme 5, a DFT study was conducted to provide a more comprehensive understanding of the nature of the involved intermediates.
DFT study
Considering that Fe(III) complex 2 is soluble in DCE and it was employed under homogeneous conditions (see above), the DFT investigation started by assessing its catalytic properties in promoting the cycloaddition reaction in solution. Iron(III) d5 metal can adopt three distinct spin states: sextet, quartet, and doublet, corresponding to five, three and one unpaired electron, respectively. The computational analysis at the B97D-DFT level of theory (further details provided in the computational details section)63 disclosed that the sextet spin state is the most stable (Fig. 7) and consequently, the following discussion and analysis will predominantly focus on this spin isomer.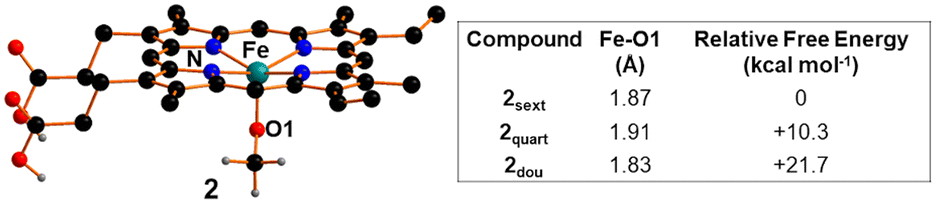 | ||
| Fig. 7 Optimized structure of compound 2 and comparison between the main structural/energetic features of the three spin states. The hydrogen atoms, except the carboxylic ones, were hidden for clarity. | ||
After establishing the most stable spin isomer, it became crucial to assess whether the methoxide axial ligand could be released in solution to act as a nucleophile in the ring-opening step. However, the possibility of methoxide dissociation from 2 forming a square planar cationic iron complex was ruled out due to the considerably high free energy cost of more than 50 kcal mol−1.
Consequently, our analysis focused on whether the energy cost of cleaving the Fe–OCH3 bond could be reduced when starting from the octahedral complex 25OMe (Fig. 8), where N-butyl-2-phenyl aziridine plays the role of the sixth axial ligand. It is worth noting that the coordination of aziridine to complex 2 occurs with a very low free energy cost of +0.2 kcal mol−1.
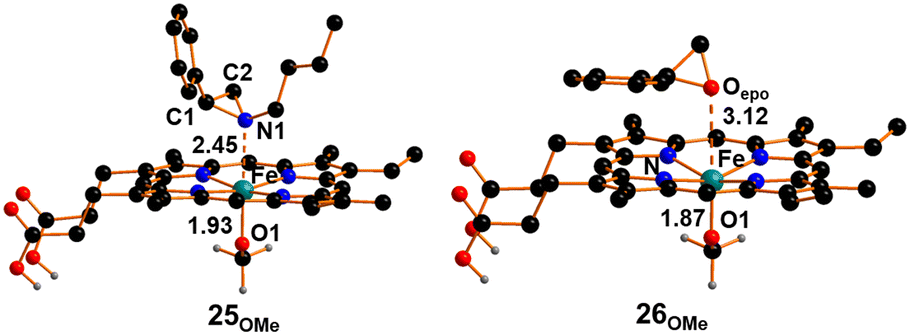 | ||
| Fig. 8 Optimized structures of compounds 25OMe and 26OMe. The hydrogen atoms, except the carboxylic ones, were hidden for clarity. | ||
The optimized structure of complex 25OMe exhibits an Fe–N1 bond length of 2.45 Å, accompanied by a subtle elongation of the trans-Fe–O distance by 0.06 Å compared to its initial value in 2. Although this effect is relatively small, it may account for the observed slight modification in the UV-vis spectrum of 2 upon its reaction with N-butyl-2-phenyl aziridine (see above). Conversely, in line with the experimental findings, the DFT analysis of the coordination of styrene epoxide to 2 revealed a somewhat hindered coordination. As illustrated in Fig. 8, the computationally isolated compound 26OMe exhibits an Fe–Oepoxide distance of 3.12 Å, and the trans Fe–O distance remains unaffected. Based on what was stated above, it is reasonable to assume that intermediate 25OMe, presenting the additional aziridine axial ligand, can release the methoxide ion into the solution. DFT analysis has shown that the cleavage of the Fe–OCH3 bond can occur with a free energy cost of +24.7 kcal mol−1, which is significantly lower than the cost of methoxy release when aziridine coordination is not considered.
The dissociation of the methoxy axial ligand leads to the formation of the ionic pair compound 27OMe reported in Fig. 9, which exhibits an O1⋯C1 distance of 3.43 Å. The release of methoxide results in a significant reduction of the Fe–N1 distance to approximately 2.13 Å, accompanied by electron depletion on the aziridine ring. Specifically, the NBO-calculated charge on the C1 center decreases by approximately 0.5 e compared to free N-butyl-2-phenyl aziridine. The electronic depletion on C1 allows the nucleophilic attack of the methoxide through a transition state (TS27-28OMe) shown in Fig. 9, with an associated free energy barrier of +6.6 kcal mol−1. The transition state nature was confirmed by the detection of a unique imaginary frequency at −356 cm−1 associated with the formation of O1⋯C1 linkage and the cleavage of the N1⋯C1 one. As shown in Fig. 9, the Fe–N1 bond in TS27-28OMe becomes shorter by 0.08 Å and a more efficient strengthening occurs in the following intermediate 28OMe (Fig. 9) up to the value of 1.93 Å. The formation of intermediate 28OMe, characterized by a fully established O1–C1 bond and a broken N1–C1 bond, occurs with a free energy gain of −40.4 kcal mol−1.
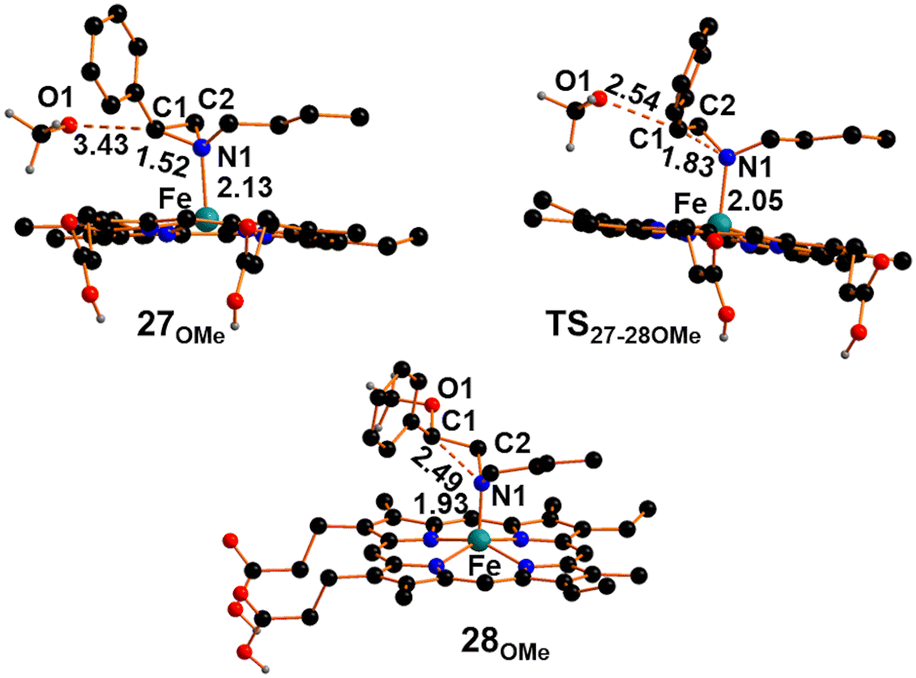 | ||
| Fig. 9 Optimized structures of intermediate 27OMe, transition state TS27-28OMe and intermediate 28OMe. The hydrogen atoms, except the carboxylic ones, were hidden for clarity. | ||
The newly acquired amidic character of the N1 atom in 28OMe may enable its reaction with CO2. This reaction results in the breaking of the Fe–N linkage, and the metal unsaturation is compensated for by coordinating one oxygen atom of the CO2 moiety. Intermediate 29OMe, as depicted in Fig. 10, was isolated with a free energy cost of +12 kcal mol−1.
 | ||
| Fig. 10 Optimized structures of intermediate 29OMe and transition state TS29-30OMe. The hydrogen atoms, except the carboxylic ones, were hidden for clarity. | ||
The CO2 moiety has lost its linearity (O2C3O3 angle is 122°) and an IR active mode has been detected for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1593 cm−1. The metal coordination of O2 reduces the available electronic density on the CO2 moiety, affecting the nucleophilic power of O3 in the ring-closure process to form oxazolidin-2-one. This limitation was partially mitigated by the elevated experimental temperature, consistent with the calculated high free energy barrier (+29.3 kcal mol−1) necessary to reach the transition state, TS29-30OMe that exhibits an aligned O3C1O1 arrangement (171°), with the methoxide ready to be released into the solution (Fig. 10). Subsequently, the ring closure forming the 30OMe intermediate (Fig. 11) is accompanied by a substantial free energy gain of −23.0 kcal mol−1. In 30OMe, the methoxide has already been released (C1⋯O1 distance exceeding 4 Å), and the oxazolidin-2-one ring has formed with a weak interaction between the nitrogen center of oxazolidin-2-one and the iron metal (Fe–N1 distance of 2.37 Å). The coordination of another aziridine molecule to the metal, in trans with respect to the oxazolidin-2-one ring, enables the formation of the octahedral complex 31OMe (Fig. 11) with a free energy cost of +8.5 kcal mol−1. The coordination of aziridine results in a significant weakening of the interaction between oxazolidin-2-one and the iron complex (with an Fe–N1 distance as large as 3.19 Å) and the dissociation of oxazolidin-2-one 14 occurs with a small free energy cost of +2.3 kcal mol−1. Then, the formation of intermediate 27OMe, the actual catalyst for the process, is ready for starting another catalytic cycle.
O stretching at 1593 cm−1. The metal coordination of O2 reduces the available electronic density on the CO2 moiety, affecting the nucleophilic power of O3 in the ring-closure process to form oxazolidin-2-one. This limitation was partially mitigated by the elevated experimental temperature, consistent with the calculated high free energy barrier (+29.3 kcal mol−1) necessary to reach the transition state, TS29-30OMe that exhibits an aligned O3C1O1 arrangement (171°), with the methoxide ready to be released into the solution (Fig. 10). Subsequently, the ring closure forming the 30OMe intermediate (Fig. 11) is accompanied by a substantial free energy gain of −23.0 kcal mol−1. In 30OMe, the methoxide has already been released (C1⋯O1 distance exceeding 4 Å), and the oxazolidin-2-one ring has formed with a weak interaction between the nitrogen center of oxazolidin-2-one and the iron metal (Fe–N1 distance of 2.37 Å). The coordination of another aziridine molecule to the metal, in trans with respect to the oxazolidin-2-one ring, enables the formation of the octahedral complex 31OMe (Fig. 11) with a free energy cost of +8.5 kcal mol−1. The coordination of aziridine results in a significant weakening of the interaction between oxazolidin-2-one and the iron complex (with an Fe–N1 distance as large as 3.19 Å) and the dissociation of oxazolidin-2-one 14 occurs with a small free energy cost of +2.3 kcal mol−1. Then, the formation of intermediate 27OMe, the actual catalyst for the process, is ready for starting another catalytic cycle.
 | ||
| Fig. 11 Optimized structures of intermediates 30OMe and 31OMe. The hydrogen atoms, except the carboxylic ones, were hidden for clarity. | ||
The formation of 14 from CO2 and N-butyl-2-phenyl aziridine, catalyzed by 27OMe, is estimated to be exergonic by −4.7 kcal mol−1. A complete free energy profile of the catalytic cycle is illustrated in Fig. 12.
 | ||
| Fig. 12 Free energy (kcal mol−1) reaction pathway for the synthesis of 14 catalyzed by the 27OMe complex. | ||
The CO2 activation by 3@CC was also computationally investigated using a model catalyst, 3(NMe4)2, which shows two peripheral ammonium cations to simulate the interaction between the iron complex and Colour Catcher®, which contains positively charged NR3+ groups used for the anchorage of hematin. The computed reaction pathway, detailed in the ESI,† revealed similar energetic features with only minor differences, none exceeding 6 kcal mol−1.
Experimental
General methods, characterization and NMR spectra of isolated compounds are reported in the ESI.†Synthesis of Fe(OMe)protoporphyrin IX (2)
1 (50.0 mg, 7.67 × 10−5 mol) was suspended in MeOH (10.0 mL) and the mixture was refluxed for two hours. At the end of the reaction, the solvent was evaporated to dryness to yield a brown solid (44.0 mg, 90% yield). LR-MS (ESI†) m/z calcd. for (C35H35FeN4O5): 647.20 found 646.27 [M–H] and 632.28 [M–CH3]. IR (Nujol) υ cm−1: 846.6, 1086.7, 1278.6, 1653.2, 2359.9. UV-vis λmax (DCE)/nm (logε) 383 (5.72), 576 (4.79), 609 (4.79)
Synthesis of sodium salt of hematin (3)
1 (20.0 mg, 3.07 × 10−5 mol) was dissolved in NaOH 0.1 M (30.0 mL) and the mixture was stirred for two hours at room temperature. At the end of the reaction, the solvent was removed under reduced pressure to yield a brown solid (18.0 mg, 94% yield). LR-MS (ESI) m/z calcd. for (C34H31FeN4Na2O5): 677.46 found 654.62 [M–Na] and 632.43 [M–2Na]. IR (Nujol) υ cm−1: 865.4, 1040.4, 1260.7, 1646.4, 2359.5, 3419.7. UV-vis λmax (NaOH 0.1 M)/nm (logε) 381 (5.66), 510 (4.72), 539 (4.72), 640 (4.43).
Preparation of 3@CC
1 (65.0 mg, 1.00 × 10−4 mol) was dissolved in 100.0 mL of NaOH 0.1 M and the mixture was stirred at room temperature until the complete dissolution of the starting hemin. The obtained solution was then placed in a coded test tube and 25 cm2 of Colour Catcher® were immersed in the solution. The sheet was left in the solution for 8 hours and to prevent porphyrin degradation, the tube was wrapped with aluminum kitchen foil. After this time, the sheet was extracted and dried on a heated plate at 85 °C. Then, it was washed twice with NaOH 0.1 M and three times with distilled water and dried at each step. No porphyrin leaching was observed. After the last wash, the sheet was dried on the heated plate and placed into a coded test tube and dried under vacuum. The obtained 3@CC was weighed and store wrapped in aluminum kitchen foil. The determination of the amount of porphyrin anchored onto the Colour Catcher® was performed by UV-vis spectroscopy by measuring the concentration of the 3-containing solution before and after the immersion of the sheet. Collected data allowed estimating an average value of anchored 3 equal to 0.054 mg cm−2.General procedure of the synthesis of cyclic carbonates
Method A: in a 2.0 mL glass liner equipped with a screw cap and glass wool, 1 (12.0 mg, 1.84 × 10−5 mol) and TBACl (10.0 mg, 3.68 × 10−5 mol) were dissolved in the desired epoxide (2.63 × 10−3 mol). The vessel was transferred into a stainless-steel autoclave and 0.6 MPa of CO2 were charged at room temperature. The autoclave was placed in a preheated oil bath at 100 °C, stirred for 4 hours and then quenched in an ice bath and slowly vented. The crude was diluted with DCM and filtered on silica to remove the catalyst. The solvent was evaporated to dryness and the residue was analyzed by 1H NMR spectroscopy by using mesitylene as the internal standard. Method B: method A was followed by using 2 (12.0 mg, 1.84 × 10−5 mol) as the catalyst. Method C: method A was followed by using 3 (12.0 mg, 1.84 × 10−5 mol) as the catalyst.General procedure of the synthesis of N-alkyl oxazolidin-2-ones
Method A: in a 2.0 mL glass liner equipped with a screw cap and glass wool, 1 (6.5 mg, 1.0 × 10−5 mol) and the desired aziridine (1.0 × 10−3 mol) were dissolved in 0.6 mL of DCE. The vessel was transferred into a stainless-steel autoclave and 1.2 MPa of CO2 were charged at room temperature. The autoclave was placed in a preheated oil bath at 125 °C, stirred for 12 hours and then quenched in an ice bath and slowly vented. The crude was diluted with DCM and filtered on silica to remove the catalyst. The solvent was evaporated to dryness and the crude was analyzed by 1H NMR spectroscopy by using mesitylene as the internal standard. Method B: method A was followed by using 2 (6.5 mg, 1.0 × 10−5 mol) as the catalyst. Method C: method A was followed by using 3 (6.9 mg, 1.0 × 10−5 mol) as the catalyst. Method D: method A was followed by using 11.5 cm2 of 3@CC (0.1% mol of anchored 3, 1.0 × 10−6 mol) as the catalyst. At the end of the reaction 3@CC was filtered off and washed three times with DCE (3 × 1.0 mL). The organic phases were collected and dried under vacuum. The obtained crude was analyzed by 1H NMR with mesitylene as the internal standard.Computational details
All the minima and transition states along the potential energy surface were isolated at the B97D-DFT level of theory63 within the Gaussian 16 package.64 The Stuttgart–Dresden pseudo-potential was used for the iron center65 and the TZVP basis set was adopted for all the other atomic species. All the optimized structures were validated as minima or transition states by the vibrational frequency calculations. All the calculations employed the CPCM model66,67 for DCE, the solvent that was experimentally used.Conclusions
In conclusion, we have presented the catalytic use of biological iron(III) protoporphyrin IX derivatives for promoting CO2 cycloaddition to three-membered rings under both homogeneous and heterogeneous conditions. This method aligns with the societal demand to transform waste, such as CO2, into valuable chemicals by maintaining a low ecological footprint. In addition, the 100% atom economical process occurs without forming by-products with a resulting reduction of costs related to product separation and purification.The synthesis of oxazolidin-2-ones is productive in the absence of any co-catalyst. This not only enhances the sustainability of the process, by reducing the necessity for additional chemicals during the reaction, but also holds significant value when the avoided reagent is a chlorinated compound, such as TBACl.
The cost-effectiveness of the protocol is further enhanced by the utilization of the heterogeneous catalyst 3@CC. This catalyst not only shows an inherent low toxicity due to the bio-characteristics of its constituents (hematin and cellulose-based material), but also exhibits exceptional thermal and chemical stability. These attributes enable the recovery and reuse of the catalyst multiple times, greatly expanding the applicability of the synthetic protocol.
In conclusion, the high availability, eco-compatibility, and cost-effectiveness of the proposed synthetic procedure pave the way for future scale-up and potential utilization on a larger scale.
Author contributions
The manuscript was written through the contributions of all authors and all authors have given approval to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Maria Vittoria Dozzi for her valuable help in performing UV-vis spectroscopy in solid state. CD, MC and EG thank Università degli Studi di Milano for the PSR 2022 grant. CD and EG thank MUR for the project PRIN 2022 “CADIVAPE” (2022FWAF2M). GM acknowledges the CINECA award under the ISCRA initiative for the availability of high-performance computing resources and support.Notes and references
- A. D. N. Kamkeng, M. Wang, J. Hu, W. Du and F. Qian, Chem. Eng. J., 2021, 409, 128138 CrossRef CAS.
- O. Akeeb, L. Wang, W. Xie, R. Davis, M. Alkasrawi and S. Toan, J. Environ. Manage., 2022, 313, 115026 CrossRef CAS PubMed.
- C. B. Agaton, Sci. Total Environ., 2021, 795, 148683 CrossRef CAS PubMed.
- A. Alok, R. Shrestha, S. Ban, S. Devkota, B. Uprety and R. Joshi, J. Environ. Chem. Eng., 2022, 10, 106922 CrossRef CAS.
- J. Zhang, C. D. Sewell, H. Huang and Z. Lin, Adv. Energy Mater., 2021, 11, 2102767 CrossRef CAS.
- N. Yusuf, F. Almomani and H. Qiblawey, Fuel, 2023, 345, 128178 CrossRef CAS.
- E. J. C. Lopes, A. P. C. Ribeiro and L. M. D. R. S. Martins, Catalysts, 2020, 10, 479 CrossRef CAS.
- T. Yan, H. Liu, Z. X. Zeng and W. G. Pan, J. CO2 Util., 2023, 68, 102355 CrossRef CAS.
- S.-L. Hou, J. Dong, X.-Y. Zhao, X.-S. Li, F.-Y. Ren, J. Zhao and B. Zhao, Angew. Chem., 2023, 62, e202305213 CrossRef CAS PubMed.
- F. Sun, E. V. Van der Eycken and H. Feng, Adv. Synth. Catal., 2021, 363, 5168–5195 CrossRef CAS.
- S. Arshadi, A. Banaei, S. Ebrahimiasl, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 19465–19482 RSC.
- F. Han, H. Li, H. Zhuang, Q. Hou, Q. Yang, B. Zhang and C. Miao, J. CO2 Util., 2021, 53, 101742 CrossRef CAS.
- A. W. Kleij, in Adv. Catal., ed. M. Diéguez and A. W. Kleij, Academic Press, 2022, vol. 70, pp. 1–28 Search PubMed.
- D. Aurbach, E. Markevich and G. Salitra, J. Am. Chem. Soc., 2021, 143, 21161–21176 CrossRef CAS PubMed.
- A. Centeno-Pedrazo, J. Perez-Arce, Z. Freixa, P. Ortiz and E. J. Garcia-Suarez, Ind. Eng. Chem. Res., 2023, 62, 3428–3443 CrossRef CAS.
- C. Foti, A. Piperno, A. Scala and O. Giuffrè, Molecules, 2021, 26, 4280 CrossRef CAS PubMed.
- A. Nazari, M. M. Heravi and V. Zadsirjan, J. Organomet. Chem., 2021, 932, 121629 CrossRef CAS.
- Q. Zhao, L. Xin, Y. Liu, C. Liang, J. Li, Y. Jian, H. Li, Z. Shi, H. Liu and W. Cao, J. Med. Chem., 2021, 64, 10557–10580 CrossRef CAS PubMed.
- S. Yuan, D.-D. Shen, Y.-R. Bai, M. Zhang, T. Zhou, C. Sun, L. Zhou, S.-Q. Wang and H.-M. Liu, Eur. J. Med. Chem., 2023, 250, 115239 CrossRef CAS PubMed.
- C. Damiano, P. Sonzini, M. Cavalleri, G. Manca and E. Gallo, Inorg. Chim. Acta, 2022, 540, 121065 CrossRef CAS.
- P. Tyagi, D. Singh, N. Malik, S. Kumar and R. Singh Malik, Mater. Today, 2023, 65, 133–165 CrossRef CAS.
- T.-d. Hu and Y.-H. Ding, Organometallics, 2020, 39, 505–515 CrossRef CAS.
- N. Panza, M. Alberti, S. Galiè, C. Damiano, F. Cargnoni, M. Italo Trioni and A. Caselli, Eur. J. Org. Chem., 2022, 2022, e202200908 CrossRef CAS.
- Q. Wang, C.-H. Guo, J. Jia and H.-S. Wu, J. Mol. Model., 2015, 21, 179 CrossRef PubMed.
- S. Kumar, M. Y. Wani, C. T. Arranja, J. d. A. e. Silva, B. Avula and A. J. F. N. Sobral, J. Mater. Chem. A, 2015, 3, 19615–19637 RSC.
- R. Luo, M. Chen, F. Zhou, J. Zhan, Q. Deng, Y. Yu, Y. Zhang, W. Xu and Y. Fang, J. Mater. Chem. A, 2021, 9, 25731–25749 RSC.
- D. Intrieri, C. Damiano, P. Sonzini and E. Gallo, J. Porphyrins Phthalocyanines, 2019, 23, 305–328 CrossRef CAS.
- J. L. S. Milani, A. M. Meireles, B. N. Cabral, W. de Almeida Bezerra, F. T. Martins, D. C. da Silva Martins and R. P. das Chagas, J. CO2 Util., 2019, 30, 100–106 CrossRef CAS.
- J. Yi, S. Sun, Z. Li, X. Gao, X. Sun, N. Wang and J. Li, Appl. Organomet. Chem., 2020, 34, e5382 CrossRef CAS.
- Y. Lu, Z. Chang, S. Zhang, S. Wang, Q. Chen, L. Feng and Z. Sui, J. Mater. Sci., 2020, 55, 11856–11869 CrossRef CAS.
- S. Jayakumar, H. Li, L. Tao, C. Li, L. Liu, J. Chen and Q. Yang, ACS Sustainable Chem. Eng., 2018, 6, 9237–9245 CrossRef CAS.
- L. Ma, Z. Su, N. Wang and J. Li, Eur. J. Inorg. Chem., 2023, 26, e202200744 CrossRef CAS.
- Z. Li, Z. Su, W. Xu, Q. Shi, J. Yi, C. Bai, N. Wang and J. Li, ChemCatChem, 2020, 12, 4839–4844 CrossRef CAS.
- Q.-J. Wu, M.-J. Mao, J.-X. Chen, Y.-B. Huang and R. Cao, Catal. Sci. Technol., 2020, 10, 8026–8033 RSC.
- D. Wang, Z. Gao, S. Duan, J. Wei, L. Ma, N. Wang and J. Li, Appl. Organomet. Chem., 2023, 37, e7121 CrossRef CAS.
- C. Maeda, J. Shimonishi, R. Miyazaki, J.-y. Hasegawa and T. Ema, Chem. – Eur. J., 2016, 22, 6556–6563 CrossRef CAS PubMed.
- X. Jiang, F. Gou, X. Fu and H. Jing, J. CO2 Util., 2016, 16, 264–271 CrossRef CAS.
- A. M. Alsharabasy, A. Pandit and P. Farràs, Adv. Mater., 2021, 33, 2003883 CrossRef CAS PubMed.
- H. C. Quadros, M. C. B. Silva and D. R. M. Moreira, Pharmaceuticals, 2022, 15, 60 CrossRef CAS PubMed.
- X. Wu, M. Wang, Y. Xie, C. Chen, K. Li, M. Yuan, X. Zhao and Z. Hou, Appl. Catal., A, 2016, 519, 146–154 CrossRef CAS.
- D. M. Rodrigues, L. G. Hunter, F. L. Bernard, M. F. Rojas, F. Dalla Vecchia and S. Einloft, Catal. Lett., 2019, 149, 733–743 CrossRef CAS.
- F. Caroleo, G. Magna, C. Damiano, M. Cavalleri, E. Gallo, C. Di Natale and R. Paolesse, Sens. Actuators, B, 2022, 364, 131900 CrossRef CAS.
- C. Damiano, M. Cavalleri, C. di Natale', R. Paolesse and E. Gallo, Chem. Eur., 2023, 1, e202300020 Search PubMed.
- G. Alberti, C. Zanoni, L. R. Magnaghi and R. Biesuz, J. Anal. Sci. Technol., 2020, 11, 30 CrossRef CAS.
- R. Biesuz, V. M. Nurchi, L. R. Magnaghi and G. Alberti, Microchem. J., 2019, 149, 104036 CrossRef CAS.
- G. Alberti, V. M. Nurchi, L. R. Magnaghi and R. Biesuz, Anal. Methods, 2019, 11, 4464–4470 RSC.
- M. Cavalleri, C. Damiano, G. Manca and E. Gallo, Chem. – Eur. J., 2023, 29, e202202729 CrossRef CAS PubMed.
- C. Damiano, P. Sonzini, G. Manca and E. Gallo, Eur. J. Org. Chem., 2021, 2021, 2807–2814 CrossRef CAS.
- P. Sonzini, C. Damiano, D. Intrieri, G. Manca and E. Gallo, Adv. Synth. Catal., 2020, 362, 2961–2969 CrossRef CAS.
- D. Carminati, E. Gallo, C. Damiano, A. Caselli and D. Intrieri, Eur. J. Inorg. Chem., 2018, 2018, 5258–5262 CrossRef CAS.
- C. Damiano, P. Sonzini, D. Intrieri and E. Gallo, J. Porphyrins Phthalocyanines, 2020, 24, 809–816 CrossRef CAS.
- C. Damiano, N. Panza, J. Nagy, E. Gallo and G. Manca, New J. Chem., 2023, 47, 4306–4312 RSC.
- P. Sonzini, N. Berthet, C. Damiano, V. Dufaud and E. Gallo, J. Catal., 2022, 414, 143–154 CrossRef CAS.
- H. Ding, B. D. Sumer, C. W. Kessinger, Y. Dong, G. Huang, D. A. Boothman and J. Gao, J. Controlled Release, 2011, 151, 271–277 CrossRef CAS PubMed.
- M. Luoni and G. Li Bassi, Non-woven Colour-Catcher fabric and method for its preparation, US Pat., US2009/0137170A1, 2009 Search PubMed; I. Tabujew and K. Peneva, Functionalization of cationic polymers for drug delivery applications, in RSC Polymer Chemistry in Regenerative Medicine, 2015, pp. 1–29 Search PubMed.
- C.-H. Zhang, Z.-L. Wu, R.-X. Bai, T.-D. Hu and B. Zhao, ACS Appl. Mater. Interfaces, 2023, 15, 1879–1890 CrossRef CAS PubMed.
- X.-R. Tian, Y. Shi, S.-L. Hou, Y. Ma and B. Zhao, Inorg. Chem., 2021, 60, 15383–15389 CrossRef CAS PubMed.
- X.-R. Tian, X.-L. Jiang, S.-L. Hou, Z.-H. Jiao, J. Han and B. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202200123 CrossRef CAS PubMed.
- M. Qiao, X.-Y. Xin, W.-M. Wang, Z.-L. Wu and J.-Z. Cui, Dalton Trans., 2023, 52, 10725–10736 RSC.
- X.-M. Kang, L.-H. Yao, Z.-H. Jiao and B. Zhao, Chem. – Asian J., 2019, 14, 3668–3674 CrossRef CAS PubMed.
- C.-S. Cao, Y. Shi, H. Xu and B. Zhao, Chem. Commun., 2021, 57, 7537–7540 RSC.
- X. Wang, W.-Y. Gao, Z. Niu, L. Wojtas, J. A. Perman, Y. S. Li, B. Aguila and S. Ma, Chem. Commun., 2018, 54, 1170–1173 RSC.
- G. Stefan, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- , M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Catalytic procedures, analytical data, NMR spectra and computational data. See DOI: https://doi.org/10.1039/d4cy00658e |
| This journal is © The Royal Society of Chemistry 2024 |