Penta- versus hexa-coordinated iridium catalysts control the reactivity of the direct reductive amination between aliphatic amines and aliphatic ketones: a DFT-guided mechanism†
Received
22nd April 2024
, Accepted 13th June 2024
First published on 13th June 2024
Abstract
Understanding reaction mechanisms of metal-catalyzed processes is of paramount importance for the design of superior catalysts that circumvent unproductive pathways, while accelerating catalyst discovery. In this respect, gaining mechanistic understanding for reactions carried out at high pressures of gas reagents remains a major limitation because special setups are typically required, which is the case for metal-catalyzed direct reductive aminations (DRA) under high H2 pressure. To overcome this issue, extensive computational calculations have been herein conducted for the iridium-catalyzed DRA between aliphatic ketones and aliphatic secondary amines. This highly atom-economic reaction delivers only water as side-product and it is relevant for the identification of active pharmaceutical ingredients. In this contribution, we highlight that the excellent reactivity encountered with very different P,P-chelating ligands results from the fact that two different mechanistic pathways operate for each system. In addition, we found that the key hydride transfer step is more accessible with a penta-coordinated iridium complex rather than with the expected hexa-coordinated iridium species using a Josiphos-type ligand when compared to the large bite-angle Xantphos. For comparison purposes, we also evaluated a related Josiphos-type ligand and a small bite-angle diphosphane.
Introduction
The fine-understanding of the exact mode of action of metal catalysts is of high relevance for the development of more powerful systems.1–3 In this regard, in-depth kinetic studies utilizing in situ or operando spectroscopy techniques are very useful to identify reaction intermediates while postulating reaction pathways.4–6 However, homogeneous catalytic reactions involving the use of gas reagents imply very demanding set-ups to monitor these processes, thus significantly limiting the gain of mechanistic insights.7–15 On the other hand, computational calculations by means of state-of-the-art density functional theory (DFT) protocols enable the identification of reaction pathways for explaining the selectivity and/or the activity reached with a given metal catalyst.16–23 This is particularly advantageous for homogeneous metal-catalysed reactions involving gaseous reagents such as H2, CO, CO2, and others, which are routinely employed in hydrogenations, hydroformylations, (alkoxy)carbonylations, carboxylations and related ones, including also the asymmetric variants.24–39
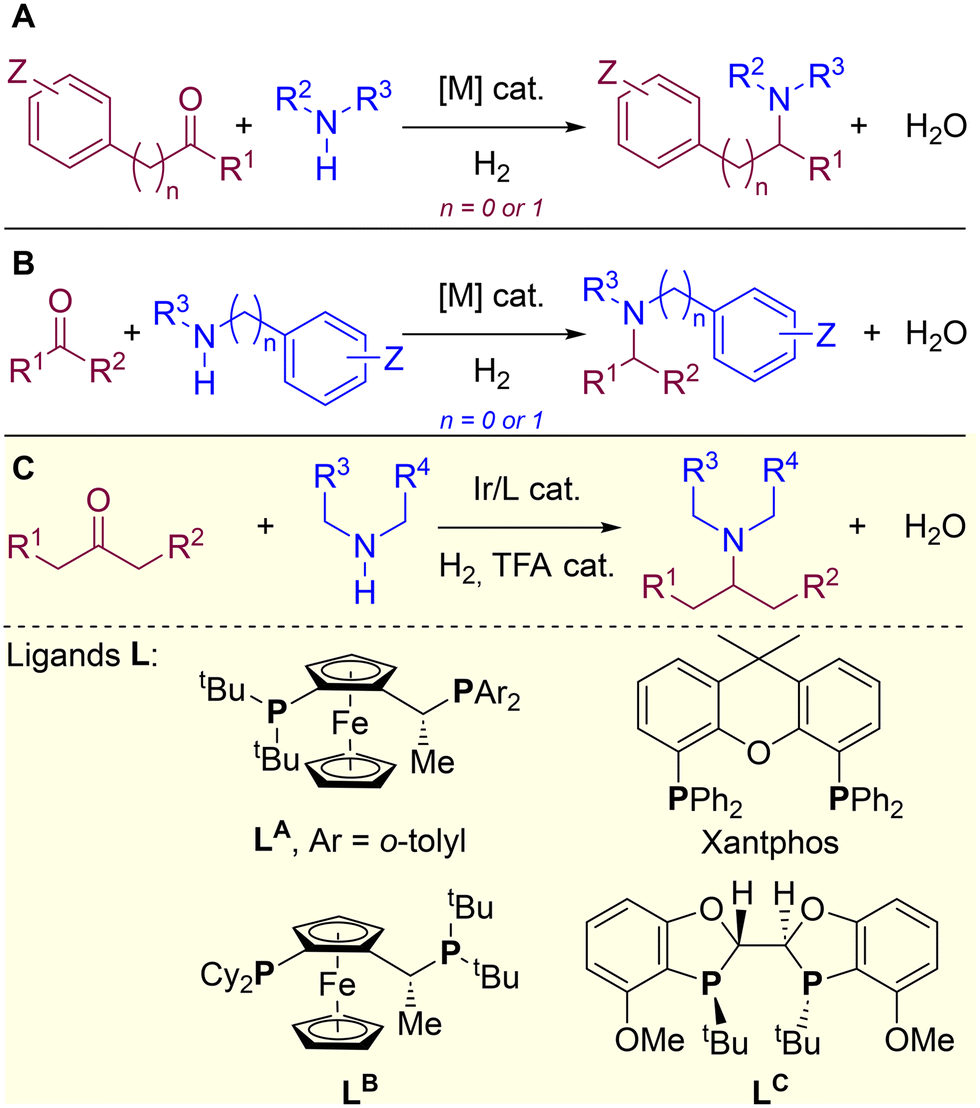
In this context, H2-mediated direct reductive aminations (DRA) catalysed by metal complexes derived mainly from Rh, Ru, Ir, Pd, Fe and Co, are very attractive as they enable the formal coupling of a ketone (or aldehyde) with a primary or secondary amine leading to the corresponding secondary or tertiary amine (Fig. 1A and B).40–44 This type of reaction meets several green chemistry principles since it is highly atom economical, benign water is formed as side-product and low catalyst loading is typically used, thus resulting in a significantly low environmental impact45–51 when compared to the use of over-stoichiometric amounts of hazardous reducing agents such as lithium, aluminium or boron hydrides, or organosilanes.52–57 Of particular importance is the access to tertiary amines via H2-mediated DRA because they are ubiquitous in materials and pharmaceutical sciences.58–64 However, the substrate scope of metal-catalysed DRA is limited to pre-activated amines and carbonyl-containing partners directly connected to an aromatic or benzylic fragment (Fig. 1A and B).65–82 In order to span H2-mediated DRA towards purely aliphatic systems, we have pioneered the very first report on the identification of an iridium/diphosphane system that overcomes this limitation, thereby enabling to successfully utilize completely unbiased aliphatic ketones and aliphatic amines (Fig. 1C).83 During this initial investigation, an important task was devoted to the identification of the most suitable ligand leading, unexpectedly, to ligands with a very different backbone structures, namely Josiphos-type ligand LA and Xantphos (Fig. 1C).83 Due to the difficulty to identify and characterize active iridium species under catalytically-relevant conditions (50 bar of H2), we turned our attention to study the whole catalytic cycle of the reaction by means of computational calculations. Herein, we present an in-depth DFT study that shows that the H2-mediated DRA between aliphatic ketones and aliphatic amines with iridium catalysis undergoes a completely different pathway depending on the nature of the ligand, being possible to reach high reactivity in both scenarios. We also found that two other ligands, commonly used for hydrogenation processes (Josiphos-type LB and BIBOP-type LC), are not appropriate for this iridium-catalysed DRA, as they promote competitive side-reactions. In addition, the role of the trifluoroacetate anion was analysed and it was found that, depending on the coordinating ability, it can lead to the stabilization of iridium species of different coordination geometry, which dramatically influence one of the key steps of the catalytic cycle, namely the hydride transfer step from the metal to the substrate. Other metal-based catalytic systems have been recently reported for the formation of purely aliphatic (chiral) amines.84–87
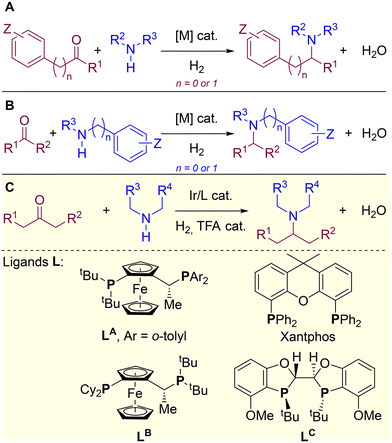 |
| | Fig. 1 Transition metal-catalysed DRA reactions: (A and B) state-of-the-art with biased substrates and (C) prior work with unbiased substrates highlighting the structure of relevant P,P-chelating diphosphane ligands, whose mechanism is herein studied by DFT calculations. R = H, alkyl or aryl. | |
Results and discussion
Initial considerations
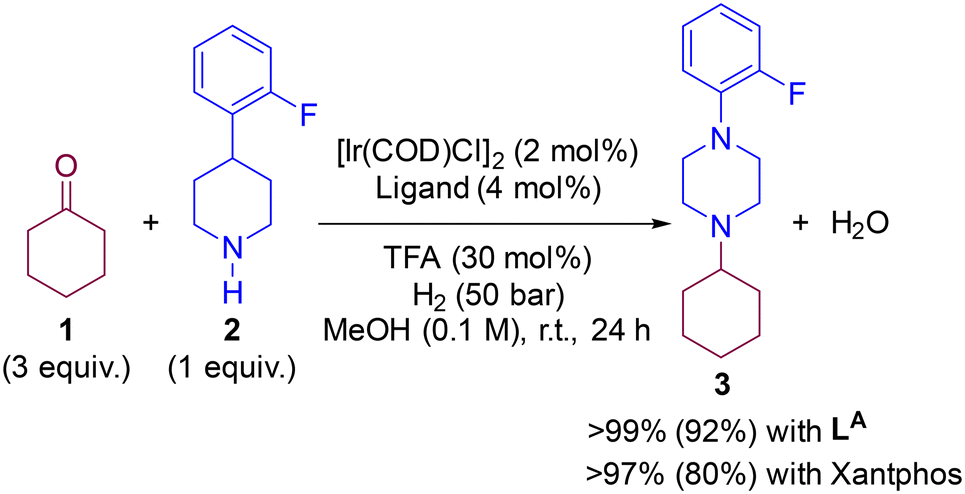
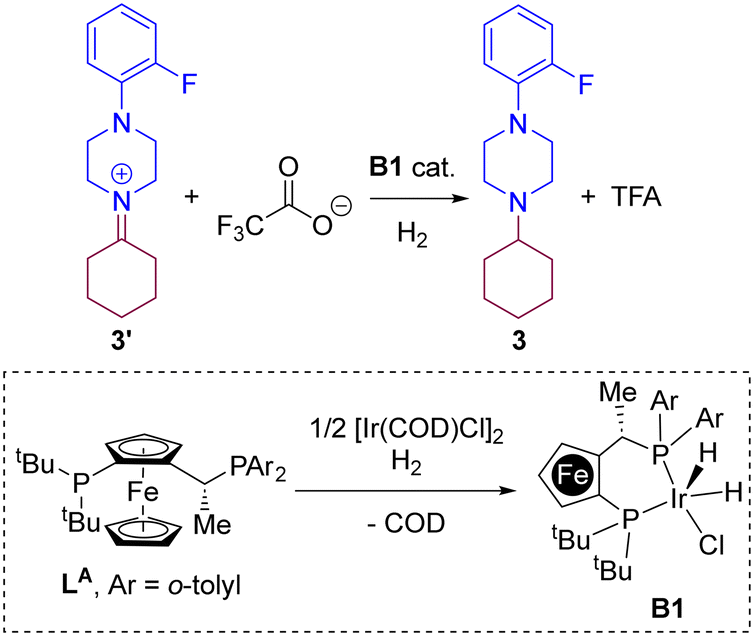
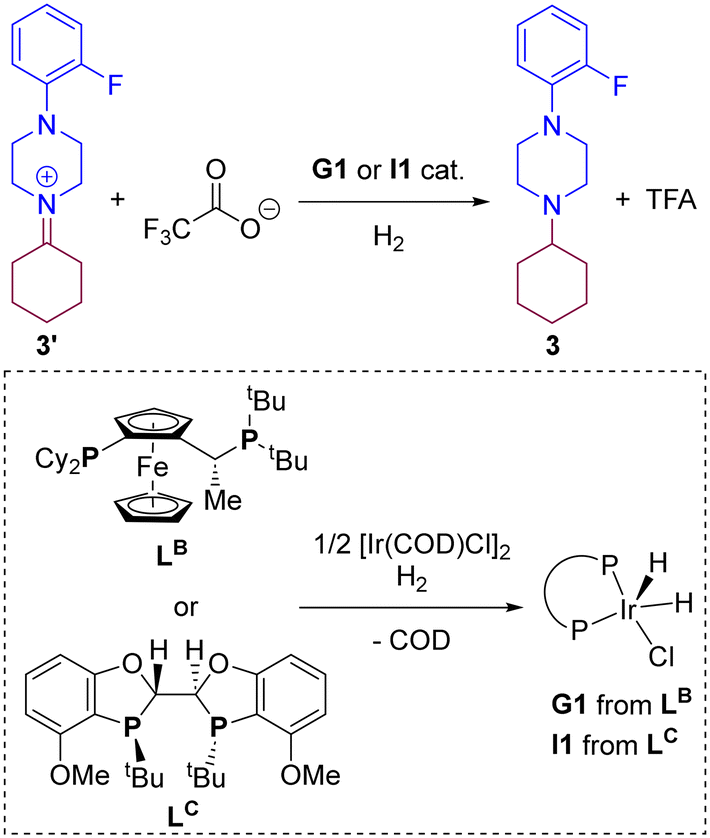
In a previous contribution, we disclosed the iridium-catalysed DRA between aliphatic ketones and aliphatic amines leading selectively to tertiary aliphatic amines under 50 bar of H2 (Scheme 1).83 After extensive screening, the optimal reaction conditions identified comprised the use of 2 mol% of [Ir(COD)Cl]2 (COD = 1,5-cyclooctadiene) dimer as the pre-catalyst and 4 mol% of a P,P-chelating ligand in the presence of 30 mol% of trifluoroacetic acid (TFA) in methanol solution at room temperature. Under these precise reaction conditions, the catalysis with the Josiphos-type ligand LA and Xantphos led, surprisingly, to equally efficient results with almost full conversion and exclusive formation of the product 3 starting from ketone 1 and the secondary amine 2. The difference in isolated yields was ascribed to the difficult, non-trivial purification of 3 during the column chromatography process. As such, for the DFT study of the reaction mechanism, we focused in the reaction involving ketone 1 and amine 2 partners, and compared the reaction coordinate trajectory by varying the nature of the ligand. Because little enantioselective induction (<20% ee) was found using pro-chiral ketones such as 2-butanone, in this study we focused on a relatively simple substrate model involving cyclohexanone (1) that leads to an achiral product 3.
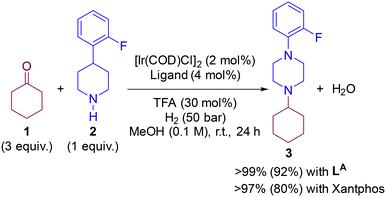 |
| | Scheme 1 Previously-developed iridium-catalysed DRA enabling formation of purely aliphatic tertiary amine 3. Isolated yields displayed in brackets.83 | |
In accordance with previous theoretical studies, we adopted the DFT ωB97X-D method88 in this study using the Gaussian 09 program.89 The geometric structures in methanol solution were optimized using BS-I basis sets, in which 6-311G(d,p) basis sets were used for non-metallic atoms, and SDD basis sets with effective core potential were used for the iridium and iron atoms. The BS-II basis sets were further used for single point energy optimization, in which 6-311++G(2d,p) basis sets were used for non-metallic atoms, and SDD basis sets with effective core potential were used for the iridium and iron atoms. Note that Ahlrichs' basis sets have been considered and led to very similar results (see ESI†). The contribution of thermal corrections and entropy to Gibbs free energies were obtained by using ωB97X-D/BS-I methods. The refined energies were then corrected to Gibbs energies at 298.15 K and 1 atm by using the ωB97X-D/BS-I harmonic frequencies. Calculations carried out at 50 atm instead of 1 atm led to very similar results (see ESI†).85,90–92 The solvent effect was evaluated using SMD (density-based solution model) solvation model.93,94 We noted that the formation of the hydrogen-bond complexes between the methanol solvent molecule and TFA derivatives are endothermic reactions, suggesting TFA derivatives cannot form stable associated complexes (see details in the ESI†). Harmonic frequency analysis is performed to verify that the optimized geometry is a minimum (no virtual frequency) or a transition state (TS, with a unique virtual frequency). All transition states were verified by employing the intrinsic reaction coordinate (IRC) procedure.95 The cartesian coordinates of all optimized structures are presented in the ESI† (Tables S1–S3).
Formation of iminium and/or enamines
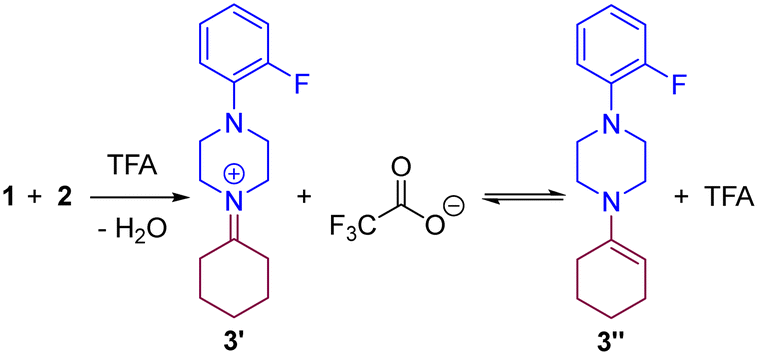
The very first step of the reaction is the condensation of ketone 1 and amine 2 assisted by TFA that may lead to the iminium 3′ and/or enamine species 3′′ according to our previous hypothesis (Scheme 2).83 To qualitatively evaluate the feasibility of the formation of the iminium 3′ and the enamine 3′′, we computed the energy landscape for the reaction of 1 with 2 in the presence of TFA (Fig. 2).
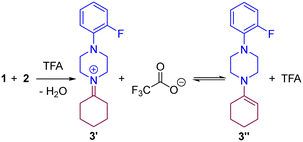 |
| | Scheme 2 Condensation of ketone 1 and amine 2 in the presence of TFA. | |
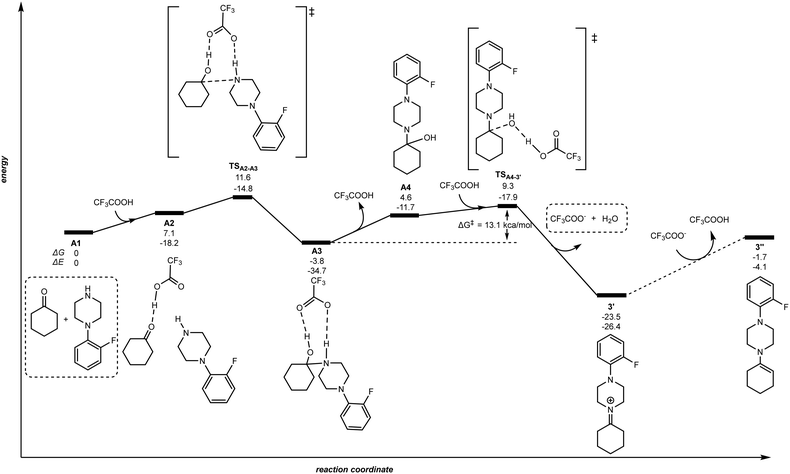 |
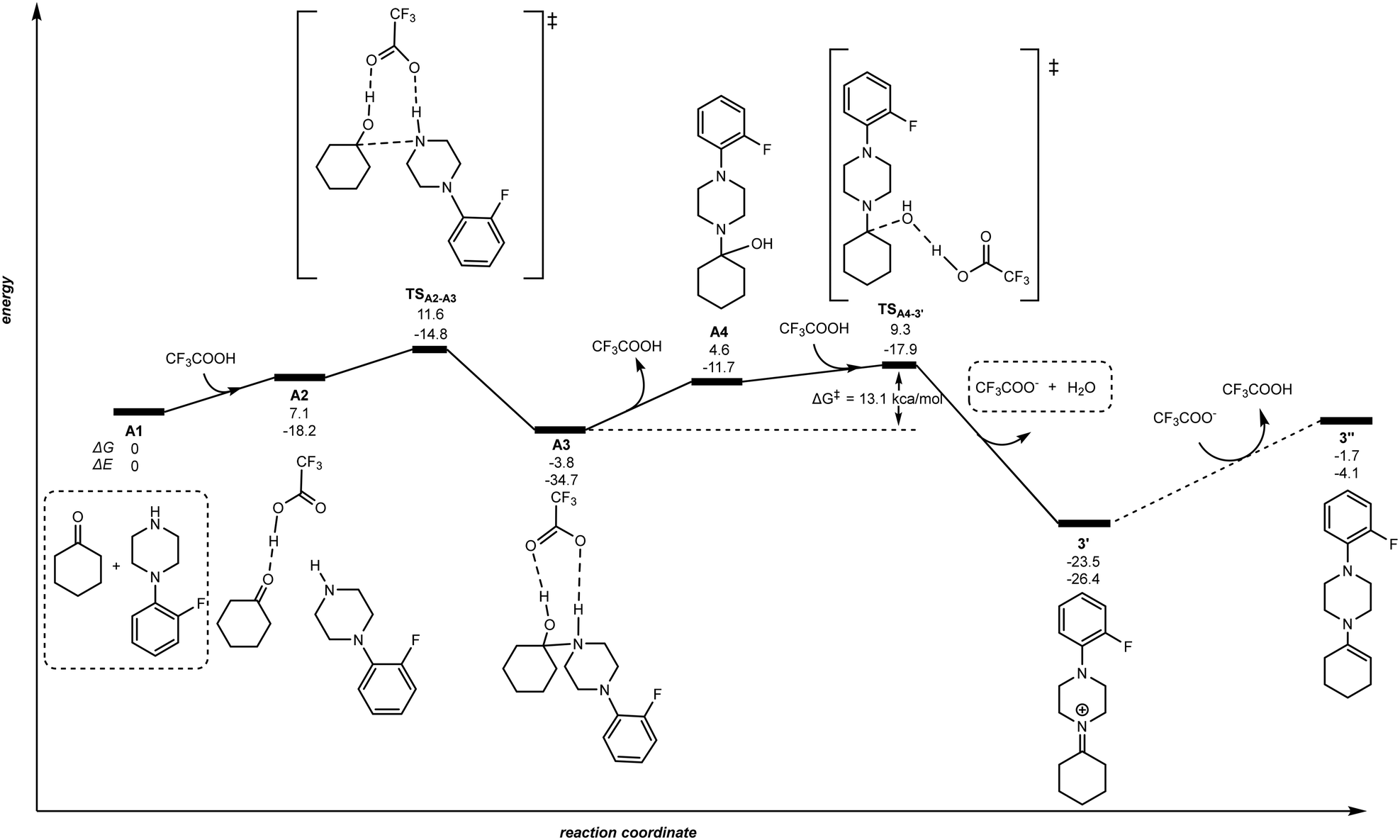
| | Fig. 2 Reaction pathway for the TFA-mediated condensation of ketone 1 and amine 2. The relative Gibbs free energy (ΔG) and potential energy (ΔE) are reported in kcal mol−1. | |
As shown in Fig. 2, the stoichiometric combination of ketone 1 and amine 2 (A1) binds a TFA molecule via hydrogen bonding between the acidic hydrogen of TFA and the carbonyl group of 1 forming the intermediate A2 with an energy increase of +7.1 kcal mol−1. The hydrogen atom from TFA is transferred to the carbonyl oxygen atom in A2via the transition state TSA2–A3, while the carbonyl carbon atom changes its hybridization from sp2 to sp3 in the bounding to the nearby amine nitrogen atom leading to species A3. The energy barrier of this intermolecular C–N coupling step is +11.6 kcal mol−1 and it is important to note an additional hydrogen bonding between the amine proton and an oxygen atom from TFA. Then, the TFA molecule is released to produce the aminoalcohol intermediate A4. In the following, A4 rebounds to TFA proceeding to the dehydration reaction through the very accessible transition state TSA4–3′. The proton in TFA is transferred to the hydroxyl oxygen atom of the aminoalcohol A4 together with the release of the trifluoroacetate anion and water producing the iminium 3′. In brief, the energy span of the transition state TSA4–3′ starting from the intermediate A3 is +13.1 kcal mol−1, and the formation of the iminium species 3′ is largely exergonic by 23.5 kcal mol−1. Note that a plausible reaction pathway from intermediate A3 to iminium 3′ considering release of trifluoroacetate anion instead of TFA before reaching the transition state TSA4–3′ was highly unfavourable with an energy span of 38.8 kcal mol−1, and 31.9 kcal mol−1 for the case in which TFA is involved in a two-fold hydrogen bonding (see Fig. S1 and S2 in the ESI†). Importantly, considering the reaction of the iminium 3′ with trifluoroacetate anion towards the formation of enamine 3′′ was strongly disfavoured with a ΔG = +21.8 kcal mol−1. As such, for purely aliphatic ketones and amines, the iminium intermediate 3′ is thermodynamically more favoured than the corresponding enamine 3′′ intermediate. Nevertheless, it is worthy to note that under the precise reaction conditions of the catalysis the presence of free TFA is unlikely because it may instantaneously react with the amine 2 and/or the product 3. As such, we cannot rule out a process from 3′ to 3′′via an acid/base reaction with the amine 2 or the product 3 acting as a base. That being said, this does not change the fact that the iminium intermediate 3′ is more favoured to form than the enamine 3′′.
Iridium-catalysed hydrogenation of the iminium intermediate 3′ and catalyst regeneration with ligand LA
Having demonstrated the high stability of iminium 3′ over the enamine 3′′, we embarked in the DFT analysis of the hydrogenation of 3′ towards the tertiary amine 3 by an iridium complex resulting from the chelation of the Josiphos-type ligand LA (Scheme 3). It is important to remind that, under the catalytic conditions, TFA is likely involved in an acid/base reaction with the product 3 at the end, leading to the protonated ammonium and triflate anion. This explains the fact that a basic work-up is required in the experimental procedure to fully recover the amine 3. From a mechanistic point of view and based on previous reports that indicate the readily formation of penta-coordinated iridium(III) species upon combination of a P,P-chelating ligand with [Ir(COD)Cl]2 under H2 pressure,30,96–102 we considered the iridium species B1 (Scheme 3) as the most reasonable catalytically active ones for the hydrogenation of iminium 3′ in the presence of trifluoroacetate anion. In the following, three different dihydride-based mechanistic pathways have been evaluated: pathways “A” (Fig. 3), “B” (Fig. 4) and “C” (Fig. 5).
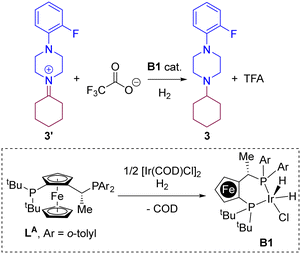 |
| | Scheme 3 Iridium-catalysed hydrogenation of iminium 3′ with the iridium B1 catalyst. | |
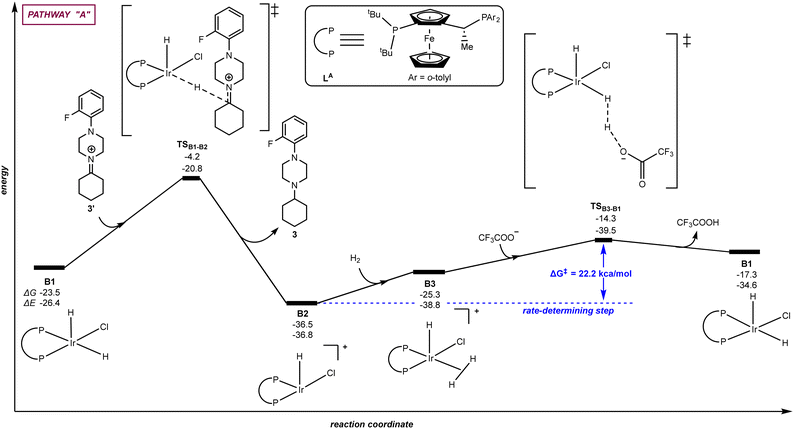 |
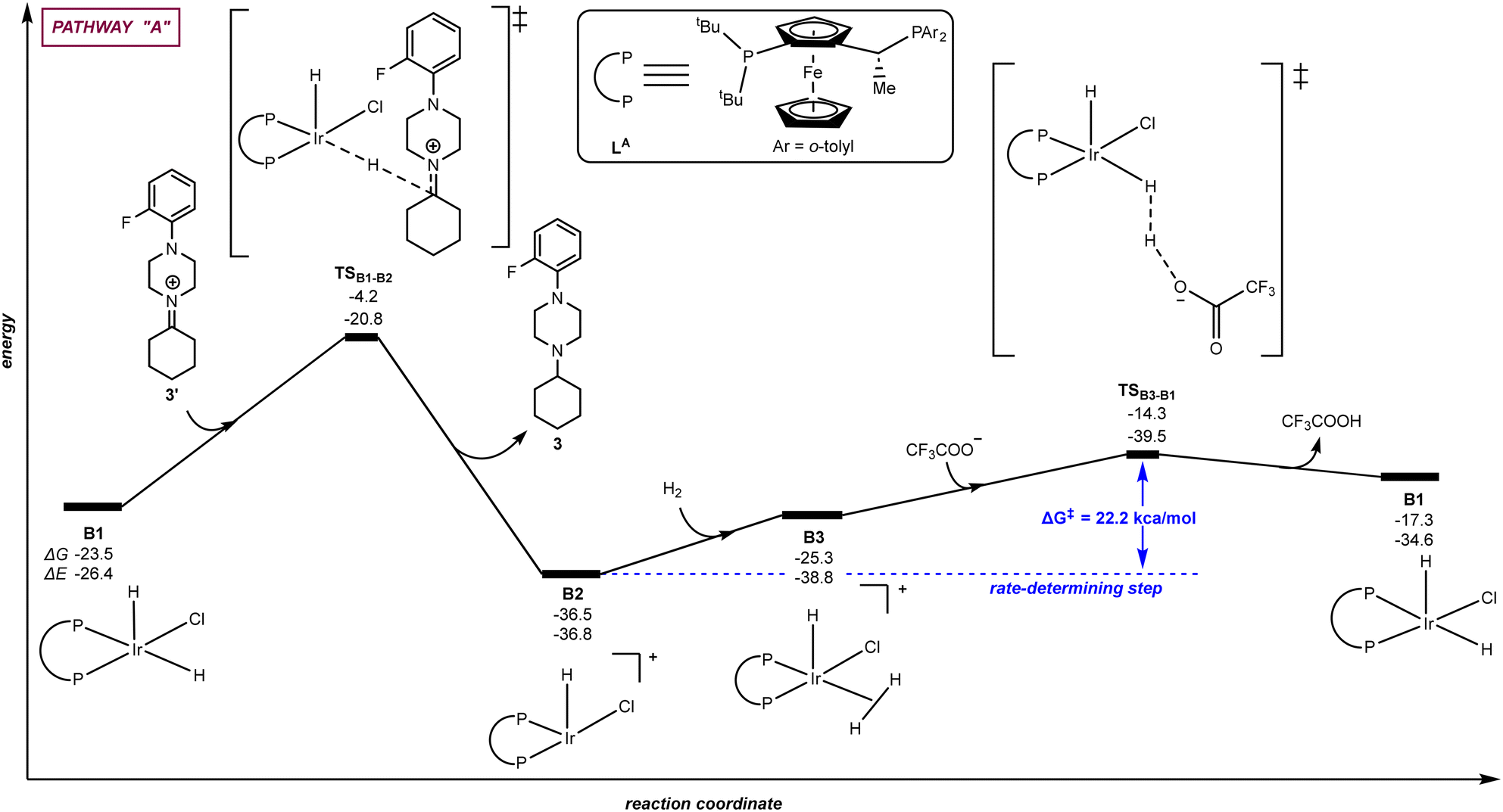
| | Fig. 3 Pathway “A”: reaction coordinate for the hydride/hydrogen transfer pathway occurring in a penta-coordinated iridium complex B1. The relative Gibbs free energy (ΔG) and potential energy (ΔE) are reported in kcal mol−1. P–P: diphosphane ligand LA. | |
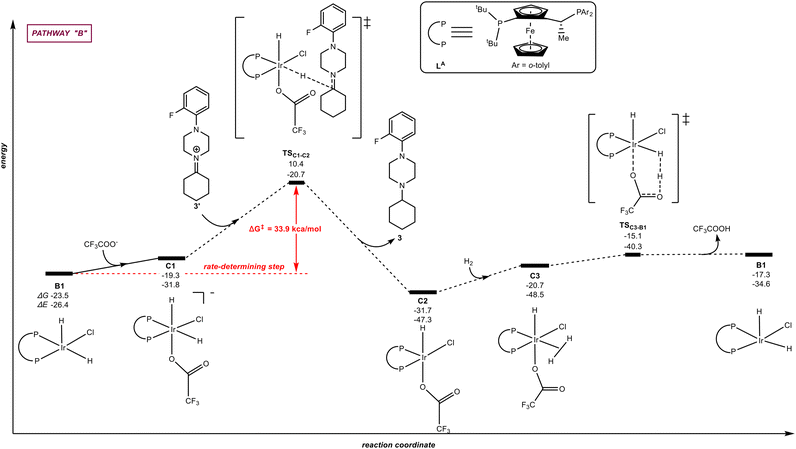 |
| | Fig. 4 Pathway “B”: reaction coordinate for the trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in a hexa-coordinated iridium complex C1. The relative Gibbs free energy (ΔG) and potential energy (ΔE) are reported in kcal mol−1. P–P: diphosphane ligand LA. | |
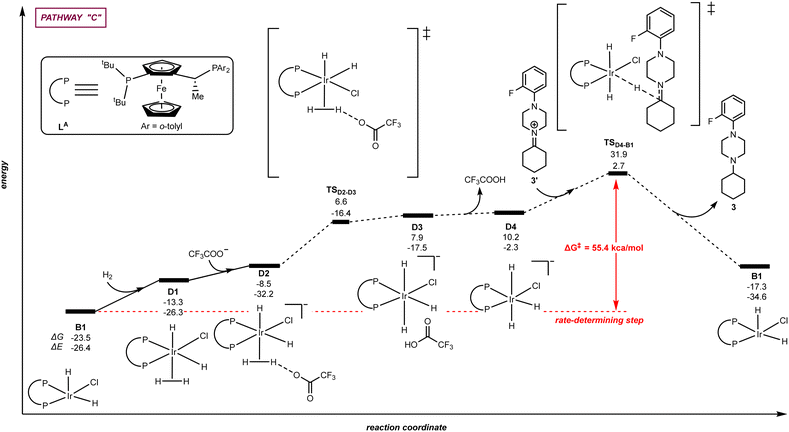 |
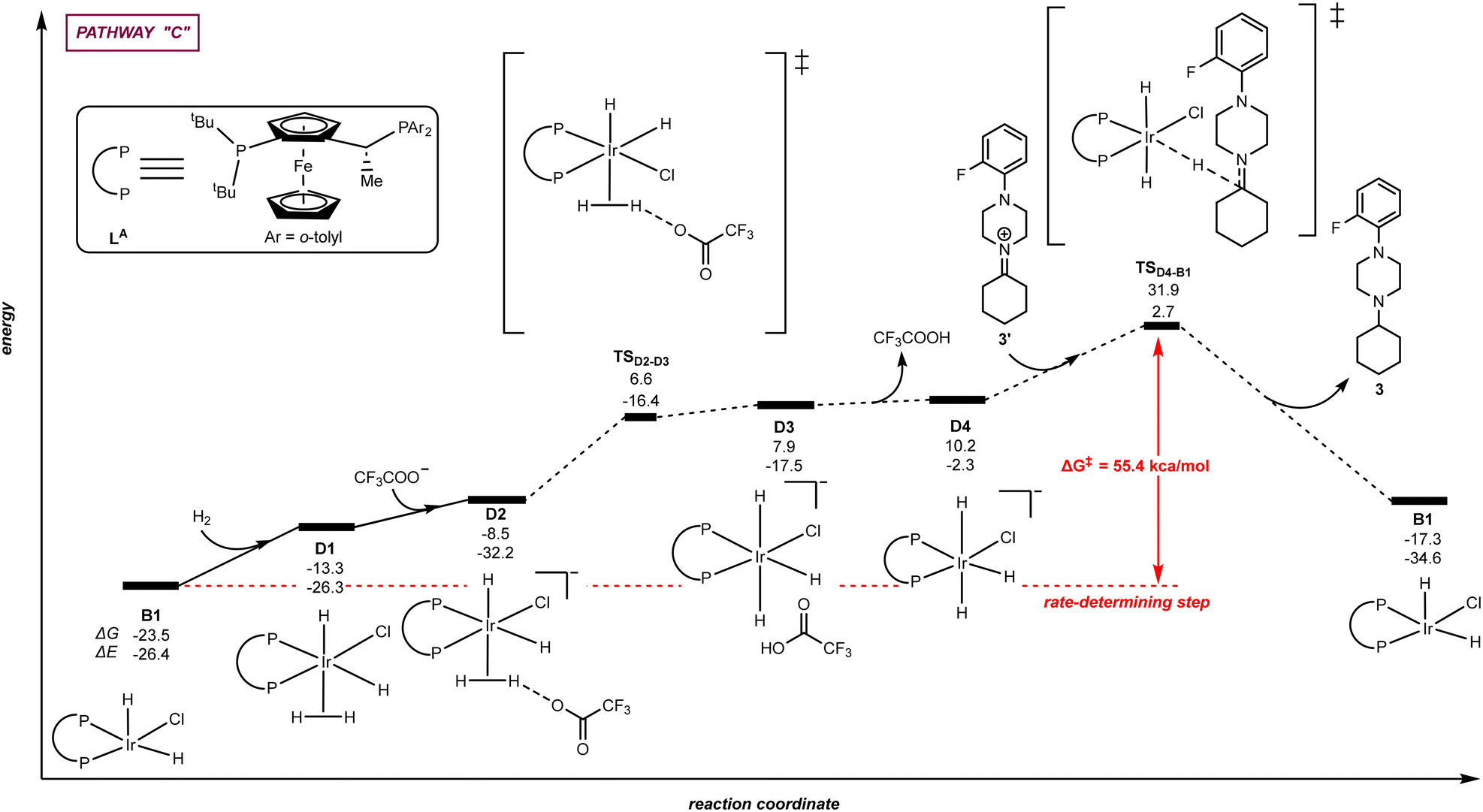
| | Fig. 5 Pathway “C”: reaction coordinate for the trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in a hexa-coordinated iridium complex D1 with initial H2 activation. The relative Gibbs free energy (ΔG) and potential energy (ΔE) are reported in kcal mol−1. P–P: diphosphane ligand LA. | |
Fig. 3 (pathway “A”) displays the hydrogenation of 3′ considering a direct hydride/hydrogen transfer with trifluoroacetate involved in the catalyst regeneration process. First, the hydride ligand of the pentacoordinate iridium complex B1 can be transferred to the unsaturated carbon atom of the intermediate iminium 3′via the transition state TSB1–B2 with an energy span of 19.3 kcal mol−1. As a result, the amine product 3 is formed. The cationic, iridium intermediate B2 may undergo reaction with H2 leading to a σ H2-ligated iridium species B3 with an energy increase of 11.2 kcal mol−1. The coordinated dihydrogen ligand on the iridium centre of B3 undergoes H–H bond heterolytic cleavage thanks to the trifluoroacetate anion through the transition state TSB3–B1, thereby releasing TFA and regenerating the catalytically active iridium species B1. From this evaluation, the H–H bond heterocyclic cleavage viaTSB3–B1 is the rate-determining step of the whole catalytic cycle with an energy span of 22.2 kcal mol−1 and the catalytic role of TFA is explained. Interestingly, it is relevant to note that these calculations did not identify any interaction between the iminium and the metal, thus ruling out any coordination to reach hexa-coordinated iridium species followed by hydride transfer.
On the other hand, we also evaluated other mechanistic scenarios, namely pathways “B” and “C” (Fig. 4 and 5, respectively). In Fig. 4 (pathway “B”) we have considered the hydride/hydrogen transfer pathway with the trifluoroacetate anion coordinating to the iridium catalyst B1via a plausible hexacoordinated iridium species C1. The trifluoroacetate anion can coordinate to B1, being this process endothermic by 4.2 kcal mol−1. The hydride ligand of the hexa-coordinated iridium complex C1 is transferred to the unsaturated carbon atom of the iminium intermediate 3′via the transition state TSC1–C2 with an energy span of 33.9 kcal mol−1. As a result, the amine product 3 and the intermediate C2, with the trifluoroacetate ligand bound to iridium, are obtained. Then, the dihydrogen molecule coordinates to the iridium centre of C2 to form the hexa-coordinated iridium complex (C3) with an energy increase of 11.0 kcal mol−1. The coordinated dihydrogen ligand on the iridium centre of C3 undergoes H–H bond heterolytic cleavage through the transition state TSC3–B1 with the trifuoroacetate ligand abstracting the hydrogen atom releasing TFA in a concerted, intramolecular manner and regenerating the catalytically active species B1. In this mechanistic scenario, the rate-determining step associated to the energy of TSC1–C2 (Fig. 4) is higher than that of TSB3–B1 (Fig. 3) by 11.7 kcal mol−1, indicating that the trifluoroacete-mediated pathway “B” is unlikely using the Josiphos-type ligand LA.
Alternatively to the trifluoroacetate-mediated pathway above-described in Fig. 4, we envisioned a different mechanistic scenario in which H2 activation occurs at the iridium species B1 leading to a hexa-coordinated iridium species with the trifluoroacetate anion behaving as an external base without coordination to iridium, namely pathway “C” (Fig. 5). More precisely, the DFT-computed pathway “C” started with the reaction of B1 with H2 in which the H2 molecule coordinates in a σ manner to the iridium centre of B1 forming the hexa-coordinated species D1 with an energy increase of +10.2 kcal mol−1. In the presence of trifluoroacetate anion, the coordinated dihydrogen ligand on the iridium centre of D1 undergoes H–H bond heterolytic cleavage via intermediate D2 and through the transition state TSD2–D3. A hydrogen atom is abstracted by the trifluoroacetate anion forming TFA as well as the anionic iridium intermediate D4. Subsequently, the hydride ligand of the hexa-coordinated iridium intermediated D4 can be transferred to the unsaturated carbon atom of the iminium intermediate 3′via the transition state TSD4–B1 with an energy barrier of +21.7 kcal mol−1 while regenerating the catalyst B1. The energy span for the hydride transfer transition state TSD4–B1 is very high (+55.4 kcal mol−1), strongly suggesting that this pathway is not accessible in the experimental conditions in which the catalysis occurs at room temperature. As such, from the three different mechanistic scenarios evaluated (Fig. 3–5), the most plausible one for the iridium-catalysed hydrogenation pathway using the ligand LA is via the iridium penta-coordinated pathway “A” displayed in Fig. 3 (vide supra).
Iridium-catalysed hydrogenation of the iminium intermediate 3′ and catalyst regeneration with Xantphos ligand
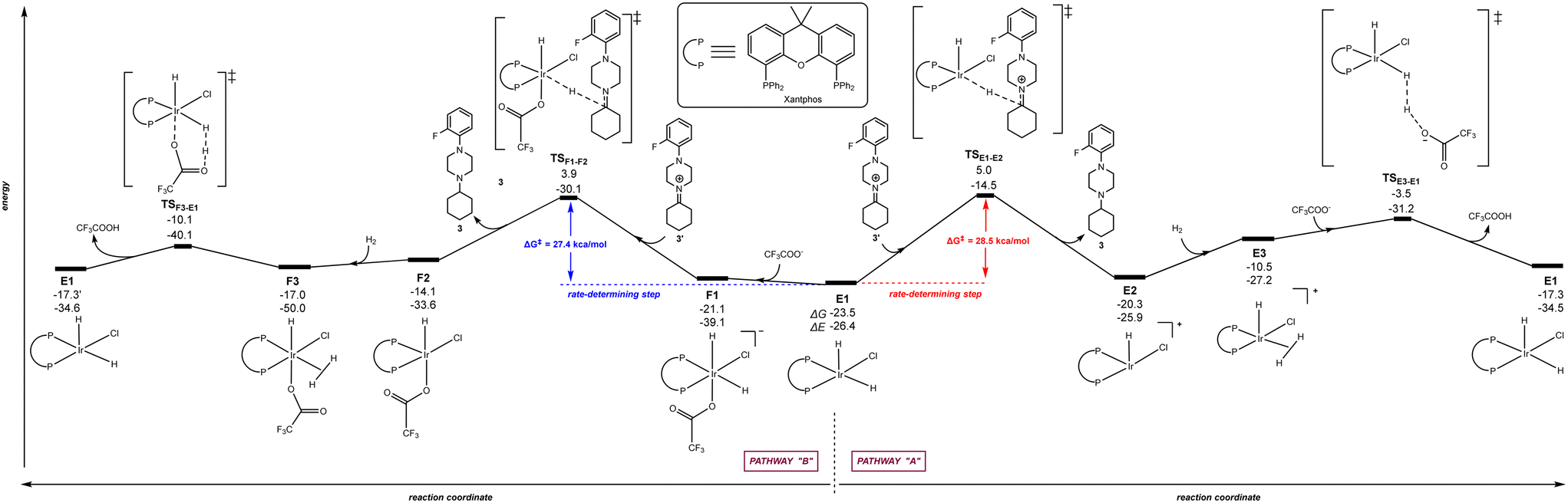
Because similar efficiency is found with the Josiphos-type ligand LA and Xantphos (Scheme 1), we were curious to see which pathway (“A”, “B” or “C”) was more favoured with Xantphos starting from the homologous catalytically active iridium species to B1, namely E1 [(Xantphos)Ir(H)2(Cl)]. Fig. 6 displays the reaction trajectory for the hydrogenation of the iminium 3′ starting from E1via a penta-coordinated iridium intermediate (right, pathway “A”) and via a hexa-coordinated iridium intermediate F1 (left, pathway “B”). The rate-determining step for pathway “A” is placed at +28.5 kcal mol−1 whereas the one for pathway “B” is at +27.4 kcal mol−1. This difference might be relevant enough for a reaction occurring at room temperature, especially considering that the key transition state (TSF1–F2) is reached via the intermediate F1 that is higher in energy than E1. Comparing both pathways after the rate-determining step, it is evident that intermediate F2 is more accessible than E2, leading to a mild trajectory for the following hydrogen activation step as well as providing an affordable driving force for the previous hydride addition step. Overall, the rate-determining step for the iridium-catalysed hydrogenation of 3′ using Xantphos appears to follow the hexa-coordinated pathway “B” and the scenario is reversed considering pathways “A” and “B” with the Josiphos-type ligand LA. Note that the pathway “C” with Xantphos, which comprised initial H2 activation followed by trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in an hexa-coordinated iridium complex led to a much higher energy barrier (+53.6 kcal mol−1) for the rate-determining step (see details in Fig. S3 in the ESI†), similarly as found in Fig. 5 for ligand LA, thereby ruling out this reaction pathway for Xantphos as well. Consequently, the Xantphos ligand, which is known to give rise to metal complexes with very large bite-angles103–105 and its phosphorus atoms are of different steric and electronic nature compared to Josiphos-type ligand LA, is a truly convenient and affordable ligand for the iridium-catalysed DRA between both aliphatic amines and aliphatic ketones.
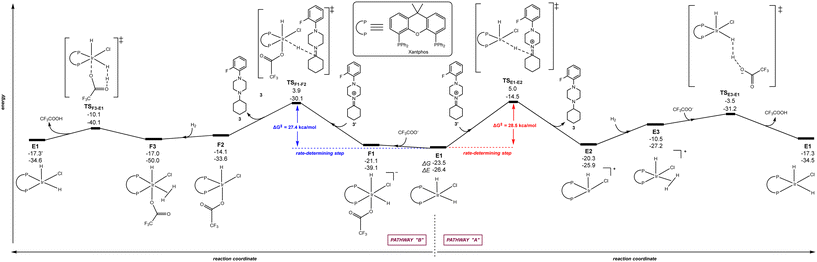 |
| | Fig. 6 Reaction coordinate for the hydride/hydrogen transfer pathway occurring in a penta-coordinated iridium complex E1 (pathway “A”, right - unfavourable) and reaction coordinate for the trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in a hexa-coordinated iridium complex F1 (pathway “B”, left - favourable). The relative Gibbs free energy (ΔG) and potential energy (ΔE) are reported in kcal mol−1. P–P: diphosphane ligand Xantphos. | |
Attempts of predicting the iridium-catalysed hydrogenation of the iminium intermediate 3′ with catalysts based on Josiphos-type ligand LB and BIBOP-type ligand LC
Based on the above-described findings, we wondered whether other type of Josiphos-type ligand, such as LB that bears different substitution patterns in the phosphorus atoms compared to LA, could eventually be used for this transformation. Moreover, due to the fact that Xantphos, in which the phosphorus atoms are wide apart over six chemical bonds, typically leads to metal complexes with a relatively large bite angle,103–105 we aimed at comparing it with a ligand whose metal complexes display a small bite angle, such as BIBOP-type ligand LC,106–110 with only three chemical bonds separating the two chelating phosphorus atoms (Scheme 4). We reasoned that these two ligands can give rise to catalytically active G1 and I1 species (Scheme 4), respectively, which we considered as the initial point for the calculations involving hydrogenation of the iminium 3′ towards amine 3.
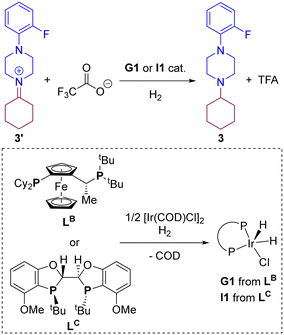 |
| | Scheme 4 Iridium-catalysed hydrogenation of iminium 3′ with the iridium G1 and I1 catalysts derived from ligands LB and LC considered for DFT computations. | |
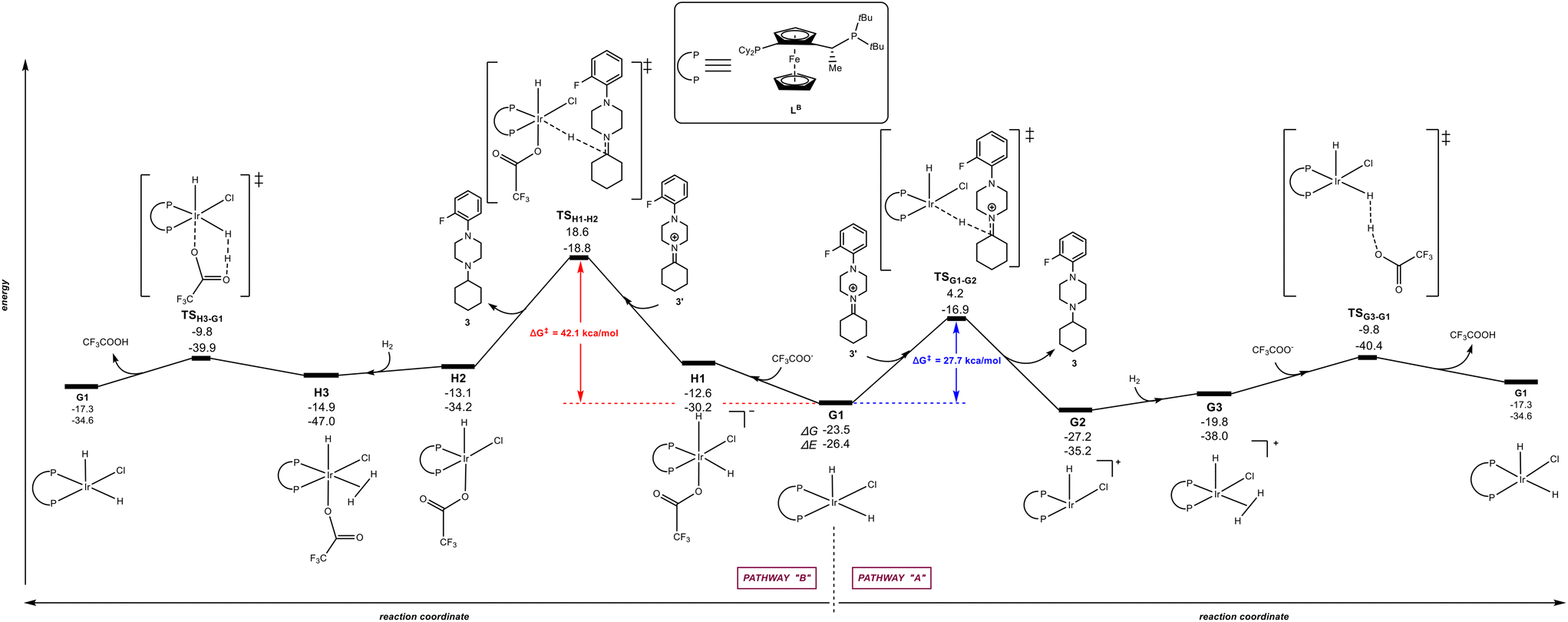
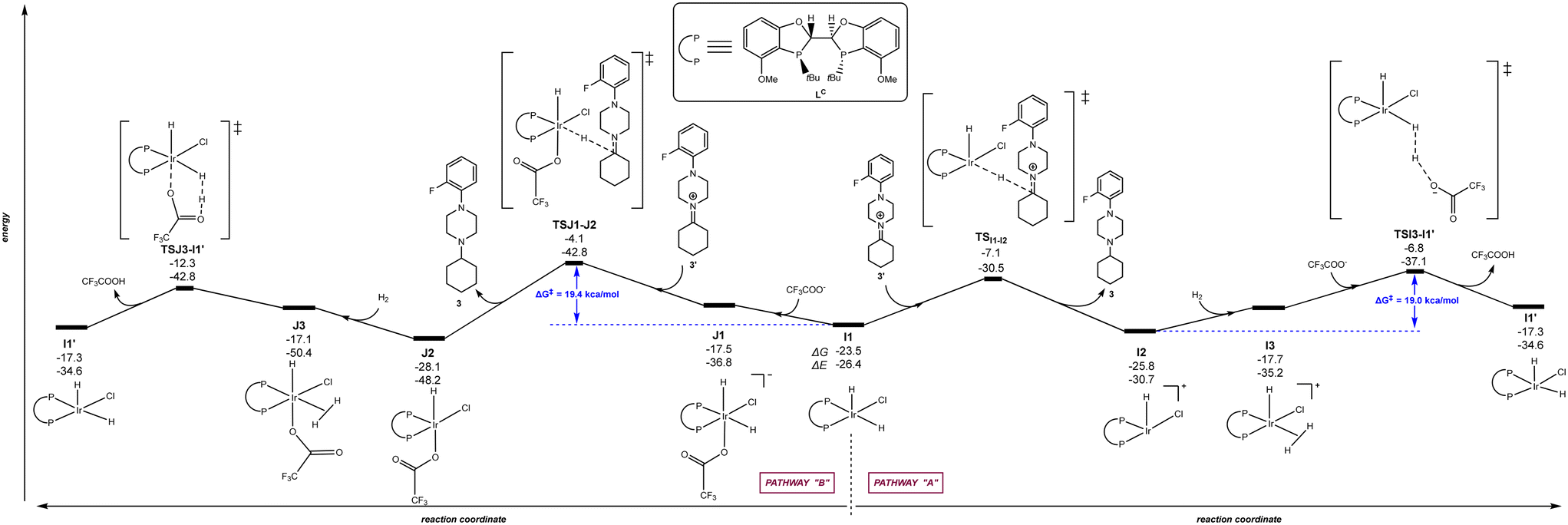
The calculations were carried out following the three pathways “A”, “B” and “C” above-considered for LA and Xantphos, respectively. Fig. 7 and 8 displays the reaction pathways “A” and “B” for LB and LC, respectively. Note that the pathway “C” with LB or LC, which comprised initial H2 activation followed by trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in an hexa-coordinated iridium complex (equivalent to Fig. 5, vide supra) led to a much higher energy barrier (+55.2 kcal mol−1 for LB and +40.0 kcal mol−1 for LC) for the rate-determining step (see details in Fig. S4 and S5 in the ESI†), similarly as found for ligands LA and Xantphos, thus ruling out this reaction pathway for LB and LC too.
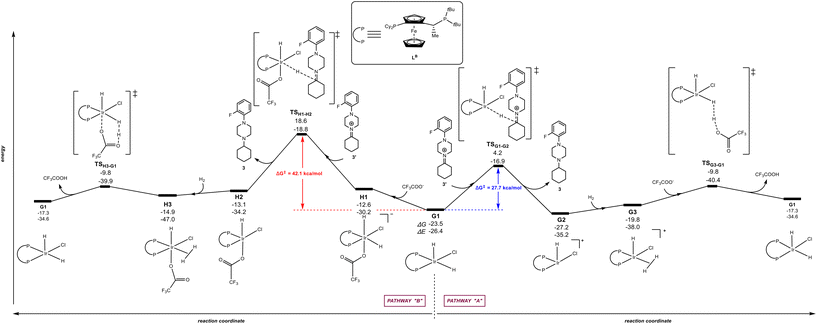 |
| | Fig. 7 Reaction coordinate for the hydride/hydrogen transfer pathway occurring in a penta-coordinated iridium complex G1 (pathway “A”, right) and reaction coordinate for the trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in a hexa-coordinated iridium complex H1 (pathway “B”, left). The relative Gibbs free energy (ΔG) and potential energy (ΔE) are reported in kcal mol−1. P–P: diphosphane ligand LB. | |
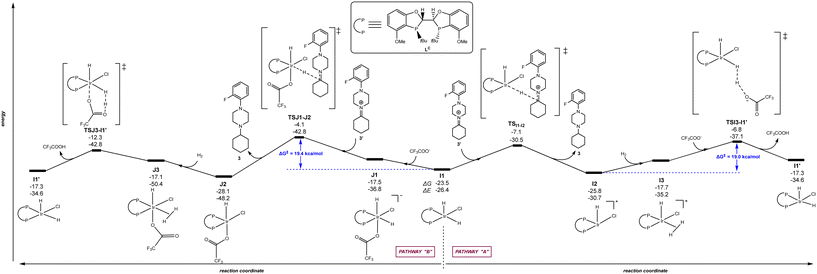 |
| | Fig. 8 Reaction coordinate for the hydride/hydrogen transfer pathway occurring in a penta-coordinated iridium complex I1 (pathway “A”, right) and reaction coordinate for the trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in a hexa-coordinated iridium complex J1 (pathway “B”, left). The relative Gibbs free energy (ΔG) and potential energy (ΔE) are reported in kcal mol−1. P–P: diphosphane ligand LC. | |
The Josiphos-type ligand LB prefers to follow the pathway “A” rather than the pathway “B” (Fig. 6) as it was also found previously for LA with the same ferrocene backbone. However, the rate-determining step for the reaction pathway “A” is 5.5 kcal mol−1 higher in energy that the one obtained with LA (27.7 kcal mol−1vs. 22.2 kcal mol−1). In agreement with a higher energetic profile associated to LB, the rate-determining step for LB in the pathway “B” reached the value of 42.1 kcal mol−1 (Fig. 7), which is 8.2 kcal mol−1 higher in energy than the one observed for pathway “B” using ligand LA. Consequently, LB appears to be less efficient than LA but similar to Xantphos, which featured a rate-determining step at a comparable 27.4 kcal mol−1 although via the pathway “B” (Fig. 6).
Regarding the behaviour of LD, we found that both reaction pathways “A” and “B” are energetically accessible with rate-determining steps associated to 19.0 kcal mol−1 and 19.4 kcal mol−1, respectively (Fig. 8). These values are very similar to those observed with LA considering the reaction pathway “A” (Fig. 3). Interestingly, the rate-determining step for pathway “B” with ligand LB correspond to the catalyst regeneration step and not the hydrogen/hydride transfer to the substrate, in stark contrast with the other three ligands analysed here. These findings indicate that, in principle, ligands LB and LC, should promote the iridium-catalysed DRA between aliphatic amines. However, we reported that when using 2-butanone instead of cyclohexanone as ketone for the reaction with the amine 2, its conversion and the isolated yields of the corresponding tertiary amine product decreased significantly compared to LA and Xantphos.83
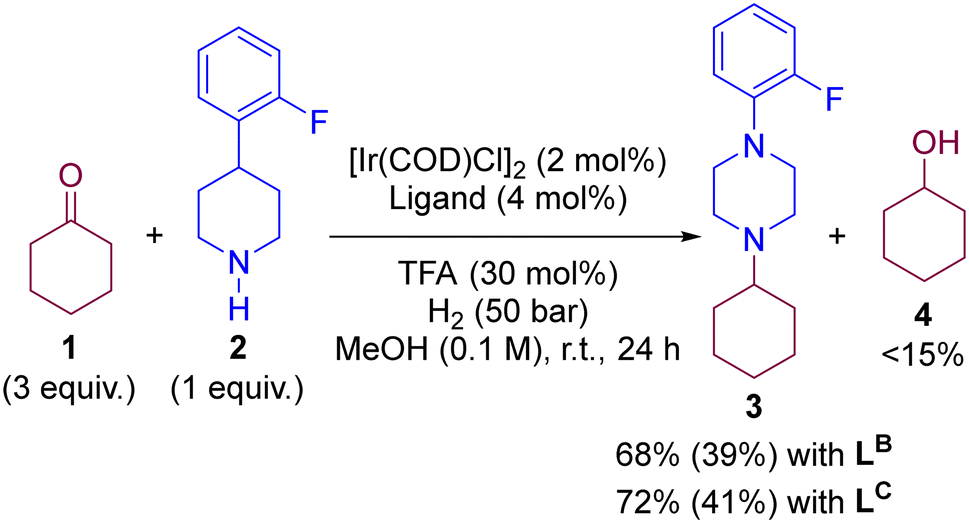
To evaluate the predicted behaviour of LB and LC, we performed experimentally the iridium-catalysed DRA between 1 and 2 under H2 atmosphere and methanol solvent at room temperature using catalytic amounts of TFA (30 mol%). Note that the reaction conditions are identical to those used with LA and Xantphos, respectively, in Scheme 1 (vide supra). After 24 hours of reaction, the conversions using LB and LC were around 70% with the tertiary amine 3 being isolated in yields close to 40% (Scheme 5). Importantly, we noted the formation of the cyclohexanol side-product 4 originating form a formal hydrogenation of the ketone 1 in yields around to 15% according to GC-MS analysis (see ESI† for details). Consequently, the performance of LB and LC for DRA predicted by DFT calculations was in competition with a ketone hydrogenation reaction, which is not the case of Josiphos-type ligand LA and Xantphos. Overall, the ligands LB and LC are not as competent as LA and Xantphos for the DRA between aliphatic ketones and amines. A summary of the key observations regarding the rate-determining step in each pathway versus the reactivity in catalysis is provided in Table 1. Finally, we envisioned to analyse by NBO analysis the key DFT-computed key transitions states involving the hydride transfer step from the metal to the substrate (Fig. S37 in the ESI†). It is found that the electronegativities of hydrides and the steric hindrance can well account for the computed energy barriers of the hydride transfer steps.
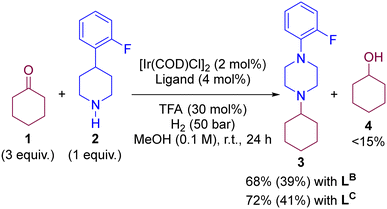 |
| | Scheme 5 Experimental results for the iridium-catalysed DRA enabling formation of purely aliphatic tertiary amine 3 using ligands LB and LC, respectively. Isolated yields displayed in brackets. | |
Table 1 Summary regarding the rate-determining step versus the activity and selectivity of the iridium-coordinated ligands
| Ligand |
Conv. |
Yield |
Yield |
Rate-determining step (kcal mol−1) |
|
2 (%) |
3 (%) |
4 (%) |
Path A |
Path B |
Path C |
|
L
A
|
>99 |
92 |
0 |
22.2 |
33.9 |
55.4 |
| Xantphos |
>97 |
80 |
0 |
28.5 |
27.4 |
53.6 |
|
L
B
|
68 |
39 |
<15 |
27.7 |
42.1 |
55.2 |
|
L
C
|
72 |
41 |
<15 |
19.0 |
19.4 |
40.0 |
Conclusions
In conclusion, by means of state-of-the-art computation calculations at the DFT level, we have demonstrated that the iridium-catalysed DRA reaction between aliphatic ketones and aliphatic amines follows very different mechanistic trajectories depending on the nature of the ligand.111,112 In particular, the already experimentally identified Josiphos-type ligands LA and Xantphos follow very different reaction pathways. Whilst LA favours the hydride/hydrogen transfer pathway occurring in a penta-coordinated iridium (pathway “A”), the large bite-angle diphosphane Xantphos is more adapted to the trifluoroacetate-mediated, hydride/hydrogen transfer pathway occurring in a hexa-coordinated iridium complex (pathway “B”). With the aim of predicting the catalytic outcome, the calculations using Josiphos-type ligand LB and the small bite-angle BIBOP-type ligand LC led to rather counterintuitive findings with both ligands appearing suitable for the iridium-catalysed DRA although via different rate-determining steps. However, the experimental results shown non-negligible formation of ketone hydrogenation as a side-reaction. As such, ligands LB and LC are engaged in undesired processes, thereby under-performing LA and Xantphos for iridium-catalysed DRA. In addition, we have shown that the iminium intermediate is more stable than the corresponding enamine for the case of purely aliphatic substrates, which is of relevance for evaluating metal-catalysed DRA or other type of (asymmetric) hydrogenations. Furthermore, we demonstrate and highlight the complex mechanistic scenarios that should have to be considered for metal-catalysed hydrogen-mediated organic transformations.
Data availability statement
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the CNRS, University of Rennes and Janssen Pharmaceutica N.V. (Johnson & Johnson) is acknowledged. The work in Hebei University is supported by the National Natural Science Foundation of China (22373030), the Excellent Youth Research Innovation Team of Hebei University (QNTD202406), and the Innovation Team Project of Hebei University (IT2023C01).
Notes and references
-
P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer, Dordrecht, 2004 Search PubMed.
-
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2009 Search PubMed.
-
M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed.
-
D. Selent and D. Heller, In-Situ Techniques for Homogeneous Catalysis in Catalysis: From Principles to Applications, ed. M. Beller, A. Renken and R. A. van Santen, Wiley-VCH, Weinheim, 2012, pp. 465–490 Search PubMed.
- M. Garland, Catal. Today, 2010, 155, 266–270 CrossRef CAS.
-
A. Haynes, The Use of High Pressure Infrared Spectroscopy to Study Catalytic Mechanisms in Mechanisms in Homogeneous Catalysis, ed. B. Heaton, Wiley-VCH, Weinheim, Germany, 2005, pp. 107–150 Search PubMed.
- N. J. Beach, S. M. M. Knapp and C. R. Landis, Rev. Sci. Instrum., 2015, 86, 104101–104109 CrossRef CAS PubMed.
- A. C. Brezny and C. R. Landis, ACS Catal., 2019, 9, 2501–2513 CrossRef CAS.
- A. Duchowny, S. A. Ortiz Restrepo, M. Adams, R. Thelen and A. Adams, Analyst, 2022, 147, 3827–3832 RSC.
- I. T. Horvath and J. M. Millar, Chem. Rev., 1991, 91, 1339–1351 CrossRef CAS.
- N. R. Jaegers, K. T. Mueller, Y. Wang and J. Z. Hu, Acc. Chem. Res., 2020, 53, 611–619 CrossRef CAS.
- J. M. Dreimann, E. Kohls, H. F. W. Warmeling, M. Stein, L. F. Guo, M. Garland, T. N. Dinh and A. J. Vorholt, ACS Catal., 2019, 9, 4308–4319 CrossRef CAS.
- E. Groppo, S. Rojas-Buzo and S. Bordiga, Chem. Rev., 2023, 123, 12135–12169 CrossRef CAS PubMed.
- T. K. Slot, N. R. Shiju and G. Rothenberg, Angew. Chem., Int. Ed., 2019, 58, 17273–17276 CrossRef CAS.
- O. Diebolt, P. W. N. M. van Leeuwen and P. C. J. Kamer, ACS Catal., 2012, 2, 2357–2370 CrossRef CAS.
- V. Butera, Phys. Chem. Chem. Phys., 2024, 26, 7950–7970 RSC.
-
F. Maseras and A. Lledós, Computational Modeling of Homogeneous Catalysis, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2002 Search PubMed.
-
K. Morokuma and D. G. Musaev, in Computational Modeling for Homogeneous and Enzymatic Catalysis: A Knowledge-Base for Designing Efficient Catalysts, Wiley-VCH, Weinheim, 2008 Search PubMed.
- A. V. Kalikadien, A. Mirza, A. N. Hossaini, A. Sreenithya and E. A. Pidko, ChemPlusChem, 2024, 89, e202300702 CrossRef.
- M. Foscato and V. R. Jensen, ACS Catal., 2020, 10, 2354–2377 CrossRef CAS.
- J. Lan, X. Li, Y. Yang, X. Zhang and L. W. Chung, Acc. Chem. Res., 2022, 55, 1109–1123 CrossRef CAS PubMed.
- W. Yang, G. A. Filonenko and E. A. Pidko, Chem. Commun., 2023, 59, 1757–1768 RSC.
- A. Lledós, Eur. J. Inorg. Chem., 2021, 2547–2555 CrossRef.
- Y. Musashi and S. Sakaki, J. Am. Chem. Soc., 2002, 124, 7588–7603 CrossRef CAS PubMed.
- Y. Luo, Z. Huang, Z. Chen, Z. Xu, J. Meng, H.-Y. Li, Q. Meng and D. Tang, J. Org. Chem., 2020, 85, 11626–11634 CrossRef CAS PubMed.
- J. Faiges, C. Borràs, I. M. Pastor, O. Pàmies, M. Besora and M. Diéguez, Organometallics, 2021, 40, 3424–3435 CrossRef CAS.
- A. M. Krieger, P. Kuliaev, F. Q. A. Hall, D. Sun and E. A. Pidko, J. Phys. Chem. C, 2020, 124, 26990–26998 CrossRef CAS PubMed.
- P. Chakraborty, B. Sundararaju, E. Manoury and R. Poli, ACS Catal., 2021, 11, 11906–11920 CrossRef CAS.
- R. P. Dias and W. R. Rocha, Organometallics, 2011, 30, 4257–4268 CrossRef CAS.
- C.-X. Cui, H. Chen, S.-J. Li, T. Zhang, L.-B. Qu and Y. Lan, Coord. Chem. Rev., 2020, 412, 213251 CrossRef CAS.
- B. Liu, N. Huang, Y. Wang, X. Lan and T. Wang, Chem. Eng. J., 2022, 441, 136101 CrossRef CAS.
- M. Besora and F. Maseras, Adv. Catal., 2021, 68, 385–426 CAS.
- S. A. Decker and T. R. Cundari, Organometallics, 2001, 20, 2827–2841 CrossRef CAS.
- S. S. Nurttila, P. R. Linnebank, T. Krachko and J. N. H. Reek, ACS Catal., 2018, 8, 3469–3488 CrossRef CAS PubMed.
- S. Zhang, Z. Li, H. Qi, Y. Zhao, Y. Tang, A. Liu, M. Pu and M. Lei, Dalton Trans., 2024, 53, 6660–6666 RSC.
- J. Fang, Z.-Q. Wang, X. Wei, Y. Ma, H. Gong, X.-Q. Gong and Z. Hou, ACS Sustainable Chem. Eng., 2021, 9, 13256–13267 CrossRef CAS.
- M. Cheong, R. Schmid and T. Ziegler, Organometallics, 2000, 19, 1973–1982 CrossRef CAS.
- F. León, A. Comas-Vives, E. Álvarez and A. Pizzano, Catal. Sci. Technol., 2021, 11, 2497–2511 RSC.
- M. Sterle, M. Huš, M. Lozinšek, A. Zega and A. E. Cotman, ACS Catal., 2023, 13, 6242–6248 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS.
- V. I. Tararov, R. Kadyrov, T. H. Riermeier and A. Börner, Chem. Commun., 2000, 1867–1868 RSC.
- K. O. Biriukov, M. M. Vinogradov, O. I. Afanasyev, D. V. Vasilyev, A. A. Tsygankov, M. Godovikova, Y. V. Nelyubina, D. A. Loginov and D. Chusov, Catal. Sci. Technol., 2021, 11, 4922–4930 RSC.
- R. R. Thakore, B. S. Takale, G. Casotti, E. S. Gao, H. S. Jin and B. H. Lipshutz, Org. Lett., 2020, 22, 632–6329 CrossRef PubMed.
-
Green Chemistry: Theory and Practice, ed. P. T. Anastas and J. C. Warner, Oxford University Press, Oxford, 1998 Search PubMed.
- B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef CAS PubMed.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- Z. Zhang, T. Ikeda, H. Murayama, T. Honma, M. Tokunaga and Y. Motoyama, Chem. – Asian J., 2022, 17, e20210124 Search PubMed.
- F. Kliuev, A. Kuznetsov, O. I. Afanasyev, S. A. Runikhina, E. Kuchuk, E. Podyacheva, A. A. Tsygankov and D. Chusov, Org. Lett., 2022, 24, 7717–7721 CrossRef CAS PubMed.
- E. Podyacheva, O. I. Afanasyev, A. A. Tsygankov, M. Makarova and D. Chusov, Synthesis, 2019, 51, 2667–2677 CrossRef CAS.
- B. Li, J.-B. Sortais and C. Darcel, RSC Adv., 2016, 6, 57603–57625 RSC.
-
Reductions by the Alumino- and Borohydrides in Organic Synthesis, ed. J. Seyden-Penne, Wiley, New York, 2nd edn, 1997 Search PubMed.
- A. F. Abdel-Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971–1031 CrossRef CAS.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- Q. Zou, F. Liu, T. Zhao and X. Hu, Chem. Commun., 2021, 57, 8588–8591 RSC.
-
Amines: Synthesis, Properties and Applications, ed. S. A. Lawrence, Cambridge University Press, Cambridge, 2004 Search PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- A. Zanasi, M. Mazzolini and A. Kantar, Multidiscip. Respir. Med., 2017, 12, 7 CrossRef PubMed.
- Y. Hirayama, M. Ikunaka and J. Matsumoto, Org. Process Res. Dev., 2005, 9, 30–38 CrossRef CAS.
-
Concise Dictionary of Pharmacological Agents: Properties and Synonyms, ed. I. K. Morton and J. M. Hall, Springer, Dordrecht, 1999 Search PubMed.
- Y. Tian, L. Hu, Y.-Z. Wang, X. Zhang and Q. Yin, Org. Chem. Front., 2021, 8, 2328–2342 RSC.
- S. H. Gilbert, S. Tin, J. A. Fuentes, T. Fanjul and M. L. Clarke, Tetrahedron, 2021, 80, 131863 CrossRef CAS.
- S. Yuan, G. Gao, L. Wang, C. Liu, L. Wan, H. Huang, H. Geng and M. Chang, Nat. Commun., 2020, 11, 621 CrossRef CAS PubMed.
- J. H. Schrittwieser, S. Velikogne and W. Kroutil, Adv. Synth. Catal., 2015, 357, 1655–1685 CrossRef CAS.
- R. Kumar, M. J. Karmilowicz, D. Burke, M. P. Burns, L. A. Clark, C. G. Connor, E. Cordi, N. M. Do, K. M. Doyle, S. Hoagland, C. A. Lewis, D. Mangan, C. A. Martinez, E. L. McInturff, K. Meldrum, R. Pearson, J. Steflik, A. Rane and J. Weaver, Nat. Catal., 2021, 4, 775–782 CrossRef CAS.
- S. Simić, E. Zukić, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Chem. Rev., 2022, 122, 1052–1126 CrossRef PubMed.
- Z. Gao, J. Liu, H. Huang, H. Geng and M. Chang, Angew. Chem., Int. Ed., 2021, 60, 27307–27311 CrossRef CAS PubMed.
- J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed.
- Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769–14827 CrossRef CAS PubMed.
- K. Källström, I. Munslow and P. G. Andersson, Chem. – Eur. J., 2006, 12, 3194–3200 CrossRef PubMed.
- N. Fleury-Brégeot, V. de la Fuente, S. Castillón and C. Claver, ChemCatChem, 2010, 2, 1346–1371 CrossRef.
- R. A. A. Abdine, G. Hedouin, F. Colobert and J. Wencel-Delord, ACS Catal., 2021, 11, 215–247 CrossRef CAS.
- A. Fabrello, A. Bachelier, M. Urrutigoïty and P. Kalck, Coord. Chem. Rev., 2010, 254, 273–287 CrossRef CAS.
- N. U. D. Reshi, V. B. Saptal, M. Beller and J. K. Bera, ACS Catal., 2021, 11, 13809–13837 CrossRef CAS.
- S. H. Gilbert, V. Viseur and M. L. Clarke, Chem. Commun., 2019, 55, 6409–6412 RSC.
- H.-U. Blaser, H.-P. Buser, H.-P. Jalett, B. Pugin and F. Spindler, Synlett, 1999, S1, 867–868 CrossRef.
- P. Cheruku, T. L. Church, A. Trifonova, T. Wartmann and P. G. Andersson, Tetrahedron, 2008, 49, 7290–7293 CrossRef CAS.
- Z. Wu, S. Du, G. Gao, W. Yang, X. Yang, H. Huang and M. Chang, Chem. Sci., 2019, 10, 4509–4514 RSC.
- M. Jouffroy, T.-M. Nguyen, M. Cordier, M. Blot, T. Roisnel and R. Gramage-Doria, Chem. – Eur. J., 2022, 28, e202201078 CrossRef CAS PubMed.
- R. Xie, H. Zhou, H. Lu, Y. Mu, G. Xu and M. Chang, Org. Lett., 2022, 24, 9033–9037 CrossRef CAS PubMed.
- I. Borthakur, S. Nandi, Y. Bilora, B. Sadhu and S. Kundu, ACS Catal., 2024, 14, 5847–5857 CrossRef CAS.
- X.-Y. Lv and R. Martin, Org. Lett., 2023, 25, 3750–3754 CrossRef CAS PubMed.
- R. Xie, C. Liu, R. Lin, R. Zhang, H. Huang and M. Chang, Org. Lett., 2022, 24, 5646–5650 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- G. J. Baker, A. J. P. White, I. J. Casely, D. Grainger and M. R. Crimmin, J. Am. Chem. Soc., 2023, 145, 7667–7674 CrossRef CAS PubMed.
- K. H. Hopmann, Chem. – Eur. J., 2015, 21, 10020–10030 CrossRef CAS PubMed.
- H. Li, M. Wen and Z.-X. Wang, Inorg. Chem., 2012, 51, 5716–5727 CrossRef CAS PubMed.
- C. J. C. A. V. Marenich and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- T. Tsutsumi, Y. Ono, Z. Arai and T. Taketsugu, J. Chem. Theory Comput., 2018, 14, 4263–4270 CrossRef CAS PubMed.
- K. H. Hopmann and A. Bayer, Organometallics, 2011, 30, 2483–2497 CrossRef CAS.
- J. M. Mwansa and M. I. Page, Catal. Sci. Technol., 2020, 10, 590–612 RSC.
- K. H. Hopmann and A. Bayer, Coord. Chem. Rev., 2014, 268, 59–82 CrossRef CAS.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS.
- V. R. Landaeta, B. K. Muñoz, M. Peruzzini, V. Herrera, C. Bianchini and R. A. Sánchez-Delgado, Organometallics, 2006, 25, 403–409 CrossRef CAS.
- M. Martín, E. Sola, S. Tejero, J. L. Andrés and L. A. Oro, Chem. – Eur. J., 2006, 12, 4043–4056 CrossRef PubMed.
- Y. Huang, S. Liu, Y. Liu, Y. Chen, M. Weisel, R. T. Williamson, I. W. Davies and X. Zhang, Tetrahedron, 2018, 74, 2182–2190 CrossRef CAS.
- P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1529 RSC.
- M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890–1901 RSC.
- W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176–179 CrossRef CAS PubMed.
- W. Tang, A. G. Capacci, A. White, S. Ma, S. Rodriguez, B. Qu, J. Savoie, N. D. Patel, X. Wei, N. Haddad, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 1104–1107 CrossRef CAS PubMed.
- S. Rodríguez, B. Qu, K. R. Fandrick, F. Buono, N. Haddad, Y. Xu, M. A. Herbage, X. Zeng, S. Ma, N. Grinberg, H. Lee, Z. S. Han, N. K. Yee and C. H. Senanayake, Adv. Synth. Catal., 2014, 356, 301–307 CrossRef.
- G. Liu, X. Liu, Z. Cai, G. Jiao, G. Xu and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 4235–4238 CrossRef CAS PubMed.
- P. Rojo, A. Riera and X. Verdaguer, Coord. Chem. Rev., 2023, 489, 215192 CrossRef CAS.
- K. A. Bunten, L. Chen, A. L. Fernandez and A. J. Poë, Coord. Chem. Rev., 2002, 233–234, 41–51 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Lanbo
Liu
a,
Zhihui
Li
a,
Thi-Mo
Nguyen
*a,
Lanbo
Liu
a,
Zhihui
Li
a,
Thi-Mo
Nguyen