
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ spectroscopic investigations on BiPhePhos modified rhodium complexes in alkene hydroformylation†
Benedict N. Leidecker a,
Dilver Peña Fuentesa,
Matthias Königb,
Jiali Liubd,
Wolfgang Baumanna,
Mathias Sawallc,
Klaus Neymeyrac,
Haijun Jiaoa,
Robert Frankebd,
Armin Börnera and
Christoph Kubis*a
a,
Dilver Peña Fuentesa,
Matthias Königb,
Jiali Liubd,
Wolfgang Baumanna,
Mathias Sawallc,
Klaus Neymeyrac,
Haijun Jiaoa,
Robert Frankebd,
Armin Börnera and
Christoph Kubis*a
aLeibniz Institute for Catalysis e.V., Albert-Einstein Str. 29a, 18059 Rostock, Germany. E-mail: christoph.kubis@catalysis.de
bEvonik Oxeno GmbH & Co. KG, Paul-Baumann-Str. 1, 45772 Marl, Germany
cInstitute of Mathematics, University of Rostock, Ulmenstr. 59, 18057 Rostock, Germany
dLehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, 44780 Bochum, Germany
First published on 14th June 2024
Abstract
Structural and dynamic properties of BiPhePhos modified rhodium complexes under hydroformylation conditions have been investigated in detail by using high-pressure (HP) in situ transmission IR- and NMR-spectroscopy. An experiment design approach which combines component/reagent perturbations, in situ-FTIR measurements and chemometric peak group analysis (PGA) led to the identification of most relevant components. The ligand coordination in the structures of the hydrido and acyl 18-VE resting state complexes has been elucidated. The hydrido complex of the type e,e-[HRh(CO)2(P∩P)] represents the dominant resting state after catalyst preformation and during the n-regioselective hydroformylation. Dimer formation only takes place to a minor extent under severe reaction conditions under hydrogen depletion. Mono- and dinuclear hydrido monocarbonyl complexes are formed at higher ligand-to-metal ratios and low partial pressures of carbon monoxide. Both stereoisomeric forms of the bisphosphite modified acyl complexes e,a-[RC(O)Rh(CO)2(P∩P)] and e,e-[RC(O)Rh(CO)2(P∩P)] are generated as an equilibrium mixture.
1. Introduction
Hydroformylation of alkenes to aldehydes is one of the most important homogeneously catalyzed processes in the chemical industry.1–3 Mononuclear rhodium complexes modified by chelating diphosphites represent a class of highly active, chemo- and regioselective as well as stable catalysts.4,5 Bidentate diphosphites generally tend to form catalyst complexes for which a lower activity is obtained compared to bulky monophosphites, but the n-regioselectivity is often significantly enhanced.6 Trivalent organic phosphite ligands can undergo several decomposition reactions such as hydrolysis, alcoholysis, trans-esterification, Arbuzov rearrangement (alkyl phosphites), O–C bond cleavage, P–O bond cleavage and reactions with aldehydes, depending on the exact ligand structure and reaction conditions.1,5In Scheme 1 a catalytic cycle based on the established Wilkinson-type dissociative mechanism with a rhodium catalyst system modified by a bidentate phosphorus ligand is displayed. Starting from a coordinatively and electronically saturated trigona-bipyramidal 18-valence electron (VE) hydrido complex [HRh(CO)2(P∩P)] 1, the dissociation of a carbonyl ligand leads to the formation of a 16 VE hydrido complex 2, which is planar and has a free coordination site. Complex 2 can activate the alkene substrate under the formation of the π-complex 3. Two isomeric coordinatively unsaturated alkyl complexes can result from complex 3 by insertion of the alkene into the H–Rh bond. The α-alkyl complex 4 is formed in case of a Rh–C1 bonding, whereas a β-alkyl complex results from a Rh–C2 bonding (4′). Eventually the formation of the iso-aldehyde takes place from the β-alkyl complex However, the β-hydride elimination from the secondary alkyl complex might lead to dissociation of the 1-alkene or 2-alkene, which might coordinate again to the catalyst. The coordination of a CO ligand to 4 lead to the coordinatively saturated alky complex 5. Through the migration of the alkyl group into the Rh–CO bond the planar 16-VE acyl complex 6 is generated. The coordination of a CO ligand forms the 18-VE acyl complex 7. Product formation might occur via oxidative addition of hydrogen to 6 followed by reductive elimination of the aldehyde and regeneration of 2. This was observed for iridium catalyst systems.7–9 However, in the literature on rhodium catalyzed hydroformylation the reaction of 6 with molecular hydrogen is also frequently denoted as hydrogenolysis of 6.4,10–12 The latter is used in Scheme 1.
 | ||
| Scheme 1 Catalytic cycle of the chelate-ligand modified rhodium catalysed hydroformylation of alkenes based on a Wilkinson-type mechanism. | ||
For the elucidation of structural and mechanistic aspects in situ/operando spectroscopy is performed at real reaction conditions. In situ HP FTIR and NMR spectroscopy are powerful techniques which allow for a nearly complete elucidation of molecular structures of catalyst complexes at relevant conditions.13 FTIR spectroscopy provides distinct spectral patterns for respective transition metal carbonyl complexes. Infrared spectroscopy in the transmission mode is very sensitive and allows for the acquisition of quantitative data in the millimolar and submillimolar concentration range. Complementary structural and dynamic information on catalyst complexes and involved equilibria can be obtained by NMR spectroscopy based on 1H, 13C, 31P and sometimes even 103Rh measurements. Generally higher molar concentrations (5–100 mM) of the catalyst complexes and/or longer acquisition times are needed for an acceptable quality of the NMR spectra. However, higher catalyst concentrations might lead to shifts in chemical equilibria between dissolved transition metal complexes under compressed gases (e.g. CO, H2) because of the varied [H2]/[M] and [CO]/[M] ratios.
Chemometric data processing tools can extract pure component spectra and corresponding concentration profiles from the collected data set.14,15 In combination with an experiment design approach based on perturbations, respective algorithms allow for the identification of major, minor and even trace components.16
The interpretation of the infrared spectra for transition metal carbonyl complexes can be improved by a vibrational mode analysis based on DFT calculation. Thus, the assignment of vibrational modes even of non-isolable intermediates becomes possible.
Time-resolved in situ spectroscopic methods coupled with a simultaneous online-product analysis such as GC or MS permit operando studies from which structure–performance relationships can be derived.
The Rh/BiPhePhos system is a very prominent catalyst for the isomerizing n-regioselective alkene hydroformylation (Fig. 1). A hydrido complex of the type [HRh(CO)2(P∩P)] is formed via an in situ formation step treating a solution of the Rh-precursor and diphosphite ligand under synthesis gas (CO/H2 mixture). Investigations by Moasser, Gladfelter and Roe revealed fundamental insights into the structural and mechanistic aspects of this catalyst system.17 In succeeding studies by several research groups additional facets such as reaction kinetics as well as catalyst stability were explored in detail.18–23 Novel separation techniques for molecular catalysts such as TMS (thermomorphic solvent systems) and organic solvent nanofiltration have been developed with the Rh/BiPhePhos catalysts.24–27 The diphosphite ligand BiPhePhos was also applied in heterogenized catalysts on basis of the SILP (supported ionic liquid phase) and POL (porous organic ligand) concepts.28–33
 | ||
| Fig. 1 a) Lewis structural formula of BiPhePhos, b): molecular structure of BiPhePhos (30% probability level, without H).34 | ||
In this present research study new results on the in situ spectroscopic characterization of this interesting hydroformylation catalyst in solution based on in situ FTIR- and NMR-spectroscopy are presented. After covering the catalytic performance behaviour for selected alkenes, a substantial component identification of the reaction system based on the combination of experiment design by multiple component/reagent perturbations and chemometric analysis of IR-spectroscopic data was carried out. Further, novel aspects on catalyst stability, the coordination of additional ligands to the [HRh(CO)2(P∩P)] catalyst and the formation and structure of the corresponding acyl complex [RC(O)Rh(CO)2(P∩P)] will be discussed.
2. Results and discussion
2.1 Hydroformylation of alkenes
The hydroformylation of butenes is an industrially relevant process. The formation of valeraldehyde from a mixture of 1-butene, E/Z-2-butene requires a rhodium catalyst which is active in double bond isomerization as well as in n-regioselective hydroformylation. The Rh/BiPhePhos catalyst fulfils the needed properties. Several model reactions with butenes, pentenes and neohexene (Scheme 2) have been performed to demonstrate the potential of this catalyst system at conditions suitable for in situ spectroscopic reaction monitoring (Table 1). The reactions were conducted at 90 °C and 2.0 MPa synthesis gas (CO/H2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), [Rh] = 1 mM (mmol L−1), [P∩P] = 1 mM, [alkene] = 1 M, solvent: cyclohexane. A transmission flow-through IR-cell with CaF2 windows and a fixed optical pathlengh of 400 μm was used for the collection of infrared spectra with good signal-to-noise ratios for rhodium concentrations [Rh] ≤ 1 mM. Further details are given in the ESI† (ESI-A).
:1), [Rh] = 1 mM (mmol L−1), [P∩P] = 1 mM, [alkene] = 1 M, solvent: cyclohexane. A transmission flow-through IR-cell with CaF2 windows and a fixed optical pathlengh of 400 μm was used for the collection of infrared spectra with good signal-to-noise ratios for rhodium concentrations [Rh] ≤ 1 mM. Further details are given in the ESI† (ESI-A).
 | ||
| Scheme 2 Isomerization–hydroformylation of a) butene, b) pentene and c) neohexene. Co-substrates CO and H2 have been left out as well as the catalyst [HRh(CO)2(P∩P)] for clarity. | ||
| Substrate | Conversion/% | Aldehyde/% | n-Regioselectivityc/% |
|---|---|---|---|
| Reaction conditions: p(H2) = 1.0 MPa, p(CO) = 1.0 MPa, ϑ = 90 °C, [Rh] = 1 mM, [P∩P] = 1 mM, [substrate] = 1 M, solvent: cyclohexane.a t = 8 h.b t = 4 h.c Via GC. | |||
| 1-Butenea | >99 | >99 | >99 |
| 2-Butenea | >99 | >99 | >99 |
| 1-Pentenea | >99 | >99 | 97.5 |
| 2-Pentenea | >99 | >99 | 97.7 |
| Neohexeneb | >99 | >99 | >99 |
Prior to the reaction start by the addition of alkene substrate, a preformation of the catalyst is carried out by treating the transition metal precursor [Rh(acac)(CO)2] with one equivalent of the diphosphite ligand at the same conditions compared to the hydroformylation experiments.
 | ||
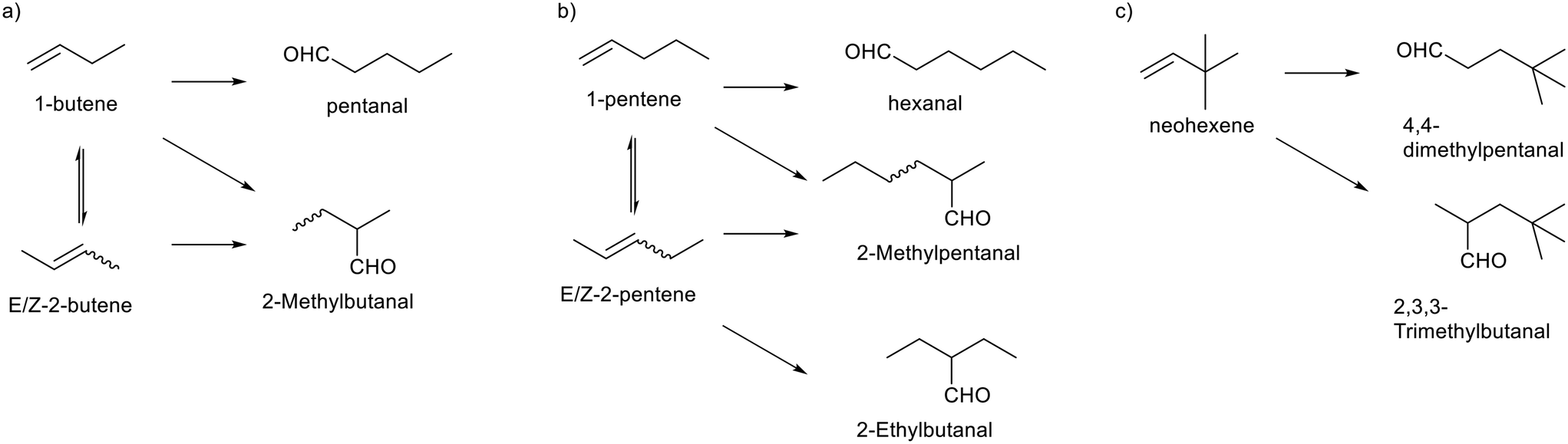
| Fig. 2 Concentration profiles of butene isomers and n-pentanal obtained for the 1-butene hydroformylation experiment. a) Absolute concentrations versus time and b) relative molar fractions of the butene isomers over time. Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, [1-butene] = 1 M, t = 8 h, p(H2) = 1.0 MPa, p(CO) = 1.0 MPa, ϑ = 90 °C, solvent: cyclohexane. | ||
The momentary content of each isomer is related to its consumption via hydroformylation. Thus, the reactivity relation is 1-butene > Z-2butene > E-2-butene, which is in accordance with their steric properties. The composition of n-butenes deviates from the thermodynamic equilibrium composition.35 The molar fraction of 1-butene after the initial conversion (10–15 min) was below the detection limit and thus lower than the equilibrium value of 0.04. Within the conversion range from 0.10–0.95 the value of Z-2-butene decreased from 0.4 to 0.2 (0.26 equilibrium), while the molar fraction of E-2-butene increased from 0.6 to 0.8 (0.69 equilibrium). It is important to note that the absolute molar concentrations of the butenes are very low at higher conversions. It can be concluded that the isomerization activity is not high enough to equilibrate the n-butenes. Formally such type of reaction system can be classified as coupled parallel reaction.36
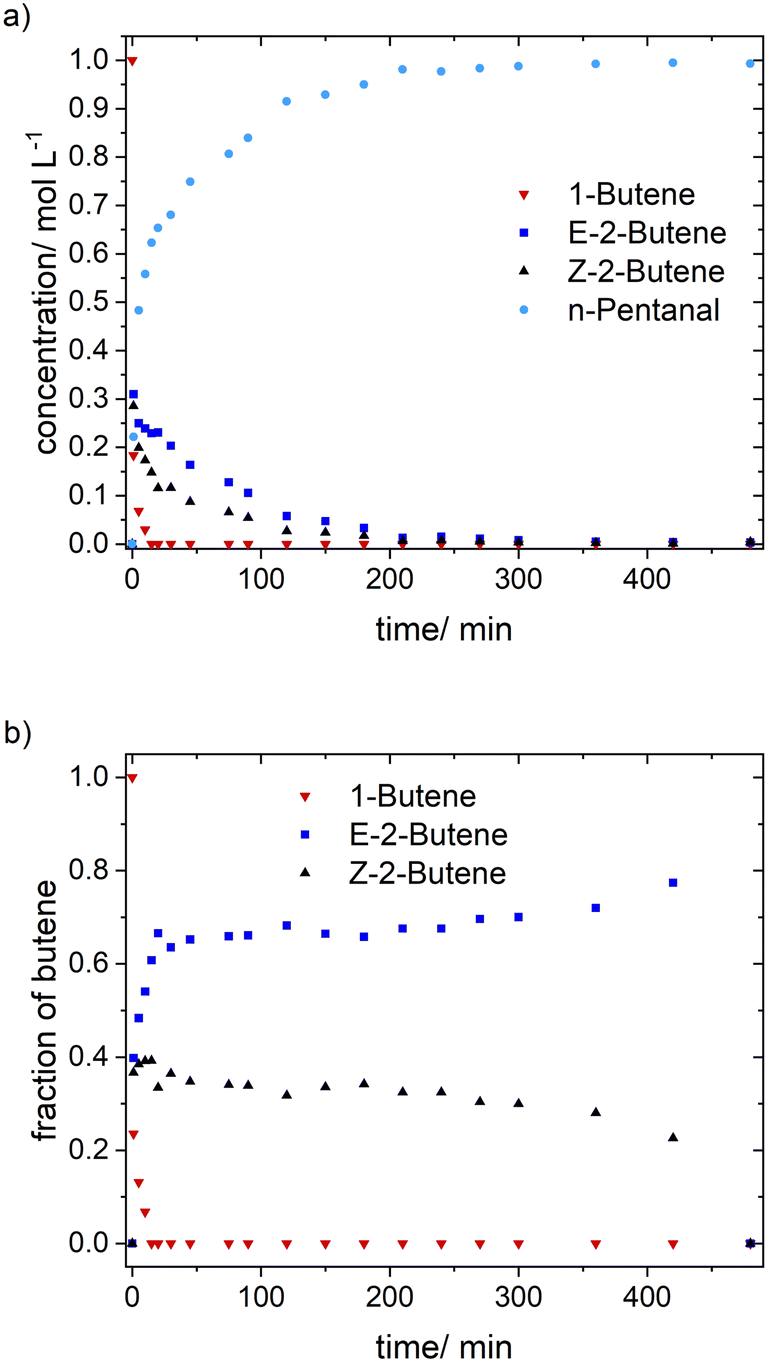
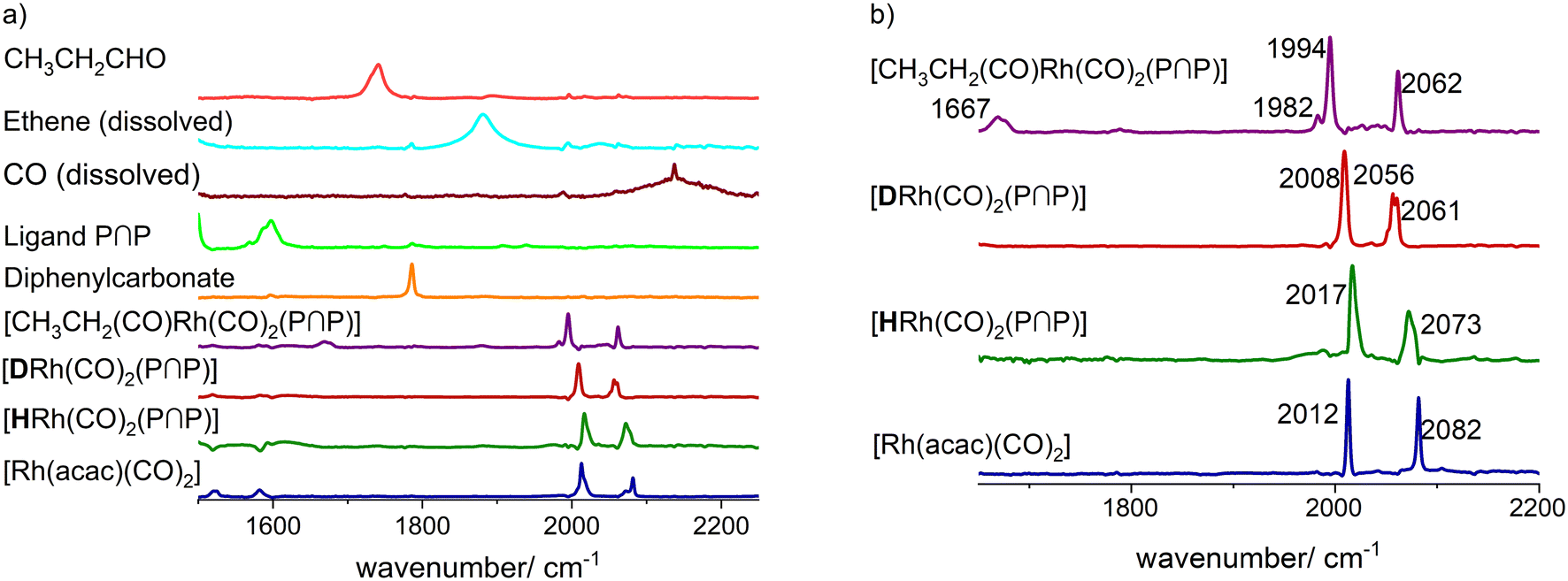
Based on the in situ FTIR spectroscopic monitoring the hydrido complex [HRh(CO)2(P∩P)] (ν(CO) = 2017, 2073 cm−1) was the only detectable rhodium species over the entire conversion range in accordance with other studies on hydroformylation with diphosphite modified rhodium catalysts (Fig. 3).6,21,37
 | ||
| Fig. 3 In situ FTIR-spectra measured during 1-butene hydroformylation. Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, [1-butene] = 1 M, t = 8 h, p(H2) = 1.0 MPa, p(CO) = 1.0 MPa, ϑ = 90 °C, solvent: cyclohexane. | ||
:E = 0.29:0.71) showed a similar behavior regarding the composition of n-pentene molar fractions during the reaction compared with the butene experiments. In both cases the fraction of 1-pentene (0.02) was only slightly lower than the value for the equilibrium composition (0.03). The fraction of the less reactive E-2-pentene increased to 0.8 (0.63 equilibrium) over the observed time while for the more reactive Z-2-pentene dropped to a value of 0.2 (equilibrium 0.33). Simultaneous in situ spectroscopic monitoring revealed that only the hydrido complex was detected over the entire conversion range. Figures of the acquired experimental data are given in ESI-C.†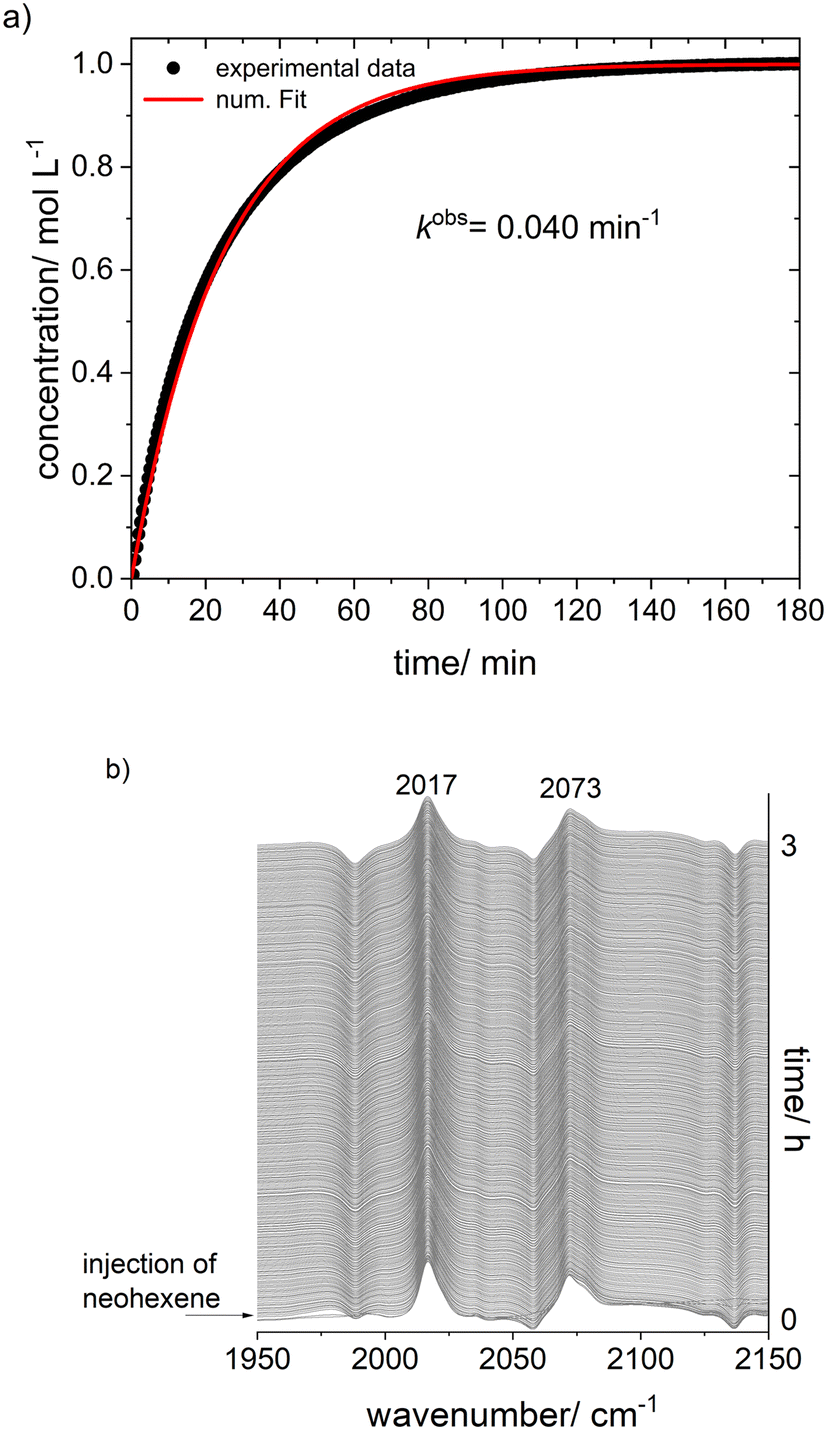 | ||
| Fig. 4 Neohexene hydroformylation using Rh/BiPhePhos catalyst. a) Concentration profile of 4,4-dimethyl-1-butene formation, b) in situ FTIR-spectra of the carbonyl region. Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, [neohexene] = 1 M, t = 3 h, p(H2) = 1.0 MPa, p(CO) = 1.0 MPa, ϑ = 90 °C, solvent: cyclohexane. | ||
2.2 Component identification based on experiment design and peak group analysis (PGA) of spectroscopic data
In this section the results from a single in situ FTIR experiment with stepwise additions of reactants aimed for the identification of all relevant components of the reaction system consisting of rhodium complexes and organic compounds are discussed. The sequential dosage of reactants at distinct intervals under continuous measurement of time-resolved infrared spectra can lead to data sets suitable for the extraction of pure component spectra using chemometric tools such as the peak group analysis (PGA).38,39 Scheme 3 gives an overview about the dosage sequence used in this experiment and in Fig. 5 the entire data set of collected infrared spectra are displayed.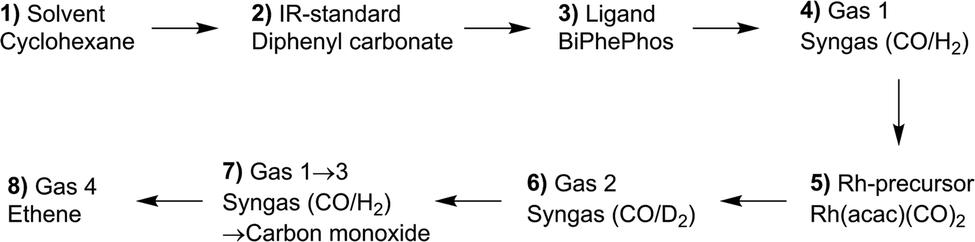 | ||
| Scheme 3 Sequence for the stepwise dosage experiment. Time interval between the steps was 10 min. Further reaction conditions: ϑ = 50 °C, [Rh] = 1 mM, [P∩P] = 1 mM, [DPC] = 1 mM, P(syngas) = 2.0 MPa, p(ethene) = 1.0 MPa. | ||
 | ||
| Fig. 5 In situ spectroscopic IR-data of a sequential dosage experiment. Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, [DPC] = 2 mM, p(H2/D2) = 1.0 MPa, p(CO) = 1.0 MPa, p(C2H4) = 1.0 MPa, ϑ = 50 °C, solvent: cyclohexane. | ||
The solvent cyclohexane, stock solutions of diphenyl carbonate as internal IR standard and BiPhePhos were injected one after the other into the reactor system (ESI-D†). Infrared spectra were registered continuously in each step within a time interval of 10 min. In the next step the reactor was charged with 2.0 MPa synthesis gas (CO/H2 = 1:1) after which the stock solution of Rh precursor [Rh(acac)(CO)2] was dosed using a syringe pump. This initiated the preformation of the hydride complex [HRh(CO)2(P∩P)] which was completed after ca. 60 min. Then, a gas-exchange for 1.0 MPa deuterium and 1.0 MPa carbon monoxide was performed to form the corresponding deuteride complex. After a repeated gas-exchange to hydrogen/carbon monoxide and then pure CO, the system was pressurized with 1.0 MPa ethene to generate acyl complexes. Due to the small size of ethene the formation of observable acyl complexes is favored under pure CO conditions.
The entire infrared spectroscopic data set was processed by the peak group analysis tool (PGA) from the FACPACK package.38–40 Peak group analysis (PGA) is a chemometric approach using Multivariate Curve Resolution (MCR) techniques to extract pure component spectra from spectral mixture data. PGA works incrementally, starting with a single peak for which it constructs the associated pure component spectrum that reproduces that peak. This process is repeated for all dominant peaks in the measured data. The resulting series of potential pure component spectra is subjected to correlation analysis to identify groups of very similar spectra. Ideally, the resulting distinct groups of spectra can be related to the pure component spectra of the reactive species.
The extracted pure component spectra are presented in Fig. 6a. There are only very minor artefacts present in the spectrum. Due to the sequential dosage experiment design linear dependencies in the data matrix related to spectra and/or concentration profiles can often be removed to a significant extent. In Fig. 6b the obtained spectra of the different rhodium complexes in an enlarged size are displayed showing detailed spectral features of the distinct components. The interpretation of the vibrational patterns and assignments to molecular structures are discussed in the following sections.
 | ||
| Fig. 6 a) Extracted pure component spectra from the PGA processing of collected in situ spectroscopic IR-data of a sequential dosage experiment, b) selection of extracted spectra for rhodium carbonyl complexes (enlarged view). Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, [DPC] = 2 mM, p(H2/D2) = 1.0 MPa, p(CO) = 1.0 MPa, p(C2H4) = 1.0 MPa, ϑ = 50 °C, solvent: cyclohexane. | ||
2.3 Spectroscopic characterization of [Rh(acac)(P∩P)]
The treatment of the rhodium precursor [Rh(acac)(CO)2] with one equivalent of BiPhePhos ligand forms the respective complex [Rh(acac)(P∩P)] under the evolution of gaseous carbon monoxide (Scheme 4). This is caused by the chelating coordination of the phosphorus atoms of BiPhePhos and substitution of the carbonyl ligands. Depending on the conditions, e.g. rhodium concentration and ligand-to-metal ratio, a certain amount of the monocarbonyl [Rh(acac)(CO)(κ1-P∩P)] can also be formed (see further below). | ||
| Scheme 4 Formation of [Rh(acac)(P∩P)] starting from [Rh(acac)(CO)2] and BiPhePhos (P∩P). The monocarbonyl complex [Rh(acac)(CO)(κ1-P∩P)] can be formed which highly depends on the reaction conditions. | ||
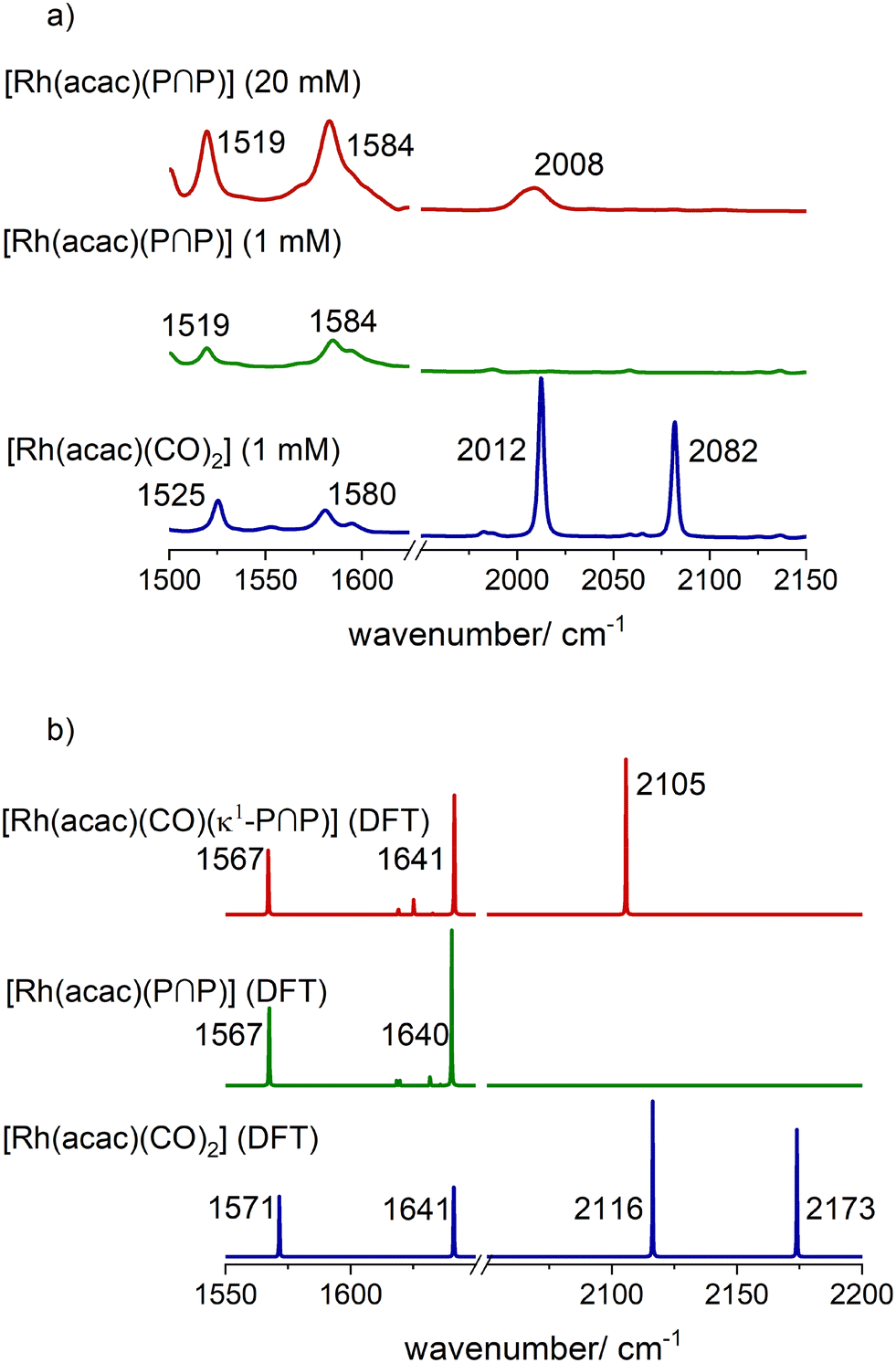
The infrared spectroscopic monitoring of this substitution reaction at [Rh] = 1 mM and [P∩P]/[Rh] = 1 shows the complete vanishing of the vibrational bands for the carbonyl ligands of [Rh(acac)(CO)2] (ν(CO) = 2012, 2082 cm−1) (Fig. 7a). Another observation is a slight shift of the positions for the vibrational bands of the acetylacetonate ligand (acac−): [Rh(acac)(CO)2] (ν(acac) = 1528, 1582 cm−1) → [Rh(acac)(P∩P)] (ν(acac) = 1520, 1584 cm−1). Such a shift is also in line with DFT-based vibrational spectra (Fig. 7b) and is characteristic for the coordination of phosphite ligands to the precursor.
 | ||
| Fig. 7 a) In situ IR-spectra of [Rh(acac)(CO)2] and [Rh(acac)(P∩P)] and b) DFT-based vibrational spectra (ESI-I†). Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, p(Ar) = 0.1 MPa, ϑ = 30 °C, solvent: cyclohexane. | ||
At higher rhodium concentrations ([Rh] = 20 mM) a band at ν(CO) = 2008 cm−1 with low intensity was observed which indicates the presence of [Rh(acac)(CO)(κ1-P∩P)] at comparatively low concentrations (Fig. 7a and b).
NMR spectroscopy on solutions with a rhodium concentration of 20 mM ([P∩P]/[Rh] = 1) allowed for the detection of the characteristic 31P doublet signals at δ(31P) = 146.3 ppm (d, 1JP,Rh = 293 Hz) for [Rh(acac)(P∩P)] as the major complex and δ(31P) = 140.3 Hz (d, 1JP,Rh = 283 Hz) for the minor monocarbonyl complex [Rh(acac)(CO)(κ1-P∩P)] with ca. 6% of the signal intensity (Fig. 8).17,41 The signal for the uncoordinated phosphite unit is expected to resonate between ca. 146–150 ppm which overlaps with the signal of [Rh(acac)(P∩P)]. The integral values for both parts of the doublet signal centered at 146.3 ppm are not identical and the difference compares approximately to the integral value of the signal at 140.3 ppm (ESI-B†). In addition, a 31P{1H}–103Rh-HMBC spectrum was measured which gave a single 103Rh-NMR signal at δ(103Rh) = 140 ppm for [Rh(acac)(P∩P)] which indicates that only one rhodium atom is coordinated in the complex. Further signals were observed at δ(103Rh) = 223 ppm for [Rh(acac)(CO)(κ1-P∩P)] and δ(103Rh) = 215 ppm for a low amount of the precursor [Rh(acac)(CO)2].
 | ||
| Fig. 8 a) 31P{1H}-NMR-spectrum of [Rh(acac)(P∩P)] and b) 31P{1H}–103Rh-NMR of [Rh(acac)(P∩P)]. Conditions: [Rh] = 20 mM, [P∩P] = 20 mM, p(Ar) = 0.1 MPa, ϑ = 25 °C, solvent: toluene-d8; spectrum b) was acquired at ϑ = 30 °C. | ||
2.4 In situ formation of [HRh(CO)2(P∩P)]
The treatment of [Rh(acac)(P∩P)] ([Rh] = 1 mM) with synthesis gas (H2/CO, 2.0 MPa) at ϑ = 50 °C to generate a corresponding hydrido complex (Scheme 5) was investigated by in situ FTIR spectroscopy. After the addition of synthesis gas the bands assigned to the dicarbonyl complex [Rh(acac)(CO)2] (ν(CO) = 2012, 2082 cm−1) appeared immediately followed by an ongoing increase of bands assigned to [HRh(CO)2(P∩P)] (ν(CO) = 2017, 2073 cm−1) (Fig. 9). The reaction was complete after ca. 50 min. A rapid partial substitution of the phosphorus atoms of the coordinated ligand by carbon monoxide at elevated pressures is a known behaviour for many diphosphites. The generation of acetylacetone in course of the hydrogenolysis of [Rh(acac)(CO)2] could not be observed clearly by IR-spectroscopy at this concentration.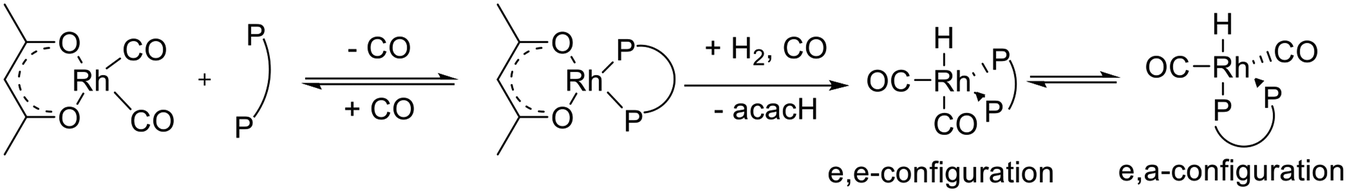 | ||
| Scheme 5 Reaction of [Rh(acac)(CO)2] with BiPhePhos (P∩P) towards possible stereoisomeric hydrido complexes. | ||
 | ||
| Fig. 9 a) In situ IR-spectra from the catalyst formation in dependency of the time; b) concentration profiles and c) pure component spectra from the peak group analysis (PGA) of IR-spectroscopic data. Conditions: [Rh] = 1 mM, [P∩P] = 1 mmol, p(CO) = 1.0 MPa, p(H2) = 1.0 MPa, ϑ = 50 °C, solvent: cyclohexane. | ||
For the elucidation of the coordination mode of the P-atoms in the hydrido dicarbonyl complex an in situ IR experiment with isotopic labeling (H/D-exchange) was performed (Fig. 10a). For further assignments of the IR-bands to vibrational modes DFT calculations based on trigonal-bipyramidal hydrido complex structures in which the hydride ligand is located in the axial position with bis-equatorial and equatorial–axial coordination of the P-ligand have been conducted (Fig. 10b and c). A shift of the respective IR-bands in the experimental spectrum of the deuterido complex to lower wavenumbers have been observed: [DRh(CO)2(P∩P)] (ν(CO) = 2009, 2058 cm−1). Such a band shift is in agreement with a bis-equatorial coordination of the P-atoms.17,42–44 Since there is a H–Rh–CO trans-arrangement in the e,e-[HRh(CO)2(P∩P)] complex the isotopic labeling with deuterium affects the corresponding combined vibrational modes (vibrational modes for an idealized geometry e,e-[HM(CO)2L2] ((Cs) → 2A′)). This is proven by the vibrational mode analysis based on DFT calculations. In the stereoisomer e,a-[HRh(CO)2(P∩P)] there are only H–Rh–CO cis-arrangements, therefore an H/D-exchange does not lead to any shift of respective carbonyl bands in the infrared spectrum (vibrational modes for an idealized geometry e,a-[HM(CO)2L2] ((Cs) → A′′ + A′)). The observed preferred formation of the e,e-[HRh(CO)2(P∩P)] stereoisomer is also in agreement with relative energies calculated by DFT computations (ESI-J†).
 | ||
| Fig. 10 a) IR spectroscopic data from H/D-exchange experiment on [H/DRh(CO)2(P∩P)] conditions: [Rh] = 1 mM, [P∩P] = 1 mM, p(CO) = 1.0 MPa, p(H2/D2) = 1.0 MPa and DFT-based IR-spectra (ESI-I†) of the H/D-exchange for b) e,e-complex and c) e,a-coordination of the ligand. Colours: black: hydrido, blue: deuteride complex. | ||
The results from an NMR-spectroscopic characterization of the hydrido complex in solution are in good accordance with the IR-spectroscopic data. Sample preparation was conducted in a 10 mL stainless steel cylinder with following conditions: [Rh] = 20 mM, [P∩P] = 20 mM, p(syngas) = 2.0 MPa, ϑ = 50 °C, t = 2 h in toluene-d8. The solution was purged with Ar and transferred after the preformation into a Young-NMR tube and measured at ϑ = 25 °C. The stability of the hydrido complex at one atmosphere of the mixture of hydrogen and carbon monoxide has been tested in a previous control experiment and is discussed further below. In the 1H-NMR spectrum a characteristic signal in the hydride region at δ(1H) = −10.6 ppm (dd, 1JH,P = 8.3 Hz, 1JH,Rh = 3.5 Hz) is observed (Fig. 11a). The small value of the coupling constant 1JH,P = 8.3 Hz indicates a cis-phosphorus-hydride coordination with only a slight distortion of the tbp complex.6,17,41,44–46 A 1H–103Rh-HMBC experiment was also conducted giving a signal at δ(103Rh) = −1146 ppm and the respective cross signals (Fig. 11b). The 31P{1H}-NMR spectrum reveals a doublet signal at δ(31P) = 174.0 ppm (d, 1JP,Rh = 237 Hz) and the 31P{1H}–103Rh-HMBC spectrum confirms the presence of a mononuclear Rh(I) complex (Fig. 12). 13C{1H}-NMR spectroscopy was performed on a 13CO-labeled (labeling degree 13CO: 50%) sample of the dissolved hydrido complex with [Rh] = 20 mM, [Rh]/[P∩P] = 1. We assigned the signals at room temperature (297 K) to an averaged doublet of triplet resonance δ(13C) = 194.6 ppm (dt, 1JC,Rh = 58.8 Hz, 2JC,P = 15.9 Hz) which corresponds to both carbonyl ligands (Fig. 13). Cooling to −60 °C (213 K) led to signal broadening. Thus, in the investigated temperature range the positions of the two carbonyl ligands are not fixed and their coordination modes could not be distinguished. Isotope effects on the shifts of 1H, 31P and 103Rh due to the 13CO labelling were detected (ESI-E†). Five-coordinate trigonal bipyramidal complexes of the type [HRh(CO)2(P∩P)] often show fluxional behavior. Intramolecular rearrangement processes are described in the literature.46–50 With respect to the carbonyl ligands also intermolecular exchange between coordinated and uncoordinated carbon monoxide might be involved.51,52 Based on the present data, no discrimination of the exact exchange mechanism is possible. A series of additional experiments would be required which go beyond the framework of the present study.
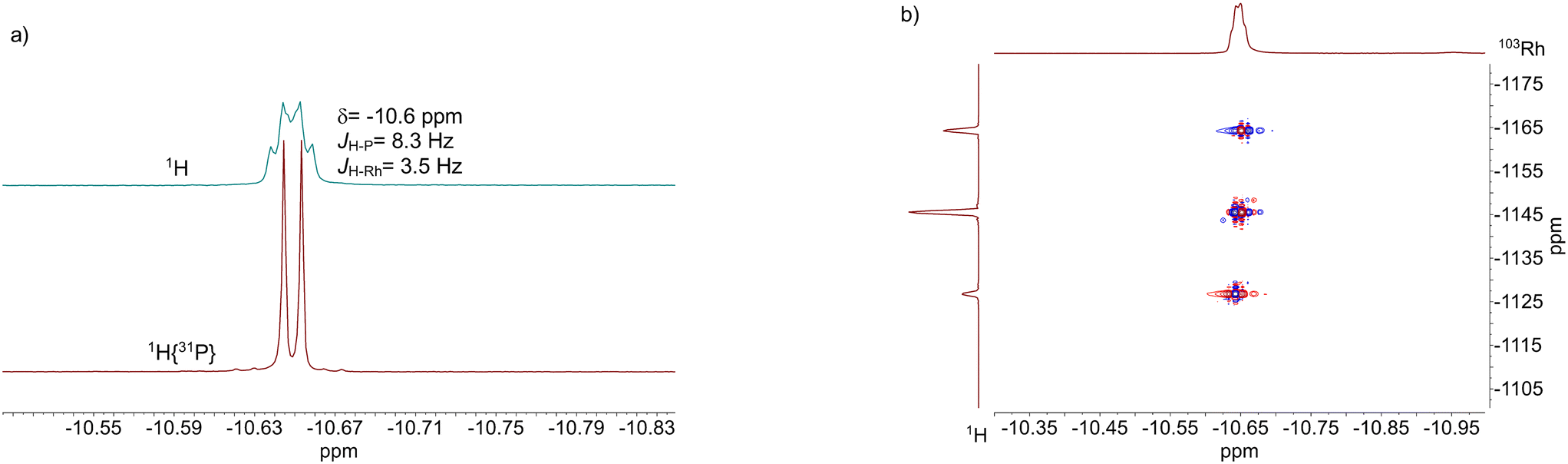 | ||
| Fig. 11 a) 1H-NMR and 1H{31P}-NMR and b) 1H–103Rh-HMBC spectrum of [HRh(CO)2(P∩P)]. Conditions: [Rh] = 20 mM, [P∩P] = 20 mM, p(Ar) = 0.1 MPa, ϑ = 25 °C, solvent: toluene-d8. | ||
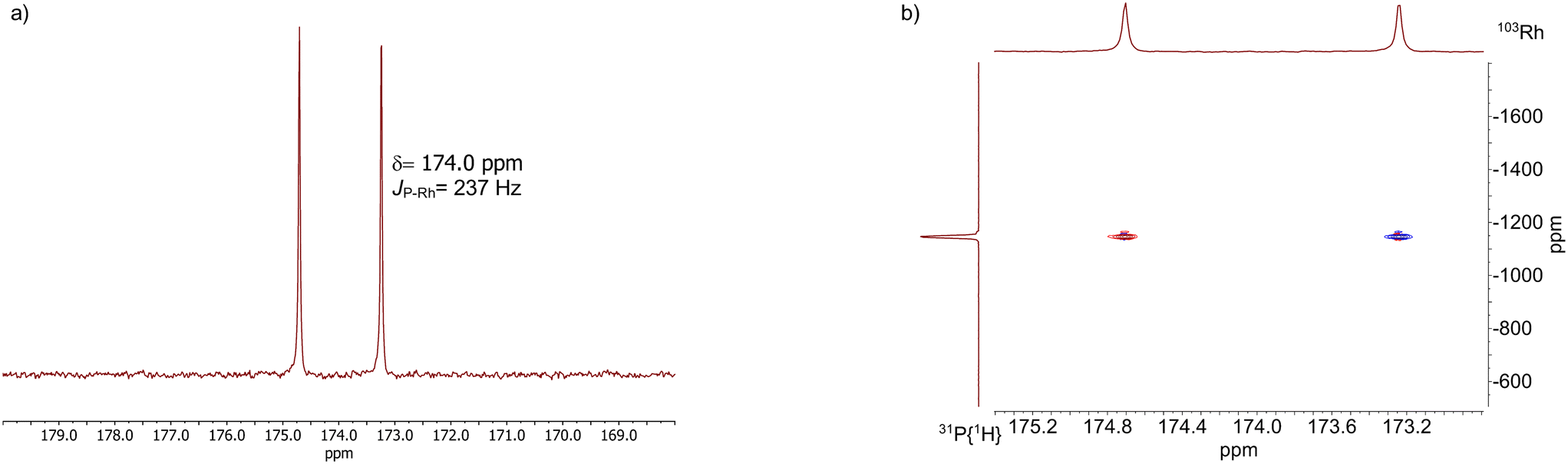 | ||
| Fig. 12 a) 31P{1H}-NMR of [HRh(CO)2(P∩P)] and b) 31P{1H}–103Rh-HMBC spectrum (no decoupling) of [HRh(CO)2(P∩P)]. Conditions: [Rh] = 20 mM, [P∩P] = 20 mM, p(CO) = 0.05 MPa, p(H2) = 0.05 MPa, ϑ = 25 °C, solvent: toluene-d8. | ||
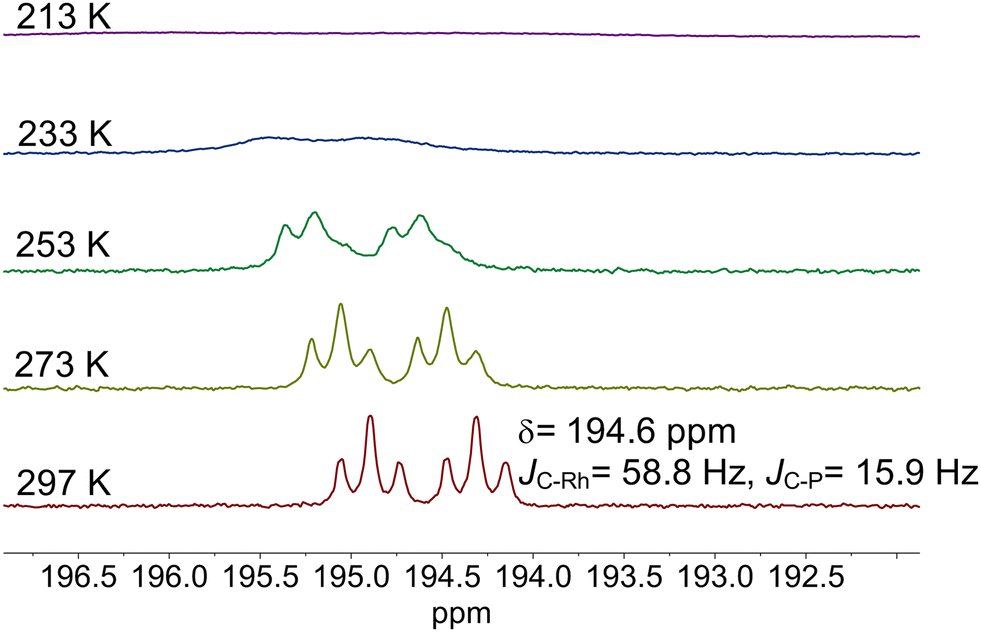 | ||
| Fig. 13 13C{1H}-NMR spectra of [HRh(CO)2(P∩P)] after isotopic labeling with 50% 13CO under variation of the temperature. Conditions: [Rh] = 20 mM, [P∩P] = 20 mM, p(12CO) = 0.025 MPa, p(13CO) = 0.025 MPa, p(H2) = 0.05 MPa, solvent: toluene-d8. | ||
2.5 Stability aspects of [HRh(CO)2(P∩P)]
Catalyst stability is a crucial issue with respect to industrial applicability and economic efficiency of a catalytic system.5,23,53,54 For diphosphite modified rhodium complexes as hydroformylation catalysts the formation of dimers and polynuclear clusters might take place at low partial pressures of hydrogen and carbon monoxide.To study the behaviour of [HRh(CO)2(P∩P)] in the absence of hydrogen and carbon monoxide, we performed an in situ IR experiment under pure inert gas at 90 °C for 9 h. The hydrido complex was generated at [Rh] = 1 mM and [P∩P]/[Rh] = 1 during the treatment with synthesis gas at 50 °C for 1 h. Then, the synthesis gas was thoroughly purged with argon (5 times gas exchange from 1.0 MPa argon → 0.1 MPa). The solution was monitored by IR-spectroscopy at 90 °C and 1.0 MPa of argon. There were no alterations in the infrared spectra within the timespan of 9 h (Fig. 14), which indicates an intrinsically good stability of the hydrido complex under inert atmospheres depleted of hydrogen and carbon monoxide.
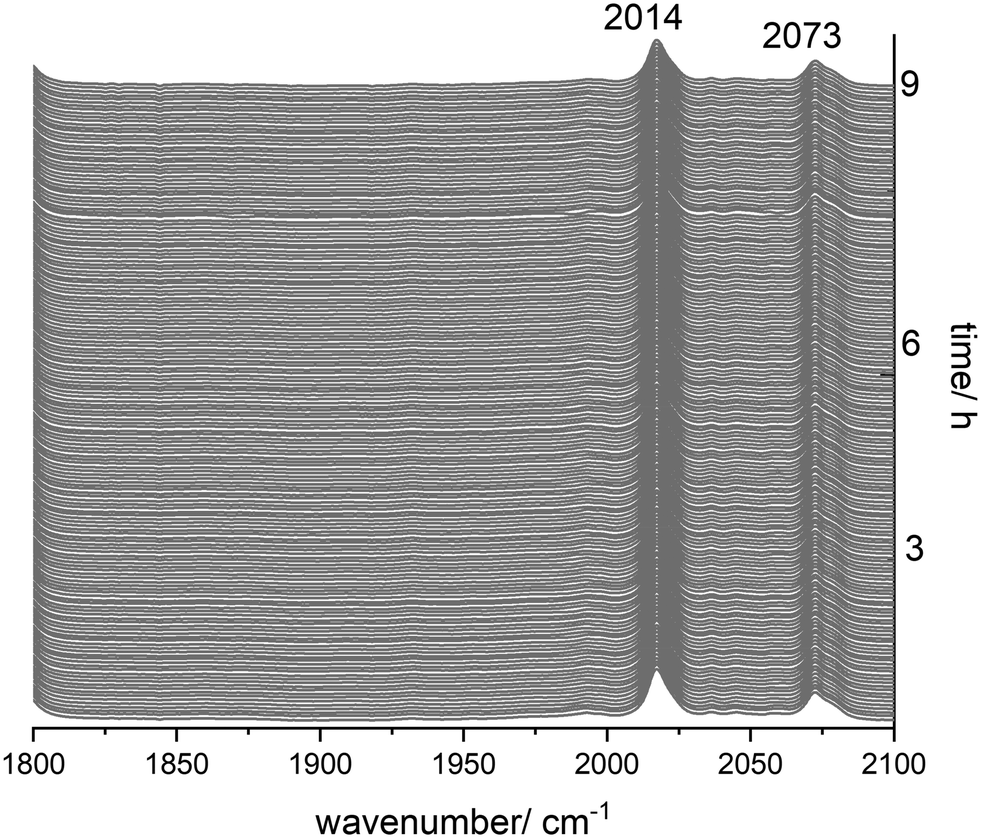 | ||
| Fig. 14 In situ IR-spectroscopic data from the stability test of [HRh(CO)2(P∩P)] under an Ar atmosphere. Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, p(Ar) = 1.0 MPa, ϑ = 90 °C, solvent: cyclohexane. | ||
Further experiments have been performed on a solution of the hydrido complex at higher rhodium concentrations ([Rh] = 20 mM and [P∩P]/[Rh] = 1, solvent: toluene-d8) analyzed by NMR-spectroscopy. After the formation of the hydrido complex at 100 °C and 2.0 MPa of synthesis gas, we released the pressure to 0.1 MPa and transferred the sample into the NMR tube. The 31P{1H}-NMR spectrum displayed the doublet signal characteristic for [HRh(CO)2(P∩P)] at δ(31P) = 174.0 ppm (d, 1JP,Rh = 237 Hz) without any additional signals (Fig. 15).
 | ||
| Fig. 15 31P{1H}-spectra of [HRh(CO)2(PP)] under syngas, CO and Ar at varying temperatures. Further conditions: [Rh] = 20 mM, [P∩P] = 20 mM, p = 0.1 MPa, solvent: toluene-d8. | ||
The removal of dissolved hydrogen by pure CO did not lead to any changes in the NMR spectrum. However, after the treatment under 1.0 MPa of carbon monoxide at 100 °C tiny signals at δ(31P) = 170.0 ppm (m, 1JP,Rh = 251 Hz) and δ(31P) = 166.8 ppm (m, 1JP,Rh = 259 Hz) appeared indicating the formation of isomeric forms of a dinuclear complex of the type [Rh2(CO)4(P∩P)2].17 The intensity of these signals further increased after the solution was further treated in inert gas (argon) at 125 °C for 2 h. In addition, a doublet signal at δ(31P) = 146.2 ppm (d, 1JP,Rh = 293 Hz) was detected which belongs to [Rh(acac)(P∩P)]. In total, ca. 9% of the signal intensity in the 31P-NMR spectrum correspond to the dinuclear complexes and ca. 6% to the precatalyst complex.
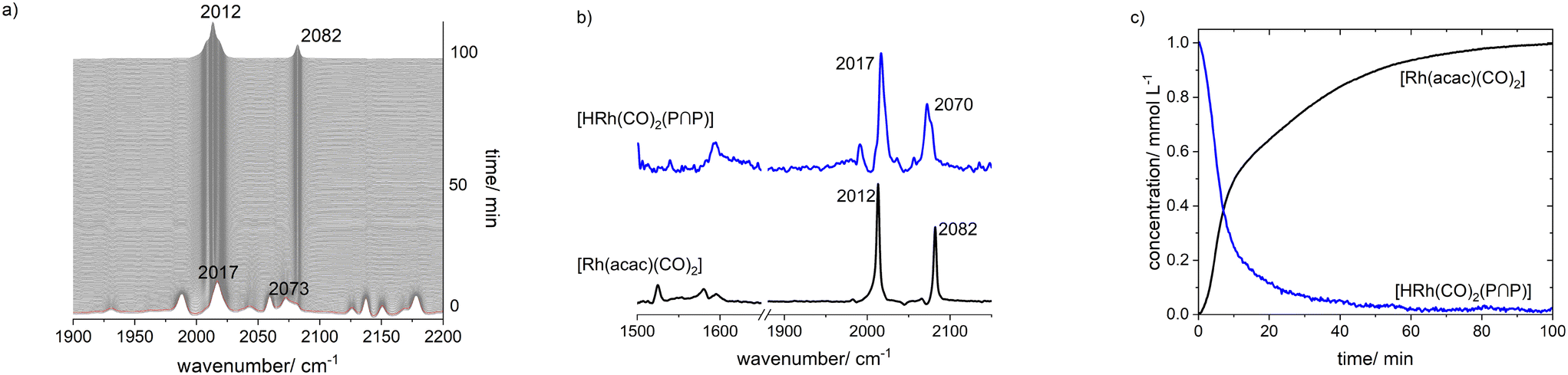 | ||
| Fig. 16 In situ IR-spectra series of the reaction of [HRh(CO)2(P∩P)] with air, b) pure component spectra extracted from a), c) corresponding relative absorbance profiles extracted with the peak group analysis. Conditions: [Rh] = 1 mM, [P∩P] = 1 mM, p(air) = 0.3 MPa, ϑ = 90 °C, t = 90 min, solvent: cyclohexane. | ||
Based on quantitative 31P-NMR spectroscopic data (Fig. 17) both samples showed a slight decrease in the fraction of the total 31P-integral of each component (0.97 initial value) directly after the addition of water: 0.87 (hydrido complex) and 0.94 (BiPhePhos).
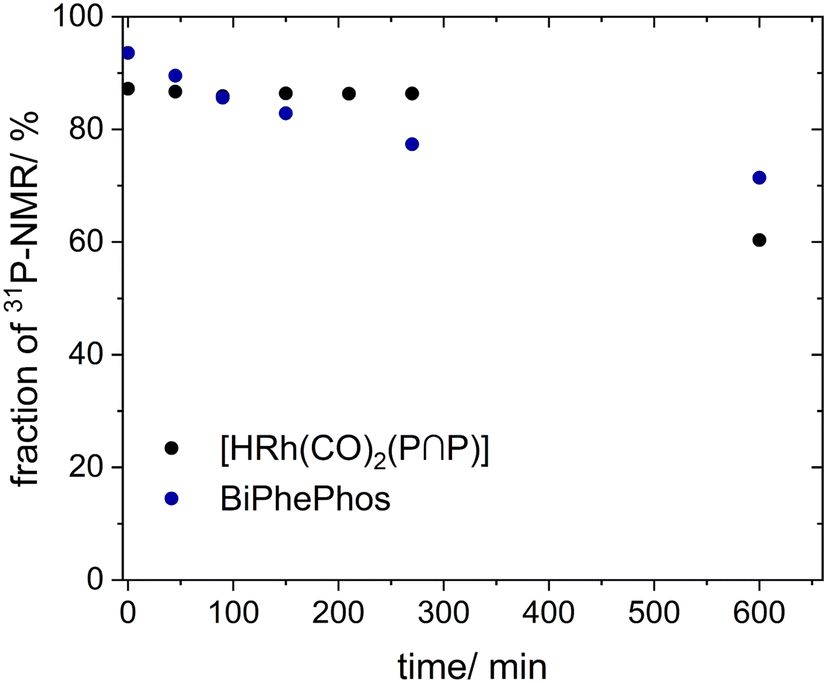 | ||
| Fig. 17 Hydrolysis control test. Integral fractions for [HRh(CO)2(P∩P)] (black) and BiPhePhos (blue) based on quant. 31P{1H}-spectra. Further conditions: [Rh] = 20 mM, [P∩P] = 20 mM, [H2O]/[P∩P] = 20, p = 0.1 MPa, solvent: 1,4-dioxane. | ||
While the fraction of [HRh(CO)2(P∩P)] remained almost constant throughout the course of the first 270 min, the fraction of BiPhePhos sample showed a succeeding decrease to 0.77 after 270 min. Longer treatments until 600 min led to an accelerated decomposition of [HRh(CO)2(P∩P)] (0.60) which might indicate an autocatalytic pathway.56,57 Such an enhanced decay was not clearly observed for BiPhePhos (0.71) within the examined time span.
2.6 Influence of the ligand-to-metal ratio on the composition of hydrido complexes
The bulkiness of a chelating diphosphite such as BiPhePhos has a tremendous impact on the molecular structure and catalytic properties of the corresponding hydrido complex. As it is outlined in this study, the P-atoms of the ligand BiPhePhos coordinate in a bis-equatorial mode in e,e-[HRh(CO)2(P∩P)] for ([P∩P]/[Rh] = 1). The coordination chemistry at larger ligand-to-metal ratios has not been described in detail in the literature so far, thus we performed comparative in situ infrared and NMR experiments for [P∩P]/[Rh] = 1, 1.5 and 2.A solution of [HRh(CO)2(P∩P)] ([Rh] = 1 mM) was prepared in the presence of the respective amount of BiPhePhos at p(CO/H2) = 2.0 MPa and ϑ = 90 °C. It is to be noticed that at elevated pressures of synthesis gas (2.0 MPa) only the infrared bands assigned to the hydrido complex were identified (Fig. 18a). During repeated gas exchange cycles (up to 12 times) with pure hydrogen, an additional band at ν(CO) = 2045 cm−1 increased until its intensity reached a constant value (Fig. 18b). As expected, the molar fraction of the putative monocarbonyl complex seems to be higher for [P∩P]/[Rh] = 2. The latter experiment was repeated for [Rh] = 20 mM in toluene-d8 (Fig. 18c) which was also used for NMR-measurements. Based on the IR-spectra the trends with respect to the population of the mononuclear complex was similar when comparing the two concentration levels of rhodium.
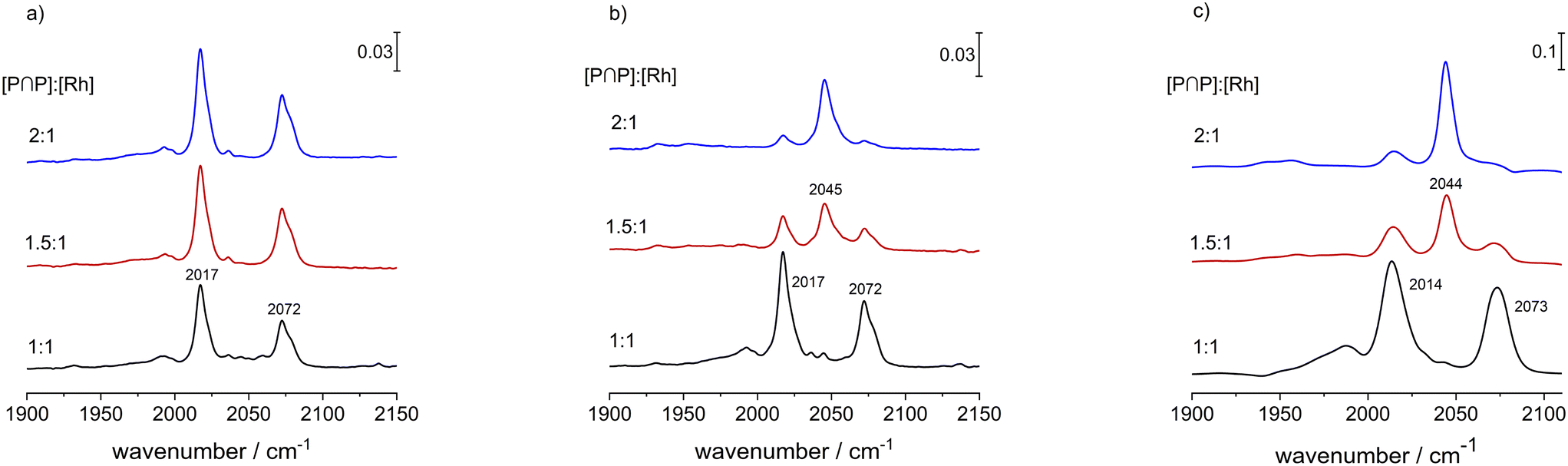 | ||
| Fig. 18 Impact of ligand-to-metal ratio on the composition of hydrido complexes. a) [Rh] = 1 mM, ϑ = 90 °C, p(CO/H2) = 2.0 MPa, solvent: cyclohexane; b) [Rh] = 1 mM, ϑ = 90 °C, p(H2) = 1.0 MPa after repeated gas exchange cycles, solvent: cyclohexane; c) [Rh] = 20 mM, ϑ = 25 °C, p(H2) = 0.1 MPa after repeated gas exchange cycles, solvent: toluene-d8. | ||
1H{31P}- and 31P{1H}-NMR spectra revealed that depending on the ligand-to-metal ratio different types of hydrido complexes were formed (Fig. 19). For [P∩P]/[Rh] = 1.5 besides the typical doublet signal for [HRh(CO)2(P∩P)] (δ(1H) = −10.6 ppm) several additional resonances in the hydride region of the 1H{31P}-NMR spectrum were present. In the 31P{1H}-NMR spectrum in addition to the doublet signal for the hydrido dicarbonyl complex (δ(31P) = 174.0 ppm) very complex coupled multiplet patterns for coordinated phosphite moieties were observed. But the spectra were without any signals in the region of uncoordinated P-atoms. Together with the results from IR-spectroscopy in which the formation of a monocarbonyl complex was observed, the spectroscopic data would be in agreement with a ligand bridging dinuclear complex of the type [HRh(CO)(P∩P)]2(P∩P) (Scheme 6).58,59 Interestingly, for [P∩P]/[Rh] = 2 new additional hydride signals were detected in the proton NMR spectrum. In the 31P-NMR spectrum also a complex multiplet pattern in the region of coordinated P-atoms was seen with additional signals in the regions of non-coordinated phosphite ligands. The normalized integral values of the signals in both frequency regions are 2:1. It was also observed in the 1H and 31P-NMR spectra that the relative signal intensities for [HRh(CO)2(P∩P)] decreased considerably. We conclude from these findings the formation of a mononuclear complex of the type [HRh(CO)(P∩P)(κ1-P∩P)] with one non-coordinated phosphorus atom (Scheme 6).60–63 The coupled multiplets result from magnetic inequivalent P-atoms and atropisomerism.45,64–66 A 1H{31P}–103Rh HMQC spectrum (Fig. 20) of this sample illustrates the involvement of several isomeric complexes. A conclusive structural assessment was impossible, but the similarity of the rhodium shifts (Fig. 20) makes a very similar coordination sphere likely.
 | ||
| Fig. 19 a) 1H{31P}-NMR and b) 31P{1H}-NMR spectra of rhodium hydride complexes in H2 atmosphere under varation of the ligand concentration (conditions: [Rh] = 20 mM, p(H2) = 0.1 MPa, ϑ = 25 °C, solvent: toluene-d8). | ||
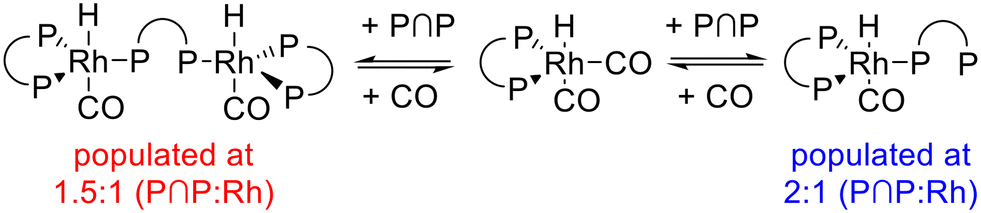 | ||
| Scheme 6 Reaction of [HRh(CO)2(P∩P)] with [P∩P] (BiPhePhos) in CO depleted atmosphere. | ||
 | ||
| Fig. 20 1H{31P}–103Rh-HMQC spectrum of stereoisomeric mixture of [HRh(CO)(P∩P)(κ1-P∩P)] type complex. Conditions: [Rh] = 20 mM, [P∩P]/[Rh] = 2, p(H2) = 0.1 MPa, ϑ = 25 °C, solvent: toluene-d8. Most correlation signals appear quartet-like along F1 as they represent RhP3 species, and the 103Rh–31P coupling is observed in this dimension. Note: the triplet signal at −1146 ppm is due to [HRh(CO)2(P∩P)]. | ||
In the next step, we conducted a H/D-isotope exchange experiment and a respective vibrational mode analysis based on DFT calculations for [HRh(CO)(P∩P)(κ1-P∩P)] to confirm the coordination mode of the phosphorus centers (Fig. 21) in the hydrido complex. For the deuterido complex a shift towards lower wavenumbers (ν(CO) = 2027 cm−1) was observed, which is in accordance with a tris-equatorial coordination of the three phosphite units.
 | ||
| Fig. 21 a) Infrared spectra of the hydrido and deuterido monocarbonyl complex of the type [HRh(CO)(P∩P)(κ1-P∩P)] for [Rh] = 1 mM and [P∩P] = 2 mM at p(H2/D2) = 0.1 MPa, ϑ = 25 °C, solvent: cyclohexane; b) calculated vibrational spectra via DFT methods (ESI-I†) based on the molecular structures with trisequatorial P-coordination. | ||
The formation of e,e,e-[HRh(CO)(P∩P)(κ1-P∩P)] with all three P-atoms coordinated in the equatorial position is likely due to sterical reasons. There are examples of e,e,e-[HRh(CO)(P∩P)L] complexes ((P∩P) = diphosphite, L = PPh3, PPh2R) with this type of coordination behavior of the P-ligands in the literature.45,64 We prepared these type of complexes within the framework of this work with (L = PPh3 and P(OPh)3) and 31P{1H}- and the 1H-NMR spectra indicated a trisequatorial P-coordination (ESI-G†).
2.7 Formation and characterization of acyl complexes of the type [RC(O)Rh(CO)2(P∩P)]
For the Rh/BiPhePhos system under catalytic reaction conditions the only detectable resting state complex is the hydrido complex [HRh(CO)2(P∩P)]. This observation can be rationalized when considering the rate control in the catalytic cycle. In the literature on hydroformylation catalysts modified with bidentate diphosphites, kinetic and spectroscopic data are reported which are in accordance with a rate control located at steps early in the catalytic cycle (CO-dissociation, alkene coordination and alkene insertion).45,67 Nevertheless, based on the established catalytic cycle, the 16 VE acyl complex is a crucial intermediate which is involved in the hydrogenolytic product formation step. In the presence of carbon monoxide, the equilibrium between the 16 and 18 VE acyl complexes lies generally far on the side of the electronically and coordinatively saturated complex.68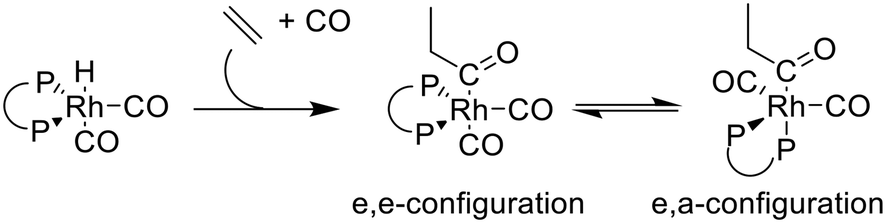 | ||
| Scheme 7 Simplified reaction scheme for the formation of stereoisomeric acyl complexes with ethene as substrate. | ||
In the first step the hydrido complex [HRh(CO)2(P∩P)] was formed at catalytic conditions ([Rh] = 1 mM, [P∩P] = 1 mM, p(CO/H2) = 2.0 MPa and ϑ = 90 °C) in cyclohexane as solvent. Then the temperature was adjusted to ϑ = 30 °C and the gas atmosphere was exchanged several times by pure carbon monoxide. In the next step, 1.0 MPa of ethene was added into the reactor. During this treatment new carbonyl vibrational bands (ν(CO) = 1670, 1995, 2061 cm−1) (Fig. 22a and ESI-H†) evolved while the bands ascribed to the hydrido complex completely vanished. The band at 1670 cm−1 lies in the spectral region expected for rhodium-acyl groups and the other bands at 1995 and 2061 cm−1 indicate two terminal carbonyl ligands.68–73 These spectral features would be in agreement with an acyl complex of the type [CH3CH2C(O)Rh(CO)2(P∩P)]. We conducted a similar experiment at higher rhodium concentrations ([Rh] = 20 mM, [P∩P]/[Rh] = 1) in toluene-d8 and the spectroscopic characteristics are comparable (Fig. 22b). Minor differences were observed such as the absence of smaller band contributions at 1677, 1983 and 2048 cm−1 observed in the experiment at [Rh] = 1 mM which stems probably from the solvent cyclohexane.
 | ||
| Fig. 22 Comparison between the in situ IR-spectra of [HRh(CO)2(P∩P)] and [CH3CH2C(O)Rh(CO)2(P∩P)] a) at lower Rh concentrations ([Rh] = 1 mM, solvent: cyclohexane), b) at higher Rh concentrations ([Rh] = 20 mM, solvent: toluene) and c) DFT-based IR-spectra (ESI-I†) of the e,a- and e,e-acyl complexes and e,e-[HRh(CO)2(P∩P)]. Experimental conditions: ϑ = 30 °C, [Rh]/[P∩P] = 1, p(CO) = 1.0 MPa, p(C2H4) = 1.0 MPa. | ||
DFT calculations of infrared spectra have been performed to elucidate the ligand coordination in stereoisomeric acyl complexes. The relative band intensity patterns for the stereoisomers e,e- and e,a-[CH3CH2C(O)Rh(CO)2(P∩P)] obtained from vibrational mode analysis have been compared to those of the experimental spectra. The experimental spectrum for the acyl complex shows an intensity ratio between both IR bands (I[ν(1995 cm−1)]/I[ν(2061 cm−1)]) of ≈1.9:1 (1 mM). Based on the calculated spectra via DFT methods an intensity ratio for corresponding band contributions of ≈1.6:1 for the e,a-isomer and of ≈1.1:1 for the e,e-isomer was obtained. This might indicate that the e,a-propionyl complexes has been formed preferentially. The relative energies of e,e- and e,a-propionyl complexes obtained from DFT computations give rise to a mixture of both stereoisomers with strong preference to the e,a-complex (ESI-J†). However, both stereoisomers have been formed which was investigated in the next step by NMR-spectroscopy.
HP NMR-spectroscopy of a solution prepared in toluene-d8 ([Rh] = 20 mM, [P∩P]/[Rh] = 1) was performed for further investigations on the molecular structure of this complex. The advantage of this method is, among others, the possibility to measure the coupling constants between 13C (acyl carbon) and 31P (phosphite ligand) directly after an isotopic labeling with 13CO (100%). A coupling constant larger than 2JC,P = ≥100 Hz indicates an e,a-ligand arrangement.46 In contrast, the coupling constant for an e,e-arrangement is significantly smaller or in some cases not detectable.6,41,44 The 13C{1H}-NMR spectrum of this sample showed two broad signals in the spectral region of acyl groups (δ(13C) = 210–235 ppm) at room temperature, that became sharper when the temperature was reduced (Fig. 23a).69,72–75 This points to occurring intra- and intermolecular exchange processes. The acyl signal at δ(13C) = 224.5 ppm appeared as a triplet of doublets at 273 K. When the temperature was further decreased to 223 K the slow exchange region was achieved and the acyl signal appeared as a doublet of doublets (1JC,Rh = 16 Hz, 2JC,P = 106 Hz). The large C–P coupling indicates an e,a-arrangement of the P-atoms. The second signal at δ(13C) = 229.7 ppm was characterized by a complex multiplet patterns with a small C–P coupling. This suggests a second acyl complex with an e,e-arrangement.
 | ||
| Fig. 23 13C{1H}-NMR (a) and 1H–13C-HMBC-NMR (b) spectra of stereoisomeric acyl complexes in a 5 mm sapphire tube after 13CO labeling. Conditions: [Rh] = 20 mM, [P∩P] = 20 mM, p(13CO) = 1.0 MPa, p(C2H4) = 1.0 MPa, solvent: toluene-d8. | ||
Due to the different sterical properties the two isomeric acyl complexes should show up different chemical shifts for the propionyl group bonded to the rhodium centre. A 1H–13C{1H}-HMBC spectrum measured at 223 K allowed us to confirm this anticipation. The HMBC-spectrum showed overall six correlation signals for the two acyl complexes (Fig. 23b). The signal at δ(13C) = 229.7 ppm corresponding to the e,e-acyl complex correlates with a signal at δ(1H) = 0.4 ppm, which is caused by a CH3-group. Other two cross-peaks for the rhodium-acyl signal at δ(13C) = 229.7 ppm and signals at δ(1H) = 1.7 and 2 ppm, reveal the two nonequivalent protons of the α-CH2 group due to the distorted symmetry. The HMBC-spectrum showed also comparable cross-peaks for the e,a-acyl complex. The signal at δ(13C) = 229.7 ppm correlates with the signal at δ(1H) = 0.5 ppm (CH3-group) as well as with δ(1H) = 1.9 and 2.5 ppm (nonequivalent protons of the α-CH2 group). Based on the correlation peaks between these CH3-groups and the acyl signals in the 13C-spectrum we were able to distinguish both complexes qualitatively.
The 31P{1H}-NMR spectra showed a very complex fine splitting due to the coupling between the 31P, 13C and 103Rh atoms. Regarding the complex structure and the molar ratios between the isomeric complexes of this sample no information was derived but clarification was achieved with a sample based on neohexene as alkene.
For further NMR-spectroscopic characterizations a sample with [Rh] = 20 mM and [P∩P]/[Rh] = 1 was prepared in toluene-d8 and labeled with 13CO. It was found that the spectroscopic characteristics did not change when the CO pressure was adjusted to 0.1 MPa which made the NMR analyses using a Young-NMR-tube possible with an improved spectra quality. 13C{1H}-NMR-measurements on this sample under variation of the temperature (298 to 203 K) showed again the formation of the two stereoisomeric e,e-/e,a-acyl complexes (ESI-H†). Cooling to 223 K of the sample lead to distinct signal patterns with a resolved doublet of doublet shape for the e,a-acyl complex. Corresponding CH-groups of the neohexyl moiety could be determined via a 1H–13C-HMBC spectrum at 253 K (ESI-H†). The tert-butyl groups for both complexes are located around 0.4 to 0.6 ppm in the 1H-NMR spectrum. To obtain further information regarding a possible intramolecular equilibrium of the acyl complexes in solution we conducted an additional 1H–1H-NOESY (EXSY) NMR experiment revealing an exchange of the –C(CH3)3-groups (Fig. 24). This result is in line with a dynamic interconversion of the configurational isomeric acyl complexes.72 The NMR data gave indications for a preferred formation of the e,a-isomer. Based on DFT computations also the e,a-complex is thermodynamically more stable for the investigated temperatures (ESI-J†).
 | ||
| Fig. 24 1H–1H-EXSY spectrum of the equilibrium between e,a- and e,e-[RC(O)Rh(CO)2(P∩P)] complexes at 253 K. Further conditions: [Rh] = 20 mM, [P∩P] = 20 mM, [neohexene] = 80 mM, p(CO) = 0.1 MPa, solvent: toluene-d8. | ||
In addition, we investigated the 31P{1H}-NMR spectrum for further details (Fig. 25). The usage of a 13CO labeled sample revealed two doublet signals of which one doublet shows a dddd fine splitting. This multiplet splitting is caused by the coupling between the 31P, 103Rh and 13C atoms. The 31P{1H}-NMR spectrum of a non 13CO-labeled sample and lower neohexene concentrations simplified the study towards an assignment of the signals to the respective stereoisomeric structures of the acyl complexes. The signal set with the observed fine splitting δ(31P) = 154.8 ppm (1JP,Rh = 262 Hz) and 171.5 ppm (1JP,Rh = 255 Hz) with a P–P coupling of 2JP,P = 147 Hz is in accordance with the e,a-complex. The two broad signals showing no distinct fine splitting at δ(31P) = 164.9 and 172.0 ppm were assigned to the e,e-acyl complex.
 | ||
| Fig. 25 31P{1H}-NMR spectra of e,a- and e,e-[RC(O)Rh(CO)2(P∩P)] complexes at 253 K (bottom: without 13C-labeling and top: after 13CO labeling). Further conditions: [Rh] = 20 mM, [P∩P] = 20 mM, [neohexene] = 80 mM, p(CO) = 0.1 MPa, solvent: toluene-d8. | ||
3. Conclusions
Rhodium complexes modified with the chelating diphosphite BiPhePhos were characterized by in situ HP IR and NMR-spectroscopy. The hydrido complex e,e-[HRh(CO)2(P∩P)] is the only observable resting state complex at conditions typical for hydroformylation. The hydrido complex is relatively stable in solution in the absence of hydrogen or synthesis gas and dinuclear complexes appear to form only after prolonged treatments at higher temperatures. In the presence of air the hydrido complex and the diphosphite are decomposed at elevated temperatures. [HRh(CO)2(P∩P)] and BiPhePhos show hydrolytic decomposition when exposed to water. The increase of the [P∩P]/[Rh] ratio to 1.5 and 2 leads to the formation of monocarbonyl complexes in CO-depleted atmospheres. One of these complexes is a ligand bridging hydrido complex (e,e,e-[HRh(CO)(P∩P)]2(P∩P)) and the other is the corresponding monomer [HRh(CO)(P∩P)(κ1-P∩P)] with an uncoordinated phosphite moiety. The population of acyl complexes of the type [RC(O)Rh(CO)2(P∩P)] is possible in H2 impoverished gas atmospheres with ethene and neohexene as alkenes. Two stereoisomers with e,e- and e,a-coordinations of both P-atoms are present in a chemical equilibrium. In Fig. 26 an overview is presented on the various rhodium carbonyl complexes being parts of a larger network of interconvertible species.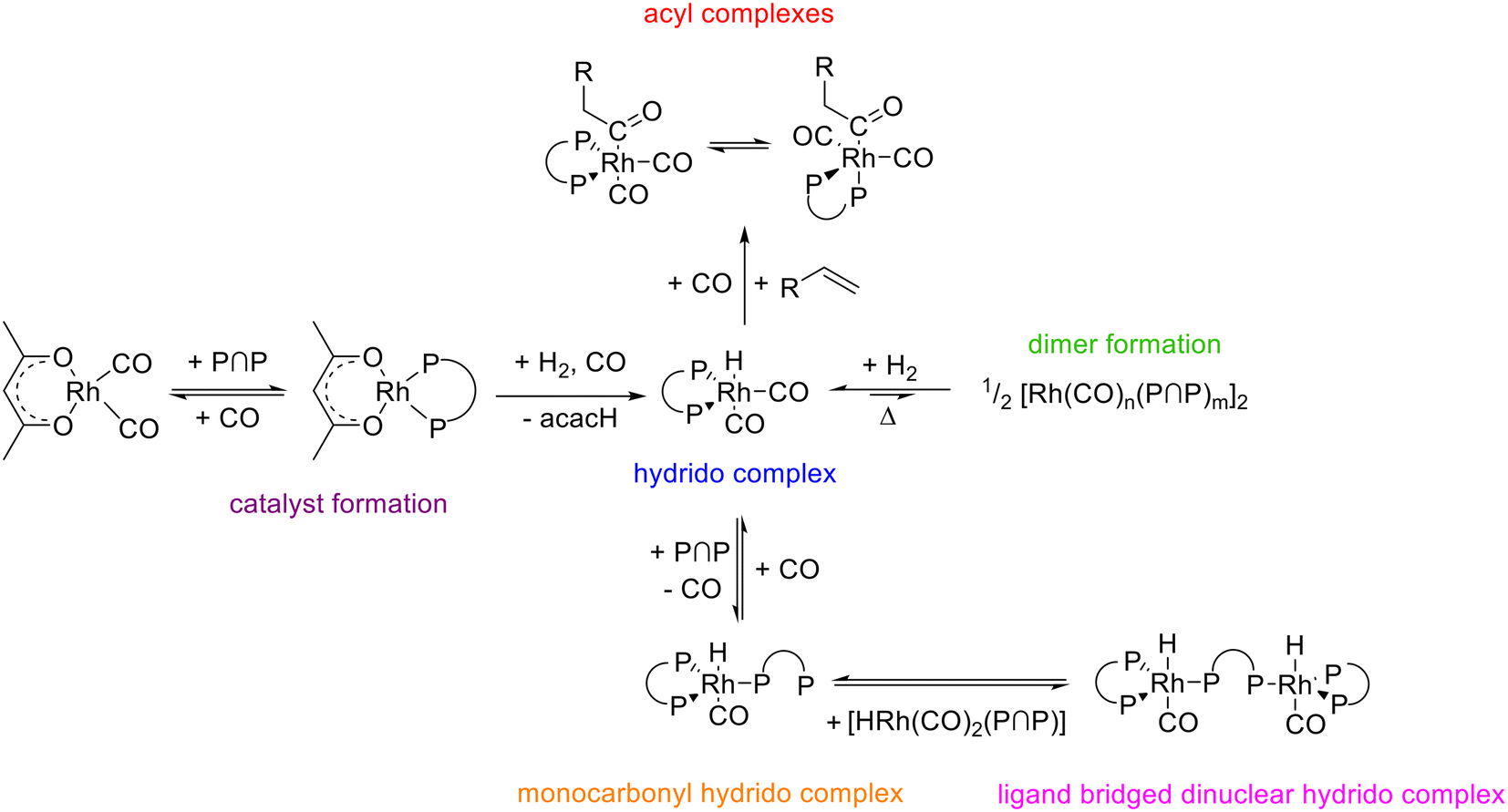 | ||
| Fig. 26 Summary of the interconversions between diphosphite (BiPhePhos) modified rhodium complexes in the alkene hydroformylation. | ||
Experimental and computational section
Further supplementary data and details on the laboratory experiments and computations are given in the ESI.†Data availability
The respective data supporting the findings of this research study is given as figures/tables of spectra and concentration–time-data in the main manuscript and in the ESI.† Original and processed data available in specific data formats can be provided on request from the corresponding author.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Jiali Liu thanks the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 859910. Christoph Kubis is thankful for the DFG Research Unit FOR 5538 IMPD4Cat (Project No. 501735683).References
- R. Franke and A. Börner, Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2016 Search PubMed.
- R. Franke, D. Selent and A. Börner, Applied Hydroformylation, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Wiley-VCH, Weinheim, 3rd edn, 2018 Search PubMed.
- Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer, Dordrecht, 2002 Search PubMed.
- P. W. N. M. van Leeuwen and J. C. Chadwick, Homogeneous Catalysts, Wiley-VCH, Weinheim, 2011 Search PubMed.
- D. Selent, R. Franke, C. Kubis, A. Spannenberg, W. Baumann, B. Kreidler and A. Börner, Organometallics, 2011, 30, 4509–4514 CrossRef CAS.
- P. P. Deutsch and R. Eisenberg, Organometallics, 1990, 9, 709–718 CrossRef CAS.
- A. B. Permin and R. Eisenberg, J. Am. Chem. Soc., 2002, 124, 12406–12407 CrossRef CAS PubMed.
- D. Guan, C. Godard, S. M. Polas, R. P. Tooze, A. C. Whitwood and S. B. Duckett, Dalton Trans., 2019, 48, 2664–2675 RSC.
- M. Garland and P. Pino, Organometallics, 1991, 10, 1693–1704 CrossRef CAS.
- A. van Rooy, E. N. Orij, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 34–43 CrossRef CAS.
- S. C. van der Slot, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. A. Iggo and B. T. Heaton, Organometallics, 2001, 20, 430–441 CrossRef CAS.
- O. Diebolt, P. W. N. M. van Leeuwen and P. C. J. Kamer, ACS Catal., 2012, 2, 2357–2370 CrossRef CAS.
- M. Garland, in Mechanisms in Homogeneous Catalysis, ed. B. Heaton, Wiley-VCH, Weinheim, 2005, pp. 151–193 Search PubMed.
- M. Garland, Catal. Today, 2010, 155, 266–270 CrossRef CAS.
- C. Li, E. Widjaja, W. Chew and M. Garland, Angew. Chem., Int. Ed., 2002, 41, 3785–3789 CrossRef.
- B. Moasser, W. L. Gladfelter and D. C. Roe, Organometallics, 1995, 14, 3832–3838 CrossRef CAS.
- A. Behr, D. Obst, C. Schulte and T. Schosser, J. Mol. Catal. A: Chem., 2003, 206, 179–184 CrossRef CAS.
- A. Behr, D. Obst and C. Schulte, Chem. Ing. Tech., 2004, 76, 904–910 CrossRef CAS.
- C. Vogl, E. Paetzold, C. Fischer and U. Kragl, J. Mol. Catal. A: Chem., 2005, 232, 41–44 CrossRef CAS.
- A. Jörke, A. Seidel-Morgenstern and C. Hamel, J. Mol. Catal. A: Chem., 2017, 426, 10–14 CrossRef.
- A. Jörke, T. Gaide, A. Behr, A. Vorholt, A. Seidel-Morgenstern and C. Hamel, Chem. Eng. J., 2017, 313, 382–397 CrossRef.
- J. M. Dreimann, E. Kohls, H. F. W. Warmeling, M. Stein, L. F. Guo, M. Garland, T. N. Dinh and A. J. Vorholt, ACS Catal., 2019, 9, 4308–4319 CrossRef CAS.
- T. Gaide, A. Jörke, K. E. Schlipköter, C. Hamel, A. Seidel-Morgenstern, A. Behr and A. J. Vorholt, Appl. Catal., A, 2017, 532, 50–56 CrossRef CAS.
- G. Kiedorf, D. M. Hoang, A. Müller, A. Jörke, J. Markert, H. Arellano-Garcia, A. Seidel-Morgenstern and C. Hamel, Chem. Eng. Sci., 2014, 115, 31–48 CrossRef CAS.
- J. M. Dreimann, M. Skiborowski, A. Behr and A. J. Vorholt, ChemCatChem, 2016, 8, 3330–3333 CrossRef CAS.
- A. Lejeune, L. Le Goanvic, T. Renouard, J.-L. Couturier, J.-L. Dubois, J.-F. Carpentier and M. Rabiller-Baudry, ChemPlusChem, 2019, 84, 1744–1760 CrossRef CAS PubMed.
- M. Haumann, M. Jakuttis, R. Franke, A. Schönweiz and P. Wasserscheid, ChemCatChem, 2011, 3, 1822–1827 CrossRef CAS.
- S. Walter, H. Spohr, R. Franke, W. Hieringer, P. Wasserscheid and M. Haumann, ACS Catal., 2017, 7, 1035–1044 CrossRef CAS.
- M. Logemann, J. M. Marinkovic, M. Schörner, E. José García-Suárez, C. Hecht, R. Franke, M. Wessling, A. Riisager, R. Fehrmann and M. Haumann, Green Chem., 2020, 22, 5691–5700 RSC.
- C. Li, K. Xiong, L. Yan, M. Jiang, X. Song, T. Wang, X. Chen, Z. Zhan and Y. Ding, Catal. Sci. Technol., 2016, 6, 2143–2149 RSC.
- C. Li, L. Yan, L. Lu, K. Xiong, W. Wang, M. Jiang, J. Liu, X. Song, Z. Zhan, Z. Jiang and Y. Ding, Green Chem., 2016, 18, 2995–3005 RSC.
- Y. Wang, L. Yan, C. Li, M. Jiang, W. Wang and Y. Ding, Appl. Catal., A, 2018, 551, 98–105 CrossRef CAS.
- B. N. Leidecker, C. Kubis, A. Spannenberg, R. Franke and A. Börner, IUCrData, 2019, 4, x191636 CrossRef CAS.
- R. A. Alberty and C. A. Gehrig, J. Phys. Chem. Ref. Data, 1985, 14, 803–820 CrossRef CAS.
- F. G. Helfferich, Kinetics of Multistep Reactions, in Comprehensive Chemical Kinetics, ed. N. J. B. Green, Elsevier Science, Amsterdam, 2nd edn, 2004, vol. 40 Search PubMed.
- C. Kubis, R. Ludwig, M. Sawall, K. Neymeyr, A. Börner, K.-D. Wiese, D. Hess, R. Franke and D. Selent, ChemCatChem, 2010, 2, 287–295 CrossRef CAS.
- https://www.math.uni-rostock.de/facpack/.
- M. Sawall, C. Kubis, E. Barsch, D. Selent, A. Börner and K. Neymeyr, J. Iran. Chem. Soc., 2016, 13, 191–205 CrossRef CAS.
- H. Schröder, M. Sawall, C. Kubis, A. Jürß, D. Selent, A. Brächer, A. Börner, R. Franke and K. Neymeyr, Chemom. Intell. Lab. Syst., 2017, 163, 55–63 CrossRef.
- A. Bara-Estaún, C. L. Lyall, J. P. Lowe, P. G. Pringle, P. C. J. Kamer, R. Franke and U. Hintermair, ChemCatChem, 2023, 15, e202201204 CrossRef.
- L. Vaska, J. Am. Chem. Soc., 1966, 88, 4100–4101 CrossRef CAS PubMed.
- C. Kubis, M. König, B. N. Leidecker, D. Selent, H. Schröder, M. Sawall, W. Baumann, A. Spannenberg, A. Brächer, K. Neymeyr, R. Franke and A. Börner, ACS Catal., 2023, 13, 5245–5263 CrossRef CAS.
- A. van Rooy, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, N. Veldman and A. L. Spek, Organometallics, 1996, 15, 835–847 CrossRef CAS.
- A. Bara-Estaún, PhD thesis, University of Bath, 2022 Search PubMed.
- P. C. J. Kamer, J. N. H. Reek and P. W. N. M. van Leeuwen, in Mechanism in Homogeneous Catalysis, ed. B. Heaton, Wiley-VCH, Weinheim, 2005, pp. 231–269 Search PubMed.
- G. J. H. Buisman, L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1997, 16, 5681–5687 CrossRef CAS.
- A. Castellanos-Páez, S. Castillón, C. Claver, P. W. N. M. van Leeuwen and W. G. J. de Lange, Organometallics, 1998, 17, 2543–2552 CrossRef.
- P. Meakin, E. L. Muetterties and J. P. Jesson, J. Am. Chem. Soc., 1972, 94, 5271–5285 CrossRef CAS.
- R. S. Berry, J. Chem. Phys., 1960, 32, 933–938 CrossRef CAS.
- L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1999, 18, 4765–4777 CrossRef CAS.
- L. A. van der Veen, P. H. Keeven, G. C. Schoemaker, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 872–883 CrossRef CAS.
- P. W. N. M. van Leeuwen, Appl. Catal., A, 2001, 212, 61–81 CrossRef CAS.
- K. Köhnke, N. Wessel, J. Esteban, J. Jin, A. J. Vorholt and W. Leitner, Green Chem., 2022, 24, 1951–1972 RSC.
- M. Gerlach, F. Jameel, A. Seidel-Morgenstern, M. Stein and C. Hamel, Catal. Sci. Technol., 2023, 13, 1788–1801 RSC.
- B. Zhang, H. Jiao, D. Michalik, S. Kloß, L. M. Deter, D. Selent, A. Spannenberg, R. Franke and A. Börner, ACS Catal., 2016, 6, 7554–7565 CrossRef CAS.
- S. Kloß, D. Selent, A. Spannenberg, R. Franke, A. Börner and M. Sharif, Catalysts, 2019, 9, 1036 CrossRef.
- U. Nettekoven, P. C. J. Kamer, M. Widhalm and P. W. N. M. van Leeuwen, Organometallics, 2000, 19, 4596–4607 CrossRef CAS.
- E. Zuidema, P. E. Goudriaan, B. H. G. Swennenhuis, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2010, 29, 1210–1221 CrossRef CAS.
- S. C. van der Slot, J. Duran, J. Luten, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 2002, 21, 3873–3883 CrossRef CAS.
- Z. Freixa and J. Carles Bayón, J. Chem. Soc., Dalton Trans., 2001, 2067–2068 RSC.
- O. R. Hughes and D. A. Young, J. Am. Chem. Soc., 1981, 103, 6636–6642 CrossRef CAS.
- B. N. Leidecker, A. Spannenberg, R. Franke, A. Borner and C. Kubis, IUCrData, 2023, 8, x230083 CrossRef CAS PubMed.
- A. van Rooy, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Organomet. Chem., 1997, 535, 201–207 CrossRef CAS.
- J. R. Briggs and G. T. Whiteker, Chem. Commun., 2001, 2174–2175 RSC.
- R. Franke, C. Borgmann, D. Hess and K.-D. Wiese, Z. Anorg. Allg. Chem., 2003, 629, 2535–2538 CrossRef CAS.
- E. Zuidema, L. Escorihuela, T. Eichelsheim, J. J. Carbo, C. Bo, P. C. Kamer and P. W. van Leeuwen, Chem. – Eur. J., 2008, 14, 1843–1853 CrossRef CAS PubMed.
- C. Kubis, M. Sawall, A. Block, K. Neymeyr, R. Ludwig, A. Börner and D. Selent, Chem. – Eur. J., 2014, 20, 11921–11931 CrossRef CAS PubMed.
- J. M. Brown and A. G. Kent, J. Chem. Soc., Perkin Trans. 2, 1987, 1597–1607 RSC.
- C. Kubis, D. Selent, M. Sawall, R. Ludwig, K. Neymeyr, W. Baumann, R. Franke and A. Börner, Chem. – Eur. J., 2012, 18, 8780–8794 CrossRef CAS PubMed.
- G. Liu, R. Volken and M. Garland, Organometallics, 1999, 18, 3429–3436 CrossRef CAS.
- S. C. van der Slot, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. A. Iggo and B. T. Heaton, Organometallics, 2001, 20, 430–441 CrossRef CAS.
- S. Güven, M. M. L. Nieuwenhuizen, B. Hamers, R. Franke, M. Priske, M. Becker and D. Vogt, ChemCatChem, 2014, 6, 603–610 CrossRef.
- E. R. Nelsen and C. R. Landis, J. Am. Chem. Soc., 2013, 135, 9636–9639 CrossRef CAS PubMed.
- P. Dingwall, J. A. Fuentes, L. E. Crawford, A. M. Z. Slawin, M. Bühl and M. L. Clarke, J. Am. Chem. Soc., 2017, 139, 15921–15932 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00481g |
| This journal is © The Royal Society of Chemistry 2024 |