
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimization of peptide foldamer-based artificial retro-aldolase†
Katarzyna
Ożga
,
Ewa
Rudzińska-Szostak
and
Łukasz
Berlicki
 *
*
Department of Bioorganic Chemistry, Faculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: lukasz.berlicki@pwr.edu.pl
First published on 12th July 2024
Abstract
Due to their predictable and controllable three-dimensional structure, peptide foldamers constitute a class of compounds beneficial for developing functional molecules. One of the most challenging applications is the construction of enzyme-like catalysts. Here, we describe the optimization of peptide foldamers composed of two 9/12/9/10-helices incorporating cis-2-aminocyclopentanecarboxylic acid residues toward retro-aldol activity. Modifications related to helix handedness, interhelical linker rigidity, and active site construction led to highly active retro-aldolase mimetics. NMR measurements confirmed the assumed arrangement of active site residues.
Introduction
Oligomers with a well-defined propensity for folding are referred to as foldamers, and they represent excellent scaffolds for the construction of functional molecules.1,2 Peptide foldamers are the most widely studied among the numerous classes of foldamers.3,4 Incorporating folding-inducing units in peptide foldamers allows rational and effective control over their folding behavior, even for very short sequences.5–7 The large number of possible sequence patterns and the variability of the stereochemical arrangements of individual amino acid residues create an enormous diversity of possible secondary structures in peptides that incorporate α- and β-amino acids.8,9 Peptides composed of β3-amino acids adopt a 14-helix conformation, similar to an α-helix in which the side chains align closer.10 Cycloalkane-containing β-amino acids are beneficial for constructing well-defined structures due to conformational rigidity combined with the availability of building blocks and the feasibility of synthesizing peptides with long sequences.11 For example, 14-helix stability can be tuned using trans-2-aminocyclohexanecarboxylic acid (ACHC).12 Sequences built with trans-2-aminocyclopentanecarboxylic acid (ACPC) adapt to the more elongated conformation of the 12-helix.13 Numerous examples of α/β-mixed sequences folding to helical conformations have been reported.14 The 9/12/9/10-helix is one of the most stable motifs in aqueous solution. Its formation is induced by the sequence motif consisting of L-α-amino acid, D-α-amino acid, cis-(1S,2R)-ACPC, cis-(1R,2S)-ACPC, where folding is induced mainly due to the rigidity of the ACPC residues.9 β-Amino acid-containing β-structures have also been successfully designed and analyzed.15,16 The design and characterization of tertiary foldameric structures is now an emerging field of study,17 and most reports are focused on the ‘foldamerization’ of already existing miniprotein structures.18,19 Studies on the GB1 domain, Trp cage, EHN, and villin, whose sequences have been alternated with β-amino acids, indicated that their folding pattern does not change if the hydrophobic core is unaffected.19–21 However, these modifications do not lead to increased conformational stability compared to their native counterparts.20 The number of de novo-designed β-amino acids containing miniproteins is small and related to structures composed of helices.22–24Foldamers have been utilized to construct molecules with various biological activities,25 such as protein–protein interaction inhibitors26–28 or antimicrobial drugs,29 and have been proven to be protease resistant, making them excellent drug candidates. However, the use of foldameric structures for the development of catalysts is still not well exploited. The high control over the arrangement of side chains in foldamers allows them to accommodate the precise geometry of catalytic residues, as was shown in recent examples of helical foldamers able to perform macrocyclization.30 In other reports, catalytic activity is highly correlated with forming a super-secondary structure via aggregation31,32 or oligomerization33,34 of short helices or β-strands. However, oligomers with tertiary structures may not only accommodate more complex catalytic sites but may also provide substrate binding sites, which is considered a basis for the high efficacy of native enzymes.35 One of the prominent successes in enzyme design that emphasized the importance of a proper binding site was to obtain highly active artificial aldolases catalyzing C–C bond formation or cleavage.36–38 However, only one study has reported a foldamer-based artificial aldolase.31 The crucial step of aldol catalysis directly leading to C–C bond cleavage is the enolization of the substrate, leading to a carbanion species. The catalysis of the retro-aldol reaction performed by native enzymes from class I strongly depends on the presence of the nucleophilic ε-amine group of the lysine residue able to form a Schiff base with the carbonyl group of the substrate39 (Fig. 1A), which facilitates enolization by lowering its pKa.
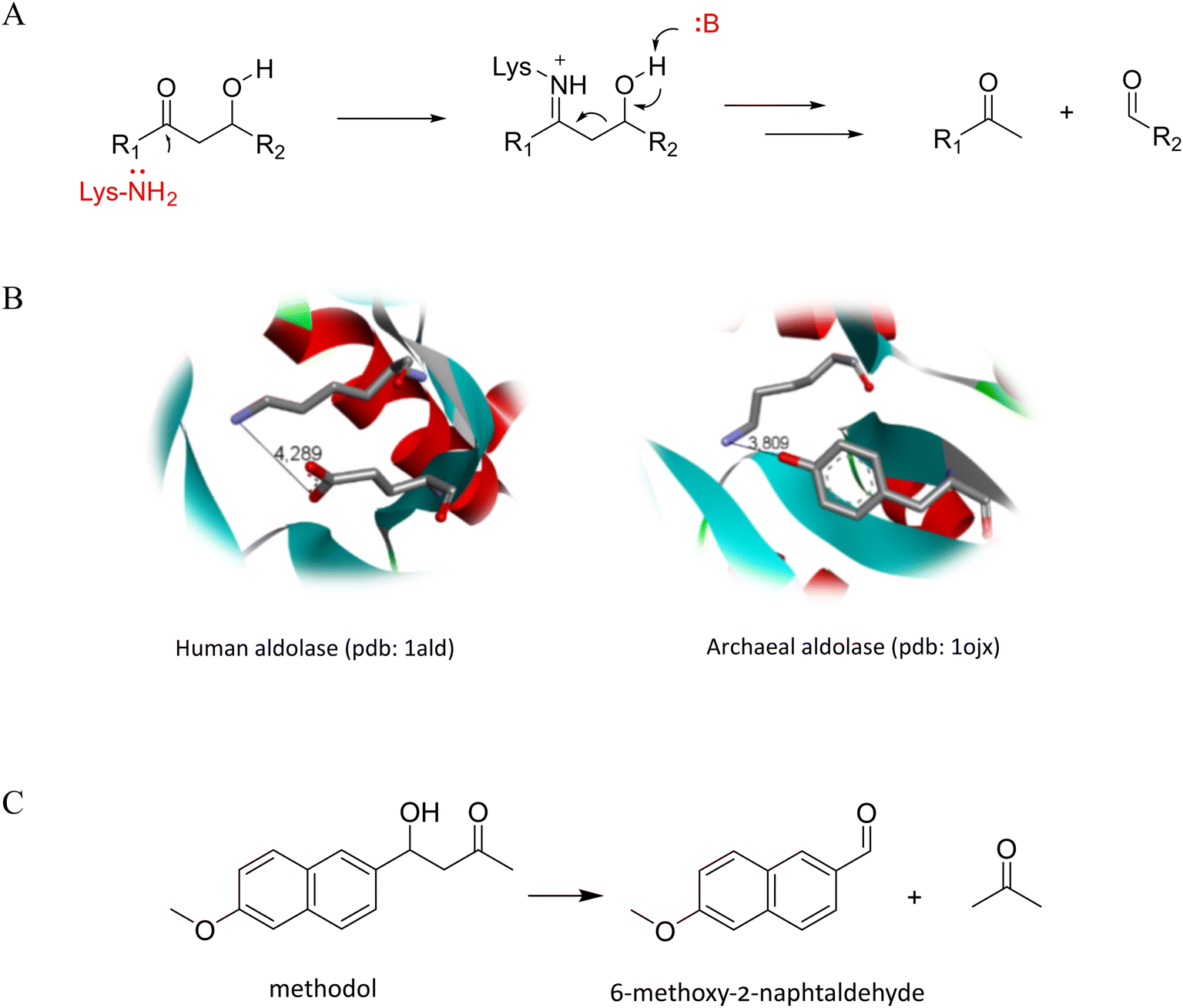 | ||
| Fig. 1 Simplified mechanism of retro-aldol catalysis of class I aldolases (A) and exemplary catalytic sites from aldolase class I (B). A retro-aldol cleavage reaction of methodol was used in this study (C). | ||
Under physiological conditions, lysine residues are not nucleophilic, and their deprotonation must be forced by destabilizing the positive charge. This may be achieved by placing the lysine residue in a hydrophobic environment or near the other positively charged residues. The hydrophobic effect on the aldol reaction conducted by the primary amine was shown based on the activity of n-butylamine dissolved in different detergents,40 and it can also be observed by introducing lysine residues into the hydrophobic cleft of previously nonactive proteins36 or in a water-free environment that emerges during aggregation.31 In native enzymes, the active site is placed in a hydrophobic region inside the TIM barrel fold and surrounded by arginine residues. Catalysis also requires acid/base catalysts to perform several proton transfers, often achieved by only one residue. The most common residues supplementing lysine are glutamate (higher organisms) or tyrosine (archaebacteria) (Fig. 1B).41–43
In this paper, we explore the possibility of developing an artificial retro-aldolase based on foldameric scaffolds. In particular, 9/12/9/10 helix, helix–loop–helix, and helix–turn–helix structures were applied for the rational stepwise construction of mimetics of class I aldolases (Schiff-base forming) that would catalyze retro-aldol cleavage of methodol (Fig. 1C).
Results and discussion
The 9/12/9/10 helix based on the ααββ sequence pattern was highly conformationally stable in water solution9 (Fig. 2) and, therefore, can be effectively used to construct higher-order structures with functionality. Taking into account artificial aldolase based on the helical peptide foldamer published by Gellman and coworkers31 with several lysine residues arranged in a row, reference peptide 1 was constructed. Peptide 1 and all subsequent oligomers were obtained using a microwave-assisted automated solid phase peptide synthesis approach with Fmoc chemistry (Table 1).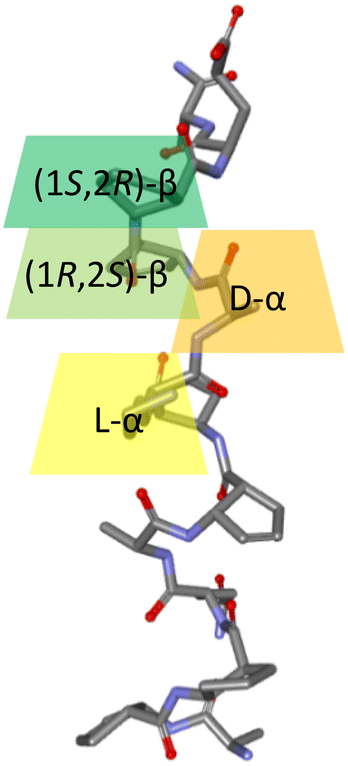 | ||
| Fig. 2 Structure of the 9/12/9/10 helix,9 with the L-α-amino acid, D-α-amino acid, cis-(1R,2S)-ACPC, and cis-(1S,2R)-ACPC positions marked in yellow (L-α), orange (D-α), light green ((1R,2S)-β) and green ((1S,2R)-β), respectively. | ||
a ![[pentagon filled]](https://www.rsc.org/images/entities/char_e13d.gif) : cis-(1R,2S)-ACPC, : cis-(1R,2S)-ACPC,  : cis-(1S,2R)-ACPC, small letters denote D-amino acid residues; catalytic lysine residues are colored in orange, other residues present in or supporting the catalytic motif are shown in purple, lysine and glutamate residues potentially forming salt bridges are colored in blue and red, respectively, residues forming a linker are shown in green and knockout residues are shown in gray. : cis-(1S,2R)-ACPC, small letters denote D-amino acid residues; catalytic lysine residues are colored in orange, other residues present in or supporting the catalytic motif are shown in purple, lysine and glutamate residues potentially forming salt bridges are colored in blue and red, respectively, residues forming a linker are shown in green and knockout residues are shown in gray.
|
|---|

|
The first series (1–5) may be treated as a control for the whole study. The sequences of helix–loop–helix (HLH)-forming peptides were designed as previously reported.44 Peptide 1 is a single helix bearing potentially active lysine residues organized on one side of the helix, and it served as a reference peptide (Fig. 2). Peptide 2 represents a helix–loop–helix peptide depleted of any primary amine group. Peptide 3 includes multiple lysine residues on both helices as the positive control. Peptides 4 and 5 have lysine residues placed on one of the helices and large hydrophobic residues (Tyr of Phe) on the other to test the impact of the hydrophobic environment on catalysis. Conformational analysis of the synthesized peptides was performed using CD spectroscopy in a phosphate-buffered solution at pH 8 (Fig. 3A). The left-handed 9/12/9/10 helix has a characteristic CD spectrum with a pronounced maximum near 205 nm. All synthesized peptides show the same shape of the spectra; however, they exhibit a more significant Cotton effect (residual molar ellipticity is higher), which indicates a higher propensity of HLH peptides to fold into helical structures. Retro-aldol activity studies were performed using previously described methodol36 as a substrate (Fig. 1C) and monitoring the fluorescent product formation (6-metoxy-2-naphtaldehyde) at 37 °C in 50 mM phosphate buffer pH 8 and the presence of 0.2 mM peptides. As expected, peptide 2 did not show any activity due to the absence of the primary amine group in the structure. The highest turnover number, kcat = 11.6 × 10−6 s−1, was measured for peptide 3 (Table 2, Fig. 3B), consistent with other reports on lysine residues bearing helices.31 Peptides 1 and 4 containing three lysine residues had similar activity; however, peptide 5, despite having the same amount of lysine residues as 1 and 4, was more active, which is correlated with the positive impact of tyrosyl residues on the reaction rate.
 | ||
| Fig. 3 CD spectra were recorded for peptides 1–11 in a phosphate-buffered solution at pH 8 (A). Michaelis–Menten plots of the methodol cleavage reaction obtained in the presence of 1–11 in solution at pH 8 at 37 °C (B). | ||
| Peptide | k cat·106 [s−1] | K M [mM] | k cat/KM·106 [s−1 mM−1] | k cat/kuncat | k cat/kuncat/na |
|---|---|---|---|---|---|
| a n, number of lysine residues in peptide sequence. b NA, not catalytically active. | |||||
| 1 | 6.3 ± 1.4 | 1.9 ± 0.7 | 3.3 | 250 | 83 |
| 2 | NAb | — | — | — | — |
| 3 | 11.6 ± 1.4 | 2.3 ± 0.4 | 5.0 | 460 | 77 |
| 4 | 6.5 ± 1.1 | 1.2 ± 0.4 | 5.4 | 260 | 87 |
| 5 | 9.8 ± 1.6 | 2.5 ± 0.4 | 4.0 | 390 | 130 |
| 6 | 9.6 ± 1.0 | 2.5 ± 0.4 | 3.8 | 380 | 190 |
| 7 | 12.4 ± 0.6 | 2.7 ± 0.2 | 4.6 | 500 | 250 |
| 8 | 13.5 ±0.6 | 3.2 ± 0.2 | 4.2 | 540 | 180 |
| 9 | 12.2 ± 0.7 | 2.9 ± 0.2 | 4.2 | 490 | 163 |
| 10 | 17.2 ± 3.4 | 4.0 ± 1.1 | 4.3 | 680 | 340 |
| 11 | 22.8 ± 1.2 | 4.2 ± 3.0 | 5.4 | 910 | 303 |
Peptides 6 and 7 were designed to reproduce the actual geometry of the active site from the class I aldolases human aldolase (PDB id 1ALD) and archaebacterial aldolase (PDB id 1OJX) (Fig. 1). The recorded CD spectra confirmed the helical fold of both (Fig. 3A). Moreover, peptides 6 and 7, which only have two lysine residues in their sequence, are also more active (the turnover number is 9.6 × 10−6 s−1 and 12.4 × 10−6 s−1, respectively, Table 2, Fig. 3B) than could be expected from the lysine residue content because they are more potent catalysts than peptides 1 and 4. Therefore, the aldolase activity, in this case, is not just proportional to the number of primary amine groups but rather arises because of the foldameric scaffold, which adequately mimics the organization of the active site. Moreover, the tyrosine residue has a better impact on catalysis than the glutamate residue because it can simultaneously be an acid/base catalyst and provide a hydrophobic environment. This observation was also supported by peptide 5 activity.
In the next round of the design (peptides 8–11), we aimed to optimize peptide 7. We placed the lysine and glutamate residues on the N- and C-termini, respectively. The favorable electrostatic interaction may help the helices stay in close contact with each other and improve the conformational stability,44 thereby improving the geometry of the active site. In sequences 9 and 10, the additional hydrophobic residue was introduced in the proximity of lysine residues that could either induce amine group deprotonation or serve as a binding site prototype that can interact with the naphthyl group of the substrate. In peptide 10, the hydrophobic residue was incorporated instead of a single lysine residue. Peptide 11 is the analog of peptide 9, although a longer glycine linker was also introduced, which may result in better fitting of both helical interfaces. The peptide helicity was confirmed by CD, as previously described (Fig. 3A). However, we did not observe an increase in the Cotton effect, which would indicate stabilization of the whole structure by salt bridge formation. The conformations of peptides 8 and 9 were further studied by 2D NMR (TOCSY, ROESY spectra, Fig. S3 and S4†), which revealed the interaction of protons between residues i and i + 2 and i and i + 3, thus confirming the folding of helical parts; however, no interhelical interaction was found, which was probably because the parallel arrangement of both helices was relatively transient (Tables S3 and S5†). Introducing a stabilizing salt bridge (peptides 8vs.7) had little effect on the activity (Table 2, Fig. 3B). A lack of effect was also observed when a phenyl residue was introduced in peptide 9. Interestingly, peptide 10 had the highest activity (turnover number equal to 17.2 × 106 s−1) among all the 3-Gly linker peptides. This peptide had one lysine residue and tyrosine residue on one of the helices (such as peptides 7–9) and another tyrosine residue on the second helix, which strongly supports the contribution of the second helix in catalysis. The highest turnover number, however, was achieved by modification of the interhelical linker (peptide 11); in this case, kcat was equal to 22.8 × 10−6 s−1 (Table 2).
In the last round of the design process, the glycine linker was replaced by a rigid linker composed of two cis-ACPC residues with the same stereochemistry. This motif is known to adopt a rigid conformation with highly restricted rotations of backbone torsion.16 Because of the very low conformational freedom of torsional angles within and adjacent to cyclic β-residue, rigidified backbones should allow for better control over the relative position of helices, resulting in more defined active site geometry. However, a limited number of helix–linker–helix combinations could create a compact structure without steric hindrance. We have already published a possible design methodology for such helix–turn–helix structures and have been successful in obtaining them, as shown in peptides 15 and 16.24 Briefly, we based on experimentally derived models of the secondary units, namely, the 9/12/9/10 helix and extended structure built from one enantiomer of cis-ACPC and the possible torsion at joints was restricted because of the cis-ACPC units. The designs varied regarding the 1) stereochemistry of the ACPC linker, 2) handedness of the helices, and 3) termination and/or beginning of the helix-forming motif. Here, we propose three additional realizations of the helix–turn–helix (HTH) structure (12–14). Next, the previously successful active site composed of lysine and tyrosine residues was grafted onto the scaffolds. Peptides 12 and 13 incorporate helices of opposite handedness; thus, their contributions to the CD spectrum cancel each other out, resulting in no observable Cotton effect. In these cases, we performed 2D NMR analysis (Tables S6–S9†). The observed interproton interactions indicate the presence of helical structures. Peptides 14–16 exhibited the expected CD spectra, with the Cotton effect observed at 205 nm specific for the 9/12/9/10-helix (Fig. 4A). Previously reported peptides 15 and 16 do not significantly accelerate the retro-aldol reaction like the first HLH peptides (Table 3, Fig. 4B).
 | ||
| Fig. 4 CD spectra were recorded for peptides 12–19 in a phosphate-buffered solution at pH 8 (A). Michaelis–Menten plots of the methodol cleavage reaction obtained in the presence of 12–19 in solution at pH 8 and 37 °C (B and C), reaction progression curves of methodol cleavage under six different concentrations (mM) in the presence of peptide 14 (D). | ||
| Peptide | k cat·106 [s−1] | K M [mM] | k cat/KM·106 [s−1 mM−1] | k cat/kuncat | k cat/kuncat/na |
|---|---|---|---|---|---|
| a n, number of lysine residues in peptide sequence. b NA, not catalytically active. | |||||
| 12 | 22.1 ± 3.2 | 3.3 ± 0.7 | 6.7 | 880 | 440 |
| 13 | 18.7 ± 1.5 | 2.2 ± 0.3 | 8.5 | 750 | 375 |
| 14 | 25.0 ± 3.0 | 5.5 ± 0.8 | 4.5 | 1000 | 1000 |
| 15 | 7.2 ± 1.6 | 2.0 ± 0.7 | 3.7 | 290 | 290 |
| 16 | 6.9 ± 0.6 | 0.9 ± 0.2 | 7.7 | 280 | 280 |
| 17 | NAb | — | — | — | — |
| 18 | 4.9 ± 1.4 | 1.8 ± 0.9 | 2.7 | 200 | 200 |
| 19 | 3.6 ± 0.2 | 1.1 ± 0.1 | 3.3 | 140 | 140 |
However, the other HTH peptides (12–14) catalyze the retro-aldol reaction of methodol with turnover number like the most active design from the previous design round 11. The most active is peptide 14, with a turnover number of 25.0 × 10−6 s−1 and a reaction acceleration rate of 1000 (Table 3, Fig. 4D).
The impact of structural complexity and well-defined conformation on the activity can be directly drawn from a comparison of peptides 1, 11, and 14, where the turnover number increases despite the decreasing amounts of lysine residues in the sequence. Although the sequence of peptide 14 contains only one lysine residue, its kcat is the highest in the whole series of peptides. To underline it, we introduced one more parameter to describe the catalytic activity, i.e., acceleration of the reaction divided by the number of lysine residues. We can conclude that, in contrast to other peptides from this study, whose activity may be non-specific and multivalent, the activity of peptide 14 is monovalent. Therefore, we performed knockout studies (peptides 17–19) in which we systematically mutated one catalytic residue to alanine. Peptide 17, which has no lysine residue in its sequence, shows no catalytic activity (Table 3, Fig. 4C). In contrast, oligomers 18 and 19, in which the catalytic triad YKY reduced to a dyad, showed significantly reduced turnover number. These observations indicate that catalytic activity results from the cooperation of all three residues of the triad. The most active peptide was also analyzed using the 2D NMR technique (Fig. 5, Table S11†).
 | ||
| Fig. 5 Regular nonsequential interproton contacts in the NOESY spectrum of peptide 14 were recorded in 10 mM phosphate buffer pH 7.5 (10% D2O) at 293 K. The contacts between the same type of protons are marked in one color. | ||
The simulated annealing protocol with restraints derived from the NMR analysis provided three significant clusters of structures (Fig. 6 and S9–S11, and Table S12†). Therefore, strong conformational preferences are observed, although the structure is not entirely rigid. The main difference in between the clusters is the relative positioning of the N-terminal helix, with the overall Cα RMSD of averaged structures equal to 2.7 (cluster 1 to cluster 2), 2.0 (cluster 3 to cluster 2) and 2.3 Å (cluster 1 to cluster 3) but less than 0.9 Å in between C-terminal helices (Fig. 6D). The origin of this conformational difference is mainly because of different torsional angles ϕ and ψ of D-Ala9 residue. The spatial arrangement of the catalytic residues (lysine and tyrosine) in calculated structures resembles that observed in the known enzymes (e.g. RA95.5-8F, PDB id 5AN7) but is less compact and less precisely defined (Table S13†).
 | ||
| Fig. 6 Averaged structures of the largest clusters were calculated for NMR-derived structures for peptide 14. Clusters 1 (A), 2 (B), and 3 (C), and all three of them superimposed by the C-terminal helix (D). | ||
Conclusions
In this work, we developed a new approach for constructing enzyme-like catalysts based on peptide foldamer scaffolds. In a few rational steps using a single helix, two helices connected by a flexible loop, and helix–turn–helix structure as scaffolds, the foldamer that exhibits enzyme-like behavior following Michaelis–Menten kinetics and significantly enhances the retro-aldol reaction rate in water was created. The control of the rigidity of the applied helical structures, the linker between them, and the retention of the catalytically active motif in an appropriate conformation was crucial for achieving high turnover number. Analysis of mutants proved that all three residues in the catalytic triad of class I aldolase work cooperatively and are indispensable for effective catalysis. The obtained activity is comparable with another report on de novo-designed retro-aldolases. The acceleration of reaction shown by peptide 14 (kcat/kuncat = 1000) is slightly lower than aggregating β-peptide foldamer (kcat/kuncat = 3000)31 and one order of magnitude worse than the computationally designed retroaldolase RA61 (kcat/kuncat = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000).37 It is worth noting that although the elaborated sequence of 19 residues is relatively short, the peptide conformation can be effectively controlled, and a working active site can be constructed. Therefore, we developed a new rational method for de novo construction of efficient catalysts that does not present the limitations of other published methods, such as the need to test a large number of catalysts45,46 and the lack of control of the three-dimensional structure.47,48 However, it should be noted that presented structures allow controlling positioning of the active triad but do not form any cavity for substrate binding; thus, no selectivity is expected. The incorporation of such features could constitute the next challenge. The wide selection of functional groups readily implemented in solid-state synthesis and the possibility of extending studied sequences provide the possibility for these new future findings and indicate the potential utility of the proposed approach in the discovery of new functional molecules.
000).37 It is worth noting that although the elaborated sequence of 19 residues is relatively short, the peptide conformation can be effectively controlled, and a working active site can be constructed. Therefore, we developed a new rational method for de novo construction of efficient catalysts that does not present the limitations of other published methods, such as the need to test a large number of catalysts45,46 and the lack of control of the three-dimensional structure.47,48 However, it should be noted that presented structures allow controlling positioning of the active triad but do not form any cavity for substrate binding; thus, no selectivity is expected. The incorporation of such features could constitute the next challenge. The wide selection of functional groups readily implemented in solid-state synthesis and the possibility of extending studied sequences provide the possibility for these new future findings and indicate the potential utility of the proposed approach in the discovery of new functional molecules.
Experimental section
All commercially available reagents and solvents were purchased from Sigma-Aldrich, Merck, Iris Biotech, or Bachem.Peptide synthesis
cis-ACPC enantiomers used for the synthesis were synthesized according to the literature,49 and Fmoc-derivatives were obtained by a standard protocol.50 The peptides were synthesized using automated solid-state synthesizer Initiator+ Alstra (Biotage) on H-Rink amide resin (ChemMatrix), loading 0.59 mmol g−1 with DMF as a solvent. Fmoc deprotection was done in 20% piperidine solution at 75°, and 0.5 M DIC and 0.5 M Oxyma Pure® were used as coupling agents. For α-amino acids (5 eq., 0.1 M) double coupling was performed (2 × 15 min in 75°) and single coupling for β-amino acids (30 min in 75°). NMP/DIPE/acetic anhydride (80:15:5) mixture was used for acetylation, and TFA/TIS/H2O (95:2.5:2.5) for deprotection (3 h, RT). The crude peptides were precipitated with cold diethyl ether and centrifuged and purified by HPLC chromatography on Knauer Prep using preparative column 50 mm × 30 mm, Thermo Scientific™ Hypersil GOLD™ (C18, 12 μm) in water/ACN as a solvent. The purity was tested using analytical column 150 mm × 4.6 mm, Kinetex 100A (C18, 5 μm).
Mass spectrometry
The high-resolution mass spectra were acquired using WATERS LCT Premiere XE System with ESI ionization and via time of flight measurement.Circular dichroism
Spectra were recorded between 250 and 190 nm on JASCO J-815 with the following parameters: 20 °C, 0.2 nm resolution, 1.0 nm bandwidth, 50 nm min−1 scanning speed, 0.02 mm cuvette path length. All the peptides were analyzed 50 mM phosphate buffer pH 8 solutions. The data was recalculated to a mean residue molar ellipticity.NMR
The 1H ROESY and TOCSY NMR measurements were performed on a Bruker Avance spectrometer operating at 600.58 MHz with TMS as the external standard. The spectra were recorded in CD3OH for peptides 8, 9, 12, and 13 and in 50 mM phosphate buffer pH 7.5 with 10% addition of D2O for peptide 14 with the ±0.1 K temperature control with presaturation of the solvent signal. The TOCSY spectra were acquired with 256 or 512 increments of 16 scans and 60–200 scans for ROESY. TOCSY spectra were recorded with 60 ms mixing time. Spin lock time of the ROESY experiments was equal to 0.2 s. The NMR data was processed and analyzed on Topspin 3.2 (Bruker BioSpin) and Sparky 3.114 software.NMR structure calculation
The NMR structure generation for peptide 14 was performed in the Xplor-NIH v. 2.41.1 program.51 Initially, 100 random conformations were generated from the sequence using seqTOpsf protocol. NMR-derived interproton contacts were classified by standard method with upper distance limits: strong 2.5 Å, medium 3.5 Å, and weak 5 Å, and the lower distance limit was set to 1.8 Å. For the β-amino acid, 3 backbone torsions were restrained to match the previous experimental data with a tolerance of 30-degree deviation. Additionally, H-bonds in between residues were introduced, known to occur in the 9/12/9/10 helix, to improve the helicity of the structure. Standard simulated annealing protocols implemented in Xplor-NIH were used and composed of the following steps: (1) high-temperature dynamics (3500 K, 800 ps, or 8000 steps), (2) simulated annealing performed from 3500 K to 25 K with 12.5 K step, at each temperature short dynamics was done (100 steps or 0.2 ps); (3) gradient minimization of the final structure. As the top energy structures did not converge, the cluster analysis in GROMACS software was performed using gmx cluster protocol with RMSD cut-off over all atoms equal to 0.25 nm, and averaged structures for the three biggest clusters were calculated.Catalytic activity measurements
The racemic mixture of methodol was synthesized according to the literature.40 The retro-aldol reactions of methodol were performed at 37 °C in 50 mM phosphate buffer pH 8 in the presence of 0.2 mM peptide (or without for uncatalyzed reaction) for 6 different concentrations of substrate: 0.0625, 0.125, 0.25, 0.5, 1 and 2 mM. DMSO was added to the final concentration of 5% for better substrate solubility. The reaction progress was monitored by the fluorescent product formation (6-methoxy-2-naphthaldehyde) for at least 2 hours using a Gemini XPS spectrofluorometer (Molecular Devices) in 96 well-flat bottom plates. The excitation and emission wavelengths equal 330 and 452 nm, and PMT gain was set to medium. All the measurements were done in triplicates and averaged. The known product concentration (up to 0.15 mM) quantitated the fluorescence signal. The data (velocities vs. concentration of substrate) were fitted to the Michaelis–Menten model for enzyme-like kinetics (presence of saturation), where Et was set to constant value 0.2 mM using Graph Pad Prism 5.0 software according to equation: v0 = Et × kcat × [S]/(KM + [S]), where the Et was set to 0.2 mM. The values were not corrected for background rate if there was a substantial difference between the reaction rate in the presence and absence of the catalysts. The kcat was calculated as the first-order reaction constant for the linear dependencies.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Science Centre, Poland (Grant No. 2016/21/B/ST5/00269, to Ł. B.).References
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- A. D. Bautista, C. J. Craig, E. A. Harker and A. Schepartz, Curr. Opin. Chem. Biol., 2007, 11, 685–692 CrossRef CAS PubMed.
- T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687–701 RSC.
- C. M. Goodman, S. Choi, S. Shandler and W. F. Degrado, Nat. Chem. Biol., 2007, 3, 252–263 CrossRef CAS PubMed.
- S. De Pol, C. Zorn, C. D. Klein, O. Zerbe and O. Reiser, Angew. Chem., Int. Ed., 2004, 43, 511–514 CrossRef CAS PubMed.
- M. Szefczyk, E. Węglarz-Tomczak, P. Fortuna, A. Krzysztoń, E. Rudzińska-Szostak and Ł. Berlicki, Angew. Chem., Int. Ed., 2017, 2087–2091 CrossRef CAS PubMed.
- E. Rudzińska-Szostak and Ł. Berlicki, Chem. – Eur. J., 2017, 23, 14980–14986 CrossRef PubMed.
- I. M. Mándity, E. Wéber, T. A. Martinek, G. Olajos, G. K. Tóth, E. Vass and F. Fülöp, Angew. Chem., Int. Ed., 2009, 48, 2171–2175 CrossRef PubMed.
- Ł. Berlicki, L. Pilsl, E. Wéber, I. M. Mándity, C. Cabrele, T. a. Martinek, F. Fülöp and O. Reiser, Angew. Chem., Int. Ed., 2012, 51, 2208–2212 CrossRef PubMed.
- D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS PubMed.
- F. Fülöp, T. A. Martinek and G. K. Tóth, Chem. Soc. Rev., 2006, 35, 323–334 RSC.
- D. H. Appella, L. A. Christianson, L. A. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071–13072 CrossRef CAS.
- E. A. Porter, X. Wang, M. A. Schmitt and S. H. Gellman, Org. Lett., 2002, 4, 3317–3319 CrossRef CAS PubMed.
- L. K. A. Pilsl and O. Reiser, Amino Acids, 2011, 41, 709–718 CrossRef CAS PubMed.
- D. Seebach, S. Abele, K. Gademann and B. Jaun, Angew. Chem., Int. Ed., 1999, 38, 1595–1597 CrossRef CAS PubMed.
- T. A. Martinek, G. K. Tóth, E. Vass, M. Hollósi and F. Fülöp, Angew. Chem., Int. Ed., 2002, 41, 1718–1721 CrossRef CAS PubMed.
- K. Ożga and Ł. Berlicki, ACS Bio Med Chem Au, 2022, 2, 316–327 CrossRef PubMed.
- G. Olajos, A. Hetényi, E. Wéber, L. J. Németh, Z. Szakonyi, F. Fülöp and T. A. Martinek, Chem. – Eur. J., 2015, 21, 6173–6180 CrossRef CAS PubMed.
- D. F. Kreitler, D. E. Mortenson, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 6498–6505 CrossRef CAS PubMed.
- Z. Reinert, G. Lengyel and W. Horne, J. Am. Chem. Soc., 2013, 135, 12528–12531 CrossRef CAS PubMed.
- M. Drewniak-Świtalska, B. Barycza, E. Rudzińska-Szostak, P. Morawiak and Ł. Berlicki, Org. Biomol. Chem., 2021, 19, 4272–4278 RSC.
- J. L. Price, E. B. Hadley, J. D. Steinkruger and S. H. Gellman, Angew. Chem., Int. Ed., 2010, 49, 368–371 CrossRef CAS PubMed.
- M. Bejger, P. Fortuna, M. Drewniak-Świtalska, J. Plewka, W. Rypniewski and Ł. Berlicki, Chem. Commun., 2021, 57, 6015–6018 RSC.
- K. Ożga, M. Drewniak-Świtalska, E. Rudzińska-Szostak and Ł. Berlicki, ChemPlusChem, 2021, 86, 646–649 CrossRef PubMed.
- C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed.
- P. Fortuna, A. Twarda-Clapa, L. Skalniak, K. Ożga, T. A. Holak and Ł. Berlicki, Eur. J. Med. Chem., 2020, 208, 112814 CrossRef CAS PubMed.
- P. Fortuna, B. M. Linhares, T. Purohit, J. Pollock, T. Cierpicki, J. Grembecka and Ł. Berlicki, Eur. J. Med. Chem., 2020, 207, 112748 CrossRef CAS PubMed.
- P. Wójcik and Ł. Berlicki, Bioorg. Med. Chem. Lett., 2016, 26, 707–713 CrossRef PubMed.
- M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2004, 126, 6848–6849 CrossRef CAS PubMed.
- Z. C. Girvin, M. K. Andrews, X. Liu and S. H. Gellman, Science, 2019, 366, 1528–1531 CrossRef CAS PubMed.
- M. Müller, M. Windsor, W. C. Pomerantz, S. Gellman and D. Hilvert, Angew. Chem., 2009, 121, 940–943 CrossRef.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, T. A. Smith, X. Hu, W. F. Degrado, I. V. Korendovych and S. Francisco, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- R. R. Araghi and B. Koksch, Chem. Commun., 2011, 47, 3544–3546 RSC.
- M. L. Zastrow and V. L. Pecoraro, Biochemistry, 2014, 53, 957–978 CrossRef CAS PubMed.
- C. Walsh, Nature, 2001, 409, 226–231 CrossRef CAS PubMed.
- L. Jiang, E. Althoff and F. Clemente, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed.
- E. A. Althoff, L. Wang, L. Jiang, L. Giger, J. K. Lassila, Z. Wang, M. Smith, S. Hari, P. Kast, D. Herschlag, D. Hilvert and D. Baker, Protein Sci., 2012, 21, 717–726 CrossRef CAS PubMed.
- R. Obexer, A. Godina, X. Garrabou, P. R. E. Mittl, D. Baker, A. D. Griffiths and D. Hilvert, Nat. Chem., 2017, 9, 50–56 CrossRef CAS PubMed.
- K. Tittmann, Bioorg. Chem., 2014, 57, 263–280 CrossRef CAS PubMed.
- J. Schmidt, C. Ehasz, M. Epperson, K. Klas, J. Wyatt, M. Hennig and M. Forconi, Org. Biomol. Chem., 2013, 11, 8419–8425 RSC.
- A. Dalby, Z. Dauter and J. Littlechild, Protein Sci., 1999, 8, 291–297 CrossRef CAS PubMed.
- E. Lorentzen, E. Pohl, P. Zwart, A. Stark, R. B. Russell, T. Knura, R. Hensel and B. Siebers, J. Biol. Chem., 2003, 278, 47253–47260 CrossRef CAS PubMed.
- E. Lorentzen, B. Siebers, R. Hensel and E. Pohl, Biochem. Soc. Trans., 2004, 32, 259–263 CrossRef CAS PubMed.
- M. Drewniak, E. Węglarz-Tomczak, K. Ożga, E. Rudzińska-Szostak, K. Macegoniuk, J. M. Tomczak, M. Bejger, W. Rypniewski and Ł. Berlicki, Bioorg. Chem., 2018, 81, 356–361 CrossRef CAS PubMed.
- J. Kofoed, T. Darbre and J. Reymond, Org. Biomol. Chem., 2006, 4, 3268–3281 RSC.
- F. Tanaka, R. Fuller and C. F. Barbas, Biochemistry, 2005, 44, 7583–7592 CrossRef CAS PubMed.
- M. Barbany, H. Gutiérrez-de-Terán, F. Sanz, J. Villà-Freixa and A. Warshel, ChemBioChem, 2003, 4, 277–285 CrossRef CAS PubMed.
- N. Singh, K. Zhang, C. A. Angulo-Pachon, E. Mendes, J. H. van Esch and B. Escuder, Chem. Sci., 2016, 7, 5568–5572 RSC.
- S. G. Davies, O. Ichihara, I. Lenoir and I. A. S. Walters, J. Chem. Soc., Perkin Trans. 1, 1994, 1411–1415 RSC.
- L. Lapatsanis, G. Milias, K. Froussios and M. Kolovos, Synthesis, 1983, 51, 671–673 CrossRef.
- C. D. Schwieters, J. J. Kuszewski and G. Marius Clore, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 47–62 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00342j |
| This journal is © The Royal Society of Chemistry 2024 |