
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi-catalyzed undirected and regioselective acceptorless dehydrogenative silylation of primary benzylic C(sp3)–H bonds†
Qing
Yu
a,
Takafumi
Yatabe
 *ab,
Takehiro
Matsuyama
a,
Tomohiro
Yabe
a and
Kazuya
Yamaguchi
*a
*ab,
Takehiro
Matsuyama
a,
Tomohiro
Yabe
a and
Kazuya
Yamaguchi
*a
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: yatabe@appchem.t.u-tokyo.ac.jp; kyama@appchem.t.u-tokyo.ac.jp; Fax: +81 3 5841 7220
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 6th March 2024
Abstract
The dehydrogenative silylation of ubiquitous inactive C–H bonds provides an eco-friendly synthetic protocol for useful organosilicon compounds. Compared with the well-established C(sp2)–H dehydrogenative silylation, C(sp3)–H dehydrogenative silylation is immature and generally requires directing groups such as heterocyclic structures and intramolecular Si–H groups. In particular, the undirected benzylic C(sp3)–H silylation has been hardly reported partly because of the difficult regioselectivity control between benzylic C(sp3)–H and aryl C(sp2)–H bonds. In this work, an undirected acceptorless dehydrogenative silylation with an excellent benzylic C(sp3)–H selectivity was successfully developed using a CeO2-supported highly dispersed Ni(0) nanocatalyst, which was prepared via reduction with pinacolborane. Surprisingly, the dehydrogenative silylation occurred preferentially at the primary benzylic positions even in the presence of secondary ones, possibly due to the steric hindrance of substrates on the Ni catalyst.
Introduction
Dehydrogenative silylation of C–H bonds is an ideal approach for the synthesis of organosilicon compounds, which are widely utilized in the fields of synthetic organic chemistry, polymer chemistry, materials chemistry, and medicinal chemistry.1–4 Compared with conventional synthetic methods for organosilicon compounds, such as the Rochow process5 and coupling reactions with organometallic compounds,6 which require prefunctionalization steps such as halogenation, the one-step dehydrogenative silylation of C–H bonds with hydrosilanes offers a high atom economy and considerably more eco-friendly routes to the desired organosilicon compounds.Although the catalytic dehydrogenative silylation of aryl and vinyl C(sp2)–H bonds has been achieved with wide substrate scopes even under mild conditions,7–11 the C(sp3)–H silylation is comparatively quite limited.4,12 In general, directing groups including heterocyclic structures and intramolecular Si–H groups are indispensable for the dehydrogenative silylation of C(sp3)–H bonds (Scheme 1a).4,12–14 In other words, undirected intermolecular C(sp3)–H dehydrogenative silylations have been hardly reported (Scheme 1b). As the first example of a catalytic undirected dehydrogenative C(sp3)–H silylation, Berry and Procopio reported a C(sp3)–H silylation of methyl groups in trimethylsilane (Scheme 1b),15 although the trace amount formation of benzylic C(sp3)–H silylated product from toluene was reported with quite low selectivity by Tanaka et al. in 1987.16 Then, Ishikawa et al. reported in 1992 a Ni-catalyzed benzylic C(sp3)–H silylation, which was limited to only mesitylene (Scheme 1b).17 In 1994, Berry et al. described a C(sp3)–H silylation of ethyl groups in triethylsilane in the presence of a Ru or Rh catalyst with tBuCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (Scheme 1b).18 Meanwhile, Tilley and Sadow reported in 2003 a silylation of methane (150 atm) and cyclopropane catalyzed by a Sc catalyst (Scheme 1b).19 Although these studies constitute pioneering examples of undirected dehydrogenative C(sp3)–H silylation, these reaction systems are impractical in organic synthetic chemistry because of their limited substrate scope and harsh reaction conditions.
CH2 (Scheme 1b).18 Meanwhile, Tilley and Sadow reported in 2003 a silylation of methane (150 atm) and cyclopropane catalyzed by a Sc catalyst (Scheme 1b).19 Although these studies constitute pioneering examples of undirected dehydrogenative C(sp3)–H silylation, these reaction systems are impractical in organic synthetic chemistry because of their limited substrate scope and harsh reaction conditions.
 | ||
| Scheme 1 Overview of the catalytic dehydrogenative silylation of C–H bonds: (a) comparison of C(sp2)–H silylation and C(sp3)–H silylation; (b) limited examples of undirected C(sp3)–H silylation; (c) the present undirected and regioselective benzylic C(sp3)–H silylation. | ||
In general, compared with aliphatic C(sp3)–H bonds, benzylic C(sp3)–H bonds can be easily and selectively functionalized using various reaction systems;20 however, reports on undirected dehydrogenative benzylic C(sp3)–H silylation are virtually nonexistent probably because of the difficulty in the dehydrogenative C(sp3)–H silylation itself and in the regioselectivity control between benzylic C(sp3)–H and aryl C(sp2)–H bonds.12 For example, in the aforementioned report by Ishikawa et al., when using p-xylene as the substrate instead of mesitylene, the selective dehydrogenative silylation of aryl C(sp2)–H bonds was observed, which suggested that aryl C(sp2)–H bonds were preferentially silylated owing to the lower steric hindrance in p-xylene compared with mesitylene (Scheme 1b).17 In fact, in metal-complex-catalyzed systems for the C–H functionalization of alkylarenes requiring C–H activation steps, including dehydrogenative silylation, the metal center can activate more easily C(sp2)–H bonds due to the interaction between the π orbitals of the substrates and the empty orbitals of the metal, whereas C(sp3)–H activation is less favorable due to the lack of π orbitals and the comparatively free bond angles.13,21–23 A more practical example of undirected dehydrogenative benzylic C(sp3)–H silylation using a Co-functionalized metal–organic framework (UiO-Co) catalyst was reported; unfortunately, extremely long reaction times (2–6 days) and solvent amounts of the alkylarene substrates were required.24 Other reports using base catalysts for undirected dehydrogenative benzylic C(sp3)–H silylation demonstrated very limited substrate scopes.25,26 Quite recently, a KOtBu-catalyzed benzylic C(sp3)–H silylation with broad substrate scopes was reported by Chauvier et al.; however, a special silylating reagent, tert-butyl-substituted silyldiazenes, was indispensable.27 Therefore, to date, undirected C(sp3)–H dehydrogenative silylation with hydrosilanes has not been firmly established even for benzylic C(sp3)–H bonds.
In contrast, dehydrogenative borylation to organoboron compounds, which possess useful properties similar to those of organosilicon compounds due to the diagonal relationship between boron and silicon,28–31 has been developed into a mature process for both directed and undirected C(sp3)–H activation processes.32,33 In fact, as one of the examples of undirected C(sp3)–H borylation, we reported a heterogeneously catalyzed highly selective benzylic C(sp3)–H borylation of alkylarenes in the presence of Ni hydroxide species supported on cerium oxide (Ni(OH)x/CeO2) using pinacolborane (HBpin) as the borylating reagent, which also acted as the in situ reductant for Ni hydroxide species to afford catalytically active ultrasmall Ni(0) nanospecies.34 The results demonstrated the potential of this heterogeneous catalytic system for realizing other regioselective undirected benzylic C(sp3)–H functionalizations different from the typical selective activation of aryl C(sp2)–H bonds using metal complex catalysts. Considering that the dehydrogenative silylation is typically less favorable kinetically and thermodynamically than the corresponding dehydrogenative borylation,35 the application of a heterogeneous Ni catalyst system to an undirected C(sp3)–H dehydrogenative silylation would require a more refined preparation of highly active Ni(0) nanocatalysts instead of our previously reported Ni(0) formation via in situ reduction.34
Herein, we developed an efficient undirected and regioselective benzylic C(sp3)–H acceptorless dehydrogenative silylation with triethoxysilane (HSi(OEt)3) using a CeO2-supported Ni(0) nanocatalyst (Ni/CeO2–HBpin), which was prepared via pretreatment under suitable conditions using HBpin as the reductant (Scheme 1c). This catalytic benzylic C(sp3)–H silylation system demonstrated a wide substrate scope of alkylarenes without dehydrogenative silylation of aryl C(sp2)–H bonds to afford various primary benzylsilanes, which are useful synthetic intermediates but inaccessible using conventional hydrosilylation routes.27,36,37 Ni/CeO2–HBpin functioned as a heterogeneous catalyst in the present silylation. Noteworthily, when both primary and secondary benzylic C(sp3)–H bonds were present, primary bonds were preferentially silylated (Scheme 1c). Since the oxidation of primary benzylic C(sp3)–H bonds is generally more difficult than that of secondary ones,38–41 such a selective dehydrogenative primary benzylic C(sp3)–H functionalization system is quite rare. Our results suggest that the corresponding C(sp3)–Si bond formation from secondary benzylic C(sp3)–H bonds on the Ni catalyst is inhibited by the steric hindrance of the substrates, leading to the unusual selectivity toward primary benzylic C(sp3)–H bonds. As summarized in Table S1,† the present catalytic undirected and acceptorless C(sp3)–H silylation more efficiently proceeds with a broader substrate scope than previously reported ones using a commercially available hydrosilane as the silylating reagent and affords benzylsilanes in a unique regioselective manner to primary benzylic positions. Furthermore, when comparing the present heterogeneously catalyzed system with the aforementioned previously reported homogeneously or heterogeneously catalyzed efficient undirected benzylic silylation systems showing comparatively broad substrate scopes24,27 in terms of green chemistry indicators like atom economy (AE), E-factor, and turnover frequency (TOF) using benzylic silylation of toluene as the representative example, all of the indicators were proved to be tremendously improved in this study as shown in Fig. S1† (AE = 99%, E-factor = 0.008, TOF = 176 h−1 at 120 °C for the silylation of toluene).
Results and discussion
Ni/CeO2–HBpin with a Ni content of 1.4 wt%, as determined by means of inductively coupled plasma atomic emission spectroscopy (ICP-AES), was prepared via a deposition–precipitation method to afford Ni(OH)x/CeO2 followed by reduction using HBpin (8 mmol HBpin for 1 g of Ni(OH)x/CeO2) (see ESI† for details). The X-ray diffraction (XRD) patterns of CeO2, Ni(OH)x/CeO2, and Ni/CeO2–HBpin exhibited no difference, indicating that large Ni species were not formed during the catalyst preparation (Fig. 1a). The Ni K-edge X-ray absorption near-edge structure (XANES) spectrum of Ni/CeO2–HBpin revealed the presence of Ni(0) species (Fig. 1b), which accounted for 57% of the Ni species in Ni/CeO2–HBpin according to a linear combination fitting (LCF) using Ni(OH)x/CeO2 and Ni foil (Fig. S2a†). The k3-weighted Fourier-transformed Ni K-edge extended X-ray absorption fine structure (EXAFS) spectrum of Ni/CeO2–HBpin showed no clear peaks of second coordination spheres, which together with the curve fitting analysis, suggested the presence of highly dispersed Ni(0) nanospecies (Fig. 1c and S3 and Table S2†). In addition, although the Ni nanospecies in Ni/CeO2–HBpin could not be clearly observed in a high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis due to the high electron density of CeO2 (Fig. 1d), the corresponding STEM energy-dispersive X-ray spectroscopy (EDS) mapping confirmed that Ni nanospecies were highly dispersed on the CeO2 surface (Fig. 1e–g). The STEM-EDS mapping of Ni/CeO2–HBpin also revealed the presence of B species derived from the HBpin reductant, which were highly dispersed on CeO2 but not localized near the Ni species (Fig. S4†). The X-ray photoelectron spectroscopy (XPS) analysis of Ni/CeO2–HBpin around the B 1s region also indicated the presence of B species attributable to boron oxide or boric acid derivatives but not to NiB species (Fig. S5a†).42,43 Thus, the B species are not likely to affect the Ni(0) nanospecies. In fact, the Ni(0) ratio to Ni(II) species observed in the XPS analysis of Ni/CeO2–HBpin around the Ni 2p region was almost the same as that determined via the LCF of the Ni K-edge XANES spectra (Fig. 1h, S2a and S6a†). Moreover, when a diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analysis was performed to evaluate the CO chemisorption on the Ni(0) nanospecies of Ni/CeO2–HBpin (Fig. 1i), a large CO chemisorption peak in the linear region and a comparatively small CO chemisorption peak in the bridged region were observed. These results indicated the existence of accessible surface sites of highly dispersed Ni(0) nanospecies in Ni/CeO2–HBpin.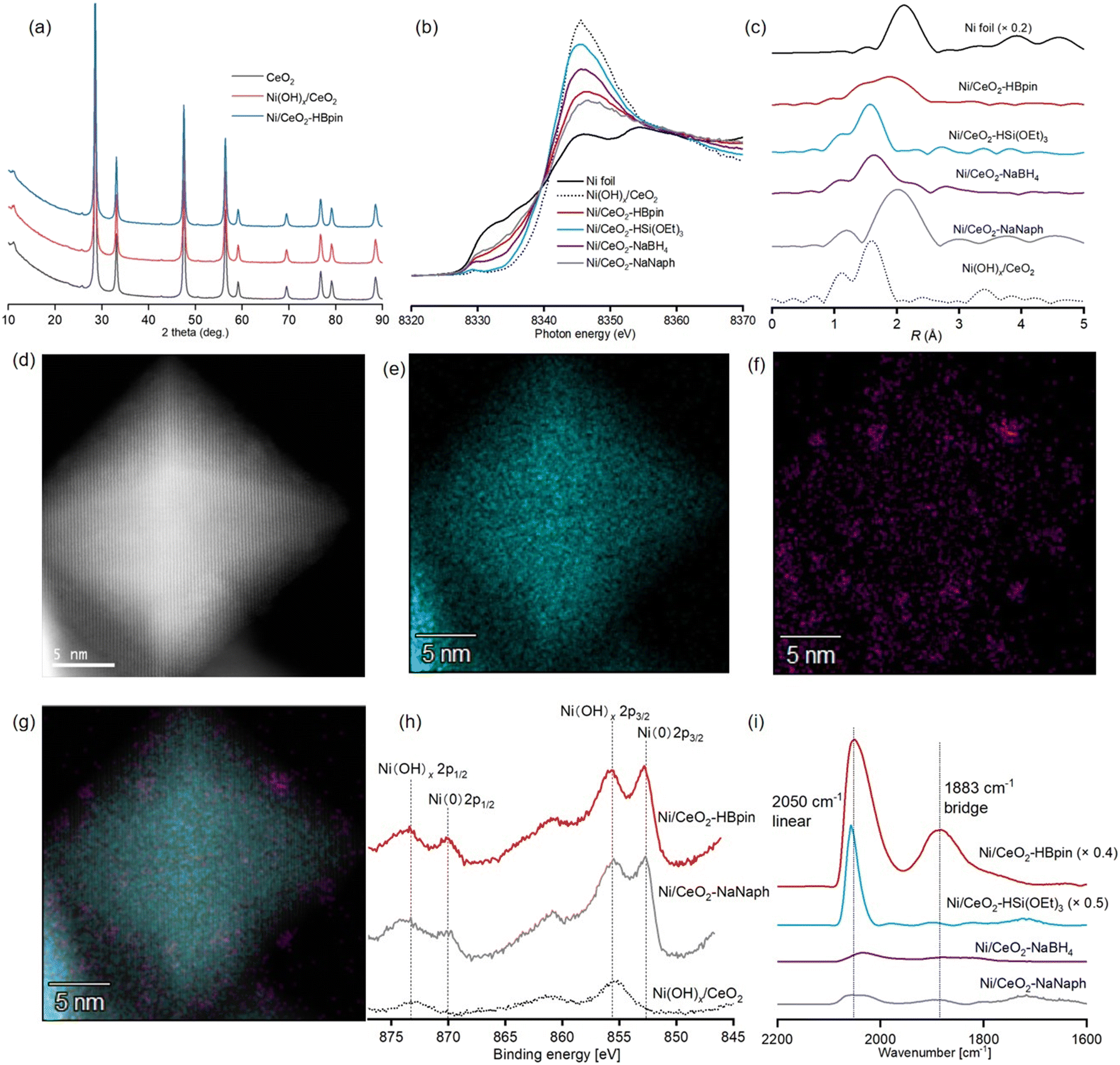 | ||
| Fig. 1 Characterization of Ni catalysts: (a) XRD patterns; (b) Ni K-edge XANES spectra; (c) Fourier-transformed k3-weighted Ni K-edge EXAFS spectra; (d) HAADF-STEM image of Ni/CeO2–HBpin; (e–g) STEM-EDS mappings of Ni/CeO2–HBpin, showing the distributions of Ce in cyan (panel (e)), Ni in magenta (panel (f)), and overlap of panels (d–f) (panel (g)); (h) XPS spectra around the Ni 2p region; (i) DRIFTS of CO chemisorption on Ni catalysts. The spectra after CO adsorption (PCO = ∼15 Torr) followed by 1 min evacuation at room temperature are shown after Kubelka–Munk transformation using the spectra in vacuo before CO adsorption as backgrounds. | ||
Initially, the dehydrogenative silylation of toluene (1a) with HSi(OEt)3 was conducted in methylcyclohexane at 120 °C under an Ar atmosphere using various supported Ni catalysts (represented as Ni/support-reductant in Table 1). On the basis of our previous report on the dehydrogenative benzylic borylation using HBpin as both the borylating reagent and reductant of Ni species in Ni(OH)x/CeO2,34 we expected that Ni(OH)x/CeO2 would be reduced by HSi(OEt)3 in the reaction system to afford the catalytically active species; however, no silylated products were detected in the presence of Ni(OH)x/CeO2 (Table 1, entry 1). The color of Ni(OH)x/CeO2 was not much changed into black during the silylation reaction compared with the case of borylation using HBpin under the same reaction conditions (Fig. S7†), suggesting that the reduction of Ni(II) species to catalytically active Ni(0) species via in situ treatment with HSi(OEt)3 was insufficient. Thus, Ni(OH)x/CeO2 was treated with various reductants, i.e., HSi(OEt)3, HBpin, sodium borohydride (NaBH4), and sodium naphthalenide (NaNaph),44 to prepare CeO2-supported Ni(0) nanocatalysts (see ESI† for details), which were characterized via Ni K-edge XAFS (Fig. 1b and c, S2 and S3 and Table S2†), DRIFTS of CO chemisorption (Fig. 1i), and XPS (Fig. 1h, S5, S6, S8 and S9†) analyses. The results confirmed the generation of Ni/CeO2–HBpin possessing highly dispersed Ni(0) nanospecies, which successfully furnished the corresponding silylated product (Table 1, entry 2). Surprisingly, this reaction afforded selectively the desired benzyltriethoxysilane (2a) as the product of the benzylic C(sp3)–H dehydrogenative silylation in 79% yield after only 15 min without any aryl C(sp2)–H silylated products (2a′)‡ (Table 1, entry 2). By contrast, the catalytic activity of Ni/CeO2–HSi(OEt)3 in the dehydrogenative silylation was quite low (Table 1, entry 3) probably because of the insufficient amount of Ni(0) species, as revealed by the Ni K-edge XANES spectrum (Fig. 1b and S2b†). Meanwhile, although the Ni K-edge XAFS and XPS spectra of Ni/CeO2–NaBH4 and Ni/CeO2–NaNaph suggested the presence of sufficient amounts of highly dispersed Ni(0) nanospecies in the catalysts (Fig. 1b, c and h, S2 and S3 and Table S2†), no silylated products were observed (Table 1, entries 4 and 5). The DRIFTS of CO chemisorption revealed that the number of surface active sites of Ni(0) nanospecies in Ni/CeO2–NaBH4 and Ni/CeO2–NaNaph was much smaller than that in Ni/CeO2–HBpin and Ni/CeO2–HSi(OEt)3 (Fig. 1i) probably due to the coverage of the active sites by reductant-derived residues like boric acid derivatives, sodium species, and naphthalene, which was supported by their XPS spectra around the B 1s, Na 1s, and C 1s regions (Fig. S5, S8 and S9†). Thus, the high catalytic activity of Ni/CeO2–HBpin to produce 2a can be attributed to the sufficient amount of accessible highly dispersed Ni(0) nanospecies that were not covered by reductant-derived residues.§ As the method without fear of the coverage by reductant-derived residues, H2 reduction of Ni(OH)x/CeO2 was also conducted, but the dehydrogenative silylation did not occur using Ni/CeO2–H2 (Table 1, entry 6) as with the case of our previous report on dehydrogenative borylation probably due to the aggregation of Ni(0) species during the reduction requiring high temperatures (∼400 °C).34 In addition, no improvement in the yield of 2a was observed when conducting the dehydrogenative silylation of 1a in the presence of Ni/CeO2–HSi(OEt)3, Ni/CeO2–NaBH4, or Ni/CeO2–NaNaph for 24 h (Table S3†). Moreover, the addition of CeO2 or CeO2–HBpin hardly affected the Ni/CeO2–HBpin-catalyzed silylation of 1a, whereas the Ni/CeO2–HBpin catalysis was considerably inhibited in the presence of Ni/CeO2–NaBH4 or Ni/CeO2–NaNaph (Table S4†). Considering that the conversion of HSi(OEt)3 in the absence of 1a or Ni catalysts was much more effectively promoted by CeO2–NaBH4 and CeO2–NaNaph than by CeO2–HBpin (Table S5†), the reductant-derived residues or the active sites of CeO2 in Ni/CeO2–NaBH4 and Ni/CeO2–NaNaph (see the XPS spectra around Ce 3d region in Fig. S10†) possibly caused decomposition of HSi(OEt)3 into SiO2 or related species, covering the Ni active sites and leading to catalyst deactivation during the silylation reaction. Therefore, the high catalytic performance of Ni/CeO2–HBpin in the present dehydrogenative silylation stems probably from its highly dispersed catalytically active Ni(0) nanospecies with few surface sites available for the side reactions of HSi(OEt)3.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield (%) | |
| 2a | 2a′ | ||
| a Reaction conditions: 1a (2 mmol), HSi(OEt)3 (0.4 mmol), catalyst (Ni: 1.8 mol%), methylcyclohexane (1 mL), Ar (1 atm), 120 °C, 15 min. Yields were determined via GC using n-hexadecane as an internal standard. b 1 h. c 1a (0.4 mmol). | |||
| 1 | Ni(OH)x/CeO2 | <1 | <1 |
| 2 | Ni/CeO2–HBpin | 79 | <1 |
| 3 | Ni/CeO2–HSi(OEt)3 | 8 | <1 |
| 4 | Ni/CeO2NaBH4 | <1 | <1 |
| 5 | Ni/CeO2–NaNaph | <1 | <1 |
| 6 | Ni/CeO2–H2 | <1 | <1 |
| 7 | Ni/TiO2–HBpin | <1 | <1 |
| 8 | Ni/ZrO2–HBpin | <1 | <1 |
| 9 | Ni/Al2O3–HBpin | <1 | <1 |
| 10 | Ni/Co3O4–HBpin | <1 | <1 |
| 11 | Ni/HAP–HBpin | 2 | <1 |
| 12b | Ni/CeO2–HBpin | 93 | <1 |
| 13b,c | Ni/CeO2–HBpin | 57 | <1 |
Next, other factors affecting the present dehydrogenative silylation, including HBpin reductant amounts (Table S6†), support effects (Table 1), and metal effects (Table S7†), were investigated. Possibly because the balance between the amount and the size of Ni(0) nanospecies was important, 8 mmol of HBpin for 1 g of Ni(OH)x/CeO2 was found to be the optimal reductant amount for this reaction (Fig. S3 and S11, Tables S2 and S6†). The support and metal effects were also prominent; no catalytic activities were observed for Ni catalysts on supports other than CeO2 (TiO2, ZrO2, Al2O3, Co3O4, or HAP) and CeO2-supported 3d metal catalysts other than Ni (Fe, Co, Cu, Zn, or Mn) (Table 1, entries 7–11, and Table S7†). Thus, only the combination of Ni and CeO2 was effective for the dehydrogenative silylation, as in our previous report on the dehydrogenative borylation using Ni(OH)x/CeO2 with HBpin, possibly because the redox properties of CeO2 promoted the formation of Ni(0) active species via reduction with HBpin.34 After optimizing the reaction solvents (Table S8†), reaction temperatures (Table S9†), and the amount of 1a (Table S10†), the desired product 2a was selectively obtained in 93% yield using Ni/CeO2–HBpin for 1 h (Table 1, entry 12). Under the optimized conditions, an H2 yield of 82% was observed via gas chromatography (GC) analysis, which is almost consistent with the yield of 2a, indicating that the reaction proceeds via acceptorless dehydrogenation. Moreover, even when using an equimolar amount of 1a to HSi(OEt)3, the desired 2a was selectively obtained albeit in a moderate yield (Table 1, entry 13).
A hot filtration test confirmed that Ni/CeO2–HBpin worked as a heterogeneous catalyst in the present dehydrogenative silylation (Fig. S12†). Also, Ni species were hardly detected via ICP-AES in the filtrate after 1a was subjected to the optimized reaction conditions for 1 h (Ni: 0.7% of the used catalyst). However, Ni/CeO2–HBpin was difficult to be reused; the yield of 2a after 1 h of the reaction was ∼20% using the 1st used catalyst, and the reaction using the 2nd used catalyst hardly proceeded. In the infrared absorption spectrometry-attenuated total reflection (IR-ATR) spectrum of the 1st used Ni/CeO2–HBpin, a strong absorbance assignable to Si–O–Si bonds was observed, suggesting that the formation of SiO2 or related species on the surface of the catalyst could be responsible for its deactivation (Fig. S13†). Furthermore, according to the Ni K-edge XANES and EXAFS spectra of the 1st used catalyst, Ni(0) species were partially oxidized to Ni(II) species during the reaction, which could be also the reason for the deactivation (Fig. S2, S3 and S11, Table S2†).
The substrate scope of the Ni/CeO2–HBpin-catalyzed dehydrogenative benzylic silylation of alkylarenes was also investigated (Scheme 2). The catalytic system afforded a variety of benzylsilanes (2) via the benzylic C(sp3)–H silylation of alkylarenes 1 with HSi(OEt)3 in a regioselective manner without any C(sp2)–H silylation product (<1% yields). The observed byproducts in the investigation of the substrate scope were summarized in Table S11.† The obtained benzylsilanes were easily isolated via simple column chromatography on silica gel. As mentioned above, 2a was efficiently obtained from 1a in a 79% isolated yield. The gram-scale synthesis of 2a was also carried out, resulting in the moderate 2a yield (Fig. S14†). When 2-, 3-, or 4-methyl-substituted toluene was used as the substrate, the corresponding monosilylated products 2b–2d were obtained. Furthermore, toluene derivatives with methoxy (2e–2g), dimethylamino (2h), trifluoromethyl (2i, 2j), and ester (2k) functional groups could be synthesized using this system. Mesitylene was also applicable to this selective benzylic C(sp3)–H monosilylation (2l). To our surprise, in the case of alkyl-substituted toluenes possessing secondary benzylic C(sp3)–H bonds, the primary benzylic C(sp3)–H bonds were regiospecifically silylated (2m–2o). This unique regioselectivity in undirected benzylic dehydrogenative silylation is unprecedented.24–27 Furthermore, considering that secondary benzylic C(sp3)–H bonds are more easily activated than primary ones in general,38–41 the present regioselectivity is quite rare even among benzylic C–H functionalization systems. Unfortunately, several alkylarenes were not applicable to this catalytic silylation system as summarized in Fig. S15.† Alkylarenes with only secondary benzylic C(sp3)–H bonds were hardly silylated in the presence of Ni/CeO2–HBpin (2p–2r), indicating its inherent low activity to secondary benzylic C(sp3)–H bonds. Moreover, silylating reagents other than HSi(OEt)3 were unsuitable for this system, as discussed below (Table S12†).
 | ||
| Scheme 2 Substrate scope of the present regioselective dehydrogenative silylation. aReaction conditions: 1 (2 mmol), HSi(OEt)3 (0.4 mmol), Ni/CeO2–HBpin (32 mg, Ni: 1.8 mol%), methylcyclohexane (1 mL), Ar (1 atm), 120 °C, 24 h. Yields were determined via GC using n-hexadecane as an internal standard. Isolated yields are shown in parentheses. The secondary C(sp3)–H silylated product (2°) yields were determined via GC. b1 h. c6 h. d8 h. eHSi(OEt)3 (0.1 mmol), 140 °C. fHSi(OEt)3 (0.1 mmol). gHSi(OEt)3 (0.2 mmol). hUsing biphenyl as an internal standard. | ||
To reveal the reaction mechanism of the present acceptorless dehydrogenative silylation, several control experiments were performed (Fig. 2). First, the addition of 3,5-di-tert-butyl-4-hydroxytoluene as a radical scavenger to the reaction of 1a (Fig. S16†) did not affect the reaction rate and 2a yield, suggesting that a radical mechanism can be excluded. Next, to investigate the C–H activation mechanism, a benzylic deuterated alkylarene, heptylbenzene-d2 (benzylic deuterated ratio = 92%), was reacted with Ni/CeO2–HBpin in methylcyclohexane at 120 °C for 4 h (Fig. 2a). As a result, the deuterated ratio of the benzylic position dropped to 26%, while the homobenzylic C(sp3)–H bond and the other C(sp3)–H bonds were considerably deuterated and the total amount of deuterium in the heptylbenzenes was maintained after the reaction, according to 1H NMR, 2H NMR, and GC–mass spectrometry (MS) analyses (Fig. S17†). This result indicated that the H/D scrambling probably occurred via oxidative addition of the benzylic C(sp3)–D bond to Ni(0) species followed by a sequence of β-hydrogen elimination producing Ni–H(D) species/insertion of Ni–H(D) species to alkenes (i.e., Ni-catalyzed chain-walking)45,46 and subsequent reductive elimination to regenerate the Ni(0) species. Additionally, when HSi(OEt)3 was reacted with toluene-d8 (1a-d8) in the presence of Ni/CeO2–HBpin, partially deuterated HSi(OEt)3 was observed in a GC-MS analysis (Fig. 2b and S18†). Therefore, both benzylic C(sp3)–H bonds and the Si–H bond of HSi(OEt)3 were possibly activated via reversible oxidative addition on the Ni(0) nanocatalyst. In addition, when dimethylphenylsilane (HSiMe2Ph) was used instead of HSi(OEt)3 in the reaction with toluene-d8 and Ni/CeO2–HBpin, no deuteration of the silane was observed (Fig. S18†), indicating that the oxidative addition of HSiMe2Ph did not occur. Thus, an electron-deficient silane like HSi(OEt)3, which can be more easily activated via oxidative addition, is indispensable for the present dehydrogenative silylation reaction.
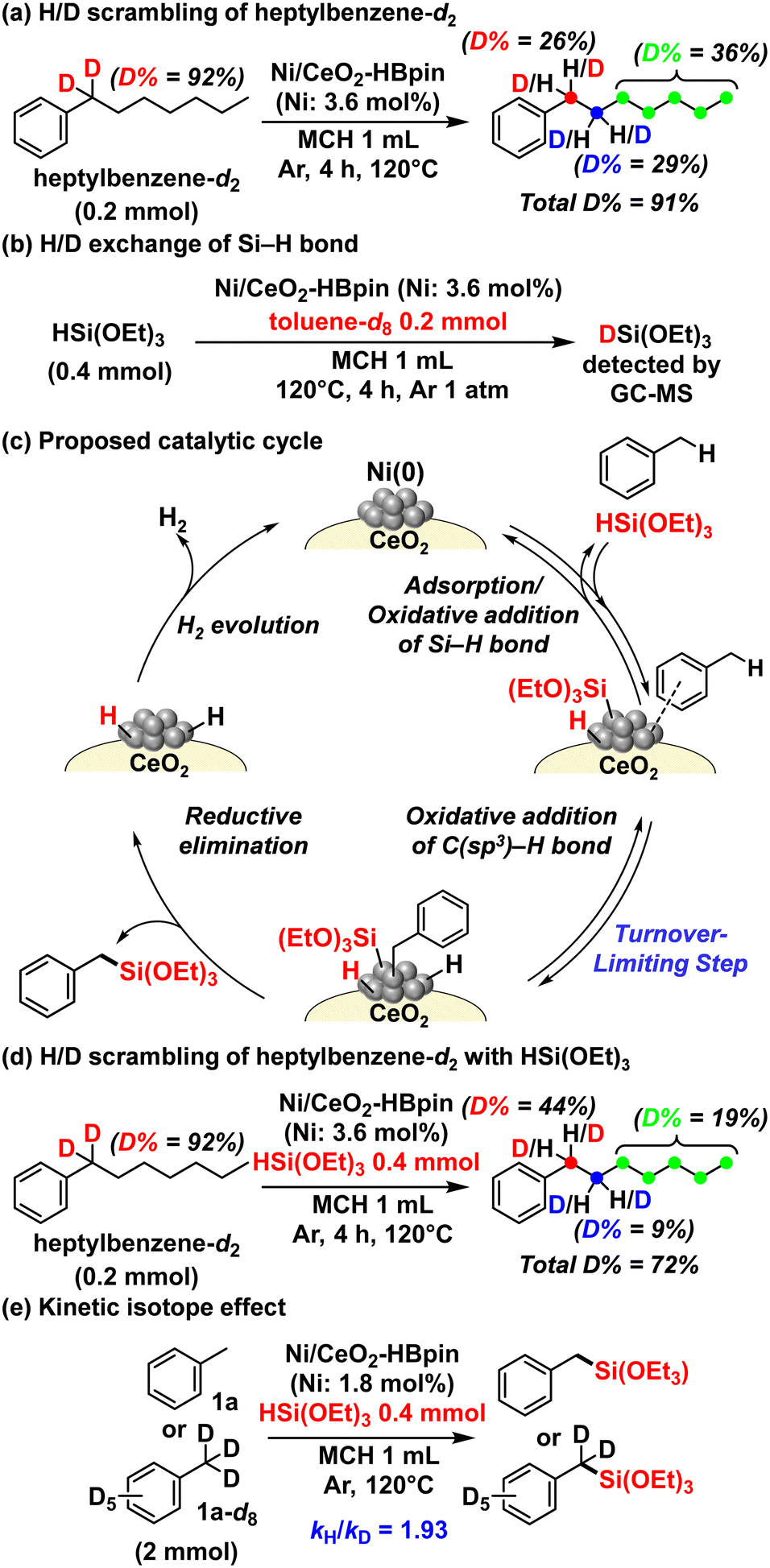 | ||
| Fig. 2 Mechanistic studies. (a) H/D scrambling in heptylbenzene-d2; (b) H/D exchange of Si–H bond in HSi(OEt)3; (c) proposed catalytic cycle; (d) H/D scrambling in heptylbenzene-d2 with HSi(OEt)3 to reveal the origin of the high regioselectivity for the secondary benzylic C(sp3)–H bonds. The deuterium percentages are based on the deuterium ratio in heptylbenzene-d2; (e) kinetic isotope effect using 1a or 1a-d8 as the substrate. | ||
On the basis of these results, the catalytic cycle shown in Fig. 2c can be proposed for this dehydrogenative benzylic silylation. First, adsorption and oxidative addition of HSi(OEt)3 and alkylarenes occur on the Ni(0) nanocatalyst. Then, reductive elimination to the desired benzylsilanes takes place, followed by H2 evolution to regenerate the Ni(0) nanocatalyst, closing the catalytic cycle. The high regioselectivity to the benzylic C(sp3)–H bond is most likely determined by the oxidative addition step of alkylarenes. Considering the aforementioned reason for the selective activation of the aryl C(sp2)–H bond catalyzed by single-site metal complexes,13,21–23 the multiple active Ni sites of the present Ni(0) nanocatalyst might provide an adsorption site for aryl rings and an oxidative addition site for benzylic C(sp3)–H bonds, enabling the unique benzylic C(sp3)–H regioselectivity. Also, the multiple active sites of Ni(0) nanospecies possibly enable the consecutive oxidative addition steps of HSi(OEt)3 and alkylarenes on different sites of the same Ni(0) nanospecies differently from general Ni(0) complexes.
In addition, to reveal the origin of the primary benzylic C(sp3)–H regioselectivity, the H/D scrambling of heptylbenzene-d2 with HSi(OEt)3 in the presence of Ni/CeO2–HBpin was examined (Fig. 2d). Although the corresponding silylated product was not obtained, the deuteration ratios of the homobenzylic C(sp3)–H bond and the other C(sp3)–H bonds except for benzylic C(sp3)–H bond were lower than those obtained in the absence of HSi(OEt)3 (Fig. 2a). The total amount of deuterium in the heptylbenzenes also decreased probably because of the H/D exchange between HSi(OEt)3 and heptylbenzenes. In other words, even in the copresence of secondary benzylic C(sp3)–H bonds and HSi(OEt)3, the Ni(0) nanospecies catalyzed the corresponding oxidative addition steps. Therefore, the secondary benzylic C(sp3)–H silylation is inhibited in this catalytic system because the steric hindrance of the alkylarenes and HSi(OEt)3 hinders the reductive elimination step on the Ni nanospecies, leading to the high regioselectivity for the primary benzylic C(sp3)–H bond.¶
Finally, to determine the turnover-limiting step, several kinetic studies were conducted. The initial production rates for the silylation of 1a with HSi(OEt)3 showed a saturation dependence on the initial concentration of 1a and HSi(OEt)3 (Fig. S19 and S20†). Then, the initial production rate of the benzylic dehydrogenative silylation of 1a was faster than that of 1a-d8 (kH/kD = 1.93) (Fig. 2e) (Fig. S21†). Moreover, the production rate under an H2 atmosphere was almost the same as that under an Ar atmosphere, suggesting that the H2 evolution step occurred easily (Fig. S22†). Consequently, the oxidative addition of alkylarenes via benzylic C–H bond cleavage is possibly the turnover-limiting step of the present acceptorless dehydrogenative silylation.
Conclusions
In summary, an undirected and regioselective acceptorless dehydrogenative silylation of primary benzylic C(sp3)–H bonds with HSi(OEt)3 affording various primary benzylsilanes without occurrence of aryl C(sp2)–H silylation was successfully developed using Ni/CeO2–HBpin as a heterogeneous catalyst. The generation of highly dispersed Ni(0) nanospecies with few surface sites for the side reactions of HSi(OEt)3 is the key to the high catalytic performance of Ni/CeO2–HBpin. Differently from general oxidation reactions of benzylic C(sp3)–H bonds, the dehydrogenative silylation of primary benzylic C(sp3)–H bonds occurred preferentially even in the presence of secondary benzylic C(sp3)–H bonds, probably because the reductive elimination step to form a C(sp3)–Si bond from secondary benzylic C(sp3)–H bonds on the Ni catalyst is inhibited by the steric hindrance of the substrates. This study paves the way for the development of novel selective undirected C–H functionalization reactions promoted by heterogeneous catalysts.Author contributions
T. Yatabe and K. Y. conceived and supervised the project. Q. Y. performed most of the experiments. T. Yabe performed the XAFS measurements. All authors contributed to data analysis and discussed the results. Q. Y. and T. Yatabe wrote the manuscript with feedback from K. Y., T. M., and T. Yabe.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI Grant No. 21K14460 and 22H04971. This work was supported by JST, PRESTO Grant Number JPMJPR227A, Japan. This work is also based on results obtained from a JPNP20004 project subsidized by the New Energy and Industrial Technology Development Organization (NEDO). We greatly appreciate Dr. Hironori Ofuchi (Japan Synchrotron Radiation Research Institute, SPring-8) for giving great support for XAFS measurements in BL14B2 (proposal no. 2022A1803, 2021A1620, and 2019B1820). A part of this work was conducted at the Advanced Characterization Nanotechnology Platform of the University of Tokyo, supported by “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. We thank Ms. Mari Morita (The University of Tokyo) for her assistance with the HAADF-STEM and EDS analyses. We also thank Dr. Daichi Yoshii and Mr. Masaya Komori for their help with preliminary experiments.Notes and references
- M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, Germany, 2000 Search PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- E. J. Corey and A. Venkateswarlu, J. Am. Chem. Soc., 1972, 94, 6190–6191 CrossRef CAS.
- C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed.
- E. G. Rochow, J. Am. Chem. Soc., 1945, 67, 963–965 CrossRef CAS.
- S. C. Richter and M. Oestreich, Trends Chem., 2020, 2, 13 CrossRef CAS.
- C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592–595 CrossRef CAS PubMed.
- C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed.
- R. Sharma, R. Kumar, I. Kumar, B. Singh and U. Sharma, Synthesis, 2015, 47, 2347–2366 CrossRef CAS.
- Z. Xu, W.-S. Huang, J. Zhang and L.-W. Xu, Synthesis, 2015, 47, 3645–3668 CrossRef CAS.
- X. Shang and Z.-Q. Liu, Org. Biomol. Chem., 2016, 14, 7829–7831 RSC.
- B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2021, 50, 5062–5085 RSC.
- C. Qin, Z. Huang, S.-B. Wu, Z. Li, Y. Yang, S. Xu, X. Zhang, G. Liu, Y.-D. Wu, L. W. Chung and Z. Huang, J. Am. Chem. Soc., 2022, 144, 20903–20914 CrossRef CAS PubMed.
- In the case of the following report on Ir-catalyzed benzylic C(sp3)–H silylation of 4-alkylpyridines with norbornene as a hydrogen acceptor, the pyridine structure is essential not for the directing group function but for the formation of the N-silylenamine intermediate. See: Y. Fukumoto, M. Hirano and N. Chatani, ACS Catal., 2017, 7, 3152–3156 CrossRef CAS.
- L. J. Procopio and D. H. Berry, J. Am. Chem. Soc., 1991, 113, 4039–4040 CrossRef CAS.
- T. Sakakura, Y. Tokunaga, T. Sodeyama and M. Tanaka, Chem. Lett., 1987, 2375–2378 CrossRef CAS.
- M. Ishikawa, S. Okazaki, A. Naka and H. Sakamoto, Organometallics, 1992, 11, 4135–4139 CrossRef CAS.
- P. I. Djurovich, A. R. Dolich and D. H. Berry, J. Chem. Soc., Chem. Commun., 1994, 1897–1898 RSC.
- A. D. Sadow and T. D. Tilley, Angew. Chem., Int. Ed., 2003, 42, 803–805 CrossRef CAS PubMed.
- Y. Zhang, T. Zhang and S. Das, Chem, 2022, 8, 3175–3201 CAS.
- A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1983, 105, 3929–3939 CrossRef CAS.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 12610 CrossRef CAS PubMed.
- T. Baba, A. Kato, H. Yuasa, F. Toriyama, H. Handa and Y. Ono, Catal. Today, 1998, 44, 271–276 CrossRef CAS.
- A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS PubMed.
- B. Neil, L. Saadi, L. Fensterbank and C. Chauvier, Angew. Chem., Int. Ed., 2023, 62, e202306115 CrossRef PubMed.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- K. Kuciński and G. Hreczycho, Green Chem., 2020, 22, 5210–5224 RSC.
- Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123–7157 CrossRef CAS.
- D. Yoshii, T. Yatabe, T. Yabe and K. Yamaguchi, ACS Catal., 2021, 11, 2150–2155 CrossRef CAS.
- J. F. Hartwig and E. A. Romero, Tetrahedron, 2019, 75, 4059–4070 CrossRef CAS PubMed.
- T. Mita, K. Michigami and Y. Sato, Org. Lett., 2012, 14, 3462–3465 CrossRef CAS PubMed.
- T. W. Reidl and J. S. Bandar, J. Am. Chem. Soc., 2021, 143, 11939–11945 CrossRef CAS PubMed.
- D. A. Powell and H. Fan, J. Org. Chem., 2010, 75, 2726–2729 CrossRef CAS PubMed.
- Z.-J. Jia, S. Gao and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 10279–10283 CrossRef CAS PubMed.
- K.-J. Liu, Z.-H. Duan, X.-L. Zeng, M. Sun, Z. Tang, S. Jiang, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 10293–10298 CrossRef CAS.
- C. Miao, H. Zhao, Q. Zhao, C. Xia and W. Sun, Catal. Sci. Technol., 2016, 6, 1378–1383 RSC.
- Y.-Z. Chen, B.-J. Liaw and S.-J. Chiang, Appl. Catal., A, 2005, 284, 97–104 CrossRef CAS.
- S. Wang, X. Xing, X. Zhang, X. Wang and X. Jing, J. Mater. Chem. A, 2018, 6, 10868–10878 RSC.
- T. Matsuyama, T. Yatabe, T. Yabe and K. Yamaguchi, ACS Catal., 2021, 11, 13745–13751 CrossRef CAS.
- F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84–88 CrossRef PubMed.
- J. Rodrigalvarez, H. Wang and R. Martin, J. Am. Chem. Soc., 2023, 145, 3869–3874 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00263f |
| ‡ Aryl silylated products were not yielded even using benzene as the substrate, indicating the quite low catalytic activity of Ni/CeO2–HBpin to aryl C(sp2)–H dehydrogenative silylation. |
| § In the Ni/CeO2–HBpin, B and C species were also observed by the STEM-EDS mapping and XPS (Fig. S4, S5 and S9†). However, the residual B species were not localized at Ni nanospecies but highly dispersed on the Ni/CeO2–HBpin catalyst (Fig. S4†), and the XPS peak intensity of the residual C species was much lower than that of Ni/CeO2–NaNaph (Fig. S9†). |
| ¶ In the case of dehydrogenative borylation using Ni/CeO2–HBpin as the catalyst and HBpin as the borylating reagent, secondary benzylic C(sp3)–H bonds can be borylated while the corresponding silylation does not proceed at all with HSi(OEt)3 (Fig. S23†). Thus, the steric hindrance of HSi(OEt)3 is also important for the suppression of secondary benzylic C(sp3)–H silylation. |
| This journal is © The Royal Society of Chemistry 2024 |