
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic cascade of DERA and ADH for lactone synthesis†
Eman
Abdelraheem‡
,
Robin
Kuijpers‡
,
Peter-Leon
Hagedoorn
 ,
Frank
Hollmann
and
Ulf
Hanefeld
*
,
Frank
Hollmann
and
Ulf
Hanefeld
*
Biocatalysis, Department of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: u.hanefeld@tudelft.nl
First published on 28th March 2024
Abstract
This study presents a three-step one pot enzymatic cascade for the synthesis of a δ-lactone. Utilising acetaldehyde, combining 2-deoxyribose-5-phosphate aldolase (DERA) with an alcohol dehydrogenase (ADH) and a cofactor regeneration system this δ-lactone is synthesised with the same stereochemistry as the statin side chain precursor. The initial stage in this cascade involves the double aldol reaction, catalysed by DERA to produce the chiral lactone precursor from the achiral substrate acetaldehyde. The main challenge at this stage is the instability of DERA in the presence of high acetaldehyde concentrations. Therefore, Lactobacillus brevis DERA with a high natural acetaldehyde tolerance was genetically engineered to further improve this property. LbDERA C42M E78K exhibited improved activity and stability (no activity loss over 2 h) compared to the wild type (20% activity loss). In the second stage of the cascade, the aldol product is selectively oxidised to the lactone. A commercially available ADH was identified to selectively catalyse this oxidation using NADP+ as electron acceptor. NADP+ regeneration was achieved using O2 as substrate in two different ways: using either photo-activated flavin or NADPH oxidase (NOX). The lactone was successfully purified from the enzymatic cascades from a preparative scale reaction in 97% purity with an optical rotation [α]D = +34.2° (c = 0.7), proving the feasibility in a multi-enzyme three-step one-pot cascade.
1. Introduction
Statins are well known as a treatment for cardiovascular diseases (CVDs). CVDs are the leading cause of death globally, taking an estimated 17.9 million lives each year, and are caused by high levels of low-density lipoprotein cholesterol (LDL-C).1 Currently, 5% of male and 4.6% of female adults have elevated LDL-C levels.2 Statins help to lower LDL-C by inhibiting 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, which is the rate-limiting enzyme in the cholesterol biosynthetic pathway (Fig. 1).2,3 Since statins were first approved by the FDA in 1987, a variety of statins with the same active side chain, similar to HMG-CoA, have become available on the market. Statins are now the largest-selling class of drugs with a global market size reaching US$ 14.9 billion in 2022.4 The need for statins is large, consequently, an efficient and environmental benign manufacturing process for the medicine is required. Because of the two chiral centres in the side-chain this remains a challenge.3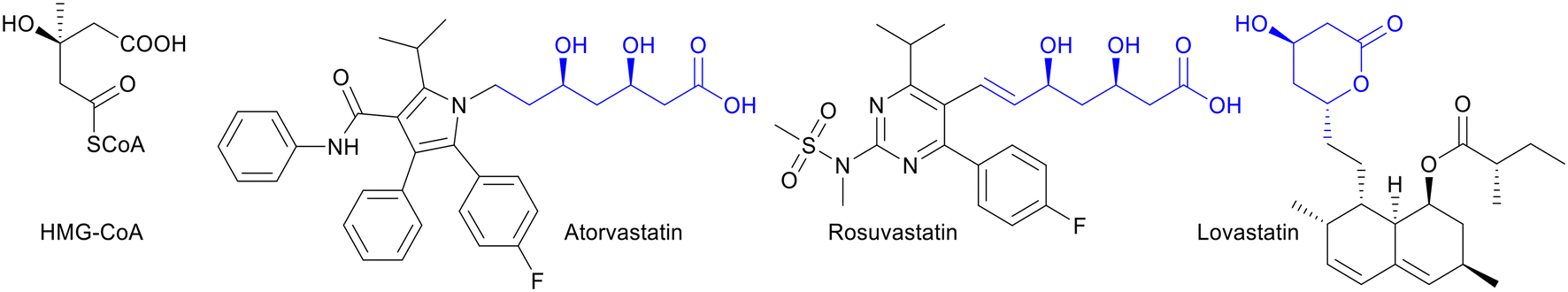 | ||
| Fig. 1 Statins are competitive inhibitors of HMG-CoA reductase. The side-chains similar to HMG-CoA are displayed in blue. | ||
Different biocatalytic methods have been developed for the production of the statin side chain all with high enantiomeric excess.3,5–8 The biocatalytic aldol reaction catalysed by 2-deoxyribose-5-phosphate aldolase (DERA, EC: 4.1.2.4) has become one of the most attractive routes for the highly stereoselective synthesis of the statin side chain precursor from the inexpensive starting material acetaldehyde (Scheme 1).3,9,10 Although the synthesis of the side chain precursor catalysed by DERA has been well characterised, it remains challenging, because of the instability of the enzyme under reaction conditions. Furthermore, the oxidation of initially formed lactol (formed in the DERA catalysed stage) to the lactone that is incorporated into the statins is often performed chemically.8,11,12 Enzymatic oxidation of the lactol to lactone is a promising step due to the mild reaction conditions with a major selectivity challenge:13–15 the hydroxyl group of the hemiacetal has to be selectively oxidised in the presence of the 3(R)-hydroxyl group. The same problem arises for the oxidation of lactol 3 to δ-lactone 4, this study's model compound. Dehydrogenases (DH) are potentially attractive catalysts for this enzymatic lactol oxidation (Scheme 1), as they showed their versatility on similar structures.13–15 Different commercial DH's have been identified and described for this process in detail. Alcohol DH (ADH) from Candida magnoliae was genetically engineered and reaction conditions were optimised in combination with NAD(P)H oxidase (NOX, EC: 1.6.3.2) for the in situ NAD(P)+ regeneration.15 The reaction utilised a crude lactol preparation from a DERA catalysed synthesis and was fully optimised on industrial scale, including O2 consumption, with an excellent mass balance. The second commercial DH, an aldehyde DH (AlDH) was part of a DERA/AlDH cascade optimised for in situ oxidation again utilising NOX for cofactor recycling.3,7,8,14 NOX is very attractive as a cofactor recycling biocatalyst as it enables the use of O2 as oxidant and formation of H2O as sole by-product; ideal from an environmental point-of-view. In the cascade process different DERAs were explored, the aldol reaction was performed with re-additions of enzyme to compensate for instability. The process then required a pH switch for the AlDH catalysed step, as the pH profiles for the DERA and AlDH were not compatible.14 Similar work with an ADH also required this pH switch.16
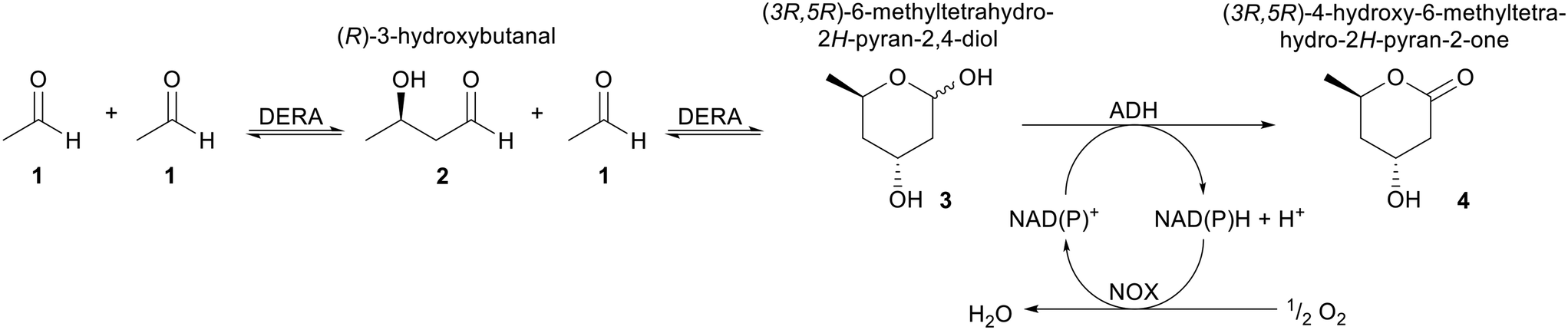 | ||
| Scheme 1 Reaction scheme for the production of the δ-lactone 4, which has the same stereochemistry as the statin side chain with acetaldehyde as starting material. | ||
Here we aim to combine the DERA catalysed double aldol reaction with the ADH catalysed oxidation with in situ cofactor recycling. One challenge in the synthesis of the statin side-chain precursor and of the δ-lactone that is the target of this study with DERA is the instability of DERA in the presence of its natural substrate, acetaldehyde.9,17,18Escherichia coli DERA (EcDERA) is irreversibly inactivated when exposed to 200–300 mM acetaldehyde due to covalent binding of but-2-enal in the active site.18 Improving the stability of DERA is essential for making the enzyme attractive for industrial applications. Numerous efforts have been made to enhance the stability, primarily through immobilisation19–22 or protein engineering.9,17,18,23,24 The abovementioned cascade studies reported the potential for a one-pot three-step, three-enzyme cascade involving genetically engineered Thermotoga maritima DERA (DERA024), an AlDH or ADH (both prozomix), and NOX (001 and 009 from prozomix) for the production of the statin side chain precursor.14,16 The primary issue with these enzymatic cascades was the instability of all the enzymes, as DERA024, AlDH or ADH, and NOX exhibited a 50% reduction in activity after 2 hours. This required intermittent enzyme addition and an undesired pH switch, increasing the buffer load of the reaction mixture. Here we report an improved cascade consisting of genetically engineered Lactobacillus brevis DERA,17 and a commercial ADH for the synthesis of δ-lactone 4 with the same stereochemistry as the statin side chain precursor. For the nicotinamide cofactor recycling two different approaches were explored; an enzymatic system using NOX and O2 with water as benign side product as also utilised in the previously reported DH work.14,16 Secondly; LED light-activated flavin mononucleotide (FMN) for cofactor regeneration was explored, again with O2 as oxidant but hydrogen peroxide as side product. Catalase was added to remove hydrogen peroxide, releasing O2 and water.25 Operating a multienzyme system requires optimising of reaction conditions and balancing factors such as temperature, pH, enzyme stability, and substrate/product toxicity. Here we report the enzymes and reaction conditions for an efficient one-pot system that contributes to a more sustainable production of δ-lactone 4.
2. Results & discussion
2.1 LbDERA C42M E78K has a high acetaldehyde tolerance
A high tolerance of DERA towards acetaldehyde is important for biotransformations under industrially relevant conditions. Various DERA enzymes from different species have been identified and engineered to improve the acetaldehyde resistance.9,24,26,27 DERA from Lactobacillus brevis ECU8302 (LbDERA) and its mutant LbDERA E78K have previously been shown to have high activity, thermostability and acetaldehyde tolerance.17LbDERA was recombinantly expressed with a C-terminally his-tag in Escherichia coli BL21(DE3), and purified using IMAC chromatography (Fig. S1, Table S3†).28LbDERA showed very good activity from pH 7.0 to 9.0 in KPi and Tris–HCl buffers and the lowest activity was measured in citrate buffer (pH 4.0 and 5.0) and in glycine NaOH buffer (pH 10.0 and 11.0). The highest activity was obtained in 100 mM KPi pH 7.5 buffer, which was used for further investigations of LbDERA variants (Fig. S2†). A direct comparison of the acetaldehyde tolerance of EcDERA and the previously reported LbDERA17 was performed. WT LbDERA is more tolerant toward high concentrations of acetaldehyde than the reported DERAs including WT EcDERA, retaining 80% of its initial activity after 120 minutes of incubation. Under the same conditions, EcDERA is completely inactivated after only 20 minutes, consistent with a previous report (Fig. 3A).18,24Two mutations have been previously reported to significantly increase the acetaldehyde resistance of DERA from different sources.17,18 The first mutation was identified in LbDERA as Glu78Lys which increased the overall protein stability by introducing two new hydrogen bonds between Lys78 and Gly71, Val96 and one salt bridge between Lys78 and Asp113 on the monomer's surface (Fig. 2), at the position where it forms a dimer.17 A different successful mutation was reported in EcDERA for Cys47. The Cys47 was demonstrated to be involved in the deactivation mechanism of DERA, by covalently binding to the aldol reaction side product but-2-enal via a Michael addition.18 The acetaldehyde resistance was improved significantly by substituting Cys47 by methionine (C47M). An amino acid sequence alignment of EcDERA and LbDERA showed that Cys47 is conserved in LbDERA as Cys42. The sequences shared identity around Cys42 and Lys78 positions (Fig. 2). To investigate the impact of the two mutations on the acetaldehyde tolerance of LbDERA they were introduced individually as well as in combination (Tables S1 and S2†). LbDERA variants were overexpressed in E. coli and purified in the same manner as the WT enzyme.28
 | ||
| Fig. 2 Alignment the crystal structures of LbDERA E78K (white, PDB ID 4XBS) and EcDERA (cyan, PDB ID 5EKY). The residue Lys78 forms hydrogen bonds with Gly71 at distances of 2.8 Å and Val96 at distances of 2.9 Å and one salt bridge with Asp113 at a distance of 2.6 Å. Sequence alignment with Cys42: 35% identity (69/200), 52% similarity (104/200) and 5% gaps (11/200). Sequence alignment with Met42: 53% identity (114/216) and 67% similarity (145/216). The image was created using PyMOL. | ||
The acetaldehyde resistance of the three mutants (single mutants E78K or C42M and the double mutant C42M E78K) was evaluated by incubating the enzyme in the presence of 100 mM and 300 mM of acetaldehyde and measuring the residual DR5P aldol cleavage activity at various time points using the coupled enzyme assay under standard conditions. In comparison to the wild-type enzyme, LbDERA E78K and C42M showed almost the same initial activity in the presence of 100 mM and 300 mM of acetaldehyde, where it retained between 60–80% of its activity after incubation for 90 minutes (Fig. 3B and C). The combination of the two mutations in one variant showed excellent stability in the presence of 100 mM and 300 mM of acetaldehyde, with no progressive activity loss observed (Fig. 3D) and the activity even tends to increase in the case of the two concentrations of the acetaldehyde 100 mM and 300 mM depending on the incubation time. This improvement of the activity to above 100% of the t = 0 value is a known artefact of the assay when this is used for the determination of DERA stability in the presence of acetaldehyde, as DERA samples introduce also acetaldehyde (1) as well as 2 and 3 into the assay. They will compete with the DR5P conversion of the activity assay and might also influence the auxiliary enzymes, as described earlier.18
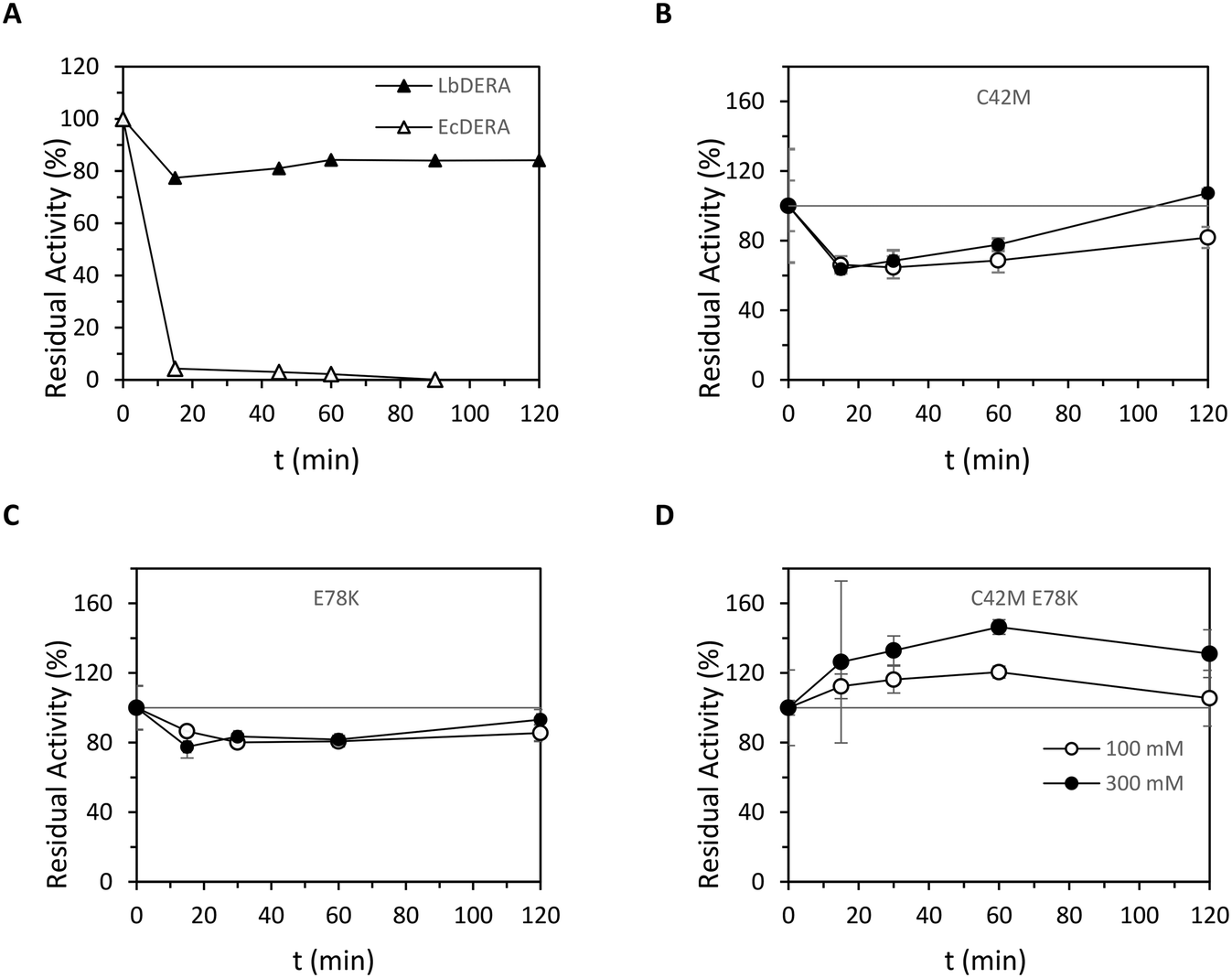 | ||
| Fig. 3 A. Acetaldehyde tolerance of LbDERA and EcDERA upon incubation with 100 mM acetaldehyde at 25 °C in 100 mM TEA buffer pH 7.0. The activity was measured using the DR5P assay: 0.4 mM DR5P, 0.2 mM NADH, 3 U GDH/TPI and 10 μL of DERA (1 mg mL−1) was mixed in 2 mL PPMA cuvettes and the absorbance at 340 nm was measured over time. Activity measurements were done in duplicates. B–D. Residual activity of different LbDERA mutants after incubation with 100 mM (○) and 300 mM (●) of acetaldehyde. 1 mg mL−1 of purified enzymes were incubated with 100 mM and 300 mM of acetaldehyde. Samples were drawn at 15, 30, 60, 120 minutes and residual activity was measured with DERA activity assay: 0.4 mM DR5P, 0.2 mM NADH, 3 U GDH/TPI and 10 μL of LbDERA (1 mg mL−1) was mixed in 2 mL PPMA cuvettes and the absorbance at 340 nm was measured over time. Activity measurements were done in duplicates. B. LbDERA C42M mutation in the active site to prevent covalent binding of butenal in the active side. C. LbDERA E78K mutation at the surface of a monomer to introduce two additional hydrogen bonds and one salt bridge to improve overall protein stability. D. Combination of the two mutations, LbDERA C42M E78K. | ||
To probe the potential of the new double mutant, the LbDERA C42M E78K variant was employed for the sequential aldol reaction to produce the lactol (3). Due to the low recovery (18%) of DERA after protein purification, the cell free extract of E. coli containing overexpressed LbDERA C42M E78K (2.5 U mL−1) was utilised for the synthesis of 3 (Table S3†). 3 was extracted with ethyl acetate and further purified with silica column purification, resulting in a yellowish oil. As previously reported the purification of lactol (3) was difficult primarily due to instability of 3 and presence of by-products.18 Even in the previously published scaled optimisation study for the separate ADH oxidation step crude lactol was used due to these difficulties.15 The purified 3 (purity of 94%) was analysed with 1H and 13C NMR, and the results are in accordance with a prior study (Fig. S7†).29 Chemical lactol oxidation produced lactone, which was fully characterised and utilised as reference material (Fig. S8†).30 With this the DERA for the first stage of the cascade and all materials to optimise the second stage were available.
2.2 Enzymatic oxidation for the production of lactone 4
The DH catalysed enzymatic oxidation of the lactol 3 produced with DERA requires only oxygen as oxidant for the NOX or light based cofactor recycling and produces water as a by-product, making the process very environmentally friendly (Scheme 1). Alcohol dehydrogenases (ADHs) are nicotinamide-cofactor dependent oxidoreductases that can catalyse the oxidation of lactol (3) to lactone (4) under mild conditions.13–16,31 An ADH panel consisting of 18 different commercial ADHs (Evoxx Technology GmbH, Monheim, Germany)32 and, Ralstonia, Lactobacillus brevis, and Lactobacillus kefir ADH was performed. The panel was screened using 3 produced with LbDERA C42M E78K as a substrate and NAD+ and NADP+ (Fig. S3†). The most active enzymes were used with higher concentrations of 3 for small scale bioconversions to 4. Six different ADHs could catalyse the conversion under non-optimised conditions, with yields varying from 8% to 51%. The highest yields of 51% and 45%, were achieved using 5 mg mL−1 ADH380 with NAD+ and ADH440 with NADP+ respectively (Table S4†). ADH380 and ADH440 were therefore further examined. The conversion of 3 to 4 after 20 hours was measured using 1 unit of ADH440, a lactone yield of 46% was achieved, while 1 unit of ADH380 (both activities determined according to suppliers instructions) resulted in a lower lactone yield of only 10% (Table S5†). As a result, ADH440 was selected for optimisation of reaction conditions. To establish the optimal temperature of ADH440, the conversion of 3 was measured after 4 and 24 hours of incubation at 30, 40 and 50 °C. A decrease in the lactone yield was observed after 24 hours at temperatures above 30 °C. Therefore, 30 °C was chosen as the optimal temperature for further experiments (Fig. S4†).2.3 Creating the cascade
Cascades have attracted considerable attention in recent years as they allow to achieve high space time yields and simplify downstream processing.5,33–35 This is also the case here. Conducting the reaction as a one-pot, three step cascade process offers the advantage of reducing the number of production steps, by eliminating the difficult purification of the lactol. In the first reaction, DERA catalyses the double aldol reaction of acetaldehyde to form lactol 3 (Scheme 1). Acetaldehyde is highly volatile and oxidises easily to acetic acid in the presence of oxygen.18,36 Therefore, it is important to perform the aldol reaction under nitrogen atmosphere. The second stage requires an oxygen-rich environment because oxygen is used as final electron acceptor during the regeneration of NADP+ (Scheme 1). To accommodate the distinct requirements of both reactions the enzymatic cascade is performed as a three-step, one-pot system. To establish the optimal reaction conditions for this one-pot system a buffer and pH screening was performed for the enzymes LbDERA C42M E78K and ADH440 independently (Fig. S5†). From the buffer and pH assay, it was found that DERA is most active in 100 mM TEA pH 7 buffer. For the buffer and pH screening, low concentrations of ADH440 were employed, leading to modest conversion yields. The conversion of 3 to 4 by ADH440 was highest (46%) in the same buffer (100 mM TEA pH 7 buffer). ADH440 shows moderate (>36%) conversion within the pH range of 6 to 7, while it shows poor conversion at pH 8 (1.6%) and no conversion at pH 9. LbDERA C42M E78K is most active in the 7–8 pH range, similar to the WT enzyme (Fig. S2 and S5†). The combination of LbDERA C42M E78K and ADH440 in 100 mM TEA at pH 7 enable thus to perform the cascade without the previously necessary pH switch.14,16Since the reactions were envisaged in one-pot, it is important to examine the acetaldehyde resistance of ADH440 and NOX, as the presence of residual acetaldehyde in the reaction could affect the performance of these enzymes. Therefore, the acetaldehyde resistance of ADH440 and NOX in the presence of 50 mM and 100 mM acetaldehyde was studied (Fig. 4). To exclude the possibility that acetaldehyde interferes with the activity assay of ADH440 and NOX, the activity in the presence of 5 mM acetaldehyde and in the absence of acetaldehyde was measured and compared. There was no significant difference between the assays.
 | ||
| Fig. 4 Acetaldehyde resistance of ADH440 and NOX. A. 4 U mL−1 ADH440 was incubated with 0, 50, and 100 mM acetaldehyde in 100 mM TEA, pH 7 buffer, 30 °C, 800 rpm, for 1, 3, 6, 24 hours. Activity = 100% at t = 0 with no acetaldehyde. Activity was measured following the activity assay in section 4.11. B. 5 mg mL−1 NOX was incubated with 0, 50, and 100 mM acetaldehyde in 100 mM TEA, pH 7 buffer, 30 °C, 800 rpm, for 1, 3, 6, 24 hours. Activity = 100% at t = 0 with no acetaldehyde. Reactions were performed in duplicates. | ||
First the activity of ADH440 in the presence of acetaldehyde was measured, no activity with acetaldehyde was observed. LeADH did exhibit activity towards acetaldehyde, indicating a lower substrate selectivity for lactol compared to ADH440.14 After six hours of exposure to acetaldehyde concentrations of 50 and 100 mM, ADH440 exhibits good acetaldehyde resistance. The relative activities compared to the reaction with zero acetaldehyde at t = 0 were 88% and 89% respectively (Fig. 4A). ADH440 incubated for 24 hours with 50 or 100 mM acetaldehyde shows relative activities of 54% and 58%, respectively, surprisingly ADH440 incubated for 24 hours with no acetaldehyde shows no activity. NOX retains 44% and 21% of its activity after 24 hours of incubation with 50 and 100 mM acetaldehyde (Fig. 4B). Incubation of NOX without acetaldehyde also shows a decrease in activity. After 24 hours, the residual activity determined for NOX was 56%.
The reduced stability of NOX led to the exploration of a second, photochemical NAD(P)+ regeneration system, utilising free flavin mononucleotide (FMN). Flavins can oxidise NAD(P)H, however, the hydride transfer from NAD(P)H to the oxidised flavin is slow.37 This rate can be enhanced through the photoexcitation of the flavin by blue LED light (465 nm).38 The reoxidation of the flavin catalyst occurs spontaneously through the reduction of oxygen to hydrogen peroxide (Scheme 2).25,38
 | ||
| Scheme 2 Oxidation of lactol 3 to lactone 4 with blue light and FMN-based regeneration system of NAD(P)+. | ||
To establish the optimal concentrations of FMN and NOX in the enzymatic cascade, lactol (3) was first produced using purified LbDERA C42M E78K yielding 54% 3 in a fed batch reaction with 300 mM acetaldehyde and 1 U mL−1LbDERA C42M E78K. Increasing the amount of enzyme did not lead to higher overall yields. This high enzyme stability is in line with earlier reported reactions and the low yield has to be attributed to the volatility of acetaldehyde.17,23,24 The reaction mixture was then utilised for the oxidation of 3 by ADH440 at different concentrations of FMN and NOX. Increasing the NOX concentration to above 5 mg mL−1 did not lead to significant increase in yield, which plateaued at circa 54%. Therefore, 5 mg mL−1 was chosen as optimal concentration of NOX (Fig. S6B†). The lactone yield did not show a significant increase with the increase of FMN concentration. The highest yield of 57% was achieved with 1 mM FMN. 1 mM FMN was chosen as optimal concentration for subsequent experiments (Fig. S6A†). As the yield showed no significant increase with the increase of FMN and NOX concentrations, it suggested that the concentration of ADH might be the limiting factor. Subsequently, different concentrations of ADH440 were tested in combination with the optimal concentrations of FMN and NOX. The enzymatic oxidation was performed using the final reaction mixture of the DERA catalysed aldol reaction, which contained 27 mM lactol. The highest lactone yield was obtained at a concentration of 40 U mL−1 ADH, resulting in high yields of 88% and 80% for NOX and FMN respectively (Fig. 5).
 | ||
| Fig. 5 Yield of lactol to lactone for different concentrations of ADH440. 27 mM crude lactol, 5 mM NADP+, 1 mM FMN/1 mg NOX, 0.5, 1, 2, 3, 4, 8, 10 U ADH440, 10 U of catalase in 100 mM TEA pH 7.0, 800 rpm, 30 °C, 20 hours. Volume: 0.2 mL. Reactions were performed in duplicates. | ||
Both systems were evaluated on a larger scale. Initially, lactol was synthesised by purified DERA C42M E78K, followed by the oxidation catalysed by ADH440, employing FMN or NOX as regeneration systems. The production of lactol was followed over time, leading to a yield of 56% of 3 after 24 hours. Following this, the reaction mixture was diluted to a concentration of 30 mM of 3 for oxidation with FMN or NOX. Both reactions were monitored over time and the highest yields were obtained after 24 hours, resulting in 70% and 77% of lactone from 3 with FMN and NOX as regeneration systems, respectively (Fig. 6). The results show that when using FMN as a regeneration system the lactol concentration decreases faster than the increase of lactone concentration within the first six hours. This could be caused by unselective side reactions of light-activated FMN, as previously observed when light-activated flavin was reported to react with amino acid residues in an enzyme's active site.39 In addition the H2O2 produced by the regeneration systems could also react with the lactone before it was converted into water and oxygen by catalase. With the NOX-based regeneration system, the lactol concentration also decreased to zero within 24 hours, without full conversion to the lactone. Earlier work with an industrial reactor, showed that a perfect mass balance for O2 and lactone can be achieved in a large, fully controlled system.15 As no side products were observed it has to be assumed that volatility and the retro-aldol reaction causes this imbalance. To further eliminate the possibility that the imbalance between 3 and 4 is a result of the oxidation of the second hydroxy group attached to the C3 atom by ADH440, the activity of ADH440 was determined with purified lactone and NADP+. There was no activity measured, indicating that ADH440 does not oxidise the second hydroxy group, as also observed for other ADHs.13,15,16 Overall the cascade consists of three steps, the aldol reactions, the oxidation and the cofactor recycling. Only the cofactor recycling is irreversible. This means that at all stages of the cascade acetaldehyde will be present and actually also formed via the retro-aldol reaction, causing loss of starting material and lowering the overall yield.
 | ||
| Fig. 6 Enzymatic cascade for the production of lactone 4. A. Fed batch synthesis of lactol 3 (●) catalysed by DERA. 1 U mL−1 of purified LbDERA C42M E78K in 45 mL 100 mM TEA pH 7.0 buffer, room temperature, stirring, under nitrogen. 3 M of acetaldehyde in 100 mM TEA pH 7.0 buffer fed with flowrate 0.5 mL h−1 to an end concentration of 300 mM and end volume of 50 mL. 500 μL of samples were taken in duplicates over time and analysed with GC. B. Oxidation of lactol 3 (●) to lactone 4 (○) catalysed by ADH440 with FMN regeneration system. The reaction mixture of A was mixed with ADH440, NADP+, FMN, and catalase. The final concentrations in the reaction were: 30 mM lactol, 40 U mL−1 ADH440, 5 mM NADP+, 1 mM FMN, and 60 U of catalase in 20 mL TEA pH 7.0 buffer in a 100 mL round bottom flask. The reaction mixture was exposed to blue light (465 nm), 30 °C, and stirred. 200 μL samples were taken over time and analysed with GC. C. Oxidation of lactol 3 (●) to lactone (○) catalysed by ADH440 with NOX as regeneration system. The reaction mixture of A was mixed with ADH440, NADP+, and NOX. The final concentrations in the reaction were: 30 mM lactol, 40 U mL−1 ADH440, 5 mM NADP+, and 5 mg mL−1 NOX in 20 mL 100 mM TEA pH 7.0 buffer in a 100 mL round bottom flask. The reaction mixture was heated to 30 °C, and stirred. 200 μL samples were taken over time and analysed with GC. | ||
The lactone was extracted and purified from the reaction mixtures, with an optical rotation of [α]D = +34.2° (c = 0.7), in agreement with the previous report.29 Isolated yields relative to acetaldehyde were low (about 5%) for the reactions using the FMN and NOX regeneration systems respectively. Previous studies also reported low purification yields (21% for lactol to lactone),13 and on a multi gram scale 45% for the two stages,12 emphasising the challenges in purification and the need for further optimisation of the downstream processing. The lactone was identified with NMR and the results were in accordance with the previous report.29 The NMR spectra of the isolated products resulted in of 97% and 92% purity for the reactions with the FMN and NOX regeneration systems respectively (Table 1). In the isolated oxidation of lactol to lactone with and industrial reactor with oxygen sparger higher yields of similar lactones could be obtained, indication that further reaction engineering is possible but requires dedicated equipment.15,40
Comparing the oxidation of 3 based on the regeneration systems, using FMN results in a more pure final product with a higher purification yield. This could be due to the initial purity of FMN being higher than the NOX preparation, a CFE. On the other hand, the use of NOX leads to a higher yield, but is also 2.5 fold more expensive than the use of FMN. Both options offer viable options for regeneration and the choice can be made based on purity, yield or costs.
3. Conclusion
Many different approaches towards the enzymatic production of the statin side chain precursor have been developed. The most concise approach is the DERA catalysed double aldol reaction, generating both stereocentres in one reaction. The drawbacks of this approach, low acetaldehyde tolerance of DERA and difficulties with isolating the lactol for a separate oxidation were addressed. The already acetaldehyde tolerant, LbDERA was engineered to further enhance the tolerance to acetaldehyde. The improved LbDERA C42M E78K was employed, together with a newly identified ADH, for the synthesis of lactone 4, with the stereochemistry of the statin side chain, in one-pot enzymatic cascade reaction. High overall selectivity and stability was achieved without the previously required pH switch between the two stages. In addition NADP+ regeneration was addressed using either a photo-activated flavin or an enzymatic method with NOX generating only environmentally benign side products. The lactone was successfully purified from both cascade reactions, showing the potential for a more efficient and sustainable production route of the lactone and possibly also for the statin side chain.4. Materials & methods
4.1 Chemicals
Acetaldehyde (>99.5%, Thermo Scientific), acetic acid (>99.8%, Honeywell), 2,3-butanedione (>97.7%, Merck), cyclohexanone (>99%, Alfa Aesar), 2,4-dinitrophenylhydrazine (TCI), D-2-deoxy-ribose-5-phosphate (DR5P) (Merck), ethyl acetate (VWR chemicals), flavin mononucleotide (73–79%, Sigma Merck), glyceraldehyde-3-phosphate (Merck), glycerol (>99.0%, Merck), imidazole (>99%, VWR chemicals), isopropyl-β-D-thiogalactopyranoside (IPTG) (>99%, CliniSciences), kanamycin sulfate (Thermo Fisher Scientific), potassium hydroxide (KOH) (Merck), magnesium chloride-hexahydrate (Boom B.V.), NAD+ (>97.5%, Carl Roth), NADP+ (98.8%, Prozomix), NADH (98.9%, Prozomix), NADPH (Prozomix), petroleum ether (VWR chemicals), phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich), silver carbonate, ca. 50 wt% on celite (Merck), sodium chloride (Central warehouse L&M), triethanolamine hydrochloride (Merck), Bacto tryptone. Bacto yeast extract.4.2 Commercial enzymes
Glycerophosphate dehydrogenase-triosephosphate isomerase (Sigma-Aldrich). NAD(P)H oxidase 001 (Prozomix). Alcohol dehydrogenase Evocatal screening kit including alcohol dehydrogenase 440 (Evoxx Technologies GmbH). Full details on these ADHs including their molecular mass and purity (SDS gel) have been described elsewhere.32 Catalase from bovine liver (3000 U mL−1) (Sigma Merck).4.3 Lactol synthesis
The fed-batch reaction was carried out with 2.5 U mL−1 cell free extract (CFE) DERA C42M E78K in 200 mL 100 mM KPi buffer pH 7.0. Before the start, the buffer with enzyme was drawn vacuum and flushed with nitrogen with a Schlenk line. 20 mL of 4.5 M acetaldehyde (99.5% purity, 5 mL ampule, one-time use) was fed with a flow rate of 0.66 mL h−1. After 40 hours the reaction mixture was divided in 15 mL batches and centrifuged at speed 3220 g for 20 minutes. The supernatant was 2× extracted with ethyl acetate (EtOAc) ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the pellets were extracted once with 10 mL EtOAc (centrifuged 3220g, 10 min). The extracted layers were combined and dried over anhydrous magnesium sulfate (MgSO4). The solvent was evaporated under reduced pressure resulting in a yellow oil (crude product). 100 mg of the crude product was dissolved in 0.5 mL EtOAc and purified with a 4 g silica column with a column volume (CV) of 6 mL using flash chromatography. The column was equilibrated with 6 CV petroleum ether (PE). The product was eluted with a linear gradient 100% PE to EtOAc 100% with a flow rate of 15 mL min−1. The lactol eluted at 70% EtOAc. The fractions were checked with thin layer chromatography (TLC) (solvent 100% EtOAc, staining: 0.5 g of potassium permanganate in 100 mL water). Pure fractions were combined. The solvent was evaporated under reduced pressure. Lactol 3 was purified as a yellowish oil, 0.3 mmol, 40 mg, 2.2% yield. 1H NMR (600 MHz, CDCl3) δ 5.32 (d, J = 4.9 Hz, 1H, H-1 α), 5.16 (d, J = 9.6 Hz, 1H, H-1 β), 4.42 (ddq, J = 12.5, 6.3, 6.3 Hz, 1H, H-5 α), 4.32 (dq, J = 5.4, 2.9 Hz, 1H, H-3 β), 4.28 (m, 1H, H-3 α), 4.21 (s, 1H, α&β OH), 4.07 (ddq, J = 12.5, 6.4, 6.4 Hz, 1H, H-5 β), 3.27 (d, 1H, OH β), 3.19 (d, 1H, OH α), 2.04–1.39 (m, 8H, H-2/4 α/β), 1.24 (d, J = 5.8 Hz, 3H, Me-6α), 1.21 (d, J = 6.1 Hz, 3H, Me-6β). 13C NMR (151 MHz, CDCl3) α-anomer: δ 21.4, 34.9, 39.8, 59.1, 64.1, 92.9. 13C NMR (400 MHz, CDCl3) β-anomer: 21.3, 39.4, 39.4, 65.5, 66.6, 92.2. NMR results are in accordance with literature (Fig. S7†).29
:1 and the pellets were extracted once with 10 mL EtOAc (centrifuged 3220g, 10 min). The extracted layers were combined and dried over anhydrous magnesium sulfate (MgSO4). The solvent was evaporated under reduced pressure resulting in a yellow oil (crude product). 100 mg of the crude product was dissolved in 0.5 mL EtOAc and purified with a 4 g silica column with a column volume (CV) of 6 mL using flash chromatography. The column was equilibrated with 6 CV petroleum ether (PE). The product was eluted with a linear gradient 100% PE to EtOAc 100% with a flow rate of 15 mL min−1. The lactol eluted at 70% EtOAc. The fractions were checked with thin layer chromatography (TLC) (solvent 100% EtOAc, staining: 0.5 g of potassium permanganate in 100 mL water). Pure fractions were combined. The solvent was evaporated under reduced pressure. Lactol 3 was purified as a yellowish oil, 0.3 mmol, 40 mg, 2.2% yield. 1H NMR (600 MHz, CDCl3) δ 5.32 (d, J = 4.9 Hz, 1H, H-1 α), 5.16 (d, J = 9.6 Hz, 1H, H-1 β), 4.42 (ddq, J = 12.5, 6.3, 6.3 Hz, 1H, H-5 α), 4.32 (dq, J = 5.4, 2.9 Hz, 1H, H-3 β), 4.28 (m, 1H, H-3 α), 4.21 (s, 1H, α&β OH), 4.07 (ddq, J = 12.5, 6.4, 6.4 Hz, 1H, H-5 β), 3.27 (d, 1H, OH β), 3.19 (d, 1H, OH α), 2.04–1.39 (m, 8H, H-2/4 α/β), 1.24 (d, J = 5.8 Hz, 3H, Me-6α), 1.21 (d, J = 6.1 Hz, 3H, Me-6β). 13C NMR (151 MHz, CDCl3) α-anomer: δ 21.4, 34.9, 39.8, 59.1, 64.1, 92.9. 13C NMR (400 MHz, CDCl3) β-anomer: 21.3, 39.4, 39.4, 65.5, 66.6, 92.2. NMR results are in accordance with literature (Fig. S7†).29
4.4 Lactone synthesis
Crude lactol (0.85 mmol) and silver carbonate on celite (1 mmol) in 10 mL heptane were heated to reflux for 6 hours. The reaction mixture was filtered over a pack of celite and washed with ethyl acetate. The solvent was evaporated under reduced pressure. 80 mg of crude lactone was dissolved in 0.5 mL EtOAc and purified with a 4 g silica column (CV of 6 mL) by flash chromatography. The column was equilibrated with 6 CV PE. The product was eluted with elution program: 100% PE to 93% PE and 7% EtOAc in 3 minutes, to 69% PE and 31% EtOAc in 3 minutes, hold for 3 minutes at 69% PE and 31% EtOAc, flush with 100% EtOAc for 4.25 minutes, with a flow rate of 15 mL min−1. The solvent was evaporated under reduced pressure. The lactone was purified as a colorless oil, 0.15 mmol, 20 mg, 18% yield. 1H NMR (400 MHz, CDCl3) δ 4.83 (ddq, J = 12.8, 6.4, 3.0 Hz, 1H, H-5), 4.38–4.28 (m, 1H, H3), 2.70 (s br, 1H, OH), 2.68 (dd, J = 17.7, 4.9 Hz, 1H, H-2), 2.58 (ddd, J = 17.7, 3.7, 1.7 Hz, 1H, H-2), 1.96 (dddd, J = 14.4, 3.8, 3.0, 1.7 Hz, 1H, H-4), 1.69 (ddd, J = 14.5, 11.2, 3.3 Hz, 1H, H-4), 1.37 (d, J = 6.4 Hz, 3H, Me-6). 13C NMR (101 MHz, CDCl3) δ 171.0, 72.5, 62.6, 38.3, 37.5, 21.3. NMR results are in accordance with literature (Fig. S8†).294.5 Cloning of Lactobacillus brevis deoC
The synthetic DERA coding gene deoC from Lactobacillus brevis, codon optimised for E. coli and with a C-terminal polyhistidine tag (His6-tag) was obtained from Bio Basic INC (Canada). pET28a-LbDERA expression vector was cloned into E. coli TOP10 to get a stable host for plasmid DNA. The sequence of the obtained plasmid was analysed by BaseClear (Leiden, The Netherlands). E. coli BL21(DE3) was transformed with the expression plasmid for the production of the His-tagged LbDERA. The nucleotide and amino acid sequences are given in the ESI,† Table S2.4.6 Site-directed mutagenesis of LbDERA
Mutants C42M and E78K were prepared using the QuikChange site-directed mutagenesis kit (Agilent). A total reaction volume of 20 μL was used, containing 5 ng template DNA, 10 μL of Q5 DNA polymerase master mix and reverse and forward mutagenic primers 10 μM (each). The mutagenic primers are listed in Table S1.† Gradient PCR program included 98 °C for 60 s; 32 cycles of 98 °C 30 s, 62–70 °C 30 s and 72 °C 7 min; 72 °C for 10 min. Ligation was carried out after PCR, 2 μL 10× T4 DNA ligase buffer and 1 μL T4 DNA ligase were added to the PCR product and incubated at room temperature for 2 h. DpnI digestion was carried out to selectively digest the naturally methylated DNA template: 2 μL 10× NEB-buffer and 1 μL DpnI enzyme were added to the ligated PCR product and incubated for 4 h at 37 °C. Afterwards, the PCR product was transformed into E. coli TOP10 and grown in 5 mL LB-kanamycine medium overnight at 37 °C. DNA plasmid was isolated using the mini prep kit (Qiagen) from four isolated colonies and further quantified using a Nanodrop spectrophotometer (Thermo Fisher). Correct introduction of the mutations was confirmed by sequencing at BaseClear (Leiden, Netherlands). The nucleotide and amino acid sequences of the different variants are given in the ESI† (Table S2).4.7 Protein expression
Chemically competent E. coli BL21 (DE3) was purchased from New England Biolabs (Beverly, MA, USA) and transformed with the different plasmids according to the heat shock method for 30 s at 42 °C. These transformants (10–100 μL) were plated on LB-agar containing 50 μg mL−1 kanamycin and incubated at 37 °C overnight. The preculture was prepared by picking individual colonies to inoculate a 10 mL LB medium containing 10 μg mL−1 kanamycin (kan) grown at 37 °C, 180 rpm overnight. The preculture was used to inoculate 1 L LB medium containing 10 μg mL−1 kanamycin. Protein expression was induced at OD600 = 1.0 (measured with the Ultrospec 2100 pro by Biochrom US) by the addition of isopropyl-β-D-thiogalactoside (IPTG) to a final concentration of 0.1 mM and cultures were grown overnight at 25 °C, 120 rpm. Cells were harvested by centrifugation at 4 °C, 10000 rpm for 10 min. The pellet was resuspended in 40 mL 100 mM KPi buffer pH 7 and centrifuged at 4 °C, 10000 rpm for 10 min then the cells were stored at −20 °C.
4.8 Protein purification
Cells were resuspended in buffer A (20 mM KPi, 100 mM NaCl, pH 7.4) to achieve an end concentration of 20% w/v with adding 5–10 mg DNAse. For cell disruption, the cell suspension was passed three times through a cell disruptor (Constant systems). Cell debris was separated from the crude extract by centrifugation at 10000g, 4 °C for 15 min and the cell free extract was obtained as the supernatant. A His-trap FF 5 mL column containing Ni2+–NTA (nickel–nitrilotriacetic acid) (Cytiva) was used for all purifications. The column was first equilibrated with 2–4 column volumes (CV) of buffer A. The cell free extract was loaded on the column. The protein of interest was eluted from the column using 20 mM KPi, 500 mM imidazole, 100 mM NaCl, pH 7.4 as buffer B in isocratic steps using 8%, 20% and 100% of buffer B. DERA eluted at 20% and 100% of buffer B. The purification was carried out on the NGC Chromatography System (BioRad). Relevant fractions of the eluted proteins were combined in separate Amicon Ultra 15 centrifugal filter Units (cut off 10 kDa) (Millipore) and centrifuged at 4 °C and 4000g till the volume was down to approximately 2.5 mL. The concentrated protein was desalted using a PD-10 column and 2.5 mL, equilibrated with 100 mM TEA buffer pH 7.0.
4.9 Protein analysis
SDS-PAGE was performed using 4× Laemmli sample buffer (Bio-Rad Laboratories), 0.5 μL 100 mM DTT 10 μL of the protein sample. The total volume of the sample is 20 μL. The samples were denatured at 95 °C for 5 min by using an Eppendorf thermomixer comfort. The 12% Bis-tris precast gel (Criterion XT) is running in 1× TGS running buffer at 200 V for 40 min. The gel was imaged with the Bio-Rad ChemiDoc MP imaging system used combined with the Blot/UV/stain-free sample tray (Fig. S1†). The protein concentration was determined using the BCA assay with BSA as a standard. The calibration curve for protein quantification was obtained by using different standards of BSA in the concentration range of 2 μg mL−1 to 50 μg mL−1.4.10 Activity assay DERA
DERA activity was measured based on a coupled enzyme assay which has been reported previously.18,24 DERA catalyses the decomposition of D-2-deoxy-ribose-5-phosphate (DR5P) to glyceraldehyde-3-phosphate and acetaldehyde. GAP is reduced to glycerol 3-phosphate by the enzymes α-glycerophosphate dehydrogenase (GDH) and triosephosphate isomerase (TPI). The reaction is monitored by the simultaneous oxidation of nicotinamide adenine dinucleotide (NADH). The activity of DERA is determined by determined the consumption of NADH where one unit of aldolase was defined as the amount of enzyme able to catalyse the cleavage of 1.0 μmol of DR5P in a minute. In a 1.5 mL semi-micro PMMA cuvette (from BRAND), 0.2 mM NADH, 0.4 mM DR5P, 3 U GDH, 11 U TPI and 10 μL of protein sample were mixed. The volume was made up to 1 mL using 100 mM triethanolamine buffer (TEA) at pH 7.0 and the absorbance of NADH consumption was monitored for 1 min at 25 °C and 700 rpm. The temperature and the stirring was kept constant by the Quantum Northwest TC1 temperature controller and absorbance measurements were detected by the Cary 60 UV-vis spectrophotometer (Agilent) at 340 nm.4.11 Activity assay ADH440
The activity of ADH440 was determined by recording the reduction of cyclohexanone. The reduction of the substrate is coupled to the oxidation of NADPH. One unit of ADH is defined as the amount of enzyme necessary to reduce 1 μmol of substrate per minute. 10 mM cyclohexanone, 0.25 mM NADPH, 3.33 mg mL−1 ADH440 in 1 mL 50 mM TEA pH 7.0 buffer were mixed in a 2.5 mL PMMA cuvette. The consumption of NADPH over time was monitored by measuring the absorbance at 340 nm, 30 °C, 700 rpm. The activity assay was adapted from the instructions provided by the supplier (Evoxx).324.12 Activity assay ADH380
Activity ADH380 was measured as the reduction of 2,3-butanedione with NADH as cofactor. The consumption of NADH was monitored by measuring the absorbance at 340 nm. One unit of ADH is defined as the amount of enzyme necessary to reduce 1 μmol of substrate per minute. To measure the activity of ADH380 a reaction with 100 mM 2,3-butanedione, 0.25 mM NADH, 0.1 mg mL−1 ADH380 in 50 mM TEA pH 7.0 buffer in 2.5 mL PMMA cuvette was measured. The consumption of NADH over time was followed by measuring the absorbance at 340 nm, 30 °C, 700 rpm. The activity assay was adapted from the instructions provided by the supplier (Evoxx).324.13 Activity assay NOX
The NOX activity was measured by monitoring the oxidation of NADH to NAD+. One unit of NOX is defined as the amount of enzyme necessary to reduce 1 μmol of NADH per minute. The measurements were performed in a 2.5 mL PMMA cuvette containing 1 mL 100 mM TEA pH 7 buffer. The measurement was started by addition of 0.1 mM NADH and 5 mg mL−1 NOX and measuring the absorbance at 340 nm at 25 °C.414.14 Buffer and pH profile of LbDERA
The optimum pH was determined using the standard activity assay of the purified enzyme (1 mg mL−1) at different buffers (100 mM): citrate (pH 4.2, 5.0 and 6.2), potassium phosphate (pH 6.2, 7.0 and 8.2), TEA (pH 7.0, 8.0 and 8.3) and Gly–NaOH (pH 8.6, 9.0, 10.0 and 10.6). The activity assay was performed as described above for the final concentrations of 0.4 mM DR5P, 0.2 mM NADH, 3 U GDH, 11 U TPI and 10 μL DERA. The volume was made up to 1 mL with different buffers. All measurements were performed in duplicate (Fig. S2†).4.15 Acetaldehyde resistance of DERA
The purified enzymes (1 mg mL−1) were incubated at 25 °C in 100 mM and 300 mM of 99.5% pure acetaldehyde obtained from Sigma-Aldrich chemicals. Samples were drawn at 15, 30, 60 and 120 min and residual activity was measured using the coupled enzyme assay as described under 4.10. The control sample was detected by incubation the enzymes at 25 °C in the absence of acetaldehyde (0 min) (Fig. 3).4.16 Screening activity of ADH with lactol (3) and NAD+
The activity of the alcohol dehydrogenases was assayed spectrophotometrically by measuring the absorbance of NAD(P)H at 340 nm. The assay was performed by using a UV star 96-well plate (a multimode spectrophotometer plate reader, Synergy 2, BioTek) with a total volume of 150 μL. The reaction mixtures contains 0.5 mg mL−1 enzyme, 2 mM lactol (3) and 1 mM NAD+ in 50 mM potassium phosphate buffer, pH 7. The samples were incubated for 1 min at 30 °C before measuring the absorbance at 340 nm. Samples were left overnight at room temperature. The activity was measured again under the same conditions. The blank measurement was done without enzyme (Fig. S3†).4.17 Screening activity of ADH with lactol (3) and NADP+
The activity of the alcohol dehydrogenases was assayed spectrophotometrically by measuring the absorbance of NAD(P)H at 340 nm. The assay was performed by using a UV star 96-well plate (a multimode spectrophotometer plate reader, Synergy 2, BioTek) with a total volume of 150 μL. The reaction mixtures contains 0.5 mg mL−1 enzyme, 2 mM lactol (3) and 1 mM NADP+ in 50 mM potassium phosphate buffer, pH 7. The samples were incubated for 1 min at 30 °C before measuring the absorbance at 340 nm. Samples were left overnight at room temperature. The activity was measured again under the same conditions. The blank measurement was done without enzyme (Fig. S3†).4.18 Screening ADH conversion lactol (3) to lactone (4)
The screening of ADH for the conversion of lactol (3) to lactone (4) was performed in 50 mM KPi, pH 7.5 buffer, containing 5 mM NAD(P)+, 1 mg mL−1 ADH, 1 mg NOX, and 20 mM 3. Total reaction volume of 200 μL. The reactions were incubated at 30 °C, for 20 hours, 800 rpm. The reaction mixture was extracted using ethyl acetate with 5 mM decane as an internal standard (3×/200 μL) and centrifuged (5 min, 13.2 krpm). The combined organic layers are dried on anhydrous MgSO4 and analysed with gas chromatography (Table S4†).4.19 Temperature assay ADH440
Temperature assay ADH440 was performed in KPi buffer (50 mM, pH 7.5), containing 5 mM of NADP+, 6 U mL−1 ADH440 and 1 mg NOX, the reactions were started by the addition of 20 mM of lactol. Total reaction volume of 200 μL. The reaction was incubated at 30 °C, 40 °C, 50 °C, 800 rpm for 4 and 24 hours, 800 rpm. The reaction mixture was extracted using ethyl acetate with 5 mM decane as an internal standard (3×/200 μL) and centrifuged (5 min, 13.2 krpm). The combined organic layers are dried on anhydrous MgSO4 and analysed with gas chromatography (Fig. S4†).4.20 Buffer and pH assay ADH440 and LbDERA C42M E78K
Reactions were performed in 2 mL Eppendorf tubes. 20 mM lactol, 5 mM NADP+, 1 mg NOX (NADPH oxidase that oxidises NADPH by reducing oxygen to water) and 1 mg ADH440 (specific activity = 1.2 U mg−1) were dissolved in 8 different buffers: 100 mM TEA pH 7/8, 100 mM KPi pH 6/7/8, 100 mM Tris–HCl pH 7/8/9, to an end volume of 200 μL. Samples were incubated for 20 hours at 30 °C, 800 rpm. Lactone is extracted from an aqueous layer with 3× 1:1 EtOAc. Samples were dried over MgSO4 and analysed with gas chromatography. For LbDERA C42M E78K the activity in the different buffers was measured following the activity assay described in 4.10 (Fig. S5†).
4.21 ADH acetaldehyde resistance
The acetaldehyde resistance of ADH440 was determined by measuring the residual activity of the enzyme after acetaldehyde incubation. 4 U mL−1 ADH440 was incubated in 0, 50, 100 mM of acetaldehyde (>99.5%, Thermo Scientific) in 100 mM TEA pH 7 buffer at 30 °C, 800 rpm, and total reaction volume of 1 mL. Samples were drawn after 1, 3, 6, and 24 hours, and residual activity of ADH440 was measured following the activity assays given in 3.11 (Fig. 4).4.22 NOX acetaldehyde resistance
The acetaldehyde resistance of NOX was determined by measuring the residual activity of the enzyme after acetaldehyde incubation. 5 mg mL−1 NOX were incubated in 0, 50, 100 mM of acetaldehyde (>99.5%, Thermo Scientific) in 100 mM TEA pH 7 buffer at 30 °C, 800 rpm, and total reaction volume of 1 mL. Samples were drawn after 1, 3, 6, and 24 hours, and residual activity of NOX was measured following the activity assays given in 3.12 (Fig. 4).4.23 Fed-batch DERA biotransformation
Reactions were performed in a 100 mL round bottom flask. 10 U of purified DERA was dissolved in 9 mL 100 mM TEA pH 7.0 buffer. The reaction was done under nitrogen. 3 M of acetaldehyde in 100 mM TEA pH 7.0 buffer was fed with a flow rate of 0.1 mL h−1 to an end concentration of 300 mM acetaldehyde. After 20 hours, 500 μL was extracted with 3× 1:1 ethyl acetate wit 5 mM decane as internal standard, dried over MgSO4, and analysed with gas chromatography.
4.24 The effect of the FMN concentration on the ADH catalysed oxidation of lactol (3)
Oxidation of lactol to lactone with ADH440 was measured for different concentrations of FMN. The reaction mixture of the fed batch reaction described in 3.20, containing 27 mM lactol was mixed with ADH440, NADP+, FMN and catalase to final concentrations of 5 mM NADP+, 2.5 U ADH440, 10 U catalase and, 0.1, 0.2, 0.5, 1, 1.5 mM FMN in 100 mM TEA pH 7 buffer. A total volume of 200 μL, 30 °C, 24 hours, 800 rpm. The reaction mixture was extracted using ethyl acetate with 5 mM decane as an internal standard (3×/200 μL) and centrifuged (5 min, 13.2 krpm). The combined organic layers are dried on anhydrous MgSO4 and analysed with gas chromatography (Fig. S6A†).4.25 The effect of the NOX concentration on the ADH catalysed oxidation of lactol (3)
Oxidation of lactol to lactone with ADH440 was measured for different concentrations of NOX. The reaction mixture of the fed batch reaction described in 3.20, containing 27 mM lactol was mixed with ADH440, NADP+, and NOX to final concentrations of 5 mM NADP+, 2.5 U ADH440, and, 1, 2, 5, 10, 15 mg mL−1 NOX in 100 mM TEA pH 7 buffer. A total volume of 200 μL, 30 °C, 24 hours, 800 rpm. The reaction mixture was extracted using ethyl acetate with 5 mM decane as an internal standard (3×/200 μL) and centrifuged (5 min, 13.2 krpm). The combined organic layers are dried on anhydrous MgSO4 and analysed with gas chromatography (Fig. S6B†).4.26 The effect of the ADH440 concentration on the oxidation of lactol (3)
Oxidation of lactol to lactone with ADH440 was measured for different concentrations of ADH with the NOX and FMN regeneration system. The reaction mixture of the fed batch reaction described in 4.23, containing 27 mM lactol was mixed with 5 mM NADP+, 0.5, 1, 2, 3, 5, 8, 10 U ADH440, and, 1 mg NOX or 1 mM FMN in 100 mM TEA pH 7 buffer. A total volume of 200 μL, 30 °C, 24 hours, 800 rpm. The reaction mixture was extracted using ethyl acetate with 5 mM decane as an internal standard (3×/200 μL) and centrifuged (5 min, 13.2 krpm). The combined organic layers are dried on anhydrous MgSO4 and analysed with gas chromatography (Fig. 5).4.27 Scale up fed batch DERA biotransformation
Fed batch synthesis of lactol catalysed by DERA. 1 U mL−1 of purified LbDERA C42M E78K in 45 mL 100 mM TEA pH 7.0 buffer, room temperature, stirring, under nitrogen. 3 M of acetaldehyde in 100 mM TEA pH 7.0 buffer fed with flowrate 0.5 mL h−1 to end concentration of 300 mM and end volume of 50 mL. 500 μL of samples were taken in duplicates over time and analysed with GC.4.28 Scale up of the FMN-driven reaction
The reaction was performed in a 100 mL round bottom flask. 20 mL of the reaction mixture of a DERA fed-batch reaction, containing 30 mM of lactol was mixed with, 5 mM NADP+, 40 U mL−1 ADH440, 60 U catalase, and 1 mM of FMN. The reaction was heated to 30 °C, and stirred for 24 hours. The reaction was exposed to blue light (λmax 465 nm). Commercially available LED lights (Paulmann, YourLED, Basic Set RGB 1.5 m) were wrapped around a thermostatted reaction vessel. 200 μL samples were taken at 1, 2, 4, 6, and 24 hours. After 24 hours the lactone was extracted from the mixture with 3× 1:1 ethyl acetate, and purified following the purification protocol in 4.4. The lactone was purified as a colorless oil, 0.23 mmol, 30 mg, 4.6% yield, [α]D = +34.2 °(c = 0.7) in agreement with [α]D = +35.0 °(c = 0.7) reported in literature.29 NMR results are in accordance with 4.4 and the literature.29
4.29 Scale up of the NOX-driven reaction
The reaction was performed in a 100 mL round bottom flask. 20 mL of the reaction mixture of a DERA fed-batch reaction, containing 30 mM of lactol was mixed with, 5 mM NADP+, 40 U mL−1 ADH440, and 5 mg mL−1 NOX. The reaction was heated to 30 °C and stirred for 24 hours. 200 μL samples were taken at 1, 2, 4, 6, and 24 hours. After 24 hours the lactone was extracted from the mixture with 3× 1:1 ethyl acetate, and purified following the purification protocol in 4.4. The lactone was purified as a colorless oil, 0.22 mmol, 28 mg, 4.4% yield, NMR results are in accordance with 4.4 and the literature.29
4.30 GC analysis screening ADH
A GC system (GC-2010 plus, Shimadzu) with column type cp wax 52 CB, column dimension of 25 m × 0.25 mm × 1.2 μm, column flow of 1 mL min−1, carrier gas: N2, injection volume of 1 μL, 50.0 split ratio and constant pressure of 84.5 kPa was used. The column oven temperature was increased over time. Temperature started at 100 °C (hold time: 3.5 min), increased with 25 °C min−1 to 180 °C (hold time: 4 min), increased wit 25 °C min−1 to 220 °C (hold time: 4 min), increased with 30 °C min−1 to 250 °C (hold time: 1 min) (Fig. S9†).4.31 GC analysis lactol and lactone
GC analysis were carried out on a gas chromatograph equipped with a FID detector and CP-sil 8 CB column (25 m × 0.25 mm × 1.2 μm). Column flow rate: 1 mL min−1, carrier gas: N2, injector temperature: 340 °C, injection mode: 1 μL split ratio 100, detector temperature 360 °C, constant pressure of 80.9 kPa. Oven programme: 80.0 °C 3 min hold, 20.0 °C min−1 to 200.0 °C 2 min hold, 20.0 °C min−1 to 215.0 2 min hold, 30 °C min−1 to 245.0 1 min hold. Rt decane = 8.9 min, rt lactol = 10.6 min, rt lactone = 12.9 min (Fig. S10†).4.32 Chiral GC analysis lactone
GC analysis were carried out on a gas chromatograph equipped with a FID detector and CP-Chirasil_DEX-CB column (25 m × 0.32 mm × 0.25 μm). Column flow rate: 1.48 mL min−1, carrier gas: He, injector temperature: 340 °C, injection mode: 1 μL split ratio 100, detector temperature 250 °C, constant pressure of 54.4 kPa. Oven programme: 100.0 °C 1 min hold, 20.0 °C min−1 to 150.0 °C 10 min hold, 25.0 °C min−1 to 230.0 1 min hold, 30 °C min−1 (Fig. S11†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for generous financial support from ERACoBiotech (grant 053.80.737; BioDiMet). They are also grateful to Tim Wesselingh, Martin Emmaneel, Jeroen Punt, Suzanne Holst, Niva Beije, Merle Meinhardt, and Annemiek Hilker for help with DERA production and optimisation.References
- M. Di Cesare, H. Bixby, T. Gaziano, L. Hadeed, C. Kabudula, D. V. McGhie, J. Mwangi, B. Pervan, P. Perel, D. Piñeiro, S. Taylor and F. Pinto, World Heart Report 2023: Confronting the World's Number One Killer, World Heart Federation, Geneva, Switzerland, 2023 Search PubMed.
- C. W. Tsao, A. W. Aday, Z. I. Almarzooq, C. A. M. Anderson, P. Arora, C. L. Avery, C. M. Baker-Smith, A. Z. Beaton, A. K. Boehme, A. E. Buxton, Y. Commodore-Mensah, M. S. V. Elkind, K. R. Evenson, C. Eze-Nliam, S. Fugar, G. Generoso, D. G. Heard, S. Hiremath, J. E. Ho, R. Kalani, D. S. Kazi, D. Ko, D. A. Levine, J. Liu, J. Ma, J. W. Magnani, E. D. Michos, M. E. Mussolino, S. D. Navaneethan, N. I. Parikh, R. Poudel, M. Rezk-Hanna, G. A. Roth, N. S. Shah, M.-P. St-Onge, E. L. Thacker, S. S. Virani, J. H. Voeks, N.-Y. Wang, N. D. Wong, S. S. Wong, K. Yaffe and S. S. Martin, Circulation, 2023, 147, e93–e621 CrossRef PubMed.
- P. Hoyos, V. Pace and A. Alcántara, Catalysts, 2019, 9, 260 CrossRef CAS.
- imarc, Statin Market Report by Type (Synthetic Statins, Natural Statins), Therapeutic Area (Cardiovascular Disorders, Obesity, Inflammatory Disorders, and Others), Drug Class (Atorvastatin, Fluvastatin, Lovastatin, Pravastatin, Simvastatin, and Others), Application (Dyslipidemia, and Others), Distribution (Hospitals, Clinics, and Others), and Region 2024–2032, 2023, Report ID: SR112024A1885, https://www.imarcgroup.com/statin-market Search PubMed.
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC.
- A. Ručigaj and M. Krajnc, Chem. Eng. J., 2015, 259, 11–24 CrossRef.
- M. M. Zhang, X. Su, E. L. Ang and H. Zhao, Pharm. Bioprocess., 2013, 1, 179–196 CrossRef.
- F. Gallou, H. Gröger and B. H. Lipshutz, Green Chem., 2023, 25, 6092–6107 RSC.
- M. Haridas, E. M. M. Abdelraheem and U. Hanefeld, Appl. Microbiol. Biotechnol., 2018, 102, 9959–9971 CrossRef CAS PubMed.
- J. Rouvinen, M. Andberg, J. Pääkkönen, N. Hakulinen and A. Koivula, Appl. Microbiol. Biotechnol., 2021, 105, 6215–6228 CrossRef CAS PubMed.
- X.-L. Tang, J.-W. Yu, Y.-H. Geng, J.-R. Wang, R.-C. Zheng and Y.-G. Zheng, Engineering, 2023, 24, 138–150 CrossRef CAS.
- W. A. Greenberg, A. Varvak, S. R. Hanson, K. Wong, H. Huang, P. Chen and M. J. Burk, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5788–5793 CrossRef CAS PubMed.
- X.-C. Jiao, Y.-J. Zhang, Q. Chen, J. Pan and J.-H. Xu, Catal. Sci. Technol., 2016, 6, 7094–7100 RSC.
- A. Švarc, Z. Findrik Blažević, Đ. Vasić-Rački, S. J. Charnock and A. Vrsalović Presečki, Chem. Eng. Res. Des., 2020, 164, 35–45 CrossRef.
- S. Bartsch, J. Brummund, S. Köpke, H. Straatman, A. Vogel and M. Schürmann, Biotechnol. J., 2020, 15, 2000171 CrossRef CAS PubMed.
- A. Švarc, M. Fekete, K. Hernandez, P. Clapés, Z. Findrik Blažević, A. Szekrenyi, D. Skendrović, Đ. Vasić-Rački, S. J. Charnock and A. V. Presečki, Chem. Eng. Sci., 2021, 231, 116312 CrossRef.
- X.-C. Jiao, J. Pan, G.-C. Xu, X.-D. Kong, Q. Chen, Z.-J. Zhang and J.-H. Xu, Catal. Sci. Technol., 2015, 5, 4048–4054 RSC.
- M. Dick, R. Hartmann, O. H. Weiergräber, C. Bisterfeld, T. Classen, M. Schwarten, P. Neudecker, D. Willbold and J. Pietruszka, Chem. Sci., 2016, 7, 4492–4502 RSC.
- D. Skendrović, A. Švarc, T. Rezić, A. Chernev, A. Rađenović and A. Vrsalović Presečki, React. Chem. Eng., 2024, 9, 82–90 RSC.
- A. Wang, M. Wang, Q. Wang, F. Chen, F. Zhang, H. Li, Z. Zeng and T. Xie, Bioresour. Technol., 2011, 102, 469–474 CrossRef CAS PubMed.
- B. Grabner, Y. Pokhilchuk and H. Gruber-Woelfler, Catalysts, 2020, 10, 137 CrossRef CAS.
- F. Subrizi, M. Crucianelli, V. Grossi, M. Passacantando, G. Botta, R. Antiochia and R. Saladino, ACS Catal., 2014, 4, 3059–3068 CrossRef CAS.
- X.-C. Jiao, J. Pan, X.-D. Kong and J.-H. Xu, Biochem. Biophys. Res. Commun., 2017, 482, 159–163 CrossRef CAS PubMed.
- M. Haridas, C. Bisterfeld, L. M. Chen, S. R. Marsden, F. Tonin, R. Medici, A. Iribarren, E. Lewkowicz, P. L. Hagedoorn, U. Hanefeld and E. Abdelraheem, Catalysts, 2020, 10, 883 CrossRef CAS.
- S. Gargiulo, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2011, 3, 338–342 CrossRef CAS.
- A. Švarc, Z. Findrik Blažević, Đ. Vasić-Rački, A. Szekrenyi, W. D. Fessner, S. J. Charnock and A. Vrsalović Presečki, J. Chem. Technol. Biotechnol., 2019, 94, 1832–1842 CrossRef.
- I. Kullartz and J. Pietruszka, J. Biotechnol., 2012, 161, 174–180 CrossRef CAS PubMed.
- R. F. Varela, A. L. Valino, E. Abdelraheem, R. Medici, M. Saye, C. A. Pereira, P. L. Hagedoorn, U. Hanefeld, A. Iribarren and E. Lewkowicz, ChemBioChem, 2022, 23, e202200147 CrossRef CAS PubMed.
- H. J. M. Gijsen and C.-H. Wong, J. Am. Chem. Soc., 1994, 116, 8422–8423 CrossRef CAS.
- G. Tojo and M. Fernández, in Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, ed. G. Tojo and M. Fernández, Springer US, Boston, MA, 2006, pp. 281–288, DOI:10.1007/0-387-25725-X_7.
- T. Vajdič, M. Ošlaj, G. Kopitar and P. Mrak, Metab. Eng., 2014, 24, 160–172 CrossRef PubMed.
- L. Mestrom, P. Bracco and U. Hanefeld, Eur. J. Org. Chem., 2017, 2017, 7019–7025 CrossRef CAS.
- A. I. Benítez-Mateos, D. Roura Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef PubMed.
- A. R. Alcántara, P. Domínguez De María, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, 15, e202102709 CrossRef PubMed.
- L. Veum and U. Hanefeld, Chem. Commun., 2006, 825–831 RSC.
- A. Scheithauer, E. von Harbou, H. Hasse, T. Grützner, C. Rijksen, D. Zollinger and W. R. Thiel, AIChE J., 2015, 61, 177–187 CrossRef CAS.
- J. B. Jones and K. E. Taylor, J. Chem. Soc., Chem. Commun., 1973, 205–206 RSC.
- M. Rauch, S. Schmidt, I. W. C. E. Arends, K. Oppelt, S. Kara and F. Hollmann, Green Chem., 2017, 19, 376–379 RSC.
- Y. Wu, C. E. Paul and F. Hollmann, ChemBioChem, 2021, 22, 2420–2423 CrossRef CAS PubMed.
- K. Mattern and S. T. Grosser, Org. Process Res. Dev., 2023, 27, 1992–2009 CrossRef CAS.
- A. V. Presečki and Đ. Vasić-Rački, Process Biochem., 2009, 44, 54–61 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00047a |
| ‡ Both authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |