
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pt-based catalysts for NOx reduction from H2 combustion engines†
Jieling
Shao
,
Phuoc Hoang
Ho
 ,
Wei
Di
,
Derek
Creaser
and
Louise
Olsson
*
,
Wei
Di
,
Derek
Creaser
and
Louise
Olsson
*
Chemical Engineering, Competence Centre of Catalysis, Chalmers University of Technology, 412 96 Gothenburg, Sweden. E-mail: louise.olsson@chalmers.se
First published on 27th April 2024
Abstract
Platinum supported on SSZ-13 zeolite has been found to be a potential catalyst for the selective catalytic reduction of NO by H2. This work has studied the effects of the H2/NO molar feed ratios (0/4.4/8.8/13.2) and the impact of water on the performance of the H2-SCR of NO on the Pt/SSZ-13 catalyst. A higher H2/NO ratio promoted the start of the reaction at lower temperatures and favoured the production of N2. The effect of Pt loadings was also studied with three loadings of 0.5/1.0/2.0 wt%. It was found that the 0.5 wt% Pt sample displayed the highest N2 selectivity of 75%. In addition, an inhibiting effect of water for H2-SCR at low temperatures was proved. Pt/SSZ-13 has shown good hydrothermal durability after 6 h in total ageing pretreatment at 800 °C and interestingly the nitrogen formation even increased. The support effect of SSZ-13, BETA and Al2O3 on H2-SCR was evaluated in terms of catalytic performance and their catalytic durabilities by hydrothermal ageing experiments, showing that zeolites are significantly better for H2 SCR. In situ DRIFT measurements helped to explore the mechanism of H2-SCR on the Pt catalyst. A careful design of the measurements was used to distinguish the overlapping peaks of the water on the DRIFT spectrum. NH4+ ions are formed and it was shown that they play a role as intermediates during the reaction to assist the NO reduction.
Introduction
Many new energy sources are being used to replace conventional fuels in vehicles to decrease the emissions of greenhouse gases (GHG). In particular, the European Commission has published the GHG-related regulation via ‘A European Green Deal’ which targets no net emissions of greenhouse gases by 2050, which presents a significant challenge. An interesting solution is therefore to use green hydrogen as a fuel. Although the exhaust gases from H2 internal combustion engines (ICE) are cleaner than those from diesel or gasoline engines, the formation of NOx is unavoidable due to the oxidation of nitrogen in the air at elevated temperatures in the chamber of the ICE. In response to this issue, selective catalytic reduction (SCR) of NOx is an essential technology to remove the NOx emission.1–3 Ammonia SCR under lean gas conditions is widely used as the industrial NOx control technology since V2O5–WO3/TiO2 was commercialized for stationary applications in the 1970s.4 Nowadays, Cu, Fe, and V-based materials are commonly used as NH3-SCR catalysts for cleaning emissions from vehicles.5–7 For ammonia SCR a urea system is needed to produce ammonia, however, in so-called hydrogen SCR, it would be beneficial to be able to use hydrogen directly as a reductant.Platinum group metals, in particular Pt and Pd, have been examined for H2-SCR catalysts due to their good water resistance.8 Pt-based catalysts exhibit superior activity in H2-SCR; however, they form large amounts of N2O,9,10 while Pd-based catalysts show better nitrogen selectivity.11–13 A variety of materials such as oxides, different oxide combinations, perovskites, etc. have been used as supports for the development of H2-SCR catalysts.14 Costa and co-workers have studied Pt supported on various metal oxides (Al2O3, SiO2, La2O3, MgO, Y2O3, CaO, CeO2, TiO2, and MgO–CeO2), and found that among different catalysts, Pt/MgO and Pt/CeO2 showed good catalytic activity and the binary oxide of 50 wt% MgO–50 wt% CeO2 was the best.15,16 However, the studies on zeolite as the support for H2-SCR catalysts are more limited. Some works have reported that Pt supported on HY,17 HZSM-5,18,19 HMCM-41,20 and HFER21 zeolites were highly active for H2-SCR due to the large surface area and the appropriate acidity, which is beneficial for the N2 selectivity.22 Additionally, Shibata et al. found that the N2 selectivity for Pt/zeolites were generally higher than those on Pt/metal oxides and were in order of Pt/MFI > Pt/BEA > Pt/Y, Pt/MOR.23 Yokota et al.24 also reported that Pt-ZSM was more active than Pt/SiO2 and Pt/γ-Al2O3. In another work, Li et al. found that Pt–Ti-MCM-41 exhibited good stability and tolerance against SO2 and CO.20
SSZ-13, a typical small-pore zeolite, is a well-known support for the development of Cu-based catalysts for NH3-SCR applications and the Cu-SSZ-13 catalysts have superior activity and N2 formation selectivity due to their extraordinary physiochemical properties.25–27 In this respect, the Cu-SSZ-13 catalyst was firstly commercialized in 2010 for NOx emission control due to its excellent performance.28,29 It not only possesses high activity at low temperatures, but its stable hydrothermal durability can cope with realistic operations.30,31 However, according to our knowledge, there are only limited published studies where Pt is supported on HSSZ-13 as the catalyst for H2-SCR. Hong et al.32 have reported that Pt/SSZ-13 exhibited an NO conversion of 81% at 100 °C with 91% N2 selectivity and this catalyst was better than Pt/ZSM-5 and Pt/SAPO-34 during stability tests in the presence of SO2 and H2O. Their activity test was performed in a microreactor (4 mm) with a powder catalyst (20–40 mesh).
Moreover, the mechanisms of H2-SCR on Pt-based catalysts have been examined by researchers. Costa et al. studied the nitrogen pathway in the reaction on Pt/MgO–CeO2 by SSITKA-MS and SSITKA-DRIFTS, in order to identify the structure-active and inactive chemisorbed NOx species.33 Zhang and co-workers proposed an N2O formation route over Pt/HY in H2-SCR, where they suggested that the N2O was mainly formed at the Pt–support interface with H spillover.34 In addition, relevant studies have also reported the effect of the interaction between Pt and the support on the reaction and the effect of some additive metals, such as Na.35,36
However, although the H2-SCR reaction is not a new reaction, most of the effect studies (H2/NO ratio, O2 concentration, water content, Pt loadings, etc.) deal with Pt/oxide catalysts. Studies on Pt/SSZ-13 are limited. Compared to Hong's work, the catalytic performances are evaluated here by a flow reactor with wash-coated monoliths instead of pure powder catalysts which is closer to a realistic after-treatment configuration. Moreover, the mechanism is examined here by using in situ DRIFT (in situ diffuse reflectance infrared Fourier transformed spectroscopy) spectroscopy. In addition, for the first time according to our knowledge, hydrothermal ageing is examined for Pt/SSZ-13 used for H2-SCR. This study aims to develop Pt/SSZ-13 catalysts as efficient catalysts for NOx reduction by H2. The effect of H2/NO ratios, Pt loadings, water concentrations and hydrothermal ageing have also been investigated by comparing the catalyst performance for the H2-SCR reaction.
Experimental
Catalyst synthesis
Three different supports BETA, SSZ-13 and Al2O3 were compared. The test results showed that the Pt supported on zeolites (SSZ-13 or BEA) exhibited significantly more nitrogen formation compared to Pt/Al2O3. Since SSZ-13 is well-known to be a superior support for ammonia SCR, compared to for example BETA, considering hydrothermal stability, it was chosen for this study. The activity results for both de-greened and aged catalyst, including the preparation of the Pt/BEA and Pt/Al2O3 catalyst can be found in the ESI.†SSZ-13 zeolite was prepared by hydrothermal synthesis from a gel with the following molar composition: 0.1Na2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1SiO2:0.025Al2O3:0.2N,N,N-trimethyl-1-adamantylammonium hydroxide (TMAdOH):44H2O.37 Sodium hydroxide (VMR Chemicals), aluminum hydroxide hydrate (Sigma-Aldrich), and fumed silica (Sigma-Aldrich) were used as sodium, aluminium, and silica sources, respectively. TMAdOH (TCI) was used as a template to help generate the Chabazite (CHA) structure. After stirring and ageing overnight, a uniform gel was formed and it was transferred to a Teflon-lined stainless-steel autoclave, heated to 160 °C and kept for 6 days. The product was washed several times with Milli-Q water and dried at 80 °C overnight. The powder was ground and calcined to remove the template at 600 °C for 8 h. The Na-SSZ-13 obtained after calcination was ion-exchanged with 0.5 M NH4NO3 (Thermo Scientific) solution (100 mL per 1 g powder) at 80 °C for 2 h, washed with Milli-Q water, and repeated twice. After that, the sample was dried overnight at 80 °C and calcined at 550 °C for 6 h to obtain the H-form of SSZ-13 (H-SSZ-13).
:1SiO2:0.025Al2O3:0.2N,N,N-trimethyl-1-adamantylammonium hydroxide (TMAdOH):44H2O.37 Sodium hydroxide (VMR Chemicals), aluminum hydroxide hydrate (Sigma-Aldrich), and fumed silica (Sigma-Aldrich) were used as sodium, aluminium, and silica sources, respectively. TMAdOH (TCI) was used as a template to help generate the Chabazite (CHA) structure. After stirring and ageing overnight, a uniform gel was formed and it was transferred to a Teflon-lined stainless-steel autoclave, heated to 160 °C and kept for 6 days. The product was washed several times with Milli-Q water and dried at 80 °C overnight. The powder was ground and calcined to remove the template at 600 °C for 8 h. The Na-SSZ-13 obtained after calcination was ion-exchanged with 0.5 M NH4NO3 (Thermo Scientific) solution (100 mL per 1 g powder) at 80 °C for 2 h, washed with Milli-Q water, and repeated twice. After that, the sample was dried overnight at 80 °C and calcined at 550 °C for 6 h to obtain the H-form of SSZ-13 (H-SSZ-13).
Three samples of Pt/SSZ-13 with different loadings of Pt (0.5 wt%, 1.0 wt%, 2.0 wt%) were prepared by an incipient wetness impregnation technique. Pt solution was prepared by mixing Pt(NO3)4 precursor (Alfa Aesar, 15 wt% Pt) and Milli-Q water to have a total volume similar to the wet volume of the zeolite support. The wet volume of H-SSZ-13 was approximately 0.6 mL g−1, close to the total pore volume determined with N2 physisorption measurement. The Pt solution was dropped slowly on 4 g of H-SSZ-13 zeolite placed in a mortar and after each drop, the mixture was conscientiously ground using a pestle to achieve a homogeneous distribution.38 After impregnation, the sample was dried overnight at 80 °C and subsequently calcined at 600 °C for 8 h to obtain the powder catalyst.
The synthesized catalyst powder after calcination was then dissolved in a solution (50 wt% ethanol and 50 wt% Milli-Q water) with a 5 wt% boehmite binder (Disperal P2, Sasol) as a slurry and dip-coated onto a honeycomb monolith (cordierite, D × L = 1.5 cm × 2.0 cm, 400 cpsi) until the desired loading (300 mg) of washcoat was reached.39 The honeycomb monolith was cut from the commercial substrate. Coated monoliths were subsequently dried in an oven at 80 °C overnight and then calcined at 500 °C for 2 h. Detailed information regarding the monolith preparation can be found elsewhere in our previous work.40,41
Activity measurements
Catalytic tests were performed with a laboratory-scale flow reactor (synthetic gas bench, SGB), where the monolith was inserted into a horizontal reactor tube (inner diameter: 16 mm) and wrapped in quartz wool to prevent the passage of gases around the outside of the monolith. The inlet gas mixture was regulated by Bronkhorst mass flow controllers (MFCs) and water vapour was produced by a controlled evaporation and mixing system (CEM). The mole fractions of all the gases at the outlet of the reactor were measured and monitored by FTIR (MKS™ Multigas 2030) and mass spectrometry (HPR-20 QIC). The flow reactor experiments are presented in Table 1 and Fig. S1.† Firstly, the catalyst monolith was de-greened using 10 mol% O2 in Ar balance at 550 °C for 4 h and pretreated using 10% O2 and 5 mol% H2O at 500 °C for 30 min with a total flow rate of 1200 NmL min−1. After the pretreatment, the test cycle of heating and cooling over a temperature range of 80 to 500 °C with a temperature ramp of 5 °C min−1 was repeated 5 times to remove initial experimental perturbations. In a typical experiment, the gas composition of the reactivity test was 10% O2, 5% H2O, 500 ppm NO, and varying H2 concentration (H2/NO = 0, 4.4, 8.8, 10, 13.2) in Ar balance. For 1 wt% Pt-SSZ-13 catalyst, another five-cycle test in the absence of water (dry test) was also performed to study the effect of water on the activity of the catalyst. The discussion of the activity test in section: H2-SCR activity tests was all based on the data of the 4th cycle. It should be noted that for 2 wt% Pt/SSZ-13, two different monoliths were prepared using the same batch of catalyst. The first monolith was used to study the effect of H2/NO ratio (after multiple other cycling experiments) and the effect of hydrothermal ageing. Monolith 2 was used to compare the activity for different Pt loadings. Other experimental details can be found in the ESI.†000 h−1 (STP))
| Step | H2-SCR experiment | Conditions |
|---|---|---|
| 1 | Degreening & pretreatment | (i) 10% O2 in Ar for 4 h at 550 °C (ii) 10% O2 and 5% H2O in Ar for 30 min at 500 °C |
| 2 | Cooling | (i) Cooling from 500 °C to 80 °C in Ar (rate: 5 °C min−1) (ii) keep at 80 °C for 30 min |
| 3 | Test cycle × 5 | (i) Continuous reaction from 80 °C to 500 °C (heating rate: 5 °C min−1) in the gas mixture of 500 ppm NO, varying H2 concentration, 10% O2, 0/5% H2O in Ar (ii) keep at 500 °C with the same gas mixture for 20 min (iii) continuous reaction from 500 °C to 80 °C (cooling rate: 5 °C min−1) in the same gas mixture |
Catalyst characterization
Powder X-ray diffraction (XRD) measurements were conducted using a Bruker AXS D8 advance operating at 40 kV and 40 mA with Cu Kα radiation to verify the materials' crystalline structure. The samples were scanned from 2θ range of 5° to 80° with a step size of 0.02° and scan time of 1 s per step. Elemental compositions (Si, Al, Pt) of the catalysts were analyzed with inductively coupled plasma sector field mass spectrometry (ICP-SFMS) analysis which was performed by ALS Scandinavia (Luleå, Sweden). N2 physisorption measurements were carried out at −196 °C with a Micromeritics Tristar II 3000 Analyzer. Before the measurement, around 0.1 g powder samples were degassed at 250 °C for 16 h under an N2 atmosphere. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method in a range of relative pressure p/p0 from 0.05 to 0.3. Additionally, the pore volume was calculated using the t-plot method.CO chemisorption measurements were performed by using an ASAP2020 Plus instrument (Micromeritics). Around 0.1 g catalyst powder sample was placed in a U-shape reactor with quartz wool. Before measurement, the sample was degassed in He and underwent 1 h of H2 reduction at 400 °C for pretreatment. After that, the powder sample was evacuated by vacuum to pressures in the range of 100–600 mmHg (at intervals of 25 mmHg) which was used for CO adsorption measurements using the double-isotherm method. The stoichiometry factor of 1 for Pt was used to calculate the amount of CO adsorption.42,43 Combining the results of CO chemisorption and ICP, the Pt active site densities with different loadings can be calculated. The estimated active site densities with units of μmol g−1, shown in Table 2, were calculated by the following equation.

W washcoat – actual washcoat mass in the monolith
0.95 – mass fraction of actual catalyst material (other than binder) in washcoat
Pt loading – mass fraction of Pt in catalyst from ICP analysis
Dispersion – from CO chemisorption measurement
M Pt – molecular weight of platinum
| Nominal loading of Pt/wt% | ICP-SFMS | CO chemisorption | TEM | BET | |||
|---|---|---|---|---|---|---|---|
| Pt/wt% | SAR (SiO2/Al2O3) | Dispersion/% | Mean particle size/nm | Estimated Pt site densitya (μmol g−1 washcoat) | Particle size/nm | Surface area/m2 g−1 | |
| a Estimated Pt site density = Wwashcoat × 0.95 × loading × dispersion/MPt/Wwashcoat. | |||||||
| 0.5 | 0.51 | 21.6 | 18.6 | 6.1 | 4.53 | 5.4 | 602 |
| 1.0 | 1.04 | 21.8 | 11.7 | 9.7 | 5.70 | 7.8 | 616 |
| 2.0 | 2.04 | 19.4 | 9.9 | 11.4 | 9.64 | 10.1 | 611 |
The morphology of catalyst particles was measured by transmission electron microscopy (TEM) using an FEI Titan 80-300 microscope equipped with a high-angle annular dark-field (HAADF) detector. The average particle size and distribution were calculated by random selection of over 100 particles using ImageJ software.
X-ray photoelectron spectroscopy (XPS) analysis was conducted using a PHI5000 VersaProbe III-Scanning XPS Microprobe™ with an X-ray source of monochromatic Al Kα radiation (Hν = 1486.6 eV). The system was aligned with Au (83.96 eV), Ag (368.21 eV) and Cu (932.62 eV). The measurements were calibrated with the adventitious carbon peak (C 1s) at 284.8 eV. Since the Pt 4f5/2 and Al 2p lines overlapped, we used the Pt 4f7/2 which could be observed clearly to distinguish them by the intensity ratio of Pt 4f7/2:Pt 4f5/2 = 4:3 and the energy separation between them is around 3.33 eV. The XPSPEAK41 software was employed for the deconvolution of experimental spectra into individual components. Background subtraction and curve fitting were processed by the Shirley model and Gauss–Lorentz functions in the software.
In situ diffuse reflectance infrared Fourier transformed spectroscopy (DRIFTS) spectra were recorded with a Bruker Vertex 70 spectrometer equipped with an MCT detector and scanning was done at 4 cm−1 resolution. A powder sample was placed into a sealed diffuse reflection chamber (Harrick Praying Mantis) equipped with a CaF2 window. A mass spectrometer (Hidden HR20) was connected to the outlet to detect the out-gases. In situ DRIFTS was conducted for three studies. The first one is the CO adsorption on Pt-SSZ13 catalysts to determine the Pt states and thus validate the XPS results, as well as provide the estimation of the stoichiometry factor for CO chemisorption on the Pt.44,45 The second DRIFTS study was to understand the reaction mechanism of H2-SCR by observing different adsorbates on the surface and their changes during the adsorption of NO followed by an introduction of H2. The last DRIFTS study aims to support the reactivity of adsorbed NH4+ species on the catalyst surface. Before each experiment, a pretreatment at 550 °C with 100 NmL min−1 of 10 vol% O2 in Ar for 30 min was performed to clean the catalyst surface. For the CO adsorption, the background was recorded at 35 °C before the measurement. Then, a flow of 100 Nml min−1 containing 1000 ppm of CO/Ar was introduced to the cell for 60 min. After that CO was cut and switched to pure Ar to flush the chamber. The spectrum was recorded every minute for 63 times from introducing CO. For the H2-SCR mechanism studies, new samples were loaded. The background was recorded at 80 °C after the pretreatment. Then, three steps were performed, firstly flowing 500 ppm NO for 60 min, flushing with Ar for 30 min, and finally introducing 5000 ppm H2 for 60 min to investigate the reaction mechanism. To study the effect of the water spectrum, an additional measurement was performed. 5000 ppm H2 and 5000 ppm O2 flowed for 60 min in the system without any NO. The water formation spectrum was recorded to compare the water peaks with the peaks in the previous spectrum. Following this, the same three steps were repeated to observe the H2-SCR reaction process. To study the different effects of the stepwise and simultaneous introduction of reacting gases, the experiment of simultaneous introduction of 500 ppm NO and 5000 ppm H2 for 60 min was also added. Moreover, 5000 ppm O2 was added afterwards to study the O2 effect on the H2-SCR reaction.
For the adsorbed NH4+ species reactivity study, de-greened 1 wt% Pt/SSZ-13 was loaded. In the same way as for the second study, the background was recorded at 80 °C after the pretreatment. The catalyst was first exposed to a flow of 500 ppm NH3 for 60 min. Then Ar was purged for 60 min to remove the gas-phase NH3 and weakly adsorbed NH3 species. After that, 500 ppm NO was introduced and kept for 60 min. For each step, 60 spectra were recorded.
Results and discussion
Catalyst characterization
Three Pt/SSZ-13 samples with 0.5 wt%, 1.0 wt%, and 2 wt% of Pt were prepared by the incipient wetness impregnation method. The Pt loadings were determined by ICP-SFMS and they were very close to the nominal values (Table 2). The SiO2 to Al2O3 molar ratio was around 20 for all samples.The XRD patterns of the lab-synthesized H-type SSZ-13 zeolite and supported Pt catalysts with different loadings are shown in Fig. 1. The XRD pattern of H-type SSZ-13 zeolite (black curve) shows the typical reflections of chabazite (CHA) structure (by citation number: #89-0735).46 The structure of the zeolite was, as expected, well preserved after loading with Pt. The XRD patterns of the three Pt/SSZ-13 catalysts, with different loadings, exhibited three characteristic peaks at 2θ = 39.8°, 46.2°, and 67.4° which correspond to (111), (200), and (220) reflections of bulk fcc Pt (by citation number: #04-0802),46 respectively. These characteristic peaks of Pt in the pattern of the 0.5 wt% Pt/SSZ-13 were weak due to the low platinum content. However, the intensity of these three peaks increased accordingly with an increase in the Pt loadings, suggesting that the crystallite size of Pt would be larger.
 | ||
| Fig. 1 X-ray diffraction patterns of H-SSZ-13 zeolite and 0.5, 1.0, and 2.0 wt% Pt/SSZ-13 catalysts. | ||
Nitrogen physisorption characterization revealed that the specific surface areas (SBET) of the three samples are similar and in the range of 600–620 m2 g−1. The Pt loadings are low and therefore do not have a great influence on the overall specific surface area.
The Pt particle sizes were determined using both STEM and CO chemisorption. The STEM images of the de-greened Pt/SSZ-13 catalysts are shown in Fig. 2 and the particle sizes determined from STEM images are shown and listed in Fig. 3 and Table 2. The average particle sizes (Fig. 3) are determined from 100 randomly selected particles in the TEM images. It should be noted that from TEM images, the observed particles have a differential distribution from small to large which lies on the external surface of zeolite. The platinum existing in ion-exchanged locations with a size smaller than the pore of zeolite (<0.4 nm) cannot be observed by TEM as well as not detected by XRD. The average particle sizes are increasing with Pt loading as expected, from 5.4 nm, 7.8 nm to 10.1 nm for 0.5, 1.0, and 2.0 wt% Pt/SSZ-13 samples. CO chemisorption measurements were performed to determine the platinum dispersion and to support the particle size determination from the TEM measurements and the results are summarized in Table 2. It should be noted that the factor of CO binding on the Pt was set as 1, based on the fact that CO was linearly adsorbed on the Pt as observed in the DRIFTS measurements (see section: In situ DRIFTS studies for CO adsorption). As the Pt loadings increased from 0.5 to 2.0 wt%, the dispersion of Pt particles decreased by almost half, from 18.6% to 9.9%. The mean particle sizes determined from the dispersions were 6.1 nm, 9.7 nm and 11.4 nm for 0.5, 1.0, and 2.0 wt% of Pt, respectively (Fig. 4). Noticeably, for each loading, the mean size of Pt determined by CO chemisorption was similar to the TEM measurements (Fig. 4). The particle sizes measured with TEM were slightly lower than those from CO chemisorption and this could be due to CO chemisorption being a bulk analysis technique in which the CO probe gas can access the whole sample, whereas TEM measurements only focus on some selected areas.
 | ||
| Fig. 2 HAADF-STEM images of Pt de-greened catalysts with different loadings (a and b) 0.5 wt% Pt/SSZ-13 (c and d) 1.0 wt% Pt/SSZ-13 (e and f) 2.0 wt% Pt/SSZ-13. | ||
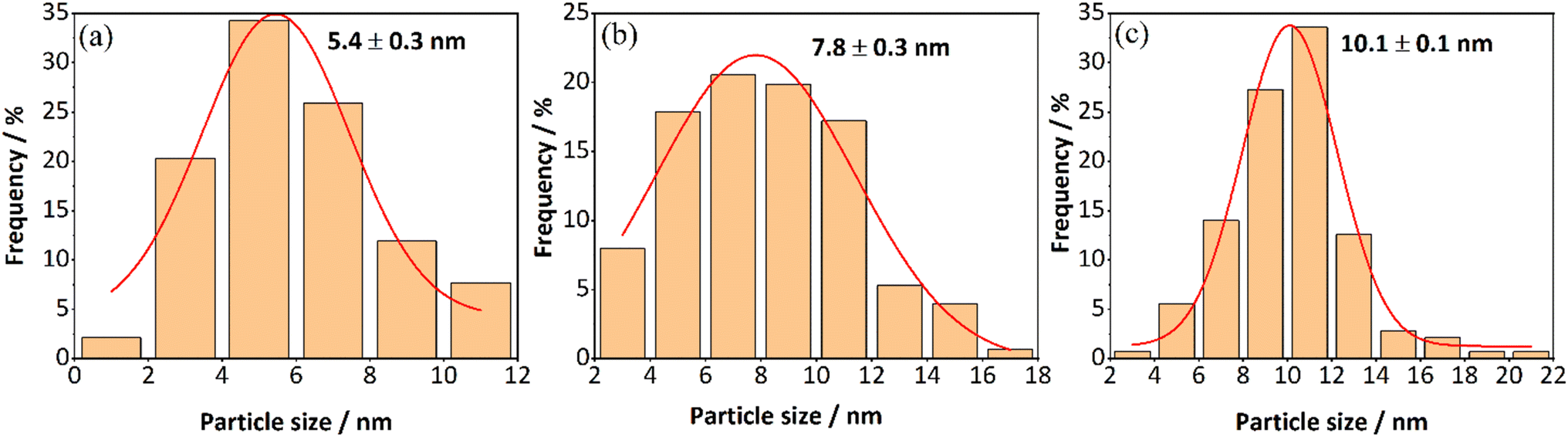 | ||
| Fig. 3 Particle size distributions of Pt/SSZ-13 de-greened catalysts with (a) 0.5 wt% (b) 1.0 wt% (c) 2.0 wt% loadings. | ||
 | ||
| Fig. 4 Particle size trends from TEM and CO chemisorption. | ||
The oxidation state was examined using XPS, and the results are shown in Fig. 5, where the spectra of Pt 4f core level of Pt/SSZ-13 de-greened catalysts with 1.0 wt% and 2.0 wt% Pt are compared. It should be noted that an overlap of binding energy between Al 2p and Pt 4f makes it difficult for the deconvolution of the spectrum of Pt 4f, in particular with a low loading of Pt. Therefore, we only show the data of the measurements for 1.0 wt% Pt/SSZ-13 and 2.0 wt% Pt/SSZ-13. For the specific data, the proportions of Pt states for 1.0 wt% and 2.0 wt% Pt/SSZ-13 are determined using the XPS PEAK4.1 software, and the distributions are shown in Table 3. Accordingly, the binding energy at 70.6/74.4 eV and 72.5/75.8 eV of the 1 wt% Pt de-greened sample can be assigned to 4f7/2/4f5/2 of Pt0 and Pt2+ species, respectively.47,48 The peaks of the 2.0 wt% Pt de-greened sample slightly shifted the binding energies, resulting from the different interactions between Pt and zeolite caused by higher Pt loading. This is related to the net electron transfer from platinum to the support, thus changing the electron density around the metal, which has been proposed by scholars in the 1970s.49 Interestingly, the 2.0 wt% Pt sample has a small amount of Pt0 at 71 eV and this peak is much less intense than for the 1.0 wt% Pt/SSZ-13 samples. It is clear that the 1.0 wt% Pt sample has a larger amount of metallic Pt (73.7%) than the 2.0 wt% Pt sample (24.8%) and a lesser amount of Pt2+ with 26.3% compared to 75.2% for the 2.0 wt% Pt sample. Additionally, Fig. S2† was plotted to study the Pt 4d5/2 spectra due to the overlapping Al 2p peak for the Pt 4f. According to the literature,50 the signal at the higher binding energy (316.0–317.1 eV) can be assigned to the Pt2+ ions, whereas the band at lower binding energy (314.3–315.5 eV) can be considered as Pt0 species. Although the signal is weak, it can be used as an aid to demonstrate that the 1.0 wt% Pt/SSZ-13 catalyst indeed has more Pt particles in the metallic state. These results are surprising since for Pt/Al2O3 larger Pt particles are usually more resistant towards platinum oxidation.51 We suggest that the reason for this surprising behaviour is that the platinum in this case is supported on a zeolite. The ion-exchanged Pt cannot be observed in our TEM images (the resolution is not high enough). Indeed, Pérez-Díaz et al. compared platinum supported on carbon or zeolite and found that the metallic platinum content was higher on Pt/X zeolite compared to Pt/C.52 Simultaneously they observed that the Pt particles were larger (11.7 nm) on Pt/C than Pt/X zeolite (7.4 nm). Thus, even though the particles on Pt/X zeolite were smaller, they still were more metallic. In our case, it is the same amount of acid sites in both 1% Pt/SSZ-13 and 2% Pt/SSZ-13, which likely results in that larger fraction of the platinum in 1% Pt/SSZ-13 being ion-exchanged. Since ion-exchanged platinum results in more metallic platinum according to Pérez-Díaz et al.52 this can explain why 1% Pt/SSZ-13 exhibit more metallic platinum even though the particles were smaller in TEM.
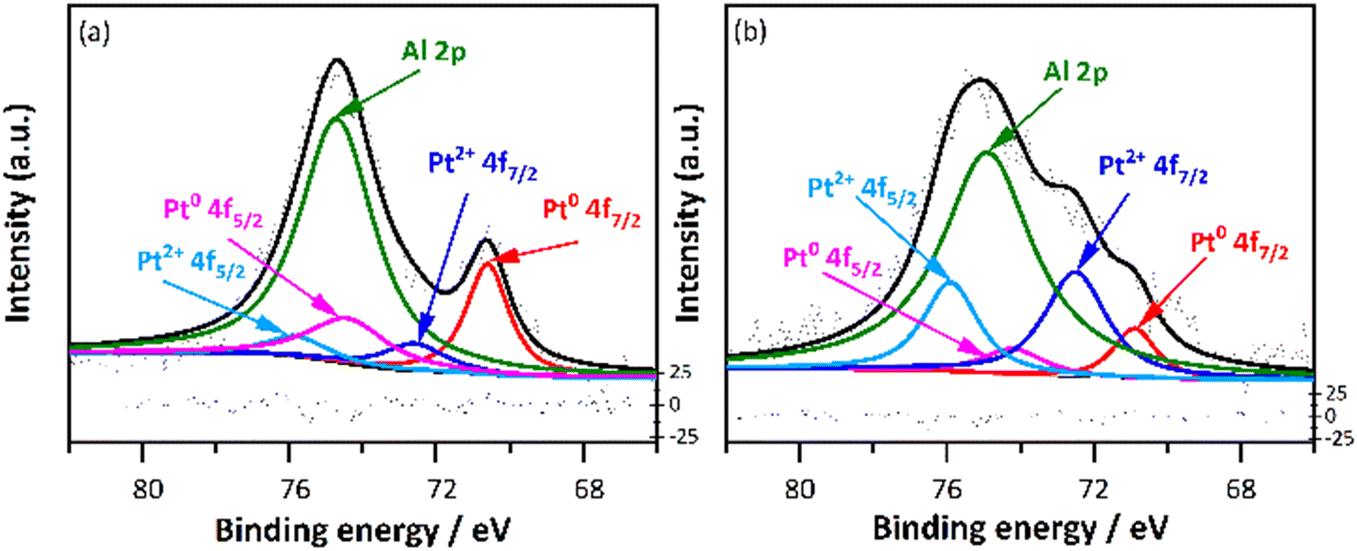 | ||
| Fig. 5 X-ray photoelectron spectra of (a) 1.0 wt% Pt/SSZ-13 (b) 2.0 wt% Pt/SSZ-13 de-greened catalysts. | ||
| Nominal Pt loading | Pt0 | Pt2+ | ||
|---|---|---|---|---|
| BE/eV (Pt 4f7/2/Pt 4f5/2) | Fraction/% | BE/eV (Pt 4f7/2/Pt4 f5/2) | Fraction/% | |
| 1.0 | 70.6/74.4 | 73.7 | 72.5/75.8 | 26.3 |
| 2.0 | 71/74.2 | 24.8 | 72.3/75.9 | 75.2 |
H2-SCR activity tests
 | ||
| Fig. 6 NOx and N2 concentration of H2-SCR reaction on 2 wt% Pt/SSZ-13 with four temperature zones ① 80–100 °C ② 100–135 °C ③ 135–330 °C ④ 330–500 °C (GHSV = 20000 h−1 (STP); gas inlet: 10% O2, 5% H2O, 500 ppm NO, 4400 ppm H2 balanced in Ar; T: 80–500 °C; heating rate: 5 °C min−1). | ||
Above 135 °C, the rate for oxidation of H2 on Pt increased further and resulted in a depletion of H2. This caused a decrease in the activity for the reduction of NO. Simultaneously, the oxidation of NO proceeded, resulting in a significant increase in NO2 concentration. In the temperature zone of 330–500 °C, H2 was completely consumed (Fig. S4†) since no reduction was observed, and NO was mainly oxidized to NO2. This resulted in the absence of N2O and N2 in the outlet (black and red curves). Finally, at high temperatures, the NO oxidation was suppressed due to the thermodynamic equilibrium.57
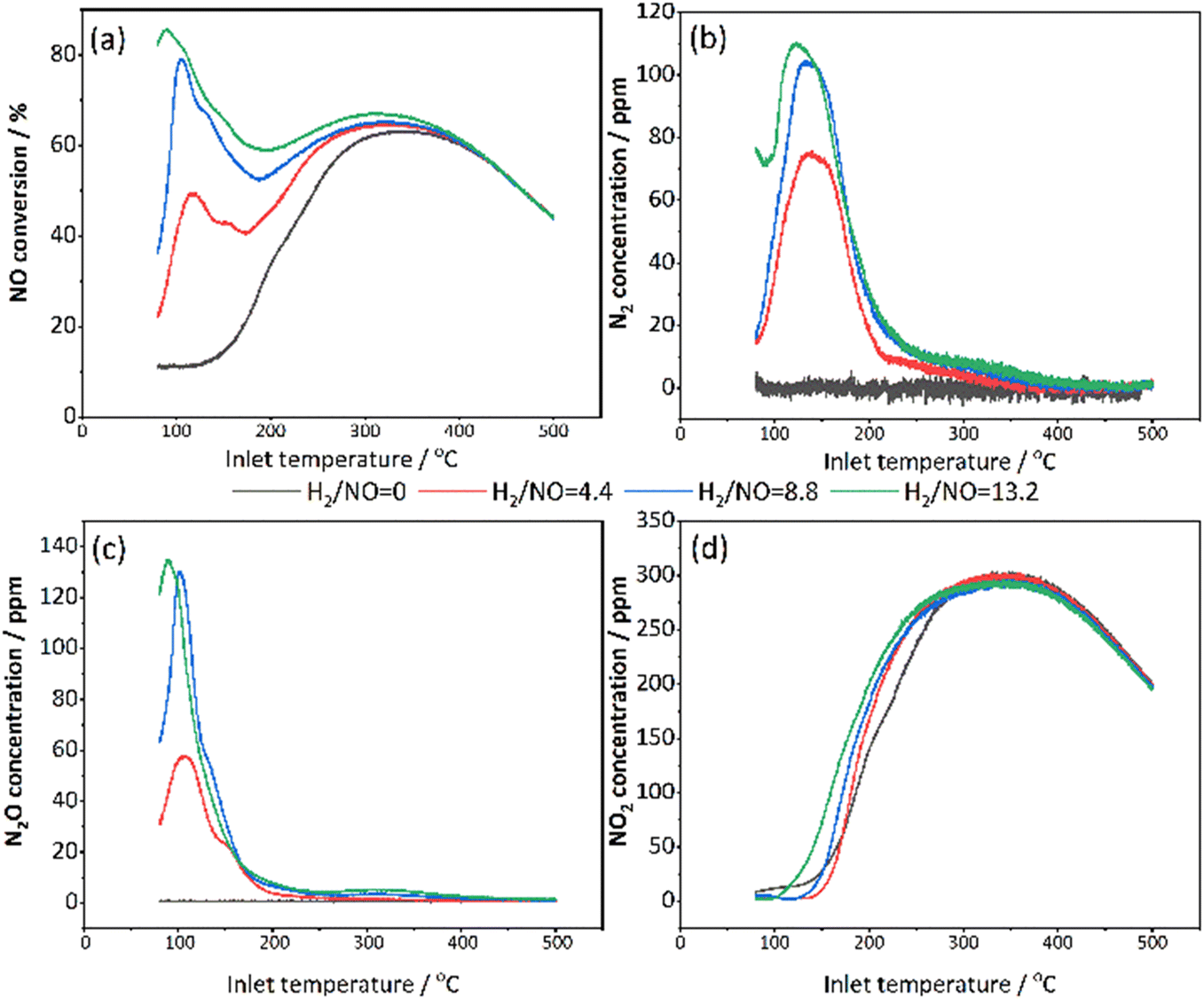 | ||
| Fig. 7 Comparison of the effect of H2/NO ratios on 2 wt% Pt/SSZ-13 catalyst, where (a) shows the conversion, and the concentration profiles are shown in (b) N2 (c) N2O (d) NO2 (GHSV = 20000 h−1 (STP); gas inlet: 10% O2, 5% H2O, 500 ppm NO, 2200/4400/6600 ppm H2 balanced in Ar; T: 80–500 °C; heating rate: 5 °C min−1). | ||
The conversion of NO at low and medium temperatures increased as the proportion of H2 increased, which is consistent with the investigation by Olympiou et al. where Pt/MgO–CeO2 was used for H2 SCR.10 They pointed out that the reduction rate of active NOx to N2 or N2O will be influenced by the surface concentration of H (θH) on Pt, thus the N2 selectivity as well. In Fig. 7a, the maximum NO conversion was approximately 85% for the H2/NO ratio of 13.2, which is close to the max. NO conversion of 80% for the H2/NO ratio of 8.8. In addition, when examining the whole temperature interval, the NO conversions were larger for the highest H2/NO ratio. Interestingly, a small shoulder is observed in connection to the first NO conversion peak for all H2/NO ratios. Moreover, a greater proportion of H2 favoured the target product N2, with a subsequent decrease in by-product N2O (Fig. 7b and c). It is reasonable to have a higher NO reduction capacity in the presence of a larger amount of hydrogen. Both N2 and N2O profiles behaved with a characteristic volcano trend at low temperatures (Fig. 7b and c), but the peaks of N2O were sharper. This means that N2 is produced over a wider temperature range than N2O. When the H2 reaches 6600 ppm, most N2 and N2O are formed (the highest active H atom density), but it was quite similar to the condition of 4400 ppm H2. Thus, the performance improvement is small when the H2 concentration rises above 8.8.
NO oxidation became dominant at high temperatures (Fig. 7d). The reason is that H2 is rapidly consumed by oxidation at high temperatures, which limited the H2-SCR reduction. Meanwhile, an interesting phenomenon is observed from Fig. 7d that as the proportion of H2 increased, the NO oxidation occurred earlier at lower temperatures and also the temperature for maximum NO2 concentration shifted to lower temperatures which both indicated that the presence of H2 promotes the NO oxidation. One of the reasons for this is an exothermic effect of the H2 combustion during the reaction, where higher H2 inlet concentrations resulted in higher temperatures inside the monoliths, which could further increase the reactions (Fig. S5†). It has been demonstrated and quantified in previous work by our group that the elevated monolith temperatures by hydrogen combustion promote NO oxidation.59 In addition, the higher H2 concentration could also result in more metallic platinum which could increase the NO oxidation.
In addition, the behaviours of H2/NO ratios remained consistent for the 1 wt% Pt/SSZ-13 catalyst in general. The gas concentrations of NO, NO2, N2O and N2 during the H2-SCR are shown in Fig. S6.† It compared the influence of H2 concentrations of 5000 ppm and 6600 ppm on 1 wt% Pt/SSZ-13 catalyst. The results show that the NO conversion was significantly higher on the 1 wt% Pt/SSZ-13 with a higher H2 concentration of 6600 ppm. Moreover, under higher concentrations of H2, the undesired by-product N2O formation was decreased. In contrast, N2 production was accelerated and enhanced at higher hydrogen concentrations. Additionally, on the 1 wt% Pt/SSZ-13 catalyst, H2 continued to assist NO oxidation, with NO production occurring at lower temperatures.
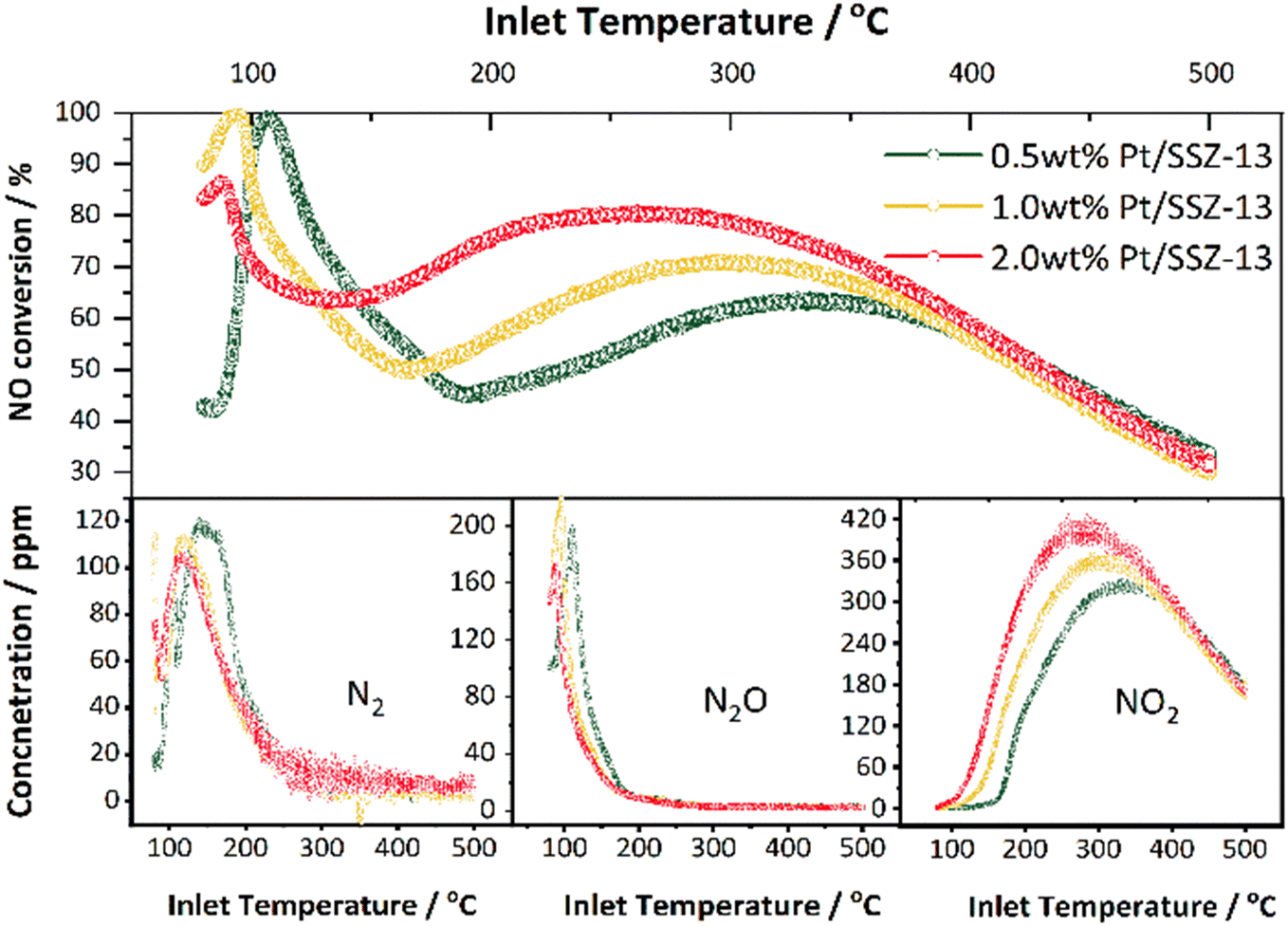 | ||
| Fig. 8 Comparison of NO conversion and N2/N2O/NO2 concentration profiles on Pt/SSZ-13 catalysts with three Pt loadings (GHSV = 20000 h−1 (STP); gas inlet: 10% O2, 5% H2O, 500 ppm NO, 5000 ppm H2 balanced in Ar; T: 80–500 °C; heating rate: 5 °C min−1). | ||
 | ||
| Fig. 9 Comparison of N2/N2O/NO2 selectivity profiles on Pt/SSZ-13 catalysts with three Pt loadings (a) 0.5 wt% Pt (b) 1.0 wt% Pt (c) 2.0 wt% Pt (d) N2 selectivities of 3 samples (GHSV = 20000 h−1 (STP); gas inlet: 10% O2, 5% H2O, 500 ppm NO, 5000 ppm H2 balanced in Ar; T: 80–500 °C; heating rate: 5 °C min−1). | ||
To summarize, 0.5 wt% Pt/SSZ-13 exhibited the best catalytic performance for H2-SCR with the highest N2 selectivity and the total amount of nitrogen produced. Furthermore, the 2 wt% Pt/SSZ-13 sample exhibited the highest oxidation activity, but lower selectivity for the NOx reduction reactions. This could be related to the H2 oxidation as discussed above, but also a particle size effect, where the 0.5 wt% Pt sample has the smallest mean particle size as observed with both CO chemisorption and TEM analysis, see Fig. 3 and Table 2. A larger number of active sites gives the reactant molecules a larger reaction area and thus a higher reactivity.61,62 However, the amount of platinum is about 4 times larger (2.04 versus 0.51%), while the dispersion is only doubled for the low-loaded platinum sample (18.6 versus 9.9%), see Table 2. This results in the 2 wt% Pt/SSZ-13 sample exhibits a factor of 2.1 times more available sites compared to the 0.5 wt% Pt/SSZ-13 sample. Olsson et al.63 observed earlier that large platinum particles are more active for NO oxidation than small particles, even though there were fewer sites. These results are in line with our results where the 2 wt% Pt/SSZ-13 was very active for oxidation reactions as seen by the higher NO2 production. This also resulted in hydrogen being oxidized faster thereby reducing the selectivity for H2-SCR. However, at very low temperatures, when the H2 was not fully consumed by its oxidation, the H2-SCR selectivity is related to the concentration of hydrogen that can reach the catalyst's active sites. In addition, the XPS results revealed (Fig. 5 and Table 3) that the lower loading sample contained a significantly larger amount of reduced platinum species, which could also be a reason for the higher selectivity for the H2-SCR reaction.
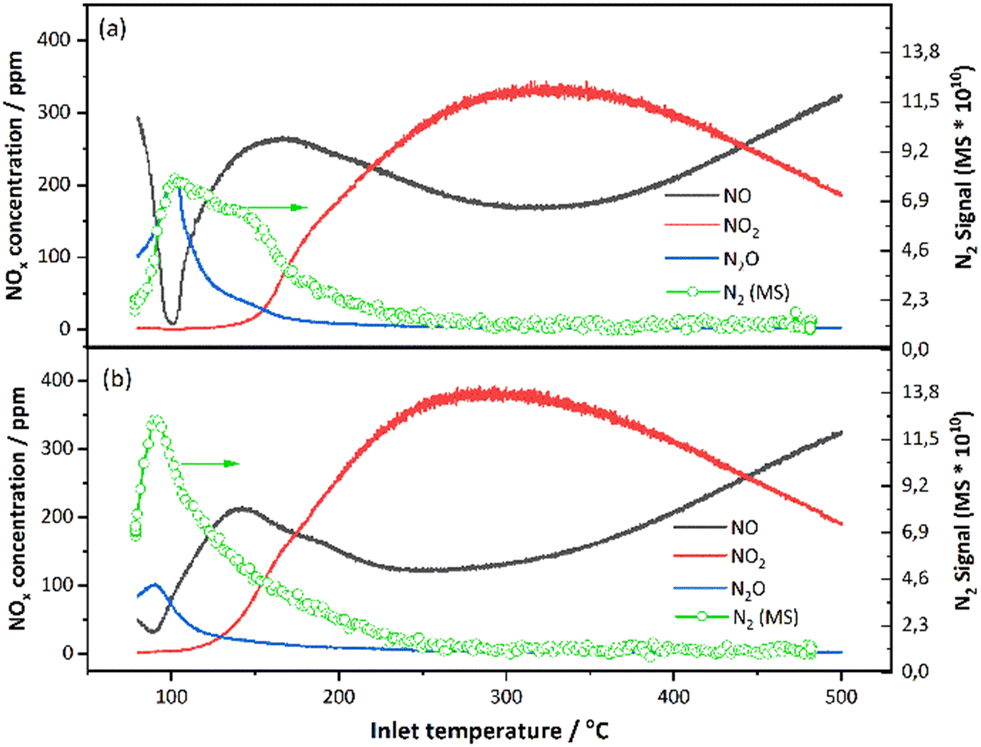 | ||
| Fig. 10 Comparison of NO/NO2/N2 concentration from FITR and N2 Signal from MS of 1 wt% Pt/SSZ-13 catalyst (a) with H2O and (b) without H2O (GHSV = 20000 h−1 (STP); gas inlet: 10% O2, 0%/5% H2O, 500 ppm NO, 5000 ppm H2 balanced in Ar; T: 80–500 °C; heating rate: 5 °C min−1). | ||
 | ||
| Fig. 11 (a) NO, NO2, (b) N2O, and N2 concentrations of H2-SCR on 2 wt% Pt/SSZ-13 (used from Fig. 7) and its hydrothermal pretreated for 2 h and 6 (2 + 4) h samples (GHSV = 20000 h−1 (STP); gas inlet: 10% O2, 5% H2O, 500 ppm NO, 4400 ppm H2 balanced in Ar; T: 80–500 °C; heating rate: 5 °C min−1). | ||
 | ||
| Fig. 12 CO DRIFTS spectra performed on 0.5 wt%/1.0 wt%/2.0 wt% de-greened Pt/SSZ-13 catalysts. | ||
Compared to Fig. S10,† the CO-adsorption profile on the fresh samples presents some differences in trend. For the higher Pt loadings, the degreening pretreatment results in more ionic Pt, which is evident by that the peak at 2098 cm−1 is declining. For Cu/SAPO-34 (also chabazite structure) similar behaviour has been seen where higher temperature results in Cu migration to ion-exchanged sites that increase the activity which could be one of the reasons to explain this result.73
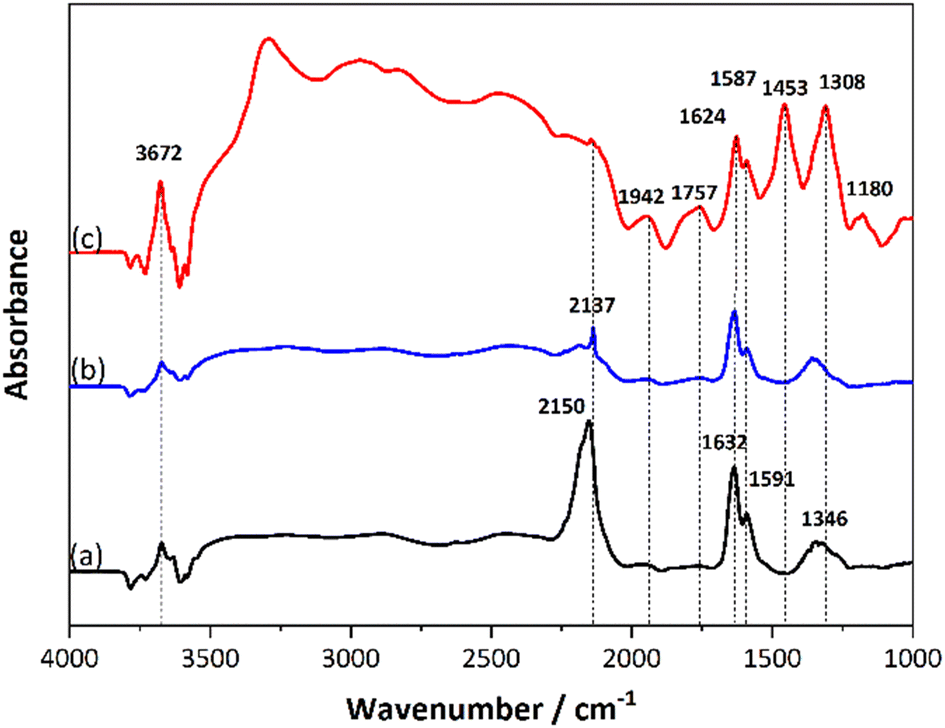 | ||
| Fig. 13 In situ DRIFTS spectra for exposure of 1 wt% Pt/SSZ-13 under (a) 500 ppm NO at 80 °C for 60 min (b) cut NO for 60 min (c) feed 5000 ppm H2 for 60 min. | ||
After introducing H2 at 80 °C, new peaks at 1180, 1308, 1453, 1757 and 1942 cm−1 appeared. Large peaks appeared above 2000 cm−1 after introducing H2 and were mainly assigned to O–H stretching from water formation. The bands at 2470 cm−1 and 2945 cm−1 referred to the OH− and O·− anions from water. Noticeably, a distinctive peak at 3303 cm−1 is shown, which can be assigned to the O–H stretching vibration of water molecules that are hydrogen bonded to the anion.75 In the range of 3580–3800 cm−1, the peaks from lower binding energies to higher binding energies are attributed to Si–OH(nest) (3590 cm−1), framework bridged hydroxyl groups-AlOHSi (3630 cm−1), extra framework AlOH groups (3670 cm−1) and terminal silanol groups – SiOHs (3750 cm−1) which are related to the OH bonds with the zeolite support.76 Moreover, it was reported that peaks belonging to the water bending mode in the gas phase and liquid phases are located at 1594 cm−1 and 1642 cm−1 on the Al2O3 surface, respectively.77 And from an FTIR study of water adsorption on H-ZSM-5 zeolite by Jobic and co-workers,78 it was verified that the peak at 1340 cm−1 could belong to bridging OH groups with deformations by different zeolite structure and composition.
Since the overlap of NO adsorption and water adsorption on the surface increased the complexity of the spectra, additional measurements were performed for verification, where only H2 and O2 were added to the system without any NO (Fig. 13a). With only H2 and O2, similar peaks as appearing in the Fig. 13c were seen, which demonstrates that the peaks belonging to the water adsorption mostly covered the peaks from NO reduction. Interestingly, a band at 1453 cm−1 was observed in Fig. 13c that could be attributed to NH4+ ions, which could be one of the intermediates of the H2-SCR reaction.79–81 After H2 + O2 reaction there is a peak at 1465 cm−1 that came from the H2O adsorption which is quite close to the peak at 1453 cm−1, which might result in partial overlaps. Moreover, according to the literature,82 peaks at 1332 cm−1 and 1625 cm−1 can be assigned to the symmetric and antisymmetric bending modes of the hydroxonium ion in the zeolite. When only flowing NO in the system (Fig. 14b), it is obvious that several peaks originally belonging to water adsorption show negative peaks due to the water removal from the surface, which also includes the peak at 1465 cm−1. For the enlarged view of the region near 1633 cm−1 in Fig. 14b, it was been mentioned in relation to Fig. 13 that the peaks at 1633 cm−1 and 1591 cm−1 are assigned to the nitrates adsorbed on the Pt and zeolite surface respectively. The intensity of these peaks became progressively stronger as the time of NO exposure increased from 10 min to 60 min. After introducing H2, it was observed that only the peak at 1633 cm−1 decreased and shifted to a lower wavenumber of 1626 cm−1 (dashed line), which illustrates that H2 adsorbed on the Pt active site, attacks the nitrates on the Pt, leading to weakened binding and a changed surface intermediate structure. The peaks above 2000 cm−1 appeared again indicating that the H2-SCR reaction began to occur, and water was formed. Most importantly, a new peak at 1454 cm−1 was formed with H2-SCR, further suggesting the presence of ammonium ions in the reaction process. It should be noted that water also has a band at 1465 cm−1 (Fig. 14a) thus it is likely that the band at 1454 cm−1 is an overlap between ammonia and water on the surface. NH3 has been reported as a by-product of H2-SCR in many publications,54,83 however the activity test in our study (section: Reaction process) did not reveal the presence of ammonia. Instead, through DRIFTS the presence of ammonium ions is shown, suggesting that the Pt/SSZ-13 could store NH4+. This is in line with the study by Giordanino et al. who studied the interaction of NH3 with Cu-SSZ-13,84 and here NH4+ could play a role as an intermediate that assists the NO reduction. As well as the work from Shibata et al., the reactions between intermediate NHx species and NO were shown as an important pathway during the H2-SCR process.23 The band recorded at 1309 cm−1 is considered a water peak in this spectrum. Although it has been reported that it may belong to nitrites (NO2−) adsorbed on the support74 due to its high peak intensity and the absence of NO flow, it is presumed to belong to water produced by the reaction. A small peak at 1180 cm−1 appears in all the spectra which also is assigned to OH groups.78
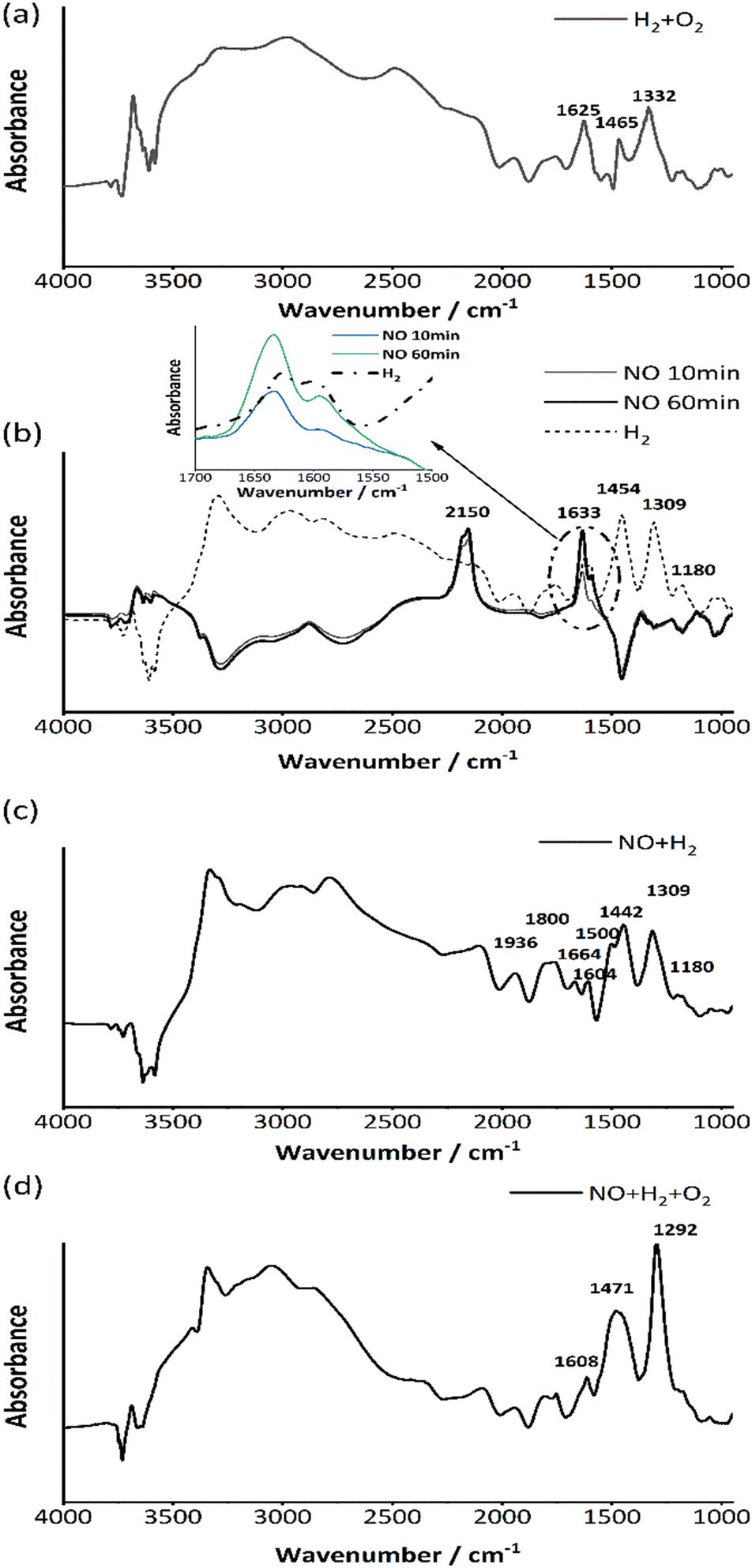 | ||
| Fig. 14 In situ DRIFTS spectra for exposure of 1 wt% Pt/SSZ-13 at 80 °C under (a) 5000 ppm H2 + 5000 ppm O2 for 60 min (b) 500 ppm NO for 60 min; flush Ar for 30 min; 5000 ppm H2 for 60 min (enlarged view of the peaks near 1633 cm−1) (c) 500 ppm NO + 5000 ppm H2 at for 60 min (d) 500 ppm NO + 5000 ppm H2 + 5000 ppm O2 for 60 min. | ||
If NO and H2 are added simultaneously (Fig. 14c), the overall spectrum is similar to Fig. 14b. However, it could be noticed that the peak at 1454 cm−1 was split into two peaks at 1442 cm−1 and 1500 cm−1, respectively. The peak at 1442 cm−1 is considered to be a shift by the H2 influence from the original peak at 1454 cm−1 of ammonium ions since the shape and intensity are quite similar.85 Due to the reaction occurring more rapidly and the competitive adsorption of H2 against NOx, the small side peak at 1500 cm−1 is assigned to different NOx adsorption species such as Pt2NO.86 Moreover, the NOx adsorption peak in the region of 1591–1633 cm−1 subsequently disappeared, which could be explained by the rapid reaction in the presence of H2. Comparing Fig. 14d with NO + O2 + H2, the peak at 1309 cm−1 is shifted to 1292 cm−1 with an extremely enhanced intensity and sharpness. The peak at 1309 cm−1 was assigned to water instead of nitrites (NO2−) with only H2 feed, but here with NO + O2 + H2, it is reasonable to consider that nitrites also contribute to the peak. Although the peaks at 1442 cm−1 and 1500 cm−1 merged into a peak around 1471 cm−1 again, this peak shape is broader, and this could be due to the overlap of multiple peaks.
 | ||
| Fig. 15 In situ DRIFTS spectra for exposure of 1 wt% Pt/SSZ-13 at 80 °C under (a) 500 ppm NH3 flowing for 60 min; Ar flowing for 60 min (b) 500 ppm NO flowing for 60 min. | ||
DRIFTS measurements have led to a more in-depth study and understanding of the key surface intermediate species involved in the reaction. Multiple tests were carried out to reduce the complexity of the system caused by the influence of water. To sum up, i) nitrosyl species weakly adsorbed on the acidic sites of the zeolite can be easily removed. ii) H2 absorbed on Pt was activated to interact with NO which was also absorbed on the Pt surface. iii) The formation of NH4+ ions was demonstrated and they can play a role as a reaction intermediate to assist in the reduction of NO, which also was shown. iv) The reaction occurred rapidly when NO and H2 entered simultaneously, where the NO storage form was changed. v) The addition of O2 to NO + H2 mixture generates more nitrite (NO2−) species on the catalyst.
Conclusions
Pt-based, lab-synthesized SSZ-13 zeolite catalysts were prepared by an incipient impregnation method with three loadings of 0.5 wt%, 1.0 wt% and 2 wt% and characterized and activity-tested in a flow reactor with wash-coated monoliths. The reaction process is complex, with H2-SCR mainly occurring at lower temperatures, and NO oxidation at higher temperatures, but with several side reactions. A higher proportion of H2 with H2/NO ratios of 4.4/8.8/13.2 promoted the reaction to start at a lower temperature and favoured the N2 formation. Compared to the higher Pt loadings, the 0.5 wt% sample has good activity and higher maximum N2 selectivity of 75%, this could be due to a lower oxidation of H2 by oxygen and the fact that the catalyst contains a higher proportion of platinum in the metallic state. Water has an inhibition effect on H2-SCR at lower temperatures due to the competitive adsorption between water and reactant molecules. Pt/SSZ-13 catalyst has shown good hydrothermal durability after 6 h in total hydrothermal ageing.In situ DRIFT studies were done to explore the reaction mechanisms. When H2 was introduced to an NO-saturated sample at 80 °C, water formation was observed showing that the SCR reaction was started. H2 is absorbed on the surface of Pt active sites and is activated to interact with nitrates that are also absorbed on the Pt surface. The bonding of nitrates on the Pt was weakened. Meanwhile, NH4+ ions were formed and shown that during the reaction they played a role as reaction intermediates to assist in the reduction of NO. Simultaneous entry of NO and H2 induces a faster reaction than sequential entry and affects the binding of surface species, especially NO. The introduction of O2 to the NO + H2 mixture generates more nitrite (NO2−) species on the catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study has been funded by Swedish Energy Agency and carried out in collaboration with Volvo AB, Scania CV and Johnson Matthey via a strategic vehicle research and innovation (FFI) project (P51458-1). We also appreciate the assistance for material characterizations of Dr Stefan Gustafsson (TEM measurements), Dr Eric Tam (XPS analysis), and Dr Andreas Schaefer (CO chemisorption). As well as Lennart Norberg and Lasse Urholm (Flow-reactor technical support).Notes and references
- J. Wang, H. Zhao, G. Haller and Y. Li, Appl. Catal., B, 2017, 202, 346–354 CrossRef CAS.
- W. Shan and H. Song, Catal. Sci. Technol., 2015, 5, 4280–4288 RSC.
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef CAS PubMed.
- F. Nakajima, Catal. Today, 1991, 10, 1–20 CrossRef CAS.
- D. Damma, P. R. Ettireddy, B. M. Reddy and P. G. Smirniotis, Catalysts, 2019, 9(4), 349 CrossRef CAS.
- I. Nova and E. Tronconi, Fundamental and Applied Catalysis Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, Springer, New York, 2014 Search PubMed.
- H. Xue, G. Li, P. Liu, F. Wang, Y. Bai and K. Wang, Adv. Mater. Res., 2012, 550, 119–123 Search PubMed.
- Z. Hu and R. T. Yang, Ind. Eng. Chem. Res., 2019, 58, 10140–10153 CrossRef CAS.
- J. Shibata, M. Hashimoto, K. I. Shimizu, H. Yoshida, T. Hattori and A. Satsuma, J. Phys. Chem. B, 2004, 108, 18327–18335 CrossRef CAS.
- G. G. Olympiou and A. M. Efstathiou, Chem. Eng. J., 2011, 170, 424–432 CrossRef CAS.
- L. Wang, H. Chen, M. H. Yuan, S. Rivillon, E. H. Klingenberg, J. X. Li and R. T. Yang, Appl. Catal., B, 2014, 152–153, 162–171 CrossRef CAS.
- J. Li, G. Wu, N. Guan and L. Li, Catal. Commun., 2012, 24, 38–43 CrossRef CAS.
- M. Mihet, M. D. Lazar, V. Almasan and V. Mirel, AIP Conf. Proc., 2012, 1425, 73–76 CrossRef CAS.
- X. Li, X. Zhang, Y. Xu, Y. Liu and X. Wang, Chin. J. Catal., 2015, 36, 197–203 CrossRef CAS.
- C. N. Costa and A. M. Efstathiou, J. Phys. Chem. C, 2007, 111, 3010–3020 CrossRef CAS.
- C. N. Costa and A. M. Efstathiou, Appl. Catal., B, 2007, 72, 240–252 CrossRef CAS.
- X. Zhang, X. Wang, X. Zhao, Y. Xu, H. Gao and F. Zhang, Chem. Eng. J., 2014, 252, 288–297 CrossRef CAS.
- Z. Hong, Z. Wang, D. Chen, Q. Sun and X. Li, Appl. Surf. Sci., 2018, 440, 1037–1046 CrossRef CAS.
- Q. Yu, M. Richter, F. Kong, L. Li, G. Wu and N. Guan, Catal. Today, 2010, 158, 452–458 CrossRef CAS.
- L. Li, P. Wu, Q. Yu, G. Wu and N. Guan, Appl. Catal., B, 2010, 94, 254–262 CrossRef CAS.
- S. Yang, X. Wang, W. Chu, Z. Song and S. Zhao, Appl. Catal., B, 2011, 107, 380–385 CrossRef CAS.
- A. Satsuma, M. Hashimoto, J. Shibata, H. Yoshida and T. Hattori, Chem. Commun., 2003, 1698–1699 RSC.
- J. Shibata, M. Hashimoto, K. I. Shimizu, H. Yoshida, T. Hattori and A. Satsuma, J. Phys. Chem. B, 2004, 108, 18327–18335 CrossRef CAS.
- K. Yokota, M. Fukui and T. Tanaka, Appl. Surf. Sci., 1997, 121, 273–277 CrossRef.
- F. Gao and J. Szanyi, Appl. Catal., A, 2018, 560, 185–194 CrossRef CAS.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- W. Hu, T. Selleri, F. Gramigni, E. Fenes, K. R. Rout, S. Liu, I. Nova, D. Chen, X. Gao and E. Tronconi, Angew. Chem., Int. Ed., 2021, 60, 7197–7204 CrossRef CAS PubMed.
- I. A. Pankin, A. Martini, K. A. Lomachenko, A. V. Soldatov, S. Bordiga and E. Borfecchia, Catal. Today, 2020, 345, 125–135 CrossRef CAS.
- H. Y. Chen, Z. Wei, M. Kollar, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2015, 329, 490–498 CrossRef CAS.
- Z. Hong, X. Sun, Z. Wang, G. Zhao, X. Li and Z. Zhu, Catal. Sci. Technol., 2019, 9, 3994–4001 RSC.
- C. N. Costa and A. M. Efstathiou, J. Phys. Chem. C, 2007, 111, 3010–3020 CrossRef CAS.
- X. Zhang, X. Wang, X. Zhao, Y. Xu, H. Gao and F. Zhang, Chem. Eng. J., 2014, 252, 288–297 CrossRef CAS.
- Z. Liu, B. Jia, Y. Zhang and M. Haneda, Ind. Eng. Chem. Res., 2020, 59, 13916–13922 CrossRef CAS.
- M. Machida and T. Watanabe, Appl. Catal., B, 2004, 52, 281–286 CrossRef CAS.
- Z. Li, M. T. Navarro, J. Martínez-Triguero, J. Yu and A. Corma, Catal. Sci. Technol., 2016, 6, 5856–5863 RSC.
- P. H. Ho, D. Yao, D. Creaser and L. Olsson, ACS Eng. Au, 2022, 2, 219–235 CrossRef CAS.
- K. Wijayanti, S. Andonova, A. Kumar, J. Li, K. Kamasamudram, N. W. Currier, A. Yezerets and L. Olsson, Appl. Catal., B, 2015, 166–167, 568–579 CrossRef CAS.
- P. H. Ho, J. Shao, D. Yao, R. F. Ilmasani, M. A. Salam, D. Creaser and L. Olsson, J. Environ. Chem. Eng., 2022, 10, 108217 CrossRef CAS.
- D. Yao, R. Feizie Ilmasani, J. C. Wurzenberger, T. Glatz, J. Han, A. Wang, D. Creaser and L. Olsson, Chem. Eng. J., 2021, 428, 132459 CrossRef.
- G. J. Freel, J. Catal., 1972, 25, 149–160 CrossRef.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS PubMed.
- E. Ivanova, M. Mihaylov, F. Thibault-Starzyk, M. Daturi and K. Hadjiivanov, J. Mol. Catal. A: Chem., 2007, 274, 179–184 CrossRef CAS.
- G. J. Freel, J. Catal., 1972, 25, 149–160 CrossRef.
- S. Gates-Rector and T. Blanton, Powder Diffr., 2019, 34, 352–360 CrossRef CAS.
- P. Bera, K. R. Priolkar, A. Gayen, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro, V. Jayaram and G. N. Subbanna, Chem. Mater., 2003, 15, 2049–2060 CrossRef CAS.
- H. H. Liu, Y. Wang, A. P. Jia, S. Y. Wang, M. F. Luo and J. Q. Lu, Appl. Surf. Sci., 2014, 314, 725–734 CrossRef CAS.
- J. C. Vedrine, M. Dufaux, C. Naccache and B. Imelik, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 440–449 RSC.
- K. Yang, L. Zhu, J. Zhang, X. Huo, W. Lai, Y. Lian and W. Fang, Catalysts, 2018, 8, 307 CrossRef.
- X. Auvray, T. Pingel, E. Olsson and L. Olsson, Appl. Catal., B, 2013, 129, 517–527 CrossRef CAS.
- P. J. Pérez-Díaz, A. Medina-Ramírez, I. R. Galindo Esquivel, G. García Ruiz and B. Ruiz-Camacho, Ionics, 2021, 27, 1813–1828 CrossRef.
- K. G. Rappé, C. DiMaggio, J. A. Pihl, J. R. Theis, S. H. Oh, G. B. Fisher, J. Parks, V. G. Easterling, M. Yang, M. L. Stewart and K. C. Howden, Emiss. Control Sci. Technol., 2019, 5, 183–214 CrossRef.
- S. Yang, X. Wang, W. Chu, Z. Song and S. Zhao, Appl. Catal., B, 2011, 107, 380–385 CrossRef CAS.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
- H. Shin, M. Choi and H. Kim, Phys. Chem. Chem. Phys., 2016, 18, 7035–7041 RSC.
- L. Olsson, B. Westerberg, H. Persson, E. Fridell, M. Skoglundh and B. Andersson, J. Phys. Chem. B, 1999, 103, 10433–10439 CrossRef CAS.
- L. Olsson, B. Westerberg, H. Persson, E. Fridell, M. Skoglundh and B. Andersson, J. Phys. Chem. B, 1999, 103, 10433–10439 CrossRef CAS.
- M. M. Azis, X. Auvray, L. Olsson and D. Creaser, Appl. Catal., B, 2015, 179, 542–550 CrossRef CAS.
- Q. Yu, M. Richter, F. Kong, L. Li, G. Wu and N. Guan, Catal. Today, 2010, 158, 452–458 CrossRef CAS.
- V. V. Pushkarev, K. An, S. Alayoglu, S. K. Beaumont and G. A. Somorjai, J. Catal., 2012, 292, 64–72 CrossRef CAS.
- L. Cao, Q. Wang and J. Yang, J. Environ. Chem. Eng., 2020, 8, 103631 CrossRef CAS.
- L. Olsson, J. Catal., 2002, 210, 340–353 CrossRef CAS.
- Z. Liu, J. Li and J. Hao, Chem. Eng. J., 2010, 165, 420–425 CrossRef CAS.
- R. Burch and M. D. Coleman, Appl. Catal., B, 1999, 23, 115–121 CrossRef CAS.
- X. Auvray and L. Olsson, Appl. Catal., B, 2015, 168–169, 342–352 CrossRef CAS.
- E. Ogel, M. Casapu, D. E. Doronkin, R. Popescu, H. Störmer, C. Mechler, G. Marzun, S. Barcikowski, M. Türk and J. D. Grunwaldt, J. Phys. Chem. C, 2019, 123, 5433–5446 CrossRef CAS.
- H. Bischofb, N. I. Jaeger, G. Schulz-Eklofp and L. Kubelkovab, J. Mol. Catal. A: Chem., 1993, 80, 95–103 CrossRef.
- E. V. Benvenutti, L. Franken, C. C. Moro and C. U. Davanzo, Langmuir, 1999, 15, 8140–8146 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- F. Azzolina-Jury and F. Thibault-Starzyk, Top. Catal., 2017, 60, 1709–1721 CrossRef CAS.
- A. Y. Stakheev, E. S. Shpiro, O. P. Tkachenko, N. I. Jaeger and G. Schulz-Ekloff, J. Catal., 1997, 169, 382–388 CrossRef CAS.
- C. Niu, X. Shi, F. Liu, K. Liu, L. Xie, Y. You and H. He, Chem. Eng. J., 2016, 294, 254–263 CrossRef CAS.
- P. G. Savva and A. M. Efstathiou, J. Catal., 2008, 257, 324–333 CrossRef CAS.
- J. Lengyel, M. Ončák, A. Herburger, C. Van Der Linde and M. K. Beyer, Phys. Chem. Chem. Phys., 2017, 19, 25346–25351 RSC.
- F. Jin and Y. Li, Catal. Today, 2009, 145, 101–107 CrossRef CAS.
- H. A. Al-Abadleh and V. H. Grassian, Langmuir, 2003, 19, 341–347 CrossRef CAS.
- H. Jobic, A. Tuel, M. Krossner and J. Sauer, J. Phys. Chem., 1996, 100, 19545–19550 CrossRef CAS.
- Y. Shi, X. Wang, L. Chen, S. Li, C. Wu, S. Shan and W. Li, Appl. Surf. Sci., 2020, 506, 144715 CrossRef CAS.
- G. Novell-Leruth, A. Valcárcel, A. Clotet, J. M. Ricart and J. Pérez-Ramírez, J. Phys. Chem. B, 2005, 109, 18061–18069 CrossRef CAS PubMed.
- S. Elzey, A. Mubayi, S. C. Larsen and V. H. Grassian, J. Mol. Catal. A: Chem., 2008, 285, 48–57 CrossRef CAS.
- H. Jobic, M. Czjzek and R. A. van Santen, J. Phys. Chem., 1992, 96, 1540–1542 CrossRef CAS.
- M. Borchers, P. Lott and O. Deutschmann, Top. Catal., 2023, 66, 973–984 CrossRef CAS.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- A. Citra and L. Andrews, J. Phys. Chem. A, 2000, 104, 8160–8172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00153b |
| This journal is © The Royal Society of Chemistry 2024 |