
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate scope driven optimization of an encapsulated hydroformylation catalyst†
Pim R.
Linnebank
a,
Alexander M.
Kluwer
b and
Joost N. H.
Reek
 *ab
*ab
aHomogeneous, Supramolecular and Bio-Inspired Catalysis, Van't Hoff Institute for Molecular Sciences University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl
bInCatT B.V, Science Park 904, 1098 XH Amsterdam, The Netherlands
First published on 16th February 2024
Abstract
Caged complexes can provide impressive selective catalysts. Due to the complex shapes of such caged catalysts, however, the level of selectivity control of a single substrate cannot be extrapolated to other substrates. Herein, the substrate scope using 41 terminal alkene substrates is investigated in the hydroformylation reaction with an encapsulated rhodium catalyst [Rh(H)(CO)3(P(mPy3(ZnTPP)3))] (CAT1). For all substrates, the amount of branched products formed was higher with CAT1 than with the unencapsulated reference catalyst [Rh(H)(CO)2(P(mPy3))2] (CAT2) (linear/branched ratio between 2.14 and 0.12 for CAT1 and linear/branched ratio between 6.22 and 0.59 for CAT2). Interestingly, the level of cage induced selectivity depends strongly on the substrate structure that is converted. Analysis of the substrate scope combined with DFT calculations suggests that noncovalent interactions between the substrate moieties and cage walls play a key role in controlling the regioselectivity. Consequently, these supramolecular interactions were further optimized by replacing the ZnTPP building block with a zinc porphyrin analog that contained OiPr substituents on the meta position of the aryl rings. The resulting caged catalyst, CAT4, converted substrates with even higher branched selectivity.
Introduction
Catalysts encapsulated in supramolecular architectures offer unique levels of selectivity that are mostly unattainable for traditional transition metal catalysts.1–10 By encapsulating a transition metal within a cage, a microenvironment is created around the active catalyst, similar to enzymes. This microenvironment enables differentiation of reactive sites on the substrate that would otherwise be indistinguishable for a traditional transition metal catalyst. Encapsulated catalysts demonstrated impressive control over the site selectivity, regioselectivity, enantioselectivity and chemoselectivity for challenging substrates for several reactions such as the hydroformylation reaction,11–20 allylic alkylation,21 substrate selective isomerization reactions,22 C–H activation,23 site selective semihydrogenation reactions,24 cyclopropanation,25,26 epoxidation,27–29 hydroboration30 and gold-catalyzed cyclization reactions.31–33A common and effective method for encapsulating transition metals is the ligand-template approach.34,35 In this approach, the ligand has a dual role as it coordinates to the catalytically active metal while it also functions as a template for the self-assembly of the capsule. One of the pioneering examples in this regard is the application of [Rh(H)(CO)3(PmPy3(ZnTPP)3)] (CAT1) as a caged hydroformylation catalyst (Fig. 1).14,15CAT1 is formed by self-assembly by combining tris(meta-pyridyl)phosphine [P(mPy3)] and three zinc meso-tetraphenylporphyrin (ZnTPP) units relying on the selective N–Zn coordination.35 The phosphine atom coordinates to rhodium when reacted with [Rh(acac)(CO)2)], and when using syngas (H2:CO), the encapsulated hydroformylation catalyst CAT1 is formed as depicted in Fig. 1.35,36
 | ||
| Fig. 1 The ligand-template approach for the formation of [Rh(H)(CO)3(P(mPy3(ZnTPP)3))] (CAT1) (DFT modeled structure). ZnTPP building blocks are depicted in yellow for clarity. | ||
In a hydroformylation reaction, an alkene reacts with syngas (a mixture of H2 and CO) in the presence of a transition metal catalyst to form an aldehyde. For terminal alkenes, often two different regioisomeric products are formed, the linear (l) and the branched (b) aldehyde (Fig. 2).37–40 The application of CAT1 in the hydroformylation of aliphatic terminal alkenes leads to enhanced regioselectivity to form predominantly the branched aldehyde, which is due to the confinement of the alkene substrate (Fig. 3a). In analogy, the hydroformylation of internal alkenes leads dominantly to the product with the aldehyde on the innermost carbon atom (Fig. 3b and c) (Fig. 4).14–17
 | ||
| Fig. 2 General scheme of the hydroformylation reaction showing that two regioisomers of the aldehyde product can be formed. | ||
 | ||
| Fig. 3 Previously reported substrate scope of aliphatic alkenes using CAT1 for the conversion of a) 1-octene, b) trans-2-octene, c) trans-3-octene, d) propene. | ||
 | ||
| Fig. 4 The substrate rotation is blocked by the ZnTPP capsule for most of the catalytic pathways that lead to the outermost aldehyde. | ||
Branched product selectivity for terminal aliphatic alkenes, without isomerization as a side reaction, is remarkable as most catalysts convert such substrates with an excess to the linear aldehyde.37,41,42 Only recently, three other catalysts have been reported that also convert aliphatic alkenes to form dominantly the branched product i.e. rhodium catalysts based on BOBPhos and TriPhos ligands.43–47 There are some catalysts that produce the branched aldehyde, even in very high selectivity, but these also show a lot of isomerization, typically leading to a mixture of branched aldehyde products.48–50 These catalysts can be very valuable in the hydroformylation of propene, as isomerization doesn't lead to other products.
The selectivity control of unfunctionalized internal alkenes by CAT1 is even more remarkable as the alkene carbon atoms of internal alkene substrates are indistinguishable in terms of electronics and sterics for traditional hydroformylation catalysts.16 These substrates do not contain a (supramolecular) directing group for differentiation between the two alkene carbon atoms51–60 and this demonstrates the power of encapsulated catalysts to control the regioselectivity.
Further improvements on CAT1 have been made by employing an analog of ZnTPP, a porpholactone, that displays a stronger zinc–pyridine interaction than ZnTPP while forming an encapsulated rhodium catalyst similar to CAT1.19
Moreover, as the space around the metal center is slightly smaller, this cage can convert propene to mainly the branched product (l/b = 0.84) (Fig. 3d), where the branched product has significant industrial potential.46,61 Recently, Dydio et al. reported a palladium based catalyst that was able to achieve exceptionally high branched selectivity for propene.61 Experiments with other substrates showed significant isomerization, making this catalyst unsuitable for branched selective reactions of other alkyl alkenes.
To date, CAT1 has only been investigated for linear terminal aliphatic alkenes such as 1-octene, 1-hexene and propene. Other terminal aliphatic alkenes of type R–CH2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C have not been explored and we were interested to what extent the substrate structure would affect the regioselectivity control of CAT1. Herein, we report the evaluation of the substrate scope of terminal alkenes using both CAT1 and a nonencapsulated analogue as a reference catalyst, CAT2 (Fig. 5).62 In a preliminary study, we have recently used part of the experimental data set to explore descriptor based approaches to understand the regioselectivity of these systems.63
C have not been explored and we were interested to what extent the substrate structure would affect the regioselectivity control of CAT1. Herein, we report the evaluation of the substrate scope of terminal alkenes using both CAT1 and a nonencapsulated analogue as a reference catalyst, CAT2 (Fig. 5).62 In a preliminary study, we have recently used part of the experimental data set to explore descriptor based approaches to understand the regioselectivity of these systems.63
 | ||
| Fig. 5 Non-encapsulated reference catalyst [Rh(H)(CO)2(P(mPy3))2] CAT2 generated by combining [P(mPy3)] and [Rh(acac)(CO)2)] under syngas conditions. | ||
Results and discussion
CAT1 has previously been studied in detail for the hydroformylation of 1-octene.16 The binding constant of the first porphyrin to the tris(meta-pyridyl)phosphine was found to be K1 = 2.5 × 103 M−1, while the second and third porphyrin displayed a higher binding constant. This cooperative binding was explained by pi–pi interactions between adjacent porphyrins, as also confirmed by the X-ray structure. This high affinity ensures that under catalytic conditions, the structure is intact. Indeed, control experiments with an excess of ZnTPP show similar selectivity to those with the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (ZnTPP/ P(mPy3)), whereas at a lower ratio the selectivity is lost.14,15,17,19 Compared to CAT2, the rhodium complex in CAT1 is encapsulated, and at the same time the coordination around rhodium has changed from bisphosphorus to monophosphorus. It is well documented that this change in a coordination mode has an impact on both the activity and the selectivity. To distinguish between these effects, detailed theoretical studies have been carried out that concluded that the difference in catalyst performance is based on a combination of both.15,64 For the remainder of the manuscript, we will refer to the encapsulation effect, which is a combination of the cage effect and change in coordination geometry.62
:1 ratio (ZnTPP/ P(mPy3)), whereas at a lower ratio the selectivity is lost.14,15,17,19 Compared to CAT2, the rhodium complex in CAT1 is encapsulated, and at the same time the coordination around rhodium has changed from bisphosphorus to monophosphorus. It is well documented that this change in a coordination mode has an impact on both the activity and the selectivity. To distinguish between these effects, detailed theoretical studies have been carried out that concluded that the difference in catalyst performance is based on a combination of both.15,64 For the remainder of the manuscript, we will refer to the encapsulation effect, which is a combination of the cage effect and change in coordination geometry.62
For the investigation of the current substrate scope, we evaluated 41 commercially available substrates using CAT1 as the encapsulated catalyst and CAT2 as the reference catalyst that is formed under the same conditions in the absence of ZnTPP. For this study, all reactions were conducted in toluene, at room temperature using 20 bar of syngas, identical to previously reported studies with CAT1 as under these conditions, both catalysts display the same kinetic behavior. Apart from the presence of ZnTPP, all conditions were the same for each catalytic entry and every reaction was run for 48 h.
Since the different alkenes investigated in this study exhibit different biases towards the formation of one of the regioisomers, comparing the absolute linear/branched ratio is less meaningful. To obtain an estimate how effective CAT1 is at enhancing branched product formation, we calculated the relative reaction barriers (ΔΔE) based on the linear/branched ratio for every substrate obtained when CAT1 or CAT2 was applied as a catalyst. For these calculations, the Boltzmann distribution was used with kB being the Boltzmann constant and T being the temperature in Kelvin at which the reaction was carried out:
 | (1) |
| Encapsulation effect = ΔΔECAT2 − ΔECAT1 | (2) |
Aliphatic alkenes with remote substituents
First, we reacted the previously reported 1-octene and other aliphatic alkenes with substitution patterns on positions 4′ and 5′ and these catalytic results are presented in Table 1. CAT1 gives full conversion after 48 h for these aliphatic substrates (1a–1g). In contrast, CAT2 gives lower conversion, which is in line with the higher activity of the caged catalyst compared to the non-encapsulated analogue.14,64 The selectivity obtained for 1-octene using CAT1 matches the previously reported regioselectivity (l/b is 0.56), whereas the uncaged catalyst CAT2 provides the aldehydes with an excess of the linear product. The regioselectivity obtained for 5-methylhexene 1b is almost equal to that for 1-octene 1a and thus this methyl group has no effect on the regioselectivity. For the substrates with a substituent on position 4, 1c–1g, the branched selectivity was significantly higher with CAT1 compared to 1-octene 1a with the l/b ranging between 0.44 and 0.27. In particular, the substrates that have a five or six membered ring on position 4, i.e. allylcyclohexane 1f and allylcyclopentane 1g, are converted with high branched selectivity with CAT1. For the substrates 1b–d, the encapsulation effect is similar to the parent 1a substrate (an encapsulation effect of 0.97–1.06 kcal mol−1). For 4,4′-dimethylpen-1-tene 1e, the encapsulation effect is lower than the 1-octene 1a encapsulation effect (0.73 kcal mol−1 for 1evs. 0.97 kcal mol−1 for 1a), which shows that the higher branched selectivity of 1e compared to 1a is mostly caused by the electronic bias of the substrate.|
|
|||
|---|---|---|---|
| Substrate/catalyst | % conva | l:bb |
Encapsulation effectc |
|
a Conversion determined by 1H NMR spectroscopy.
b The linear:branched ratio determined by GC.
c The encapsulation effect calculated by subtracting the relative energy barriers of the encapsulated reaction outcome (using CAT1) from the unencapsulated reaction outcome (using CAT2).
|
|||
| 1a/CAT1 | 100 | 0.56 | 0.97 kcal mol−1 |
| 1a/CAT2 | 70 | 2.88 | |
| 1b/CAT1 | 100 | 0.54 | 1.01 kcal mol−1 |
| 1b/CAT2 | 75 | 3.04 | |
| 1c/CAT1 | 100 | 0.44 | 0.99 kcal mol−1 |
| 1c/CAT2 | 44 | 2.46 | |
| 1d/CAT1 | 100 | 0.42 | 1.06 kcal mol−1 |
| 1d/CAT2 | 41 | 2.51 | |
| 1e/CAT1 | 100 | 0.44 | 0.74 kcal mol−1 |
| 1e/CAT2 | 46 | 1.52 | |
| 1f/CAT1 | 100 | 0.27 | 1.40 kcal mol−1 |
| 1f/CAT2 | 96 | 2.86 | |
| 1g/CAT1 | 100 | 0.39 | 1.11 kcal mol−1 |
| 1g/CAT2 | 84 | 2.55 | |

Aliphatic alkenes with substituents close to the alkene
The second set of substrates for the scope evaluation focused on compounds that contain an additional alkyl substituent on position 3 (Table 2). These substrates display a wide variation of selectivity control with CAT1 (l/b between 0.49 and 2.11). In contrast, the non-encapsulated catalyst CAT2 converts all these substrates with a comparable regioselectivity (l/b of around 5.5), which is common for aliphatic alkenes bearing a substituent on position 3.68,69 Interestingly, the encapsulation effect on 2a and 2b is higher than that observed for the parent substrate 1a, showing that the presence of 5- and 6-membered rings close to the alkene increases the encapsulation effect and results in higher selectivity control. In contrast, when the ring size increases further, as is the case for 2d, the regioselectivity control is lower.|
|
|||
|---|---|---|---|
| Substrate/catalyst | % conva | l:bb |
Encapsulation effectc |
|
a Conversion determined by 1H NMR spectroscopy.
b The linear:branched ratio determined by GC.
c The encapsulation effect calculated by subtracting the relative energy barriers of the encapsulated reaction outcome (using CAT1) from the unencapsulated reaction outcome (using CAT2).
|
|||
| 2a/CAT1 | 100 | 0.49 | 1.33 kcal mol−1 |
| 2a/CAT2 | 80 | 4.62 | |
| 2b/CAT1 | 100 | 0.83 | 1.19 kcal mol−1 |
| 2b/CAT2 | 53 | 6.22 | |
| 2c/CAT1 | 100 | 1.33 | 0.88 kcal mol−1 |
| 2c/CAT2 | 73 | 5.91 | |
| 2d/CAT1 | 100 | 2.11 | 0.59 kcal mol−1 |
| 2d/CAT2 | 69 | 5.75 | |

Aliphatic alkenes with oxygen atoms in the chain
We next evaluated how the caged catalyst controls the regioselectivity of aliphatic alkenes that contain an ether (3a–e), ester (3f–g) or ketone (3h) substituent (Table 3).|
|
|||
|---|---|---|---|
| Substrate/catalyst | % conva | l:bb |
Encapsulation effectc |
|
a Conversion determined by 1H NMR spectroscopy.
b The linear:branched ratio determined by GC.
c The encapsulation effect calculated by subtracting the relative energy barriers of the encapsulated reaction outcome (using CAT1) from the unencapsulated reaction outcome (using CAT2).
|
|||
| 3a/CAT1 | 89 | 0.59 | 0.49 kcal mol−1 |
| 3a/CAT2 | 22 | 1.34 | |
| 3b/CAT1 | 100 | 0.57 | 0.50 kcal mol−1 |
| 3b/CAT2 | 64 | 1.27 | |
| 3c/CAT1 | 100 | 0.32 | 0.36 kcal mol−1 |
| 3c/CAT2 | 57 | 0.59 | |
| 3d/CAT1 | 82 | 0.55 | 0.24 kcal mol−1 |
| 3c/CAT2 | 78 | 0.82 | |
| 3e/CAT1 | 100 | 0.34 | 1.20 kcal mol−1 |
| 3e/CAT2 | 30 | 2.60 | |
| 3f/CAT1 | 100 | 0.21 | 1.07 kcal mol−1 |
| 3f/CAT2 | 98 | 1.27 | |
| 3g/CAT1 | 100 | 0.47 | 0.90 kcal mol−1 |
| 3g/CAT2 | 84 | 2.14 | |
| 3h/CAT1 | 100 | 0.50 | 0.95 kcal mol−1 |
| 3h/CAT2 | 84 | 2.50 | |

For the allylether type substrates 3a–d, there is only a minor difference between the regioselectivity displayed by the encapsulated catalyst CAT1 and the unencapsulated catalyst CAT2, albeit that CAT1 converts these substrates with higher branched selectivity than CAT2. This is also reflected in the low encapsulation effects ranging between 0.24 kcal mol−1 and 0.50 kcal mol−1. For these substrates, the branched selectivity is already high for CAT2 due to the polarization of the CC bond.70,71 In contrast, 4-methoxybut-1-ene (3e), a substrate in which the ether moiety is one atom position farther away from the alkene, displays an encapsulation effect of 1.21 kcal mol−1. Apparently, the precise position of an ether group in the substrate has a large effect on the selectivity control (Fig. 6). For the ketone and ester substrates 3f–h, the encapsulation effects are similar to the parent 1-octene, 1a. Methyl 3-butenoate 3f is converted with a high branched selectivity with CAT1 (l/b = 0.21). The encapsulation effect is similar to the parent substrate 1a, showing that the high regioselectivity of 3f is partly caused by the polarization of the alkene.
 | ||
| Fig. 6 Position of the ether moiety crucial for obtaining high regioselectivity control. | ||
Allylbenzene derivatives
Allylbenzene derivatives are the next class of substrates that was investigated (Table 4). Allylbenzene 4a forms a higher proportion of the branched product with the encapsulated CAT1 and exhibits a higher encapsulation effect compared to 1-octene, 1a (1.13 kcal mol−1vs. 0.97 kcal mol−1). In contrast, 3-buten-1-ylbenzene 4b was converted with a similar regioselectivity as observed for 1-octene 1a showing that the distal phenyl group does not affect the selectivity.|
|
|||
|---|---|---|---|
| Substrate/catalyst | % conva | l:bb |
Encapsulation effectc |
|
a Conversion determined by 1H NMR spectroscopy.
b The linear:branched ratio determined by GC.
c Encapsulation calculated by subtracting the relative energy barriers of the encapsulated reaction outcome (using CAT1) from the unencapsulated reaction outcome (using CAT2).
|
|||
| 4a/CAT1 | 100 | 0.36 | 1.13 kcal mol−1 |
| 4a/CAT2 | 73 | 2.42 | |
| 4b/CAT1 | 100 | 0.52 | 1.01 kcal mol−1 |
| 4b/CAT2 | 57 | 2.86 | |
| 4c/CAT1 | 100 | 0.32 | 1.21 kcal mol−1 |
| 4c/CAT2 | 44 | 2.46 | |
| 4d/CAT1 | 100 | 0.50 | 1.00 kcal mol−1 |
| 4d/CAT2 | 41 | 2.72 | |
| 4e/CAT1 | 100 | 0.27 | 1.38 kcal mol−1 |
| 4e/CAT2 | 46 | 2.79 | |
| 4f/CAT1 | 100 | 0.18 | 1.48 kcal mol−1 |
| 4f/CAT2 | 95 | 2.20 | |
| 4g/CAT1 | 100 | 0.31 | 1.26 kcal mol−1 |
| 4g/CAT2 | 84 | 2.62 | |
| 4h/CAT1 | 80 | 0.71 | 0.77 kcal mol−1 |
| 4h/CAT2 | 62 | 2.62 | |
| 4i/CAT1 | 100 | 0.12 | 1.83 kcal mol−1 |
| 4i/CAT2 | 33 | 2.66 | |
| 4j/CAT1 | 100 | 0.53 | 0.82 kcal mol−1 |
| 4j/CAT2 | 41 | 2.11 | |
| 4k/CAT1 | 100 | 0.29 | 1.23 kcal mol−1 |
| 4k/CAT2 | 83 | 2.30 | |

Interestingly, the presence of a methyl group on the phenyl ring, i.e. in substrates 4c–4e, significantly changes the regioselectivity outcome with CAT1. A methyl group on the ortho position (4c (l/b = 0.32)) or the para position (4e (l/b = 0.27)) of the allylbenzene derivatives results in a higher branched selectivity than the parent allylbenzene 4a. In contrast, a methyl group on the meta position, 4d, leads to lower branched selectivity (l/b = 0.50). Control experiments with CAT2 displayed similar regioselectivity for the allyltoluene substrates 4c–4e, indicating that the variation in regioselectivity control is primarily caused by CAT1.
Since the presence of one methyl group significantly affects the regioselectivity, we also explored allylbenzene derivatives with two or three methyl groups on the phenyl ring (4f–4i). Analogous to the allyltoluene substrates 4c–4e, the presence of methyl groups on the ortho and/or para positions results in high branched selectivity for 4f–4g compared to 4a. Two methyl groups on the meta position (4h) provide lower branched selectivity when converted with CAT1 compared to 4a. It is noteworthy that the variations in the regioselectivity are amplified with two methyl groups compared to the substrates with a single methyl group (Fig. 7).
 | ||
| Fig. 7 Clear regioselectivity trends for allylbenzene derivatives result in the identification of privileged allylmesitylene substrates for CAT1. | ||
Allylmesitylene 4i, which has two methyl groups on the ortho positions and one methyl group on the para position, leads to an exceptionally high branched selectivity of l/b = 0.12 and displays an encapsulation effect of 1.83 kcal mol−1. It is noteworthy that the branched selectivity for allylmesitylene 4i is even higher than the branched selectivity for allylbenzene derivatives previously reported.46
Next, allylnaphthalene substrates 4j and 4k were investigated. In this class of substrates, the regioselectivity varies in the relative position of the naphthalene group with respect to the allyl reactive group. That is, 4k is converted with higher branched selectivity with CAT1 (l/b = 0.29), whereas 4j reacts with decreased branched selectivity (l/b = 0.53) compared to the parent allylbenzene 4a.
Next, the substrate scope was extended to allylbenzene derivatives with heteroatom substituents. We commenced our investigations with the substrates 2-allylanisole, 3-allylanisole and 4-allylanisole (5a–5c) (Table 5). The substrate with the methoxy moiety on the ortho (5a) or meta (5b) position is converted by CAT1 with a significantly lower branched selectivity (l/b = 0.53 and 0.49 respectively) than that we observed for the parent allylbenzene 4a (l/b = 0.36). In contrast, when the methoxy substituent is on the para position (5c), the branched selectivity was higher (l/b = 0.29). Again, the control experiments using CAT2 give comparable levels of regioselectivity for all three substrates (5a–c). In addition, the hydroformylation of 4-allyl-1,2-dimethoxybenzene (5d) and 5-allyl-1,2,3-trimethoxybenzene (5e), bearing two and three methoxy moieties on the phenyl ring, respectively, displays lower branched selectivity (l/b = 0.53 for 5d and l/b = 0.53 for 5e) than that of the parent allylbenzene 4a.
|
|
|||
|---|---|---|---|
| Substrate/catalyst | % conva | l:bb |
Encapsulation effectc |
|
a Conversion determined by 1H NMR spectroscopy.
b The linear:branched ratio determined by GC.
c Encapsulation calculated by subtracting the relative energy barriers of the encapsulated reaction outcome (using CAT1) from the unencapsulated reaction outcome (using CAT2).
|
|||
| 5a/CAT1 | 100 | 0.53 | 0.97 kcal mol−1 |
| 5a/CAT2 | 53 | 2.73 | |
| 5b/CAT1 | 58 | 0.49 | 0.93 kcal mol−1 |
| 5b/CAT2 | 14 | 2.39 | |
| 5c/CAT1 | 100 | 0.29 | 1.38 kcal mol−1 |
| 5c/CAT2 | 47 | 2.96 | |
| 5d/CAT1 | 100 | 0.53 | 0.94 kcal mol−1 |
| 5d/CAT2 | 45 | 2.61 | |
| 5e/CAT1 | 99 | 0.53 | 0.91 kcal mol−1 |
| 5e/CAT2 | 25 | 2.46 | |

The final subset of substrates that was explored was allylbenzene derivatives with halogen substituents (Table 6). Both 1-allyl-4-(trifluoromethyl)benzene 6a and 1-allyl-4-fluorobenzene 6b were converted by the caged catalyst CAT1 with high branched selectivity (l/b = 0.15 for 6a and l/b = 0.25 for 6b). These results show that a substituent on the para position of the allylbenzene derivative results in a more branched selectivity, as improved branched selectivity is also observed for the substrates with methyl groups (4c) and methoxy groups (5c) on the para position with CAT1 (vide supra). The allylbenzene derivative that contained a halogen atom on the meta position reacts with a lower regioselectivity compared to allylbenzene 4a. In particular, 3-chloro-1-allylbenzene 6d exhibits lower branched selectivity (l/b = 0.41), whereas the corresponding fluorine analog, 3-fluoro-1-allylbenzene 6c, gives a similar encapsulation effect and regioselectivity compared to the parent allylbenzene 4a. Substrates that have a methyl or methoxy group on this position also display a lower encapsulation effect compared to 4a (vide supra).
|
|
|||
|---|---|---|---|
| Substrate/catalyst | % conva | l:bb |
Encapsulation effectc |
|
a Conversion determined by 1H NMR spectroscopy.
b The linear:branched ratio determined by GC.
c The encapsulation effect calculated by subtracting the relative energy barriers of the encapsulated reaction outcome (using CAT1) from the unencapsulated reaction outcome (using CAT2).
|
|||
| 6a/CAT1 | 100 | 0.15 | 1.46 kcal mol−1 |
| 6a/CAT2 | 52 | 1.76 | |
| 6b/CAT1 | 100 | 0.25 | 1.32 kcal mol−1 |
| 6b/CAT2 | 42 | 2.33 | |
| 6c/CAT1 | 100 | 0.33 | 1.07 kcal mol−1 |
| 6c/CAT2 | 53 | 2.00 | |
| 6d/CAT1 | 100 | 0.41 | 0.95 kcal mol−1 |
| 6d/CAT2 | 47 | 1.84 | |
| 6e/CAT1 | 100 | 0.31 | 1.08 kcal mol−1 |
| 6e/CAT2 | 43 | 1.93 | |
| 6f/CAT1 | 100 | 0.42 | 0.56 kcal mol−1 |
| 6f/CAT2 | 54 | 1.09 | |

The hydroformylation of 2-bromo-1-allylbenzene 6e yields a similar encapsulation effect to that of allylbenzene 4a. Allylpentafluorobenzene 6f is converted with a branched selectivity comparable to allylbenzene 4a. However, the control experiment with CAT2 reveals that the CC polarization of the substrate results in more branched product (l/b = 1.09) formation compared to that of 4a, which is reflected in a low encapsulation effect (0.56 kcal mol−1).
Optimization of reaction conditions
The exploration of the large substrate scope with CAT1 shows a large variation in the regioselectivity control as evidenced by the large variation in encapsulation effects (0.24–1.83 kcal mol−1). To shed light on this, we conducted DFT calculations (ADF, BLYP-D3, DZP) with CAT1 and replaced one CO moiety with allylbenzene coordinated to rhodium (Fig. 8).72–76 The lowest energy structures of the alkene coordination complex show that the phenyl ring of the substrate is in close contact (2.6 Å) with a phenyl ring of the ZnTPP building block, which is also observed in previous DFT calculations on this system.17 | ||
| Fig. 8 DFT-optimized structure of [Rh(H)(CO)2(allylbenzene)(P(mPy3(ZnTPP)3))]. The ZnTPP building block is colored yellow. The allylbenzene substrate is colored red. Hydrogens are removed for clarity apart from the relevant phenyl rings, which display CH–π interactions. | ||
Based on the large variation in selectivity control, which cannot be explained on the basis of sterics, we propose that such interactions play a role in determining the regioisomeric outcome. Moreover, for several other hydroformylation catalyst systems, it has been established that CH–π interactions between the substrate and the catalysts play a crucial role in controlling the regioselectivity.45,77–80
With this in mind, we intended to optimize the regioselectivity displayed by these types of caged catalysts by using analogues of the ZnTPP building block. Previous studies have shown that the shape of CAT1 type cages is only preserved with ZnTPP building blocks that are functionalized with a single substituent at the meta position of the phenyl rings of zinc porphyrin.17,20,81–83 When the phenyls were functionalized with two meta substituents and/or with a substituent on the ortho or para position, steric hindrance disrupts crucial CH–π interactions between the different ZnTTP building blocks required for the formation of the cage.17 As a result, the branched selectivity is lost.
Hence, we used two porphyrins to generate novel catalysts; one contains an electron withdrawing substituent, CF3, coined mCF3ZnTPP (Fig. 9, left)84 and one contains an electron donating isopropoxide substituent (OiPr), coined mOiPrZnTPP (Fig. 9, right), on the meta position. Molecular modeling using DFT shows that capsules based on these building blocks (mCF3ZnTPP and mOiPrZnTPP) provide cages CAT3 and CAT4, respectively (Fig. 10) with shapes that are similar in structure to the parent CAT1.
 | ||
| Fig. 9 ZnTPP analogs with a single substituent on the meta position of all phenyl rings. | ||
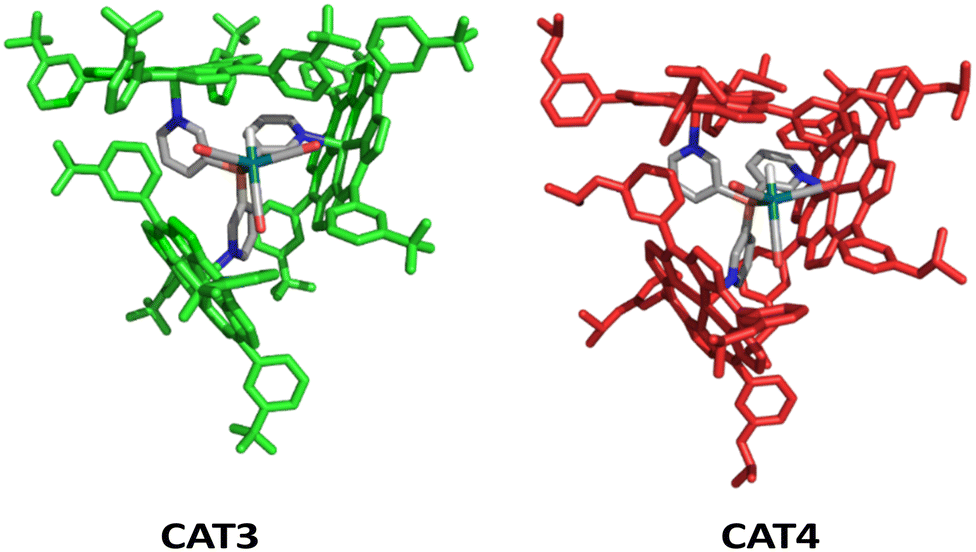 | ||
| Fig. 10 DFT-optimized structures of ([Rh(H)(CO)3(P(mPy3(mCF3ZnTPP)3))] (CAT3) (left) and [Rh(H)(CO)3(P((mPy3(mOiPrZnTPP)3))] (CAT4)) (right). Porphyrin building blocks were colored green and red for clarity. | ||
CAT3 and CAT4 were used as hydroformylation catalysts for several substrates that were evaluated in the substrate scope of CAT1 (Table 7). We used parents 1-octene 1a and allylbenzene 4a and several privileged substrates that display large encapsulation effects with CAT1; allylcyclohexane 1f, vinylcyclopentane 2a, 4-methoxybut-1-ene 3e, allylmesitylene 4i, 4-trifluoromethyl-1-allylbenzene 6a and 4-fluoro-1-allylbenzene 6b.
|
|
|||
|---|---|---|---|
| Substrate/catalyst | % conva | l:bb |
Encapsulation effectc |
|
a Conversion determined by 1H NMR spectroscopy.
b The linear:branched ratio determined by GC.
c The encapsulation effect calculated by subtracting the relative energy barriers of the encapsulated reaction outcome (using CAT1, CAT3 or CAT4) from the unencapsulated reaction outcome (using CAT2).
|
|||
| 1a/CAT1 | 100 | 0.56 | 0.97 |
| 1a/CAT3 | 100 | 0.44 | 1.11 |
| 1a/CAT4 | 100 | 0.42 | 1.14 |
| 4a/CAT1 | 100 | 0.36 | 1.13 |
| 4a/CAT3 | 99 | 0.31 | 1.24 |
| 4a/CAT4 | 100 | 0.26 | 1.34 |
| 1f/CAT1 | 100 | 0.27 | 1.40 |
| 1f/CAT3 | 100 | 0.25 | 1.44 |
| 1f/CAT4 | 100 | 0.21 | 1.55 |
| 2a/CAT1 | 100 | 0.49 | 1.33 |
| 2a/CAT3 | 66 | 0.32 | 1.58 |
| 2a/CAT4 | 96 | 0.33 | 1.56 |
| 3e/CAT1 | 100 | 0.34 | 1.21 |
| 3e/CAT3 | 70 | 0.35 | 1.19 |
| 3e/CAT4 | 100 | 0.29 | 1.30 |
| 4i/CAT1 | 100 | 0.12 | 1.83 |
| 4i/CAT3 | 100 | 0.19 | 1.56 |
| 4i/CAT4 | 100 | 0.11 | 1.89 |
| 6a/CAT1 | 100 | 0.16 | 1.42 |
| 6a/CAT3 | 66 | 0.14 | 1.50 |
| 6a/CAT4 | 100 | 0.11 | 1.64 |
| 6b/CAT1 | 100 | 0.25 | 1.32 |
| 6b/CAT3 | 72 | 0.26 | 1.30 |
| 6b/CAT4 | 100 | 0.18 | 1.52 |

In general, CAT3 and CAT4 give high branched selectivity characteristic for the caged catalyst for all substrates investigated, which confirms that these porphyrins form cages under catalytic conditions. Importantly, the branched selectivity was higher when CAT4 was used as the catalyst compared to the results obtained with CAT1 for all substrates evaluated. Previous studies show that electron donating substituents at the meta position of the porpyhrin destabilize the cage formation due to weaker CH–π interactions between the three porpyhrins, and therefore the improved regioselectivity is not caused by stronger porphyrin assembly. More likely, this is caused by favorable noncovalent interactions between the substrate moieties and the more electron rich phenyl ring of the mOiPrZnTPP building block for the branched product. For allylmesitylene 4i and 4-trifluoromethyl-1-allylbenzene 6a, the branched selectivity is l/b = 0.11 when CAT4 is used as the catalyst. This is the most branched-selective hydroformylation reaction of allylbenzene derivatives known to date.43,46,47
In particular, the encapsulation effect of 1-octene 1a is 0.23 kcal mol−1 higher for CAT4 than for CAT1. Interestingly, the substrates 1a, 2a, 4a, 1f and 6a also give a higher branched selectivity with CAT3, which shows that the branched selectivity of these substrates increases by using an electron withdrawing substituent or an electron donating substituent on the phenyl rings of the ZnTPP building block.
CAT3 displayed lower conversions than CAT1 and CAT4 for several substrates. Previous studies have shown that the presence of an electron withdrawing substituent (i.e. NO2) on the meta position of the phenyl ring of the Zn porphyrin lowers the conversion compared to CAT1 due to a lower dynamicity of the cage as a result of stronger CH–π interactions between the porphyrin building blocks of the cage.15,17
Conclusions
In summary, we have demonstrated that CAT1 is able to convert a wide range of substrates with high selectivity to the branched product. Analysis of the substrate scope with CAT1 reveals a large variation in the catalytic outcome, even when corrected for inherent substrate biases determined from the control experiments with CAT2. This investigation identified substrates that are converted with a high degree of selectivity enhancement to the branched product. For aliphatic substrates such as allylcyclohexane, allylcyclopentane, vinylcyclopentane, and vinylcyclohexane, large encapsulation effects are observed compared to the parent 1-octene, which shows that such cyclic shapes enhance the branched selectivity with CAT1. In contrast, allylether substrates display remarkably low encapsulation effects, which appear to be affected by the relative position of oxygen with respect to the alkene. Clear regioselectivity trends are also established for allylbenzene derivatives. For these substrates, substituents on the ortho and para positions improved the branched product formation and substituents on the meta position lowered the branched product formation compared to the unsubstituted analogue. These trends led us to identify an allylbenzene derivative allylmesitylene as a substrate that is converted by the cage catalyst CAT1 with exceptionally high regioselectivity (l/b = 0.12).Analysis of the substrate scope suggests that noncovalent interactions between the substrate and the walls of the encapsulated CAT1 contribute to the regioselectivity outcome. With this in mind, we optimized these interactions using ZnTPP analogs that have a single electron donating substituent (OiPr) or electron withdrawing substituent (CF3) on the meta position of all phenyl rings of the porphyrin as cage building blocks to generate two new encapsulated catalysts: CAT3 and CAT4. Both encapsulated catalysts were able to form caged structures around the active rhodium site, and displayed the typical branched selectivity for such cages. In particular, CAT4 gave higher branched selectivity than CAT1 for all substrates investigated. This study shows that the exploration of a larger substrate scope with various structural elements provides new insights into how encapsulated catalysts steer the regioselectivity in the hydroformylation. Based on this insight, the cage building blocks were redesigned, leading to further improvements of the regioselectivity displayed by these encapsulated catalysts.
Author contributions
PL and JR devised the project. PL conducted the catalytic experiments, performed DFT calculations and wrote the manuscript. JR and SK supervised the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Bin Sun is kindly acknowledged for supplying the [P(mPy3)] ligand used in this study. Ed Zuidinga is acknowledged for his HR-MS measurements. NWO, the Dutch science foundation, is acknowledged for financial support (LIFT-project 731.015.419). We also would like to thank InCatT for financial support and useful discussions. Dr. Rosalba Bellini is kindly acknowledged for supplying the mCF3ZnTPP building block. Dr. Xiaowu Wang is kindly acknowledged for supplying the mOiPrZnTPP building block.References
- D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed.
- S. H. A. M. Leenders, R. Gramage-Doria, B. De Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- L. Catti, Q. Zhang and K. Tiefenbacher, Chem. – Eur. J., 2016, 22, 9060–9066 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- R. J. Severinsen, G. J. Rowlands and P. G. Plieger, J. Inclusion Phenom. Macrocyclic Chem., 2019, 96, 29–42 CrossRef.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- T. Gadzikwa, R. Bellini, H. L. Dekker and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 2860–2863 CrossRef CAS PubMed.
- C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680–2687 CrossRef PubMed.
- S. S. Nurttila, W. Brenner, J. Mosquera, K. M. van Vliet, J. R. Nitschke and J. N. H. Reek, Chem. – Eur. J., 2019, 25, 609–620 CrossRef CAS PubMed.
- V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 4271–4274 CrossRef CAS PubMed.
- V. F. Slagt, P. C. J. Kamer, P. W. N. M. Van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 1526–1536 CrossRef CAS PubMed.
- M. Kuil, T. Soltner, P. W. N. M. Van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2006, 128, 11344–11345 CrossRef CAS PubMed.
- V. Bocokić, A. Kalkan, M. Lutz, A. L. Spek, D. T. Gryko and J. N. H. Reek, Nat. Commun., 2013, 4, 2670 CrossRef PubMed.
- M. Jouffroy, R. Gramage-Doria, D. Armspach, D. Sémeril, W. Oberhauser, D. Matt and L. Toupet, Angew. Chem., Int. Ed., 2014, 53, 3937–3940 CrossRef CAS PubMed.
- X. Wang, S. S. Nurttila, W. I. Dzik, R. Becker, J. Rodgers and J. N. H. Reek, Chem. – Eur. J., 2017, 23, 14769–14777 CrossRef CAS PubMed.
- L. J. Jongkind and J. N. H. Reek, Chem. – Asian J., 2020, 15, 867–875 CrossRef CAS PubMed.
- C. Gibson and J. Rebek, Org. Lett., 2002, 4, 1887–1890 CrossRef CAS PubMed.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746–2747 CrossRef CAS PubMed.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 9781–9797 CrossRef CAS PubMed.
- T. A. Bender, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 11806–11810 CrossRef CAS PubMed.
- M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanovi
![[c with combining umlaut]](https://www.rsc.org/images/entities/char_0063_0308.gif) -Burmazovi, J. N. H. Reek and B. De Bruin, Chem. – Eur. J., 2014, 20, 4880–4884 CrossRef CAS PubMed.
-Burmazovi, J. N. H. Reek and B. De Bruin, Chem. – Eur. J., 2014, 20, 4880–4884 CrossRef CAS PubMed. - M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanovi-Burmazovi, J. N. H. Reek and B. De Bruin, Chem. – Eur. J., 2013, 19, 10170–10178 CrossRef CAS PubMed.
- P. F. Kuijpers, M. Otte, M. Dürr, I. Ivanović-Burmazović, J. N. H. Reek and B. De Bruin, ACS Catal., 2016, 6, 3106–3112 CrossRef CAS.
- M. L. Merlau, M. D. P. Mejia, S. T. Nguyen and J. T. Hupp, Angew. Chem., Int. Ed., 2001, 40, 4239–4242 CrossRef CAS PubMed.
- J. L. Suk, S. H. Cho, K. L. Mulfort, D. M. Tiede, J. T. Hupp and S. B. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 16828–16829 CrossRef PubMed.
- P. Zhang, J. Meijide Suárez, T. Driant, E. Derat, Y. Zhang, M. Ménand, S. Roland and M. Sollogoub, Angew. Chem., Int. Ed., 2017, 56, 10821–10825 CrossRef CAS PubMed.
- R. Gramage-Doria, J. Hessels, S. H. A. M. Leenders, O. Tröppner, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2014, 53, 13380–13384 CrossRef CAS PubMed.
- Q.-Q. Wang, S. Gonell, S. H. a. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J.-P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213–7218 CrossRef CAS PubMed.
- A. W. Kleij and J. N. H. Reek, Chem. – Eur. J., 2006, 12, 4218–4227 CrossRef CAS PubMed.
- L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed.
- S. Das, G. W. Brudvig and R. H. Crabtree, Chem. Commun., 2008, 413–424 RSC.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- S. S. Nurttila, P. R. Linnebank, T. Krachko and J. N. H. Reek, ACS Catal., 2018, 8, 3469–3488 CrossRef CAS PubMed.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the art, Springer Netherlands, Dordrecht, 2004, vol. 1 Search PubMed.
- P. W. N. M. van Leeuwen, C. P. Casey and G. T. Whiteker, Rhodium catalyzed hydroformylation, Kluwer Academic Publishers, Dordrecht, 2000 Search PubMed.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- C. P. Casey, G. T. Whiteker, M. G. Melville, L. M. Petrovich, J. A. Gavney and D. R. Powell, J. Am. Chem. Soc., 1992, 114, 5535–5543 CrossRef CAS.
- A. Phanopoulos and K. Nozaki, ACS Catal., 2018, 8, 5799–5809 CrossRef CAS.
- G. M. Noonan, J. A. Fuentes, C. J. Cobley and M. L. Clarke, Angew. Chem., Int. Ed., 2012, 51, 2477–2480 CrossRef CAS PubMed.
- P. Dingwall, J. A. Fuentes, L. Crawford, A. M. Z. Slawin, M. Bühl and M. L. Clarke, J. Am. Chem. Soc., 2017, 139, 15921–15932 CrossRef CAS PubMed.
- L. Iu, J. A. Fuentes, M. E. Janka, K. J. Fontenot and M. L. Clarke, Angew. Chem., Int. Ed., 2019, 58, 2120–2124 CrossRef CAS PubMed.
- Z. Yu, M. S. Eno, A. H. Annis and J. P. Morken, Org. Lett., 2015, 17, 3264–3267 CrossRef CAS PubMed.
- M. Y. S. Ibrahim, J. A. Bennett, D. Mason, J. Rodgers and M. Abolhasani, J. Catal., 2022, 409, 105–117 CrossRef CAS.
- L. A. Van Der Veen, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 336–338 CrossRef CAS PubMed.
- D. Selent, K. D. Wiese, D. Röttger and A. Börner, Angew. Chem., Int. Ed., 2000, 39, 1639–1641 CrossRef CAS PubMed.
- T. E. Lightburn, M. T. Dombrowski and K. L. Tan, J. Am. Chem. Soc., 2008, 130, 9210–9211 CrossRef CAS PubMed.
- C. L. Joe, T. P. Blaisdell, A. F. Geoghan and K. L. Tan, J. Am. Chem. Soc., 2014, 136, 8556–8559 CrossRef CAS PubMed.
- C. U. Grünanger and B. Breit, Angew. Chem., Int. Ed., 2008, 120, 7456–7459 CrossRef.
- C. U. Grünanger and B. Breit, Angew. Chem., Int. Ed., 2010, 49, 967–970 CrossRef PubMed.
- T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 311–315 CrossRef PubMed.
- T. Šmejkal, D. Gribkov, J. Geier, M. Keller and B. Breit, Chem. – Eur. J., 2010, 16, 2470–2478 CrossRef PubMed.
- W. Fang, F. Bauer, Y. Dong and B. Breit, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- W. Fang and B. Breit, Angew. Chem., Int. Ed., 2018, 57, 14817–14821 CrossRef CAS PubMed.
- P. R. Linnebank, S. F. Ferreira, A. M. Kluwer and J. N. H. Reek, Chem. – Eur. J., 2020, 26, 8214–8219 CrossRef CAS PubMed.
- J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed.
- M. Sigrist, Y. Zhang, C. Antheaume and P. Dydio, Angew. Chem., Int. Ed., 2022, 61, e202116406 CrossRef CAS PubMed.
- With the selection of CAT2 as the model system for the caged catalysts, we have a reference system in which all the experimental details are the same, with the exception of the presence of the specified porphyrin. The generation of monoligated complexes, similar to CAT1, is only possible with ligands that differ largely in electronic and steric properties. Such variation in ligand properties does not allow for a direct comparison where cage formation is the sole variable under study. These factors will significantly complicate the analysis by introducing multiple variables that affect the catalytic outcome. In contrast, the use of CAT2 offers a more controlled and consistent framework, where the effects of porphyrin addition can be isolated and evaluated, and interpretation is supported by previous results and theoretical calculations.
- P. R. Linnebank, D. A. Poole, A. M. Kluwer and J. N. H. Reek, Faraday Discuss., 2023, 244, 169–185 RSC.
- I. Jacobs, B. De Bruin and J. N. H. Reek, ChemCatChem, 2015, 7, 1708–1718 CrossRef CAS.
- P. C. J. Kamer, A. Van Rooy, G. C. Schoemaker and P. W. N. M. Van Leeuwen, Coord. Chem. Rev., 2004, 248, 2409–2424 CrossRef CAS.
- S. C. Van der Slot, P. C. J. Kamer, P. W. N. M. Van Leeuwen, J. A. Iggo and B. T. Heaton, Organometallics, 2001, 20, 430–441 CrossRef CAS.
- A. Van Rooy, J. N. H. De Bruijn, K. F. Roobeek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, J. Organomet. Chem., 1996, 507, 69–72 CrossRef CAS.
- L. Diab, T. Šmejkal, J. Geier and B. Breit, Angew. Chem., Int. Ed., 2009, 48, 8022–8026 CrossRef CAS PubMed.
- I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 280–284 CrossRef CAS PubMed.
- R. I. McDonald, G. W. Wong, R. P. Neupane, S. S. Stahl and C. R. Landis, J. Am. Chem. Soc., 2010, 132, 14027–14029 CrossRef CAS PubMed.
- N. Ruiz, A. Polo, S. Castillón and C. Claver, J. Mol. Catal. A: Chem., 1999, 137, 93–100 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 391–403 Search PubMed.
- E. J. Baerends, T. Ziegler, J. Autschbach, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. F. Guerra and M. Franchini, ADF2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2017.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- E. van Lenthe, A. Ehlers and E. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- M. Kumar, R. V. Chaudhari, B. Subramaniam and T. A. Jackson, Organometallics, 2014, 33, 4183–4191 CrossRef CAS.
- Y. Dangat, S. Popli and R. B. Sunoj, J. Am. Chem. Soc., 2020, 142, 17079–17092 CrossRef CAS PubMed.
- M. Kumar, R. V. Chaudhari, B. Subramaniam and T. A. Jackson, Organometallics, 2014, 33, 4183–4191 CrossRef CAS.
- H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC.
- A. W. Kleij, M. Kuil, D. M. Tooke, A. L. Spek and J. N. H. Reek, Inorg. Chem., 2005, 44, 7696–7698 CrossRef CAS PubMed.
- A. W. Kleij, M. Lutz, A. L. Spek, P. W. N. M. Van Leeuwen and J. N. H. Reek, Chem. Commun., 2005, 3661–3663 RSC.
- M. Kuil, P. E. Goudriaan, A. W. Kleij, D. M. Tooke, A. L. Spek, P. W. N. M. Van Leeuwen and J. N. H. Reek, Dalton Trans., 2007, 2311–2320 RSC.
- R. Bellini and J. N. H. Reek, Chem. – Eur. J., 2012, 18, 13510–13519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00051j |
| This journal is © The Royal Society of Chemistry 2024 |