
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormoxyboranes as hydroborane surrogates for the catalytic reduction of carbonyls through transfer hydroboration†‡
Gabriel
Durin§
 ,
R. Martin
Romero§
,
Timothé
Godou
,
Clément
Chauvier
,
Pierre
Thuéry
,
Emmanuel
Nicolas
and
Thibault
Cantat
*
,
R. Martin
Romero§
,
Timothé
Godou
,
Clément
Chauvier
,
Pierre
Thuéry
,
Emmanuel
Nicolas
and
Thibault
Cantat
*
NIMBE, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette Cedex, France. E-mail: thibault.cantat@cea.fr
First published on 16th February 2024
Abstract
A new class of Lewis base stabilized formoxyboranes demonstrates the feasibility of catalytic transfer hydroboration. In the presence of a ruthenium catalyst, they have shown broad applicability for reducing carbonyl compounds. Various borylated alcohols are obtained in high selectivity and yields up to 99%, tolerating several functional groups. Computational studies enabled to propose a mechanism for this transformation, revealing the role of the ruthenium catalyst and the absence of hydroborane intermediates.
Introduction
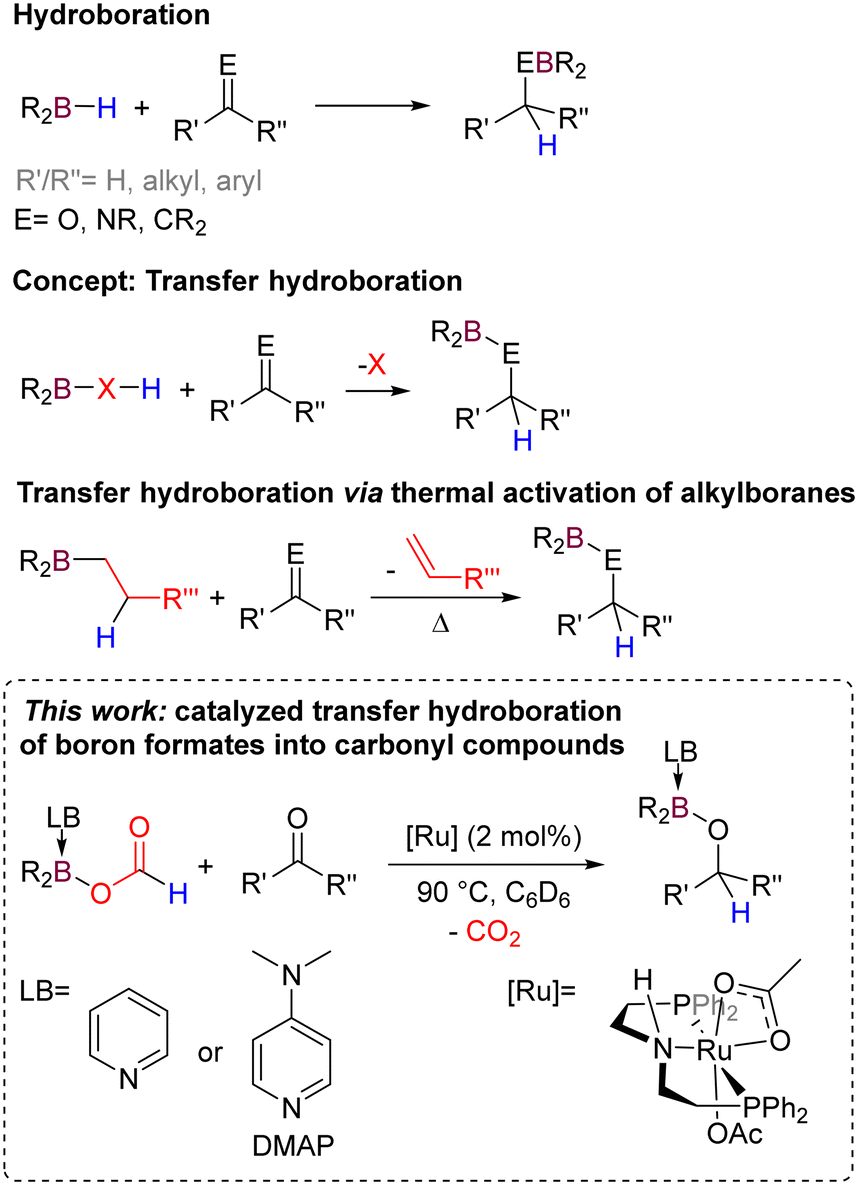
Hydroboranes are highly efficient reductants towards unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds and are widely used to reduce functional groups such as aldehydes, ketones, esters, or amides (Scheme 1).1 However, they are still considered disposable hydrides and are produced by an energy-intensive process known as the Schlesinger or Bayer process.2 Surrogates of hydroboranes that could be recycled are therefore of particular interest for improving these hydroboration procedures. Hydroelementation reactions traditionally require the presence of a well-defined E–H bond in the reagent. In related silicon chemistry, recent investigations have shown the possibility of performing transfer hydrosilylation reactions circumventing the use of genuine hydrosilanes.3 The potential of formates and their derivatives in reduction chemistry has attracted high interest due to their availability and renewable nature.4 Indeed, formates can be efficiently and selectively produced by hydrogenation or electroreduction of CO2.5 Nevertheless, the concept of transfer hydroboration chemistry has not been investigated so far. Previously, we have shown that bisformoxyboranes could yield methoxyboranes upon thermal decomposition, suggesting the implication of borohydride intermediates, which in turn could be trapped by adding aldehydes.6 Similarly, alkyl boranes decompose thermally at high temperatures (>150 °C) to produce well-defined B–H intermediates for hydroboration reactions (Scheme 1),7 with some exceptions that can proceed at lower temperatures through a 6-member ring transition state.8
C and CO bonds and are widely used to reduce functional groups such as aldehydes, ketones, esters, or amides (Scheme 1).1 However, they are still considered disposable hydrides and are produced by an energy-intensive process known as the Schlesinger or Bayer process.2 Surrogates of hydroboranes that could be recycled are therefore of particular interest for improving these hydroboration procedures. Hydroelementation reactions traditionally require the presence of a well-defined E–H bond in the reagent. In related silicon chemistry, recent investigations have shown the possibility of performing transfer hydrosilylation reactions circumventing the use of genuine hydrosilanes.3 The potential of formates and their derivatives in reduction chemistry has attracted high interest due to their availability and renewable nature.4 Indeed, formates can be efficiently and selectively produced by hydrogenation or electroreduction of CO2.5 Nevertheless, the concept of transfer hydroboration chemistry has not been investigated so far. Previously, we have shown that bisformoxyboranes could yield methoxyboranes upon thermal decomposition, suggesting the implication of borohydride intermediates, which in turn could be trapped by adding aldehydes.6 Similarly, alkyl boranes decompose thermally at high temperatures (>150 °C) to produce well-defined B–H intermediates for hydroboration reactions (Scheme 1),7 with some exceptions that can proceed at lower temperatures through a 6-member ring transition state.8
 | ||
| Scheme 1 Examples of hydroboration and transfer hydroboration of unsaturated functional groups. | ||
To our knowledge, a catalytic strategy that allows the use of a hydroborane surrogate has yet to be demonstrated. In this communication, we report the synthesis of Lewis base-stabilized formoxyboranes and their reactivity for catalytic transfer hydroboration of aldehydes and ketones without the formation of intermediates containing B–H bonds. This methodology gives access to a variety of borylated alcohols with high yields and selectivities.
Results and discussion
Formoxyboranes are scarce in the literature, being only observed as products or intermediates in the hydroboration of CO2,9 and their reactivity remains elusive. To probe the feasibility of using formoxyboranes as hydroborane surrogates, we decided to synthesize such compounds and explore their reactivity in the reduction of aldehydes and ketones as model substrates. Two possible synthetic routes can be envisaged for the formation of formoxyboranes: (i) the dehydrogenative coupling of a hydroborane with formic acid or (ii) the addition of a formate salt to the corresponding chloroborane. When Cy2BH is placed in the presence of formic acid in THF at room temperature for 16 h, we were pleased to observe that a reaction did take place, but that instead of the targeted bis-alkylformoxyborane Cy2BOCHO, an oligomer was obtained: a crystalline cyclic hexamer was observed, formed through Lewis adduct formation between the terminal oxygen atom of the formate group and the borane (see ESI,‡ Section S2). It is reminiscent of the tetramer reported by Hazari et al. as a product of CO2 hydroboration with 9-BBN.9 To avoid this oligomerization, we considered using an additional Lewis base, which could also help modulate the reactivity of the formoxyborane.10 Thus, we considered adding pyridine to synthesize formoxyboranes to lead to monomeric formate species. We were pleased to see that a solution of 9-BBN (9-borabicyclo[3.3.1]nonane), pyridine, and formic acid in toluene led, after 18 h at room temperature, to the desired formoxyborane (1a) in 96% yield and H2 as the only by-product (Scheme 2). This method is very efficient and enables the isolation of four different Lewis-base stabilized formoxyboranes 1a–d in good to high yield (82 to 98%), with varying substituents at boron (BBN vs. Cy2B) and Lewis bases (pyridine vs. DMAP). To our knowledge, they are the first monomeric formoxyboranes and were fully characterized by NMR spectroscopy and X-ray diffraction. In contrast, poor yields were obtained for the synthesis of quinoline analogs, and, despite our efforts, we could not purify such compounds (see ESI,‡ Section S3). | ||
| Scheme 2 Synthesis of formoxyboranes from hydroboranes and formic acid. | ||
Instead of dehydrogenating formic acid with hydroboranes, an alternative synthesis involves a salt metathesis starting from chloroboranes and sodium formate (Scheme 3). Interestingly, chloroboranes can be directly generated from the reaction between BCl3 and boronic anhydrides,2 the latter being typical byproducts of hydroboration reactions, allowing a route towards circular boron usage. The reaction between Cy2BCl and NaOCHO in the presence of pyridine in acetonitrile has been first investigated. Under these conditions, no reaction occurred even at an increased temperature of 130 °C for 48 h. However, using LiCl as a phase transfer catalyst (20 mol%) improved the efficiency of the reaction, and Cy2BOCHO·pyridine (1c) was obtained in 94% yield after 20 h at 100 °C.
 | ||
| Scheme 3 Synthesis of formoxyboranes from chloroborane and sodium formate. | ||
The reactivity of formoxyboranes was then probed in the reduction of carbonyl groups to demonstrate their ability to mimic the behavior of hydroboranes. Acetophenone was chosen as a model substrate. A mixture of 1a and acetophenone (3a) heated at 130 °C in C6D6 did not lead to any reaction (Table 1, entry 1). Since Cy2BH readily reacts with ketones,11 this reaction suggests that 1a does not generate a B–H intermediate by direct decarboxylation at 130 °C. [(PN(H)P)Ru(OAc)2] (2a) (PN(H)P = HN–(CH2CH2P(Ph)2)2) was introduced as a catalyst due to its ability to promote the decarboxylation of formates and the subsequent CO bond reduction by a Ru–H intermediate.12 The use of 2 mol% of 2a with a mixture of BBNOCHO·pyridine (1a) and acetophenone (3a) at 90 °C led to the desired borylated alcohol product 4aa in 99% yield after 4 h (Table 1, entry 2). Lowering the temperature from 90 to 70 °C (Table 1, entry 3) decreased the catalytic performance to 71% after 40 h, and no conversion was observed at room temperature (Table 1, entry 4). Switching the Lewis base from pyridine to DMAP gave similar results (Table 1, entry 5). A change of substituents on boron, e.g., using 1c instead of 1a, has no significant influence on this reaction, obtaining a similar yield of ca. 98% of borylated alcohol 4ac (Table 1, entry 6). However, 1d was less effective (4ad was obtained in 63% yield, Table 1, entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | Formoxyborane | T (°C) | t (h) | Yieldb (%) |
| a 0.1 mmol scale. b Yields are determined by 1H NMR with 1,3,5-trimethoxybenzene as the internal standard. See ESI‡ for more details. c This reaction was run without any catalyst. | ||||
| 1c | 1a | 130 | 48 | 0 |
| 2 | 1a | 90 | 4 | 99 |
| 2 | 49 | |||
| 3 | 1a | 70 | 40 | 71 |
| 4 | 1a | r.t. | 40 | 0 |
| 5 | 1b | 90 | 3 | 96 |
| 2 | 93 | |||
| 6 | 1c | 90 | 3 | 98 |
| 2 | 95 | |||
| 7 | 1d | 90 | 3 | 63 |

As the performances of formates 1a, 1b, and 1c were very similar (>95% yield within a few hours), their reactivity in the catalytic transfer hydroboration of pentan-3-one was tested (Table 2). After 5 h at 90 °C, the borylated alcohol products 4ba, 4bb, and 4bc were obtained in 50, 92, and 27% yield, respectively (Table 2, entries 1 to 3). The most reactive 9-BBN-based formoxyboranes 1b was chosen for the rest of the study.
|
|
||||
|---|---|---|---|---|
| Entry | Formoxyborane | R2 | LB | Yieldb (%) |
| a General conditions: 0.1 mmol of substrate, 0.15 mmol of formoxyborane, 2 μmol of catalyst, 0.5 mL of C6D6. b NMR yields were determined by 1H NMR integration of the –CH signals versus an internal standard (1,3,5-trimethylbenzene). | ||||
| 1 | 1a | 9-BBN | Pyridine | 50 |
| 2 | 1b | 9-BBN | DMAP | 92 |
| 3 | 1c | Cy2 | Pyridine | 27 |

The transfer hydroboration could be extended to a wide variety of ketones and aldehydes (Scheme 4). Electron-donating (4c–e) and electron-withdrawing groups (4f–i) were tolerated with different substitution patterns, giving a quantitative yield in a short reaction time (<3 h) in most cases. We noted a drop in the yield when a nitro group (4g) was present, presumably due to its interaction with the boron or its reduction. The presence of heterocycles, such as pyridine or furane, was perfectly tolerated by the system obtaining the hydroborated products 4j–l quantitatively. The same behavior was observed for ketones bearing a longer alkyl chain (4m) or diarylketones (4n). Aliphatic ketones also reacted in excellent yields (4o). More challenging α,β-unsaturated ketones were also tested, and the formal 1,2-addition of the hydroborane was observed in moderate to excellent yields for 4p–r (62–93%). In all these cases, the unsaturation remained untouched, and no 1,4-addition product or enol ether was observed. Finally, aldehydes were easily reduced into their corresponding borylated alcohols (4s, 4t) within 30 minutes.
 | ||
| Scheme 4 Scope of the catalytic transfer hydroboration of carbonyl compounds. | ||
All the related free alcohols were obtained upon aqueous workup, except for 4k. This particular case can be explained by the chelating effect of the nearby nitrogen of the pyridine moiety, which stabilizes the borylated product. To highlight the potential applicability of the method, the free alcohol resulting from the hydrolysis of 4i was isolated upon purification in 97% yield (see ESI,‡ Section S10).
To gain insights into the mechanism of the reaction, we searched for the active species involved in the catalytic transfer of hydroboration. First, the formation of CO2 as a by-product of the reaction was confirmed by GC analysis of the gas phase of the reaction (see ESI,‡ Section S12). Then, we observed that the recorded 1H NMR spectrum shows a main ruthenium hydride species at −16.7 ppm during catalysis. This signal is closely related to the previously reported carbonyl complex [(PN(H)P)RuH(CO)(OAc)] (2b) (δ = −16.6 ppm).13 This species 2b was identified as a ruthenium complex generated from the reaction with silyl formates. Therefore, it is plausible that the same pathway exists in the presence of formoxyboranes. In addition, the presence of a ruthenium carbonyl complex was also confirmed by IR spectroscopy, showing a νCO frequency of 1923 cm−1 (see ESI,‡ Section S13). To prove this hypothesis, 2b was synthesized and used as a precatalyst (Scheme 5). A similar catalytic activity was obtained for the borylated product 3a (99%) after 3 h, supporting the conversion of 2a to 2b under catalytic conditions. Finally, the replacement of [(PN(H)P)Ru(OAc)2] by the methylated derivative [(PN(Me)P)Ru(OAc)2] as the catalyst suppressed almost completely the reaction, showing the critical role of the N–H moiety in the ligand for this reaction (see ESI,‡ Sections S13–S15).
 | ||
| Scheme 5 Plausible precatalyst pathway from 2a to 2b (top) and precatalyst test with 2b (bottom). | ||
Density functional theory (DFT) calculations were performed to understand better the mechanism of the reaction (Scheme 6). A multi-step initiation pathway can be proposed as described in Scheme 5, starting from 2a to generate 2b. A ligand exchange from acetate to formate is needed to enter the catalytic cycle from [(PN(H)P)RuH(CO)OCHO] (Ia) species. The formate ligand in Ia first switches from η1-O to η1-H coordination, leading to Ib (ΔG = +14.8 kcal mol−1) through TSa–b at 22.0 kcal mol−1, with H-bonding between one oxygen atom in the formate and the N–H moiety in the ligand. Decarboxylation can then occur viaTSb–c (ΔΔG‡ = +6.8 kcal mol−1) to form ruthenium bis-hydride complex Ic, lying 17.0 kcal mol−1 above the starting reactants. The latter complex is reminiscent of intermediates found in the mechanisms of formic acid dehydrogenation, as described by the groups of Schneider and Hazari for iron complexes,14 or ourselves in the case of cobalt.15 Hydrogen bonding between the N–H ligand and the ketone favors the barrierless hydride transfer of [Ru]–H to form the η1-H ruthenium alkoxide complex Id (ΔG = 23.3 kcal mol−1). The reorganization of this complex from η1-H to η1-O ruthenium alkoxide Ie (ΔG = 7.2 kcal mol−1) is also barrierless. Formoxyborane 1b can be coordinated through TSe–f (ΔG‡ = +15.5 kcal mol−1), leading to If, 11.4 kcal mol−1 higher in energy than Ia. Complex If features a formate ligand bridging between ruthenium and boron, and the alkoxy moiety is H-bonded to the N–H moiety of the PNP ligand. The decoordination of DMAP from boron is barrierless and leads to high-energy intermediate Ig (ΔG = 28.7 kcal mol−1). A nucleophilic attack of the alkoxide on the boron atom can occur through TSg–h (ΔG‡ = +32.5 kcal mol−1), leading to Ih (ΔG = +3.7 kcal mol−1). Complex Ih features a formate bridging between Ru and B and an alkoxide bridging between B and the N–H moiety of the PNP ligand on Ru. The release of the product through transition state TSh–a (ΔG‡ = +5.6 kcal mol−1) followed by the coordination of DMAP regenerates Ia, thereby closing the catalytic cycle. The total energetic span of the catalytic cycle, 32.5 kcal mol−1, is slightly higher than expected compared to the reaction conditions (4 h at 90 °C) but consistent within the expected uncertainty range of 3 kcal mol−1.16 It is defined by Ia and the transition state for the transmetallation TSg–h, which is the rate-determining transition state of the reaction.
 | ||
| Scheme 6 Computed mechanism for the catalytic transfer of hydroboration of acetophenone 3a with 1b catalyzed by Ia at B3LYP-D3/Def2SVP level of theory and SMD model to account for solvent effect (C6D6). | ||
Other mechanistic pathways were also considered but discarded because of their high energy demand (see ESI,‡ Section S15). In particular, forming the B–H bond is not thermodynamically possible under the reaction conditions (BBNOCHO·DMAP → BBNH + CO2 + DMAP: ΔG = +30.7 kcal mol−1). It is interesting to note that compared to silyl formates, formoxyboranes are thermodynamically more difficult to decarboxylate (Et3SiOCHO → Et3SiH + CO2: ΔG = +5.8 kcal mol−1; BBNOCHO → BBNH + CO2: ΔG = +15.2 kcal mol−1; BBNOCHO·DMAP → BBNH·DMAP + CO2: ΔG = +13.9 kcal mol−1).
Conclusions
In summary, the synthesis of Lewis base-stabilized formoxyboranes is reported. The combination of formoxyboranes with a suitable ruthenium catalyst enables mimicking the reactivity of hydroboranes, as demonstrated with the formal hydroboration of ketones and aldehydes. Mechanistic studies have highlighted the crucial role of the ligand during the catalytic process and indicated that the rate-determining transition state is the transmetallation of a formate ligand from boron to ruthenium. This work demonstrates the potential of hydroborane surrogates as hydroborating reagents in challenging transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by CEA, CNRS, the University Paris-Saclay, CINES (HPC Computing time on Occigen, grant no. A0080806494), and the European Research Council (ERC Consolidator Grant Agreement no. 818260). The authors thank Dr Lucile Anthore-Dalion for her help in performing the GC analysis of the gas phase.Notes and references
- (a) T. Yamakawa, M. Masaki and H. Nohira, Bull. Chem. Soc. Jpn., 1991, 64, 2730–2734 CrossRef CAS; (b) A. S. B. Prasad, J. V. B. Kanth and M. Periasamy, Tetrahedron, 1992, 48, 4623–4628 CrossRef CAS; (c) H. C. Brown, J. V. B. Kanth and M. Zaidlewicz, J. Org. Chem., 1998, 63, 5154–5163 CrossRef CAS; (d) H. C. Brown and N. Ravindran, J. Am. Chem. Soc., 1976, 98, 1785–1798 CrossRef CAS; (e) S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed; (f) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS; (g) C. J. Barger, A. Motta, V. L. Weidner, T. L. Lohr and T. J. Marks, ACS Catal., 2019, 9, 9015–9024 CrossRef CAS.
- R. J. Brotherton, C. J. Weber, C. R. Guibert and J. L. Little, Boron compounds, in Ullmann's encyclopedia of industrial chemistry, John Wiley & Sons, Ltd, Ed., 2000, https://onlinelibrary.wiley.com/doi/10.1002/14356007.a04_309 Search PubMed.
- (a) A. Studer and S. Amrein, Angew. Chem., Int. Ed., 2000, 39, 3080–3082 CrossRef CAS; (b) S. Amrein, A. Timmermann and A. Studer, Org. Lett., 2001, 3, 2357–2360 CrossRef CAS PubMed; (c) S. Amrein and A. Studer, Helv. Chim. Acta, 2002, 85, 3559–3574 CrossRef CAS; (d) A. Simonneau and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 11905–11907 CrossRef CAS PubMed; (e) S. Keess, A. Simonneau and M. Oestreich, Organometallics, 2015, 34, 790–799 CrossRef CAS; (f) C. Chauvier, P. Thuery and T. Cantat, Angew. Chem., Int. Ed., 2016, 55, 14096–14100 CrossRef CAS PubMed.
- (a) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed; (b) R. F. Nie, Y. W. Tao, Y. Q. Nie, T. L. Lu, J. S. Wang, Y. S. Zhang, X. Y. Lu and C. C. Xu, ACS Catal., 2021, 11, 1071–1095 CrossRef CAS; (c) D. Wei, R. Sang, P. Sponholz, H. Junge and M. Beller, Nat. Energy, 2022, 7, 438–447 CrossRef CAS.
- (a) M. W. Farlow and H. Adkins, J. Am. Chem. Soc., 2002, 57, 2222–2223 CrossRef; (b) E. Graf and W. Leitner, J. Chem. Soc., Chem. Commun., 1992, 623–624 RSC; (c) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS; (d) W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed; (e) P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS PubMed; (f) L. Chen, Z. Guo, X. G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabehere-Mallart, K. C. Lau, T. C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed; (g) A. Taheri and L. A. Berben, Chem. Commun., 2016, 52, 1768–1777 RSC; (h) R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed; (i) R. M. Bullock, A. K. Das and A. M. Appel, Chem. – Eur. J., 2017, 23, 7626–7641 CrossRef CAS PubMed; (j) S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- C. Chauvier, P. Thuery and T. Cantat, Chem. Sci., 2016, 7, 5680–5685 RSC.
- (a) H. C. Brown, P. K. Jadhav and K. S. Bhat, J. Am. Chem. Soc., 1985, 107, 2564–2565 CrossRef CAS; (b) B. M. Mikhailov, Y. N. Bubnov, A. V. Tsyban and M. S. Grigoryan, J. Organomet. Chem., 1978, 154, 131–145 CrossRef CAS; (c) B. M. Mikhailov, V. G. Kiselev and Y. N. Bubnov, Russ. Chem. Bull., 1965, 14, 865–867 CrossRef; (d) W. Fenzl and R. Koster, Angew. Chem., Int. Ed. Engl., 1971, 10, 750–751 CrossRef CAS; (e) M. M. Midland, J. E. Petre, S. A. Zderic and A. Kazubski, J. Am. Chem. Soc., 1982, 104, 528–531 CrossRef CAS.
- (a) N. Miyaura, M. Itoh, A. Suzuki, H. C. Brown, M. M. Midland and P. Jacob, J. Am. Chem. Soc., 1972, 94, 6549–6550 CrossRef CAS; (b) M. M. Midland, A. Tramontano and S. A. Zderic, J. Organomet. Chem., 1978, 156, 203–211 CrossRef CAS.
- M. R. Espinosa, D. J. Charboneau, A. G. de Oliveira and N. Hazari, ACS Catal., 2019, 9, 301–314 CrossRef CAS.
- M. Horn, L. H. Schappele, G. Lang-Wittkowski, H. Mayr and A. R. Ofial, Chem. – Eur. J., 2013, 19, 249–263 CrossRef CAS PubMed.
- H. C. Brown, S. Krishnamurthy and N. M. Yoon, J. Org. Chem., 1976, 41, 1778–1791 CrossRef CAS.
- R. M. Romero, N. Thyagarajan, N. Hellou, C. Chauvier, T. Godou, L. Anthore-Dalion and T. Cantat, Chem. Commun., 2022, 58, 6308–6311 RSC.
- C. Chauvier, A. Imberdis, P. Thuery and T. Cantat, Angew. Chem., Int. Ed., 2020, 59, 14019–14023 CrossRef CAS PubMed.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Wurtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- N. Lentz, A. Aloisi, P. Thuery, E. Nicolas and T. Cantat, Organometallics, 2021, 40, 565–569 CrossRef CAS.
- M. Bursch, J. M. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed.
Footnotes |
| † This article was previously posted as a preprint on ChemRxiv: DOI: https://doi.org/10.26434/chemrxiv-2023-6ml06. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2284250–2284254. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy01702h |
| § These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |