
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective linear ethylene oligomerization over nickel-containing zeotypes with tetravalent framework heteroatoms†
Yunfei
Bai
ab,
Tomás
Cordero-Lanzac
 c,
Ainara
Nova
c,
Unni
Olsbye
c,
Esben
Taarning
a and
Juan S.
Martinez-Espin
*a
c,
Ainara
Nova
c,
Unni
Olsbye
c,
Esben
Taarning
a and
Juan S.
Martinez-Espin
*a
aTopsoe A/S, Haldor Topsøes Allé 1, 2800 Kongens Lyngby, Denmark. E-mail: jsme@topsoe.com
bAarhus University, Nordre Ringgade 1, 8000 Aarhus C, Denmark
cCentre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, Blindern, 0315 Oslo, Norway
First published on 5th March 2024
Abstract
Heterogeneously catalyzed ethylene oligomerization to higher olefins remains an industrial challenge due to the difficulties to effectively activate ethylene and simultaneously ensure high product selectivities. Nickel aluminosilicate zeolites have shown promise as alternatives to homogeneous catalysts, but the strong Brønsted acidity of these materials typically results in a broad product distribution and the accumulation of heavy oligomers within the channels and cages of porous zeolites. Herein, we report the positive impact on selectivity to linear olefinic products with the replacement of Brønsted acidic zeolites by a family of Lewis acidic Beta zeotypes as supports for nickel. Our findings reveal that the acidic nature of the zeotypes is fundamental to direct the successful incorporation of nickel into active species. Sn-, Hf- and Zr-Beta stand out as compared to Ti- and Ge-Beta in terms of acid strength, nickel ion-exchange capacity, and ultimately, catalytic activity. All active materials yield remarkable selectivities (>90%) to linear olefins. In contrast, conventional Ni/Al-Beta, having Brønsted acidity, provides a complex product spectrum due to the promotion of competitive reaction pathways (e.g., skeletal isomerization, cracking and hydrogen transfer reactions). Furthermore, the retained hydrocarbon species need higher oxidation temperatures to be removed over Ni/Al-Beta as compared to Ni-containing Lewis acidic zeotypes, hinting towards a milder coking deactivation of the latter materials. These insights show the potential for improving ethylene oligomerization selectivity and catalyst stability with the utilization of a new class of zeotypes without strong Brønsted acidity, but with the ability to anchor transition metals.
1. Introduction
Short olefins, such as butenes and hexenes, serve as fundamental building blocks in the chemical industry, being indispensable feedstocks and intermediates for a diverse array of chemical products and materials, including polymers, detergents, and fuel additives.1–3 The growing demand for these olefins necessitates the exploration of more sustainable production methods.4,5 Historically, naphtha reforming has been the primary source of C2–C6 alkenes. Ethane steam cracking is another important source of ethylene, but leads to less propene and butene.6–8 In recent years, rapid advancements in second-generation bio-ethanol production have opened up promising avenues for the sustainable generation of ethylene through the dehydration of ethanol.9–11 This bio-derived ethylene can serve as a promising alternative to traditional fossil-based feedstocks and can subsequently be subjected to oligomerization to yield longer-chain olefins. Consequently, there has been growing interest in the development of efficient and environment-friendly processes for ethylene oligomerization, aimed at enhancing the value creation for the production of fuels, polymers, and platform chemicals.1,3Homogeneous catalysis, utilizing transition metal catalysts in organic solvents, is predominantly employed in commercial ethylene oligomerization processes.12–14 Historically, metal complexes of various elements, including Ni, Ti, Zr, Cr, Co and Fe, have been used as efficient homogeneous catalysts for this reaction.14 Currently, only a few of these processes have been successfully commercialized and industrialized. Major industrial processes employ catalysts such as trialkylaluminum (Chevron and Ineos) and nickel complexes (Shell, UOP, and Phillips). These technologies are capable of producing linear alpha-olefins (LAOs) or 2-butenes with high selectivity.3,9,15 However, homogeneous processes present several limitations, including product separation, constraints on catalyst reusability, and the use of co-catalysts and harmful solvents. Heterogeneous catalysis, as a potential solution to these challenges, has thus received significant attention in recent years.1,15 Focus areas of current research include solid acids, nickel complexes supported on oxides, metal–organic frameworks (MOFs), and Ni-aluminosilicates.16–25 Among these, nickel-containing aluminosilicates, such as mesoporous amorphous silica-alumina and zeolites, are considered promising candidates due to their robustness in a wide temperature range, ease of regeneration, and their cost-effectiveness and accessibility.26–29 However, an industrialized process for ethylene oligomerization based on heterogeneous catalysis has yet to be realized. In comparison with the currently industrialized homogeneous processes, there remains a significant need to optimize heterogeneous catalysts, especially in terms of enhancing product selectivity. This includes improvements in product linearity, the distribution of longer olefins, and the positioning of the double bond. Furthermore, effectively addressing catalyst deactivation is critical to satisfy the viable commercialization of the technology.3,9,30
Zeolites, as a family of microporous aluminosilicate materials, are built from TO4 tetrahedra units. Incorporation of trivalent metal atoms (e.g., Al, Ga) in place of silicon in the framework imparts Brønsted acidity, while tetravalent metal atom substitution (e.g., Sn, Zr, Hf) results in Lewis acidity.31–33 Brønsted acidic zeolites have been intensively explored as catalysts for ethylene oligomerization.34 Nickel ion exchanged aluminum-containing microporous zeolites, such as X, Y, and MCM-22, generally undergo rapid deactivation due to the accumulation of heavy oligomers within their channels and cages.26,35,36 Nickel-containing Al-Beta zeolites with small crystal sizes have been reported to exhibit higher resistance to deactivation compared to other zeolites. However, strong Brønsted acidity, induced by trivalent metals, can trigger side reactions such as skeletal isomerization and cracking, leading to a broad product range that complicates downstream separation processes.30 For instance, Martínez et al. investigated Ni/Al-Beta catalysts prepared through both ion-exchange and impregnation methods.27 Their work revealed that the formation of branched oligomers and odd-numbered products was catalyzed by Brønsted acid sites. Similarly, Henry et al. explored the use of nickel-containing nano- or micron-sized Beta zeolites.29 Their findings demonstrated that the generation of branched products could be limited when employing nano-sized Ni/Al-Beta. However, the complete elimination of these side products remained challenging, and a decline in butene selectivity was observed with increasing conversions.
Zeolites containing strong Brønsted acid sites may catalyze the formation of higher alkene products, through acid-catalyzed oligomerization, potentially leading to pore blockage and subsequent catalyst deactivation.37–39 Furthermore, numerous publications have reported the formation of small amounts of alkanes, such as isobutane, during ethylene oligomerization.35,40–42 Such species are formed via hydrogen transfer reactions, and to satisfy the stoichiometry, polyunsaturated and/or unsaturated cyclic hydrocarbons are formed alongside them.42 Such molecules have been classified as coke precursors in other reactions involving hydrocarbon conversion over zeolites.43,44
By varying the heteroatoms in the framework, the properties of Beta zeolites can be adjusted, directly impacting catalyst performance. Meloni et al. demonstrated the potential for modifying ethylene oligomerization activity over Ni/M-Beta catalysts by introducing different trivalent metals in the zeolitic framework. As a result, nickel species supported on Beta zeotypes with Fe or Ga incorporation exhibited higher turnover frequencies (TOFs) towards ethylene oligomerization as compared to conventional Al-Beta, and the authors correlated it directly to the changes in the nickel electron density, influenced by the framework heteroatoms.45 Considering the ability to tune nickel electron densities by the zeolitic support, our study introduces the unexplored use of tetravalent substituted heteroatoms in the Beta framework (Sn, Hf, Zr, Ti, Ge). Lewis acid sites, generated from tetravalent atom substitution, are acknowledged due to their capacity to activate carbonyl groups and coordinate with hydroxyl functionalities, thereby being widely investigated for carbohydrate conversion.46,47 Such materials have been proven to have ion exchange capabilities in water rich environments.48,49 This property may originate from the different coordinations of the tetravalent metals in the zeolitic network leading to “open” (hydrated) and “closed” (dehydrated) sites, which are exemplified in Scheme 1. The resulting protons connected to the metals and those in close proximity may be susceptible for ion exchange. In contrast, Al-Beta contains Brønsted sites known to enable nickel ion exchange.30,50–52
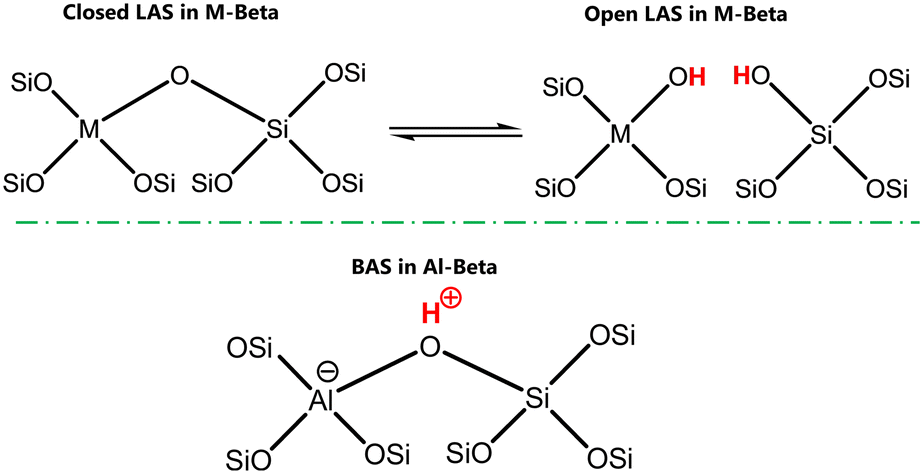 | ||
| Scheme 1 Simplified structures of acid sites in zeolitic materials. M represents the framework tetravalent heteroatom. LAS is for Lewis acid sites and BAS is for Brønsted acid sites. | ||
Recently, Sun et al. synthesized and investigated Ni-containing Sn-Beta zeotypes via mild dealumination followed by nickel incorporation.53 They observed a larger fraction of the open tin sites due to the presence of nickel, which led to increased activity in both glucose isomerization and the retro-aldol cleavage process yielding methyl lactate. These results are in line with previous studies using alkali and alkaline-earth cations exchanged over Sn-Beta,48,49,54 and expand the known ion-exchange capacity of the materials to transition metals.
In this study, we explored the use of nickel-containing tetravalent metal-substituted Beta zeolites as catalysts in the context of ethylene oligomerization. We investigated the determinant role of the Lewis acid sites and their strength in guiding the formation of active nickel species. The catalytically active catalysts, containing Sn, Hf and Zr, showed very high selectivity to linear alpha olefins (>90%), opening the door for the utilization of a new family of promising solid acids for ethylene oligomerization.
2. Experimental section
2.1. Catalyst synthesis
The heteroatom substituted Beta zeotypes were hydrothermally synthesized using a previously reported method.55 In a typical synthesis procedure, 30.6 g of tetraethyl orthosilicate (TEOS, Sigma-Aldrich, 98%) was mixed with 33.1 g of tetraethylammonium hydroxide (TEAOH, Sigma-Aldrich, 35% in water) under stirring for 60–120 minutes, until a homogenous solution was formed. Afterwards, for Sn-Beta, a specific amount of tin(IV) chloride pentahydrate (SnCl4·5H2O, Sigma-Aldrich, 98%) was dissolved in 2 mL of demineralized water and added to the solution dropwise to control the nominal Si to Sn ratios to 100, 150 and 200. For Al-Beta, Zr-Beta and Hf-Beta, the metal sources were changed into aluminum(III) chloride hexahydrate (AlCl3·6H2O, Fluka, 99%), zirconyl(IV) chloride octahydrate (ZrOCl2·8H2O, Sigma-Aldrich, 98%) and hafnium(IV) chloride (HfCl4, Sigma-Aldrich, 98%), respectively. For Ge-Beta, the metal source germanium(IV) oxide (GeO2, Sigma-Aldrich, 99%) was dissolved into TEAOH directly, followed by adding 2 mL of demineralized water into the solution. For Ti-Beta, tetraethyl orthotitanate ((CH3CH2O)4Ti, Sigma-Aldrich, technical grade) was used. This titanium source was dissolved in a mixture of hydrogen peroxide and water before being used. Moreover, one Si-Beta sample without any framework heteroatom was also prepared by dropping 2 mL of demineralized water into the synthesis gel.The mixture was maintained under stirring until a viscous gel formed. Subsequently, 3.1 g of hydrofluoric acid (HF, Fluka, 47–51%) was diluted with 1.6 g of demineralized water and added to the synthesis gel, forming a brittle solid precursor with the approximate composition of 1.0Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(1/x)M:0.55TEA+:0.55F−, in which M represents the framework heteroatom in the synthesis gel, and x represents the nominal Si to heteroatom molar ratio. The resulting precursor was then crushed and transferred into an autoclave with a Teflon liner. The hydrothermal process was conducted at 140 °C until full crystallization was achieved. The obtained precursors were filtered, washed with abundant water, dried at 80 °C overnight and calcined in air at 550 °C for 6 hours at a temperature ramp of 5 °C min−1. The as-synthesized samples were named xM-Beta. In the final step, Ni was loaded onto the M-Beta zeotypes using the incipient wetness method with nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, Sigma-Aldrich, 99%) solution to achieve specific Ni loadings of 0.5, 1.0, and 2.0 wt%. The impregnated Ni/M-Beta precursors were then dried at 80 °C overnight and calcined at 550 °C (3 °C min−1) for 6 hours to complete the synthesis.
:(1/x)M:0.55TEA+:0.55F−, in which M represents the framework heteroatom in the synthesis gel, and x represents the nominal Si to heteroatom molar ratio. The resulting precursor was then crushed and transferred into an autoclave with a Teflon liner. The hydrothermal process was conducted at 140 °C until full crystallization was achieved. The obtained precursors were filtered, washed with abundant water, dried at 80 °C overnight and calcined in air at 550 °C for 6 hours at a temperature ramp of 5 °C min−1. The as-synthesized samples were named xM-Beta. In the final step, Ni was loaded onto the M-Beta zeotypes using the incipient wetness method with nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, Sigma-Aldrich, 99%) solution to achieve specific Ni loadings of 0.5, 1.0, and 2.0 wt%. The impregnated Ni/M-Beta precursors were then dried at 80 °C overnight and calcined at 550 °C (3 °C min−1) for 6 hours to complete the synthesis.
Additionally, a control sample by loading NiO over Sn-Beta was prepared, named NiO/Sn-Beta. NiO was obtained by calcining nickel(II) nitrate at 550 °C for 6 hours in an air atmosphere. The preparation targeted 1 wt% nickel loading over 150Sn-Beta following incipient wetness impregnation. Therefore, 0.038 g NiO was dispersed in 2.73 mL water and the solution was then thoroughly mixed with 3 g 150Sn-Beta, dried overnight and calcined as described above for the Ni/M-Beta catalysts.
2.2. Characterization
Elemental analysis of M-Beta was conducted to measure the heteroatom and silicon contents by the X-Ray Fluorescence (XRF) technique, using a Supermini 200 (Rigaku) instrument. N2 adsorption–desorption experiments were carried out using Belsorp Mini II equipment at −196 °C. Prior to the experiments, the calcined catalysts were outgassed under vacuum for 0.5 h at 80 °C, followed by a period of 3 h at 300 °C. From the isotherms, specific surface areas were calculated using the BET method, micropore volumes and external surface areas were calculated using the t-plot method and mesopore volumes were calculated by difference with the total adsorbed volume. The morphology of the zeotypes was observed using an XL30 field emission scanning electron microscope (SEM).Transmission FTIR experiments were carried out using a Vertex 70 spectrometer for CO and CD3CN adsorption. Before the experiment, the samples were prepared by pressing around 25 mg of sample powder into pellets, and then placing them into copper envelopes made for the quartz testing cell with KBr windows. The samples were pretreated under vacuum at 30 °C overnight, heated up to 450 °C with a ramp of 5 °C min−1, held for 1 hour, then cooled down to room temperature before tests. For CO adsorption experiments, the blank spectra were acquired under vacuum with liquid nitrogen cooling of the test cell. Afterwards, CO was introduced to the test cell and adsorbed at liquid nitrogen temperature. To start the experiments, the samples were firstly saturated with high pressure of the probe molecule (3–5 mbar), and the desorption pressure was controlled stepwise from saturation pressure to around 10−2 mbar, while CO desorption spectra were taken during this process. For CD3CN adsorption experiments, the samples were saturated with the probe molecule (above 3 mbar), and the desorption was carried out stepwise. Under 0.25 mbar, the characteristic peaks of the gas phase and physically adsorbed CD3CN are not as intense, while Lewis and Brønsted acid sites and silanols remain fully titrated and are easier to deconvolute. The spectra in this work were recorded in the range of 4000–500 cm−1, by accumulating 128 scans at 2 cm−1 resolution. The obtained spectra were background subtracted with a blank spectrum (before adsorption). FTIR data were analyzed using Bruker OPUS software, Origin 8.5.1, and OriginPro 2023.
NH3 Temperature-Programmed Desorption (NH3-TPD) tests were conducted using Mettler Toledo TGA/DSC 1 equipment over the zeolitic materials. Around 40 mg of sample was placed into a crucible for each test. The pretreatment of the sample was conducted in a He/Ar (inert flow, 31%/69%, volume ratio) mixture flow at 500 °C for 110 minutes, with a ramp of 20 °C min−1. Afterwards, the samples were cooled down to room temperature under an inert flow. The NH3 adsorption was conducted by switching the gas flow to 2 vol% NH3/inert and allowing the sample to remain at around 35 °C (room temperature) for 30 minutes. Subsequently, the samples were flushed at the same temperature using a He/Ar (31/69%, volume ratio) flow for 30 minutes to remove the remaining physical adsorbates. The desorption of the tested samples was conducted by increasing the temperature of the cell from room temperature to 500 °C with a ramp of 10 °C min−1 under flushing of an inert flow. The mass change during the test procedure was recorded using a built-in balance. To elucidate the NH3 desorption profile as a function of temperature during the TPD process, the derivative of the mass change was computed. The initial step involved normalizing the absolute mass change observed during the desorption phase by the sample weight at the beginning of the heating ramp. Subsequently, the first derivative of the normalized mass change with respect to temperature was calculated, thereby yielding the desorption rate throughout the heating ramp. Thereafter, the profiles of heteroatom-containing samples were normalized by subtracting the corresponding profile of Si-Beta (with negligible uptake) to avoid too much interference from physisorbed NH3 due to the very low temperatures at which it was required to perform the adsorption over these weakly acidic materials. Data analysis was conducted using Origin 8.5.1 software.
The reverse ion exchange experiments of the Ni/M-Beta samples were conducted by treating 0.2 g sample with 5 mL of 2 mol L−1 ammonium nitrate (NH4NO3, Sigma-Aldrich, ≥98.0%) solution, aiming to dissolve the exchangeable nickel species in a highly concentrated NH4NO3 solution. The solution was kept under magnetic stirring at 80 °C for 6 hours. After finishing, the obtained liquid was filtered to remove the remaining solids, and analyzed by Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-OES) on Perkin Elmer model Optima 3000 (Varian Vista) equipment.
2.3. Catalyst testing
Catalytic tests were carried out in a fixed bed stainless steel reactor in a Microactivity Effi setup (PID Eng & Tech). Before each experiment, 500 mg of catalyst was pressed, crushed and sieved to 250–425 μm grain size. The catalyst was loaded in the reactor and activated in situ at 300 °C overnight under 1 bar of N2 gas (40 mL min−1). In particular, for two experiments conducted with an extended time on stream (TOS), the pretreatment protocol was modified; the catalyst was heated to 500 °C and maintained for one hour under a N2 atmosphere at 1 bar and a flow rate of 40 mL min−1. The total pressure was then increased in Ar (up to 30 bar) and controlled by a back-pressure regulator before running the reaction. During the reaction, the partial pressures were controlled by adjusting the individual flows of ethylene (provided by Praxair, 3.5 grade) and inert gas (Ar, Praxair, 5.0), keeping the total pressure constant at 30 bar. Typical reaction conditions were: 250 °C with a total pressure of 30 bar and 20 mL min−1 Ar mixed with 10 mL min−1 ethylene (contact time of 0.017 min gcat mL−1). An additional test of 1-butene isomerization was carried out using the same experimental setup to assess the activity of the Ni-free 150Sn-Beta zeotype. Prior to the experiment, 200 mg catalyst was pre-treated following the same protocol used in the experiments with an extended TOS (500 °C for one hour). The isomerization reaction was conducted at 250 °C and atmospheric pressure. The feed flow was regulated to 20 mL min−1 Ar mixed with 4 mL min−1 1-butene.A small portion of the reactor effluent was led through heated lines to an online gas chromatograph (Scion 456-GC) for the on-line quantification of products. Briefly, two effluent aliquots were sampled in valve loops for the quantification of permanent gasses and hydrocarbons. The first one was separated in HayeSep Q, HayeSep N and MolSieve 13X, after which it was analyzed in a TCD (in this case, N2 and Ar were the only product analyzed). The second aliquot was sent to an Rt-Stabilwax, and then split through a dean switch to an Rt-Alumina/MAPD and an Rtx-1, both connected to FIDs. The three first columns were maintained isothermal at 80 °C, while the last three were ramped up from 40 to 250 °C. All effluent products were analyzed simultaneously, without the need to condense the heavy products. Ethylene conversion and product selectivity were calculated on a carbon basis by using the following formulas:



where xi is the product i carbon fraction; RFi is the response factor normalized on a carbon basis; xethylene is the ethylene carbon fraction in the products; Si is the selectivity to product i on a carbon basis; X is the ethylene conversion; Yi is the yield of product i on a carbon basis.
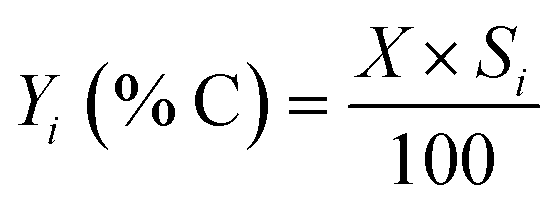
2.4. Temperature-Programmed Oxidation over spent catalysts
The Temperature-Programmed Oxidation (TPO) experiments were conducted using a Netzsch STA 449 F3 Jupiter instrument. The spent catalysts evaluated were subjected to a comparable TOS in order to enable reliable comparisons. The total TOS was 195 minutes for Ni/Sn-Beta and Ni/Hf-Beta, and 150 minutes for Ni/Al-Beta (operated both at 250 and 300 °C). As a control experiment, fresh Ni/Sn-Beta was tested. For each test, around 10 mg of sample was placed into an alumina crucible. The samples were heated with a ramp of 5 °C per minute and up to 600 °C under a synthetic air flow. The mass change during the experiment was recorded on a percentage basis to conduct derivative thermogravimetry (DTG) analysis.3. Results and discussion
3.1. Catalyst characterization
The heteroatom contents of the zeolitic supports, determined by XRF, are presented in Table 1, featuring a nominal Si/M ratio of 100 in the synthesis gel for most samples in the list. The Si/M ratios show a close correspondence with the nominal values, implying a negligible loss of metal sources during the hydrothermal synthesis. Furthermore, XRD confirmed the Beta framework in all zeotypes (Fig. S1†). N2 adsorption–desorption showed similar textural properties in all zeolitic materials (Table S1 and Fig. S2†), and the SEM images (Fig. S3†) illustrated the micron-sized crystals of the materials typically obtained using the hydrothermal synthetic methodology selected.55 Hence, the zeolitic materials exhibit comparable Si/M ratios, textural properties and crystal sizes, while the identity of the framework heteroatom incorporated in the zeotypes is the primary difference among them.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching of Si-Beta and heteroatom substituted Beta zeotypes
N stretching of Si-Beta and heteroatom substituted Beta zeotypes
| Entry | Sample | Heteroatom (M) loadinga (wt%) | Heteroatom (M) loadinga (mmol g−1) | Si/M (molar ratio) | NH3 uptakeb (mmol g−1) | CN stretching wavenumber for CD3CN adsorption (cm−1) |
|
|---|---|---|---|---|---|---|---|
| Open Lewis acid | Closed Lewis acid | ||||||
| a Measured by XRF. b Measured by NH3 TPD/TGA. c n.a., not available. d n.m., not measured. | |||||||
| 1 | Sn-Beta | 2.1 | 0.174 | 94 | 0.192 | 2316 | 2308 |
| 2 | Hf-Beta | 2.5 | 0.140 | 114 | 0.215 | 2310 | 2306 |
| 3 | Zr-Beta | 1.3 | 0.143 | 110 | 0.175 | 2303 | n.a.c |
| 4 | Ti-Beta | 0.6 | 0.131 | 123 | 0.054 | n.m.d | n.m.d |
| 5 | Ge-Beta | 1.2 | 0.169 | 95 | 0.012 | n.m.d | n.m.d |
| 6 | Si-Beta | 0 | 0 | — | 0.020 | n.m.d | n.m.d |
| 7 | Al-Beta | 0.109 (nominal) | 150 (nominal) | 0.231 | Brønsted acid | Lewis acid | |
| 2300 | 2323 | ||||||
 | ||
| Fig. 1 NH3-TPD/TGA profiles for M-Beta zeolitic materials. | ||
To complement the assessment of the acidity of the zeolitic materials, the adsorption of CD3CN was tracked via infrared spectroscopy (Fig. S4†), and Table 1 shows the assignments for the CN stretching upon the interaction with different types of acid sites. This technique confirmed the introduction of the heteroatoms within the Beta framework generating open/closed Lewis acid sites over Sn-, Hf- and Zr-Beta, while strong Brønsted acid sites were observed only over Al-Beta. The assignments are in line with the literature57–60 and reinforce the differences in acid strength between M-Beta materials observed via NH3-TPD.
 | ||
| Fig. 2 Nickel-ion exchange capacities of different catalysts, measured from NH4+ reverse ion exchange. Ni/M ratios represent the molar fraction of nickel-exchanged species by the total metal content. | ||
In the case of Ni/Si-Beta, devoid of any heteroatom substitution in the support framework, a low concentration of exchangeable Ni species is observed, which is similar to that of Ni/Ti-Beta. These results show that catalysts without acidity or very weak acidity, as reflected by NH3-TPD, did not lead to a high density of exchanged Ni species, implying a poor capacity for ion exchange. For the catalyst samples containing Sn and Zr within the framework, with higher acid strength observed from the stronger interaction with NH3, a markedly higher concentration of exchangeable Ni species is detected, which corresponds to 0.31–0.39 Ni/M molar ratios. These results confirm the ion-exchange capacity of these Lewis acidic zeotypes, which have been shown for alkali metals,48 but also highlight the importance of the zeotype acid strength to enable the incorporation of Ni in ion-exchangeable positions. Notably, we confirmed over the Sn-Beta samples with different Sn contents that Ni-exchanged species correlate well with the heteroatom content, thereby implying that the acid sites and/or their surroundings serve as essential anchoring points for Ni (Fig. S5(a)†). However, increasing the Ni loading over a sample with similar acidity led to lower levels of exchanged species (Fig. S5(b)†). As a result, a larger fraction of non-exchangeable Ni species is likely formed, probably in the form of nickel oxides. To elucidate whether these Ni species have potential for ion exchange, the NiO/Sn-Beta sample was subjected to the same treatment. A low concentration of nickel species was detected in the liquid after ion exchange, corresponding to 0.02 Ni/Sn substitution, thereby confirming that most exchangeable Ni species are cationic in nature. In the case of Ni/Al-Beta, a 0.39 Ni/Al molar ratio was estimated based on ion-exchanged species. In contrast to Lewis acidic zeotypes, Ni is exchanged over the Brønsted acidic protons. We hypothesize that the different natures of the heteroatoms interacting with Ni can impact the electronic environment around the metal species and therefore, its catalytic properties, which will be evaluated in the following sections.
O bond vibration is sensitive to both metal and acidic sites, and enables a direct comparison of the strength within those interactions.61Fig. 3 shows the evolution of the FTIR-CO spectra during desorption over the selected Ni/M-Beta and M-Beta samples. Additionally, as control experiments, the spectra of heteroatom-free Si-Beta, with and without Ni, are shown in Fig. S6.† Ni/Al-Beta is among the most studied catalyst in the literature, and the CO band assignments are well accepted. Ni2+ carbonyls in Al-Beta zeolites emerge at wavenumbers of >2200 cm−1, with bands at 2215 and 2205 cm−1 associated with isolated Ni2+ cations exchanged in different positions of the Beta framework.50 On defect-rich Al-Beta materials, an additional band is reported to be observed at 2194 cm−1. However, this band is not observed in our sample, likely due to the low-defective nature of the hydrothermally prepared Al-Beta zeolite (Fig. 3a and b). Additional bands are reported in the literature due to CO interactions with Ni-free Al-Beta at 2175, 2166, 2157, 2143 and 2132 cm−1, and are assigned to the interaction with Brønsted sites, aluminols, silanols, physisorbed CO, and CO interacting with the framework oxygen ions, respectively.50,61 All these bands are observed to a different extent over Ni/Al-Beta (Fig. 3b), including the band at 2175 cm−1, which implies that there are free Brønsted sites remaining after Ni loading, and supports the nature of Ni exchanged species as divalent Ni2+ on the catalyst.
 | ||
| Fig. 3 FTIR spectra within the CO vibration region upon desorption over Si/M-Beta catalysts. (a) and (b): Al-Beta without and after nickel loading; (c) and (d): Sn-Beta without and after nickel loading. The boxes filled with color represent different species upon CO adsorption. Spectra of Zr-Beta and Hf-Beta are shown in (e) and (f), with the overlay plots to show the shift. | ||
Comparing Ni/Al-Beta to Ni/Sn-Beta (Fig. 3b and d, respectively), the bands associated to silanols, physisorbed CO and CO interacting with framework oxygen are common in both samples. Most redshifted bands in Ni/Sn-Beta emerge after nickel incorporation at around 2192 and 2183 cm−1. In line with the Ni/Al-Beta assignments, we tentatively attribute this band to the interaction of CO with Ni2+ ion exchange over Lewis acid sites or their proximity within Sn-Beta. In these measurements, no clear bands were associated to the interaction of CO with Sn sites. In contrast, Zr-Beta and Hf-Beta (Fig. 3e and f, respectively) showed an additional band at 2188 cm−1. This band has been assigned in the literature to the interaction of CO with Zr Lewis open sites,63 which is a plausible hypothesis for our samples. After Ni loading, a slight shift towards a higher wavenumber region is observed in the same area (from 2188 cm−1 to 2192 cm−1) for Ni/Hf- and Ni/Zr-Beta, which may be attributed to the overlap of the CO interacting with both Ni exchanged in the environments of the Lewis acid sites, and also with the “free” Lewis acid sites. We speculate whether the broader range of acidity of Zr-Beta and Hf-Beta as compared to Sn-Beta (shown previously by NH3-TPD) is responsible for the observed differences between the Lewis acidic supports. Table 2 summarizes the proposed assignments for the interaction of CO over the Ni/M-Beta catalysts. Overall, these results support the presence of Ni2+ species over the nickel-doped Sn-, Hf- and Zr-Beta catalysts, but reflect different electronic environments for nickel as compared to Ni/Al-Beta.
| Surface group interacting with CO | Frequency shift (cm−1) | Samples | Ref. |
|---|---|---|---|
| Ni2+ exchanged in Brønsted sites | 2215, 2205 | Ni/Al-Beta | 50, 61 |
| Ni2+ exchanged in Lewis sites | 2192, 2183 (tentative) | Ni/Sn-Beta, Ni/Zr-Beta, Ni/Hf-Beta | This work |
| Hydrated Lewis sites | 2188 | Ni/Zr-Beta, Zr-Beta, Ni/Hf-Beta, Hf-Beta | 63 |
| Non-hydrated Lewis sites | 2177 | Ni/Zr-Beta, Zr-Beta, Ni/Hf-Beta, Hf-Beta | 63 |
| Brønsted sites | 2176 | Ni/Al-Beta, Al-Beta | 50, 61 |
| Silanols | 2154 | All | 50, 61 |
| Physisorbed CO | 2142 | All | 50, 61 |
| Framework oxygen ions | 2136 | All | 50, 61 |
3.2. Ethylene oligomerization over Ni/M-Beta with different framework heteroatoms
As shown in the previous section, the Ni/M-Beta catalysts with different trivalent and tetravalent heteroatoms display distinct characteristics in relation to both nickel species and acid sites, which are likely to influence their catalytic performance towards ethylene conversion. The series of Ni-exchanged zeolitic materials was compared in gas-phase ethylene oligomerization using a fixed bed reactor with an ethylene partial pressure of 10 bar and a contact time of 0.017 min gcat−1 mL−1 at temperatures above 250 °C.The rate of ethylene conversion versus time on stream (TOS) for all catalysts is shown in Fig. 4. Most of the experiments were conducted at 250 °C, and an additional experiment over Ni/Sn-Beta was carried out at 300 °C. Carbon balances displayed good stability with values consistently nearing 100% (Fig. S7†). In a control experiment, Ni/Si-Beta was found to be inactive for ethylene oligomerization. Ni/Ti-Beta and Ni/Ge-Beta also showed no apparent activity. All the other catalysts were active for converting ethylene under similar reaction conditions. These results align well with the characterization discussed earlier, reflecting that a certain level of acid strength is needed to ion-exchange significant amounts of Ni into active species. Importantly, Ni-free Sn-Beta was also found to be inactive towards ethylene conversion, implying that nickel is fundamental over all these materials to convert ethylene under the tested conditions (Fig. S8†).
 | ||
| Fig. 4 Rate of ethylene conversion versus time on stream for all catalysts. Reference reaction conditions: 30 bar total pressure, Pethylene = 10 bar, T = 250 °C and contact time of 0.017 min gcat mL−L. An additional experiment is presented using Ni/Sn-Beta at 300 °C. | ||
Furthermore, all active catalysts exhibited a relatively rapid decrease in activity during the initial stage, followed by a period of slower deactivation under the tested conditions. This phenomenon, observed in other published studies, has been associated with the formation of long-chain products and the following deposition, blocking the access to the active sites of the catalyst.26,29 Notably, Ni/Al-Beta, the only catalyst with strong Brønsted acid sites, shows a faster deactivation profile compared to the other samples characterized with Lewis acidity. We speculate that the presence of strong Brønsted sites in Ni/Al-Beta may lead to a strong interaction with reactants/products and/or the formation of long-chain products, thereby hindering their desorption. This could be in contrast to the behavior observed with Lewis acidic zeotypes, which possess weaker acidity and presumably facilitate an easier desorption of products. After stabilization, the TOF of the tested catalysts follows the order Ni/Hf-Beta ≈ Ni/Zr-Beta > Ni/Sn-Beta (300 °C) > Ni/Sn-Beta (250 °C) > Ni/Al-Beta. However, determining the exact reason for the activity differences remains challenging. These differences could be attributed to varying degrees of deactivation, distinct catalytic activity inherent to the exchanged Ni sites, or possibly non-optimized nickel-to-metal (Ni/M) ratios across the range of materials studied. Indeed, we employed the Ni/M ratio of 1:1 (mol:mol) across all other Lewis acidic catalysts, observed as optimal for Ni/Sn-Beta (as shown in Fig. S9†), but the ideal ratio may vary for each catalyst. Nevertheless, the results indicated that variations in Ni/Sn ratios did not significantly affect product selectivity, leading us to focus our comparative analysis primarily on this aspect.
The product distributions obtained among the active catalysts are compared in Fig. 5 at similar ethylene conversion levels. As it was not possible to reach similar conversions for Ni/Al-Beta at 250 °C to directly compare to the rest of the catalysts, additional experiments were carried out at 300 °C, thereby enabling a reliable product distribution comparison. At ∼10–15% ethylene conversion and 250 °C, the three tetravalent metal substituted samples exhibit comparable product distributions, in which linear butenes are the main products (above 70%). The selectivity of different butene isomers is also very similar over these catalysts (Fig. S10†), where the α-olefin, 1-butene, represents approximately 20–25% of all butenes. The rest are also exclusively linear, 2-butenes (cis- and trans-), derived from double bond migration isomerization. These numbers approach thermodynamic equilibrium.64 Only two other families of products are observed over the Ni exchanged Lewis acidic catalysts, saturated products (exclusively ethane) and hexenes. Saturated product formation is derived from hydrogen transfer reactions, as there is no other source of hydrogen. Hexenes are formed following the Schulz–Flory distribution based on the chain growth probability.30 When comparing to Ni/Al-Beta, larger differences in product selectivity are observed. Despite the lower ethylene conversion level at 250 °C (around 5%), and the potential faster deactivation of the most active sites, a broader range of products is yet observed over this catalyst with the presence of hydrocarbons derived from skeletal isomerization (i-butene) and cracking (pentenes). In order to enable a fair comparison, the product distributions for Ni/Sn-Beta and Ni/Al-Beta are compared at similar conversion levels at 300 °C. The product distribution over Ni/Sn-Beta remains almost unaltered and only a slight increase in ethane generation is observed, likely associated to favored hydrogen transfer reactions with higher temperature. In contrast, the product distribution over Ni/Al-Beta becomes more complex. Scheme 2 depicts the proposed reaction pathways for the different product families. While linear butenes remain the main product, different catalytic pathways are observed: further oligomerization (hexenes, C7+), skeletal isomerization (i-butene, i-butane, i-pentane), hydrogen transfer (ethane, i-butane, n-butane, i-pentane), and cracking (propene, pentenes, i-pentane). As cracking and skeletal isomerization are conventional reactions catalyzed over Brønsted sites, we associate the formation of these products with the presence of free Brønsted sites over Ni/Al-Beta.65 Therefore, we conclude that Lewis acid sites or exchanged Ni on the Lewis acidic supports are not that active under the same conditions as Brønsted acid sites to catalyze skeletal isomerization and cracking reactions, resulting in high selectivity towards linear alkene products. Interestingly, we addressed the isomerization capabilities of the Lewis acidic zeotypes by feeding 1-butene over Sn-Beta and confirmed that double bond migration isomerization can be catalyzed by this family of materials (Fig. S11†). Hence, Lewis acid sites, present in Ni-containing zeotypes, contribute to shape the product distribution during ethylene oligomerization, while maintaining exclusively linear oligomeric products.
 | ||
| Fig. 5 Product selectivity (bars) and ethylene conversion (black squares) obtained from ethylene oligomerization over Ni/M-Beta catalysts. Reference reaction conditions: 30 bar total pressure, Pethylene = 10 bar and contact time of 0.017 min gcat mL−L. | ||
 | ||
| Scheme 2 Proposed reaction pathways for ethylene oligomerization and subsequent reactions over the Ni/M-Beta catalysts. | ||
The evolution of different product families with ethylene conversion is shown in Fig. 6 to assess the effect of conversion levels on product selectivity. Two distinguished trends can be observed based on the acidic nature of the zeolitic supports. Focusing first on products derived only from ethylene oligomerization, linear butenes and hexenes, shown in Fig. 6a, a very high and stable selectivity (>90%) for all three nickel-containing Lewis acidic zeotypes is observed. In contrast, Ni/Al-Beta shows a rapid decrease in selectivity with increasing conversion levels. The only other family of products observed over the catalysts containing Sn, Hf and Zr is the saturated products (Fig. 6b), which in this case is exclusively due to ethane formation. The Brønsted acidic catalyst shows relatively similar selectivity to the saturated products at 250 °C (exclusively ethane), but their formation increases considerably with conversion and temperature. In the case of Ni/Al-Beta at 300 °C, butanes and pentanes are identified, while Ni/Sn-Beta still shows only ethane in this product family (Fig. 5). Hence, the formation of saturated products is promoted with temperature and the presence of Brønsted acid sites, likely occurring over the acidic sites via hydrogen transfer reactions, which have been identified to occur specially in the beginning of the reaction.40,66 Therefore, longer studies under steady-state conditions would be required to reliably assess the activity of our catalytic systems towards hydrogen transfer. Theoretically, the formation of saturated products could lead to polyunsaturated or cyclic hydrocarbons due to stoichiometry; however, we did not detect such products in the outlet product stream. We speculate whether those compounds might be retained on the catalysts or promote coke formation, as it has been shown in the methanol-to-hydrocarbons reaction over zeolitic materials.43,44 The carbon balances (Fig. S7†) support this hypothesis as the experiments for Ni/Sn-Beta at 300 °C and Ni/Al-Beta at 250 °C show the lowest carbon recovery (approximately 95%), and those tests also show the highest formation of saturated products.
 | ||
| Fig. 6 Selectivity towards (a) linear butenes and hexenes, (b) saturated products, (c) cracking products and (d) isomerization products over Ni/M-Beta catalysts during ethylene oligomerization. Reference reaction conditions: 30 bar total pressure, Pethylene = 10 bar and contact time of 0.017 min gcat mL−L. | ||
To assess the nature of the retained hydrocarbons, TPO experiments were carried out over the spent catalysts from tests conducted over comparable TOS (Ni/Al-Beta for 155 min; Ni/Sn-Beta and Ni/Hf-Beta for 195 min, Fig. S12†). In all cases, 10–12% weight loss was observed upon oxidation (Table S3 and Fig. S13†), but the DTG profile changed based on the nature of the framework heteroatom (Fig. 7). The Lewis acidic catalysts showed a “softer” nature of the carbon species retained with maxima at 355, 375 and 415 °C, whereas Ni/Al-Beta showed an additional oxidation maximum at 445 °C. These latter species are harder to oxidize, suggesting the presence of a “harder” coke over the Brønsted acidic catalyst. Therefore, we conclude that the presence of strong Brønsted acid sites within the Ni/Al-Beta catalyst leads to more severe coking/retention of hydrocarbons, while the Ni-containing Lewis acidic zeotypes mitigate it.
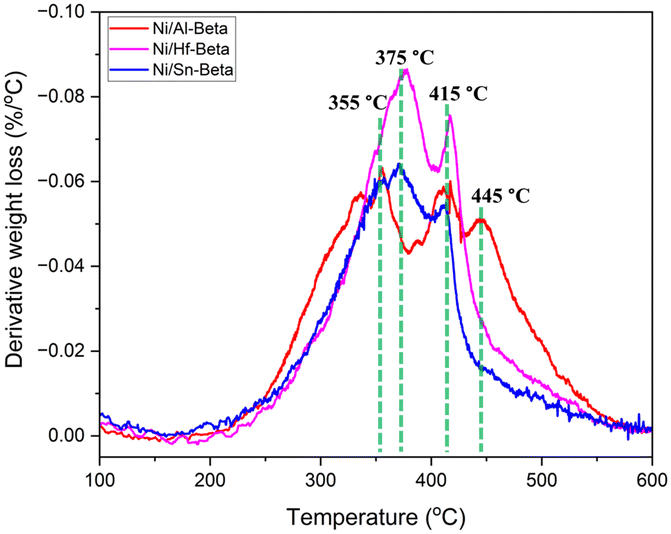 | ||
| Fig. 7 Derivative weight loss profiles versus temperature during the TPO test of spent catalysts. | ||
As shown in Fig. 6c and d, a more obvious difference in product distribution is observed with respect to cracking (propene and pentenes/pentanes) and skeletal-isomerized products (i-butene/i-butane/i-pentene/i-pentane). These reaction pathways are completely absent over the Ni-containing Lewis acidic zeotypes. However, in the case of Ni/Al-Beta, these products are formed via Brønsted-catalyzed reactions.
An interesting difference in the ratio between butenes and hexenes was also observed for all catalysts (Fig. 8). This ratio is potentially correlated to the propagation and termination probabilities in the conventional Schulz–Flory distribution observed during ethylene oligomerization,67 although higher ethylene conversion levels than those reported here and the detection of larger oligomer products (C8, C10…) are required to confirm it. Nevertheless, these differences may indicate the possibility to tailor the catalyst towards lower or higher olefins depending on the heteroatom substituted within the zeolitic framework. At similar conversion levels, the highest ratios are observed for Ni/Al-Beta, but they sharply decrease upon conversion, likely due to the larger extent of the additional reaction pathways aforementioned (e.g., cracking). Consistent ratios, however, are observed for the Ni-containing Lewis acidic zeotypes at conversion levels between 5 and 15% (Fig. 8). Interestingly, Ni/Sn-Beta showed the lowest ratios with higher hexene yields obtained as compared to the Ni/Hf-Beta and Ni/Zr-Beta catalysts. These results suggest intrinsic differences in the rates of chain propagation over termination. Furthermore, they indicate that a certain level of selectivity control in the ethylene oligomerization reaction might be achievable by employing various metal framework zeotypes as supports for nickel ions.
 | ||
| Fig. 8 Butene to hexene ratios as a function of ethylene conversion over Ni/M-Beta catalysts during ethylene oligomerization. Reference reaction conditions: 30 bar total pressure, Pethylene = 10 bar and contact time of 0.017 min gcat mL−L. | ||
Overall, this study on the use of Ni-containing Lewis acidic zeotypes for ethylene oligomerization shows promising results in various aspects: 1) high selectivity towards linear olefinic products; 2) the possibility to tune the composition of the linear products with the nature of the tetravalent metal in the zeotypes, and; 3) low selectivity to hydrogen transfer and “softer” coke composition, which may provide higher catalyst stability. Nevertheless, further studies are needed to assess reliably the catalytic activity, and the use of sub-micron-sized materials is envisioned to enable stable operation for longer times.
Conclusions
This work uncovers the use of a novel class of nickel-containing zeotypes and their potential as catalysts for ethylene oligomerization. The materials are characterized by the presence of Lewis acid sites and the absence of strong Brønsted characteristics. Furthermore, the zeotypes show their capability towards nickel ion exchange, resulting in the generation of catalytically active fundamental nickel species to convert ethylene. The interaction between exchanged nickel species and the different framework heteroatoms substituted in the Beta structure leads to the possibility of tuning the electronic environments around the metal, affecting the catalytic performance in ethylene oligomerization.Nickel-containing zeotypes with tetravalent substituted heteroatoms (Sn, Hf, Zr) consistently yielded a remarkable and stable selectivity (>90%) towards linear butene and hexene products, where oligomerization and double bond migration isomerization are the main catalytic pathways. Conversely, Ni/Al-Beta was found to catalyze additional skeletal isomerization, cracking and hydrogen transfer reactions, forming side products such as i-butene, propene, pentenes and alkanes. We associate the formation of these undesired products to the presence of strong Brønsted sites, which are absent in the nickel-containing Lewis acidic zeotypes. Interestingly, differences in C4/C6 ratios are observed between the catalysts, reflecting their ability towards chain propagation and chain termination during ethylene oligomerization, a parameter that could be used to assist catalyst design for the desired product distribution. Furthermore, the low selectivity to hydrogen transfer reactions is linked to the mitigation of coking and/or retention of hydrocarbons.
Author contributions
Conception and design: Esben Taarning, Juan S. Martinez-Espin, Yunfei Bai, Tomás Cordero-Lanzac; data acquisition: Yunfei Bai, Tomás Cordero-Lanzac; data analysis: Juan S. Martinez-Espin, Yunfei Bai, Tomás Cordero-Lanzac; drafting the article: Juan S. Martinez-Espin, Yunfei Bai; revising the article: all; supervision: Esben Taarning, Ainara Nova, Unni Olsbye.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 859910. T. C. L., A. N. and U. O. acknowledge the Norwegian Research Council through contract no. 288331 (CO2LO) and 314321 (CO2PCAT).References
- A. Finiels, F. Fajula and V. Hulea, Catal. Sci. Technol., 2014, 4, 2412–2426 RSC.
- E. Camara Greiner and Y. Inoguchi, Chemical Economics Handbook, IHS Chemical, 2010, pp. 1–78 Search PubMed.
- H. Olivier-Bourbigou, P. A. R. Breuil, L. Magna, T. Michel, M. F. Espada Pastor and D. Delcroix, Chem. Rev., 2020, 120, 7919–7983 CrossRef CAS PubMed.
- J. Ding and W. Hua, Chem. Eng. Technol., 2013, 36, 83–90 CrossRef CAS.
- R. A. Weusthuis, J. M. Aarts and J. P. Sanders, Biofuels, Bioprod. Biorefin., 2011, 5, 486–494 CrossRef CAS.
- Y. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. Li, ACS Catal., 2019, 9, 8592–8621 CrossRef CAS.
- P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed.
- J. N. Armor, Catal. Today, 2014, 236, 171–181 CrossRef CAS.
- V. Hulea, ACS Catal., 2018, 8, 3263–3279 CrossRef CAS.
- J. A. Posada, A. D. Patel, A. Roes, K. Blok, A. P. Faaij and M. K. Patel, Bioresour. Technol., 2013, 135, 490–499 CrossRef CAS PubMed.
- J. Rass-Hansen, H. Falsig, B. Jørgensen and C. H. Christensen, J. Chem. Technol. Biotechnol., 2007, 82, 329–333 CrossRef CAS.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed.
- J. Skupinska, Chem. Rev., 1991, 91, 613–648 CrossRef CAS.
- A. Al-Jarallah, J. Anabtawi, M. Siddiqui, A. Aitani and A. Al-Sa'doun, Catal. Today, 1992, 14, 1–121 CrossRef.
- W. Keim, Angew. Chem., Int. Ed., 2013, 52, 12492–12496 CrossRef CAS PubMed.
- C. O'connor and M. Kojima, Catal. Today, 1990, 6, 329–349 CrossRef.
- M. Peuckert and W. Keim, J. Mol. Catal., 1984, 22, 289–295 CrossRef CAS.
- E. Angelescu, M. Che, M. Andruh, R. Zãvoianu, G. Costentin, C. Miricã and O. D. Pavel, J. Mol. Catal. A: Chem., 2004, 219, 13–19 CrossRef CAS.
- J. Canivet, S. Aguado, Y. Schuurman and D. Farrusseng, J. Am. Chem. Soc., 2013, 135, 4195–4198 CrossRef CAS PubMed.
- M. I. Gonzalez, J. Oktawiec and J. R. Long, Faraday Discuss., 2017, 201, 351–367 RSC.
- E. Rozhko, A. Bavykina, D. Osadchii, M. Makkee and J. Gascon, J. Catal., 2017, 345, 270–280 CrossRef CAS.
- J. R. Sohn, W. C. Park and S.-E. Park, Catal. Lett., 2002, 81, 259–264 CrossRef CAS.
- J. Heveling, C. Nicolaides and M. Scurrell, Appl. Catal., A, 1998, 173, 1–9 CrossRef CAS.
- V. Hulea and F. Fajula, J. Catal., 2004, 225, 213–222 CrossRef CAS.
- J. Heveling, A. van der Beek and M. de Pender, Appl. Catal., 1988, 42, 325–336 CrossRef CAS.
- M. Lallemand, O. A. Rusu, E. Dumitriu, A. Finiels, F. Fajula and V. Hulea, Appl. Catal., A, 2008, 338, 37–43 CrossRef CAS.
- A. Martínez, M. A. Arribas, P. Concepción and S. Moussa, Appl. Catal., A, 2013, 467, 509–518 CrossRef.
- R. Y. Brogaard and U. Olsbye, ACS Catal., 2016, 6, 1205–1214 CrossRef CAS.
- R. Henry, M. Komurcu, Y. Ganjkhanlou, R. Y. Brogaard, L. Lu, K.-J. Jens, G. Berlier and U. Olsbye, Catal. Today, 2018, 299, 154–163 CrossRef CAS.
- E. Koninckx, P. S. F. Mendes, J. W. Thybaut and L. J. Broadbelt, Appl. Catal., A, 2021, 624, 118296 CrossRef CAS.
- G. Yang, L. Zhou and X. Han, J. Mol. Catal. A: Chem., 2012, 363, 371–379 CrossRef.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Phys. Chem. C, 2013, 117, 3976–3986 CrossRef CAS.
- G. Sastre and A. Corma, Chem. Phys. Lett., 1999, 302, 447–453 CrossRef CAS.
- N. V. Choudary and B. L. Newalkar, J. Porous Mater., 2011, 18, 685–692 CrossRef.
- M. Lallemand, A. Finiels, F. Fajula and V. Hulea, Appl. Catal., A, 2006, 301, 196–201 CrossRef CAS.
- L. Bonneviot, D. Olivier and M. Che, J. Mol. Catal., 1983, 21, 415–430 CrossRef CAS.
- O. Kresnawahjuesa, G. Kühl, R. J. Gorte and C. Quierini, J. Catal., 2002, 210, 106–115 CrossRef CAS.
- H. J. Kim, J.-W. Kim, N. Kim, T.-W. Kim, S. H. Jhung and C.-U. Kim, Mol. Catal., 2017, 438, 86–92 CrossRef CAS.
- M. Sanati, C. Hornell and S. G. Jaras, Catalysis, 2007, 14, 236–288 Search PubMed.
- A. Martínez Gómez-Aldaraví, C. Paris, M. Moliner and C. Martínez, J. Catal., 2023, 426, 140–152 CrossRef.
- G. Chen, H. Liu, S. Fadaeerayeni, J. Shan, A. Xing, J. Cheng, H. Wang and Y. Xiang, Catal. Sci. Technol., 2020, 10, 4019–4029 RSC.
- F. Jin, Y. Yan and G. Wu, Catal. Today, 2020, 355, 148–161 CrossRef CAS.
- J. S. Martínez-Espín, K. De Wispelaere, T. V. W. Janssens, S. Svelle, K. P. Lillerud, P. Beato, V. Van Speybroeck and U. Olsbye, ACS Catal., 2017, 7, 5773–5780 CrossRef.
- S. Müller, Y. Liu, F. M. Kirchberger, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, J. Am. Chem. Soc., 2016, 138, 15994–16003 CrossRef PubMed.
- M. Meloni and R. C. Runnebaum, Catal. Sci. Technol., 2021, 11, 3393–3401 RSC.
- M. S. Holm, Y. J. Pagán-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702–706 RSC.
- L. Botti, S. A. Kondrat, R. Navar, D. Padovan, J. S. Martinez-Espin, S. Meier and C. Hammond, Angew. Chem., Int. Ed., 2020, 59, 20017–20023 CrossRef CAS PubMed.
- S. G. Elliot, I. Tosi, S. Meier, J. S. Martinez-Espin, S. Tolborg and E. Taarning, Catal. Sci. Technol., 2019, 9, 4339–4346 RSC.
- S. Tolborg, I. Sádaba, C. M. Osmundsen, P. Fristrup, M. S. Holm and E. Taarning, ChemSusChem, 2015, 8, 613–617 CrossRef CAS PubMed.
- S. Moussa, M. A. Arribas, P. Concepción and A. Martínez, Catal. Today, 2016, 277, 78–88 CrossRef CAS.
- A. Penkova, S. Dzwigaj, R. Kefirov, K. Hadjiivanov and M. Che, J. Phys. Chem. C, 2007, 111, 8623–8631 CrossRef CAS.
- K. Góra-Marek, A. Glanowska and J. Datka, Microporous Mesoporous Mater., 2012, 158, 162–169 CrossRef.
- P. Sun, C. Liu, H. Wang, Y. Liao, X. Li, Q. Liu, B. F. Sels and C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215737 CrossRef CAS PubMed.
- L. Botti, R. Navar, S. Tolborg, J. S. Martínez-Espín and C. Hammond, ACS Sustainable Chem. Eng., 2022, 10, 4391–4403 CrossRef CAS PubMed.
- S. Tolborg, A. Katerinopoulou, D. D. Falcone, I. Sádaba, C. M. Osmundsen, R. J. Davis, E. Taarning, P. Fristrup and M. S. Holm, J. Mater. Chem. A, 2014, 2, 20252–20262 RSC.
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef CAS PubMed.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- B. A. Johnson, J. R. Di Iorio and Y. Román-Leshkov, J. Catal., 2021, 404, 607–619 CrossRef CAS.
- V. L. Sushkevich, A. Vimont, A. Travert and I. I. Ivanova, J. Phys. Chem. C, 2015, 119, 17633–17639 CrossRef CAS.
- K. Chakarova, P. Nikolov and K. Hadjiivanov, Catal. Commun., 2013, 41, 38–40 CrossRef CAS.
- S. Moussa, P. Concepción, M. A. Arribas and A. Martínez, ACS Catal., 2018, 8, 3903–3912 CrossRef CAS.
- I. V. Elev, B. N. Shelimov and V. B. Kazansky, J. Catal., 1984, 89, 470–477 CrossRef CAS.
- V. L. Sushkevich, D. Palagin and I. I. Ivanova, ACS Catal., 2015, 5, 4833–4836 CrossRef CAS.
- M. Betz, C. Fuchs, T. A. Zevaco, U. Arnold and J. Sauer, Biomass Bioenergy, 2022, 166, 106595 CrossRef CAS.
- E. A. Uslamin, H. Saito, N. Kosinov, E. Pidko, Y. Sekine and E. J. M. Hensen, Catal. Sci. Technol., 2020, 10, 2774–2785 RSC.
- H. O. Mohamed, V. K. Velisoju, I. Hita, O. Abed, R. K. Parsapur, N. Zambrano, M. B. Hassine, N. Morlanes, A.-H. Emwas, K.-W. Huang and P. Castaño, Chem. Eng. J., 2023, 475, 146077 CrossRef CAS.
- G. J. P. Britovsek, R. Malinowski, D. S. McGuinness, J. D. Nobbs, A. K. Tomov, A. W. Wadsley and C. T. Young, ACS Catal., 2015, 5, 6922–6925 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01736b |
| This journal is © The Royal Society of Chemistry 2024 |