
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 chemisorption and activation on carbon nitride with less amino groups boost CO2 photoreduction†
Debin
Zeng‡
 ,
Xiting
Wang‡
,
Chunguang
Kuai
,
Zhuo
Jiang
* and
Yuzheng
Guo
*
,
Xiting
Wang‡
,
Chunguang
Kuai
,
Zhuo
Jiang
* and
Yuzheng
Guo
*
School of Electrical Engineering and Automation, Wuhan University, Wuhan, 430072, PR China. E-mail: zhuojiang@whu.edu.cn; yguo@whu.edu.cn
First published on 30th January 2024
Abstract
CO2 chemisorption and activation are two crucial factors that can significantly impact the overall CO2 conversion efficiency. However, achieving effective modulation of these processes in metal-free photocatalysts has proven to be challenging. In this study, we present a novel approach for activating CO2 molecules by selectively removing some amino groups at the boundary of carbon nitride (CN). By doing so, we observed a concentration of photogenerated electrons at the nitrogen atoms neighboring the eliminated –NH2 group. This electron concentration enhances the chemisorption and activation of CO2, resulting in lowered energy barriers for the rate-determining step from CO2 to *COOH. Interestingly, we found that only the nitrogen atoms adjacent to the removed –NH2 group in CN serve as catalytic sites, in contrast to all the nitrogen atoms neighboring the –NH2 group in CN–NH2. As a result, CN exhibits superior catalytic efficiency, with a turnover frequency (TOF) for CO that is 23.11 times higher than that of CN–NH2 under identical conditions.
1 Introduction
Solar-driven CO2 reduction is a promising way to address both environmental and energy shortage issues.1–3 Despite significant efforts over the past few decades, the conversion efficiency of this process has remained unsatisfactory. A key challenge in developing catalysts with high CO2 conversion efficiency lies in the limited chemisorption ability of CO2, coupled with the high activation energy required for CO2 molecules to react on catalyst surfaces.4–6 Consequently, addressing and enhancing these two aspects play crucial roles in promoting CO2 photoreduction reactions.Recently, various strategies have been employed to enhance the photoelectric conversion efficiency by improving the separation efficiency of photogenerated carriers. These strategies include the loading of co-catalysts and the construction of heterojunctions. However, the chemisorption and activation of CO2, which are influenced by the size and type of transition metals, are often overlooked due to the challenges associated with detecting these processes at the molecular level. Green et al. reported that the size of Ptn− clusters plays a dominant role in both CO2 chemisorption and activation efficiency.7 Additionally, Weber et al. found that metal anions can affect CO2 chemisorption and activation performance through gas-phase complexation between CO2 and metal anions.8
Apart from the size and type of transition metals, electron redistribution on catalytic sites can also modify the CO2 chemisorption and activation process. For instance, in a study by Cao et al., electron transfer from an Au atom to a Cd vacancy led to the redistribution of electrons in CdS, resulting in a strong CO2 chemisorption and activation ability. On the other hand, when an Au atom was anchored at an S vacancy, this electron transfer process was prevented, causing CO2 molecules to only physically adsorb at the catalytic sites, even under the same CO2 atmosphere.9 Therefore, the modulation of the charge distribution of catalytic sites is significant for improving CO2 chemisorption and activation, particularly in the context of non-metallic catalysts.
Since the initial report by Wang et al. on the ability of C3N4 to generate hydrogen under light illumination,10 this material has attracted significant research attention in subsequent decades. The interest in C3N4 stems from its favourable band structure and adjustable functionalities.11–14 In this particular study, we synthesized carbon nitride (CN–NH2) and its derivative, CN, by precisely removing certain –NH2 groups from CN–NH2. This coordination defect caused electron accumulation around the nitrogen catalytic sites adjacent to the lost –NH2 group, resulting in a much higher negative charge density in these sites. Due to electron redistribution, the catalytic sites were regarded as Lewis basic sites for CO2 chemisorption and activation, thus boosting CO2 photoreduction under mild conditions.
2 Experimental details
Synthesis of CN–NH2
Specifically, 1 g of C3H6N6 was placed into a muffle furnace, heated to 500 °C at a heating rate of 2 °C min−1, and maintained at this temperature for 4 h. The obtained material was termed as CN–NH2.Synthesis of CN
In a typical synthesis, 2 g of hexadecyl trimethyl ammonium bromide (CTAB) was dissolved in a mixed solution of cyclohexane, 1-pentanol and deionized water, and its total volume was 60 mL. This mixture was stirred for 30 min. Next, 1 g melamine was added into the mixed solution, and the pH was adjusted to 2 using HNO3 with stirring. The resulting mixture was stirred for 1 h and transferred to a 100 mL Teflon autoclave, where it was heated at 160 °C for 16 h. The synthesized prepolymer was washed with anhydrous ethanol and deionized water 10 times each, subsequently dried at 60 °C for 10 h, and finally placed in a tube furnace. The prepolymer was heated to 500 °C at a heating rate of 2 °C min−1 under a 100 sccm argon flow and maintained at this temperature for 4 h. The obtained material was termed as CN. In CN, no bromine element was detected in X-ray photoelectron spectroscopy (XPS), suggesting the absence of residual bromine (Fig. S1†). Additionally, the results obtained from energy dispersive X-ray spectroscopy (EDS) showed negligible levels of bromine, further confirming its absence (Fig. S2†). To investigate the influence of CTAB, pH condition, solvothermal time, and temperature on the properties of CN, controlled experiments were conducted. CTAB, a cationic surfactant, carries a positive charge in aqueous solution. Due to its strong interaction, CTAB cations can react with the –NH2 groups of melamine, resulting in the partial removal of –NH2 groups of melamine.15,16Characterization
X-ray diffraction (XRD) patterns were recorded using an XPert Pro X-ray diffractometer with Cu Kα radiation (λ = 1.54 Å). Fourier transform infrared (FTIR) spectra were obtained on a Thermo FTIR5700 Spectrometer. Raman spectra were recorded on a RENISHAW inVia Raman Microscope equipped with a 780 nm laser. Room temperature electron paramagnetic resonance (EPR) spectra were obtained on a JEOL ESR spectrometer (JES-FA200). XPS analyses were conducted using an ESCALAB250Xi with Al Kα radiation. The molar loss of nitrogen and hydrogen elements was detected using a vario EL III elemental analyzer. Field-emission scanning electron microscopy (FESEM) images were obtained on a Zeiss SIGMA. Scanning transmission electron microscopy (STEM) and elemental mapping were conducted on a JEM-F200 transmission electron microscope operating at an acceleration voltage of 200 kV. The N2 adsorption/desorption isotherms, porosity and CO2 adsorption isotherms were studied using an automatic analyzer (ASAP 2020). UV-vis diffuse reflectance spectra (DRS) were obtained using a UV-vis spectrophotometer (Shimadzu UV-2600) equipped with an integrating sphere detector. The EIS Nyquist plots, transient photocurrent response and Mott–Schottky curves were obtained on an electrochemical workstation (CHI760E). Time-resolved fluorescence spectra were recorded on a single photon counting module (PicoQuant, PMA Hybrid), which was excited using a 405 nm pulse laser. The zeta potentials of the synthesized materials were collected using a Zetasizer Nano ZSP.In situ FTIR spectra of CO2 photoreduction
In situ FTIR spectra of CO2 photoreduction were acquired using a Thermo Scientific Nicolet iS50 spectrometer equipped with an in situ diffuse reflectance accessory cell. The sample was filled and compacted in the sample cell, and argon gas was introduced into the in situ cell to remove impurities. CO2 and H2O were subsequently introduced into the in situ cell, and the system was allowed to equilibrate for 30 min to ensure that the adsorption and desorption of the gases reached equilibrium. Prior to light irradiation, an FTIR spectrum was collected to establish the background, including catalyst and CO2/H2O information.In situ FTIR spectra of CO2 chemisorption
A 100 mg sample was subjected to 500 °C in a 5% H2/Ar air flow for 1 h to desorb the adsorbed impurities. The sample was subsequently cooled to 100 °C, followed by the introduction of argon gas into the in situ cell to purge H2. A background spectrum of the catalyst information was obtained prior to collecting the spectra upon the introduction of a 10% CO2/Ar air flow into the in situ cell.CO2 temperature-programmed desorption and mass spectra
A 100 mg sample was heated to 500 °C in a helium air flow for 1 h to remove adsorbed impurities. After cooling to 50 °C, the sample was subjected to CO2 in a temperature-programmed chemisorption instrument for 30 min, allowing the chemical adsorption of CO2 on the catalyst. Subsequently, gaseous CO2 was purged from the system with a helium air flow for 30 min. The temperature of the sample was then increased at a rate of 10 °C min−1 while collecting the CO2 temperature-programmed desorption signal. The desorbed CO2 from the temperature-programmed chemisorption instrument was further analyzed by mass spectrometry.CO2 photoreduction test
Photocatalytic tests for all samples were conducted using a Labsolar-6A system (Beijing Perfectlight) equipped with a 300 W Xe lamp (Beijing Perfectlight, PLS-SXE300, 320–780 nm, 1.50 W cm−2). For the photocatalytic performance test, 20 mg of photocatalyst was dispersed in deionized water and dried in a quartz reactor at 60 °C overnight. To provide protons and act as a hole scavenger, 2 mL of deionized water was injected into the quartz reactor. Prior to irradiation, the reactor was pumped to a vacuum three times, and then filled with high-purity CO2 gas to 70 kPa. Following a 3 h irradiation period, gas products were collected and analyzed using a gas chromatograph (Fuli, GC 9790 II) equipped with flame ionization and thermal conductivity detectors.Calculation details
The first-principles calculations were performed on the Vienna Ab initio Simulation Package (VASP).17 The interaction between ions and valance electrons was described by utilizing projector augmented wave (PAW) potentials while the exchange correlation energy was obtained by generalized gradient approximation.18,19 Furthermore, the DFT-D2 correction was included to evaluate the van der Waals interactions. The cutoff energy and Brillouin zone were 400 eV and 2 × 2 × 1 sheet k-point mesh, respectively. The convergence process of structure relaxations would be ceased when the conventional energy and force were 10−5 eV and 0.03 eV Å−1, respectively. CN–NH2 and CN had a vacuum layer of 15 Å placed above the surface slab.The adsorption energy (Ead) defined the interaction between intermediates and the surface, which was defined as Ead = Eads + Esur − Esys, where Eads is the adsorbate energy prior to adsorption, Esur is the surface energy prior to adsorption and Esys is the system energy after adsorption. Gibbs free energies for gaseous and adsorbed intermediates were calculated at 298.15 K based on the expression GE = EDFT + EZEP − TS, where EDFT is the electronic energy calculated by VASP, EZEP is the zero-point energy and TS is entropy contribution.20
3 Characterization results
Structural characterization
The X-ray diffraction (XRD) patterns of both CN–NH2 and CN showed a good match with that of the hexagonal g-C3N4 (JCPDS card No. 087-1526), which corresponds to carbon nitride with a graphitic-like phase (Fig. 1a and S3†). In the XRD pattern, the diffraction peak centered at 2θ = 12.9° corresponds to the (100) plane, which represents the intralayer repeating units of heptazine heteroatoms with the –NH2 group.21 Additionally, another diffraction peak observed at 2θ = 27.2° corresponds to the (002) plane, which indicates the interlayer periodic stacking similar to graphite. Importantly, it was observed that both the (100) and (002) planes of CN were shifted to lower angles compared to CN–NH2. This shift can be attributed to the absence of the –NH2 group, which weakens the interlayer stacking, as reported in previous studies.22,23 | ||
| Fig. 1 Phase and structural characterization of synthesized samples. a) X-ray diffraction (XRD) patterns. b) Fourier transform infrared reflection (FTIR) spectra. c) Electron paramagnetic resonance (EPR) spectra. d) X-ray photoelectron spectra (XPS) of N 1s. e) Solid-state 13C magic angle spinning nuclear magnetic resonance (MAS NMR) spectra. f) Solid-state 1H MAS NMR spectra. g) The elemental content comparison between CN and CN–NH2 for confirming the –NH2 loss. The minimum units of atomic structures of h) CN–NH2 and i) CN. | ||
To confirm the absence of the –NH2 group in CN, Fourier transform infrared reflection (FTIR) spectra were used to analyze CN–NH2 and CN in the spectral range of 3600–500 cm−1 (Fig. 1b). In the FTIR spectra, clear N–H stretching vibration signals were observed at 3428 and 3491 cm−1, indicating the presence of the –NH2 group in CN–NH2.21,24 However, in the CN spectra, these signals were significantly weakened, suggesting the loss of the –NH2 group. This observation confirms that the –NH2 groups were successfully removed from the CN material.
Furthermore, other characteristic peaks were analyzed in the FTIR spectra. The peak at 809 cm−1 corresponds to the stretching vibration of the triazine ring, while the peaks at 1240, 1324, 1414, and 1560 cm−1 represent the stretching vibration of aromatic heterocycles.25–27 The presence of these peaks in the CN spectra indicates that the removal of the –NH2 group did not damage the main structure of the material. This suggests that the structural integrity of the CN material was maintained even after the removal of the –NH2 groups. These FTIR spectral analyses provide strong evidence for the absence of –NH2 groups in the CN material and demonstrate that the removal of these groups did not adversely affect the main structure of the material. The preservation of aromatic heterocycles in CN was also confirmed by Raman spectroscopy, which displayed the increased signal intensities of aromatic heterocycles relative to CN–NH2 (Fig. S4†). This unambiguously revealed that the removal of the –NH2 group did not damage the main structure. Moreover, electron paramagnetic resonance (EPR) confirmed a strong EPR signal at g = 2.003 for CN compared with CN–NH2, unambiguously unveiling the presence of unpaired electron signals caused by the removal of the –NH2 group (Fig. 1c and S5†).28,29
Previous studies revealed that the removal of the –NH2 group in carbon nitride would boost the electron migration from the bulk phase to the surface.22,30 Therefore, accurately quantifying the loss percentage of –NH2 groups in the CN structure was crucial. As shown in the XPS spectra (Fig. 1d and Table S1†), the content of C–N–H in CN decreased obviously compared with CN–NH2, demonstrating the successful removal of –NH2 groups. Moreover, magic angle spinning nuclear magnetic resonance (MAS NMR) experiments, employing C-labeled samples, confirmed the preservation of the primary structure as evidenced by the unaltered N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (∼163.5 ppm) and NC–N2 (∼154.5 ppm) groups (Fig. 1e).31 In contrast, MAS NMR experiments utilizing H-labeled samples demonstrated a substantial decrease in the peaks corresponding to the NH (∼8.9 ppm) and NH2 (∼4.6 ppm) groups after the removal of the –NH2 group (Fig. 1f).31 By precisely fitting the peak areas of CN–NH2 and CN independently in the MAS spectra, the percentage of NH2 was observed to decrease from 59.45% (CN–NH2) to 26.58% (CN) (Table S2†), surpassing the reduction observed in the XPS results, likely attributable to the surface and bulk disparities. Additionally, elemental analysis was performed to quantitatively determine the nitrogen and hydrogen element losses in CN. The loss of N and H in the CN sample was determined to be 27.41 and 58.73 μmol, respectively, with a nitrogen-to-hydrogen ratio close to 1
N (∼163.5 ppm) and NC–N2 (∼154.5 ppm) groups (Fig. 1e).31 In contrast, MAS NMR experiments utilizing H-labeled samples demonstrated a substantial decrease in the peaks corresponding to the NH (∼8.9 ppm) and NH2 (∼4.6 ppm) groups after the removal of the –NH2 group (Fig. 1f).31 By precisely fitting the peak areas of CN–NH2 and CN independently in the MAS spectra, the percentage of NH2 was observed to decrease from 59.45% (CN–NH2) to 26.58% (CN) (Table S2†), surpassing the reduction observed in the XPS results, likely attributable to the surface and bulk disparities. Additionally, elemental analysis was performed to quantitatively determine the nitrogen and hydrogen element losses in CN. The loss of N and H in the CN sample was determined to be 27.41 and 58.73 μmol, respectively, with a nitrogen-to-hydrogen ratio close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, affirming the loss of –NH2 groups (Fig. 1g). Notably, the –NH2 loss in CN was calculated to be 14.07% based on the –NH2 content criterion in CN–NH2. These results, in conjunction with the aforementioned measurements, provided a detailed atomic structure of CN–NH2 and CN, as depicted in Fig. 1h and i, respectively.
:2, affirming the loss of –NH2 groups (Fig. 1g). Notably, the –NH2 loss in CN was calculated to be 14.07% based on the –NH2 content criterion in CN–NH2. These results, in conjunction with the aforementioned measurements, provided a detailed atomic structure of CN–NH2 and CN, as depicted in Fig. 1h and i, respectively.
Morphology characterization
The synthesis of CN with a porous framework was accomplished through a solvothermal treatment and subsequent one-step calcination process (Fig. 2a). The elemental mapping images in Fig. 2b and c confirmed the uniform distribution of carbon and nitrogen elements in CN after the removal of the –NH2 group. The CN construct exhibited a porous framework structure, as revealed by field-emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), and scanning transmission electron microscopy (STEM) analysis (Fig. 2d–g and S6–S8†). The pore size (4.5 to 52 nm) measured from the TEM image indicated the presence of mesopores in CN (Fig. S9†). The nitrogen adsorption–desorption isotherms and pore size distribution analysis suggested that both CN–NH2 and CN exhibited mesopores. Moreover, it was found that the pore size of CN is larger than that of CN–NH2, further confirming the successful removal of –NH2 in CN (Fig. S10†). Notably, the inserted figure showed that the pore size of CN ranged from 3 to 50 nm, consistent with the pore size distribution results measured in the TEM image. The nanoparticle size of CN ranged from 160 to 710 nm, which was in sharp contrast with the nanoparticle size of CN–NH2, ranging from 1030 to 4160 nm (Table S3†). This significant difference in size can be attributed to the presence of –NH2 groups on the CN–NH2 surface. Additionally, the porous structure and smaller nanoparticle size of CN contributed to its significantly improved BET specific surface area of 10.51 m2 g−1, which was 5.2 times greater than that of CN–NH2 (Table S4†). Furthermore, the removal of the –NH2 group enhanced the CO2 adsorption capacity, with CN demonstrating nearly 242% higher CO2 adsorption compared to CN–NH2, as depicted in Fig. S11.† | ||
| Fig. 2 Physical morphology of CN with a porous frame. a) The synthesis process of CN with a porous frame. Elemental mapping: b) carbon element and c) nitrogen element. d) Field-emission scanning electron microscopy (FESEM) image. e and f) Transmission electron microscopy (TEM) images. g) Scanning transmission electron microscopy (STEM) image. | ||
CO2 photoreduction performance
The CO2 photoreduction performance of CN–NH2 and CN was assessed in a gas–solid system without the use of sacrificial agents or photosensitizers. To investigate the dependence of photocatalytic performance on catalyst mass, we conducted photocatalytic tests using CN with catalyst masses ranging from 0 to 65 mg (Fig. S12†). The results indicated a gradual enhancement of photocatalytic performance with increasing catalyst mass. We determined that a catalyst mass of 20 mg was appropriate, as it struck a balance between photocatalytic performance and the rationalized utilization cost of the catalyst. As depicted in Fig. 3a and S13.† CN–NH2 exhibited CH4 and CO production rates of 1.67 and 18.07 μmol g−1 h−1. In contrast, CN displayed CH4 and CO production rates of 1.44 and 58.83 μmol g−1 h−1. The CO production rate of CN (58.83 μmol g−1 h−1) was 3.26 times higher than that of CN–NH2 (18.07 μmol g−1 h−1), while the CO selectivity of CN (97.61%) exceeded that of CN–NH2 (91.52%) (Fig. S14†). As shown in Fig. 3b, under visible light illumination, the CO2 reduction performance of CN was enhanced, resulting in a CO production rate of 21.61 μmol g−1 h−1, which was 5.61 times higher than that of CN–NH2 (3.85 μmol g−1 h−1). Notably, its conversion rate and selectivity surpassed those of some recently reported photocatalysts in CO2 photoreduction (Table S5†), underscoring the superior application potential of the photocatalyst investigated in this study. Additionally, we evaluated the photocatalytic performance of CN by substituting CO2 with argon (Fig. 3a). The results indicated the presence of only trace amounts of CO, confirming that the products may have originated from the catalytic process. This finding was further supported by an isotope tracer experiment using 13CO2 instead of 12CO2, which solely detected signals of 13CH4 (m/z = 17) and 13CO (m/z = 29) under identical conditions, providing clear evidence that both CH4 and CO originated from CO2 molecules rather than the decomposition of the photocatalyst itself (Fig. 3c). The detection of the 16O2 and 18O2 signals indicated that the generated holes were involved in the oxidation of H2O, leading to the production of O2 (Fig. 3c and S15†). This process was accompanied by the generation of ˙OH (hydroxyl radicals) as well (Fig. S16†). Both O2 and ˙OH contribute to balancing the consumption of electrons and holes in the photocatalytic system. In addition to its significantly improved CO2 reduction performance, the stability of CN was investigated after five consecutive cycles. Only a slight deactivation of 16.64% was observed during the final cycle, indicating reliable photocatalytic stability (Fig. 3d). This finding was consistent with the unchanged XRD peaks observed in Fig. S17,† clearly demonstrating that CN could be regarded as an exceptional catalyst for CO2 reduction. To investigate the influence of light intensity, the apparent quantum yield (AQY) was further explored. The AQYs at 350, 365, 420, and 550 nm were determined to be 0.37, 0.19, 0.072, and 0.019%, respectively (Fig. S18†), aligning well with its light absorption curve, thereby indicating that the photocatalytic performance stemmed from its light absorption capacity.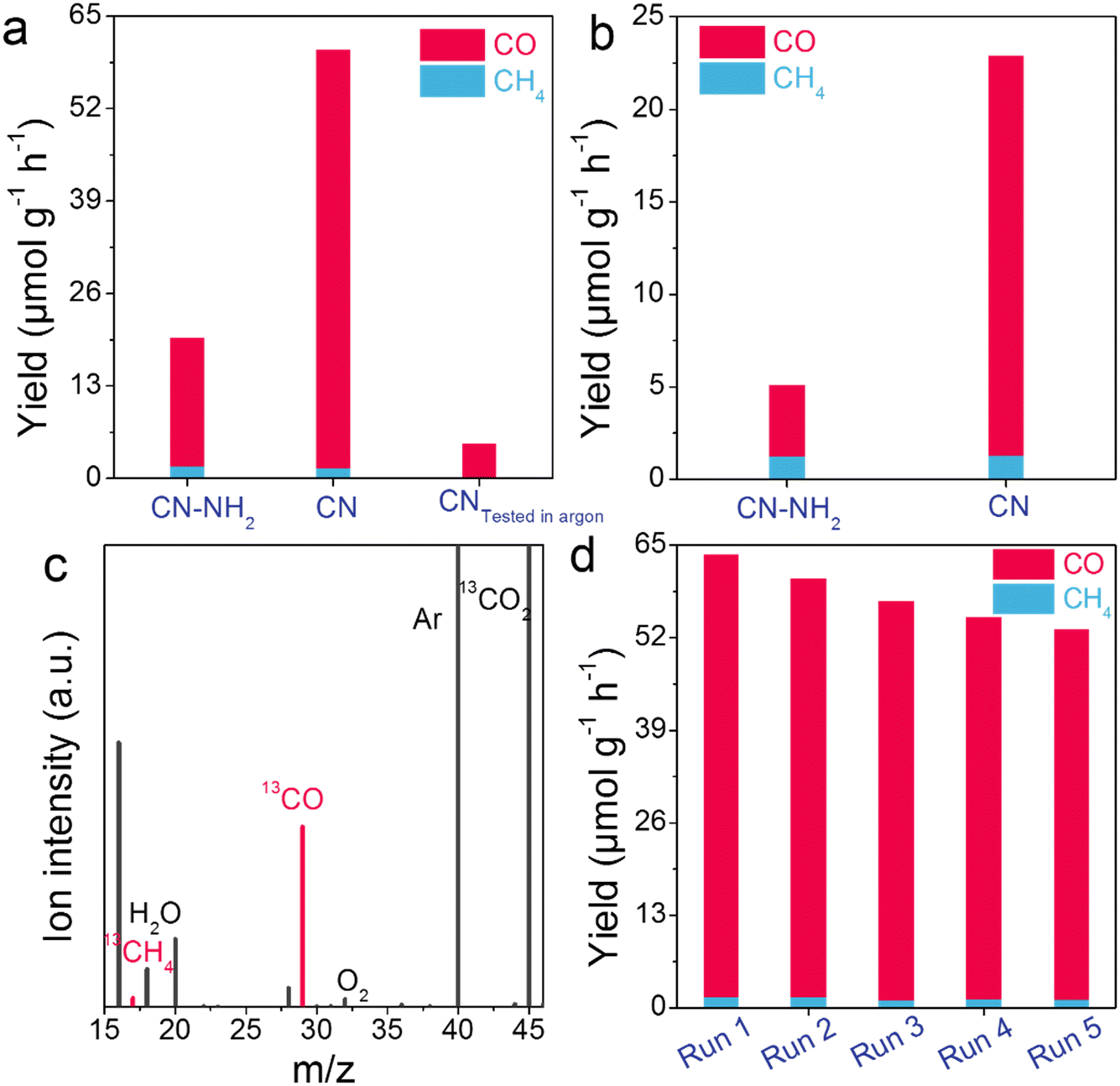 | ||
| Fig. 3 CO2 photoreduction performance. a) Photocatalytic performance of CN–NH2 and CN. b) Photocatalytic performance obtained under visible light. c) The isotope tracer experiment of 13CO2 photoreduction. d) Cycling tests of CO2 photoreduction on CN (the reactor was evacuated and refilled with new H2O and CO2 for the next cycling test). | ||
Mechanism discussion of CO2 photoreduction performance enhancement
The band structures of CN–NH2 and CN were investigated using Mott–Schottky analysis and UV-vis diffuse reflectance (UV-vis DRS) spectroscopy, as shown in Fig. S19† and 4a, respectively. Both materials exhibited redox potentials that were suitable for CO2 reduction and H2O oxidation. Previous studies have indicated that the efficiency of CO2 chemisorption and activation is influenced by the electron density at the catalytic site.9,32–35 Therefore, it is crucial to comprehensively evaluate the electron density at the catalytic sites of CN–NH2 and CN. This evaluation requires an examination of the light absorption capacity and charge separation efficiency, as these factors govern the electron density in the catalytic sites. In the following section, we will gradually investigate these aspects. | ||
| Fig. 4 CN showed superior light harvesting and photoelectric conversion performance, which could enhance the electron density at catalytic sites. a) UV-vis diffuse reflectance spectra. b) Time-resolved fluorescence spectra. c) Transient photocurrent response. d) Zeta potential test. e) The charge density difference (isosurfaces with the yellow zone reflected electron enrichment, while isosurfaces with the green zone reflected the electron depletion). | ||
As depicted in Fig. 4a and S20,† CN exhibited superior light harvesting ability in both UV and visible light regions, accompanied by a noticeable reduction in the band gap from 2.64 eV (CN–NH2) to 2.38 eV (CN). This signified that the removal of the –NH2 group enhanced the light harvesting ability compared to the original constructs, which may be attributed to the multiple reflections of light within the porous structure.36,37
The charge separation efficiency of CN–NH2 and CN was assessed using electrochemical impedance spectroscopy (EIS), time-resolved fluorescence (TRPL), and Bode-phase spectroscopy. The contact resistance values are presented in Fig. S21 and S22,† with the Nyquist curve of CN exhibiting a much smaller semicircle compared to CN–NH2, indicating lower contact resistance for CN. The reduction in contact resistance had a direct impact on significantly improving the charge transfer efficiency, as supported by the electronic lifetimes obtained from time-resolved transient fluorescence decay spectra. The average electronic lifetimes provided a comprehensive reflection of both defect-related nonradiative recombination processes (electron trapping by defects) and radiative recombination processes (excited-state electrons recombining with holes). The average electronic lifetime of CN (6.23 ns) exhibited a 136% increase compared to CN–NH2 (4.58 ns), indicating a substantial improvement in charge transfer efficiency (Fig. 4b and S23†). Bode-phase spectra further supported this enhanced charge transfer efficiency, with electronic lifetimes of 0.34 and 0.28 μs observed for CN and CN–NH2, respectively (Fig. S24†). These prolonged excited state lifetimes contributed to the substantially higher photocurrent in transient photocurrent response spectroscopy (Fig. 4c and S25†), as well as enhanced photovoltage (Fig. S26†). All these results indicated that the removal of the –NH2 group effectively enhanced the electron density at catalytic sites.
The charged characteristics of the samples were further examined by analysing the zeta potential. Zeta potential measurements revealed that CN exhibited a negative charge of −30.73 mV, indicating a higher electronegativity. This negative charge suggests that CN can contribute electrons more easily to activate CO2 (Fig. 4d). In comparison, CN–NH2 exhibited a zeta potential of only 0.34 mV. The difference in zeta potential indicates a higher concentration of electrons enriched around the catalytic sites of CN compared to CN–NH2 (Fig. 4e). This finding highlights the presence of Lewis basic sites on CN, which could contribute electrons to activate CO2, while the adsorbed CO2 acts as an electron acceptor. Consequently, the activation of CO2 relies on the Lewis base/acid interaction between the negatively charged CN and the CO2 electron acceptor.
As mentioned earlier,9,32–35 the electron concentration at catalytic sites plays a significant role in the chemisorption and activation of CO2 molecules, which are crucial steps in CO2 reduction. To assess the CO2 chemisorption ability of CN and CN–NH2, we conducted CO2 temperature-programmed desorption (CO2-TPD) analysis. As shown in Fig. 5a, CN exhibited two distinct and intense CO2-TPD signals, while negligible signals were observed in CN–NH2, indicating the superior efficiency of CO2 chemisorption in CN. Furthermore, within CN, the CO2 chemisorption signal at approximately 427 °C surpassed the signal at around 202 °C, suggesting stronger and more stable chemisorption at higher temperatures. These two signals indicated the presence of two different CO2 chemisorption sites in CN: the nitrogen atoms adjacent to the –NH2 group and the nitrogen atoms adjacent to the removed –NH2 group (Fig. 5a). Importantly, the temperature of the obtained CO2 chemisorption peak at around 200 °C in CN was slightly lower than that of CN–NH2, highlighting the crucial role of removing the –NH2 group at the boundary of the CN host. The decomposition temperature of CN (∼451 °C) exceeded that of the CO2 chemisorption signal at ∼427 °C, confirming that the latter signal originated from adsorbed CO2 rather than catalyst decomposition (Fig. S27†). Mass spectrometry analysis of the desorbed CO2 from CO2-TPD further supported the conclusion that CN adsorbed a significantly larger quantity of CO2 (Fig. 5b). XPS analysis of the O 1s region revealed the presence of adsorbed H2O, carbonates, and adsorbed CO2 on both CN–NH2 and CN (Fig. 5c).38–41 Importantly, the peak area corresponding to adsorbed CO2 on CN was 3.58 times higher than that on CN–NH2, underscoring the superior CO2 chemisorption performance of CN (Fig. 5c and Table S6†). Collectively, these results clearly demonstrated the enhanced CO2 chemisorption and activation achieved through electron enrichment at catalytic sites.
 | ||
| Fig. 5 Electron enrichment at the catalytic sites of CN enhanced the CO2 chemisorption and activation. a) CO2 temperature-programmed desorption (CO2-TPD) for revealing CO2 chemisorption. b) The collected CO2 mass spectra during the TPD process (the mass signals were collected at m/z = 44; m/z is the mass to charge ratio). c) XPS of O 1s for revealing CO2 chemisorption. d) Free energy of CO2 reduction revealed that CO2 activation lowered the energy barriers of CO2 reduction. | ||
The activation of CO2 was evaluated using density functional theory (DFT) calculations, which served to calculate the rate-determining step of CO2 reduction from CO2 to *COOH as a standard (Fig. 5d). The energy barrier for this rate-determining step in CN (0.96 eV) was substantially lower compared to CN–NH2 (2.16 eV), clearly indicating that the electron concentration at catalytic sites significantly reduces the CO2 activation energy, thus promoting the CO2 conversion rate under mild conditions.
Taking into account that the higher CO2 conversion efficiency may be attributed to the larger BET surface area, we have calculated the normalized performance, which is based on the surface area to CO2 reduction performance (Table S7†), to gain insight into the factors influencing the catalyst. The normalized performance of CN and CN–NH2 is 5.59 and 8.94 μmol h−1 m−2, respectively. This trend rules out the effect of the surface area and further emphasizes the significance of the –NH2 vacancy in CO2 adsorption and activation.
Both the higher CO2 chemisorption and activation abilities will lead to the generation of key intermediates during CO2 conversion. Hence, we employed in situ Fourier-transform infrared (in situ FTIR) spectroscopy during CO2 photoreduction. Interestingly, both CN and CN–NH2 constructs exhibited the presence of two distinct intermediates, namely *CHO and *CH3O,42 which were spectroscopically identified at wavenumbers of 1050 and 1075 cm−1, respectively (Fig. S28†). Notably, a strong bridge-bonded *CO signal was detected in CN–NH2,43 while a negligible *CO signal was detected in CN, suggesting that CN could desorb the *CO to produce CO efficiently.
The CO2 reduction pathways were examined to elucidate the correlation between the efficiency of CO2 reduction and catalytic sites (Fig. 6a and b). In the case of CN–NH2, initial adsorption of CO2 occurred on nitrogen atoms neighbouring the –NH2 group, followed by a reaction with a proton–electron pair to generate an intermediate species, *COOH. Subsequently, the *COOH species engaged in a further reaction with another proton–electron pair, resulting in the formation of the *CO intermediate and H2O. Finally, the *CO species were desorbed from the nitrogen atoms adjacent to the –NH2 group. CN exhibited an analogous CO2 reduction pathway to CN–NH2. Notably, only the nitrogen atoms adjacent to the removed –NH2 group served as catalytic sites in CN, whereas the nitrogen atoms adjacent to the –NH2 group functioned as catalytic sites in CN–NH2. Consequently, by considering the disparity in catalytic sites, it was possible to discern the relationship between CO2 reduction efficiency and catalytic sites. The CO production rates of CN and CN–NH2 were 58.83 and 18.07 μmol g−1 h−1, respectively. Thus, the production rate of CO achieved by CN was 1.00 × 106 μmol gcatalytic site−1 h−1, while CN–NH2 exhibited a production rate of 4.33 × 104 μmol gcatalytic site−1 h−1 for CO (Tables S8 and S9†). Moreover, the turnover number (TON) and turnover frequency (TOF) of CN were 23.11 times higher than those of CN–NH2 (Table S10†). These findings collectively suggested that the modulation of the –NH2 group enhanced the efficiency of CO2 reduction.
 | ||
| Fig. 6 Density functional theory (DFT) calculation revealing CO2 reduction pathways on: a) CN–NH2 and b) CN. | ||
Conclusions
In this study, we successfully fabricated a carbon nitride material with partially removed –NH2 groups (referred to as CN). Remarkably, CN exhibited superior light harvesting and photoelectric conversion capabilities compared to CN–NH2, leading to higher electron population enrichment at catalytic sites. This electron enrichment at catalytic sites played a pivotal role in enhancing the chemisorption and activation of CO2, effectively lowering the energy barrier associated with CO2 reduction. As a result of the enhanced CO2 chemisorption and activation, CN demonstrated exceptional photocatalytic performance surpassing CN–NH2. This investigation not only emphasized the significance of CO2 chemisorption and activation through catalytic site modulation, but also established a promising avenue for the development of novel catalysts aimed at boosting CO2 reduction.Author contributions
Debin Zeng: resources, software, methodology, validation, writing-original draft, and writing-review & editing. Xiting Wang: software. Chunguang Kuai: software. Zhuo Jiang: conceptualization, methodology, validation, and writing-review & editing. Yuzheng Guo: conceptualization, methodology, validation, and writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFA1502902) and the National Natural Science Foundation of China (No. 62174122, No. 22101217). The numerical calculations in this article were done on the supercomputing system in the Supercomputing Center of Wuhan University.References
- Y. L. Chen, Y. Qu, X. Zhou, D. Z. Li, P. Xu and J. M. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 41527–41537 CrossRef CAS PubMed.
- Y. L. Chen, X. Q. Liu, L. Hou, X. R. Guo, R. W. Fu and J. M. Sun, Chem. Eng. J., 2020, 383, 123132 CrossRef CAS.
- Y. L. Chen, Y. Qu, P. Xu, X. Zhou and J. M. Sun, J. Colloid Interface Sci., 2021, 601, 326–337 CrossRef CAS PubMed.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, E. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- A. E. Green, J. Justen, W. Schöllkopf, A. S. Gentleman, A. Fielicke and S. R. Mackenzie, Angew. Chem., 2018, 130, 15038–15042 CrossRef.
- J. M. Weber, Int. Rev. Phys. Chem., 2014, 33, 489–519 Search PubMed.
- Y. H. Cao, L. Guo, M. Dan, D. E. Doronkin, C. Q. Han, Z. Q. Rao, Y. Liu, J. Meng, Z. A. Huang, K. B. Zheng, P. Chen, F. Dong and Y. Zhou, Nat. Commun., 2021, 12, 1675 Search PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- R. R. Wang, P. J. Yang, S. B. Wang and X. C. Wang, J. Catal., 2021, 402, 166–176 CrossRef CAS.
- G. C. Huang, Q. Niu, J. W. Zhang, H. M. Huang, Q. S. Chen, J. H. Bi and L. Wu, Chem. Eng. J., 2022, 427, 131018 CrossRef CAS.
- G. J. Chen, F. Wei, Z. R. Zhou, B. Su, C. Yang, X. F. Lu, S. B. Wang and X. C. Wang, Sustainable Energy Fuels, 2023, 7, 381–388 RSC.
- G. J. Chen, Z. R. Zhou, B. F. Li, X. H. Lin, C. Yang, Y. X. Fang, W. Lin, Y. D. Hou, G. G. Zhang and S. B. Wang, J. Environ. Sci., 2023 DOI:10.1016/j.jes.2023.05.028.
- W. Chananchana, P. Nuengmatcha and S. Chanthai, Orient. J. Chem., 2017, 33, 2920 CrossRef CAS.
- H. W. Gu, Y. B. Guo, S. Y. Wong, Z. Zhang, X. P. Ni, Z. X. Zhang, W. X. Hou, C. B. He, V. P. W. Shim and X. Li, Microporous Mesoporous Mater., 2013, 170, 226–234 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS PubMed.
- J. P. Perdew and K. Burke, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- Z. A. Lan, G. G. Zhang and X. C. Wang, Appl. Catal., B, 2016, 192, 116–125 CrossRef CAS.
- Y. Y. Kang, Y. Q. Yang, L. C. Yin, X. D. Kang, L. Z. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- S. E. Guo, Z. P. Deng, M. X. Li, B. J. Jiang, C. G. Tian, Q. J. Pan and H. G. Fu, Angew. Chem., 2016, 128, 1862–1866 CrossRef.
- G. Q. Li, N. Yang, W. L. Wang and W. F. Zhang, J. Phys. Chem. C, 2009, 113, 14829–14833 CrossRef CAS.
- J. J. Chen, Z. Y. Mao, L. X. Zhang, D. J. Wang, R. Xu, L. J. Bie and B. D. Fahlman, ACS Nano, 2017, 11, 12650–12657 CrossRef CAS PubMed.
- W. N. Xing, W. G. Tu, M. Ou, S. Y. Wu, S. M. Yin, H. J. Wang, G. Chen and R. Xu, ChemSusChem, 2019, 12, 2029–2034 CrossRef CAS PubMed.
- W. Liu, C. J. Song, M. P. Kou, Y. Y. Wang, Y. Deng, T. Shimada and L. Q. Ye, Chem. Eng. J., 2021, 425, 130615 CrossRef CAS.
- M. H. Liu, C. G. Wei, H. Y. Zhuzhang, J. M. Zhou, Z. M. Pan, W. Lin, Z. Y. Yu, G. G. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113389 CrossRef CAS PubMed.
- C. D. Lv, Y. M. Qian, C. S. Yan, Y. Ding, Y. Y. Liu, G. Chen and G. H. Yu, Angew. Chem., 2018, 130, 10403–10407 CrossRef.
- Y. H. Li, M. L. Gu, X. M. Zhang, J. J. Fan, K. L. Lv, S. A. C. Carabineiro and F. Dong, Mater. Today, 2020, 41, 270–303 CrossRef CAS.
- F. Y. Liu, Y. Tong, C. F. Li and X. L. Liu, J. Phys. Chem. Lett., 2021, 12, 10359–10365 CrossRef CAS PubMed.
- H. Li, B. C. Zhu, S. W. Cao and J. G. Yu, Chem. Commun., 2020, 56, 5641–5644 RSC.
- Y. Y. Sun, G. H. Li, W. Sun and X. X. Zhou, J. CO2 Util., 2023, 67, 102344 CrossRef CAS.
- U. J. Etim, C. C. Zhang and Z. Y. Zhong, Nanomaterials, 2021, 11, 3265 CrossRef CAS PubMed.
- L. L. Lin, K. Wang, K. Yang, X. Chen, X. Z. Fu and W. X. Dai, Appl. Catal., B, 2017, 204, 440–455 CrossRef CAS.
- D. B. Zeng, X. T. Wang, Y. S. Liu, D. M. Liu, Z. Y. Zhang, L. F. Fei, J. Robertson, C. G. Kuai and Y. Z. Guo, ACS Sustainable Chem. Eng., 2022, 10, 5758–5769 CrossRef CAS.
- D. B. Zeng, L. T. Wang, R. A. Du, Z. Wu, X. T. Wang, C. G. Kuai, Y. Z. Guo and C. L. Yu, Mater. Chem. Phys., 2023, 128021 CrossRef CAS.
- M. Favaroa, H. Xiao, T. Cheng, W. A. Goddard III, J. Yano and E. J. Crumlin, Proc. Natl. Acad. Sci., 2017, 114, 6706–6711 CrossRef PubMed.
- X. Y. Deng, A. Verdaguer, T. Herranz, C. Weis, H. Bluhm and M. Salmeron, Langmuir, 2008, 24, 9474–9478 CrossRef CAS PubMed.
- C. Zhang, M. Y. Zhang, Y. Li and D. M. Shuai, Appl. Catal., B, 2019, 248, 11–21 CrossRef CAS.
- B. Eren, C. Heine, H. Bluhm, G. A. Somorjai and M. Salmeron, J. Am. Chem. Soc., 2015, 137, 11186–11190 CrossRef CAS PubMed.
- X. D. Li, Y. F. Sun, J. Q. Xu, Y. J. Shao, J. Wu, X. L. Xu, Y. Pan, H. X. Ju, J. F. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- T. Wang, L. Chen, C. Chen, M. T. Huang, Y. J. Huang, S. J. Liu and B. X. Li, ACS Nano, 2022, 16, 2306–2318 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS of Br 3d, XRD spectra, Raman spectra, EPR spectra, table of peak areas of C–N–H, N–(C)3 and C–NC based on XPS, table of peak areas of NH and NH2 groups based on 1H MAS NMR, SEM, TEM, HRTEM, STEM, pore size distribution, nitrogen adsorption–desorption isotherm, summary table of BET specific surface areas, pore sizes, and nanoparticle sizes, CO2 adsorption isotherms, photocatalytic performance, comparison table of photocatalytic performance, redox potential, UV-vis DRS, EIS, time-resolved transient fluorescence decay spectra, transient photocurrent response, photovoltage, thermogravimetry analysis, CO2 mass spectra, and table of peak areas of adsorbed H2O, carbonates and adsorbed CO2 based on XPS. See DOI: https://doi.org/10.1039/d3cy01585h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |