
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into size effects of Pt/Al2O3 catalysts on hydrogen production from methylcyclohexane dehydrogenation†‡
Yiqing
Wu
a,
Yuanyuan
Li
a,
Xinbin
Yu
a,
Xiaohan
Ma
b,
Matthew
Boebinger
b,
Juliane
Weber
a and
Zili
Wu
 *ab
*ab
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. E-mail: wuz1@ornl.gov
bCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA
First published on 20th December 2023
Abstract
Liquid organic hydrogen carriers (LOHCs) have been considered as a promising technique for hydrogen storage and transportation. In this study, we investigated the impacts of Pt size on the catalytic performance of Pt/Al2O3 catalysts for the abstraction of H2via dehydrogenation of methylcyclohexane (MCH), one of the most desired LOHC candidates. As the Pt loading increases from 0.05% to 5%, Pt species transition from nanoclusters to well-defined nanoparticles with the Pt size increasing from ∼0.7 nm to 1.7 nm. During MCH dehydrogenation, the turnover frequency (TOF) displays a volcano dependence on the Pt size, as the 0.1% Pt/Al2O3 sample with medium Pt size exhibits the highest atomic efficiency. Furthermore, the catalyst stability of Pt/Al2O3 samples is also dependent on the Pt size with the small Pt size in the 0.05% Pt/Al2O3 sample leading to fast catalyst deactivation due to the accumulation of coke on the Pt active sites. In summary, this study underscores the importance of Pt size to the activity and stability of Pt/Al2O3 in MCH dehydrogenation, providing new insights into the catalyst design for LOHC applications.
1. Introduction
In the global energy transition from fossil fuels to renewable energy, wind and solar energies have been steadily increasing their shares.1,2 Due to their intermittent characteristics and remote production location, efficient storage and transportation of renewable energy has become the prerequisite to fully unlock their potential towards the zero carbon target.1 A variety of energy storage approaches, such as batteries, hydrogen, hydro-reservoirs, thermal reservoirs, flywheels, etc., have been proposed to tackle this challenge.3 Among these solutions, hydrogen has long been considered as an efficient, clean energy carrier for renewable energy with water the only product from direct combustion or conversion to electricity through fuel cells.1 Despite an excellent gravimetric energy density of 120 MJ kg−1, three times that of gasoline, hydrogen has a rather low volumetric energy density at ambient conditions due to its low volume density, slowing the development of hydrogen transportation and storage.4,5Current commercial techniques are based on high-pressure compression and cryogenic liquefaction, both of which are highly energy-intensive and pose significant safety concerns. These drawbacks prompt the development of physical storage of hydrogen in mediums like porous carbon and metal organic frameworks (MOFs), and chemical storage in metal hydride, liquid organic hydrogen carriers (LOHCs), methanol, NH3, etc.6–8 Among them, LOHCs have emerged as a promising approach due to their relatively low cost and excellent compatibility with current fossil fuel infrastructure.1,9 LOHCs, consisting of a pair of hydrogen-rich organic compound and its corresponding dehydrogenated hydrogen-lean counterpart, such as cyclohexane/benzene, methylcyclohexane (MCH)/toluene and perhydro-dibenzyltoluene/dibenzyltoluene, remain liquid in the normal operation temperature range, allowing for the utilization of existing fossil fuel infrastructures and hence significantly reducing implementation cost.
Hydrogen is stored in LOHCs during an exothermic hydrogenation process and released in an endothermic dehydrogenation process, both of which are catalytic processes. The hydrogen storage capacity of LOHCs is generally above 6 wt% and up to 7.2 wt% in the cyclohexane–benzene pair, making them economically feasible for hydrogen storage and transportation.2,9,10 Among various LOHC candidates, the methylcyclohexane (MCH)/toluene pair with 6.2 wt% hydrogen storage capacity shows a great potential due to its desirable physical properties, including abundant production capability, low melting point and low toxicity.9,11
Compared to the exothermic hydrogenation of MCH, its dehydrogenation is endothermic, requiring high energy input, and is prone to fast catalyst deactivation. A variety of catalysts have been explored to address these issues, which can be categorized into supported noble metal and transition metal catalysts. Noble metals, such as Pt, Pd and Ir, supported on various supports have been employed for MCH dehydrogenation due to their high MCH conversion and excellent selectivity to toluene.12–14 To reduce the amount of noble metal, a second metal can be added to form bimetallic catalysts, such as Pt–Mo, Pt–Fe and Pt–Cu, enabling enhanced resistance to coking and higher selectivity to desired toluene.12,15,16 Transition metals, notably Ni, have also been explored, which generally show inferior activity and lower toluene selectivity compared to noble metal catalysts.17–19 Among them, Pt supported on γ-alumina (Pt/γ-Al2O3) often demonstrates the highest activity and stability. γ-Alumina is a versatile non-reducible support commonly employed at both industrial and laboratory scale for its high surface area and robust thermal stability. Depending on the loading, treatment conditions and specific properties of the alumina support, a variety of Pt species can exist on the γ-alumina, including single atoms, nanoclusters, and nanoparticles, as depicted in Scheme 1.20–23
 | ||
| Scheme 1 Pt supported on Al2O3 as single atoms, 2D nanoclusters and 3D nanoparticles. | ||
In recent years, intensive studies on single-atom catalysts have demonstrated their excellent catalytic properties for many chemical reactions,24–27 including hydrocarbon dehydrogenation. For example, during the propane dehydrogenation reaction, the turnover frequency (TOF) of Pt species atomically dispersed on the Al2O3 support is 3-fold and 7-fold higher than that of subnanometer-sized clusters and nanoparticles, respectively.23 On the other hand, the latest studies suggest that compared to Pt single atoms, 2D Pt nanoclusters supported on graphene or Pt nanoparticles supported on Al2O3 exhibit better activity and stability in LOHC dehydrogenation.20,28 These discrepancies prompt us to study the elusive effects of Pt size on the catalytic performance of industry-relevant Pt/Al2O3 catalysts in the MCH dehydrogenation. Herein, we characterized the nature of Pt active sites on the Al2O3 support using high-resolution electron microscopy and in situ DRIFTS. Both the activity and the stability of Pt/Al2O3 catalysts are found to be dependent on the Pt size, opening new avenues to optimize the catalyst design for MCH dehydrogenation.
2. Experiments
2.1 Catalyst preparation
Pt/Al2O3 catalysts were prepared via an incipient impregnation method with tetraamine platinum nitrate (Sigma Aldrich no. 267740, >99.5 wt%) as the Pt precursor. Al2O3 with a surface area of ∼120 m2 g−1 was purchased from Sigma Aldrich. In a typical synthesis, a certain amount of tetraamine platinum nitrate was dissolved in 0.2 mL deionized water aided by sonication for 30 mins. The solution was then added dropwise into 1 g Al2O3 support under continuous stirring with a spatula to disperse the Pt precursors. The resulting wet sample was dried at 100 °C overnight and calcined in air at 500 °C for 4 hours. A series of Pt/Al2O3 samples with various Pt loadings were synthesized using this method and are named by their respective Pt loading. For example, 1 wt% Pt supported on Al2O3 is designated as 1Pt.2.2 Catalyst characterization
The Pt concentration in these samples was determined via inductively coupled plasma atomic emission spectroscopy (ICP-AES) performed at Galbraith Inc. (Knoxville, TN). Nitrogen physisorption at 77 K was conducted using a Micromeritics Gemini 275 instrument with the surface area determined via the Brunauer–Emmett–Teller (BET) method. Powder X-ray diffraction (XRD) measurements were performed on a Shimadzu powder X-ray diffractometer with CuKα radiation (k = 1.5406 Å). The morphology and particle size of Pt species on the Al2O3 support were examined with transmission electron microscopy. A FEI Titan aberration-corrected transmission electron microscope was used in high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) mode at 300 kV. Pt/Al2O3 samples were prepared as suspensions in isopropanol, sonicated for 10 mins and drop casted on lacy carbon grids. To reduce sample contamination from isopropanol, the samples were baked overnight at 150 °C. The average diameter of the Pt species, namely the Pt size, was measured by counting over 50 particles.The structure and types of Pt species were examined using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) with CO as the probe molecule, conducted on a Bruker Tensor 27 FTIR spectrometer equipped with an in situ reaction chamber (Harrick Scientific Products Inc.). Prior to the adsorption of CO, the Pt/Al2O3 catalysts were pretreated under a flow of 4% H2/Ar for one hour at 400 °C and then cooled to 25 °C, at which temperature the background spectrum was acquired. Adsorption of CO was carried out by flowing 2% CO/He to the sample at a flow rate of 30 mL min−1 until saturation, followed by a purge for 30 mins to remove the physisorbed and weakly adsorbed CO. IR spectra were periodically acquired during the CO adsorption and purge process, from which the pre-acquired background was subtracted to obtain the IR spectra of adsorbed species on the surface.
The types of coke formed in the MCH dehydrogenation reaction were determined by temperature programmed oxidation (TPO) and Raman spectroscopy. The TPO was conducted after the reaction with the residual MCH and products were purged by an Ar flow (50 mL min−1) for 2 hours. The spent catalyst was heated from room temperature to 700 °C at a ramp rate of 10 °C min−1 under a flow of 5% O2/He at a flow rate of 50 mL min−1. The oxidation products were continuously monitored using a mass spectrometer (GSD-300, OmniStar). Raman spectra were collected on a multiwavelength Raman system (Princeton Instruments Acton Trivista 555) at room temperature. A 244 nm laser excitation and a UV-enhanced liquid N2-cooled CCD detector (Princeton Instrument) were used. For each measurement, the exposure time was 30 seconds and the number of accumulations was 2 on a moving stage.
2.3 Catalytic tests
MCH dehydrogenation was carried out on an AMI 200 apparatus. In a typical experiment, 30 mg catalyst was loaded into a quartz tube with the catalyst stabilized by quartz wool on both the upper and lower sides of the sample. Prior to the reaction, the catalyst was pretreated under a flow of 30 mL min−1 4% H2/Ar at 400 °C for one hour at a ramping rate of 10 °C min−1. Upon cooling to 200 °C, MCH was injected at a flow rate of 0.006 mL min−1 to a flow of 6.5 mL min−1 Ar (14 kPa MCH) and the resulting mixed gas was introduced to the catalysts to initiate the reaction. The catalyst temperature was kept at 200 °C for 3 hours to reach steady-state conversion and was then increased stepwise to 400 °C at intervals of 40 °C, staying at each step for one hour. The MCH reactant and products were monitored by a gas chromatograph (GC7920, Agilent) equipped with both FID and TCD detectors, using an HP-5 column (Agilent) and a ShinCarbon packed column (Restek), respectively. For the long-time stability test, the catalyst was pretreated using the same method but cooled to 320 °C, at which temperature the MCH dehydrogenation reaction was carried out continuously for 20 hours. As shown in the calculation in the ESI,‡ both mass and heat transfer limitations are minimal in our reaction tests.3. Results and discussion
3.1 Morphology and texture properties of Pt/Al2O3 catalysts
The Pt concentration and surface area of Pt/Al2O3 catalysts are shown in Table 1. The concentrations of Pt in these samples determined via ICP-AES are 0.04 wt%, 0.10 wt%, 0.45 wt%, 0.94 wt% and 4.50 wt% for the 0.05Pt, 0.1Pt, 0.5Pt, 1Pt and 5Pt samples, respectively, matching well with the nominal loading amounts of Pt and thus indicating negligible loss during the sample preparation and calcination processes. The surface areas of these catalysts are similar at 120–130 m2 g−1, reflecting high dispersion of Pt species without severe pore blocking in the porous Al2O3 support.| Sample | 0.05Pt | 0.1Pt | 0.5Pt | 1Pt | 5Pt |
|---|---|---|---|---|---|
| Pt concentration (wt%) | 0.04 | 0.10 | 0.45 | 0.94 | 4.50 |
| Bet surface area (m2 g−1) | 124 | 122 | 124 | 130 | 122 |
| Pt dispersion (%) by STEM | ∼100 | ∼100 | ∼100 | 97 | 67 |
Fig. 1 shows the XRD patterns of as-calcined Pt/Al2O3 samples. Only the feature peaks of γ-alumina are observed while those associated with bulk PtO2 (33.9° 2θ) or bulk Pt (39.8° 2θ) are not detected,29,30 indicating the absence of Pt or PtO2 nanoparticles or their sizes are less than the detection limit (around 2–3 nm), which further corroborates the high dispersion of Pt on the Al2O3 support.
 | ||
| Fig. 1 XRD patterns of as-calcined Pt/Al2O3 samples. | ||
For Pt/Al2O3 samples after calcination at elevated temperature in air, Pt can exist as single atoms, nanoclusters, and nanoparticles.31,32 However, upon H2 activation at elevated temperatures, the Pt species are expected to change. The HAADF-STEM images in Fig. 2 show the size and dispersion of Pt on the Al2O3 support after H2 activation at 400 °C for 1 hour. For all samples, only Pt clusters or particles can be observed. For the 0.05Pt and 0.1Pt samples shown in Fig. 2a and b, Pt atoms on the Al2O3 support display no clear lattice structure and loosely spread on the support, which can be categorized as nanoclusters.31,32 By measuring the diameter of these Pt nanoclusters, the Pt size is determined to be ∼0.6 nm for the 0.05Pt sample and ∼0.7 nm for the 0.1Pt sample (Fig. 2e), with the latter sample displaying a higher density of nanoclusters on the support due to its higher Pt loading. As the Pt loading increases to 1%, Pt nanoparticles with well-defined lattice are readily observed, as displayed in the red circle in Fig. 2c, and the average Pt size increases to 1.2 nm. A further increase of Pt loading to 5% leads to more well-defined nanoparticles with the average Pt size increasing to 1.7 nm. The size of most Pt species, even in the 5Pt sample, stays below 3 nm, in accordance with the absence of Pt or PtO2 feature peaks in their XRD patterns, confirming the high dispersion of Pt species on the Al2O3 support. Based on the STEM images, we estimated the Pt dispersion in different samples and the results are summarized in Table 1. For the 1Pt and 5Pt samples, by assuming a spherical model for the well-defined Pt nanoparticles, the Pt dispersion was calculated to be 97% and 67%, respectively.33 Given the smaller Pt size in the 0.05Pt and 0.1Pt samples, the Pt dispersions in these samples are anticipated to be ∼100%, implying that almost all of the Pt atoms are available for reaction. Although the Pt size in the 0.5Pt sample was not measured, its Pt dispersion is expected to be higher than that of the 1Pt sample, close to 100%.
 | ||
| Fig. 2 HAADF-STEM images of 0.05Pt (a), 0.1Pt (b), 1Pt (c) and 5Pt (d) samples after H2 activation at 400 °C for one hour and the Pt size distribution determined using these images (e) with the error bar representing the standard deviation of the size measurement. Note that the red circles in (c) and (d) exhibit the well-defined Pt crystal lattice. | ||
To further identify Pt species and examine the interaction between Pt and the Al2O3 support, CO-DRIFTS was carried out for the Pt/Al2O3 samples, as shown in Fig. 3. CO has been extensively employed as a probe molecule to reveal the dispersion and structure of supported Pt species. Upon H2 activation, two broad CO IR bands are observed, one centered at 2053 cm−1 and the other one centered at 1810 cm−1 (Fig. 3a and b). Within the first IR band, a shoulder peak is observed at ∼2110 cm−1 for all Pt/Al2O3 samples, which can be assigned to CO adsorbed on Pt single atoms supported on Al2O3.23,34 Given the rather weak feature of this IR band, the percentage of Pt single atoms is expected to be very small, which is in accordance with their absence under STEM as shown in Fig. 2(a–d). In the literature, the first IR band centered at 2053 cm−1 has been frequently assigned to linear adsorbed CO on reduced Pt species, with the second IR band centered at 1810 cm−1 to bridge-adsorbed CO.23,35–37 Although the correlation between the wavenumber of adsorbed CO species and the Pt species, e.g. nanoclusters and nanoparticles, remains under debate, the IR band at 1810 cm−1 has often been reported to originate from the CO bridge-adsorbed on large Pt nanoparticles.23,35,36 For the 0.05Pt and 0.1Pt samples, the IR band at 1810 cm−1 is rather weak (Fig. 3b), which increases substantially and becomes rather evident for the 1Pt and 5Pt samples. This trend is consistent with the observation under STEM that with increasing Pt loading, Pt transitions from nanoclusters with a small Pt size to Pt nanoparticles with a large Pt size.
 | ||
| Fig. 3 CO-DRIFTS spectra of Pt/Al2O3 samples (a) and enlarged spectra for the two low loading samples (b). | ||
Compared to ex situ STEM that may miss key features observable only under real reaction conditions, the CO-DRIFTS experiment with the in situ reduction pretreatment complements the microscopy study by confirming the observation of nanoclusters and nanoparticles as well as demonstrating the presence of Pt single atom sites not observed by STEM. In summary, the Pt size and morphology are studied for Pt/Al2O3 samples with Pt loading ranging between 0.05 wt% and 5 wt%. At low Pt loadings of 0.05 wt% and 0.1 wt%, the Pt species present as Pt nanoclusters loosely spread on the Al2O3 support with Pt size around 0.6–0.7 nm, leading to full exposure of Pt atoms to reactants. In comparison, at high loading, well-defined Pt nanoparticles emerge on the Al2O3 surface with Pt size reaching 1.2–1.7 nm, leading to a portion of Pt atoms enclosed in the particles.
3.2 MCH dehydrogenation reactions
Light-off MCH dehydrogenation tests were carried out over these Pt/Al2O3 samples with different Pt sizes in the temperature range of 200–400 °C, which are shown in Fig. 4a together with the equilibrium conversion. The reaction plot can be categorized into two temperature regimes: 200–320 °C, where the MCH conversion ramps up with the increase of temperature, and 320–400 °C, where the MCH conversion either plateaus or declines with increasing temperature. In the first temperature regime, MCH conversion increases with the increase of Pt concentration until reaching 1% Pt loading, as the 1Pt sample shows the same MCH conversion as the 5Pt sample. It is worth noting that in this temperature range, the MCH conversion of Pt/Al2O3 samples, even for the high Pt loading samples, is significantly lower than the equilibrium conversion. In the second temperature regime (320–400 °C), MCH conversion is 100% for 0.5Pt, 1Pt and 5Pt samples, equal to the equilibrium conversion. For the 0.1Pt sample, the MCH conversion reaches ∼83% at 320 °C but decreases slightly to 75% at 400 °C, indicating a minor deactivation at high temperature. In contrast, the MCH conversion of the 0.05Pt sample increases to 50% at 320 °C but then rapidly decreases to 20% at 400 °C, reflecting a severe loss of available active sites due to catalyst sintering or coking. Such an evident deactivation is also observed for the Pt/Al2O3 sample with 0.025% Pt loading (Fig. S1‡). Note that the absence of conversion decrease for the 0.5Pt, 1Pt and 5Pt samples does not imply no loss of active sites. Compared to 0.1Pt and 0.05Pt samples with low Pt loading, the relatively large amount of Pt in these high loading samples can maintain the complete conversion of MCH despite any potential loss of available active Pt sites.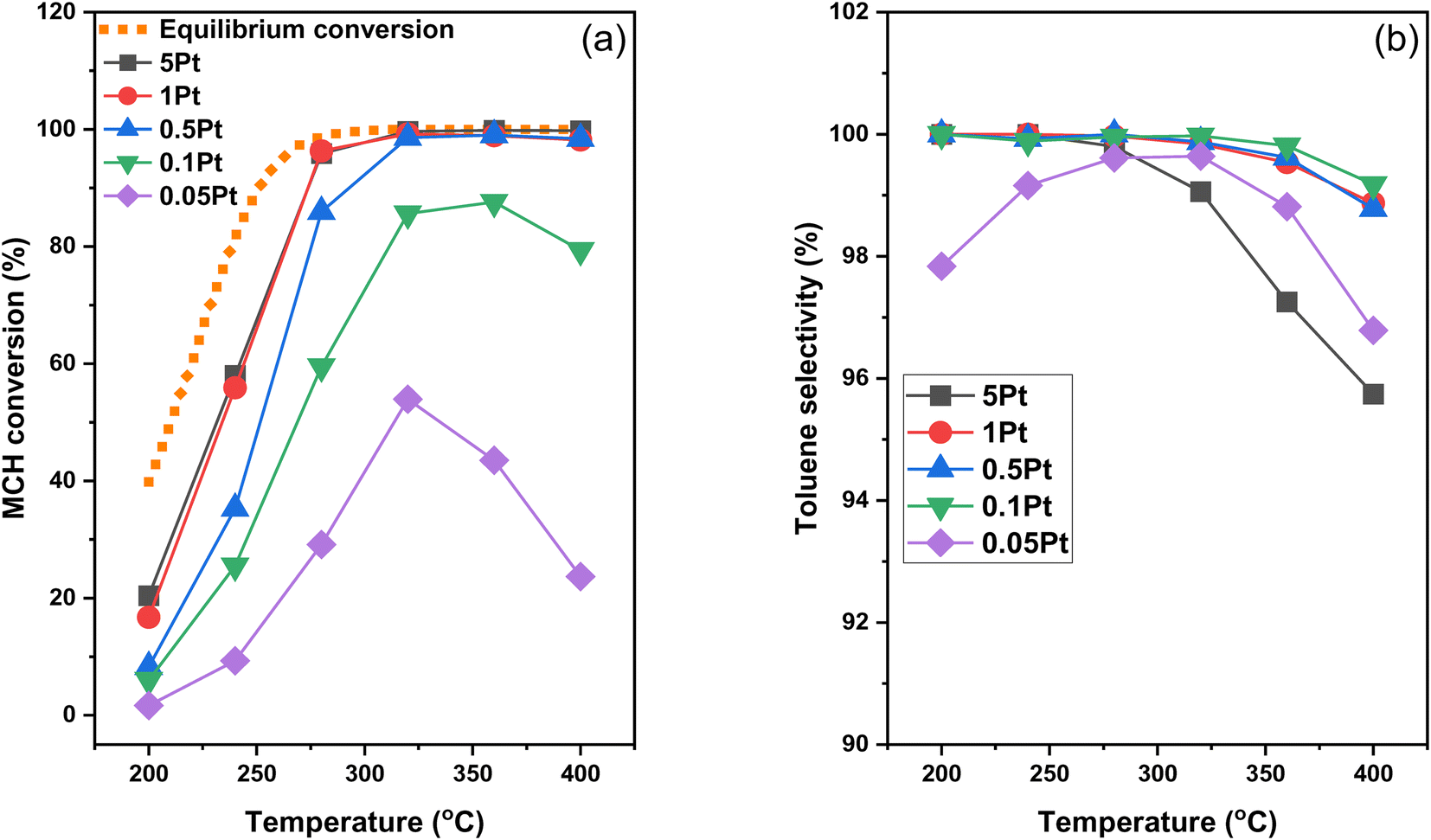 | ||
| Fig. 4 MCH conversion (a) and toluene selectivity (b) over Pt/Al2O3 catalysts with different Pt loadings at different reaction temperatures. Reaction conditions: 30 mg catalyst, 6.5 mL min−1 Ar and 0.006 mL min−1 MCH injected using a syringe pump. The equilibrium conversion is calculated based on the equilibrium parameters obtained from previous literature38 with the equilibrium constant of 3.6 × 109 kPa3 at 650 K for the dehydrogenation of MCH to toluene and hydrogen. | ||
Fig. 4b shows the selectivity to toluene in the MCH dehydrogenation, which is essential for the MCH–toluene pair to serve as durable LOHCs in hydrogen transportation and storage. This is because practical LOHCs need to sustain hundreds of hydrogenation–dehydrogenation cycles without being converted into side products, which otherwise can affect the dehydrogenation/hydrogenation performances and require high energy input to be separated out. The toluene selectivity is higher than 99% for 0.1Pt, 0.5Pt and 1Pt samples across the whole temperature range. In comparison, the 0.05Pt sample shows relatively low toluene selectivity at both ends of this temperature range, yielding side products such as methylcyclohexene, benzene and xylene,11,39 whereas the 5Pt sample exhibits slightly low selectivity at high temperature.
To quantify the atomic efficiency of Pt active sites, the turnover frequency (TOF) of Pt/Al2O3 catalysts was calculated at 200 °C and 240 °C based on the Pt dispersion determined by the STEM images (Table 1). Note that the MCH conversion at 200 °C is controlled to less than 20% for all samples to ensure that the reaction occurs under the kinetic differential regime, allowing the evaluation of the intrinsic activity of Pt active species. At 200 °C, TOF increases from 1766 mol H2 (mol Pt h)−1 for the 0.05Pt sample to 3211 mol H2 (mol Pt h)−1 for the 0.1Pt sample, and then decreases substantially to 922 and 326 mol H2 (mol Pt h)−1, respectively, for the 1Pt and 5Pt samples. The same trend is observed at 240 °C, although the MCH conversion at this temperature is higher than 20% for most samples, likely out of the kinetic differential regime. Nevertheless, the volcano trends of TOF at both 200 and 240 °C clearly demonstrate the dependence of atomic efficiency on the Pt loading as the 0.1Pt sample with the medium Pt size shows the highest atomic efficiency. Since the TOF is referenced against the exposed Pt sites determined by STEM, the availability of Pt sites cannot account for the huge TOF difference among them as 0.1Pt samples exhibit around 3 and 9 times higher TOF than the 1Pt and 5Pt samples, respectively.
Recent studies on graphene supported Pt and Pd in LOHC dehydrogenation suggest that the increase of Pt size leads to stronger binding of dehydrogenated products on the active sites, which blocks the access of reactants, resulting in fast deactivation.28,40 To check whether this blocking effect applies in this study, we carried out MCH dehydrogenation at 200 °C with both MCH reactant and toluene product in the feed at a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio over 0.1Pt and 1Pt samples (Fig. S2‡). The co-feed of toluene leads to ∼35% activity loss in the first 2 hours for both samples, after which the MCH conversion becomes relatively stable, reflecting similar impacts of toluene on the Pt/Al2O3 catalysts. Hence, the poisoning effect of toluene is likely not the reason for the much higher TOF of the 0.1Pt sample compared to the 1Pt sample.
:1 ratio over 0.1Pt and 1Pt samples (Fig. S2‡). The co-feed of toluene leads to ∼35% activity loss in the first 2 hours for both samples, after which the MCH conversion becomes relatively stable, reflecting similar impacts of toluene on the Pt/Al2O3 catalysts. Hence, the poisoning effect of toluene is likely not the reason for the much higher TOF of the 0.1Pt sample compared to the 1Pt sample.
Given the different morphologies and Pt sizes within our samples, the heterogeneity of Pt sites cannot be ignored, which is a common cause for the activity difference observed on nanoparticles with different sizes and morphologies as the activity of Pt sites depends on their specific local environments, such as the edge, step and terrace.37,41,42 For the dehydrogenation of perhydro-dibenzyltoluene (H18-DBT), another commonly studied LOHC, the heterogeneity of Pt sites was reported to exhibit substantial impacts on the catalyst activity with the dehydrogenation TOF decreasing with the increase of Pt nanoparticle size.20 This size effect is attributed to the larger amount of highly active Pt sites on the small nanoparticles compared to the large nanoparticles. It is reasonable to assume that the heterogeneity effect is also significant for the Pt/Al2O3 samples studied in this work. Therefore, we postulate that the medium Pt size favoring the presence of a highly active Pt site contributes to the higher TOF observed in the 0.1Pt sample. The exact nature of this presumed Pt active site warrants further investigation including computational studies to understand the size effect.
At the other end of the Pt size spectrum, it is surprising that the atomic efficiency of the 0.1Pt sample almost doubles that of the 0.05Pt sample at 200 °C. The comparison between the 0.1Pt and 0.05Pt samples points to the fact that the MCH dehydrogenation likely requires multiple Pt sites simultaneously to catalyze the C–H bond cleavage, the commonly recognized rate-determining step, as reported recently.28,40 Compared to the 0.1Pt sample, the smaller Pt size in the 0.05Pt sample implies fewer Pt atoms in the nanoclusters, making it less efficient to break the six C–H bonds in MCH and thus leading to lower TOF. When the Pt loading further decreases to 0.025 wt% and 0.01 wt%, the TOF decreases disproportionately, with the 0.01 wt% sample showing negligible activity even at 320 °C (Fig. S1‡), underscoring the importance of multiple Pt sites in the MCH dehydrogenation. The light-off MCH dehydrogenation results clearly indicate that by tailoring the Pt size, a balance can be achieved between the activity of Pt sites and the number of active sites to optimize the performance of Pt/Al2O3 catalysts.
3.3 Stability of Pt/Al2O3 in MCH dehydrogenation
While light-off tests enable the evaluation of catalysts across a wide temperature range, LOHCs normally operate at a fixed temperature in practical applications. The catalyst stability at such a temperature is another crucial factor in addition to activity. Herein, we selected three Pt/Al2O3 catalysts, 0.05Pt, 0.1Pt and 1Pt samples, as they display a volcano trend in TOF, shown in Fig. 5, and studied their stability during a 20 h test at 320 °C (Fig. 6). The reason for choosing 320 °C as the test temperature is that all three catalysts reach the highest activity without exhibiting severe deactivation at this temperature in the light-off tests. To make a fair comparison, the total amount of Pt in these samples is maintained at the same level by using different amounts of catalyst. This is achieved by using 60 mg 0.05Pt sample, 30 mg 0.1Pt sample, and 3 mg 1Pt sample mixed with 27 mg quartz sand for the MCH dehydrogenation tests. The 0.1Pt and 1Pt samples show the same conversion profile as a function of reaction time, with the MCH conversion reaching ∼65% at time-on-stream (TOS) of 2 h and then gradually decreasing to ∼40% at the end of 20 h reaction. In comparison, the 0.05Pt sample shows a slightly lower MCH conversion at the initial stage, which drops more rapidly compared to that of the other two catalysts, reaching only ∼10% at the end of 20 h reaction. Through linear fitting of the MCH conversion as a function of TOS, the deactivation rate follows the order 0.1Pt > 1Pt > 0.05Pt, losing 1.1%, 1.6% and 2.5% MCH conversion per hour, respectively, in Fig. S3,‡ implying a much faster deactivation rate for the 0.05Pt sample. This notable difference clearly indicates that the 0.05Pt sample with smaller Pt size is much more vulnerable to deactivation compared to the other two samples, albeit with the same amount of Pt. With the increase of Pt size, the deactivation rate becomes irrelevant with the Pt loading, as evidenced by the same reaction rate profile between 0.1Pt and 1Pt samples. With respect to the toluene selectivity, 0.1Pt and 1Pt samples maintain the selectivity to toluene close to 100% throughout the 20 h reaction, whereas that of the 0.05Pt sample slowly decreases from 100% at the beginning of the test to 98% at TOS of 10 h, and then rapidly declines to 85% at TOS of 20 h. As discussed above, the heterogeneity of Pt nanoclusters and nanoparticles leads to Pt active sites with different activity and selectivity. It has been reported that during the catalyst deactivation, the Pt active sites that favor the reaction path to produce toluene deactivate faster than other types of active sites, leading to the decline in toluene selectivity,39 which is consistent with our observations. | ||
| Fig. 5 TOF of Pt over Pt/Al2O3 catalysts at 200 °C (a) and 240 °C (b) in MCH dehydrogenation. | ||
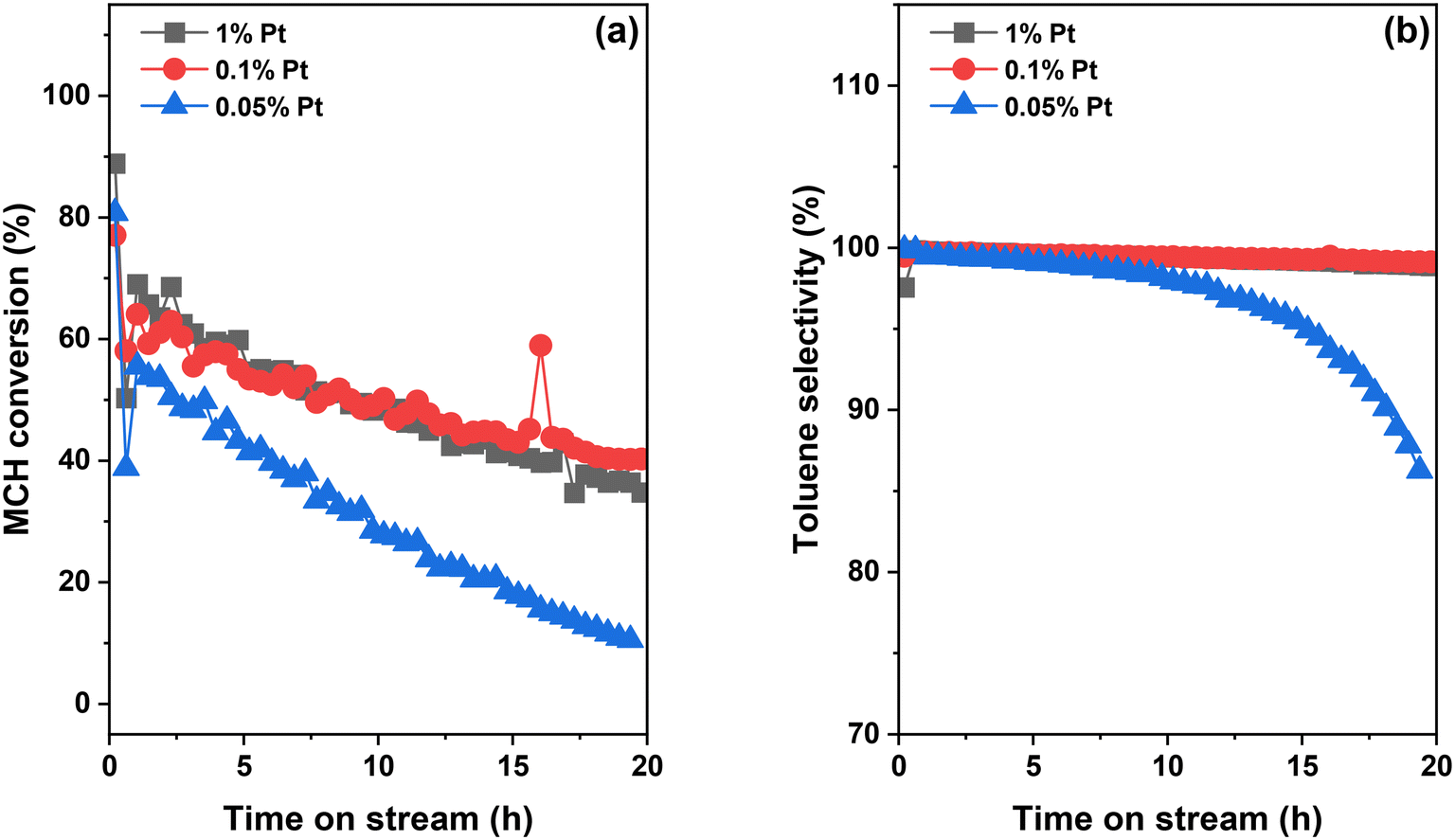 | ||
| Fig. 6 MCH conversion (a) and toluene selectivity (b) over Pt/Al2O3 catalysts during 20 h MCH dehydrogenation tests at 320 °C. Amounts of catalysts used for the reaction: 30 mg 0.1Pt sample, 60 mg 0.05Pt sample, and 3 mg 1Pt sample mixed with 27 mg quartz sand. Reaction conditions: 6.5 mL min−1 Ar and 0.006 mL min−1 MCH injected using a syringe pump. | ||
One of the common causes of Pt catalyst deactivation is the sintering of Pt to form large Pt species under the reaction conditions.43,44 To examine the Pt size in the spent samples after 20 h test, HAADF-STEM was carried out as exhibited in Fig. 7. Compared to fresh samples shown in Fig. 2, significant aggregation was not observed for all samples, with Pt species maintaining similar morphology and size before and after reactions. Note that for the 0.05Pt sample, the Pt size increased slightly from ∼0.6 to ∼0.7 nm without notable aggregation. Therefore, the sintering of Pt species is apparently not the major cause for the fast deactivation observed for the 0.05Pt sample.
 | ||
| Fig. 7 HAADF-STEM images of spent 0.05Pt (a), 0.1Pt (b) and 1Pt (c) samples after 20 h MCH dehydrogenation at 320 °C with their corresponding Pt size distribution (d). | ||
In addition to Pt sintering, coke deposition is another common cause of catalyst deactivation in the dehydrogenation reaction. To identify coke species and study their impacts on catalyst stability, TPO was carried out over spent Pt/Al2O3 samples right after the 20 h MCH reaction at 320 °C, as shown in Fig. 8. The major product from TPO is CO2 with no CO detected. The 1Pt sample displays a large coke oxidation peak at 580 °C with a shoulder peak at around 350 °C. The TPO profile of the 0.1Pt sample is similar but the major oxidation peak shifts down to 550 °C with the shoulder peak more pronounced than that of the 1Pt sample. The 0.05Pt sample, in contrast, presents a rather different TPO profile with the high temperature oxidation peak and the medium temperature peak blended into a broad oxidation peak. The high temperature peak increases from 410 °C of the 0.05Pt sample to 550 °C of the 0.1Pt sample, and finally to 580 °C of the 1Pt sample. This notable increase in the oxidation temperature indicates that compared to the 0.05Pt sample, 1Pt and 0.1Pt samples favor coke that is more resistant to oxidation. The total amount of coke was also analyzed by integrating their respective oxidation peaks, following the order 0.1Pt > 1Pt > 0.05Pt.
 | ||
| Fig. 8 TPO profiles of spent 0.05Pt, 0.1Pt and 1Pt samples after 20 h reaction at 320 °C. | ||
It is commonly accepted that larger amounts of coke and hard-to-burn coke lead to more severe catalyst deactivation,45,46 which is opposite to our observations in this study. The contrast between the least amount of coke that can be easily burned off and the highest deactivation rate for the 0.05Pt sample likely points to the notion that the type of coke has a larger impact than the amount of coke on the Pt/Al2O3 catalyst deactivation. In propane dehydrogenation (PDH) over Pt/SBA-15 catalysts, Kumar et al. reported that the addition of Sn to form a Pt–Sn bimetallic catalyst leads to three times higher amount of coke, yet considerably improves the PDH activity and stability.47 They attributed this counter-intuitive behavior to the enhanced migration of coke precursors from the Pt sites to the support aided by the Sn sites, which leads to a higher amount of coke while keeping the Pt active sites clean and thus improving the catalyst activity and stability. It has been reported that during the PDH reaction, coke species deposited on the Pt metal sites and the Al2O3 support differ considerably in terms of their resistance to oxidation. During the TPO process, the coke deposited on Pt metal sites is oxidized at around 200–300 °C, while that on the Al2O3 support is oxidized above 400 °C.48–50 According to these studies, the coke on the Pt sites causes more severe deactivation as it blocks the Pt active sites. Although MCH dehydrogenation is different from propane dehydrogenation, the impacts of coke on the same Pt/Al2O3 catalyst are expected to be similar. Indeed, their coke analysis is consistent with our observations48–50 that for the 1Pt and 0.1Pt samples, the minor shoulder peak is centered at around 325 °C with the major oxidation peak over 550 °C, indicating that the shoulder peak can be assigned to the coke deposited on Pt sites and the latter on the Al2O3 support. As for the 0.05Pt sample, the two types of coke are blended to a broad peak centered at 410 °C. This qualitatively indicates that the 0.1Pt and 1Pt samples contain a larger amount of coke on the support, while the 0.05Pt sample contains a comparable amount of coke deposited on the Pt metal sites and the support. As the coke on the Pt sites is more detrimental, the 0.05Pt sample suffers a higher catalyst deactivation rate, albeit with a smaller amount of coke, compared to the other two samples.
Raman spectroscopy was conducted to further study the nature of coke species after 20 h MCH dehydrogenation at 320 °C, as shown in Fig. 9. Two notable peaks are observed for these samples at 1609 and 1385 cm−1, which can be assigned to the G-band and D-band of carbonaceous species, respectively.48–51 The G-band peak is reported to originate from perfect graphite-like coke while the D-band peak is linked to disordered graphite-like coke. Therefore, the peak area ratio G/D is always used as a measure of the graphitization degree of carbon materials, with a higher value indicating a high degree of graphitization and therefore higher resistance to oxidation. For the three Pt/Al2O3 materials studied here, the G/D ratio follows the order 0.05Pt (1.1) < 0.1Pt (1.2) < 1Pt (1.6), indicating that the increase of Pt size leads to a higher degree of graphitization. This trend further corroborates the observation from the TPO profiles that the 0.1Pt and 1Pt samples contain a higher amount of coke that is resistant to oxidation, which is likely deposited on the Al2O3 support. In contrast, the 0.05Pt sample contains a larger amount of coke deposited on the Pt sites, leading to a more severe deactivation compared to the 0.1Pt and 1Pt samples.
 | ||
| Fig. 9 Raman spectra of spent Pt/Al2O3 samples aiming to determine the coke species deposited on these samples during the 20 h stability tests. The laser excitation wavelength is 244 nm. | ||
4. Conclusion
In this work, we studied the impacts of Pt size on the catalytic performance of industrially relevant Pt/Al2O3 catalysts for the dehydrogenation of MCH to toluene, a promising LOHC pair for hydrogen transportation and storage. A series of Pt/Al2O3 samples were prepared via an incipient wetness impregnation method and characterized collectively using STEM, XRD and CO-DRIFTS. In the light-off MCH dehydrogenation tests, the TOF of Pt/Al2O3 catalysts exhibits a volcano trend over the Pt size, with the medium Pt size in the 0.1Pt sample showing the highest atom efficiency. This indicates that a balance between the activity of Pt sites and the number of neighboring sites needs to be achieved for an optimized catalytic performance. In addition, small Pt size, as in the 0.05Pt sample, promotes the formation of coke deposited on the Pt active sites, which is more detrimental to the catalyst stability than the coke deposited on the support as in the 0.1Pt and 1Pt samples. We believe that these insights from the current study can help guide the design of efficient Pt/Al2O3 catalysts in MCH dehydrogenation as well as other industrially important reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of the Center for Understanding & Controlling Accelerated and Gradual Evolution of Materials for Energy (UNCAGE-ME), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award no. DE-SC0012577. The synthesis and characterization (DRIFTS, Raman spectroscopy, STEM, and nitrogen physisorption) were conducted as part of a user project at the Center for Nanophase Materials Sciences (CNMS), which is a U.S. Department of Energy, Office of Science User Facility at Oak Ridge National Laboratory.References
- M. Niermann, S. Drunert, M. Kaltschmitt and K. Bonhoff, Energy Environ. Sci., 2019, 12, 290–307 RSC.
- P. T. Aaldto-Saksa, C. Cook, J. Kiviaho and T. Repo, J. Power Sources, 2018, 396, 803–823 CrossRef.
- K. M. Tan, T. S. Babu, V. K. Ramachandaramurthy, P. Kasinathan, S. G. Solanki and S. K. Raveendran, J. Energy Storage, 2021, 39, 102591 CrossRef.
- T. E. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- A. Sánchez, Q. Zhang, M. Martín and P. Vega, Energy Convers. Manage., 2022, 252, 115056 CrossRef.
- Z. Abdin, C. Tang, Y. Liu and K. Catchpole, iScience, 2021, 24, 102966 CrossRef CAS.
- J. Andersson and S. Grönkvist, Int. J. Hydrogen Energy, 2019, 44, 11901–11919 CrossRef CAS.
- K. Sisáková, N. Podrojková, R. Oriňaková and A. Oriňak, Energy Fuels, 2021, 35, 7608–7623 CrossRef.
- P. M. Modisha, C. N. M. Ouma, R. Garidzirai, P. Wasserscheid and D. Bessarabov, Energy Fuels, 2019, 33, 2778–2796 CrossRef CAS.
- P. C. Rao and M. Yoon, Energies, 2020, 13, 6040 CrossRef CAS.
- J. Meng, F. Zhou, H. Ma, X. Yuan, Y. Wang and J. Zhang, Top. Catal., 2021, 64, 509–520 CrossRef CAS.
- A. Nakano, S. Manabe, T. Higo, H. Seki, S. Nagatake, T. Yabe, S. Ogo, T. Nagatsuka, Y. Sugiura, H. Iki and Y. Sekine, Appl. Catal., A, 2017, 543, 75–81 CrossRef.
- D. K. Cromwell, P. T. Vasudevan, B. Pawelec and J. L. G. Fierro, Catal. Today, 2016, 259, 119–129 CrossRef CAS.
- S. Horikoshi, M. Kamata, T. Sumi and N. Serpone, Int. J. Hydrogen Energy, 2016, 41, 12029–12037 CrossRef CAS.
- X. Zhang, N. He, L. Lin, Q. Zhu, G. Wang and H. Guo, Catal. Sci. Technol., 2020, 10, 1171–1181 RSC.
- S. Kobayashi, S. Kaneko, M.-a. Ohshima, H. Kurokawa and H. Miura, Appl. Catal., A, 2012, 417–418, 306–312 CrossRef CAS.
- A. H. Al-ShaikhAli, A. Jedidi, D. H. Anjum, L. Cavallo and K. Takanabe, ACS Catal., 2017, 7, 1592–1600 CrossRef CAS.
- N. Boufaden, R. Akkari, B. Pawelec, J. L. G. Fierro, M. Said Zina and A. Ghorbel, Appl. Catal., A, 2015, 502, 329–339 CrossRef CAS.
- S. Yolcular and Ö. Olgun, Catal. Today, 2008, 138, 198–202 CrossRef CAS.
- F. Auer, A. Hupfer, A. Bösmann, N. Szesni and P. Wasserscheidpeter, Catal. Sci. Technol., 2020, 10, 6669–6678 RSC.
- E. Ogel, M. Casapu, D. E. Doronkin, R. Popescu, H. Störmer, C. Mechler, G. Marzun, S. Barcikowski, M. Türk and J. D. Grunwaldt, J. Phys. Chem. C, 2019, 123, 5433–5446 CrossRef CAS.
- C. Dessal, A. Sangnier, C. Chizallet, C. Dujardin, F. Morfin, J.-L. Rousset, M. Aouine, M. Bugnet, P. Afanasiev and L. Piccolo, Nanoscale, 2019, 11, 6897–6904 RSC.
- W. Zhang, H. Wang, J. Jiang, Z. Sui, Y. Zhu, D. Chen and X. Zhou, ACS Catal., 2020, 10, 12932–12942 CrossRef CAS.
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 RSC.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- L. Chen, P. Verma, K. Hou, Z. Qi, S. Zhang, Y.-S. Liu, J. Guo, V. Stavila, M. D. Allendorf, L. Zheng, M. Salmeron, D. Prendergast, G. A. Somorjai and J. Su, Nat. Commun., 2022, 13, 1092 CrossRef CAS PubMed.
- Y. Deng, Y. Guo, Z. Jia, J.-C. Liu, J. Guo, X. Cai, C. Dong, M. Wang, C. Li, J. Diao, Z. Jiang, J. Xie, N. Wang, H. Xiao, B. Xu, H. Zhang, H. Liu, J. Li and D. Ma, J. Am. Chem. Soc., 2022, 144, 3535–3542 CrossRef CAS PubMed.
- J. Lipp, R. Banerjee, M. F. Patwary, N. Patra, A. Dong, F. Girgsdies, S. R. Bare and J. R. Regalbuto, Chem. Mater., 2022, 34, 8091–8111 CrossRef CAS.
- J. Lee, E. J. Jang, D. G. Oh, J. Szanyi and J. H. Kwak, J. Catal., 2020, 385, 204–212 CrossRef CAS.
- W. Sinkler, S. I. Sanchez, S. A. Bradley, J. Wen, B. Mishra, S. D. Kelly and S. R. Bare, ChemCatChem, 2015, 7, 3779–3787 CrossRef CAS.
- S. A. Bradley, W. Sinkler, D. A. Blom, W. Bigelow, P. M. Voyles and L. F. Allard, Catal. Lett., 2012, 142, 176–182 CrossRef CAS.
- G. Bergeret and P. Gallezot, Particle Size and Dispersion Measurements, Handbook of Heterogeneous Catalysis, 2008, pp. 738–765 Search PubMed.
- Z. L. Zhang, Y. H. Zhu, H. Asakura, B. Zhang, J. G. Zhang, M. X. Zhou, Y. Han, T. Tanaka, A. Q. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS.
- F. C. Meunier, J. Phys. Chem. C, 2021, 125, 21810–21823 CrossRef CAS.
- F. Faisal, C. Stumm, M. Bertram, T. Wähler, R. Schuster, F. Xiang, O. Lytken, I. Katsounaros, K. J. J. Mayrhofer, M. A. Schneider, O. Brummel and J. Libuda, Phys. Chem. Chem. Phys., 2018, 20, 23702–23716 RSC.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- T. Schildhauer, E. Newson and S. Müller, J. Catal., 2001, 198, 355–358 CrossRef CAS.
- F. Alhumaidan, D. Tsakiris, D. Cresswell and A. Garforth, Int. J. Hydrogen Energy, 2013, 38, 14010–14026 CrossRef CAS.
- C. Dong, Z. Gao, Y. Li, M. Peng, M. Wang, Y. Xu, C. Li, M. Xu, Y. Deng, X. Qin, F. Huang, X. Wei, Y.-G. Wang, H. Liu, W. Zhou and D. Ma, Nat. Catal., 2022, 5, 485–493 CrossRef CAS.
- A. Liu, X. Liu, L. Liu, Y. Pu, K. Guo, W. Tan, S. Gao, Y. Luo, S. Yu, R. Si, B. Shan, F. Gao and L. Dong, ACS Catal., 2019, 9, 7759–7768 CrossRef CAS.
- M. Pan, J. Wang, W. Fu, B. Chen, J. Lei, W. Chen, X. Duan, D. Chen, G. Qian and X. Zhou, Green Energy Environ., 2020, 5, 76–82 CrossRef.
- E. D. Goodman, J. A. Schwalbe and M. Cargnello, ACS Catal., 2017, 7, 7156–7173 CrossRef CAS.
- T. W. Hansen, A. T. DeLaRiva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- E. T. C. Vogt, D. Fu and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2023, 62, e202300319 CrossRef CAS PubMed.
- J. Zhou, J. Zhao, J. Zhang, T. Zhang, M. Ye and Z. Liu, Chin. J. Catal., 2020, 41, 1048–1061 CrossRef CAS.
- M. Santhosh Kumar, D. Chen, A. Holmen and J. C. Walmsley, Catal. Today, 2009, 142, 17–23 CrossRef CAS.
- Z. Lian, C. Si, F. Jan, S. Zhi and B. Li, ACS Catal., 2021, 11, 9279–9292 CrossRef CAS.
- H.-Z. Wang, L.-L. Sun, Z.-J. Sui, Y.-A. Zhu, G.-H. Ye, D. Chen, X.-G. Zhou and W.-K. Yuan, Ind. Eng. Chem. Res., 2018, 57, 8647–8654 CrossRef CAS.
- Q. Li, Z. Sui, X. Zhou, Y. Zhu, J. Zhou and D. Chen, Top. Catal., 2011, 54, 888–896 CrossRef CAS.
- J. J. H. B. Sattler, A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2013, 15, 12095–12103 RSC.
Footnotes |
| † Notice: this manuscript has been authored by UT-Battelle, LLC, under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01568h |
| This journal is © The Royal Society of Chemistry 2024 |