
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring structure, temperature and activity correlations in the selective oxidation of lower olefins over Bi–Mo–Co–Fe–O catalysts by spatial reactor profile measurements†
Linda
Klag
 a,
Sebastian
Weber
ab,
Raimund
Horn
c,
Thomas L.
Sheppard‡
ab and
Jan-Dierk
Grunwaldt
*ab
a,
Sebastian
Weber
ab,
Raimund
Horn
c,
Thomas L.
Sheppard‡
ab and
Jan-Dierk
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany
cInstitute of Chemical Reaction Engineering, Hamburg University of Technology, 21073 Hamburg, Germany
First published on 16th January 2024
Abstract
Improving process efficiency in selective oxidation of lower olefins over mixed metal oxide catalysts requires profound knowledge of the dynamic behaviour of exothermic reactions along the reactor. For this purpose, structure–activity correlations of two Bi–Mo–Co–Fe–O model catalysts were investigated by means of structure, temperature and activity profiling in selective propylene and isobutene oxidation. Both catalysts showed pronounced differences in selectivity, which strongly affected the temperature and gas phase concentration gradients along the reactor, and thus the reaction network of each olefin oxidation process. Complementary structure profiling by synchrotron XRD identified the evolution of crystalline metal oxide phases after testing in propylene oxidation. Molybdate-based structures (e.g., α-Bi2Mo3O12, Bi3FeMo2O12) were found to moderate oxygen mobility during catalytic reaction and increase selectivity towards acrolein/methacrolein, while particularly single metal oxides (i.e., Co3O4, Fe3O4) enhanced oxygen mobility drastically and favoured total oxidation. Comparison of selective propylene and isobutene oxidation revealed the metal oxide phase ensembles within each catalyst had comparable effects on both reaction networks. Hence, the spatially-resolved testing and characterization allowed a systematic study of the catalytic processes along the reactor, showing great promise for knowledge-based optimization of selective oxidation processes.
Introduction
The selective oxidation of lower olefins over bismuth–molybdate based mixed metal oxides (MMO) is of great interest in chemical industry for the production of various organic intermediates (e.g., acrolein and methacrolein).1,2 Although these processes are well-established, neither selective propylene nor isobutene oxidation runs at maximum selectivity but rather produce undesired by-products such as CO2.3,4 Enhancing process efficiency by improving catalytic performance and lowering CO2 emissions requires the combined efforts of industry and academia,5,6 including a fundamental understanding of the working principles of MMO catalysts.7–10 The mechanism and kinetics in selective propylene (amm)oxidation over bismuth molybdates were recently reviewed by Bell,11 reporting general acceptance of the Mars–van-Krevelen mechanism and identifying C–H bond activation in the methyl group of propylene as a rate-limiting step. Notably, most research has focused on selective propylene oxidation over bismuth molybdate catalysts, while different mechanisms have been proposed for isobutene oxidation.12–15 In parallel, the characterization landscape has evolved significantly,16–21 making it more feasible to tackle the structural complexity of such catalysts.22 For example, operando characterization has produced conclusive data on structure–activity correlations of simplified model systems (e.g., 2-component Bi–Mo–O).23 Further structural insights into more active multicomponent systems (e.g., 4-component Bi–Mo–Co–Fe–O) were gained recently, therefore unravelling the complex phase mixtures present, and the role of individual metal oxide phases.24–26 Parallels between the selective oxidation of propylene to acrolein and isobutene to methacrolein were also found.27,28Most studies of selective olefin oxidation on bismuth molybdates have been conducted in an integral manner, by correlating reactor in- and outlet measurements and deconvoluting the catalyst structure at a fixed bed position. However, selective oxidation of both propylene and isobutene are highly exothermic reactions. This can lead to temperature gradients along the reactor, which can in turn cause catalyst structural gradients and affect local reactant and product concentrations. For this reason, recent developments in literature have moved towards in situ/operando spatially-resolved techniques, which are sensitive to gradients in temperature, structure, and/or concentration within the catalyst bed.29–31 Touitou et al. published one of the first setups for combined temperature and concentration profiling along powdered catalyst beds,32,33 which was later extended to simultaneous X-ray absorption spectroscopy (XAS) profiling by Goguet et al.34,35 Moreover, Horn et al. reported on spatially-resolved experiments in a lab-scale compact profile reactor (CPR) under various pressures,36 temperatures,37 and with simultaneous spectroscopy38,39 and diffraction studies.40
Especially from an industrial point of view, it is essential to understand the dynamics along the reactor to e.g., prevent by-product formation. Apart from CO and CO2, acrylic acid is a common by-product in selective propylene oxidation, which is mainly undesired due to its tendency to polymerize and subsequent difficulties in removing it from the product stream.10 Improving process selectivity requires profound knowledge on the complex reaction networks, which in turn allows further optimization of process parameters and reactor design.41 On a fundamental level, such reaction networks were mainly predicted from simulations and kinetic studies in the past.42–47 Since the assumptions for such kinetic models (e.g., low conversion, isothermal conditions) are often rather simplified, more realistic parameters need to be considered to optimise the catalytic processes under operating conditions. For this purpose, non-isothermal spatial profiling experiments can enable new insights beyond the conventional “black box” approach and can validate or complement theoretical predictions about catalyst behaviour. In this context, Ganzer and Freund derived a detailed reaction network for selective propylene oxidation from spatially-resolved data over an industrial multicomponent bismuth molybdate catalyst in a pilot plant.48 Stehle et al. investigated a Bi–Mo–Co–Fe-oxide catalyst by activity and temperature profiling under industrially relevant conditions in a fixed-bed lab-reactor, which led to validation of the proposed reaction networks in literature.49 However, previous spatially-resolved studies in selective propylene oxidation did not combine catalytic testing and characterization simultaneously. Consequently, these studies did not consider the influence of temperature and concentration profiles on the local catalyst structure, and thus on the reaction network. Instead, variations in the profiles were rather induced by adapting the process parameters (e.g., coolant temperature, oxygen-to-propylene ratio).48,50 Hence, it is equally important to rationalize the structure and performance of different catalysts together with resulting gradients along the reactor. Such an approach can deliver profound insights into the roles of various metal oxide phases in the selective oxidation of lower olefins.
In this study, we report on spatial profiling experiments during both the selective oxidation of propylene and isobutene over Bi–Mo–Co–Fe-oxides in a CPR setup. Two flame-spray prepared catalysts differing strongly in selectivity28 were investigated with respect to temperature and gas phase concentration gradients along the catalyst bed. Flame spray pyrolysis is advantageous for the systematic study of such complex mixed metal oxides in terms of easy adjustment of elemental composition including high reproducibility and accessibility to certain metastable phases in a single step.24,27,51 On the one hand, a selective catalyst towards acrolein/methacrolein was chosen, on the other hand a more unselective system mostly forming total oxidation products. The spatially-resolved data obtained for selective propylene oxidation was complemented by investigations on possible structural gradients of both catalysts using ex situ synchrotron XRD after testing. Together with the activity and temperature profiles obtained for isobutene oxidation, the datasets allowed a better comparison of both olefin oxidations and gave new insights into their reaction networks. In this way, we aimed for a deeper understanding of the catalytic behaviour of Bi–Mo–Co–Fe–O catalysts in two potentially similar oxidation reactions by studying the spatial evolution of their metal oxide phases, gas phase concentration and temperature, and thus the dynamics along the reactor.
Experimental
Catalyst synthesis and characterization
Two Bi–Mo–Co–Fe–O catalysts (denoted as FSP-Co and FSP-U) were prepared by flame spray pyrolysis (FSP) as described in ref. 28. The catalysts differed in their elemental composition (Table 1), with FSP-Co containing the highest amount of cobalt (40 mol%) and FSP-U containing the highest amount of molybdenum (50 mol%), referring to an elemental composition firstly reported and recommended by Moro-Oka and Ueda.7 Both catalysts were characterised by various complementary techniques, including Raman spectroscopy and X-ray diffraction (XRD) to provide information on both the amorphous and crystalline metal oxide phase mixtures present.| Sample | Metal ratio/mol% | |||
|---|---|---|---|---|
| Bi | Mo | Co | Fe | |
| FSP-Co | 5.0 ± 0.1 | 35.0 ± 0.9 | 40.1 ± 0.8 | 19.9 ± 0.5 |
| FSP-U | 4.2 ± 0.1 | 50.2 ± 1.3 | 33.3 ± 0.6 | 12.3 ± 0.3 |
Spatially-resolved catalytic testing in a compact profile reactor (CPR)
The spatially-resolved catalytic testing in selective propylene and isobutene oxidation was performed in a CPR setup (REACNOSTICS GmbH). The spatial profiling technique along packed beds of powdered catalysts was conducted in a similar way as in previous work.38,40 Further information on the CPR setup is given in the ESI† (section S1). In this way, the gas concentration and catalyst temperature along the catalyst bed were recorded within a single experiment. In addition, the CPR allows recording structure profiles by e.g., XAS (ESI,† section S6) and synchrotron XRD (see results and discussion section).For each profile measurement, the catalysts FSP-Co and FSP-U were ground, pressed, and sieved (sieve fraction of 300–450 μm). In the case of selective propylene oxidation, 600 mg of the sieve fraction was filled in a fused silica tubular reactor, while for selective isobutene oxidation 100 mg of the sieved catalyst was diluted with 300 mg SiC (450–600 μm) to avoid thermal runaway. This resulted in a catalyst bed length of 25 mm in selective isobutene oxidation, and 31 mm (FSP-U) or 43 mm (FSP-Co) in selective propylene oxidation. The variations in bed lengths for the latter were related to a slight difference in thickness of the reactor tubes and their wall thickness. The catalyst bed was fixed by quartz wool plugs and a sampling capillary was vertically positioned through the centre of the catalyst bed. For preconditioning, the catalysts were heated to 180 °C (5 °C min−1) in synthetic air (N2/O2 = 80/20 vol%). Subsequently, each catalyst was heated stepwise (2 °C min−1) up to the respective ignition temperature under reaction conditions (N2/O2/CnH2n/H2O = 70/14/8/8 vol%, ∼1 bar). An overview of the chosen reaction conditions for each reaction is given in Table 2.
| Propylene oxidation (He/O2/C3H6/H2O = 70/14/8/8 vol%) | |||||
|---|---|---|---|---|---|
| Catalyst | Total flow mL min−1 | T oven/°C | m cat/g | Dilution cat.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SiC :SiC |
GHSV/h−1 |
| FSP-Co | 75 | 380 | 600 | — | 10877 |
| FSP-U | 75 | 380 | 600 | — | 10877 |
| Isobutene oxidation (He/O2/C4H8/H2O = 70/14/8/8 vol%) | |||||
|---|---|---|---|---|---|
| FSP-Co | 50 | 400 | 100 | 1:3 |
12473 |
| FSP-U | 50 | 450 | 100 | 1:3 |
12473 |
Prior to each profile acquisition, the catalyst was stabilized for ∼2 h under reaction conditions as monitored by gas chromatography (GC) measurements behind the catalyst bed. These measurements were repeated after profile acquisition to confirm that the integral catalytic performance remained constant during each experiment. Additionally, the oxygen content of the integral product stream was constantly monitored by an on-line oxygen sensor, showing stable oxygen consumption. Preliminary tests in a lab-scale testing unit further supported stable catalytic performance of FSP-Co and FSP-U during several days on stream and showed the same integral olefin conversions as observed within this study in the CPR.
For each profile, the reactor/oven unit was moved stepwise along the sampling capillary from outlet to inlet of the catalyst bed. Typically, a step size of 3–4 mm was chosen, but smaller steps (1–2 mm) were applied in the beginning and end of the catalyst bed to properly detect the transition zone between catalyst and quartz wool. This resulted in at least 10 individual measurement points for each profile. GC data acquisition at each measurement point lasted for 20 min, resulting in a total profile acquisition time of ∼4 h. From the resulting chromatograms, olefin and oxygen conversion, yield and selectivity towards the reaction products was calculated, as described in the ESI,† section S2. Based on the resulting concentration profiles, local reaction rates were calculated (ESI,† section S2), which report the conversion or formation of species between two consecutive measurement points.
Spatially-resolved characterization by synchrotron XRD
Spatially-resolved synchrotron XRD experiments were performed ex situ at the P21.1 beamline (DESY, Hamburg, Germany). For this purpose, the catalysts were cooled down to room temperature in inert atmosphere after catalytic testing in propylene oxidation, and sent to the beamline. XRD patterns were acquired using a Perkin Elmer XRD1621 detector and monochromatic beam (λ = 0.12224 Å, 700 × 700μm2). Azimuthal integration of the acquired 2D images was done with the pyFAI package.52 XRD patterns were recorded along the spent catalyst bed with a step size of 2 mm (FSP-Co) or 0.912 mm (FSP-U) and 60 s acquisition time. Additionally, a LaB6 reference was measured for sample to detector distance calibration and to retrieve an instrumental profile function. Sequential Rietveld refinement (2θ = 1–6°) was performed using TOPAS (v.6, Bruker AXS),53 with references available in the Inorganic Crystal Structure Database (ICSD, see ESI† section S3). From Rietveld refinement crystalline phase amounts and crystallite sizes were obtained. More details on the sequential Rietveld refinement can be found in the ESI† section 3.2.Results and discussion
Catalysts and process conditions for spatial profiling in selective olefin oxidation
Two FSP-prepared Bi–Mo–Co–Fe–O catalysts with different elemental composition were investigated in a compact profile reactor (CPR) during the selective oxidation of propylene and isobutene, respectively. The corresponding metal oxide phase composition of the “as-prepared” catalysts FSP-Co and FSP-U is summarized in Table 3. Both catalysts were chosen as representative model systems for providing high catalytic activity and enabling either a rather selective (FSP-U) or unselective (FSP-Co) reaction pathway in propylene and isobutene oxidation.| Catalyst | Raman spectroscopy | Synchrotron XRD | Crystalline phase amounts/wt% |
|---|---|---|---|
| a An additional phase, which was not conclusively identified, was detected but disappeared during reaction. | |||
| FSP-Co | α-CoMoO4 | α-CoMoO4 | 30.5 ± 0.3 |
| β-CoMoO4 | β-Co0.7Fe0.3MoO4 | 37.3 ± 0.3 | |
| Co3O4/Fe3O4 | Co3O4 | 6.9 ± 0.3 | |
| Fe3O4 | 19.0 ± 0.6 | ||
| Bi3FeMo2O12 | 6.0 ± 1.3 | ||
| FSP-Ua | α-CoMoO4 | α-CoMoO4 | 12.9 ± 0.6 |
| β-CoMoO4 | β-Co0.7Fe0.3MoO4 | 58.6 ± 0.8 | |
| α-Bi2Mo3O12 | α-Bi2Mo3O12 | 16.5 ± 0.4 | |
| Fe2Mo3O12 | 12.0 ± 0.8 | ||
For each reaction, same catalyst masses of FSP-Co and FSP-U were chosen for comparability. However, selective isobutene oxidation required a higher GHSV compared to propylene oxidation due to a generally higher reactivity. Hence, both catalysts were diluted with SiC to compromise between controllable and comparable reaction conditions during isobutene oxidation while still achieving an ideal flow pattern (e.g., catalyst bed length to reactor diameter ratio).54 This resulted in different bed lengths for each lower olefin oxidation. For better comparison, we discuss the profiles with respect to their relative catalyst bed position compared to the full catalyst bed length (“0” at the beginning, “1” at the end). Moreover, each condition (catalyst composition, hydrocarbon species) required different temperatures to initiate the reaction. For this reason, the oven temperature was adapted to the respective ignition temperature of the reaction. An overview of the chosen reaction conditions for each experiment is shown in Table 2.
Selective propylene oxidation: influence of elemental composition on activity and temperature profiles
The temperature and activity profiles of FSP-U and FSP-Co with corresponding product amounts and formation/reaction rates measured during selective propylene oxidation are given in Fig. 1. Under identical reaction conditions, FSP-U showed an integral propylene conversion of ∼60% (Fig. 1a), while FSP-Co was significantly more active showing ∼80% propylene conversion (Fig. 1b). The high activity of FSP-Co was linked to increased oxygen consumption of ∼90% and high selectivity towards the total oxidation products CO and CO2 (SCOx ∼40%). Notably, only low selectivity (∼3%) towards acrylic acid was obtained in the case of FSP-Co. In contrast, the selectivity towards COx reached only ∼13% in the case of FSP-U, while other reaction products included acrolein (SACR ∼68%) and acrylic acid (SAA ∼14%). Other by-products were minor for both catalysts and are thus not further discussed. The calculated carbon balance closed within 97 ± 3% in the case of selective propylene oxidation over FSP-U, while larger deviations were found over FSP-Co (91 ± 5%). | ||
| Fig. 1 Catalytic activity, selectivity and temperature profiles (a and b), concentration profiles of the reaction products (c and d) and formation/reaction rates to the products (e and f) for FSP-U (left) and FSP-Co (right) during selective propylene oxidation. Profiles were acquired under reaction conditions (N2/O2/C3H6/H2O = 70/14/8/8 vol%; GHSV 10877 h−1) at respective ignition temperature (380 °C). Position 0 mm refers to the beginning and position 31 mm (FSP-U) and 43 mm (FSP-Co) to the end of the catalyst bed. | ||
Moreover, different temperature profiles were obtained for both systems. For FSP-U, a hot-spot was observed in the first third (1/3) of the catalyst bed with a maximum at ∼405 °C and pos. 4.9 mm, correlating to the highest observed acrolein formation rate (Fig. 1e). Hence, the heat load was highest where the highest reaction rate of the selective oxidation of propylene to acrolein occurred. In contrast, lower temperatures correlated with enhanced by-product formation. Thus, CO, CO2 and acrylic acid formation rates increased along the catalyst bed, with highest acrylic acid formation in the second third (2/3) and highest COx formation in the last third (3/3) of the catalyst bed. This temperature dependency of the acrolein and acrylic acid formation is comparable to those in literature.48 The increase in acrolein formation rate at 29 mm in Fig. 1e may be due to inhomogeneities in the catalyst bed or to a local measurement error at the end of the catalyst bed.
Interestingly, similar temperature regimes detected in the first third of the catalyst bed of both FSP-U and FSP-Co resulted in significantly lower acrolein but higher CO2 formation rate for FSP-Co (Fig. 1f). This emphasizes that solely adapting the catalyst temperature to an optimum is not sufficient to guarantee catalyst selectivity. Even though the reaction rate to acrolein in FSP-Co further increased with higher temperatures (T > 405 °C), CO2 and CO formation drastically increased towards the end of the bed of FSP-Co. This was accompanied by a pronounced hot-spot (∼450 °C) in the last third of the bed and very high oxygen conversions (up to 90%, Fig. 1b). This led also to a higher CO2-production towards the end of the catalyst bed (crossover point of CO and CO2-production at about 30 mm of the catalyst bed).
Notably, the acrolein yields obtained for FSP-Co and FSP-U dependent on propylene conversion were nearly identical within the entire conversion range (Fig. 2) and almost stoichiometric at conversions <25% (in the first third of the bed). This indicates that both catalysts follow the same kinetics with respect to the first reaction step. As FSP-Co was less selective than FSP-U, the integral catalytic performance of each 4-component system was therefore defined by side reactions. Such consecutive or parallel side reactions were further indicated by the slopes of the acrolein yields measured along the catalyst bed (see ESI,† Fig. S1), which flattened from the second third of the catalyst bed at further increasing propylene conversion. Similar behavior was also reported in literature,48–50 with an even more pronounced flattening of the acrolein yield curves that reached an optimum at propylene conversions >80%. Still, acrolein yields and byproduct formation are correlated, as byproducts are partly formed from acrolein.
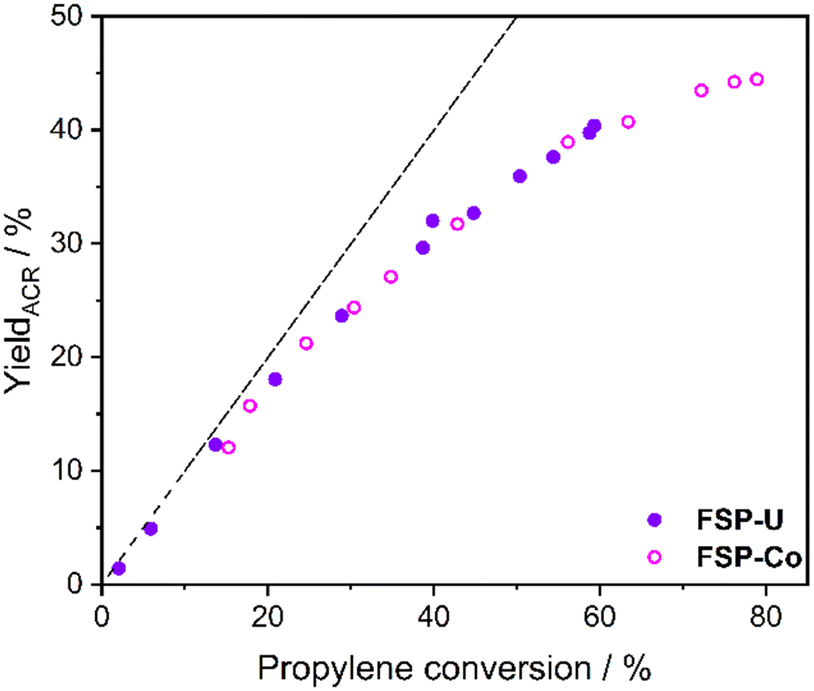 | ||
| Fig. 2 Acrolein yield over propylene conversion for FSP-Co (light) and FSP-U (dark) measured during selective propylene oxidation. Dotted line indicates the progress of the stoichiometric reaction. | ||
In order to unravel the reaction network for selective propylene oxidation, the (by-)product yield ratios obtained along the catalyst bed are given in Fig. 3. Particularly acrylic acid was found to be formed consecutively from acrolein (Fig. 3a), which agrees with previous reports on the reaction networks in propylene oxidation over bismuth molybdate based systems.48,49 However, similar acrolein amounts formed in both catalysts resulted in different acrylic acid yields. According to this, the high hot-spot temperature measured in FSP-Co seemed to either suppress the partial oxidation of acrolein to acrylic acid, or accelerate the acrylic acid decomposition to COx.
 | ||
| Fig. 3 Yield ratios of acrolein/acrylic acid (a), acrolein/COx (b), CO/CO2 (c) and acrylic acid/COx (d) of FSP-Co (light) and FSP-U (dark) along the catalyst bed during selective propylene oxidation. | ||
For both catalysts, CO formation was detected starting from relative bed position 0.35 (Fig. 3b and c). This was accompanied by a bed temperature of ∼400 °C in both cases. However, with further increasing CO formation from this position, the catalyst temperature increased in the case of FSP-Co while it decreased for FSP-U. Thus, no clear trend in temperature dependency was observed, which hinted to different reaction pathways for CO formation within each system. In the case of FSP-U, CO formation started together with the maximum of YACR/YCO2 at decreasing propylene reaction rate (Fig. 3b), indicating that CO is rather formed consecutively from acrolein. Since similar acrolein yields were found for FSP-U and FSP-Co in dependence of propylene conversion, the lower YACR/YCO ratios in FSP-Co suggest that CO is rather formed from acrylic acid or propylene directly, which is also supported by the increased propylene conversion. Notably, in both cases no CO was detected in the first third of the catalyst bed at similar oxygen over propylene conversions (see ESI,† Fig. S2). Hence, CO formation was suppressed at XO2/XC3H6 < 0.5. In contrast, CO2 was already detected in the first third of the bed, indicating its parallel formation to acrolein directly from propylene for both catalysts. In addition, the YACR/YCO2 ratios decreased in the second and last third of the bed for both catalysts, indicating that CO2 was additionally formed from acrolein (Fig. 3b) and acrylic acid (Fig. 3d). Notably, CO2 and CO formation were more favored along the entire catalyst bed of FSP-Co (Fig. 3b–d), even though temperature regimes were partly similar to FSP-U.
Hence, the pronounced differences in terms of catalytic performance are probably related to the different metal oxide phase composition of FSP-Co and FSP-U. Their opposite selectivity trends in turn resulted in distinct temperature gradients along the catalyst bed, leading to extensive combustion at the end of the bed in FSP-Co coupled to a hot-spot. This further correlated with different oxygen/propylene ratios along the reactor (Fig. 4), although the same gas feed mixture was dosed for the selective oxidation over FSP-Co and FSP-U. Notably, the same catalyst temperature did not involve the same oxygen to propylene ratio for each catalyst. This indicates that the metal oxide phases within FSP-Co and FSP-U induce a different oxygen mobility within the catalytic reaction.
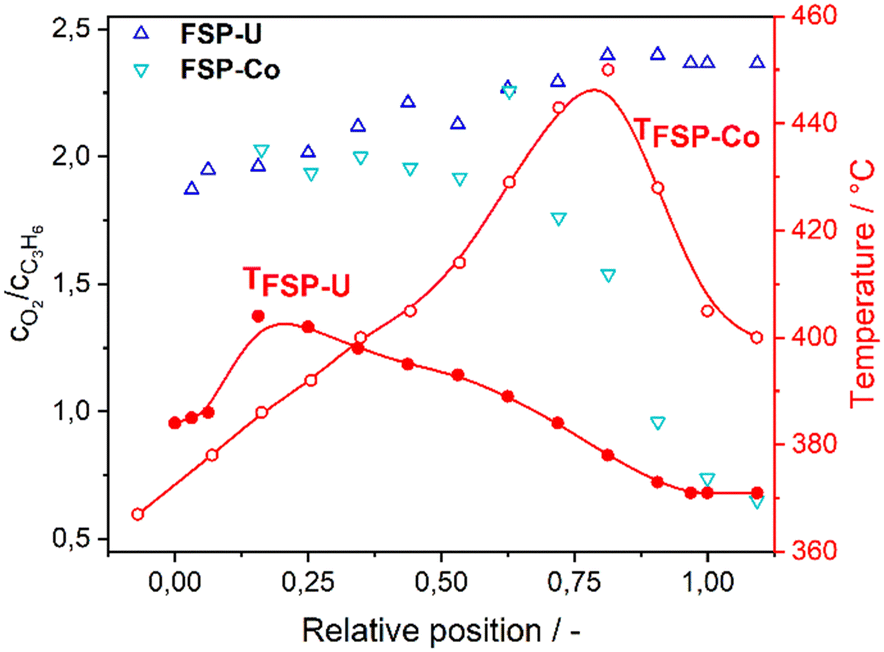 | ||
| Fig. 4 Molar ratio of oxygen to propylene for FSP-Co (light) and FSP-U (dark) together with temperature profiles along the catalyst bed during selective propylene oxidation. | ||
While it is well-known that the selective oxidation of propylene proceeds via the Mars–van-Krevelen mechanism,11 implying that nucleophilic oxygen species from the bulk form acrolein, with subsequent re-oxidization by gas phase oxygen, the role of the metal oxide phases and the exposed surfaces is still not fully understood. Bielański and Haber postulated the concept of nucleophilic and electrophilic oxygen species, which describes the partial or total oxidation pathway over transition metal oxides based on the reactivity of their metal centers.55 Accordingly, molybdate structures induce the formation of nucleophilic oxygen species which participate in the selective pathway, while e.g., Co3O4 generates electrophilic oxygen species which adsorb on the metal oxide surface and lead to total oxidation. Hence, the different metal oxide phases within FSP-Co and FSP-U seem to trigger a different shift in the equilibrium between the nucleophilic and electrophilic oxygen species. Consequently, both catalysts enabled different reaction pathways within the complex reaction network of propylene oxidation over Bi–Mo–Co–Fe–O catalysts (Fig. 5). At the same time, the selective pathway further includes several mechanistic steps,10,11,56 including olefin adsorption, H-abstraction, O-insertion and aldehyde desorption. However, different metal centers contributing to the redox cycle were described.56–59 Generally, a multifunctionality of the active sites is suggested,8 with the selective oxidation being claimed to take place on bismuth molybdate sites, while the activation of gas phase oxygen to nucleophilic O2− is suggested to proceed on iron molybdate sites.7,26 Since both α-Bi2Mo3O12 and Fe2Mo3O12 were detected in FSP-U, this supports that both phases are crucial for the selective reaction pathway. To further investigate the influence of catalyst structure on concentration and temperature distribution, their structural composition was unraveled by synchrotron XRD profiling after selective propylene oxidation.
 | ||
| Fig. 5 Schematic illustration of the reaction network for selective propylene oxidation over FSP-Co and FSP-U. Bold arrows indicate the most favoured reaction pathway and dashed arrows the least favoured. The arrows depict the general pathways but do not imply more detailed mechanistic information. | ||
Ex situ synchrotron XRD profiling after selective propylene oxidation
The characterization of the 4-component Bi–Mo–Co–Fe–O system requires advanced and complementary techniques, including Raman spectroscopy, synchrotron XRD and multi-edge X-ray absorption spectroscopy (XAS).22 Due to the complex metal oxide phase mixtures, we used synchrotron-based methods for spatially-resolved characterization. Note that access is limited and thus, we report on selected ex situ synchrotron XRD profiles of FSP-Co and FSP-U after testing in selective propylene oxidation (Fig. 6a and b). The spatial profiling of the catalyst structure with respect to crystalline metal oxide phase amounts and crystallite sizes (via Rietveld refinement) allowed to relate these to the strong temperature and concentration gradients, thus giving insights on structure–activity correlations along the catalyst bed. Note that experiments in a microreactor setup showed that the crystalline metal oxide phase composition of each catalyst remained stable after catalytic testing with cooling in He atmosphere. | ||
| Fig. 6 Ex situ XRD profiles of FSP-U (a, left) and FSP-Co (b, right) acquired after catalytic testing in selective propylene oxidation with subsequent cooling in inert He atmosphere. Distribution of crystalline phases (c and d) and crystallite sizes (e and f) derived from Rietveld refinement. The results were averaged and plotted over the relative catalyst bed position. Non-averaged data points are given together with additional Rietveld refinement results in the ESI,† section S5. Note that crystallite sizes of certain metal oxides could not be stably refined. | ||
Similar to the initial compositions of both catalysts (see Table 3), also the (in situ) activated states of each catalyst strongly differed from each other. Notably, each metal oxide was detected along the entire catalyst bed of both systems, but several quantitative differences were found (Fig. 6c and d). In the case of FSP-U, gradients in the amount of β-CoMoO4, α-CoMoO4 and Mo18O52 were observed (Fig. 6c). The strongest gradient was detected in the first third of the catalyst bed, with an increasing amount of β-CoMoO4 (from ∼54 to 59 wt%) together with decreasing α-CoMoO4 (∼6 to 4 wt%) and Mo18O52 (∼11 to 7 wt%) amounts. Thus, the hot-spot found in the first third of the bed of FSP-U was accompanied by the transformation from α-CoMoO4 to β-CoMoO4, indicating that both phases are more temperature sensitive compared to crystalline Fe2Mo3O12, Fe3O4 and α-Bi2Mo3O12, which remained stable.
In contrast to the other metal oxides, Mo18O52 was formed during catalytic reaction for FSP-U and was more prominent in the last third of the catalyst bed. The formation of Mo18O52 is probably associated to the reduction of Fe2Mo3O12 to FeMoO4 and MoO3.60 Since Ressler et al. observed Mo18O52 as an intermediate during TPR of MoO3 in propylene atmosphere,61 this might indicate a more reduced or less re-oxidized catalyst with lower bed temperatures. This is probably attributed to the lower oxygen concentration (Fig. 1a) and would further explain the increasing molar ratio of oxygen/propylene towards the end of the bed (Fig. 4). Moreover, it is possible that the higher temperatures in the first third of the bed promoted the incorporation of Mo18O52 together with iron oxide into β-CoMoO4, forming β-Co0.7Fe0.3MoO4. Since also highest acrolein formation was found in the first third of the bed, at constant α-Bi2Mo3O12 amount, this suggests a selective role of β-Co0.7Fe0.3MoO4. The phase interaction between β-Co0.7Fe0.3MoO4 and α-Bi2Mo3O12 is generally assumed to enhance catalytic performance.62,63 Due to their isostructural nature, β-CoMoO4 and β-Co0.7Fe0.3MoO4 could not be clearly distinguished even by synchrotron XRD. Still, the corresponding crystallite sizes of β-CoMoO4 increased significantly in the first third of the bed, which could indicate that more iron was incorporated at higher temperatures (Fig. 6e). Notably, the higher crystallite sizes of β-CoMoO4/β-Co0.7Fe0.3MoO4 directly correlated with increasing acrolein amounts in relation to propylene conversion in both catalysts (see ESI,† Fig. S8), further strengthening its relevance for high selectivity. Notably, crystallite sizes of β-CoMoO4/β-Co0.7Fe0.3MoO4 were in a comparable regime for FSP-U and FSP-Co, and those of Fe3O4 and Co3O4 remained nearly constant, while an increase in crystallite sizes of γ-Bi2MoO6 was detected in the last third of the bed of FSP-Co (Fig. 6e and f). Hence, the different temperature gradients and hot-spot positions within each catalyst bed affected metal oxide phases differently in both systems.
In contrast to FSP-U, α-CoMoO4 was completely transformed to β-CoMoO4 in FSP-Co, without showing any pronounced gradients in β-CoMoO4 amounts (∼68–70 wt%, Fig. 6d). Even though this implied a relatively high amount of tetrahedrally coordinated Mo6+ sites, which were for example discussed within scheelite-structured bismuth molybdates (e.g., Bi3FeMo2O12, α-Bi2Mo3O12) to enhance catalytic performance,64 FSP-Co was less selective. This again points to the relevance of phase cooperation65 as well as to the donor and/or acceptor properties of the respective metal oxide phases.66 Notably, the amount of crystalline Co3O4 increased in the last half of the catalyst bed, which correlated with an increased oxygen consumption, higher COx formation and thus higher hot-spot temperature. At the same time, this correlated with a decreased Bi3FeMo2O12 amount, which was in turn higher in the first half of the bed. Hence, these results support the selective role of Bi3FeMo2O12 and the unselective role of Co3O4 found in the context of selective isobutene oxidation.28 Moreover, Bi3FeMo2O12 might moderate the oxygen mobility through the catalyst bulk, while Co3O4 enhances the oxygen mobility drastically. This supports the findings of Liotta et al., who attributed an increased activity of Co3O4 to the higher mobility of lattice oxygen forming mainly CO2 in total oxidation experiments.67 Consequently, the local insights into catalyst structure could be correlated to distinct features in catalytic activity and selectivity, thus providing information on the roles and cooperation of the metal oxide phases in catalytic reaction.
In the end, the phase mixture within FSP-U led to a more moderate oxygen mobility compared to FSP-Co, resulting in a better balance of the oxygen/propylene concentration along the reactor and thus a less pronounced hot-spot. Hence, the molybdate-based structures in FSP-U (i.e., α-Bi2Mo3O12, Fe2Mo3O12) revealed better redox properties compared to the increased amounts of single metal oxides (i.e., Fe3O4, Co3O4) in FSP-Co, which rather favoured overoxidation. As a next step, the CPR setup further allows simultaneous structure, activity and temperature profiling of the catalyst under working conditions (operando). For demonstration of this capability, spatially-resolved XAS results (Mo K-edge) measured during selective isobutene oxidation over FSP-U are given in the ESI,† section S6.
Selective isobutene oxidation: influence of elemental composition on activity and temperature profiles
Spatially-resolved measurements of temperature and activity were further conducted during the selective oxidation of isobutene over FSP-U and FSP-Co (Fig. 7), again showing strong differences in catalytic activity. Notably, at the same GHSV (12473 h−1) different oven temperatures were required to ignite the reaction. In the case of FSP-U, the reaction started at 450 °C and an integral isobutene conversion of ∼80% with high selectivity to methacrolein (SMAC ∼80%) was detected (Fig. 7a). For FSP-Co, a lower temperature (400 °C) was sufficient to ignite the reaction, reaching an integral isobutene conversion of ∼70% (Fig. 7b). However, isobutene conversion was limited by the gas phase oxygen, as an oxygen consumption of ∼97% was measured. This was accompanied by a comparably high selectivity towards total oxidation products (SCOx ∼35%). Notably, for both catalysts no significant total amount (<0.002 μmol) of methacrylic acid was measured. Similar to selective propylene oxidation, only other minor by-products were detected and were not further followed up. The calculated carbon balance ranged within 89 ± 5% over FSP-U and 92 ± 6% over FSP-Co. This was attributed to carbon deposits as well as product polymerisation and depositions, which were inevitably observed at high olefin conversions.
 | ||
| Fig. 7 Catalytic activity, selectivity and temperature profiles (a and b), concentration profiles of the reaction products (c and d) and formation/reaction rates of the products (e and f) of FSP-U (left) and FSP-Co (right) during selective isobutene oxidation. Profiles were acquired under reaction conditions (N2/O2/C4H8/H2O = 70/14/8/8 vol%; GHSV 12473 h−1) at respective ignition temperatures and catalysts were diluted with SiC (1:3). Position 0 mm refers to the beginning and position 25 mm to the end of the catalyst bed. | ||
For both catalysts, hot-spot formation (∼475 °C) was observed within the first third of the catalyst bed. Considering the lower ignition temperature for FSP-Co, the heat release was much more pronounced over FSP-Co than over FSP-U. This resulted in a stronger gradient within the first third of the catalyst bed of FSP-Co. The temperature gradient was rather constant (∼410–390 °C) within the last part of the reactor (9–25 mm) considering the lower oxygen content (Fig. 7b), which probably even led to carbon formation. Such carbon formation has been typically observed for propylene oxidation under reducing atmosphere.23 Notably, oxygen consumption exceeded isobutene conversion from the position of the hot-spot onwards (∼2 mm). In this hot-spot regime, the highest reaction rates were detected and decreased with lower temperature (Fig. 7f). Moreover, the formation rate of CO2 was higher than for methacrolein at each bed position measured. Hence, the origin of the strong hot-spot in the case of FSP-Co was clearly due to the unfavoured total oxidation of isobutene as evidenced by the increased CO2 concentration at the hot-spot position, which remained high along the entire catalyst bed (Fig. 7d).
For FSP-U, temperature and concentration gradients were more evenly distributed along the entire catalyst bed (0–25 mm), not showing any increased oxygen consumption. In contrast to FSP-Co, the hot-spot in FSP-U correlated to the highest reaction rate to methacrolein (at ∼4 mm, Fig. 7e). COx formation was enhanced in the second and last third of the catalyst bed, accompanied by decreased temperature. Hence, a temperature dependency for methacrolein formation was found, which was underlined by similar slopes of the temperature and methacrolein formation rate along the entire catalyst bed. Still, similar temperature regimes in FSP-Co resulted in enhanced oxygen consumption and thus higher CO2 formation, emphasizing again that catalyst selectivity cannot solely be controlled by temperature. At the same time, the absolute methacrolein amounts formed at similar conversion were nearly identical for both catalysts (Fig. 7c and d), thus underlining the relevance of catalyst temperature for the product formation.
The methacrolein yields measured over isobutene conversion showed nearly stoichiometric behaviour for FSP-U in the first third of the catalyst bed (up to Xisobutene ∼40%). For FSP-Co, such stoichiometric behaviour was only observed up to Xisobutene ∼10% and the methacrolein yields in dependence of isobutene conversion remained generally lower at increasing conversion (Fig. 8). This indicates that for FSP-Co by-products were already formed at low conversion directly from isobutene. Notably, for both catalysts the methacrolein yields increased rather linearly over isobutene conversion, which suggests that no significant product amounts were formed consecutively from methacrolein. Hence, in both cases by-products were mostly formed in parallel to methacrolein directly from isobutene.
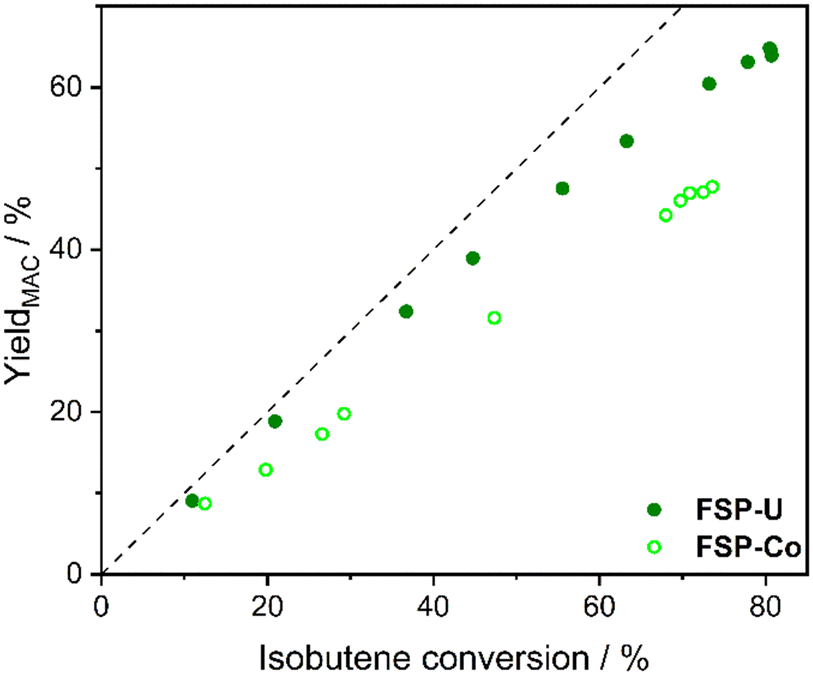 | ||
| Fig. 8 Methacrolein yield over isobutene conversion for FSP-Co (light) and FSP-U (dark) measured during selective isobutene oxidation. Dotted line indicates the progress of the stoichiometric reaction. | ||
In the case of FSP-U, CO formation was detected from relative bed position 0.25 onwards (Fig. 9a). Since this correlated with the maximum of YMAC/YCO2 at increasing oxygen/isobutene ratio, CO might have been additionally formed consecutively from methacrolein in FSP-U. However, CO formation remained rather constant in the second and last bed third, suggesting low temperature dependency and a rather low consecutive formation from methacrolein. Instead, CO formation might be rather dependent on the XO2/XC4H8 ratio, as also suggested for propylene oxidation. Again, CO was only detected at XO2/XC4H8 ratios higher than ∼0.5 (see ESI,† Fig. S4).
 | ||
| Fig. 9 Yield ratios of methacrolein/COx (a) and CO/CO2 (b) for FSP-Co (light) and FSP-U (dark) along the catalyst bed during selective isobutene oxidation. | ||
For FSP-Co, YMAC/YCO2 and YMAC/YCO remained nearly constant along the entire catalyst bed supporting that COx was mainly formed from isobutene directly. This was further underlined by the constant product concentrations from the hot-spot onwards. For both catalysts, CO2 formation was higher than CO formation, as also represented by the yield ratios of COx obtained for both catalysts (YCO/YCO2 < 1, see Fig. 9b). Hence, the reaction rate of isobutene to CO2 was significantly faster than for the oxidation to CO on both catalysts. The lower YCO/YCO2 ratios in the case of FSP-Co underlined its higher tendency of COx formation. Moreover, the spatially-resolved insights revealed that the reaction in FSP-Co took place entirely within the first third of the catalyst bed, pointing out the high catalyst activity and lower methacrolein selectivity due to total oxidation.
Similar to propylene oxidation, both catalysts enabled different reaction pathways within the complex reaction network of isobutene oxidation over Bi–Mo–Co–Fe–O catalysts (see Fig. 10), which showed comparable trends for both hydrocarbon reactants. Hence, the catalyst structures had similar effects on the catalytic behaviour during both propylene and isobutene oxidation.
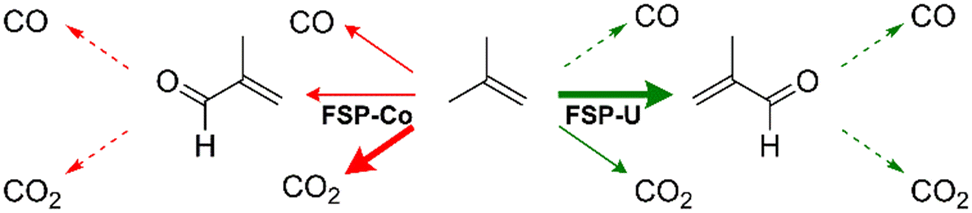 | ||
| Fig. 10 Schematic illustration of the reaction network for selective isobutene oxidation over FSP-Co and FSP-U. Bold arrows indicate the most favoured reaction pathway and dashed arrows the least favoured. The arrows depict the general pathways but do not imply more detailed mechanistic information. | ||
Comparison of reaction networks in selective olefin oxidation
To draw conclusions on the catalytic behavior and reaction networks in lower olefin oxidation, we directly compared the profiles acquired for FSP-Co and FSP-U during selective propylene and isobutene oxidation. Since both reactions required different conditions (e.g., catalyst dilution or reaction temperature, see Table 2), Thotspot/Toven ratios were calculated for better comparison of the released heat. Remarkably, these ratios revealed similar values for FSP-U and FSP-Co during both reactions (Fig. 11a). In both cases, FSP-Co favored more the total oxidation. Notably, the relative position of the hot-spot differed, being shifted more towards the end of the catalyst bed during propylene oxidation. This might be attributed to the catalyst composition resulting in a slower reaction or total oxidation of propylene compared to isobutene. The higher amount of unselective reactions over FSP-Co in isobutene oxidation culminated even in nearly full oxygen consumption at lower olefin conversions, despite the catalyst was more diluted compared to propylene oxidation. Hence, the gas phase oxygen was more easily converted to CO and CO2 in the presence of a second methyl group. | ||
| Fig. 11 Ratios of Thotspot/Toven (a) and oxygen/olefin concentration (b) along the catalyst bed of FSP-U and FSP-Co during selective propylene (filled) or isobutene (empty) oxidation. | ||
As discussed in the previous sections, the released heat during each reaction correlated with the oxygen to hydrocarbon ratio, with low ratios resulting in increased hot-spot temperatures (Fig. 11a and b). Notably, the oxygen to hydrocarbon concentration was nearly identical for FSP-U over both propylene and isobutene conversion (see ESI,† Fig. S6). This went along with similar amounts of acrolein and methacrolein formed in each reaction. Remarkably, the reaction rate achieved in the first third of the bed for FSP-U was about twice as high for methacrolein compared to acrolein. Thus, the selective oxidation of isobutene proceeded faster than the selective propylene oxidation, which is most likely attributed to its additional methyl group. This further resulted in a higher integral conversion of isobutene at higher GHSV compared to propylene.
For both reactions, similar CO amounts were detected, emphasizing its previously discussed dependency on the oxygen to hydrocarbon ratio. Notably, CO formation was found slower in isobutene oxidation than CO2 formation, but equally fast in propylene oxidation. This could indicate that the olefins rather tend to form CO2, while acrolein or methacrolein oxidation rather results in CO formation, supported by the apparently greater stability of methacrolein compared to acrolein. The consecutive formation of CO and CO2 from acrolein competed with its partial oxidation to acrylic acid, which resulted in similar amounts of all three by-products over FSP-U. In contrast, methacrolein was not found to be partially oxidized to methacrylic acid. Hence, the partial oxidation of acrolein proceeded faster than the total oxidation of propylene over FSP-U, while the opposite trend was observed for the partial oxidation of methacrolein. However, with higher temperatures in propylene oxidation over FSP-Co subsequent acrylic acid formation could not be observed. Regardless of the catalyst, it was found that for propylene oxidation the consecutive oxidation of acrolein was more critical for the overall catalytic performance in terms of selectivity, while for isobutene the parallel reactions to its selective oxidation seemed to determine the performance.
Overall, the results show that both catalysts enabled comparable reaction pathways (selective vs. unselective) in both selective propylene and isobutene oxidation. Hence, the catalytic performance and released heat were rather predominated by the catalyst structure than by the applied process conditions. In particular, the metal oxide phase ensemble of each catalyst was found to affect oxygen mobility during lower olefin oxidation in a similar way regardless of feedstock, indicating that a certain optimum in the oxygen to olefin ratio and catalyst temperature is needed for high selectivity. Even though it was reported that temperature profiles in selective propylene oxidation strongly depend on the experimental conditions (e.g., temperature, pressure, oxygen-to-propylene or water-to-propylene ratio),48 it seems that such parameters rather influence the temperature and concentration distribution along the reactor than the overall integral catalytic performance. Finally, the spatial profiles of FSP-Co and FSP-U represented a suitable reflection of the selective and unselective reaction pathways in lower olefin oxidation.
Conclusions
Two FSP-prepared Bi–Mo–Co–Fe–O catalysts with different elemental composition were investigated during selective oxidation of propylene and isobutene by spatially-resolved experiments in a compact profile reactor. The catalysts were chosen as model systems exhibiting high selectivity towards acrolein/methacrolein (FSP-U), or unselectively forming other by-products such as CO and CO2 (FSP-Co). Spatial profiling revealed strong temperature and concentration gradients along the reactor with additional structural insights into metal oxide phase distribution.The initial composition of the as-prepared catalyst directly influenced the reaction network of both selective propylene and isobutene oxidation. This was attributed to a different oxygen mobility of specific metal oxide phases when cooperating with further phases in the 4-component system. In particular, single metal oxide phases detected in FSP-Co (i.e., Fe3O4, Co3O4) lead to unselective reactions, while molybdate structures (e.g., α-Bi2Mo3O12, Fe2Mo3O12) within FSP-U were found to be more selective. Probably, they moderate oxygen availability and thus improve the redox properties and decrease total oxidation. Consequently, the more active and unselective catalyst FSP-Co additionally showed increased oxygen consumption compared to FSP-U. This further resulted in a more distinct hot-spot and high CO2 formation. In contrast, temperature and concentration gradients were more uniform along the catalyst bed for FSP-U.
Notably, both catalysts showed several parallels in selective propylene and isobutene oxidation, e.g., similar oxygen/olefin concentration gradients. Moreover, the heat release during each reaction was found nearly identical for both catalysts, although the hot-spot positioned differed for FSP-Co. This was attributed to the higher activity and thus faster reaction rates found in selective isobutene oxidation. Overall, these results show that the catalyst composition affected the temperature and concentration profiles during each reaction in a comparable way.
The distribution of crystalline metal oxide phases of FSP-U and FSP-Co were obtained by spatially-resolved synchrotron XRD with Rietveld refinement after testing in propylene oxidation. For FSP-U, strongest gradients were observed for crystalline β-CoMoO4, α-CoMoO4 and Mo18O52, which resulted in different amounts of tetrahedral and octahedral coordinated Mo along the catalyst bed. An increased amount of tetrahedrally coordinated Mo detected in the first third of the bed correlated with an increased acrolein formation and was further attributed to the formation of selectively acting β-Co0.7Fe0.3MoO4, with larger crystallite sizes of β-CoMoO4/β-Co0.7Fe0.3MoO4 in the hot-spot regime. Increased amounts of Mo18O52 towards the end of the catalyst bed of FSP-U indicated a higher degree of catalyst reduction, which correlated with a lower oxygen concentration and higher COx formation instead. For FSP-Co, the phase mixture differed from FSP-U. An increased amount of Co3O4 probably enhanced the total oxidation, while Bi3FeMo2O12 performed more selectively. In addition, the high temperature in the catalyst bed led to larger crystallite sizes of γ-Bi2MoO6. Although the phase interplay of the entire phase mixture including the surface structure in such complex 4-component systems is crucial for the overall catalytic performance, especially ternary phases (i.e., β-Co0.7Fe0.3MoO4, Bi3FeMo2O12) were found beneficial for enhancing selectivity, while single metal oxides (i.e., Fe3O4, Co3O4) favoured the highly exothermic total oxidation in both selective oxidation reactions.
Overall, the spatially-resolved studies of Bi–Mo–Co–Fe-oxides in selective olefin oxidation have provided a further understanding of structure–activity correlations, which is crucial for knowledge-based catalyst design. In future, the spatial profiling approach can be extended towards e.g., long-term stability tests or kinetic modelling for a more profound understanding of the reaction mechanisms. Moreover, spatially-resolved studies on multi-zone catalyst beds with systems clearly differing in activity appear promising to further understand industrially-relevant multicomponent catalysts. Finally, the approach to combine spatially-resolved activity/performance, temperature and structural studies should be extended to further operando techniques, such as XAS, XRD/PDF or Raman spectroscopy.
Author contributions
Conceptualization: LK, JDG, TS; data curation: LK; formal analysis: LK; funding acquisition: JDG; investigation: LK; methodology: LK, JDG, RH, TS; project administration: LK, JDG; resources: JDG; supervision: JDG, TS; validation: LK, SW, JDG, TS; visualization: LK; writing – original draft preparation: LK; writing – review and editing: LK, SW, RH, TS, JDG.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge SOLEIL (Saint-Aubin, France) for provision of synchrotron radiation facilities and we would like to thank Dr. Camille La Fontaine and Dr. Valérie Briois for assistance in using beamline ROCK for XAS measurements (proposal number 20201100). The work at ROCK was supported by a public grant overseen by the French National Research Agency (ANR) as part of the “Investissements d'Avenir” program (reference: ANR-10-EQPX-45). We acknowledge DESY (Hamburg, Germany), a member of Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at beamline P21.1 (PETRA III) and we would like to thank Dr. Ann-Christin Dippel and Dr. Ida Gjerlevsen Nielsen for assistance in XRD measurements. We acknowledge KIT and DFG for financing the Raman spectrometer system (INST 121384/73-1). We are grateful to Dr. Birte Wollak for the support with profile measurements and beamtime, as well as to Dr. Abhijeet Gaur, Dr. Bidyut Bikash Sarma and Dr. Matthias Stehle for assistance during beamtimes. We thank Dr. Maik Finsel and Michael Schmidt from Reacnostics GmbH for the technical support with the profile reactor. We thank KIT and the CRC1441 “TrackAct” (under project-ID 426888090) for financial support.Notes and references
- J. Haber, Fundamentals of Hydrocarbon Oxidation, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 3359–3384 Search PubMed.
- R. K. Grasselli and J. D. Burrington, Oxidation of Low-Molecular-Weight Hydrocarbons, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 3479–3489 Search PubMed.
- G. Centi, F. Cavani and F. Trifirò, Trends and Outlook in Selective Oxidation, Selective Oxidation by Heterogeneous Catalysis, Springer, Boston, MA, 2001, pp. 1–24 Search PubMed.
- D. Arntz, A. Fischer, M. Höpp, S. Jacobi, J. Sauer, T. Ohara, T. Sato, N. Shimizu and H. Schwind, Acrolein and Methacrolein, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2007, vol. 1, pp. 329–346 Search PubMed.
- U. Bornscheuer, R. W. Fischer, L. J. Gooßen, R. Schlögl, R. Schomäcker and S. Schunk, Katalytische Oxidationsreaktionen als Schlüsseltechnologie, Katalytische Oxidationsreaktionen als Schlüsseltechnologie, German Catalysis Society, 2015, ISBN: 978-3-89746-176-5, https://dechema.de/dechema_media/Downloads/Positionspapiere/Positionspapier_GeCatS_2015_final.pdf, (accessed 15.01.2024) Search PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- Y. Moro-Oka and W. Ueda, Adv. Catal., 1994, 40, 233–273 CAS.
- R. K. Grasselli, Top. Catal., 2002, 21, 79–88 CrossRef CAS.
- J. F. Brazdil, Appl. Catal., A, 2017, 543, 225–233 CrossRef CAS.
- P. Sprenger, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2017, 7, 5628–5642 CrossRef CAS.
- A. T. Bell, J. Catal., 2022, 408, 436–452 CrossRef CAS.
- Z. Zhai, X. Wang, R. Licht and A. T. Bell, J. Catal., 2015, 325, 87–100 CrossRef CAS.
- R. S. Mann and D. W. Ko, J. Catal., 1973, 30, 276–282 CrossRef CAS.
- J. Liu, G. Wang, X. Zhu, C. Li and H. Shan, Appl. Surf. Sci., 2019, 470, 846–853 CrossRef CAS.
- F. Benyahia and A. M. Mearns, Appl. Catal., 1990, 66, 383–393 CrossRef CAS.
- B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4351–4360 RSC.
- J.-D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037–3047 RSC.
- J.-D. Grunwaldt and C. G. Schroer, Chem. Soc. Rev., 2010, 39, 4741–4753 RSC.
- A. M. Beale, S. D. Jacques and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4656–4672 RSC.
- H. Topsøe, J. Catal., 2003, 216, 155–164 CrossRef.
- A. Chakrabarti, M. E. Ford, D. Gregory, R. Hu, C. J. Keturakis, S. Lwin, Y. Tang, Z. Yang, M. Zhu, M. A. Bañares and I. E. Wachs, Catal. Today, 2017, 283, 27–53 CrossRef CAS.
- L. Klag, T. L. Sheppard and J.-D. Grunwaldt, ChemCatChem, 2023, 15, e202201276 CrossRef CAS.
- P. Sprenger, M. Stehle, A. Gaur, A. M. Gänzler, D. Gashnikova, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2018, 8, 6462–6475 CrossRef CAS.
- P. Sprenger, T. L. Sheppard, J.-P. Suuronen, A. Gaur, F. Benzi and J.-D. Grunwaldt, Catalysts, 2018, 8, 356–376 CrossRef.
- P. Sprenger, M. Stehle, A. Gaur, J. Weiß, D. Brueckner, Y. Zhang, J. Garrevoet, J.-P. Suuronen, M. Thomann, A. Fischer, J.-D. Grunwaldt and T. L. Sheppard, ChemCatChem, 2021, 13, 2483–2493 CrossRef CAS.
- K. Amakawa, J. M. Mauss, P. Müller, B. Hinrichsen, S. Hirth, A. Bader, S. W. T. Price, S. D. Jacques and J. Macht, Sci. Adv., 2023, 9, eadh5331 CrossRef CAS PubMed.
- M. Stehle, A. Gaur, S. Weber, T. L. Sheppard, M. Thomann, A. Fischer and J.-D. Grunwaldt, J. Catal., 2022, 408, 339–355 CrossRef CAS.
- L. Klag, A. Gaur, M. Stehle, S. Weber, T. L. Sheppard and J.-D. Grunwaldt, ACS Catal., 2023, 13, 14241–14256 CrossRef CAS.
- K. Morgan, J. Touitou, J.-S. Choi, C. Coney, C. Hardacre, J. A. Pihl, C. E. Stere, M.-Y. Kim, C. Stewart and A. Goguet, ACS Catal., 2016, 6, 1356–1381 CrossRef CAS.
- A. Urakawa, N. Maeda and A. Baiker, Angew. Chem., Int. Ed., 2008, 47, 9256–9259 CrossRef CAS PubMed.
- A. Goguet, C. Stewart, J. Touitou and K. Morgan, In situ spatially resolved techniques for the investigation of packed bed catalytic reactors: Current status and future outlook of Spaci-FB, in Advances in Chemical Engineering, ed. A. G. Dixon and O. Deutschmann, Academic Press, 2017, vol. 50, pp. 131–160 Search PubMed.
- J. Touitou, K. Morgan, R. Burch, C. Hardacre and A. Goguet, Catal. Sci. Technol., 2012, 2, 1811–1813 RSC.
- J. Touitou, R. Burch, C. Hardacre, C. McManus, K. Morgan, J. Sá and A. Goguet, Analyst, 2013, 138, 2858–2862 RSC.
- C. Stewart, E. K. Gibson, K. Morgan, G. Cibin, A. J. Dent, C. Hardacre, E. V. Kondratenko, V. A. Kondratenko, C. McManus, S. Rogers, C. E. Stere, S. Chansai, Y. C. Wang, S. J. Haigh, P. P. Wells and A. Goguet, ACS Catal., 2018, 8, 8255–8262 CrossRef CAS PubMed.
- D. Decarolis, A. H. Clark, T. Pellegrinelli, M. Nachtegaal, E. W. Lynch, C. R. A. Catlow, E. K. Gibson, A. Goguet and P. P. Wells, ACS Catal., 2021, 11, 2141–2149 CrossRef CAS PubMed.
- R. Horn, O. Korup, M. Geske, U. Zavyalova, I. Oprea and R. Schlögl, Rev. Sci. Instrum., 2010, 81, 064102 CrossRef CAS PubMed.
- O. Korup, S. Mavlyankariev, M. Geske, C. F. Goldsmith and R. Horn, Chem. Eng. Process., 2011, 50, 998–1009 CrossRef CAS.
- B. Wollak, D. E. Doronkin, D. Espinoza, T. Sheppard, O. Korup, M. Schmidt, S. Alizadefanaloo, F. Rosowski, C. Schroer, J.-D. Grunwaldt and R. Horn, J. Catal., 2022, 408, 372–387 CrossRef CAS.
- M. Geske, O. Korup and R. Horn, Catal. Sci. Technol., 2013, 3, 169–175 RSC.
- D. Espinoza, B. Wollak, T. L. Sheppard, A. C. Dippel, M. Sturm, O. Gutowski, M. Schmidt, O. Korup and R. Horn, ChemCatChem, 2022, 14, e202200337 CrossRef CAS.
- R. Schlögl, Top. Catal., 2016, 59, 1461–1476 CrossRef.
- J. Haber and W. Turek, J. Catal., 2000, 190, 320–326 CrossRef CAS.
- H. Tan, J. Downie and D. Bacon, Can. J. Chem. Eng., 1989, 67, 412–417 CrossRef CAS.
- H. Redlingshöfer, A. Fischer, C. Weckbecker, K. Huthmacher and G. Emig, Ind. Eng. Chem. Res., 2003, 42, 5482–5488 CrossRef.
- L. Bui, R. Chakrabarti and A. Bhan, ACS Catal., 2016, 6, 6567–6580 CrossRef CAS.
- S. Breiter and H.-G. Lintz, Catal. Lett., 1994, 24, 343–348 CrossRef CAS.
- S. Breiter and H.-G. Lintz, Chem. Eng. Sci., 1995, 50, 785–791 CrossRef CAS.
- G. Ganzer and H. Freund, Ind. Eng. Chem. Res., 2019, 58, 1857–1874 CrossRef CAS.
- M. Stehle, T. L. Sheppard, M. Thomann, A. Fischer, H. Besser, W. Pfleging and J.-D. Grunwaldt, Catal. Sci. Technol., 2021, 11, 5781–5790 RSC.
- H. Redlingshöfer, O. Kröcher, W. Böck, K. Huthmacher and G. Emig, Ind. Eng. Chem. Res., 2002, 41, 1445–1453 CrossRef.
- K. Schuh, W. Kleist, M. Høj, V. Trouillet, A. D. Jensen and J.-D. Grunwaldt, Chem. Commun., 2014, 50, 15404–15406 RSC.
- J. Kieffer, V. Valls, N. Blanc and C. Hennig, J. Synchrotron Radiat., 2020, 27, 558–566 CrossRef CAS PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- A. Bielański and J. Haber, Catal. Rev.: Sci. Eng., 1979, 19, 1–41 CrossRef.
- R. K. Grasselli, J. D. Burrington, D. J. Buttrey, P. DeSanto, C. G. Lugmair, A. F. Volpe and T. Weingand, Top. Catal., 2003, 23, 5–22 CrossRef CAS.
- N. Arora, G. Deo, I. E. Wachs and A. M. Hirt, J. Catal., 1996, 159, 1–13 CrossRef CAS.
- R. B. Licht and A. T. Bell, ACS Catal., 2016, 7, 161–176 CrossRef.
- A. B. Getsoian, V. Shapovalov and A. T. Bell, J. Phys. Chem. C, 2013, 117, 7123–7137 CrossRef CAS.
- A. Gaur, M. Stehle, K. V. Raun, J. Thrane, A. D. Jensen, J.-D. Grunwaldt and M. Høj, Phys. Chem. Chem. Phys., 2020, 22, 11713–11723 RSC.
- T. Ressler, J. Wienold, R. E. Jentoft and T. Neisius, J. Catal., 2002, 210, 67–83 CrossRef CAS.
- D.-H. He, W. Ueda and Y. Moro-Oka, Catal. Lett., 1992, 12, 35–44 CrossRef CAS.
- H. Ponceblanc, J.-M. M. Millet, G. Coudurier and J. C. Védrine, Synergy Effect of Multicomponent Co, Fe, and Bi Molybdates in Propene Partial Oxidation, in Catalytic Selective Oxidation, ed. S. T. Oyama and J. W. Hightower, ACS Publications, Washington, DC, 1993, pp. 262–272 Search PubMed.
- J. F. Brazdil, Catal. Sci. Technol., 2015, 5, 3452–3458 RSC.
- R. K. Grasselli, Catal. Today, 2014, 238, 10–27 CrossRef CAS.
- L. T. Weng and B. Delmon, Appl. Catal., A, 1992, 81, 141–213 CrossRef CAS.
- L. F. Liotta, M. Ousmane, G. Di Carlo, G. Pantaleo, G. Deganello, G. Marcì, L. Retailleau and A. Giroir-Fendler, Appl. Catal., A, 2008, 347, 81–88 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01445b |
| ‡ Current address: Institute of Chemical Technology, Leipzig University, 04103 Leipzig, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |