
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient iron-catalyzed direct acylation of amines with carboxylic acids and esters under oxygenated conditions†
Maria
Obieta
,
Garazi
Urgoitia
,
María Teresa
Herrero
and
Raul
SanMartin
 *
*
Department of Organic and Inorganic Chemistry, Faculty of Science and Technology, University of the Basque Country (UPV-EHU), 48940 Leioa, Spain. E-mail: raul.sanmartin@ehu.eus
First published on 3rd January 2024
Abstract
Amides are ubiquitous in natural and synthetic compounds, and amidation is by far the most prevalent reaction in medicinal chemistry. In addition, atom-economical procedures for the direct amidation of esters or acids with amines are in high demand. Encouraged by the abundance and low toxicity of iron compounds, we envisaged that a new iron-catalyzed protocol for the acylation of amines with both esters and carboxylic acids could be designed if the iron catalyst was combined with dioxygen. Several experiments were carried out in order to define the iron source and to evaluate the effect of molecular oxygen, additives and different reaction media. A number of substrates were then reacted under optimized conditions, and experimental studies (kinetic, radical trapping and electron paramagnetic resonance experiments) were conducted to shed light on the reaction mechanism. As a result, a new use for dioxygen as an inducer of the direct amidation between amines and carboxylic acids or esters has been found. Thus, an earth-abundant first-row metal catalyst (Fe(acac)3) at low loading combined with pivalic acid and molecular oxygen at atmospheric pressure triggers the reaction in a biodegradable greener solvent such as diethyl carbonate. More than 65 high-yielding examples prove the generality of the procedure, which also resulted to be scalable. In addition, insight into the mechanism behind this reaction taking place under oxygenated conditions is provided as well as an explanation for the results obtained in the absence of dioxygen.
Introduction
There is a growing interest in the development of new iron-catalyzed reactions, stimulated by the scarcity of precious metals and the abundance and comparatively much lower toxicity of iron compounds.1 This interest, reflected in the 12-fold increase of the number of publications with the term “iron-catalyzed” in the title in the past 20 years,2 has led to the discovery of a number of procedures based on iron catalysts that rival and even exceed the performance of other metal-catalyzed protocols.3 However, there is much room for improvement and many unexplored applications for iron catalysts are yet to be discovered.4Due to the need for activating agents, coupling reagents and additives to perform amidation reactions, which are the most prevalent reactions in medicinal chemistry,5 amide formation with high atom economy has been regarded as a key goal.6
In this respect, unlike other amidation reactions that require some of the aforementioned activating agents in stoichimetric amounts (e.g. carbodiimides,7 benzotriazoles,8 carbon tetrachloride9 α,α-dichlorodiphenylmethane10 or tert-butyl isocyanide and S-phenyl benzenethiosulfonate11), direct acylation of amines with esters or carboxylic acids has been scarcely explored using iron catalysts.12,13 Indeed, most of the reports on this atom-economical process (Scheme 1) show a scope limited to primary amines and/or aliphatic acid derivatives,12b,13 and no general method for the iron-catalyzed direct acylation of amines using both carboxylic acids and esters as acylating agents has been reported to date.
 | ||
| Scheme 1 Iron-catalyzed direct amidation between carboxylic acids and amines. | ||
As part of our ongoing research on iron catalysts,14 we became interested in the iron-catalyzed acylation of amines with esters and unactivated acids. From the initial experiments we noticed a curious difference between reactions under inert conditions (Ar) and those conducted open to the atmosphere. Interestingly, in spite of the plethora of iron-catalyzed oxidative reactions found in the literature (oxidation of alcohols and methylene compounds to carbonyl compounds and carboxylic acids,15 oxidative cross-dehydrogenative coupling,16 oxidative alkenylation of benzylic C–H bonds with diazo compounds,17 oxidative homo-coupling of alkenyllithiums,18 alkene syn-dihydroxylation19 or 1,2-alkylarylation of styrenes with α-carbonyl alkyl bromides and indoles,20inter alia), no oxidant has ever been employed in this field of iron-catalyzed direct amidation, let alone molecular oxygen, which is abundant, inexpensive and completely harmless to the environment.
Several reports have shown the beneficial effect of molecular oxygen in reactions not directly related to classical oxidation processes, like the molecular oxygen-induced activation of certain bonds,21 of molybdenum-based Lewis acids22 and of gold catalysts.23
In addition, activation by dioxygen has resulted to be essential for certain challenging reactions such as skeletal rearrangements24 photoisomerization between the cis and the trans states of azobenzenes25 and other oxygen-driven photoisomerization reactions of alkenes and polyenes,26 redox-neutral nucleophilic aromatic substitutions leading to seven-membered ring formation,27 conversion of pincer metal complexes into metallabenzenes,28 and iron-catalyzed dearomatization of 2-naphthols.29
Accordingly, we decided to investigate the influence of dioxygen on iron-catalyzed amidation. In this paper we present (i) a highly efficient and general procedure for the direct acylation of amines applicable to both carboxylic acids and esters and (ii) a mechanistic insight into the role of the iron catalyst system and the aerobic reaction conditions.
Results and discussion
Phenyl benzoate (1a) and acetic acid (2a) were chosen as model substrates to react with benzylamine (3a). A variety of iron sources were tested as catalysts in different solvents in a preliminary screening of reaction conditions,30 and the catalyst loading was limited to 10−2 mol% in order to maximize efficiency. Most of these initial assays were carried out with acid 2a as the acylating agent, and only some promising reaction conditions were tested on benzoate 1a. From these preliminary results FeCl3, FeBr2 and Fe(acac)3 were chosen as catalysts for further optimization as well as pivalic acid as an additive (Table 1).|
|
|||
|---|---|---|---|
| Entry | 1a/2a | Conditionsa | Productb (%) |
| a Reaction conditions: 1a or 2a (0.54 mmol), amine 3a (1.5 equiv.), PivOH (1 equiv.), [Fe] (0.01 mol%), solvent (1 mL mmol−11a/2a), Ar, air or O2 (1 atm), T, 24 h. See ref. 34 and the ESI† for potential safety hazards associated with the combination of DEC and O2. b Isolated yield. c Amine 3a (1.0 equiv.). d Amine 3a (2.0 equiv.). e 0.5 equiv. of PivOH were added. f No PivOH was added. | |||
| 1 | 2a | FeCl3, PhMe, Ar, 100 °C | 4ab (8) |
| 2 | 2a | FeBr2, dioxane, Ar, 100 °C | 4ab (11) |
| 3 | 2a | Fe(acac)3, DMF, Ar, 100 °C | 4ab (7) |
| 4 | 2a | FeCl3, DMSO, Ar, 100 °C | 4ab (10) |
| 5 | 2a | Fe(acac)3, EtOH, Ar, 100 °C | 4ab (7) |
| 6 | 2a | Fe(acac)3, DMA, Ar, 100 °C | 4ab (9) |
| 7 | 2a | Fe(acac)3, DEC, Ar, 100 °C | 4ab (15) |
| 8 | 2a | Fe(acac)3, DEC, air, 100 °C | 4ab (83) |
| 9 | 2a | Fe(acac)3, DEC, O2, 100 °C | 4ab (91) |
| 10 | 2a | Fe(acac)2, DEC, O2, 100 °C | 4ab (89) |
| 11 | 2a | FeCl3, DEC, O2, 100 °C | 4ab (59) |
| 12 | 1a | Fe(acac)3, DEC, O2, 100 °C | 4aa (90) |
| 13 | 1a | Fe(acac)3, DEC, Ar, 100 °C | 4aa (50) |
| 14 | 1a | Fe(acac)3, DEC, O2, 80 °C | 4aa (90) |
| 15 | 1a | Fe(acac)3, DEC, O2, 60 °C | 4aa (84) |
| 16c | 1a | Fe(acac)3, DEC, O2, 80 °C | 4aa (36) |
| 17 | 2a | Fe(acac)3, DEC, O2, 80 °C | 4ab (39) |
| 18d | 2a | Fe(acac)3, DEC, O2, 100 °C | 4ab (98) |
| 19e | 1a | Fe(acac)3, DEC, O2, 80 °C | 4aa (92) |
| 20d,e | 2a | Fe(acac)3, DEC, O2, 100 °C | 4ab (57) |
| 21f | 1a | Fe(acac)3, DEC, O2, 80 °C | 4aa (52) |
| 22d,f | 2a | Fe(acac)3, DEC, O2, 100 °C | 4ab (12) |
| 23 | 1a | DEC, O2, 80 °C | 4aa (15) |
| 24d | 2a | DEC, O2, 100 °C | 4ab (9) |

After trying different solvents for the amidation of 2a, we found that diethyl carbonate (DEC) provided slightly better yields if combined with Fe(acac)3 (entry 7 vs. 1–6). To our surprise, the yield rose significantly from 15% to 83% when the reaction mixture in the latter solvent was heated in an open vessel (entry 7 vs. 8). Furthermore, a slight increase was observed when the reaction was carried out in an oxygen atmosphere (entry 9). Remarkably worse results were obtained from other iron(III) catalysts under the latter oxygenated conditions (entry 11). Interestingly, Fe(acac)2 showed a similar catalytic profile (entry 10). On the basis of the wider availability and lower cost of Fe(acac)3, we chose this iron(III) source to continue the optimization of reaction conditions. An excellent yield (entry 12) was obtained when benzoate 1a was subjected to the reaction conditions displayed in entry 9, but attempts to further reduce the catalyst loading were unsuccessful.30 In contrast to 2a, a moderate yield was obtained for the reaction from 1a under argon (entry 13). No change was observed when the reaction temperature was decreased to 80 °C in the case of 1a but the yield dropped to 39% from 2a (entries 14 and 16). With regard to the stoichiometry of the reaction, optimal results were obtained with 1.5 equiv. of 3a, although the yield was improved to almost quantitative values when 2 equiv. of the amine were reacted with 2a (entry 18). After adjusting the optimal amount of the additive for each substrate (entries 18–19), blank experiments proved the need for pivalic acid, especially for 2a (entries 21–22) and of the iron catalyst (entries 23–24).
The scope of the reaction was initially explored by reacting benzoate 1a with a number of primary amines 3. As shown in Table 2, the corresponding secondary benzamides 4aa–4au were isolated with moderate to good yields regardless of the structure of the amine (alkylamine, arylamine, propargylamine or allylamine) employed. However, a substantial decrease in the yield of N-arylamides 4an, 4ao and 4aq was observed in comparison with their structural analogs 4am, 4at and 4au, probably due to the steric hindrance associated to O-substituted 2-ethylaniline, 2-methoxyaniline and 2,5-dimethylaniline. This steric drawback became a serious limitation with O,O′-disubstituted anilines, and the reaction with 2,6-dimethylaniline provided only 7% of the corresponding amide 4ap. With regard to N-benzylamides 4aa, 4ac and 4ad, in addition to the good yield obtained, it should be pointed out that the amidation reaction of 4ad took place with complete retention of configuration.

This fact was confirmed by comparison of the optical rotation of (S)-(−)-α-methylbenzylamine and (S)-N-(1-phenylethyl)benzamide (4ad) with literature data.31a–c
Next, different cyclic and linear secondary amines were reacted with ester 1a to provide tertiary benzamides 4av–4bd (Table 3). No clear trend was observed for the results from cyclic amines (amides 4av–4ba), but it is noteworthy that the reaction tolerated the presence of halogen substituents (4bc) and that steric hindrance might be the reason for the moderate yield from dibenzylamine (4bd).

Table 4 shows the results from the reaction of benzylamine 3a with readily available30 esters 1b–1r. The results from the preparation of acetamide 4ab indicate that although phenyl esters (R2 = Ph) provided better yields, the procedure is also amenable to methyl, ethyl and trimethylsilyl esters. A similar trend was observed for phenylacetamide 4be. Other arylacetamides 4bf, 4bg and 4bh were easily prepared from the corresponding methyl and phenyl esters. Good yields were also obtained from phenylbenzoates 1k–1n, and the presence of sterically hindering or reactive groups (o-methyl and o-hydroxy for 1m and 1k, respectively) did not affect the reaction yield. Ethyl 2-cyanoacetate (1o), butyl formate (1p) and phenyl pivaloylacetate (1q) were successfully reacted with benzylamine (3a) under optimized conditions, and a good yield for prolinamide 4bp was obtained from phenyl N-tosylprolinate (1r).

Interestingly, the reaction between commercially available ethyl 4-bromobutanoate (1s) and benzylamine (3a) provided selectively 1-benzylpyrrolidin-2-one (4bq) as a result of a one-pot amidation/nucleophilic substitution process (Scheme 2). Although the conversion was relatively low (unreacted starting materials were obtained along with cyclized product 4bq), it should be pointed out that no substitution by-product (e.g. ethyl 4-(benzylamino)butanoate) was detected in the reaction crude.
 | ||
| Scheme 2 One-pot amidation/nucleophilic substitution. | ||
Regarding the use of optically active compounds and following our previous observation on the preparation of (S)-N-(1-phenylethyl)benzamide (4ad, Table 2), the reaction between phenyl (S)-2-methoxy-2-phenylacetate (1j) and (S)-1-phenylethan-1-amine (3c) took place with complete stereoselectivity, as target (S)-2-methoxy-2-phenyl-N-((S)-1-phenylethyl)acetamide (4br) was obtained with excellent yield as a single diastereoisomer and enantiomer (Scheme 3). A complete retention of configuration was also observed for the reaction, leading to 4bh (Table 4).31d–g X-ray diffractometry of both 4bh and 4br confirmed32 the displayed stereochemistry, which is shown in Fig. 1.
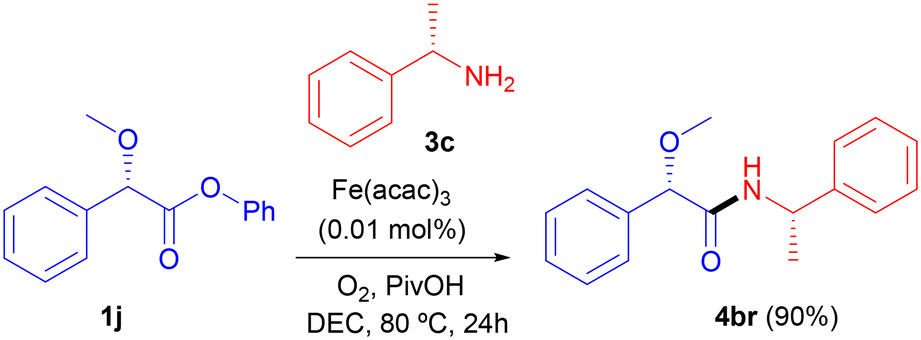 | ||
| Scheme 3 Stereoselective amidation. | ||
 | ||
| Fig. 1 ORTEP diagrams of chiral amides 4bh and 4br with thermal ellipsoids given at 50% probability level. | ||
Finally, the model reaction was conveniently scaled up. Thus, 2 grams of starting benzoate 1a were reacted with benzylamine (3a) to provide benzyl benzamide (4aa) with 95% yield. Furthermore, the fact that this scalable reaction is carried out in diethyl carbonate (DEC) makes the process more sustainable. Indeed, diethyl carbonate is an environment-friendly and biodegradable solvent with mild toxicity presently used in pharmaceutical formulations, in green liquid–liquid extractions, as a component of electrolytes in lithium batteries and as a fuel additive.33 However, even considering the atmospheric pressure employed and the low volatility of this solvent (b.p.: 126 °C, vapor pressure at 80 °C: 0.2 bar), its flammability in an oxygen-rich atmosphere should not be ignored,34 and therefore a hazard warning note is included in the experimental procedures.30 In spite of that, the use of pure dioxygen as an oxidant and organic carbonates as solvents in reactions even at high oxygen pressures has been reported without any safety issues.33b,35
The reaction conditions optimized for the aminolysis of acetic acid (2a) with benzylamine (3a) were then applied to a variety of carboxylic acids 2 and amines 3 (Table 5). The structural diversity of both acid and amine reagents should be pointed out, and most of the 23 secondary amides prepared in this way were obtained with good yields, although slightly inferior results were achieved from unsaturated acids (amides 4cd, 4cg, 4ci and 4cj). The presence of a bulky o-ethyl group in 2-ethylaniline or the use of branched carboxylic acids such as 2-(prop-2-yn-1-yl)octanoic acid did not have a deleterious effect on the reaction outcome (amides 4ca and 4ck, respectively). In addition, as with the aminolysis of esters 1, the acylation of amines 3 with acids 2 occurred with complete retention of configuration, and both amides 4bh and 4ce were isolated as single enantiomers.31,32

To conclude the scope studies of this amidation reaction, piperidine, pyrrolidine, morpholine, indoline and tetrahydroisoquinoline were reacted with a handful of structurally dissimilar carboxylic acids 2, thus providing the tertiary amides 4 shown in Table 6. Good yields were obtained in all cases, irrespective of the structure of the amidation partner.
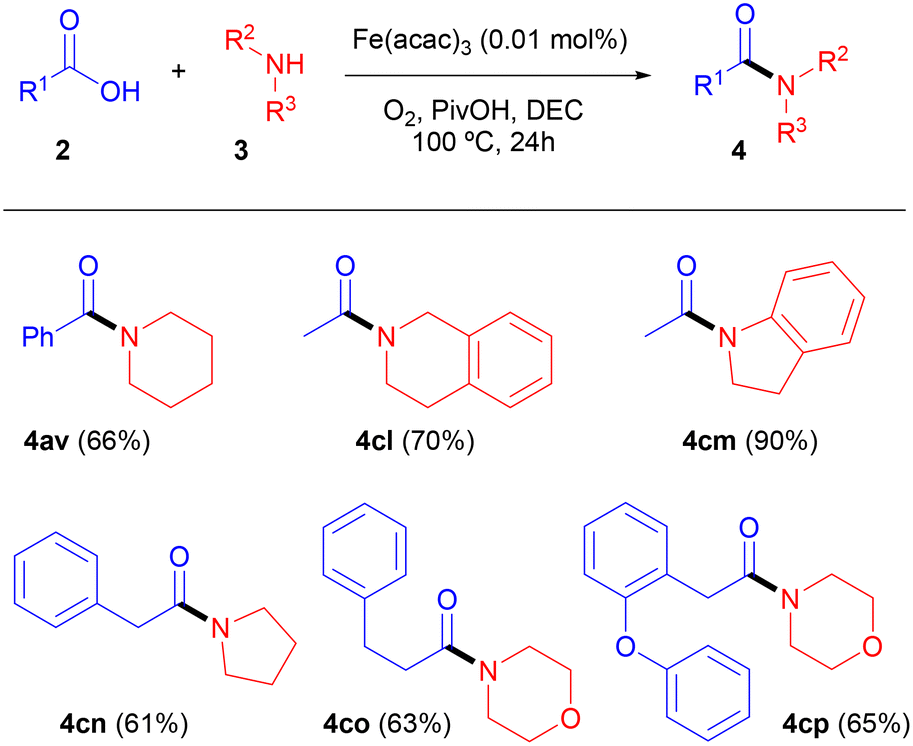
On another note, it should be mentioned that in addition to the morpholinamide derived from 2-(2-phenoxyphenyl)acetic acid (4cp), several N-benzylamides derived from acids 2 (4ci and 4ck, Table 5) and from esters 1 (4bh, 4br, 4bo, and 4bp, Table 4 and Scheme 3) have been synthetized for the first time in this work.
Since the presented protocol can use esters and acids as acylating agents for amines, we decided to compare the results in those cases where the same amide had been prepared. No difference could be found for acetamide 4ab (99% vs. 98%), but for secondary benzamides 4aa, 4af, 4ag and 4am and tertiary benzamide 4av significantly higher yields were obtained from esters 1 than from acids 2 (92%, 89%, 93%, 88% and 94% vs. 78%, 74%, 68%, 75% and 66%, respectively). However, the convenience of both atom-economical processes and the release of water as a by-product in the case of acid precursors cannot be ignored.
A number of experiments were then carried out to shed light on the reaction mechanism. We had observed that aminolysis of esters 1 and acids 2 took place with similar kinetics although acids required a higher temperature to react. A meaningful example of this subtle difference was the selective monoamidation of 1,1-cyclopropanedicarboxylic acid monomethyl ester (5) when treated with one equivalent of amine 3a under optimized reaction conditions (Scheme 4). Besides, as predicted, the conversion rate vs. time curves for 1a → 4aa and 2a → 4ab are very similar, both of hyperbolic shape, which probably accounts for homogeneous catalysis.36 Although the reaction temperature is 20 °C higher in the case of 2a, the reaction from 1a shows slightly faster kinetics (Fig. 2).
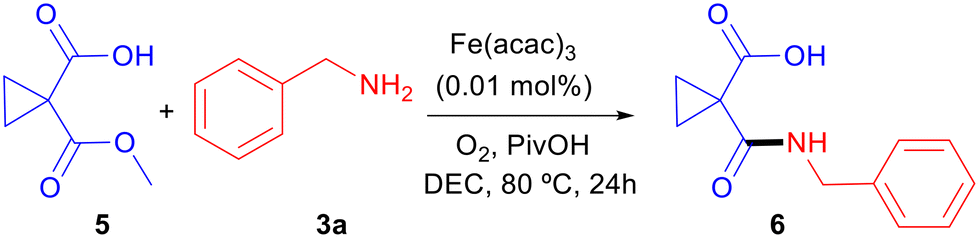 | ||
| Scheme 4 Selective amidation of 1-(methoxycarbonyl)cyclopropane-1-carboxylic acid (5). | ||
 | ||
| Fig. 2 Aminolysis of phenyl benzoate (1a) and of acetic acid (2a) with benzylamine (3a) as a function of time. | ||
UPLC-ESI-QTOF analysis of the crude (reaction time: 12 h) of the reaction between ester 1a and amine 3a revealed the presence of acetylacetone, N-benzyl-1-phenylmethanimine, phenylmethanimine, ethyl benzylcarbamate, phenol and 2,2-dimethylpropaneperoxoic acid (perpivalic acid). A small amount of both imines, along with ethyl benzylcarbamate and phenol, was also detected by GC-MS of the reaction crude. As shown in Table 1, an excess of the amine is required for both ester and acid substrates, as the use of equimolecular amounts of the amine results in moderate to low yields. Therefore we propose that the excess amine is one of the main driving forces of the reaction and that ethyl benzylcarbamate, easily formed by reaction between DEC and 3a, acts as a reservoir for 3a, as the acidic medium derived from pivalic acid releases 3a by ethanolysis of the carbamate. In fact, the reaction between ester 1a and ethyl benzylcarbamate (1.5 equiv.) under optimized reaction conditions provided target amide 4aa with good yield (83%).
Instead of benzylamine (3a), commercially available N-benzyl-1-phenylmethanimine (N-benzylidenebenzylamine) was reacted with ester 1a under optimized conditions in order to check if this imine could be a reaction intermediate. However, only 12% of 4aa was obtained.
Additionally, in order to define the role of dioxygen in this reaction, other oxidants such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and potassium peroxymonosulfate (Oxone®) were assayed under anaerobic conditions (Ar) in the the above reaction between ester 1a and amine 3a. Only target 4aa was isolated in the case of potassium peroxymonosulfate, but with a poor 9% yield.
In another experiment carried out in the absence of the acylating agent 1 or 2, the aforementioned acetylacetone, N-benzyl-1-phenylmethanimine, phenylmethanimine and ethyl benzylcarbamate were detected by UPLC-ESI-QTOF. Furthermore, after performing the same experiment, water was added, the mixture was extracted with dichloromethane and the resulting aqueous solution was acidified with H2SO4. Potassium permanganate titration provided evidence for the presence of Fe2+ ions in the latter solution and revealed that almost half of the initial amount of Fe(acac)3 had been reduced to Fe(II) species.
A significant number of iron-catalyzed reactions mediated by molecular oxygen are based on radical pathways.15c,37 Therefore, the participation of radical species was investigated by adding several radical scavengers and trapping agents to the reaction between ester 1a and amine 3a. As shown in Table 7, little or no inhibition was observed even when this radical interception was attempted using stoichiometric amounts of the aforementioned trapping reagents. Sodium azide, a typical quenching agent for singlet oxygen, had no effect on the reaction outcome either.
|
|
||
|---|---|---|
| Entry | Radical trapping reagenta | 4aa (%) |
| a Reaction conditions: ester 1a (0.54 mmol), amine 3 (0.81 mmol), radical trapping reagent (0.54 mmol), PivOH (0.27 mmol), Fe(acac)3 (0.01 mol%), DEC (1 mL mmol−11a), O2 (1 atm), 80 °C, 24 h. b Isolated yields. TEMPO: 2,2,6,6-tetramethylpiperidine 1-oxyl; BHT: butylated hydroxytoluene; DPPH: 2,2-diphenyl-1-picrylhydrazyl; PBN: N-tert-butyl-α-phenylnitrone. | ||
| 1 | TEMPO | 92 |
| 2 | Galvinoxyl | 66 |
| 3 | BHT | 93 |
| 4 | DPPH | 94 |
| 5 | PBN | 95 |
| 6 | NaN3 | 93 |

However, electron paramagnetic resonance (EPR) analysis of the reaction at t = 1 h showed two overlapping signals corresponding to two relatively stable radicals (Fig. 3a).
 | ||
| Fig. 3 Experimental (blue line) and least squares fit (red line) EPR spectra of the reaction (1a + 3a → 4aa) at t = 1 h before (a) and after (b) addition of N-tert-butyl-α-phenylnitrone (PBN). | ||
On the basis of the observed splitting pattern and g-factors, these stable radicals are O-centered radicals. After addition of PBN to the reaction mixture at t = 1 h, the EPR signals of the generated spin-adducts confirmed this hypothesis (Fig. 3b). Accordingly, it seemed that radical species (probably O-centered radicals) were formed during the progress of the reaction but they did not intervene as intermediates and did not affect the reaction outcome.
Pivalic acid has emerged as a crucial proton shuttle in various metal-catalyzed reactions. Its unique properties, demonstrated through its effectiveness in sub- and overstoichiometric amounts and even as a solvent, have been extensively studied. Replacing pivalic acid with other carboxylic acids, such as benzoic or acetic acid, often leads to significant changes in the results of the reaction, underscoring its distinct role in facilitating proton transfer.38 As mentioned before, in our case a small excess of the amine was required to obtain reasonable yields. However, the presence of pivalic acid as an additive (0.5–1.0 equiv.) had a positive impact on the reaction outcome. We therefore proposed that pivalic acid would act as a ligand facilitating the interaction of the iron center with dioxygen.
With the results of all these studies in hand, we proposed the mechanism depicted in Scheme 5 to explain the formation of 4aa by aminolysis of 1a with 3a under oxygenated conditions. After protonation of the acetylacetonate ligands of Fe(acac)3 by pivalic acid, the resulting Fe(III) ions would partially oxidize benzylamine to the corresponding imine or N-benzyl-1-phenylmethanimine in the presence of oxygen, as described by Castro39 and Xu.40 This process would release catalytic amounts of Fe(II) species, which are essential for the next step. Additionally, as a result of the interaction between Fe(acac)3 and pivalic acid, the so-formed trinuclear pivalate complex–pivalic acid adduct [Fe3O(OPiv)6(PivOH)3] contains a high-spin Fe(II) core and shows a temperature-dependent mixed-valence state.41 These Fe(II) species would readily interact with dioxygen to generate ferric superoxo species A and/or iron(III) peroxo intermediate B, which in the presence of hydrogen atom donors (e.g. starting amine 3a, pivalic acid or later released phenol) would transform into iron(III)-hydroperoxo intermediate C.42 Nucleophilic attack at this intermediate C by 1a or protonated 1a′ would form iron(III) acylperoxo complex D, which would undergo addition by amine 3a to the electrophilic carbonyl center. After prototropic tautomerism E–F, condensation would release phenol and generate key intermediate G. A nucleophilic attack by pivalic acid, akin to that experienced by iron(III)-hydroperoxo species C, would liberate N-benzylacetimidic acid 4aa′, in equilibrium with target amide 4aa. As a result, iron(III)-pivaloylperoxo complex H would form. The thermodinamically favourable homolytic cleavage of the Fe(III)–O bond of high-spin Fe(III)–alkylperoxo complexes to generate Fe(II) complexes and alkylperoxyl radicals has been discussed by Solomon and Que.43 This process would take place at complex H so that Fe(II) species that would reinitiate the catalytic cycle would be released, along with unstable acyl peroxy radical I.
 | ||
| Scheme 5 A mechanistic proposal for the iron-catalyzed aminolysis of phenyl benzoate 1a in the presence of dioxygen. | ||
Reaction of the latter with previously liberated phenol would generate persistent phenoxyl radical,44 one of the aforementioned radical species detected by EPR. Considering the radical scavenging ability of pivalic acid,45 formation of pivaloxyl radical cannot be discarded. A similar mechanism would account for the formation of 4ab.46 At this point, and although all the mechanistic steps are based on experimental results or literature reports, it should be pointed out that our proposal is purely tentative. Indeed, our assumptions that the aminolysis step takes place on intrinsically unstable peroxide species (E–G) and about the subsequent homolysis of the Fe–O bond at complex H are somewhat questionable. However, the kinetic, radical trapping, detection by UPLC-ESI-QTOF analysis, electron paramagnetic resonance and additional experiments conducted along with the literature precedents on related processes offer tentative support for our proposal.
Conclusions
To sum up, a highly efficient procedure for the iron-catalyzed direct acylation of amines with carboxylic esters and acids has been developed. This scalable and atom-economical process requires the use of molecular oxygen as an inducer of the reaction along with pivalic acid as an additive. The generality of the procedure, applicable to a wide range of primary and secondary amines, is demonstrated by more than 65 examples presented in this paper. In addition, the reaction is carried out in a greener solvent, diethyl carbonate. A main mechanism based on a Fe(II)/Fe(III) cycle and the interaction of the former ion with dioxygen has been proposed to explain the role of the components in the reaction. On the basis of the experimental studies carried out, a subsidiary mechanism responsible for the results in the absence of oxygen is also suggested.Author contributions
MTH, GU and RS contributed to searching and collating of the relevant literature and the proof-reading of the document. MO and GU carried out the investigation, experiments, and analysis. MTH and RS conceptualized and supervised the study and wrote the body of the article. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basque Government (IT1583-22). G. U. and M. O. thank the University of the Basque Country (UPV/EHU) for a postdoctoral and predoctoral scholarship, respectively. Finally, technical and human support provided by SGIker is gratefully acknowledged.Notes and references
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2014, 104, 6217–6254 CrossRef PubMed; (b) H. Du, F. Deng, R. R. Kommalapati and A. S. Amarasekara, Renewable Sustainable Energy Rev., 2020, 134, 110292 CrossRef; (c) P. DaBell and S. P. Thomas, Synthesis, 2020, 52, 949–963 CrossRef CAS; (d) Y. Hong, L. Jarrige, K. Harms and E. Meggers, J. Am. Chem. Soc., 2019, 141, 4569–4572 CrossRef CAS PubMed.
- Data derived from Clarivate Web of Science, © Copyright Clarivate, 2023. All rights reserved, https://www.webofscience.com/wos/woscc/basic-search, (accessed August 2023).
- (a) M. M. Beromi, C. R. Kennedy, J. M. Younker, A. Carpenter, S. J. Mattler, J. A. Throckmorton and P. J. Chirik, Nat. Chem., 2021, 13, 156–162 CrossRef PubMed; (b) L. Yang, D. Cheng, H. Xu, X. Zeng, X. Wan, J. Shui, Z. Xiang and D. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6626–6631 CrossRef CAS PubMed; (c) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- (a) A. Fürstner, ACS Cent. Sci., 2016, 2(11), 778–789 CrossRef PubMed; (b) F. Moccia, L. Rigamonti, A. Messori, V. Zanotti and R. Mazzoni, Molecules, 2021, 26, 2728 CrossRef CAS PubMed; (c) H. Fujisaki, T. Ishizuka, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Nature, 2023, 616, 476–481 CrossRef CAS PubMed; (d) J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, T. P. Senftle, W. Liu, J. Wang, Y. Wang, A. Zhang, Q. Fu, C. Song and X. Guo, Sci. Adv., 2022, 8, eabm3629 CrossRef CAS PubMed; (e) C. Empel, S. Jana and R. M. Koenigs, in Iron Catalysis: Design and Applications, ed. J. M. Palomo, World Scientific, Singapore, 2021, ch. 7, pp. 203–252 Search PubMed.
- (a) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed; (b) J. Magano, Org. Process Res. Dev., 2022, 26, 1562–1689 CrossRef CAS.
- (a) J. S. Carey, D. Laffan, C. Thomsonc and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (b) R. Marcia de Figueiredo, J. S. Suppo and J. M. Campagn, Chem. Rev., 2016, 116, 12029–12122 CrossRef PubMed; (c) A. Taussat, R. Marcia de Figueiredo and J. M. Campagne, Catalysts, 2023, 13, 366 CrossRef CAS.
- (a) P. Xiao, Z. Zhang, J. Ge, Y. Deng, X. Chen, J.-R. Zhang, Z. Deng, Y. Kambe, D. V. Talapin and Y. Wang, Nat. Commun., 2023, 14, 49 CrossRef CAS PubMed; (b) Z.-C. Liu, Z.-Q. Wang, X. Zhang and L. Yin, Nat. Commun., 2023, 14, 2187 CrossRef CAS PubMed.
- (a) F. Rey-Tarrio, R. Rodriguez, E. Quinoa and F. Freire, Nat. Commun., 2023, 14, 1742 CrossRef CAS PubMed; (b) B. C. Sanders, S. Pokhrel, A. D. Labbe, I. I. Mathews, C. J. Cooper, R. B. Davidson, G. Phillips, K. L. Weiss, Q. Zhang, H. O'Neill, M. Kaur, J. G. Schmidt, W. Reichard, S. Surendranathan, J. Parvathareddy, L. Phillips, C. Rainville, D. E. Sterner, D. Kumaran, B. Andi, G. Babnigg, N. W. Moriarty, P. D. Adams, A. Joachimiak, B. L. Hurst, S. Kumar, T. R. Butt, C. B. Jonsson, L. Ferrins, S. Wakatsuki, S. Galanie, M. S. Head and J. M. Parks, Nat. Commun., 2023, 14, 1733 CrossRef CAS PubMed.
- C. Chu and R. Liu, Appl. Catal., B, 2011, 101, 343–347 CrossRef CAS.
- C.-H. Lee, S.-M. Lee, B.-H. Min, D.-S. Kim and C.-H. Jun, Org. Lett., 2018, 20, 2468–2471 CrossRef CAS PubMed.
- Y.-P. Zhu, S. Sergeyev, P. Franck, R. V. A. Orru and B. U. W. Maes, Org. Lett., 2016, 18, 4602–4605 CrossRef CAS PubMed.
- (a) B. D. Mkhonazi, M. Shandu, R. Tshinavhe, S. B. Simelane and P. T. Moshapo, Molecules, 2020, 25, 1040 CrossRef CAS PubMed; (b) S.-S. Weng, C.-S. Ke, F.-K. Chen, Y.-F. Lyu and G.-Y. Lin, Tetrahedron, 2011, 67, 1640–1648 CrossRef CAS.
- (a) K. V. N. S. Srinivas and B. Das, J. Org. Chem., 2003, 68, 1165–1167 CrossRef CAS PubMed; (b) P. Mäki-Arvela, J. Zhu, N. Kumar, K. Eränen, A. Aho, J. Linden, J. Salonen, M. Peurla, A. Mazur, V. Matveev and D. Y. Murzin, Appl. Catal., A, 2017, 542, 350–358 CrossRef; (c) H. Basavaprabhu, K. Muniyappa, N. R. Panguluri, P. Veladi and V. V. Sureshbabu, New J. Chem., 2015, 39, 7746–7749 RSC; (d) J. Kothandapani, A. Ganesan and S. S. Ganesan, Synthesis, 2017, 49, 685–692 CAS; (e) Y. Terada, N. Ieda, K. Komura and Y. Sugi, Synthesis, 2008, 2318–2320 CAS; (f) B. Sreedhar, V. Bhaskar, Ch. Sridhar, T. Srinivas, L. Kótai and K. Szentmihályi, J. Mol. Catal. A: Chem., 2003, 191, 141–147 CrossRef CAS.
- J. Diaz de Sarralde, E. Astobieta, A. Sevilla, Y. Rincón, M. T. Herrero, G. Urgoitia and R. SanMartin, Environ. Chem. Lett., 2022, 20, 3421–3427 CrossRef.
- (a) B. Stanje, P. Traar, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Dalton Trans., 2018, 47, 6412–6420 RSC; (b) L. Xu, Y. Chen, Z. Shen, Y. Wang and M. Li, Tetrahedron Lett., 2018, 59, 4349–4354 CrossRef CAS; (c) P. Hu, M. Tan, L. Cheng, H. Zhao, R. Feng, W.-J. Gu and W. Han, Nat. Chem., 2019, 10, 2425–2434 Search PubMed; (d) H. Yu, Q. Zhao, Z. Wei, Z. Wu, Q. Li, S. Han and Y. Wei, Chem. Commun., 2019, 55, 7840–7843 RSC.
- (a) K. Qiao, D. Zhang, K. Zhang, X. Yuan, M.-W. Zheng, T.-F. Guo, Z. Fang, L. Wan and K. Guo, Org. Chem. Front., 2018, 5, 1129–1134 RSC; (b) Z.-L. Li, K.-K. Sun, P.-Y. Wu and C. Cai, J. Org. Chem., 2019, 84, 6830–6839 CrossRef CAS PubMed.
- J.-L. Shi, Q. Luo, W. Yu, B. Wang, Z.-J. Shi and J. Wang, Chem. Commun., 2019, 55, 4047–4050 RSC.
- Z. Zhong, Z.-Y. Wang, S.-F. Ni, L. Dang, H. K. Lee, X.-S. Peng and H. N. C. Wong, Org. Lett., 2019, 21, 700–704 CrossRef CAS PubMed.
- M. Borrell and M. Costas, ACS Sustainable Chem. Eng., 2018, 6, 8410–8416 CrossRef CAS.
- X.-H. Ouyang, R.-J. Song, M. Hu, Y. Yang and J.-H. Li, Angew. Chem., Int. Ed., 2016, 55, 3187–3191 CrossRef CAS PubMed.
- P. Peng, Q. Lu, L. Peng, C. Liu, G. Wang and A. Lei, Chem. Commun., 2016, 52, 12338–12341 RSC.
- (a) Y. Yamashita, M. M. Salter, K. Aoyama and S. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 3816–3819 CrossRef CAS PubMed; (b) S. Huo, L. Meng, Y. Zeng and X. Li, Inorg. Chem., 2022, 61, 4714–4724 CrossRef CAS PubMed.
- (a) H. Kitahara, I. Kamiya and H. Sakurai, Chem. Lett., 2009, 38, 908–909 CrossRef CAS; (b) M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, Chem. Commun., 2010, 46, 145–147 RSC.
- Y.-F. Wang, F.-L. Zhang and S. Chiba, Org. Lett., 2013, 15, 2842–2845 CrossRef CAS PubMed.
- K. Kuntze, J. Isokuortti, A. Siiskonen, N. Durandin, T. Laaksonen and A. Priimagi, J. Phys. Chem. B, 2021, 125(45), 12568–12573 CrossRef CAS PubMed.
- O. Turque, A. Greer and O. R. Wauchope, Org. Biomol. Chem., 2020, 18, 9181–9190 RSC.
- Z. Huang, X. Ji and J.-P. Lumb, Org. Lett., 2021, 23, 236–241 CrossRef CAS PubMed.
- K. Masada, S. Kusumoto and K. Nozaki, Angew. Chem., Int. Ed., 2022, 61, e202117096 CrossRef CAS PubMed.
- T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2012, 134, 20017–20020 CrossRef CAS PubMed.
- See the ESI† for a more comprehensive survey of reaction conditions, the preparation of carboxylic acid derivatives and a mechanistic proposal for the formation of 4ba.
- (a) J. Li, M. A. Siegler, M. Lutz, A. L. Spek, R. J. M. K. Gebbink and G. van Koten, Organometallics, 2009, 28, 5323–5332 CrossRef CAS; (b) M. Figlus, A. C. Tarruella, A. Messer, S. L. Sollis and R. C. Hartley, Chem. Commun., 2010, 46, 4405–4407 RSC; (c) F. F. Flores, M. S. Rivadeneyra and S. Bernès, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76, 1229–1233 CrossRef PubMed; the optical purity of the obtained chiral amides 4ad, 4br, 4bh and 4ce was confirmed by comparison of the optical rotation with literature values. See: (d) D. C. Leustra, D. T. Nguyen and J. Mecinovic, Tetrahedron, 2015, 71, 5547–5553 CrossRef; (e) S. Porto, J. M. Seco, J. F. Espinosa, E. Quiñoá and R. Riguera, J. Org. Chem., 2008, 73, 5714–5722 CrossRef CAS PubMed; (f) R. García, J. M. Seco, S. A. Vázquez, E. Quiñoá and R. Riguera, J. Org. Chem., 2006, 71, 1119–1130 CrossRef PubMed; (g) C. E. Rye and D. Barker, J. Org. Chem., 2011, 76, 6636–6648 CrossRef CAS PubMed. See also the ESI† for further details.
- Analysis of the absolute structure using likelihood methods (Hooft, Straver and Spek, 2008) was performed using PLATON (Spek, 2010). The Friedel pair coverage of the experiment is almost complete (99–100%). The results indicated that the absolute structure had been correctly assigned. The method calculated that the probability that the structure is inverted is smaller than 10–53 for 4fa and smaller than 10–16 for 4fc. The absolute structure parameter y was calculated using PLATON. The resulting value was y = −0.02(11), which together with the Flack parameter value, indicates that the absolute structure has probably been determined correctly. CCDC deposition numbers for 4fa and 4fc are 2290369 and 2290372, respectively, and the corresponding checkCIF reports are provided as ESI.† See: (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS; (b) R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96–103 CrossRef CAS PubMed; (c) A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2010 Search PubMed; (d) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- (a) K. Shukla and V. C. Srivastava, RSC Adv., 2016, 6, 32624–32645 RSC; (b) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- (a) D. R. Stull, Ind. Eng. Chem., 1947, 39, 517–540 CrossRef CAS; (b) V. Pokorný, V. Štejfa, M. Fulem, C. Červinka and K. Růžička, J. Chem. Eng. Data, 2017, 62, 3206–3215 CrossRef; (c) P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1537–1543 CrossRef CAS PubMed; (d) C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRef CAS PubMed.
- (a) D.-H. Liu, H.-L. He, Y.-B. Zhang and Z. Li, ACS Sustainable Chem. Eng., 2020, 8, 14322–14329 CrossRef CAS; (b) C. Nunes de Melo, Y. Blanc Rodrigues and P. A. Robles-Azocar, Inorg. Chim. Acta, 2021, 517, 120192 CrossRef CAS; (c) J.-L. Wang, L.-N. He, C.-X. Miao and Y.-N. Li, Green Chem., 2009, 11, 1317–1320 RSC.
- (a) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS; (b) E. Bayram, J. C. Linehan, J. L. Fulton, J. A. S. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Ozkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889–18902 CrossRef CAS PubMed.
- (a) R.-M. Hu, D.-Y. Han, N. Li, J. Huang, Y. Feng and D.-Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 3876–3880 CrossRef CAS PubMed; (b) X. Xie, J. Cao, Y. Xiang, R. Xie, Z. Suo, Z. Ao, X. Yang and H. Huang, Appl. Catal., B, 2022, 309, 121235 CrossRef CAS.
- (a) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed; (b) Y. Moon, D. Kwon and S. Hong, Angew. Chem., Int. Ed., 2012, 51, 11333–11336 CrossRef CAS PubMed; (c) P. Wen, Y. Li, K. Zhou, C. Ma, X. Lan, C. Ma and G. Huang, Adv. Synth. Catal., 2012, 354, 2135–2140 CrossRef CAS; (d) D. Kim, G. Choi, W. Kim, D. Kim, Y. K. Kang and S. H. Hong, Chem. Sci., 2021, 12, 363–373 RSC.
- C. E. Castro, M. Jamin, W. Yokoyama and R. Wade, J. Am. Chem. Soc., 1986, 108, 4179–4187 CrossRef CAS.
- (a) E. Zhang, H. Tian, S. Xu, X. Yu and Q. Xu, Org. Lett., 2013, 15, 2704–2707 CrossRef CAS PubMed; See also (b) J. P. Saucedo-Vázquez, P. M. H. Kroneck and M. E. Sosa-Torres, Dalton Trans., 2015, 44, 5510–5519 RSC.
- K. Asamaki, T. Nakamoto, S. Kawata, H. Sano, M. Katada and K. Endo, Inorg. Chim. Acta, 1995, 236, 155–161 CrossRef CAS.
- (a) Y.-M. Lee, S. Hong, Y. Morimoto, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2010, 132, 10668–10670 CrossRef CAS PubMed; (b) Y. Jiang, J. Telser and D. P. Goldberg, Chem. Commun., 2009, 6828–6830 RSC; (c) F. Oddon, Y. Chiba, J. Nakazawa, T. Ohta, T. Ogura and S. Hikichi, Angew. Chem., Int. Ed., 2015, 54, 7336–7339 CrossRef CAS PubMed; (d) M. A. Dedushko, J. H. Pikul and J. A. Kovacs, Inorg. Chem., 2021, 60, 7250–7261 CrossRef CAS PubMed.
- (a) N. Lehnert, R. Y. N. Ho, L. Que Jr. and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 12802–12816 CrossRef CAS PubMed; (b) J. Kim, Y. Zang, M. Costas, R. G. Harrison, E. C. Wilkinson and L. Que Jr., J. Biol. Inorg. Chem., 2001, 6, 275–284 CrossRef CAS PubMed.
- (a) Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CrossRef CAS; (b) B. Dellinger, S. Lomnicki, L. Khachatryan, Z. Maskos, R. W. Hall, J. Adounkpe, C. McFerrin and H. Truong, Proc. Combust. Inst., 2007, 31, 521–528 CrossRef PubMed.
- T. Nauser and R. E. Bühler, J. Chem. Soc., Faraday Trans., 1994, 90, 3651–3656 RSC.
- For an explanation of the results obtained under argon, see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, UPLC-ESI-QTOF-MS identification of intermediates and 1H and 13C NMR spectra of isolated compounds. CCDC 2290369 and 2290372. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy01429k |
| This journal is © The Royal Society of Chemistry 2024 |