
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperando UV-vis spectroscopy for real-time monitoring of nanoparticle size in reaction conditions: a case study on rWGS over Au nanoparticles†
Chiara
Negri
 a,
Riccardo
Colombo
a,
Mauro
Bracconi
a,
Cesare
Atzori
b,
Alessandro
Donazzi
a,
Andrea
Lucotti
c,
Matteo
Tommasini
c and
Matteo
Maestri
*a
a,
Riccardo
Colombo
a,
Mauro
Bracconi
a,
Cesare
Atzori
b,
Alessandro
Donazzi
a,
Andrea
Lucotti
c,
Matteo
Tommasini
c and
Matteo
Maestri
*a
aLaboratory of Catalysis and Catalytic Processes, Dipartimento di Energia, Politecnico di Milano, Via la Masa 34, 20156 Milano, Italy. E-mail: matteo.maestri@polimi.it
bEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, CS 40220, Grenoble, France
cDepartment of Chemistry, Materials and Chemical Engineering, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133, Milano, Italy
First published on 7th February 2024
Abstract
We propose the use of surface plasmon resonance (SPR) as a distinctive marker for real-time monitoring in reaction conditions of gold nanoparticles supported on α-Al2O3. The study leverages the SPR shape-and-size dependency to monitor metal nanoparticles in reaction conditions, evidencing an influence of both dimensions and agglomerations on the SPR peak position. Operando measurements, coupling UV-vis spectroscopy and catalytic testing, allows to follow the dynamics during nanoparticle formation (Au3+ to Au0 reduction) and during the reverse water gas shift reaction (CO2 + H2 → CO + H2O). The catalyst structure and stability in reaction conditions was further confirmed by operando X-ray spectroscopy and PXRD data. Overall, this approach enables the direct acquisition of information on the structure–activity relationship of metal-based supported catalysts under actual reaction conditions.
1 Introduction
Heterogeneous catalysis nowadays plays a pivotal role in the chemical sector, in energy applications and for environmental protection. The constant demand for more efficient and sustainable processes and resource exploitation calls for increasing efforts to improve the catalytic processes for a more viable future. This is a key-challenge in reaction engineering and requires a deep knowledge of the catalyst functionality at atomistic level, to enable a rational design of both optimized processes and new materials.1,2 In this scenario it is fundamental to acknowledge the “living character” of heterogeneous catalyst under the reacting environment, that is, the dynamic change in its structure during the reaction as a function of the operating conditions.3,4 Specifically, this is crucial for supported metal-based catalysts, where nanoparticles can change their size and shape depending on the applied conditions, thus varying the available active sites and affecting the overall catalytic performances.5,6 As a consequence, to gain a full comprehension of the catalyst functioning under real working conditions, it is necessary to assess the actual nanoparticles morphology and to correlate it with the observed macroscopic kinetic results.7,8 Currently, the state-of-the-art approach to assess the structure of supported metal-based catalysts applies techniques operating at conditions significantly different from the reacting environment of kinetic analysis (i.e. high vacuum, low temperature vs. high pressure, high temperature). These ex situ characterization methods (i.e. HR-TEM, H2 and CO chemisorption, FT-IR spectroscopy with probe molecules9,10) allow for the study of nanoparticle morphology11–13 prior to and after the catalytic testing.5,9,10,14,15 Thus, we are in presence of a gap between morphology and catalytic performances.8,16 To contribute in resolving this, the current study exploits the surface plasmon resonance (SPR) of metal nanoparticles in operando conditions, to characterize their size and structure in reaction conditions. In fact, since SPR originates from coherent excitations of the electronic density of metallic nanostructures, it is intrinsically strongly dependent upon size and shape of the metal nanoparticles.17–19 In the growing field of nanoplasmonics, plasmons are used to enhance the electromagnetic field from an external source, and metal nanoparticles are synthetized ad hoc to allow local field effects at specific wavelengths of interest.20,21 If this correspondence is univocal, the SPR shape-and-size dependency becomes a fingerprint to monitor the metal nanoparticles morphological changes in reacting conditions. So far, this approach has been applied ex situ, to monitor the presence supported metal nanoparticles (i.e. for gold-based systems) on reducible (i.e. TiO2, CeO2) and non-reducible (i.e. Al2O3, SiO2) oxides.22 In this playground, the possibility to monitor in operando metal nanoparticles formation and their size and shape via Surface Plasmon Resonance on heterogeneous catalysts represent a novelty. In this article, we present an UV-vis spectroscopy study of Au-based catalysts, targeting the in-operando characterization of gold nanoparticles structure. In fact, nanostructured gold is characterized by a large number of conduction electrons easily polarized by an external electromagnetic field, i.e. induced by an UV-vis light source.23 Hence, we employed an ad hoc developed UV-vis setup, to monitor the catalysts optical properties, aiming at gaining information on the size, shape, and stability of Au nanoparticles in a packed bed reactor, that is, with an operando approach. Furthermore, since gold nanoparticles have proven to be active for the reverse water gas shift (rWGS) reaction (CO2 + H2 ↔ CO + H2O) when highly dispersed on a support,24–26 we chose this reaction as a case study of the possibility to monitor the nanoparticle stability in reaction conditions. Thus, with the developed operando setup, we probed the behaviour of the formed Au nanoparticles in operando, exposing the catalyst to rWGS reaction conditions. X-ray absorption spectroscopy and synchrotron-based XRD are used as complementary techniques to witness the applicability of our experimental approach in studying the catalyst structure and behaviour. In parallel, HR-TEM and field-emission SEM (FESEM) allowed us to assess the shape of the formed Au nanoparticles, confirming our SPR-based methodology. On a general basis, these findings are of interest since direct information on the structure of metal sites at temperature and chemical conditions relevant for the catalytic application are obtained, with a novel approach to the field of metal-based supported catalysts characterization.2 Experimental and methods
2.1 Materials
The Au-based materials studied in this paper were in-house synthetized. The AuAl2O3_30 and AuAl2O3_60 samples were prepared by direct impregnation of an α-Al2O3 support (Puralox, Sasol) with commercial colloidal suspensions of Au NPs with nominal diameter of 30 nm and 60 nm, respectively (HiQ-Nano, 1 mgAu mL−1). In total, 20 impregnations with the not diluted suspension were performed to obtain the 0.4% Au wt% loading. The 4 wt% AuAl2O3_4 catalyst was obtained by impregnation of the α-Al2O3 with a HAuCl4 solution (Sigma-Aldrich, 99.99% au, 30 wt% in dilute HCl). The resulting powder was dried overnight at room temperature and was subsequently washed carefully to remove clorine. For each washing step (three performed with NH3 (Sigma-Aldrich, 1 M) and five with H2O, to restore the pH to 7) the powder was mechanically stirred until homogenization and centrifuged for 5 minutes at 4350 rpm. The washed powder was dried at room temperature and stored in dark conditions.2.2 UV-vis spectroscopy operando setup
The ex situ and operando UV-vis spectra were measured coupling a powder reactor (Fig. 1) with a UV-vis diffuse reflectance probe, having six radiating optical fibers and one reading fiber. The data were recorded with the fiber always perpendicular to the investigated surface. The operando spectra were measured combining in an oven a high-temperature UV-vis reflectance probe with a powder reactor, to measure UV-vis spectra in reaction conditions. Thus, the experimental setup we developed allows us to measure the spectroscopic data while performing the targeted catalytic testing, with controlled temperature and gas feed. At variance with the spectroscopic cells usually utilized, our system allows for the acquisition of UV-vis spectra with the targeted sample directly placed in a tubular fixed-bed flow quartz reactor (5 mm internal diameter), designed to minimize the pressure drops and to avoid the mass transfer limitations. The reactants flow is passing through the entire catalytic bed height (from top to bottom), and the reactor can be vertically translated during the reaction, to monitor the optical properties of the sample in different positions along the catalytic bed. To exclude contributions from the specular reflectance (i.e. from the reactor outer walls) and to compensate for the possible absorption from quartz SiO and/or SiOH groups, the reference spectrum for the operando experiments is measured at room temperature (RT) in the same optical configuration. Thus, barium sulphate (chosen as white reference for both the ex situ and operando spectra) has been loaded in the quartz reactor, and placed inside the testing gas rig oven. This allows for the measurement of UV-vis data in reaction conditions without any modification to the reactor geometry and cross-section, thus avoiding any possible effect on the fluid dynamics. The ex situ and operando UV-vis spectra were recorded in the 200–1000 nm range at 1 nm resolution on a Exemplar Plus BTC655N spectrometer by B&W Tek, equipped with a black-thinned charge-coupled device (CCD) detector and B&W Tek BDS130A (deuterium−halogen) light source. Spectra are reported as relative reflectance (R%) defined as:| R% = Rsample/Rreference |
 | ||
| Fig. 1 Pictorial representation of the developed operando UV-vis setup combining the packed-bed reactor and the UV-vis probe. | ||
Prior to the measurement, the sample is pelletized with a hydraulic press, successively chopped and sieved, selecting for the measurement the fraction between 75 and 106 μm. We have validated that the selected diameter range avoids any mass transfer limitations. The quartz reactor is loaded with ∼200 mg of powder, and the catalyst bed is hold in place by a quartz wool support and by a sieved fraction of inert quartz (850 and 1160 μm). The temperature is controlled by a thermocouple (500 μm) placed inside the catalytic bed. The operando UV-vis experiments were performed during the reduction pretreatment (50 Ncc min−1 − 5% H2/N2 balanced) and during the rWGS catalytic testing (50 Ncc min−1, CO2/H2 = 1, GHSV = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1). The reaction was monitored over time, until the steady state was reached both in the spectra and in CO2 conversion. Measurements were carried out from RT to 200/600 °C with a heating ramp of 2/5 °C min−1, flowing the desired gases (H2/N2 or CO2/H2/N2) with an ad hoc built gas flow setup. The setup consisted of four channels, each of them connected to a specific gas bottle and to a dedicated mass flow controller. The reaction products are monitored by means of an on-line gas cromatograph (Micro Gas Chromatograph GCX, Pollution).
000 h−1). The reaction was monitored over time, until the steady state was reached both in the spectra and in CO2 conversion. Measurements were carried out from RT to 200/600 °C with a heating ramp of 2/5 °C min−1, flowing the desired gases (H2/N2 or CO2/H2/N2) with an ad hoc built gas flow setup. The setup consisted of four channels, each of them connected to a specific gas bottle and to a dedicated mass flow controller. The reaction products are monitored by means of an on-line gas cromatograph (Micro Gas Chromatograph GCX, Pollution).
2.3 X-ray absorption spectroscopy
X-ray Absorption Spectroscopy (XAS) data were collected at the BM31 beamline30 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) using a quartz capillary with 1 mm external diameter, optimized for XAS data collection in transmission mode. The AuAl2O3_4 catalyst was pressed and sieved, and the 75 and 106 μm fraction was selected for the measurement. Au L3-edge XAS measurements were performed in transmission mode, employing a double-crystal Si (111) monochromator for the incident energy scan, and ionization chambers to detect incident (I0) and transmitted (I1,2) photons. An Au metal foil was measured simultaneously using a third ionization chamber I2, for energy calibration purposes.31 XAS spectra of ∼5 min each (energy range 11800–12950 eV; energy step = 0.5 eV; acquisition time of 70 ms per point in the whole energy range) were measured during the pretreatment in H2 (10 Ncc min−1 − 5% H2/He balanced) and during the rWGS reaction (10 Ncc min−1, CO2/H2 = 1). The gas composition from the reactor outlet has been continuously monitored by means of a mass spectrometer (data not reported). The reaction was monitored over time, until the steady state was reached.
The Athena software (Demeter package)32 was used to align XAS data by using the corresponding Au metal foil spectra and for normalization to unity of the edge jump. The same program was used for the extraction of the χ(k) function. R-Space FT-EXAFS spectra were obtained by calculating the Fourier transform of the k2χ(k) functions in the (3.0–12.9) Å−1k-range.
2.4 Multivariate curve resolution-alternate least squares (MCR-ALS)
The time-resolved spectra are analyzed through the multivariate curve resolution-alternate least squares whose purpose is the resolution of the underlying contributions in unknown unresolved datasets.33–35First, the identification of the number of pure species is obtained by Principal Component Analysis (PCA) of the experimental spectral dataset.36,37 The number of correct principal components is evaluated by qualitative analysis, i.e. scree plot and R-factor, as well as by considering statistical tests, i.e. imbedded error function (IE-test), factor indicator function (IND-function) and Malinowski F-Test.38
MCR-ALS requires an initial estimate of either the concentration profiles or the spectra of pure components. Here, the simple-to-use interactive self-modeling mixture analysis (SIMPLISMA)39 has been employed for obtaining the initial estimates of the pure spectra.
Once initial estimates are evaluated, MCR-ALS solves iteratively eqn (S10) and (S11) (as detailed in ESI† section 3) by alternating least square algorithm calculating the concentration profiles and the pure spectra optimally fitting the experimental data matrix. The optimization terminates when the difference between the model fit between two consecutive iterations does not significantly improve. During the optimization, to suppress the effect of rotational and scale ambiguities and fostering the physical/chemical meaningfulness, some constraints are introduced. In each of the iterative cycle, the computed profiles (concentrations, spectra) are modified to force to obey to the conditions imposed by some constraints.40 Heren the non-negativity and mass conservation have been employed.
Additional details on the method and on the implementation are reported in ESI† sections 3 and 4.
2.5 PXRD, HRTEM and FESEM
HR-TEM analyses were performed by means of the high-resolution TEM JEOL JEM 3010-UHR, equipped with lanthanum hexaboride (LaB6) light source, a nominal operating voltage of 300 kV and a theoretical resolution of 0.17 nm. The samples were prepared depositing the catalyst powder on a 3 mm copper grid. The HR-TEM images were analyzed using ImageJ, an open-source software. The particles size distribution was calculated counting on average more than 200 nanoparticles per sample. The FESEM analyses were carried out with a TESCAN S9000G microscope, which features a Schottky field emission gun and a resolution of 0.7 nm.
3 Results and discussion
3.1 Operando UV-vis spectroscopy
Using ex situ UV-vis diffuse reflectance (DR) spectroscopy, we validated the sensitivity of the SPR peak position to anchored nanoparticles dimensions. On the basis of this SPR experimental approach, we subsequently focused on monitoring in situ the formation of gold nanoparticles, and in operando their stability in reaction conditions.It is here worth recalling that the SPR phenomenon arises from the light absorbed and scattered by a metal nanoparticle and it is quantitively evaluated through the respective cross section coefficient σabs and σscat.42,43 The extinction cross section σext is the sum of these contributions (σext = σabs + σscat), and it shows a strong dependency on particle size.44 This dependency results in a significative increase in absorbed light for even a moderate increase in the cross section values, that is, for an increase in the nanoparticles particle size, and in a damping when decreasing the nanoparticle dimensions.17 Furthermore, the SPR peak position (i.e., the maximum of the extinction cross-section) is also sensitive to the size of the nanoparticles. A red shift of the SPR peak accounts for increased dimensions of the nanoparticles, whereas a blue-shift corresponds to a nanoparticle size decrease.5,17 The collective oscillations of electrons in the conduction band of gold atoms are thus responsible for the SPR peaks present in the spectra reported in Fig. S1,† measured for two Au/Al2O3 samples, obtained by direct impregnation of an α-Al2O3 with commercial suspensions of gold nanoparticles, having nominal diameter of 30 (AuAl2O3_30) and 60 (AuAl2O3_60) nm, respectively. The spectra show a minimum in the R% (i.e. the maximum of the extinction cross section) at different wavenumbers, as well as differences in the bandwidth. The AuAl2O3_60 curve presents a broad band centered at 540 nm, while the spectrum obtained for the AuAl2O3_30 sample, prepared with nanoparticles with a smaller diameter, presents a narrower band, sharper and more defined, centered at 530 nm. Since the position and intensity of Au SPR peaks depend on the size and shape of gold nanoparticles, and on the dielectric properties of the medium (i.e. the support), the observable differences between the experimental curves can be rationalized in terms of nanoparticles dimensions. In fact, we can rule out a major contribution on the SPR peak position from the dielectric constant of the surrounding means (air and α-Al2O3), as they are equal for both samples. The resolution the acquired spectra is ±1 nm, thus this experimental evidence highlights the potentiality of correlating the plasmon peak with metal nanoparticles dimensions, when anchored on a solid medium. The HR-TEM images (Fig. S2†) highlight that, in both samples, gold nanoparticles are mainly spherical. Furthermore, on average, gold nanoparticles are larger and with a broader distribution than the nominal one (38 ± 3 nm vs. 30 and 124.9 ± 8 nm vs. 60). Thus, as an increase in bandwidth and a red-shift of the SPR maximum is expected with the growth of nanoparticles size. The observed SPR λmax shift between the AuAl2O3_30 and AuAl2O3_60 spectra can consequently be correlated with the differences in the average nanoparticle diameters obtained from HR-TEM. The observed shift in the SPR λmax proved that monitoring the SPR peak position as a function of nanoparticle size is suitable to study metal nanoparticles on supported catalysts. Anchoring the metal nanoparticles on the Al2O3 support does not hinder the sensitivity of the SPR peak on the nanoparticles size.
With this approach, we studied a 4 wt% Au/Al2O3 sample (hereafter AuAl2O3_4) during the thermal treatment in H2 (5% H2/N2, 50 Ncc min−1) from RT to 200 °C, which has been reported as optimal to obtain active gold catalysts.45 Indeed, the H2-reduction treatment results in the formation of gold nanoparticles, highlighted by the presence of a visible SPR peak in the spectrum measured at 200 °C (red curve in Fig. 2). Comparing the SPR λmax centered at 540 nm and the bandwidth with the spectra measured ex situ, we can infer that the formed gold nanoparticles present a broad size distribution, and/or are present as aggregates (i.e. by comparison with the AuAl2O3_60 spectrum). The broadening of the SPR peak in the in situ spectrum, might be related to the concomitant thermal excitation of the free electrons of gold, in parallel with the generation of the plasmon wave when working at high temperature. The FESEM results reported in Fig. 3a show that the sample is characterized by small, isolated gold nanoparticles and larger agglomerates, which present a raspberry-like structure. These agglomerates have a size ranging between 15 and 100 nm and are formed by nanoparticles that are still separated and have not collapsed and/or coalesced (see Fig. 3c). From the HR-TEM images in Fig. 3b and c, we can observe that Au nanoparticles in the agglomerates have a slightly different shape when compared to the isolated ones. In fact, it is clearly visible that the agglomerated nanoparticles present a more spherical shape (Fig. 3c), while the majority of the observed isolated nanoparticles is characterized by a well-defined cubo-octahedral shape (Fig. 3b), which has been reported to be the most energetically favoured structure for gold nanoparticles with diameters higher that 3 nm.5,6,46,47
 | ||
| Fig. 2 In situ UV-vis DR spectra of AuAl2O3_4 during H2-pretreatment from RT to 200 °C (blue to red curves); blue dotted curve: spectrum at ca. 135 °C. | ||
 | ||
| Fig. 3 (a) FESEM and (b) and (c) HRTEM images of AuAl2O3_4; (d) magnification of gold nanoparticles in (c) with highlighted lattice planes; (e) NPs size distribution of AuAl2O3_4. | ||
Furthermore, we observed that Au particles on average expose [2,0,0] and [1,1,1] lattice planes, as visible in Fig. 3d, which are the main expected crystallographic reflexes for nano-structured gold.45 These findings are in agreement with the PXRD data (Fig. S3†) measured for the AuAl2O3_4. The diffractograms present very sharp peaks clearly ascribable to the support (crystalline α-Al2O3 or corundum (ICSD entry no. 9770)48) which is mostly unperturbed by the H2 pre-treatment. The observable minor peak shifts and intensity changes are due to the lattice thermal expansion and the increase of the atom thermal motions. In parallel, we observe the appearance of a new phase, that is, fcc metallic Au (ICSD entry no. 64701), presenting the characteristic reflexes at 6.6° for the [1,1,1] lattice plane, at 7.63° for [2,0,0], and at 10.08° for [2,2,0]. Minor signals related to the [3,1,1] and [2,2,2] lattice planes are also visible at higher θ values. The interplanar distances (0.204 nm and 0.238 nm) calculated from HR-TEM images (Fig. 3d) are in accordance with the peaks visible in the PXRD pattern and correspond to the most abundant [2,0,0] and [1,1,1] facets. The average nanoparticles size has been evaluated from HR-TEM images, resulting to be of 5.99 ± 0.13 nm in agreement with literature values (Fig. 3e),5,25,46,49 with a quite narrow distribution (less than 10% nanoparticles have diameters between 8–10 nm and less than 8% have an average size of 10 ≤ d ≤ 16). Thus, the general intense broadening of all diffraction peaks is due to the size of the Au nanoparticles, as highlighted by TEM imaging. Despite the narrow particle size distribution, the sample has an intrinsic heterogeneity (i.e. the presence of larger agglomerates) mirrored by the observed SPR spectral features, which can then be considered representative of the average gold speciation.
It is now noteworthy to mention that initially gold is present on the alumina surface as isolated Au3+ ions, resulting from the impregnation of the α-Al2O3 with the gold precursor. Indeed, the initial spectrum measured in H2 at RT (dark blue curve in Fig. 2) is characterized by a broad absorption below 600 nm, which is likely to be related to the Ligand-to-Metal (L → M) Charge Transfer transitions, in agreement with features observed on similar Au3+ complexes.50 Specifically, it is reasonable to assign the band at 450 nm (measured at half height) to electronic transitions from the O atoms present in the gold coordination sphere (O → Au3+). The observed charge–transfer transitions are at lower energy when compared to those reported for [AuCl4]− complexes in literature,5 in agreement with the position of oxygen and OH groups in the spectrochemical series of ligands. The observable features below 270 nm, which falls in the spectral region where the electronic transitions of Au+ are expected,51 are in our case instead related to the absorption from the quartz wall reactor.
When increasing the temperature from RT to 200 °C in H2 feed, we can observe a progressive rearrangement of the chemical species on the catalyst surface. In fact, the progressive broadening of the LMCT transitions band and the decrease in reflectance can possibly stem for the formation of some multimerics Au clusters on the Al2O3 surface. With the progressive temperature increase, an inflection point at about 550 nm starts to appear (Fig. 2, blue dotted curve, ca. 135 °C) which suggests the initial formation of gold nanoparticles (i.e. around 2 nm).17 These nanoparticles act as nuclei for the growth of larger ones, as the Au3+ → Au0 reduction proceeds, until all the gold is present on the Al2O3 surface as Au0. At high temperature, the changes observed in the 200–300 nm range are related to a worse background compensation.
3.2 X-ray absorption spectroscopy and MCR-ALS analysis
To further corroborate the developed experimental approach in monitoring the formation of gold nanoparticles, we followed Au3+ → Au0 reduction by means of X-ray absorption spectroscopy. The resulting Au L3 XANES spectra, measured during the 5% H2-pretreatment, are reported in Fig. 4a (from dark blue to red curves). The Au L3 XANES are known to be sensitive not only to the oxidation state of the X-ray absorber atom, but also to the electronegativity, the type and the symmetry of the ligands, thus being a complementary technique to the applied UV-vis DR. The first spectrum (dark blue curve) presents an intense white line feature around 11920 eV, typical of the Au3+ oxidized state of gold. The Au3+ white line is shifted to lower energies, at variance with the higher energies usually measured for oxidized species. This is ascribable to the 2s → 5d dipole-allowed transitions, that anticipate the excitation of the bulk electrons, and correlates with the number of holes in Au 5d band.52–54 Duggar at about 100 °C, the intensity in the white line region started to decrease and the edge position shifts to higher energies, stemming for a change in Au3+ oxidation state. The changes in the white line area might be associated with changes in the ionicity and/or electron transfer between the absorbing atom and the ligands. Moreover, between 11930 eV and 11955 eV, we can observe the growth of new features, while those between 11955 eV and 11980 eV tends to decrease. The observed behaviour is associated with the progressive conversion of the oxidized Au3+ species to Au0. The spectrum at 200 °C (red curve) closely resemble that of metallic gold, with less intense features,52,53,55 accounting for a lower Au average coordination number. The absence of the white line in the spectrum of metallic gold results from its electronic configuration, as Au0 (5d10, 6s2) has the d level completely filled.56,57
 | ||
| Fig. 4 (a) Operando XANES and (b) EXAFS during H2-pretreatment from RT to 200 °C (dark blue to red curves; yellow curve in (b): EXAFS spectrum of bulk gold); (c) XANES spectra of pure components (μipure(E)) derived from MCR-ALS analysis of temperature-dependent XANES dataset during H2-pretreatment from RT to 200 °C, assuming NPC = 3 and (d) concentration profiles of the Au-species corresponding to the pure XANES spectra. | ||
To clarify the dynamics of the Au3+ → Au0 reduction, we performed MCR-ALS analysis on the XANES spectra (additional details in ESI† section 4). The first step of the analysis involved the determination of the number of pure components. To this aim, we performed principal component analysis (PCA) of the XANES dataset (see ESI† section 3). From qualitative analysis based on the abstract components (see Fig. S6†) and the R-factor (see Fig. S7†) as well as statistical analysis, we identified the presence of three principal components (see Fig. S8†). The selection of Npure could strongly influence the MCR-ALS results. Hence, we repeated the analysis for downsizing the component space equal to 2. The results are reported in ESI† section 5. The analysis revealed that the number of pure components identified by statistical analysis, Npure = 3, represents an optimal value-resolution. The results from MCR-ALS analysis are reported in Fig. 4, in terms of pure XANES spectra and concentration profiles (Fig. 4c and d, respectively). Considering the observable XANES features of the MCR spectra, we can identify the presence of three Au species, characterized by different oxidation states. We assign the blue spectrum in Fig. 4b, characterized by an intense white line feature and by the edge position at lower energies to an Au3+ oxidized species. The red curve in Fig. 4c shows similar features to that of metallic gold and is assigned to Au0. The third component is characterized by a white line feature of lower intensity and by an edge position shifted to higher energy. We hypothesize that this species is an Auδ+ intermediate, which is formed during the progressive reduction and rearrangements of Au atom on the Al2O3 surface. This is in agreement with the behaviour observed in the UV-vis spectra, which are characterized by progressive changes in the LMCT region, stemming for changes in the ligand coordination sphere and bond character of the absorber atoms, as a result of the progressive Au3+ → Au0 reduction. The corresponding relative concentration profiles of the formed species (see Fig. 4c) further underline the formation and the consumption of the Auδ+ intermediate (grey line in Fig. 4d) as the reduction of Au3+ proceeds. In fact, we can observe that the concentration of Au3+ species starts to decrease shortly after feeding the reactive gas feed (see blue line in Fig. 4d), with the concomitant growth of the Auδ+ intermediate. This species reaches a maximum around 125 °C, with a subsequent sharp decrease, related to its progressive reduction to Au0. The temperature at which the concentration of Au0 starts to increase at the expenses of Auδ+ (ca. 135 °C) corresponds to the temperature at which we observed the appearance of the initial SPR feature in the UV-vis spectrum (blue dotted line in Fig. 2). This results in a fair agreement in the description of the evolution of the investigated system between the two applied experimental approaches. Above 135 °C, Au0 becomes the most prevalent species anchored on Al2O3.
Fourier transformed (FT) Extended X-ray Absorption Fine Structure (EXAFS) provides additional information on the formed gold nanoparticles. EXAFS spectra measured during H2-pretreatment are reported in Fig. 4b. The dark blue spectrum (Fig. 4b) corresponds to the catalyst precursor just before the activation. In accordance with literature,53 it closely resembles the spectrum of Au2O3, further confirming that, prior to the thermal treatment, the as-prepared sample contain isolated Au3+ species. Specifically, the absence of any peaks associated to Au–Au backscattering features excludes the presence of Au0, while the peak at 1.6 Å is related to the presence of oxygen or nitrogen atoms in the first coordination shell of Au. During the H2-pretreatment we can observe the progressive decrease of the first shell feature, with the growth of two peaks assigned to Au–Au backscattering amplitude. Thus, the reduction in hydrogen led to the formation of Au–Au shell at expense of the initial Au–O or Au–N one and the final spectrum (red curve in Fig. 4b) is similar to the one of bulk gold (yellow curve in Fig. 4b). The EXAFS spectrum of bulk gold presents two intense peaks at around 2.5 Å and 3 Å, respectively linked to the first Au coordination shell, characterized by the presence of 12 Au neighbouring atoms (approximate Au–Au distance 2.85 Å) and to the second shell of 6 atoms (approximate Au–Au distance 4.08 Å). The different intensity in the EXAFS features between the yellow and red curves proves the lower coordination number for Au in our sample, confirming the average small dimensions of the gold nanoparticles formed, in line with the HR-TEM and PXRD findings. The results reported so far allow for the description of the average gold speciation, highlighting the possibility to exploit the SPR to monitor the formation of gold nanoparticles with well-defined shapes and sizes.
3.3 SPR monitoring in operando during CO2 activation
To showcase the SPR monitoring in conditions relevant for the catalysis, we exposed the AuAl2O3_4 catalyst to a mixture of CO2 and H2. We selected the reverse water gas shift reaction (rWGS, CO2 + H2 ↔ CO + H2O)), since supported gold nanoparticles are active hydrogenation catalysts.58,59 This reaction on Au-based catalysts proceeds via associative mechanism, that is, H2 is dissociatively chemisorbed on Au0 while CO2 is activated on the Al2O3 surface. The spillover of H* species on the support allows for the formation of reaction intermediates (i.e. formates) that decompose to CO and H2O.25
Fig. 5 shows CO2 conversion as a function of the reaction temperature and time-on-stream for four rWGS tests under stoichiometric conditions (5% H2, 5% CO2 and 90% N2, GHSV = 15000 Nl kgcat−1 h−1), from RT to 600 °C. In all the performed tests, CO2 conversion increased with temperature and remained well below the thermodynamic value (see orange curve in Fig. 5). The reported catalytic results allowed us to observe that the catalyst was active, with CO2 conversion values at 600 °C that slightly oscillate around a value of 9.5%.25,60 Throughout the reaction CO was the sole product measured, with no traces of CH4 or other oxygenated side products, thus highlighting a high CO selectivity on Au-based catalysts.25,60Fig. 6a reports the UV-vis DR spectra measured during the RT-600 °C temperature ramp under stochiometric rWGS conditions (from red to light blue curves). Feeding the reactive gas mixture on the sample does not alter the SPR peak priorly formed (see red curve in Fig. 6a), which remains well-defined. As the reaction temperature increases, the operando spectra are characterized by a progressive broadening and red-shift of the observable SPR peak (from 540 nm, after the in situ H2-pretreatment, to 550 nm). Once reached 600 °C, we do not observe any further changes in the UV-vis spectra over time (see dashed purple line in Fig. 6a, measured after 230 minutes). The SPR peak remains stable and symmetric during the reaction, stemming for a preserved regular morphology of gold nanoparticles and for the absence of major agglomeration phenomena. In fact, the observed SPR peak broadening and the consequent λmax SPR red-shift might be primarily related to the temperature increase, which induces a strong excitation in the Au nanoparticles surface electrons and a thermal dilatation of Au lattice.17 These hypotheses have been confirmed by the HR-TEM analysis on the spent catalyst (Fig. S5†), which is characterized by small, isolated nanoparticles, with many neighbouring agglomerates with the peculiar raspberry-like shape as the fresh catalyst. The average nanoparticle sizes between the two samples are comparable (6.67 ± 0.10 nm vs. 5.99 ± 0.13 nm) as the size distributions, since 90% of nanoparticles have a dimension lower than 10 nm. These findings allowed us to prove the feasibility of the developed experimental approach in monitoring metal nanoparticles size in reaction conditions.
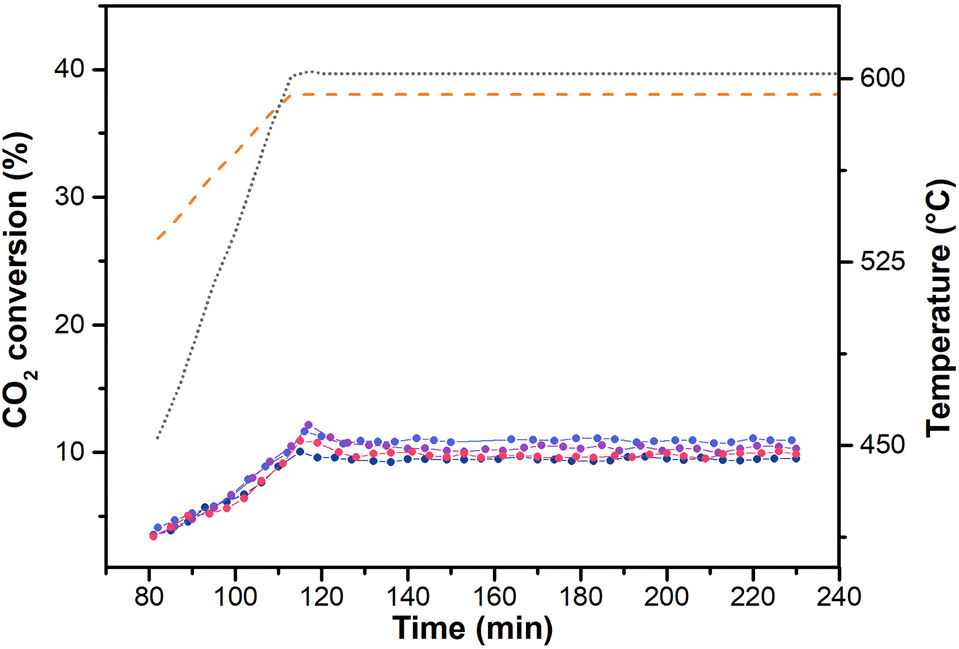 | ||
| Fig. 5 CO2 conversion on AuAl2O3_4; (colour code: test 1, light blue; test 2; test 3, purple; test 4, black; dashed orange line: equilibrium); temperature profile reported as grey dotted line. | ||
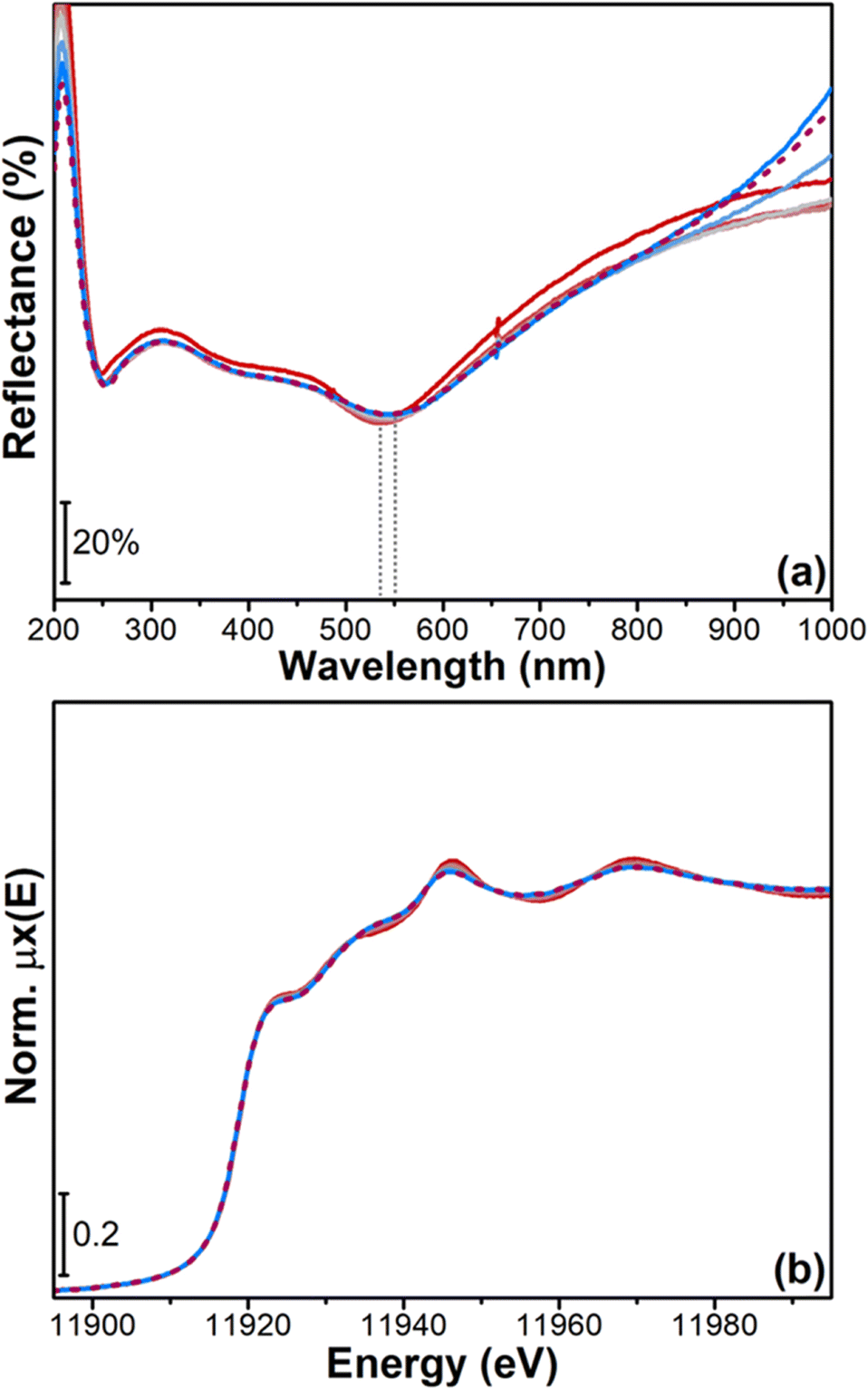 | ||
| Fig. 6 (a) Operando UV-vis DR spectra during rWGS from RT to 600 °C (from red to light blue curves) and after 230 minutes in isotherm at 600 °C (dashed purple curve); (b) operando XANES spectra during rWGS from RT to 600 °C (from red to light blue curves) and after 230 minutes in isotherm at 600 °C (dashed purple curve). The grey dotted lines highlight the shift in the SPR peak position. | ||
Moreover, the reactivity towards CO2/H2, monitored in parallel by operando XANES (Fig. 6b), resulted to be in agreement with the UV-vis data. The spectra measured during the temperature ramp from RT to 600 °C and in isotherm at 600 °C are reported in Fig. 6b. The changes visible in Au L3 post-edge features during the rWGS ramp from RT to 600 °C (from red to light blue curves) can be again associated with the temperature increase, which promotes electron mobility and causes the broadening of the observed XANES features. There are no changes in the pre-edge and white line region, thus confirming that, during reaction, the sample preserves its Au0 oxidation state. Furthermore, from a qualitative analysis of the XANES spectra, we can infer that sintering does not occur significantly, since this phenomenon would change the average coordination number of Au atoms, thus modifying the features visible in the XANES spectra towards those of bulk gold.52 In agreement with the operando UV-vis data, the XANES spectrum measured after 230 minutes (purple dashed line in Fig. 6b) show the catalyst stability over time in the applied experimental conditions.
Indeed, from the spectroscopic data reported it emerges the feasibility of using the SPR peak as a marker of the metal oxidation state and of the nanoparticles size, with the possible further support of advances characterization techniques such as operando XAS. The results obtained validating the technique on gold nanoparticles can be applied to any metals presenting the SPR phenomenon in the selected spectral range.
4 Conclusions
In this study, we have introduced surface plasmon resonance (SPR) as a distinctive marker for the morphology of supported metal nanoparticles in conditions pertinent to catalysis. Utilizing ex situ UV-vis spectroscopy, we examined the SPR characteristics of gold nanoparticles on α-Al2O3, demonstrating the capability of SPR to track changes in nanoparticle size under reaction conditions. Operando UV-vis spectroscopy allowed us following the dynamics of nanoparticles formation and of their reactivity during the reverse water gas shift reaction. We monitored the progressive change in coordination of Au3+ ions through the development of the SPR peak, and the subsequent stability of the formed gold nanoparticles in reaction conditions. Namely, the SPR peak position of supported gold nanoparticles was found to be affected both by the nanoparticle dimensions and the presence of agglomerates. The catalytic results measured during the operando UV-vis experiment evidenced a good activity for rWGS of the Au catalyst, with a 100% CO selectivity. Operando XAS and PXRD data confirmed the experimental evidences from UV-vis spectroscopy, showing that Au nanoparticles are formed with very small size and are stable in reacting feed. Our findings validate the potential of using SPR for studying the morphology of catalysts. This method could be effectively applied to other supported-metal catalysts that exhibit SPR phenomena, such as Pt, Ag, and Cu. Furthermore, the technique can be applied as a design-of-experiment methodology to properly select and screen the measurements to be performed at large scale facilities (synchrotron), aiming at optimizing data acquisition and material characterization. Overall, the developed approach allows for the direct investigation of metal-based supported catalysts, gaining direct information on the structure–activity relation under catalysis relevant conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from MIUR (Italy) – FARE (Framework per l'attrazione e il rafforzamento delle eccellenze per la ricerca in Italia) – project SPOON (Surface Plasmon resOnance for the in-Operando characterization of shape and size of supported metallic Nanoparticles), prot. number R16RNP82WW and from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 966758 (ERC-PoC project SPECTROKIN). The European Synchrotron Radiation Facility (ESRF, Grenoble, France) is acknowledged for beamtime allocation on BM31 beamline and we would like to thank Dr. Stoian for the assistance and support in using BM31 beamline. The authors are deeply indebted to Dr. Valsania and Dr. Rebba for the HR-TEM and FESEM measurements. The authors are thankful to F. Alleva for the support in the development of the UV-vis setup, and for the UV-vis spectra acquisition and data elaboration.References
- M. P. Dudukovic, Science, 2009, 325, 698–701 CrossRef CAS PubMed.
- T. Van Gerven and A. Stankiewicz, Ind. Eng. Chem. Res., 2009, 48, 2465–2474 CrossRef CAS.
- G. Gert, Nobel Lecture in Chemistry, 2007 Search PubMed.
- Z. Hu, J. Han, Y. Wei and Z. Liu, ACS Catal., 2022, 12, 5060–5076 CrossRef CAS.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, G. M. Veith, D. Wang, M. Manzoli, L. Prati and G. J. Hutchings, Chem. Soc. Rev., 2016, 45, 4953–4994 RSC.
- H. Yoshida, Y. Kuwauchi, J. R. Jinschek, K. J. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta and S. Takeda, Science, 2012, 335, 317–319 CrossRef CAS PubMed.
- B. Weckhuysen, in In situ Spectroscopy of Catalysts, American Scientific Publishers, 2004 ch. 11, pp. 1–11 Search PubMed.
- A. Chakrabarti, M. E. Ford, D. Gregory, R. Hu, C. J. Keturakis, S. Lwin, Y. Tang, Z. Yang, M. Zhu, M. A. Bañares and I. E. Wachs, Catal. Today, 2017, 283, 27–53 CrossRef CAS.
- M. Manzoli, F. Boccuzzi, A. Chiorino, F. Vindigni, W. L. Deng and M. Flytzani-Stephanopoulos, J. Catal., 2007, 245, 308–315 CrossRef CAS.
- M. Manzoli, A. Chiorino and F. Boccuzzi, Surf. Sci., 2003, 532, 377–382 CrossRef.
- F. Vindigni, M. Manzoli, A. Chiorino and F. Boccuzzi, Gold Bull., 2009, 42, 106–112 CrossRef CAS.
- F. Menegazzo, F. Pinna, M. Signoretto, V. Trevisan, F. Boccuzzi, A. Chiorino and M. Manzoli, ChemSusChem, 2008, 1, 320–326 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- M. Shekhar, W. S. Lee, M. C. Akatay, L. Maciel, W. J. Tang, J. T. Miller, E. A. Stach, M. Neurock, W. N. Delgass and F. H. Ribeiro, J. Catal., 2022, 405, 475–488 CrossRef CAS.
- M. Shekhar, J. Wang, W.-S. Lee, W. D. Williams, S. M. Kim, E. A. Stach, J. T. Miller, W. N. Delgass and F. Ribeiro, J. Am. Chem. Soc., 2012, 134, 4700–4708 CrossRef CAS PubMed.
- A. Urakawa, Curr. Opin. Chem. Eng., 2016, 12, 31–36 CrossRef.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Marago and M. A. Iati, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- V. Amendola, S. Polizzi and M. Meneghetti, J. Phys. Chem. B, 2006, 110, 7232–7237 CrossRef CAS PubMed.
- A. M. Watson, X. Zhang, R. A. de la Osa, J. M. Sanz, F. Gonzalez, F. Moreno, G. Finkelstein, J. Liu and H. O. Everitt, Nano Lett., 2015, 15, 1095–1100 CrossRef CAS PubMed.
- V. Myroshnychenko, J. Rodriguez-Fernandez, I. Pastoriza-Santos, A. M. Funston, C. Novo, P. Mulvaney, L. M. Liz-Marzan and F. J. G. de Abajo, Chem. Soc. Rev., 2008, 37, 1792–1805 RSC.
- C. M. Cobley, S. E. Skrabalak, D. J. Campbell and Y. N. Xia, Plasmonics, 2009, 4, 171–179 CrossRef CAS.
- Y. Borensztein, L. Delannoy, A. Djedidi, R. Barrera and C. Louis, J. Phys. Chem. C, 2010, 114, 9008–9021 CrossRef CAS.
- V. Amendola and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 4277–4285 CrossRef CAS.
- M. Ziemba, J. Weyel and C. Hess, Appl. Catal., A, 2022, 301, 120825 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- T. V. W. Janssens, B. S. Clausen, B. Hvolbaek, H. Falsig, C. H. Christensen, T. Bligaard and J. K. Norskov, Top. Catal., 2007, 44, 15–26 CrossRef CAS.
- C. Negri, M. Signorile, N. G. Porcaro, E. Borfecchia, G. Berlier, T. V. W. Janssens and S. Bordiga, Appl. Catal., A, 2019, 578, 1–9 CrossRef CAS.
- F. C. Jentoft, Adv. Catal., 2009, 52, 129–211 CAS.
- F. C. Meunier, React. Chem. Eng., 2016, 1, 134–141 RSC.
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet, S. Gatla, R. Hino, I. Kantor, T. Mairs, M. Munoz, S. Pasternak, F. Perrin and S. Pascarelli, J. Synchrotron Radiat., 2015, 22, 1548–1554 CrossRef CAS PubMed.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. de Juan and R. Tauler, Anal. Chim. Acta, 2003, 500, 195–210 CrossRef CAS.
- A. de Juan and R. Tauler, Crit. Rev. Anal. Chem., 2006, 36, 163–176 CrossRef CAS.
- R. Tauler, Chemom. Intell. Lab. Syst., 1995, 30, 133–146 CrossRef CAS.
- C. Ruckebusch, Data Handl. Sci. Technol., 2016, 30, 1–4 Search PubMed.
- J. Timoshenko and A. I. Frenkel, ACS Catal., 2019, 9, 10192–10211 CrossRef CAS.
- A. Martini and E. Borfecchia, Crystals, 2020, 10, 664 CrossRef CAS.
- W. Windig, C. E. Heckler, F. A. Agblevor and R. J. Evans, Chemom. Intell. Lab. Syst., 1992, 14, 195–207 CrossRef CAS.
- J. Jaumot, R. Gargallo, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef CAS PubMed.
- S. A. Maier, Plasmonics: fundamentals and applications, Springer, 2007 Search PubMed.
- U. Kreibig and M. Vollmer, in Optical properties of metal clusters, Springer, 1995, pp. 13–201 Search PubMed.
- T. Biwa, M. Yui, T. Takeuchi and U. J. M. T. Mizutani, Mater. Trans., 2001, 42, 939–950 CrossRef CAS.
- A. C. Gluhoi, X. Tang, P. Marginean and B. E. Nieuwenhuys, Top. Catal., 2006, 39, 101–110 CrossRef CAS.
- P. D. Srinivasan, H. D. Zhu and J. J. Bravo-Suarez, Mol. Catal., 2021, 507, 111572 CrossRef CAS.
- A. S. Barnard, X. M. Lin and L. A. Curtiss, J. Phys. Chem. B, 2005, 109, 24465–24472 CrossRef CAS PubMed.
- L. W. Finger and R. M. Hazen, J. Appl. Phys., 1978, 49, 5823–5826 CrossRef CAS.
- J. Hernandez, J. Solla-Gullon, E. Herrero, J. M. Feliu and A. Aldaz, J. Nanosci. Nanotechnol., 2009, 9, 2256–2273 CrossRef CAS PubMed.
- T. M. Salama, T. Shido, R. Ohnishi and M. Ichikawa, Zh. Fiz. Khim., 1996, 100, 3688–3694 CAS.
- J. L. Margitfalvi, A. Fasi, M. Hegedus, F. Lonyi, S. Gobolos and N. Bogdanchikova, Catal. Today, 2002, 72, 157–169 CrossRef CAS.
- E. Bus, R. Prins and J. van Bokhoven, Phys. Chem. Chem. Phys., 2007, 9, 3312–3320 RSC.
- L. Delannoy, N. Weiher, N. Tsapatsaris, A. M. Beesley, L. Nchari, S. L. Schroeder and C. Louis, Top. Catal., 2007, 44, 263–273 CrossRef CAS.
- J. W. Watkins, R. C. Elder, B. Greene and D. W. Darnall, Inorg. Chem., 1987, 26, 1147–1151 CrossRef CAS.
- N. Weiher, E. Bus, L. Delannoy, C. Louis, D. E. Ramaker, J. Miller and J. van Bokhoven, J. Catal., 2006, 240, 100–107 CrossRef CAS.
- G. Corro, S. Cebada, U. Pal and J. L. G. Fierro, J. Catal., 2017, 347, 148–156 CrossRef CAS.
- H. Duggal, P. Rajput, I. Alperovich, T. Asanova, D. Mehta, S. N. Jha and S. Gautam, Vacuum, 2020, 176, 109294 CrossRef CAS.
- J. Guzman and B. C. Gates, Am. Ethnol., 2003, 115, 714–717 Search PubMed.
- S. A. Jimenez-Lam, M. G. Cardenas-Galindo, B. E. Handy, S. A. Gomez, G. A. Fuentes and J. C. Fierro-Gonzalez, J. Phys. Chem. C, 2011, 115, 23519–23526 CrossRef CAS.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, React. Chem. Eng., 2021, 6, 954–976 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01392h |
| This journal is © The Royal Society of Chemistry 2024 |