
Strategies for the proton-coupled multi-electron reduction of CO2 on single-atom catalysts
Zhiyuan
Zheng
ab,
Yiming
Yue
a,
Hongying
Zhuo
a,
Qinggang
Liu
*a and
Yanqiang
Huang
 a
a
aCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, PR China. E-mail: liuqg@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
First published on 17th November 2023
Abstract
Catalytic reduction of CO2 to high value-added chemicals is an important approach for tackling the rising CO2 concentration in the atmosphere. Recently, a range of heterogeneous and potentially low-cost single-atom catalysts (SACs) have emerged as promising candidates for the reduction of CO2. However, in comparison to conventional metal nanoparticle catalysts, SACs have long faced challenges in reactions involving multiple reactants and multiple reaction steps due to the limitation of isolated metal sites. This review presents the most recent research advances on the development of single-atom catalysis for deep reduction of CO2. Based on the approaches proposed for proton-coupled multi-electron transfer, detailed introductions and summaries were classified into three categories: 1) strengthen the metal–support interaction to achieve a synergistic catalysis; 2) rational design and regulation of the coordination environment of isolated metal atoms; 3) development of SACs with multi-atom active sites. Finally, the main challenges and future research directions in the field of SACs for CO2 reduction are proposed.
1. Introduction
With the escalating consumption of fossil fuels, the rapid global industrial revolution has resulted in a significant surge in greenhouse gas emissions, causing irreversible damage to the Earth's ecological environment. Since the 1990s, numerous nations have commenced collaboration in tackling the issue of global climate change. In 1997, the Kyoto Protocol achieved a momentous milestone by constructing an international institutional framework to address climate change, effectively restraining emissions of greenhouse gases.1–3Utilizing CO2 as a sustainable C1 feedstock to produce valuable chemical products via catalytic reduction is an important approach for CO2 mitigation. Despite its potential advantages, there are still technological difficulties in its practical application. The intrinsic thermodynamic inertness of CO2 (with a standard Gibbs free energy of −394.38 kJ mol−1) presents the most fundamental challenge.4 CO2 is the ultimate byproduct of fossil fuel combustion, and it possesses the lowest energy density among all carbon-containing species. Transition of CO2 requires significant energy input to overcome the energy barrier of the first C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation. As a result, the activity and selectivity of catalysts for CO2 reduction are relatively low.5,6
O bond dissociation. As a result, the activity and selectivity of catalysts for CO2 reduction are relatively low.5,6
Presently, the techniques for CO2 conversion can be divided into three categories: electrocatalysis, photocatalysis and thermocatalysis.7,8 The electrocatalytic reduction of CO2 is mainly driven by sustainable energy sources such as solar and wind power. Nevertheless, the electrochemical CO2 reduction competes with hydrogen evolution reaction in aqueous solution.9,10 The sluggish dynamics of CO2 activation and the limited selectivity of this technology hinder its practical applications. Photocatalysis technology offers a solution to alleviate the emission of CO2 by harnessing solar energy as a driving force.11 However, regulating the dynamics of photoinduced charge transfer is still a significant scientific issue. Additionally, quantum efficiency is also a significant factor influencing the efficiency of a photocatalyst.12 Thermocatalysis converts CO2 into more valuable products through heat energy. Thermocatalysis often grapples with the challenges of high temperature-induced side reactions and catalyst sintering. However, it possesses the capacity to convert large amounts of CO2 concurrently, rendering it more amenable to large-scale application.13–15
In the past decades, there has been considerable focus on the single-atom catalysts (SACs). SACs offer unique distinctiveness, primarily due to their unsaturated binding sites, the highest atomic utilization efficacy, and the explicitly defined active sites.16,17 In 1979, Yates et al. utilized nuclear magnetic resonance (NMR) spectroscopy and 13C relaxation time distribution to discover that Rh species can be stably distributed in atomic form on the surface of Al2O3.18 Subsequently, Heiz et al.19 employed the soft-landing approach to deposit Pd atoms onto the surface of MgO. They found that the single-atom Pd catalyst can drive acetylene cyclo-trimerization at 300 K. In 2011, Zhang et al.17 reported a Pt1/Fe2O3 SAC prepared with the wet chemistry method. They demonstrated that this catalyst has excellent catalytic performance in the catalytic oxidation of CO. Subsequently, research into SACs has entered an intense development stage and a significant body of literature in the field of CO2 conversion has been produced.
Li et al.20 first reported the use of Ni SACs for the electrocatalytic reduction of CO2 to CO. The single-atom Ni–N–C catalysts, synthesized via a ZIF-8-assisted approach, demonstrated superior performance compared to Ni nanoparticles. Recent density functional theory (DFT) studies revealed that using graphitic carbon nitride (g-C3N4) as support, the single-atom Pd sites can reduce CO2 into HCOOH, whereas the single-atom Pt sites favour the production of CH4. Carbon nanotube (CNT) supported Ni- and Ru-based SACs were also reported for CO2 hydrogenation. Both catalysts were capable of CO2 hydrogenation at 210 °C, with Ni SACs exhibiting higher selectivity for CO, while Ru SACs exhibiting higher selectivity for CH4.
Despite the significant success achieved by SACs, they face significant challenges in catalyzing reactions that involve multiple reactants.21 In a SAC, only the single isolated atom acts as the active site for the adsorption of reactant. Nevertheless, in most catalytic reactions, co-adsorption of multiple reactants is involved on the catalyst surface. In addition, single atoms often have a positive charge and are in a state of electron deficiency, resulting in weaker electron transfer capabilities when compared to clusters and particles. Consequently, SACs tend to convert CO2 into CO, as the electronic state limitations of the active center result in limited product diversity. This issue is illustrated by the difficulty in achieving six-electron transfer and methane or methanol production during CO2 hydrogenation with SACs.22,23
This review summarizes the latest breakthroughs in single-atom catalysis, with the aim of achieving deep reduction of CO2via electrocatalysis, thermocatalysis, and photocatalysis. The ways to regulate the activity of CO2 reduction on SACs can be summarized as follows: 1) strengthen the metal–support interaction to achieve synergistic catalysis; 2) rational design and regulation of the coordination environment of isolated metal atoms; 3) development of SACs with multi-atom active sites. Furthermore, the reasons for the change in catalytic activity, from single atoms up to clusters, are also explored in this article.
2. Overview of catalytic CO2 reduction paths
2.1 Mechanism of thermocatalytic CO2 hydrogenation
Methanol is an important C1 product in thermal catalytic CO2 hydrogenation, while it is often accompanied by the co-production of several other byproducts (CO/CH4/HCOOH). This paragraph will analyse the mechanism of C1 product generation from the perspective of the methanol production pathway. Numerous reaction pathways have been proposed for the methanol synthesis through CO2 hydrogenation, owing to variations in the catalysts system and reaction conditions. The common reaction pathways are the formate pathway, the reverse water gas shift (RWGS) pathway, and the trans-COOH pathway (Fig. 1).24 In the formate pathway, CO2 is hydrogenated to form formate (HCOO*), formyl (H2COO*), formaldehyde (H2CO*), methoxy (H3CO*), and eventually methanol (CH3OH). The formation of HCOO* and H2COO* in the formate pathway is the rate-controlling step.25,26 In the RWGS pathway, CO2 undergoes hydrogenation to generate a *COOH intermediate, which subsequently dissociates into CO. The CO is then further hydrogenated to produce formyl (HCO*), H2CO*, H3CO*, and CH3OH.27–29 The trans-COOH pathway generates a variety of intermediates, including carboxyl groups (COOH*), dihydroxy methylene (COHOH*) and hydroxymethyl (COH*). Further hydrogenation leads to the formation of hydroxyformyl (HCOH*) and hydroxymethyl (H2COH*) intermediates, ultimately resulting in the production of CH3OH.30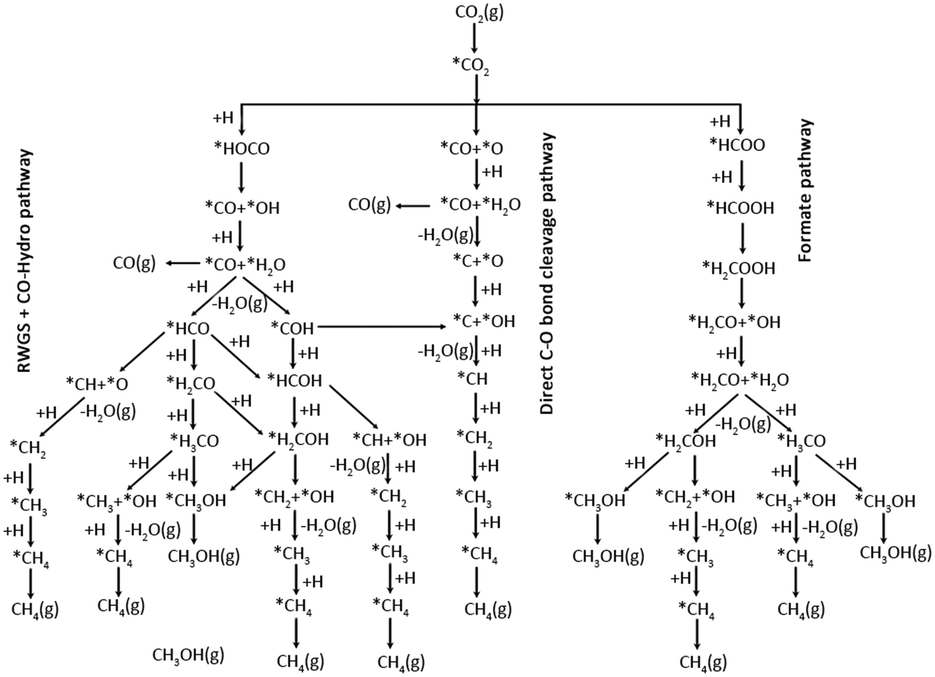 | ||
| Fig. 1 Possible reaction pathways of CO2 hydrogenation to CO, CH3OH, and CH4. *(X) indicates adsorbed species. Reproduced from ref. 24 with permission from American Chemical Society, copyright 2017. | ||
The direct hydrogenation of CO2 to produce C2 products is a complex process involving multiple steps of bond breaking and carbon coupling, generating various C1 products such as CH3OH, CO, and CH4. There remains controversy over the reaction mechanism of CO2 hydrogenation due to the presence of different concentrations of surface species. Arakawa et al. proposed a mechanism involving CO as an intermediate, combining the RWGS and synthesis gas-to-ethanol mechanisms. This involves CO generation, CO and H2 dissociation promoting carbon chain growth to form CxHy, followed by undissociated CO insertion (CO insertion into alkyl or alkyl insertion into CO), and finally hydrogenation to produce high-carbon alcohols. C–O dissociation-insertion and C–C coupling occur at two active sites and require the distance between the active sites to be close enough to increase the possibility of the reaction occurring.31,32
2.2 Mechanism of electrocatalytic CO2 reduction
CO2 electrocatalytic reduction is a multistep proton–electron coupled transfer reaction involving the transfer of 2e−, 4e−, 6e−, and 8e−. As the CO2 electroreduction reaction occurs at the interface between a solid-phase electrocatalyst electrode and a CO2 saturated electrolyte, the heterogeneous reaction at this interface mainly involves the following three steps: (1) CO2 chemisorption on the surface of the electrocatalyst; (2) electron transfer or proton–electron coupled transfer causing the cleavage of the CO bond and/or the formation of C–H bond; (3) the product desorbing from the surface of the electrocatalyst and diffusing into the electrolyte (Fig. 2).33
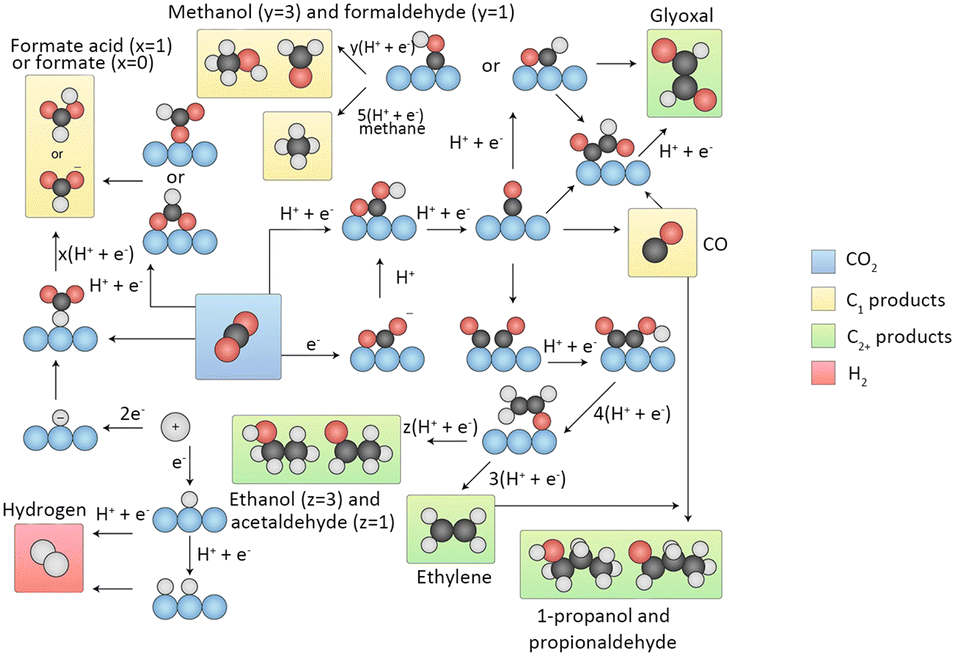 | ||
| Fig. 2 Overview of reaction pathways for CO2RR towards different products. Black spheres, carbon; red spheres, oxygen; white spheres, hydrogen; blue spheres, (metal) catalyst. The arrows indicate whether proton, electron or CPETs take place. Reproduced from ref. 33 with permission from Springer Nature, copyright 2019. | ||
Two pathways are involved in the formation of C1 products. The first pathway is known as the formate pathway, which occurs when hydrogen reacts with the C-site of CO2 to form an *OCHO. This intermediate is further hydrogenated to produce HCOOH. The second pathway is referred to as the carboxyl pathway, which occurs when hydrogen reacts with an O-site of CO2 to form the *COOH intermediate. This intermediate is then hydrogenated to form *CO. CO is a crucial intermediate that can desorb from the surface of the catalyst. Moreover, it can be reduced to *CHO or *COH and proceed through various proton–electron transfers to generate CH4 and CH3OH ultimately. Along with the other pathway, HCOOH can also be formed.
In the process of generating C2 products, CO is a crucial intermediate produced by the reduction of CO2. The process of C2 product generation involves two primary paths of C–C coupling. In the first path, two CO molecules undergo dimerization, giving rise to *OC–CO, which then couples. In the second path, CO undergoes hydrogenation to produce *CHO/*COH, which subsequently couples with another CO molecule to form *OC–CHO/*OC–COH. Crucial intermediates, such as *OCCO, *OCCHO, and *OCCOH, undergo multiple hydrogenation steps to generate C2 products like C2H4 and C2H5OH.33
The generation of C3 products, including propanol (C3H7OH), starts with the CH3CHO* intermediate. *CO insertion triggers the formation of the intermediate CH3COCHO*, which then undergoes four H+/e− transfers, leading to the intermediate CH3CH2CHO*. Further two H+/e− transfer steps eventually yield C3H7OH.34
2.3 Mechanism of photocatalytic CO2 reduction
The CO2–H2O reaction on a semiconductor surface results in several pathways, such as the formaldehyde, carbene, and glyoxal pathways, leading to the production of high-value-added products. Activation of CO2 in the formaldehyde pathway (Fig. 3a) is achieved by combining the active site of the catalyst with one O atom from CO2. The transfer of an electron to CO2 results in the formation of a ·CO2− radical, followed by a proton addition, resulting in a ·COOH radical. The coupling of a proton and an electron in the ·COOH ultimately gives rise to the production of formic acid. Finally, the formic acid accepts two protons, producing formaldehyde and H2O. CH3OH and CH4 can also be synthesized via this pathway if adequate electrons and protons are present, with CH4 formation occurring through the ·CH3 radical intermediate.35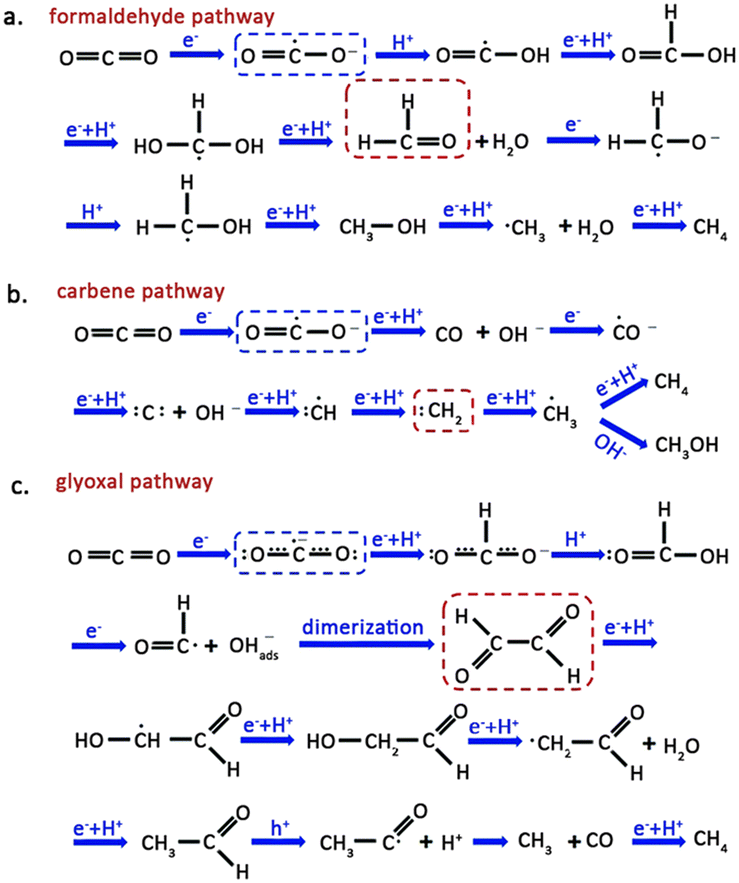 | ||
| Fig. 3 Schematic illustration for the (a) formaldehyde pathway, (b) carbene pathway and (c) glyoxal pathway for CO2 reduction. The blue and red dashes highlight the starting CO2˙− species and key intermediates involved in the pathways. Reproduced from ref. 35 with permission from Royal Society of Chemistry, copyright 2020. | ||
The carbene pathway initially consumes two electrons to generate CO, which serves as an intermediate for subsequent reactions to yield either CH3OH or CH4 (Fig. 3b). The adsorption strength between CO and the catalyst determines whether CO quickly desorbs from the surface or accepts electrons and protons to produce subsequent products. Subsequently, the ·CH3 radical undergoes two reaction pathways: one involves its reaction with OH− to form CH3OH, while the other involves its combination with a proton to produce CH4.36,37
The glyoxal pathway for CO2 transformation involves both of its O atoms coordinating with the active site in a bidentate manner (Fig. 3c). Subsequently, ·CO2− radical interacts with H+ to form formate. Coupling of formate with another H+ generates formic acid. The formyl radical (·HCO) dimerizes to produce glyoxal, a precursor to C2 and C3 products. Like the reaction pathway mentioned above, the combination of ·CH3 radical with a proton generates CH4, with CO acting as a byproduct of the reaction. The appearance of other reaction intermediates in the catalytic system may also cause polymerization of ·CH3 into C2 products.38
3. Transforming CO2 into deep reduction products over SACs
3.1 SACs for CO2 hydrogenation by thermocatalysis
Heterogeneous catalytic hydrogenation is one of the most viable industrial pathways for CO2 conversion. CO2 exhibits high inertness, which requires the catalytic hydrogenation to occur at elevated temperatures. High-pressure reaction conditions are necessary for producing high-valued products; however, the associated high energy consumption and cost pose significant challenges to the large-scale production.39 Harsh reaction conditions also present a heightened challenge in preserving the stability of SACs. The study of thermal catalytic CO2 hydrogenation endeavours to enhance catalyst durability, diminish energy consumption, and generate value-added products.40Fan et al.42 employed a vacuum filtration technique to immobilize Ru atoms onto a porous hexagonal boron nitride substrate (Table 1). Under the conditions of 350 °C and 1 MPa, a remarkable CO2 conversion of 28.7% was attained, with CH4 selectivity as high as 93.5%. The high specific surface area and abundant surface defects of boron nitride facilitate a strong anchoring effect for Ru atoms, thereby ensuring atomic dispersion as well as high catalyst stability. The Ru K-edge X-ray absorption near edge structure (XANES) reveals the formation of a coordinated structure between the Ru and N atoms, leading to a substantial decrease in the oxidation state of Ru. According to the DFT results (Fig. 4), low-valence Ru is identified as the primary active center in CO2 methanation. Camila Rivera-Cárcamo et al.43 found that Ru single atoms on carbon carriers were difficult to maintain atomic dispersion, while those on TiO2 were more stable. The obtained SAC exhibited remarkable differences in the CO2 methanation reaction. X-ray photoelectron spectroscopy (XPS) revealed that the carbon-supported Ru atoms manifest an electron-deficient state, in contrast to those supported by TiO2. The Ru species, abundant in electrons, foster CH4 formation more favorably.
| Catalyst | Temp. (°C) | P (MPa) | CO2 Con. (%) | CH4 Sel. (%) | HCOOH (TON) | CH3OH Sel. (%) | C2H5OH (TOF/h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Ru/pBN-0.58%F | 350 | 1 | 28.7 | 93.5 | — | — | — | 42 |
| RuSA/TiO2 | 210 | 0.61 | 15.6 | 97.3 | — | — | — | 43 |
| Rh1/ZrO2 | 440 | 1 | 62 | 99 | — | — | — | 48 |
| Ir/AP-POP | 120 | 6 | — | — | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 135 135 |
— | — | 44 |
| Ru@MCM-3 | 90 | 3.5 | — | — | >2000 | — | — | 49 |
| Ir1/In2O3 | 200 | 6 | — | — | — | — | 481 | 45 |
| Cu/ZrO2 | 180 | 3 | ∼2.7 | — | — | 100 | — | 51 |
| 7.5% Pt/MoS2 | 150 | 3.2 | — | — | — | 81.3 | — | 52 |
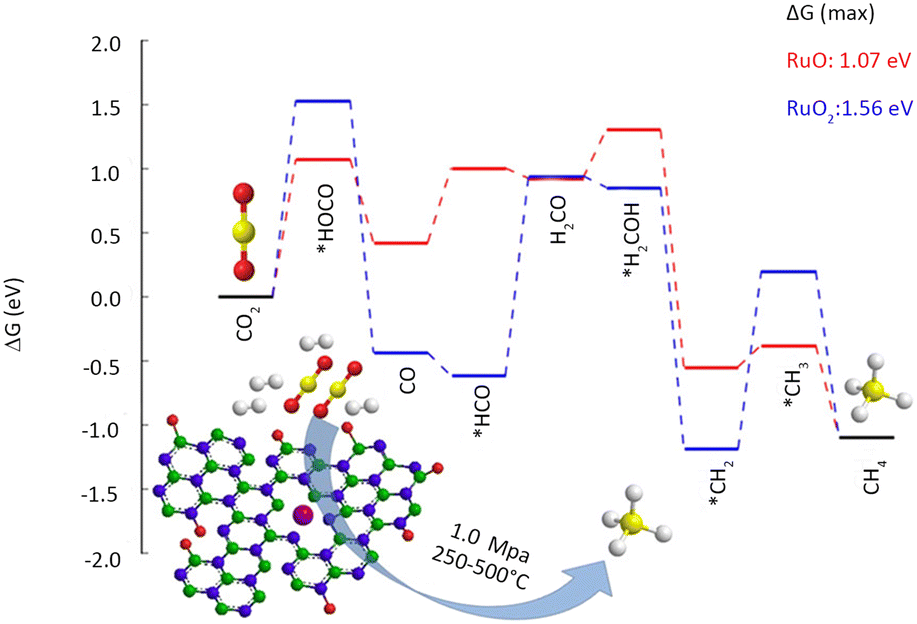 | ||
| Fig. 4 Reaction free-energy diagram for CO2 hydrogenation at 350 °C (623 K). Top view of RuO and RuO2 at the BN vacancy. Reproduced from ref. 42 with permission from American Chemical Society, copyright 2019. | ||
The liquid-phase hydrogenation reaction of CO2 occurs in organic solvents, wherein the metal sites are susceptible to loss. Substrate materials such as nitrogen-rich organic polymers can effectively stabilize metal atoms. Shao et al.44 fabricated an amino pyridine-functionalized porous organic polymer for the construction of Ir SAC. (Fig. 5a), which demonstrated excellent activity for the hydrogenation of CO2 into formate under liquid-phase conditions. XPS analysis indicated that the Ir atoms are primarily in a trivalent oxidation state, which is attributed to the coordination of the aminopyridine with Ir atom. DFT calculations demonstrated that this unique structure facilitates hydrogen dissociation, thereby promoting formate production.
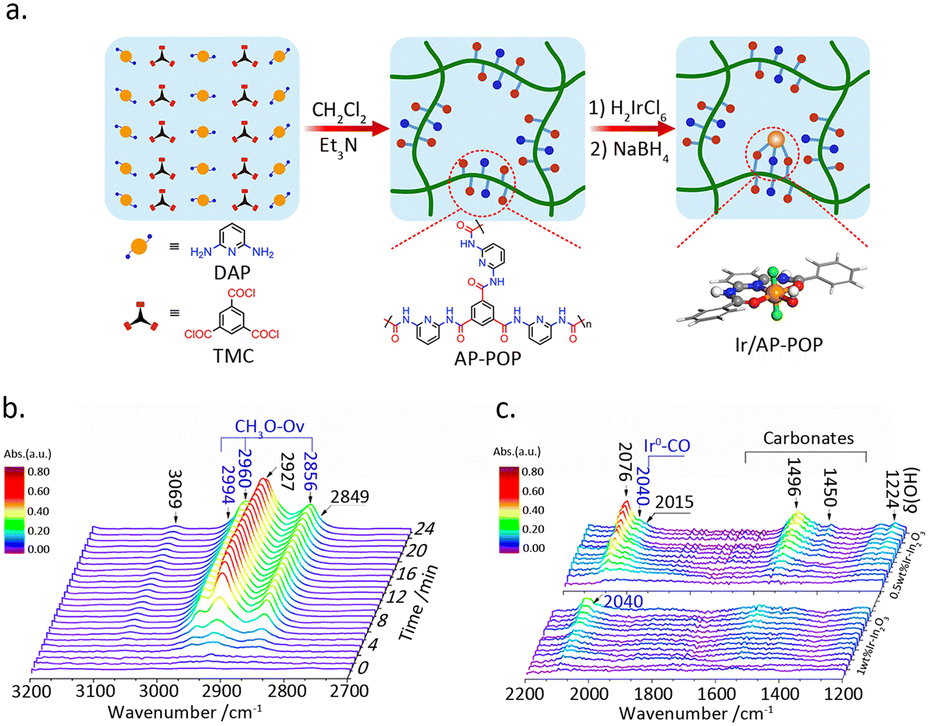 | ||
| Fig. 5 (a) A schematic illustration of the synthesis of Ir/AP-POP SAC. Reproduced from ref. 44 with permission from Elsevier, copyright 2019. (b) In situ FTIR spectra of CO2 hydrogenation with Ir1–In2O3 catalyst bubbled in aqueous solution. (c) Ir–In2O3 samples with different Ir loadings after exposure to CO2 at 200 °C. Reproduced from ref. 45 with permission from American Chemical Society, copyright 2020. | ||
Lately, the controlled growth of the carbon chain via CO2 hydrogenation to yield high-value C2 products has attracted considerable interest. However, the pursuit of attaining remarkable selectivity for ethanol by single-atom catalysis continues to pose a challenge. Ye et al.45 engineered a bifunctional Ir1/In2O3 SAC by anchoring single-atom Ir onto an In2O3 support. By integrating two active catalytic centres, this Ir SAC proved effective for the hydrogenation of CO2 in the liquid-phase, presenting high selectivity for ethanol (>99%) and a high initial turn-over frequency (481 h−1). Detailed characterization studies revealed that isolated Ir atoms couple with neighboring oxygen vacancies to form Lewis acid–base pairs, activating CO2 and producing a carbonyl (CO*) intermediate species adsorbed on the Ir atom. This CO* then couples with the methoxy to form a C–C bond (Fig. 5b and c). This efficacious bifunctional SAC introduces a novel strategy for designing catalysts for single-atom catalysis, through the synergistic utilization of single-atom sites and the roles of substrates.
Büchel et al.47 used the double-nozzle flame pyrolysis method to prepare Rh SACs supported on Al2O3. When different promoters were added, the catalyst activity significantly varied. Potassium-promoted catalysts primarily produced CO, while barium-promoted catalysts could convert CO2 into CH4. The CO-FTIR results indicated that promoters had a significant impact on the valence state of Rh. The presence of barium resulted in the Rh catalyst exhibiting a valence state closer to zero. The valence state distribution of Rh atoms was the main cause of the selectivity difference. The research conducted by Li et al.48 further demonstrated that tailoring both the electronic properties of metal atoms and carriers through alkali ion doping can serve as an effective strategy for tuning the selectivity of CO2 hydrogenation. They found that by doping Na+ into Rh1/ZrO2, even at high temperatures of up to 440 °C and a high CO2 conversion of 43%, the reaction pathway can be switched from methanation to RWGS. Mechanistic studies indicated that Na+ enhances the adsorption of CO2 and COOH* on the ZrO2 carrier through Na–O interactions, thereby inducing the formation of CO. Additionally, Na+ endows the Rh single atom with more electron deficiency, which greatly suppresses the activation of H2 on the catalyst surface and promotes the desorption of CO. Wang et al.49 employed molecular sieve synthesis technology to encapsulate Ru atoms within the inner walls of MCM-41. The resultant structure imparted the essential structural stability required for the dispersion of Ru at the atomic level (Fig. 6a). The catalyst facilitated the conversion of CO2 into formate during the liquid-phase hydrogenation process. DFT calculations unveiled that the highly dispersed Ru atoms served as the active sites for H2 dissociation (Fig. 6b).
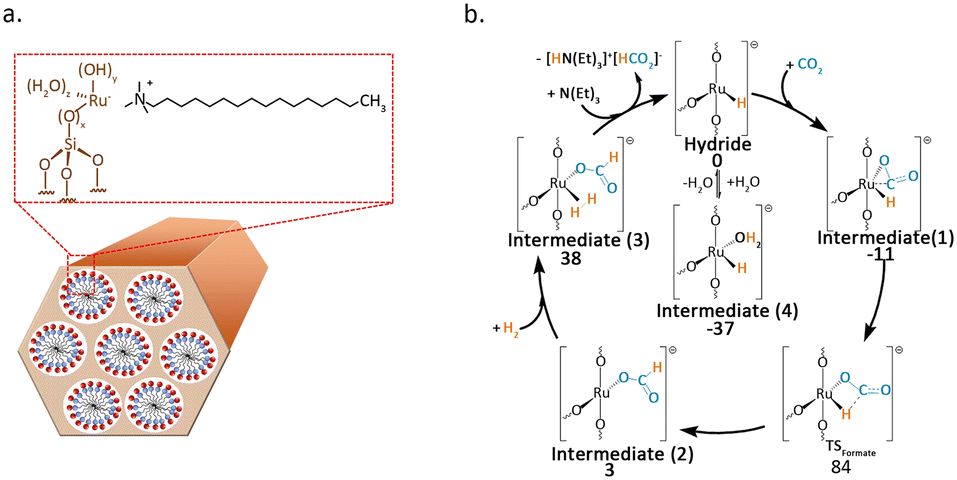 | ||
| Fig. 6 (a) Scheme of the synthesis of Ru@MCM catalyst. (b) Catalytic cycle and respective Gibbs free energies (kJ mol−1) for the proposed mechanism for Ru@MCM-3. DFT calculations performed with PBE0/TZP//PBE0/SVP at 298 K. Reproduced from ref. 49 with permission from Elsevier, copyright 2021. | ||
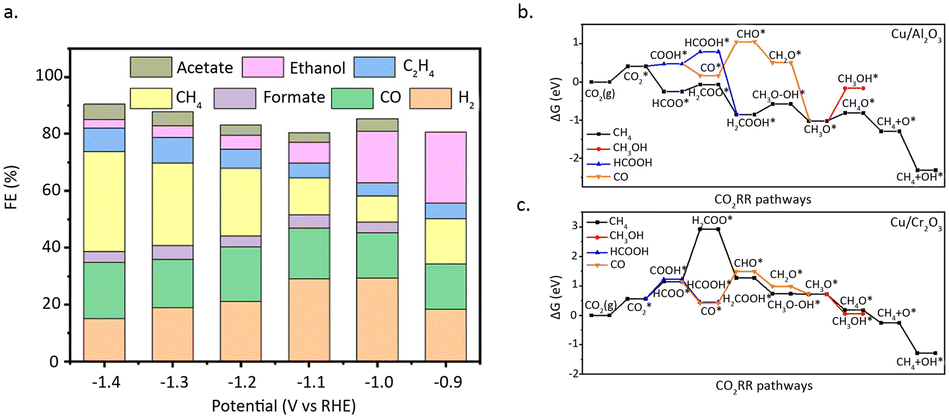
Zhao et al.51 reported a copper-based catalyst featuring isolated copper sites for the conversion of CO2 into methanol. Detailed characterization studies revealed that the catalyst, comprised of single-atom Cu–Zr units with Cu1–O3 units, exclusively facilitates the synthesis of methanol at approximately 180 °C. In contrast, the presence of small copper clusters or nanoparticles leads to the formation of the by-product, CO (Fig. 7a and b). The exceptional activity of these isolated copper sites, coupled with the discernible structural patterns identified in the study, broadens the prospects of single-atom catalysis in thermal catalytic CO2 hydrogenation. Li et al.52 reported that the synergistic interaction between adjacent Pt monomers on MoS2 markedly enhances the catalytic activity for CO2 hydrogenation, compared with isolated single atoms. Adjacent Pt monomers were achieved by increasing the Pt mass loading, while maintaining the dispersion of Pt at the atomic level. As shown in Pt L3-edge XANES profiles, with the increase of Pt loading, the white line intensity for Pt/MoS2 decreased which indicated the difference of electron density. Mechanistic studies indicated that the synergies between adjacent Pt single-atom sites can effectively reduce the reaction barrier and follows a reaction pathway distinct from that of isolated Pt SAC (Fig. 7c). Isolated Pt sites tend to catalytically convert CO2 directly into methanol, whereas adjacent Pt SAC hydrogenates CO2 stepwise into formic acid and methanol (Fig. 7d and e).
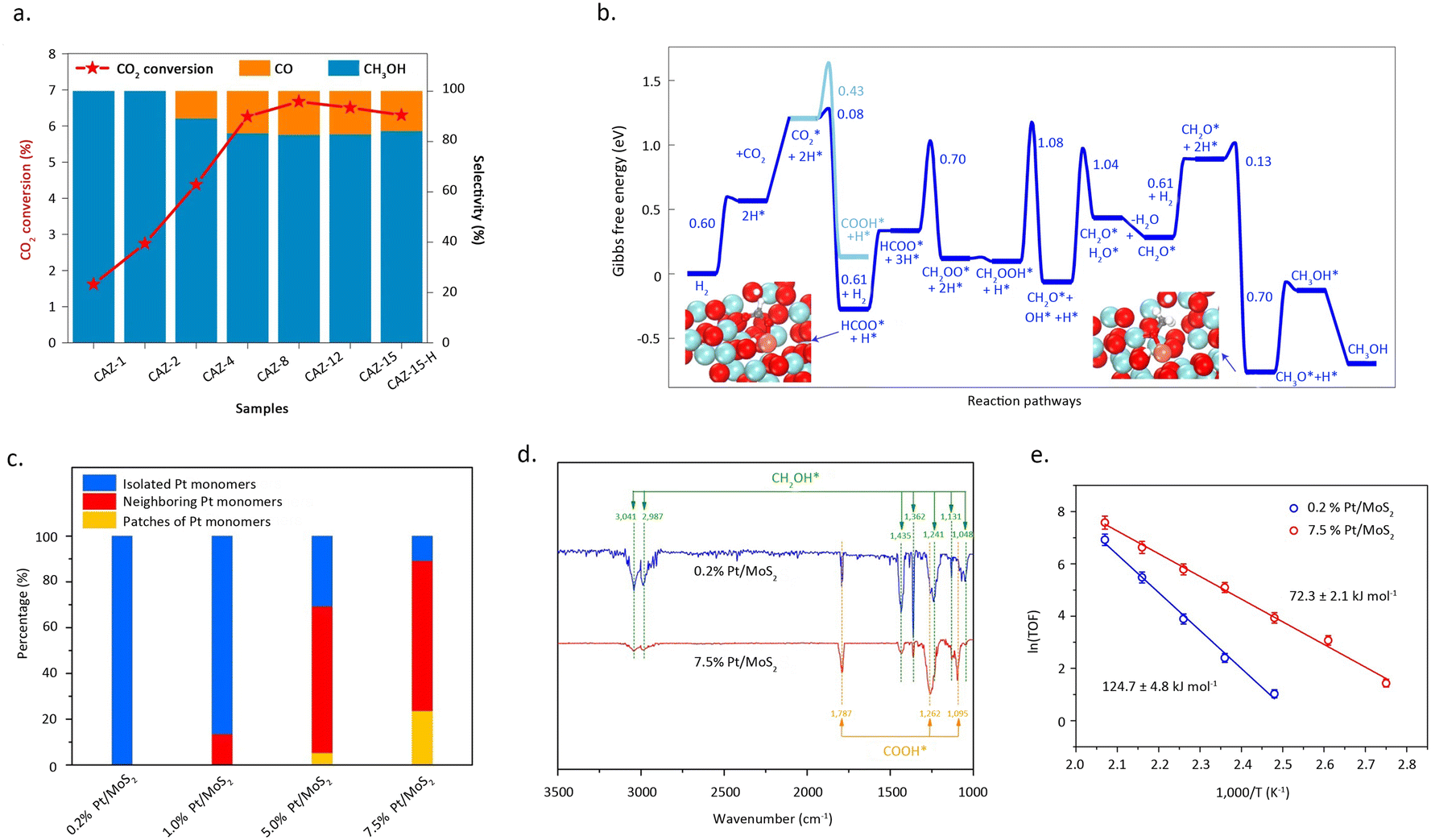 | ||
| Fig. 7 (a) The catalytic activity of CAZ-x and CAZ-15–H. (b) Gibbs free-energy profile of CO2 hydrogenation to CH3OH/CO. Reproduced from ref. 51 with permission from Springer Nature, copyright 2022. (c) Histogram of the contents of isolated Pt monomers, neighbouring Pt monomers, and patches of Pt monomers for atomically dispersed Pt/MoS2 with different Pt loadings. (d) In situ DRIFT spectra of 0.2% Pt/MoS2 and 7.5% Pt/MoS2. The spectra were obtained after exposure of the samples to the mixed gas (CO2:H2 = 1:3, 1 bar) at 150 °C for 0.5 h. (e) The Arrhenius plots of 0.2% Pt/MoS2 and 7.5% Pt/MoS2. Reproduced from ref. 52 with permission from Springer Nature, copyright 2018. | ||
3.2 SACs for CO2 reduction by electrocatalysis
Electrocatalytic CO2 reduction typically occurs in inorganic salt solutions, accompanied by hydrogen evolution reaction.53,54 During the process of electrocatalysis, the electrochemical workstation acts as an electron pump, providing the requisite electrons. CO2 is adsorbed onto a cathode electrode coated with a catalyst, forming adsorbed products following the dissociation of CO2 and the transfer of protons and electrons. The difficulty of obtaining electrons during the CO2 reduction process, as well as the ability of inorganic salt to provide carbon, result in different CO2 reduction products.55,56 Moreover, a significant advantage of CO2 reduction by electrocatalysis lies in their ability to take place under mild reaction conditions. Currently, the catalytic reduction of CO2 using SACs has garnered considerable attention. Important progress has been made in understanding the dynamic evolution of catalyst structures during the reaction process, reaction mechanisms, and the development of novel SACs.Genovese et al.58 successfully achieved the high-selectivity conversion of CO2 to acetic acid under low-potential conditions using Fe SAC supported on carbon–nitrogen. HAADF-STEM images (Fig. 8a) confirmed the isolated Fe single-atom sites in the catalyst. Temperature-programmed XPS (Fig. 8b) measurements confirmed that the Fh–FeOOH phase transitions from an Fe(III) phase to a mixed Fe(III)/Fe(II) phase. Combining with the results from EXAFS spectra, it can be seen that CO2 primarily adsorbs onto the Fe(II) sites and bonds with the pyridine N. N dopants on carbon have a dual function: they coordinate with CO2-related species and stabilize these Fe(II) species, hindering their further reduction. The excellent C–C coupling performance is due to the synergistic effect between the surface nitrogen species of the support and Fe–OOH.
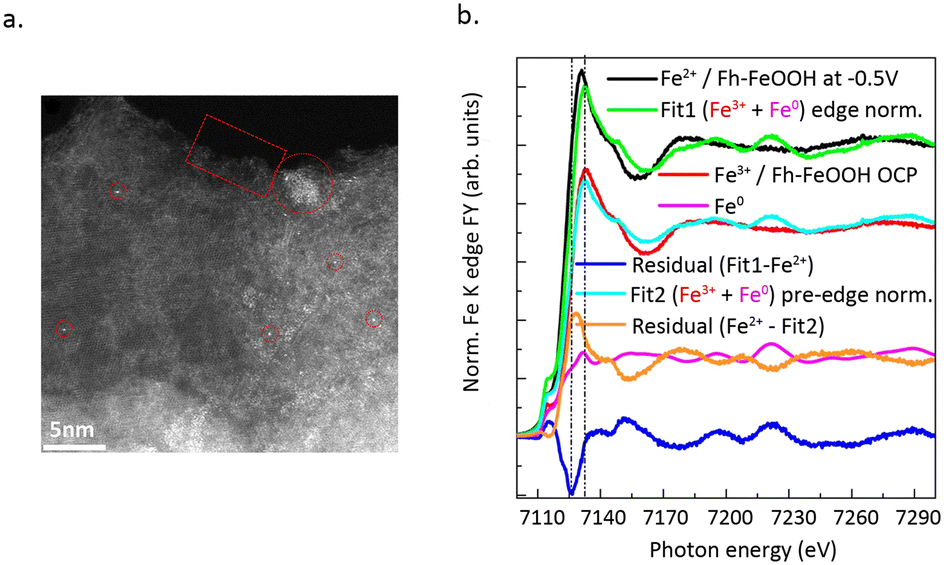 | ||
| Fig. 8 (a) HAADF STEM micrographs of sample Fe/N–C (scale bar 5 nm). (b) Normalized Fe K edge spectra of Fe/N–C at −0.5 V in 0.05 M KHCO3 (black line); Fe/N–C at OCP in 0.05 M KHCO3 component of the fit (red line); Fe foil component of the fit (magenta line); envelope of Fe/N–C at −0.5 V with Fe(III) component (Fe/N–C at OCP) and Fe0 component (Fe foil) in fit 1 (green line). Reproduced from ref. 58 with permission from Springer Nature, copyright 2018. | ||
Metal oxides frequently contain oxygen vacancies, which are a key factor in improving their catalytic performance.59 Oxygen vacancies typically contain extra electrons, thereby facilitating the adsorption and activation of CO2 reactants. Consequently, the overpotential of the reaction decreases while its activity increases. Chen et al.60 utilized a porous monolayer of Al2O3 as a substrate for the deposition of individual copper atoms, enabling the highly selective electrochemical reduction of CO2 to CH4 (Fig. 9a). Bader charge analysis indicated that copper atoms on the Al2O3 surface had higher oxidation states. The Lewis acid sites on the surface of the Al2O3 facilitated the transfer of electrons between the copper atoms and the support. DFT calculations (Fig. 9b) suggested that Cu atoms with a low electron density favor bonding with CO2, producing electron-rich intermediates that facilitate the production of CH4. Hung et al.61 synthesized a copper-loaded Fe SAC for the electrochemical conversion of CO2 to CH4. In situ X-ray absorption spectroscopy (XAS) results indicated that the Fe single atoms remain highly dispersed during the reaction process. DFT calculations showed that the Fe single atoms efficiently stabilize CO species while promoting the formation of *COH intermediate species. The formation of *COH leads to a decreased energy barrier during the conversion of CO2 to CH4. On the other hand, Wang et al.62 designed a CeO2 supported Cu SAC to reduce CO2 to CH4. The strong metal–support interaction between CeO2 and Cu ensures a high dispersion of Cu on the CeO2 surface. Theoretical simulations revealed the presence of three oxygen vacancies surrounding the Cu atoms. A high concentration of surface defects aids in promoting the adsorption of CO2 and carbon-containing intermediates, thereby enhancing the efficiency of CO2 conversion into CH4.
 | ||
| Fig. 9 (a) CO2RR performance of Cu/C–Al2O3 SAC. (b) Calculated free-energy diagrams for the CO2RR over Cu/Al2O3 SAC and (c) Cu/Cr2O3 SAC. Reproduced from ref. 60 with permission from American Chemical Society, copyright 2021. | ||
Zhao et al.64 designed a carbon–nitrogen coated Cu SAC for the electrochemical reduction of CO2 to multi-carbon products. The reaction achieved a selectivity of 36.7% for acetone. The interaction between Cu and pyrrole nitrogen is elucidated through EXAFS and XPS measurements. DFT calculations (Fig. 10a) confirmed that the Cu atoms, which coordinated with four pyrrole nitrogen, serve as the active sites, and can decrease the free energy required for CO2 activation and carbon–carbon coupling. Meanwhile, Yang et al.65 developed a through-hole carbon nanofiber-loaded Cu SAC, a flexible material (CuSAc/TCNFs) (Fig. 10b). This catalyst can significantly promote the electrocatalytic reduction of CO2 to methanol. DFT calculations show (Fig. 10c) that Cu atoms coordinated with different nitrogen species exhibit different adsorption abilities to intermediate species. Specifically, Cu atoms coordinated with four pyridine nitrogen have strong adsorption capabilities to the *CO intermediate species, allowing CO2 to be transformed into CH3OH instead of desorbing to produce gaseous CO.
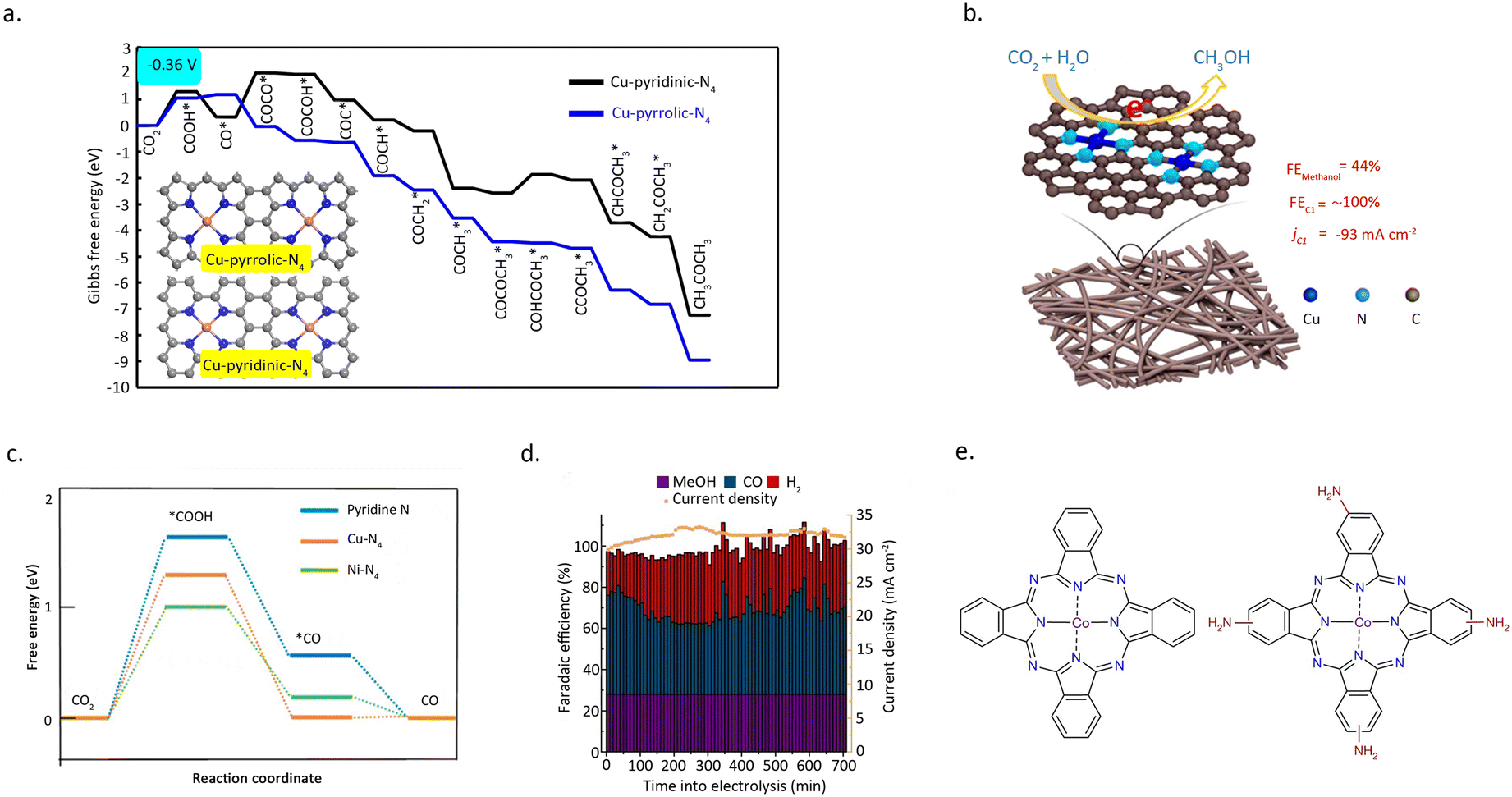 | ||
| Fig. 10 (a) Free energy diagrams calculated at a potential of −0.36 V for CO2 reduction to CH3COCH3 on Cu–pyridinic-N4 and Cu–pyrrolic-N4 sites of Cu–SA/NPC. Reproduced from ref. 64 with permission from Springer Nature, copyright 2020. (b) The schematic and methanol faradaic efficiencies of CuSAs/TCNFs. (c) Free energy diagram of CO2 to CO on pyridine N, Ni–N4, and Cu–N4 structure. Reproduced from ref. 65 with permission from American Chemical Society, copyright 2019. (d) Structural comparison between CoPc and CoPc–NH2. (e) Product selectivity (FE) and total current density for a 12 h electrolysis of CO2 reduction catalysed by CoPc–NH2/CNT at −1.00 V versus RHE. Reproduced from ref. 67 with permission from Springer Nature, copyright 2019. | ||
In addition to catalytic activity, regulating the N-coordination environment around the metal centres can also have a significant impact on the stability of the SACs.66 Wu et al.67 prepared a single-atom Co catalyst by absorbing cobalt phthalocyanine onto carbon nanotubes (CoPc/CNT), which demonstrated remarkable efficacy in catalyzing the reduction of CO2 into CH3OH (Fig. 10d). However, CoPc is prone to reduction during the reaction, leading to poor stability of the catalyst. Intriguingly, the introduction of an amino functional group on the benzene ring can significantly enhance the stability of the catalyst. Amino, acting as an electron-donating group, diminishes the electron transfer from Co to the benzene ring, effectively maintaining the stability of Co during the reaction process. (Fig. 10e) This inference can be substantiated by the UV-vis spectra of CoPc–NH2/CNT before and after the reaction, as the amino-functionalized CoPc catalyst shows no significant structural changes under the reaction conditions.
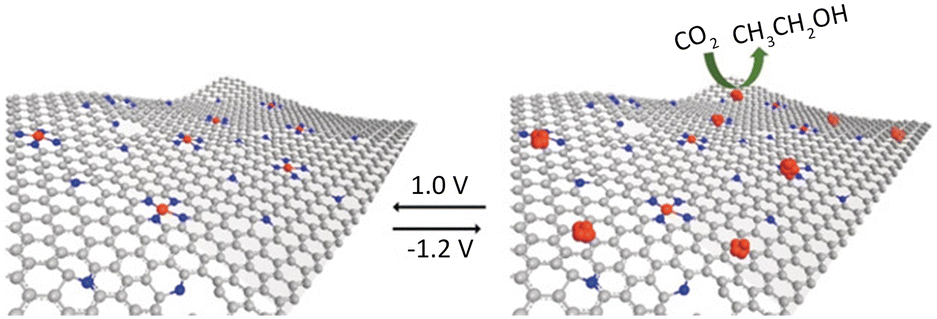 | ||
| Fig. 11 N-doped carbon containing isolated Cu atoms has highly selective for CO2 electroreduction to ethanol. Reproduced from ref. 69 with permission from John Wiley and Sons, copyright 2019. | ||
Guan et al.70 employed a nitrogen coordination strategy to load Cu single atoms on carbon–nitrogen materials and investigated their performance in electrocatalytic CO2 reduction. Interestingly, at low Cu loading (2.1 mol%), Cu primarily existed in the form of single atoms, coordinated with four nitrogen atoms, which predominantly catalyzed the reduction of CO2 to CH4. At high Cu loading (4.9 mol%), the active sites transformed into adjacent Cu–N2 structures. DFT calculations confirmed that the adjacent Cu–N2 structures exhibited stronger adsorption towards CO species, promoting the formation of OC–COH intermediates. This elucidates the high selectivity observed in the catalytic reduction of CO2 to C2H4 by adjacent Cu–N2 centres. Furthermore, constructing bimetallic SACs is another effective approach to enhance catalytic activity. Apart from regulating the distance between metal centres by adjusting the metal loadings, the interaction between bimetallic species also plays a crucial role in determining catalyst performance.
Xie et al.71 reported an efficacious atomic pair electrocatalyst on a carbon nanosheet array electrode with a hierarchical integration, demonstrating a synergistic effect in prompting the activity and selectivity of the CO2 reduction towards formate formation. The atomic pair catalyst comprises adjacent Ni and Sn, each surrounded by four nitrogen atoms (N4–Ni–Sn–N4). The synthesized NiSn atomic pair catalyst manifests exceptional activity for the CO2 reduction towards formate with a turnover frequency of 4752 h−1, outperforming previously reported SACs. Both experimental data and DFT calculations corroborated the electron redistribution of Sn influenced by the neighbouring Ni, which diminishes the energy barrier of the formation of *OCHO intermediate and renders this crucial step thermodynamically spontaneous. This synergistic catalysis offers a paradigm for the rational design and fabrication of atomic pair electrocatalysts with enhanced efficacy.
3.3 SACs for CO2 reduction by photocatalysis
Photo-driven CO2 conversion involves the reduction of CO2 and the oxidation reactions of H2O. The reaction process involves the transfer of electrons and holes and happens in three steps: (1) when the incident light energy (hv) is greater than or equal to the forbidden band width (Eg), electrons in the semiconductor catalyst valence band (VB) absorb energy from photons and are excited to the conduction band (CB), while holes are left in the VB simultaneously. (2) Photo-generated electrons in the CB and the holes in the VB are then transferred from the bulk to the surface of the catalyst. (3) The electrons and holes that have moved to the surface react with molecules adsorbed on the catalyst surface, such as CO2 and H2O.72,73 Semiconductor materials (such as TiO2, ZnS) or heterojunction structures composed of two materials are commonly used as traditional photocatalysts. Traditional photocatalytic materials are prone to decreased stability due to light corrosion. Metal–organic frameworks (MOFs) materials are increasingly used in photocatalysis due to their high specific surface area and porosity, which promote charge and mass transfer. Metal sites in MOFs materials are typically in a coordination unsaturated state, which have a stronger adsorption capacity for CO2.74,75 Currently, the main research directions are in the development of efficient MOFs-based photocatalysts and the study of the mechanism of the reaction process.Sun et al.76 prepared a MOF-253 material and used it as a carrier to prepare Ru-based photocatalysts. Upon loading of Ru–carbonyl complex (MOF-253-Ru(CO)2Cl2), MOF-253 exhibits remarkable photocatalytic activity for CO2 reduction under visible light irradiation. It is worth noting that the performance can be further enhanced by the simultaneous loading of a photosensitizer. Detailed characterization reveals that MOF-253 not only acts as a solid ligand for constructing a supported photocatalyst, but also serves as a platform for constructing composite photocatalytic systems, facilitating charge transfer between the photo-sensors and the surface-anchored photocatalysts.
Ye et al.77 synthesized a novel catalyst using a MOF as the matrix, wherein coordinatively unsaturated single Co atoms were incorporated. This newly developed catalyst exhibits remarkable capability in capturing and photo catalytically reducing CO2 under visible-light irradiation. Detailed mechanistic studies revealed that the presence of single Co atoms in the MOF greatly enhances the electron–hole separation efficiency in the porphyrin units, allowing the photo-generated excitons to selectively migrate from the porphyrin to the single Co atom center, thereby providing a long-lived electron supply to the adsorbed CO2 molecules. As a result, the Co-containing porphyrin-MOF exhibits a significantly enhanced CO2 photoreduction rate compared to the unmodified MOF, with a 3.13-fold increase in CO produce rate and a 5.93-fold increase in CH4 generation rate.
In addition to MOFs, the covalent organic framework (COF) material, a category of crystalline porous materials formed through the covalent bonding of organic building blocks, has garnered increasing interest in the field of photocatalytic CO2 conversion. Kou et al.78 integrated the advantages of micropore structure and single atom catalysis by introducing MoN2 single-atom sites into COF, realizing the first application of COF-based catalyst in reducing CO2 to CxHy under visible light, with the C2H4 selectivity up to 42.92%. The experimental and theoretical findings elucidated that the integration of a single Mo atom within the COF enhances the adsorption and activation of CO, thereby promoting its subsequent hydrogenation reaction. Concurrently, the energy barriers for the production of C2H4 is lowered. This research provides a novel concept for the photochemical reduction of CO2 to C2 derivatives utilizing metal single-atom modified COFs.
Sharma et al.79 reported a simple synthesis strategy toward single-atom Ru catalyst entrapped in porous C3N4 (RuSA–C3N4). Remarkably, by employing this RuSA–C3N4 composite under ambient conditions, an efficient photocatalytic conversion of CO2 into CH3OH was achieved, boasting an impressively high yield of 1500 μmol g−1. XPS measurements disclosed the formation of two distinct Ru moieties: Ru4+ and Ru2+, without any metallic Ru species (Fig. 12a). XANES and Raman analysis confirmed the bonding of Ru to N/C sites (Fig. 12b). These sites serve as a bridge, facilitating the electron transfer and increasing the charge density on Ru, thereby diminishing the barrier for photocarrier transfer. Additionally, the introduction of Ru single atoms extended the lifetime of charge carriers, with the average lifetime on RuSA–C3N4 being 7.6 ns compared to 7.1 ns onC3N4. This, in turn, facilitates the heightened photocatalytic efficacy of RuSA–C3N4 in the aqueous reduction of CO2 to methanol.
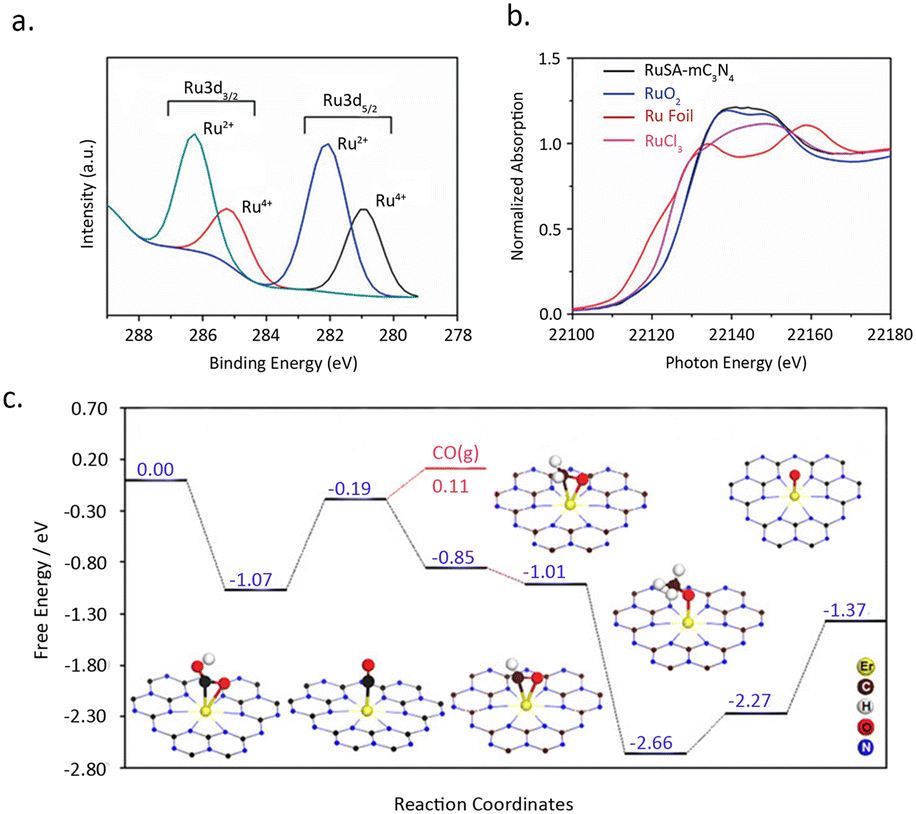 | ||
| Fig. 12 (a) Ru 3d high-resolution XPS spectra of RuSA–mC3N4. (b) Ru K edge XANES of RuSA–mC3N4 (black), RuO2 standard (blue), RuCl3 standard (pink) and Ru foil (red). Reproduced from ref. 79 with permission from John Wiley and Sons, copyright 2021. (c) Calculated free-energy diagram for CO2 reduction to CO and CH4 on the Er1/CN-NT catalyst, as well as the adsorption configurations of key intermediates. The Er, C, H, O, and N atoms are given in yellow, brown, white, red, and blue, respectively. Reproduced from ref. 80 with permission from John Wiley and Sons, copyright 2020. | ||
In addition, introducing rare-earth atoms into carbon–nitrogen materials can also improve its photocatalytic performance. Ji et al.80 developed a Er1–CN-NT catalyst by employing the atom-confinement and coordination (ACC) strategy, where Er single atoms are distributed uniformly on CN nanotubes. The catalyst exhibited superior activity in the photoreduction of CO2 to CH4 in pure water solutions. In situ XANES spectra of Er1/CN-NT revealed that Er was oxidized to a higher valence state by CO2, confirming that Er played a crucial role in the adsorption and conversion of CO2. DFT calculations (Fig. 12c) indicated that CO2 through the –COOH pathway generated CO and CH4.
Zhao et al.81 incorporated Dy single atoms into the heterojunction of CdS and C3N4, which resulted in a six-fold increase in catalytic activity for the photoreduction of CO2 to CH4 (Fig. 13a). The rise in activity is primarily attributed to the function of Dy as a charge transfer bridge, which elevates the separation efficiency of electrons and holes. HAADF-STEM measurements revealed that Dy is dispersed on the catalyst surface in a monatomic form. Furthermore, XPS results for C 1s, N 1s, and Dy 4d indicated that the introduction of Dy leads to the disappearance of COOR and C–NC species, while a new peak appears at 155.6 eV in the Dy 4d spectrum, suggesting coordination between Dy3+ and C and N (Fig. 13b). DFT calculations showed that the introduction of Dy reduces the bond length of Cd–N at the heterojunction interface, indicating a significant promoting effect of Dy single atom on the formation of the heterojunction structure.
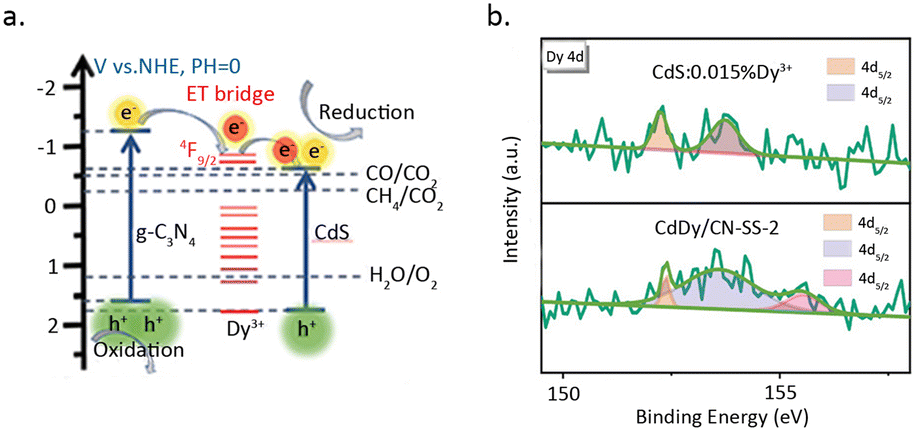 | ||
| Fig. 13 (a) The proposed mechanism diagram of pCO2RR. (b) Dy 4d XPS spectra of CdS. Reproduced from ref. 81 with permission from John Wiley and Sons, copyright 2021. | ||
Using UiO-66–NH2 as support, Wang et al.82 developed a Cu SAC (Cu–SAs/UiO-66–NH2) through a photoreduction method, which significantly promotes the photocatalytic reduction of CO2. Importantly, the developed Cu–SAs/UiO-66–NH2 facilitated the solar-driven CO2 conversion to methanol and ethanol, with producing rates of 5.33 and 4.22 μmol h−1 g−1, respectively. These yields markedly surpass those of UiO-66–NH2 and Cu nanoparticle counterpart. Theoretical calculations indicated that introducing Cu single atoms on UiO-66–NH2 reduces the reaction barrier for the conversion of CO2 into CHO* and CO* intermediates, thereby demonstrating excellent selectivity for methanol and ethanol.
Ma et al.83 utilized a thermolysis-induced gasification approach to prepare a Co/g-C3N4 SAC with a significant metal loading of up to 24.6 wt%. The obtained Co SAC exhibited remarkable activity for the photo-reduction of CO2 to methanol and dimethyl ether (Fig. 14a). HAADF-STEM and EDX mapping images unveiled the atomic dispersion of Co, and no Co–Co bonds were detected by the EXAFS measurements. The XPS results show that the electron density of carbon and nitrogen significantly increases with the increase in Co loading, effectively promoting the capture of photocarriers. Additionally, the generated Co–N2C structure alters the band structure of g-C3N4. The valence band maximum (VBM) and conduction band minimum (CBM) of Co/g-C3N4 moved up 0.99 eV and 0.78 eV as compared to those of g-C3N4, respectively. Interestingly, this substantial change in the band gap can only be achieved when the density of single atom sites is sufficiently high. The formation of high-density Co–N2C sites facilitates the transfer of photocarriers and simultaneously suppresses the recombination of electrons and holes.
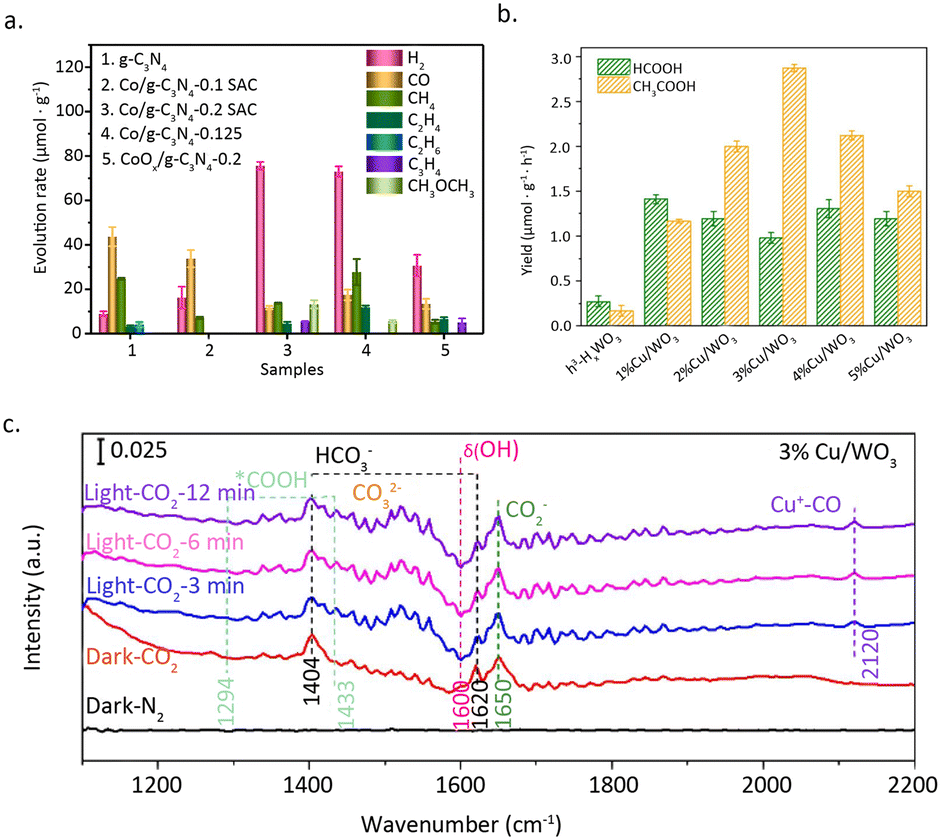 | ||
| Fig. 14 (a) H2, CO, CH4, C2H4, C2H6, C3H6, CH3OCH3 products at 4 h over g-C3N4, Co/g-C3N4-0.1 SAC, Co/g-C3N4-0.2 SAC, Co/g-C3N4-0.25, and CoOx/g-C3N4-0.2. Reproduced from ref. 83 with permission from Elsevier, copyright 2022. (b) Photocatalytic activity of the different Cu loading catalysts. (c) In situ DRIFTS spectra of 3%Cu/WO3. Reproduced from ref. 84 with permission from Elsevier, copyright 2023. | ||
In addition, appropriate atomic density is vital for the generation of C2 products. In a study conducted by Zeng et al.,84 it was found that two-dimensional hexagonal tungsten oxide (h′-HxWO3) with an appropriate loading of Cu atoms can promote the photocatalytic reduction of CO2 to acetic acid (Fig. 14b). HAADF-STEM and EDX measurements confirmed the atomic dispersion of Cu. This inference is further supported by the linear adsorption of CO on Cu at 2120 cm−1 (Fig. 14c). XPS results indicated that the oxidation state of Cu is +1, while electron transfer from W to Cu results in an increase in the oxidation state of W from +5 to +6. Compared to h′-HxWO3, 3%Cu/WO3 with appropriate atomic spacing exhibited stronger adsorption capabilities for intermediate species (HCO3−, COOH, CO), promoting the carbon–carbon coupling between COOH and CO to produce acetic acid. In addition to its impact on the key step of carbon–carbon coupling, the loading of Cu single atoms also plays an important role in the adsorption of CO2 on the catalyst surface. CO2 temperature-programmed desorption (TPD) results showed that three CO2 adsorption configurations, including HCO3−, bidentate carbonate (b-CO32−), and monodentate carbonate (m-CO32−), can form on h′-HxWO3. The introduction of an appropriate amount of Cu single atoms enhanced the binding abilities of all three adsorbed species, which explains the promoting effect of Cu single atoms on the catalytic performance.
4. Conclusions and perspectives
This article provides an overview of the pathways for achieving multi-electron transfer for CO2 reduction by single-atom catalysis. In addition, detailed discussions are presented on the strategies for tuning the geometry and electronic structure of single-atom metal sites, as well as the relationship between local coordination structures and the catalytic activity. This study presents new insights into the atomic-level design of highly active SACs for CO2 reduction. To accelerate research on efficient SACs for CO2 reduction, we have identified the critical challenges that necessitate attention in this field.1) The long-term stability of SACs
SACs have emerged as a promising avenue for CO2 reduction owing to its precise coordination structure, complete atomic efficiency, and high catalytic activity. However, catalyst deactivation poses a major hurdle for its industrial implementation. At low metal loading, catalytic activity falls short of production requirements, while increasing loading leads to aggregation of active metal sites, even under mild electrochemical and photocatalytic conditions as discussed above. Therefore, to achieve stable production by single atom catalysis, stability under reaction conditions needs to be addressed. Stability can be improved by selecting suitable metal precursors, such as metal phthalocyanines, with N anchoring ability and steric hindrance effect that inhibits metal atom aggregation. Additionally, engineering metal oxide carriers with increased oxygen vacancy concentration or introducing N and S atoms for carbon carriers can enhance anchoring ability. Finally, confinement of metal sites using MOFs, molecular sieves, and hierarchical zeolites, combined with structural matching between carriers and metal precursors, can greatly improve catalyst stability.2) In situ characterization techniques for single-atom catalysis
During the reaction process catalysts are typically not static. Regardless of the cause, such as adsorption and desorption of reaction molecules or induction of energy from the reaction environment, metal atoms on the catalyst's surface are subject to disturbance. For instance, copper phthalocyanine exists in a dispersed single-atom state before and after electrochemical CO2 reduction, but in situ testing reveals the truth of Cu atom aggregation. This highlights the need for characterization of SACs, particularly under catalytic conditions. Although electron microscopy, synchrotron-based X-ray absorption spectroscopy, infrared spectroscopy, Raman spectroscopy, and XPS are currently employed techniques for observing catalysts during reaction, time and spatial resolution limitations prevent obtaining specific details such as oxidation state or complex coordination environments. Given that reactions occur at the molecular and even atomic level, featuring timescales of nanoseconds or even picoseconds, current methods prove insufficient for detecting.3) Exploring multi-atomic catalysts
In traditional SACs, each metal center exists independently as individual active sites. This reduces the catalytic ability when multiple reactants and multiple-step reactions are involved. To overcome this limitation, dual-atom catalysts were developed by introducing metal atoms at adjacent sites. These catalysts possess two neighbouring metal sites that can work synergistically during complex reactions, while still maintaining the advantages of SACs. Furthermore, dual-atom catalysts can alter intermediate adsorption configurations to lower the elementary reaction energy barrier. In the presence of two adjacent active sites, the adsorption of intermediate species typically undergoes significant changes. Dual-atom catalysts can also facilitate the conversion of CO2 to C2 products. Typically, SACs during the CO2 hydrogenation process mainly produce C1 products because of the high dispersion of active sites. Dual-atom catalysts, however, can close the gap between active metals to allow for C–C coupling.4) Novel synthetic methodologies for SACs
In recent years, numerous methodologies have been developed for the synthesis of SACs. However, the pursuit of novel synthetic strategies to achieve precise control over the coordination structure of active sites remains a pivotal task. For instance, although various methods have been reported for the preparation of carbon-based SACs, selective modulation of the active site structure through dopants such as P and N species still proves to be exceedingly challenging. Furthermore, due to the diversity of active sites in heterogeneous SACs, identifying true active sites and synthesizing SACs with identical structures pose significant challenges. Theoretically, this task can be solved via the efforts of combining advanced synthetic methodologies, characterization techniques, and DFT calculations. However, all the above tools currently present several shortcomings. For example, the current characterization techniques are even difficult to determine the atomic composition of the active site in SACs, let alone determine the number of coordinating atoms. As the design of SACs based on detailed synthetic understanding remains a long-term objective, it is essential to develop novel strategies that can circumvent these limitations for achieving visible breakthroughs in single-atom catalysis.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the CAS Project for Young Scientists in Basic Research (YSBR-022), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB36030200), the National Natural Science Foundation of China (21925803, U19A2015, 22008136).References
- M. Ronda-Lloret, G. Rothenberg and N. R. Shiju, ChemSusChem, 2019, 12, 3896–3914 CrossRef CAS.
- W. W. Ingwersen, A. S. Garmestani, M. A. Gonzalez and J. J. Templeton, Clean Technol. Environ. Policy, 2013, 16, 719–730 CrossRef.
- A. V. Eliseev, M. M. Arzhanov, P. F. Demchenko and I. I. Mokhov, Izv., Acad. Sci., USSR, Atmos. Oceanic Phys., 2009, 45, 271–283 CrossRef.
- Q. Liu, X. Yang, L. Li, S. Miao, Y. Li, Y. Li, X. Wang, Y. Huang and T. Zhang, Nat. Commun., 2017, 8, 1407 CrossRef.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- Q. Liu, J. Ma and C. Chen, Chin. J. Catal., 2022, 43, 898–912 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS.
- D. H. Nam, P. De Luna, A. Rosas-Hernandez, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS.
- J. Zhang, G. Zeng, L. Chen, W. Lai, Y. Yuan, Y. Lu, C. Ma, W. Zhang and H. Huang, Nano Res., 2022, 15, 4014–4022 CrossRef CAS.
- A. Alvarez, M. Borges, J. J. Corral-Perez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, ChemPhysChem, 2017, 18, 3135–3141 CrossRef CAS.
- M. U. Khan, L. Wang, Z. Liu, Z. Gao, S. Wang, H. Li, W. Zhang, M. Wang, Z. Wang, C. Ma and J. Zeng, Angew. Chem., Int. Ed., 2016, 55, 9548–9552 CrossRef PubMed.
- L. Zeng, Z. Wang, Y. Wang, J. Wang, Y. Guo, H. Hu, X. He, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 142, 75–79 CrossRef.
- K. Chen, J. Yu, B. Liu, C. Si, H. Ban, W. Cai, C. Li, Z. Li and K. Fujimoto, J. Catal., 2019, 372, 163–173 CrossRef CAS.
- Y. Li, S. H. Chan and Q. Sun, Nanoscale, 2015, 7, 8663–8683 RSC.
- J. Peng, S. Wang, H.-J. Yang, B. Ban, Z. Wei, L. Wang and B. Lei, Fuel, 2018, 224, 481–488 CrossRef CAS.
- J. Wang, Q. Hao, H. Zhong, K. Li and X. Zhang, Nano Res., 2022, 15, 5816–5823 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- J. T. Yates, S. D. Worley, T. M. Duncan and R. W. Vaughan, J. Chem. Phys., 1979, 70, 1225–1230 CrossRef CAS.
- A. S. S. Abbet, U. Heiz, W.-D. Schneider, A. M. Ferrari, G. Pacchioni and N. Rösch, J. Am. Chem. Soc., 2000, 122, 3453–3457 CrossRef.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS.
- L. Wang, D. Wang and Y. Li, Carbon Energy, 2022, 4, 1021–1079 CrossRef CAS.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef PubMed.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- B. Yan, B. Zhao, S. Kattel, Q. Wu, S. Yao, D. Su and J. G. Chen, J. Catal., 2019, 374, 60–71 CrossRef CAS.
- Y. Kim, T. S. B. Trung, S. Yang, S. Kim and H. Lee, ACS Catal., 2016, 6, 1037–1044 CrossRef CAS.
- P. Panagiotopoulou and X. E. Verykios, J. Phys. Chem. C, 2017, 121, 5058–5068 CrossRef CAS.
- L. Liu, F. Fan, M. Bai, F. Xue, X. Ma, Z. Jiang and T. Fang, Mol. Catal., 2019, 466, 26–36 CrossRef CAS.
- C. Liu and P. Liu, ACS Catal., 2015, 5, 1004–1012 CrossRef CAS.
- Y. F. Zhao, Y. Yang, C. Mims, C. H. F. Peden, J. Li and D. H. Mei, J. Catal., 2011, 281, 199–211 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Appl. Organomet. Chem., 2000, 14, 836–840 CrossRef CAS.
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Chem, 2021, 7, 849–881 CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M. Zhou, W. Kong, M. Xue, H. Li, M. A. Khan, B. Liu, F. Lu and X. Zeng, Catal. Sci. Technol., 2023, 2044–4761 CAS.
- T. Kong, Y. Jiang and Y. Xiong, Chem. Soc. Rev., 2020, 49, 6579–6591 RSC.
- Z. Sun, H. Wang, Z. Wu and L. Wang, Catal. Today, 2018, 300, 160–172 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539–573 CrossRef.
- S. Wang, L. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2023, 16, 2759–2803 RSC.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2094–2100 CrossRef CAS.
- M. Fan, J. D. Jimenez, S. N. Shirodkar, J. Wu, S. Chen, L. Song, M. M. Royko, J. Zhang, H. Guo, J. Cui, K. Zuo, W. Wang, C. Zhang, F. Yuan, R. Vajtai, J. Qian, J. Yang, B. I. Yakobson, J. M. Tour, J. Lauterbach, D. Sun and P. M. Ajayan, ACS Catal., 2019, 9, 10077–10086 CrossRef CAS.
- C. Rivera-Cárcamo, C. Scarfiello, A. B. García, Y. Tison, H. Martinez, W. Baaziz, O. Ersen, C. Le Berre and P. Serp, Adv. Mater. Interfaces, 2020, 8, 2001777 CrossRef.
- X. Shao, X. Yang, J. Xu, S. Liu, S. Miao, X. Liu, X. Su, H. Duan, Y. Huang and T. Zhang, Chem, 2019, 5, 693–705 CAS.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- Y. Chen, H. Li, W. Zhao, W. Zhang, J. Li, W. Li, X. Zheng, W. Yan, W. Zhang, J. Zhu, R. Si and J. Zeng, Nat. Commun., 2019, 10, 1885 CrossRef PubMed.
- R. Büchel, A. Baiker and S. E. Pratsinis, Appl. Catal., A, 2014, 477, 93–101 CrossRef.
- S. Li, Y. Xu, H. Wang, B. Teng, Q. Liu, Q. Li, L. Xu, X. Liu and J. Lu, Angew. Chem., 2023, 135, e202218167 CrossRef.
- Q. Wang, S. Santos, C. A. Urbina-Blanco, W. Y. Hernández, M. Impéror-Clerc, E. I. Vovk, M. Marinova, O. Ersen, W. Baaziz, O. V. Safonova, A. Y. Khodakov, M. Saeys and V. V. Ordomsky, Appl. Catal., B, 2021, 290, 120036 CrossRef CAS.
- M.-M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlögl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS PubMed.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nat. Catal., 2022, 5, 818–831 CrossRef CAS.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed.
- X. Wang, X. Sang, C. L. Dong, S. Yao, L. Shuai, J. Lu, B. Yang, Z. Li, L. Lei, M. Qiu, L. Dai and Y. Hou, Angew. Chem., 2021, 60, 11959–11965 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. K. Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y. Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS PubMed.
- S. F. Hung, A. Xu, X. Wang, F. Li, S. H. Hsu, Y. Li, J. Wicks, E. G. Cervantes, A. S. Rasouli, Y. C. Li, M. Luo, D. H. Nam, N. Wang, T. Peng, Y. Yan, G. Lee and E. H. Sargent, Nat. Commun., 2022, 13, 819 CrossRef CAS PubMed.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- J. Zhang, C. Zheng, M. Zhang, Y. Qiu, Q. Xu, W.-C. Cheong, W. Chen, L. Zheng, L. Gu, Z. Hu, D. Wang and Y. Li, Nano Res., 2020, 13, 3082–3087 CrossRef.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- J. Gu, C. S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS.
- X. Liu, Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, J. Mater. Chem. A, 2019, 7, 3648–3654 RSC.
- D. Karapinar, N. T. Huan, N. R. Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. G. Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., 2021, 60, 7382–7388 CrossRef CAS PubMed.
- M. Tawalbeh, R. M. N. Javed, A. Al-Othman and F. Almomani, Energy Convers. Manage., 2023, 279, 116755 CrossRef CAS.
- Y. Wang, X. Cui, J. Zhang, J. Qiao, H. Huang, J. Shi and G. Wang, Prog. Mater. Sci., 2022, 128, 100964 CrossRef CAS.
- H. Chen, K. Shen, Q. Mao, J. Chen and Y. Li, ACS Catal., 2018, 8, 1417–1426 CrossRef CAS.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- D. Sun, Y. Gao, J. Fu, X. Zeng, Z. Chen and Z. Li, Chem. Commun., 2015, 51, 2645–2648 RSC.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- M. Kou, W. Liu, Y. Wang, J. Huang, Y. Chen, Y. Zhou, Y. Chen, M. Ma, K. Lei, H. Xie, P. K. Wong and L. Ye, Appl. Catal., B, 2021, 291, 120146 CrossRef CAS.
- P. Sharma, S. Kumar, O. Tomanec, M. Petr, J. Z. Chen, J. T. Miller, R. S. Varma, M. B. Gawande and R. Zboril, Small, 2021, 17, e2006478 CrossRef PubMed.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- Y. Zhao, Z. Han, G. Gao, W. Zhang, Y. Qu, H. Zhu, P. Zhu and G. Wang, Adv. Funct. Mater., 2021, 31, 2104976 CrossRef CAS.
- G. Wang, C. T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS.
- M. Ma, Z. Huang, D. E. Doronkin, W. Fa, Z. Rao, Y. Zou, R. Wang, Y. Zhong, Y. Cao, R. Zhang and Y. Zhou, Appl. Catal., B, 2022, 300, 120695 CrossRef CAS.
- D. Zeng, H. Wang, X. Zhu, H. Cao, Y. Zhou, W. Wang, L. Zhang and W. Wang, Chem. Eng. J., 2023, 451, 138801 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |