
Towards catalytic redox-active iridium polypyridyl complex by in situ photosubstitution†
Yi Zhen
Tan
,
Xiangyang
Wu
,
Yunpeng
Lu
,
Shunsuke
Chiba
 and
Edwin K. L.
Yeow
*
and
Edwin K. L.
Yeow
*
School of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 21 Nanyang Link, 636371, Singapore. E-mail: edwinyeow@ntu.edu.sg
First published on 10th October 2023
Abstract
In this study, we show that the hydrodehalogenation of reductively inert aryl halides is facilitated by the heteroleptic Ir(III) complex [Ir(dF(CF3)ppy)2(dtbbpy)]+ (1) in the presence of N-(tert-butoxycarbonyl)-proline and cesium carbonate under irradiation with blue light. We observed in situ modification of 1 to yield intact (Ir-int) and degraded (Ir-deg) complexes. Ir-int complexes are formed through the functionalization of both the C^N and N^N ligands with α-amino radicals, formed via single-electron-oxidation of cesium carboxylate salt (N-Boc-Pro-OCs) derived from N-(tert-butoxycarbonyl)-proline by the photoexcited Ir complex. In this functionalization, electron-withdrawing fluorine atoms on the dF(CF3)ppy ligands are substituted. The destabilization of the HOMO of the structurally modified Ir-int results in a bathochromic shift of both the excited triplet state absorption and phosphorescence bands when compared to pristine 1. In the presence of excess N-Boc-Pro-OCs, the free Ir-int undergo rapid quenching via excited-state charge-transfer complex formation. The Ir-int˙−, after radical ion separation, are responsible for the hydrodehalogenation reaction of aryl halides.
1. Introduction
Highly phosphorescent iridium(III) complexes display good emission quantum yields and radiative rates, making them excellent candidates as chemosensors,1,2 dyes in organic light-emitting diodes and light-emitting electrochemical cells,3,4 and photocatalysts in CO2 reduction, water splitting, polymerization and organic synthesis.5–11 In particular, the photoinduced dehalogenative functionalization of reductively inert aryl halides were performed, to varying degrees of success, using several Ir(III) complexes such as the homoleptic fac-[Ir(ppy)3] (ppy = 2-phenylpyridine),7 and heteroleptic [Ir(ppy)2(dtbbpy)]+ (dtbbpy = 4,4′-di-tert-butyl-2,2′-bypyridine),8 [Ir(dF(CF3)ppy)2(dtbbpy)]+ (dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine)9 and Ir(ppy)2(NacNac) containing an electron-rich ancillary β-diketiminate ligand (NacNac).10,11 In this work, we will focus on [Ir(dF(CF3)ppy)2(dtbbpy)]+ (1) (Fig. 1) and its photocatalytic role in the hydrodehalogenation reaction of aryl halides (Ar–X).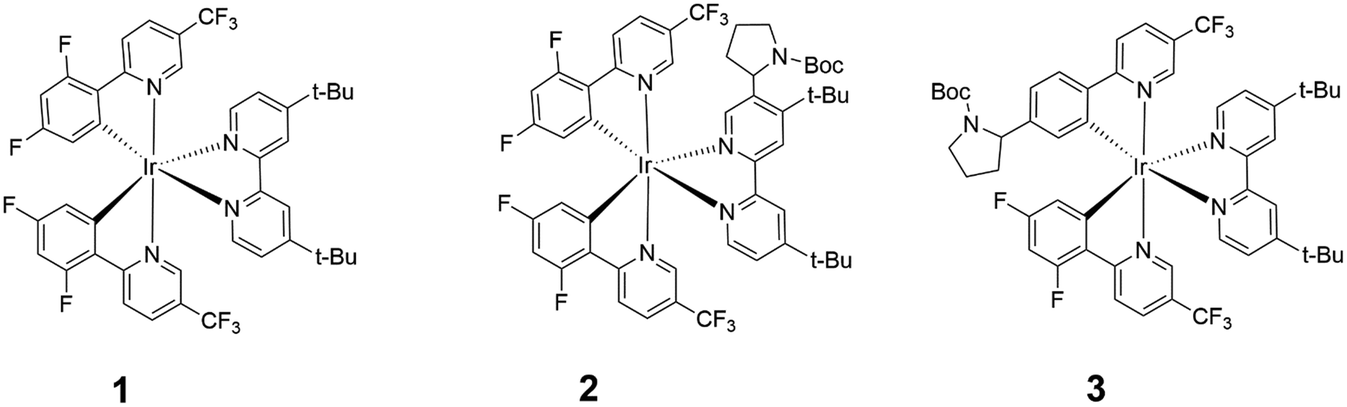 | ||
| Fig. 1 Chemical structures of [Ir(dF(CF3)ppy)2(dtbbpy)]+ (1), 2 and 3. The calculated molecular weights of 1 and 2 are 997.0 and 1146.2 g mol−1, respectively. | ||
Photocatalytic single-electron-reduction of Ar–X (X = I, Br, Cl), in general, occurs via two plausible mechanisms. In the oxidative quenching mechanism, the photoexcited Ir(III) complex (Ir(III)*) directly donates an electron to Ar–X to yield an anion radical [Ar–X]˙− that undergoes mesolytic cleavage of the Ar–X bonds (Scheme 1).11–13 The resulting aryl radical Ar˙ can then abstract a hydrogen atom to form the hydrodehalogenated arenes (Ar–H). The ground-state Ir(III) complex is subsequently regenerated when the oxidized Ir complex (Ir(III)˙+) is reduced by a sacrificial electron donor (D). In the reductive quenching mechanism, the photoexcited Ir(III)* is first reduced by D to form Ir(III)˙− that transfers an electron to Ar–X; yielding [Ar–X]˙− and regenerating the Ir(III) complex.8 However, a conundrum arises when the reductive quenching mechanism is invoked. Given that the reduction potential (Ered) of most aryl halides is more negative than −2 V (e.g., Ered = −2.42 V vs. SCE for 4-bromobenzotrifluoride),14 and the oxidation potential (Eox) of the ground state of commonly utilized Ir(III)˙− is >1.6 V (e.g., E1/2(IrIII/II) = −1.37 V vs. SCE for 1),15 the resulting Gibbs free energy of electron transfer from Ir(III)˙− to Ar–X, based on the Rehm–Weller equation, is positive (i.e., ΔG° = Eox − Ered > 0). This means that charge-transfer is thermodynamically unfavorable. Apart from the models in Scheme 1, a multiphoton mechanism involving the ejection of an electron following photooxidation of the triplet state of a water-soluble Ir complex has also been demonstrated.16,17
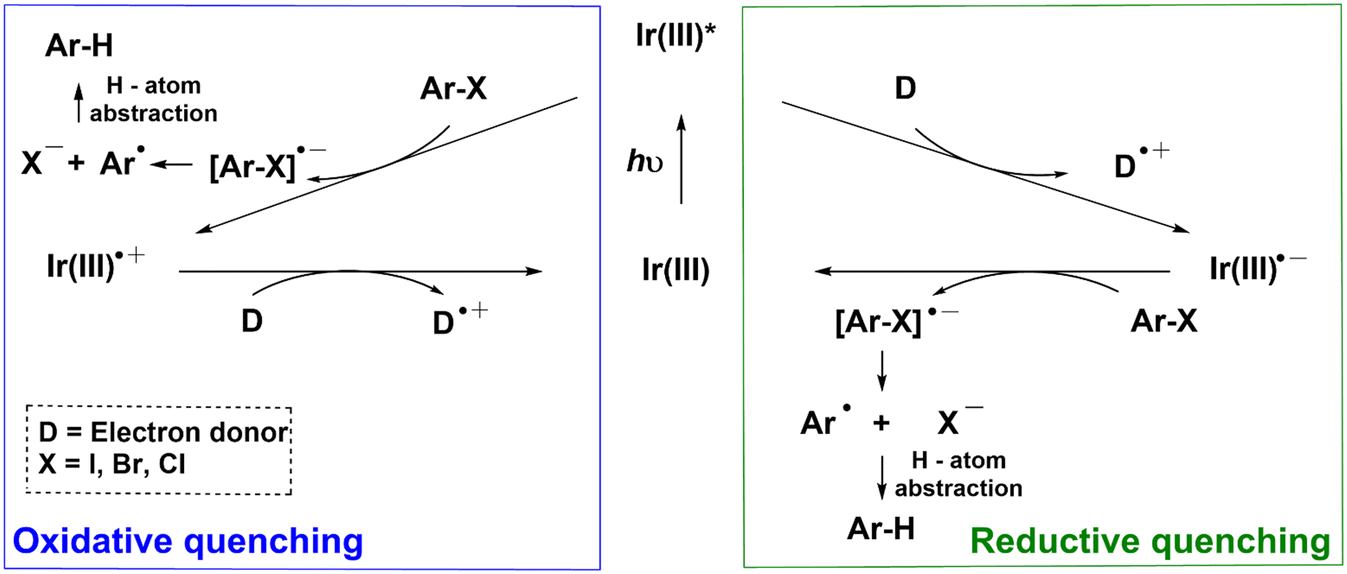 | ||
| Scheme 1 Conventional photoredox catalytic mechanisms (oxidative quenching and reductive quenching) for the hydrodehalogenation of organic halides (Ar–X). D is the sacrificial electron donor. | ||
An important issue that is often neglected when using iridium-based photocatalysts in organic synthesis is the effect of continuous light irradiation on the dye molecule. At room temperature, photoexcited Ir(III) complex can undergo thermal population of the excited metal-centered triplet state (3MC) from the lowest excited triplet metal-to-ligand charge-transfer state (3MLCT).18 In the 3MC state, Ir(III) complexes that are unstable experience breakage of an Ir–N coordinating bond between the metal and ligand; paving the way for possible photodechelation of the ligand and coordination of solvent or surrounding molecules to the iridium.19–21 Complete dissociation of the ligand can be further achieved after triplet–triplet annihilation (TTA) between two 3MC states to a reactive vibrational state.18 The degraded compounds act as luminescence quenching sites and are responsible for the detrimental effect on the operational lifetime of blue phosphorescence organic light emitting diodes (PhOLEDs).22–24
Compromising the structure integrity of Ir(III) photocatalysts during photochemical processes has been shown to play an important role in their catalytic activity. The in situ photoinduced modification of the ligand(s) of several types of Ir(III) complexes with a sacrificial redox agent or substrate has previously been reported. For example, the reductive quenching of excited [Ir(ppy)2(dtbbpy)]+ by a tertiary amine electron donor (e.g., trimethylamine) yielded an Ir(III) complex analogue with a semi-saturated dtbbpy ligand.25,26 The modified Ir(III) complex was found to be capable of reducing even aryl chlorides. In a separate study, in situ functionalization of the 2-phenylpyridine ligands of fac-[Ir(ppy)3] by an alkyl radical derived from ethyl bromoacetate occurred during light exposure.27 While the catalytic activity of fac-Ir(ppy)3 is deactivated due to its transformation to the alkylated complexes, the latter behave as reactive photocatalysts affording good product yield of the alkylated indole.
The photocatalyst 1 in combination with a Ni catalyst was previously employed to drive the decarboxylative cross-coupling reaction between α-amino acid derivatives such as N-(tert-butoxycarbonyl)-proline (N-Boc proline) and aryl halides (Scheme 2A).28 Single-electron-oxidation of the α-amino acid by photoexcited 1 led to decarboxylative formation of the corresponding α-amino radical, which underwent single-electron-oxidative addition to the Ni complex. Interestingly, in our study reported here, we observed that in the absence of the Ni complex, dehalogenation of reductively recalcitrant aryl bromides and chlorides took place when a dimethylformamide (DMF) solution of the aryl halides, 1, N-Boc proline and Cs2CO3 was irradiated with blue light (Scheme 2B). Furthermore, 1 was found to undergo photo-induced degradation and structural modification involving step-wise functionalization of its ligands with α-amino radical. The modified analogues clearly displayed exceptional photoexcited reducing capability. We report the structure-redox activity relationship of the modified Ir complexes and propose a plausible mechanism behind the dehalogenation process.
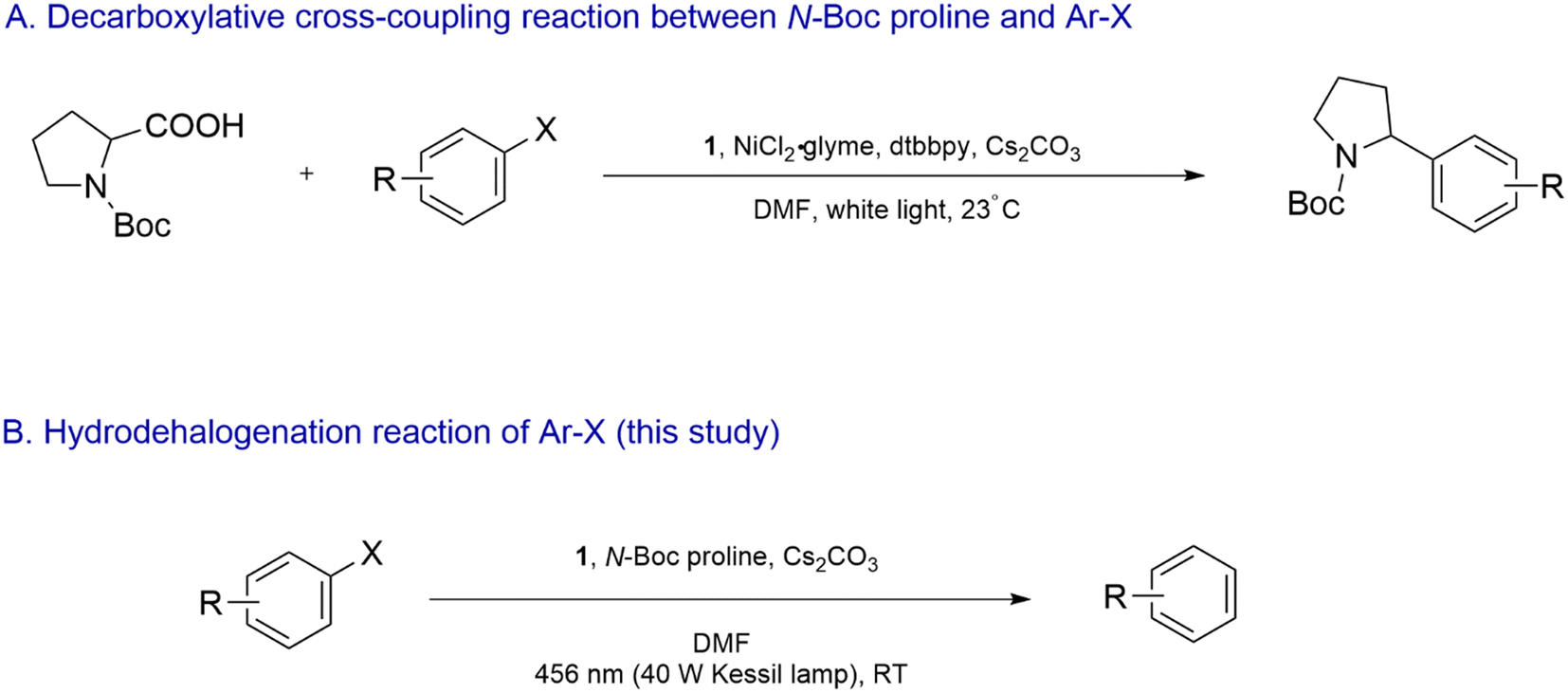 | ||
| Scheme 2 (A) 1/Ni dual-catalyzed decarboxylative cross-coupling reaction between N-Boc proline and Ar–X as reported by Zuo et al. in ref. 28. (B) Hydrodehalogenation of Ar–X in the presence of photoexcited 1 and 2 as reported in this study. | ||
2. Results and discussion
2.1 In situ ligand photosubstitution
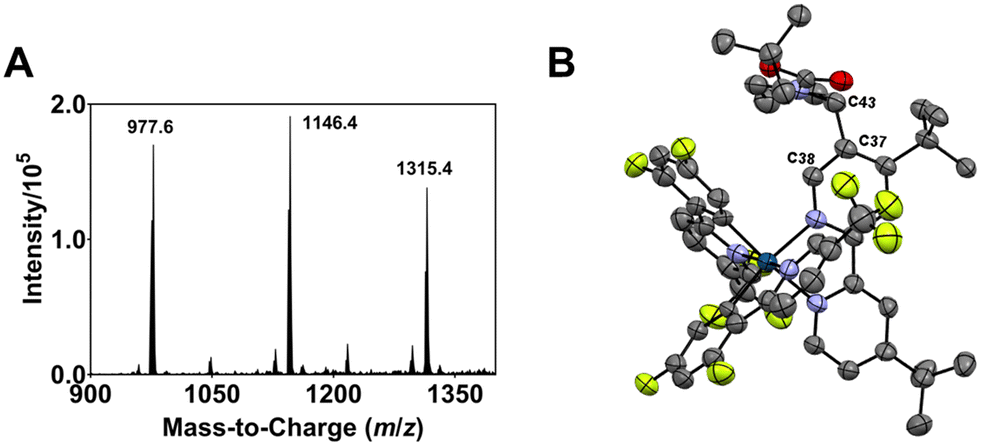 | ||
| Fig. 2 (A) ESI-MS spectrum of the modified Ir complexes formed after irradiating a degassed DMF solution of 1, N-Boc proline and Cs2CO3 with blue light (456 nm, 40 W lamp) for 30 min. (B) ORTEP molecular structure of Ir complex 2 (refer also to the structure represented by ChemDraw in Fig. 1). The acetate counter anions are removed for clarity. | ||
The m/z 1146.4 solute eluted between retention times tR = 132 and 147 min (Fig. S2†) afforded X-ray-quality single crystals via slow diffusion of pentane into a dichloromethane solution of the compound (see Fig. S3† for the ESI-MS spectrum of the isolated compound). An orange crystal with an orthorhombic crystal structure is used for the X-ray crystallographic analysis. Fig. 2B depicts the Oak Ridge Thermal Ellipsoid Plot (ORTEP) drawing of the mono N-Boc pyrrolidine (NBP)-substituted (n = 1) Ir complex 2 (see also Fig. 1). The α-carbon (C43) of a NBP substituent is attached to the C37 position of the ancillary bipyridine (N^N) ligand. The interaction between C43⋯C37 has a bond length of 1.504 Å, typical of a covalent bond, and a C38⋯C37⋯C43 bond angle of 116.211°. The average Ir–NN^N, Ir–CC^N and Ir–NC^N bond lengths are 2.123 ± 0.011 Å, 2.019 ± 0.002 Å, and 2.049 ± 0.003 Å, respectively; close to the corresponding values for 1 (i.e., 2.126 ± 0.003 Å, 2.015 ± 0.001 Å, and 2.042 ± 0.001 Å, respectively) (Fig. S4†). The average C–Ir–N bite angle and N–Ir–N bite angle of 2 are 80.7 ± 0.4° and 76.6°, respectively; comparable to the values for 1 (i.e., 80.1 ± 0.1° and 76.8°, respectively). Acetate ions from the HPLC eluant (ammonium acetate in water), acting as counter anions, were replaced by hexafluorophosphate (PF6−) via an ion exchange prior to utilization of the isolated Ir complex in further experiments.
The m/z 1146 solute collected between tR = 110 and 127 min (Fig. S2†) does not yield single crystals for X-ray crystallographic analysis; suggesting an Ir complex with a NBP substituted at a different position compared to 2. While the integrated intensities of the 1H nuclear magnetic resonance (NMR) signals in the aromatic region for 1 and 2 indicate the correct number of protons (i.e., 16 for 1 and 15 for 2), a similar observation is not noted for the solute collected between tR = 110 and 127 min (Fig. S5;†13C NMR in Fig. S6†); suggesting that the eluate contains regioisomers of mono-NBP substituted Ir complexes not separated by HPLC. The solute with the m/z 1315.4 peak is ascribed to an Ir complex with two NBP units functionalized on the ligands (n = 2) [1 + 2NBP − 2H]. The non-singular peak feature in the HPLC chromatogram (Fig. S2†) indicates a mixture of di-NBP substituted isomers formed.
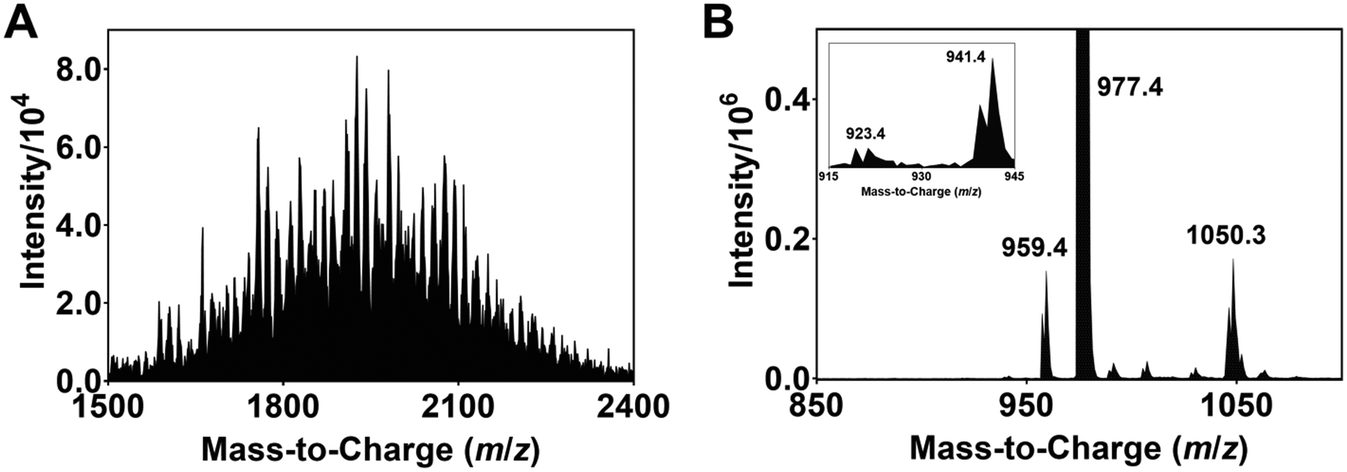 | ||
| Fig. 3 ESI-MS spectra of modified Ir complexes formed after irradiating a degassed DMF solution of (A) 1, N-Boc proline and Cs2CO3 with blue light (456 nm, 40 W lamp) for 8 h, and (B) 1 with blue light for 6 h. The inset in (B) is the zoom-in image of the spectrum from m/z 915 to 945. | ||
The isolation and determination of the exact chemical structures of individual modified complexes are challenging. Nonetheless, plausible identities can be proposed based on the ESI-MS data. Tables S1 and S2† lists the assignments for all discernible peaks observed after irradiation for 8 h. The compounds are divided into two groups, namely modified Ir(III) complexes where none of the ligands are dissociated (Ir-int, Table S1†) and degraded complexes where photodechelation of ligand takes place (Ir-deg, Table S2†). In the case of Ir-int, between six and eight NBP units are substituted on the Ir(III) complex. An example of an Ir-int complex is m/z 2038.8 which has seven NBP substituents attached, and one CF3 group and three F atoms eliminated (cf.1). On the other hand, Ir-deg complexes have i) a C^N or N^N ligand dissociated (e.g., m/z 573.1 and 1715.8), or ii) a DMF/NBP molecule bound to an iridium with either a monodentate ligand, resulting from the breakage of one of the Ir–N coordinating bonds (e.g., m/z 1740.8, 1926.1), or a ligand completely removed (e.g., m/z 1773.7). Similar peaks within 1500 < m/z < 2400 are also seen in the ESI-MS spectra of the solutions for tirra = 12 and 14 h (Fig. S1E and F†). Since no extra peaks with m/z > 2400 are observed, this suggests that further light exposure does not create heavier iridium complexes and an equilibrium has been reached.
To demonstrate that photoinduced defluorination and ligand dechelation occurs, a degassed DMF solution of 1 is irradiated with a 456 nm light and the ESI-MS spectrum of the resulting solution recorded. Peaks associated with complexes with the dissociation of one, two and three fluorine atoms at m/z 959.4 ([1 − F + H]), 941.4 ([1 − 2F + 2H]) and 923.4 ([1 − 3F + 3H]), respectively, are observed (Fig. 3B). In addition, the peak at m/z 1050.3 is ascribed to a complex with the decoordination of an Ir–N bond, and the coordination of a DMF solvent molecule to the Ir ([1 + DMF]).19–21 These peaks, however, are not observed for a non-irradiated sample of 1 (Fig. S1A†). Previous studies have shown that blue OLEDs comprising of iridium-based complexes with multifluorinated ligands are often unstable and undergo ligand dechelation and defluorination during device operation and light/heat treatment.22,29–31 In particular, Yang et al. utilized NMR spectroscopy to detect the decomposition of 1 following blue light irradiation.32 The electron withdrawing effect of the fluorine substituents polarizes the C–F bonds and thus allows the former to be susceptible to substitution by NBP radicals formed after oxidation of the cesium carboxylate salt of N-Boc proline (see discussion below).33,34
The data here clearly show that during initial light exposure, 1 is first converted to mono- and di-NBP substituted Ir complexes (e.g., 2) which are then further transformed, upon prolonged irradiation, into Ir-int with a higher number of NBP substituents, at possibly both the C^N and N^N ligands, and degraded Ir-deg compounds. Similar observations are also seen when the aryl halide 4-bromobenzotrifluoride is included in the reaction mixture during light irradiation (Fig. S8, ESI†). In the photophysical study below, the eluate containing Ir-int and Ir-deg complexes with m/z > 1400, collectively called Ir-mod, is used for further analysis.
2.2 Photophysical study
The ultraviolet (UV)-visible absorption spectra of DMF solutions of 1 (30 μM), 2 and the modified complexes Ir-mod formed after tirra = 8 h (i.e., Ir-mod (tirra = 8 h)) and isolated using a silica column are shown in Fig. 4A. The intense UV absorption bands at ∼266 and ∼302 nm are attributed to intraligand ππ* transitions at the C^N ligand and N^N ligand, respectively.35 The relatively weak band with maximum intensity at ∼ 381 nm for 1 and 2 is attributed to spin-allowed mixed 1MLCT and ligand (C^N)-to-ligand (N^N) charge-transfer (1LLCT) transitions (i.e., 1MLLCT).36,37 This peak becomes less apparent for Ir-mod (tirra = 8 h). Bands visible at wavelengths >450 nm are often attributed to spin-forbidden CT transitions (i.e., 3MLLCT).37,38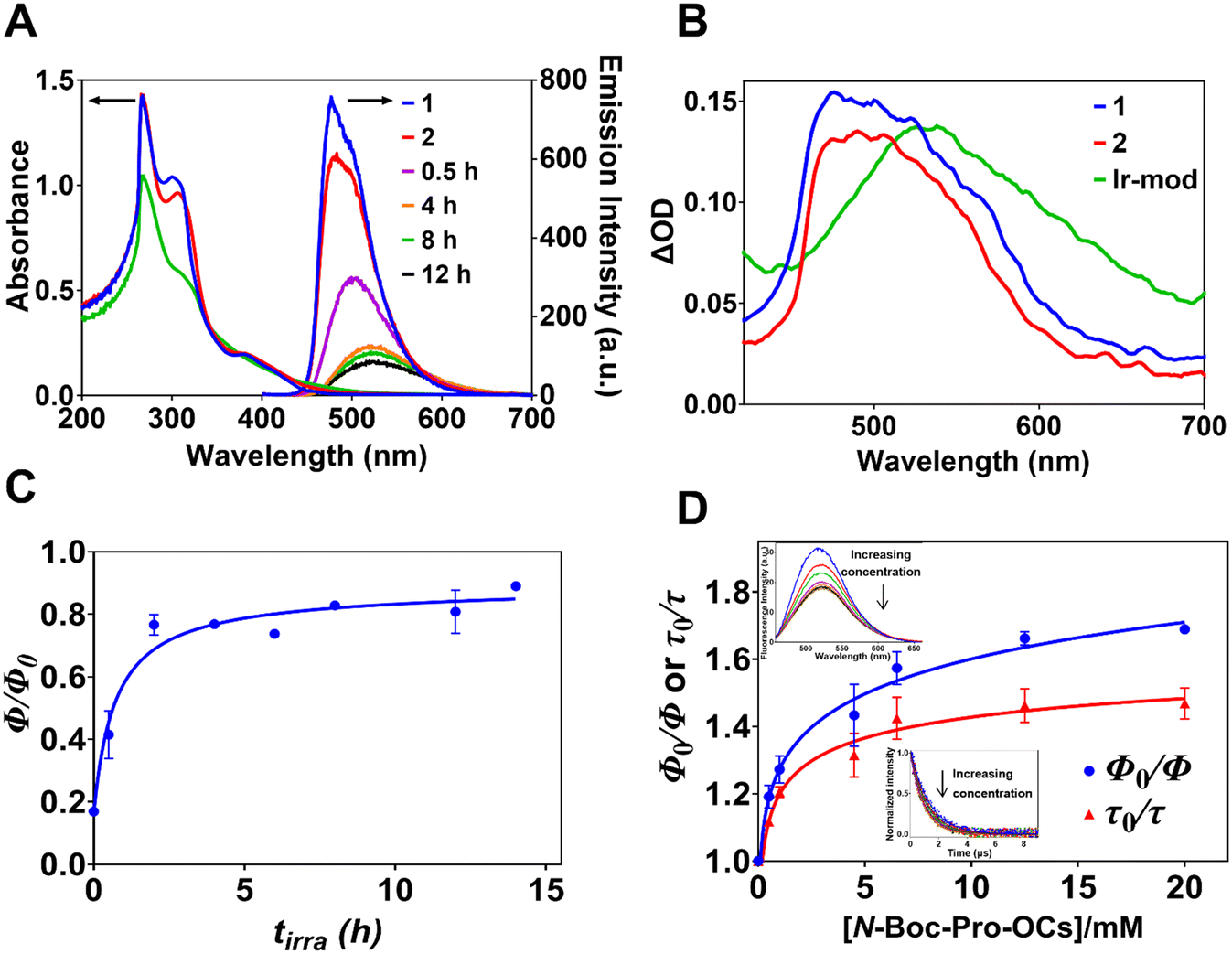 | ||
| Fig. 4 (A) The absorption and emission spectra of 1 (30 μM), 2 and Ir-mod (tirra = 8 h) in degassed DMF. The emission spectra of the isolated modified Ir complexes formed after 456 nm light exposure for tirra = 30 min, 4 h, 8 h and 12 h are also given. The absorbance at the excitation wavelength (i.e., λex = 340 nm) is kept constant at 0.33 for all samples. (B) ns-Transient absorption spectra of degassed DMF solutions of 1, 2 and Ir-mod (tirra = 8 h) at a delay time of 0.068 μs after 355 nm pulsed excitation. (C) The relationship between Φ/Φ0vs. tirra for isolated Ir-mod prepared after various tirra. (D) The relationship between Φ0/Φ and τ0/τ vs. [N-Boc-Pro-OCs] for Ir-mod (tirra = 8 h). The insets show the emission spectra (λex = 340 nm) and lifetime decay profiles (λex = 355 nm and λem = 530 nm) for different [N-Boc-Pro-OCs]. The lifetime profiles are described using a single-exponential decay function. | ||
The emission spectra of 1 and 2 in degassed DMF display maximum intensities at 477 and 481 nm, respectively, with a slight shoulder at 501 nm (Fig. 4A). The corresponding emission quantum yields (Φ) of 1 and 2 are 0.70 ± 0.02 and 0.59 ± 0.02, respectively. The broadened emission spectra of Ir-mod, isolated using a silica column, after different light irradiation times tirra display lower intensities and are red-shifted due to the formation of Ir-int (Fig. 4A). The maximum emission intensity of Ir-mod (tirra > 2 h) occurs at ca. 524 nm. Our density functional theory (DFT) calculations show that since both the metal d orbital and C^N ligand orbital contribute to the HOMO, the addition of electron-donating NBP to and removal of electron-withdrawing fluorine atoms from the cyclometalated ligand destabilizes the HOMO (e.g., compound 3 in Fig. 1 with a NBP attached onto one of the C^N ligands that has both F atoms removed is chosen as a model compound in the computational chemistry calculation; see Tables S3 and S9†).30 Therefore, the HOMO–LUMO gap of Ir-int is reduced, resulting in the observed bathochromic shift. The reduced luminescence is due to the degradation process forming Ir-deg compounds. This is in line with the observation that when a degassed DMF solution of 1 is exposed to a 456 nm light, its emission is also gradually reduced with irradiation time due to the formation of degraded iridium complexes (Fig. S10† and 3B).
The phosphorescence lifetime of 1 (τ0 = 0.98 ± 0.05 μs) experiences only a slight increment when transformed to 2 (τ0 = 1.29 ± 0.03 μs) (Fig. S11†). The Ir(III) complex with a NBP unit substituted at a different position compared to 2 (i.e., m/z 1146 solute collected between tR = 110 and 127 min in Fig. S2†) also displays a similar lifetime of 1.32 ± 0.02 μs (Fig. S12†). In addition, the emission lifetime of a mixture of Ir(III) complexes with two NBP substituents (m/z 1315.4 solute) has a lifetime of 1.42 ± 0.05 μs (Fig. S12†). The data suggests that the substitution of NBP and the number of substituents do not significantly affect the luminescence lifetimes of the modified Ir(III) complexes. The lifetime of Ir-mod (tirra = 8 h) is 1.22 ± 0.07 μs (Table 1 and Fig. S11†) arising from free Ir-int.
| Φ | τ 0 (μs) | τ (μs) | K SS (M−1) | k SS ÷ 108a (M−1 s−1) | |
|---|---|---|---|---|---|
| K TR (M−1) | k TR ÷ 108b (M−1 s−1) | ||||
| a kss = KSS/τ0. b k TR = KTR/τ0. c Isolated modified Ir complexes formed after tirra = 8 h. | |||||
| 1 | 0.70 ± 0.02 | 0.98 ± 0.05 | 0.188 ± 0.006 | 1218.3 ± 0.5 | 12.8 ± 0.0 |
| 1088.7 ± 66.0 | 11.1 ± 0.7 | ||||
| 2 | 0.59 + 0.02 | 1.29 ± 0.03 | 0.236 ± 0.080 | 1247.5 ± 137.5 | 12.2 ± 1.3 |
| 1305.3 ± 75.0 | 10.1 ± 0.6 | ||||
| Ir-mod | — | 1.22 ± 0.07 | 1.07 ± 0.06 | — | — |
Nanosecond-transient absorption spectroscopy (ns-TA) was performed to investigate the kinetics of photoexcited Ir complexes. Both 1 and 2 in degassed DMF show a transient absorption band centered at ca. 495 nm (excited at λex = 355 nm) that is ascribed to the absorption of the excited 3MLLCT state (Fig. 4B).39,40 In the case of Ir-mod (tirra = 8 h), the TA band is red-shifted to 530 nm (Fig. 4B); in agreement with the trend observed in the steady-state emission spectrum (Fig. 4A). The transient kinetic profiles of 1, 2 and Ir-mod (tirra = 8 h), probed at the band maxima over different delay times, decay mono-exponentially with a lifetime component of 1.25, 1.52 and 1.46 μs, respectively (Fig. S13 and S14†). The lifetime values obtained are consistent with the phosphorescence lifetimes of the Ir complexes (Table 1).
While 1 and 2 are unquenched by N-Boc proline (see Fig. S15† for 1), their luminescence intensities are significantly reduced in the presence of the corresponding cesium carboxylate salt (N-Boc-Pro-OCs) (Fig. S16†). The emission quenching dynamics of 1 and 2 by N-Boc-Pro-OCs is studied using the Stern–Volmer analysis. In this case, both the steady-state emission intensity and phosphorescence lifetime are quenched by increasing concentrations of N-Boc-Pro-OCs ([N-Boc-Pro-OCs]) added. For example, the emission lifetimes of 1 and 2 are reduced to 0.188 ± 0.006 and 0.236 ± 0.080 μs, respectively, by 4.5 mM N-Boc-Pro-OCs (Table 1). In this case, the mol ratio N-Boc-Pro-OCs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 is similar to N-Boc proline:1 used in the hydrodehalogenation reaction discussed below. The linear Stern–Volmer plots (Fig. S17†), described by either Φ0/Φ = 1 + KSS[N-Boc-Pro-OCs] or τ0/τ = 1 + KTR[N-Boc-Pro-OCs], where Φ0(τ0) and Φ(τ) are the emission quantum yields (lifetimes) in the absence and presence of N-Boc-Pro-OCs, respectively, yield relatively similar Stern–Volmer constants KSS and KTR, and corresponding quenching constants kSS and kTR (Table 1). Therefore, the 3MLLCT state of 1 and 2 undergoes quenching by accepting an electron from N-Boc-Pro-OCs.28,41
:1 is similar to N-Boc proline:1 used in the hydrodehalogenation reaction discussed below. The linear Stern–Volmer plots (Fig. S17†), described by either Φ0/Φ = 1 + KSS[N-Boc-Pro-OCs] or τ0/τ = 1 + KTR[N-Boc-Pro-OCs], where Φ0(τ0) and Φ(τ) are the emission quantum yields (lifetimes) in the absence and presence of N-Boc-Pro-OCs, respectively, yield relatively similar Stern–Volmer constants KSS and KTR, and corresponding quenching constants kSS and kTR (Table 1). Therefore, the 3MLLCT state of 1 and 2 undergoes quenching by accepting an electron from N-Boc-Pro-OCs.28,41
In order to understand the interaction between Ir-mod and N-Boc-Pro-OCs, we plotted the emission quantum yield ratios (Φ/Φ0) of Ir-mod formed after different tirra in the presence (Φ) and absence (Φ0) of [N-Boc-Pro-OCs] = 4.5 mM. It is observed that Φ/Φ0 increases with tirra before levelling-off at tirra ∼ 5 h. (Fig. 4C). We did not observe any band shift or broadening in the UV-visible absorption spectrum of Ir-mod (tirra = 8 h) in the presence of a high concentration of N-Boc-Pro-OCs (e.g., 20 mM) (Fig. S18†) indicating that no ground-state complexes are formed between the two moieties. On the other hand, the corresponding emission spectrum of Ir-mod (tirra = 8 h) is red-shifted in the presence of the N-Boc-Pro-OCs (e.g., by 6 nm when [N-Boc-Pro-OCs] = 20 mM, see inset of Fig. 4D). This is likely due to the formation of excited-state charge transfer complexes.42,43 It is worth noting that in the case of 1, no emission peak shift is observed at high concentrations of N-Boc-Pro-OCs (Fig. S19†). Interestingly, the luminescence lifetime of Ir-mod (tirra = 8 h) is only slightly quenched in the presence of N-Boc-Pro-OCs (inset of Fig. 4D). The non-linear Stern–Volmer plots in Fig. 4D reveals the existence of two groups of Ir-int.44 The first group, contributing to the initial fast increase in Φ0/Φ, is attributed to the quenching of free Ir-int by N-Boc-Pro-OCs. We therefore propose that an excited-state charge-transfer complex is likely formed between the free triplet Ir-int and N-Boc-Pro-OCs: 3Ir-int + N-Boc-Pro-OCs → [Ir-int˙−/NBP·]. The second group, responsible for the plateauing of Φ0/Φ at higher [N-Boc-Pro-OCs], is made up of Ir-int that are not quenched by N-Boc-Pro-OCs.44 Steric hindrance arising from some of the heavily NBP-substituted Ir-int could prevent N-Boc-Pro-OCs from approaching close enough to create a charge-transfer complex.
In summary, during the initial stages of blue light irradiation of 1 with N-Boc-Pro-OCs, charge-transfer results in the formation of N-Boc pyrrolidine radicals that undergo substitution onto the metal complex ligands. With prolonged light excitation, 1 is modified to Ir-int where portion of the excited 3Ir-int is rapidly quenched by forming excited-state charge-transfer complexes.
2.3 Electrochemical characterization
The reduction potentials of 1 in DMF are determined to be Ered = −1.31 and −1.64 V vs. SCE, corresponding to the single-electron transfer to the N^N ligand and C^N ligand, respectively (Fig. 5 and Table 2).15 Compared to the cyclic voltammogram measured in acetonitrile (ACN), the Ered values obtained are not significantly different (i.e., −1.35 and −1.73 V vs. SCE in ACN, Fig. S20†). In addition, the reversible oxidative peak of 1 in ACN yields an oxidation potential Eox = +1.73 V vs. SCE, in line with the literature value.15 On the other hand, the oxidative wave in DMF is not observed within the limited electroactive window of the solvent (∼−2 V to +1.5 V for DMF). The cyclic voltammogram of 2 (Fig. 5) displays similar reduction potentials to that of 1 (Table 2). This suggests that the NBP substituted on the ancillary dtbbpy ligand does not significantly perturb the energy of the LUMO. This result is in line with the computational DFT calculations (Table S3 and Fig. S9†). Unlike 1 and 2 where the oxidative wave is not observed, Ir-mod (tirra = 8 h) complexes clearly show an electrochemically irreversible oxidative peak at +1.00 V (Fig. 5). It should be noted that the anodic peak is not attributed to the oxidation of NBP itself (i.e., +0.80 V vs. SCE in DMF, Fig. S21†). The cyclic voltammogram of Ir-mod (tirra = 8 h) shows a single reversible reductive peak with a potential of −1.41 V vs. SCE.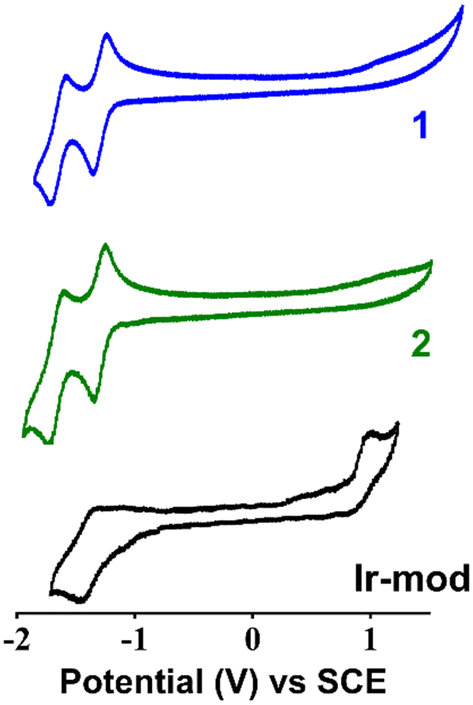 | ||
| Fig. 5 Cyclic voltammograms of 1, 2 and Ir-mod (tirra = 8 h) in degassed DMF. | ||
| Compound | E ox, V vs. SCE | E red, V vs. SCE |
|---|---|---|
| 1 | — | −1.31; −1.64 |
| 2 | — | −1.31; −1.65 |
| Ir-mod | +1.00 | −1.41 |
2.4 Hydrodehalogenation of aryl halides
We investigate the hydrodehalogenation of a series of aryl halides using the Ir complexes discovered in this study. We embarked on our investigation by using 4-bromobenzotrifluoride as a model substrate and first examined its debromination in degassed DMF using 1. In the absence of 1 and N-Boc proline, no product yield is detected after exposing the sample to a 456 nm light (40 W, Kessil lamp) for 24 h (entry 1, Table 3). The formation of α,α,α-trifluorotoluene in a small amount is observed when 1 (1 mol%) is added in the reaction mixture without N-Boc proline (entry 2, Table 3). On the other hand, full conversion of 4-bromobenzotrifluoride is achieved when a solution including 1 (1 mol%), N-Boc proline (0.03 M) and Cs2CO3 (0.03 M) is irradiated with blue light for 24 h, affording α,α,α-trifluorotoluene in 93% yield (entry 3, Table 3). Although a chain-propagation process involving the α-amino radical formed after reduction of the carboxylate salt N-Boc-Pro-OCs by photoexcited 1 might be responsible for the dehalogenation reaction,11 the measured photochemical quantum yield of α,α,α-trifluorotoluene is low (i.e., ∼0.4%). In addition, based on an alternating light on/off cycle experiment, it is noted that no debromination occurs when the light is turned off (Fig. S22†). Therefore, dark radical chain reaction as a primary mechanism can be ruled out.| Entry | Base | N-Boc proline | Ir(III) complex | 456 nm light | Product yield (%) | Conversion yield (%) |
|---|---|---|---|---|---|---|
| a From gas-chromatography with flame ionization detection (GC-FID). b From 19F NMR. | ||||||
| 1 | Cs2CO3 | − | − | + | 0a (0)b | 0a (3)b |
| 2 | Cs2CO3 | − | 1 | + | 5a (5)b | 13a (8)b |
| 3 | Cs2CO3 | + | 1 | + | 93a (91)b | 100a (91)b |
| 4 | Cs2CO3 | + | 2 | + | 91a (94)b | 100a (91)b |
| 5 | Cs2CO3 | + | Ir-mod | + | 85a (84)b | 83a (89)b |
| 6 | Cs2CO3 | + | Ir-mod | − | 0a (0)b | 2a (2)b |
| 7 | Cs2CO3 | − | Ir-mod | + | 12a (9)b | 21a (12)b |
Highly efficient hydrodebromination is observed using 2 (1 mol%) under similar reaction conditions, providing α,α,α-trifluorotoluene in 91% yield (entry 4, Table 3). In the absence of both N-Boc proline and Cs2CO3, the product yield is only 2% when complex 2 is used. The absence of N-Boc proline and presence of Cs2CO3 afford the formation of α,α,α-trifluorotoluene in 10% yield when 2 is used, comparable to that of 1.
The activity of the modified complexes in Ir-mod (tirra = 8 h) is assessed by assuming an average molecular weight of 1915 g mol−1 from the ESI-MS data. The yield of α,α,α-trifluorotoluene is 85% in the presence of 1.5 mol% Ir-mod (tirra = 8 h) (entry 5, Table 3). The absence of light results in a detrimental effect on the hydrodebromination process (entry 6, Table 3); clearly showing that a photoinduced reaction is required. In addition, removing N-Boc proline while retaining Cs2CO3 in the reaction results in inefficient debromination of 4-bromobenzotrifluoride by Ir-mod (tirra = 8 h) (entry 7, Table 3). The substrate scope of the hydrodehalogenation is further expanded to include a series of bromoarenes and chloroarenes, and good to excellent yields of the dehalogenated products are detected in the presence of 1 and Ir-mod (tirra = 8 h) (Scheme 3). It is clearly demonstrated here that the modified Ir complexes behave as active photocatalysts.
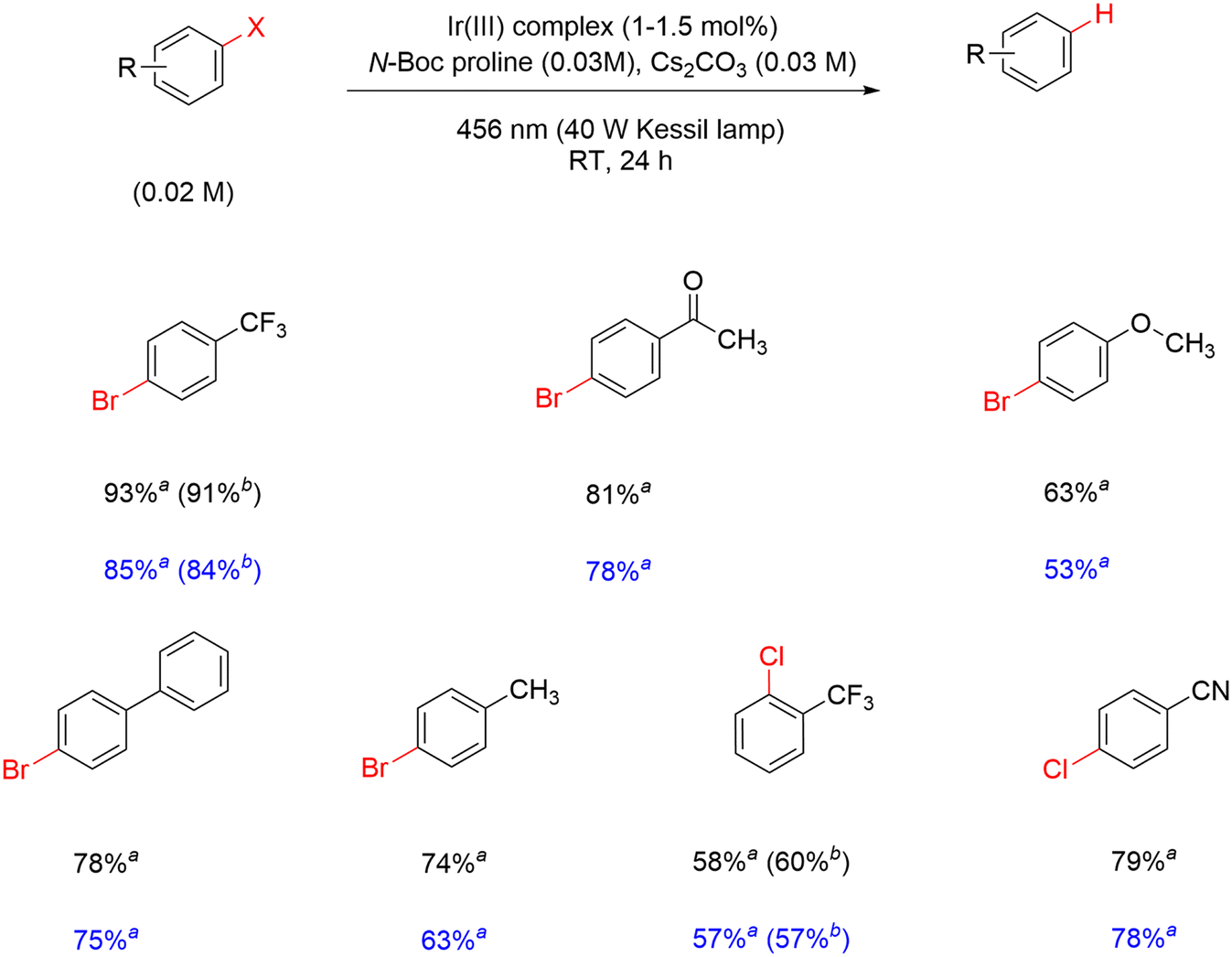 | ||
| Scheme 3 Hydrodehalogenation of aryl halides using 1 (yields reported in black) and Ir-mod (tirra = 8 h) (yields reported in blue). aFrom GC-FID; bfrom 19F NMR. | ||
We propose that the modified Ir complexes with NBP substituent(s) are responsible for the hydrodehalogenation process. 1 is converted to catalytically active Ir-int complexes with reaction time. The Ir(III) complex 1 becomes more substituted with NBP as the reaction progresses, and those that do not suffer from ligand dissociation (i.e., Ir-int) are most likely responsible for the catalytic action. Oxidative quenching of Ir-int by aryl halides is inefficient as verified experimentally by the lack of phosphorescence quenching of Ir-mod (tirra = 8 h) in the presence of 4-bromobenzotrifluoride (Fig. S23†). Since ground-state Ir-mod (tirra = 8 h) is unable to catalyze the reaction (entry 6, Table 3), abstraction of the halogen from the aryl halide by ground-state Ir-int, a mechanism previously observed in pincer-type Ir complexes, is also not applicable.45
Based on the above results, we propose a mechanism as illustrated in Scheme 4. 3Ir-int form charge-transfer complexes (i.e., [Ir-int˙−/NBP·]).46 After radical ion separation, Ir-int˙− transfers an electron to an aryl halide (Ar–X) and promotes the hydrodehalogenation reaction. Despite having an oxidation potential that is seemingly insufficient to reduce Ar–X (Fig. 5), we observed that Ir-Int˙− is still capable of interacting with several aryl halides (Scheme 3). This behavior is not unique to our system. Previous studies have shown that Ir complexes consisting of a NacNac ancillary ligand may undergo a reductive quenching mechanism to form reduced Ir(III) complexes that are able to catalyze the dehalogenation of organic halides.10,11 As discussed by Connell et al., errors from the cathodic peak potential of substrates lead to uncertainty when considering thermodynamic factor.25,26 In addition, processes such as the tendency of α-amino radicals to act as a halogen-atom transfer (XAT) agent may effectively lower the reduction potentials of the substrates.26,47 The NBP radicals formed may subsequently undergo several reaction pathways. For example, the NBP radical abstracts a hydrogen atom from the solvent to generate N-Boc pyrrolidine or forms a dimer by coupling with another NBP radical. Both N-Boc pyrrolidine and the dimer were observed in the gas chromatography-MS (GC-MS) analysis of the crude reaction mixture (Fig. S24†).
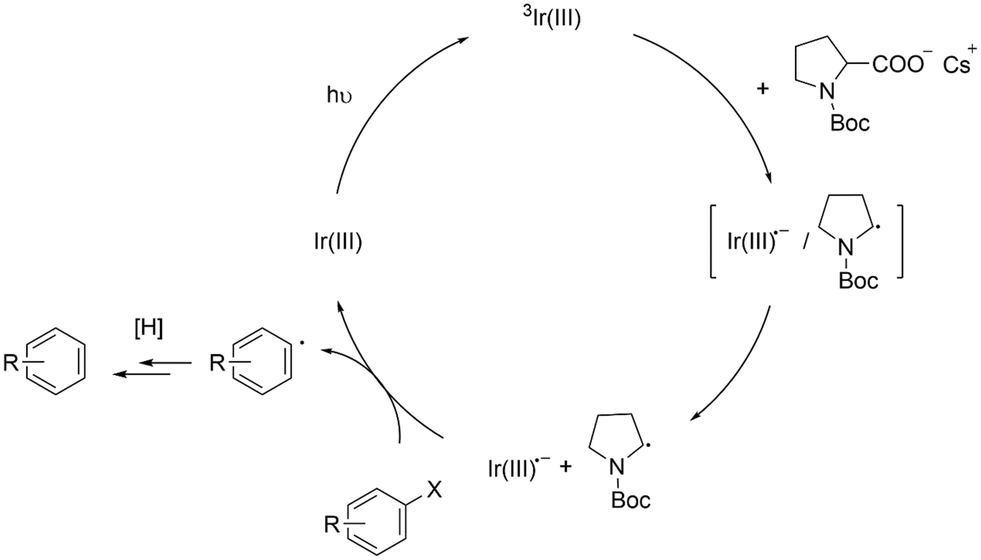 | ||
| Scheme 4 Proposed mechanism for the hydrodehalogenation of aryl halides. | ||
Finally, we tested the catalytic activity of Ir-mod (tirra = 8 h) together with Ni to drive the decarboxylative cross-coupling reaction between 2 and 4-bromobenzotrifluoride under blue light irradiation (Scheme 2A). An 88% product yield is obtained, indicating that the modified Ir complexes catalyse the reaction.
Conclusions
In summary, we have demonstrated that 1 is unstable when exposed to blue light in the presence of the α-amino acid (e.g., N-Boc proline) and Cs2CO3. In particular, N-Boc pyrrolidine are substituted onto the ligands of 1 in a stepwise manner, first forming mono- and di-NBP substituted Ir complexes at short light irradiation times before higher order substitution takes place upon prolonged excitation. At the same time, electron-withdrawing fluorine atoms and CF3 on the C^N ligands may be removed resulting in modified Ir-int complexes with destabilized HOMO, lower oxidation potential, and red-shifted exited triplet state absorption and phosphorescence bands. At the same time, 1 also undergoes degradation to compounds with photodechelated ligand and metal center with surrounding molecules (e.g., solvent) bound to it. The modified Ir complexes are subsequently used to facilitate hydrodehalogenation of a series of aryl halides with good to excellent yields via excited-state charge-transfer complexes formation. This study underscores the importance of investigating how an initial Ir(III) photocatalyst is modified in situ during long light exposure (e.g., duration of reaction), and the consequences on the overall efficiency of the photochemical reaction.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Pharma Innovation Programme Singapore PIPS (A*STAR, SERC A19B3a0014) and the Singapore Ministry of Education MoE Tier 1 grants (RG83/22 and RG83/20). We would also like to thank Dr. Yongxin Li for assistance in the X-ray crystallography measurements.Notes and references
- K. K.-W. Lo, S. P.-Y. Li and K. Y. Zhang, New J. Chem., 2011, 35, 265–287 RSC.
- D.-L. Ma, S. Lin, W. Wang, C. Yang and C.-H. Leung, Chem. Sci., 2017, 8, 878–889 RSC.
- I. N. Mills, J. A. Porras and S. Bernhard, Acc. Chem. Res., 2018, 51, 352–364 CrossRef CAS PubMed.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7094 RSC.
- T. M. Monos and C. R. J. Stephenson, Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons Ltd., 1st edn, 2017, pp. 541–581 Search PubMed.
- F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- J. D. Nguyen, E. A. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS.
- V. Mdluli, S. Diluzio, J. F. Kowalewski, T. U. Connell, D. Yaron, T. Kowalewski and S. Bernhard, ACS Catal., 2020, 10, 6977–6987 CrossRef CAS.
- M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2019, 3, 40–47 CrossRef.
- J.-H. Shon, S. Sittel and T. S. Teets, ACS Catal., 2019, 9, 8646–8658 CrossRef CAS.
- J.-H. Shon, D. Kim, M. D. Rathnayake, S. Sittel, J. Weaver and T. S. Teets, Chem. Sci., 2021, 12, 4069–4078 RSC.
- N. Takeda, P. V. Poliakov, A. R. Cook and J. R. Miller, J. Am. Chem. Soc., 2004, 126, 4301–4309 CrossRef CAS PubMed.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. J. von Wangelin, Chem. – Eur. J., 2015, 21, 15496–15501 CrossRef CAS PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023–11029 RSC.
- D. Jacquemin and D. Escudero, Chem. Sci., 2017, 8, 7844–7850 RSC.
- S. Schmidbauer, A. Hohenleutner and B. König, Beilstein J. Org. Chem., 2013, 9, 2088–2096 CrossRef PubMed.
- A. Soupart, F. Alary, J.-L. Heully and I. M. Dixon, Inorganics, 2020, 8, 15 CrossRef CAS.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliot and I. M. Dixon, Coord. Chem. Rev., 2020, 408, 213184 CrossRef CAS.
- R. Seifert, I. Rabelo de Moraes, S. Scholz, M. C. Gather, B. Lüssem and K. Leo, Org. Electron., 2013, 14, 115–123 CrossRef CAS.
- R. Ragni, E. A. Plummer, K. Brunner, J. W. Hofstraat, F. Babudri, G. M. Flarinola, F. Naso and L. De Cola, J. Mater. Chem., 2006, 16, 1161–1170 RSC.
- D. Wang, C. Cheng, T. Tsuboi and Q. Zhang, CCS Chem., 2020, 2, 1278–1296 CrossRef CAS.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658 CrossRef CAS.
- J. C. Bawden, P. S. Francis, S. DiLuzio, D. J. Hayne, E. H. Doeven, J. Truong, R. Alexander, L. C. Henderson, D. E. Gómez, M. Massi, B. I. Armstrong, F. A. Draper, S. Bernhard and T. U. Connell, J. Am. Chem. Soc., 2022, 144, 11189–11202 CrossRef CAS PubMed.
- J. J. Devery III, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers II and C. R. Stephenson, Chem. Sci., 2015, 6, 537–541 RSC.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS.
- I. R. de Moraes, S. Scholz, B. Lüssem and K. Leo, Org. Electron., 2011, 12, 341–347 CrossRef.
- V. Sivasubramaniam, F. Brodkorb, S. Hanning, H. P. Loebl, V. van Elsbergen, H. Boerner, U. Scherf and M. Kreyenschmidt, J. Fluorine Chem., 2009, 130, 640–649 CrossRef CAS.
- Y. Zheng, A. S. Batsanov, R. M. Edkins, A. Beeby and M. R. Bryce, Inorg. Chem., 2011, 51, 290–297 CrossRef.
- C. Yang, F. Mehmood, T. L. Lam, S. L.-F. Chan, Y. Wu, C.-S. Yeung, X. Guan, K. Li, C. Y.-S. Chung, C.-Y. Zhou, T. Zou and C.-M. Che, Chem. Sci., 2016, 7, 3123–3136 RSC.
- A. F. Henwood and E. Zysman-Colman, Top. Curr. Chem., 2016, 374, 25–65 CrossRef PubMed.
- D. Tordera, J. J. Serrano-Pérez, A. Pertegás, E. Ortí, H. J. Bolink, E. Baranoff, M. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 3391–3397 CrossRef CAS.
- K. P. Zanoni, B. K. Kariyazaki, A. Ito, M. K. Brennaman, T. J. Meyer and N. Y. Murakami Iha, Inorg. Chem., 2014, 53, 4089–4099 CrossRef CAS PubMed.
- F. Neve, M. La Deda, A. Crispini, A. Bellusci, F. Puntoriero and S. Campagna, Organometallics, 2004, 23, 5856–5863 CrossRef CAS.
- K. Hasan, A. K. Bansal, I. D. W. Samuel, C. Roldán-Carmona, H. J. Bolink and E. Zysman-Colman, Sci. Rep., 2015, 5, 12325 CrossRef CAS PubMed.
- K. P. Zanoni, R. L. Coppo, R. C. Amaral and N. Y. Murakami Iha, Dalton Trans., 2015, 44, 14559–14573 RSC.
- M. Teders, C. Henkel, L. Anhäuser, F. Strieth-Kalthoff, A. Gómez-Suárez, R. Kleinmans, A. Kahnt, A. Rentmeister, D. Guldi and F. Glorius, Nat. Chem., 2018, 10, 981–988 CrossRef CAS PubMed.
- A. Kaga, X. Wu, J. Y. Lim, H. Hayashi, Y. Lu, E. K. Yeow and S. Chiba, Beilstein J. Org. Chem., 2018, 14, 3047–3058 CrossRef CAS PubMed.
- M. Jiang, H. Yang, Y. Jin, L. Ou and H. Fu, Synlett, 2018, 29, 1572–1577 CrossRef CAS.
- H. Hayashi, B. Wang, X. Wu, S. J. Teo, A. Kaga, K. Watanabe, R. Takita, E. K. L. Yeow and S. Chiba, Adv. Synth. Catal., 2020, 362, 2223–2231 CrossRef.
- M. Austin, C. Covell, A. Gilbert and R. Hendrickx, Liebigs Ann./Recl., 1997, 943–946 CrossRef CAS.
- C. V. Kumar, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1985, 107, 5518–5523 CrossRef CAS.
- M. C. Haibach, B. M. Stoltz and R. H. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 15123–15126 CrossRef CAS PubMed.
- S. L. Mattes and S. Farid, Science, 1984, 226, 917–921 CrossRef CAS PubMed.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2240788 and 2240794. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy00942d |
| This journal is © The Royal Society of Chemistry 2024 |