
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Active template synthesis
Romain
Jamagne
a,
Martin J.
Power
a,
Zhi-Hui
Zhang
b,
Germán
Zango
a,
Benjamin
Gibber
a and
David A.
Leigh
 *ab
*ab
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: david.leigh@manchester.ac.uk
bSchool of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, P. R. China
First published on 5th September 2024
Abstract
The active template synthesis of mechanically interlocked molecular architectures exploits the dual ability of various structural elements (metals or, in the case of metal-free active template synthesis, particular arrangements of functional groups) to serve as both a template for the organisation of building blocks and as a catalyst to facilitate the formation of covalent bonds between them. This enables the entwined or threaded intermediate structure to be covalently captured under kinetic control. Unlike classical passive template synthesis, the intercomponent interactions transiently used to promote the assembly typically do not ‘live on’ in the interlocked product, meaning that active template synthesis can be traceless and used for constructing mechanically interlocked molecules that do not feature strong binding interactions between the components. Since its introduction in 2006, active template synthesis has been used to prepare a variety of rotaxanes, catenanes and knots. Amongst the metal–ion-mediated versions of the strategy, the copper(I)-catalysed alkyne–azide cycloaddition (CuAAC) remains the most extensively used transformation, although a broad range of other catalytic reactions and transition metals also provide effective manifolds. In metal-free active template synthesis, the recent discovery of the acceleration of the reaction of primary amines with electrophiles through the cavity of crown ethers has proved effective for forming an array of rotaxanes without recognition elements, including compact rotaxane superbases, dissipatively assembled rotaxanes and molecular pumps. This Review details the active template concept, outlines its advantages and limitations for the synthesis of interlocked molecules, and charts the diverse set of reactions that have been used with this strategy to date. The application of active template synthesis in various domains is discussed, including molecular machinery, mechanical chirality, catalysis, molecular recognition and various aspects of materials science.
 Romain Jamagne | Romain Jamagne was born in Brussels, Belgium and received his BSc from the University of Geneva and his MSc from the University of Strasbourg. In 2019 he moved to the group of Prof. David A. Leigh at the University of Manchester to pursue his PhD on mechanically interlocked molecules, which he completed in 2023. Currently, he is a postdoctoral researcher with Prof. Michel Rickhaus at the University of Geneva. His research interests include molecular machines and supramolecular materials. |
 Martin J. Power | Martin Power is from Tramore, Ireland and holds a BA in Chemistry from Trinity College Dublin and an MSc in Complex Systems Chemistry from the University of Strasbourg. Since 2020, he has been a Marie Skłodowska-Curie PhD student in the group of Prof. David A. Leigh as part of the ArtMoMa network. His research interests include molecular machines, mechanically interlocked molecules, and their applications to organic synthesis. |
 Zhi-Hui Zhang | Zhi-Hui Zhang, originally from Yunnan, China, obtained her MChem degree from Sun Yat-sen University, where she began her research journey in Supramolecular Chemistry. In 2015, she commenced her doctoral studies as a PhD candidate under the supervisor of Prof. Stephen M. Goldup at the University of Southampton, focusing on rotaxanes. Returning to China in October 2019, she joined the lab of Prof. David A. Leigh and Prof. Liang Zhang at East China Normal University in Shanghai. Her current research focuses on molecular nanotopology. |
 Germán Zango | Germán Zango was born in Madrid, Spain. He holds a degree in Chemistry and a PhD in Organic Chemistry from the Universidad Autónoma de Madrid, as well as a Master's degree in Nanoscience and Nanotechnology. He worked as a Marie Skłodowska-Curie Postdoctoral Research Fellow in the group of Prof. David A. Leigh at the University of Manchester between 2018 and 2021. His research interests range from the design and development of materials with applications in photovoltaics, to new advanced supramolecular materials and molecular machines. |
 Benjamin Gibber | Benji Gibber grew up in London, before receiving his MChem from the University of Oxford. In 2018, he moved to the University of Manchester to join the group of Prof. David Leigh to undertake his PhD on allosteric control of topology and chirality in supramolecular chemistry. After obtaining his PhD in 2022, he took a position in Cambridge to qualify as a UK and European patent attorney in chemistry, materials, and life sciences. His research interests include molecular topology and functional nanoparticles. |
 David A. Leigh | David Leigh obtained his BSc and PhD from the University of Sheffield. After postdoctoral research at the National Research Council (NRC) in Ottawa he was appointed to a Lectureship at the University of Manchester Institute of Science and Technology (UK). After holding Chairs at the Universities of Warwick and Edinburgh, he returned to Manchester in 2012, where he currently holds the Sir Samuel Hall Chair of Chemistry and is a Royal Society Research Professor. His research interests include chemical topology and molecular ratchets. |
Introduction
The synthesis of mechanically interlocked molecules (e.g. catenanes, rotaxanes and knots) has been explored since the middle of the 20th century.1,2 Early methods for their synthesis, such as the statistical approach of Wasserman3,4 (catenanes) and Harrison and Harrison (rotaxanes)5 or the directed synthesis strategies of Lüttringhaus and Schill,6 resulted in very low yields and/or required long synthetic schemes. In 1983, Sauvage and co-workers introduced a sea change in synthetic approaches to interlocked molecules by combining macrocycle 1 and diol 2 with Cu(I) to form complex 3-Cu(I), which subsequently underwent macrocyclisation of the threaded phenanthroline unit with a oligoethylene glycol chain to form metallo-catenane 4-Cu(I) in 42% yield (Fig. 1a).7,8 The key feature of this synthesis was the use of the metal ion as a template to hold the organic components in a threaded arrangement such that connecting the end groups together resulted in a mechanically linked molecule (Fig. 1). | ||
| Fig. 1 Classic examples of the passive metal template synthesis of interlocked molecules. (a) The Sauvage group's synthesis of catenane 4-Cu(I) in two sequential steps: coordination of the bidentate ligands to the metal in a tetrahedral geometry, followed by capturing of the threaded structure through covalent bond formation.7 (b) The Leigh group's synthesis of benzylic imine catenanes by imine formation about an octahedral Zn(II) template, followed by reduction of the imines and extraction of the metal ions to give wholly organic catenane 5.9 Although the Zn(II) ions promote formation of the imine bonds in addition to holding the ligands in place, the metal cannot be removed from the imine catenate without a subsequent reduction step to stabilise the covalent framework in the absence of the metal. The two-step synthesis of 5 is therefore an example of passive metal template synthesis. | ||
Since this seminal work, numerous strategies for positioning molecular components for interlocking have been developed, utilising diverse metal coordination modes and geometries10–12 and/or noncovalent interactions based on aromatic stacking interactions,13,14 hydrogen bonding,15 and/or hydrophobic effects16,17 (Fig. 2).18 The classic metal template approach7,9–12 to interlocked molecules involves two distinct processes which are generally accomplished step-wise (Fig. 2a):
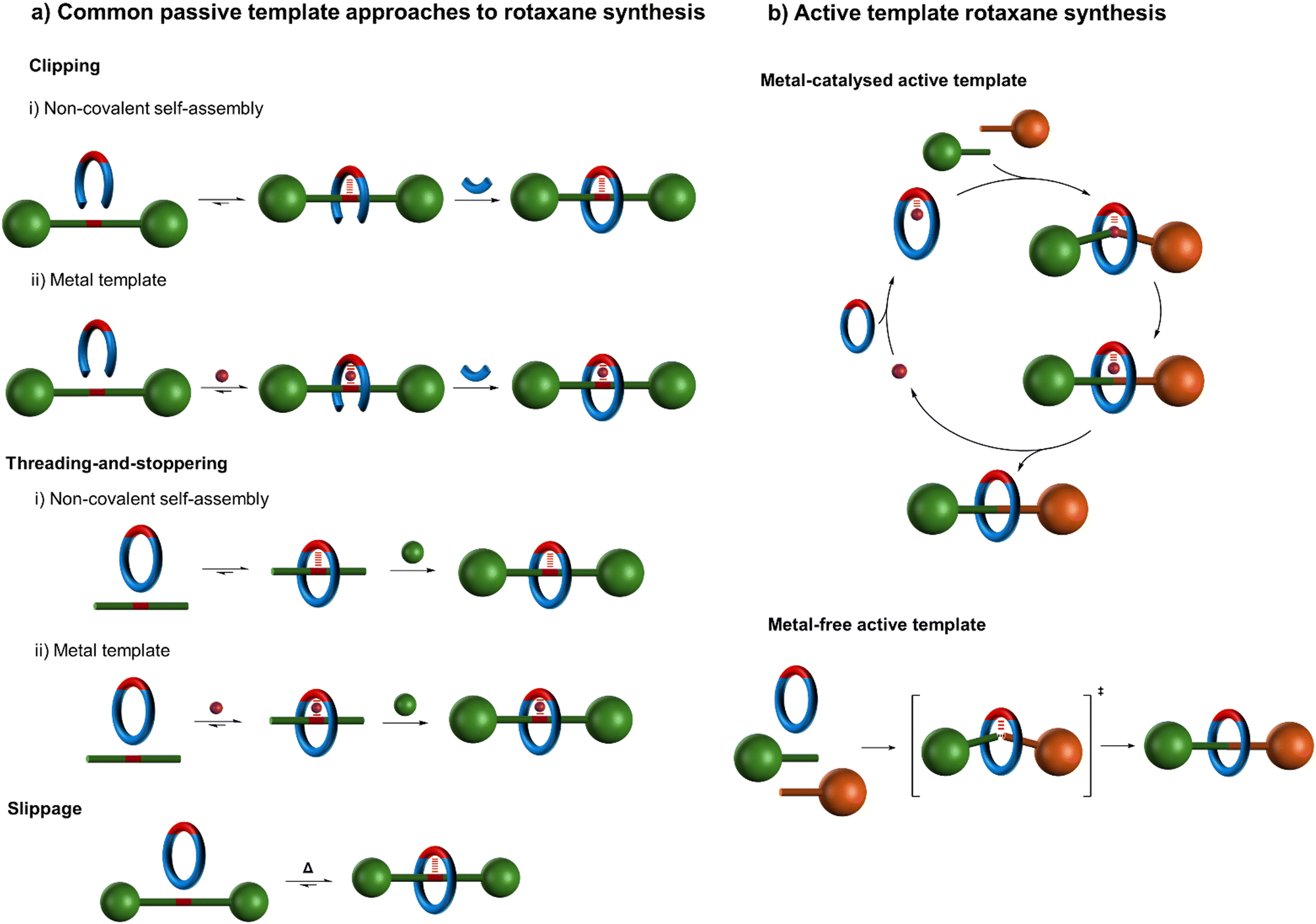 | ||
| Fig. 2 Schematic representation of (a) various common passive template strategies for rotaxane synthesis18–20 and (b) active template synthesis of rotaxanes.19,20,22 (a) The ‘clipping’ and ‘threading-and-stoppering’ approaches involve thermodynamically driven assembly processes followed by capturing of the interlocked structure by covalent bond formation.18 The ‘slipping’ or ‘slippage’ approach18 involves the macrocycle passing over the stoppers at an elevated temperature; at lower temperatures the macrocycle is then kinetically trapped on the axle. As a covalent bond does not need to be broken to disassemble such a threaded structure, such supramolecular assemblies are better described as kinetically stable pseudo-rotaxanes rather than as rotaxanes (see footnote 194 in ref. 21). (b) Active template synthesis is kinetically driven; component assembly and the covalent capture of the interlocked structure occur contemporaneously. | ||
(i) Component positioning: in which the well-defined coordination geometry of the metal is used to arrange ligands such that joining them together favours the formation of a mechanically interlocked structure.
(ii) Component connecting: a subsequent step in which the threaded arrangement present in the supramolecular complex is captured through one or more covalent bond-forming reactions.
As the component positioning step only exploits the coordination geometry of the metal ion, rather than any ability of the metal to promote the formation of covalent bonds that persist once the metal has been removed, this approach can be categorised as ‘passive’ metal template synthesis.10 In contrast, ‘active’ metal template synthesis19,20,22 (Fig. 2b) proceeds in a single step that exploits both the coordination geometry of a metal (in acting as a template to organise ligands into a threaded or entwined intermediate), and the metal's ability to catalyse covalent bond-forming reactions (in order to covalently capture the interlocked structure).
Passive and active template methodologies diverge in the principles that underpin them (Fig. 2).19 The passive template approach generally involves the formation of a thermodynamically favourable threaded or entwined intermediate complex (Fig. 2a).18 This minimum energy arrangement of building blocks is subsequently captured, typically by macrocyclisation or stoppering, in a second, bond-forming, process. In contrast, active template synthesis (Fig. 2b) does not involve a stable intermediate, but rather depends upon a catalysed bond-forming reaction occurring faster through a macrocyclic cavity than outside of it, irrespective of whether any attractive noncovalent interactions between the components are present in the final product.19,20 The active template process is thus kinetically driven and offers the prospect of assembling thermodynamically unfavourable interlocked structures.
Active template synthesis offers a number of distinctive features and potential advantages compared to passive template strategies:
(i) The absence of recognition motifs between the components of the final product provides an opportunity for the traceless synthesis of so-called ‘impossible’ rotaxanes.23–26
(ii) Sub-stoichiometric amounts of the metal can often be used,22,27 which turn over during their catalysis of the covalent bond-forming reaction(s).
(iii) The formation of a sterically shielded threaded structure can kinetically stabilise otherwise transient intermediates, aiding the elucidation of reaction mechanisms.28
(iv) The kinetically driven nature of active template synthesis allows for the formation of kinetically stable (because of the mechanical bond), but thermodynamically unstable, interlocked structures.19,20
Active template Cu-catalysed alkyne–azide cycloaddition
The concept of catalytically forming mechanical bonds
The concept of combining metal catalysis with template organisation for the assembly of interlocked molecules was introduced by Leigh and co-workers in 2006.22 A macrocycle pyridine group was used to bind Cu(I) within the macrocyclic cavity such that transiently coordinated azide and alkyne ligands were forced to attach to the tetrahedral Cu(I) centre through opposite faces of the macrocycle. The endotopic Cu(I) ion (and a second Cu(I) ion28) then promotes the Cu-catalysed alkyne–azide cycloaddition (CuAAC) of the attached ligands to form the threaded axle. The reaction of azide 4, alkyne 5, pyridine macrocycle 6 and Cu(MeCN)4PF6 produced [2]rotaxane 7 in up to 94% yield (Fig. 3).22 A sub-stoichiometric amount of the Cu(I) catalyst could be used, with loadings as low as 4 mol% still resulting in 82% yield of rotaxane 7. A 2007 study investigated the influence of macrocycle structure, and found that a strongly coordinating mono- or bidentate motif (pyridine or bypyridine) and minimal steric bulk or rigidity around the metal centre were most effective for rotaxane formation.28 | ||
| Fig. 3 Introduction of active metal template synthesis by Leigh and co-workers.22 | ||
In 2011, the Goldup group found that smaller bipyridine macrocycles gave higher [2]rotaxanes yields, particularly with short axles.29 The Crowley group subsequently demonstrated that for exo-alcohol-functionalised pyridyl macrocycles, even smaller macrocycles could be used,30 including 24-membered rings which begin to approach the limit possible31 for accommodating the thickness of a threaded saturated hydrocarbon chain.
Insights into the mechanism of CuAAC catalysis
The pathway of CuAAC catalysis begins with Cu(I) acetylide formation, followed by [3+2] cycloaddition with an azide, creating a Cu(I)-triazolizide species as a key intermediate.32In the early-2000s various intermediates for the CuAAC reaction had been postulated in experimental and computational studies.33–35 However, unambiguous evidence for various intermediates, and in particular whether one or two Cu(I) atoms were involved, remained unclear. In 2007 Leigh and co-workers reported unexpected [3]rotaxane formation during active metal template synthesis with a pyridine macrocycle (Fig. 4).28 The trapping of two rings threaded onto an axle produced by the formation of one triazole group, using macrocycles that could clearly bind to only one metal ion each, provided clear experimental evidence that two distinct Cu(I) atoms are involved in catalysing the [3+2] cycloaddition, at least with this type of ligand system.
 | ||
| Fig. 4 Mechanism of the active template CuAAC reaction leading to [2]- or [3]rotaxanes depending on the ratio of macrocycle to Cu(I).28 | ||
In the proposed reaction pathway (Fig. 4), macrocycle 6 and the half-axle components bind to Cu(I) to form 7-Cu(I). This intermediate then undergoes the Cu(I)-catalysed 1,3-cycloaddition leading to interlocked complex 8-Cu(I) and, ultimately, metal-free [2]rotaxane 9. Alternatively, the second metal ion of 10-[Cu(I)]2 can be bound to another macrocycle prior to the azide–alkyne cycloaddition occurring, leading to [3]rotaxane 11. The formation of [3]rotaxane is enhanced by increasing the macrocycle![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu(I) ratio, consistent with this mechanistic interpretation. Goldup and coworkers later found that high temperatures, non-coordinating solvents and larger macrocycles can also increase the amount of [3]rotaxane formed.36
:Cu(I) ratio, consistent with this mechanistic interpretation. Goldup and coworkers later found that high temperatures, non-coordinating solvents and larger macrocycles can also increase the amount of [3]rotaxane formed.36
Goldup and coworkers have isolated a Cu(I)-triazolizide rotaxane complex from an active template CuAAC reaction.37 The complex was remarkably stable, even under aqueous conditions, with the tightly threaded macrocycle sterically inhibiting protonation of the Cu–C bond. This was the first triazolide intermediate isolated from a CuAAC reaction.
These findings illustrate well how mechanical bonding can provide mechanistic insights into the catalysis manifolds used for active template synthesis.
CuAAC-mediated active metal template synthesis of catenanes and knots
CuAAC-mediated active template synthesis has been used to synthesise a broad range of rotaxanes, including [2]rotaxanes via either threading22,28 or clipping,38 heterocircuit [3]rotaxanes,39 doubly threaded [3]rotaxanes40 and triply threaded [4]rotaxanes.41 Its application to other topologies has been somewhat less explored. In 2009, the Leigh group used CuAAC active template synthesis to produce homocircuit and heterocircuit [2]catenanes in up to 53% yield.42 The Goldup group subsequently improved the scope and yields, making catenanes with smaller macrocycles.43 Their improvement is particularly notable as the earlier active template syntheses of catenanes42 required high dilution and long reaction times (typically 1–7 days), whereas Goldup's system gave yields of up to 98% in 4 hours. Otsuka and co-workers have reported the synthesis of [3]catenanes using CuAAC active template rotaxane formation followed by macrocyclisation of the bis(2,2,6,6-tetramethyl-piperidin-1-yl) disulfide stoppers into a catenane.44Leigh and co-workers have assembled a trefoil knot 12 using a combination of passive and active template processes (Fig. 5).45 The trefoil knot is formed in 24% yield from a single molecular strand, using one Cu(I) ion to coordinate to both bipyridine units, forming a loop in the strand, while a second Cu(I) ion acts as the active template site, catalysing the CuAAC reaction of the alkyne and azide termini through the loop.45
 | ||
| Fig. 5 Molecular trefoil knot 12 synthesised through active template CuAAC synthesis through the macrocyclic loop formed by Cu(I)-coordination to the two bipyridine groups.45 | ||
Active template Glaser–Hay oxidative homocoupling of terminal alkynes
The Leigh group's introduction of the active template synthesis concept22 (Fig. 3) was followed about a year later by Saito and co-workers use of stoichiometric quantities of a preformed macrocyclic Cu(I)-phenanthroline complex, 13-Cu(I), to promote the Glaser–Hay oxidative homocoupling of alkyne 14 (Fig. 6a).46 This generated [2]rotaxane 15 in 72% yield. The reaction likely proceeds through the pathway shown in Fig. 6a. First, alkyne 14 adds to 13-Cu(I) to form Cu(I)-acetylide intermediate 16-Cu(I). The Cu(I) centre is then oxidised by I2 and a second alkyne adds to the resulting Cu(III) species (or possibly a bimetallic Cu(II) intermediate) on the opposite face of the macrocycle. Reductive elimination generates the covalent bond that captures the threaded structure. The Cu(I) is then removed with KCN to give metal-free [2]rotaxane 15. | ||
| Fig. 6 (a) Proposed mechanism for the active template synthesis of [2]rotaxanes through the Cu(I)-mediated Glaser–Hay homocoupling of terminal alkynes.46 (b) Selected examples of interlocked structures synthesised by active template Glaser couplings.47–49 (c) Saito's post-assembly modification a diyne rotaxane to an aryl pyrrole rotaxane.50 | ||
Active template Glaser and Glaser-type couplings have subsequently been used to synthesise a variety of interlocked molecules, including [2]rotaxanes,47,50–64 a [2]catenane,65 and doubly threaded [3]rotaxanes such as 18 (Fig. 6b).48,66 The approach can be combined with passive template approaches to create more elaborate architectures such as 19.49 Its suitability for forming π-conjugated rotaxanes has been utilised to synthesise an array of ‘molecular wires’ (see section on sheathed ‘molecular wires’).
Although most active template Glaser couplings use Cu(I)-phenanthroline complexes, an exception is [2]rotaxane 17 (Fig. 6b), synthesised from a macrocyclic Hamilton-type receptor, albeit in modest (9%) yield.67
Saito and co-workers have investigated the effects of macrocycle size, axle length and stoichiometry on the yields of singly threaded [2]- and doubly threaded [3]rotaxanes made via active template Glaser couplings. The ratio of [3]rotaxane to [2]rotaxane was increased using larger macrocycles and longer thread building blocks.48
Interlocked molecules prepared by Glaser couplings are readily modified by reaction of the diyne, increasing the diversity of structures accessible using this active template reaction. Saito and co-workers described the post-modification of rotaxane 20 to [2]rotaxane 21 bearing an aryl pyrrole moiety in the axle (Fig. 6c).50 This allowed the shuttling dynamics of rotaxanes with different levels of steric bulk in the axle to be studied,54 as well as chiral HPLC separation of mechanically planar chiral rotaxanes with rotationally asymmetric macrocycles.53
Active template Cadiot–Chodkiewicz heterocoupling of terminal alkynes with alkyne halides
Active template Glaser–Hay homocoupling reactions result in rotaxanes with a diyne axle with mirror plane symmetry. Unsymmetrical diyne axle rotaxanes are accessible through the active template Cadiot–Chodkiewicz heterocoupling of alkynes and alkyne halides. An unsymmetrical axle is useful for switchable rotaxane-based molecular shuttles, pumps and other types of molecular devices.The Cadiot–Chodkiewicz active template synthesis of [2]rotaxanes was introduced by Leigh and co-workers in 2008 (Fig. 7).67 This reaction can be particularly sensitive to the order in which the building blocks, reagents and catalyst are combined: mixing a terminal alkyne, alkyne halide and bipyridine macrocycle 22 with an amine base and CuI resulted in a mixture of desired heterocoupling and undesired homocoupling products.67 In contrast, preforming copper acetylide 23-Cu(I), followed by subsequent addition of the macrocycle and alkyne halide 24 increased the rotaxane yield up to 85% with almost exclusive selectivity for the heterocoupled [2]rotaxane 25. The mechanism appears to proceed through the formation of Cu(I)-acetylide complex 26-Cu(I), which undergoes oxidative addition of 24 through the opposite face of the macrocycle to form 27-Cu(III). Reductive elimination affords the heterocoupled product, rotaxane 25. The strategy was later extended to the active template synthesis of heterocircuit [2]catenanes.42
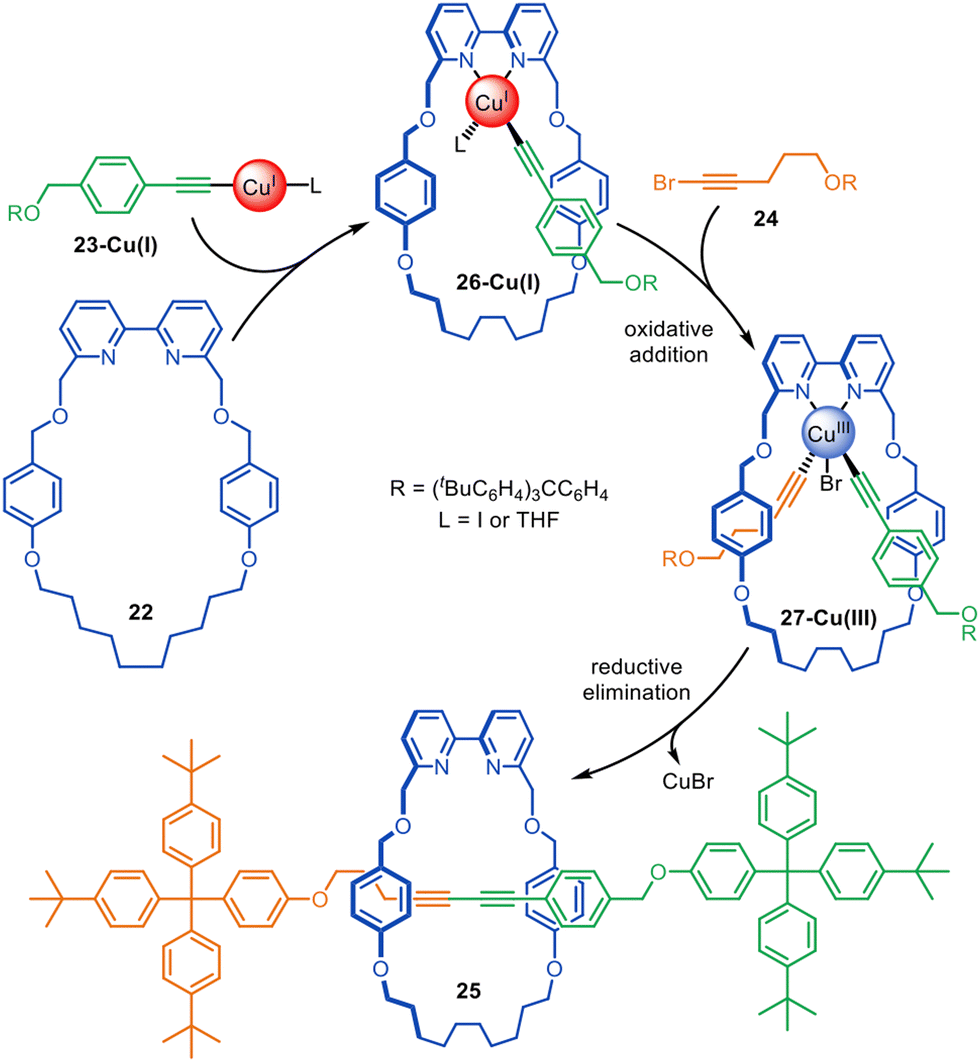 | ||
| Fig. 7 Proposed mechanism for the active template synthesis of [2]rotaxanes through the Cu(I)-mediated Cadiot–Chodkiewicz heterocoupling of terminal alkynes and alkyne halides.67 | ||
The Cadiot–Chodkiewicz active template reaction has been used for the synthesis of a variety of interlocked structures,68–71 often with better yields than the comparative Glaser couplings or enabling the formation of rotaxanes that were not accessible through alkyne homocouplings.68 It has proved particularly well-suited for the synthesis of polyyne and cumulene [2]rotaxanes, which generally show improved stability over their non-interlocked equivalents (see section on sheathed ‘molecular wires’).
Jasti and coworkers have made an impressive range of fluorescent ‘nanohoop’ [2]rotaxanes and [c2]daisy-chain rotaxanes through Cadiot–Chodkiewicz active template synthesis. The structures feature a shape-persistent macrocyclic belt containing a 2,6-connected pyridine residue (e.g.26) to endotopically coordinate the metal that promotes rotaxane formation. The [c2]daisy-chain rotaxane 27 has a relatively compact structure, apparent from the space-filling representation of the X-ray structure (Fig. 8).69,70
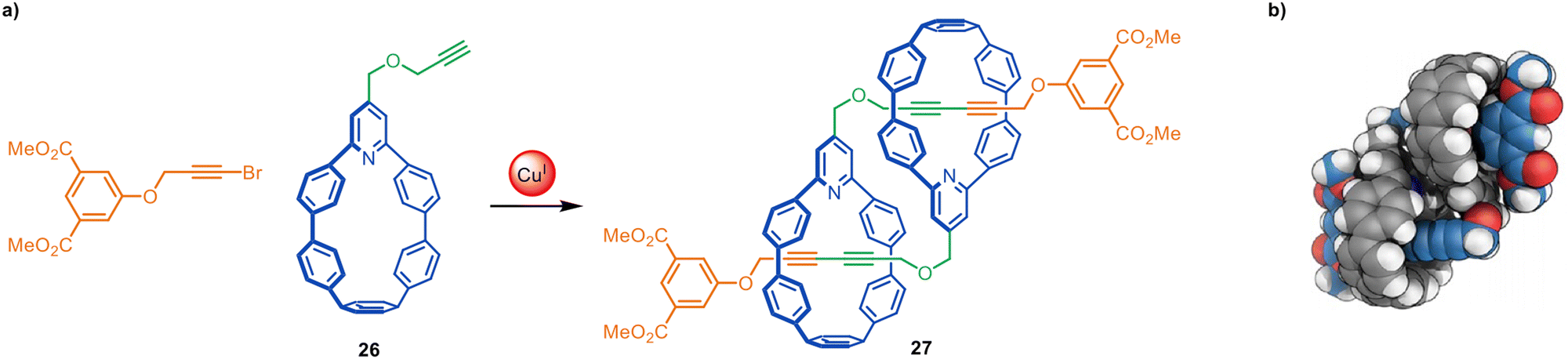 | ||
| Fig. 8 (a) Jasti's synthesis of a [c2]daisy-chain rotaxane 27 from an active template Cadiot–Chodkiewicz reaction. (b) Space-filling representation of 27.69 | ||
Other copper-mediated active metal template reactions
In addition to using 13-Cu(I) in Glaser–Hay active template synthesis, Saito and co-workers used the phenanthroline complex in a copper-mediated Ullmann C–S heterocoupling of aryl iodide 28 and thiol 29 (10-fold excess of each) to give [2]rotaxane 30 in a modest (27%) yield (Fig. 9).46 | ||
| Fig. 9 Saito's active template synthesis of a [2]rotaxane by Ullmann C–S coupling of a thiol and aryl iodide.46 | ||
The same Cu(I)-phenanthroline macrocycle has also been used in a Sonogashira-type active template heterocoupling of aryl iodides and terminal alkynes, generating rotaxanes in up to 52% yield (Fig. 10).72 Concurrent oxidative homocoupling of the terminal alkynes occurs in such reactions and so the corresponding diyne rotaxanes were formed as side-products in each case. The mechanism is presumed to proceed via the formation of Cu(I)-acetylide 31-Cu(I), followed by oxidative addition of aryl iodide 32 to form Cu(III)-complex 33-Cu(III). The latter undergoes reductive elimination through the macrocycle cavity to form [2]rotaxane 34. The best yields and selectivity for the heterocoupling product were obtained using the ortho-isomer of 32.
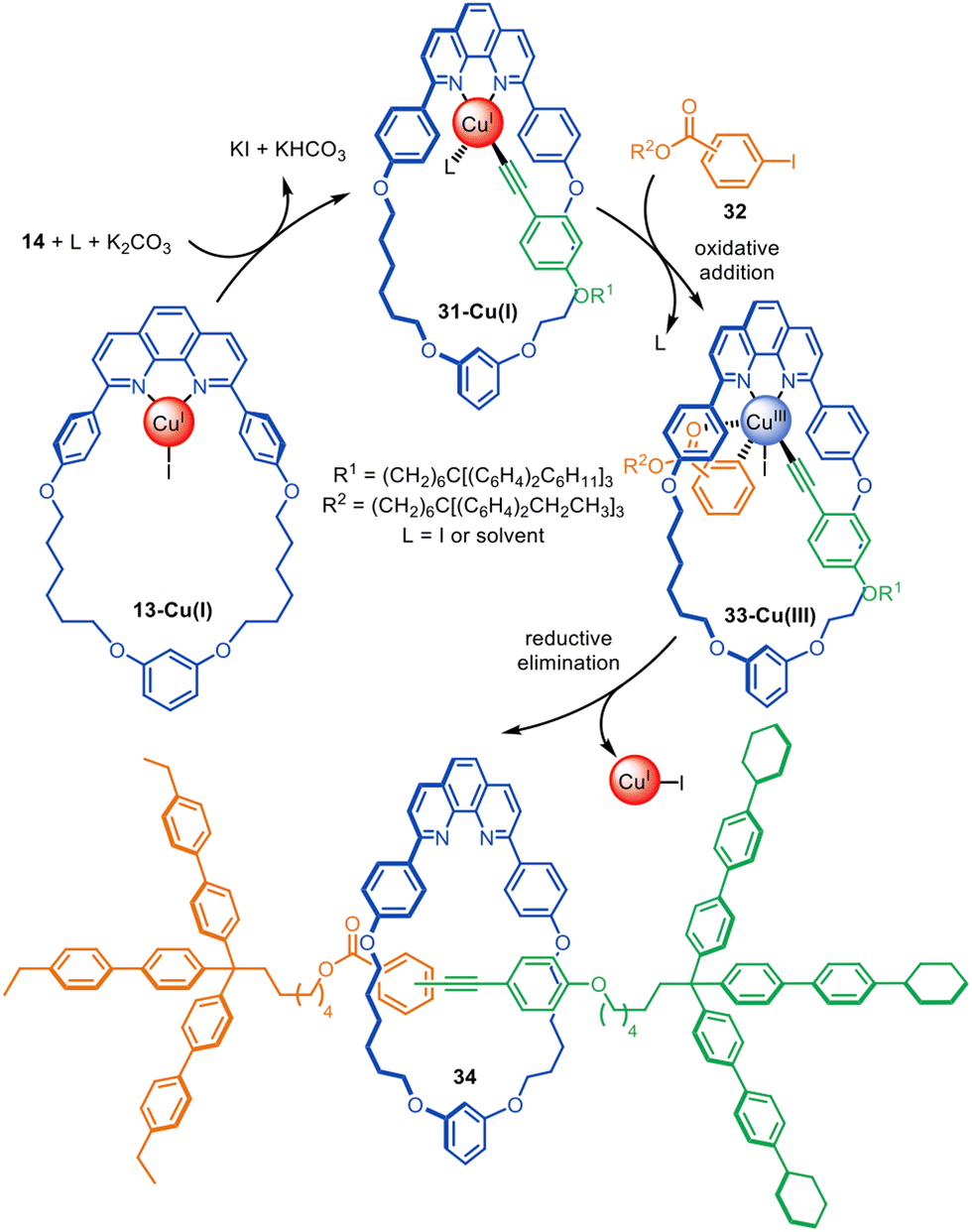 | ||
| Fig. 10 Saito's active template synthesis of [2]rotaxanes through the Cu(I)-mediated Sonogashira-type coupling of terminal alkynes and aryl iodides.72 | ||
Leigh and co-workers have used Cu(I) catalysis in the active template Goldberg amidation of aryl halides to form [2]rotaxanes with chiral C2-symmetric cyclohexyldiamine macrocycles.73 The mechanism is similar to the active template Cu(I)-mediated Sonogashira-type coupling (Fig. 10), with an aryl amide rather than an alkyne undergoing ligation with 21-Cu(I). The resulting chiral rotaxanes were subsequently investigated as ligands for enantioselective metal catalysis (see section on asymmetric, diastereoselective and other types of catalysts).
Other transition-metal-mediated active template reactions
Copper-catalysed reactions have proved to be particularly successful and robust catalytic manifolds for active template synthesis. This is because copper catalysis is often promoted by particular ligand types (which can form the basis of a suitable macrocycle) and because of the well-defined 3D coordination geometries of Cu(I) and Cu(III) which aid ligation of the axle building blocks through opposite faces of the macrocycle. However, copper is somewhat limited in its ability to activate sp3-hybridised carbons compared to other metals such as palladium,74 nickel,75 rhodium76 and ruthenium.77 In the next sections we give examples of how reactions catalysed by these, and a number of other transition metals, have also proved amenable for active template synthesis.Palladium-catalysed active template synthesis
Palladium-catalysed cross-coupling transformations are extensively employed in organic synthesis for the construction of C–C bonds. Although Pd(II) has a 2D square planar coordination geometry, which is poorly suited for endotopic binding within a macrocyclic cavity compared to the 3D tetrahedral geometry of Cu(I), palladium offers high catalytic activity in numerous reactions. Attempts by the Leigh group to use cross-couplings that feature transient Pd(0) in the catalytic cycle proved unsuccessful for active template synthesis, apparently because of the poor coordination of Pd(0) leading to detachment of the metal from various macrocycle types during key steps of the catalytic cycle.27Leaning on previously reported Pd(II)-mediated passive template syntheses of rotaxanes78–81 and catenanes for the macrocyclic ligand design,82,83 Leigh and co-workers showed that, despite its square planar coordination geometry, Pd(II) can be a suitable metal for the active template synthesis of [2]rotaxanes through alkyne homocoupling (Fig. 11).84 The reaction proceeds with the replacement of the trans-chloride ligands of 35-Pd(II) by the Cu(I)-acetylide unit 36 to form a threaded trans-37-Pd(II) complex. Isomerisation to cis-38-Pd(II) occurs without breaking the Pd–acetylide bond, preserving the threaded architecture. Reductive elimination occurs from the cis configuration to yield diyne [2]rotaxane 39 after decomplexation of the weakly coordinating Pd(0). Only a catalytic amount of Pd(II) (5 mol%) is required, together with I2 or O2 to reoxidise the Pd(0) formed in situ, giving rotaxane yields up to 90%.
 | ||
| Fig. 11 Active template synthesis of [2]rotaxanes through the Pd(II)-mediated oxidative homocoupling of terminal alkynes.84 | ||
The oxidative Heck cross-coupling reaction85 is a variant of palladium cross-couplings that does not feature Pd(0) in the mechanism. Accordingly, it is more suitable as the basis for active template synthesis. Leigh and co-workers demonstrated its efficacy in the synthesis of a variety of [2]rotaxanes using a bipyridine macrocycle and boronic acid and electron-poor alkene axle building blocks (Fig. 12).27 The catalytic loading could be as low as 1 mol% while still furnishing a rotaxane yield of 66%, the largest catalytic turnover number reported to date for an active template reaction.
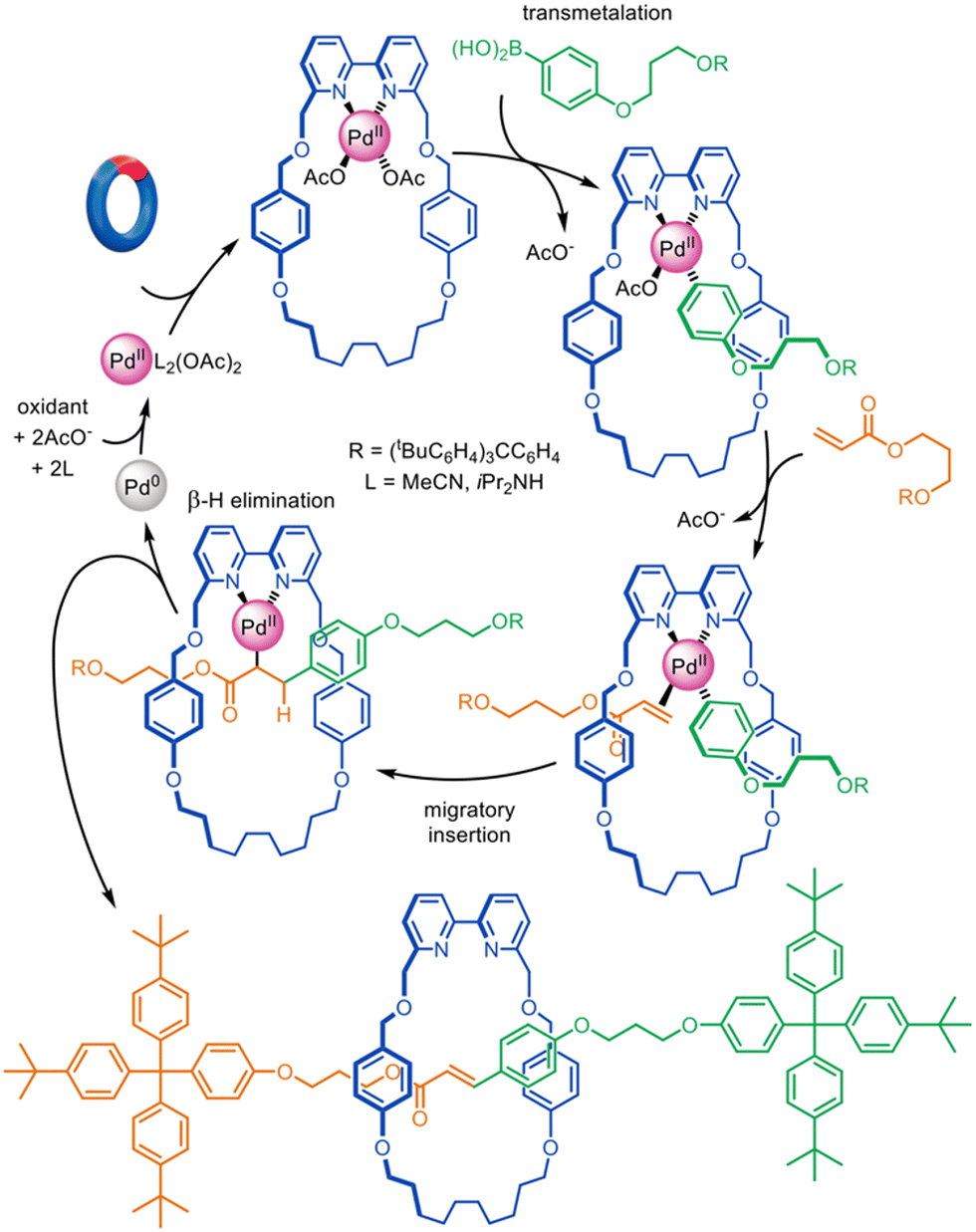 | ||
| Fig. 12 Active template synthesis of [2]rotaxanes through Pd(II)-mediated oxidative Heck cross-coupling.27 | ||
The examples in Fig. 11 and 12 demonstrate the significant potential of palladium for active template synthesis, particularly if macrocyclic ligands can be found that can overcome the lability of Pd(0) so that it remains coordinated to the components during various important steps in the mechanism.
Very high yielding four-component synthesis of rotaxanes by successive Pd(II)-promoted Michael additions of α-cyano esters to alkyl vinyl ketones has been reported using tridentate pyrroloimidazolone or bisoxazoline pincer macrocycles (Fig. 13).86 The formation of complex 40-Pd(II) enables two consecutive 1,4-conjugate additions to α,β-unsaturated ketone 41 from opposite macrocycle faces to quantitatively form [2]rotaxane 42-Pd(II). In this example the template interactions between thread and macrocycle remain in the rotaxane product, a rare feature for active template synthesis. Accordingly, the intercomponent interactions were used to good effect for the synthesis of rotaxanes with well-defined switching of the macrocycle position on the axle (molecular shuttles).
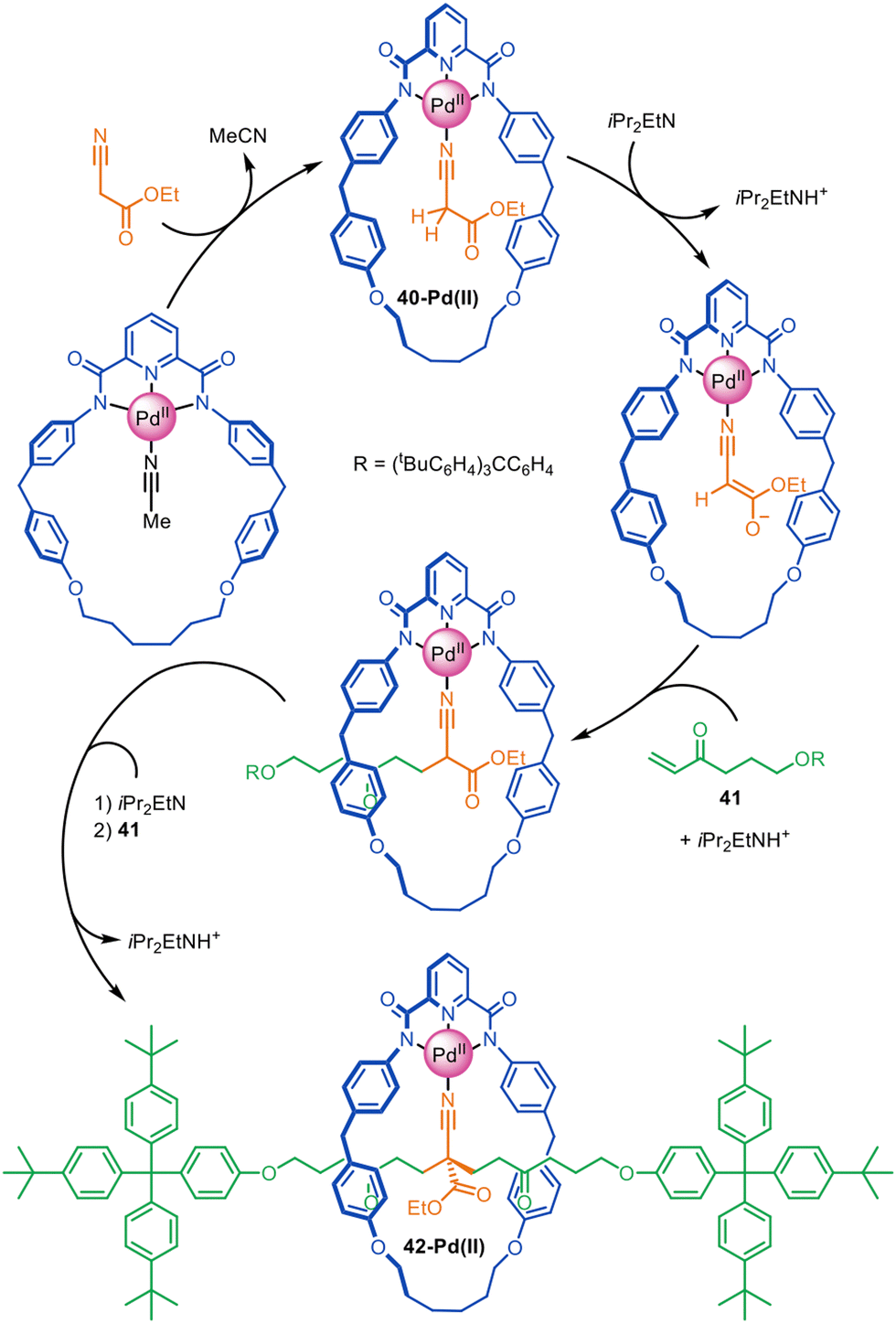 | ||
| Fig. 13 Active template synthesis of [2]rotaxanes through successive Pd(II)-mediated Michael additions of four components.86 | ||
Nickel-mediated active template synthesis
An unusual dual Ni-/Cu-mediated alkyne homocoupling active template reaction has been reported.87 The system uses an octahedral Ni(II) macrocyclic complex followed by addition of CuI which results in reductive elimination of the half-axle ligands to yield [2]rotaxanes. The synthesis was high yielding (up to 95%), although stoichiometric quantities of NiCl2 and CuI are required.Active template synthesis that deletes atoms from the building blocks while joining them to form a hydrocarbon region of a threaded axle is a particularly appealing target because it is completely traceless. The main obstacle to doing so lies in achieving effective and catalytic formation of C(sp3)–C(sp3) bonds using organometallic chemistry. This is generally difficult due to slow rates of oxidative addition of metal centres to C(sp3)–heteroatom bonds, concomitant with facile β-hydride elimination of the resulting organometallic intermediate.88–91 Most successful C(sp3)–C(sp3) coupling protocols involve alkyl halides activated towards oxidative addition and lacking β-hydrogens.92,93
Adapting the work of Fu and co-workers on the coupling of unactivated alkyl halides,94–98 the active template synthesis of alkyl chain axle-region rotaxanes was achieved by Leigh and co-workers through the Ni(0)-catalysed C(sp3)–C(sp3) homocoupling of bromoalkanes (Fig. 14).99 The reaction was initially thought to likely proceed through a Negishi-type mechanism,91 however mechanistic studies indicate a pathway similar to the Ni-mediated coupling of aryl halides.100 Oxidative addition of alkyl bromide 43 to 44-Ni(0) is followed by the one-electron reduction of Ni(II), allowing a second oxidative addition of 43, ligating the axle building blocks through opposite faces of the macrocycle. Reductive elimination then captures threaded architecture 44, while Zn(0) reduces the resulting Ni(I) species to catalytically active Ni(0).
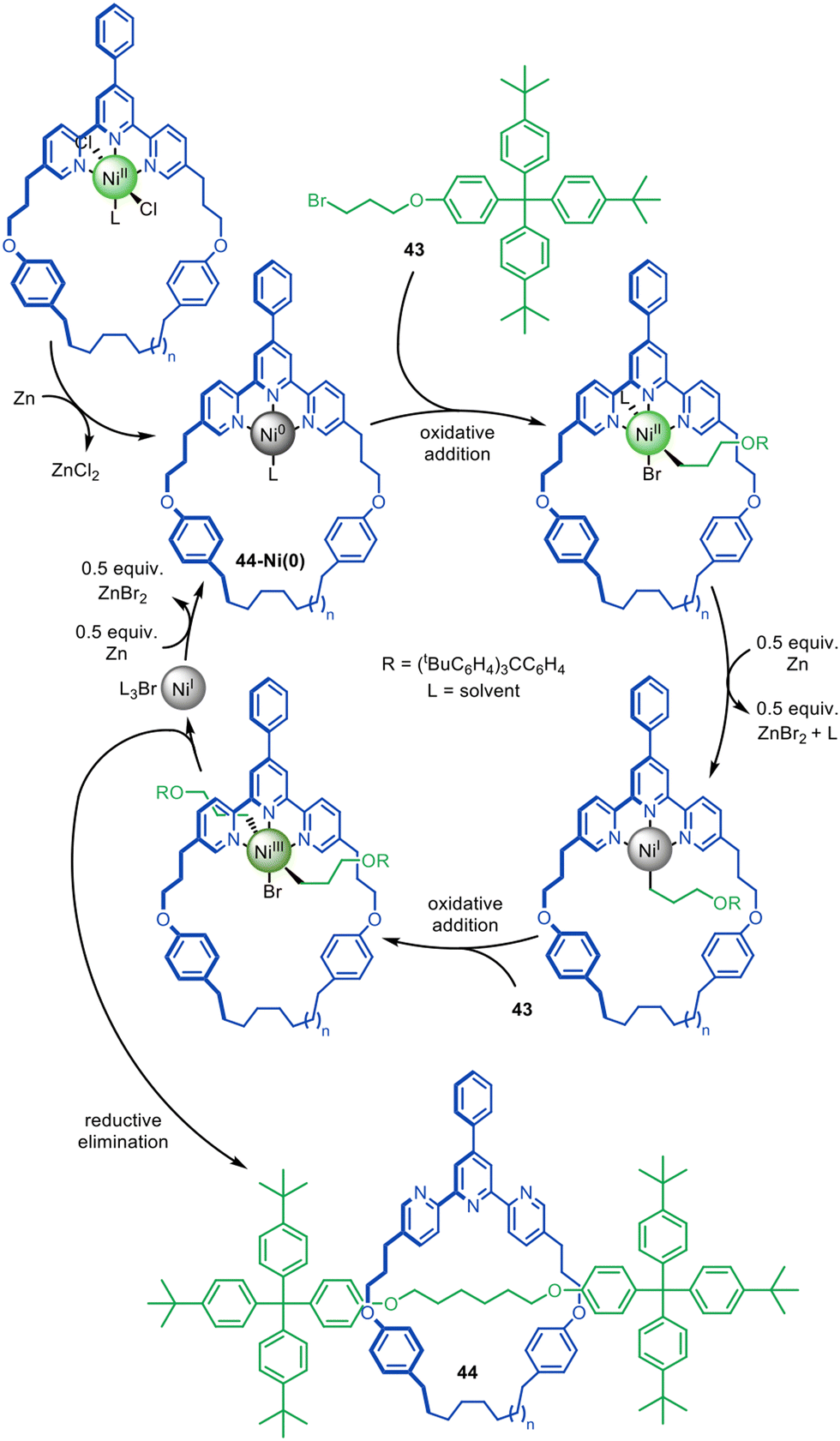 | ||
| Fig. 14 Active template synthesis of traceless [2]rotaxanes through the Ni(0)-mediated homocoupling of unactivated alkyl bromides.99 | ||
Saito and co-workers have carried out the cross-coupling of iodoarenes mediated by a dibenzodihydrophenanthroline–Ni complex.101 This was used for the synthesis of [2]rotaxanes with up to ten para-phenylene units in the axle.
Active template synthesis by Ni-catalysed homocouplings of alkyl bromides can produce multiply threaded rotaxanes. Leigh and co-workers isolated doubly threaded rotaxane 45 in up to 51% yield using a terpyridine 35-membered macrocycle (Fig. 15a).102 During the process, [2]rotaxane 44 was also produced but the ratio of doubly- over singly-interlocked product could be increased by using 43 in large excess. The mechanism appears to proceed in a stepwise manner in which the [2]rotaxane is formed first, followed by binding of a second set of axle building blocks to the metal coordinated in the [2]rotaxane to yield [3]rotaxane 45 after a second reductive elimination.
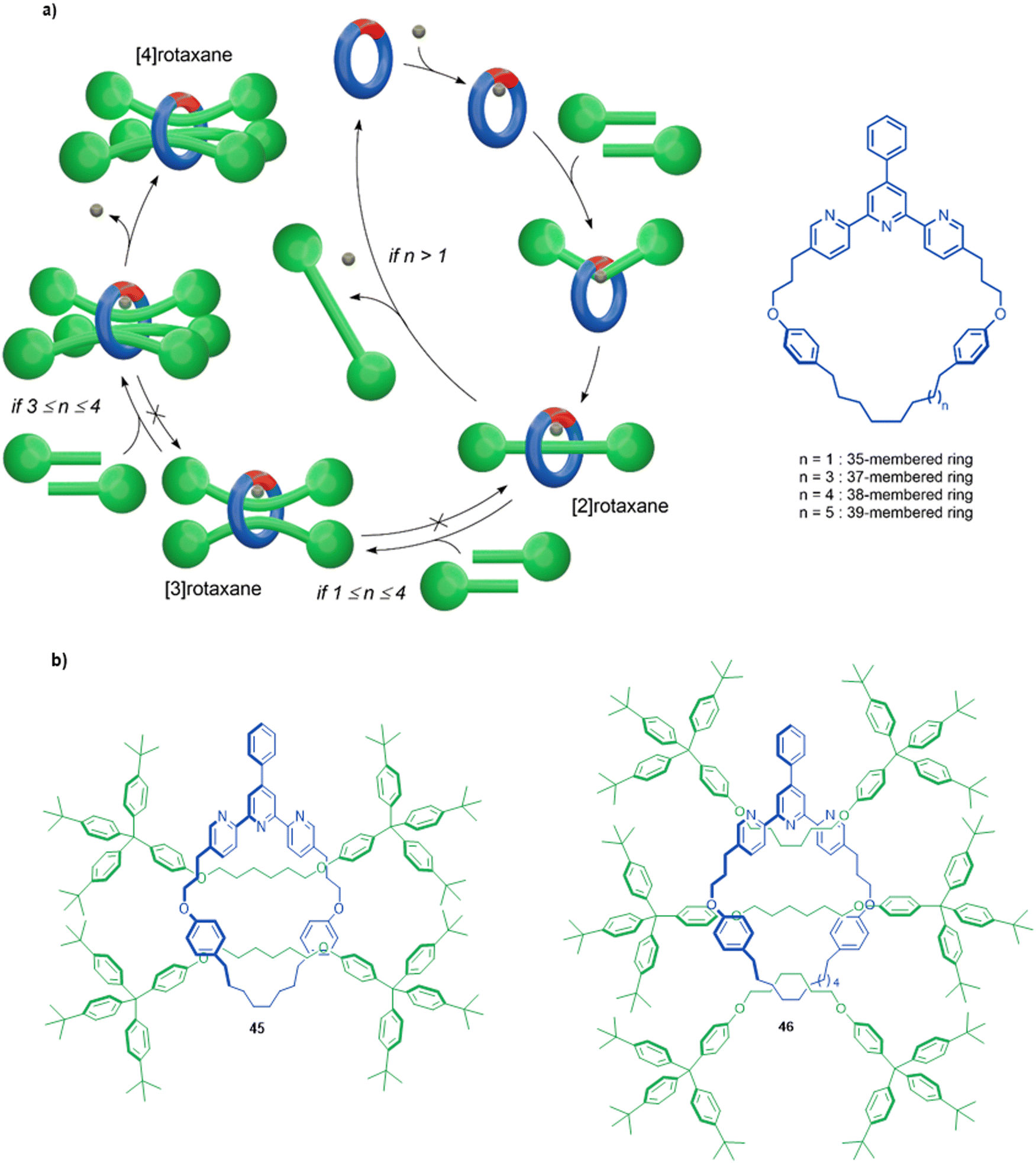 | ||
| Fig. 15 (a) Mechanism of the active template Ni(0)-mediated active template synthesis of rotaxanes with one, two or three axles threaded through a single macrocycle. (b) Doubly and triply threaded rotaxanes formed using this method.102 | ||
A triply threaded [4]rotaxane 46 was obtained using the same strategy by increasing the size of the macrocycle from a 35-membered ring to either a 37- or a 38-membered ring (Fig. 15b).103 The increased size meant the macrocycle could accommodate the formation of a third axle within the cavity. [2]Rotaxane was not observed in the product distribution using the larger ring systems, only the doubly- and triply-threaded products. This suggests that the [2]rotaxane does not have stoppers large enough to prevent dethreading of the larger macrocycles unless the metal remains coordinated (which reduces the effective size of the cavity, and the coordinated metal can then promote the formation of the second and third axle; Fig. 15a). Also consistent with this, when a slightly larger 39-membered macrocycle was used only non-interlocked coupled products were observed. To date, rotaxane 46 is the sole example of a rotaxane containing more than two axles threaded through a single macrocycle.
The formation of traceless rotaxanes by active template synthesis was later extended by the Leigh group to the preparation of unsymmetrical axle [2]rotaxanes by the Ni-catalysed C(sp3)–C(sp3) cross-coupling of alkylzinc with redox-active esters (Fig. 16),104 a reaction introduced by Baran and co-workers.105 Transmetallation of Ni(I)-bipyridine macrocycle 47-Ni(I) with alkylzinc 48, followed by single electron transfer (SET) from the Ni(I) to the phthalimide group of redox-active ester 49 and consecutive decarboxylative fragmentation, leads to cationic intermediate 50-Ni(II).106 Radical recombination renders Ni(III) intermediate 51-Ni(III), which undergoes reductive elimination through the macrocycle cavity to form [2]rotaxane 52, while regenerating the catalytic Ni(I) species. [2]Rotaxane 53 was also formed, resulting from the active template homocoupling of alkylzinc 48.
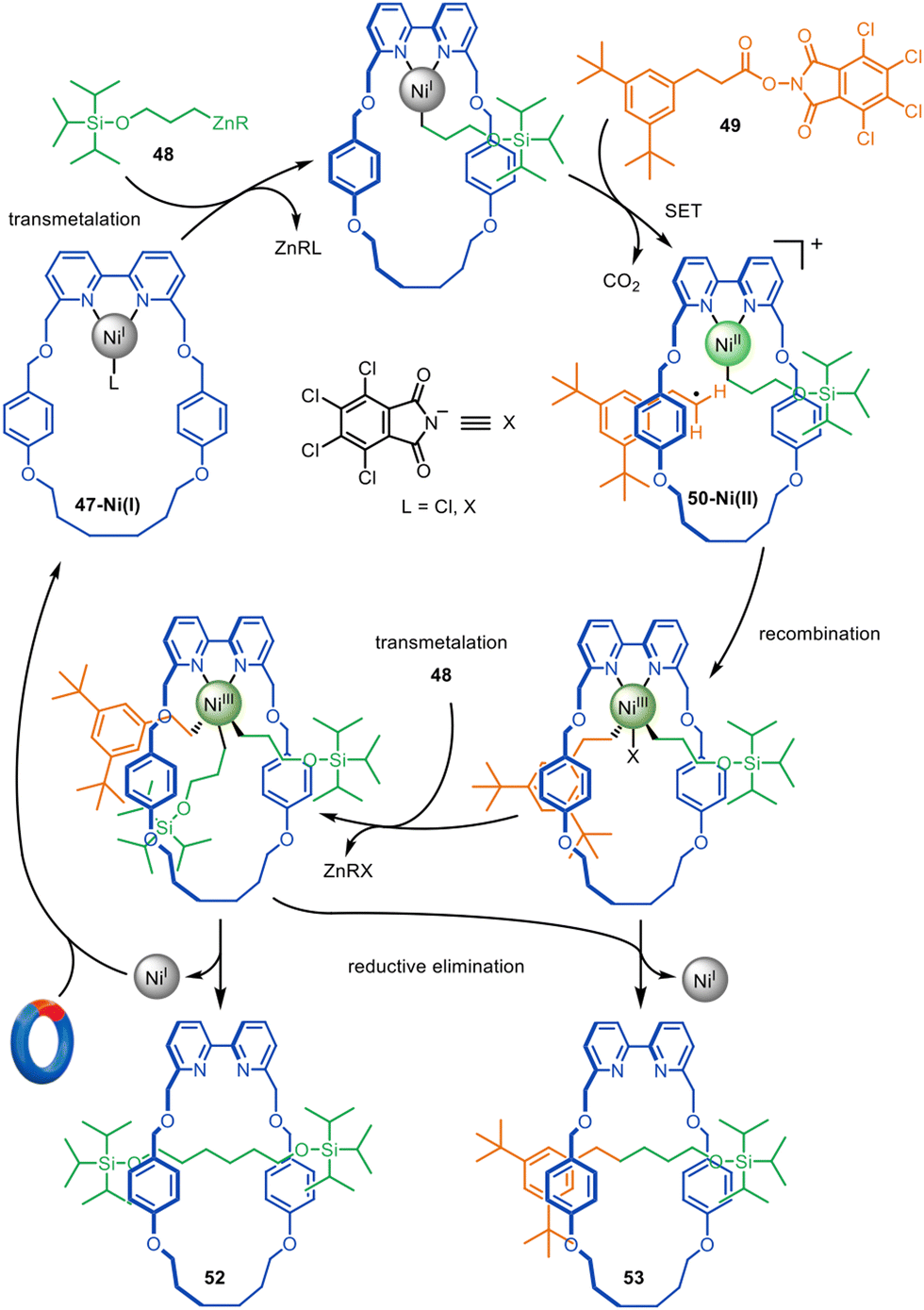 | ||
| Fig. 16 Mechanism of the active template synthesis of traceless [2]rotaxanes through the Ni(I)-mediated heterocoupling of alkylzinc and redox-active esters.104 | ||
Recently, Kimizuka, Yagi and Itami reported a novel Ni-mediated homocoupling active template reaction employing a similar bipyridine macrocycle in the synthesis of catenane 54 (Fig. 17).107 The threading unit is based on Itami's previous synthesis of catenanes and a trefoil knot through a covalent scaffold approach,108 and features cyclohexa-1,4-diene units109 that are converted to phenyl groups to form the all-benzene ‘nanobelt’ of the final catenane.
 | ||
| Fig. 17 Kimizuka, Yagi, and Itami's Ni(0)-mediated active template synthesis of [8]cycloparaphenylene catenane 54.107 | ||
Zinc-, cobalt-, ruthenium- and rhodium-mediated active template synthesis
Although reactions based on copper, palladium and nickel catalysis are the most popular manifolds for active template synthesis, reactions promoted by other metals have also been explored. Zinc(II) has been used for the active template synthesis of [2]rotaxanes by the Lewis acid-catalysed Diels–Alder cycloaddition of imidazolidones with cyclopentadiene derivatives.110 Rotaxane 55 forms in up to 91% yield using either Zn(II) or Cu(II) ions (Fig. 18). Upon coordination of dienophile 56 to macrocycle 57-Zn(II), the activated threaded complex 58-Zn(II) is formed. Cycloaddition with cyclopentadiene 59 produces 60-Zn(II), with subsequent demetallation affording metal-free [2]rotaxane 55. Only one of the two interconverting isomers of 59 engages in the active template reaction with a 9:1 preference for the endo adduct observed.
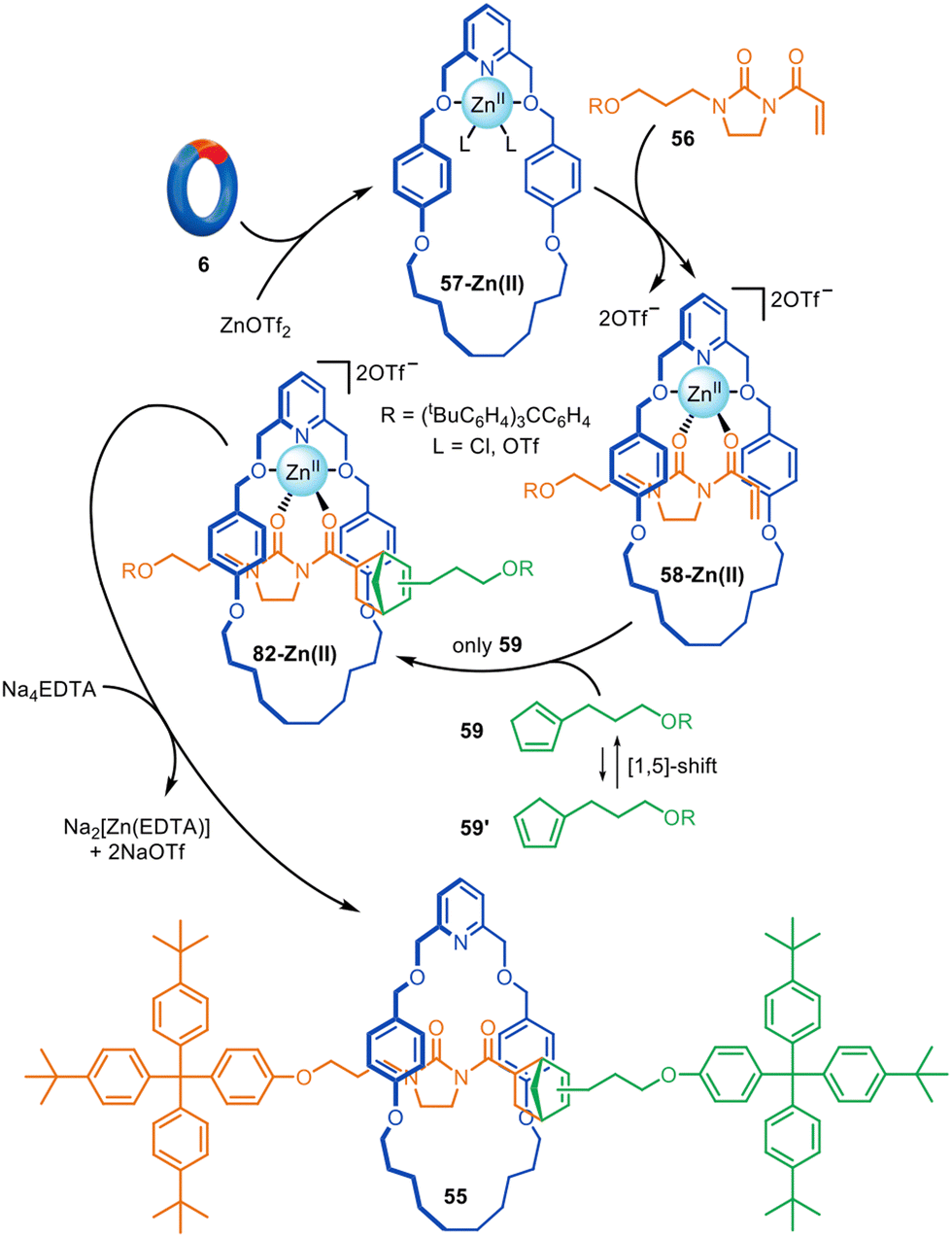 | ||
| Fig. 18 Active template synthesis of [2]rotaxanes through the Zn(II)-mediated Diels–Alder of imidazolidones and cyclopentadiene derivatives.110 | ||
A small number of active metal template reactions have been developed that proceed through radical mechanisms involving a redox process between the metal and the ligand. Co(II)porphyrinates form nucleophilic radical carbenes that react with olefins to form cyclopropanes.111–113 Megiatto and co-workers exploited this chemistry for the active template synthesis of rotaxanes with macrocycle 60-Co(II).114 The mechanism involves a radical carbene transfer reaction between diazo and styrene half-threads (Fig. 19). Coordination of 3,5-diphenylpyridine ligand 61 to the exo-face of the Co(II)porphyrinate (it is too bulky to coordinate to the endo-face) causes diazo-acetate 62 to react with macrocycle 63-Co(II) inside the cavity, leading to Co(III)-carbene radical 64-Co(III). Subsequent carbene transfer with styrene derivative 65 generates rotaxane 66-Co(II). In the absence of 61 the yield of [2]rotaxane decreased from 95% to 74%,115 demonstrating the importance of coordination of an exo-ligand for promoting formation of the interlocked product over the free thread.
 | ||
| Fig. 19 Megiatto's active template synthesis of [2]rotaxanes through the Co(II)-mediated radical carbene transfer between diazo and styrene derivatives.114 | ||
The same group reported that a Ru(II)-porphyrinate macrocycle promotes N–H carbene insertion between amines and diazo half-threads in an active template fashion (Fig. 20).116 Ru(II)-porphyrinate 67-Ru(II) differs from 60-Co(II) in that it is only composed of aromatic C–H bonds and therefore chemically inert to Ru(II)-carbenoids. Introduction of a bulky carbene ligand exo- to the macrocycle forces reaction of diazo 69 to occur inside the cavity. Addition of amine 68 generates [2]rotaxane 70-Ru(II) in quantitative yield.
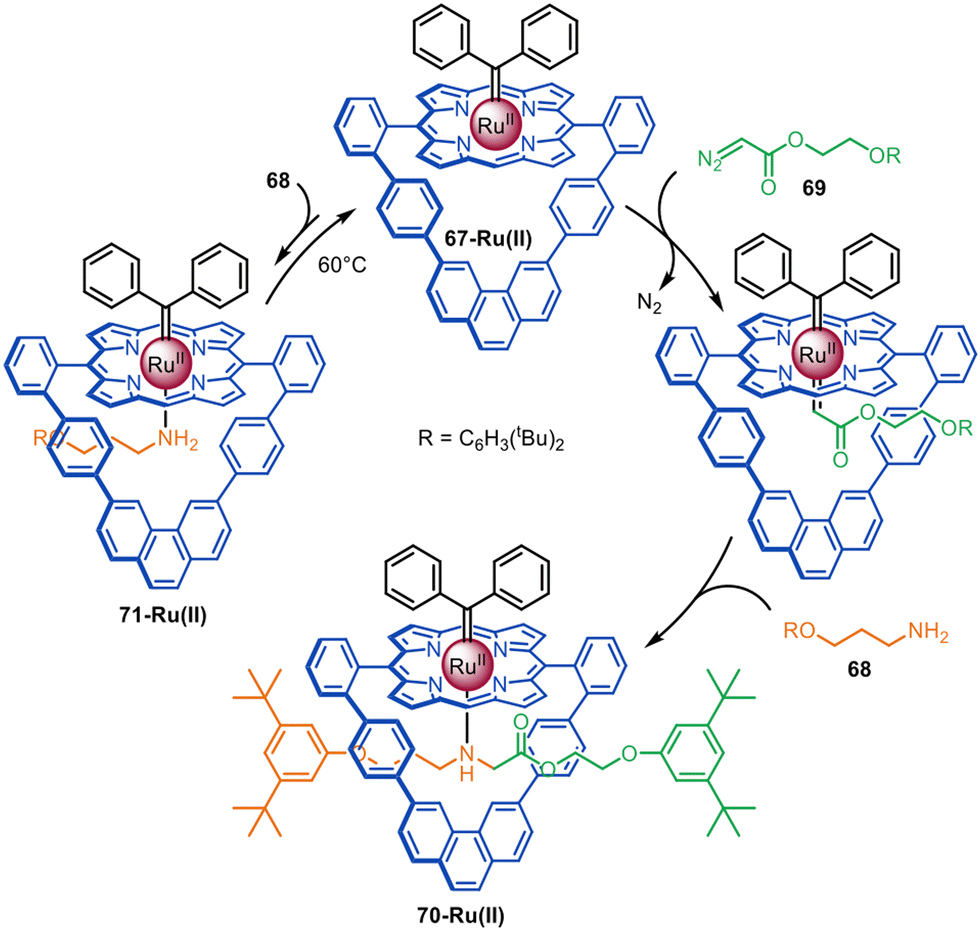 | ||
| Fig. 20 Megiatto's active template synthesis of [2]rotaxanes via Ru(II)-mediated N–H carbene insertion.116 | ||
The distinctive reactivity and catalytic properties of rhodium have been exploited for active template synthesis by the Chaplin group.117–120 They first described the formation of pseudorotaxane 71-Rh(I) by a Rh(I)-promoted terminal alkyne coupling reaction that forms an enyne axle (Fig. 21).117 Ligand exchange between terminal alkyne 72 and Rh(I) ethylene complex 73-Rh(I) leads to Rh(I)-alkyne complex 74-Rh(I), which undergoes oxidative addition to give 75-Rh(III). A second equivalent of 72 inserts into the Rh–H bond in a ‘head-to-tail’ type mechanism to form a Rh(I) gem-alkenyl complex 76-Rh(III) that is sufficiently stable to be isolated. However, upon heating to 80 °C, 76-Rh(III) reductive eliminates and does not afford the expected Rh(I) gem-enyne pseudorotaxane 77-Rh(I), but instead quantitatively forms E-enyne pseudorotaxane 71-Rh(I). This suggests a mechanism in which β-hydride abstraction of 76-Rh(III) occurs and the equilibrium is driven towards the formation of E-enyne Rh(III) complex 78-Rh(III), which can rapidly undergo reductive elimination to form 71-Rh(I). Only the gem-enyne product is formed when the reaction is performed with an acyclic CNC ligand. This study illustrates how active template synthesis can provide insights on ligand tuning to change reaction regioselectivity.118
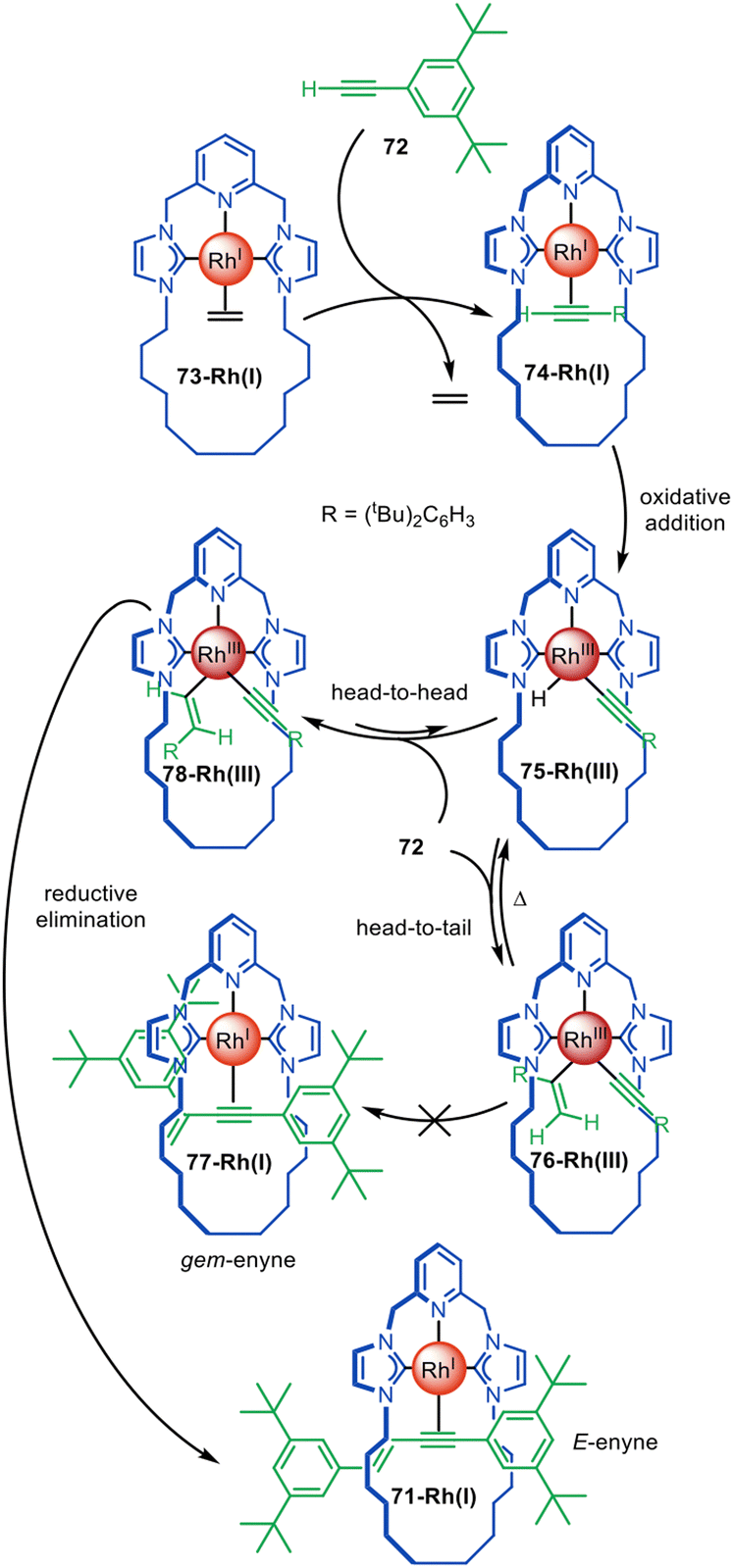 | ||
| Fig. 21 Chaplin's active template synthesis through the Rh(I)-mediated homocoupling of terminal alkynes to form a threaded E-enyne.117,118 | ||
The Chaplin group developed a Rh-mediated active template synthesis of diyne rotaxanes from alkynyl Grignard reagents (Fig. 22).119 Transmetallation of alkynyl Grignard 79 with a macrocyclic phosphinite pincer complex 80-Rh(III) leads to 81-Rh(III). Subsequent decarbonylation promoted by Me3NO affords diyne [2]rotaxane 82-Rh(I) through reductive elimination (81% yield over 2 steps). The group later reported a similar pincer complex with bulkier stoppers that could be converted to a hydrocarbon-based rotaxane.120 Demetallation followed by conversion of the phosphine ligands to thiophosphenes gave [2]rotaxanes, while a subsequent clipping reaction was used to synthesise a [2]catenane.
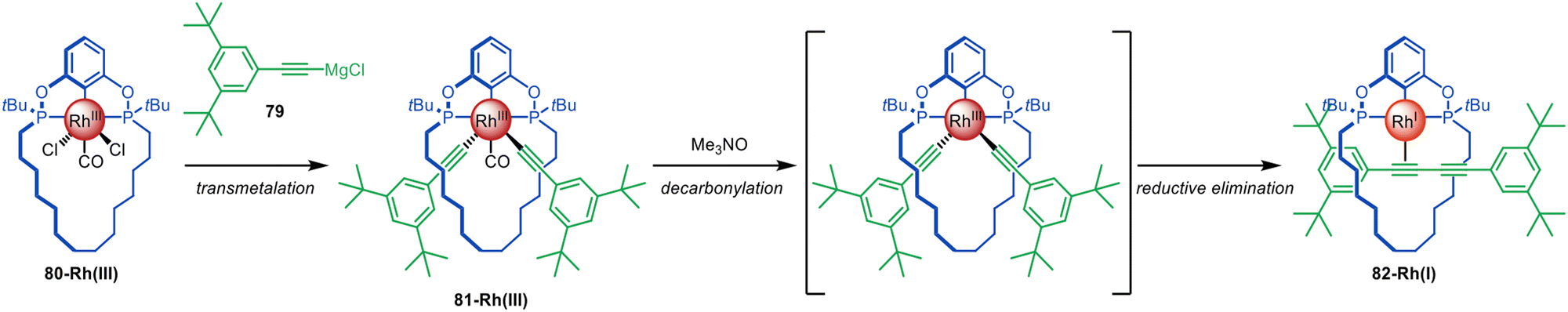 | ||
| Fig. 22 Chaplin's active template synthesis of diyne rotaxanes through Rh(III)-mediated homocoupling of alkynyl Grignard reagents.119 | ||
Metal-free active template synthesis
A key feature of active template synthesis is that the reaction between the thread building blocks is substantially accelerated through a macrocycle's cavity compared to outside of it. This can also be achieved without metal catalysis. As long as the threaded product resulting from such a process is kinetically stable, it can provide a useful and effective means of spontaneously assembling rotaxanes under kinetic control. (Note: if the threaded structures are not kinetically stable under the reaction conditions, any rotaxane present is the result of conventional ‘passive template’ synthesis under thermodynamic control.)Mock's cucurbituril-promoted Huisgen azide–alkyne cycloaddition
In 1989 Mock and co-workers carried out a rotaxane-forming reaction that can be considered a combination of passive ammonium ion template synthesis and metal-free active template synthesis.121 In a mechanistic investigation of the copper-free (Huisgen) azide–alkyne cycloaddition reaction of ammonium substrates catalysed by cucurbituril macrocycles, [2]rotaxane product 83 was formed by accelerated axle formation within the cucurbituril cavity (Fig. 23). The bulky substituents of the azide and alkyne reactants mean that the triazole axle cannot dethread. The cycloaddition was shown to be substantially accelerated (ca. 105-fold rate increase) due to favourable orientation of the reactants within the cavity and strain-promoted reactivity. | ||
| Fig. 23 Mock's synthesis of a [2]rotaxane through cucurbituril-promoted Huisgen azide–alkyne cycloaddition of ammonium-functionalised axle building blocks.121 | ||
Similar complexation-driven effective molarity increases to accelerate axle-forming reactions have been used to assemble (poly)rotaxanes122 and for ‘cooperative capture’ synthesis.123 In addition to cucurbiturils, other 3D ‘molecular barrels’, such as cyclodextrins, pillararenes and calixarenes, have been used in such approaches. For example, Arduini, Credi and co-workers reported that a tri-urea-calix[6]arene accelerates the alkylation of a complexed pyridylpyridinium guest to form a rotaxane (Fig. 24).124,125 Macrocycle 84 forms a 1:1 complex with 85 in toluene, and upon addition of stoppering components 86 and 87, [2]rotaxane 88 is formed in up to 70% yield. The macrocycle accelerates alkylation of the pseudorotaxane 16× faster than the axle is alkylated in the absence of the macrocycle. The authors ascribe the rate enhancement to the increase of pyridine nucleophilicity inside the electron-rich cavity of 84 which contains multiple aromatic rings, as well as urea groups that could stabilize the transition state. No formation of 89 is observed due to the steric preferences of the non-symmetrical macrocycle. The authors later carried out the selective synthesis of all three possible orientational isomers of related calix[6]arene-based [3]rotaxanes.126
 | ||
| Fig. 24 Credi and Arduini's cooperative capture synthesis of a [2]rotaxane.124,125 | ||
Metal-free active synthesis of rotaxanes from crown ether-accelerated reactions of amines and nucleophiles
In 2017, the Leigh group described the synthesis of [2]rotaxanes with a bifunctional macrocycle designed to stabilise the transition state of the axle-forming reaction (Fig. 25).127 The intention was that macrocycle 90, with a hydrogen bond donor pyridyl-2,6-dicarboxyamide unit at one end and an hydrogen bond acceptor oligo(ethylene glycol) chain at the other, would be able to promote the addition reaction of a primary amine 91 to a cyclic sulfate 92 through the cavity by stabilising the reaction transition state. The [2]rotaxane 93 was formed in 70% yield. Host–guest studies confirmed that the macrocycle does not strongly bind to either of the half-thread components, but rather stabilises the transition state of the reaction proceeding through the cavity. | ||
| Fig. 25 Metal-free active template synthesis of a [2]rotaxane through transition state stabilisation.127 | ||
The control experiments used to interrogate the mechanism of formation of rotaxane 93 included the use of analogues of macrocycle 90 that were missing either the two amide groups (intended to stabilise the forming anion in the transition state) or the oligoethylene glycol chain (intended to stabilise the forming cation in the transition state). However, although replacing the glycol units with a hydrocarbon chain led to no rotaxane formation, a small amount (5%) of rotaxane was still formed if the macrocycle amide groups were changed to esters. This suggested that crown ethers might be able to promote the alkylation of a primary amine through the crown ether cavity. This is somewhat counterintuitive as amines bind only very weakly to crown ethers and, when protonated as ammonium groups (which do bind strongly to crown ethers) then there is no lone pair with which to act as a nucleophile. Nevertheless, this is exactly what happens (Fig. 26).128
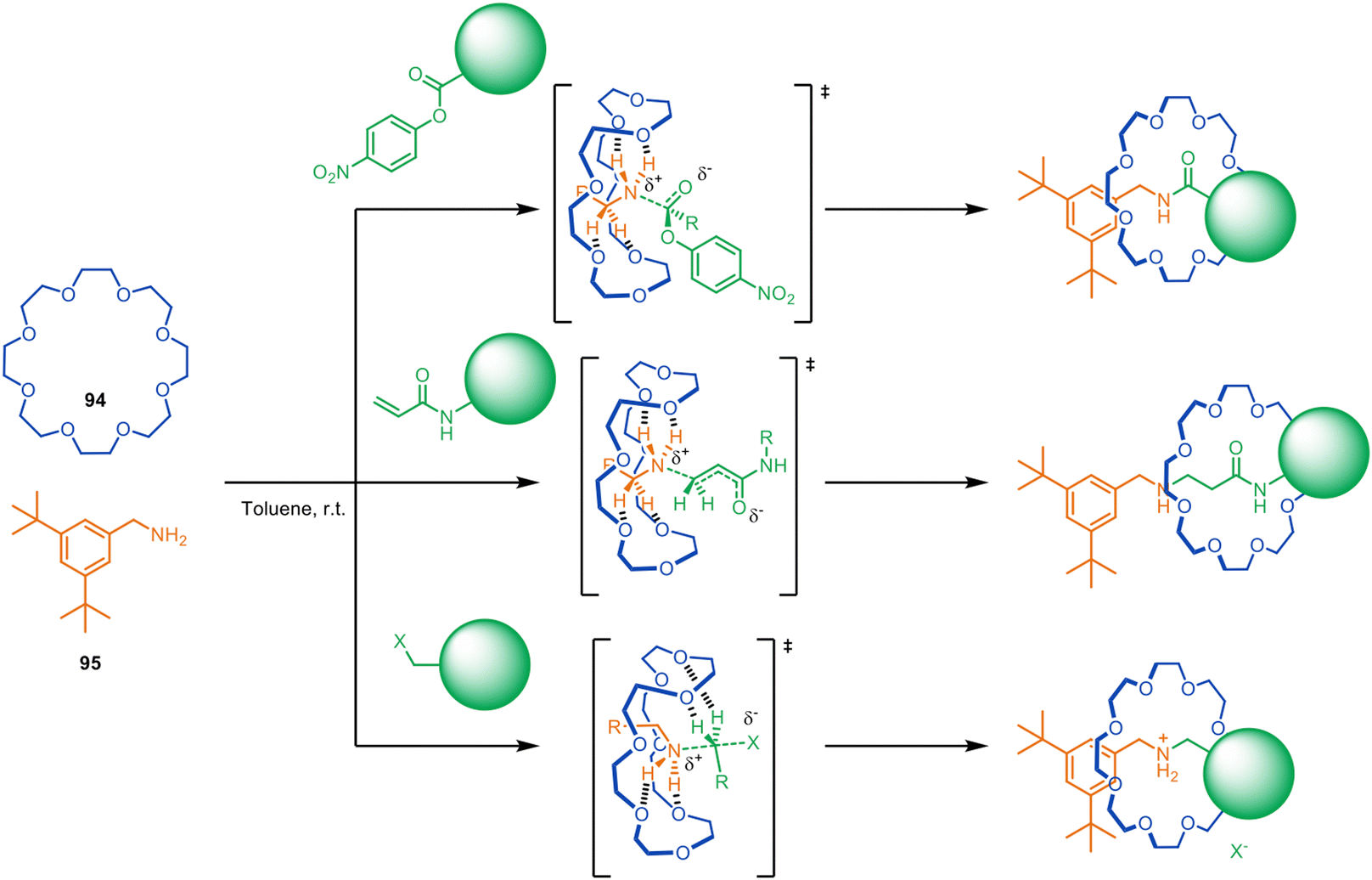 | ||
| Fig. 26 Metal-free active template synthesis of rotaxanes from primary amines, electrophiles and crown ethers.128,129 | ||
Despite having modest binding constants for the thread building blocks, crown ethers such as 94 accelerate the reaction of primary alkyl- and benzylamines such as 95 with a series of electrophiles to form rotaxanes (Fig. 26).128 Kinetic studies show rotaxane formation with a benzylamine is ∼26× faster than the background thread-forming reaction. The process allows the reagent-less formation of crown ether rotaxanes in a matter of hours at room temperature, accomplished by simply mixing together the macrocycle and the two thread-forming building blocks in toluene.
The rotaxane formation is driven by crown ether hydrogen bonding stabilising the partial positive charges formed in the transition state, a process that has previously been observed in ethylene-glycol-oligomer-catalysed aminolysis reactions.130,131 The utility of the metal-free active template reaction was demonstrated by the one-step synthesis of (i) pH-switchable molecular shuttles via aza-Michael addition and (ii) crown ether-peptide rotaxanes via N-acylation (Fig. 26).128
The system was subsequently extended to other electrophiles, producing crown ether rotaxanes based on (thio)urea, carbamate, sulfonamide and phosphoramidate/phosphinamide-functionalised axles in up to 95% yield and >100:1 rotaxane-to-free-thread selectivity.129,132 It also proved possible to use crown ether catalysed N-arylation of primary amines with electron-deficient aryl halides to produce rotaxanes with aniline threads. This metal-free active template N-acylation strategy has been used for the single-step enantioselective synthesis of mechanically planar chiral [2]rotaxanes (see section on mechanical and topological chirality).133
In an important recent development, the principle of enhancing the nucleophilicity of primary amines by hydrogen bonding to a macrocycle has been expanded from crown ethers to cyclic peptides.134 The Leigh group have previously threaded cyclo(proline) macrocycles such as 96 onto diammonium axles to form rotaxanes of cyclic peptides by passive template synthesis in up to 63% yield.135 Numata and coworkers found that this macrocycle-ammonium passive template motif could be adapted to metal-free macrocycle-amine active template synthesis in a similar manner to the crown ether-ammonium/amine system.134 With a glycine-derived amine nucleophile and phenylalanine-derived nitrophenol ester electrophile metal-free active template synthesis generated wholly peptidic rotaxanes (Fig. 27).134 The yields reported are very modest (∼1%), which the authors ascribe to the tendency of cyclic peptide oxygens to point out of the cavity and the presence of competing intermolecular hydrogen bonds, both of which limit the ability of the macrocycle to form strong hydrogen bonds with the amine. As well as being a strategy to mechanically interlocked peptides, the extension of metal-free active template synthesis from crown ethers to ammonium-binding cyclic peptides suggests that other ammonium-binding macrocycles (e.g. cucurbiturils, pillararenes, etc) may prove efficacious at accelerating the reaction of amines through a macrocyclic cavity for active template synthesis.
 | ||
| Fig. 27 Numata's metal-free active template synthesis of wholly peptidic rotaxanes from a glycine nucleophile, a phenylalanine-nitrophenol ester electrophile and cyclo(proline) macrocycle.134 | ||
Protein metal-free active template synthesis
The metal-free active template concept has also been applied to protein catenane synthesis.136 The Zhang group used protein engineering to design a catenane-forming reaction based on a SpyTag–SpyCatcher complex137 (Fig. 28a) where the catalytic unit embedded in a cyclic protein promotes isopeptide bond formation of a telechelic protein through its cavity (Fig. 28b). These entangled topologies displayed enhanced stability against proteolytic digestion, thermal and mechanical denaturing, without compromising their biological function. The group also demonstrated that introducing mutations in the catalytically active SpyStapler protein (SpyStapler003; Fig. 28c) led to improved efficacy of the active template reaction, which could be exploited for the synthesis of higher-order protein [n]catenanes in good yields (Fig. 28c).138 | ||
| Fig. 28 Zhang's metal-free active template synthesis of protein heterocatenanes.136–138 (a) SpyStapler-mediated isopeptide bond formation between SpyTag and BDTag.137 (b) Active template synthesis of a protein [2]catenane from a cyclic protein incorporating the SpyStapler sequence (c-SpyStapler-POI) and a linear protein terminated with the BDTag and SpyTag sequences at either end.136 (c) Higher order protein [n]catenanes (n = 2–5) assembled using a mutated SpyStapler sequence (SpyStapler003).138 | ||
The group have also developed a novel protein (‘AT-Snoop’) for active template protein synthesis.139 AT-Snoop and AT-Spy both catalyse isopeptide bond-formation but react orthogonally using different proteins domains. The orthogonality was exploited for the synthesis of [n]heterocatenanes, including an asymmetric protein olympiadane.138
Active template assembly of interlocked nanoscale toroids
A remarkable spontaneous assembly of nanoscale interlocked supramolecular toroids arises from a process closely related to metal-free active template synthesis (Fig. 29).140 Yagai and co-workers observed that molecules consisting of a barbiturate head group, a rigid central core and non-polar tail self-assemble into supramolecular hydrogen bonded rosettes. The rosettes stack in an off-set manner, leading to curvature that ultimately leads to the formation of toroids of ∼13 nm diameter. Nucleation of toroid assembly is quicker from sites within the toroid cavity than outside it (the fundamental principle behind active template synthesis; see Introduction and Fig. 2b) resulting in interlocking of the toroids. A range of interlocked materials with topologies were obtained, ranging from Hopf link [2]catenanes, to [5]ring olympiadanes and poly[n]catenanes.140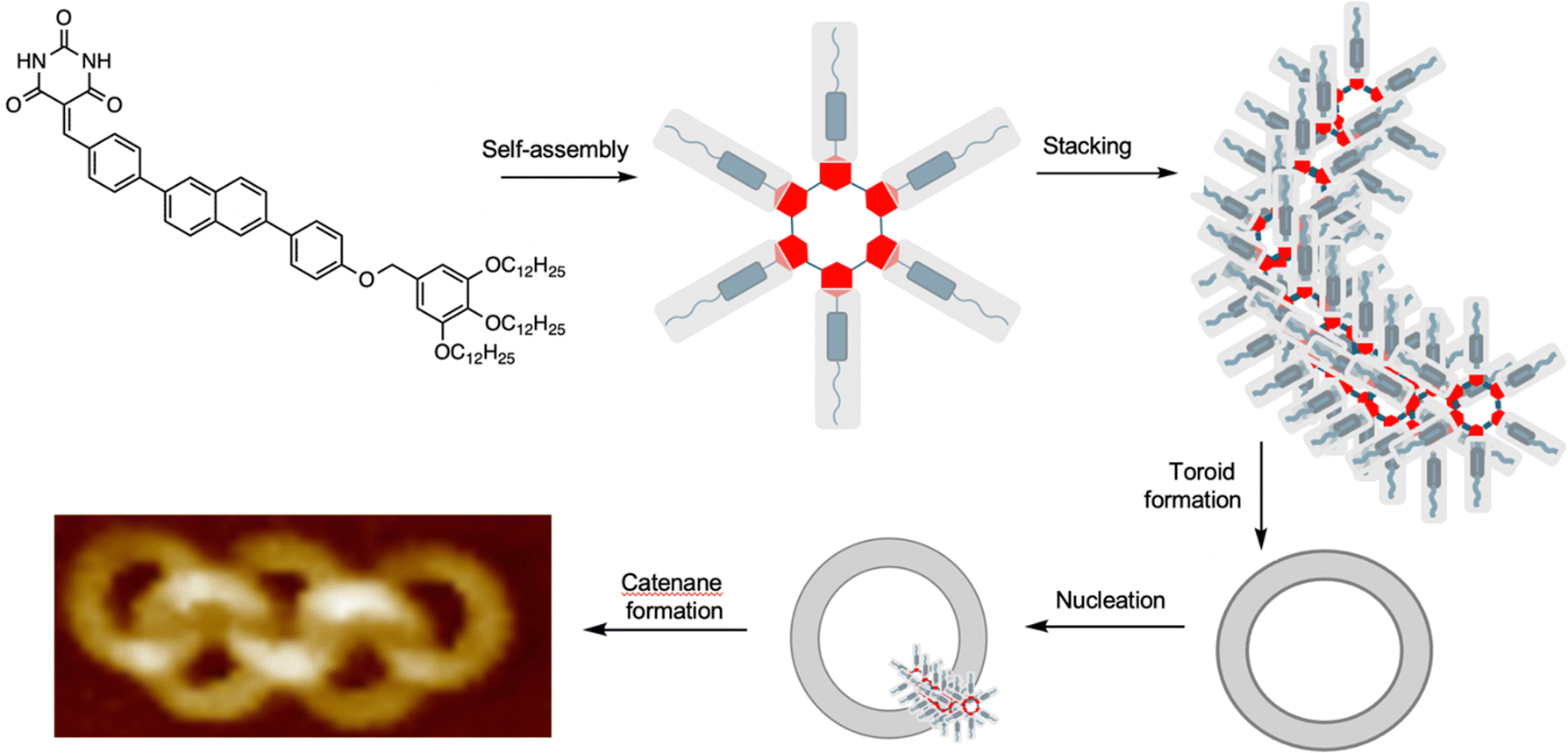 | ||
| Fig. 29 Yagai's self-assembling polycatenanes consisting of ∼13 nm diameter supramolecular rings.140 The molecules with a polar head group, rigid segment and non-polar tail assemble into rosettes that stack to form helical strands and toroids. The internal surface of the toroids seed the formation of new stacks, leading to supramolecular polycatenanes.140 | ||
Applications of active template synthesis
The special characteristics of active template synthesis include the lack of a requirement for persistent recognition motifs between the components in the interlocked product, and that the threaded structures need not be thermodynamically stable. This opens the way for the synthesis of interlocked molecules that are inaccessible through passive template synthesis. These features have led to active template synthesis being explored to make molecules for fields as diverse as molecular machinery, drug delivery systems, sensing devices, catalysis and materials science.Molecular shuttles
Active template synthesis has been used to prepare a number of rotaxane-based molecular shuttles and switches; that is, rotaxanes in which the macrocycle incessantly moves from one well-defined site on the thread to another (resulting in a net change in ring position in the case of switches).28,67,86,110 The dynamic aspects of these systems have been reviewed elsewhere,141,142 but some advantages of using active template synthesis for their assembly are illustrated by switchable molecular shuttle 97 (Fig. 30).67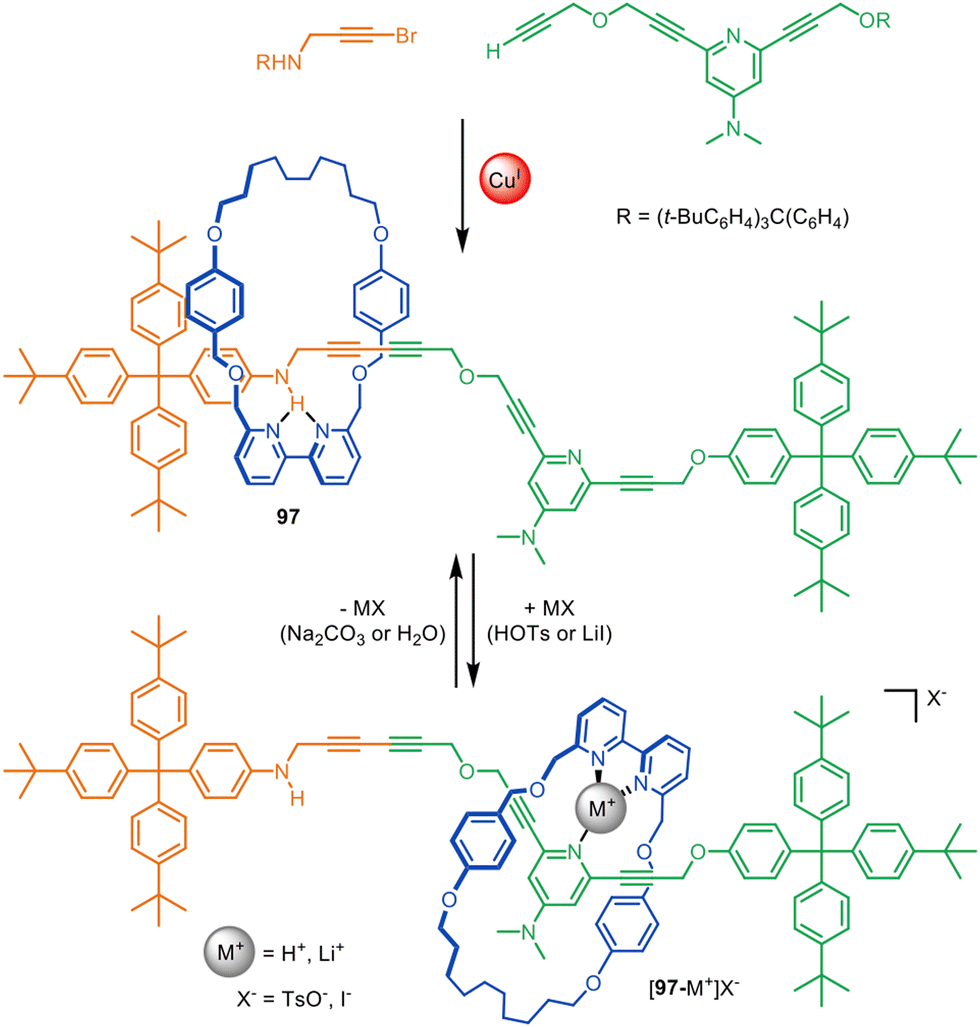 | ||
| Fig. 30 Active template Cadiot–Chodkiewicz synthesis of molecular shuttle 97. The position of the macrocycle on the axle is governed by relatively weak intercomponent interactions.67 | ||
Molecular shuttle 97 was synthesised through an active template Cadiot–Chodkiewicz heterocoupling, with one alkyne unit containing an aniline and the other a dimethylaminopyridine (DMAP) group (Fig. 30).67 In its unprotonated form the macrocycle of 97 preferentially coordinates to the aniline site on the axle through a single hydrogen bond with an intercomponent binding energy of −3.9 kcal mol−1, which would be far too weak to thermodynamically drive rotaxane synthesis. Protonation or Li+ coordination of the DMAP group of 97 triggers the translocation of the macrocycle to the DMAP axle site. This illustrates the efficacy of active template synthesis for the preparation of rotaxane-based molecular shuttles with much weaker intercomponent interactions (and correspondingly faster dynamics) than those required for passive template methods.143
The active template Cadiot–Chodkiewicz reaction has been used to develop other mechanically interlocked structures without strong intercomponent binding interactions, such as the [c2]daisy chain rotaxane 27 described by Jasti and co-workers (Fig. 8). This structure can be switched from a comparatively stable contracted form at temperatures lower than −40 °C to an extended state at higher temperatures.59
Goldup and co-workers used active template CuAAC synthesis to prepare molecular shuttles in which the shuttling of a bipyridine ring between bidentate and tridentate coordinating sites144 could be controlled by metal ions with different preferential coordination geometries.145
Weiss, Guldi, Hayashi and co-workers have reported light-switchable molecular shuttling in a [2]rotaxane based on an electron donor–acceptor system composed of a Zn–porphyrin-based macrocycle and a fullerene-stoppered thread.146 An active template CuAAC reaction installs a triazole moiety in the axle that forms one binding site in the resulting shuttle.
Beer and co-workers have also reported porphyrin-based molecular shuttles synthesised by active template protocols. In those examples the position of a pyridine macrocycle (used to direct the CuAAC reaction) is controlled via the addition of a Lewis base or anion.147
Mechanical and topological chirality
Active template synthesis has been used to make a range of mechanically and topologically chiral interlocked molecules, in which the chirality arises solely from the orientation of the interlocked components as opposed to point-chirality embedded in their structures. Active template synthesis is well-suited for this purpose as, like enantioselective synthesis, it is inherently under kinetic control. The Goldup group have made extensive use of active template CuAAC reactions to prepare mechanically planar chiral rotaxanes and catenanes (Fig. 31).148 A rotationally asymmetric bipyridine macrocycle (98), a sugar-based azide (99) and an alkyne (100) reacted in the presence of Cu(I) to produce a diastereomeric mixture of [2]rotaxane 101. Although the reaction did not impart stereoselectivity in the reaction outcome, it enabled simple separation of both diastereomers by chromatography. The chiral sugar could subsequently be replaced by an achiral amine nucleophile, removing the element of point chirality from the interlocked structures resulting in the isolation of both [2]rotaxane enantiomers (Smp)- and (Rmp)-102.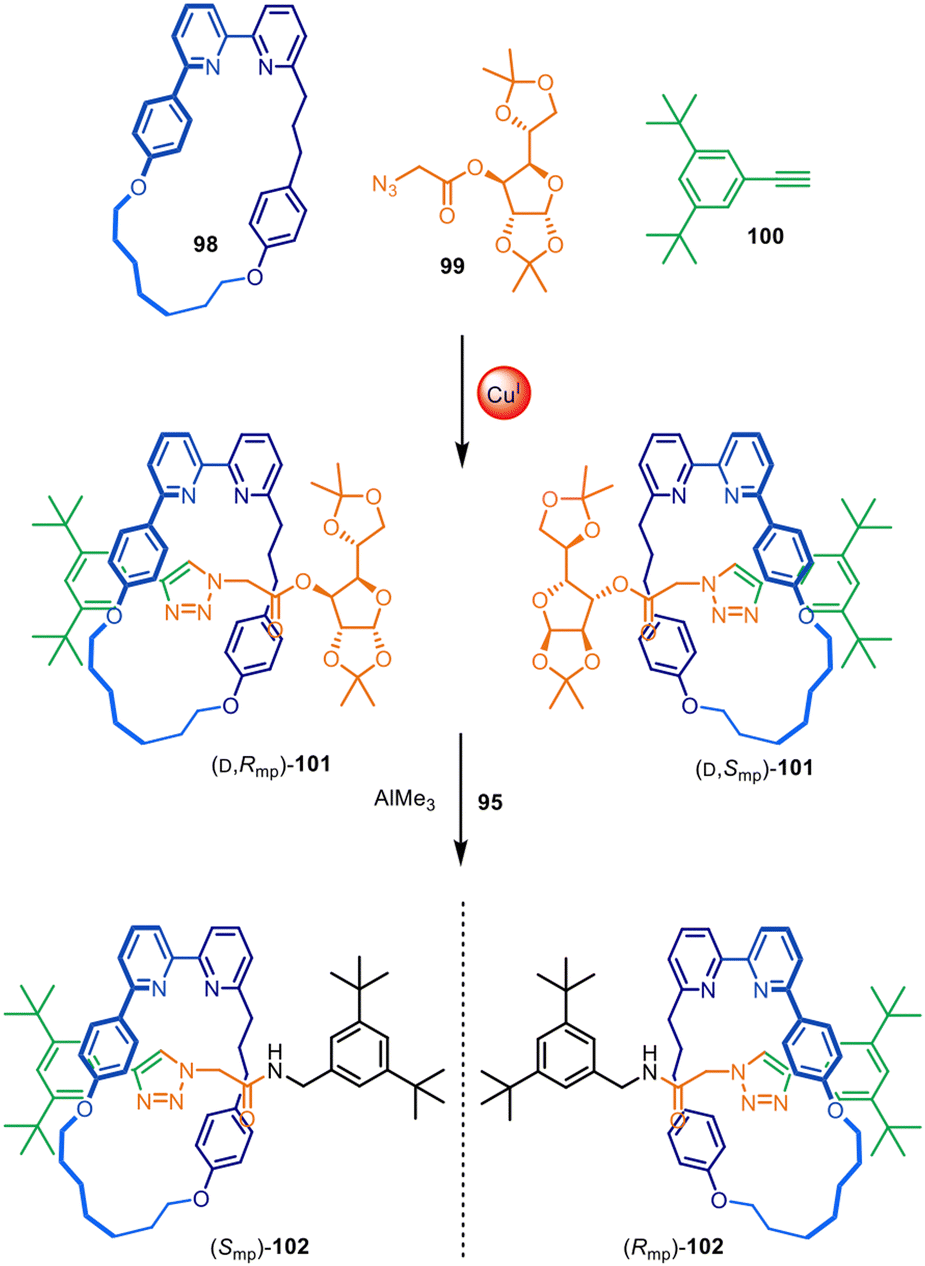 | ||
| Fig. 31 Goldup's active template synthesis of mechanically planar chiral rotaxanes (Smp)- and (Rmp)-102.148 | ||
The approach was later expanded to diastereoselective synthesis of mechanically planar chiral rotaxanes by modifying the stereodirecting moiety in the half-thread building block (Fig. 32).149,150 Excellent diastereomeric ratios of 98:2 were obtained using chiral amino acid-derived azide 102. Post-assembly removal of the point chirality source by symmetrisation of the amino acid stereocentre rendered the enantioenriched mechanically planar chiral rotaxane (Smp)-103. Similar methodology was later applied to the stereoselective preparation of enantiopure topologically chiral catenanes.151
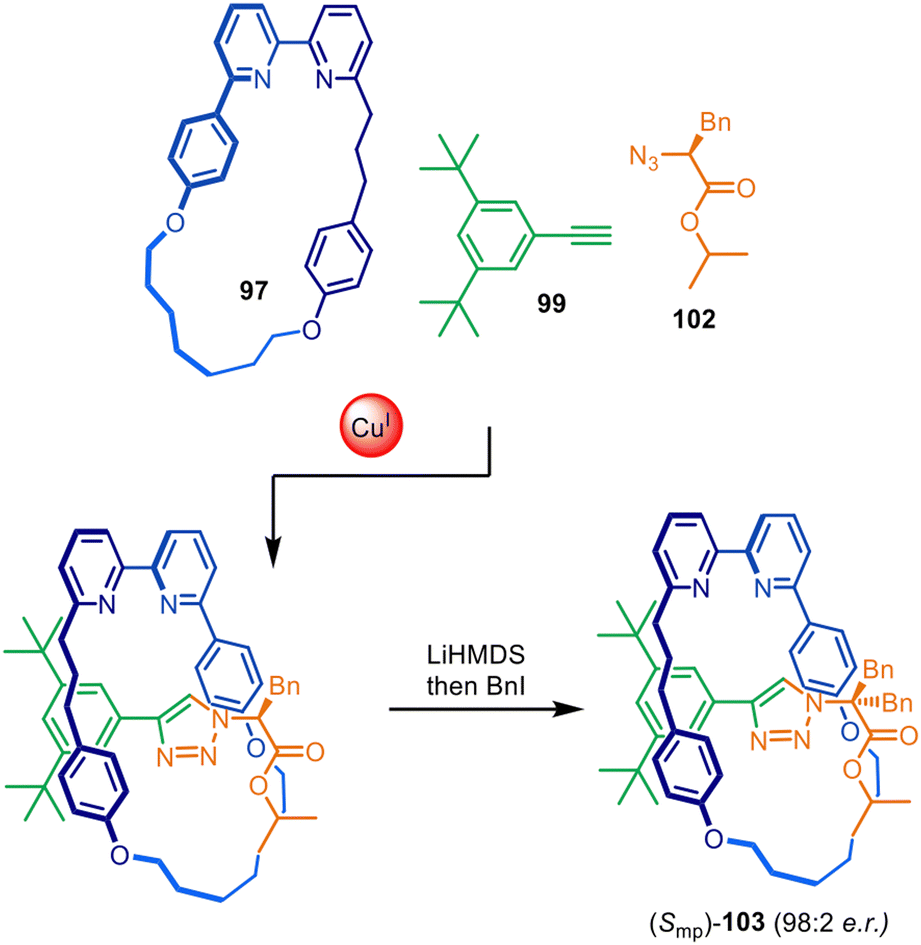 | ||
| Fig. 32 Goldup's diastereoselective active template synthesis of mechanically planar chiral rotaxane (Smp)-103.149 | ||
The Goldup group have also demonstrated that this chiral auxiliary can act as an mechanical interlocking auxiliary,23–25i.e. a group that facilitates the formation of the mechanical bond but is subsequently cleaved from the structure after shuttling of the macrocycle to a different part of the axle (Fig. 33).25 The auxiliary was removed to leave ‘impossible’ rotaxane23,24 (Smp)-104, with no recognition motif between the axle and macrocycle. The group also investigated attaching the chiral auxiliary to the macrocycle, rather than an axle-forming component.152 This could be used to direct the formation of both mechanically planar chiral rotaxanes and topologically chiral catenanes with good stereoselectivity (typically >90% ee).
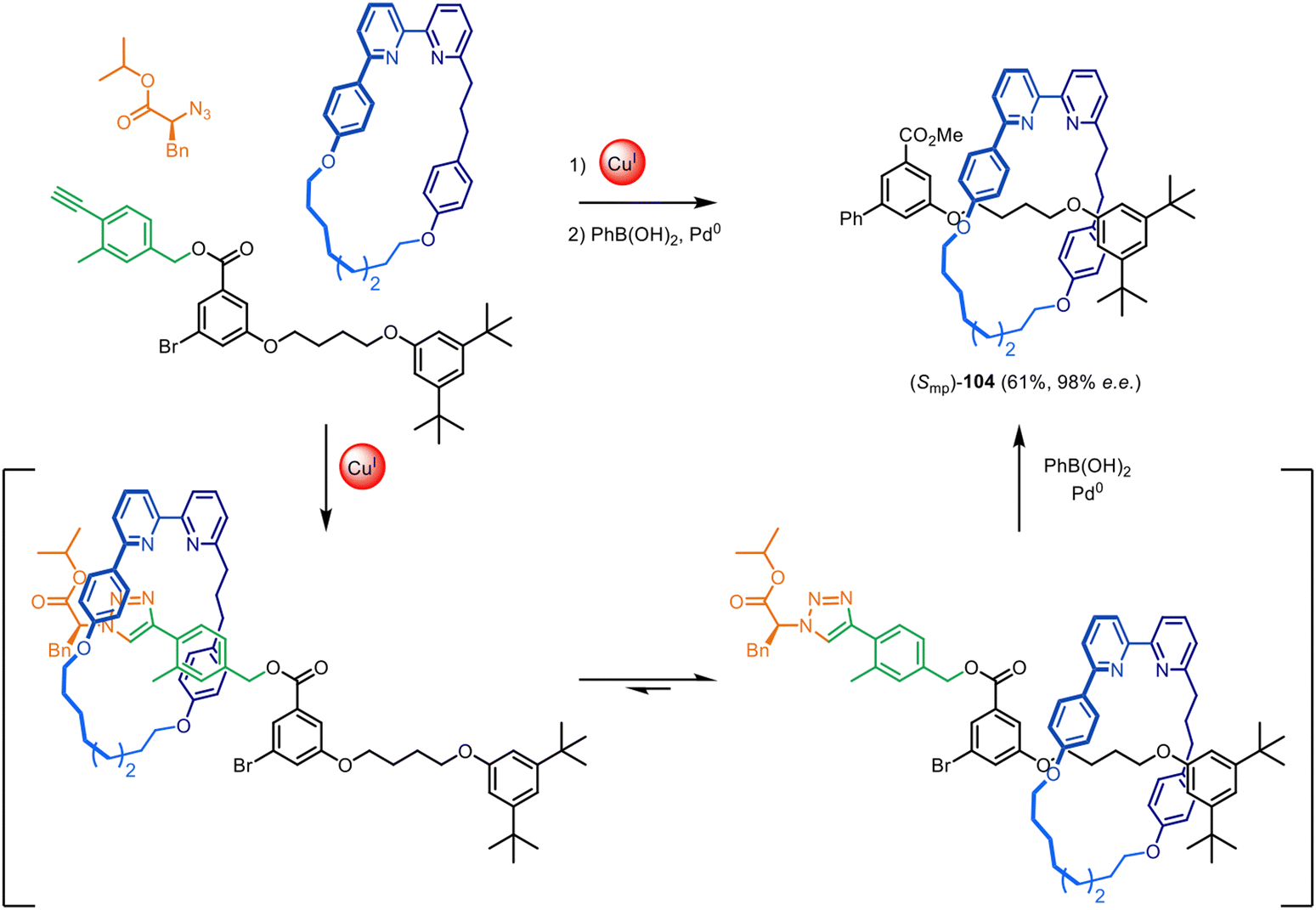 | ||
| Fig. 33 Goldup's mechanical interlocking chiral auxiliary for the active template synthesis of ‘impossible’ mechanically planar chiral rotaxanes.25 | ||
CuAAC active template synthesis has also been used by the Papot group for the diastereoselective synthesis of [1]rotaxanes (‘molecular lassos’).153,154 The point-chiral stereogenic unit has not yet been removed, meaning that a mechanically planar chiral [1]rotaxane with chirality solely arising from the mechanical bond remains to be achieved.
The active template mechanical interlocking chiral auxiliary systems developed by the Goldup group have been used to make a number of catenanes and rotaxanes with other, previously unexplored, types of chirality, including co-conformationally ‘pseudo-topologically’ chiral catenanes,155 mechanically (but not topologically) planar catenanes,156 mechanically axially chiral catenanes and noncanonical mechanically axially chiral rotaxanes.157–159 The topological aspects of these fascinating systems has recently been reviewed elsewhere.160 The versatility and efficiency of the active template approach has facilitated the exploration of new types of chiral interlocked molecules, leading to a better understanding of aspects of molecular asymmetry.
The Leigh group reported a single-step asymmetric synthesis of mechanically planar chiral rotaxanes by using primary amine 105, activated ester 106a–b with a chiral leaving group and achiral crown ether macrocycle 107 with Cs symmetry in an application of metal-free active template synthesis (Fig. 34).161 The incorporation of cinchonidine pseudoenantiomers as directing groups enabled the synthesis of either enantiomer of [2]rotaxane 108 in 40–50% ee. X-Ray crystal structures and computational modelling indicated π-stacking between the naphthalene ring of the macrocycle and the electron-deficient aromatic stopper helps to direct the reaction stereoselectivity.
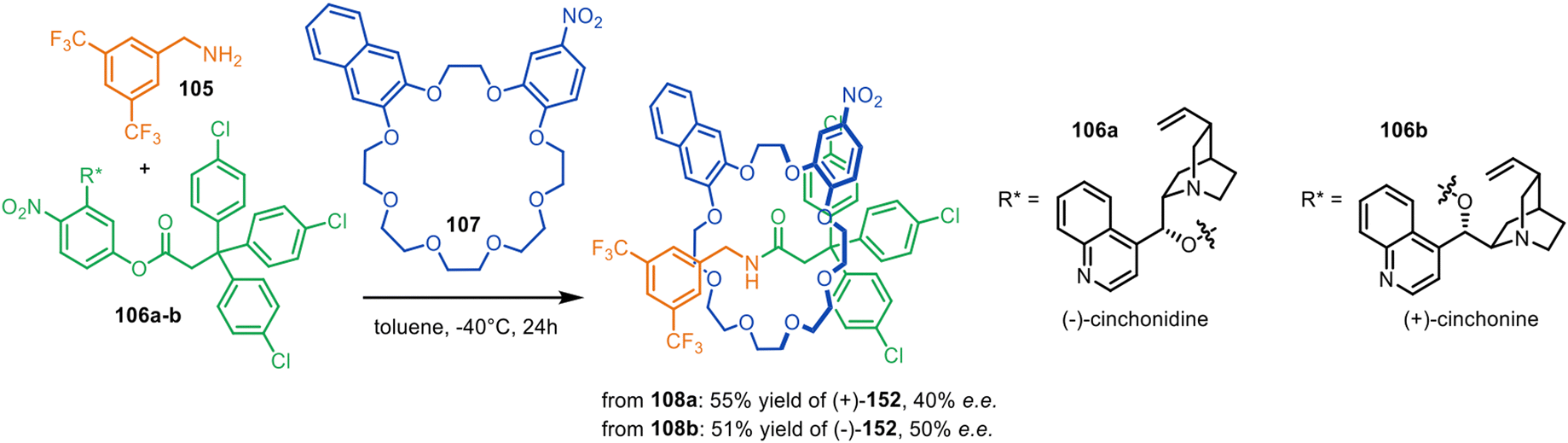 | ||
| Fig. 34 Single-step asymmetric metal-free active template synthesis of mechanically planar [2]rotaxanes.161 | ||
A different approach to mechanically planar chiral rotaxanes, also exploiting metal-free active template synthesis, was developed by Tian and Zhu.162 Modified bis(chloroaryl) crown ethers were used as catalysts for the spontaneous formation of [2]rotaxanes using a range of electrophiles and amine nucleophiles. The prochiral interlocked molecule (with an unsymmetrical axle but a rotationally symmetric ring) then underwent catalytic desymmetrisation through a Pd-catalysed asymmetric Suzuki–Miyaura reaction to yield the chiral rotaxanes in up to 97:3 er.
Asymmetric, diastereoselective and other types of catalysts
In 2015, Leigh and co-workers reported the first use of a mechanically chiral [2]rotaxane ligand for enantioselective catalysis, synthesised via active template methodology.73 [2]Rotaxane 109 (Fig. 35) was prepared through Cu(I)-mediated Goldberg active template synthesis. The chiral interlocked structure has an endotopic binding site on the macrocycle that can bind various transition metals. The chiral rotaxane ligand was used for the Ni(II)-catalysed enantioselective Michael addition of diethyl malonate 110 to various nitrostyrene derivatives 111a–f, affording products 112a–f in good yield and enantiomeric enrichment (typically >98% yield and 93:7 er). Chiral induction with the rotaxane ligand was significantly more effective than with a comparative acyclic chiral ligand (68:32 er), although considerably longer reaction times were needed with the rotaxane (27 days compared to 2 days). Both the strong enantioselectivity and the slow reaction times can be attributed to the sterically hindered binding pocket of the rotaxane which results in a catalytic site where the chirality is well-expressed in all three dimensions.
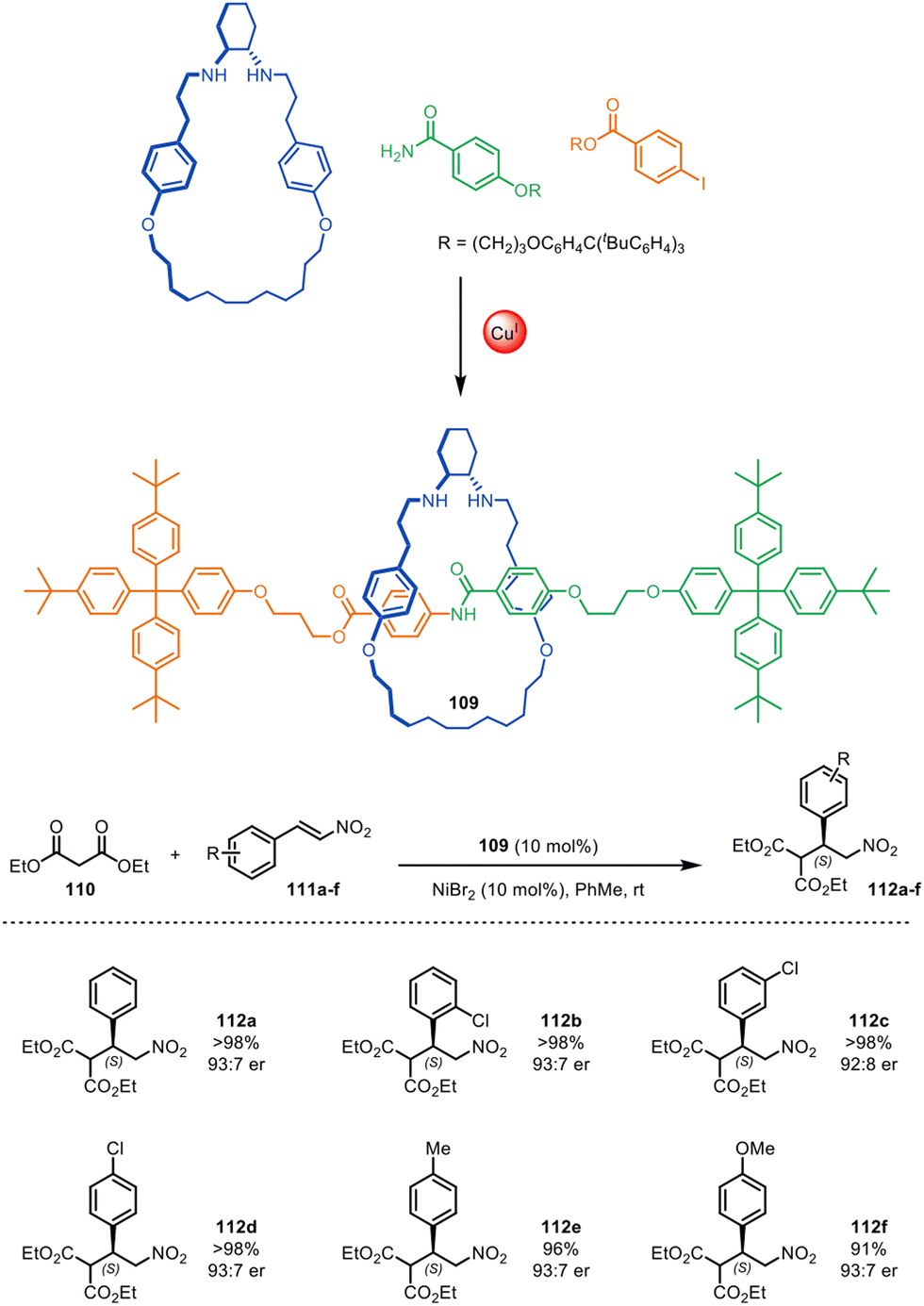 | ||
| Fig. 35 Mechanically chiral [2]rotaxane 109, synthesised through a Goldberg active template reaction, and its scope as a ligand for the nickel-catalysed enantioselective Michael addition of diethyl malonate and trans-β-nitrostyrenes.73 | ||
The Goldup group have demonstrated that chiral rotaxanes can be used as ligands for diastereoselective catalysis. An active template CuAAC reaction was used to synthesise the interlocked ligand for [2]rotaxane-gold catalyst 113-Au(I).163 The catalytic activity could be switched on by the addition of catalytically innocent metal cations (Fig. 36). 113-Au(I) was shown to catalyse Toste's Au(I)-mediated modification of the Ohe–Uemura cyclopropanation reaction.164,165
 | ||
| Fig. 36 Goldup's [2]rotaxane precatalyst 113-Au(I) prepared by an active template CuAAC reaction, and the effect of additives on its catalytic efficacy in the Toste–Ohe–Uemura cyclopropanation reaction.163 | ||
Building on this system, Goldup and co-workers described a catalyst for asymmetric synthesis that relies on the mechanically planar chirality of the rotaxane ligand as the sole source of chiral induction.166 Ligand (Rmp)-114 was synthesised via a diastereoselective active template CuAAC reaction149 (Fig. 37a) (see section on mechanical and topological chirality). Upon coordination to Au(I) and activation with a cofactor, (Rmp)-114-Au(I) proved an effective catalyst for the Au(I)-mediated Toste–Ohe–Uemura cyclopropanation, generating a range of cyclopropanes (e.g.115) in good yields and stereoselectivities (62–92% de for the cis diastereomer, 9–77% ee; Fig. 37b). Modelling and transition state calculations suggest that (Rmp)-114-Au(I) possesses a well-defined and rigid chiral environment around the metal centre (Fig. 37c).
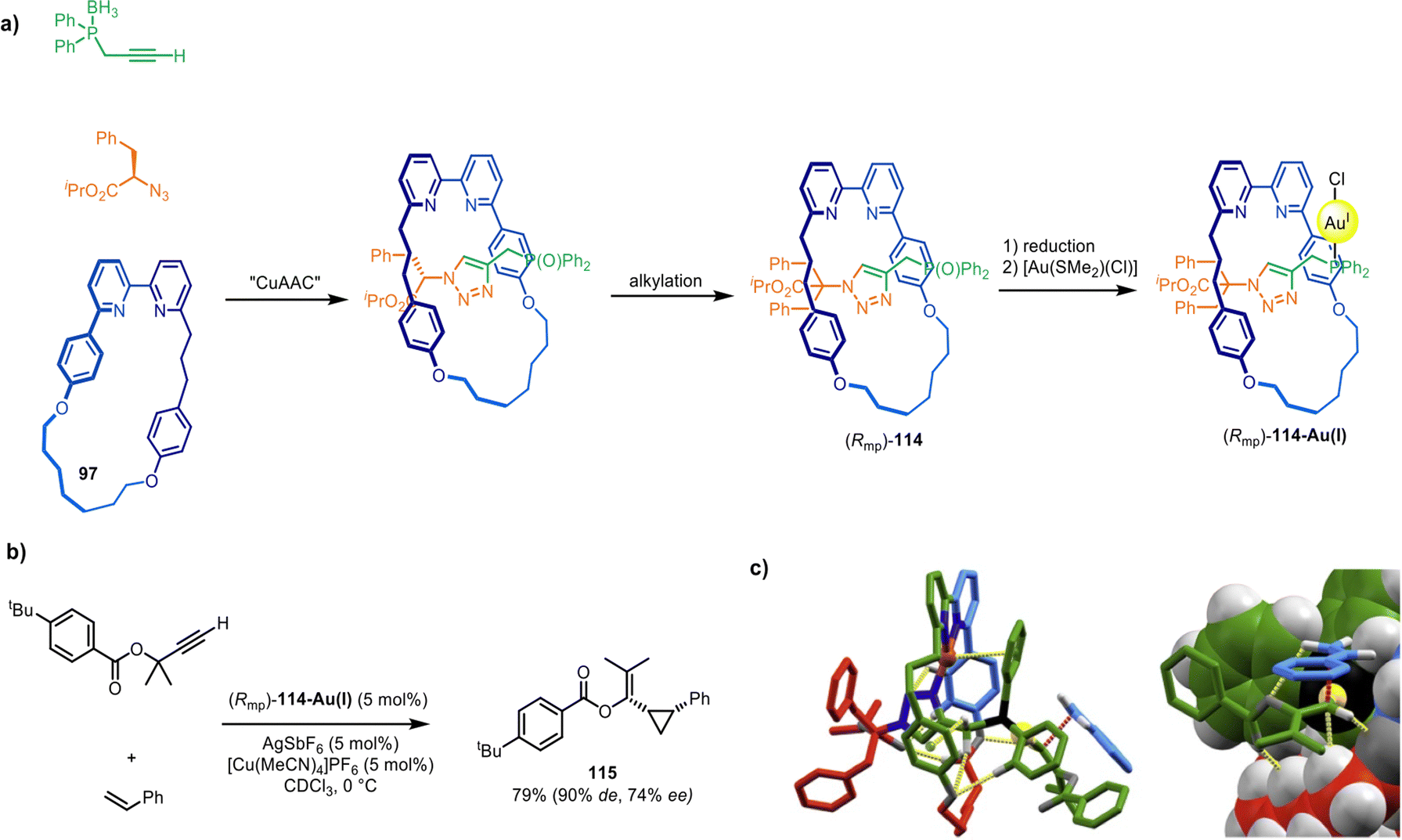 | ||
| Fig. 37 (a) Goldup's active template synthesis of mechanically planar chiral rotaxane precatalyst (Rmp)-114-Au(I). (b) Example of a diastereoselective and enantioselective Toste–Ohe–Uemura cyclopropanation reaction promoted by 114-Au(I). (c) Computationally modelled transition state.166 | ||
The Goldup group have also reported [2]- and [3]rotaxane organocatalysts, synthesised via CuAAC active template reactions, that promote the anion–π catalysed Michael addition of malonic acid monothioester 116 to β-nitrostyrene 117 (Fig. 38). The selectivity of 62:1 with [3]rotaxane catalyst 118 for addition/decarboxylation product 119 was comparable with the most effective non-interlocked catalysts previously reported.167 Computational studies suggested that the high selectivity observed may be due to π-stacking between the protonated bipyridine group and the naphthalene diimide.
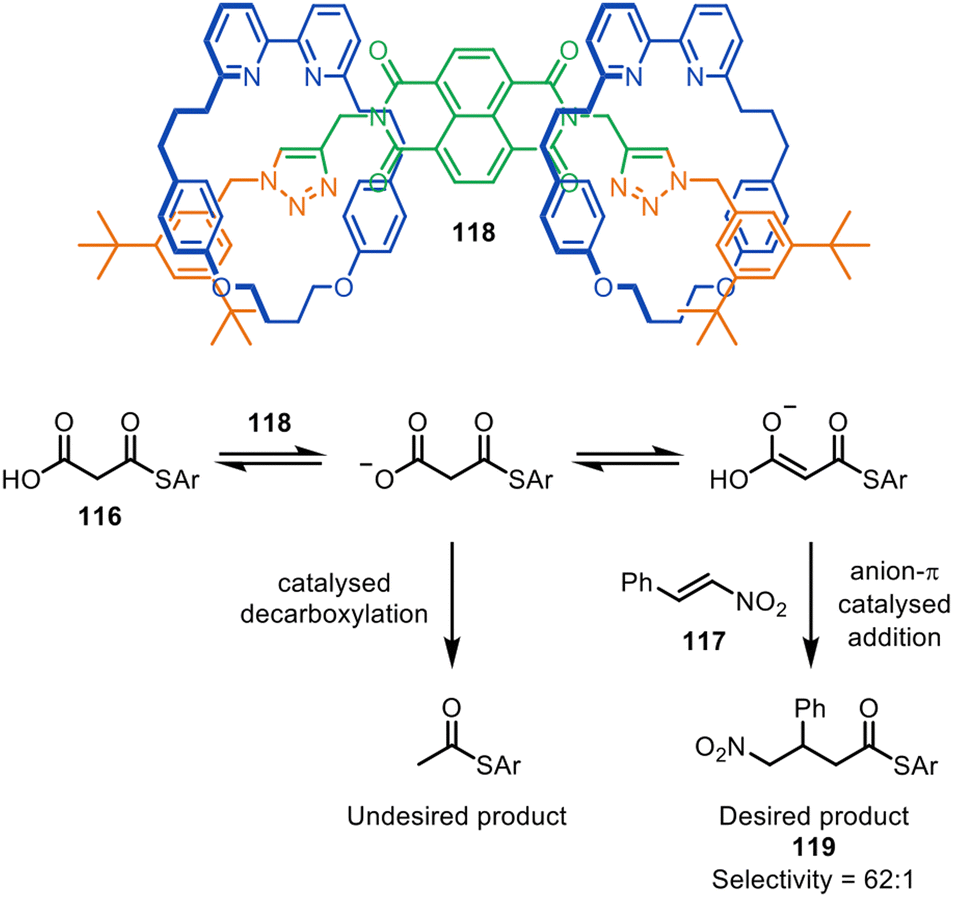 | ||
| Fig. 38 Goldup's [3]rotaxane anion–π organocatalyst 118 for the Michael addition of malonic acid monothioester to β-nitrostyrene.167 | ||
In catalysis applications other than stereoselective catalysis, Schmittel and co-workers have exploited the triazole units formed during CuAAC active template reactions to create binding sites for an allosterically regulated silver(I) catalyst.168 Catalysis by the [2]rotaxane was regulated by steric restrictions on the conformational mobility of the components inhibiting the formation of catalytically active species. They have also described the design of a three-input logic AND gate with catalytic output using a [2]rotaxane shuttle prepared by active template synthesis.169
Sensing and molecular recognition
Beer and co-workers have used active template synthesis to assemble [2]rotaxane hosts for molecular recognition and sensing.170 Based on their earlier halogen bonding host molecules,171,172 halogen bond donating units were incorporated in both the macrocycle and axle components of a rotaxane to generate a 3D anion binding pocket. Modification of the active template CuAAC reaction installed a iodotriazole ring as part of the rotaxane axle173 with a bis-iodotriazole macrocycle, generating [2]rotaxane 120 in 25% yield (Fig. 39a). Exotopic coordination of a Re(I) atom to the triazole ligands was then used to orient the iodine atoms towards the inner cavity of the macrocycle. Host 120-Re(I) preferentially bound size- and shape-complementary Cl− anions over other halide and acetate anions. The group have subsequently explored structurally related rotaxanes that bind halides in the presence of water,174,175 hydrogen and halogen bonding rotaxanes for alkali metal-halide ion-pair sensing,176,177 and a ferrocene-functionalised rotaxane for electrochemical sensing of bromide.178 Rotaxanes 121-Zn(II) and 122 (Fig. 39b) proved to be effective fluorescent sensors179 and chiral anion receptors,180 respectively, and several related rotaxanes have been prepared that can switch between cationic and anionic binding modes.181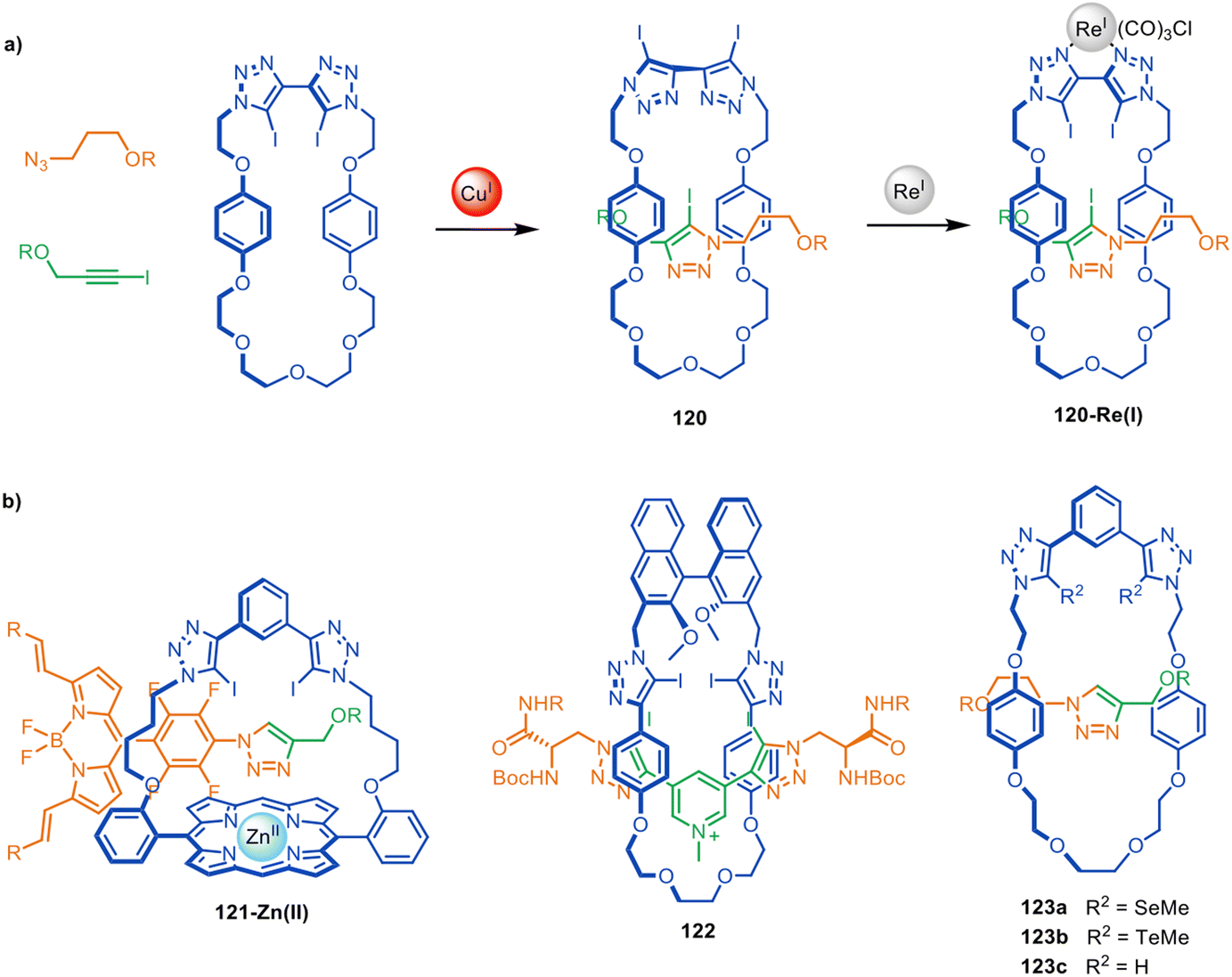 | ||
| Fig. 39 (a) Beer's active template synthesis of [2]rotaxane 120-Re(I) for anion sensing. (b) Other examples from the Beer group of halogen- and chalcogen-bonding rotaxane hosts prepared by active template synthesis.170,179,180,182 | ||
The Beer group have also demonstrated that Cu(I) binding to alkyl-selenium or alkyl-tellurium moieties in a macrocycle can be used to direct CuAAC active template synthesis through the macrocycle to form chalcogen-derivatised rotaxanes such as 123a–b (Fig. 39b).182 The resulting rotaxanes bind hard and soft anions in a broad range of environments.
A range of hydrogen bonding motifs unrelated to the active template assembly process have been incorporated into mechanically interlocked hosts for the recognition of small molecules and anions. McClenaghan, Tucker and co-workers prepared [2]rotaxane systems containing barbital recognition sites, such as 17 (Fig. 6b), via active template copper-catalysed CuAAC and Glaser reactions.47,183 Beer et al. constructed a neutral [2]rotaxane host for anionic guests using a hydrogen bond donating indolocarbazole unit as part of the axle.184 A [2]rotaxane host including an amide binding site was used by the Ghosh group for the electrochemical sensing of anions.185 Jasti and co-workers used a Cu-catalysed active template reaction for a family of [2]rotaxanes in which fluoride-70 or thiolate-186 induced dethreading results in the switching on of fluorescence.
The Goldup group have used CuAAC active template synthesis to build a number of rotaxanes for anion187 and cation188–190 binding. Some of the interlocked ligands stabilise distorted metal coordination geometries with unusual coordination numbers due to the sterically restricted 3D shape of the binding pocket.189 This is exploited in a Pt(II)-rotaxane complex, in which the metal is stabilised towards oxidation and acts as a reversible stimuli-responsive phosphorescent sensor for Ag(I) ions.190
Prototypes of drug delivery systems
The lack of need for specific functional groups on the axle of rotaxanes using active template synthesis led to the design of enzyme-sensitive [2]rotaxane 124 by Papot, Leigh and co-workers for the controlled release of the anticancer drug paclitaxel in tumour cells (Fig. 40).191 Prodrug stopper 125 and hydrophilic fragment 126 were coupled through a self-immolative macrocycle (127) containing a Cu(I)-coordinating pyridine group. While the non-interlocked thread undergoes rapid esterase hydrolysis in plasma, the macrocycle in 124 acts as a shield, protecting the ester group in the axle from enzymatic degradation. However, cancer cells overexpress β-galactosidase, which triggers a cascade of reactions that culminate in the opening of the macrocycle and release of the active form of the drug. | ||
| Fig. 40 Papot and Leigh's active template CuAAC synthesis of β-galactosidase-cleavable [2]rotaxane 124, which releases paclitaxel in tumour cells.191 | ||
Besides providing temporary shielding of an axle, rotaxane formation can be used to transiently modify the solubility and absorption properties of molecular components. Lewis et al. reported the use of an a CuAAAC active template [2]rotaxane as a triggerable cage for G-quadruplex (G4) DNA binders.192 The [2]rotaxane has better cell permeability than the analogous non-interlocked DNA binder, but lower cytotoxicity (probably due to the macrocycle inhibiting access to the G4 DNA binder). Enzymatic or light-triggered cleavage of one of the rotaxane stoppers liberates the G4 DNA binder within the cell, restoring its binding properties and cytotoxicity with high spatiotemporal control. Goldup, Tavassoli and coworkers have designed a triggered Curtius rearrangement immolative system where macrocycles can be functionalised at a late stage to incorporate a range of different cargos and trigger units.193 The approach was demonstrated with bipyridine macrocycles for CuAAC active template reactions and, notably, with crown ethers for metal-free active template synthesis.
Sheathed ‘molecular wires’: rotaxanes and catenanes with extended-conjugated components
Active template alkyne homocoupling and alkyne–alkyne heterocoupling reactions have been used to prepare a wide range of polyyne rotaxanes with π-conjugated axles. The groups of Anderson, Tykwinski,51,61,62,71 and Gladysz52,58,59 have explored the synthesis of polyyne rotaxanes using active template Glaser and Cadiot–Chodkiewicz couplings. Long polyyne chains are prone to degradation and a threaded sheath structure can provide substantially improved stability.61–63 Sheathed polyynes also show some modifications in photophysical behaviour compared to the non-interlocked components,64,68 leading to their investigation as molecular wires.The Anderson group have used active template Glaser couplings for the synthesis of π-conjugated rotaxanes with Zn(II)-porphyrin stoppers at either end (Fig. 41).60 Porphyrin [2]rotaxane 128 was elaborated into [4]catenane 129 in 62% yield using 130 as a radial template for the interlocked architecture. [7]Catenane 131 was also formed as a Vernier template by-product of the synthesis.
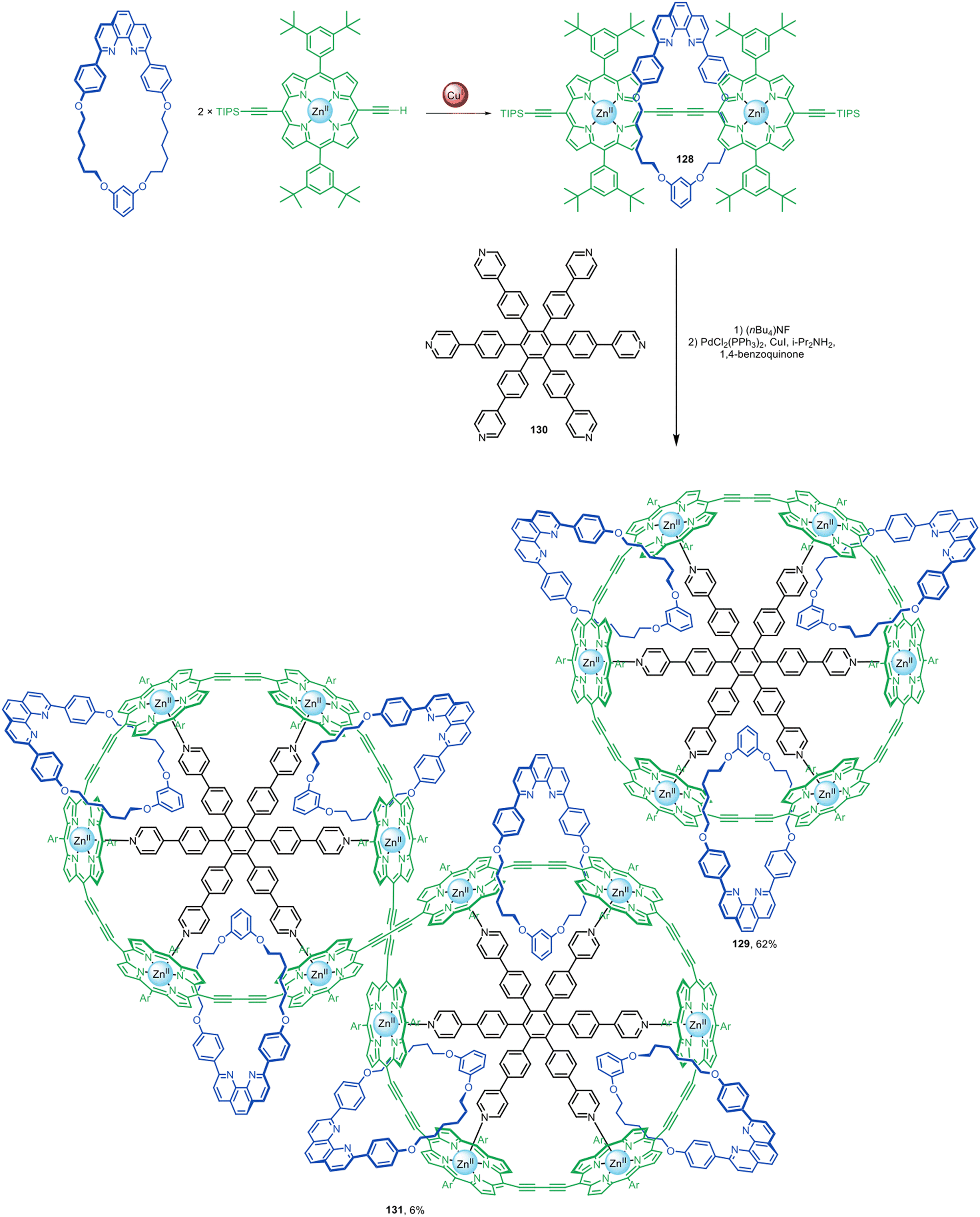 | ||
| Fig. 41 Anderson's assembly of [4]- and [7]catenanes from [2]rotaxanes prepared by active template Glaser homocouplings.60 | ||
The Anderson and Jasti groups have both used masked phenyl and alkyne units to extend the number of π-conjugated units in the axle and increase the diversity of accessible polyyne rotaxane and catenane structures. Jasti and co-workers used cyclic precursors to [n]cycloparaphenylene nanohoops for the synthesis of π-conjugated rotaxanes and catenanes via active template Cadiot–Chodkiewicz heterocouplings (Fig. 42).194 The bent cyclohexadiene moieties in 132 facilitate endotopic metal binding, which is difficult to achieve with cylindrical [n]cycloparaphenylenes. Reductive aromatisation of the cyclohexadienes furnishes the nanohoops in [3]rotaxane 133 and catenanes 134–135. The group later expanded the methodology to CuAAC active template synthesis, producing triazole catenanes in high yields.195
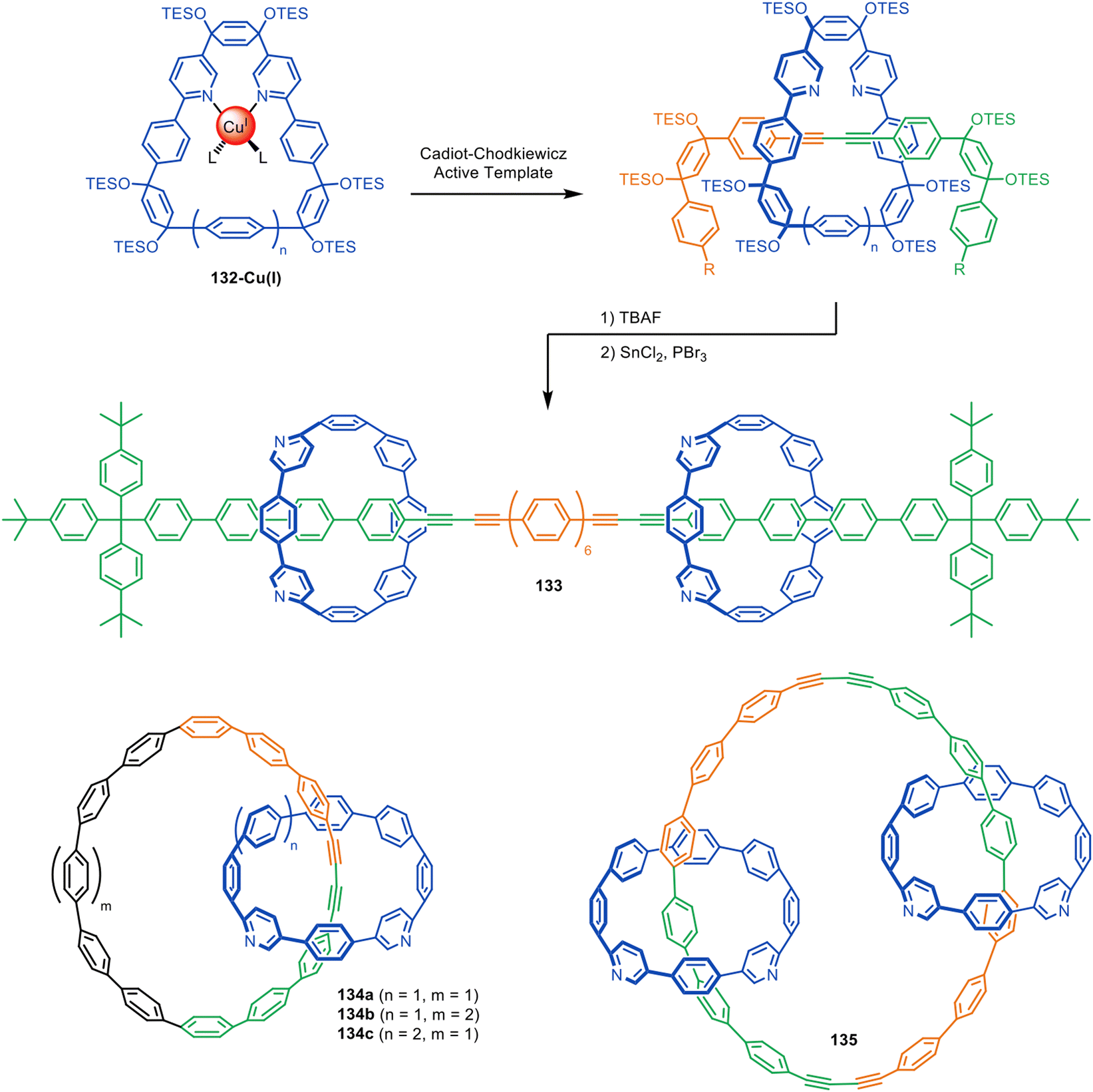 | ||
| Fig. 42 Jasti's assembly of interlocked nanocarbons via active template Cadiot–Chodkiewicz synthesis.194 | ||
Anderson and co-workers have employed indane196 and bicyclo[4.3.1]decatriene197 as photolabile masked-alkyne equivalents as well as dicobalt carbonyl complexes that can be unmasked by oxidative decomplexation.198 These synthons have been used in active template Glaser and Cadiot–Chodkiewicz syntheses of rotaxanes and catenanes, including polyyne [3]rotaxane 136 (Fig. 43) which has 14 contiguous alkyne units in the axle stabilised by the nanohoop sheath. The strategy was later used to prepare a polyrotaxane with an extraordinary 34(!) contiguous triple bonds in the axle and four threaded macrocycles.199 This polyyne rotaxane is in a length regime where the electronic properties of polyynes converge to those of carbyne. The rotaxane displayed electronic properties similar to those predicted for an infinite carbyne, illustrating the potential of polyyne rotaxanes to act as molecular wires.
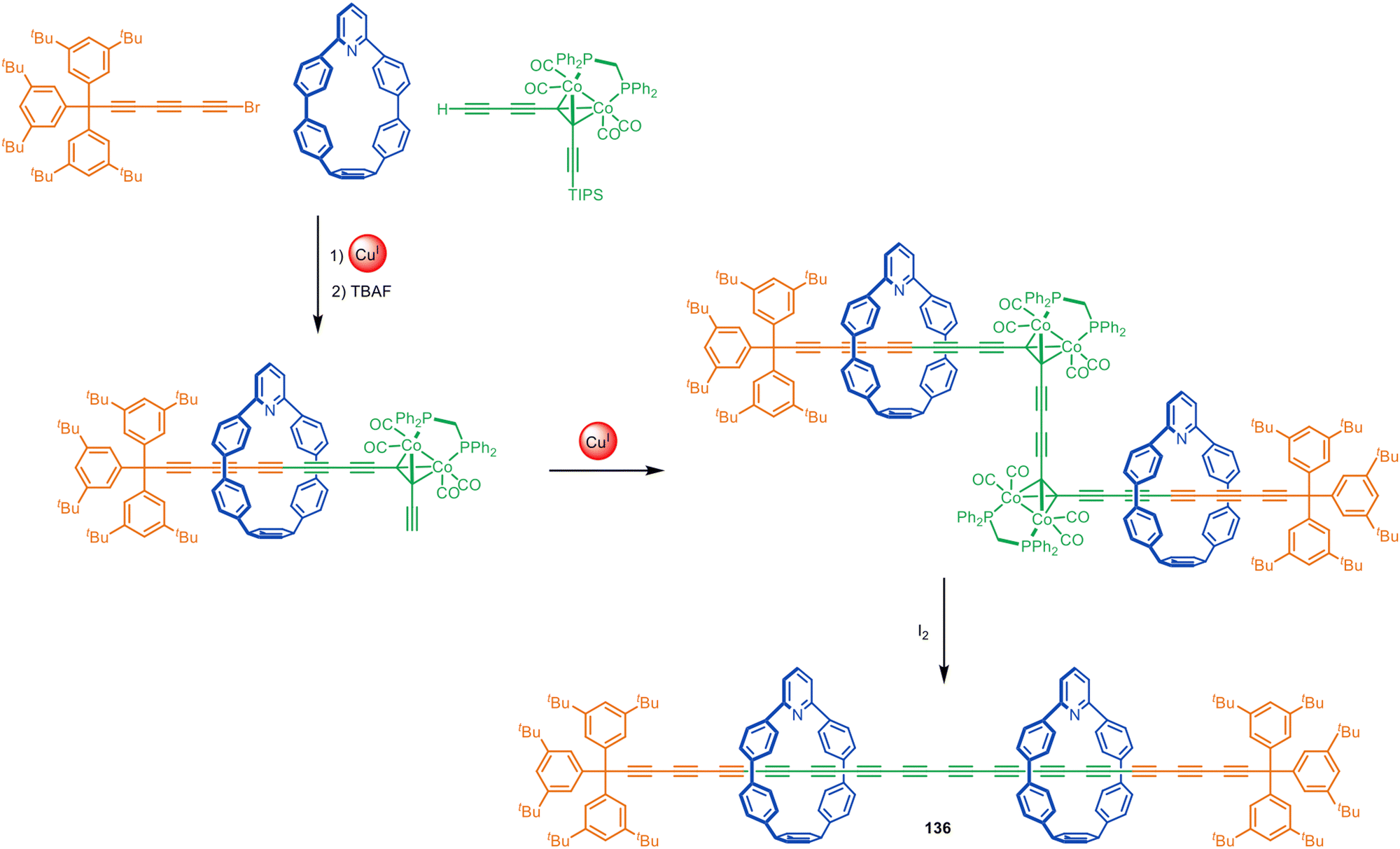 | ||
| Fig. 43 Anderson's extended polyyne rotaxane 136 synthesised via a Cadiot–Chodkiewicz active template reaction followed by oxidative unmasking of the bridging alkynes.198 | ||
The synthesis of catenanes with this method has also been reported, however, unmasking of the alkynes resulted in a complex mixture of products that could not be purified.
The CuAAC active template reaction has been used by the groups of Ngo, D’Souza and Goldup for the synthesis of porphyrinoid rotaxanes.200,201 These compounds displayed similar photophysical properties to the free thread component, but the threaded macrocycles inhibit intermolecular interactions between axles including π-stacking-driven aggregation and ligand-driven dimerisation.
Modification of chemical and physical properties
Mechanically interlocking molecular components can significantly alter both chemical and physical properties.202 Active template synthesis is particularly useful in this regard as it enables components with otherwise weak binding affinities to be locked in close proximity, a situation that is hard to achieve in other ways.Leigh and co-workers have used metal-free active template synthesis to make compact amine-crown ether rotaxanes that exhibit exceptional basicity (Fig. 44).203 The super-basicity results from three main factors: (1) the formation of strong hydrogen bonding and electrostatic interactions in the protonated salt of the rotaxane; (2) the forced proximity (and lack of conformational and co-conformational freedom) of the amine and ethylene glycol functional groups, resulting in electrostatic repulsion between the interlocked macrocycle and thread; (3) a high degree of preorganisation of the components in the free-base rotaxane that favours amine protonation.
 | ||
| Fig. 44 Spontaneous assembly by metal-free active template synthesis of compact rotaxane superbase 137.203 | ||
A range of compact rotaxane superbases was prepared by metal-free active template in 1–3 steps from commercially available starting materials. The compact rotaxane superbases had pKaH+ values as high as 32.2 for 137 in acetonitrile (13 pKaH+ units higher than the non-interlocked components, substantially larger than that of typical crown ether-amine rotaxanes), which are comparable to some phosphazene superbases. The highest basicities were generated with the smallest macrocycle (21-crown-7) and the most electron-rich dibenzylamine axles. The most efficacious compact rotaxane superbases showed minimal nucleophilicity and excellent hydrolytic stability in comparison to conventional commercial superbases.
Rotaxane formation, either by active or passive synthesis, can also be used to tune photophysical properties. An illustration is the Goldup and Zysman-Colman groups use of CuAAC active template synthesis to prepare carbazole-benzophenone-based rotaxanes that display thermally activated delayed fluorescence (TADF).204 A large increase in photoluminescence quantum yield results from threading one or two rings around the TADF emitting unit. The interlocked structure also increases photostability of the emitters and altered their emission wavelength, shifting it slightly to the red compared to the non-interlocked thread. These properties arise from the close proximity between the macrocycle and axle and resulting weak interactions that finely tune the HOMO energy level, the singlet–triplet energy gap, and the energy gap between the charge transfer states of the emitters.
A [2]rotaxane system capable of reversible electronic energy transfer (REET) between chromophores attached to the axle and macrocycle of a rotaxane has been reported by the McClenaghan and Goldup groups.205 Delayed luminescence and long luminescence lifetimes result from the close proximity of the chromophores.
Interlocked systems accessible through active template synthesis have also been investigated as single-ion magnets. The rigid coordination environment present in compact rotaxanes was used to design cobalt(II) ligands that form complexes that show magnetic anisotropy and field-induced single-ion magnet behaviour.206
The restriction of interlocked component (co-)conformational freedom in relatively compact rotaxanes accessible by active template synthesis can manifest itself in effects observable by EPR207 and/or NMR spectroscopy.208 The restricted dynamics influences EPR resolution207 and hydrogen bonding interactions208 can be directly probed through 1H–15N multiple quantum coherence NMR experiments.
Oligomers, polymers and other materials
Mechanically interlocked polymers are appealing materials due to the structural stability conferred by the mechanical bond and their potential for stimuli-responsive behaviour.209 Most poly[n]rotaxanes are prepared under thermodynamic control (i.e. conventional passive template synthesis) or by the polymerisation of pre-formed rotaxane monomers. However, Goldup and co-workers have shown that well-defined oligo[n]rotaxanes can be prepared through iterative CuAAC active template synthesis (Fig. 45).210 A series of monodispersed rotaxane oligomers of controlled length was prepared using small bipyridine macrocycles, such as 138, and functionalised monomer 139 in a series of sequential coupling and deprotection steps. Homo[n]rotaxanes such as 140 and 141, as well as hetero[n]rotaxanes incorporating different macrocycles threaded in sequence, were synthesised in high overall yield (90–92% per step). The reduced reactivity of sequential active template reactions around a branched central unit due to the steric bulk of incoming macrocycles was later harnessed for the stepwise synthesis of [3]- and [4]rotaxanes from a 1,3,5-triethynylbenzene core.41 | ||
| Fig. 45 Goldup's synthesis of homo[6]rotaxane 134 through iterative high-yielding active template CuAAC reactions.210 | ||
The Goldup and Tavassoli groups have prepared oligonucleotide-based rotaxanes by adapting click DNA ligation211,212 to active template CuAAC synthesis.213 Using a bipyridine macrocycle and oligonucleotide strands functionalised with an alkyne or azide group in the 3′ and 5′ positions in their chain termini, two different mechanically interlocked DNA strands could be prepared in good yield. The threaded macrocycle had a shielding effect on duplex formation, inhibiting DNA hybridisation and PCR amplification of the oligonucleotide rotaxanes compared to non-interlocked analogues.
Hybrid organic–inorganic interlocked structures offer an alternative class of materials. Szyszko and co-workers have used CuAAC active template synthesis for the assembly of a series of hybrid organic–inorganic rotaxanes incorporating cubic silesquioxanes as one of the stoppering elements.214 The hybrid rotaxanes demonstrated improved thermal stability in the solid state compared to wholly organic analogues.
Mullen and co-workers have reported the CuAAC active template synthesis of [2]rotaxanes on polymer resins.215 This generated higher yields of interlocked products than comparable passive template solid phase synthesis.
Molecular machinery
The well-defined large amplitude dynamics possible in rotaxane and catenane architectures, together with active template synthesis being under kinetic control, has led to its exploitation in both the synthesis and operation of artificial molecular machines. Leigh and co-workers have used CuAAC active template synthesis to assemble rotaxane machines (e.g.142) that perform sequence-selective synthesis in a manner reminiscent of the ribosome (Fig. 46).216–221 The rotaxane design features a catalyst-bearing macrocycle that moves along the threaded axle, removing the amino acid barriers that block its path, transferring them to the growing peptide chain in the sequence it encounters them along the track (Fig. 46). Active template synthesis was also used to make a rotaxane synthon that could be added to longer tracks, resulting in higher overall yields than the original strategy.217 The concept was later extended to connect other types of building blocks in sequence to form oligomers and polymers, including nonproteinogenic β3-amino acids,218 leucine-ester-derivatised polystyrene chains,219 and oligomers joined through iterative carbon–carbon bond formation.220 Parallel operation of two molecular peptide synthesisers in the same pot was used to assemble a decapeptide.221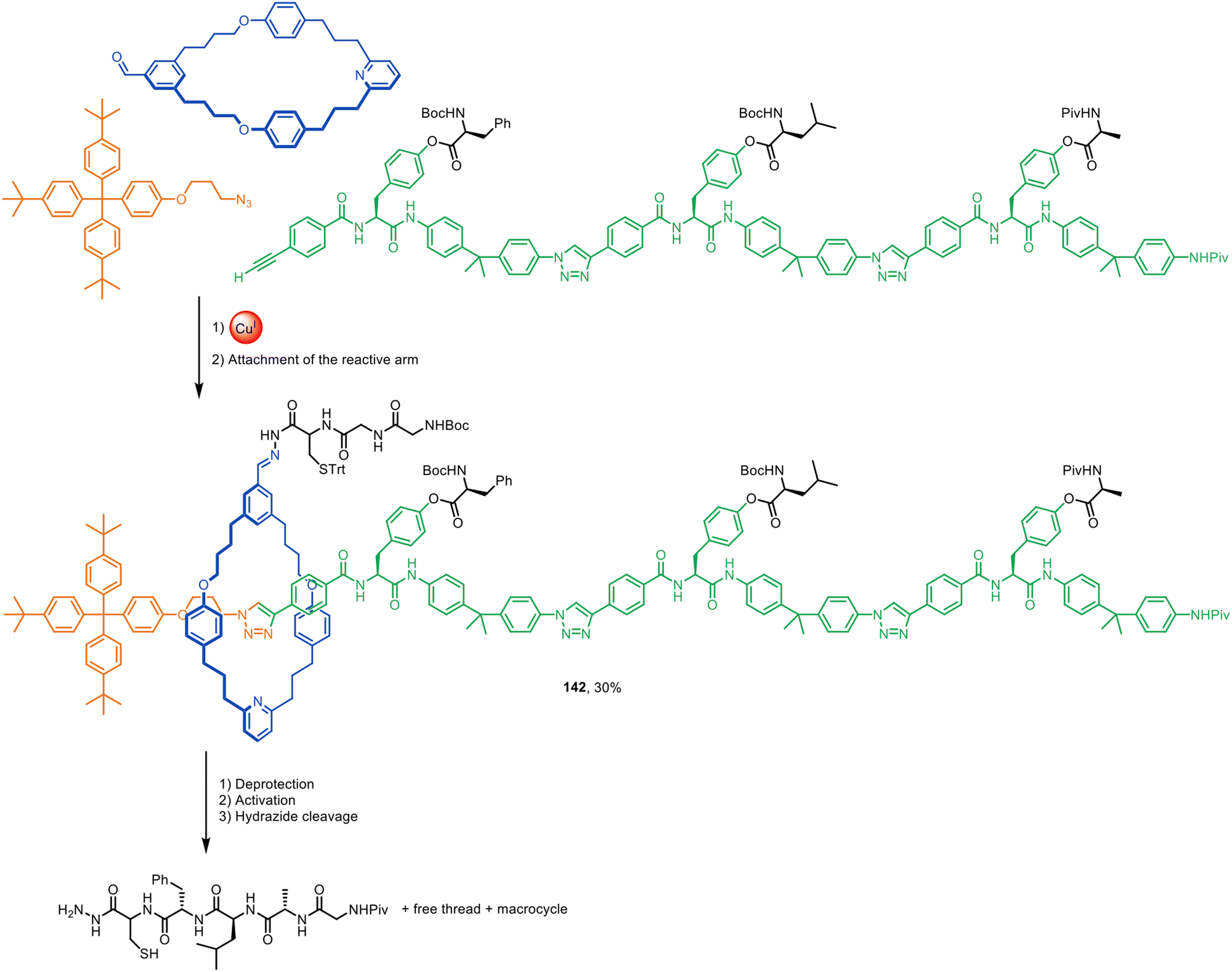 | ||
| Fig. 46 A rotaxane-based peptide synthesiser 135, assembled through CuAAC active template synthesis.216 | ||
Catalysis-driven metal-free active template synthesis has been used to demonstrate the mechanism of chemically fuelled molecular pumps. Molecular pump 143 contains an activated ester electrophile that causes 24-crown-8 (94) to thread through its accelerated reaction with benzylamine 105, forming amide rotaxane 144 (Fig. 47).222 Subsequent addition of a bulky N-Boc group forms 145, with the macrocycle shuttling to the oligo(ethylene glycol) collection thread. Substitution of the amide to form phenolic ester 146, followed by another crown ether threading active template reaction, generates [3]rotaxane 147.
 | ||
| Fig. 47 (a) A stepwise-operated molecular pump that makes use of successive metal-free active template transamidation reactions.222 Macrocycles are pumped onto the axles by a sequence of: (1) an active template transamidation reaction, (2) Boc-addition, and (3) substitution by an activated ester. (b) [5]Rotaxane 148 formed after two pumping cycles from a thread with pump motifs at either terminus. X-Ray crystal structure of 148. (c) [4]Rotaxane 149, in which the macrocycles have been threaded onto the axle in a controlled sequence. | ||
Every pumping cycle can add an additional macrocycle to the thread for each activated ester present, and so [5]rotaxane 148 is obtained after only two transamidation pumping cycles from a thread with pumping modules at both ends. In this high energy, kinetically locked, structure an array of weak CH–O hydrogen bonds between the components is apparent from X-ray crystallography (Fig. 47b). By carrying out pumping cycles with different crown ether derivatives, it was also possible to synthesise rotaxanes with specific sequences of threaded macrocycles, such as 149 (Fig. 47c).
Metal-free active template synthesis also enables the design of molecular pumps that operate autonomously, in which macrocycles are continuously pumped onto an axle, away from a state of equilibrium, powered by a chemical fuel.223 Catalysis-driven molecular pump 150 features a free benzylic amine at one end and a bulky stopper at the other (Fig. 48). The poor binding affinity of crown ether macrocycles for triazoles means that a vanishingly small proportion of macrocycles are threaded on the axle at equilibrium. However, an Fmoc-derived chemical fuel, 151, acts as an electrophile for a crown ether metal-free active template reaction, forming [2]rotaxane car-152 (the italicised prefix refers to the position of the ring on the axle). The Fmoc-carbamate group prevents macrocycle dethreading and the restricted steric environment around the carbamate site results in the macrocycle being displaced to the catchment region of the thread (tri-152). The presence of i-Pr2NH base in the reaction mixture triggers decomposition of the Fmoc-carbamate group faster when the bulky macrocycle is distant from the carbamate group. The trifluoromethyl group (shown in red in Fig. 48) is sufficiently large (a molecular ‘speed bump’) that it slows dethreading of the macrocycle from the catchment region off the open amine-terminated end of the thread.224 Therefore pumping of another crown ether onto the axle occurs faster than dethreading of the ring already on the axle, leading to the formation of [4]rotaxane 153. Macrocycles are thus continuously pumped onto the axle until all of the Fmoc-fuel is depleted, after which the rings dethread to regenerate the original ring equilibrium distribution in solution.
 | ||
| Fig. 48 An autonomous catalysis-driven artificial molecular pump 150 which operates continuously in the presence of chemical fuel 151.223 | ||
Unusual reactivity and mechanistic insights
As noted earlier, active template synthesis can provide mechanistic information about the catalytic manifolds employed, for example when more than one ring is unexpectedly threaded onto an axle or multiple strands are encircled by a single ring.28,36,39,40,48,61,102,103 Sometimes the mechanical bond can lead to other unexpected reaction outcomes. Chaplin et al. reported the selective formation of E-enyne products in their Rh(I)-mediated active template formation of pseudorotaxanes (Fig. 21)117 and have observed unusual C–C σ-bond oxidative additions118 (see section on zinc-, cobalt-, ruthenium- and rhodium-mediated active template synthesis).Goldup and co-workers found an unusual tandem process occurred during the active template CuAAC synthesis of [2]rotaxanes involving propargylic alcohol 154 as the alkyne substrate. When small bipyridine macrocycle 155 was used, a post-assembly rearrangement of the stabilised Cu(I)-triazolide interlocked derivative was triggered rendering the unexpected acrylamide rotaxane 156 (Fig. 49).225 Mechanistic studies suggest that mechanical bond stabilisation of the triazolide intermediate and the steric constraints imposed during rotaxane formation alters the chemoselectivity of the metal-catalysed reaction.
 | ||
| Fig. 49 Proposed mechanism of the tandem active template CuAAC rearrangement reported by Goldup and co-workers.225 | ||
Conclusions
The active template synthesis of entwined and interlocked structures is an efficacious and versatile strategy that complements conventional passive template synthesis. In addition to often requiring only sub-stoichiometric quantities of metal salts (or no reagents whatsoever in the case of metal-free active template synthesis), other key advantages include that its use can be traceless, without the need for binding sites in both components to be threaded or interlocked. Furthermore, the assembly process is carried out under kinetic control, enabling the synthesis of interlocked products that are not necessarily thermodynamically stable. Accordingly, threaded structures can be generated with functional groups held in close proximity to each other in (co)-conformations that might not be accessible through other synthetic strategies. Active template synthesis often proceeds in high yields, with simple experimental procedures and readily available starting materials. Rotaxanes formed through active template chemistries are currently being investigated for potential applications in fields as diverse as sensing, catalysis, drug delivery, materials and molecular machinery.Much of the early work in this field focussed on expanding the toolbox of catalytic reactions suitable for active template synthesis. A particularly attractive target remains finding a way to maintain the coordination of Pd(0) to a component throughout the catalytic cycle so that Pd(0)-mediated cross-coupling reactions can be used. Other challenges arise from functional group tolerance, and the ligand requirements of particular metal-catalysed reactions for active template synthesis if the preparation or handling of the building blocks is problematic.
Finally, while substantial progress has been made in the synthesis of interlocked molecules over the last four decades, practical applications of rotaxanes and catenanes remain scarce. The lack of requirements for permanent binding motifs may make active template synthesis more amenable for making practically useful interlocked structures than other synthetic strategies. Active template synthesis appears a promising route to rotaxanes and catenanes with applications in nano-electronics and biotechnology. Further exploration of different metal-mediated and metal-free active template methodologies, coupled with potential applications of newly accessible structure types, augurs well for the continued advancement18 of the chemistry of mechanically interlocked molecules.
Author contributions
This Review was written through the contributions of all authors.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
We thank the EPSRC Centre for Doctoral Training in Integrated Catalysis (EP/S023755/1, studentship to R. J.), the European Union Marie Skłodowska-Curie Actions Innovation Training Network ArtMoMa 860434 (studentship to M. P.), the Engineering and Physical Sciences Research Council (EPSRC; EP/P027067/1) and the European Research Council (ERC Advanced grant 786630) for funding.Notes and references
- H. Frisch, I. Martin and H. Mark, Monatsh. Chem., 1953, 84, 250–256 CrossRef CAS.
- H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS.
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS.
- A. S. Baluna, A. Galan, D. A. Leigh, G. D. Smith, J. T. J. Spence, D. J. Tetlow, I. J. Vitorica-Yrezabal and M. Zhang, J. Am. Chem. Soc., 2023, 145, 9825–9833 CrossRef CAS PubMed.
- I. T. Harrison and S. Harrison, J. Am. Chem. Soc., 1967, 89, 5723–5724 CrossRef CAS.
- G. Schill and A. Lüttringhaus, Angew. Chem., Int. Ed. Engl., 1964, 3, 546–547 CrossRef.
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J.-M. Kern, J. Am. Chem. Soc., 1984, 106, 3043–3045 CrossRef CAS.
- D. A. Leigh, P. J. Lusby, S. J. Teat, A. J. Wilson and J. K. Y. Wong, Angew. Chem., Int. Ed., 2001, 40, 1538–1543 CrossRef CAS PubMed.
- J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- E. R. Kay and D. A. Leigh, Top. Curr. Chem., 2005, 262, 133–177 CrossRef CAS.
- M. Fujita, Acc. Chem. Res., 1999, 32, 53–61 CrossRef CAS.
- F. B. L. Cougnon, K. Caprice, M. Pupier, A. Bauzá and A. Frontera, J. Am. Chem. Soc., 2018, 140, 12442–12450 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Hoboken, NJ, 2017 Search PubMed.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS.
- D. A. Leigh, F. Zerbetto and E. R. Kay, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef PubMed.
- V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed.
- J. S. Hannam, S. M. Lacy, D. A. Leigh, C. G. Saiz, A. M. Z. Slawin and S. G. Stitchell, Angew. Chem., Int. Ed., 2004, 43, 3260–3264 CrossRef CAS PubMed.
- S. Chao, C. Romuald, K. Fournel-Marotte, C. Clavel and F. Coutrot, Angew. Chem., Int. Ed., 2014, 53, 6914–6919 CrossRef CAS PubMed.
- A. de Juan, D. Lozano, A. W. Heard, M. A. Jinks, J. M. Suarez, G. J. Tizzard and S. M. Goldup, Nat. Chem., 2022, 14, 179–187 CrossRef CAS PubMed.
- A. Saura-Sanmartin, Chem. – Eur. J., 2024, 30, e202304025 CrossRef CAS PubMed.
- J. D. Crowley, K. D. Hänni, A. L. Lee and D. A. Leigh, J. Am. Chem. Soc., 2007, 129, 12092–12093 CrossRef CAS PubMed.
- V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed.
- H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS PubMed.
- A. Noor, W. K. C. Lo, S. C. Moratti and J. D. Crowley, Chem. Commun., 2014, 50, 7044–7047 RSC.
- S. Dasgupta and J. Wu, Chem. Sci., 2012, 3, 425–432 RSC.
- V. D. Bock, H. Hiemstra and J. H. Van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS.
- S. I. Presolski, V. Hong, S. H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, 8593–8599 Search PubMed.
- C. Iacobucci, A. Lebon, F. De Angelis and A. Memboeuf, Chem. – Eur. J., 2016, 22, 18690–18694 CrossRef CAS PubMed.
- E. A. Neal and S. M. Goldup, Chem. Sci., 2015, 6, 2398–2404 RSC.
- J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318–13321 CrossRef CAS PubMed.
- C. C. Anghel, T. A. Cucuiet, N. D. Hădade and I. Grosu, Beilstein J. Org. Chem., 2023, 19, 1776–1784 CrossRef CAS PubMed.
- E. A. Neal and S. M. Goldup, Angew. Chem., Int. Ed., 2016, 55, 12488–12493 CrossRef CAS PubMed.
- S. M. Goldup, D. A. Leigh, P. R. McGonigal, V. E. Ronaldson and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 315–320 CrossRef CAS PubMed.
- J. E. M. Lewis, J. Winn and S. M. Goldup, Molecules, 2017, 22, 1–12 CrossRef PubMed.
- S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symes and J. Wu, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS PubMed.
- J. E. M. Lewis, F. Modicom and S. M. Goldup, J. Am. Chem. Soc., 2018, 140, 4787–4791 CrossRef CAS PubMed.
- R. Takashima, D. Aoki, A. Takahashi and H. Otsuka, Org. Biomol. Chem., 2023, 22, 927–931 RSC.
- P. E. Barran, H. L. Cole, S. M. Goldup, D. A. Leigh, P. R. McGonigal, M. D. Symes, J. Wu and M. Zengerle, Angew. Chem., Int. Ed., 2011, 50, 12280–12284 CrossRef CAS PubMed.
- S. Saito, E. Takahashi and K. Nakazono, Org. Lett., 2006, 8, 5133–5136 CrossRef CAS PubMed.
- A. Tron, P. J. Thornton, B. Kauffmann, J. H. R. Tucker and N. D. McClenaghan, Supramol. Chem., 2016, 28, 733–741 CrossRef CAS.
- Y. Yamashita, Y. Mutoh, R. Yamasaki, T. Kasama and S. Saito, Chem. – Eur. J., 2015, 21, 2139–2145 CrossRef CAS PubMed.
- R. Hayashi, K. Wakatsuki, R. Yamasaki, Y. Mutoh, T. Kasama and S. Saito, Chem. Commun., 2014, 50, 204–206 RSC.
- Y. Matsuoka, Y. Mutoh, I. Azumaya, S. Kikkawa, T. Kasama and S. Saito, J. Org. Chem., 2016, 81, 3479–3487 CrossRef CAS PubMed.
- L. D. Movsisyan, D. V. Kondratuk, M. Franz, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, Org. Lett., 2012, 14, 3424–3426 CrossRef CAS PubMed.
- N. Weisbach, Z. Baranov, S. Gauthier, J. H. Reibenspies and J. A. Gladysz, Chem. Commun., 2012, 48, 7562–7564 RSC.
- Y. Mochizuki, K. Ikeyatsu, Y. Mutoh, S. Hosoya and S. Saito, Org. Lett., 2017, 19, 4347–4350 CrossRef CAS PubMed.
- Y. Yamashita, Y. Saito, S. Kikkawa, Y. Mutoh, S. Hosoya, I. Azumaya and S. Saito, Eur. J. Org. Chem., 2019, 3412–3420 CrossRef CAS.
- Y. Kawasaki, S. Rashid, K. Ikeyatsu, Y. Mutoh, Y. Yoshigoe, S. Kikkawa, I. Azumaya, S. Hosoya and S. Saito, J. Org. Chem., 2022, 87, 5744–5759 CrossRef CAS PubMed.
- S. Rashid, T. Murakami, H. Koganezawa, Y. Yoshigoe, S. Hosoya and S. Saito, New J. Chem., 2022, 47, 900–909 RSC.
- S. Saito, Y. Hirano, Y. Mutoh and T. Kasama, Chem. Lett., 2015, 44, 1509–1511 CrossRef CAS.
- H. Sahnoune, Z. Baranová, N. Bhuvanesh, J. A. Gladysz and J. F. Halet, Organometallics, 2013, 32, 6360–6367 CrossRef CAS.
- Z. Baranová, H. Amini, N. Bhuvanesh and J. A. Gladysz, Organometallics, 2014, 33, 6746–6749 CrossRef.
- M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC.
- L. D. Movsisyan, M. Franz, F. Hampel, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, J. Am. Chem. Soc., 2016, 138, 1366–1376 CrossRef CAS PubMed.
- M. Franz, J. A. Januszewski, D. Wendinger, C. Neiss, L. D. Movsisyan, F. Hampel, H. L. Anderson, A. Görling and R. R. Tykwinski, Angew. Chem., Int. Ed., 2015, 54, 6645–6649 CrossRef CAS PubMed.
- D. C. Milan, M. Krempe, A. K. Ismael, L. D. Movsisyan, M. Franz, I. Grace, R. J. Brooke, W. Schwarzacher, S. J. Higgins, H. L. Anderson, C. J. Lambert, R. R. Tykwinski and R. J. Nichols, Nanoscale, 2017, 9, 355–361 RSC.
- L. D. Movsisyan, M. D. Peeks, G. M. Greetham, M. Towrie, A. L. Thompson, A. W. Parker and H. L. Anderson, J. Am. Chem. Soc., 2014, 136, 17996–18008 CrossRef CAS PubMed.
- Y. Sato, R. Yamasaki and S. Saito, Angew. Chem., Int. Ed., 2009, 48, 504–507 CrossRef CAS PubMed.
- R. Hayashi, Y. Mutoh, T. Kasama and S. Saito, J. Org. Chem., 2015, 80, 7536–7546 CrossRef CAS PubMed.
- J. Berná, S. M. Goldup, A. L. Lee, D. A. Leigh, M. D. Symes, G. Teobaldi and F. Zerbetto, Angew. Chem., Int. Ed., 2008, 47, 4392–4396 CrossRef PubMed.
- D. R. Kohn, L. D. Movsisyan, A. L. Thompson and H. L. Anderson, Org. Lett., 2017, 19, 348–351 CrossRef CAS PubMed.
- J. M. Van Raden, N. N. Jarenwattananon, L. N. Zakharov and R. Jasti, Chem. – Eur. J., 2020, 26, 10205–10209 CrossRef CAS PubMed.
- J. M. Van Raden, B. M. White, L. N. Zakharov and R. Jasti, Angew. Chem., Int. Ed., 2019, 58, 7341–7345 CrossRef CAS PubMed.
- M. Franz, J. A. Januszewski, F. Hampel and R. R. Tykwinski, Eur. J. Org. Chem., 2019, 3503–3512 CrossRef CAS.
- K. Ugajin, E. Takahashi, R. Yamasaki, Y. Mutoh, T. Kasama and S. Saito, Org. Lett., 2013, 15, 2684–2687 CrossRef CAS PubMed.
- S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, J. Am. Chem. Soc., 2015, 137, 7656–7659 CrossRef CAS PubMed.
- H. Li, B. J. Li and Z. J. Shi, Catal. Sci. Technol., 2011, 1, 191–206 RSC.
- T. Iwasaki and N. Kambe, Top. Curr. Chem., 2016, 374, 66 CrossRef PubMed.
- S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304–8329 CrossRef CAS PubMed.
- Ruthenium in catalysis, Top. Organomet. Chem., ed. C. Bruneau and P. H. Dixneuf, Springer, 2014, vol. 48 Search PubMed.
- L. Hogg, D. A. Leigh, P. J. Lusby, A. Morelli, S. Parsons and J. K. Y. Wong, Angew. Chem., Int. Ed., 2004, 43, 1218–1221 CrossRef CAS PubMed.
- A. M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914–3918 CrossRef CAS PubMed.
- Y. Furusho, T. Matsuyama, T. Takata, T. Moriuchi and T. Hirao, Tetrahedron Lett., 2004, 45, 9593–9597 CrossRef CAS.
- D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Angew. Chem., Int. Ed., 2005, 44, 4557–4564 CrossRef CAS PubMed.
- A.-M. L. Fuller, D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, J. Am. Chem. Soc., 2005, 127, 12612–12619 CrossRef CAS PubMed.
- D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Chem. Commun., 2005, 4919–4921 RSC.
- J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, A. L. Lee and D. A. Leigh, Angew. Chem., Int. Ed., 2007, 46, 5709–5713 CrossRef PubMed.
- X. Du, M. Suguro, K. Hirabayashi, A. Mori, T. Nishikata, N. Hagiwara, K. Kawata, T. Okeda, H. F. Wang, K. Fugami and M. Kosugi, Org. Lett., 2001, 3, 3313–3316 CrossRef CAS PubMed.
- S. M. Goldup, D. A. Leigh, P. J. Lusby, R. T. McBurney and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 3381–3384 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, N. D. Gowans, D. A. Leigh, V. E. Ronaldson and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 6243–6248 CrossRef CAS PubMed.
- D. J. Cárdenas, Angew. Chem., Int. Ed., 2003, 42, 384–387 CrossRef PubMed.
- D. J. Cárdenas, Angew. Chem., Int. Ed., 1999, 38, 3018–3020 Search PubMed.
- F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 8347–8349 CrossRef CAS PubMed.
- M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525–1532 CrossRef CAS.
- M. D. Tzirakis, M. N. Alberti, H. Weissman, B. Rybtchinski and F. Diederich, Chem. – Eur. J., 2014, 20, 16070–16073 CrossRef CAS PubMed.
- J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed.
- J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726–14727 CrossRef CAS PubMed.
- C. Fischer and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 4594–4595 CrossRef CAS PubMed.
- F. O. Arp and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 10482–10483 CrossRef CAS PubMed.
- B. Saito and G. C. Fu, Chemtracts, 2008, 21, 308–309 Search PubMed.
- S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 12645–12647 CrossRef CAS PubMed.
- S. M. Goldup, D. A. Leigh, R. T. McBurney, P. R. McGonigal and A. Plant, Chem. Sci., 2010, 1, 383–386 RSC.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS.
- M. Ohta, A. Okuda, S. Hosoya, Y. Yoshigoe and S. Saito, Chem. – Eur. J., 2024, 30, e202304309 CrossRef CAS PubMed.
- H. M. Cheng, D. A. Leigh, F. Maffei, P. R. McGonigal, A. M. Z. Slawin and J. Wu, J. Am. Chem. Soc., 2011, 133, 12298–12303 CrossRef CAS PubMed.
- J. J. Danon, D. A. Leigh, P. R. McGonigal, J. W. Ward and J. Wu, J. Am. Chem. Soc., 2016, 138, 12643–12647 CrossRef CAS PubMed.
- J. Echavarren, M. A. Y. Gall, A. Haertsch, D. A. Leigh, V. Marcos and D. J. Tetlow, Chem. Sci., 2019, 10, 7269–7273 RSC.
- J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C. M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS PubMed.
- T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- H. Ishibashi, M. Rondelli, H. Shudo, T. Maekawa, H. Ito, K. Mizukami, N. Kimizuka, A. Yagi and K. Itami, Angew. Chem., Int. Ed., 2023, 62, e202310613 CrossRef CAS PubMed.
- Y. Segawa, M. Kuwayama, Y. Hijikata, M. Fushimi, T. Nishihara, J. Pirillo, J. Shirasaki, N. Kubota and K. Itami, Science, 2019, 365, 272–276 CrossRef CAS PubMed.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- J. D. Crowley, K. D. Hänni, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 5309–5314 CrossRef CAS PubMed.
- L. Huang, Y. Chen, G. Y. Gao and X. P. Zhang, J. Org. Chem., 2003, 68, 8179–8184 CrossRef CAS PubMed.
- W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. De Bruin, J. Am. Chem. Soc., 2010, 132, 10891–10902 CrossRef CAS PubMed.
- C. te Grotenhuis, N. van den Heuvel, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2018, 57, 140–145 CrossRef CAS PubMed.
- A. F. P. Alcântara, L. A. Fontana, V. H. Rigolin, Y. F. S. Andrade, M. A. Ribeiro, W. P. Barros, C. Ornelas and J. D. Megiatto, Angew. Chem., Int. Ed., 2018, 57, 8979–8983 CrossRef PubMed.
- A. F. P. Alcântara, L. A. Fontana, M. P. Almeida, V. H. Rigolin, M. A. Ribeiro, W. P. Barros and J. D. Megiatto, Chem. – Eur. J., 2020, 26, 7808–7822 CrossRef PubMed.
- L. A. Fontana, M. P. Almeida, A. F. P. Alcântara, V. H. Rigolin, M. A. Ribeiro, W. P. Barros and J. D. Megiatto, Nat. Commun., 2020, 11, 6370 CrossRef CAS PubMed.
- C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–12006 CrossRef CAS PubMed.
- C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Chem. – Eur. J., 2020, 26, 14715–14723 CrossRef CAS PubMed.
- B. Leforestier, M. R. Gyton and A. B. Chaplin, Angew. Chem., Int. Ed., 2020, 59, 23500–23504 CrossRef CAS PubMed.
- T. M. Hood, S. Lau and A. B. Chaplin, RSC Adv., 2024, 14, 7740–7744 RSC.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, J. Org. Chem., 1989, 54, 5302–5308 CrossRef CAS.
- D. Tuncel and J. H. G. Steinke, Chem. Commun., 1999, 1509–1510 RSC.
- X. Hou, C. Ke and J. F. Stoddart, Chem. Soc. Rev., 2016, 45, 3766–3780 RSC.
- G. Orlandini, G. Ragazzon, V. Zanichelli, A. Secchi, S. Silvi, M. Venturi, A. Arduini and A. Credi, Chem. Commun., 2017, 53, 6172–6174 RSC.
- V. Zanichelli, G. Ragazzon, G. Orlandini, M. Venturi, A. Credi, S. Silvi, A. Arduini and A. Secchi, Org. Biomol. Chem., 2017, 15, 6753–6763 RSC.
- M. Bazzoni, L. Andreoni, S. Silvi, A. Credi, G. Cera, A. Secchi and A. Arduini, Chem. Sci., 2021, 12, 6419–6428 RSC.
- G. De Bo, G. Dolphijn, C. T. McTernan and D. A. Leigh, J. Am. Chem. Soc., 2017, 139, 8455–8457 CrossRef CAS PubMed.
- S. D. P. Fielden, D. A. Leigh, C. T. McTernan, B. Pérez-Saavedra and I. J. Vitorica-Yrezabal, J. Am. Chem. Soc., 2018, 140, 6049–6052 CrossRef CAS PubMed.
- S. D. P. Fielden, ChemSystemsChem, 2024, 6, e202300048 CrossRef CAS.
- N. Basilio, L. García-Río, J. C. Mejuto and M. Pérez-Lorenzo, J. Org. Chem., 2006, 71, 4280–4285 CrossRef CAS PubMed.
- J. C. Hogan and R. D. Gandour, J. Org. Chem., 1992, 57, 55–61 CrossRef CAS.
- C. Tian, S. D. P. Fielden, G. F. S. Whitehead, I. J. Vitorica-Yrezabal and D. A. Leigh, Nat. Commun., 2020, 11, 744 CrossRef CAS PubMed.
- C. Tian, S. D. P. Fielden, B. Pérez-Saavedra, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2020, 142, 9803–9808 CrossRef CAS PubMed.
- T. Kurita, M. Higashi, J. Gimenez-Dejoz, S. Fujita, H. Uji, H. Sato and K. Numata, Biomacromolecules, 2024, 25, 3661–3670 CrossRef CAS PubMed.
- V. Aucagne, D. A. Leigh, J. S. Lock and A. R. Thomson, J. Am. Chem. Soc., 2006, 128, 1784–1785 CrossRef CAS PubMed.
- X. Da and W.-B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 11097–11104 CrossRef CAS PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, e690–e697 CrossRef CAS PubMed.
- F. Zhang, Y. Liu, Y. Shao and W.-B. Zhang, CCS Chem., 2024, 6, 377–389 CrossRef CAS.
- F. Zhang, Y. Liu, X.-D. Da and W.-B. Zhang, CCS Chem., 2024, 6, 1047–1059 CrossRef CAS.
- S. Datta, Y. Kato, S. Higashiharaguchi, K. Aratsu, A. Isobe, T. Saito, D. D. Prabhu, Y. Kitamoto, M. J. Hollamby, A. J. Smith, R. Dagleish, N. Mahmoudi, L. Pesce, C. Perego, G. M. Pavan and S. Yagai, Nature, 2020, 583, 400–405 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, A. L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS.
- B. Hesseler, M. Zindler, R. Herges and U. Lüning, Eur. J. Org. Chem., 2014, 3885–3901 CrossRef CAS.
- J. P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed.
- J. E. M. Lewis, R. J. Bordoli, M. Denis, C. J. Fletcher, M. Galli, E. A. Neal, E. M. Rochette and S. M. Goldup, Chem. Sci., 2016, 7, 3154–3161 RSC.
- M. Wolf, A. Ogawa, M. Bechtold, M. Vonesch, J. A. Wytko, K. Oohora, S. Campidelli, T. Hayashi, D. M. Guldi and J. Weiss, Chem. Sci., 2019, 10, 3846–3853 RSC.
- J. T. Wilmore, Y. Cheong Tse, A. Docker, C. Whitehead, C. K. Williams and P. D. Beer, Chem. – Eur. J., 2023, 29, e202300608 CrossRef CAS PubMed.
- R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed.
- M. A. Jinks, A. de Juan, M. Denis, C. J. Fletcher, M. Galli, E. M. G. Jamieson, F. Modicom, Z. Zhang and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 14806–14810 CrossRef CAS PubMed.
- M. A. J. Koenis, C. S. Chibueze, M. A. Jinks, V. P. Nicu, L. Visscher, S. M. Goldup and W. J. Buma, Chem. Sci., 2020, 11, 8469–8475 RSC.
- M. Denis, J. E. M. Lewis, F. Modicom and S. M. Goldup, Chem, 2019, 5, 1512–1520 CAS.
- S. Zhang, A. Rodríguez-Rubio, A. Saady, G. J. Tizzard and S. M. Goldup, Chem, 2023, 9, 1195–1207 CAS.
- N. Pairault, A. Bessaguet, R. Barat, L. Frédéric, G. Pieters, J. Crassous, I. Opalinski and S. Papot, Chem. Sci., 2021, 12, 2521–2526 RSC.
- A. R. Puente, A. Bessaguet, N. Pairault, G. Pieters, J. Crassous, P. L. Polavarapu, I. Opalinski and S. Papot, Chirality, 2021, 33, 773–782 CrossRef CAS PubMed.
- A. Rodríguez-Rubio, A. Savoini, F. Modicom, P. Butler and S. M. Goldup, J. Am. Chem. Soc., 2022, 144, 11927–11932 CrossRef PubMed.
- N. Pairault, F. Rizzi, D. Lozano, E. M. G. Jamieson, G. J. Tizzard and S. M. Goldup, Nat. Chem., 2023, 15, 781–786 CrossRef CAS PubMed.
- J. R. J. Maynard, P. Gallagher, D. Lozano, P. Butler and S. M. Goldup, Nat. Chem., 2022, 14, 1038–1044 CrossRef CAS PubMed.
- A. Savoini, P. R. Gallagher, A. Saady and S. M. Goldup, J. Am. Chem. Soc., 2024, 146, 8472–8479 CrossRef CAS PubMed.
- P. R. Gallagher, A. Savoini, A. Saady, J. R. J. Maynard, P. W. V. Butler, G. J. Tizzard and S. M. Goldup, J. Am. Chem. Soc., 2024, 146, 9134–9141 CrossRef CAS PubMed.
- S. M. Goldup, Acc. Chem. Res., 2024, 57, 1696–1708 CrossRef CAS PubMed.
- C. Tian, S. D. P. Fielden, B. Pérez-Saavedra, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2020, 142, 9803–9808 CrossRef CAS PubMed.
- M. Li, X. L. Chia, C. Tian and Y. Zhu, Chem, 2022, 8, 2843–2855 CAS.
- M. Galli, J. E. M. Lewis and S. M. Goldup, Angew. Chem., Int. Ed., 2015, 54, 13545–13549 CrossRef CAS PubMed.
- K. Miki, K. Ohe and S. Uemura, J. Org. Chem., 2003, 68, 8505–8513 CrossRef CAS PubMed.
- M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 18002–18003 CrossRef CAS PubMed.
- A. W. Heard and S. M. Goldup, Chem, 2020, 6, 994–1006 CAS.
- J. R. J. Maynard, B. Galmés, A. D. Stergiou, M. D. Symes, A. Frontera and S. M. Goldup, Angew. Chem., Int. Ed., 2022, 61, e202115961 CrossRef CAS PubMed.
- S. Kundu, D. Mondal, E. Elramadi, I. Valiyev and M. Schmittel, Org. Lett., 2022, 24, 6609–6613 CrossRef CAS PubMed.
- S. Kundu, D. Mondal, V. V. Rajasekaran, A. Goswami and M. Schmittel, Inorg. Chem., 2022, 61, 17007–17011 CrossRef CAS PubMed.
- M. J. Langton, Y. Xiong and P. D. Beer, Chem. – Eur. J., 2015, 21, 18910–18914 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893–11895 CrossRef CAS PubMed.
- J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021 CrossRef CAS PubMed.
- J. Y. C. Lim, T. Bunchuay and P. D. Beer, Chem. – Eur. J., 2017, 23, 4700–4707 CrossRef CAS PubMed.
- X. Li, J. Y. C. Lim and P. D. Beer, Faraday Discuss., 2017, 203, 245–255 RSC.
- V. K. Munasinghe, J. Pancholi, D. Manawadu, Z. Zhang and P. D. Beer, Chem. – Eur. J., 2022, 28, e202201209 CrossRef CAS PubMed.
- H. Min Tay, A. Docker, Y. Cheong Tse and P. D. Beer, Chem. – Eur. J., 2023, 29, e202301316 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Eur. J. Org. Chem., 2019, 3433–3441 CrossRef CAS.
- Y. Cheong Tse, R. Hein, E. J. Mitchell, Z. Zhang and P. D. Beer, Chem. – Eur. J., 2021, 27, 14550–14559 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228–12239 CrossRef CAS PubMed.
- X. Li, J. Y. C. Lim and P. D. Beer, Chem. – Eur. J., 2018, 24, 17788–17795 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, A. L. Thompson, K. E. Christensen, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 3122–3133 CrossRef CAS PubMed.
- M. Douarre, V. Martí-Centelles, C. Rossy, A. Tron, I. Pianet and N. D. McClenaghan, Supramol. Chem., 2020, 32, 546–556 CrossRef CAS.
- A. Brown, T. Lang, K. M. Mullen and P. D. Beer, Org. Biomol. Chem., 2017, 15, 4587–4594 RSC.
- M. Nandi, S. Bej, S. Bhunia and P. Ghosh, ChemElectroChem, 2020, 7, 1038–1047 CrossRef CAS.
- C. E. Otteson, C. M. Levinn, J. M. Van Raden, M. D. Pluth and R. Jasti, Org. Lett., 2021, 23, 4608–4612 CrossRef CAS PubMed.
- M. Denis, L. Qin, P. Turner, K. A. Jolliffe and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5315–5319 CrossRef CAS PubMed.
- M. Denis, J. Pancholi, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5310–5314 CrossRef CAS PubMed.
- M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879–889 CrossRef CAS PubMed.
- Z. Zhang, G. J. Tizzard, J. A. G. Williams and S. M. Goldup, Chem. Sci., 2020, 11, 1839–1847 RSC.
- R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- T. Kench, P. A. Summers, M. K. Kuimova, J. E. M. Lewis and R. Vilar, Angew. Chem., Int. Ed., 2021, 60, 10928–10934 CrossRef CAS PubMed.
- A. Saady, G. K. Malcolm, M. P. Fitzpatrick, N. Pairault, G. J. Tizzard, S. Mohammed, A. Tavassoli and S. M. Goldup, Angew. Chem., Int. Ed., 2024, 63, e202400344 CrossRef CAS PubMed.
- J. H. May, J. M. Van Raden, R. L. Maust, L. N. Zakharov and R. Jasti, Nat. Chem., 2023, 15, 170–176 CrossRef CAS PubMed.
- J. H. May, J. M. Fehr, J. C. Lorenz, L. N. Zakharov and R. Jasti, Angew. Chem., Int. Ed., 2024, e202401823 CAS.
- S. L. Woltering, P. Gawel, K. E. Christensen, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2020, 142, 13523–13532 CrossRef CAS PubMed.
- P. Gawel, S. L. Woltering, Y. Xiong, K. E. Christensen and H. L. Anderson, Angew. Chem., Int. Ed., 2021, 60, 5941–5947 CrossRef CAS PubMed.
- C. W. Patrick, J. F. Woods, P. Gawel, C. E. Otteson, A. L. Thompson, T. D. W. Claridge, R. Jasti and H. L. Anderson, Angew. Chem., Int. Ed., 2022, 61, e202116897 CrossRef CAS PubMed.
- C. W. Patrick, Y. Gao, P. Gupta, A. L. Thompson, A. W. Parker and H. L. Anderson, Nat. Chem., 2024, 16, 193–200 CrossRef CAS PubMed.
- T. H. Ngo, J. Labuta, G. N. Lim, W. A. Webre, F. D’Souza, P. A. Karr, J. E. M. Lewis, J. P. Hill, K. Ariga and S. M. Goldup, Chem. Sci., 2017, 8, 6679–6685 RSC.
- H. T. Ngo, J. E. M. Lewis, D. T. Payne, F. D’Souza, J. P. Hill, K. Ariga, G. Yoshikawa and S. M. Goldup, Chem. Commun., 2020, 56, 7447–7450 RSC.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- M. J. Power, D. T. J. Morris, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2023, 145, 8593–8599 CrossRef CAS PubMed.
- P. Rajamalli, F. Rizzi, W. Li, M. A. Jinks, A. K. Gupta, B. A. Laidlaw, I. D. W. Samuel, T. J. Penfold, S. M. Goldup and E. Zysman-Colman, Angew. Chem., Int. Ed., 2021, 60, 12066–12073 CrossRef CAS PubMed.
- S. Yu, A. Kupryakov, J. E. M. Lewis, V. Martí-Centelles, S. M. Goldup, J. L. Pozzo, G. Jonusauskas and N. D. McClenaghan, Chem. Sci., 2021, 12, 9196–9200 RSC.
- M. Cirulli, E. Salvadori, Z. H. Zhang, M. Dommett, F. Tuna, H. Bamberger, J. E. M. Lewis, A. Kaur, G. J. Tizzard, J. van Slageren, R. Crespo-Otero, S. M. Goldup and M. M. Roessler, Angew. Chem., Int. Ed., 2021, 60, 16051–16058 CrossRef CAS PubMed.
- L. Gualandi, P. Franchi, E. Mezzina, S. M. Goldup and M. Lucarini, Chem. Sci., 2021, 12, 8385–8393 RSC.
- M. A. Jinks, M. Howard, F. Rizzi, S. M. Goldup, A. D. Burnett and A. J. Wilson, J. Am. Chem. Soc., 2022, 144, 23127–23133 CrossRef CAS PubMed.
- S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC.
- J. E. M. Lewis, J. Winn, L. Cera and S. M. Goldup, J. Am. Chem. Soc., 2016, 138, 16329–16336 CrossRef CAS PubMed.
- A. H. El-Sagheer and T. Brown, J. Am. Chem. Soc., 2009, 131, 3958–3964 CrossRef CAS PubMed.
- M. Kukwikila, N. Gale, A. H. El-Sagheer, T. Brown and A. Tavassoli, Nat. Chem., 2017, 9, 1089–1098 CrossRef CAS PubMed.
- A. Acevedo-Jake, A. T. Ball, M. Galli, M. Kukwikila, M. Denis, D. G. Singleton, A. Tavassoli and S. M. Goldup, J. Am. Chem. Soc., 2020, 142, 5985–5990 CrossRef CAS PubMed.
- R. A. Grzelczak, A. Władyczyn, A. Białońska, Ł. John and B. Szyszko, Chem. Commun., 2023, 59, 7579–7582 RSC.
- S. W. Hewson and K. M. Mullen, Org. Biomol. Chem., 2018, 16, 8569–8578 RSC.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D’Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- G. De Bo, S. Kuschel, D. A. Leigh, B. Lewandowski, M. Papmeyer and J. W. Ward, J. Am. Chem. Soc., 2014, 136, 5811–5814 CrossRef CAS PubMed.
- G. De Bo, M. A. Y. Gall, M. O. Kitching, S. Kuschel, D. A. Leigh, D. J. Tetlow and J. W. Ward, J. Am. Chem. Soc., 2017, 139, 10875–10879 CrossRef CAS PubMed.
- G. De Bo, M. A. Y. Gall, S. Kuschel, J. De Winter, P. Gerbaux and D. A. Leigh, Nat. Nanotechnol., 2018, 13, 381–385 CrossRef CAS PubMed.
- C. T. McTernan, G. De Bo and D. A. Leigh, Chem, 2020, 6, 2964–2973 CAS.
- J. Echavarren, M. A. Y. Gall, A. Haertsch, D. A. Leigh, J. T. J. Spence, D. J. Tetlow and C. Tian, J. Am. Chem. Soc., 2021, 143, 5158–5165 CrossRef CAS PubMed.
- L. Binks, C. Tian, S. D. P. Fielden, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2022, 144, 15838–15844 CrossRef CAS PubMed.
- S. Amano, S. D. P. Fielden and D. A. Leigh, Nature, 2021, 594, 529–534 CrossRef CAS PubMed.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- F. Modicom, E. M. G. Jamieson, E. Rochette and S. M. Goldup, Angew. Chem., Int. Ed., 2019, 58, 3875–3879 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |