
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent synthetic strategies for the functionalization of fused bicyclic heteroaromatics using organo-Li, -Mg and -Zn reagents†
Vasudevan
Dhayalan
 *a,
Vishal S.
Dodke
b,
Marappan
Pradeep Kumar
a,
Hatice Seher
Korkmaz
c,
Anja
Hoffmann-Röder
c,
Pitchamuthu
Amaladass
d,
Rambabu
Dandela
b,
Ragupathy
Dhanusuraman
e and
Paul
Knochel
*c
*a,
Vishal S.
Dodke
b,
Marappan
Pradeep Kumar
a,
Hatice Seher
Korkmaz
c,
Anja
Hoffmann-Röder
c,
Pitchamuthu
Amaladass
d,
Rambabu
Dandela
b,
Ragupathy
Dhanusuraman
e and
Paul
Knochel
*c
aDepartment of Chemistry, National Institute of Technology Puducherry, Karaikal-609609, Union Territory Puducherry, India. E-mail: dhaya.chem@nitpy.ac.in; Web: https://vasudeva49.wixsite.com/catalysislab
bDepartment of Industrial and Engineering Chemistry, Institute of Chemical Technology, Indian Oil Odisha Campus, IIT, Kharagpur extension Centre, Mouza Samantpuri, Bhubaneswar-751013, Odisha, India
cDepartment of Chemistry, Ludwig-Maximilians-University München, Butenandtstrasse 5–13, Haus F, 81377 Munich, Germany. E-mail: paul.knochel@cup.uni-muenchen.de
dDepartment of Chemistry, Madanapalle Institute of Technology & Science, Madanapalle 517325, Andhra Pradesh, India
eCentral Instrumentation Facility (CIF), School of Physical, Chemical and Applied Sciences, Pondicherry University, Puducherry-605014, India
First published on 23rd September 2024
Abstract
This review highlights the use of functionalized organo-Li, -Mg and -Zn reagents for the construction and selective functionalization of 5- and 6-membered fused bicyclic heteroaromatics. Special attention is given to the discussion of advanced syntheses for the preparation of highly functionalized heteroaromatic scaffolds, including quinolines, naphthyridines, indoles, benzofurans, benzothiophenes, benzoxazoles, benzothiazoles, benzopyrimidines, anthranils, thienothiophenes, purine coumarins, chromones, quinolones and phthalazines and their fused heterocyclic derivatives. The organometallic reagents used for the desired functionalizations of these scaffolds are generally prepared in situ using the following methods: (i) through directed selective metalation reactions (DoM), (ii) by means of halogen/metal exchange reactions, (iii) through oxidative metal insertions (Li, Mg, Zn), and (iv) by transmetalation reactions (organo-Li and Mg transmetalations with ZnCl2 or ZnO(Piv)2). The resulting reactive organometallic reagents allow a wide range of C–C, C–N and C–X cross-coupling reactions with different electrophiles, employing in particular Kumada or Negishi protocols among other transition metal (Pd, Ni, Co, Cu, Cr, Fe, etc.)-catalyzed processes. In addition, key developments concerning selective metalation techniques will be presented, which rely on the use of RLi, LDA and TMP metal bases. These methods are now widely employed in organic synthetic chemistry and have proven to be particularly valuable for drug development programs in the pharmaceutical industry. New and improved protocols have resulted in many Li, Mg and Zn organyls now being compatible with functionalized aryl, heteroaryl, alkenyl, alkynyl and alkyl compounds even in the presence of labile functional groups, making these reagents well-suited for C(sp2)–C(sp2), C(sp2)–C(sp) and C(sp2)–C(sp3) cross-coupling reactions with fused heteroaryl halides. In addition, the use of some transition metal-catalyzed processes occasionally allows a reversed role of the reactants in cross-coupling reactions, providing alternative synthetic routes for the preparation of fused heteroaromatic-based bioactive drugs and natural products. In line with this, this article points to novel methods for the functionalization of bicyclic heteroaromatic scaffolds by organometallic reagents that have been published in the period 2010–2023.
 Vasudevan Dhayalan | Dr Vasudevan Dhayalan obtained his MSc in Organic Chemistry (2005) and his PhD in Organic Chemistry (2011) from the University of Madras, India, under the supervision of Prof. A. K. Mohanakrishnan. He then joined the group of Prof. Masahiko Hayashi at Kobe University as a postdoctoral researcher (2011–2012). Later, he worked with Prof. Paul Knochel at Ludwig-Maximilians-University Munich (Germany) for three years. He also worked with Prof. Anat Milo at Ben-Gurion University of the Negev (Beer Sheva, Israel) on a PBC fellowship (2016–2020). Currently, he is a Ramanujan Faculty member at NIT Puducherry (Karaikal, India). His research focuses on the design and synthesis of drugs and natural products via catalysis. He has authored more than 80 publications, book chapter, and holds one patent. |
 Vishal S. Dodke | Vishal S. Dodke received an MSc degree in Organic Chemistry from Savitribai Phule Pune University, India in 2010. He was a Doctoral Student of Chemistry at Jacobs University Bremen, Germany where he focused on the organocatalysis. Subsequently, he worked with the private companies and earned experience in organic synthesis. He joined as a PhD student under the supervision of Dr Rambabu Dandela at Institute of Chemical Technology, Indian Oil Bhubaneswar Campus, Odisha, India in September 2023. Within this group, he explores advancements in synthetic methods and their application in the synthesis of some pharmaceutically relevant small molecules and their biological studies. |
 Marappan Pradeep Kumar | Marappan Pradeep Kumar, originally from Tiruppur, Tamil Nadu, holds a BSc in Chemistry from Bharathiar University (2018–2021) and an MSc in Chemical Sciences from Pondicherry University (2021–2023). He gained research experience through a summer research fellowship at IIT Madras under the guidance of Prof. G. Sekar. Currently, he serves as a Project Assistant in the OMC research group at the National Institute of Technology Puducherry, Karaikal, Puducherry, working under the supervision of Dr Vasudevan Dhayalan. His research interests encompass organocatalysis and green chemistry, reflecting his commitment to sustainable and environmentally friendly chemical processes. |
 Hatice Seher Korkmaz | Hatice Seher Korkmaz was born 1996 in İzmir (Turkey). In 2021, she obtained her bachelor's degree at the Bilkent University (Turkey). In 2023, she completed her master's degree at the same university. Her master thesis focused on inverse electron-demand Diels–Alder reactions of 1,2-diazines. In 2024, she moved to the Munich (Germany) and she is currently PhD candidate at the Ludwig-Maximilians-University in Munich (Germany) by Profs. A. Hoffmann-Röder and P. Knochel. Her research interest focuses on preparing organometallic reagents and methodologies for application in organic synthesis and the synthesis of natural products. |
 Anja Hoffmann-Röder | Anja Hoffmann-Röder received her diploma in chemistry from the Rheinische Friedrich-Wilhelms University Bonn (1999) and her PhD degree (2003, N. Krause) from the Technical University Dortmund in Germany. After a postdoctoral stay with F. Diederich at ETH Zürich in Switzerland (2003–2005), she returned to Germany to start her independent research as a Liebig Scholar at Johannes-Gutenberg University Mainz with H. Kunz. In 2006, she became an Emmy Noether Research Group Leader there and in 2009, she was also appointed junior professor for bioorganic chemistry. Since 2011 she has been working as associate professor of organic chemistry at Ludwig-Maximilians University Munich, Germany. Her research interests mainly focus on the synthesis of carbohydrate and glycopeptide mimetics for synthetic vaccines as well as on novel polyfunctional small molecules for biological applications. |
 Pitchamuthu Amaladass | Pitchamuthu Amaladass obtained his PhD, in Organic Chemistry (year: 2007) from the University of Madras, Department of Organic Chemistry under the supervision of Dr A. K. Mohanakrishnan (Prof. & Head of Organic Chemistry Department, University of Madras). His area of research is in synthetic organic chemistry. He did his post-doctoral research on synthesis of organic functional materials from Weizmann institute of science (Israel), Nanyang Technological University (Singapore) and Seoul National University (South korea) (2008–2015). After completing his post-doctoral research, he worked as research professor at Korea University in South Korea (2017–2018). Currently, he is working as Assistant professor in the department of Chemistry from Madanapalle Institute of Technology & Science (MITS). Now, he is focussing on the synthesis of organic functional materials. |
 Rambabu Dandela | Dr Rambabu Dandela obtained his PhD from Dr Reddy's Institute of Life Sciences, University of Hyderabad campus, in 2013. After a postdoctoral stay with Prof. Michael M. Meijer at Ben-Gurion University of the Negev (2013–2017), he returned to India and joined as Ramanujan Faculty Fellow at CSIR-NCL, Pune. Since 2018 he has been working as assistant professor of chemistry at ICT-IOC, Bhubaneswar. His research interests lie at the interface of chemistry and biology with particular focuses on structure-based drug design and polymorphism in pharmaceutical solids. He has authored more than 190 publications, a number of book chapters, has 8 patents issued/pending. |
 Ragupathy Dhanusuraman | Dr Ragupathy Dhanusuraman is working as a Professor in School of Chemical, Physical & Applied Sciences, Pondicherry University, India. Previously, he worked as Associate Professor in the Department of Chemistry, National Institute of Technology Puducherry, Karaikal. He received his doctoral degree in Chemistry from Kyungpook National University Daegu, South Korea (2009). He has 109 Peer-reviewed International Journals, 4 Patents, 5 Book-Chapters and more than 50 papers in Conference Proceedings. He has guided six PhDs and several MSc students. He is a life fellow member in various scientific societies. His areas of research include; Nanomaterials, Organic Polymers, Energy & Electrochemistry. |
 Paul Knochel | Paul Knochel has been a full professor for organic chemistry at the Ludwig-Maximilians-University Munich/Germany (LMU) since 1999. He did his undergraduate studies at the University of Strasbourg (France) and his PhD at the ETH-Zürich with Prof. D. Seebach. He spent 4 years at the CNRS at the University Pierre and Marie Curie in Paris with Prof. J.-F. Normant and one year of post-doctoral studies at Princeton University in the laboratory of Prof. M. F. Semmelhack. In 1987, he accepted a position as Assistant Professor at the University of Michigan at Ann Arbor, MI. In 1991, he became Full Professor at this university and in 1992, he moved to Philipps-University at Marburg (Germany) as C4-Professor in Organic Chemistry. His research interests include the development of novel organometallic reagents and methods for use in organic synthesis, asymmetric catalysis and natural product synthesis. |
1 Introduction
Organometallic reagents (R-M; M = Li, Mg, Zn; R = alkyl, alkenyl, alkynyl, aryl, heteroaryl) are widely used in synthetic organic chemistry and various areas of catalysis.1–10These reagents play an important role in drug discovery and development, in the synthesis of natural products and bioactive compounds,11–17 as well as in programs dedicated to medicinal and materials chemistry.18–23 A variety of synthetic methods have been developed for the preparation of organometallic reagents such as organolithium, organomagnesium, and organozinc reagents and their derived cognates. Their potential use in catalytic applications has been investigated by several leading research groups such as those of Snieckus,24,25 Knochel,26–30 Harutyunyan,31–35 Aggarwal,36–40 Feringa,41–44 Marek,45–50 Mongin,51–53 Kürti,54–56 Maes,57–59 Buchwald,60 Smith,61 Li,62 and others.63–65 All of these methods most likely utilize one of the following procedures depicted in Fig. 1: (i) directed oxidative metal insertions in the presence of LiCl salts or InCl3-Lewis acid (Mg, Zn, Al, Mn, In),66,67 (ii) halogen–metal exchange reactions (X/M, X = Br, I; M = Li, Mg, Zn, Sm, La, Mn) using Turbo Grignard reagents (i-PrMgCl·LiCl) or other organometallic reagents,68–72 and (iii) chemo- and regioselective direct metalations using Knochel–Hauser bases (TMPLi, -Mg and -Zn).73–75
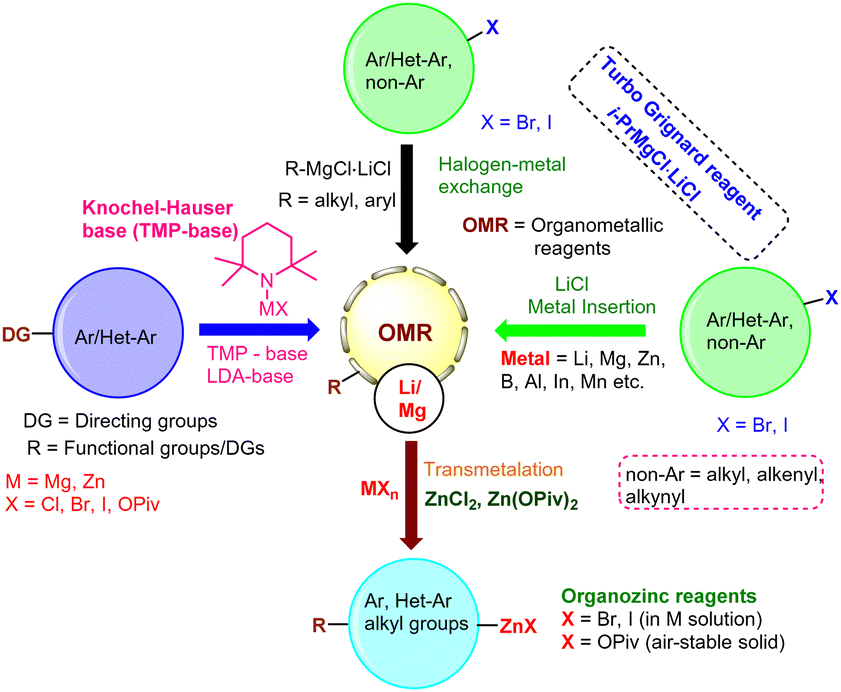 | ||
| Fig. 1 Reported methods for the preparation of organometallic reagents (RMX). | ||
In the last two decades, organolithium, magnesium and zinc reagents derived from TMP-H (2,2,6,6-tetramethylpiperidinyl) have played an important role in organic synthesis. These organometallic reagents have proven to be very effective as they facilitate the chemo- and regioselective metalation of various aromatic and non-aromatic heterocyclic scaffolds.73–75 The various alkylamine bases, including TMP-M, R1R2N-M (M = Li, Mg, Zn; R1 & R2 = alkyl) and LDA, are synthesized from commercially available secondary amine sources such as TMPH, DIPA and their derivatives employing readily available alkyl lithium reagents. These methods, developed by researchers such as Knochel, Snieckus, Mongin and others, have demonstrated the broad applicability of TMP-bases in numerous catalytic and synthetic transformations.73–75 Recent reports have repeatedly emphasized the influence of ligands on the reactivity and stability of organometallic reagents. For example, organozinc pivalates (R-ZnOPiv) exhibit higher stability than halogenated organometallic compounds (RMX, M = Mg, Zn; X = Cl, Br, I). In addition, recent kinetic and mechanistic reports have described that salt-stabilized organozinc pivalates show a significant counterion effect due to –OPiv coordination, making them easy to handle even under an oxygen-containing atmosphere. Since these reagents can be stored with no noticeable degradation or loss of yield for up to 48 h under air, they are ideally suited for various transition metal-catalyzed Negishi cross-coupling reactions.76–83
Another major synthetic challenge is the regio- and enantioselective synthesis and functionalization of small organic molecules, as some of these N-heterocyclic systems can serve as chiral ligands or catalysts in synthetic transformations to produce chiral organic intermediates (Fig. 2).84,85
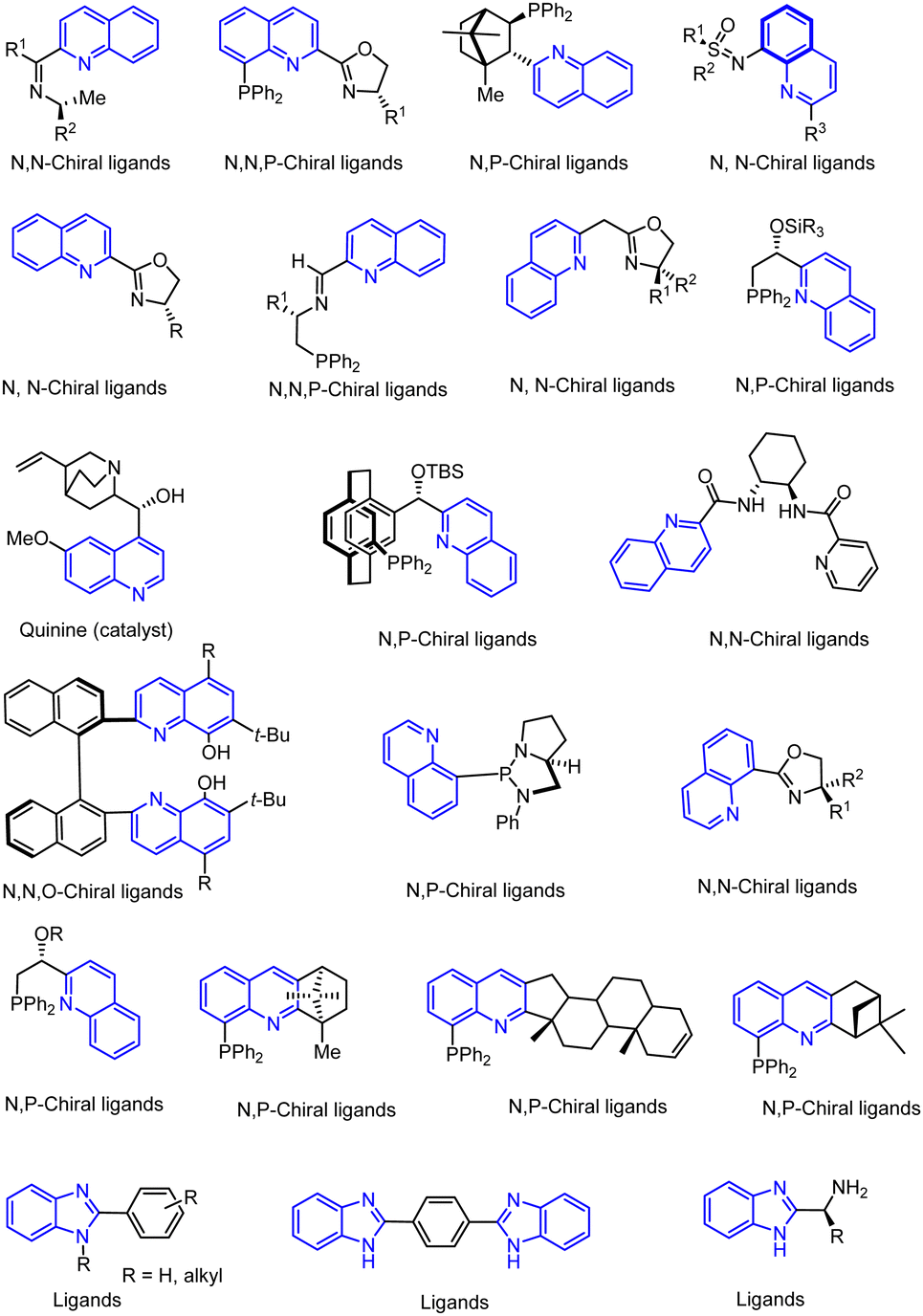 | ||
| Fig. 2 N-Heterocyclic aromatic compounds for use as ligands and catalysts. | ||
Highly selective and reactive aryl and alkyl organometallic reagents (Li, Mg, Zn) can be attached to electrophilic carbonyl or imine scaffolds to generate complex tetrasubstituted chiral alcohols and amines by catalysis.86–90 The preferred organometallic compounds for these transformations are organo-Li, -Mg and -Zn reagents, due to their accessibility, low cost and non-toxicity, which favours their use in research laboratories and the pharmaceutical industry. Extensive work has shown that many functionalized fused bicyclic heteroaromatic molecules are preferred structural motifs, due to their (potential) biological activity. Thus, such compounds are of particular interest for drug development (Fig. 3) but also for the production and modification of novel catalysts, as well as for material science applications.
 | ||
| Fig. 3 Selective examples of bicyclic fused heteroaromatic natural products and bioactive compounds. | ||
Such applications require access to substituted scaffolds of diverse heteroaromatic core structures such as quinoline, quinoxaline, naphthyridine, indole, benzofuran, benzimidazole, benzothiophene, benzoxazole, benzothiazole, benzopyrimidine, anthranile, thienothiophene, triazole, purine, coumarin, but also chromones, quinolones and phthalazines.91–95
Fused heterocyclic scaffolds have been decorated by various catalytic methods such as metal-free and metal-catalyzed C–H activation reactions, radical transformations, photocatalytic processes, etc.96–98 However, only a few cases require stoichiometric amounts of transition metal catalysts or excess amounts of organometallic reagents to perform these transformations. Moreover, transition metal-catalyzed processes mediated by organometallic reagents can be associated with undesirable side effects, including β-hydride elimination and homocoupling reactions.99 The functionalization of bicyclic fused heteroaromatic compounds using transition metal catalysts (Co, Fe, Ni, Cr) and organometallic reagents has therefore attracted considerable attention in organic synthesis, particularly in the pharmaceutical industry and sustainable catalysis.
2 Functionalization of fused bicyclic hetero-aromatic compounds with TMPLi, -Mg, -Zn
In 2010, one of the best strategies for the functionalization of quinoline scaffolds 1 utilizing TMPMgCl·BF3-mediated selective metalations of N-heterocycles was reported by Knochel's group, leading to the preparation of type 2 intermediates.100 Subsequent transmetalation with ZnCl2 (1.1 equiv.) followed by Pd-catalyzed Negishi cross-coupling with aryl iodide afforded the 2-arylated quinoline 3a in 79% yield. Similarly, a rapid transmetalation of the magnesium species with CuCN·2LiCl and subsequent acylation reaction with acid chloride led to the corresponding heteroaryl ketone in 94% yield (Scheme 1).101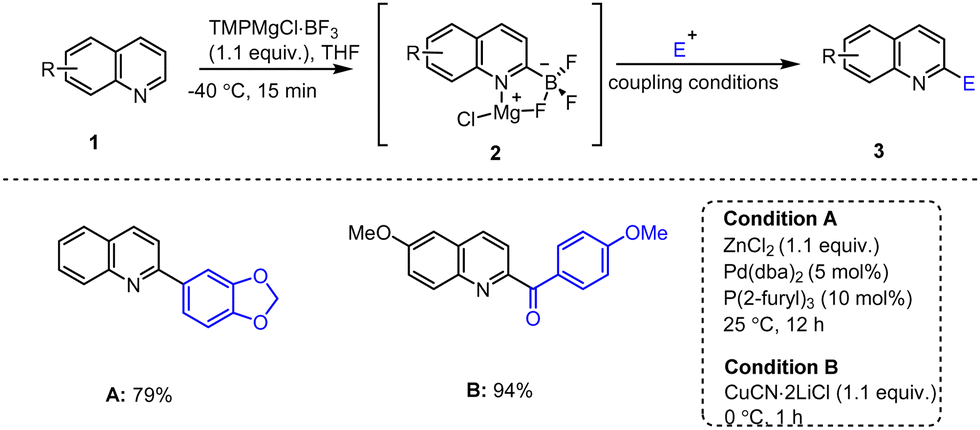 | ||
| Scheme 1 TMPMgCl·BF3-mediated regioselective metalation of quinoline for subsequent cross-coupling reactions. | ||
Rohbogner et al. developed a method for the preparation of pharmaceutically active quinoline scaffolds such as Talnetant 7 and the P-selectin inhibitor 9 using TMP-M-mediated selective metalations of quinoline derivative 4. Cross-coupling reactions, deprotection, saponification and amination describe the reaction sequences that provide the expected target quinoline scaffolds 7 and 9 in a few steps under mild conditions and in excellent yields (Scheme 2).102
 | ||
| Scheme 2 TMP-Metal base-mediated synthesis of biologically active Talnetant and a P-selectin inhibitor scaffold. | ||
Furthermore, to obtain the reactive magnesium intermediates 11, many N-heterocyclic phosphorodiamidate derivatives of type 10 including quinoline and quinoxaline molecules were subjected to a directed ortho-metalation (DoM) procedure with TMPMgCl·LiCl or TMP2Mg·2LiCl. Later, the resulting organometallic reagents 11 were transmetalated with ZnCl2 or CuCN·2LiCl and subsequently reacted with various electrophiles to give the corresponding arylation, acylation, thiolation and allylation products providing e.g., the highly functionalized quinoline and quinoxaline scaffolds 12 in good yields of up to 87% (Scheme 3).102,103
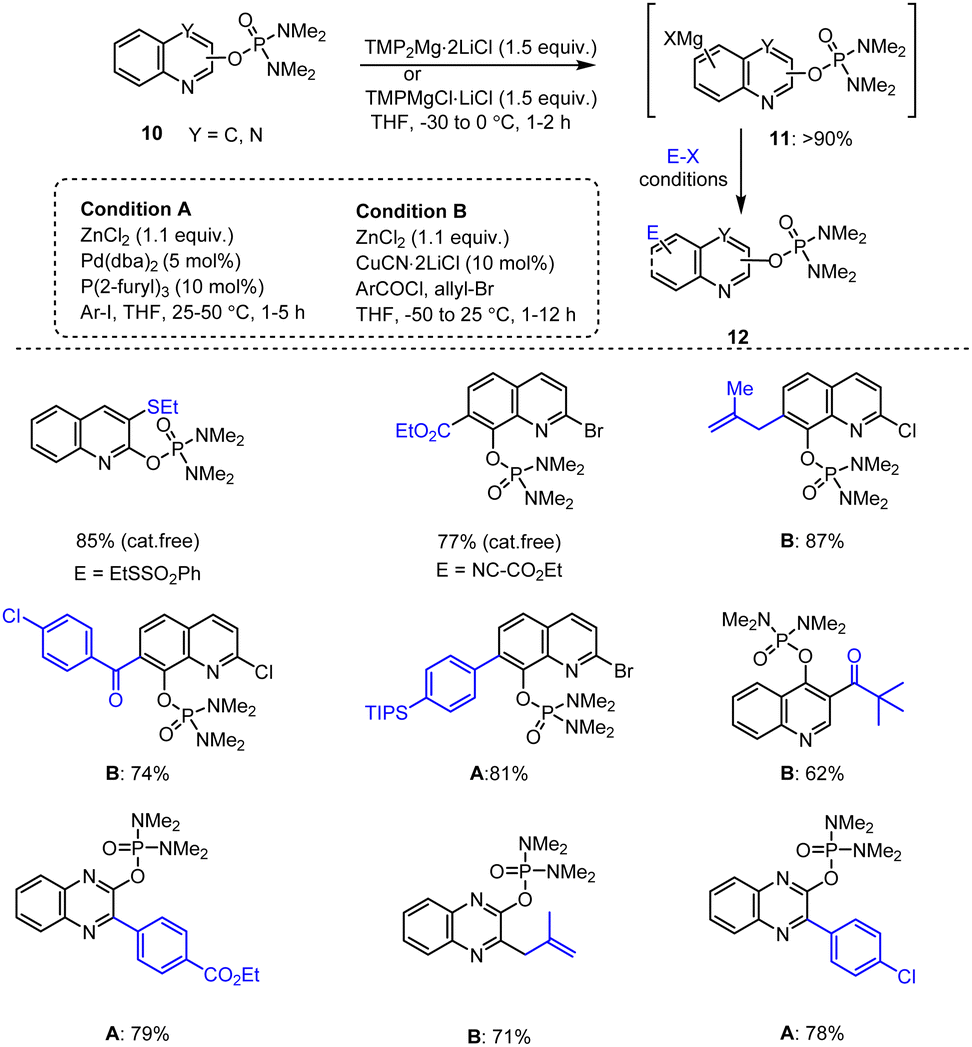 | ||
| Scheme 3 TMP-Base-mediated metalation of quinolines and quinoxalines for subsequent functionalization with different electrophiles. | ||
A facile and efficient palladium-catalyzed direct benzylation of methylquinoline derivatives using TMPZnX-derived bases was accomplished in 2011 by Duez et al.104 The desired zinc-containing quinoline derivatives were prepared by direct TMPZnCl-mediated metalations of 2/4-methylquinoline 13 in THF at 25 °C for 1 h. Subsequently, palladium-catalyzed Negishi cross-couplings of the resulting benzylic zinc reagents with a variety of aryl bromides (0.8 equiv.) were performed. When using Pd(OAc)2 (2 mol%) and SPhos (4 mol%) as the catalyst, functionalized quinoline scaffolds 15 were obtained in excellent yields of up to 97%, with this method also tolerating sensitive functional groups such as OH, NH2, CN and CF3 (Scheme 4).
 | ||
| Scheme 4 Negishi cross-coupling of 2/4-methylpyridine after zincation using TMPZnCl. | ||
In 2016, Mongin, Halauko and co-workers showed that a series of chloroquinolines 16 could be deprotometalated with a TMEDA-based mixed n-BuLi and TMPLi combination. Good regioselectivities were observed with a corresponding lithium-copper bimetallic combination and were confirmed by consistent trapping with reactive acid chlorides in THF at r.t. The resulting carbonyl compounds 17 were produced in moderate yields (Scheme 5).105
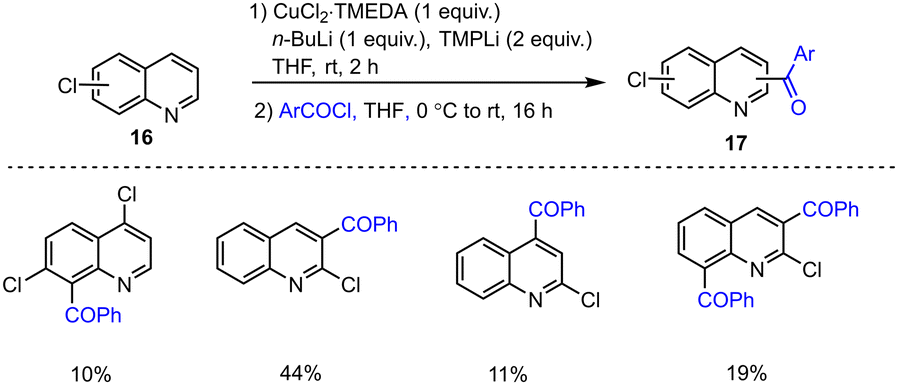 | ||
| Scheme 5 Deproto-metalation of chloroquinolines with amido-based bimetallic species and subsequent quenching with acid chlorides. | ||
Jaric et al. developed a successful method for the functionalization of bioactive quinine cores 18 to be used as organocatalysts by employing MeLi and TMPMgCl in the presence of BF3·OEt2. The subsequent trapping reactions such as Pd-catalyzed Negishi couplings, Cu-catalyzed allylations, and direct quenching with bromo- and iodo-electrophiles furnished the corresponding C-3 functionalized quinoline heterocycles 19–21 in good yields of 41–66% (Scheme 6).106
 | ||
| Scheme 6 C-3- or C-2-Functionalization of quinine using TMPMgCl·LiCl. | ||
Knochel described an efficient TMP-base-mediated protocol for the synthesis of functionalized aminoquinolines 24via transition metal-free secondary amination of quinoline 2/8-sulfonamides and 8-napthylsulfonyl chlorides. This was accomplished using R2NMgCl·LiCl in THF at 25 °C for 1–8 h. The selective magnesiation of quinoline-2/8-sulfonamides 22 was also described using TMPMgCl·LiCl. A variety of quinoline derivatives 24 functionalized at the 2/8 position were prepared by successive C–N couplings with various amine-based organometallic reagents (R2NMgCl·LiCl) under mild conditions to afford the expected amino quinolines in good yields (Scheme 7).107 In addition, the possible mechanism of this metal-free direct amination was described based on two possible scenarios (Scheme 7). According to mechanistic pathway A, the first step is the selective addition of the magnesium reagents  to the C
to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the quinoline skeleton 23, which leads to the formation of the key intermediate A, ultimately providing aminated product 24 after elimination of R2NSO2MgCl. Alternatively, addition of R2NMgCl·LiCl to the 2-quinolinylsulfonamide group was proposed first, resulting in species B, which can undergo an intramolecular transfer reaction of the amino group to magnesiated intermediate A. After elimination, the latter then also affords the desired aminated quinoline 24 (route B, Scheme 7).
N bond of the quinoline skeleton 23, which leads to the formation of the key intermediate A, ultimately providing aminated product 24 after elimination of R2NSO2MgCl. Alternatively, addition of R2NMgCl·LiCl to the 2-quinolinylsulfonamide group was proposed first, resulting in species B, which can undergo an intramolecular transfer reaction of the amino group to magnesiated intermediate A. After elimination, the latter then also affords the desired aminated quinoline 24 (route B, Scheme 7).
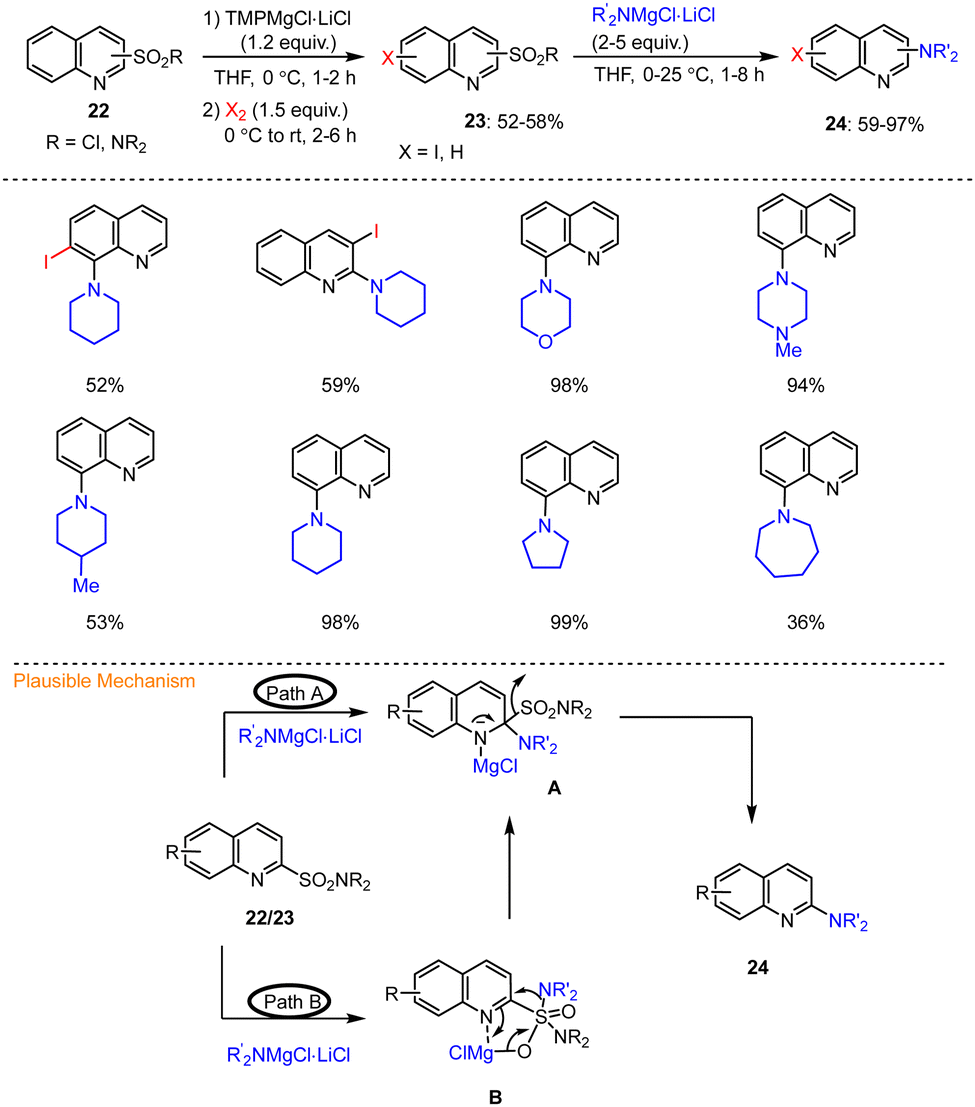 | ||
| Scheme 7 Functionalization of quinoline 2- and 8-sulfonamides by TMPMgCl·LiCl followed by desulfonylation and addition of R2NMgCl. | ||
Manolikakes et al. demonstrated a simple method for the preparation of pyridylmethyl alcohols using TMPMgCl·BF3, a frustrated Lewis pair that mediates selective metalations to organotrifluoroborates. The latter readily adds to a variety of aromatic aldehydes and ketones in the absence of a transition metal catalyst to afford diarylmethyl alcohols 26 in good yields of 65% (Scheme 8).108
 | ||
| Scheme 8 Metalation of quinoline followed by 1,2-addition to aromatic aldehydes and ketones. | ||
Knochel, Zipse and co-workers reported a protocol for the full functionalization of pyrazolo[1,5-a]pyrimidine scaffolds 27, 30, 33 at positions 2, 3 and 7 using TMP-M bases. Reaction of 5-chloro-pyrazolo[1,5-a]pyrimidine with TMPZn under mild conditions afforded heterocyclic zinc intermediates 28, 31, 34. Subsequent reactions with various readily accessible electrophiles in the presence of palladium or copper catalysts afforded the corresponding fused pyrazolo[1,5-a]pyrimidine derivatives 29, 32, 35 in good to excellent yields (Scheme 9–11).109 These functionalized heterocyclic compounds are often used in pharmaceutical applications, and the described catalytically active system tolerates both electron-rich and electron-poor functional groups such as Me, SPh, OMe, Cl, I, CN, NO2, and CO2Et.110,111
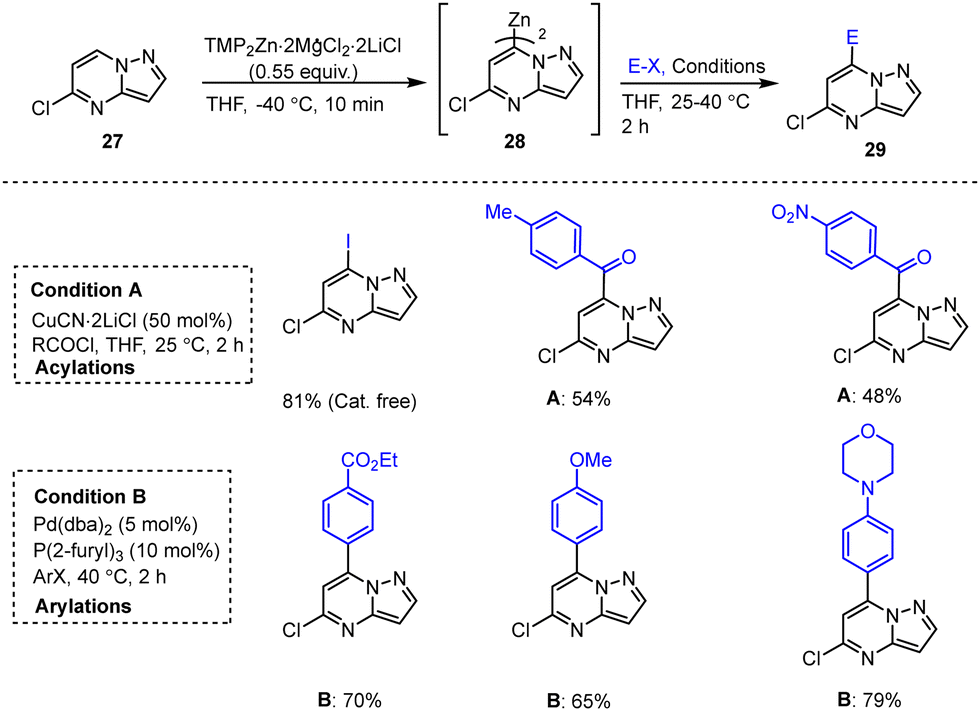 | ||
| Scheme 9 Selective zincation at position 7 of the pyrazolo[1,5-a]pyrimidine core for subsequent cross-coupling and acylation reactions. | ||
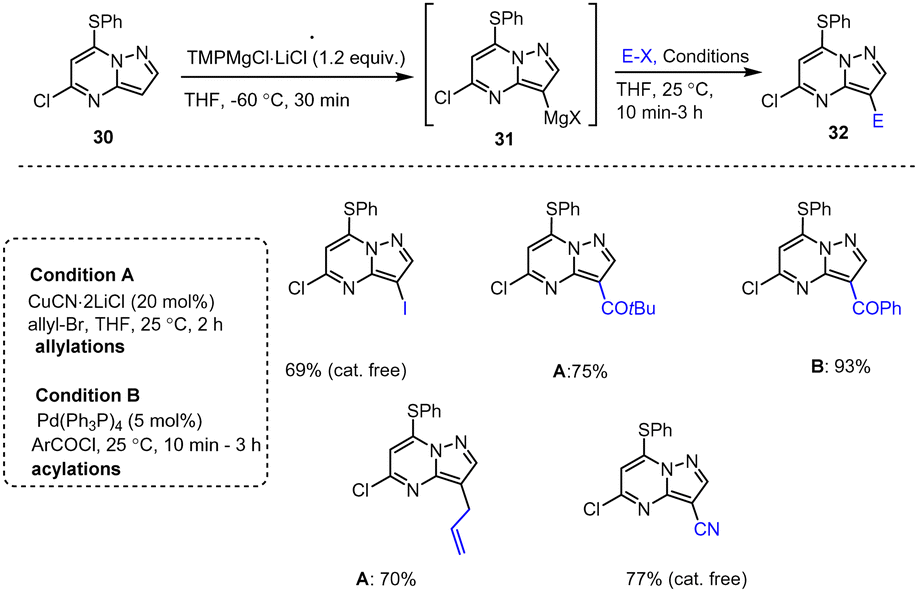 | ||
| Scheme 10 Selective C2-metalation of the pyrazolo[1,5-a]pyrimidine scaffold followed by electrophilic trapping reactions. | ||
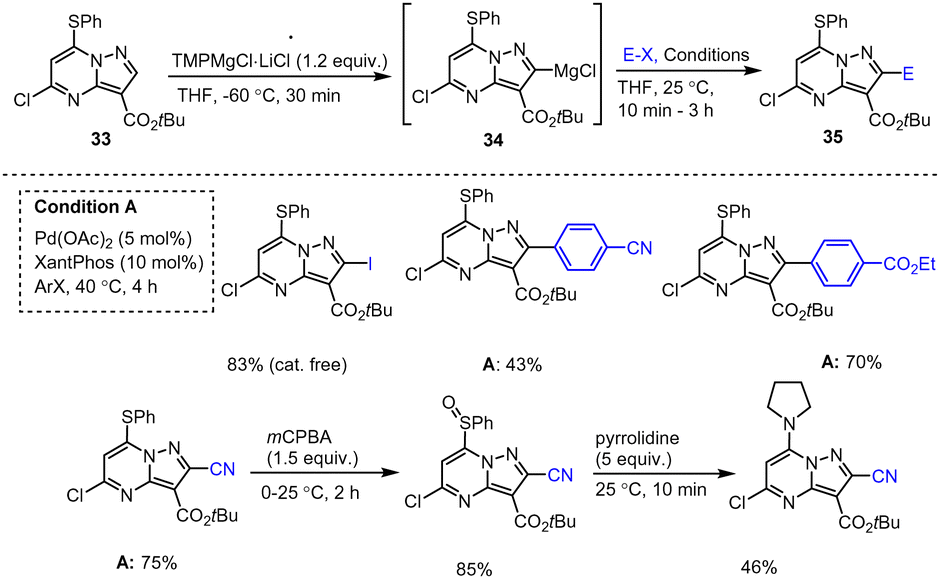 | ||
| Scheme 11 Selective metalation of pyrazolo[1,5-a]pyrimidine at 2-position using TMPMgCl·LiCl and quenching with electrophiles. | ||
Unsinn et al. investigated a simple, mild, and efficient protocol for the regioselective C-3-metallation of 1H-indazoles 36 with TMP2Zn. The resulting indazolylzinc reagents of type 37 could be smoothly arylated by Pd-catalyzed Negishi cross-coupling reactions in THF at 50 °C with various aryl iodides. The process took 8–24 h to afford the indazolyl analogs 38 in good yields. In addition, copper-mediated acylations and allylations are also applicable under these mild conditions (Scheme 12).112 This synthetic method is suitable for the preparation of various biologically active N-heterocycles.
 | ||
| Scheme 12 Pd-Catalyzed Negishi cross-coupling reactions of indazolyl zinc reagents. | ||
In 2012, Duez et al. developed an efficient method for the Pd-catalyzed arylation of different 2-methyl-5-membered fused heterocycles of type 39 with TMPLi bases. This innovative synthetic process involves TMPLi-mediated selective metalation at the benzylic position in THF at −78 °C, followed by transmetalation with ZnCl2 to form the corresponding organozinc intermediate 40. Subsequent Pd-catalyzed Negishi cross-coupling reaction under mild conditions provided the desired arylated fused bicycles 41 with indole, benzothiophene and benzofuran heterocyclic cores in good to excellent yields of up to 98% (Scheme 13).113 Under the optimized reaction conditions, fused bicyclic heteroaromatic compounds containing different functional groups with electron-poor and electron-rich substituents were obtained, making this approach widely applicable for the synthesis of pharmaceutically active molecules (API).
 | ||
| Scheme 13 Pd-Catalyzed benzylation of 2-methyl-5 membered fused heterocycles using TMPLi base. | ||
Unsinn et al. presented an improved strategy for the synthesis of bis-heteroaryl zinc reagents 43 and reported on their subsequent reaction with various electrophiles. The refined procedure using a TMP-Mg base in the presence of ZnCl2 is preferable to the methods previously developed by Knochel et al. in which zinc bases are prepared from commercially available 2,2,6,6-tetramethylpiperidinyl. Most importantly, this novel protocol enables the isolation of heterocyclic products in high yields under mild conditions and with shorter reaction times. This is particularly important for the synthesis of organozinc reagents on an industrial scale and for the subsequent arylation and acylation reactions. Remarkably, the organozinc reagents not only exhibited excellent reactivity, but also tolerated a wide range of sensitive functional groups. Consequently, a direct conversion to the corresponding highly desirable organometallic zinc reagents 43 was possible, which could then be readily reacted with various electrophiles to generate functionalized heterocyclic compounds 44 in high yields (Scheme 14).114
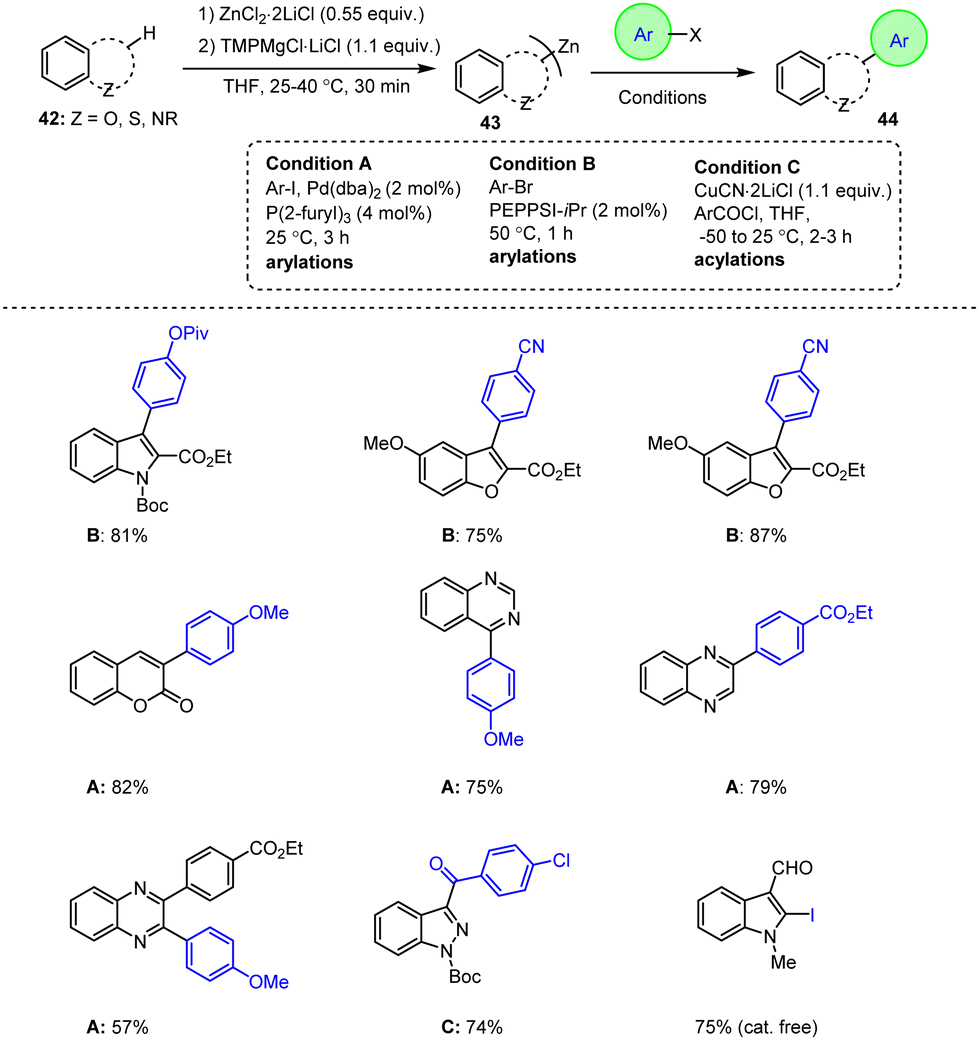 | ||
| Scheme 14 Functionalization of fused heteroaromatic indole, benzofuran, coumarin, quinoxaline and benzothiophene derivatives. | ||
Klier et al. presented a Lewis acid that triggered the regioselective metalation of several chromones and quinolones 45. In the absence of the Lewis acid MgCl2, zincation is observed at the C-3 position, whereas in the presence of MgCl2, zincation is observed at the C-2 position. Subsequent Pd-catalyzed Negishi C(sp2)–C(sp2)-coupling under mild conditions afforded the corresponding desired functionalized fused 6-membered heterocycles 47 in good to excellent yields in the range of 63–90% (Scheme 15).115
 | ||
| Scheme 15 TMP-Base mediated functionalization of chromones, quinolones and thiochromones using Pd-catalysis. | ||
Stathakis et al. used a wide range of interesting organozinc pivalates 50 prepared by TMPZnOPiv-mediated selective metalation methods. These organozinc reagents showed high stabilities and good reactivities in C–C cross-coupling reactions with various readily available electrophiles. In addition to CuCN-mediated acylation and allylation reactions, Pd-catalyzed Negishi couplings were performed in the presence of catalytic amounts of Pd(dba)2 and the ligand TFP in THF. The resulting solid organozinc pivalates of type 50 are easy to handle in industrial experiments because their stability is maintained (>90%) for 4–6 h. As shown in Scheme 16, a broad spectrum of aryl and alkenyl zinc pivalates efficiently reacted with different electrophiles, such as aryl halides, acid chlorides, allyl bromides and iodine, leading to the production of functionalized heterocyclic compounds 51 in good to excellent yields.116
 | ||
| Scheme 16 Pd- and Cu-Catalyzed cross-couplings of organozinc pivalates. | ||
A simple and efficient cobalt-catalyzed Negishi-type cross-coupling reaction of heteroaromatic zinc reagents 53 with secondary and primary alkyl iodides or bromides using a THF-soluble homogeneous catalytic system of CoCl2·2LiCl (20 mol%) and TMEDA (30 mol%) allowed production of the desired alkylated heterocycles 54 in yields of up to 88%. As shown in Scheme 17, the required organozinc reagents were prepared from readily available heteroaromatic compounds 52via a TMPZn-mediated selective metalation reaction.117
 | ||
| Scheme 17 Cobalt-catalyzed cross-coupling reactions of heteroaryl zinc reagents with alkyl halides. | ||
Bresser et al. reported a wide range of functionalized heteroaryl-zinc reagents synthesized by directed zincation of the sensitive and weakly deactivated heteroaromatic compounds 55 with TMPZnCl under distinct optimized conditions. The resulting heteroaryl-zinc organometallics 56 further exhibited excellent reactivity in various electrophilic addition reactions, and afforded the corresponding heteroaromatic compounds 57 in moderate to high yields (Scheme 18).118
 | ||
| Scheme 18 Regio- and chemo-selective zincation of fused heteroaromatics using TMPZnCl. | ||
Crestey et al. described a strategy for the functionalization of phthalazine scaffolds via regioselective zincation of chlorophthalazines using TMPZnCl·LiCl under microwave irradiation. This approach led to novel polysubstituted phthalazine derivatives of type 60 after trapping the resulting organozinc reagents with various electrophiles. In addition, Negishi cross-coupling reactions between chlorophthalazines and organometallic reagents were conducted using a Pd catalyst, providing highly functionalized phthalazine scaffolds 59 in excellent yields of up to 94% (Scheme 19).119
 | ||
| Scheme 19 Synthesis of polyfunctionalized phthalazines using TMPZnCl·LiCl. | ||
Zimdars et al. demonstrated the selective metalation of positions 4 and 7 of the benzo[c][1,2,5]thiadiazole scaffold 61 using TMP-bases in THF at −40 °C for 14 h. The corresponding reactive Mg-intermediate 62 was readily transmetalated with ZnCl2 followed by electrophilic quenching. In this way, Pd catalysis enabled the preparation of functionalized asymmetric disubstituted benzothiadiazole derivatives 64 in high yields, as shown in Scheme 20.120
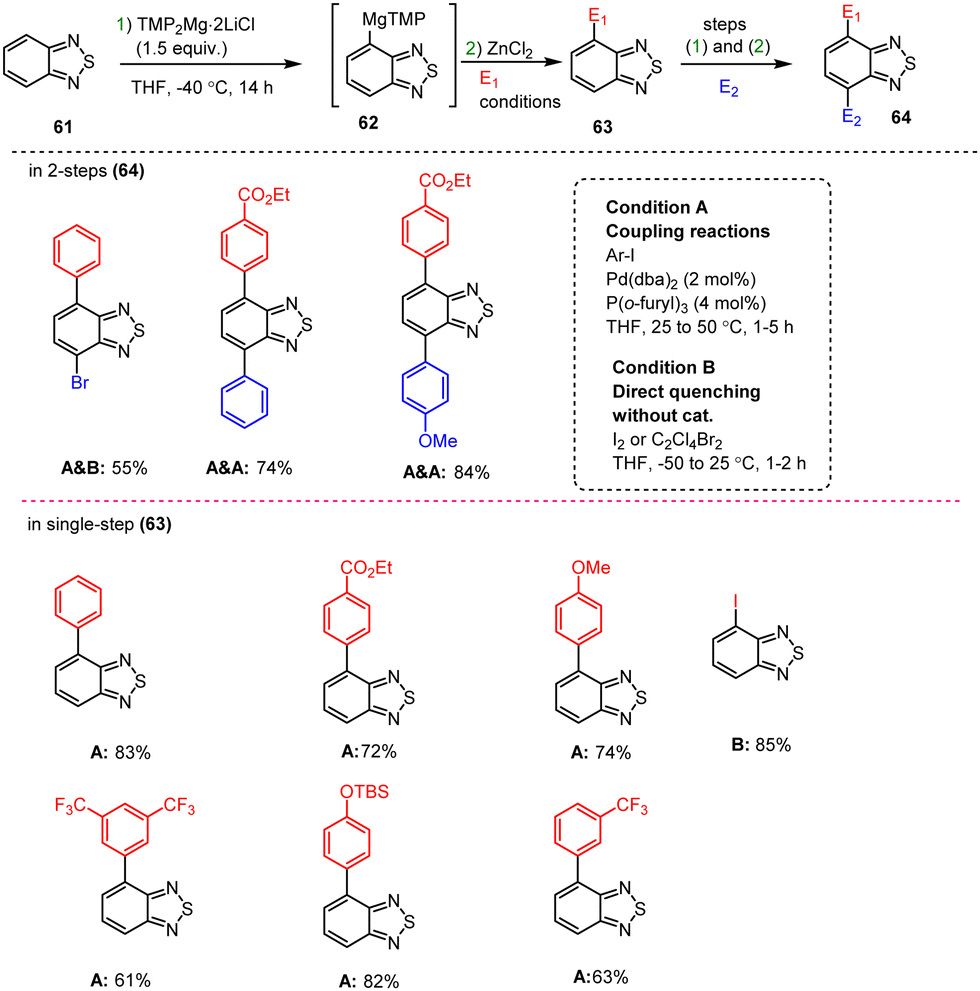 | ||
| Scheme 20 Double functionalization of benzo[c][1,2,5]thiadiazole via TMPMg-mediated coupling reactions. | ||
In 2020, Balkenhohl et al. reported a powerful model for predicting site-selective metalation approaches with TMPZnCl·LiCl. The pKa values of the functionalized condensed N-heterocycles were calculated and compared with experimental results of deprotonations. Thus, the fused heteroaromatic bicycles 65 such as pyrido[2,3-b]pyrazine, imidazo[1,2-b]pyridazine, [1,2,4]triazolo[4,3-a]pyrazine, [1,2,4]triazolo[1,5-a]pyrimidine, and imidazo[1,5-a]pyridine as well as quinazoline were smoothly deprotonated at the predicted positions (pKa = 24.6–39.7), leading to the corresponding aryl zinc reagents 66 in 40–70% yield. After iodolysis or palladium-catalyzed Negishi cross-coupling reactions, the expected highly functionalized N-heterocycles of type 67 were obtained in 42–59% yields (Scheme 21).121
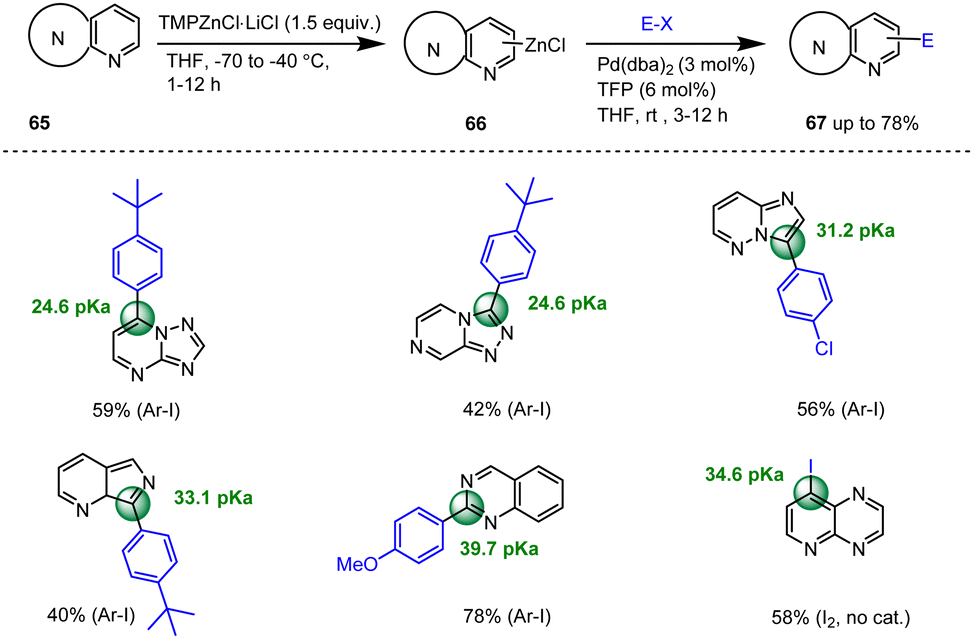 | ||
| Scheme 21 TMPZnCl·LiCl-promoted regioselective deprotonations and subsequent functionalizations of N-heterocycles. | ||
Regioselective magnesiation of benzothiophene and benzofuran scaffolds 68 with TMPMgCl·LiCl in THF at 0 °C for 2 h, followed by a 12 h trapping reaction using the thio-electrophile (Me2NC(S)S)2 afforded the desired benzothienyl dithiocarbamates 70 in excellent yields of 76–90%. Kienle et al. also prepared substituted 2-aryl and 3-aryl benzothiophene derivatives under similar reaction conditions (Scheme 22).122 This method allows polycyclic heteroaromatic compounds such as dibenzothiophenes and dibenzothieneothiophenes scaffolds to be prepared in good yields. Derivatives of benzothiophenes, dibenzothiophenes, dibenzothieno–thiophenes and their S-heterocyclic congeners are broadly applied in various fields such as in agriculture, for pharmaceuticals and dyes, as well as in building blocks for conductive polymers.
 | ||
| Scheme 22 Preparation of heteroaryl dithiocarbamates using TMPMgCl·LiCl for metalation. | ||
Kunz et al. showed that direct magnesiation with TMPMgCl·LiCl enables highly regioselective and complete functionalization of the thieno[3,2-b]thiophene core 71 under mild conditions. A wide variety of sensitive functional groups can be successfully introduced as substituents, yielding various polyfunctionalized fused thienothiophenes 74 and 76 (Scheme 23),123 which are otherwise difficult to process into promising heterocyclic scaffolds. This protocol could allow the fine-tuning of material properties of such S-heterocycles (e.g. absorption bands, overlap of frontier orbitals) by introducing specific side chains into monomeric building blocks. The conjugated heterocyclic aromatic compounds represent a new class of S-containing condensed bicyclic heterocycles and their polymers, which may also be of interest as materials for OLEDs or solar cells.
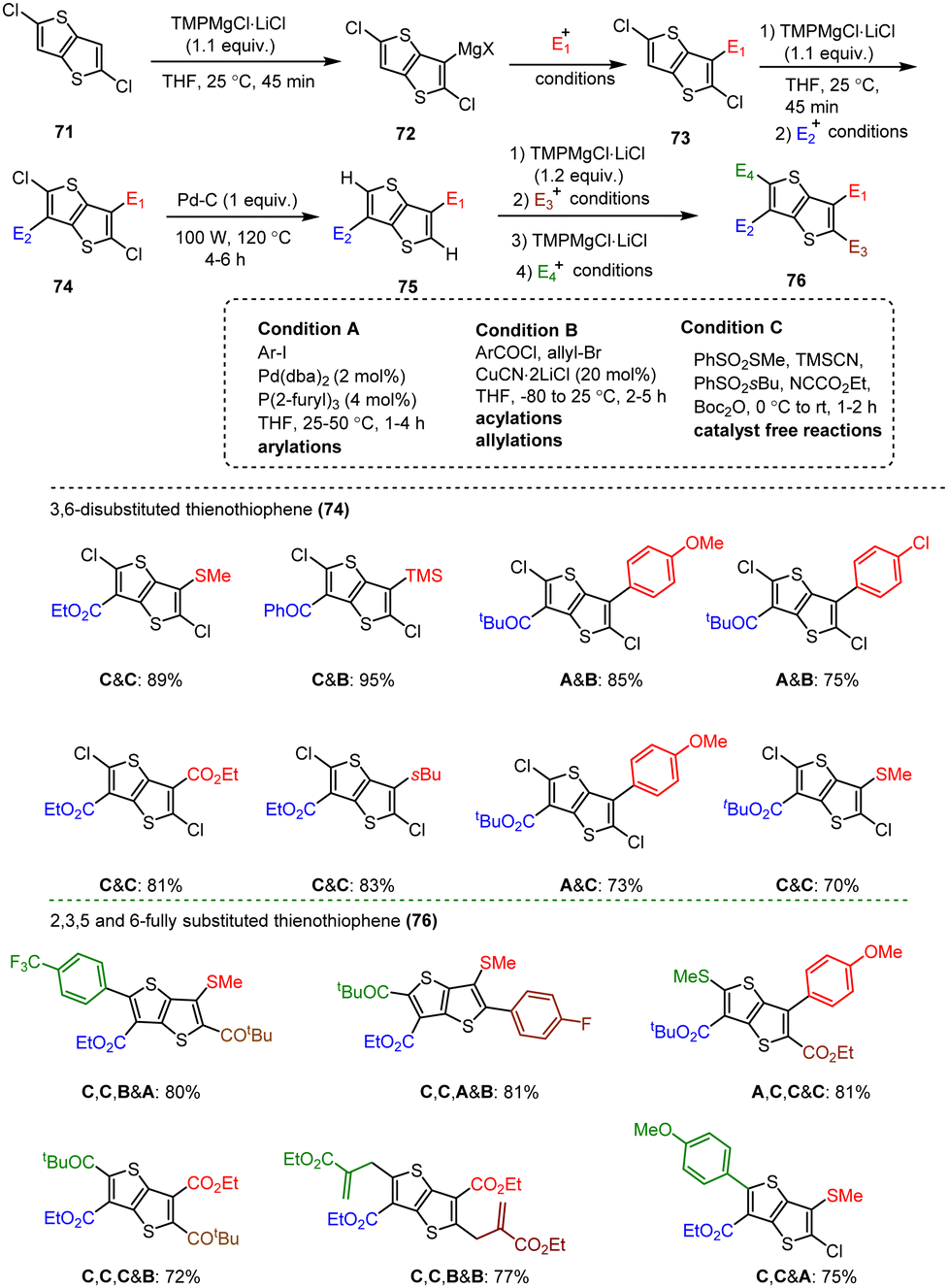 | ||
| Scheme 23 Synthesis of 2,3,5 and 6-substituted thienothiophenes. | ||
Knochel and co-workers developed a practical magnesiation protocol for trifluoromethylated pyrazinamide 77, which is carried out at 0 °C and is compatible with carbonyl functionalities. Subsequent quenching with heteroaryl halides led to satisfactory yields of arylated indole 79. After deprotection of the pyrazinamide with K2CO3 under green conditions, the targeted free heterocyclic amine 80 was obtained in good yield (Scheme 24).124
 | ||
| Scheme 24 Synthesis of GSK-3 and Lck protein kinase inhibitor via chemoselective magnesiation, followed by Pd-catalyzed coupling reaction and deprotection. | ||
Starting from alkynyl(aryl)thioethers 81, Kunz et al. developed a novel intramolecular carbomagnesiation protocol for the synthesis of magnesiated benzothiophene intermediate 82. Other heteroaromatic Mg species also reacted with readily accessible electrophiles to give the highly functionalized benzo[b]thieno[2,3-d]thiophenes of type 83 in excellent yields (Scheme 25).125 The method tolerates a wide range of functional groups, and the authors further elaborated the cyclization process to produce highly diverse condensed benzothiophene scaffolds as well as new complex heterocyclic analogs under mild conditions.
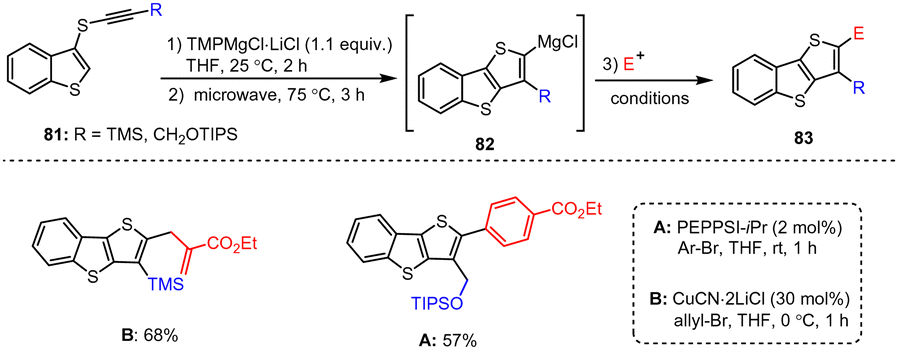 | ||
| Scheme 25 Functionalized benzo[b]thiophenes obtained by magnesiation and subsequent quenching with electrophiles. | ||
Frischmuth et al. prepared a wide range of polyfunctional SF5-substituted indole analogs 86 using TMPMgCl·LiCl. This was accomplished via the formation of organomagnesium intermediate 85 through the reaction between indole 84 and the TMP base over a period of 0.5–2 h at moderate conditions (Scheme 26).126 A library of SF5-substituted heteroaromatic compounds could be accessed with the help of this organometallic strategy to enable the discovery of new biologically active indole compounds.
 | ||
| Scheme 26 Functionalization at the position C-2 and C-3 of protected SF5-substituted indole derivative using TMPMgCl·LiCl. | ||
Groll et al. showed that the combination of BF3·OEt2 with TMPZn bases enables regioselective functionalization of fused heterocycles and thieno-pyrimidines. Remarkably, the use of BF3·OEt2 together with TMPZn bases enabled efficient zincation of the C-2 position, whereas metalation at the C-6 position was observed in the absence of the Lewis acid. The pre-formed organozinc species subsequently reacted with different electrophiles smoothly to produce the desired functionalized heterocycles 88 and 89 in good yields (Scheme 27).127
 | ||
| Scheme 27 Switchable, regioselective metalation of thieno-pyrimidines with TMPZn-base. | ||
The regioselective functionalization of a broad spectrum of N-protected purine scaffolds was successfully performed by Crestey et al. For this purpose, they applied a TMP base to substituted purine derivatives 90 for forming a zinc or magnesium intermediate 91. This is furnished after trapping with various electrophiles such as I2, Br2, Ar–I, and allyl bromide the desired highly substituted purine analogs 92 (Scheme 28). Subsequent arylation reaction was achieved with Pd(dba)2 (2 mol%) in combination with the ligand TFP (4 mol%) at 45 °C for 14 h, while for the corresponding allylation reaction CuCN·2LiCl (10 mol%) in THF at −60 to 25 °C for 2 h was used.128
 | ||
| Scheme 28 Regioselective functionalization of purine scaffolds with TMP metal bases. | ||
Klatt et al. reported a new and efficient protocol for the regioselective metalation of the condensed heteroaromatic cinnoline backbone using two complementary methods. While the use of BF3·OEt2 and TMP2Mg·2LiCl (method A) allowed magnesiation at the C-3 position, application of TMPMgCl·LiCl (method B) enabled selective zincation at the C-8 position of the cinnoline skeleton. By using the TMP-Mg base, this reaction allowed the formation of reactive Mg intermediates of both simple and substituted cinnoline derivatives. Subsequent coupling reactions with various electrophiles such as X2, aryl halides, allyl bromide and acid chlorides in the presence of Pd(dba)2 (2 mol%) with TFP (4 mol%) or CuCN·2LiCl (1.1 equiv.) furnished the desired polyfunctionalized heterocycles 94 and 95 in good to excellent yields (Scheme 29).129
 | ||
| Scheme 29 Regioselective metalations for subsequent functionalization of the cinnoline scaffold. | ||
By using a flow process, highly sensitive, electron-poor benzothiazoles 96 can be efficiently transmetalated upon treatment with TMPLi in the presence of MgCl2 or ZnCl2·2LiCl to yield the corresponding organomagnesium or organozinc reagents such as 97 (Scheme 30). According to Becker et al.,130 these flow reactions take place under significantly milder conditions than in the batch process within 40 s (0 °C instead of −78 °C). The resulting heteroaromatic metalation intermediates can then be reacted with various electrophiles, whereby the reaction scope of the flow metalations is also significantly larger than in the corresponding batch processes. In addition, these flow reactions can be easily scaled up by extending the reaction time without further optimization steps. As a result, easily modified benzothiazole scaffolds such as 98 can be produced in high yields.
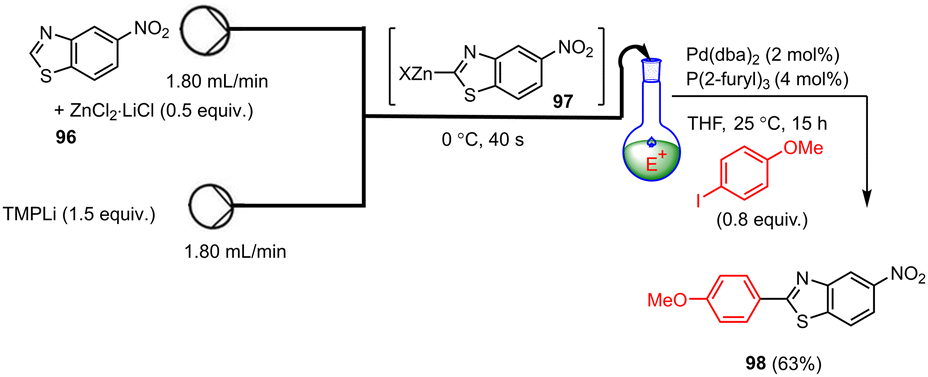 | ||
| Scheme 30 Continuous-flow zincation of benzothiazole followed by Pd-catalyzed trapping with Ar–I. | ||
An efficient method for the functionalization of substituted quinoxalines by metalation at the C-6 and C-8 positions was developed by Nafe et al. using TMPLi and 2,3-dichloroquinoxalines. This protocol enables the synthesis of interesting, highly functionalized quinoxaline scaffolds via subsequent Pd-catalyzed cross-couplings (Scheme 31).131
 | ||
| Scheme 31 Synthesis of substituted quinoxalines using TMPLi, ZnCl2, and electrophiles. | ||
In addition, the resulting functionalized products can be used further to construct expanded O- or S-heterocyclic compounds via anellation reactions. Such expanded quinoxaline scaffolds are characterized by robust photoluminescence with high molecular extinction coefficients within the blue and green spectral range and thus represent interesting potential fluorescent imaging tools with fine-tuned optical properties (Scheme 32).132
 | ||
| Scheme 32 Functionalization of quinoxalines at C-2 and C-3 for anellation reactions. | ||
Balkenhohl et al. developed regioselective metalation and functionalization reactions of the condensed 1,5-naphthyridine scaffold 103 mediated by TMP–metal bases. A clever combination of TMPMg and TMPZn bases allowed regioselective bisfunctionalization of the 1,5-naphthyridine core, which is an important heteroaromatic scaffold. Furthermore, the C-8 substituted 1,5-naphthyridine 105 allows additional regioselective functionalization at the C-4 position by using TMPMg or TMPZn bases under mild conditions (Scheme 33).133 Thus, this reaction method is not only a key technique for the design and synthesis of OLED materials, but can also be used for pharmaceutical applications, such as the production of potentially antibacterial and antiviral agents.
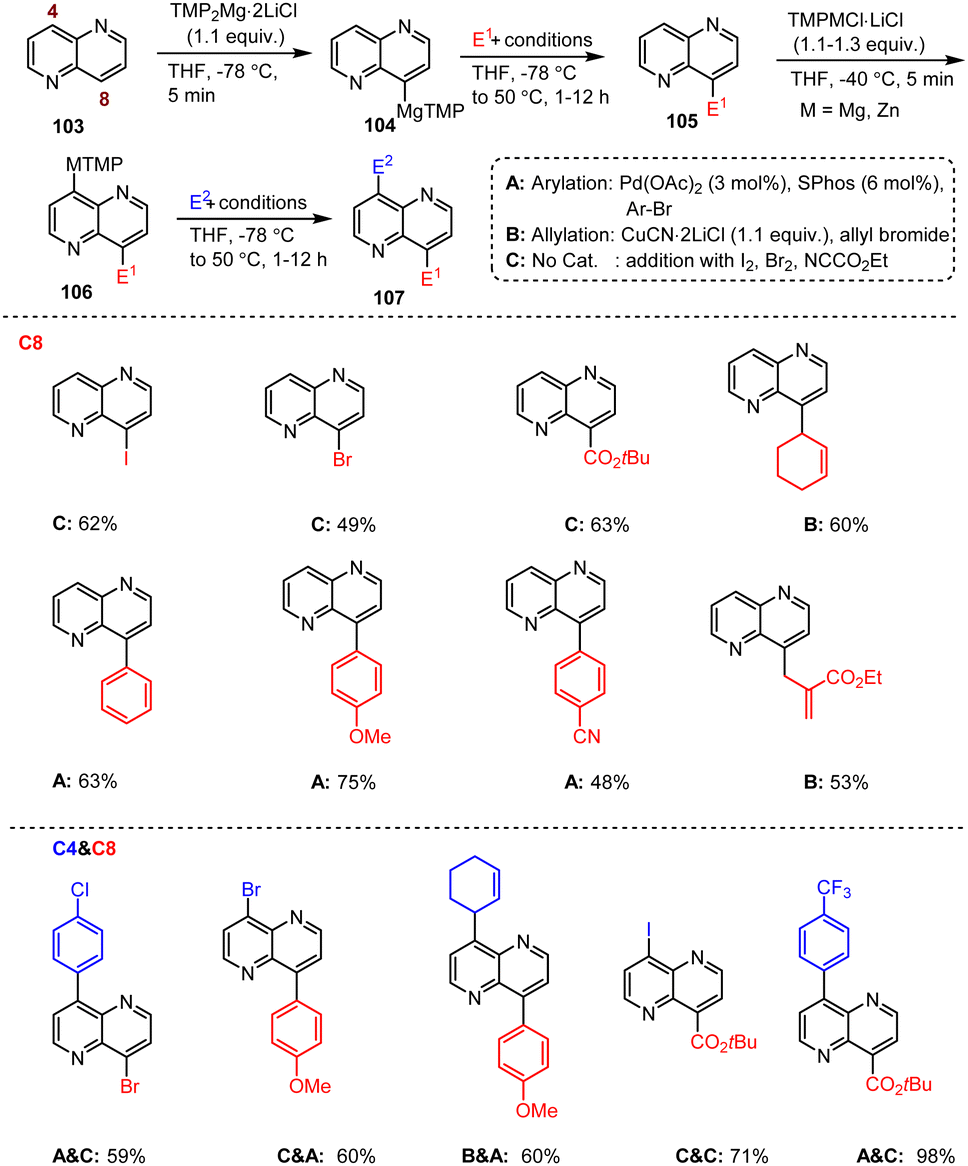 | ||
| Scheme 33 Regioselective metalation and functionalization of 1,5-naphthyridines. | ||
A recent discovery of a highly regioselective functionalization of pyrazolo[1,5-a]pyridine by TMPMgCl·LiCl at the C-2 and C-7 positions, guided solely by the presence or absence of BF3·OEt2, was also described by Balkenhohl et al. A wide range of functionalized pyrazolo[1,5-a]pyridine derivatives 109 and 110 thus obtained under moderate metalation conditions with suitable regioselectivities. The organomagnesium reagents prepared in situ reacted smoothly with various electrophiles, such as I2, C2Cl4Br2, NCCO2Et, MeSSO2Me, and Tietze's reagent. Moreover, CuCN-mediated acylation and allylation reactions were also successfully carried out (Scheme 34).134
 | ||
| Scheme 34 Metalation of pyrazolo[1,5-a]pyridine for functionalization at C-2 and C-7. | ||
Knochel, Wagschal et al. reported a highly regioselective metalation of various aryl-substituted azoles by using a sterically hindered TMPMgBu base. In this reaction, arylazole 111 for example, was allowed to react with TMPMgBu in a toluene/hexane combination at room temperature for 1–6 h before it was subjected to Negishi cross-coupling reactions with various (hetero)aryl halides in the presence of organozinc reagents and a suitable palladium catalyst. The resulting polyfunctionalized arylazole scaffolds 112 were isolated in excellent yields of up to 91% (Scheme 35).135,136 This protocol could be useful for preparing key intermediates of active pharmaceutical ingredients (API), as well as for several late-stage modifications of drug-related aromatic compounds. Mechanistic studies emphasize the key role of the TMPMg base for the observed selectivity, which could be exploited for different cross-coupling reactions and synthetic applications in organic chemistry.
 | ||
| Scheme 35 Regioselective metalation and functionalization of various aryl substituted azole systems using a sterically hindered TMPMgBu base. | ||
Furthermore, Knochel, Bein and co-workers reported a selective functionalization sequence of readily available 7-substituted SEM-protected 1H-imidazo[1,2-b]pyrazole scaffold. Successive functional group installations in the 3- and 2-positions were achieved through consecutive metalations using metal amides followed by quenching reactions with suitable electrophiles. For example, the cyano-substituted 1H-imidazo[1,2-b]pyrazole 116 was selectively metalated at C-3 with TMPMgCl·LiCl to generate the magnesiated intermediate 117, which was then successfully reacted with different electrophiles to provide e.g., allylated, acylated, thiolated as well as Negishi-type arylation products 118 in 65–84% yields (Scheme 36).137
 | ||
| Scheme 36 Selective metalation and quenching reactions of 7-CN substituted 1H-imidazo[1,2-b]pyrazole derivatives. | ||
Further functionalization in position 2 of the 3-ester substituted N-heterocycle 119 was achieved via a bis-organozinc species 120 generated upon treatment with bis-base TMP2Zn·MgCl2·2LiCl at 0 °C in THF. Subsequent Cu-catalyzed acylations with various types of acylchlorides and Negishi-type cross-couplings proceeded smoothly to afford the desired trisubstituted heterocycles 121 in good yields (Scheme 37).137 The high chemoselectivity of the reactive intermediate zinc species even allowed the use of electrophiles containing sensitive functional groups such as an ester or a nitro group.
 | ||
| Scheme 37 Selective metalation and functionalization of the 7-CN and 3-CO2Et disubstituted 1H-imidazo[1,2-b]pyrazoles. | ||
Moreover, this consecutive functionalization sequence was applied to the synthesis of a non-classical isostere 126 of the indolyl drug pruvanserin (Scheme 38). The latter is a selective 5-HT2A serotonin receptor antagonist suffering from low solubility under physiological conditions. Comparative assays between the original drug and the isostere revealed a significantly improved solubility in aqueous media due to the substitution of the indole ring with the 1H-imidazo[1,2-b]pyrazole core. Fused five-membered N-heterocyclic scaffolds such as 1H-imidazo[1,2-b]pyrazoles recently attracted much attention as key structural elements for many pharmaceutical, agrochemical and material science applications.138
 | ||
| Scheme 38 Consecutive functionalization of 1H-imidazo[1,2-b]pyrazole within the synthesis of the pruvanserin isostere 126. | ||
Melzig et al. established an efficient two-step protocol for the 2,3-difunctionalization of benzofuran scaffolds 127 (Scheme 39). Here in the first step, a sulfoxide group acts as a metalation-directing group (DoM) in the presence of TMPMgCl·LiCl to allow smooth ortho-magnesiation and electrophile trapping. In the second step, the sulfoxide group of 128 works instead as a leaving group enabling a sulfoxide-magnesium exchange in the presence of commercially available Turbo-Grignard (i-PrMgCl·LiCl). Upon further reaction of the in situ prepared novel organomagnesium species with electrophiles, highly functionalized heterocyclic compounds 129 were obtained in good yields. The chemoselective TMPMgCl·LiCl and i-PrMgCl·LiCl reagents are compatible with a wide range of functional groups (e.g. F, Cl, CF3, CN, CO2tBu, alkynyl, ether, thioether), so that this method is particularly suitable for gram-scale syntheses in standard laboratories.139
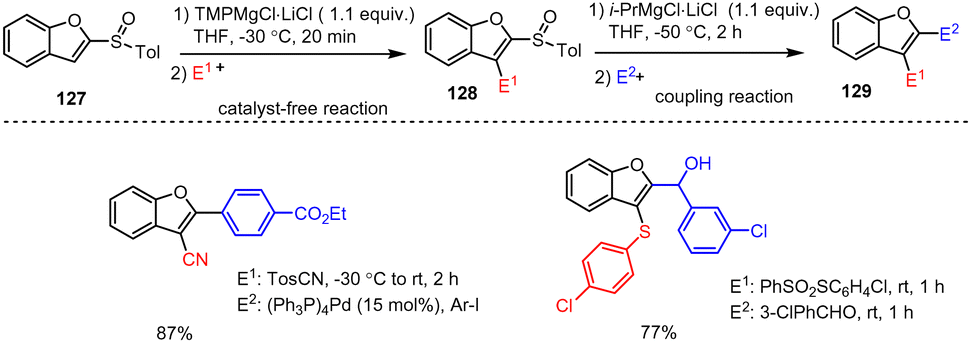 | ||
| Scheme 39 TMPMgCl-Mediated metalation of benzofuran followed by a sulfoxide–Mg exchange reaction and trapping with various electrophiles. | ||
Mongin, Halauko, Chevallier and co-workers developed a deproto-metalation method for several N-arylated 1H-benzotriazoles 130 with TMP-bases. The in situ prepared organometallic reagents reacted readily with commercially available iodine reagents and gave mono- or diiodinated heterocyclic compounds 132 in good to high yields. Varying amounts of the TMP-base (1.5–3 equiv.) were used with substrates bearing electron-rich and electron-poor substituents (Scheme 40).140
 | ||
| Scheme 40 Deproto-metalation of benzotriazole by TMP-base for subsequent iodination. | ||
The same group also succeeded in functionalizing indole derivatives 134 under very similar conditions, i.e., in the presence of TMPLi and ZnCl2·TMEDA. For example, selective deproto-metalation of the protected indoles followed by quenching with I2 led to the formation of the iodinated indole derivatives 135 in satisfactory yields (Scheme 41).141
 | ||
| Scheme 41 Deproto-metalation of protected indole by TMP-base for iodination. | ||
In addition, a strategy for the direct metalation of methylated quinoline and quinoxaline scaffolds 136 using LDA and TMPLi as bases was reported in 2023. Subsequent capture reactions with various electrophiles at low temperatures led to quinolinyl alcohol and amine derivatives 137 in moderate yields (Scheme 42).142
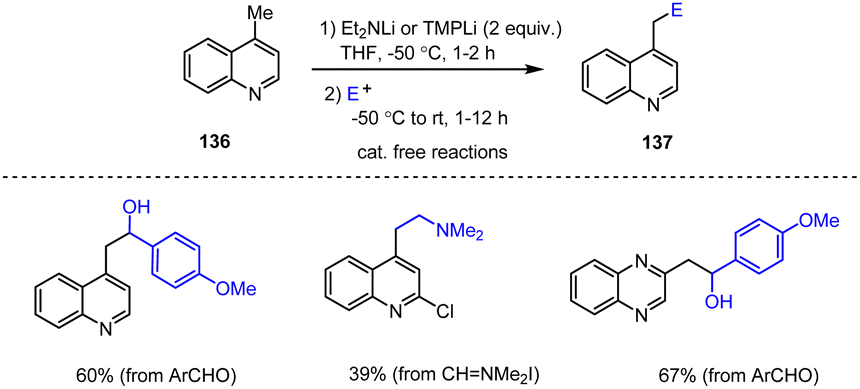 | ||
| Scheme 42 Deproto-metalation of methylated quinolines and quinoxalines using LDA or TMPLi bases for subsequent electrophilic trapping reactions. | ||
Clososki et al. described the straightforward C-2 and C-5 functionalization of indolizine motifs 138 with an ester group at C-1. The directed metalation process took place under mild conditions using the organometallic bases LDA or TMPMgCl·LiCl, whereby the reaction of the corresponding organometallic intermediates (A and B) with different electrophiles enabled the production of difunctionalized indolizines 139 and 140 in high yields. While LDA favoured C-5 functionalization, the TMPMg base yielded mostly C-2 functionalized derivatives via selective ortho-metalation. However, the regioselectivity of these reactions was not only dependent on the choice of the base. Rather, in the case of the TMPMgCl·LiCl-mediated reaction, electrophile-controlled regioselectivity was observed due to a dynamic equilibrium between the two reactive C-2/C-5-organomagnesium species. The scope of applicable substrates for this process is summarized in Scheme 43143 affording a wide range of potentially biologically active heterocyclic compounds in good yields and with high functional group tolerance.
 | ||
| Scheme 43 LDA- and TMPMgCl-mediated selective directed metalation of Indolizine derivatives followed by reaction with various electrophiles. | ||
3 Functionalization of fused bicyclic heteroaromatics with organolithium reagents RLi
Snieckus and co-workers presented a general method for preparing air-stable ortho-boropinacolato aryl sulfonamides 142via the directed ortho-metalation (DoM) method. Metalation of aryl sulfonamides of type 141 with LDA at −78 °C generates highly reactive aryl-lithium species. Subsequent trapping with i-PrOBPin boron reagents (4 equiv.) produced the corresponding ortho-boropinacolato aryl(heteroaryl) sulfonamides 142 in good yields. Moreover, 142 was subjected to a Pd-catalyzed C(sp2)–C(sp2) Suzuki–Miyaura cross-coupling reaction with readily accessible aryl and heteroaryl halides in DMF/H2O at 80 °C providing biaryl and heterobiaryl sulfonamides and in particular functionalized indole scaffolds 143 (Scheme 44).144 This method overcomes previous failings in the preparation of aryl sulfonamide boronic acids and should be of value for medicinal chemistry programs centered on the sulfonamide functional group.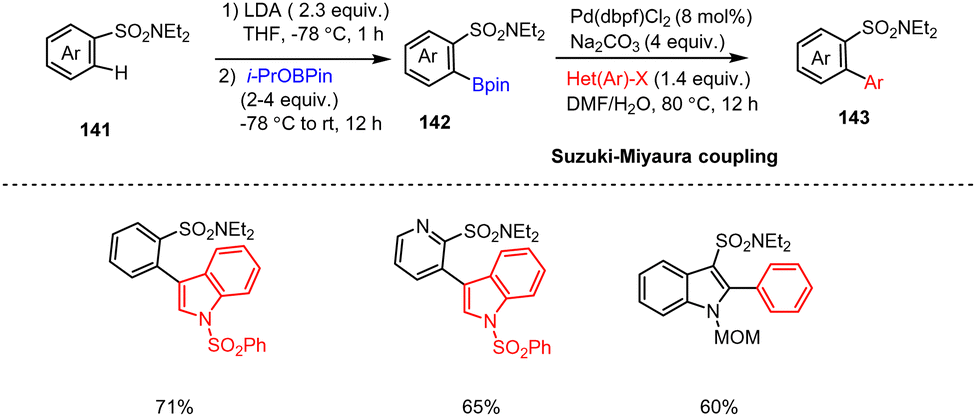 | ||
| Scheme 44 LDA-Mediated one-pot directed ortho-metalation of aryl sulfonamides followed by Suzuki Miyaura cross-couplings. | ||
Recently, Houk, Smith and co-workers developed an efficient protocol for the palladium-catalyzed cross-coupling reaction of readily available aryl and pyridyllithium reagents 145 at room temperature using a reusable siloxane transfer reagent 144. The crystalline, bench-stable siloxane reagent is easily prepared in a one-step protocol, and its use eliminates the need for pre-functionalization, as well as isolation of organometallic cross-coupling partners. Importantly, this reagent can be recovered and reused without sacrificing reactivity. Both electron-rich and electron-poor substrates can be efficiently cross-coupled, and a variety of common functional groups on the electrophilic partner were well tolerated (i.e. esters, nitriles, azaheterocycles, fluorinated aromatics and quinolines) as well as sterically burdened aryl chlorides. Hence, functionalized quinoline and benzoxazole derivatives 146 can be obtained in acceptable yields by C(sp2)–C(sp2) cross-coupling reactions of aryl lithium and aryl chlorides in the presence of Pd pre-catalyst (1–5 mol%) and the XPhos ligand (Scheme 45).145 The Pd-catalyzed cross-coupling reaction of the heteroaryl chlorides with aryllithium reagents comprises the following typical steps: oxidative addition to the intermediate A, followed by transmetallation with ArLi to form intermediate B. Reductive elimination finally yields the desired heterocyclic product 146 in good yield and regenerates the active Pd catalyst for the next cycle run.
 | ||
| Scheme 45 Pd-Catalyzed cross-coupling reactions of heteroaryl chlorides with aryl lithium reagents in the presence of siloxane transfer reagent 144. | ||
Efficient regioselective functionalization of fused 7-azaindole heterocycles using LDA and amide bases was reported by Kitching, Snieckus and co-workers. Carbamoyl-protected 7-azaindole 147 underwent a rapid LDA-mediated regioselective metalation followed by trapping with various electrophiles to generate C-2-substituted 7-azaindole derivatives 149 in excellent yields (Scheme 46).146
 | ||
| Scheme 46 Regioselective functionalization of Carbamoyl-protected 7-azaindoles using LDA. | ||
Under similar mild conditions, the regioselective functionalization of 7-azaindole by controlled ring isomerization using directed metalation group (DMG) migration was also achieved (Scheme 47).147 By using a TMPLi base, azaindole 150 was first regioselectively metalated at the C-6 position and then quenched with an electrophile to obtain a C-6-substituted derivative 151. Subsequently, a carbamoyl group shift (a dance from N7 to N1) was performed in the presence of a catalytic amount of ClCONR2, leading to the formation of product 152. A second directed metalation and electrophile quench sequence then provided 2,6-substituted azaindoles 153. Overall, the controlled migration of the carbamoyl group enables multiple functionalization events of the bioactive azaindole scaffold, bypassing the removal and introduction of another DMG and instead allowing the same DMG group to direct functionalization at a new, distant site. Furthermore, the use of the directed metalation group dance strategy could be applied to a late-stage deuteration of an antipsychotic compound (L-745870) demonstrating a new strategy for the functionalization of the bioactive azaindole scaffold and related N-heterocycles.
 | ||
| Scheme 47 Regioselective functionalization of 7-azaindole at C-6 and C-2 positions via controlled DMG migration. | ||
The same research group also reported a method for a rather unusual C-4 peri-metalation of NH-free azaindoles 154, in which CONEt2 and SO2NEt2 groups as DMG on C-3 allowed the highly regioselective synthesis of 4-substituted derivatives via anionic shielding of C-2. The reaction is robust and scalable. If such anionically shielding DMG is introduced at C-4 (156), successive ortho-metalations (DoM) of C-2 (157) and C-5 (158) are possible. The multiple, sequential DoM reactions (i.e., ring-walk metalation sequences) provide a rational and generally regioselective route to polysubstituted 7-azaindoles 159 and other heterocycles of potential pharmaceutical significance (Scheme 48).148
 | ||
| Scheme 48 Organolithium-mediated C-4-peri-metalation of substituted 7-azaindole derivatives followed by electrophilic additions. | ||
4 Functionalization of fused bicyclic heteroaromatics by organo-Mg reagents (RMgX)
In 2017, Provot and co-workers developed an efficient Kumada coupling reaction between chlorovinyl benzothiophene 160 and Grignard reagent 161 in the presence of Fe(acac)3 (10 mol%) in THF at −30 °C leading to the highly substituted benzothiophene 162 in good yields (Scheme 49).149 | ||
| Scheme 49 Fe-Catalyzed Kumada coupling for the synthesis of 2,3-disubsituted benzothiophene derivatives. | ||
Knochel and co-workers have shown that similar to Fe(III) catalysis, chemoselective cross-coupling reactions of the Kumada type can also be carried out using catalytic amounts of chromium(II) chloride (Scheme 50).150 Thus, substrates considered challenging such as isoquinoline and quinoline derivatives 163 were successfully coupled with a wide range of functionalized aryl(heteroaryl)-Grignard reagents 164 in the presence of CrCl2 (3 mol%). These reactions, which led exclusively to the formation of the α-arylated heterocycles 165 without significant amounts of homocoupling products, proceed rapidly within minutes at room temperature in cyclopentyl methyl ether (CPME). Several functional groups, including esters and acetals, are tolerated by this method and the required chromium salts can be successfully separated after the reaction using solid supports.
 | ||
| Scheme 50 CrCl2-Catalyzed cross-couplings of 2-quinolinyl halides with ArMgX. | ||
Later, the research group also developed a strategy for the transition metal-free amination of 8-quinoline sulfonic acids 166 using magnesium amides of the type R2NMgCl·LiCl. Thus, numerous cyclic secondary amines were first successfully converted by i-PrMgCl·LiCl into the corresponding magnesium amides, which in turn reacted readily with the quinoline sulfonic acids under mild conditions and provided the desired aminoquinolines 167 in excellent yields (Scheme 51).151 Aminoquinoline scaffolds derived from heteroaryl sulfonic acids are of major interest in pharmaceutical chemistry and drug development.
 | ||
| Scheme 51 Catalyst-free amination reaction of 8-quinoline sulfonic acid using magnesium amides. | ||
Steib et al. reported on a ligand-free Cr-catalyzed amination reaction of various N-heterocyclic quinoline and quinoxaline chlorides. The catalytic regioselective amination of 2-chloroquinolines, 1-chloroisoquinolines and 2,3-dichloroquinoxalines 168 with a wide range of aliphatic, benzylic, and saturated (hetero-)cyclic magnesium amides were described. The C–N coupling reactions were carried out in THF at 50 °C for 2–24 h and led to the desired aminated bicyclic heteroaromatic compounds 169 in 54–95% yields (Scheme 52).152
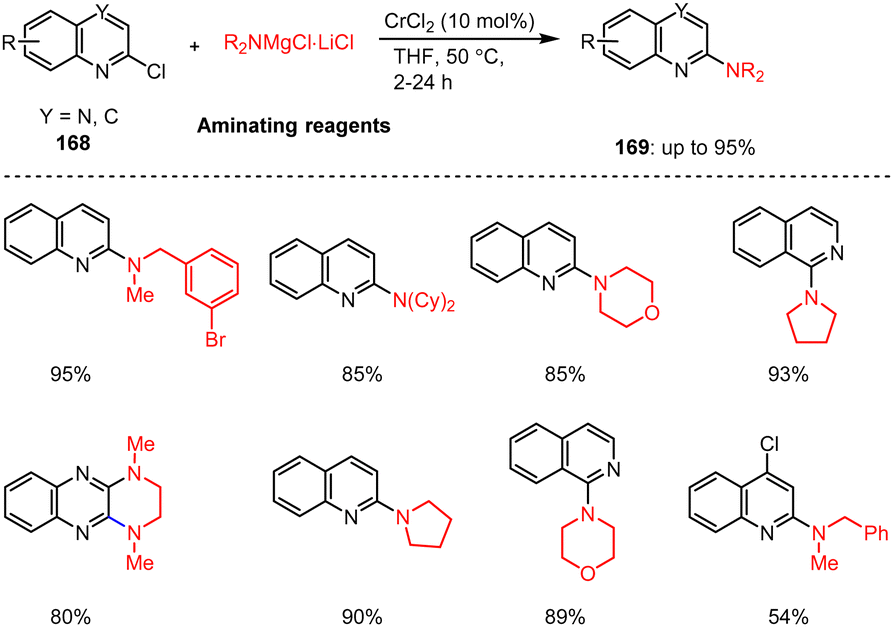 | ||
| Scheme 52 Cr(II)-Catalyzed amination of 2-quinonyl chlorides with magnesium amides. | ||
The highly regioselective synthesis of functionalized S-heterocycles 172 under mild conditions and with the use of five-membered fused 2-thienyl-magnesium intermediates 171 was presented by Sämann et al. For this purpose, an efficient Br/Mg exchange reaction between unsymmetrically substituted dibromothienothiophenes 170 and i-PrMgCl·LiCl in THF at 0 °C was carried out in good yields. Ring substituents, such as thioether or trimethylsilyl groups, as well as pyridyl and thienyl groups or ortho-substituted aryl groups of the thienothiophenes directed the Br/Mg exchange at position C5 with excellent regioselectivity of up to >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 53).153 Subsequently, these heterocyclic magnesium derivatives were selectively functionalized with electrophiles such as aldehydes, aryl iodides, acyl chlorides or aryl sulfinyl chlorides, providing the targeted thienothiophene derivatives. Finally, the resulting bromo heterocycles can be readily subjected to a second Br/Mg exchange, followed by further electrophilic functionalizations.
:1 (Scheme 53).153 Subsequently, these heterocyclic magnesium derivatives were selectively functionalized with electrophiles such as aldehydes, aryl iodides, acyl chlorides or aryl sulfinyl chlorides, providing the targeted thienothiophene derivatives. Finally, the resulting bromo heterocycles can be readily subjected to a second Br/Mg exchange, followed by further electrophilic functionalizations.
 | ||
| Scheme 53 Preparation of functionalized thienothiophenes by reaction of heteroaryl magnesium reagents with electrophiles. | ||
A simple and practical method was developed by Kuzmina et al. using a non-toxic iron catalyst. This new approach for 2-substituted quinoline and isoquinoline scaffolds allowed smooth C(sp2)–C(sp2) cross-couplings of N-heterocyclic halides 173 with various electron-poor and electron-rich aryl magnesium reagents (ArMgX, 164) in the presence of FeBr3 (3 mol%) (Scheme 54).154 The inexpensive and efficient iron cross-coupling reaction was carried out in a mixture of THF and tBuOMe at 20 °C and turned out to be a key procedure for the preparation of highly functionalized N-heterocycles 174 in excellent yields with no formation of undesired homocoupling by-products. Thus, organomagnesium reagents with a variety of sensitive functional groups such as F, Cl, CF3, OPiv, OBoc, OMe, Me, and NMe2 were successfully employed in this method. The resulting quinolines and heterocyclic derivatives thereof have shown major promise for the treatment of several diseases, including inflammation, cancer, diabetes, and malaria, as well as for various viral infections.
 | ||
| Scheme 54 FeBr3-Catalyzed cross-coupling reactions of quinoline- and isoquinoline-based halides with functionalized organomagnesium reagents. | ||
In 2013, Knochel and co-workers published a simple and efficient catalytic process for the preparation of 2- or 4-substituted heterocyclic quinoline motifs 176. Using CrCl2 (3 mol%) catalyst, N-heterocyclic chlorides 175 could be successfully used in a Kumada cross-coupling reaction with aryl(het)-magnesium reagents 164 under sustained conditions. When operated for 15 min up to 2 h in THF at 25 °C, this technique provided substituted quinolines 176 with good yields of up to 89% (Scheme 55).155
 | ||
| Scheme 55 CrCl2-Catalyzed coupling reaction of quinoline-based chlorides with arylmagnesium reagents. | ||
In addition, the same group reported the serendipitous discovery that the addition of quinoline or isoquinoline dramatically increased the rate and yield of Fe- and Co-catalyzed cross-coupling reactions. This ligand acceleration allowed the general scope of the described cross-coupling reactions to be extended to complex functional groups and the formation of heteroaryl–heteroaryl bonds, where the desired products were previously obtained at very low yields. For example, by using CoCl2 (3 mol%) as a catalyst in the presence of isoquinoline (10 mol%), functionalized aryl and heteroaryl Grignard reagents 164 were successfully coupled with Br-, Cl-substituted quinolines 177 in reasonable yields (Scheme 56).156
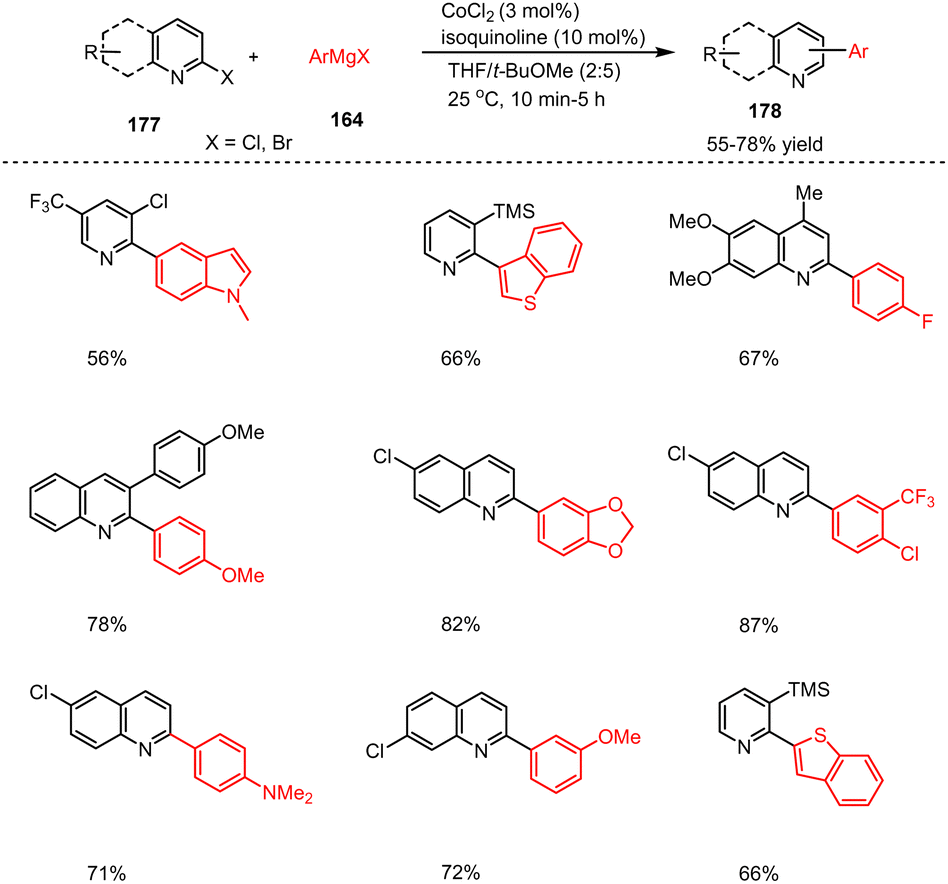 | ||
| Scheme 56 Co-Catalyzed Kumada coupling of 2-quinonyl halides. | ||
Starting from easily accessible bromoaniline derivatives 179, Frischmuth et al. investigated a mild and efficient intramolecular Cu-mediated carbomagnesiation method for the preparation of functionalized 4-azaindoles of type 181. Subsequent further functionalization of these 4-azaindoles with various electrophiles thus provided access to highly functionalized N-heterocycles of type 184 in excellent yields of 74–85%. The preparation of key magnesium intermediates 180 for intramolecular cyclization was carried out by halogen–metal exchange reactions using i-PrMgCl·LiCl, tolerating a broad spectrum of functional groups in the substrate. Starting from TMS-containing heterocyclic precursor 181, which is available in a few steps, the 3-iodoazaindole 182 was prepared by treatment with ICl, and then subjected to the halogen–metal exchange followed by an electrophilic scavenging reaction with acid chlorides. The corresponding, highly functionalized 4-azaindole ketones 184 were obtained in reasonable amounts (Scheme 57).157
 | ||
| Scheme 57 Synthesis of 2,3-disubstituted 7-azaindoles using i-PrMgCl. | ||
Barl et al. reported the complete functionalization of the 7-azaindole scaffold 188 using the following reaction sequence: halogenation, cyclization, directed metalation, and halogen/Mg as well as sulfoxide/Mg exchange. By using this procedure, a complex and fully functionalized 7-azaindole 197 was obtained starting from the commercially available aniline derivative 185via sequential transformation of the corresponding substituted key intermediate 188 (Scheme 58).158 Using n-BuLi, TMPLi and i-PrMgCl as key metalating reagents, this multistep protocol afforded the desired key azaindole structure 197 with moderate yields and high tolerance of functional groups.
 | ||
| Scheme 58 Method for full functionalization of the 7-azaindole core by selective metalation and sulfoxide/Mg exchange reaction. | ||
In 2013, Knochel and co-workers developed a formal regioselective C(sp2)–C(sp3) cross-coupling of substituted pyridines and quinoline derivatives 198via mild BF3·OEt2-mediated nucleophilic addition reaction of Grignard or organozinc reagents, followed by chloranil-mediated oxidative aromatization. The high regioselectivity and broad tolerance towards functional groups make this method very valuable for the preparation of polyfunctional pyridines and quinolines 199 (Scheme 59).159 Moreover, these reactions can be easily carried out on a larger scale with no reduction in yield.
 | ||
| Scheme 59 Lewis acid-promoted direct alkylation of quinoline derivatives with alkylmagnesium reagents. | ||
A general and mild intramolecular method for a copper-mediated carbomagnesiation reaction was presented by Nickel et al., which can be used to synthesize functionalized N-heterocycles such as pyrrolo[2,3-d]pyrimidines 201 and azaindole derivatives 208. In this work, pyrrolo[2,3-d]pyrimidines were prepared from metalated pyrrolo[2,3-d]pyrimidines, which in turn were accessible by treatment of N-alkynyl-5-iodo-6-sulfamido-pyrimidines with i-PrMgCl·LiCl, followed by transmetalation with CuCN·2LiCl and intramolecular carbocupration. Finally, the desired polyfunctional pyrrolo[2,3-d]pyrimidines 200 were obtained after an electrophilic quenching reaction with allyl halides or acid chlorides. In addition, subsequent reaction with ICl, followed by Negishi cross-coupling in the presence of PEPPSI-iPr as the catalyst, allows these compounds to be further functionalized and converted into fully substituted N-heterocycles 204 (Schemes 60 and 61).160,161
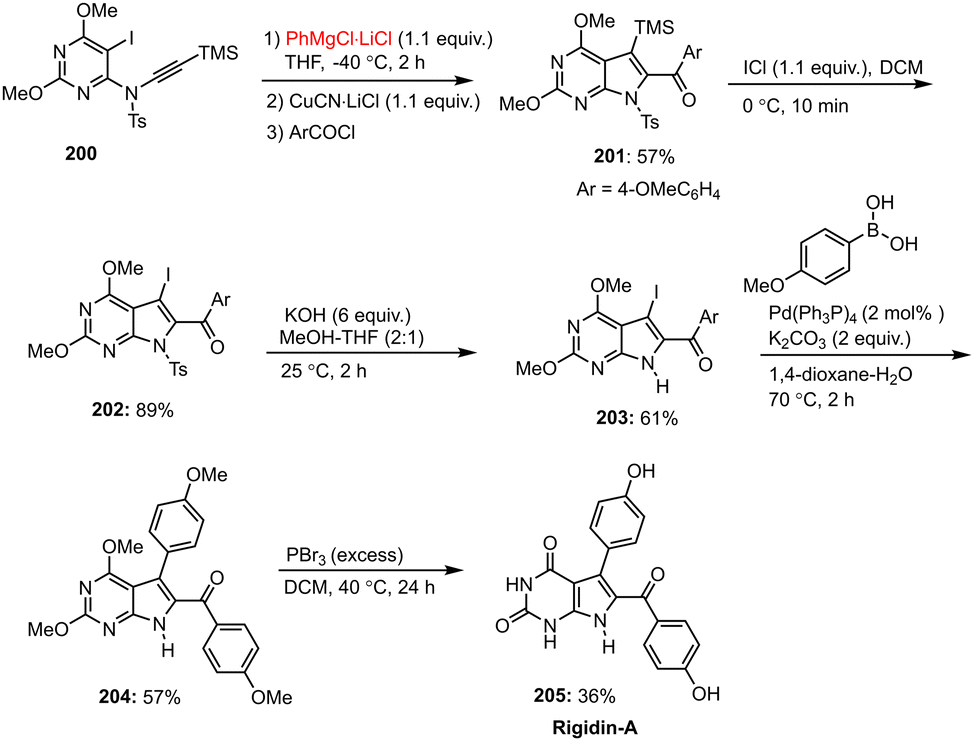 | ||
| Scheme 60 Synthesis of rigidin-A using Grignard reagents. | ||
 | ||
| Scheme 61 Synthesis of 7-azaserotonin analog 211 using RMgX. | ||
The methods described could offer short synthetic routes to biologically relevant molecules, as shown by the formal synthesis of the marine alkaloid rigidin-A 205 and of 7-azaserotonin 211, a closely related serotonin derivative (Schemes 60 and 61).160,161
Greiner et al. reported a practical method for the modification of 1,8-disubstituted 2,7-naphthyridine derivatives by iron-catalyzed C(sp2)–C(sp3) cross-couplings with alkyl Grignard reagents. To incorporate alkyl substituents into the naphthyridine backbone, the 3,6-chloro substituents of 2,7-naphthyridine 212 have been replaced through cross-coupling reactions with various organomagnesium compounds 164a resulting in tetrasubstituted symmetric and unsymmetric naphthyridine motifs 213 (Scheme 62).162
 | ||
| Scheme 62 Fe-Catalyzed cross-couplings of the 1,8-dimethylnaphthyridine with alkyl Grignard reagents. | ||
Similarly, iodoisoquinolines and chloroquinolines 214 can be alkylated with alkylmagnesium reagents 164b using the tetrahydrofuran-soluble chromium(III) complex CrCl3·3THF. This efficient chromium(III)-catalyzed C(sp2)–C(sp3) cross-coupling protocol proceeds within minutes at room temperature and yields the desired products 215 without the formation of homocoupling by-products that are commonly observed in the related iron-, cobalt- or manganese-catalyzed reactions (Scheme 63).163
 | ||
| Scheme 63 Cr(III)-Catalyzed C(sp2)–C(sp3) Kumada coupling reactions of alkylmagnesium reagents with 2-chloroquinoline. | ||
In addition, Ziegler et al. developed a new halogen–Mg exchange reagent 217 using the alkylmagnesium alkoxide s-BuMgOR·LiOR (R = 2-ethylhexyl), which undergoes very fast Br/Mg and Cl/Mg exchange reactions in toluene. These processes are not only about 30 times faster than those of the previously used exchange reagent sBu2Mg·2LiCl, but also about 110 times faster than those with Turbo-Grignard (i-PrMgCl·LiCl). In addition, the resulting Grignard reagents of the type ArMgOR·LiOR or HetArMgOR·LiOR can be easily added to ketones and acyl chlorides to form 218-type reaction products under mild conditions (Scheme 64).164 The synthesis of Grignard reagents in hydrocarbons or toluene is of great interest, as these weakly coordinated Grignard reagents can exhibit unusual reactivity and are also considered as industry-friendly reagents.
 | ||
| Scheme 64 Functionalized fused heteroarenes via Br/Mg-exchange reaction and quenching with electrophiles. | ||
Aminated N-heterocycles play an important role in modern pharmaceutical chemistry, which makes the development of synthetic protocols for their efficient preparation very attractive. In 2018, Knochel and colleagues presented the amination of phosphorodiamidate-substituted quinolines and quinoxalines with the magnesium amides R2NMgCl·LiCl under catalyst-free conditions. Here, 2-, 4- and 8-hydroxyquinoline and 2-hydroxyquinoxaline derivatives were converted to the corresponding phosphorodiamidates 219 and subjected to an amination reaction using various pharmaceutically active amines to give the fused N-heterocycles of type 220 (Scheme 65).165,166
 | ||
| Scheme 65 Synthesis of 2-amino quinoline and quinoxalines with R2NMgCl·LiCl. | ||
Since the phosphorodiamidate function is a strong direct metalating group (DMG), the amination can be combined with an ortho-functionalization step. Thus, several phosphoro-diamidate-substituted N-heterocycles 221 were treated at 0 °C for 1 h with TMPMgCl·LiCl or TMP2Mg·2LiCl in THF. The type 222 Mg species formed were then either quenched with electrophiles such as I2 or (BrCl2C)2 or were successfully employed in Cu-catalyzed acylation reactions and Pd-catalyzed Negishi cross-couplings after Zn transmetalation steps. The resulting functionalized heterocycles 223 were finally subjected to the amination reaction that led to 2,3-difunctionalized quinoline derivatives 224 in 35–66% yields over two steps (Scheme 66).165
 | ||
| Scheme 66 TMP-base mediated directed ortho-metalation and functionalization of various aryl phosphorodiamidates, followed by amination with R2NMgCl·LiCl. | ||
Sulfur-containing organic molecules are often useful building blocks in organic synthesis, which is why, transition metal-catalyzed desulfinative cross-coupling reactions are used to produce heterobiaryl products, for example. As these processes usually require high temperatures due to catalyst deactivation, milder and transition metal-free desulfination protocols are desirable. Along this line, Wei et al. presented a cross-coupling reaction of heteroaryl sulfinates with Grignard reagents for the synthesis of heterobiaryls. Here, fused N-heteroaryl sulfinates 225 were reacted with functionalized aryl and alkyl Grignard reagents 164c to produce quinoline-based biaryl heterocycles 226 under mild conditions (Scheme 67).167 The transformation demonstrates great potential for the preparation of functionalized fused heteroaromatic compounds by utilizing quinoline and pyridine sulfinates as electrophilic starting materials for C–N cross-coupling reactions with no need for additional catalysts or bases.
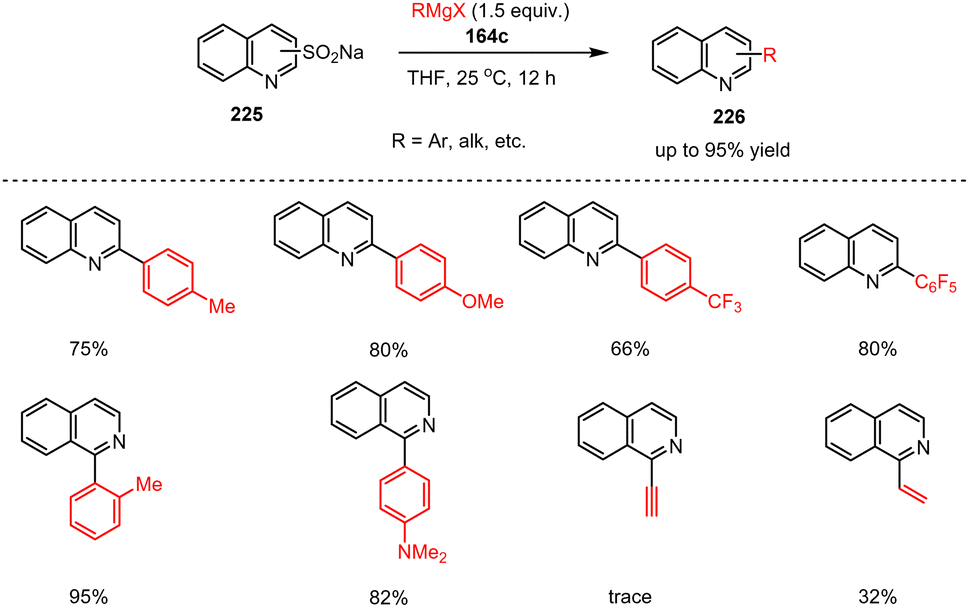 | ||
| Scheme 67 Transition-metal-free functionalization of heterocycles via desulfinative cross-coupling of quinolinyl sulfinates with aryl and alkyl Grignard reagents. | ||
A selective acylation protocol developed by the Knochel group for readily available heteroaryl magnesium reagents 227 and commercially available ester derivatives run at favourable temperatures (−5 to 25 °C) and short reaction times (2–10 min) under continuous flow conditions. The flow conditions prevent premature collapse of the hemiacetal intermediates despite non-cryogenic conditions, thereby providing satisfactory yields for heteroaryl ketones 229. The coordination ability of the ester and aryl(het)magnesium reagents was decisive for the outcome of the acylation reaction (Scheme 68).168
 | ||
| Scheme 68 Synthesis of heteroaryl ketone using ester scaffolds and Grignard reagents. | ||
Knochel, Bein and co-workers reported the selective functionalization of the 5-Br-substituted 1H-imidazo[1,2-b]pyrazole scaffold 230 using a Br/Mg exchange reaction with i-PrMgCl in THF at r.t. The resulting reactive Mg intermediate 231 reacted with various commercially available electrophiles, such as by direct quenching with highly reactive electrophiles or by Pd-catalyzed cross-coupling as well as Cu-mediated acylation or allylation reactions. Overall, these sequences led to the formation of functionalized condensed N-heterocycles 232 in good to excellent yields (Scheme 69).137,169
 | ||
| Scheme 69 Selective functionalization of the brominated fused aromatic 1H-imidazo [1,2-b]pyrazole via Br/Mg exchange reaction with i-PrMgCl. | ||
Kürti, Ess and co-workers have described an intramolecular amination of arene C(sp2)–H bonds at low temperature, without the use of transition metals and with high regioselectivity. The reaction is operationally simple and scalable (1–10 mmol) and allows the formation of fused N-heterocycles 234 under mild conditions, using readily available 2-nitrobiaryls 233 and PhMgBr (Scheme 70).170 Initially, the Grignard reagent attacks the nitro group of 233, whereby a reactive aryl nitroso intermediate B is formed after elimination of magnesium phenolate. This species then reacts with a second equivalent of PhMgBr to form an aryl nitrene intermediate D, which cyclizes intramolecularly to the desired carbazole derivative 234. This method also allowed the synthesis of the two bioactive carbazole alkaloids Clausin V and Glycoborin.
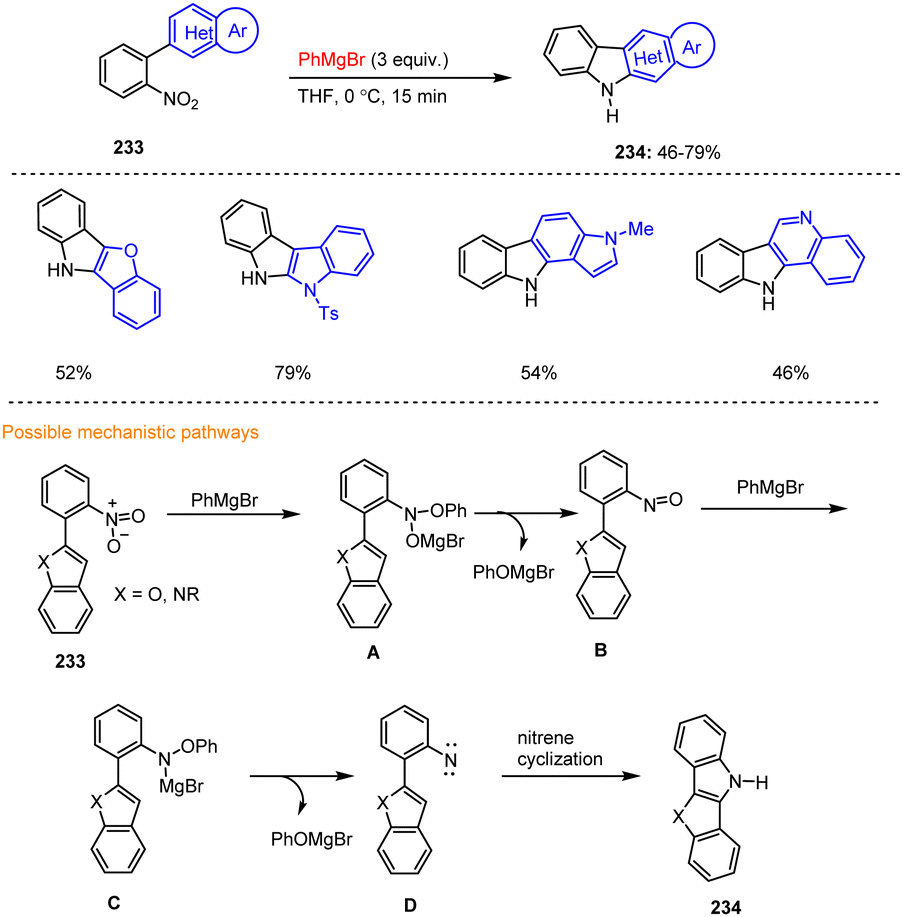 | ||
| Scheme 70 Highly regioselective synthesis of fused carbazoles using Grignard reagents. | ||
In 2010, the group of Knochel and co-workers developed a simple and extremely versatile one-pot strategy for the preparation of organo-fluorine compounds by converting functionalized aryl halides 235 into aryl and heteroaryl fluorides 237. This method enables efficient and direct, metal-free syntheses of fluorinated isoquinoline and benzothiophene derivatives as well as arylated fluorine compounds, which are otherwise difficult to prepare by the conventional catalytic methods (Scheme 71).171
 | ||
| Scheme 71 Electrophilic fluorination of heteroaryl magnesium reagents using NFSI. | ||
The dearomative functionalization of heteroaromatics is one of the most straightforward approaches for the synthesis of chiral heterocyclic systems, key building blocks for both synthetic chemistry and drug discovery. Yan et al. have recently presented a catalytic system that enables the nucleophilic dearomatization of quinolines 238 in combination with organometallic compounds very efficiently. A synergistic combination of Lewis acid, chiral copper(I) catalyst, and Grignard reagent allows to overcome the energy barrier of dearomatization and to obtain chiral C-4 addition products 240 with almost absolute regio- and stereo-control. Remarkably, in a preparative reaction, the amount of chiral copper catalyst can be reduced to 0.1 mol%, leading to the highest turnover number (TON) reported so far in any enantioselective reaction with Grignard reagents. Molecular modelling suggests that the role of the Lewis acid is not only to activate the substrate towards potential nucleophilic addition, but also to subtly direct the regiochemistry, by preventing C-2 addition (Scheme 72).172
 | ||
| Scheme 72 Dearomative functionalization of quinoline position C-4 to access tetrahydroquinolines. | ||
A convenient method for the synthesis of functionalized quinolines is based on site-selective modification by C–H functionalization of easily accessible quinoline scaffolds. An illustrative example of such an approach involving Ni-catalyzed C–H bond arylation of 8-aminoquinoline motifs at the distant C-4 position was presented by Qiu, Kambe and co-workers. The authors proposed the following reaction mechanism for this catalytic direct C–H arylation at C-4 of 8-aminoquinoline scaffolds: first, deprotonation of the quinolinylamide 241 with t-BuOK takes place, resulting after isomerization, in the formation of intermediate B. Its coordination to an in situ generated Ni(0) species leads to the key intermediate C, which then undergoes a nucleophilic addition of ArMgX to afford intermediate D. After HMgX elimination and subsequent protonation, the desired functionalized quinolines 242 are formed. The protocol shows a broad range of functional group tolerance to various functionalized aryl Grignard reagents 164 and aminoquinoline motifs 241 providing the desired arylated products 242 in good to excellent yields (Scheme 73).173 As a result, rapid access to bioactive and multi-substituted aminoquinolines is made possible.
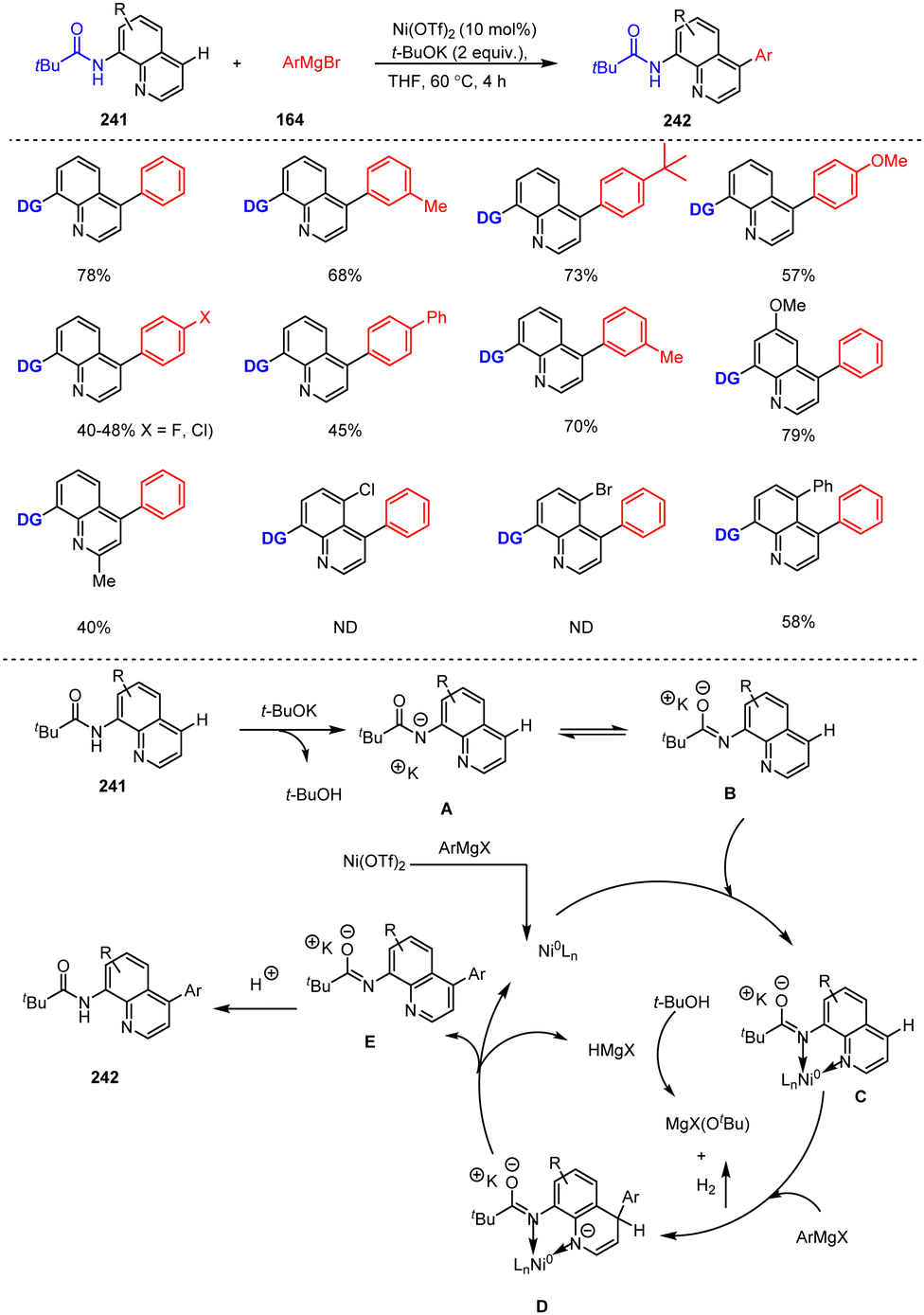 | ||
| Scheme 73 Ni-Catalyzed remote C(sp2)–H bond arylation of 8-amino protected quinolines. | ||
A practical and efficient solution for the direct amination of heteroaryl Li, Mg and Zn reagents, which has long been considered difficult, was developed in Kürti's group. Heteroaryl organometallics such as quinoline derivatives 243 could be converted to the desired amination products in the absence of directing groups by using an oxaziridine or hydroxylamine reagent 244 in the presence of Cu(I) salts. This new approach for direct electrophilic amination thus seems to represent a general yet simple method that could be used for the efficient production of many structurally complex active pharmaceutical compounds and natural products (Scheme 74).174
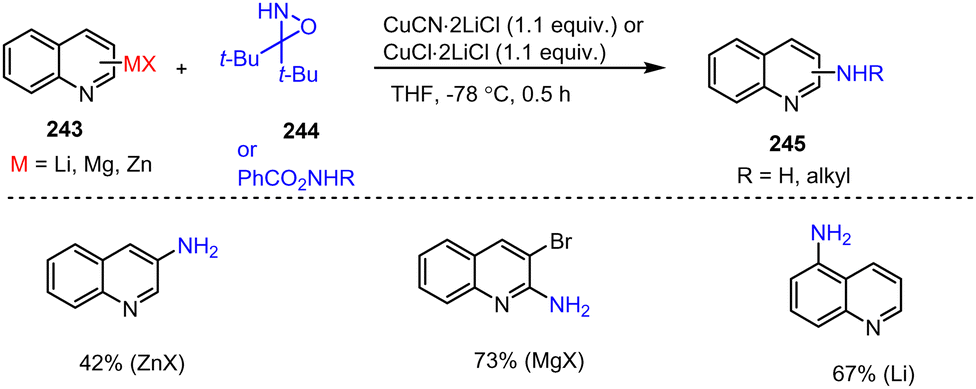 | ||
| Scheme 74 Amination of structurally diverse heteroaryl-based organometallic reagents. | ||
Recently, Harutyunyan's group developed a simple and chemoselective asymmetric addition protocol for the efficient construction of chiral heterocyclic molecules of type 247. Using readily available Grignard reagents 164b and an activating Lewis acid (BF3·OEt2), a wide range of β-substituted conjugated alkenyl-N-heteroaromatics 246 could be chemo- and enantioselectively alkylated by copper-catalyzed conjugate addition. This synthetic protocol is of particular interest for applications in medicinal chemistry as it allows the introduction of linear, branched, and cyclic alkyl chains as well as a phenyl group at the β-carbon position of the alkenyl N-heteroaromatic compounds. The overall synthetic success depends largely on the interplay of the substrate-activating Lewis acid and the use of highly reactive Grignard reagents in the presence of a diphosphine-stabilized copper catalyst (Scheme 75).175
 | ||
| Scheme 75 Catalytic asymmetric conjugate addition of alkyl Grignard reagents to alkenyl-substituted benzothiazole and benzoxazole. | ||
The same group subsequently reported a related reaction in which alkyl Grignard reagents 161 are added enantioselectively under mild conditions to vinylogous imines synthesized in situ from sulfonylindoles 248. In the presence of a copper(I) salt and a chiral phosphoramidite ligand, high yields, and enantiomeric ratios of chiral 3-sec-alkyl-substituted indoles 249, which are important structural elements of several drugs and alkaloids, can be obtained (Scheme 76).176 In addition, the reaction can also be conducted on a larger scale with only 1 mol% of the catalyst and with no loss of yield or enantiomeric purity of the product.
 | ||
| Scheme 76 Cu-Catalyzed enantioselective addition of ethyl Grignard reagents to indole-derived vinylogous imines. | ||
In addition to indole motifs, substituted benzofuran scaffolds, e.g.251, are also important structural elements in pharmacologically active compounds, for instance in protein tyrosine phosphatase inhibitors. Thus, Baralle et al. successfully demonstrated an efficient cross-coupling reaction between branched alkyl Grignard reagents 164b and 2-thiomethylbenzofurans 250 employing a Ni-NHC (Ni-N-heterocyclic carbene) complex as the catalyst. Even 3-(4-biphenylyl)-2-alkylbenzofurans can be assembled in one step employing reaction, rendering such compounds accessible in a diversity-oriented manner to be used as intermediates for the preparation of PTP 1B inhibitors (Scheme 77).177
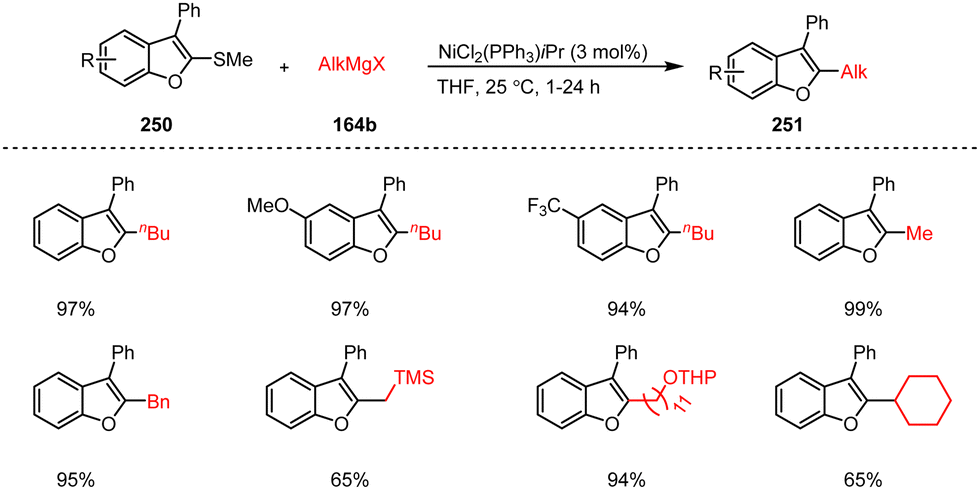 | ||
| Scheme 77 Ni-Catalyzed Kumada cross-coupling of 2-thiomethyl benzofurans with alkyl Grignard reagents. | ||
5 Organozinc-mediated functionalization of fused bicyclic heteroaromatics and their scaffolds
Organozinc reagents are well known for their transmetalation abilities and excellent tolerance towards functional groups. Organozinc pivalates (RZnOPiv) have been reported as salt-stabilized solid aryl, heteroaryl and benzyl organozinc compounds, which not only exhibit improved resistance to air and moisture, but also show excellent reactivity in Negishi cross-coupling and carbonyl addition reactions. These organozinc compounds are prepared in a one-pot process under mild conditions from aryl and heteroaryl halides (Br, Cl) as well as benzyl chlorides 252 using Mg and Zn(OPiv)2·2LiCl (Scheme 78).178 The solid materials 253 obtained after evaporation of the solvent can be stored at room temperature under argon for months and may even be exposed to air for short periods. | ||
| Scheme 78 Pd-catalyzed C(sp2)–C(sp2)-cross-coupling of solid aryl, heteroaryl and benzylic organozinc pivalates with aryl electrophiles. | ||
In a subsequent study, the pool of solid organozinc pivalates was further expanded and their stability in air was improved while their reactivity in Negishi cross-coupling reactions was maintained. The decisive factor here was the use of a directed metalation procedure of arenes and heteroarenes with TMPMgCl·LiCl followed by transmetalation with Zn(OPiv)2 (Scheme 79).179 The Pd-catalyzed Negishi cross-coupling reaction of aryl halides 257 with heteroaryl-zinc reagents generates the depicted intermediates A–C by means of the well-known reaction sequence of initial oxidative addition to B, subsequent transmetalation with HetAryl–ZnOPiv 256 to form the key intermediate C and final reductive elimination to the desired cross-coupling product 258. This protocol also yielded fine powdered organozinc pivalates 256 that were easy to handle after evaporation of the solvent and that retained most of their activity (>85%), even when exposed to air for 4 h. Moreover, they could be readily used in Negishi cross-coupling reactions with a wide range of electrophiles 257 employing technical grade solvents, which is particularly important for industrial applications.
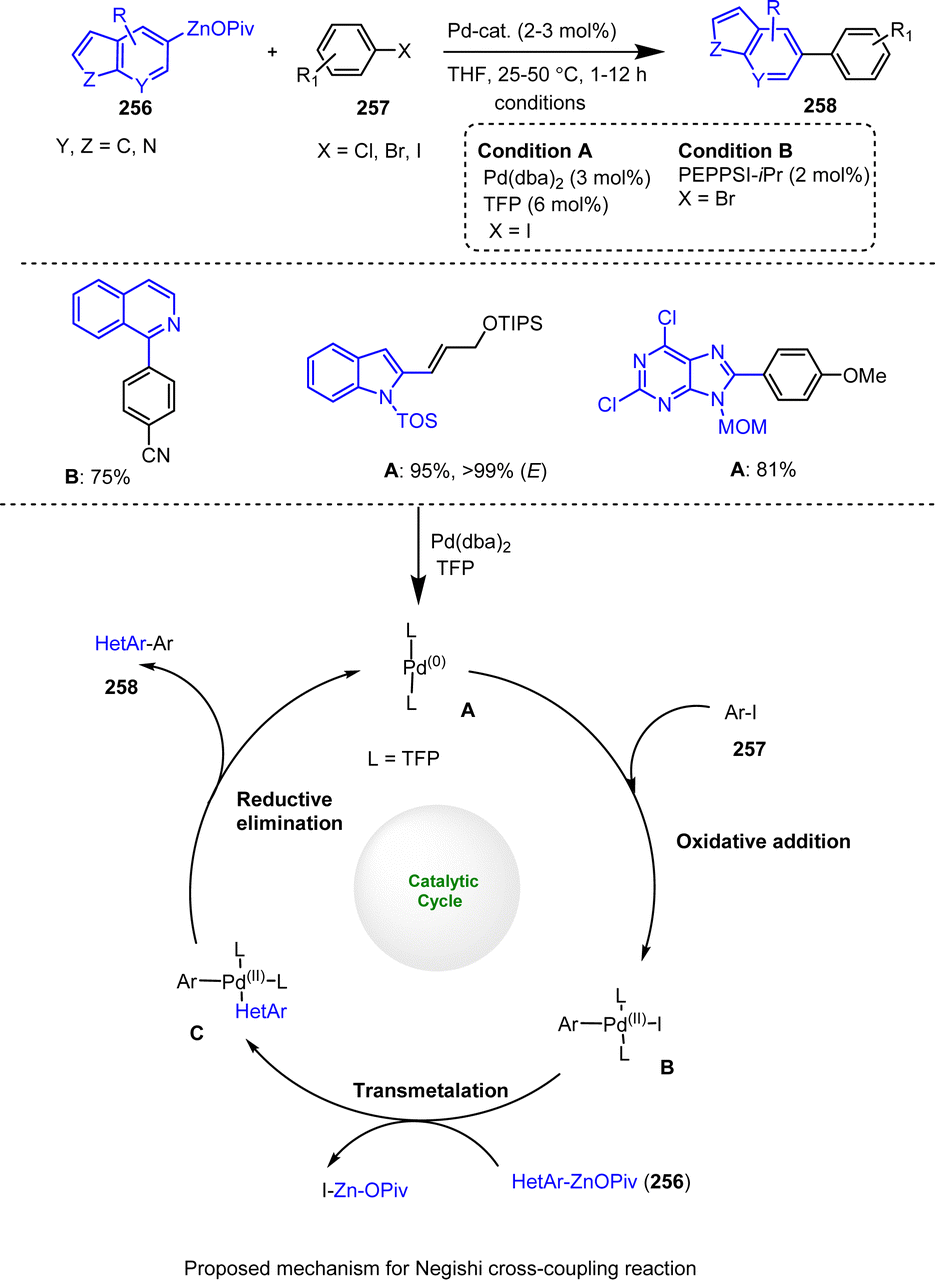 | ||
| Scheme 79 C(sp2)–C(sp2) cross-coupling reaction of solid and air-stable organozinc reagents and addition of electrophiles enabled by palladium catalysts. | ||
Barl et al. introduced a method for the functionalization of 7-azaindole via a cross-coupling reaction with alkylzinc reagents. Thus, by using a Pd-catalyzed Negishi reaction between the Reformatsky-type reagents 259 and silyl protected 3-bromo-7-azaindoles 260, targeted 7-azaindole carboxylic acid ester derivatives 261 were formed. Final TBAF-mediated removal of the TBDMS protecting group in THF at 0 °C afforded 2-(7-azaindolyl) carboxylic acid esters 261 in high yields of up to 94% (Scheme 80).180
 | ||
| Scheme 80 Pd-Catalyzed C(sp2)–C(sp3)-cross-coupling of 7-azaindoles and Reformatsky-type reagents. | ||
A simple, cobalt-catalyzed procedure for the carbon–carbon coupling of halogenated ketones and N-heterocyclic chlorides/bromides 262 with various (hetero)aryl zinc reagents 263 was developed by Haas et al. (Scheme 81).181 Different electron-poor and electron-rich functionalized aryl zinc reagents were successfully coupled within a few hours at room temperature under the reported conditions. Moreover, the use of sodium formate HCO2Na (50 mol%) proved to be decisive for the success of these cross-coupling reactions.
 | ||
| Scheme 81 Co-Catalyzed cross-coupling reaction of aryl halides and organozinc reagents. | ||
According to Hammann et al., the previously described air- and moisture-stable aryl and heteroaryl zinc pivalates 265 can also be successfully subjected to Co-catalyzed cross-coupling reactions with various aryl and heteroaryl halides 266. The proposed method is again characterized by its broad applicability, as both electron-rich and electron-poor electrophiles are tolerated in the presence of CoCl2. Thus, the corresponding reactions of 5-indolylzinc pivalate with 2-bromopyrimidine or 2-chloropyrazine, for example, proceeded in good yields (Scheme 82).182
 | ||
| Scheme 82 Broadly applicable and robust Co-catalyzed cross-coupling reaction of functionalized organozinc pivalates with aryl halides. | ||
Cobalt-catalyzed electrophilic amination reactions of organozinc pivalates with O-benzoylhydroxylamines under mild and sustained reaction conditions were first published by Chen et al. Under the described reaction conditions, C–N cross-couplings between N-hydroxylamine benzoates and heteroaryl zinc pivalates 270 could be carried out at 25 °C within 2–4 h in the presence of CoCl2·2LiCl (2.5–5.0 mol%), yielding the corresponding tertiary (hetero-) arylated amines in good amounts (Scheme 83).183 In addition, this method provides access to pharmacologically relevant diarylamine and aryl(heteroaryl)amine units. For example, a clinical candidate for the treatment of tuberculosis was synthesized using this Co-catalyzed amination protocol as the key step.
 | ||
| Scheme 83 Co catalyzed amination of heteroaryl zinc pivalates with various O-benzoyl-hydroxylamines and anilines. | ||
In 2017, Hammann et al. delineated an improved method for the preparation of air-stable silyl group-protected alkynyl zinc reagents 273 that can be successfully employed in CoCl2-catalyzed C(sp2)–C(sp) coupling reactions with various heteroaryl halides 272. The described process can be used to functionalize various biologically important heterocyclic scaffolds, such as quinoxalines and quinoline analogs 274 under sustainable conditions with acceptable yields, while incorporating synthetically beneficial alkyne residues (Scheme 84).184
 | ||
| Scheme 84 CoCl2-catalyzed C(sp2)–C(sp)-cross-coupling reaction of silyl-protected alkynylzinc pivalates with heteroaryl halides. | ||
In 2019, Ni-catalyzed C(sp2)–C(sp2) and C(sp2)–C(sp) cross-coupling reactions between functionalized heteroaryl zinc pivalates 275 and various heteroaryl triflates as well as nonaflates 276 were reported with good to excellent yields. It is noteworthy that only 0.5 mol% of the Ni catalyst was required for these Negishi cross-couplings in THF at 40 °C (Scheme 85).185
 | ||
| Scheme 85 Ni-Catalyzed Negishi cross-coupling of aryl and heteroaryl zinc pivalates with heteroaryl triflates. | ||
Organozinc pivalates are also useful reagents for the late-stage functionalization of small peptides and cyclopeptides by means of Negishi cross-coupling reactions. For example, Leroux et al. were able to successfully couple peptides 278 containing tyrosine or phenylalanine scaffolds with highly functionalized aryl and heteroaryl organozinc reagents under Pd catalysis. For this purpose, the corresponding readily available iodotyrosine- or iodophenylalanine-containing peptides 279 were site-specifically reacted with the respective organozinc pivalates 280 in the presence of the Pd catalyst in THF at 25 °C for 4–8 h to provide the modified target peptides 281 (Scheme 86).186
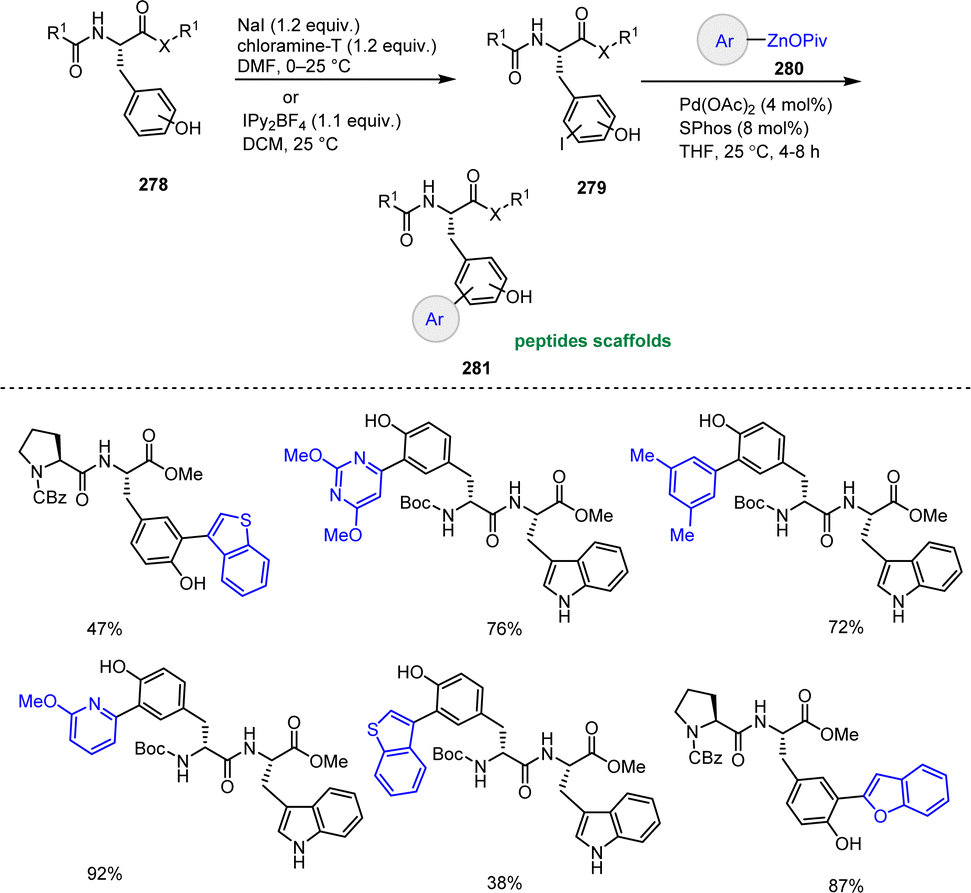 | ||
| Scheme 86 Pd-Catalyzed late-stage functionalization of cyclopeptides and peptides with (hetero)aryl zinc reagents. | ||
Cobalt-catalyzed electrophilic amination reactions of anthranilic derivatives 282 with functionalized aryl, heteroaryl, alkenyl and alkyl zinc pivalates 280 can be used to produce complex, condensed N-heterocyclic scaffolds 286 under mild conditions. Li et al. were able to show that the aniline derivatives 283 initially resulting from Co-catalyzed amination reactions can be cyclized under acidic conditions within a cascade reaction to form new complex, condensed quinolines (Scheme 87),187 some of which are of interest for materials science applications such as organic light-emitting diodes or semiconductors in perovskite solar cells due to their promising properties.
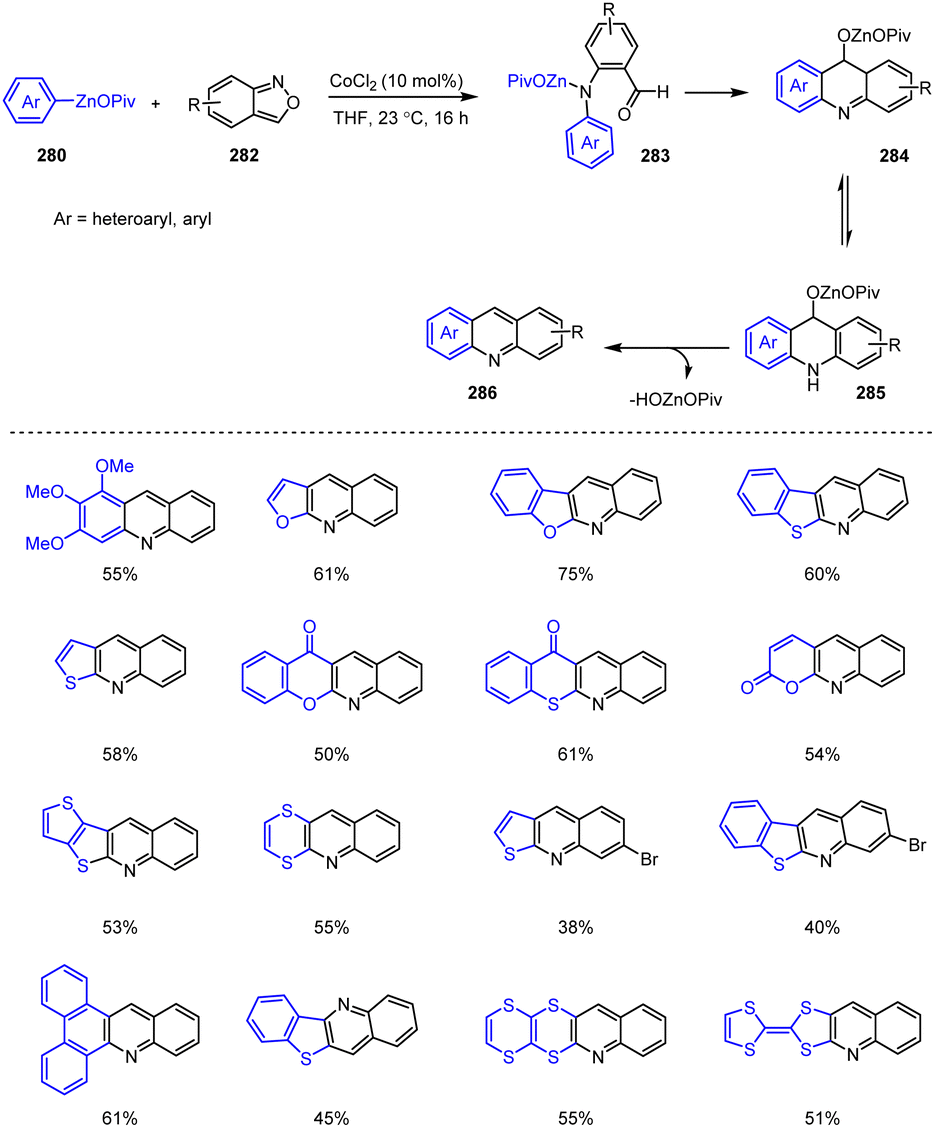 | ||
| Scheme 87 Co-Catalyzed domino cascade amination reactions of arylzinc reagents with anthranil derivatives. | ||
Recently, Lei, Li and co-workers presented a method for the regioselective difluoroalkylarylation of alkenes and 1,3-dienes using functionalized aryl zinc pivalates 280 and difluoroalkyl bromides 288 in the presence of a cobalt catalyst. This one-pot three-component cross-coupling reaction can be used to form difluoroalkylarylated products with predictable regioselectivity and high diastereoselectivity by cascade-like C(sp3)–C(sp3) and C(sp3)–C(sp2) bond formations. With activated and non-activated alkenes 287, the reaction proceeds under remarkably mild conditions with a broad range of substrates using user-friendly solid zinc reagents of excellent functional group tolerance. The process is therefore of particular interest for pharmaceutical applications and late-stage functionalization of drug candidates, as it is easily scalable and allows access to e.g., difluorinated indole scaffolds 289 (Scheme 88a).188a
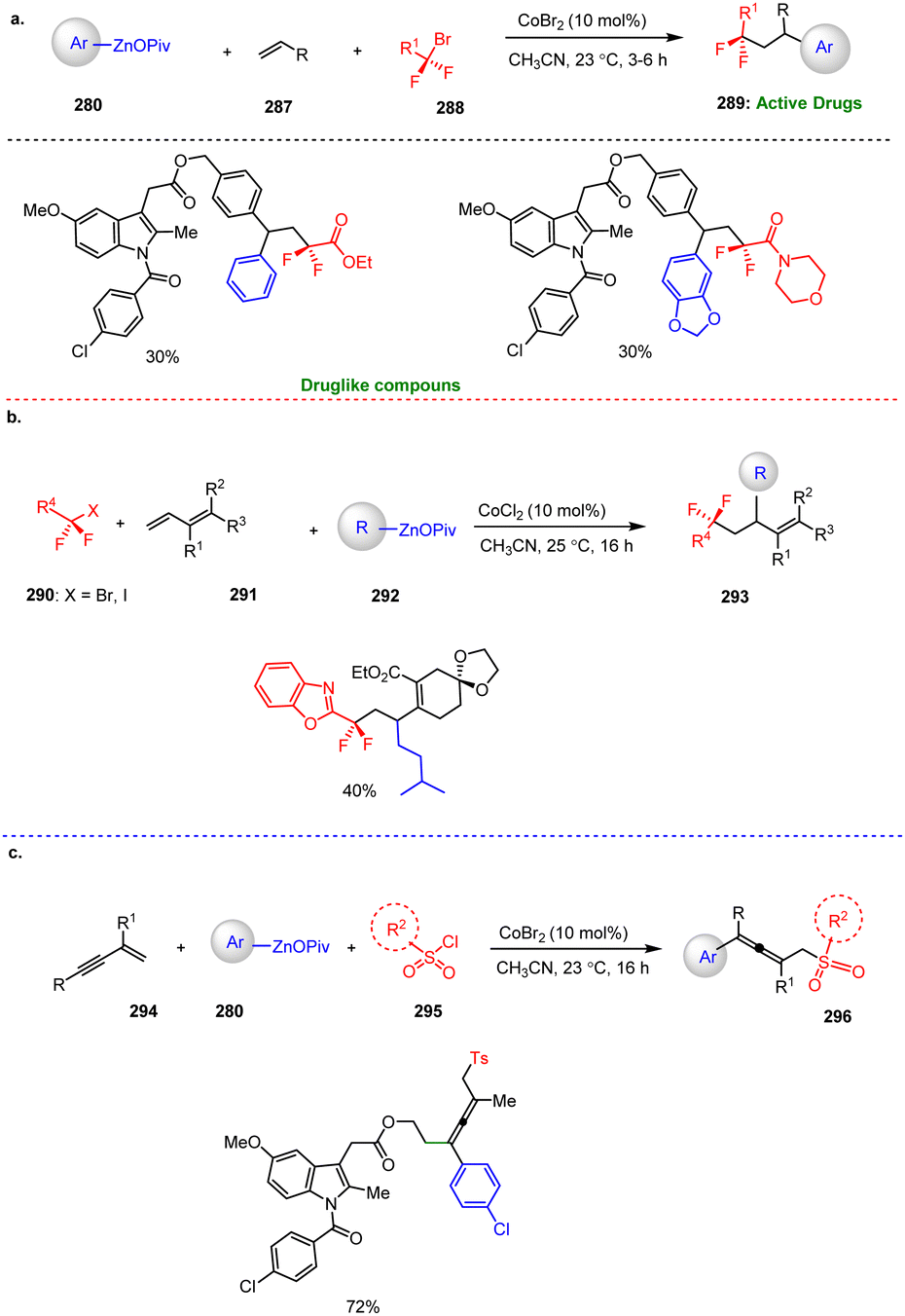 | ||
| Scheme 88 Fluoroalkylarylation of drugs scaffolds by cobalt catalysis and organozinc pivalates. | ||
In addition, the preparation of solid, salt-stabilized branched alkyl–zinc reagents of the alkyl–ZnOPiv and R3Si–ZnOPiv type was reported. Due to OPiv coordination, these reagents exhibited not only enhanced stability but also increased reactivity in Co-catalyzed difluoroalkylation reactions of dienoates, allowing modular, site-selective installation of CF2 and C(sp3) groups to the double bonds. The twofold C(sp3)–C(sp3) cross-couplings of 291 proceeded under mild conditions and were characterized by a broad substrate spectrum and high compatibility with functional groups, thereby enabling an efficient late-stage functionalization of bioactive molecules and fluorinated products 293 (Scheme 88b).188b
Furthermore, both salt-stabilized aryl and alkyl zinc pivalates 280 show superior reactivity for the cobalt-catalyzed 1,4-carbosulfonylation of 1,3-enynes 294 with sulfonyl chlorides 295 compared to conventional halide-supported organozinc reagents. In this way, highly functionalized α-allene sulfones 296 can be easily and selectively produced via the cascade formation of C–C/C–S bonds leading inter alia to functionalized bioactive indole scaffolds (Scheme 88c).188c
A new method for the preparation of solid, air-stable silyl zinc pivalates from the corresponding silyl lithium reagents by transmetalation with Zn(OPiv)2 was recently presented by Li and co-workers. The resulting organosilyl zinc pivalates 298 could be successfully used for the silylative difunctionalization of alkenes. Using a convenient chelate-assisted Ni-catalyzed regioselective alkyl- and benzyl-silylation of 8-aminoquinoline-functionalized alkenes 297, the corresponding highly functionalized alkyl silanes 300 could be generated in very good yields. As before, OPiv coordination proved to be crucial for improving the reactivity of the silyl zinc pivalates and provided access to alkyl silanes with a broad substrate spectrum and a high tolerance towards functional groups. The synthetic utility of this alkene carbosilylation sequence was again demonstrated by late-stage functionalizations of natural products and drug-like molecules as well as by simple, subsequent transformations of the resulting alkyl silanes (Scheme 89).189
 | ||
| Scheme 89 Catalytic applications of air-stable silylated zinc pivalates in nickel-catalyzed carbosilylation reactions of various olefins and alkyl halides. | ||
A protocol for converting various readily available N-heteroaryl chlorides 301 into the corresponding organozinc reagents by Co-catalyzed zinc dust insertion in the presence of zinc pivalates in benzonitrile was reported by Kremsmair et al. The resulting organozinc pivalates 302 were successfully reacted with a wide range of electrophiles, such as commercially available aryl halides or acid chlorides, employing Pd or Cu catalysts to give the targeted functionalized heteroarenes 303, as presented in Scheme 90.190
 | ||
| Scheme 90 Synthesis of heteroaryl organozinc reagents enabled by cobalt catalysis for cross-coupling reactions. | ||
Recently, Šebesta and co-workers presented a mechanochemical Negishi C(sp2)–C(sp2) cross-coupling reaction of aryl zinc pivalates 304 with aryl(heteroaryl) bromides 305 at room temperature under air. Using this simple Pd-catalyzed process, quinoline and benzothiophene derivatives 306 were prepared in good yields and within a short time by ball milling. The reaction tolerates various functional groups and can be carried out on a useful preparative scale (Scheme 91).191
 | ||
| Scheme 91 Mechanochemical Pd-catalyzed Negishi coupling reaction of aryl halides and organozinc pivalates. | ||
Melzig et al. reported the functionalization of various thiomethyl-substituted fused N-heterobicycles 307 (e.g., based on isoquinolines, quinazolines, benzothiazoles, or benzoxazole scaffolds) by nickel-catalyzed cross-coupling reactions with functionalized alkyl, aryl, heteroaryl, and benzylic zinc reagents 308. These reactions can be carried out using an inexpensive catalytic system of Ni(acac)2 and the DPE-Phos ligand at 25 °C, as shown in Scheme 92.192
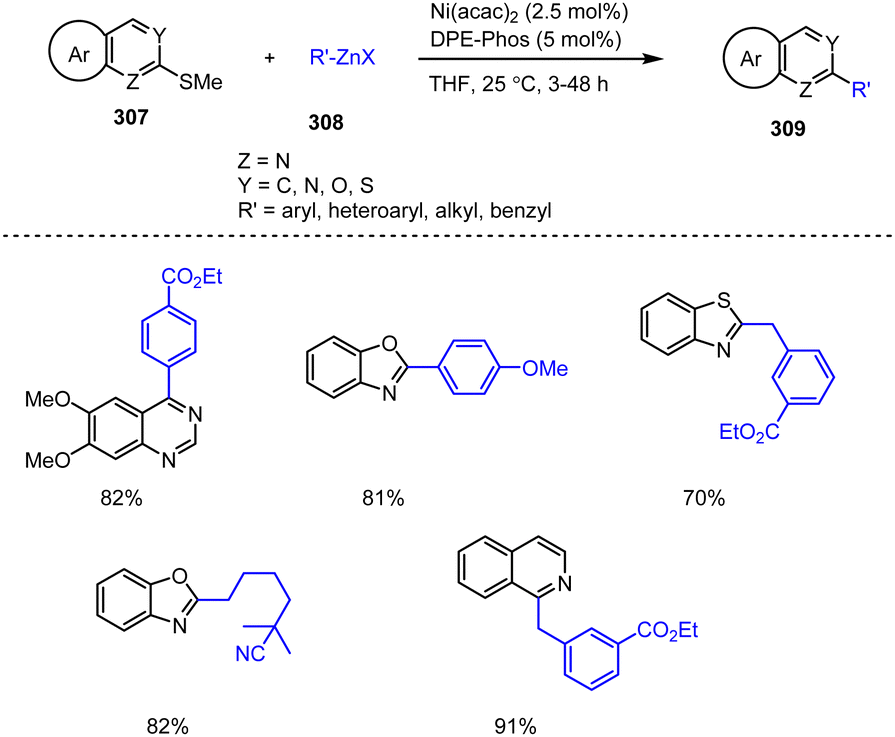 | ||
| Scheme 92 Ni-Catalyzed C(sp2)–C(sp2) and C(sp2)–C(sp3) cross-coupling reactions of thiomethyl-substituted bicyclic heterocycles with organozinc reagents. | ||
A mild Negishi cross-coupling of 2-pyridylorganozinc reagents 311 and heteroaryl chlorides 312 was described by Luzung, Patel, and Yin. Using catalytic amounts of Pd2(dba)3 (2 mol%) and XPhos as the ligand, many examples of the desired heterocycles 313 were accessible in high yields, complementing the existing coupling reactions for 2-heterocyclic organometallic reagents. The organozinc reagents 311 required for this purpose can be generated in situ under Knochel conditions from 2-pyridyl bromides 310 and i-PrMgCl at room temperature (Scheme 93).193
 | ||
| Scheme 93 Pd-Catalyzed cross-coupling of 2-pyridyl organometallic reagents and heteroaryl chlorides with X-Phos ligand. | ||
Knochel's group has also developed a Ni-catalyzed cross-coupling of heteroaryl halides, including indole, quinoline and quinoxaline derivatives, with aminoalkyl zinc reagents 314 at room temperature. This method allows a convenient one-step preparation of various heterocyclic aminoalkyl products 315 (Scheme 94), which are structural components of numerous biologically active compounds. The required aminoalkyl zinc bromides were readily obtained from the corresponding aminoalkyl chlorides by Mg insertion and transmetalation with ZnCl2. Suitable electrophilic coupling partners comprise aryl, heteroaryl and alkenyl iodides as well as the corresponding -bromides, -chlorides and -triflates. The viability of this method was impressively demonstrated by short total syntheses of the natural products (±)-galipinin and (±)-cusparin.194
 | ||
| Scheme 94 Direct amino alkylation of heteroarenes via Ni-catalyzed Negishi cross-coupling reactions. | ||
Hyodo et al. have developed an oxidative atom-economic alternative to the traditional cross-coupling reactions between organohalides and organometallic species enabling a one-step arylation of electron-deficient N-heteroarenes. In this process, functionalized N-heterocycles 316, such as quinolines, indoles and quinoxalines, were arylated at the most electrophilic site in the presence of a nickel catalyst and PCy3 as ligand using diaryl zinc reagents 317 as shown in Scheme 95.195
 | ||
| Scheme 95 Ni-Catalyzed arylation of quinoline scaffolds using Ar2Zn reagents. | ||
Buchwald and co-workers reported the Negishi cross-coupling of polyfunctionalized heteroaryl halides (Cl, Br, I) with 3,3-disubstituted allylzinc reagents using a previously established precatalytic palladacycle system with remarkable functional group tolerance. The method is characterized by an exceptionally low catalyst loading and mild reaction conditions. In addition, it enables the coupling of aryl/vinyl halides 266 with a wide range of complex heteroaromatic zinc compounds 319 that are often considered difficult. Many of the reported heterocyclic compounds 320 can be found as structural elements in pharmaceuticals (Scheme 96).196 Moreover, Negishi couplings of 3,3-disubstituted allyl zinc reagents are of particular interest for the preparation of biologically active heteroaromatic compounds with prenyl-like side chains. A successful application of this method was demonstrated with the synthesis of the naturally occurring anti-HIV agent siamenol.
 | ||
| Scheme 96 Pd-Catalyzed Negishi couplings of hetero(aryl) zinc reagents with heteroaryl halides. | ||
In 2013, Colombe et al. developed two protocols for the synthesis of solid and air-stable 2-pyridyl zinc reagents 322/323, which were designed for bench-top handling and served as practical alternatives to organoboronates in cross-coupling reactions.197 The organozinc reagents described were subsequently employed in Negishi couplings with aryl halides under Pd catalysis using SPhos as the ligand (Scheme 97). Moreover, the influence of additional stabilizing ligands on the manageability of the organozinc reagents 323 was investigated, with dioxane proving to be particularly suitable for enhancing stability under atmospheric conditions.
 | ||
| Scheme 97 Preparation of solid 2-pyridyl zinc reagents for Negishi cross-couplings. | ||
Giri and co-workers reported a strategy for the bisfunctionalization of non-activated olefins with two carbon-based units in the 1,2-position using substituted organozinc reagents. For this purpose, suitable alkyl/arylzinc reagents 326 were first prepared and reacted in radical cyclization reactions in situ with LiCl in DMF in the presence of CuI (2 mol%) to give C(sp3)–Cu complexes. The latter were then captured in a second tandem step by a ligand-free Cu-catalyzed Negishi cross-coupling reaction with aryl and heteroaryl iodides 326. The resulting (arylmethyl-)carbocycles and heterocycles 327 were obtained in moderate to high yields and allow the rapid construction of complex biologically active molecular fragments with e.g., indanyl, dihydrofuranyl and indolinyl rings from simple and readily available chemical starting materials (Scheme 98).198
 | ||
| Scheme 98 Cu-Catalysed cross-coupling of aryl and alkyl zinc reagents with various heteroaryl iodides. | ||
A report was published in 2017 by Greiner et al. on the preparation of polyfunctionalized naphthyridine derivatives 330 by Co-catalyzed Negishi cross-coupling reactions of halogenated naphthyridines 328 with alkyl/aryl magnesium halides, as well as functionalized aryl/heteroaryl zinc reagents 329 in the presence of CoCl2·2LiCl (5 mol%) and sodium formate as an additive (50 mol%). Some of these naphthyridine derivatives 330 are of interest for materials science applications due to their strong fluorescent properties (PLQE = 20–95%) with tuneable emissions from blue to yellow and long excited state lifetimes (3.8–12.0 ns, Scheme 99).199
 | ||
| Scheme 99 Co-Catalyzed arylation of aryl(heteroaryl) zinc reagents with iodo-/chloro-naphthyridines. | ||
Knochel and co-workers demonstrated that cobalt-catalyzed electrophilic aminations of heteroaryl zinc reagents 332 and aminating reagents (R1R2N-OBZ) can proceed under moderate reaction conditions. This electrophilic process also enabled access to various C–N-coupled highly substituted amine products 334 in good to excellent yields using CoCl2 (5 mol%) in THF at room temperature (Scheme 100).200 Furthermore, a candidate for a new anti-tuberculosis drug (Q203) was produced in a few steps and in very good yields with this method.183
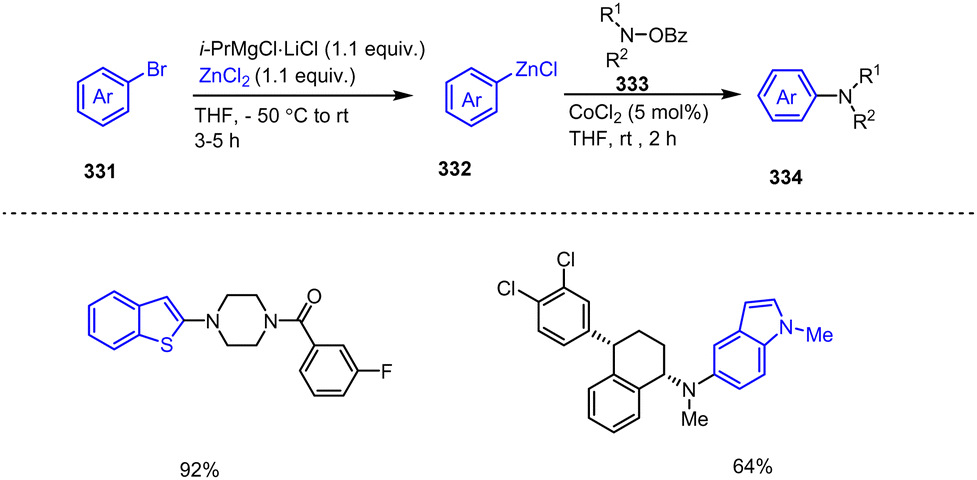 | ||
| Scheme 100 Amination of solid heteroaryl zinc pivalates with O-benzoylhydroxylamines. | ||
Knochel, Hevia and co-workers developed a method for the preparation of polyfunctional diaryl and diheteroaryl zinc species by I/Zn or Br/Zn exchange reactions with bimetallic reagents of the general formula  (R′ = s-Bu, t-Bu, p-tol). With the help of these atom economic I/Zn and Br/Zn exchange reactions, even highly sensitive functional groups such as triazines, keto and aldehyde groups, as well as nitro groups could be tolerated in the substrate. Thus, this protocol allows access to many functionalized (hetero)arenes after scavenging reactions with different electrophiles. Scheme 101 shows an example of such a reaction sequence, in which alcohol 335 is first converted by Et2Zn into the organozinc species 336. Subsequent treatment with s-BuLi generates the reactive bimetallic reagent 337, which then allows functionalization of the iodoquinoline backbone via nucleophile 338, leading to the desired modified heteroaromatic compound 339 by Cu-catalyzed 1,4-addition to methyl vinyl ketone. Structural and spectroscopic studies revealed the necessity of forming highly reactive bimetallic lithium bis(alkyl)bis(alkoxy) zincates to generate the polyfunctional aryl and heteroaryl zinc reagents from the corresponding organic iodides or bromides.201
(R′ = s-Bu, t-Bu, p-tol). With the help of these atom economic I/Zn and Br/Zn exchange reactions, even highly sensitive functional groups such as triazines, keto and aldehyde groups, as well as nitro groups could be tolerated in the substrate. Thus, this protocol allows access to many functionalized (hetero)arenes after scavenging reactions with different electrophiles. Scheme 101 shows an example of such a reaction sequence, in which alcohol 335 is first converted by Et2Zn into the organozinc species 336. Subsequent treatment with s-BuLi generates the reactive bimetallic reagent 337, which then allows functionalization of the iodoquinoline backbone via nucleophile 338, leading to the desired modified heteroaromatic compound 339 by Cu-catalyzed 1,4-addition to methyl vinyl ketone. Structural and spectroscopic studies revealed the necessity of forming highly reactive bimetallic lithium bis(alkyl)bis(alkoxy) zincates to generate the polyfunctional aryl and heteroaryl zinc reagents from the corresponding organic iodides or bromides.201
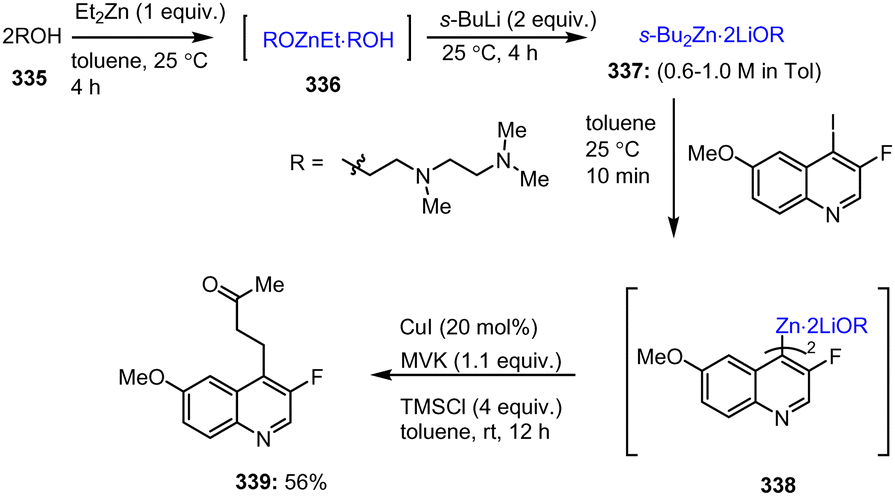 | ||
| Scheme 101 Example for the reparation of a mixed-metal reagent and its application. | ||
Graßl et al. were able to show that a broad spectrum of alkyl, aryl and heteroaryl zinc halides can be successfully aminated with highly functionalized alkyl, aryl, and heterocyclic azides in the presence of FeCl3 (0.5 equiv.) and without further ligand addition. The reaction proceeded at 50 °C within 1 h in good yields and afforded highly functionalized secondary amines such as diarylamine 342 (Scheme 102).202 This non-toxic Fe-mediated electrophilic amination protocol is particularly suitable for the synthesis of pharmaceutically active amine scaffolds and permits the successful use of peptidic azides.
 | ||
| Scheme 102 Fe-Mediated electrophilic amination of functionalized aryl zinc reagents with heteroaromatic azides. | ||
The Knochel group has developed an efficient approach to various polyfunctionalized 5-, 6- and 7-membered heterocycles, such as furans, pyrroles, quinolines, benzo[b]thieno-[2,3-b]pyridines, naphthyridines, fused pyrazoles and 2,3-dihydrobenzo[c]azepines, using conjugated β-silylated organometallic reagents. The required conjugated alkenyl Li, Mg and Zn reagents 347–349 combine an electrophilic acetal function with two 1,1-bimetallic nucleophilic moieties of well-differentiated reactivity and are readily accessible from propargylic alcohols 343 (Scheme 103). In addition, the silyl group can be converted into various carbon–carbon bonds during the construction of the heterocycles.203
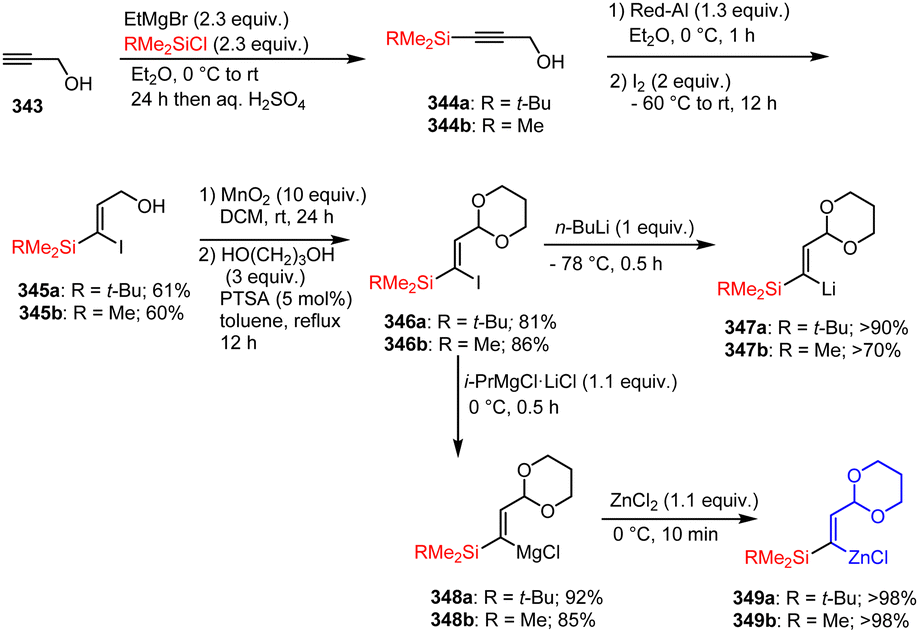 | ||
| Scheme 103 Preparation of acetal-protected organometallic Li-, Mg-, and Zn-reagents from propargylic alcohols. | ||
Examples of the preparation of fused 6-membered heterocycles such as quinolines, benzo[b]thieno-[2,3-b]pyridine and naphthyridines relevant for pharmaceutical applications are depicted in Scheme 104. For this purpose, the alkenyl zinc compounds 349 were subjected to Pd-catalyzed Negishi cross-coupling reactions with various 1-halo-2-nitroarenes 350, yielding the corresponding alkenylated nitroarenes of type 351. Subsequent In- or Zn-mediated reduction and acidic acetal cleavage afforded the desired condensed pyridines and quinoline 352 in 41–70% yields. The use of 3-bromo-2-nitrobenzo[b]thiophene or bromo-nitro-pyridines as coupling reagents thus enabled the rapid preparation of the valuable benzo[b]thieno[2,3-b]pyridines and the corresponding 1,5- and 1,6-naphthyridines.203
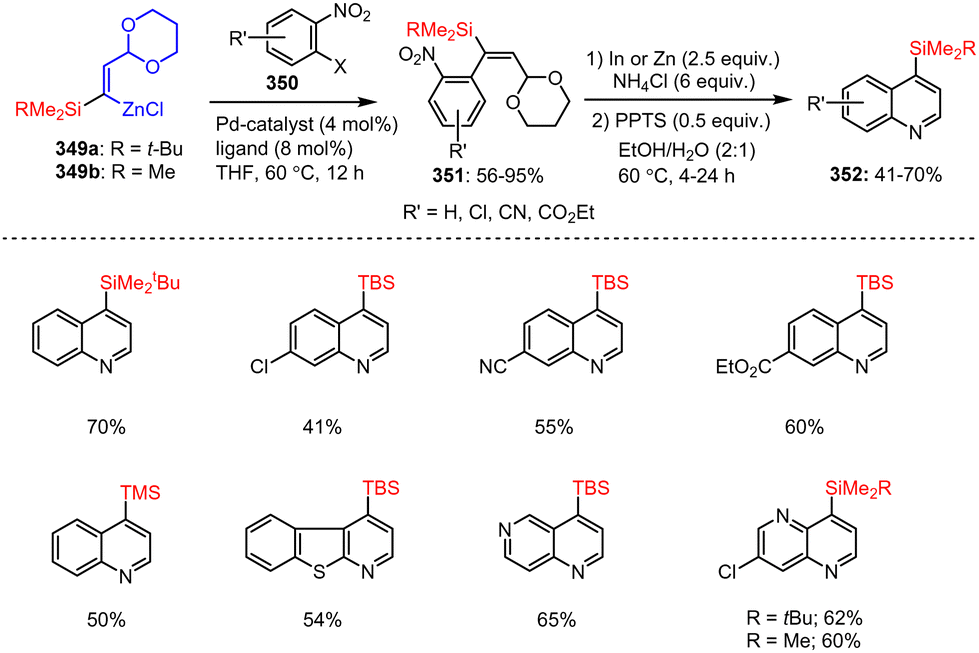 | ||
| Scheme 104 Synthesis of fused hetero aromatic compounds via sequences of Pd-catalyzed cross-coupling, reduction, acidic hydrolysis, and cyclization. | ||
Further functionalization of the quinoline scaffolds through I/Si exchange proved to be extremely difficult for both the 4-TMS and the 4-TBS substituted derivatives 352a/b due to the electron deficiency in the pyridyl ring. However, the 1,2-addition of an organolithium reagent (nBuLi or PhLi) to this pyridyl ring, followed by a scavenging reaction with ClCO2Et, led to its de-aromatization to form the alkenyl silane 353. Subsequent iodination with NIS and Ag2CO3 afforded the 4-iodo derivatives 354 in 95–96% yields, which were then successfully subjected to Negishi cross-coupling reactions with phenylzinc chloride or 1-methyl-1H-indol-5-yl-zinc bromide, leading to the coupling products 355. Finally, re-aromatization with KOH/N2H4 in ethylene glycol (190 °C, 2 h) afforded the desired 2,4-difunctionalized quinolines 356 (Scheme 105).203
 | ||
| Scheme 105 Functionalization of the quinolines scaffolds via I/Si exchange, Negishi-coupling and subsequent re-aromatization. | ||
Since the development of organozinc pivalates as versatile organometallic reagents with high reactivity and excellent air and moisture stability, these compounds have experienced an enormous surge in popularity for organic synthesis applications in recent years. The ease of handling and the absence of sophisticated inert gas techniques make these reagents widely applicable even in traditional (school) synthesis laboratories. In addition, novel protocols that guarantee increased tolerance even to highly sensitive functional groups facilitate the use of these reagents as tools for the construction of complex functional molecules in almost all areas of synthetic chemistry.
6 Selective examples of biologically active bicyclic fused heteroaromatic compounds
Many of the fused heterocyclic aromatic compounds presented so far in this review are fundamental scaffolds of biologically active natural products and drugs. While several key heterocyclic structures can be isolated from natural sources or built up stepwise using classical synthetic routes, over the last three decades research groups have been increasingly engaged in the development of synthetic methods that allow rapid, modular access to these scaffolds. Highly functionalized condensed heteroaromatic compounds are characterized by diverse biological activities and are therefore of central importance as fundamental lead structures in various fields of medicinal chemistry and the pharmaceutical industry. For example, active substances for the treatment of malaria, tuberculosis, HIV, cancer, and depression, to name just a few applications, are based on different suitably functionalized heteroaromatic scaffolds. Synthetic methods that facilitate the construction of heteroaromatics and their modification through late-stage functionalization are therefore of great importance for the further development of drugs and for medical advances. The following section provides an overview of various condensed heteroaromatic motifs found in active ingredients, along with their pharmaceutical and biological applications.204–270 (Table 1)| S. no. | Compound | Biological activity | Ref. no. |
|---|---|---|---|
| 1 |

|
Neuroprotective agent | 204 |
| 2 |

|
Inhibitor of MRSA biofilms | 205 |
| 3 |

|
Anti-RSV | 206 |
| 4 |

|
Antibacterial | 207 |
| 5 |

|
Antimicrobial | 208 |
| 6 |
|
Inhibitor of bacterial enzyme | 209 |
| 7 |

|
Inhibitor of RSK2 | 210 |
| 8 |
|
Inhibitor of 12-LOX | 211 |
| 9 |

|
Antitumor activity | 212 |
| 10 |
|
Inhibitor of SIRT3 | 213 |
| 11 |

|
Disruptor of biofilm formation in V. cholerae bacteria | 214 |
| 12 |

|
Inhibitor of pan-phosphodiesterase family | 215 |
| 13 |

|
Anti-castration resistant prostate cancer and CYP17 lyase-selective inhibitor | 216 |
| 14 |

|
Antiviral | 217 |
| 15 |
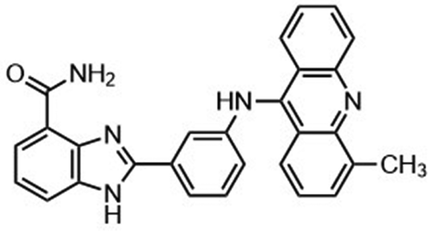
|
Inhibitor of Topo and PARP-1 | 218 |
| 16 |

|
Anti-HIV | 219 |
| 17 |
|
Autofluorescent antimalarial | 220 |
| 18 |

|
Anticancer | 221 |
| 19 |

|
Anticancer | 222 |
| 20 |

|
Anticancer | 223 |
| 21 |

|
Anticancer | 224 |
| 22 |

|
Anticancer | 225 |
| 23 |

|
Anticancer | 226 |
| 24 |
|
Antiproliferative | 227 |
| 25 |

|
Anti-breast cancer | 228 |
| 26 |

|
Anti-influenza virus activity | 229 |
| 27 |

|
Potent inhibitor of ALK mutants | 230 |
| 28 |
|
Anticancer | 231 |
| 29 |

|
Anticancer | 232 |
| 30 |

|
Anticancer | 233 |
| 31 |

|
Anticancer | 234 |
| 32 |

|
Telomerase inhibitor | 235 |
| 33 |

|
Antiproliferative | 236 |
| 34 |

|
Antifungal | 237 |
| 35 |

|
Antiproliferative | 238 |
| 36 |
|
Antitumor | 239 |
| 37 |
|
Inhibitor of microtubule formation | 240 |
| 38 |

|
Antimitotic activity | 241 |
| 39 |

|
c-Met inhibitor | 242 |
| 40 |

|
Leishmanicidal activity | 243 |
| 41 |

|
Antioxidant | 244 |
| 42 |
|
Anti-inflammatory | 245 |
| 43 |

|
Antidepressant | 246 |
| 44 |

|
Anti-HIV | 247 |
| 45 |
|
Human TLR7 agonist | 248 |
| 46 |

|
Human TLR8 agonist | 249 |
| 47 |

|
Human TLR8 agonist | 250 |
| 48 |
|
Antifungal | 251 |
| 49 |
|
Antibacterial | 252 |
| 50 |
|
Antibacterial | 253 |
| 51 |
|
Antifungal | 254 |
| 52 |
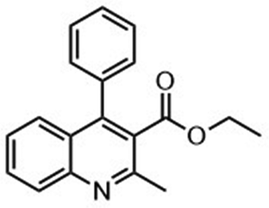
|
Anti-tubercular | 255 |
| 53 |

|
Anti-protozoal | 256 |
| 54 |

|
Antimalarial | 257 |
| 55 |
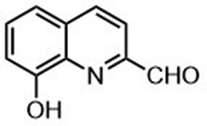
|
Anticancer | 258 |
| 56 |

|
Anticancer | 259 |
| 57 |

|
Antimicrobial agent | 260 |
| 58 |

|
Potent antagonist | 261 |
| 59 |
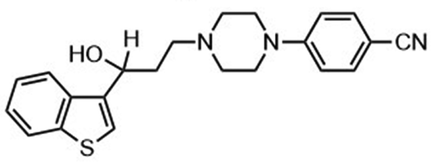
|
Antidepressant | 262 |
| 60 |

|
Bactericidal | 263 |
| 61 |

|
Dual CDK2/CDK9 inhibitor and antitumor | 264 |
| 62 |

|
CDK9 inhibitor | 265 |
| 63 |

|
Antiangiogenic and antitumor | 266 |
| 64 |

|
PI3K HDAC inhibitor and antiproliferative | 267 |
| 65 |
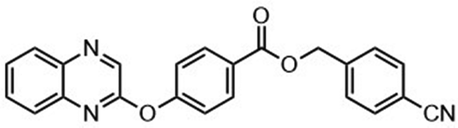
|
Fungicidal | 268 |
| 66 |

|
Anti-enteroviral | 269 |
| 67 |

|
Antibacterial | 270 |
7 Selective examples for bicyclic heteroaromatic system-based material applications
Functionalized condensed heteroaromatic compounds are not only of interest due to their prevalence in biologically active compounds. Rather, highly π-conjugated sulfur-, oxygen- and nitrogen-based fused heterocycles show great application potential for materials sciences and can be used for instance in the development of organic light-emitting diodes (OLEDs), polymer LEDs, organic semiconductors, field-effect transistors, photoelectrically luminescent materials and solar cells. Further potential areas of application can be derived from the table compiled in this section.271–319 (Table 2)| S. no. | Compound structure | Activity/properties | Ref. no. |
|---|---|---|---|
| 1 |
|
Photochromo-phore | 271 |
| 2 |

|
OLED | 272 |
| 3 |
|
OLED | 273 |
| 4 |

|
PLED | 274 |
| 5 |
|
Electrochromic devices | 275 |
| 6 |

|
LED | 276 |
| 7 |

|
Optoelectronics | 277 |
| 8 |

|
Optoelectronics | 278 |
| 9 |

|
White light emission | 279 |
| 10 |

|
Fluorescent material | 280 |
| 11 |

|
Organic electronics | 281 |
| 12 |

|
Optoelectronics | 282 |
| 13 |

|
Fluorescent sensor | 283 |
| 14 |
|
Optoelectronics | 284 |
| 15 |

|
Photovoltaics | 285 |
| 16 |

|
Optoelectronics | 286 |
| 17 |
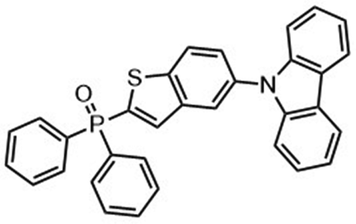
|
OLEDS | 287 |
| 18 |
|
Liquid crystalline semiconductors | 288 |
| 19 |

|
Organic semiconductor | 289 |
| 20 |
|
Electrochromic and optical properties | 290 |
| 21 |

|
Field effect transistors | 291 |
| 22 |

|
Optoelectronics | 292 |
| 23 |

|
Solid state fluorescence | 293 |
| 24 |
|
Chromophores | 294 |
| 25 |

|
Chromophores | 295 |
| 26 |
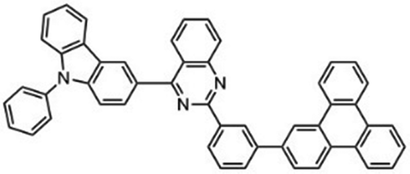
|
OLEDS | 296 |
| 27 |

|
OLEDS | 297 |
| 28 |

|
OLEDS | 298 |
| 29 |

|
OLEDS | 299 |
| 30 |

|
Fluorophores | 300 |
| 31 |

|
Fluorescent material | 301 |
| 32 |

|
Optoelectronics | 302 |
| 33 |
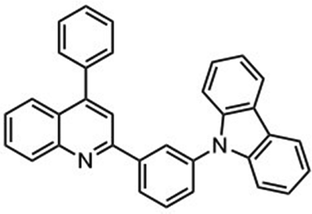
|
OLEDS | 303 |
| 34 |

|
Dye for bioimaging | 304 |
| 35 |

|
Light emitting diodes | 305 |
| 36 |
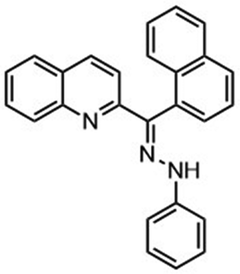
|
Photo-switches in photonics | 306 |
| 37 |
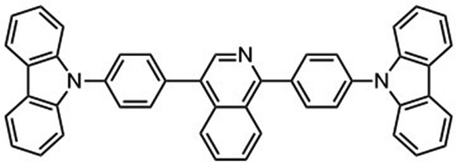
|
OLEDS | 307 |
| 38 |

|
Organic field effect transistors | 308 |
| 39 |

|
Optical chromophores | 309 |
| 40 |

|
Field effect transistors | 310 |
| 41 |

|
Solar cells | 311 |
| 42 |
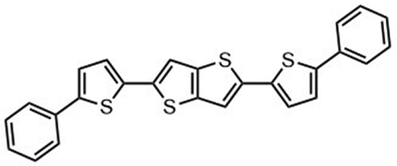
|
P-type organic semi-conductors | 312 |
| 43 |
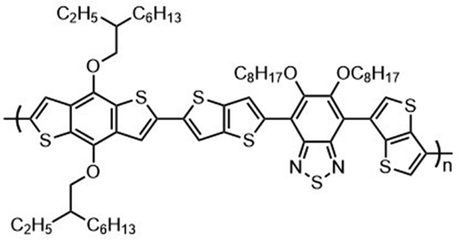
|
Photovoltaics | 313 |
| 44 |

|
N-type molecular semi-conductors | 314 |
| 45 |

|
Electric device applications | 315 |
| 46 |
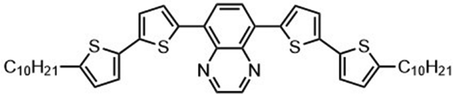
|
Organic field transistors | 316 |
| 47 |

|
Photo-luminescence | 317 |
| 48 |

|
Electro-luminescent materials | 318 |
| 49 |

|
OLEDS | 319 |
8 Conclusions
This review highlights important synthetic advances in the functionalization of 5- or 6-membered ring-fused bicyclic heteroaromatic frameworks using organo-Li, Mg and Zn reagents. The syntheses presented include complex sequences based on direct selective metalations, halogen/metal exchange reactions, oxidative metal insertions and transmetalation processes. Protocols for efficient preparations of different functionalized organometallic reagents with aryl, heteroaryl, alkenyl, alkynyl and alkyl groups allow their use in high-performance transition metal-catalyzed cross-coupling reactions with various electrophiles, which makes such reaction sequences widely applicable in synthesis programs and for the development of active pharmaceutical ingredients. Thus, selective functionalization of important heterocyclic scaffolds such as quinoline, indole, benzofuran, benzothiophene, benzoxazole, benzothiazole, benzopyrimidine, anthranil, thienothiophene, coumarin, chromones, quinolones, phthalazines and their condensed heterocyclic scaffolds can now be carried out in almost any desired position. This overview thus underlines the importance of modern synthetic technology with regard to functional group tolerance, cost efficiency and sustainable reaction conditions in the production of heterocyclic bioactive drugs and natural products.Data availability
The authors confirm that the data supporting the findings of this study are available in the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Vasudevan Dhayalan is thankful to the DST-SERB for the Ramanujan Fellowship (grant no. RJF/2020/000038), and the Core research grant (CRG/2022/001855) for financial support and the National Institute of Technology Puducherry, Karaikal, India for their assistance with research. Rambabu Dandela thanks to DST-SERB Core research grant (CRG/2023/001402). Dr P. Amaladass expresses gratitude to the Research and Development Cell of Madanapalle Institute of Technology & Science for their unwavering support. Prof. Anja Hoffmann-Röder thanks the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, SFB 1309-325871075) for financial support. We thank Aksa S. Annie for fruitful discussions and support.Notes and references
- V. Dhayalan, D. Sharma, R. Chatterjee and R. Dandela, Eur. J. Org. Chem., 2023, e202300285 CrossRef CAS.
- M. Balkenhohl and P. Knochel, Chem. – Eur. J., 2020, 26, 3688–3697 CrossRef CAS PubMed.
- D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Chem. Rev., 2014, 114, 1207–1257 CrossRef CAS PubMed.
- (a) O. M. Kuzmina, A. K. Steib, A. Moyeux, G. Cahiez and P. Knochel, Synthesis, 2015, 1696–1705 CAS; (b) A. Desaintjean, T. Haupt, L. J. Bole, N. R. Judge, E. Hevia and P. Knochel, Angew. Chem., Int. Ed., 2021, 60, 1513–1518 CrossRef CAS PubMed.
- (a) P. Knochel, T. Thaler and C. Diene, Isr. J. Chem., 2010, 50, 547–557 CrossRef CAS; (b) H. Ila, J. T. Markiewicz, V. Malakhov and P. Knochel, Synthesis, 2013, 2343–2371 CAS; (c) K. Schwärzer, H. Zipse, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 20235–20241 CrossRef PubMed.
- B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS PubMed.
- R. P. Jumde, F. Lanza, T. Pellegrini and S. R. Harutyunyan, Nat. Commun., 2017, 8, 2058 CrossRef PubMed.
- M. Rodríguez-Fernández, X. Yan, J. F. Collados, P. B. White and S. R. Harutyunyan, J. Am. Chem. Soc., 2017, 139, 14224–14231 CrossRef PubMed.
- M. Fañanás-Mastral, M. Pérez, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Angew. Chem., Int. Ed., 2012, 51, 1922–1925 CrossRef PubMed.
- S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed.
- S. R. Harutyunyan, Z. Zhao, T. D. Hartog, K. Bouwmeester, A. J. Minnaard, B. L. Feringa and F. Govers, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8507–8512 CrossRef CAS PubMed.
- (a) M. A. Ganiek, M. R. Becker, M. Ketels and P. Knochel, Org. Lett., 2016, 18, 828–831 CrossRef CAS PubMed; (b) L. Klier, E. Aranzamendi, D. Ziegler, J. Nickel, K. Karaghiosoff, T. Carell and P. Knochel, Org. Lett., 2016, 18, 1068–1071 CrossRef CAS PubMed; (c) D. Haas, D. Sustac-Roman, S. Schwarz and P. Knochel, Org. Lett., 2016, 18, 6380–6383 CrossRef CAS PubMed.
- J. Skotnitzki, L. Spessert and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 1509–1514 CrossRef CAS PubMed.
- (a) J. Skotnitzki, A. Kremsmair, B. Kicin, R. Saeb, V. Ruf and P. Knochel, Synthesis, 2020, 873–881 CAS; (b) M. Ellwart, G. Höfner, A. Gerwien, K. T. Wanner and P. Knochel, Synthesis, 2017, 5159–5166 CAS.
- I. S. Makarov, C. E. Brocklehurst, K. Karaghiosoff, G. Koch and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 12774–12777 CrossRef CAS PubMed.
- Y. B. Malysheva, S. Combes, A. Y. Fedorov, P. Knochel and A. E. Gavryushin, Synlett, 2012, 1205–1208 CAS.
- V. Morozova, J. Skotnitzki, K. Moriya, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 5516–5519 CrossRef CAS PubMed.
- H. Langhals, P. Knochel, A. Walter and S. Zimdars, Synthesis, 2012, 3465–3476 CrossRef CAS.
- T. Schlücker, V. Dhayalan, H. Langhals, C. Sämann and P. Knochel, Asian J. Org. Chem., 2015, 4, 763–769 CrossRef.
- (a) J. Nafe, S. Herbert, F. Auras, K. Karaghiosoff, T. Bein and P. Knochel, Chem. – Eur. J., 2015, 21, 1102–1107 CrossRef CAS PubMed; (b) J. Nafe, F. Auras, K. Karaghiosoff, T. Bein and P. Knochel, Org. Lett., 2015, 17, 5356–5359 CrossRef CAS PubMed.
- V. Dhayalan, F. R. Alcañiz, V. Werner, K. Karaghiosoff and P. Knochel, Synthesis, 2015, 3972–3982 CAS.
- S. Fernandez, M. A. Ganiek, M. Karpacheva, F. C. Hanusch, S. Reuter, T. Bein, F. Auras and P. Knochel, Org. Lett., 2016, 18, 3158–3161 CrossRef CAS PubMed.
- S. I. Druzhinin, S. R. Dubbaka, P. Knochel, S. A. Kovalenko, P. Mayer, T. Senyushkina and K. A. Zachariasse, J. Phys. Chem. A, 2008, 112, 2749–2761 CrossRef CAS PubMed.
- (a) J. J. Patel, M. Laars, W. Gan, J. Board, M. O. Kitching and V. Snieckus, Angew. Chem., Int. Ed., 2018, 57, 9425–9429 CrossRef CAS PubMed; (b) R. E. Miller, T. Rantanen, K. A. Ogilvie, U. Groth and V. Snieckus, Org. Lett., 2010, 12, 2198–2201 CrossRef CAS PubMed; (c) D. Wu, A. Dasgupta, K.-H. Chen, M. Neuber-Hess, J. Patel, T. E. Hurst, J. D. Mewburn, P. D. A. Lima, E. Alizadeh, A. Martin, M. Wells, V. Snieckus and S. L. Archer, FASEB J., 2020, 34, 1447–1464 CrossRef CAS PubMed.
- (a) M. A. J. Miah, M. P. Sibi, S. Chattopadhyay, O. B. Familoni and V. Snieckus, Eur. J. Org. Chem., 2018, 440–446 CrossRef CAS; (b) J. Fässler, J. A. McCubbin, A. Roglans, T. Kimachi, J. W. Hollett, R. W. Kunz, M. Tinkl, Y. Zhang, R. Wang, M. Campbell and V. Snieckus, J. Org. Chem., 2015, 80, 3368–3386 CrossRef PubMed; (c) K. Groom, S. M. S. Hussain, J. Morin, C. Nilewski, T. Rantanen and V. Snieckus, Org. Lett, 2014, 16, 2378–2381 CrossRef CAS PubMed; (d) X. Wang, J.-M. Fu and V. Snieckus, Helv. Chim. Acta, 2012, 95, 2680–2694 CrossRef CAS.
- (a) B. Wei, Q. Ren, T. Bein and P. Knochel, Angew. Chem., Int. Ed., 2021, 60, 10409–10414 CrossRef CAS PubMed; (b) P. Knochel and K. P. Cole, Org. Process Res. Dev., 2021, 25, 2188–2191 CrossRef CAS; (c) S. H. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 9717–9720 CrossRef CAS PubMed.
- (a) A. B. Bellan and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 1838–1841 CrossRef CAS PubMed; (b) T. Thaler, B. Haag, A. Gavryushin, K. Schober, E. Hartmann, R. M. Gschwind, H. Zipse, P. Mayer and P. Knochel, Nat. Chem., 2010, 2, 125–130 CrossRef CAS PubMed.
- G. Berionni, V. Morozova, M. Heininger, P. Mayer, P. Knochel and H. Mayr, J. Am. Chem. Soc., 2013, 135, 6317–6324 CrossRef CAS PubMed.
- K. Groll, T. D. Blümke, A. Unsinn, D. Haas and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 11157–11161 CrossRef CAS PubMed.
- T. D. Blümke, T. Klatt, K. Koszinowski and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 9926–9930 CrossRef PubMed.
- S. Somprasong, M. C. Reis and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2023, 62, e202217328 CrossRef CAS PubMed.
- Y. Guo, M. Castiñeira Reis, J. Kootstra and S. R. Harutyunyan, ACS Catal., 2021, 11, 8476–8483 CrossRef CAS PubMed.
- (a) M. Zurro, L. Ge and S. R. Harutyunyan, Org. Lett., 2022, 24, 6686–6691 CrossRef CAS PubMed; (b) Y. Guo and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2019, 58, 12950–12954 CrossRef CAS PubMed; (c) A. V. R. Madduri, A. J. Minnaard and S. R. Harutyunyan, Org. Biomol. Chem., 2012, 10, 2878–2884 RSC.
- C. Boldrini, M. C. Reis and S. R. Harutyunyan, J. Org. Chem., 2022, 87, 12772–12782 CrossRef CAS PubMed.
- X. Yan and S. R. Harutyunyan, Nat. Commun., 2019, 10, 3402 CrossRef PubMed.
- V. Fasano, R. C. Mykura, J. M. Fordham, J. J. Rogers, B. Banecki, A. Noble and V. K. Aggarwal, Nat. Synth., 2022, 1, 902–907 CrossRef.
- K. J. Chambers, P. Sanghong, D. Carter Martos, G. Casoni, R. C. Mykura, D. Prasad Hari, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2023, 62, e202312054 CrossRef CAS PubMed.
- H. Wang, W. Han, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2022, 61, e202207988 CrossRef CAS PubMed.
- (a) J. Wu, P. Lorenzo, S. Zhong, M. Ali, C. P. Butts, E. L. Myers and V. K. Aggarwal, Nature, 2017, 547, 436–440 CrossRef CAS PubMed; (b) D. P. Hari, R. Madhavachary, V. Fasano, J. Haire and V. K. Aggarwal, J. Am. Chem. Soc., 2021, 143, 7462–7470 CrossRef CAS PubMed.
- (a) A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117–122 CrossRef CAS PubMed; (b) D. P. Hari, J. C. Abell, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 5515–5520 CrossRef CAS PubMed.
- M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667–672 CrossRef CAS PubMed.
- M. Pérez, M. Fañanás-Mastral, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Nat. Chem., 2011, 3, 377–381 CrossRef PubMed.
- (a) S. R. Harutyunyan, F. López, W. R. Browne, A. Correa, D. Peña, R. Badorrey, A. Meetsma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 9103–9118 CrossRef CAS PubMed; (b) C. Vila, M. Giannerini, V. Hornillos, M. Fañanás-Mastral and B. L. Feringa, Chem. Sci., 2014, 5, 1361–1367 RSC; (c) D. Heijnen, J.-B. Gualtierotti, V. Hornillos and B. L. Feringa, Chem. – Eur. J., 2016, 22, 3991–3995 CrossRef CAS PubMed.
- (a) B. Mao, K. Geurts, M. Fañanás-Mastral, A. W. van Zijl, S. P. Fletcher, A. J. Minnaard and B. L. Feringa, Org. Lett., 2011, 13, 948–951 CrossRef CAS PubMed; (b) P. H. Bos, A. Rudolph, M. Pérez, M. Fañanás-Mastral, S. R. Harutyunyan and B. L. Feringa, Chem. Commun., 2012, 48, 1748–1750 RSC.
- Y. Cohen and I. Marek, Acc. Chem. Res., 2022, 55, 2848–2868 CrossRef CAS PubMed.
- Y. Cohen, A. Cohen and I. Marek, Chem. Rev., 2021, 121, 140–161 CrossRef CAS PubMed.
- D. S. Müller and I. Marek, Chem. Soc. Rev., 2016, 45, 4552–4566 RSC.
- V. Lanke and I. Marek, J. Am. Chem. Soc., 2020, 142, 5543–5548 CrossRef CAS PubMed.
- F.-G. Zhang and I. Marek, J. Am. Chem. Soc., 2017, 139, 8364–8370 CrossRef CAS PubMed.
- (a) T. Pavlíčková, Y. Stöckl and I. Marek, Org. Lett., 2022, 24, 8901–8906 CrossRef PubMed; (b) C. Tugny, F.-G. Zhang and I. Marek, Chem. – Eur. J., 2019, 25, 205–209 CrossRef CAS PubMed; (c) Y. Cohen and I. Marek, Org. Lett., 2019, 21, 9162–9165 CrossRef CAS PubMed.
- W. Erb, L. Kadari, K. Al-Mekhlafi, T. Roisnel, V. Dorcet, P. R. Krishna and F. Mongin, Adv. Synth. Catal., 2020, 362, 832–850 CrossRef CAS.
- (a) M. Hedidi, J. Maillard, W. Erb, F. Lassagne, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet, M. Hamzé, Z. Fajloun, B. Baratte, S. Ruchaud, S. Bach, G. Bentabed-Ababsa and F. Mongin, Eur. J. Org. Chem., 2017, 5903–5915 CrossRef CAS; (b) M. Hedidi, G. Bentabed-Ababsa, A. Derdour, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, F. Chevallier, T. Roisnel, V. Dorcet and F. Mongin, Tetrahedron, 2016, 72, 2196–2205 CrossRef CAS.
- M. Hedidi, D. Gandrath, Y. Kitazawa, Y. Tatsuya, M. Kimura, W. Erb, G. Bentabed-Ababsa, F. Chevallier, M. Uchiyama, P. Gros and F. Mongin, New J. Chem., 2019, 43, 14898–14907 RSC.
- N. E. Behnke, K. Lovato, M. Yousufuddin and L. Kürti, Angew. Chem., Int. Ed., 2019, 58, 14219–14223 CrossRef CAS PubMed.
- P. V. Kattamuri, J. Yin, S. Siriwongsup, D.-H. Kwon, D. H. Ess, Q. Li, G. Li, M. Yousufuddin, P. F. Richardson, S. C. Sutton and L. Kürti, J. Am. Chem. Soc., 2017, 139, 11184–11196 CrossRef CAS PubMed.
- (a) Z. Zhou, Z. Ma, N. E. Behnke, H. Gao and L. Kürti, J. Am. Chem. Soc., 2017, 139, 115–118 CrossRef CAS PubMed; (b) N. E. Behnke, R. Kielawa, D.-H. Kwon, D. H. Ess and L. Kürti, Org. Lett., 2018, 20, 8064–8068 CrossRef CAS PubMed.
- T. Verhelst, S. Verbeeck, O. Ryabtsova, S. Depraetere and B. U. W. Maes, Org. Lett., 2011, 13, 272–275 CrossRef CAS PubMed.
- P. Mampuys, T. D. Moseev, M. V. Varaksin, J. De Houwer, C. M. L. Vande Velde, O. N. Chupakhin, V. N. Charushin and B. U. W. Maes, Org. Lett., 2019, 21, 2699–2703 CrossRef CAS PubMed.
- O. Ryabtsova, T. Verhelst, M. Baeten, C. M. L. Vande Velde and B. U. W. Maes, J. Org. Chem., 2009, 74, 9440–9445 CrossRef CAS PubMed.
- (a) M. A. Oberli and S. L. Buchwald, Org. Lett., 2012, 14, 4606–4609 CrossRef CAS PubMed; (b) H. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2017, 139, 11590–11594 CrossRef CAS PubMed; (c) W. Shu and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 5355–5358 CrossRef CAS PubMed; (d) W. Shu, L. Pellegatti, M. A. Oberli and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 10665–10669 CrossRef CAS PubMed.
- (a) K. T. O’Brien and A. B. Smith, III, Org. Lett., 2019, 21, 7655–7659 CrossRef PubMed; (b) Y. Chen, X. Zhang, F. Liu, G. He, J. Zhang, K. N. Houk, A. B. Smith and Y. Liang, Chin. Chem. Lett., 2021, 32, 441–444 CrossRef CAS PubMed; (c) M. H. Nguyen and A. B. Smith, III, Org. Lett., 2013, 15, 4872–4875 CrossRef CAS PubMed.
- (a) X. Liu, J. Wang and J. Li, Synlett, 2022, 1688–1694 CrossRef; (b) Z. Duan, S. Dong and J. Li, Org. Biomol. Chem., 2023, 21, 5506–5510 RSC; (c) B. Hu, X. Cheng, Y. Hu, X. Liu, K. Karaghiosoff and J. Li, Angew. Chem., Int. Ed., 2021, 60, 15497–15502 CrossRef CAS PubMed.
- (a) A. N. Baumann, A. Music, J. Dechent, N. Müller, T. C. Jagau and D. Didier, Chem. – Eur. J., 2020, 26, 8382–8387 CrossRef CAS PubMed; (b) F. Reiners, E. Joseph, B. Nißl and D. Didier, Org. Lett., 2020, 22, 8533–8537 CrossRef CAS PubMed; (c) M. Eisold, A. Müller-Deku, F. Reiners and D. Didier, Org. Lett., 2018, 20, 4654–4658 CrossRef CAS PubMed.
- (a) J. L. Jeffrey and R. Sarpong, Org. Lett., 2012, 14, 5400–5403 CrossRef CAS PubMed; (b) S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed; (c) Y. Fu, B.-L. Gou, C.-Z. Shi, Z. Du and T. Shen, ChemCatChem, 2018, 10, 4253–4257 CrossRef CAS; (d) N. Tezuka, K. Shimojo, K. Hirano, S. Komagawa, K. Yoshida, C. Wang, K. Miyamoto, T. Saito, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2016, 138, 9166–9171 CrossRef CAS PubMed; (e) S. Panda, A. Coffin, Q. N. Nguyen, D. J. Tantillo and J. M. Ready, Angew. Chem., Int. Ed., 2016, 55, 2205–2209 CrossRef CAS PubMed; (f) Y. Nagashima, R. Takita, K. Yoshida, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2013, 135, 18730–18733 CrossRef CAS PubMed; (g) B. M. Trost and C. A. Kalnmals, Chem. – Eur. J., 2018, 24, 9066–9074 CrossRef CAS PubMed.
- (a) R. Chinchilla, C. Nájera and M. Yus, Chem. Rev., 2004, 104, 2667–2722 CrossRef CAS PubMed; (b) H. K. Khartabil, P. C. Gros, Y. Fort and M. F. Ruiz-López, J. Am. Chem. Soc., 2010, 132, 2410–2416 CrossRef CAS PubMed; (c) G. Rouquet, D. C. Blakemore and S. V. Ley, Chem. Commun., 2014, 50, 8908–8911 RSC; (d) Y. Nassar, F. Rodier, V. Ferey and J. Cossy, ACS Catal., 2021, 11, 5736–5761 CrossRef CAS.
- (a) T. Blümke, Y.-H. Chen, Z. Peng and P. Knochel, Nat. Chem., 2010, 2, 313–318 CrossRef PubMed; (b) P. Knochel, M. A. Schade, S. Bernhardt, G. Manolikakes, A. Metzger, F. M. Piller, C. J. Rohbogner and M. Mosrin, Beilstein J. Org. Chem., 2011, 7, 1261–1277 CrossRef CAS PubMed; (c) Z. Peng and P. Knochel, Org. Lett., 2011, 13, 3198–3201 CrossRef CAS PubMed; (d) A. D. Benischke, G. Le Corre and P. Knochel, Chem. – Eur. J., 2017, 23, 778–782 CrossRef CAS PubMed.
- (a) C. Sämann, M. A. Schade, S. Yamada and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 9495–9499 CrossRef PubMed; (b) C. Sämann, V. Dhayalan, P. R. Schreiner and P. Knochel, Org. Lett., 2014, 16, 2418–2421 CrossRef PubMed; (c) V. Dhayalan and P. Knochel, Synthesis, 2015, 3246–3256 CAS; (d) V. Dhayalan, C. Sämann and P. Knochel, Chem. Commun., 2015, 51, 3239–3242 RSC; (e) D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS; (f) T. D. Blümke, T. Klatt, K. Koszinowski and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 9926–9930 CrossRef PubMed.
- T. Brückl, I. Thoma, A. J. Wagner, P. Knochel and T. Carell, Eur. J. Org. Chem., 2010, 6517–6519 CrossRef.
- A. D. Benischke, L. Anthore-Dalion, G. Berionni and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 16390–16394 CrossRef CAS PubMed.
- M. Ketels, M. A. Ganiek, N. Weidmann and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 12770–12773 CrossRef CAS PubMed.
- D. S. Ziegler, B. Wei and P. Knochel, Chem. – Eur. J., 2019, 25, 2695–2703 CrossRef CAS PubMed.
- L. Anthore-Dalion, A. D. Benischke, B. Wei, G. Berionni and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 4046–4050 CrossRef CAS PubMed.
- M. Balkenhohl and P. Knochel, SynOpen, 2018, 02, 0078–0095 CrossRef CAS.
- (a) N. M. Brikci-Nigassa, G. Bentabed-Ababsa, W. Erb and F. Mongin, Synthesis, 2018, 3615–3633 CAS; (b) S. M. Manolikakes, N. M. Barl, C. Sämann and P. Knochel, Z. Naturforsch., 2013, 68b, 411–422 CrossRef.
- J. Board, J. L. Cosman, T. Rantanen, S. P. Singh and V. Snieckus, Platin. Met. Rev., 2013, 57, 234–258 CrossRef.
- Y.-H. Chen, M. Ellwart, V. Malakhov and P. Knochel, Synthesis, 2017, 3215–3223 CAS.
- F. H. Lutter, S. Graßl, L. Grokenberger, M. S. Hofmayer, Y.-H. Chen and P. Knochel, ChemCatChem, 2019, 11, 5188–5197 CrossRef CAS.
- A. Kremsmair, J. H. Harenberg, K. Schwärzer, A. Hess and P. Knochel, Chem. Sci., 2021, 12, 6011–6019 RSC.
- (a) A. B. Charette, C. Molinaro and C. Brochu, J. Am. Chem. Soc., 2001, 123, 12160–12167 CrossRef CAS PubMed; (b) T. J. Greshock, K. P. Moore, R. T. McClain, A. Bellomo, C. K. Chung, S. D. Dreher, P. S. Kutchukian, Z. Peng, I. W. Davies, P. Vachal, M. Ellwart, S. M. Manolikakes, P. Knochel and P. G. Nantermet, Angew. Chem., Int. Ed., 2016, 55, 13714–13718 CrossRef CAS PubMed; (c) Y.-H. Chen, C. P. Tüllmann, M. Ellwart and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 9236–9239 CrossRef CAS PubMed.
- W. A. Herrmann, A. M. J. Rost, J. K. M. Mitterpleininger, N. Szesni, S. Sturm, R. W. Fischer and F. E. Kühn, Angew. Chem., Int. Ed., 2007, 46, 7301–7303 CrossRef CAS PubMed.
- M. Ellwart and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 10662–10665 CrossRef CAS PubMed.
- A. Hernán-Gómez, E. Herd, E. Hevia, A. R. Kennedy, P. Knochel, K. Koszinowski, S. M. Manolikakes, R. E. Mulvey and C. Schnegelsberg, Angew. Chem., Int. Ed., 2014, 53, 2706–2710 CrossRef PubMed.
- J. Li and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 11436–11440 CrossRef CAS PubMed.
- (a) V. Dhayalan, R. Dandela, K. B. Devi and R. Dhanusuraman, SynOpen, 2022, 06, 31–57 CrossRef CAS; (b) B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502–10566 CrossRef CAS PubMed; (c) V. Dhayalan and M. Hayashi, Synthesis, 2012, 2209–2216 CAS; (d) C. A. Caputo and N. D. Jones, Dalton Trans., 2007, 4627–4640, 10.1039/B709283K.
- (a) G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC; (b) H.-L. Kwong, H.-L. Yeung, C.-T. Yeung, W.-S. Lee, C.-S. Lee and W.-L. Wong, Coord. Chem. Rev., 2007, 251, 2188–2222 CrossRef CAS; (c) M. Hechavarría Fonseca and B. König, Adv. Synth. Catal., 2003, 345, 1173–1185 CrossRef; (d) A. Pfaltz, Acc. Chem. Res., 1993, 26, 339–345 CrossRef CAS.
- J. Rong, J. F. Collados, P. Ortiz, R. P. Jumde, E. Otten and S. R. Harutyunyan, Nat. Commun., 2016, 7, 13780 CrossRef PubMed.
- U. Bhakta, P. V. Kattamuri, J. H. Siitonen, L. B. Alemany and L. Kürti, Org. Lett., 2019, 21, 9208–9211 CrossRef CAS PubMed.
- P. Ortiz, J. F. Collados, R. P. Jumde, E. Otten and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2017, 56, 3041–3044 CrossRef CAS PubMed.
- (a) J. F. Collados, R. Solà, S. R. Harutyunyan and B. Maciá, ACS Catal., 2016, 6, 1952–1970 CrossRef CAS; (b) F. Caprioli, A. V. R. Madduri, A. J. Minnaard and S. R. Harutyunyan, Chem. Commun., 2013, 49, 5450–5452 RSC.
- (a) J. Rong, T. Pellegrini and S. R. Harutyunyan, Chem. – Eur. J., 2016, 22, 3558–3570 CrossRef CAS PubMed; (b) P. Ortiz, A. M. del Hoyo and S. R. Harutyunyan, Eur. J. Org. Chem., 2015, 72–76 CrossRef CAS; (c) Z. Wu, S. R. Harutyunyan and A. J. Minnaard, Chem. – Eur. J., 2014, 20, 14250–14255 CrossRef CAS PubMed.
- K. N. Venugopala, V. Rashmi and B. Odhav, Biomed. Res. Int., 2013, 2013, 963248 CAS.
- P.-Y. Chung, Z.-X. Bian, H.-Y. Pun, D. Chan, A. S.-C. Chan, C.-H. Chui, J. C.-O. Tang and K.-H. Lam, Future Med. Chem., 2015, 7, 947–967 CrossRef CAS PubMed.
- (a) Salahuddin, M. Shaharyar and A. Mazumder, Arab. J. Chem., 2017, 10, S157–S173 CrossRef CAS; (b) J. Farhat, L. Alzyoud, M. Alwahsh and B. Al-Omari, Cancers, 2022, 14, 2196 CrossRef CAS PubMed; (c) T. Van de Walle, L. Cools, S. Mangelinckx and M. D'Hooghe, Eur. J. Med. Chem., 2021, 226, 113865 CrossRef CAS PubMed; (d) I. Briguglio, S. Piras, P. Corona, E. Gavini, M. Nieddu, G. Boatto and A. Carta, Eur. J. Med. Chem., 2015, 97, 612–648 CrossRef CAS PubMed.
- (a) B. S. Matada, R. Pattanashettar and N. G. Yernale, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS PubMed; (b) S. Samanta, S. Kumar, E. K. Aratikatla, S. R. Ghorpade and V. Singh, RSC Med. Chem., 2023, 14, 644–657 RSC; (c) V. Yadav, J. Reang, V. Sharma, J. Majeed, P. C. Sharma, K. Sharma, N. Giri, A. Kumar and R. K. Tonk, Chem. Biol. Drug Des., 2022, 100, 389–418 CrossRef CAS PubMed.
- (a) D. Sharma, V. Dhayalan, R. Chatterjee, M. Khatravath and R. Dandela, ChemistrySelect, 2022, 7, e202104299 CrossRef CAS; (b) S. M. Umer, M. Solangi, K. M. Khan and R. S. Z. Saleem, Molecules, 2022, 27, 7586 CrossRef CAS PubMed; (c) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed; (d) C. Teixeira, N. Vale, B. Pérez, A. Gomes, J. R. B. Gomes and P. Gomes, Chem. Rev., 2014, 114, 11164–11220 CrossRef CAS PubMed.
- (a) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (b) G. A. Ramann and B. J. Cowen, Molecules, 2016, 21, 986 CrossRef PubMed; (c) T. Iwai and M. Sawamura, ACS Catal., 2015, 5, 5031–5040 CrossRef CAS; (d) P. Kannaboina, K. Mondal, J. K. Laha and P. Das, Chem. Commun., 2020, 56, 11749–11762 RSC; (e) J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723–1736 CrossRef CAS PubMed.
- (a) A. Corio, C. Gravier-Pelletier and P. Busca, Molecules, 2021, 26, 5467 CrossRef CAS PubMed; (b) M. Wasa, B. T. Worrell and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 1275–1277 CrossRef CAS PubMed; (c) A. K. Dhiman, A. Thakur, R. Kumar and U. Sharma, Asian J. Org. Chem., 2020, 9, 1502–1518 CrossRef CAS; (d) M. Monika and S. Selvakumar, Synthesis, 2019, 4113–4136 CAS.
- (a) D. Morgan, S. J. Yarwood and G. Barker, Eur. J. Org. Chem., 2021, 1072–1102 CrossRef CAS; (b) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Adv. Synth. Catal., 2021, 363, 2723–2739 CrossRef CAS; (c) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed.
- (a) G. Cahiez, A. Moyeux, J. Buendia and C. Duplais, J. Am. Chem. Soc., 2007, 129, 13788–13789 CrossRef CAS PubMed; (b) E. J. Alexanian and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 15627–15635 CrossRef CAS PubMed; (c) F. Rekhroukh, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 11920–11929 CrossRef CAS PubMed; (d) Z.-Y. Wang, X.-S. Peng and H. N. C. Wong, Asian J. Org. Chem., 2020, 9, 1834–1840 CrossRef CAS; (e) M. Karimzadeh-Younjali and O. F. Wendt, Helv. Chim. Acta, 2021, 104, e2100114 CrossRef CAS; (f) Y. Zhu, T. Xiong, W. Han and Y. Shi, Org. Lett., 2014, 16, 6144–6147 CrossRef CAS PubMed.
- P. Knochel, M. C. P. Yeh, S. C. Berk and J. Talbert, J. Org. Chem., 1988, 53, 2390–2392 CrossRef CAS.
- M. Jaric, B. A. Haag, A. Unsinn, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 5451–5455 CrossRef CAS PubMed.
- C. J. Rohbogner, S. Wirth and P. Knochel, Org. Lett., 2010, 12, 1984–1987 CrossRef CAS PubMed.
- C. J. Rohbogner, S. Wirth and P. Knochel, Synfacts, 2010, 0936 Search PubMed.
- S. Duez, A. K. Steib, S. M. Manolikakes and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 7686–7690 CrossRef CAS PubMed.
- K. Snégaroff, T. T. Nguyen, N. Marquise, Y. S. Halauko, P. J. Harford, T. Roisnel, V. E. Matulis, O. A. Ivashkevich, F. Chevallier, A. E. H. Wheatley, P. C. Gros and F. Mongin, Chem. – Eur. J., 2011, 17, 13284–13297 CrossRef PubMed.
- M. Jaric, B. A. Haag, S. M. Manolikakes and P. Knochel, Org. Lett., 2011, 13, 2306–2309 CrossRef CAS PubMed.
- M. Balkenhohl, C. François, D. Sustac Roman, P. Quinio and P. Knochel, Org. Lett., 2017, 19, 536–539 CrossRef CAS PubMed.
- S. M. Manolikakes, M. Jaric, K. Karaghiosoff and P. Knochel, Chem. Commun., 2013, 49, 2124–2126 RSC.
- S. Kumar Rout, A. Kastrati, H. Jangra, K. Schwärzer, A. S. Sunagatullina, M. Garny, F. Lima, C. E. Brocklehurst, K. Karaghiosoff, H. Zipse and P. Knochel, Chem. – Eur. J., 2022, 28, e202200733 CrossRef CAS PubMed.
- R. N. Rao and K. Chanda, Bioorg. Chem., 2020, 99, 103801 CrossRef CAS PubMed.
- C. Le Manach, D. Gonzàlez Cabrera, F. Douelle, A. T. Nchinda, Y. Younis, D. Taylor, L. Wiesner, K. L. White, E. Ryan, C. March, S. Duffy, V. M. Avery, D. Waterson, M. J. Witty, S. Wittlin, S. A. Charman, L. J. Street and K. Chibale, J. Med. Chem., 2014, 57, 2789–2798 CrossRef CAS PubMed.
- A. Unsinn and P. Knochel, Chem. Commun., 2012, 48, 2680–2682 RSC.
- S. Duez, A. K. Steib and P. Knochel, Org. Lett., 2012, 14, 1951–1953 CrossRef CAS PubMed.
- A. Unsinn, S. H. Wunderlich and P. Knochel, Adv. Synth. Catal., 2013, 355, 989–995 CrossRef CAS.
- L. Klier, T. Bresser, T. A. Nigst, K. Karaghiosoff and P. Knochel, J. Am. Chem. Soc., 2012, 134, 13584–13587 CrossRef CAS PubMed.
- C. I. Stathakis, S. M. Manolikakes and P. Knochel, Org. Lett., 2013, 15, 1302–1305 CrossRef CAS PubMed.
- J. M. Hammann, D. Haas and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 4478–4481 CrossRef CAS PubMed.
- T. Bresser, M. Mosrin, G. Monzon and P. Knochel, J. Org. Chem., 2010, 75, 4686–4695 CrossRef CAS PubMed.
- F. Crestey and P. Knochel, Synthesis, 2010, 1097–1106 CAS.
- S. Zimdars, H. Langhals and P. Knochel, Synthesis, 2011, 1302–1308 CAS.
- M. Balkenhohl, H. Jangra, I. S. Makarov, S.-M. Yang, H. Zipse and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 14992–14999 CrossRef CAS PubMed.
- M. Kienle, A. Unsinn and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 4751–4754 CrossRef CAS PubMed.
- T. Kunz and P. Knochel, Chem. – Eur. J., 2011, 17, 866–872 CrossRef CAS PubMed.
- G. Monzón, I. Tirotta and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 10624–10627 CrossRef PubMed.
- T. Kunz and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 1958–1961 CrossRef CAS PubMed.
- A. Frischmuth, A. Unsinn, K. Groll, H. Stadtmüller and P. Knochel, Chem. – Eur. J., 2012, 18, 10234–10238 CrossRef CAS PubMed.
- K. Groll, S. M. Manolikakes, X. M. du Jourdin, M. Jaric, A. Bredihhin, K. Karaghiosoff, T. Carell and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 6776–6780 CrossRef CAS PubMed.
- F. Crestey, S. Zimdars and P. Knochel, Synthesis, 2013, 3029–3037 CAS.
- T. Klatt, D. S. Roman, T. León and P. Knochel, Org. Lett., 2014, 16, 1232–1235 CrossRef CAS PubMed.
- M. R. Becker and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 12501–12505 CrossRef CAS PubMed.
- J. Nafe, S. Herbert, F. Auras, K. Karaghiosoff, T. Bein and P. Knochel, Chem. – Eur. J., 2015, 21, 1102–1107 CrossRef CAS PubMed.
- S. Achelle, C. Baudequin and N. Plé, Dyes Pigm., 2013, 98, 575–600 CrossRef CAS.
- M. Balkenhohl, R. Greiner, I. S. Makarov, B. Heinz, K. Karaghiosoff, H. Zipse and P. Knochel, Chem. – Eur. J., 2017, 23, 13046–13050 CrossRef CAS PubMed.
- M. Balkenhohl, B. Salgues, T. Hirai, K. Karaghiosoff and P. Knochel, Org. Lett., 2018, 20, 3114–3118 CrossRef CAS PubMed.
- F. H. Lutter, L. Grokenberger, L. A. Perego, D. Broggini, S. Lemaire, S. Wagschal and P. Knochel, Nat. Commun., 2020, 11, 4443 CrossRef CAS PubMed.
- A. Hess, J. P. Prohaska, S. B. Doerrich, F. Trauner, F. H. Lutter, S. Lemaire, S. Wagschal, K. Karaghiosoff and P. Knochel, Chem. Sci., 2021, 12, 8424–8429 RSC.
- K. Schwärzer, S. K. Rout, D. Bessinger, F. Lima, C. E. Brocklehurst, K. Karaghiosoff, T. Bein and P. Knochel, Chem. Sci., 2021, 12, 12993–13000 RSC.
- S. Grosse, V. Mathieu, C. Pillard, S. Massip, M. Marchivie, C. Jarry, P. Bernard, R. Kiss and G. Guillaumet, Eur. J. Med. Chem., 2014, 84, 718–730 CrossRef CAS PubMed.
- L. Melzig, C. B. Rauhut, N. Naredi-Rainer and P. Knochel, Chem. – Eur. J., 2011, 17, 5362–5372 CrossRef CAS PubMed.
- E. Nagaradja, F. Chevallier, T. Roisnel, V. Dorcet, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and F. Mongin, Org. Biomol. Chem., 2014, 12, 1475–1487 RSC.
- M. Y. Messaoud, G. Bentabed-Ababsa, M. Hedidi, A. Derdour, F. Chevallier, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, L. Picot, V. Thiéry, T. Roisnel, V. Dorcet and F. Mongin, Beilstein J. Org. Chem., 2015, 11, 1475–1485 CrossRef CAS PubMed.
- M. Hasyeoui, P. M. Chapple, F. Lassagne, T. Roisnel, M. Cordier, A. Samarat, Y. Sarazin and F. Mongin, Eur. J. Org. Chem., 2023, e202300555 CrossRef CAS.
- M. F. Z. J. Amaral, A. A. Baumgartner, R. Vessecchi and G. C. Clososki, Org. Lett., 2015, 17, 238–241 CrossRef CAS PubMed.
- C. Schneider, E. Broda and V. Snieckus, Org. Lett., 2011, 13, 3588–3591 CrossRef CAS PubMed.
- D. Martinez-Solorio, B. Melillo, L. Sanchez, Y. Liang, E. Lam, K. N. Houk and A. B. Smith, III, J. Am. Chem. Soc., 2016, 138, 1836–1839 CrossRef CAS PubMed.
- M. E. Dalziel, J. J. Patel, M. K. Kaye, J. L. Cosman, M. O. Kitching and V. Snieckus, Angew. Chem., Int. Ed., 2019, 58, 7313–7317 CrossRef CAS PubMed.
- N. D. Adams, J. L. Adams, J. L. Burgess and A. M. Chaudhari, et al. , J. Med. Chem., 2010, 53, 3973–4001 CrossRef CAS PubMed.
- C. Schneider, E. David, A. A. Toutov and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 2722–2726 CrossRef CAS PubMed.
- G. Zhao, M. Alami and O. Provot, RSC Adv., 2017, 7, 46007–46013 RSC.
- A. K. Steib, O. M. Kuzmina, S. Fernandez, S. Malhotra and P. Knochel, Chem. – Eur. J., 2015, 21, 1961–1965 CrossRef CAS PubMed.
- M. Balkenhohl, V. Valsamidou and P. Knochel, Eur. J. Org. Chem., 2019, 5165–5168 CrossRef CAS.
- A. K. Steib, S. Fernandez, O. M. Kuzmina, M. Corpet, C. Gosmini and P. Knochel, Synlett, 2015, 1049–1054 CAS.
- C. Sämann, B. Haag and P. Knochel, Chem. – Eur. J., 2012, 18, 16145–16152 CrossRef PubMed.
- O. M. Kuzmina, A. K. Steib, D. Flubacher and P. Knochel, Org. Lett., 2012, 14, 4818–4821 CrossRef CAS PubMed.
- A. K. Steib, O. M. Kuzmina, S. Fernandez, D. Flubacher and P. Knochel, J. Am. Chem. Soc., 2013, 135, 15346–15349 CrossRef CAS PubMed.
- O. M. Kuzmina, A. K. Steib, J. T. Markiewicz, D. Flubacher and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 4945–4949 CrossRef CAS PubMed.
- A. Frischmuth and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 10084–10088 CrossRef CAS PubMed.
- N. M. Barl, E. Sansiaume-Dagousset, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 10093–10096 CrossRef CAS PubMed.
- Q. Chen, X. M. du Jourdin and P. Knochel, J. Am. Chem. Soc., 2013, 135, 4958–4961 CrossRef CAS PubMed.
- J. Nickel, M. Fernández, L. Klier and P. Knochel, Chem. – Eur. J., 2016, 22, 14397–14400 CrossRef CAS PubMed.
- M. Tsuda, K. Nozawa, K. Shimbo and J. I. Kobayashi, J. Nat. Prod., 2003, 66, 292–294 CrossRef CAS PubMed.
- R. Greiner, R. Blanc, C. Petermayer, K. Karaghiosoff and P. Knochel, Synlett, 2016, 231–236 CAS.
- A. B. Bellan, O. M. Kuzmina, V. A. Vetsova and P. Knochel, Synthesis, 2017, 188–194 CAS.
- D. S. Ziegler, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 6701–6704 CrossRef CAS PubMed.
- M. Balkenhohl, B. Heinz, T. Abegg and P. Knochel, Org. Lett., 2018, 20, 8057–8060 CrossRef CAS PubMed.
- P. R. Graves, J. J. Kwiek, P. Fadden, R. Ray, K. Hardeman, A. M. Coley, M. Foley and T. A. J. Haystead, Mol. Pharmacol., 2002, 62, 1364–1372 CrossRef CAS PubMed.
- J. Wei, H. Liang, C. Ni, R. Sheng and J. Hu, Org. Lett., 2019, 21, 937–940 CrossRef CAS PubMed.
- B. Heinz, D. Djukanovic, M. A. Ganiek, B. Martin, B. Schenkel and P. Knochel, Org. Lett., 2020, 22, 493–496 CrossRef CAS PubMed.
- A. T. Baviskar, C. Madaan, R. Preet, P. Mohapatra, V. Jain, A. Agarwal, S. K. Guchhait, C. N. Kundu, U. C. Banerjee and P. V. Bharatam, J. Med. Chem., 2011, 54, 5013–5030 CrossRef CAS PubMed.
- H. Gao, Q.-L. Xu, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., Int. Ed., 2014, 53, 2701–2705 CrossRef CAS PubMed.
- S. Yamada, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 2215–2218 CrossRef CAS PubMed.
- X. Yan, L. Ge, M. Castiñeira Reis and S. R. Harutyunyan, J. Am. Chem. Soc., 2020, 142, 20247–20256 CrossRef CAS PubMed.
- L. Zhu, X. Sheng, Y. Li, D. Lu, R. Qiu and N. Kambe, Org. Lett., 2019, 21, 6785–6789 CrossRef CAS PubMed.
- Z. Zhou, Z. Ma, N. E. Behnke, H. Gao and L. Kürti, J. Am. Chem. Soc., 2017, 139, 115–118 CrossRef CAS PubMed.
- R. P. Jumde, F. Lanza, M. J. Veenstra and S. R. Harutyunyan, Science, 2016, 352, 433–437 CrossRef CAS PubMed.
- L. Ge, M. Zurro and S. R. Harutyunyan, Chem. – Eur. J., 2020, 26, 16277–16280 CrossRef CAS PubMed.
- A. Baralle, S. Otsuka, V. Guérin, K. Murakami, H. Yorimitsu and A. Osuka, Synlett, 2015, 327–330 CAS.
- S. Bernhardt, G. Manolikakes, T. Kunz and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9205–9209 CrossRef CAS PubMed.
- C. I. Stathakis, S. Bernhardt, V. Quint and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 9428–9432 CrossRef CAS PubMed.
- N. M. Barl, V. Malakhov, C. Mathes, P. Lustenberger and P. Knochel, Synthesis, 2015, 692–700 CAS.
- D. Haas, J. M. Hammann, F. H. Lutter and P. Knochel, Angew. Chem., Int. Ed., 2016, 55, 3809–3812 CrossRef CAS PubMed.
- J. M. Hammann, F. H. Lutter, D. Haas and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 1082–1086 CrossRef CAS PubMed.
- Y.-H. Chen, S. Graßl and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 1108–1111 CrossRef CAS PubMed.
- J. M. Hammann, L. Thomas, Y.-H. Chen, D. Haas and P. Knochel, Org. Lett., 2017, 19, 3847–3850 CrossRef CAS PubMed.
- M. S. Hofmayer, F. H. Lutter, L. Grokenberger, J. M. Hammann and P. Knochel, Org. Lett., 2019, 21, 36–39 CrossRef CAS PubMed.
- M. Leroux, T. Vorherr, I. Lewis, M. Schaefer, G. Koch, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 8231–8234 CrossRef CAS PubMed.
- J. Li, E. Tan, N. Keller, Y.-H. Chen, P. M. Zehetmaier, A. C. Jakowetz, T. Bein and P. Knochel, J. Am. Chem. Soc., 2019, 141, 98–103 CrossRef CAS PubMed.
- (a) X. Cheng, X. Liu, S. Wang, Y. Hu, B. Hu, A. Lei and J. Li, Nat. Commun., 2021, 12, 4366 CrossRef CAS PubMed; (b) J. Lin, K. Chen, J. Wang, J. Guo, S. Dai, Y. Hu and J. Li, Chem. Sci., 2023, 14, 8672–8680 RSC; (c) S. Dong, Z. Tian, J. Wang, L. He and J. Li, J. Catal., 2023, 425, 350–358 CrossRef CAS.
- J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202202379 CrossRef CAS PubMed.
- A. Kremsmair, S. Graßl, C. J. B. Seifert, E. Godineau and P. Knochel, Synthesis, 2021, 4068–4074 CAS.
- T. Čarný, T. Peňaška, S. Andrejčák and R. Šebesta, Chem. – Eur. J., 2022, 28, e202202040 CrossRef PubMed.
- L. Melzig, A. Metzger and P. Knochel, J. Org. Chem., 2010, 75, 2131–2133 CrossRef CAS PubMed.
- M. R. Luzung, J. S. Patel and J. Yin, J. Org. Chem., 2010, 75, 8330–8332 CrossRef CAS PubMed.
- L. Melzig, T. Dennenwaldt, A. Gavryushin and P. Knochel, J. Org. Chem., 2011, 76, 8891–8906 CrossRef CAS PubMed.
- I. Hyodo, M. Tobisu and N. Chatani, Chem. – Asian J., 2012, 7, 1357–1365 CrossRef CAS PubMed.
- Y. Yang, N. J. Oldenhuis and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 615–619 CrossRef CAS PubMed.
- J. R. Colombe, S. Bernhardt, C. Stathakis, S. L. Buchwald and P. Knochel, Org. Lett., 2013, 15, 5754–5757 CrossRef CAS PubMed.
- S. Thapa, A. Kafle, S. K. Gurung, A. Montoya, P. Riedel and R. Giri, Angew. Chem., Int. Ed., 2015, 54, 8236–8240 CrossRef CAS PubMed.
- R. Greiner, D. S. Ziegler, D. Cibu, A. C. Jakowetz, F. Auras, T. Bein and P. Knochel, Org. Lett., 2017, 19, 6384–6387 CrossRef CAS PubMed.
- S. Graßl, Y.-H. Chen, C. Hamze, C. P. Tüllmann and P. Knochel, Org. Lett., 2019, 21, 494–497 CrossRef PubMed.
- M. Balkenhohl, D. S. Ziegler, A. Desaintjean, L. J. Bole, A. R. Kennedy, E. Hevia and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 12898–12902 CrossRef CAS PubMed.
- S. Graßl, J. Singer and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 335–338 CrossRef PubMed.
- Z.-L. Shen, V. Dhayalan, A. D. Benischke, R. Greiner, K. Karaghiosoff, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2016, 55, 5332–5336 CrossRef CAS PubMed.
- M. Anzini, A. Chelini, A. Mancini, A. Cappelli, M. Frosini, L. Ricci, M. Valoti, J. Magistretti, L. Castelli, A. Giordani, F. Makovec and S. Vomero, J. Med. Chem., 2010, 53, 734–744 CrossRef CAS PubMed.
- W. M. Huggins, W. T. Barker, J. T. Baker, N. A. Hahn, R. J. Melander and C. Melander, ACS Med. Chem. Lett., 2018, 9, 702–707 CrossRef CAS PubMed.
- X. Zheng, C. Liang, L. Wang, K. Miao, B. Wang, W. Zhang, D. Chen, G. Wu, W. Zhu, L. Guo, S. Feng, L. Gao, H. C. Shen and H. Yun, MedChemComm, 2019, 10, 970–973 RSC.
- Ž. Skok, M. Barančoková, O. Benek, C. D. Cruz, P. Tammela, T. Tomašič, N. Zidar, L. P. Mašič, A. Zega, C. E. M. Stevenson, J. E. A. Mundy, D. M. Lawson, A. Maxwell, D. Kikelj and J. Ilaš, ACS Med. Chem. Lett., 2020, 11, 2433–2440 CrossRef PubMed.
- G. Catinella, L. M. Mattio, L. Musso, S. Arioli, D. Mora, G. L. Beretta, N. Zaffaroni, A. Pinto and S. Dallavalle, Int. J. Mol. Sci., 2020, 21, 2168 CrossRef CAS PubMed.
- A. E. Cotman, M. Durcik, D. Benedetto Tiz, F. Fulgheri, D. Secci, M. Sterle, Š. Možina, Ž. Skok, N. Zidar, A. Zega, J. Ilaš, L. Peterlin Mašič, T. Tomašič, D. Hughes, D. L. Huseby, S. Cao, L. Garoff, T. Berruga Fernández, P. Giachou, L. Crone, I. Simoff, R. Svensson, B. Birnir, S. V. Korol, Z. Jin, F. Vicente, M. C. Ramos, M. De La Cruz, B. Glinghammar, L. Lenhammar, S. R. Henderson, J. E. A. Mundy, A. Maxwell, C. E. M. Stevenson, D. M. Lawson, G. V. Janssen, G. J. Sterk and D. Kikelj, J. Med. Chem., 2023, 66, 1380–1425 CrossRef CAS PubMed.
- A. Costales, M. Mathur, S. Ramurthy, J. Lan, S. Subramanian, R. Jain, G. Atallah, L. Setti, M. Lindvall, B. A. Appleton, E. Ornelas, P. Feucht, B. Warne, L. Doyle, S. E. Basham, I. Aronchik, A. B. Jefferson and C. M. Shafer, Bioorg. Med. Chem. Lett., 2014, 24, 1592–1596 CrossRef CAS PubMed.
- D. K. Luci, J. B. Jameson, A. Yasgar, G. Diaz, N. Joshi, A. Kantz, K. Markham, S. Perry, N. Kuhn, J. Yeung, E. H. Kerns, L. Schultz, M. Holinstat, J. L. Nadler, D. A. Taylor-Fishwick, A. Jadhav, A. Simeonov, T. R. Holman and D. J. Maloney, J. Med. Chem., 2014, 57, 495–506 CrossRef CAS PubMed.
- S. Y. Han, C. O. Lee, S. H. Ahn, M. O. Lee, S. Y. Kang, H. J. Cha, S. Y. Cho, J. Du Ha, J. W. Ryu, H. Jung, H. R. Kim, J. S. Koh and J. Lee, Invest. New Drugs, 2012, 30, 518–523 CrossRef CAS PubMed.
- H. S. Salo, T. Laitinen, A. Poso, E. Jarho and M. Lahtela-Kakkonen, Bioorg. Med. Chem. Lett., 2013, 23, 2990–2995 CrossRef CAS PubMed.
- B. León, J. C. N. Fong, K. C. Peach, W. R. Wong, F. H. Yildiz and R. G. Linington, Org. Lett., 2013, 15, 1234–1237 CrossRef PubMed.
- J. J. Cui, H. Shen, M. Tran-Dubé, M. Nambu, M. McTigue, N. Grodsky, K. Ryan, S. Yamazaki, S. Aguirre, M. Parker, Q. Li, H. Zou and J. Christensen, J. Med. Chem., 2013, 56, 6651–6665 CrossRef CAS PubMed.
- C. Padmakar Darne, U. Velaparthi, M. Saulnier, D. Frennesson, P. Liu, A. Huang, J. Tokarski, A. Fura, T. Spires, J. Newitt, V. M. Spires, M. T. Obermeier, P. A. Elzinga, M. M. Gottardis, L. Jayaraman, G. D. Vite and A. Balog, Bioorg. Med. Chem. Lett., 2022, 75, 128951 CrossRef CAS PubMed.
- A. Carta, G. Sanna, I. Briguglio, S. Madeddu, G. Vitale, S. Piras, P. Corona, A. T. Peana, E. Laurini, M. Fermeglia, S. Pricl, A. Serra, E. Carta, R. Loddo and G. Giliberti, Eur. J. Med. Chem., 2018, 145, 559–569 CrossRef CAS PubMed.
- Z. Yuan, S. Chen, C. Chen, J. Chen, C. Chen, Q. Dai, C. Gao and Y. Jiang, Eur. J. Med. Chem., 2017, 138, 1135–1146 CrossRef CAS PubMed.
- S. B. Patel, B. D. Patel, C. Pannecouque and H. G. Bhatt, Eur. J. Med. Chem., 2016, 117, 230–240 CrossRef CAS PubMed.
- L. Herrmann, M. Leidenberger, A. Sacramento de Morais, C. Mai, A. Çapci, M. da Cruz Borges Silva, F. Plass, A. Kahnt, D. R. M. Moreira, B. Kappes and S. B. Tsogoeva, Chem. Sci., 2023, 14, 12941–12952 RSC.
- D. J. Fu, P. Li, B. W. Wu, X. X. Cui, C. Bin Zhao and S. Y. Zhang, Eur. J. Med. Chem., 2019, 165, 309–322 CrossRef CAS PubMed.
- T. G. Kraljević, A. Harej, M. Sedić, S. K. Pavelić, V. Stepanić, D. Drenjančević, J. Talapko and S. Raić-Malić, Eur. J. Med. Chem., 2016, 124, 794–808 CrossRef PubMed.
- R. An, Z. Hou, J. T. Li, H. N. Yu, Y. H. Mou and C. Guo, Molecules, 2018, 23, 2281 CrossRef PubMed.
- B. Kahveci, F. Yılmaz, E. Menteşe and S. Ülker, Arch. Pharm. Chem. Life Sci., 2017, 350, e1600369 CrossRef PubMed.
- M. Holiyachi, S. L. Shastri, B. M. Chougala, L. A. Shastri, S. D. Joshi, S. R. Dixit, H. Nagarajaiah and V. A. Sunagar, ChemistrySelect, 2016, 1, 4638–4644 CrossRef CAS.
- R. Goel, V. Luxami and K. Paul, RSC Adv., 2015, 5, 37887–37895 RSC.
- H. Dai, M. Huang, J. Qian, J. Liu, C. Meng, Y. Li, G. Ming, T. Zhang, S. Wang, Y. Shi, Y. Yao, S. Ge, Y. Zhang and Y. Ling, Eur. J. Med. Chem., 2019, 166, 470–479 CrossRef CAS PubMed.
- S. Dhawan, N. Kerru, P. Awolade, A. Singh-Pillay, S. T. Saha, M. Kaur, S. B. Jonnalagadda and P. Singh, Bioorg. Med. Chem., 2018, 26, 5612–5623 CrossRef CAS PubMed.
- M. Wang, G. Zhang, Y. Wang, J. Wang, M. Zhu, S. Cen and Y. Wang, Bioorg. Med. Chem. Lett., 2020, 30, 127143 CrossRef CAS PubMed.
- S. Mah, J. Jang, D. Song, Y. Shin, M. Latif, Y. Jung and S. Hong, Org. Biomol. Chem., 2019, 17, 186–194 RSC.
- S. K. J. Shaikh, M. S. Sannaikar, M. N. Kumbar, P. K. Bayannavar, R. R. Kamble, S. R. Inamdar and S. D. Joshi, ChemistrySelect, 2018, 3, 4448–4462 CrossRef CAS.
- R. R. Manda, R. V. Nadh, T. L. Viveka, G. Angajala and V. Aruna, J. Mol. Struct., 2023, 1285, 135453 CrossRef CAS.
- B. M. Chougala, S. Samundeeswari, M. Holiyachi, N. S. Naik, L. A. Shastri, S. Dodamani, S. Jalalpure, S. R. Dixit, S. D. Joshi and V. A. Sunagar, ChemistrySelect, 2017, 2, 5234–5242 CrossRef CAS.
- K. M. Hosamani, D. S. Reddy and H. C. Devarajegowda, RSC Adv., 2015, 5, 11261–11271 RSC.
- S. Xue, L. Ma, R. Gao, Y. Li and Z. Li, Acta Pharm. Sin. B, 2014, 4, 313–321 CrossRef PubMed.
- D. S. Reddy, M. Kongot, V. Singh, M. A. Siddiquee, R. Patel, N. K. Singhal, F. Avecilla and A. Kumar, Arch. Pharm., 2021, 354, e2000181 CrossRef PubMed.
- M. Mahapatra, P. Mohapatra, S. K. Sahoo, A. K. Bishoyi, R. N. Padhy and S. K. Paidesetty, J. Mol. Struct., 2023, 1283, 135190 CrossRef CAS.
- L. Yang, Z. Hu, J. Luo, C. Tang, S. Zhang, W. Ning, C. Dong, J. Huang, X. Liu and H. B. Zhou, Bioorg. Med. Chem., 2017, 25, 3531–3539 CrossRef CAS PubMed.
- M. T. Gabr, Heterocycl. Commun., 2018, 24, 243–247 CrossRef CAS.
- P. Mutai, G. Breuzard, A. Pagano, D. Allegro, V. Peyrot and K. Chibale, Bioorg. Med. Chem., 2017, 25, 1652–1665 CrossRef CAS PubMed.
- O. Galayev, Y. Garazd, M. Garazd and R. Lesyk, Eur. J. Med. Chem., 2015, 105, 171–181 CrossRef CAS PubMed.
- D. Lu, A. Shen, Y. Liu, X. Peng, W. Xing, J. Ai, M. Geng and Y. Hu, Eur. J. Med. Chem., 2016, 115, 191–200 CrossRef CAS PubMed.
- J. C. Coa, W. Castrillón, W. Cardona, M. Carda, V. Ospina, J. A. Muñoz, I. D. Vélez and S. M. Robledo, Eur. J. Med. Chem., 2015, 101, 746–753 CrossRef CAS PubMed.
- N. A. Liberto, J. B. Simões, S. de Paiva Silva, C. J. da Silva, L. V. Modolo, Â. de Fátima, L. M. Silva, M. Derita, S. Zacchino, O. M. P. Zuñiga, G. P. Romanelli and S. A. Fernandes, Bioorg. Med. Chem., 2017, 25, 1153–1162 CrossRef CAS PubMed.
- M. P. Pinz, A. S. Reis, R. L. de Oliveira, G. T. Voss, A. G. Vogt, M. do Sacramento, J. A. Roehrs, D. Alves, C. Luchese and E. A. Wilhelm, Regul. Toxicol. Pharmacol., 2017, 90, 72–77 CrossRef CAS PubMed.
- P. Zajdel, K. Marciniec, A. Maślankiewicz, K. Grychowska, G. Satała, B. Duszyńska, T. Lenda, A. Siwek, G. Nowak, A. Partyka, D. Wróbel, M. Jastrzębska-Więsek, A. J. Bojarski, A. Wesołowska and M. Pawłowski, Eur. J. Med. Chem., 2013, 60, 42–50 CrossRef CAS PubMed.
- J. F. Mouscadet and D. Desmaële, Molecules, 2010, 15, 3048–3078 CrossRef CAS PubMed.
- E. Yoo, B. M. Crall, R. Balakrishna, S. S. Malladi, L. M. Fox, A. R. Hermanson and S. A. David, Org. Biomol. Chem., 2013, 11, 6526–6545 RSC.
- D. B. Salunke, E. Yoo, N. M. Shukla, R. Balakrishna, S. S. Malladi, K. J. Serafin, V. W. Day, X. Wang and S. A. David, J. Med. Chem., 2012, 55, 8137–8151 CrossRef CAS PubMed.
- M. Beesu, G. Caruso, A. C. D. Salyer, K. K. Khetani, D. Sil, M. Weerasinghe, H. Tanji, U. Ohto, T. Shimizu and S. A. David, J. Med. Chem., 2015, 58, 7833–7849 CrossRef CAS PubMed.
- Y. M. Fang, R. R. Zhang, Z. H. Shen, H. K. Wu, C. X. Tan, J. Q. Weng, T. M. Xu and X. H. Liu, J. Heterocycl. Chem., 2018, 55, 240–245 CrossRef CAS.
- M. Alagumuthu and S. Arumugam, Bioorg. Med. Chem., 2017, 25, 1448–1455 CrossRef CAS PubMed.
- N. C. Desai, B. Y. Patel and B. P. Dave, Med. Chem. Res., 2017, 26, 109–119 CrossRef CAS.
- G. Z. Yang, J. K. Zhu, X. D. Yin, Y. F. Yan, Y. L. Wang, X. F. Shang, Y. Q. Liu, Z. M. Zhao, J. W. Peng and H. Liu, J. Agric. Food Chem., 2019, 67, 11340–11353 CrossRef CAS PubMed.
- B. Tanwar, A. Kumar, P. Yogeeswari, D. Sriram and A. K. Chakraborti, Bioorg. Med. Chem. Lett., 2016, 26, 5960–5966 CrossRef CAS PubMed.
- J. McNulty, R. Vemula, C. Bordón, R. Yolken and L. Jones-Brando, Org. Biomol. Chem., 2014, 12, 255–260 RSC.
- S. Vandekerckhove, S. De Moor, D. Segers, C. De Kock, P. J. Smith, K. Chibale, N. De Kimpe and M. D’Hooghe, MedChemComm, 2013, 4, 724–730 RSC.
- S. H. Chan, C. H. Chui, S. W. Chan, S. H. L. Kok, D. Chan, M. Y. T. Tsoi, P. H. M. Leung, A. K. Y. Lam, A. S. C. Chan, K. H. Lam and J. C. O. Tang, ACS Med. Chem. Lett., 2013, 4, 170–174 CrossRef CAS PubMed.
- K. Li, Y. Li, D. Zhou, Y. Fan, H. Guo, T. Ma, J. Wen, D. Liu and L. Zhao, Bioorg. Med. Chem., 2016, 24, 1889–1897 CrossRef CAS PubMed.
- T. Barbier, A. Barbry, J. Magand, C. Badiou, F. Davy, A. Baudouin, Y. Queneau, O. Dumitrescu, G. Lina and L. Soulère, Biomolecules, 2022, 12, 131 CrossRef CAS PubMed.
- C. Bai, S. Wu, S. Ren, M. Zhu, G. Luo and H. Xiang, Bioorg. Med. Chem., 2021, 47, 116395 CrossRef CAS PubMed.
- L. Berrade, B. Aisa, M. J. Ramirez, S. Galiano, S. Guccione, L. R. Moltzau, F. O. Levy, F. Nicoletti, G. Battaglia, G. Molinaro, I. Aldana, A. Monge and S. Perez-Silanes, J. Med. Chem., 2011, 54, 3086–3090 CrossRef CAS PubMed.
- P. J. Masih, T. Kesharwani, E. Rodriguez, M. A. Vertudez, M. L. Motakhaveri, T. K. Le, M. K. T. Tran, M. R. Cloyd, C. T. Kornman and A. M. Phillips, Pharmaceuticals, 2022, 15, 39 CrossRef CAS PubMed.
- U. Singh, G. Chashoo, S. U. Khan, P. Mahajan, A. Nargotra, G. Mahajan, A. Singh, A. Sharma, M. J. Mintoo, S. K. Guru, H. Aruri, T. Thatikonda, P. Sahu, P. Chibber, V. Kumar, S. A. Mir, S. S. Bharate, S. Madishetti, U. Nandi, G. Singh, D. M. Mondhe, S. Bhushan, F. Malik, S. Mignani, R. A. Vishwakarma and P. P. Singh, J. Med. Chem., 2017, 60, 9470–9489 CrossRef CAS PubMed.
- M. A. Qhobosheane, L. J. Legoabe, B. Josselin, S. Bach, S. Ruchaud and R. M. Beteck, Chem. Biol. Drug Des., 2020, 96, 1395–1407 CrossRef CAS PubMed.
- B. Daydé-Cazals, B. Fauvel, M. Singer, C. Feneyrolles, B. Bestgen, F. Gassiot, A. Spenlinhauer, P. Warnault, N. Van Hijfte, N. Borjini, G. Chevé and A. Yasri, J. Med. Chem., 2016, 59, 3886–3905 CrossRef PubMed.
- K. Zhang, F. Lai, S. Lin, M. Ji, J. Zhang, Y. Zhang, J. Jin, R. Fu, D. Wu, H. Tian, N. Xue, L. Sheng, X. Zou, Y. Li, X. Chen and H. Xu, J. Med. Chem., 2019, 62, 6992–7014 CrossRef CAS PubMed.
- X. Tang, Q. Zhou, W. Zhan, D. Hu, R. Zhou, N. Sun, S. Chen, W. Wu and W. Xue, RSC Adv., 2022, 12, 2399–2407 RSC.
- R. Ibba, P. Corona, F. Nonne, P. Caria, G. Serreli, V. Palmas, F. Riu, S. Sestito, M. Nieddu, R. Loddo, G. Sanna, S. Piras and A. Carta, Pharmaceuticals, 2023, 16, 429 CrossRef CAS PubMed.
- N. D. Gaikwad, S. V. Patil and V. D. Bobade, Bioorg. Med. Chem. Lett., 2012, 22, 3449–3454 CrossRef CAS PubMed.
- Z. Sun, H. Li, G. Liu, C. Fan and S. Pu, Dyes Pigm., 2014, 106, 94–104 CrossRef CAS.
- C. Martin, C. Borreguero, K. Kennes, M. Van Der Auweraer, J. Hofkens, G. De Miguel and E. M. García-Frutos, ACS Energy Lett., 2017, 2, 2653–2658 CrossRef CAS.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M. A. Kakimoto, Chem. Mater., 2008, 20, 2532–2537 CrossRef CAS.
- H. Zhu, H. Tong, Y. Gong, S. Shao, C. Deng, W. Z. Yuan and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2172–2181 CrossRef CAS.
- M. Ileri, S. O. Hacioglu and L. Toppare, Electrochim. Acta, 2013, 109, 214–220 CrossRef CAS.
- S. Kumar, T. Marcato, S. I. Vasylevskyi, J. Jagielski, K. M. Fromm and C. J. Shih, J. Mater. Chem. C, 2019, 7, 8938–8945 RSC.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- A. P. Chafin, M. C. Davis, W. W. Lai, G. A. Lindsay, D. H. Park and W. N. Herman, Opt. Mater., 2011, 33, 1307–1315 CrossRef CAS.
- F. Lu, R. Hu, S. Wang, X. Guo and G. Yang, RSC Adv., 2017, 7, 4196–4202 RSC.
- C. H. Chen, Z. H. Luo and K. Tan, Macromol. Chem. Phys., 2018, 219, 1–7 Search PubMed.
- G. K. Dutta, S. Guha and S. Patil, Org. Electron., 2010, 11, 1–9 CrossRef CAS.
- A. G. Dikundwar, G. K. Dutta, T. N. G. Row and S. Patil, Cryst. Growth Des., 2011, 11, 1615–1622 CrossRef CAS.
- G. R. Suman, S. G. Bubbly, S. B. Gudennavar and V. Gayathri, J. Photochem. Photobiol., A, 2019, 382, 111947 CrossRef CAS.
- N. P. Thekkeppat, M. Lakshmipathi, A. S. Jalilov, P. Das, A. M. P. Peedikakkal and S. Ghosh, Cryst. Growth Des., 2020, 20, 3937–3943 CrossRef CAS.
- A. Iwan, M. Palewicz, M. Krompiec, M. Grucela-Zajac, E. Schab-Balcerzak and A. Sikora, Spectrochim. Acta, Part A, 2012, 97, 546–555 CrossRef CAS PubMed.
- S. Pu, M. Li, C. Fan, G. Liu and L. Shen, J. Mol. Struct., 2009, 919, 100–111 CrossRef CAS.
- C. Seo, J. M. Choi, S. S. Hong, J. Y. Lee and S. Y. Seo, Dyes Pigm., 2017, 136, 145–149 CrossRef CAS.
- Y. J. Zhong, K. Q. Zhao, B. Q. Wang, P. Hu, H. Monobe, B. Heinrich and B. Donnio, Dyes Pigm., 2020, 173, 107964 CrossRef CAS.
- A. Tanimoto and T. Yamamoto, Macromolecules, 2006, 39, 3546–3552 CrossRef CAS.
- E. Kaya, A. Balan, D. Baran, A. Cirpan and L. Toppare, Org. Electron., 2011, 12, 202–209 CrossRef CAS.
- S. Yum, T. K. An, X. Wang, W. Lee, M. A. Uddin, Y. J. Kim, T. L. Nguyen, S. Xu, S. Hwang, C. E. Park and H. Y. Woo, Chem. Mater., 2014, 26, 2147–2154 CrossRef CAS.
- Y. Chen, Q. Zhang, M. Du, G. Li, Z. Li, H. Huang, Y. Geng, X. Zhang and E. Zhou, ACS Appl. Polym. Mater., 2019, 1, 906–913 CrossRef CAS.
- C. Carayon and S. Fery-Forgues, Photochem. Photobiol. Sci., 2017, 16, 1020–1035 CrossRef CAS PubMed.
- T. Uchacz, P. Szlachcic, A. A. Fedorchuk, E. Gondek, A. Danel, B. Jarosz, A. M. ElNaggar, A. A. Albassam, I. V. Kityk, G. Lakshminarayana, P. Czaja and P. Karasiński, J. Mol. Struct., 2018, 1173, 531–540 CrossRef CAS.
- K. Erden and C. Dengiz, J. Org. Chem., 2022, 87, 4385–4399 CrossRef CAS PubMed.
- Z. Zhang, J. Xie, H. Wang, B. Shen, J. Zhang, J. Hao, J. Cao and Z. Wang, Dyes Pigm., 2016, 125, 299–308 CrossRef CAS.
- D. Y. Kim, J. Kang, S. E. Lee, Y. K. Kim and S. S. Yoon, Luminescence, 2017, 32, 1180–1185 CrossRef CAS PubMed.
- B. Li, Z. Wang, S. J. Su, F. Guo, Y. Cao and Y. Zhang, Adv. Opt. Mater., 2019, 7, 1–8 Search PubMed.
- R. Keruckiene, S. Vekteryte, E. Urbonas, M. Guzauskas, E. Skuodis, D. Volyniuk and J. V. Grazulevicius, Beilstein J. Org. Chem., 2020, 16, 1142–1153 CrossRef CAS PubMed.
- Z. Wang, H. Li, Z. Peng, Z. Wang, Y. Wang and P. Lu, RSC Adv., 2020, 10, 30297–30303 RSC.
- Z. Mao, L. Hu, X. Dong, C. Zhong, B. F. Liu and Z. Liu, Anal. Chem., 2014, 86, 6548–6554 CrossRef CAS PubMed.
- R. Flores-Noria, R. Vázquez, E. Arias, I. Moggio, M. Rodríguez, R. F. Ziolo, O. Rodríguez, D. R. Evans and C. Liebig, New J. Chem., 2014, 38, 974–984 RSC.
- G. Sych, D. Volyniuk, O. Bezvikonnyi, R. Lytvyn and J. V. Grazulevicius, J. Phys. Chem. C, 2019, 123, 2386–2397 CrossRef CAS.
- P. Tarnow, C. Zordick, A. Bottke, B. Fischer, F. Kühne, T. Tralau and A. Luch, Chem. Res. Toxicol., 2020, 33, 742–750 Search PubMed.
- X. Shao, W. Liu, R. Guo, J. Chen and N. Zhou, Dyes Pigm., 2021, 188, 10919 CrossRef.
- B. Mravec, Š. Budzák, M. Medved’, L. F. Pašteka, C. Slavov, T. Saßmannshausen, J. Wachtveitl, J. Kožíšek, L. Hegedüsová, J. Filo and M. Cigáň, J. Org. Chem., 2021, 86, 11633–11646 CrossRef CAS PubMed.
- Q. Xie, Y. Qu, G. Wang, X. Luo, D. Zhang, H. Zhou, L. Wang, L. Wang, Y. Miao and J. Huang, Dyes Pigm., 2022, 205, 110559 CrossRef CAS.
- Y. Y. Noh, R. Azumi, M. Goto, B. J. Jung, E. Lim, H. K. Shim, Y. Yoshida, K. Yase and D. Y. Kim, Chem. Mater., 2005, 17, 3861–3870 CrossRef CAS.
- A. B. Marco, R. Andreu, S. Franco, J. Garín, J. Orduna, B. Villacampa and R. Alicante, Tetrahedron, 2013, 69, 3919–3926 CrossRef CAS.
- Q. Wu, M. Wang, X. Qiao, Y. Xiong, Y. Huang, X. Gao and H. Li, Macromolecules, 2013, 46, 3887–3894 CrossRef CAS.
- G. Li, M. Liang, H. Wang, Z. Sun, L. Wang, Z. Wang and S. Xue, Chem. Mater., 2013, 25, 1713–1722 CrossRef CAS.
- M. O. Ahmed, W. Pisula and S. G. Mhaisalkar, Molecules, 2012, 17, 12163–12171 CrossRef CAS PubMed.
- X. Wang, Y. Sun, S. Chen, X. Guo, M. Zhang, X. Li, Y. Li and H. Wang, Macromolecules, 2012, 45, 1208–1216 CrossRef CAS.
- M. Durso, D. Gentili, C. Bettini, A. Zanelli, M. Cavallini, F. De Angelis, M. Grazia Lobello, V. Biondo, M. Muccini, R. Capelli and M. Melucci, Chem. Commun., 2013, 49, 4298–4300 RSC.
- M. Yasa, S. Surmeli, T. Depci, L. Toppare and S. O. Hacioglu, Macromol. Chem. Phys., 2020, 221, 1–10 CrossRef.
- G. K. Dutta and S. Patil, Org. Electron., 2012, 13, 1266–1276 CrossRef CAS.
- F. S. Mancilha, B. A. DaSilveira Neto, A. S. Lopes, P. F. Moreira, F. H. Quina, R. S. Gonçalves and J. Dupont, Eur. J. Org. Chem., 2006, 4924–4933 CrossRef CAS.
- K. R. Justin Thomas, M. Velusamy, T. Lin Jiann, C. H. Chuen and Y. T. Tao, Chem. Mater., 2005, 17, 1860–1866 CrossRef.
- C. Martín, K. Kennes, M. Van der Auweraer, J. Hofkens, G. de Miguel and E. M. García-Frutos, Adv. Funct. Mater., 2017, 27, 1–10 CrossRef.
Footnote |
| † Dedicated to Prof. Bert Maes. |
| This journal is © The Royal Society of Chemistry 2024 |