
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible photons as ideal reagents for the activation of coloured organic compounds
Lorenzo
Di Terlizzi
 ,
Luca
Nicchio
,
Stefano
Protti
* and
Maurizio
Fagnoni
*
,
Luca
Nicchio
,
Stefano
Protti
* and
Maurizio
Fagnoni
*
PhotoGreen Lab, Department of Chemistry, University of Pavia, Viale Taramelli 12, 27100 Pavia, Italy. E-mail: stefano.protti@unipv.it; fagnoni@unipv.it
First published on 10th April 2024
Abstract
In recent decades, the traceless nature of visible photons has been exploited for the development of efficient synthetic strategies for the photoconversion of colourless compounds, namely, photocatalysis, chromophore activation, and the formation of an electron donor/acceptor (EDA) complex. However, the use of photoreactive coloured organic compounds is the optimal strategy to boost visible photons as ideal reagents in synthetic protocols. In view of such premises, the present review aims to provide its readership with a collection of recent photochemical strategies facilitated via direct light absorption by coloured molecules. The protocols have been classified and presented according to the nature of the intermediate/excited state achieved during the transformation.
 Lorenzo Di Terlizzi | Lorenzo Di Terlizzi obtained his master's degree in organic chemistry in 2020 with a thesis on the development of aggressive diradicals starting from chloroaryl substituted carboxylic acids in the PhotoGreen lab under the supervision of Prof. Fagnoni and Prof. Protti. After that, he started his PhD in the same group, focusing on the use of arylazo sulfones to generate reactive intermediates under visible-light irradiations. He is currently a post-doc in the PhotoGreen Lab dealing with visible-light induced polymerizations, visible-light photoacid generators and photochemical synthesis. |
 Luca Nicchio | Luca Nicchio received his master's degree in chemistry from the University of Pavia (Italy) in 2022, under the supervision of Prof. Maurizio Fagnoni (Photogreen Lab). Luca is currently a second year PhD student in a joint PhD program between Université Paris-Saclay and University of Pavia (Université Paris-Saclay International Joint PhD Program (“Cotutelle”) 2022), under the supervision of Profs. Luc Neuville and Prof. Stefano Protti. His research subject focuses on the development of new and green synthetic approaches through visible-light generation of radical intermediates from arylazo sulfones and arylazo sulfonates. |
 Stefano Protti | Stefano Protti completed his PhD in Pavia (2007). Later he moved to the LASIR Laboratory (Lille, France) at the iBitTec-S laboratory (CEA Saclay, France) carrying out studies on photocatalyzed oxidation reactions for energy storage, he moved again to the University of Pavia, where has been serving as an Associate Professor since 2018. He currently serves as an editor for the SPR in Photochemistry (Royal Society of Chemistry), member of the International Advisory Board of EuJOC and an associate editor of the Photochemical and Photobiological Sciences. His work is mainly focused on the development of visible light mediated synthetic strategies. |
 Maurizio Fagnoni | Maurizio Fagnoni is currently a Full Professor in Organic Chemistry at the University of Pavia (PhotoGreen Lab, Italy). His research interests are mainly focused on the photoinduced or photocatalytic generation of reactive intermediates such as (bi)radicals, (phenyl) cations and radical ions and their application in eco-sustainable synthesis or in photolithography as well as biological application. He is currently a member of the committees of the Interdivisional group of Green Chemistry and Photochemistry of the Italian Society of Chemistry. In 2022, he received the Theresian Medal from the University of Pavia. |
1. Introduction
If only Giacomo Ciamician, while exposing an ethanolic solution of benzophenone to the Italian sun, had imagined what would happen a century later… come on, he predicted exactly that photochemistry would acquire a predominant role in organic synthesis!1–7 The capabilities of photons as reactants,8–13 capable of promoting the chemical conversion of a substrate without leaving traces in the reaction vessel, have been fully exploited in the last two decades through the overwhelming achievements of visible-light photocatalysis (in most cases, of photoredox catalysis) in synthesis.14–19 Such success has enabled the design of a plethora of versatile synthetic strategies, which have found wide application ranging from the synthesis of polyfunctionalized materials to the late-stage functionalization of bioactive molecules.20 Along with chemical reactions, new photochemical devices and reactors have been proposed, thanks to the widespread availability of unexpensive visible-light sources, including compact fluorescent lamps and LEDs, apart from natural sunlight.21,22In these strategies, the photocatalyst plays the role of absorbing photons and promoting, through its excited state, a chemical modification of (the) substrate(s) via different pathways that include electrons, atoms (e.g., hydrogen23 and halogens24) or energy transfer25 (a representative example in Scheme 1a).
 | ||
| Scheme 1 (a) A photocatalytic reaction (the coloured PC promotes the transformation upon light absorption). Colouring strategies to allow visible photons to induce a chemical reaction in a colourless compound through (b) and (c) chromophore activation by a Brønsted/Lewis acid, (d) and (e) the in situ formation of either an EDA complex or a coloured reactive species through nucleophilic activation and (f) the incorporation of a dyedauxiliary group in the colourless compound. The ideal case is (g) the direct photolysis of a coloured compound. Blue color represents the species that absorb visible photons. | ||
Obviously, a photocatalyst is not even needed if a photoreactive molecule, prone to undergo chemical conversion through direct visible-light absorption, is available; however, several organic compounds are colorless and absorb exclusively in the UV region. To overcome this limitation, alternative strategies have been designed to generate a coloured species in situ (or at least to enable the use of a longer wavelength to promote the process).
The first approach involves chromophore activation mainly through a complexing agent (e.g., Lewis and Brønsted acids).26 In this case, the chromophore (e.g., an enone A) may enhance its ε value by complexing with a Rh complex, thus facilitating the [2+2] cycloaddition onto a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (Scheme 1b). Alternatively, the protonation of colourless dithiane B generates a coloured thionium ion C, which is prone to forming a cyclobutane ring upon irradiation (Scheme 1c).27 Honestly, the latter two routes have not found a widespread application and have been applied only to selected cases.
C bond (Scheme 1b). Alternatively, the protonation of colourless dithiane B generates a coloured thionium ion C, which is prone to forming a cyclobutane ring upon irradiation (Scheme 1c).27 Honestly, the latter two routes have not found a widespread application and have been applied only to selected cases.
The formation of an electron donor acceptor (EDA) complex via electronic interaction of two (or more) colourless reaction partners (Scheme 1d) is a quite common strategy.28–33 The thus formed complex has a distinct absorption profile (a bathochromic shift occurred) different from that of the starting materials. The main drawback of this approach is the requirement for markedly different electronic characteristics of the reaction partners, necessitating the use of both a good electron-rich (e.g., the indole D) and an electron-poor (a benzyl bromide E) derivative to promote the EDA formation. An intriguing alternative is the generation of the coloured species H through the in situ chemical reaction of the starting (colourless) substrate F (having a leaving group LG) with an organic catalyst (in most cases, a nucleophile G, Scheme 1e).34 However, this approach is still in its infancy and only a few examples have been reported. These “colouring” strategies have been successfully applied either to a small range of substrates that bears a complexing site or to reactants having electronic properties conducive to the formation of an EDA complex. Another emerging approach involves the incorporation of a dyedauxiliary group in a colourless compound to impart both color and photoreactivity. An exemplary case is that of arylazo sulfones J (Scheme 1f), obtained in two steps from anilines I and prone to generating several radical intermediates upon photolysis.35,36 Saying so, the ideal case is the photochemical conversion of a (purposely designed) coloured compound into a valuable product without any additives, as it is the case for α-diazoesters K, which are widely used as precursors of carbenes L (Scheme 1g).37
Nonetheless, the fact that a given organic compound is coloured does not guarantee that it is photochemically active upon visible-light absorption. As an example, C-nitroso compounds (e.g., nitrosoaromatics) are green/blue coloured species but photochemically inactive due to their easy conversion to Z- and E-azodioxy dimers.38 In rare instances, such as in the case of 2-methyl-2-nitrosopropane (λmax at 680 nm due to an nπ* transition), photodissociation occurs.39
At the same time, the photoreactivity of a coloured substance may lead to interesting applications, as in the case of diazoarenes, discovered in 1883, probably the most well-known (and investigated) class of coloured compounds. The significant absorption in the visible region (430–450 nm, ε = 500–1500 M−1 cm−1) is largely due to the partially forbidden nπ* electronic transition which leads to E/Z isomerization,40 the phenomenon mostly applied in fields different from organic synthesis such as the development of photoswitches.41
A selection of compounds having photoreactive chromophores whose absorption lies in the visible region is briefly described below and sketched in Fig. 1.
 | ||
| Fig. 1 Light absorption range in the visible region of selected coloured photoreactive organic compounds. | ||
Carbonyls . It is well known that simple ketones do not absorb in the visible region, with the remarkable exception of some aromatic derivatives such as thioxanthone,42 9-fluorenone42 and 5,7,12,14-pentacenetetrone,43 which may find application as photoorganocatalysts.18 In contrast, α-diketones are slightly coloured and undergo the typical reactions of the carbonyl group (via the nπ* excited state) upon visible-light irradiation44–46 including [2+2]-heterocycloadditions.47 Quinones exhibit an absorption band in the 400–450 nm range,48 and have been employed in valuable reactions including cycloadditions,49 single electron transfer (SET) processes or photooxidations. Anthraquinones and naphthoquinones are well-investigated chromophores, capable of absorbing blue light and acting as triplet sensitizers (via the ππ* state) as well as photoredox and HAT photocatalysts.50,51
Thioketones mainly absorb in the 500–600 nm region, thanks to a dipole-forbidden n → π* transition, but they are rather rarely used in photochemistry compared to the corresponding ketones.52 The main application of these compounds is the synthesis of thietanes via a thia-Paternò-Büchi but mostly under UV radiation.47,53
The photochemistry of cyanoarenes (and in particular the significant reduction potential in the singlet excited state (e.g., >3.00 V vs. SCE for 1,2,4,5-tetracyanobenzene)54 was fundamental in the development of electron transfer processes.54 Some compounds belonging to this class (such as 9,10-dicyanoanthracene, DCA and 1,2,3,5-tetrakis(carbazole-9-yl)-4,6-dicyanobenzene, 4CzIPN) have found application in photoredox catalysis.55 Among aromatic derivatives, polycyclic aromatic hydrocarbons having at least four fused aromatic rings are coloured and known to undergo [4+4] photocyclodimerization.56–58
Two main classes are known to deliver carbenes upon photolysis, namely, acylsilanes and diazoderivatives. Acylsilanes exhibit an absorption band in the 350–420 nm region, making such compounds ideal precursors of α-siloxy carbenes upon visible-light irradiation via 1,2-photo Brook rearrangement.59,60 Similarly, diazocompounds including α-diazoesters and α-diazoketones are characterized by an absorption band in the 400–500 nm region, imparted by the NN moiety.61,62
A few other classes of compounds are known to release radicals by photohomolysis.63 The long wavelength absorption band of 1,4-dihydropyridines (such as the calcium-channel blocking drug Nifedipine64,65) located around 360 nm (ε = 7000 M−1 cm−1) extends beyond 400 nm and is attributed to a ππ* internal charge transfer transition. Recently, such compounds have been employed as electron and proton donors66 as well as alkyl radical precursors67 in photoredox catalyzed processes but the direct photocleavage of a C–C bond is an option.
Arylazo sulfones (Scheme 1f)35,36 and arylazo sulfonates68 show an absorption band in the 400–450 nm range, arising from a nπ* transition. Irradiating these sulfones with blue light induces the homolytic cleavage of the N–S bond, resulting in the subsequent formation of aryldiazenyl, sulfonyl and aryl radicals,35 making these compounds ideal candidates for the uncatalyzed formation of C–C bonds.69
N-Iodo and N-bromo amides and imides have been reported to absorb light above 400 nm and undergo N-halogen bond cleavage, releasing a halogen atom.70,71 Among hypervalent iodine(III) reagents, benziodoxoles are the only ones reported as photoreactive under visible-light irradiation and employed in alkynylation processes.72 Finally, both diselenides and ditellurides have a significant absorption tail in the visible region (400–550 nm), which has been assigned to the nσ* transition band,73–75 which is responsible for inducing Se–Se and Te–Te bond cleavage. In some instances, visible-light is an important tool for the uncaging of functional groups.76
As hinted above, all these classes were found to undergo chemical transformations when irradiated with visible-light (via the formation of either a reactive intermediate or a concerted reaction). We then recognized that in the last few years, the interest in the use of coloured organic compounds (whether commercially available or purposely prepared) in synthesis dramatically increased through the adoption of visible-light uncatalyzed procedures.77 We thus prepared the present review with the aim to provide the reader with a realistic portrait of the capabilities of the photochemical approach and to encourage interest in a new era of photocatalyst-free processes. The procedures described have been classified based on the reaction mechanism and mostly are referred to the 2015–2023 period along with previous seminal examples.
2. Intermediate free reactions
The intermediate free reactions involve a CX (XC, O) or a NN bond and mainly belong to two categories, namely, E/Z isomerization and cycloaddition.
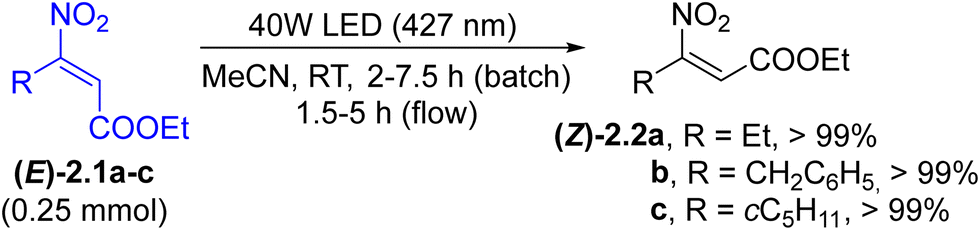
E/Z isomerization. Photochemical and photocatalyzed E/Z isomerization of double bonds40,78 is considered as the preferred approach for changing the stereochemical configuration under mild conditions. Usually, the approach is used to obtain the less stable Z isomer. In this context, (heteroaryl)azo derivatives can undergo E/Z isomerization when irradiated with an appropriate visible wavelength. However, despite its application in several fields, azo group isomerization has limited synthetic importance.40 On the other hand, only few classes of olefins exhibit a significant absorption in the visible region. These mainly include nitro alkenes and stilbenes. Thus, poorly available and rather unstable (Z)-β-nitroacrylates ((Z)-2.2a–c) have been prepared in quantitative yields from the corresponding E-isomers ((E)-2.1a–c) upon blue light irradiation (Scheme 2). The transformation was performed under both batch and flow conditions. The process allowed the use of Z-derivatives in cycloaddition reactions in a stereocontrolled manner.79 Similarly, the E to Z isomerization of styrylpyridin-2(1H)-ones has been described to occur under blue light (420 nm) irradiation under an argon atmosphere.80 The conversion of E-stilbenes to Z-stilbenes took place efficiently in a MeCN–NaOH 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture again upon blue light irradiation.81
:1 mixture again upon blue light irradiation.81
 | ||
| Scheme 2 Visible-light promoted selective E/Z conversion of β-nitroacrylates. | ||
The E to Z isomerization of amino cinnamates at 405 nm in the presence of a chiral phosphoric acid catalyst was used to induce the formation of atropisomeric N-aryl quinolones with low rotational barriers, yielding up to 94% and up to >99% ee, thus opening an interesting route to axially chiral materials.82
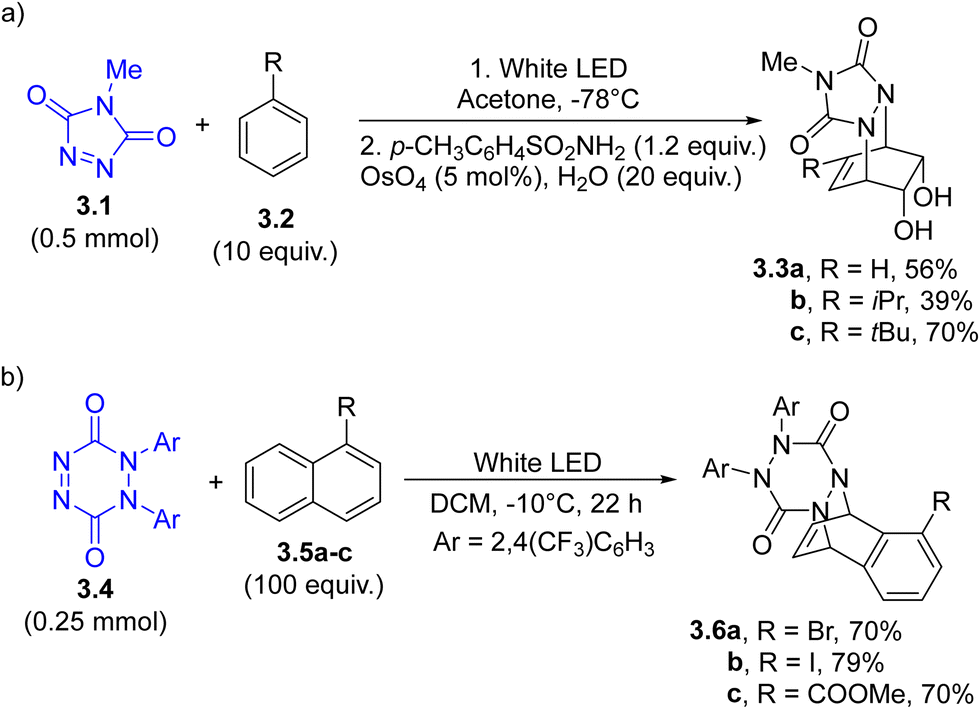
Cycloaddition. As in the case of E/Z isomerization, cycloadditions represent a research field where photochemistry always plays a key role as a powerful alternative to thermal and catalyzed approaches.83 As an example, the NN double bond may act as an arenophile in [4+2] photocycloadditions, enabling the chemical dearomatization of arenes and naphthalenes.84 Thus, N-methyl-1,2,4-triazole-3,5-dione 3.1 (Scheme 3a) was employed in the photoinduced dearomative dihydroxylation and diaminodihydroxylation of simple arenes to produce substituted cyclohexenes 3.3a–c. The protocol was applied in the synthesis of, among the others, carbasugar MK7607.85
 | ||
| Scheme 3 Photoinduced dearomatization of (a) benzenes and (b) naphthalenes. | ||
1,2-Dihydro-1,2,4,5-tetrazine-3,6-diones (3.4, an example in Scheme 3b) have been recently investigated as dearomative agents for naphthalenes (3.5a–c), arenes and nitrogen-based heterocycles. The so obtained cycloadducts 3.6a–c were efficiently functionalized by utilizing different approaches, including palladium- or copper catalyzed allylic substitution processes and non-catalytic hydroboration reaction.86
The photoinduced arene–alkyne [3+2] cycloaddition of 1-alkynylnaphthalen-2-ols (4.1a–c, Scheme 4) under photocatalyst-free conditions followed by air mediated oxidation of the obtained hexacycles 4.2a–c was exploited for the efficient and stereoselective preparation of complex three-dimensional hexacyclic architectures 4.3a–c in good yields and complete stereoselectivities.87
 | ||
| Scheme 4 Photoinduced [3+2] cycloaddition of 1-alkynylnaphthalen-2-ols. | ||
The light-driven [2+2] cycloaddition of olefins has been often exploited as an atom efficient tool to achieve cyclobutane scaffolds. The preparation of syn-cyclobutanes (5.2a–c, Scheme 5) was recently achieved by irradiating a solid-state suspension of 5.1a–c in water, efficiently furnishing the desired products with excellent diastereoselectivity.88 The supramolecular complexes 5.3a–c played a key role in driving the regioselectivity of the [2+2] addition. Interestingly, the head-to-head species 5.3a–c can be smoothly converted to the corresponding head to tail form through melt crystallization, allowing for a divergent and versatile strategy.
 | ||
| Scheme 5 Purple light mediated synthesis of cyclobutanes 5.2a–c. | ||
The achievements in visible-light photocatalysis in the last few decades have also brought traditional photochemical processes into focus, including Paternò–Büchi cycloadditions.89–92 However, the visible-light mediated, photocatalyst-free synthesis of oxetanes has only been sparsely reported in the literature. Benzyls (6.1a and b, Scheme 6), which exhibit a weak absorption in the visible region (400–450 nm), were successfully employed in both regio- and stereoselective [2+2] cycloaddition with simple olefins (e.g., 6.2) to produce the desired oxetanes 6.3a and b, along with a minor amount of 1,4-dioxanes derived from a [4+2] cycloaddition. The reaction was optimized under continuous flow conditions and the products were obtained with similar satisfactory selectivity and yield.93 Analogously, visible-light mediated Paternò–Büchi reactions on simple olefins have been performed by using anthraquinones94 or α-ketoesters95 as carbonyl donors.
 | ||
| Scheme 6 Visible-light promoted preparation of oxetanes. | ||
2-(4-Hydroxyphenyl)-substituted aldehydes and ketones (7.4a–c) bearing a diaryl-substituted quaternary stereocenter have been prepared via a visible-light-assisted and zinc triflate-catalyzed multicomponent reaction of benzoquinone (7.1), alkynes 7.2a–c, and a nitrogen or sulfur containing nucleophilic partner such as an indole 7.3 or a thiol.96 In the suggested mechanism illustrated in Scheme 7, 7.2a–c underwent a Paterno–Buchi [2+2] cycloaddition with the photoexcited 17.1, forming 1,4-singlet biradicals 17.5a–c (paths a and b) which in turn are converted to spiro-oxetene derivatives 7.6a–cvia intramolecular coupling (path c). These rather unstable species are smoothly converted to p-quinone methides 7.7a–cvia retroelectrocyclization (path d). Coordination of 7.7a–c with Zn(OTf)2 enables the nucleophilic addition of 7.3 (path e) to afford, after protonation of intermediates 7.8a–c the desired adducts 7.4a–c with the concomitant release of the Lewis acid catalyst (path f).96 An asymmetric version of this protocol has been recently developed using chiral phosphoric acid catalysis.97 An analogous approach involves CuCl as the Lewis acid and alcohols as nucleophiles and it was applied in the synthesis of alkoxysubstitued aldehydes, a set of compounds exhibiting interesting antifungal activity.98 Dihydroquinoxalines were instead formed by irradiating a mixture of p-quinones, alkynes and ortho-phenylenediamines under basic conditions (NaOAc, 20 mol%).48
 | ||
| Scheme 7 Three-component reaction for the preparation of 2-(4-hydroxyphenyl)-substituted aldehydes and ketones. | ||
In Scheme 8, a rare example of 6π-photocyclization promoted by visible-light under photocatalyst-free conditions is described. Thus, ortho-biaryl-appended β-ketoesters 8.1a–d (in equilibrium with the enol tautomers 8.3a–d) underwent an efficient 6-endo-trig cyclization/1,5-H shift to yield 9,10-dihydrophenanthren-9-ols 8.2a–d. The authors postulated a conrotatory ring closure followed by a suprafacial 1,5-hydrogen shift to explain the reactivity.99
 | ||
| Scheme 8 Visible-light-induced 6π-photocyclization. | ||
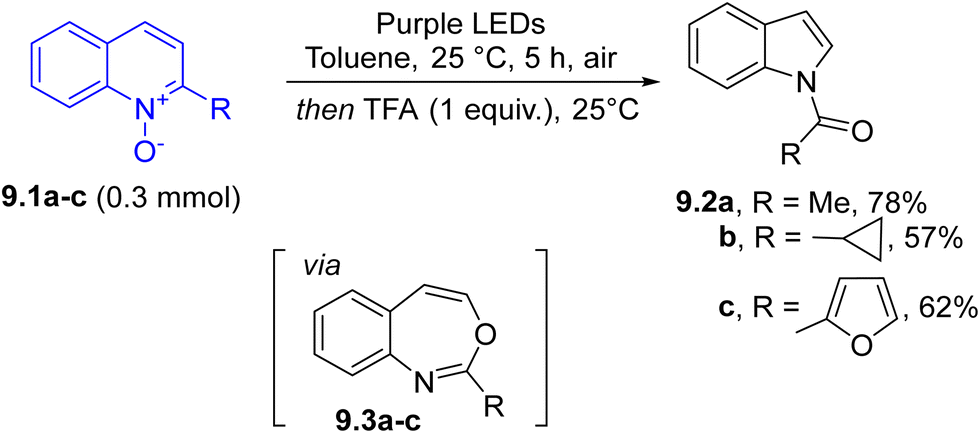
Photochemistry was also used for the skeleton rearrangement of azaarenes. Thus, under purple LED irradiation, quinoline N-oxides 9.1a–c promoted the formation of 3,1-benzoxazepines 9.3a–c, which upon acid treatment gave N-acylindoles 9.2a–c in variable yields (Scheme 9). The process can be described as a C2-selective, net carbon deletion of azaarenes and the mildness of this approach facilitated the late-stage elaboration of compounds of pharmaceutical interest.100
 | ||
| Scheme 9 Photoinduced ring contraction of quinoline-N-oxides for the synthesis of N-acylindoles. | ||
Noteworthily, when 3,1-benzoxazepines underwent an oxidative cleavage (via ozonolysis at −78 °C) followed by heating at 90 °C, a direct conversion of quinolines into quinazolines took place. This transformation has important implications in drug discovery since it replaces an aromatic carbon atom with a nitrogen atom through a C-to-N transmutation.101
3. Reactions via carbon-based radicals
3.1. C(sp3)-based radicals
N-(Acyloxy)-2-thiopyridones are a class of coloured compounds also known as Barton esters, which represent a promising alternative in the generation of radicals under tin-free conditions.102 The use of such derivatives pertains to the dyedauxiliary strategy (Scheme 1f) since a (photo)labile N–O bond is installed upon esterification of the corresponding carboxylic acid with N-hydroxythiopyridone.
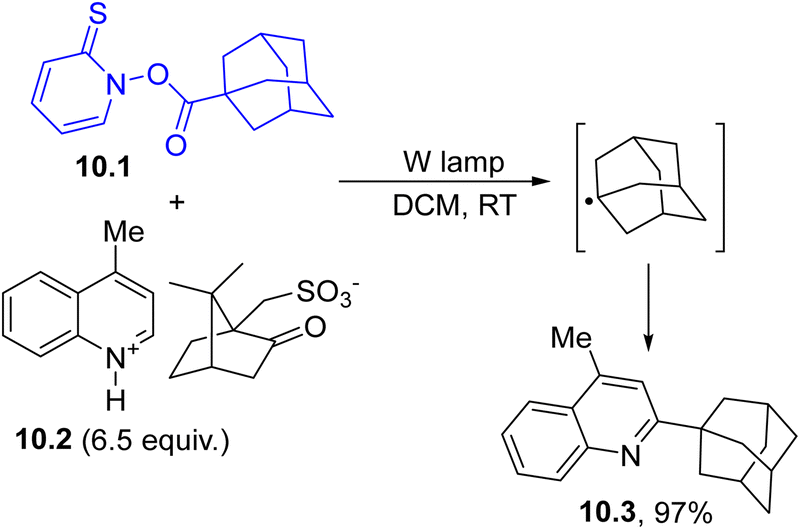
In particular, the photoinduced cleavage of the N–O bond followed by CO2 loss resulted in the formation of an alkyl radical. This behavior has found several synthetic applications, including the Minisci reaction103 reported in Scheme 10. In this case, adamantyl radical (in turn generated via visible-light mediated decarboxylative decomposition of ester 10.1) was trapped by lepidinium camphorsulphonate 10.2 (a heteroaromatic protonated base) and led to 2-substituted lepidine 10.3 in almost quantitative yield.104
 | ||
| Scheme 10 Visible-light generation of alkyl radicals for the functionalization of lepidine. | ||
Various aliphatic nitriles have been smoothly prepared from the corresponding Barton esters. As an example, oleic acid derivative 11.1 was successfully used to obtain the corresponding nitrile 11.3 in a high yield in the presence of methanesulfonyl cyanide 11.2 (Scheme 11).105 The same approach was used to achieve thiocyanates by the photolysis of Barton esters in the presence of methanesulfonyl (or p-toluensulfonyl) isothiocyanate.106
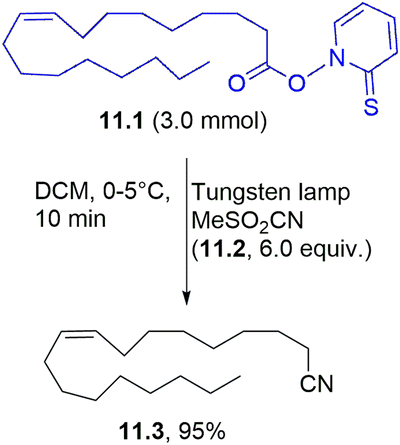 | ||
| Scheme 11 Visible-light promoted synthesis of nitriles. | ||
In the last decade, Hantzsch esters (HEs) have been recognized as stable and easily available reservoirs of carbon-based radicals under photochemical conditions.66 The construction of a dihydropyridine scaffold makes HEs coloured and photoreactive (Scheme 1f). Alkyl-1,4-dihydropyridines 12.1a–c have been employed in the regioselective late-stage alkylation of nitrogen-based heterocycles (including quinazolin-4(3H)-one 12.2, Scheme 12) under blue light irradiation.107 The radical pair is formed by direct C–R cleavage in compounds 12.1a–c. The photochemistry of such compounds has been elegantly combined with Ni-based catalysis, to develop a protocol for promoting a C(sp2)–C(sp3) cross-coupling between aryl bromides and benzyl radicals generated from HEs.108 In this case, the excitation of HEs promoted an electron transfer with a Ni(I) species and the resulting radical cation released the desired radical through fragmentation. More recently, such derivatives were also employed as neutral precursors of alkyl radicals for the preparation of trifluoromethylated amino acids under continuous microflow conditions, by using in situ generated α-CF3 ketimine esters as the radical acceptor.109
 | ||
| Scheme 12 Hantzsch esters as a source of alkyl radicals. | ||
Boracene-based borates (absorption wavelength with a tail up to 430 nm) have been recently reported as a source of tertiary, secondary, and primary alkyl radicals.110 Such derivatives (in analogy with HEs) may release the radical via direct excitation or by acting as a strong SET reductant where the radical is released from the oxidized form of the boracene. The boracene dyedauxiliary group here has been introduced by reaction with a Grignard or an organolithium reagent. Recently, 2,2′-(pyridine-2,6-diyl)diphenol-based borates 13.1a–c have been employed for the development of several synthetic protocols including decyanoalkylation of pyridines, acylation and Giese-type hydroalkylation of electron-poor alkenes (13.2, Scheme 13a).111 An NHC carbene catalyzed three-component process involving boronate 13.4, an alkene (mainly styrenes, 13.5) and acyl imidazoles 13.6 has been proposed as a method for the alkylacylation of olefins to form highly functionalized ketones 13.8 (Scheme 13b).112 The reaction was promoted by the initial formation of an acyl azolium intermediate through the reaction of the NHC carbene (generated in situ by deprotonation of triazolium 13.7) and 13.6, which engaged in a SET reaction with excited 13.4.
 | ||
| Scheme 13 Use of boracene-based borates as a source of alkyl radicals for (a) Giese reaction and (b) for the preparation of unsymmetrical ketones. | ||
Ethynylbenziodoxolones (EBXs) are traditionally employed as alkyne donors in photoredox catalyzed alkynylation reactions. However, the use of aryl substituted EBXs 14.1 as visible-light alkynylating agents in the presence of monoalkyl oxalate salts 14.2a and b (Scheme 14) has been recently described.113 The excited EBX may oxidize 14.2a and b, allowing the release of the desired alkyl radical, which is capable of reacting with 14.1, resulting in the formation of a C(sp3)–C(sp) bond to give 14.3a and b. The versatility of the method is evident from the alternative use of carboxylic acid, alkyl trifluoroborates and even N-substituted aldimines as radical precursors in place of the oxalate salt.113 It should be noticed, however, that when the reaction took place under 4-CzIPN photocatalyzed conditions, a lower excess of 14.1 was required resulting in a better overall yield.
 | ||
| Scheme 14 Blue light induced synthesis of internal alkynes. | ||
Photogenerated alkyl radicals have been also employed in the formation of C–heteroatom bonds. A protocol for the photochemical remote borylation of unactivated C(sp3)–H bonds tethered to an O-oxalate hydroxamic ester (15.1a–c, Scheme 15) was applied in the preparation of linear and cyclic tertiary and secondary borylation products 15.2a–c.114 The reaction started from the homolysis of the N–O bond followed by a 1,5-HAT step where the resulting carbon radical was trapped by bis(catecholato)diboron. The method was also applied for the C–H borylation of biologically relevant compounds.
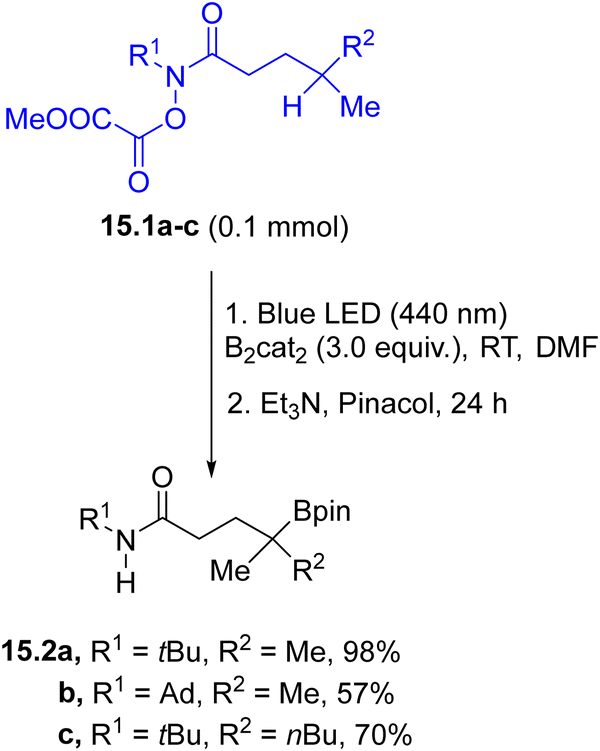 | ||
| Scheme 15 Visible-light driven remote C–H borylation protocol. | ||
The irradiation of a Barton ester in the presence of thionitrites led to a decarboxylative nitrosation resulting in the formation of trans-nitroso dimers,115 whereas alkyl azides have been obtained in the presence of ethanesulfonyl azide.116 Another route for forming a carbon-nitrogen bond involved a Barton nitrite ester-type cyclization. Thus, photoactive N-ethyl-N-nitrosobenzamides 16.1a–c (Scheme 16) underwent a cyclization to afford substituted benzo[d][1,2]oxazin-1-ones 16.2a–c. The key-intermediates are alkyl radicals 16.4a–c, generated via remote C–H atom bond abstraction occurring in N-centered radical 16.3a–c. Radicals 16.4a–c intercepted the previously released persistent nitric oxide radical (NO), forming the corresponding oximes, which upon cyclization formed 16.2a–c.117
 | ||
| Scheme 16 Preparation of benzo[d][1,2]oxazin-1-ones upon visible-light photohomolysis of the N–N bond in N-ethyl-N-nitrosobenzamides 16.1a–c. | ||
C–P bonds can instead be achieved by trapping a photogenerated alkyl radical (in turn obtained via photolysis of derivative 17.1) using white phosphorous (P4) followed by treatment with 30% H2O2 to afford the corresponding aliphatic phosphonic acid (17.2, Scheme 17).118
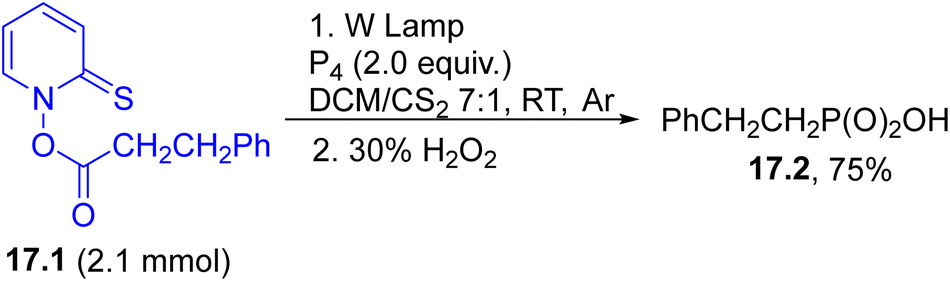 | ||
| Scheme 17 Synthesis of a phosphonic acid. | ||
Synthetic interest in photolabile esters also arises from the conversion of carboxylic acids into alcohols, which can be achieved by using modified Barton esters, namely, N-hydroxy-4-methyl-2-thiazolinethiones (e.g.18.1, Scheme 18). In this case, the alkyl radical generated from 18.1 reacted with oxygen and a thiol giving an hydroperoxide that was, in turn, reduced by PPh3 to afford alcohol 18.2 in a good yield.119
 | ||
| Scheme 18 Decarboxylative hydroxylation via modified Barton esters. | ||
Also, C–S bonds can be easily formed using Barton esters. Thiosulfonates (RSO2SPy; Py = pyridine) can be formed in up to 90% yield by trapping of the photogenerated alkyl radical with liquid SO2 (used as cosolvent with DCM).120 The products are, however, easily converted into sulfones and sulfonamides. A Barton ester may likewise be prepared from a modified glycine appended to a Wang type resin. The ensued irradiation enabled the synthesis of peptides containing modified amino acids.121
The carbon-based radical generated by the photochemical decomposition of a Barton ester (again following the strategy described in Scheme 1f) can be used in the generation of a further reactive radical via group transfer reaction. The strategy was applied in the synthesis of nucleoside antibiotic showdomycin (19.6, Scheme 19). In this case, the photocleavage of the Barton ester 19.1 released a methyl radical that in turn reacted with organotellurium 19.2 (path a) to form the α-oxy radical intermediate 19.4. Trapping of 19.4 by 19.3 followed by oxidation afforded maleimide 19.5 (path b). Treatment of the latter imide under acidic conditions resulted in the formation of the desired 19.6 (path c).122
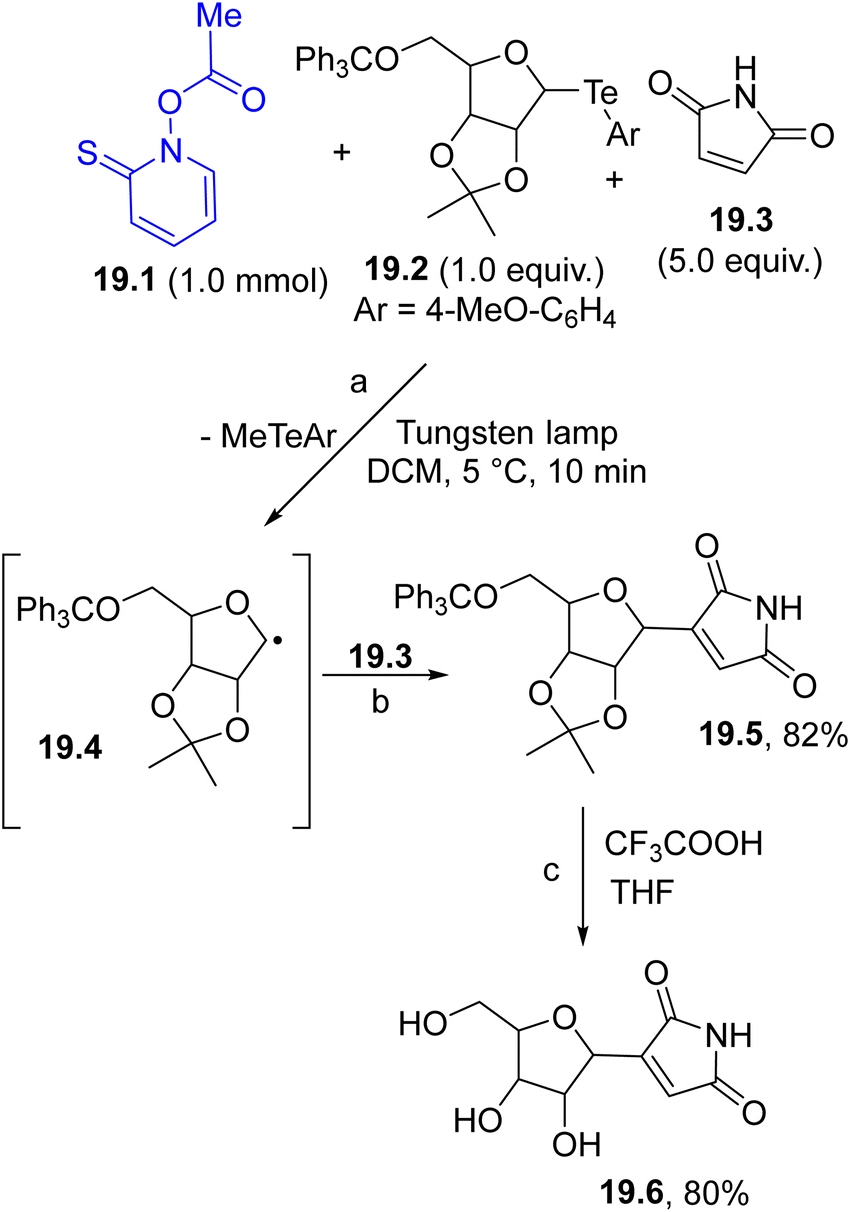 | ||
| Scheme 19 Preparation of showdomycin 19.6via the photogenerated α-oxy radical 19.4. | ||
The photohomolysis of the O–NO bond in acyloxy nitroso compounds (nitrite esters) has recently been described to promote the generation of α-(acyloxy)alkyl radicals along with nitric oxide. Both generated radicals were used in the 1,2-functionalization of electron-poor olefins to form α-oximinoketones.123
α-Oxy radicals are the intermediates for the reductive alkylation of 1,2-diketones using visible-light and organic halides (mainly iodides, Scheme 20). The SET reaction between the excited phenanthrene-9,10-dione 20.1 and triethylamine led to the concomitant formation of the α-oxy radical 20.4 and an α-aminoalkyl radical, that promoted the C–X bond cleavage in halides 20.2a–evia a halogen atom transfer process (XAT). The coupling with the resulting alkyl radicals and 20.4 led to the formation of the desired α-hydroxyl ketones 20.3a–e in a very good yield. The reaction worked to some extent even when aryl halides were used.46
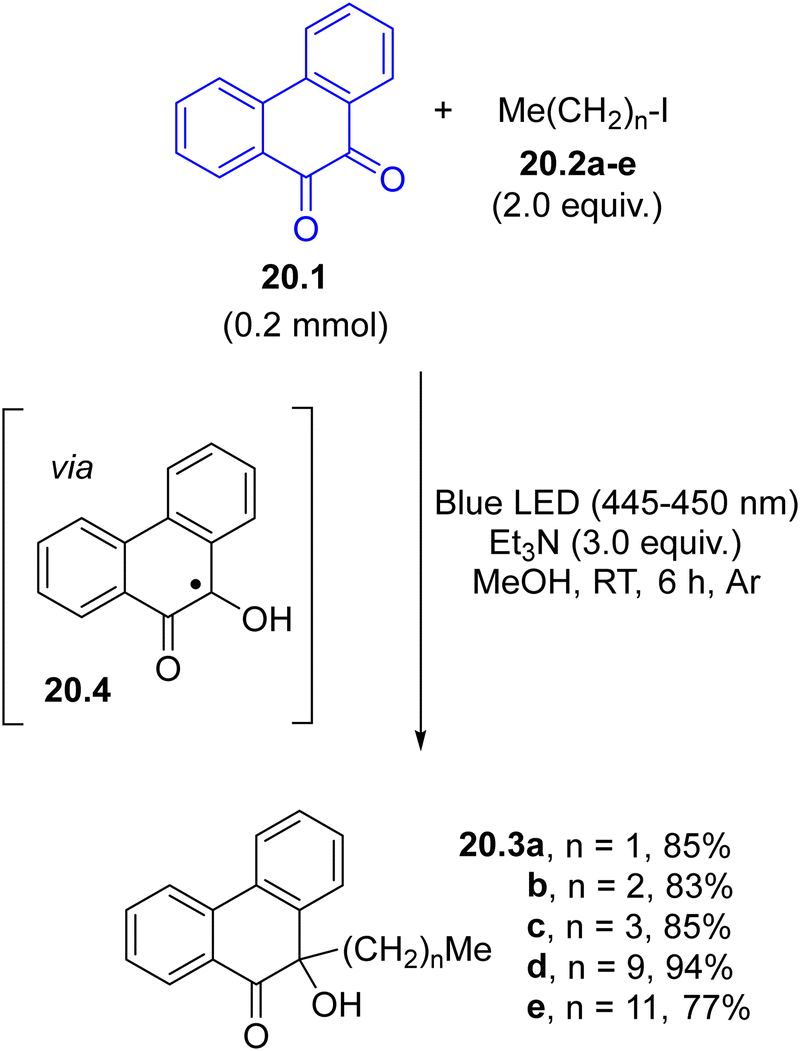 | ||
| Scheme 20 Photoinduced coupling between a 1,2-diketone and alkyl iodides. | ||
Some substituted alkyl halides may show a weak absorption in the visible region and thanks to the photolability of the C–X bond (mainly C–I), they are an interesting source of carbon-based radicals capable of attacking double or triple C–C bonds.
As an example, ethyl difluoroiodoacetate 21.1 (Scheme 21a) exhibits weak absorption in the blue region (400–500 nm), enough to promote the homolysis of the C–I bond, resulting in the release a difluoroalkyl radical/iodine radical pair that has been exploited in the 1,2-functionalization of alkynes 21.2a–c to form styrenes 21.3a–c.124 When bromodifluoroacetamides were irradiated in the presence of (iodoethynyl)arenes, alkynyldifluoroacetamides were obtained in good yields via a formal C(sp)–C(sp3) bond cross-coupling.125 Similarly, photolysis of ICH2CN (21.4, an example in Scheme 21b) resulted in the formation of an α-cyanomethyl radical (path a) readily trapped by cinnamic acid (21.5, path b). The resulting benzyl radical 21.7 underwent oxidation (path c) and decarboxylation (path d), forming nitrile 21.6 in ca. 80% yield.126
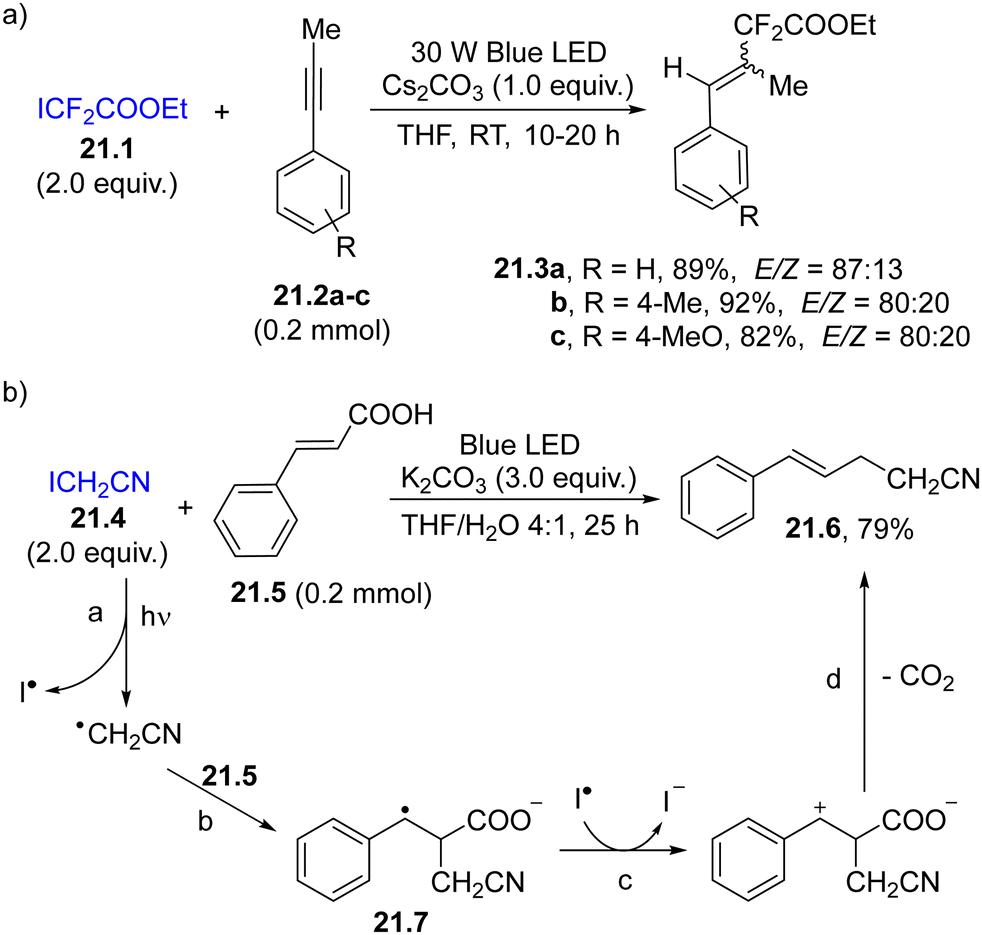 | ||
| Scheme 21 Photohomolysis of a C–I bond for the carbon-based radical addition onto (a) phenyl acetylenes and (b) cinnamic acid. | ||
A wide range of electrophilic carbon-based intermediates including, among the others, α-cyanomethyl, acetonyl radical and dichloromethyl radicals have been generated via hydrogen atom transfer between an aryl radical (in turn generated via visible-light irradiation of an arylazo sulfone) and acetonitrile, acetone and dichloromethane, respectively.127 The resulting radicals have been applied in the synthesis of indolinones from the corresponding N-aryl acrylamides.127
The blue light photolysis of α-halo boronic esters (e.g., α-iodo BPin) and the subsequent formation of α-boryl radicals was recently employed for the preparation of allyl boronic esters via reaction with substituted styrenes.128
3-Substituted chroman-4-ones 22.5a–c have been accessed via a carbon–carbon bond-forming cyclization cascade promoted by a C(sp3)–Br homolytic cleavage occurring under visible-light irradiation (Scheme 22). In the proposed mechanism, the first formed radicals 22.2a–c (path a) were involved in a cascade radical cyclization, to form alkoxy radicals 22.3a–c (path b) and α-hydroxy radicals 22.4a–c from them after a 1,2-hydrogen shift (path c). The oxidation of 22.4a–c by bromo atoms afforded, upon deprotonation (path d), the desired products 22.5a–c with high regio- and diastereoselectivity (dr always >20:1).129
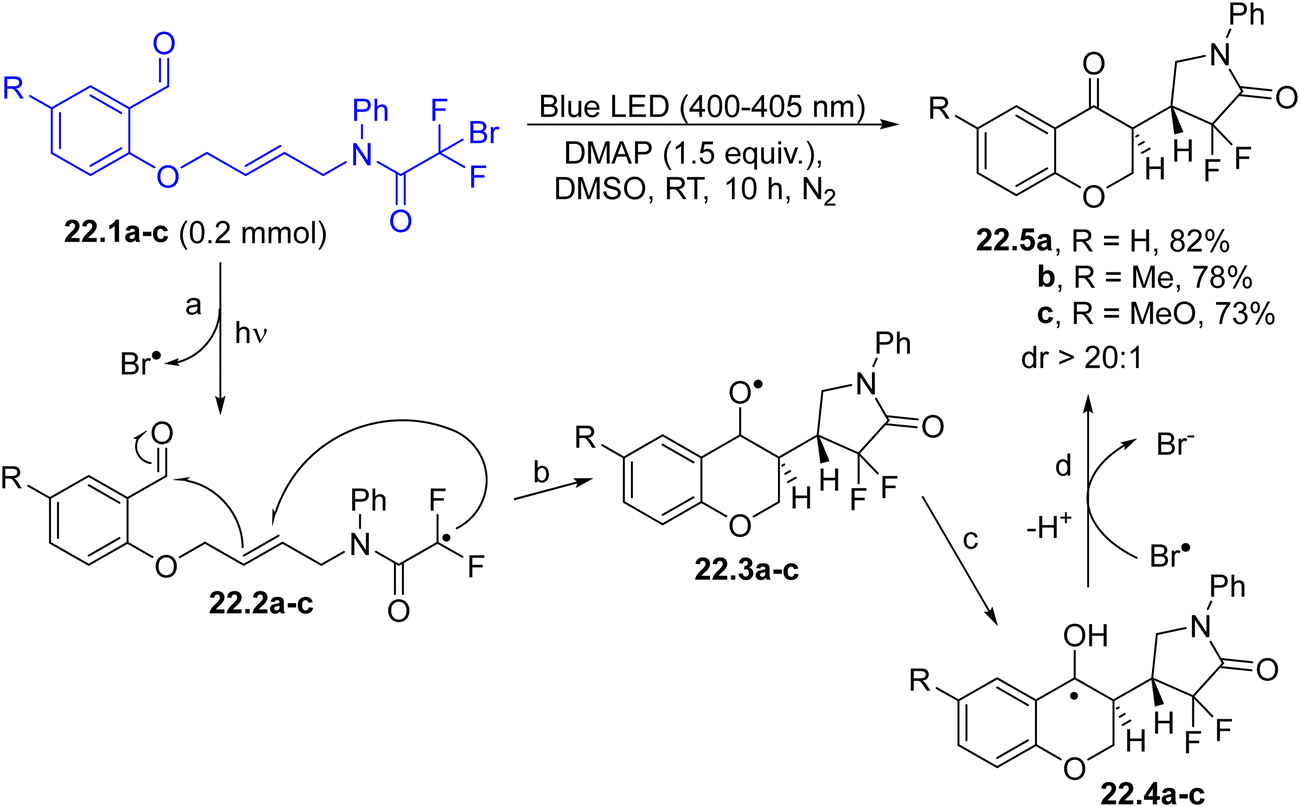 | ||
| Scheme 22 3-Substituted chroman-4-ones via photoinduced cascade radical cyclization. | ||
The synthesis of gem-dihaloenones from tetrahalomethanes (e.g., tetrabromomethane 23.1, Scheme 23) and arylacetylenes 23.2a–c has been described to occur in organic/aqueous solvent upon blue light irradiation.130 As depicted in Scheme 23, the photoinduced homolysis of the C–Br bond in 23.1 promoted the formation of a Br˙/CBr3˙ radical pair (path a). The carbon radical was trapped by 23.2a–c, producing vinyl radicals 23.4a–c (path b), which then coupled with Br˙ to form the Kharasch adduct 23.5a–c (path c). Allylic substitution between 23.5a–c and water gave the final enones 23.3a–c after HBr release (paths d and e). A similar approach was also proposed by using aqueous DMSO or neat alcohols as reaction media.131 The generation of a trihalomethyl radical was also exploited in the iron(0) catalysed benzannulation of allylarenes to form 1-halonaphthalenes.132
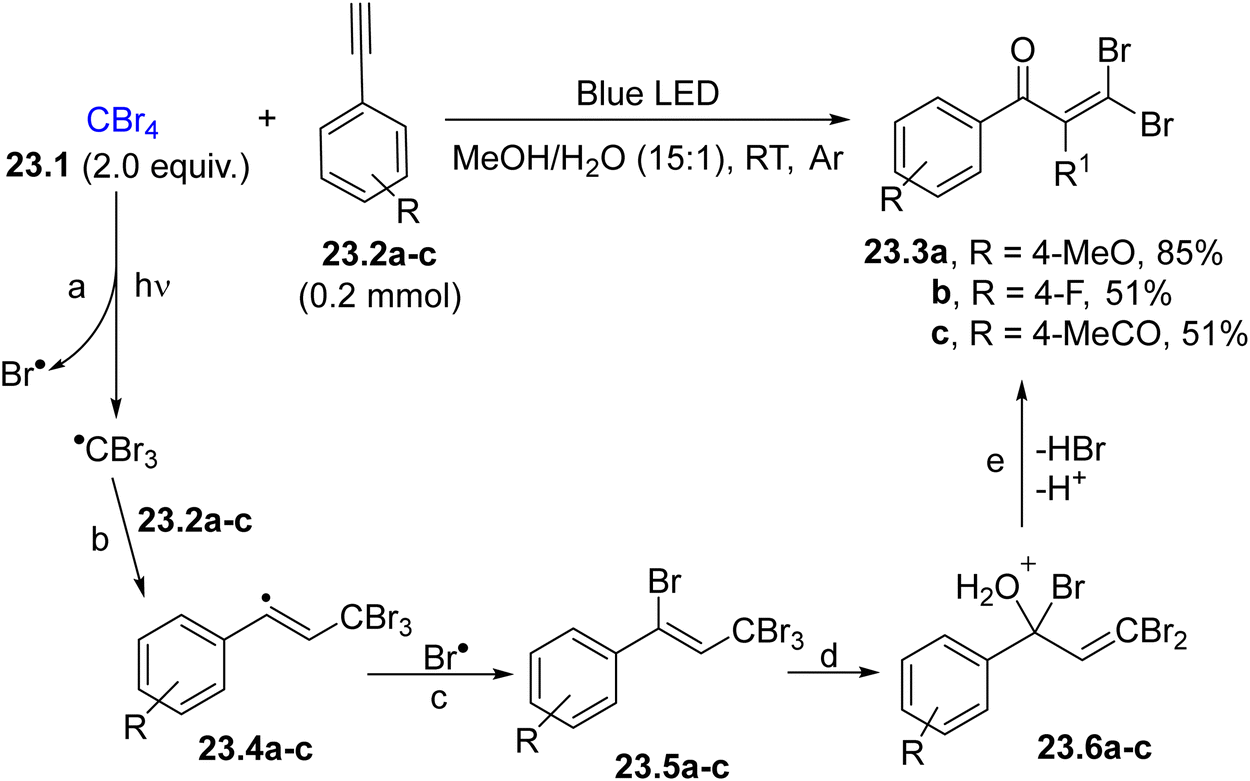 | ||
| Scheme 23 Synthesis of gem-dihaloenones. | ||
Umemoto's reagent 24.1 (Scheme 24) has traditionally been employed in photoredox catalysed trifluoromethylation. Recently, the possibility of generating a trifluoromethyl radical via direct irradiation of 24.1 has been identified and exploited for the preparation of β-trifluoromethyl enamides 24.3a–c133 and in the trifluoromethylation-arylation of N-acrylamides to produce oxindoles.134
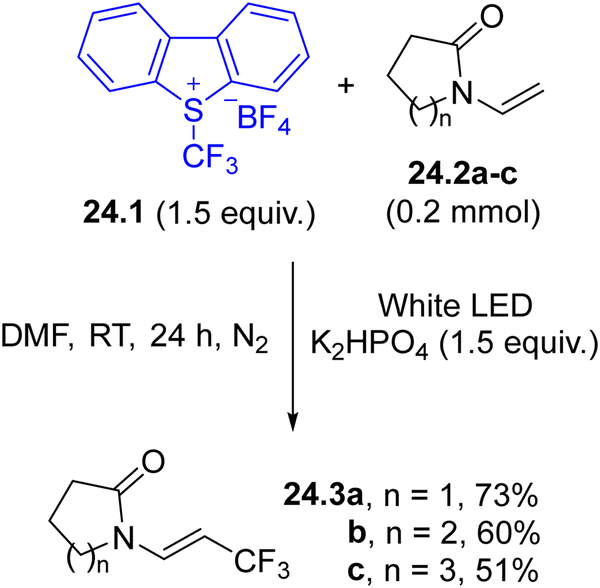 | ||
| Scheme 24 Umemoto’s reagent for the light-driven uncatalyzed trifluoromethylations. | ||
3.2. C(sp2)-based radicals
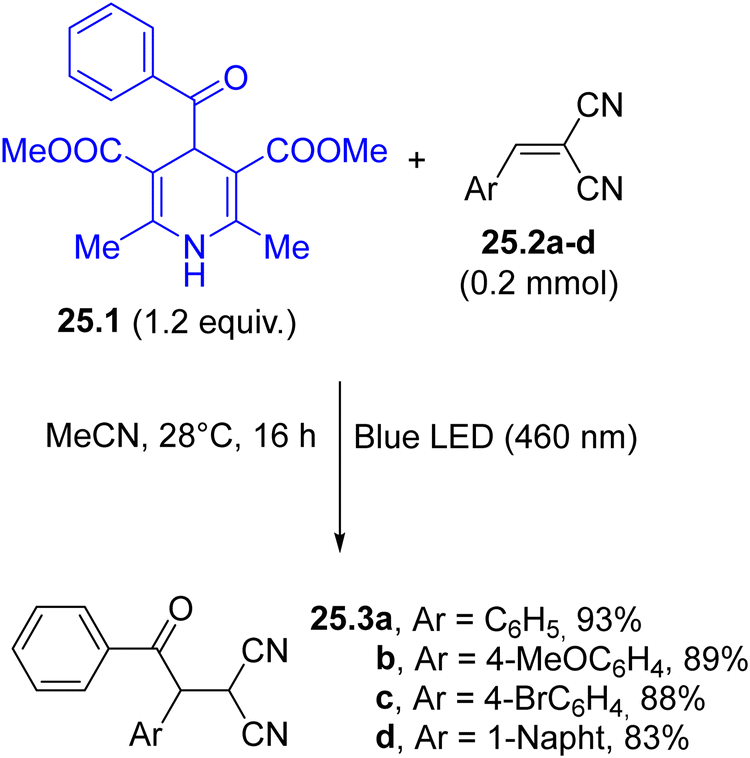 | ||
| Scheme 25 Hantzsch esters for visible-light driven acylation reactions. | ||
The preparation of acyl trifluoromethylthio and trifluoromethylseleno esters has been made possible by irradiating 4-acyl substituted Hantzsch esters with visible-light in the presence of S-(trifluoromethyl) 4-methylbenzenesulfonothioate and Se-(trifluoromethyl) 4-methylbenzenesulfonoselenoate as the radical traps.138
The hydroacylation of azoarenes 26.1a–e (some examples in Scheme 26) has been achieved by using α-ketoacids as acyl radical precursors. In the suggested mechanism, photoexcited 26.1*a–e underwent reductive quenching by 26.2, leading to the formation of, after proton and CO2 loss, the corresponding acyl radical 26.4. The latter intermediate coupled with radical anions 26.5a–e, resulting in the formation of acyl hydrazines 26.3a–e through the protonation of the anions 26.6a–e.139
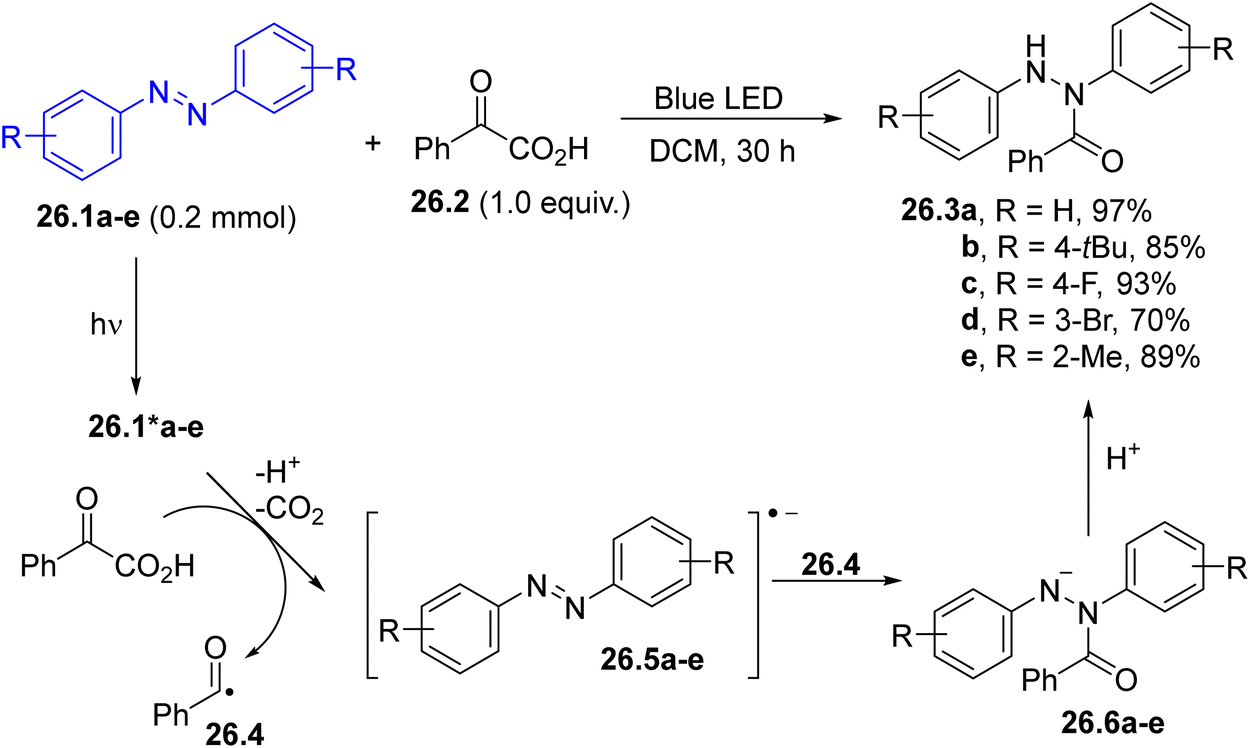 | ||
| Scheme 26 Photoinduced hydroacylation of azoarenes. | ||
Acyl radicals arising from α-ketoacids have also been employed for the synthesis of multi-functionalized cyclopentane scaffolds via tandem radical addition on terminal alkynes, translocation and cyclization in water.140
N–SO2R moiety, capable of imparting both colour and photoreactivity to the starting compound (Scheme 1f).35,36,141 Accordingly, upon visible-light irradiation, a N–S bond homolytic cleavage occurred from the 1nπ* singlet excited state of 27.1, producing a diazenyl radical 27.2 and the sulfonyl radical 27.3. Aryl radical 27.4 is then released upon nitrogen loss from 27.2, and such an intermediate has been applied in the development of a plethora of protocols for the formation of both C–C and C–heteroatom bonds.
 | ||
| Scheme 27 (a) Photocleavage of the N–S bond in arylazo sulfones. (b) Arylation of heterocycles via photogenerated aryl radicals. | ||
As summarized in Scheme 27b, aryl radicals generated from arylazo sulfones 27.5 may be trapped by heterocycles including furans (path a), thiophenes142 or xanthines (path b)143 yielding the corresponding (hetero)biarenes 27.6 and 27.7 in high regioselectivity. The latter compounds have been likewise isolated in comparable yields upon natural sunlight exposition in a sun-flow reactor, enabling a significant reduction in the residence time (down to 4 h).144 The visible-light mediated arylation of quinoxalin-2(1H)-ones was described by using both arylazo sulfones145 and aryl acyl peroxides as sources of aryl radicals.146
Arylation of aryl boronic acids using arylazo sulfones to yield biaryls took place in modest yield under gold(I) catalysis in the presence of a base, where the ipso-substitution observed was directed by the presence of the B(OH)2 moiety.147 However, in the absence of an aryl boronic acid, the irradiation induced the selective formation of the corresponding (hetero)biaryls 28.2a–e (Scheme 28) in up to quantitative yield, with impressive functional group tolerance. The protocol was likewise successfully applied in the synthesis of binaphthyls and 2,2′-bipyridines.148
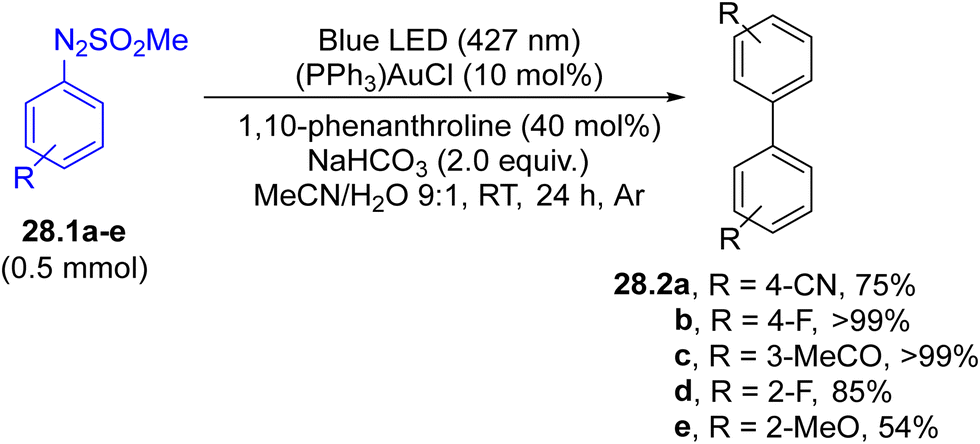 | ||
| Scheme 28 Blue LED driven preparation of biaryls. | ||
The generation of aryl radicals from arylazo sulfones has been investigated in the presence of different π-bond systems. As a result, valuable compounds, including α-aryl carbonyls 29.3a–d (Scheme 29, path a, by reaction with enol silyl ethers 29.2)149 and allyl arenes 29.5a–d (in the presence of allyl sulfones 29.4, Scheme 29, path b)150 have been smoothly prepared. The trapping of a photogenerated aryl radical by an isonitrile in aqueous solvent was also found to be useful in promoting the synthesis of aroyl amides.151
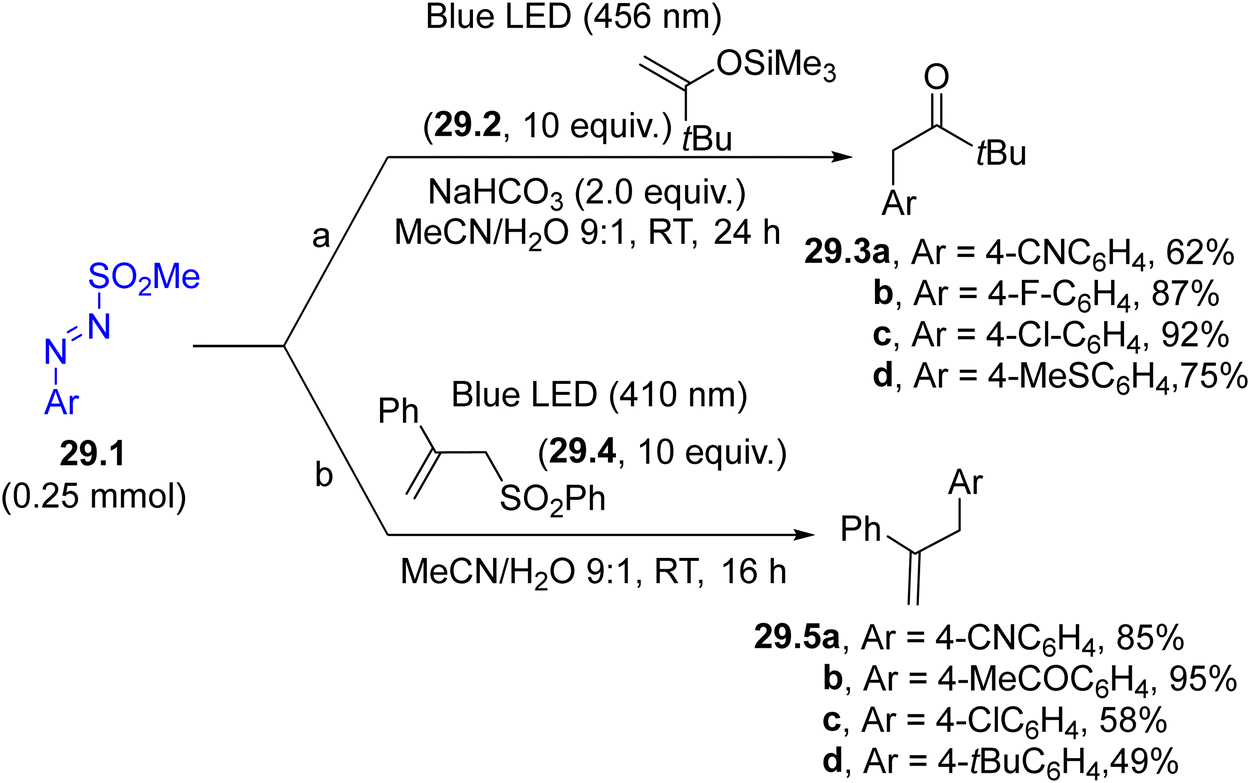 | ||
| Scheme 29 Aryl radicals for the synthesis of α-aryl carbonyls and allyl arenes. | ||
Arylazo sulfones have been largely applied in the formation of aryl-heteroatom bonds, giving access, among the others, to aryl boronates and aryl trifluoroborate salts,152 aryl stannanes 30.3a–c (in the presence of an hexaalkylstannane 30.2, Scheme 30, path a),153 aryl phosphonates,154 unsymmetrical aryl selenides 30.5a–c and tellurides (Scheme 30, path b155) and aryl trifluoromethylthioethers 30.7a–c (Scheme 30, path c).156
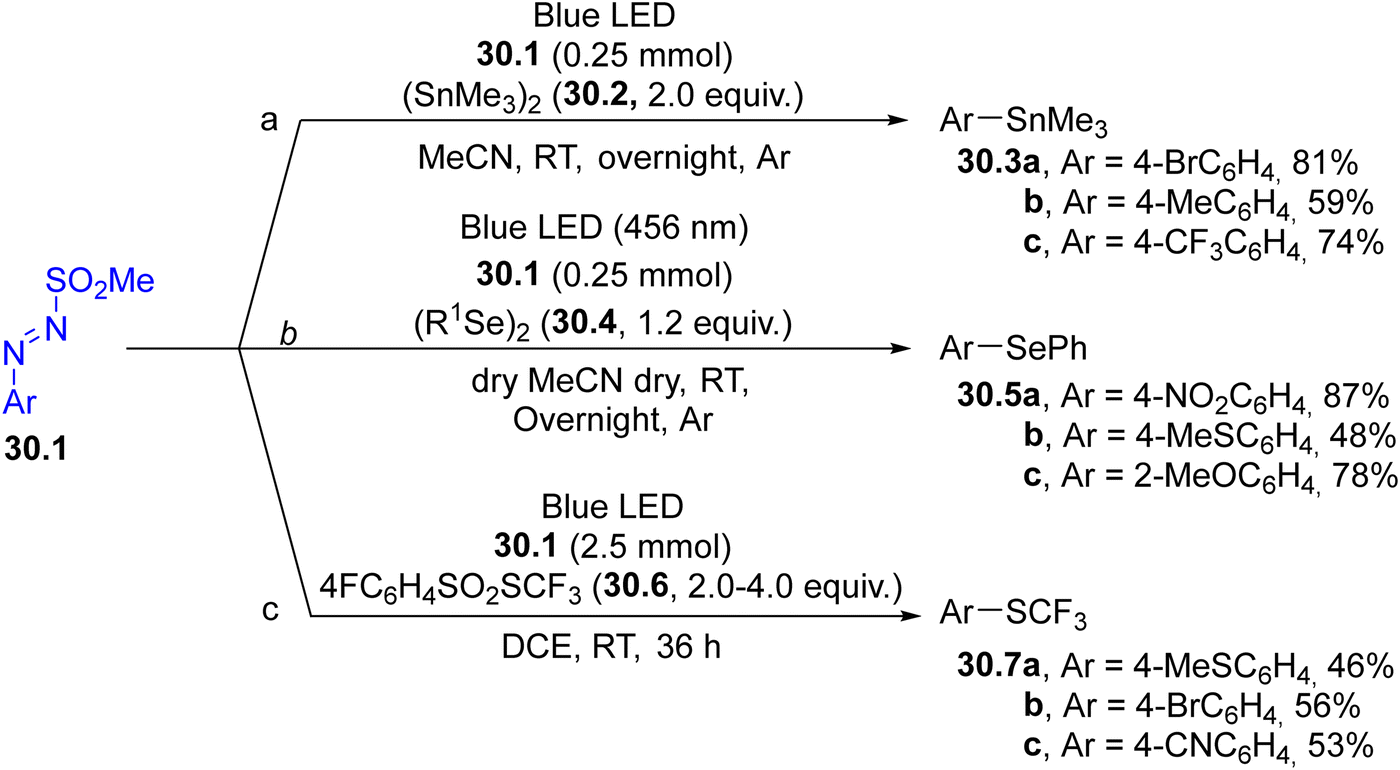 | ||
| Scheme 30 Formation of an Ar–heteroatom bond via photogenerated aryl radicals. | ||
Finally, visible-light irradiation of arylazo sulfones in deuterated media (isopropanol-d8/H2O 9:1) resulted in a deutero-deamination process, yielding the corresponding monodeuterated arene.157
Aryl radicals may be generated by a visible-light driven atom transfer. This is a rare case where a coloured organic compound absorbed the light, but it was not incorporated in the end product. In fact, the photocleavage of the labile S–S bond (BDE ca. 50 kcal−1 mol−1)158 in disulfide 31.2 generated a thiyl radical, which promoted the desilylation of arylsilanes 31.1. This resulted in the formation of aryl radicals 31.4 and desilylated derivatives 31.3 from it through a HAT step (Scheme 31).159
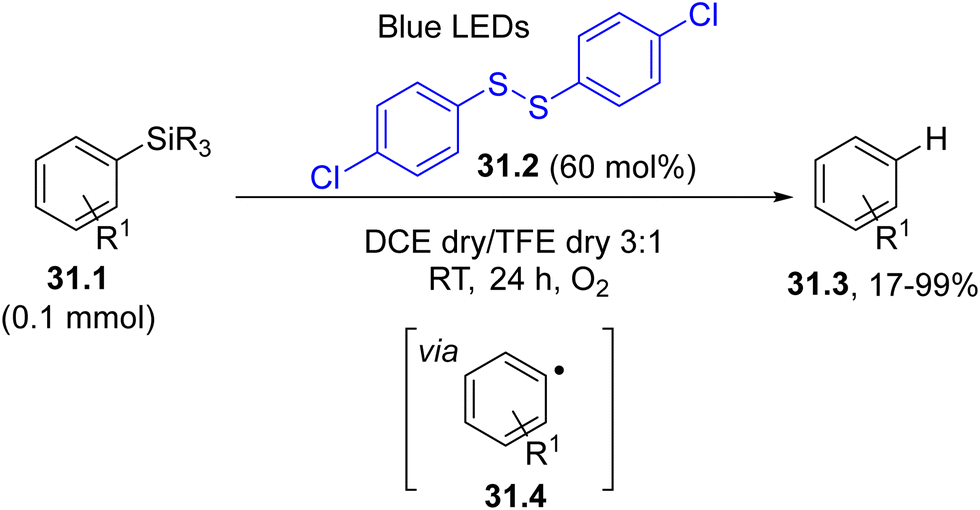 | ||
| Scheme 31 Aerobic visible-light induced desilylation of arylsilanes. | ||
4. Reactions via heteroatom-based radicals
4.1. Boron-based
A rare case involving the use of boryl radicals is exemplified in Scheme 32 in the context of the pyridylation of several carbon/heteroatom-hydrogen bonds. In fact, the visible-light homolysis of the N–O bond in N-methoxypyridinium salts 32.1a–c generated a methoxy radical, which then abstracted a hydrogen atom from an amine-borane (e.g.32.2) to release a boron-based radical 32.3. Addition of 32.3 to 32.1a–c led to the assembly of various pyridines (32.4a–c) with the concomitant release of a methoxy radical, which further propagated the reaction.160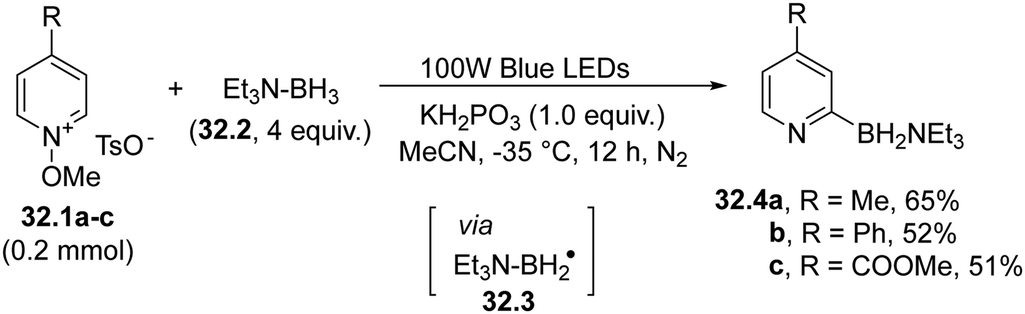 | ||
| Scheme 32 Visible-light formation of an Ar–B bond via photolysis of N-methoxypyridinium salts. | ||
4.2. Nitrogen-based
Nitrogen centered radicals are, in most cases, formed via the visible-light induced homolysis of a N-heteroatom bond present in purposely designed derivatives. An exception is the dated example presented in Scheme 33, where the C–N bond in N-acyl PTOC (N-hydroxy-pyridine-2-thione) carbamate 33.1 was found to be sufficiently labile for cleavage upon tungsten lamp irradiation, resulting in the release of amidyl radical 33.2. Radical cyclization (in a 5-exo mode) onto the tethered double bond followed by the incorporation of the –SPy group led to the synthesis of N-acyl pyrrolidine 33.3 in ca. 70% yield.161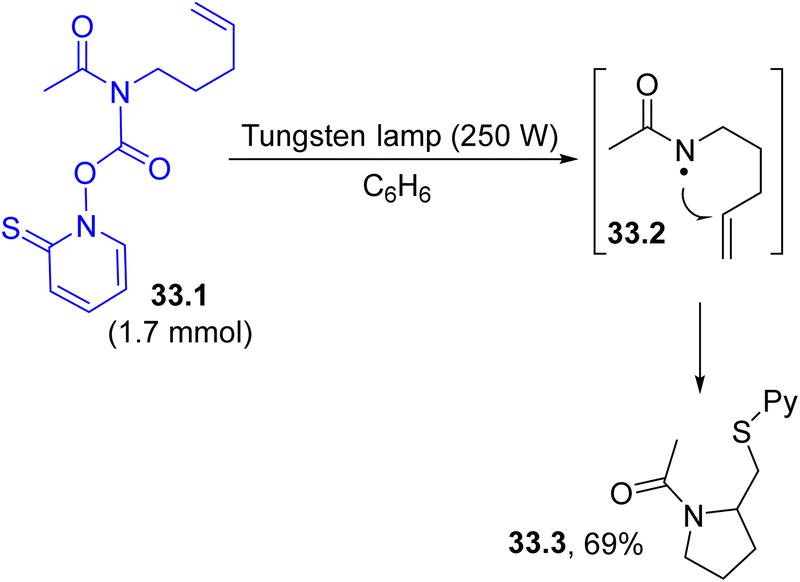 | ||
| Scheme 33 Preparation of N-acyl pyrrolidines via photogenerated amidyl radicals. | ||
Nitrogen-centered radicals may also be formed starting from a carbamate (a protected primary aliphatic amine having leaving groups bearing labile N–O bonds) under photoenzymatic conditions. Thus, the radicals generated under blue LED irradiation were engaged in an enantioselective intermolecular radical hydroamination of styrenes, driven by an ene-reductase.162
The amidyl radical was likewise generated from a stable and isolable N-dithiocarbamate 34.3 (in turn obtained from a carboxylic acid (34.1) and a thiocarmabamylsulfenamide (34.2)) through an amido coupling reaction (Scheme 34). The photolysis of 34.3 led to the formation of two radical species: the amidyl 34.4 and the sulfur-centered radical 34.5. Intramolecular HAT promoted by the nitrogen radical enabled the site specific derivatization of a remote C–H bond. The resulting carbon radical was then able to couple with 34.5 to complete a selective and efficient dithiocarbamylation (product 34.6).163
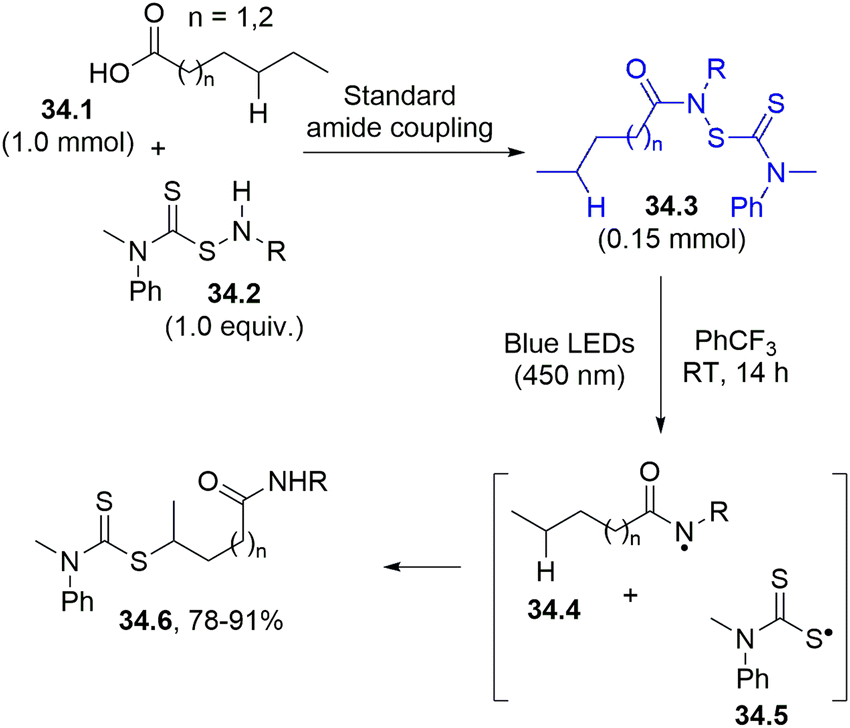 | ||
| Scheme 34 Synthesis of alkyl dithiocarbamates. | ||
The generation of a nitrogen-based radical can also be achieved by cleaving a N-halogen bond such as in the case of N-bromosaccharin 35.1 (Scheme 35). Ambient light exposure caused the homolytic cleavage of the N–Br bond in 35.1. Then, the saccharin nitrogen radical 35.2 promoted the imidation of different (hetero)arenes Ar–H in a good regio- and chemoselective fashion.164
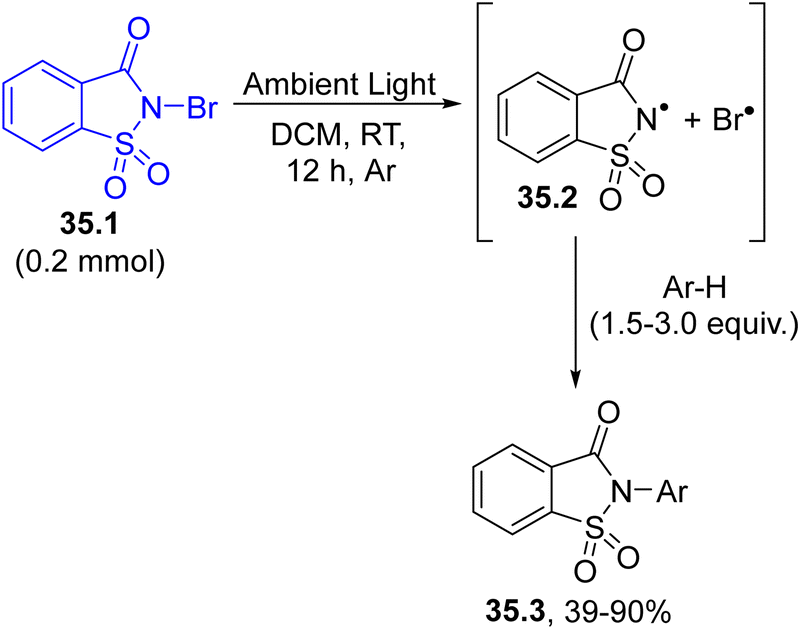 | ||
| Scheme 35 Visible-light driven imidation of arenes employing N-bromosaccharin. | ||
Hypervalent iodine derivatives have been successfully employed for the α-ketoacylation of sulfoximines by using aryl alkynes. The excitation of the hypervalent iodine compound 36.1 was carried out using blue light and a sulfoximidoyl radical 36.4 was then formed. The trapping of 36.4 by aryl alkyne 36.2 released a reactive vinyl radical 36.5, which was subsequently trapped by dioxygen, forming peroxyl radical 36.6. This intermediate evolved into the unstable 1,2-dioxete 36.9, which ultimately rearranged to yield the desired N-α-ketoacylatedsulfoximine 36.3 (Scheme 36).165
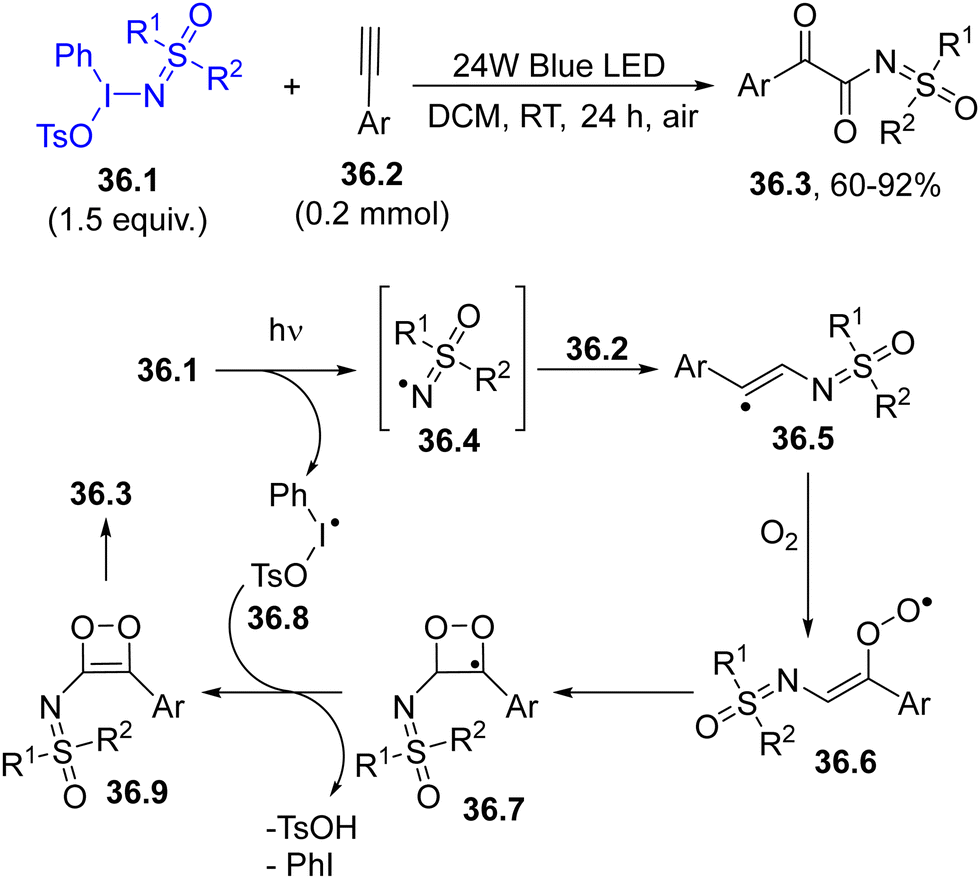 | ||
| Scheme 36 Visible-light driven α-ketoacylation of sulfoximines. | ||
The in situ-performed oxidation of alkylamines (e.g.37.1) under aerobic conditions was adopted to obtain N-nitrosoalkylamines 37.2, a photoreactive class of molecule, which, after irradiation in acidic media with blue or purple light gave two reactive species from the corresponding N-nitrosammonium ion 37.5, namely, an aminium radical cation 37.7 and nitric oxide 37.6. The former species attacked a CC bond in alkenes (e.g., 37.3) generating a carbon radical 37.8 which recombined with nitric oxide, yielding 37.9 that readily tautomerized to oxime 37.4. (Scheme 37).166 The reaction was also effective when a mixture of 37.2 and 37.3 was photolyzed under blue LED irradiation, although the authors preferred to carry out most of the reactions by using purple LED (395–405 nm).
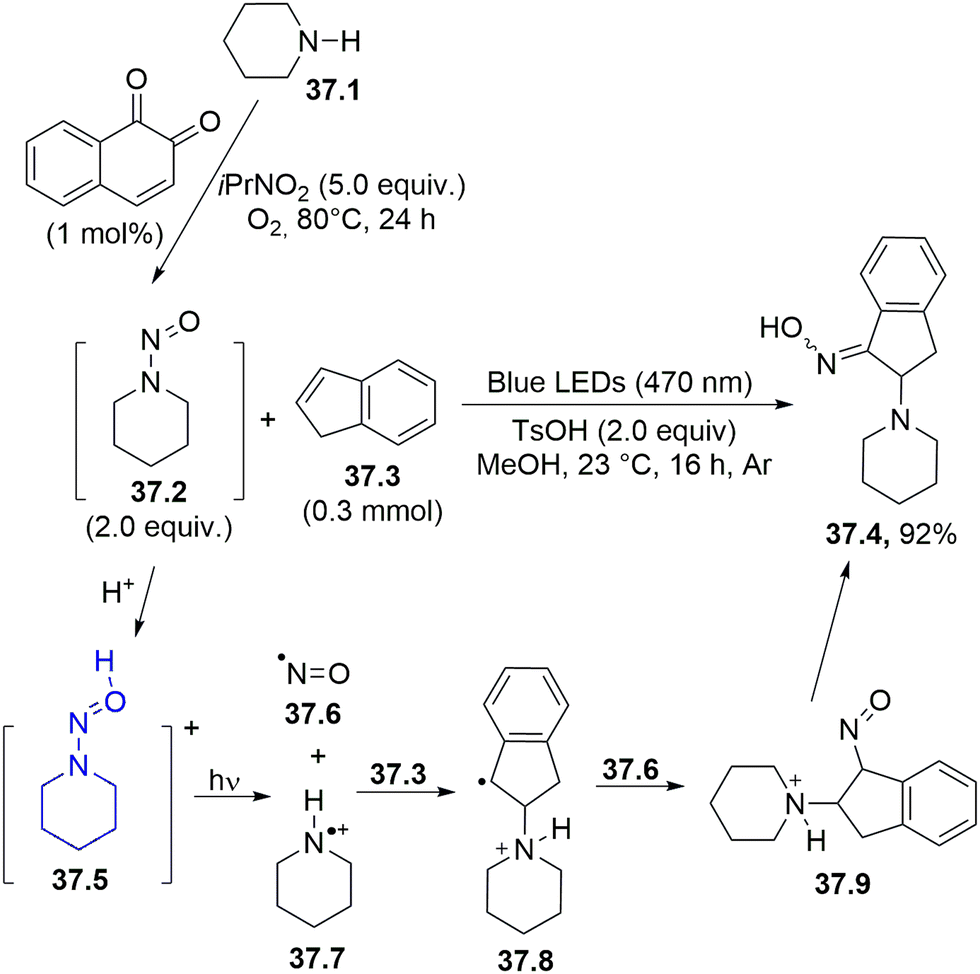 | ||
| Scheme 37 Visible-light induced one-pot tandem 1,2-diamination of alkenes. | ||
A different method to form a C–N bond is by employing the dyedauxiliary strategy (Scheme 1f), making use of aryldiazenyl radicals 38.4 photogenerated from arylazo sulfones 38.1. Exposing arylazo sulfone to blue light in the presence of enol silyl ethers of 1,3-diketones 38.2 led to the diazenylation of the double bond, forming aryldiazenyl-1,3-diketones 38.3 (Scheme 38).167
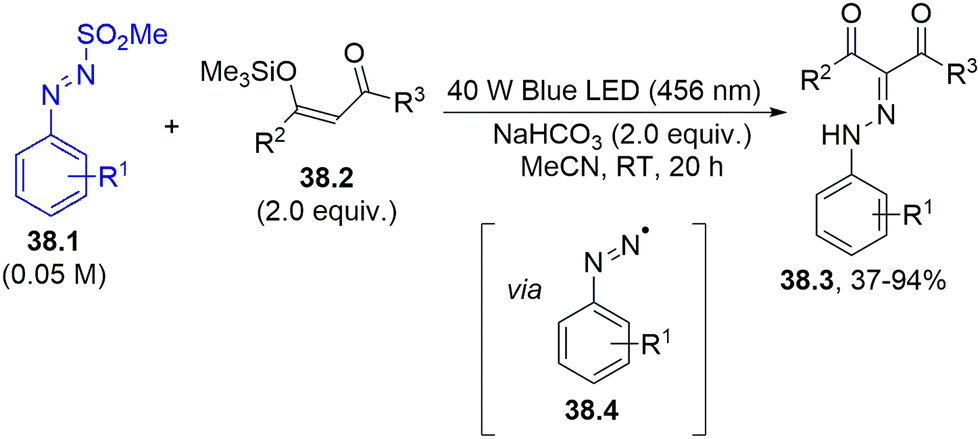 | ||
| Scheme 38 Synthesis of aryldiazenyl-1,3-diketones via photogenerated aryldiazenyl radicals. | ||
The photohomolysis of a N–O bond was indeed used to form iminyl radicals 39.2, thanks to the incorporation of a nitroaromatic as dyedauxiliary group. For example, the near-visible-light irradiation of o-nitrobenzyl oxime ethers 39.1 in a mixture of DMSO and a sodium phosphate buffered aqueous solution smoothly released iminyl radicals 39.2. The mechanism involved an initial intramolecular hydrogen atom transfer at the benzylic position by the nitro group, which promoted the cleavage of the N–O bond present in the oxime moiety. The cyclization of the thus formed radical onto the adjacent aromatic ring gave access to differently substituted phenanthridines 39.3 (Scheme 39).168 Noteworthily, this process may proceed even in a cellular environment where a bioactive derivative may be released.
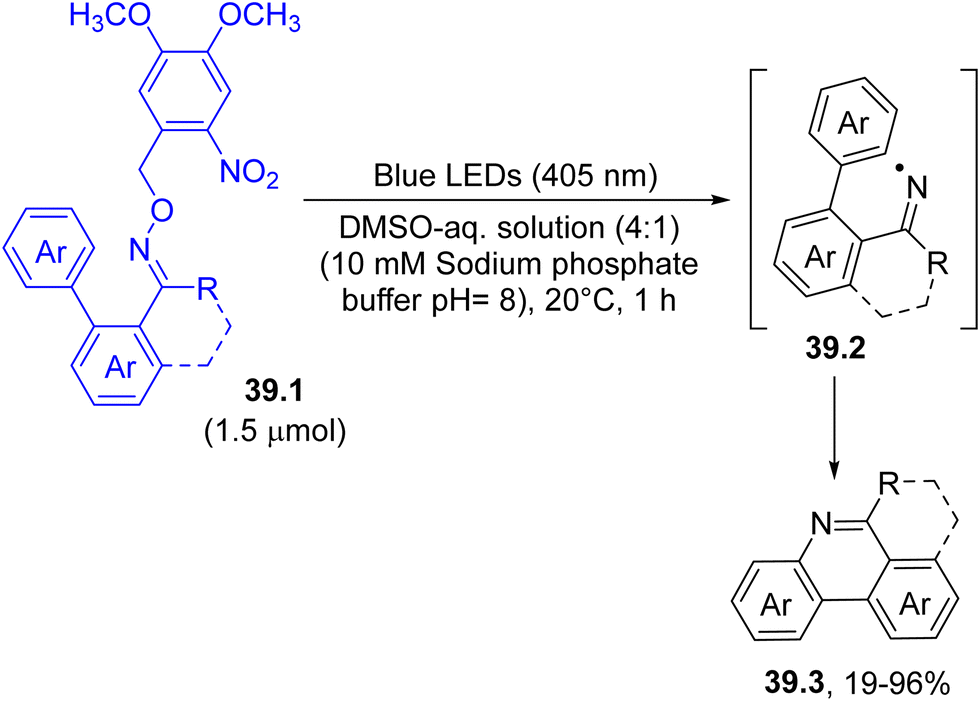 | ||
| Scheme 39 Synthesis of phenanthridine from the photolysis of o-nitrobenzyl oxime ethers. | ||
4.3. Oxygen and sulfur-based
Chalcogen-based radicals were widely used in synthesis. These radicals may be formed from various precursors exploiting the lability of a Ch–X bond (often a Ch–Ch bond).The generation of oxygen-based radicals is well known. However, the direct visible-light generation is not trivial and in selected cases, photohomolysis of the N–O bond can be a solution even though the resulting radicals (mostly carbonyloxy) are prone to fragmentation, giving access to other reactive radical species.169 On the other hand, sulfur-based radicals are readily accessed by the photocleavage of (aryl) disulfides. In fact, the lability of the S–S bond (the BDES–S for PhSSPh is ca. 50 kcal−1 mol−1)158 coupled with the absorption in the visible region (at least for aromatic disulfides) made these compounds the elective substrates for the preparation of thiyl radicals. As a matter of fact, radical routes to C–S bonds are rather common.170,171
This approach was adopted for the photoinduced intramolecular annulation of 2-alkynylbiphenyls 40.2 with various disulfides 40.1. Thus, blue light irradiation of 40.1 in acetonitrile for 8 h released two thiyl radicals, which upon addition to the triple bond of 40.2 led to the formation of 9-sulfenylphenanthrenes 40.4 upon vinyl radical (40.3) cyclization. (Scheme 40).172
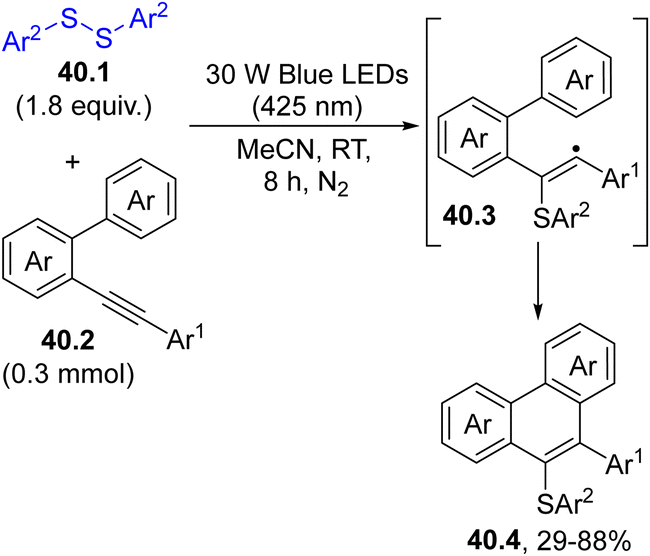 | ||
| Scheme 40 Photochemical access to 9-sulfenylphenanthrenes. | ||
The photocleavage of a PhS–SPh bond was used for a multicomponent radical cross-coupling reaction initiated with the sulfur radical addition onto [1.1.1]propellane that, in the presence of N-heterocycles, formed potentially bioactive 1-arylthiol-3-heteroaryl bicyclo[1.1.1]pentanes.173
The direct cleavage of an S–H bond to generate a thiyl radical in the presence of an alkene (or an alkyne) is quite common under UV irradiation to perform the so-called thiol–ene (or a thiol–yne) reaction.174–176 In rare instances, the reaction may be promoted by visible-light as depicted in Scheme 41. Thus, the naturally abundant Euphorbia factor L3 (41.2) was functionalized by attack of a visible-light-generated arylthiyl radical (from differently substituted thiophenols 41.1) on the exocyclic double bond. The resulting radical 41.3 underwent cyclization onto the endocyclic double bond, forming a seven-membered ring and a five-membered ring in one step. The new compounds obtained (41.4) belong to a new group of biorelevant sulfur-containing premyrsinane-type diterpenes (Scheme 41).177
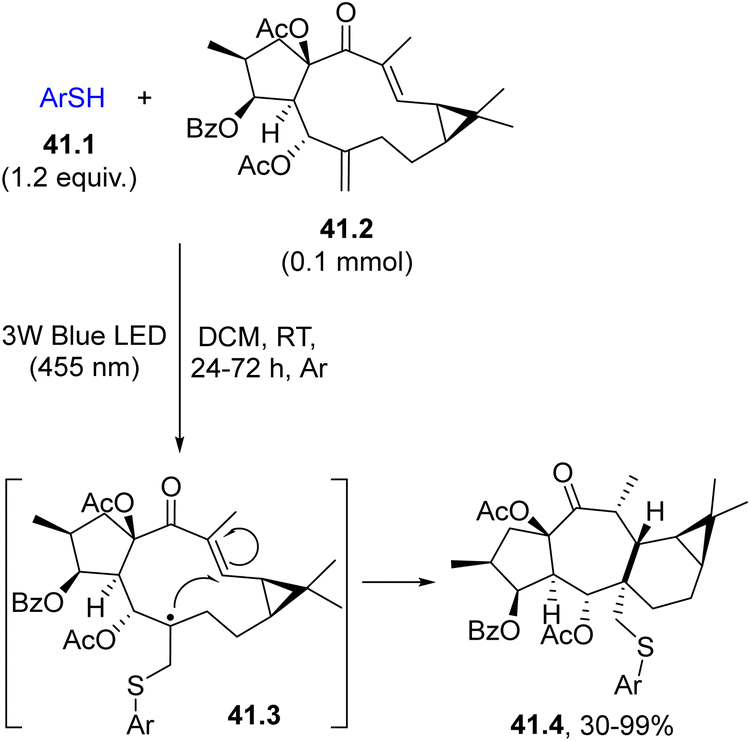 | ||
| Scheme 41 Thiol–ene reaction for the preparation of sulfur-containing premyrsinane-type diterpenes. | ||
A thiol–ene reaction was applied to 1,3-butadiene (an important feedstock). The thiyl radical generated from a diaryldisulfide added to the diene and the resulting allyl radical was trapped by a Ti(III)-based catalyst, and the formed adduct in turn reacted with an aldehyde to give several allylic 1,3-thioalcohols with exceptional regio- and diastereoselectivity.178
The generation of a sulfur centered radical can be efficiently achieved by employing coloured N-xanthylamides. As an example, the visible-light exposure of 42.1 caused the N–S cleavage and the resulting amidyl radical was capable of abstracting a hydrogen atom from a cycloalkane 42.2a–d. The resulting cycloalkyl radical may now couple with the previously formed sulfur radical, enabling a valuable C–H xanthylation process to yield O-ethyl carbonodithioates 42.3a–d (Scheme 42).179
 | ||
| Scheme 42 C–H xanthylation of cycloalkyls. | ||
The versatility of aryl azosulfones (see Schemes 27–30) in sulfonylation processes is witnessed by their use even as sulfonyl radical generators. Scheme 43 presents some examples.
 | ||
| Scheme 43 Visible-light-promoted functionalization of differently substituted styrenes through arylazo sulfones. | ||
The sulfonyl radical 43.3 liberated from 43.1 added onto unsubstituted styrenes and the resulting benzyl radical reacted with molecular oxygen, releasing β-oxo sulfone 43.4 (Scheme 43, path a).180 The same oxysulfonylation products were also formed by the reaction of photogenerated 43.3 with phenyl alkynes under aerated conditions.181 On the other hand, the fate of the reaction was different when the sulfonyl radical attacked cinnamic acid (Scheme 43, path b). The benzyl radical intermediate was oxidized to a cation and captured by an iodide anion. The concomitant loss of the iodide anion and carbon dioxide assured the regeneration of the double bond and an (E)-vinyl sulfone 43.5 was isolated in a good yield.182 The competitive addition of the aryl radical onto the double bond was prevented by its conversion to phenol, thanks to the presence of water and air in the reaction mixture. A related reaction involved the addition of 43.3 onto β-nitrostyrenes where again an (E)-vinylsufone 43.6 was formed by the elimination of the NO2 group (Scheme 43, path c).183 More recently, a procedure for the 1,2-difunctionalization of styrenes via arylazo sulfones and the subsequent synthesis of α-sulfonyl arylhydrazones in a 100% atom economy fashion has been proposed. Also, in this case, the initial step is the regioselective addition of the photogenerated sulfonyl radical onto the double bond of styrenes.184
The sulfonyl radical produced from arylazo sulfones (Scheme 1f) was also employed for the functionalization of 1,6-enynes (e.g., N-phenyl-N-(prop-2-yn-1-yl)methacrylamides), delivering a range of sulfonylated γ-butyrolactams.185 Interestingly, if the sulfonyl radical 44.2 is not trapped in the reaction it is converted into an acid depending on the reaction conditions, namely, methanesulfinic acid 44.3 (a weak acid) under an inert atmosphere or methansulfonic acid 44.4 (a strong acid) in oxygenated media (Scheme 44). Accordingly, sulfones 44.1 may behave as PhotoAcid Generators (PAGs).186
 | ||
| Scheme 44 Visible-light promoted generation of acidic species by irradiation of arylazo sulfones. | ||
This behaviour prompted the application of these new PAGs to catalyze different chemical transformations in an eco-sustainable way with the advantage of the slow release of the acid in solution during the irradiation event (Scheme 45). In doing so, the protection of alcohols as THP acetals has been successfully carried out employing arylazo sulfone 45.2 as a PAG (Scheme 45a).186 In particular, alcohol 45.1 was converted into the corresponding acetal 45.3 (an orthogonally protected diol) in quantitative yield with no competitive deprotection of the silyl ether.186
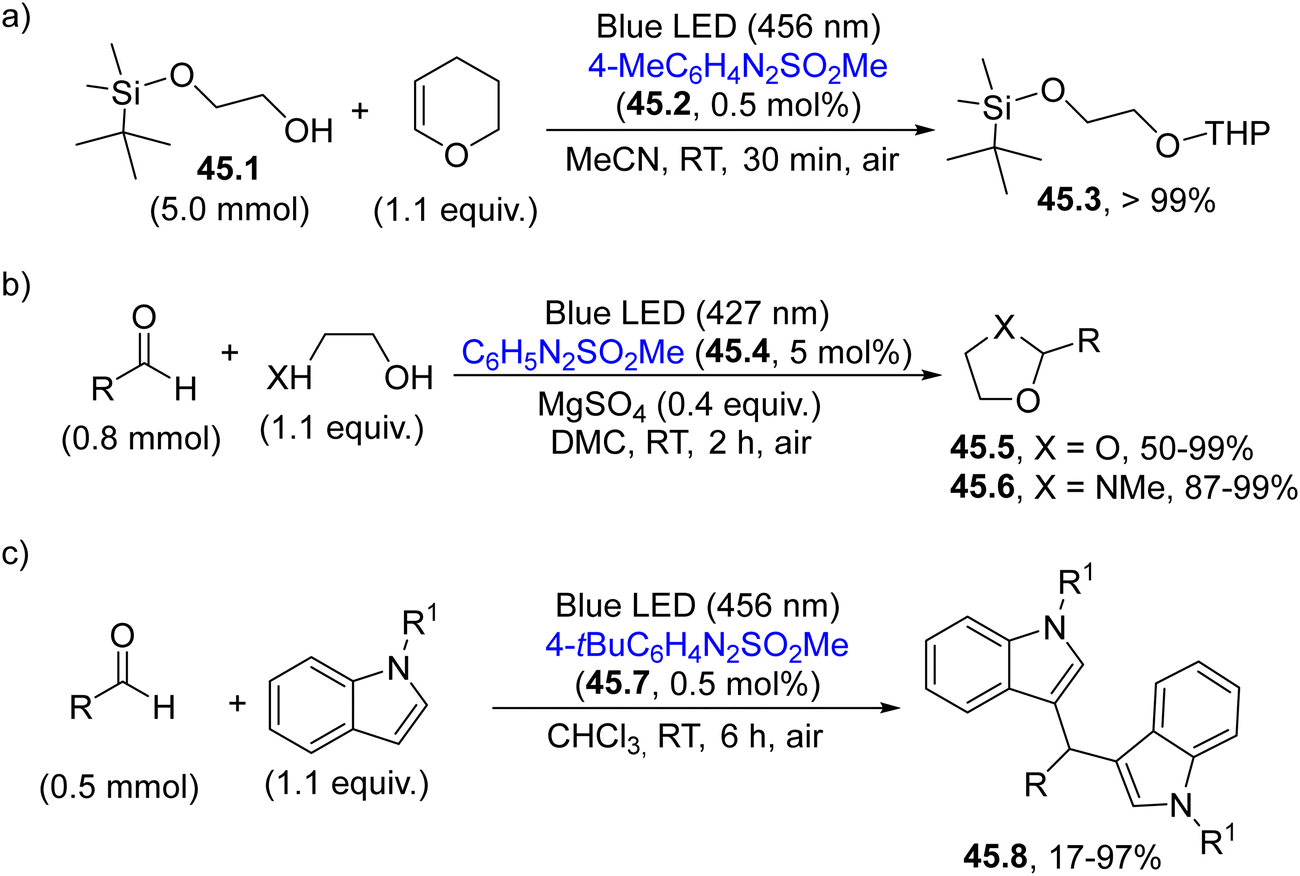 | ||
| Scheme 45 Application of arylazo sulfones as visible-light absorbing PAGs for the (a) protection of alcohols (b) protection of carbonyls and (c) preparation of diarylmethanes. | ||
Moreover, the acidity resulting from the photolysis of 45.4, was able to catalyze the protection of aldehydes (or ketones) as dioxolanes (45.5) or N-methyl oxazolidines (45.6) in up to 99% yield in a benign DMC solvent (Scheme 45b).187 In the latter case, due to its structure, the PAG served as a traceless promoter since the byproducts nitrogen, benzene and the sulfonic acid may be easily eliminated from the final mixture, enabling the isolation of the protected aldehydes through a simple filtration on silica gel. Finally, arylazo sulfones proved to be efficient PAGs also in the visible-light mediated Friedel–Crafts reaction of aldehydes on differently substituted indoles for the smooth preparation of diarylmethanes 45.8 (Scheme 45c).188
The photocleavage of a Se–S bond in selenosulfonates 46.1 enabled the functionalization of different vinyldiazo compounds 46.2. The tosyl radical formed in the irradiation added to the double bond of 46.2 causing a nitrogen loss from intermediate 46.4. Recombination with the PhSe˙ radical completed the radical 1,3-selenosulfonylation (Scheme 46).189 The advantage of the method is the high atom economy and the use of an inexpensive light source (Blue LED) and of a green solvent.
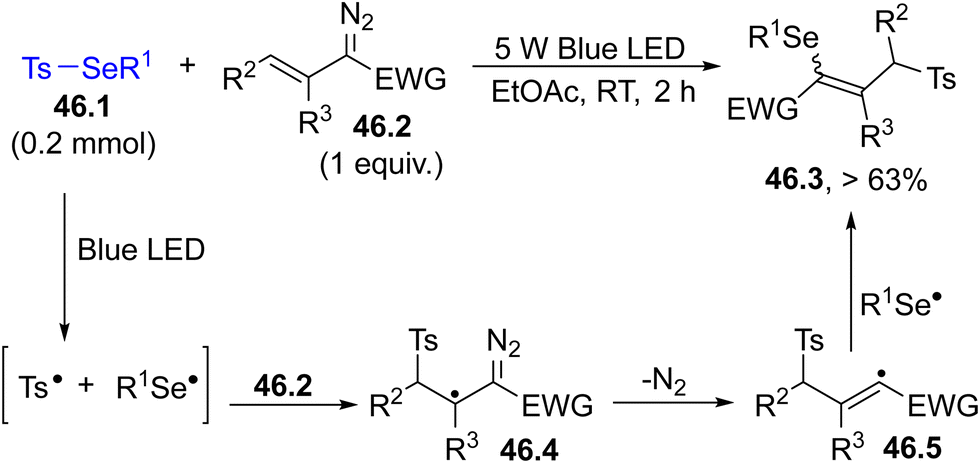 | ||
| Scheme 46 Visible-light radical 1,3-selenosulfonylation of vinyldiazo derivatives. | ||
The same TsSePh derivative was likewise used for the preparation of valuable 8-membered sulfonyl-benzo[b]azocines via a radical 8-endo sulfonylcyclization.190 The tosyl radical may derive from the photohomolysis of the S–I bond in Ts–I under blue LED irradiation, inducing a radical cyclization in 1,6-diynes.191
The sulfonyl radical may be generated by carbon radical addition onto a SO2 surrogate (DABSO = DABCO·(SO2)2). A devised strategy, involved the photolysis of PhI(OCOCH2F)2 to liberate the CH2F radical, which was promptly trapped by DABSO to generate a monofluoromethylsulfonyl radical, which was used for the preparation of oxindoles from N-arylacrylamides in up to 97% yields.192
The synthesis of 1,2,4-dithiazoles 47.2 was carried out starting from thioamides 47.1 by simply employing visible-light sources and oxygen as the oxidant (Scheme 47). The mechanism hypothesized indicated a PET reaction between 47.1 and oxygen to deliver the thioamidyl radical 47.3 that was captured by another molecule of 47.1, generating the heterocycle via an intermolecular nucleophilic addition and desulfurization.193
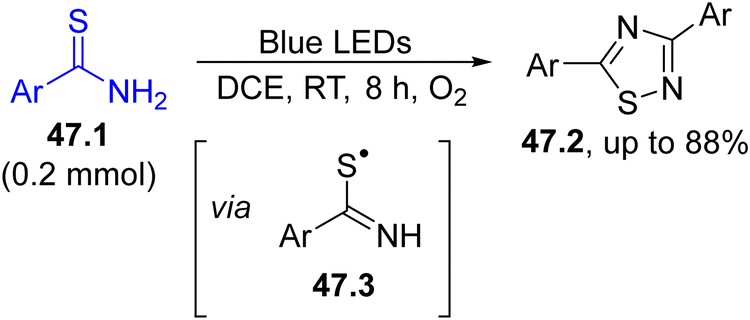 | ||
| Scheme 47 Aerobic visible-light induced synthesis of 1,2,4-thiadiazoles from thioamides. | ||
4.4. Selenium-based
The adoption of mild routes to the formation of Se–X bonds is desirable in the frame of green and mild approaches. The use of a photochemical reaction to promote a selenylation event is an appealing choice. Usually, the selenylating agent is a diselenide RSeSeR (mostly R is an aryl group)75 that showed an absorption tail in the visible region (λ < 500 nm), attributed to the n → σ* transition, coupled with the labile nature of the Se–Se bond (the BDESe–Se for PhSeSePh is ca. 41 kcal−1 mol−1).158 Thus, the species derived from the cleavage of the Se–Se bonds may be used for the functionalization of alkenes, alkynes and (hetero)aromatics.As an example, Scheme 48 shows an example of the selenofunctionalization of alkenes. The reaction of PhSe˙ (released from the photolysis of diselenide 48.1) with CBr4 to yield PhSeBr was invoked as the key intermediate. Derivative 48.3 was obtained in an 85% yield, which upon elaboration gave the amaryllidaceae alkaloid (±)-γ-lycorane 48.4.194
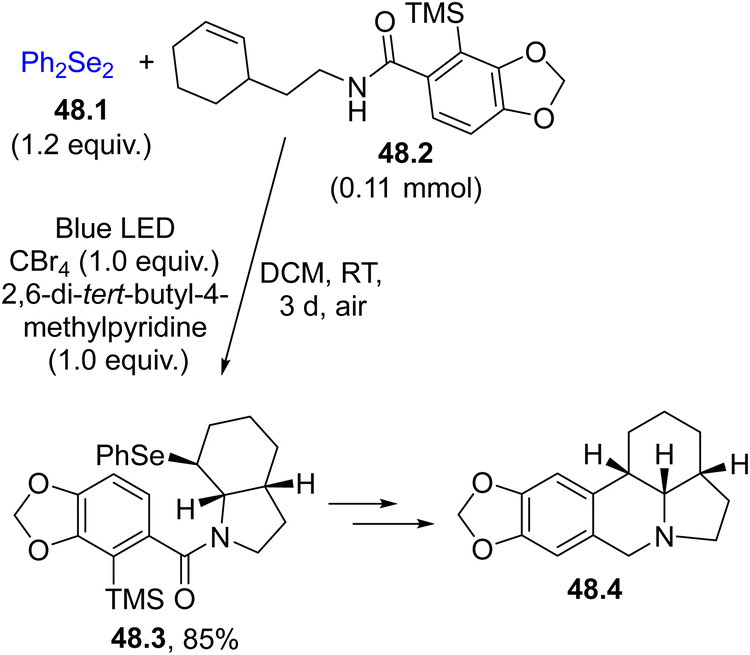 | ||
| Scheme 48 Selenofunctionalization of a CC bond as the key step in the preparation of the alkaloid (±)-γ-lycorane 48.4. | ||
The peculiar photochemistry of diselenide 49.1 has been exploited to achieve the oxoselenylation of vinylarene 49.2 (Scheme 49). The so-formed α-aryl selenomethyl ketone 49.3 resulted from the attack of the selenium-based radical onto 49.2 followed by reaction with oxygen. Although the reaction gave good yields under blue LED irradiation, a CFL lamp was mainly adopted.195
 | ||
| Scheme 49 Photochemical route to α-aryl selenomethyl ketones. | ||
A related reaction was carried out in the presence of a sulfonimide (e.g., saccharin 50.2) where a visible-light-induced three component reaction took place resulting in an intermolecular aminoselenation of alkenes. The mechanism is depicted in Scheme 50. Thus, the selenyl radical 50.4 generated by the blue LED irradiation of diselenide 50.1 added to styrene 50.3, forming the β-seleno alkyl radical 50.5, which further reacted with 50.1 affording an unstable 1,2-bis(diselenide) 50.6. An intramolecular nucleophilic attack of selenium at the benzylic position took place generating an aryl-stabilized seleniranium ion 50.7 and the phenylselenyl anion 50.8−, which deprotonated saccharin 50.2, releasing neutral phenylselenol 50.8 and the saccharin anion 50.2−. Saccharin derivative 50.9 was then obtained by coupling between 50.2− and 50.7. A broad variety of aminoselenation products was accessible in good yields with excellent functional group compatibility again by adopting a CFL lamp as the light source.196
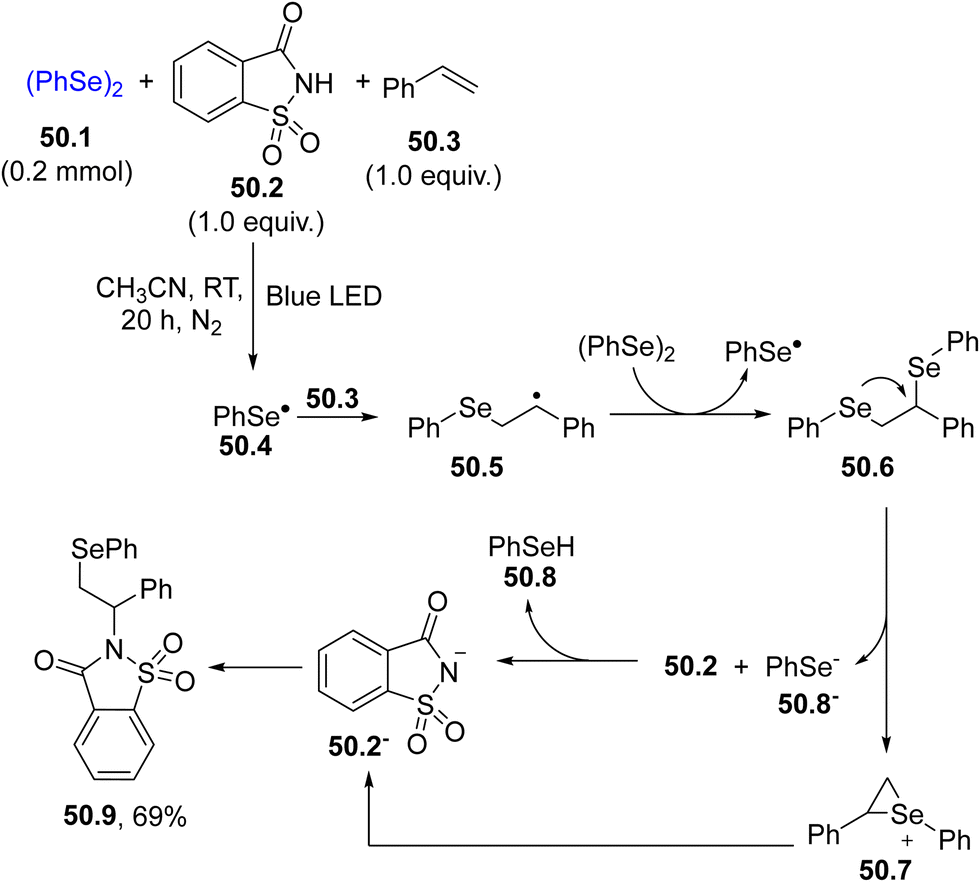 | ||
| Scheme 50 Intermolecular aminoselenation of alkenes. | ||
Selenium-based radicals were likewise engaged in the selenylation/cyclization of enaminones 51.1 for the practical synthesis of 3-selanyl-4H-chromen-4-ones 51.2/51.4. Two main approaches have been followed (Scheme 51). The first one was performed under (photo)catalyst free conditions and no additional oxidants were needed to obtain the final product 51.2. The selenium source was derived again from diaryl selenides (path a).197 In this case the PhSe˙ formed was oxidized by air to the corresponding PhSe+, which promoted a ring closure upon addition onto the CC bond in 51.1 (Scheme 51, path a).197 The second approach is a green and practical protocol, using trifluoromethyl tolueneselenosulfonate 51.3 and enaminones 51.1 to synthesize a wide range of chromones 51.4 under photocatalyst and oxidant free conditions (Scheme 51, path b). The first step was the photogeneration of CF3Se˙ which smoothly reacted with the double bond of 51.1 and the thus obtained radical was oxidized to the corresponding cation by oxygen to generate iminium 51.5. The intramolecular attack of the OH group followed by N,N-dimethyl group loss, yielded chromones 51.4 in up to 91% yield, where various functional groups were tolerant to the photoirradiation conditions.198
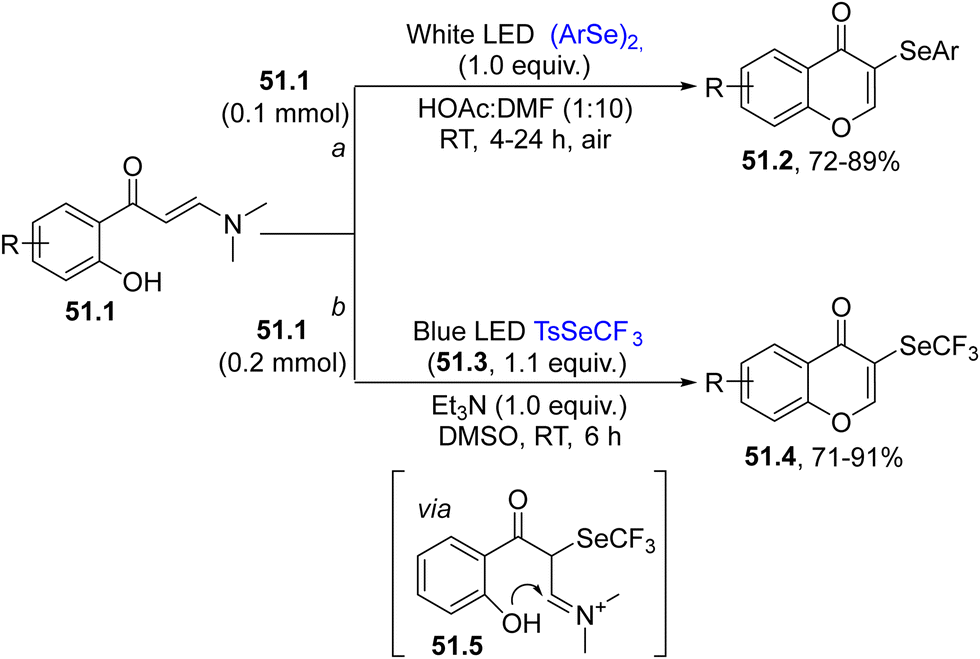 | ||
| Scheme 51 Visible-light synthesis of chromones employing selenium-based radicals. | ||
An alternative methodology for the construction of heterocycles is presented in Scheme 52 where two C–Se bonds and one C–C bond are constructed simultaneously. Following this approach, a dichalcogenide 52.1 serves as the source of a selenium centered radical. Upon irradiation, the radical formed by the homolytic cleavage of the chalcogen–chalcogen bond, underwent a radical domino reaction leading to a [2+2+1] heteroannulation of 1,7-enynes 52.2a, b. The chalcogen-based radical reacted readily with the electron-poor olefin, forming the radical intermediates 52.3a, b, which cyclized onto the triple bond affording vinyl radicals 52.4a, b. The latter closed the cycle by attacking the chalcogen moiety and, after few more steps, selenopheno[3,4-c]quinolin-4(5H)-ones 52.7a, b (or even thieno[3,4-c]quinolin-4(5H)-ones) were formed.199 Moreover, the selenium radical addition onto the C–C triple bond in methyl(2-(phenylethynyl)phenyl)sulfanes was developed as a novel route for synthesizing 3-arylselanyl benzothiophenes.200
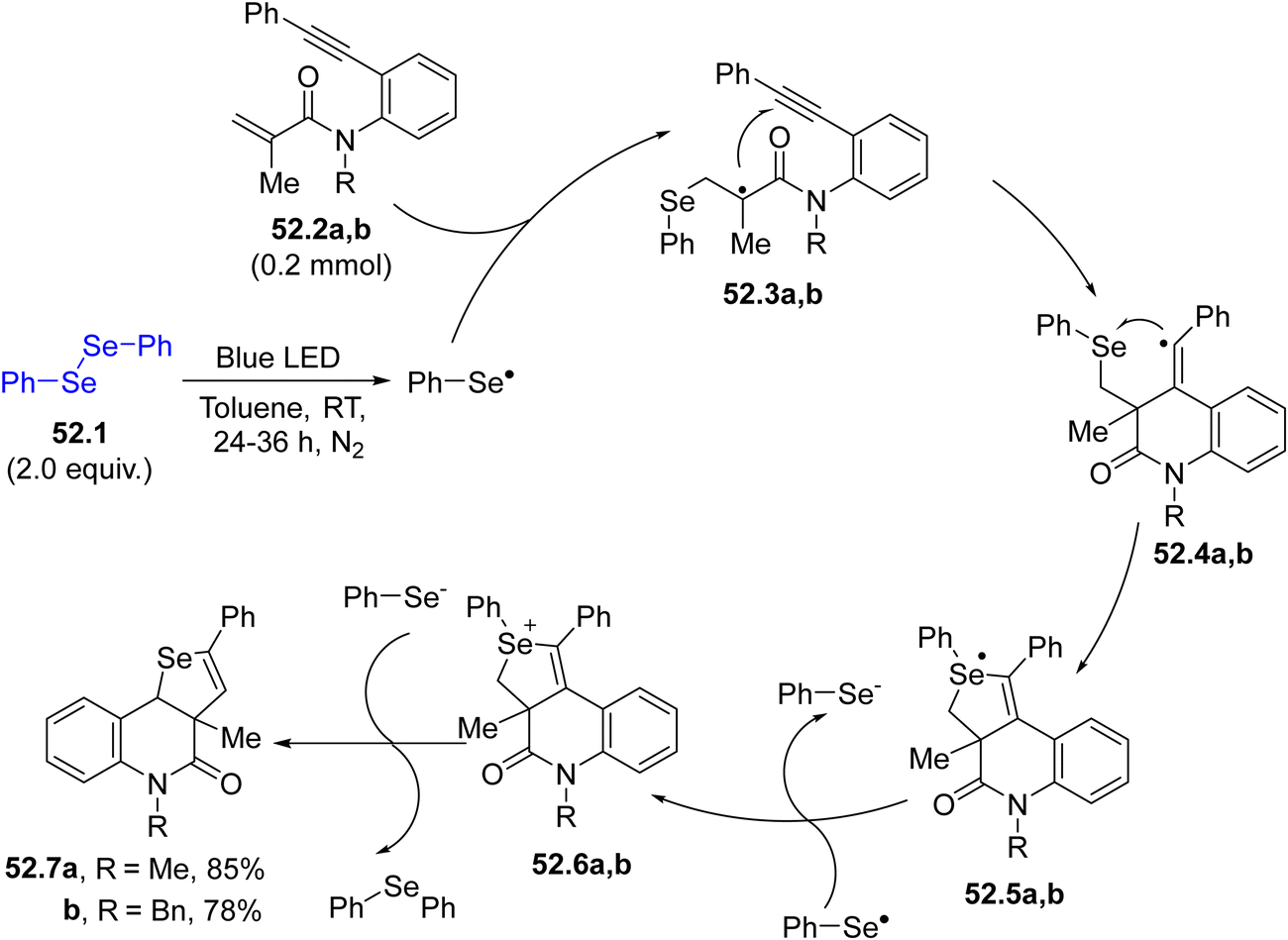 | ||
| Scheme 52 Visible-light mediated photocatalytic synthesis of selenopheno[3,4-c]quinolin-4(5H)-ones. | ||
Seleno-benzo[b]azepines were readily formed upon photohomolysis of Ts-SePh in the presence of N-allyl-4-methyl-N-(2-(1-phenylvinyl)phenyl)benzenesulfonamide. Despite the first step being the generation of the PhSe˙ radical, the authors postulated the addition onto the CC double bond of PhSe+ derived from the oxidation of the latter radical by dioxygen.201
Another important process involving selenium radicals is their addition onto C–C triple bonds for the formation of substituted double bonds. Scheme 53 shows that phenyl acetylenes are ideal reaction partners. Thus, the regioselective addition of the phenylselenyl radical (from 53.1) onto the C–C triple bond of 53.2 generated an alkenyl radical that attacked the cyclohexadienone scaffold intramolecularly. The capture of another phenylselenyl radical by the resulting α-keto radical led to the formation of heterocycle 53.3. The action of basic water on 53.3 resulted in a nucleophilic displacement, forming chrom-6(5H)-enone 53.4, a potent cytotoxic agent against HepG-2 cell lines.202
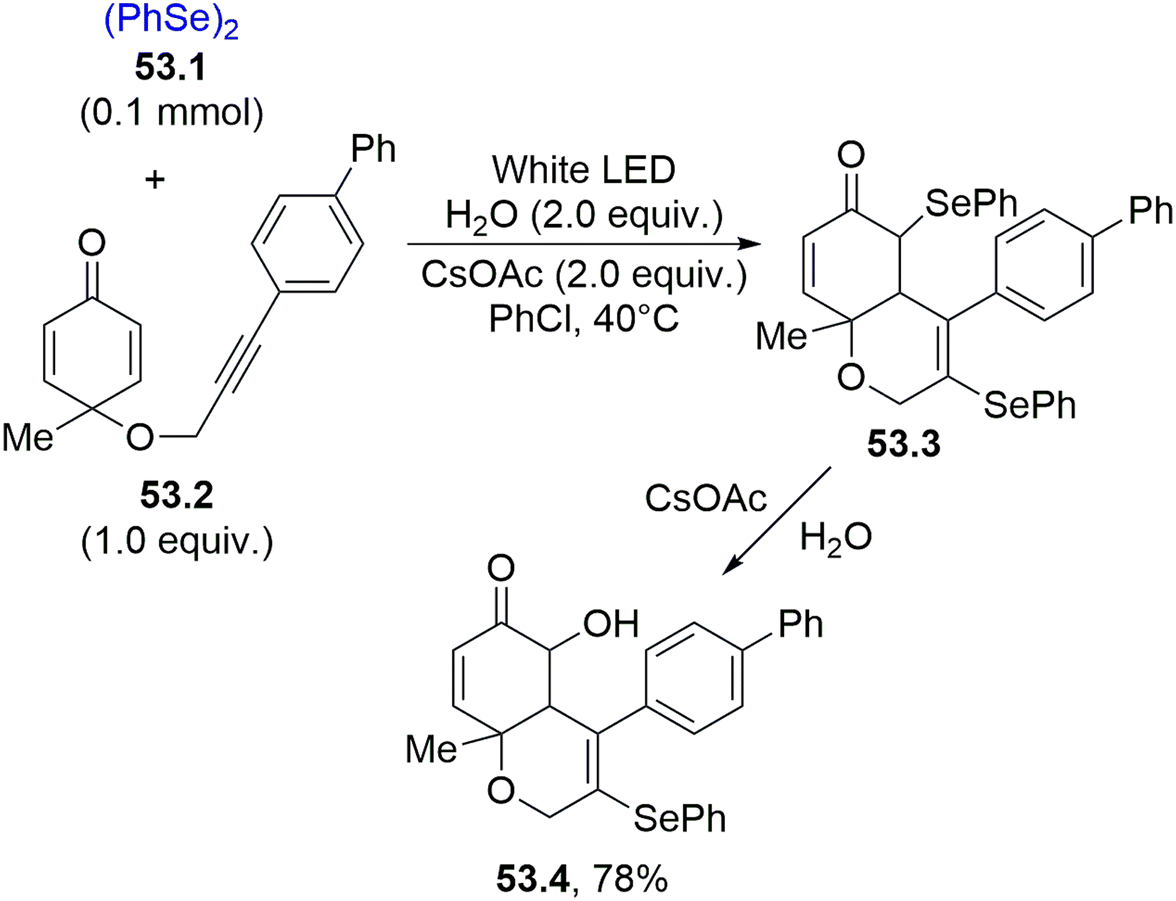 | ||
| Scheme 53 Phenylselenyl radical addition onto C–C triple bond for the synthesis of chromen-6(5H)-ones. | ||
The construction of multiple C–Se and C–S bonds was achieved in the three component reaction between a diselenide, phenyl acetylene, and DABCO·(SO2)2, resulting in the formation of β-sulfonylvinylselanes (mostly in the E configuration).203
Electron-poor alkynes are likewise good radical traps for the selenyl radicals. As an example, ynones 54.2a, b were used in the preparation of 3-selenospiroindolenines (54.4a, b, having a promising anticancer activity, Scheme 54). The attack of PhSe˙ onto the triple bond followed by the cyclization on the indole ring released the adduct radicals 54.3a, b, which upon oxidation and deprotonation of the hydrogen atom of the NH group yielded the spirocompounds 54.4a, b in moderate to good yields.204
 | ||
| Scheme 54 Preparation of 3-selenospiroindolenines. | ||
Similarly, spiro-cyclohexadienones were obtained by a dearomative selenylative carbo-spirocyclisation promoted by the phenylselenyl radical addition onto aromatic homologated-ynones.205 Again, a dearomative cascade cyclization of biaryl ynones with diselenides yielded a series of selenated spiro[5.5]trienones in moderate to good yields.206 If the same approach was applied to N-aryl-N-(2-hydroxyethyl)propiolamides 55.2 as reaction partners (under oxygenated conditions), a dearomative oxo-spirocyclization took place affording a series of selenium-containing benzo[b]pyrrolo[2,1-c][1,4]oxazine-3,9-diones 55.4. In the mechanism, the selenium radical triggered an ipso-cyclization which was followed by an oxo-Michael addition onto the cyclohexadienone 55.3 (Scheme 55).207
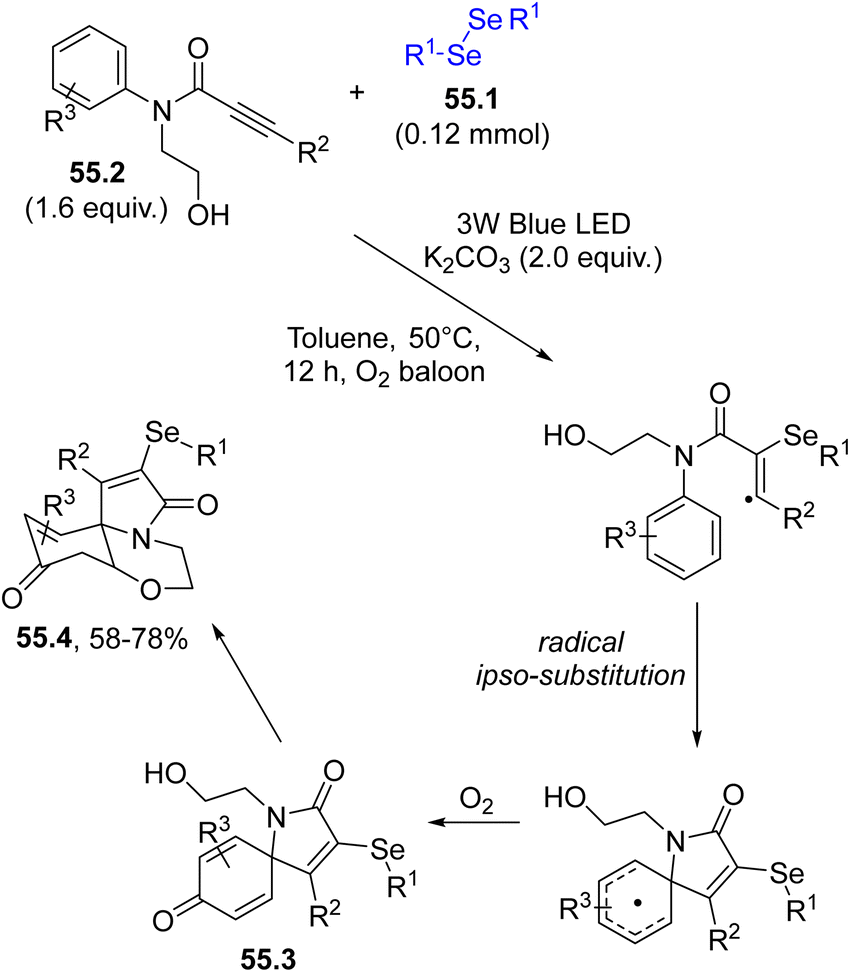 | ||
| Scheme 55 Visible-light dearomative oxo-spirocyclization of N-aryl-N-(2-hydroxyethyl)propiolamides. | ||
In some instances, aryl alkynoates were used in spirocyclization reactions, such as in the domino reaction leading to 1,1-diselenidealkenes 56.7 (Scheme 56). The usual addition of the selenyl radical onto 56.2 promoted a domino reaction and after a 1,4-migration (on intermediate 56.4), decarboxylation and selenyl radical recombination, 56.7 was isolated as a result of a double selenium radical addition on aryl alkynoate 56.2.208
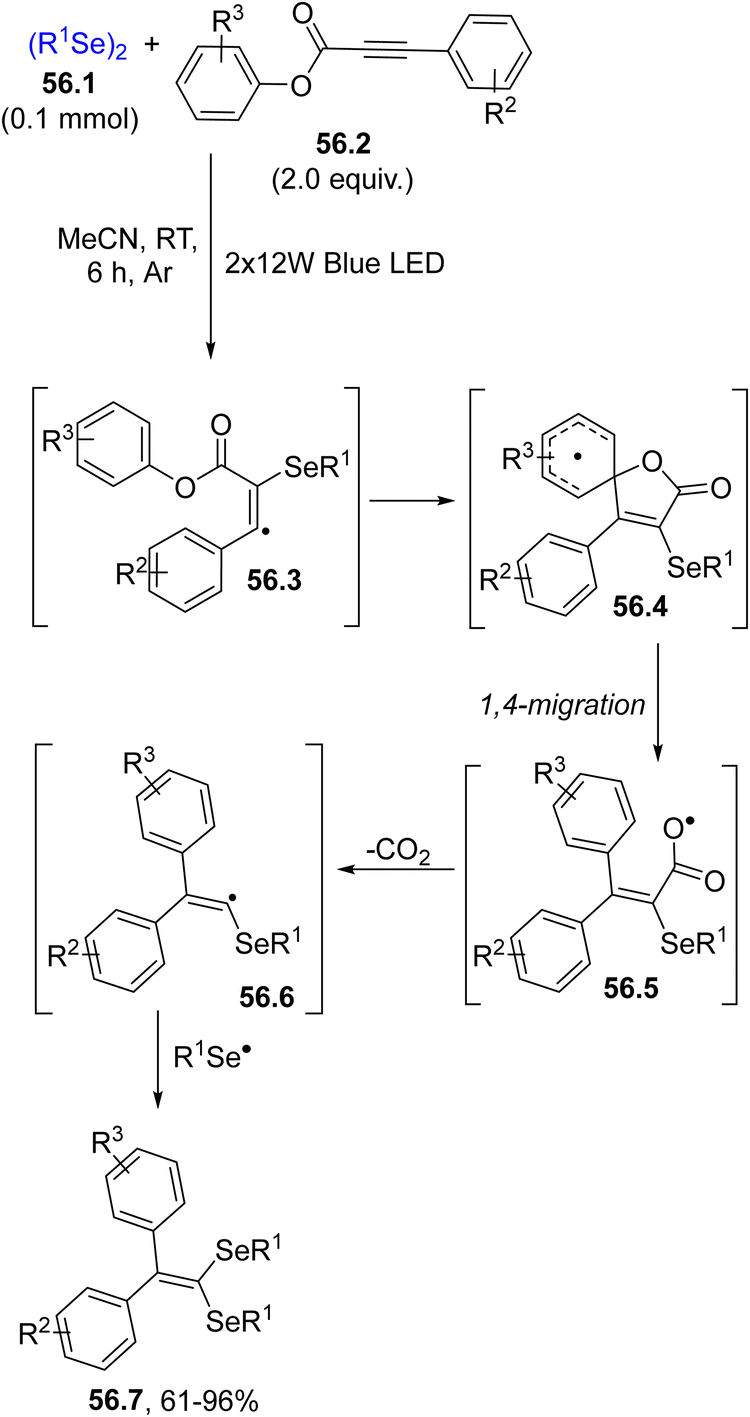 | ||
| Scheme 56 Visible-light synthesis of 1,1-diselenidealkenes. | ||
The usefulness of selenium-based radicals can be also appreciated in the functionalization of electron-rich (hetero)aromatics (e.g., indoles) under mild conditions. Thus, dialkyldiselenide 57.1 was easily broken under blue light irradiation, generating 3-selenylindoles 57.3 in a good yield upon reaction with unsubstituted indole 57.2 (Scheme 57a). The protocol was also effective in the derivatization of anilines, phenols or other heterocycles while maintaining EtOH as the benign solvent (Scheme 57b).209
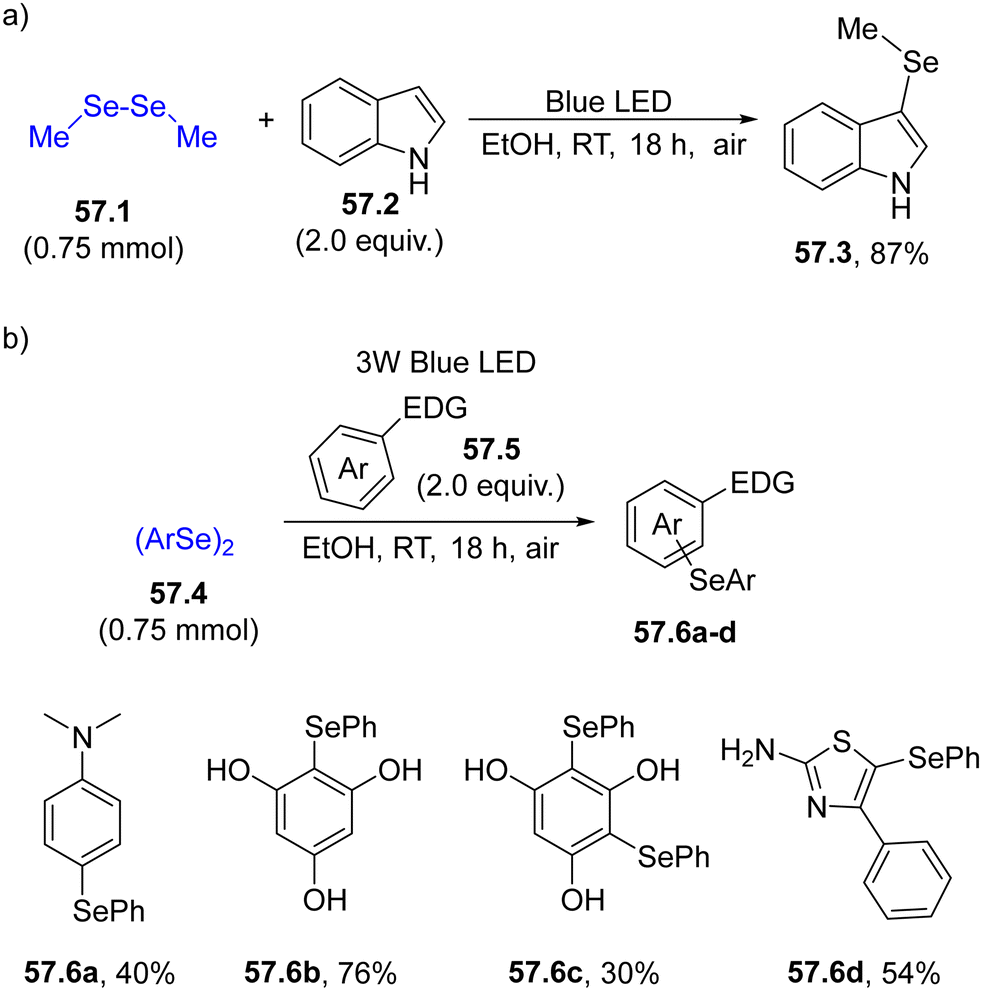 | ||
| Scheme 57 Photoinduced forging of an Ar–Se bond for the preparation of (a) 3-selenylindoles and (b) for the derivatization of electron-rich benzenes. | ||
4.5. Tellurium-based
In the frame of chalcogen-based radicals, the use of tellurium derivatives is sparsely reported. In analogy with selenium radicals, diaryl ditellurides are the elective radical sources due to their absorption up to 550 nm. Terminal alkynes may be easily functionalized to give bis(phenyltelluro)alkenes by the formation of two C–Te bonds.210 Irradiation of 58.1 induced the ditelluration of alkyne 58.2, resulting in the formation of trifunctionalized alkene 58.4 in a good yield but with poor E selectivity (Scheme 58).74 The initial homolysis of the Te–Te bond liberated a PhTe˙ radical which upon addition onto 58.2 generated vinyl radicals 58.3 and 58.4 from it through reaction with another molecule of 58.1.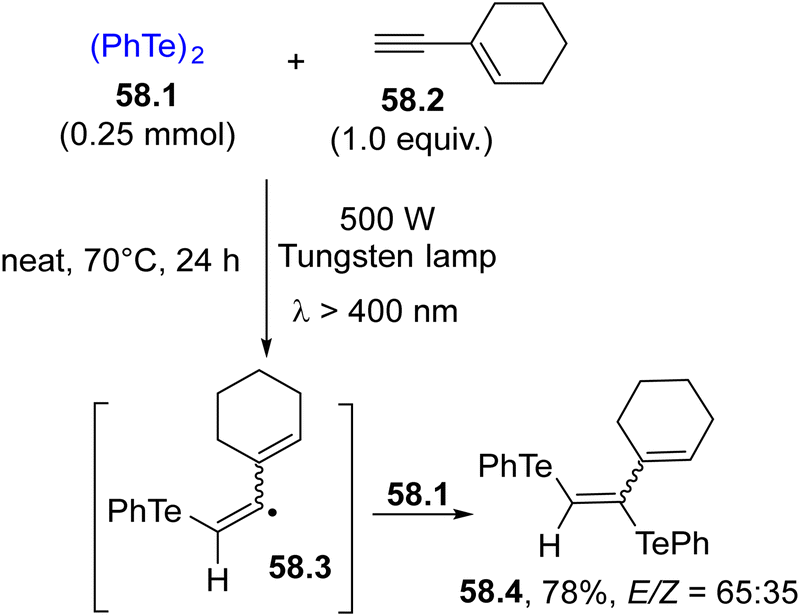 | ||
| Scheme 58 Visible-light induced ditelluration of an alkyne. | ||
In some cases, ditellurides were used in combination with other (ArCh)2 (with Ch = chalcogen) derivatives. In fact, the photolysis of 59.1 in the presence of isonitrile 59.3 resulted in an unproductive reaction. However, when 59.2 was added to the reaction mixture, a thiotelluration reaction took place where product 59.4 was easily isolated in a good yield (Scheme 59).211 Lower yields were however detected when the aryl isocyanide did not bear strong electron-withdrawing groups.
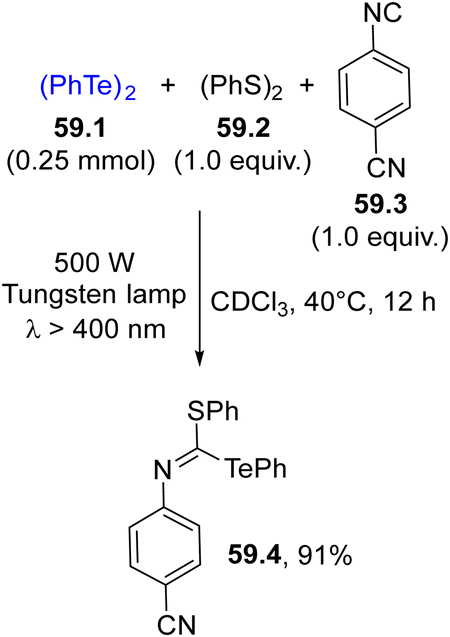 | ||
| Scheme 59 Thiotelluration of aryl isocyanides. | ||
4.6. Halogen atoms
Halogen atoms may serve as useful intermediates for eco-sustainable halogenation reactions but they are rarely used. The generation of these radicals has been proposed for the preparation of α-halomethyl ketones starting from terminal alkynes 60.2 (Scheme 60). Irradiation of N-bromo- or N-iodosuccinimides 60.1a, b with 450 nm light liberated the corresponding halogen radical that readily added to 60.2 and the resulting vinyl radical was oxidized by oxygen, yielding ketones 60.3a–d.71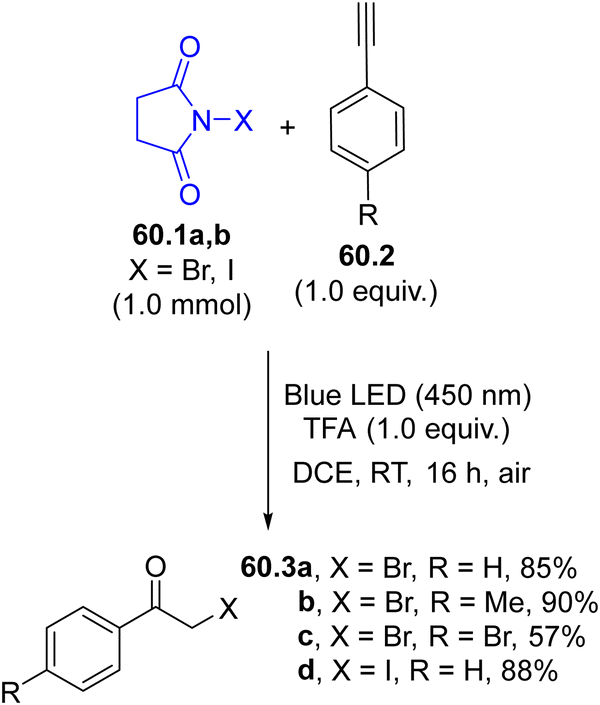 | ||
| Scheme 60 Functionalization of terminal alkynes using NBS or NIS as the source of halogen atoms. | ||
A bromo atom may be incorporated into complex structures, namely, the terpenoid natural product (+)-sclareolide (61.2), by the photolysis of a N-bromoamide (61.1, Scheme 61). The visible-light induced (using a 100 W tungsten lamp) cleavage of the N–Br bond in 61.1 resulted in the liberation of a reactive amidyl radical that abstracted a hydrogen atom from 61.2 in a selective fashion (out of 26 aliphatic C–H bonds). The combination of the resulting carbon radical with the bromine atom afforded the C2-equatorial bromination product 61.3 as a single regio- and stereoisomer in a satisfying yield.70
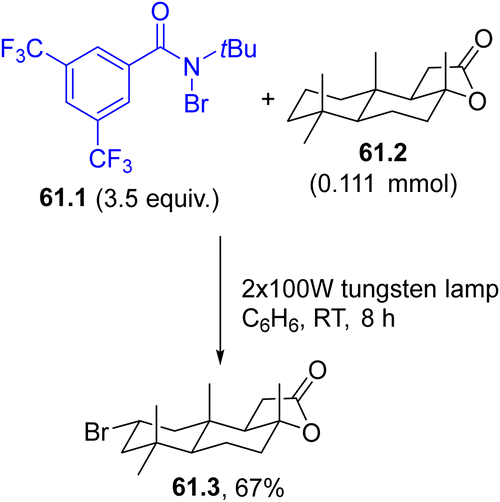 | ||
| Scheme 61 Photoinduced bromination of (+)-sclareolide. | ||
An intriguing reaction is the concise and efficient ring-opening difluorination strategy developed for the synthesis of highly functionalized hydroxy-containing α,α-difluoro-β-ketoamides 62.3a–c through derivatization of 4-aminocoumarins 62.2a–c in dimethyl carbonate (DMC) as a green solvent (Scheme 62). The target was smoothly achieved under visible-light irradiation in air at room temperature without the addition of any other external photocatalysts. With this protocol, compounds 62.3a–c were successfully synthesized under mild conditions in up to 91% yield.212
 | ||
| Scheme 62 Photochemical synthesis of α,α-difluoro-β-ketoamides. | ||
The first photochemical event was the release of a fluorine atom through the photocleavage of the N–F bond in NFSI 62.1, which smoothly added to 62.2a–c. The N-centered radical 62.4 underwent a HAT reaction with 62.5, yielding 62.7. A further radical addition/HAT sequence led to the formation of imine intermediate 62.9. Finally, the water hydrolysis of 62.9 produced the primary amine 62.11 that promoted the aminolysis of the lactone moiety on the resulting ketone 62.10, thus leading to the desired α,α-difluoro-β-ketoamides.212
5. Reactions via nitrenes
Photogenerated nitrenes are less used intermediates with respect to carbenes (see Section 6) although they can lead to valuable intramolecular insertion reactions onto C(sp2)–H or N–H bonds. The photolysis of an azido group is often used for the generation of nitrenes. As an example, nitrene 63.3 (formed from vinyl azide 63.1 by photoinduced nitrogen loss) underwent an intramolecular attack to yield 2H-azirine 63.4 (Scheme 63). Then, the attack of a cyanamide anion (obtained by deprotonation of 63.2) opened the three-membered ring, releasing 2-aminoimidazole 63.6 (useful for the preparation of anti-infective and anti-cancer derivatives) in a good yield.213 Curiously, the same reaction took place by adopting microwave irradiation in place of light.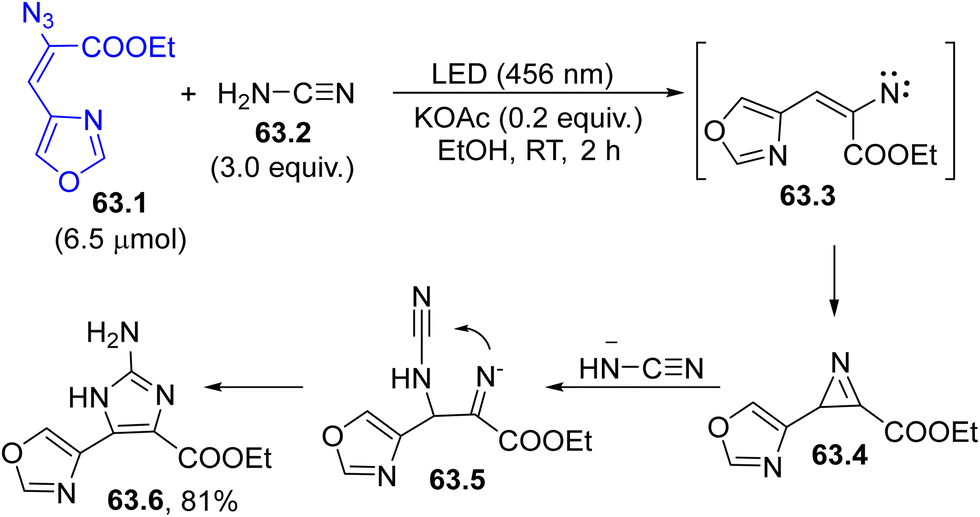 | ||
| Scheme 63 Photogenerated nitrenes for the synthesis of 2-aminoimidazoles. | ||
A similar intermediate, 2H-azirine, was reported in the synthesis of (E)-stilbenes through the irradiation of 3-(2-aminoaryl)-2-azido-1-arylprop-2-en-1-ones, followed by an 1,2-acyl migration214 and in the synthesis of pyrroles from α-keto vinyl azides.215
Nitrenes may be easily formed starting even from aryl azides. Thus, the visible(solar) light-driven photocyclization of functionalized aryl azides 64.1a–c yielded 2H-indazole-3-carboxamides 64.3a–c (Scheme 64). A N–N bond coupling was achieved starting from 64.1a–c upon a photodenitrogenation step followed by nitrene, 64.2a–c, insertion and by the aromatization of the resulting intermediate.216
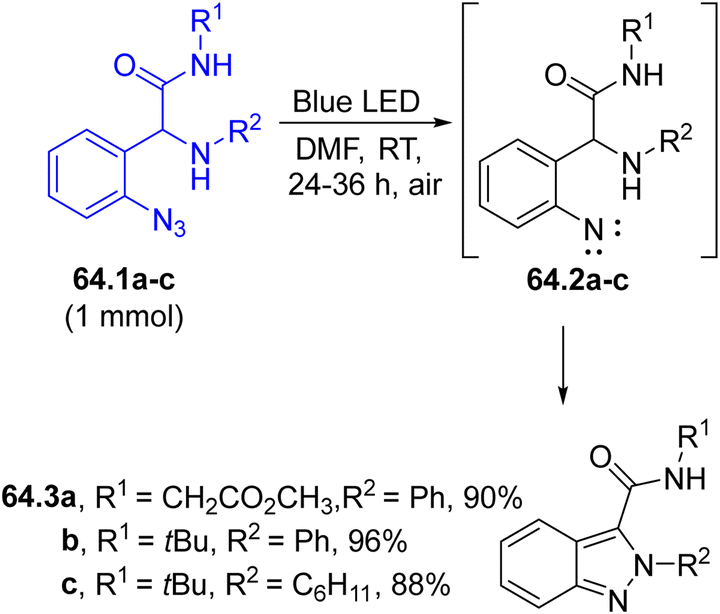 | ||
| Scheme 64 Photochemical synthesis of 2H-indazole-3-carboxamides via nitrene generation. | ||
The generation of nitrenes from aryl azides, a well-known phenomenon, has been recently rediscovered. An aryl nitrene, photogenerated by Blue LED exposure of azide 65.1, formed a strained 2H-azirine through nitrogen insertion onto the C1–C2 arene π-bond (Scheme 65). The resulting strained three-membered ring underwent a thermal 6π electrocyclic opening, forming a seven-membered electrophilic ketenimine that was intercepted by amine 65.2 to yield azepine 65.3. The latter compound could be isolated or treated with photogenerated singlet oxygen (by acenaphthylene sensitization) to produce endoperoxide 65.4 that evolved into 2-aminopyridine 65.5.217 If the reaction is carried out by using ethylaminoehanol as the secondary amine, the resulting azepine can be treated with N-bromocaprolactam to produce a pyridyl derivative through an ipso-selective nitrene internalization.218
 | ||
| Scheme 65 Conversion of aryl azides into 2-aminopyridines. | ||
A similar process involved irradiation of aryl azides in the presence of alcohols followed by treatment with trifluoroacetic anhydride (TFAA), resulting in the formation of ortho-aminophenols via a dearomative-rearomative sequence.219
Azides, however, are known hazardous compounds. Accordingly, coloured aryl sulfilimines (66.1, Scheme 66) were devised as safer alternatives for aryl nitrene generation. Thus, visible-light exposure of 66.1 promoted the intramolecular C–H insertion of an aryl ring of the resulting nitrene, producing Clausine C (66.2 a natural compound) in a very good yield on a 5 mmol scale.220
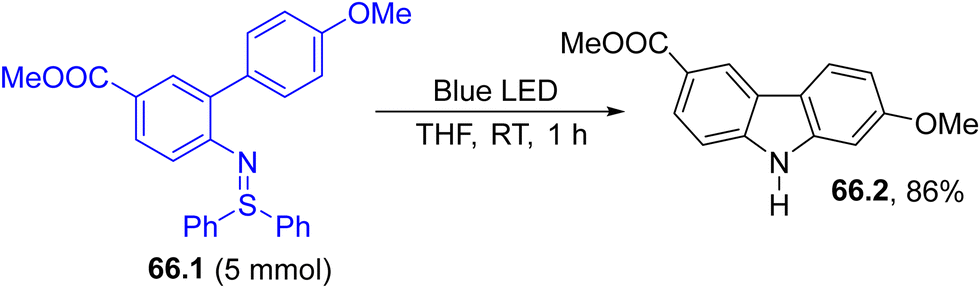 | ||
| Scheme 66 Aryl sulfilimines as precursors for the preparation of Clausine C (66.2). | ||
Aminoiodinanes 67.1a–c are a class of compounds capable of generating nitrenes after visible-light exposure. Utilizing these compounds, a two-step protocol allowing the C–H amination of cyclic ethers followed by the reduction of the resulting products 67.3a–c has been developed for the preparation of amino alcohols 67.4a–c (Scheme 67). The initial C–H functionalization was accelerated by visible-light, improving the reactivity compared to the thermal process performed in the dark. The key step was the formation of the nitrene intermediates 67.2a–c, which can interconvert between the singlet and the triplet state. Both states reacted selectively towards the methylene group of THF present at the α position with respect to the oxygen atom (C–H insertion for the singlet and hydrogen atom transfer for the triplet), both leading to 67.3a–c.221
 | ||
| Scheme 67 Photoinduced C–H amination of THF. | ||
Triplet nitrenes formed from compounds 67.1 may be used for the formation of C–N bonds in the C–H functionalization of allylic methylene groups (e.g., in α-methyl styrene). Interestingly, the reactivity shifted when a photocatalyst (e.g., a Ru(III) complex) was present since a nitrene radical anion was formed, leading to the aziridination of the olefin.222 In the same way, the reaction of aminoiodinanes 68.1 with sulfides 68.2 or allyl sulfides 68.3 can yield two different reaction outcomes although, in both cases, the first step was the singlet nitrene addition onto the sulfur atom (Scheme 68). In the case of sulfides 68.2, sulfilimines 68.4a–c, were readily formed, but the allyl-substituted sulfilimine generated from 68.3 underwent a [2,3]-sigmatropic rearrangement to furnish allylic sulfenamides 68.5a–d as final products.223
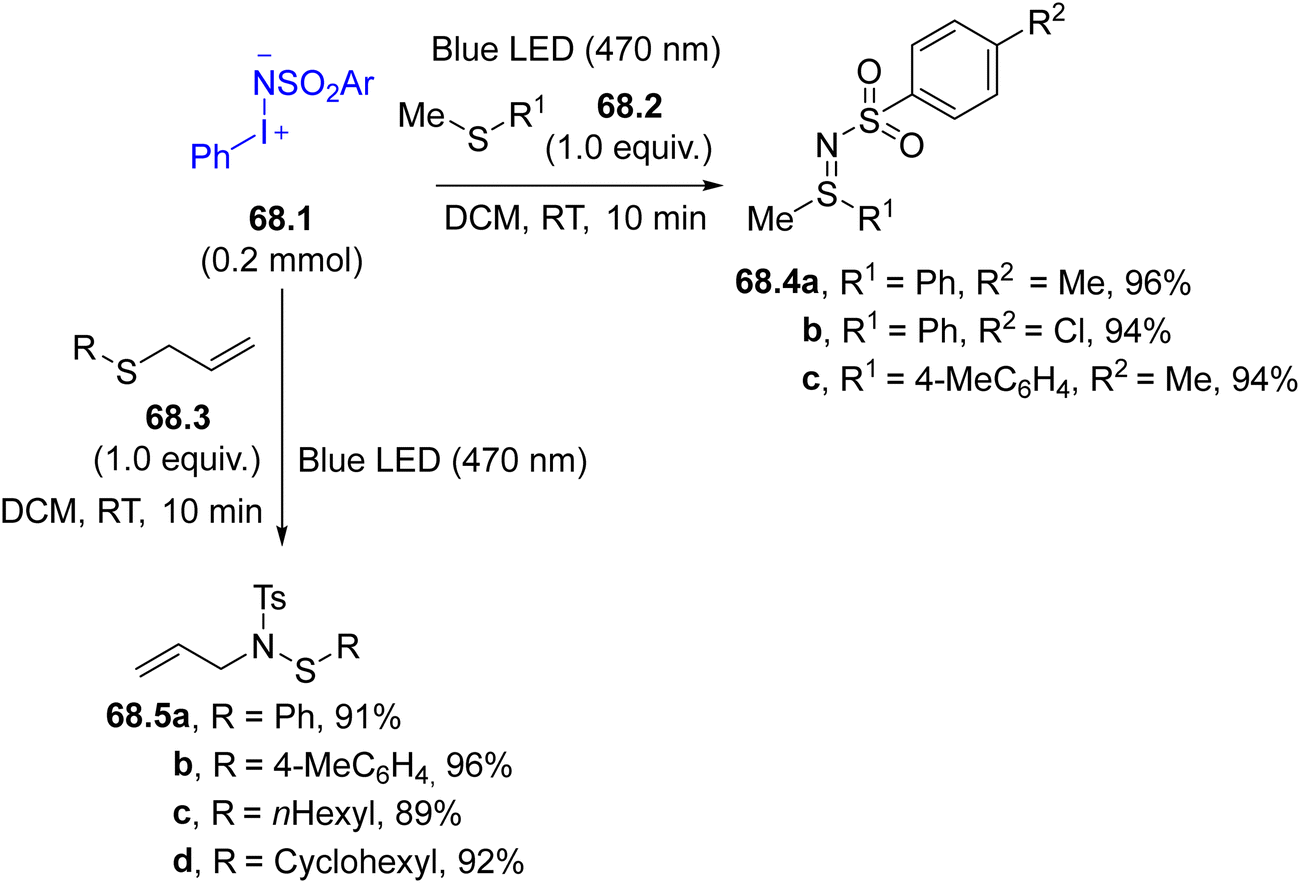 | ||
| Scheme 68 Synthesis of sulfilimines and allylic sulfenamides. | ||
Recently, there has been an increased interest in the photochemistry of nitroarenes.224 As a matter of fact, aryl nitrenes were also obtained through the visible-light photolysis of nitroaromatics (e.g., 69.1, Scheme 69). Thus, the excited 69.1 underwent deoxygenation with P(OiPr)3 to form nitrene 69.4 and the corresponding aminoazepine from it. Treatment with TFAA facilitated the conversion of the seven-membered ring to ortho-phenylenediamine 69.3 in a modest yield.225 Otherwise, photolysis of the initially formed 3H-azepin-2-amine may induce an excited-state-4π electrocyclization, which upon hydrogenolysis generated bicyclic amine derivatives with full diastereocontrol.226
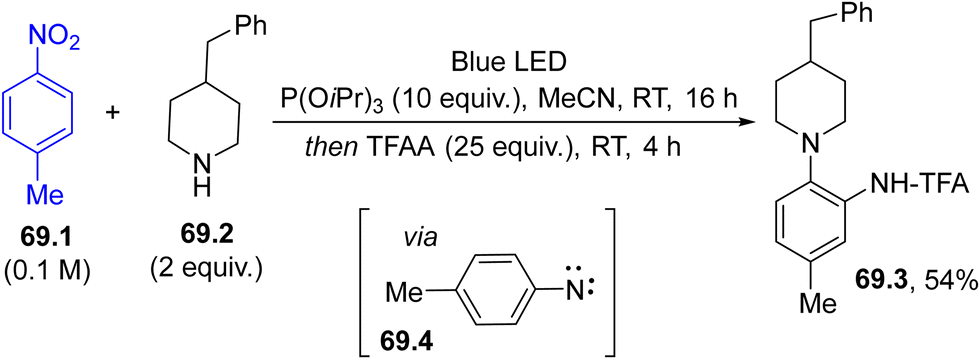 | ||
| Scheme 69 Preparation of ortho-phenylenediamines via dearomative rearomative coupling of nitrobenzenes and amines. | ||
6. Reactions via carbenes
Among the possible intermediates that can be formed, carbenes are probably the most easily generated species under visible-light irradiation, thus expanding their role in syntheses based on carbene transfer reactions.227–230 These carbenes may exist as singlets or triplets and the relative stability strongly depends on the structure of the precursors. The presence of a lone pair adjacent to the vacant 2p-orbital at the carbenic site stabilizes singlets rather than triplets. As a matter of fact, the different multiplicity of carbenes may affect their reactivity and chemoselectivity.231,232As emphasized in the Introduction section (Scheme 1g), diazo derivatives (especially α-diazo ketones (esters) 70.1, Scheme 70) are the preferred species for carbene photogeneration.62,233–237 Visible-light irradiation of compounds 70.1 (R1 ≠ OR) generates an α-keto carbene that rapidly rearranges to a ketene 70.2, via a Wolff rearrangement (Scheme 70, path a). However, the photolysis of α-diazo esters led a diverted chemistry since carbene 70.3 is exclusively formed (path b). Another class of coloured carbene precursors is acylsilanes (70.4).238 These compounds readily undergo a photoinduced 1,2-rearrangement (the so-called Brook rearrangement, path c) to siloxy carbenes 70.5.59,230
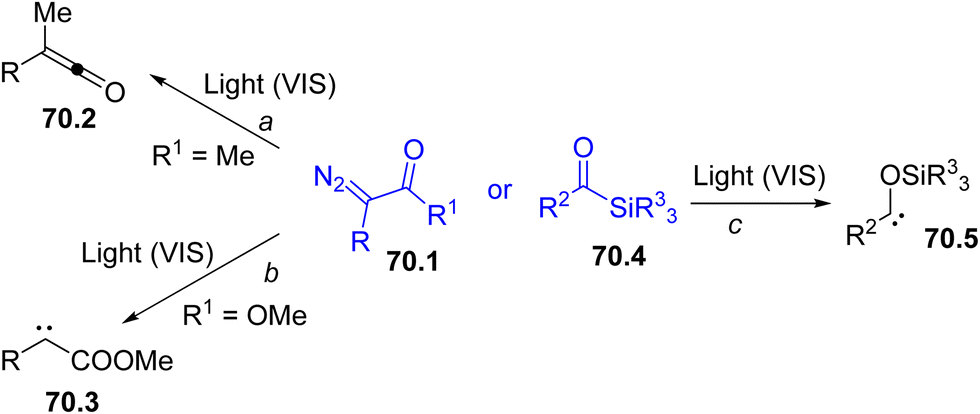 | ||
| Scheme 70 Main approaches for the photochemical generation of carbenes. | ||
The synthetic applications of these carbenes range from X–H and C–H insertion, cycloaddition onto unsaturated bonds, addition onto a heteroatom nucleophile to generate ylide intermediates, often inducing Doyle–Kirmse rearrangement like reactions.
6.1. Formation of a three-membered ring
A three-membered ring may be easily formed by the addition of a carbene onto an electron-rich double or a triple C–C bond (Scheme 1g). A typical example is described in Scheme 71a where a cyclic triene 71.3 was exclusively formed upon a regio- and stereoselective carbene addition onto a polyunsaturated carbocycle (cyclooctatetraene 71.2). The usefulness of compound 71.3 was demonstrated by its facile derivatization via a 6π-electrocyclization/Diels–Alder reaction cascade.37 Allyl alcohols have been likewise cyclopropanated starting from aryldiazoacetates to yield valuable cyclopropane-fused bicyclic lactones. In this case, the OH group was involved in the transesterification event with no competitive O–H insertion in the reaction with the carbene.239 | ||
| Scheme 71 Synthesis of cyclopropanes via addition of a photogenerated carbene onto (a) a tetraene (b) styrenes and (c) alkenes. | ||
When siloxy carbenes add to a CC bond, a cyclopropanol derivative is formed. As an example, in Scheme 71b, trifluoroacetylsilane 71.4 was converted into the corresponding carbene (of triplet multiplicity as proved by calculations) that smoothly added to both aromatic and aliphatic alkenes (e.g., styrenes 71.5a–d), resulting in a cis-selective [2+1] cyclization reaction yielding silyl ethers 71.6a–d, that can be converted to cyclopropanols through simple treatment with TBAF.240
The reaction can be applied to aromatic acylsilanes having a tethered olefin moiety on the aromatic ring. Upon visible-light absorption an intramolecular [2+1]-cycloaddition took place releasing bicyclo[3.1.0]hexanes and bicyclo[4.1.0]heptanes in a good yield. Owing to the nucleophilicity of the photogenerated carbene, the addition onto electron-poor olefins such as acrylates was also effective.241
Rarely, carbene may be formed by the photolysis of a C,Se-selenonium ylide (71.7, Scheme 71c). The resulting disubstituted carbene led to cyclopropanes 71.9a–c by reaction with electron-rich (71.8a, b) and electron-poor (71.8c) olefins.242
The reactivity of the carbenes is so high that even a CC bond included in a (hetero)aromatic ring may be used for the construction of a three-membered ring. Indole is the elective heteroaromatic ring that enables the synthesis of cyclopropane-fused indolines. Thus, the straightforward addition of a carbene onto indoles 72.2a–d led to derivatives 72.3a–d with satisfactory yields and diastereoselectivity (Scheme 72a).243 An intramolecular version of this reaction was applied to a diazoacetate prepared from a tryptamine leading to azepino[4,5-b]indoles.244
 | ||
| Scheme 72 Photogenerated carbenes for the (a) cyclopropanation of indoles and (b) synthesis of spiro[norcaradiene-7,3′-indolin]-2′-ones. | ||
Even benzene (used as the reaction solvent) can serve as a reaction partner to form the corresponding norcaradiene derivatives through cyclopropanation.245 A useful application of this strategy is shown in Scheme 72b. 3-Diazooxindoles 72.4a–d smoothly reacted with benzene under blue LED irradiation to form spiro[norcaradiene-7,3′-indolin]-2′-ones 72.5a–d in variable yields. The resulting cyclohexadienes were easily converted by means of rearrangement and epoxidation reactions.246
The trapping of a carbene with a triple C–C bond yields a cyclopropene. Thus, photolysis of a diazoderivative in the presence of phenyl acetylene easily formed the corresponding phenyl cyclopropenes.247 The carbene addition was efficient even when decorated alkynes such as propargylic alcohols were used (73.2a–d, Scheme 73a). The metal-free conditions adopted here showed an orthogonal behavior with respect to related metal-catalyzed carbene transfer reactions where the OH group was the reactive site. However, the outcome of the photochemical reaction depended on the substitution of the carbinol carbon since an O–H insertion product was formed when primary alcohols were tested.248
 | ||
| Scheme 73 Preparation of cyclopropenes via photogenerated carbenes from (a) diazoalkanes and (b) diazirines. | ||
Diazo compounds are not the exclusive precursors of carbenes. In fact, diazirine 73.4 easily liberated a carbene upon purple LED irradiation to yield 3-trifluoromethyl-3-aryl-cyclopropene 73.6 with yields up to 97% when trapped by diphenylacetylene 73.5 (Scheme 73b). Such a cyclopropenation occurred more efficiently under continuous flow conditions and adduct 73.6 was formed in 73% yield when the reaction was run at a 10 mmol scale for a residence time of 50 min. Diazirines was more effective than the corresponding diazo compounds as demonstrated by the control experiment although a diazo compound was formed to some extent from the diazirine during the reaction.249
In one instance, a three-membered ring heterocycle was obtained starting from an α-diazo ester (Scheme 74a). The reaction is a formal [2+1] cycloaddition between the carbene photogenerated from 74.1 and a formaldimine (74.4) resulting from the decomposition of hexahydro-1,3,5-triazine 74.2 in DMSO. Noteworthily, a solvent-controlled divergent cycloaddition took place when the reaction was performed in DCM, leading instead to the formation of imidazolidine 74.5via the intermediate 74.6 (Scheme 74b).250
 | ||
| Scheme 74 Solvent dependent preparation of (a) three-membered and (b) five-membered heterocycles. | ||
6.2. C–C bond formation
An intriguing approach in C–C bond formation via carbenes is the insertion into a C(spn)–H bond as shown in Scheme 75. The generation of a carbene in cycloalkanes as reaction media led to C(sp3)–H insertion although with modest yield.251 Nevertheless, photolysis of phenyl NHPI diazoacetates in a DCM/cycloalkane 1:1 mixture yielded the corresponding C–H insertion products in a very good yield.252 However, the intramolecular insertion led to better results as in the irradiation of 75.1 where the photogenerated carbene 75.2 cleanly gave lactone 75.3 in more than 90% yield (Scheme 75a).
 | ||
| Scheme 75 Carbene insertion onto (a) a C(sp3)–H, (b) a C(sp2)–H, and (c) a C(sp)–H bond. | ||
A related strategy was applied for the synthesis of valuable spiro-β-lactones and -lactams.253 The insertion into aromatic or alkynyl C–H bonds required metal catalysis. To promote C–H insertion into a naphthtyl amine, a quinoline unit must be incorporated, playing the role of a directing group (see compound 75.5 in Scheme 75b). The presence of two nitrogen atoms in 75.5 allowed for the formation of a Pd(II) complex that underwent a carbene addition and the desired naphthyl acetate 75.6 was formed through reductive elimination from the resulting Pd(IV) complex. The reaction did not require any reaction media since diazoacetate 75.4 functioned as the solvent.254 The C–H insertion into a C(sp)–H bond required the use of a copper catalyst and a modified bisoxazoline SaBox ligand (e.g.75.9, Scheme 75c). The process was based on the trapping of the α-siloxy carbene onto the SaBox/Cu(I) catalyst, resulting in the release an α-siloxy Fischer metal carbene that ultimately led to alkynyl alcohol 75.10 in a high yield showing a good broad substrate scope and a remarkable heterocycle tolerance.255
In rare instances, a photogenerated carbene may be engaged in a formal C–C bond insertion. Again, a diazoderivative (76.1, Scheme 76) was the ideal substrate for the carbene liberation. In this case, the addition of the carbene on the enolic form of 1,3-diketone 76.2, generated the cyclopropanol derivative 76.4, which upon C–C cleavage enabled the isolation of 1,4-diketone 76.3.256
 | ||
| Scheme 76 Synthesis of a 1,4-diketone via C–C bond insertion. | ||
A related free carbene transfer reaction of α-diazoesters with γ-diketones was developed by using a chiral bifunctional guanidine-amide organocatalyst to prepare δ-diketones having a quaternary carbon chiral center.257 The same kind of compounds were prepared through a formal carbene insertion reaction of 1,3-diketones with diazoesters. The enantioselectivity of the thus formed quaternary center was assured by the presence of a chiral phosphoric acid catalyst.258
In other instances, the cyclopropane obtained via [2+1] cycloaddition was not stable, thus leading to intriguing chemistry. As an example, carbene addition onto enaminones led to unstable cyclopropyl amine that upon nitrogen atom-driven ring opening followed by elimination of a secondary amine formed an α-substituted γ-ketoester.259 Carbene addition onto electron-poor dienes such as dicyano-2-methylenebut-3-enoates induced a cyclopropanation–vinyl cyclopropane rearrangement sequence, yielding decorated cyclopentenes via a formal [4+1] cycloaddition.260
Another interesting application of photogenerated carbenes is the preparation of olefins. A possible strategy is illustrated in Scheme 77a. A carbene formed from aryl diazoacetate 77.1 was trapped by the sulfur ylide 77.2. The negatively polarized carbon atom initiated a nucleophilic attack on the electrophilic carbene site, yielding intermediate 77.4 and trisubstituted olefin 77.3 from it via dimethyl sulfide elimination. The approach was used for the formal synthesis of an ETA receptor radioligand.261
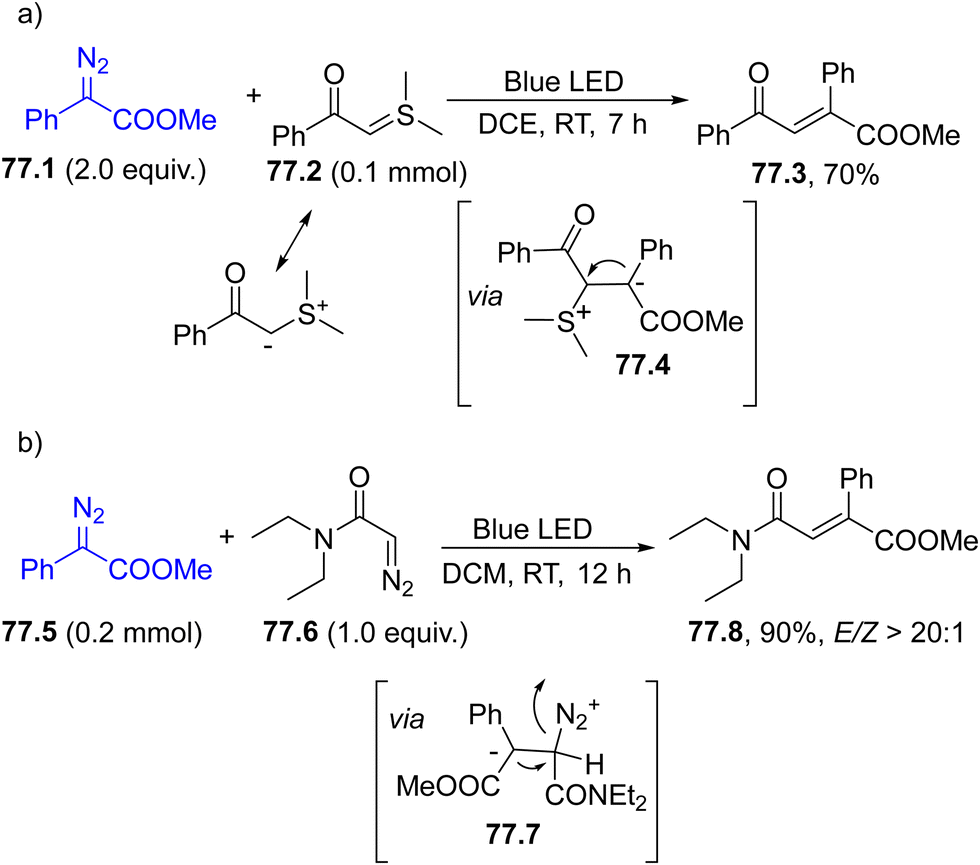 | ||
| Scheme 77 Preparation of alkenes via visible-light reaction of carbenes with (a) a sulfur ylide and (b) a α-diazoamide. | ||
Diphenylmethanethiones can also be used as reaction partners. In fact, addition of the carbene (from a diazoindolone) on the sulfur atom of the thione resulted in the formation of a sulphonium ylide that ultimately led to substituted 3-arylidene oxindoles via an electro-cyclization reaction followed by elemental sulfur elimination from the resulting episulfide.262
All-carbon tetra-substituted olefins were instead formed in moderate to good yields through the visible-light induced coupling between a diazo compound and an iodonium ylide.263 Moreover, densely substituted itaconimides were easily prepared under solvent-free conditions through a multicomponent reaction by adopting (substituted) pyridines, aryl diazoesters and N-alkylmaleimides as the reaction partners.264
The selective use of the wavelength enabled the coupling of two diazoesters (77.5 and 77.6), where a single carbene was formed (from 77.5), and the subsequent addition onto 77.6 led to intermediate 77.7, which upon dinitrogen extrusion formed the trisubstituted alkene 77.8 (mostly in the E configuration, Scheme 77b).265
As mentioned previously, acylsilanes are converted into siloxy carbenes upon photolysis. However, in rare cases, the carbonyl group of the starting acyl derivative may be restored at the end of the reaction. In fact, the siloxy carbenes generated from acyl silane 78.1 was engaged in the nucleophilic addition onto aldehydes 78.2a–d (activated by the Lewis acid ZnI2). A zwitterionic intermediate was formed, which upon 1,4-silyl migration, yielded α-siloxyketones 78.3a–d in variable yields in what was considered a sort of formal cross benzoin-type condensation (Scheme 78a).266 The reaction achieved some success even in the absence of any Lewis acids.267 The electrophilicity of fluorinated ketones was also sufficient, enabling them to function as nucleophilic carbene trapping reagents, resulting in the formation of trifluoromethylated benzoin-type adducts in almost quantitative yields.268 The addition of siloxy carbenes onto a CO group may take place intramolecularly. Accordingly, benzoyl silane was coupled with an N-phenyl maleimide via Rh catalysis and the resulting adduct led to the formation of interesting tricyclic γ-lactams upon visible-light irradiation.269
 | ||
| Scheme 78 Synthesis of (a) α-siloxyketones and (b) α-ketoesters via photogenerated siloxy carbenes. | ||
An intriguing application of siloxy carbenes involves their trapping with carbon dioxide. Thus, irradiation of silanes 78.4a–d under 1 atm CO2 released the corresponding α-ketocarboxylates that can be easily converted onto the corresponding methyl esters 78.5a–d through the in situ addition of TMSCHN2 (Scheme 78b). Mechanistic investigations highlighted the involvement of the singlet state of the siloxy carbene as the intermediate.270
Siloxy carbenes may release a ketone as the final product even when reacting with double or triple C–C bonds. In the first case, acylsilanes were used for a light promoted copper-catalyzed enantioselective allylic acylation. Thus, the carbene was intercepted first by a chiral Cu(I) complex, which upon addition of an allylic phosphate formed an acylcopper(I)–alkene complex, which upon excitation liberated an allyl ketone in up to >99% ee.271
Finally, the silylacylations of an alkyne may take place in an inter- or intra-molecular manner to give functionalized enonyl silanes (in the former case)272 or silylated chromones (in the latter)273 in a satisfactory yield via a cyclopropene intermediate. The intermolecular version of the reaction demonstrated synthetic significance only when using alkynes having electron-withdrawing substituents.
The carbene generated from cyclic diazo imides exhibited solvent-dependent reactivity when thiols were present in the reaction mixture. In fact, in DCM, the carbene was formed in the triplet state and was involved in a C–H functionalization/thiolation process, yielding indane-fused pyrrolidines via an intramolecular cyclization. On the other hand, in MeCN as the reaction medium, a clean reaction took place and hydrazones were formed where the thiols acted as the reductant.274
In rare instances, the addition of a carbene onto a heteroatom led to a C–C bond formation. Thus, the photogenerated α-carboxymethyl carbenes 79.4a–c added to the boron atom of an arylboronic acid 79.2 (paths, a and b). The ylides 79.5a–c formed underwent a 1,2-sigmatropic shift (path c) and the hydrolysis of the obtained boronates 79.6a–c (path d) yielded α-substituted esters 79.3a–c in a discrete yield (Scheme 79). This approach was applied for the synthesis of different pharmaceutics, including, among the others, muscarinic antagonists Benactyzine and Aprophen.275
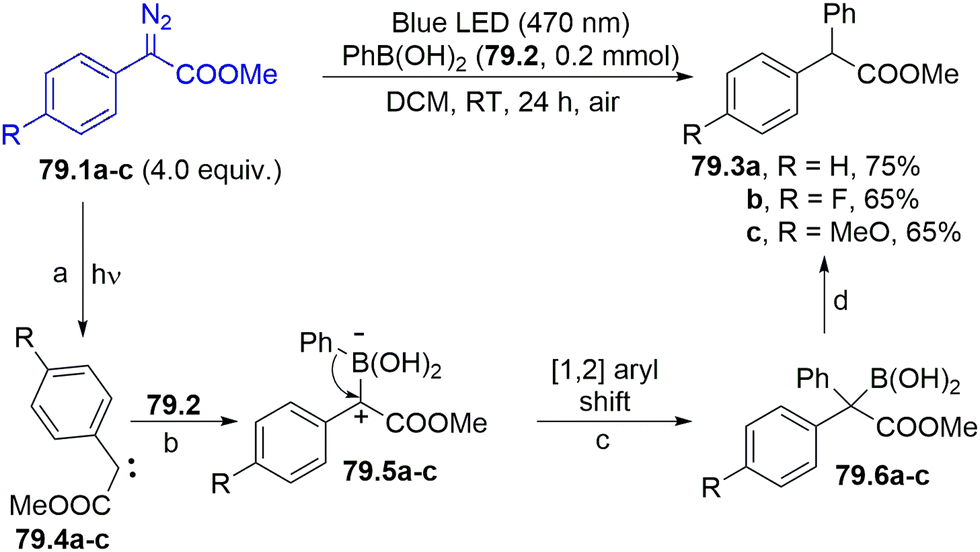 | ||
| Scheme 79 Preparation of α-substituted esters 79.3a–c. | ||
6.3. C–N bond formation
The main method for the formation of a C–N bond via photogenerated carbenes (whether from diazocompounds or acylsilanes) involves N–H insertion into N-heterocycles always under metal-free conditions. Scheme 80a illustrates the case of a pyrazole. The carbene derived from 80.1 underwent a nucleophilic attack by the azarene 80.2 and the N–H inserted derivative 80.3 was obtained in a good yield.276 When the same protocol was applied to 1,2,3-triazoles, the reaction proceeded successfully even in the absence of the base.276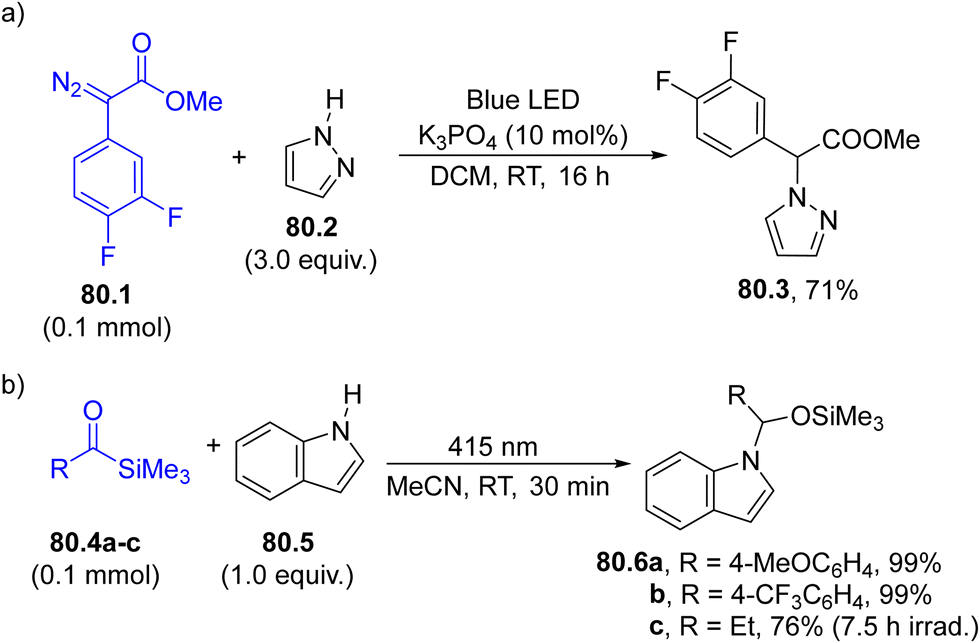 | ||
| Scheme 80 Photoinduced release of carbenes for the N–H insertion into (a) pyrazole and (b) indole. | ||
However, the most investigated N-heterocycle is indole. The N–H insertion may take place with the most used photogenerated carbenes. As an example, Scheme 80b shows the insertion of siloxycarbenes (from silanes 80.4a–c) into indole 80.5, yielding stable silylated N,O-acetals 80.6a–c in almost quantitative yield in a very short time (30 min for benzoyl silanes).277 The same approach was used for the functionalization of the indole core in tryptophan-containing oligopeptides.278
The carbene generated from azidophenyl diazoacetate was reacted with indole to form a key compound for the preparation of the natural product (−)-psychotrimine. In this work, it was found that the efficiency of the synthesis of the N-alkylated products depended on the nature of the indoles (high for 3-substituted indoles having electron-withdrawing groups and low for those having electron-donating groups).61 As a matter of fact, carbenes readily inserted into the N–H bond of carbazole,279 dibenzoazepines,279 and even purine derivatives, causing a site-selective N1-alkylation for the construction of acyclic nucleoside analogues.280
Another approach for the C–N bond formation involves the addition of the carbene onto a nitrile (used as the reaction medium). The carbene, in turn generated from visible-light photolysis of diazoacetate 81.1, was trapped by MeCN to generate the corresponding nitrile ylide 81.4, which upon proton transfer with a carboxylic acid (81.2) yielded a nitrilium ion 81.5, which was prone to undergo a nucleophilic attack by the carboxylate anion. The resulting intermediate 81.6 was then converted into diacylglycine ester 81.3via a Mumm rearrangement (Scheme 81).281,282
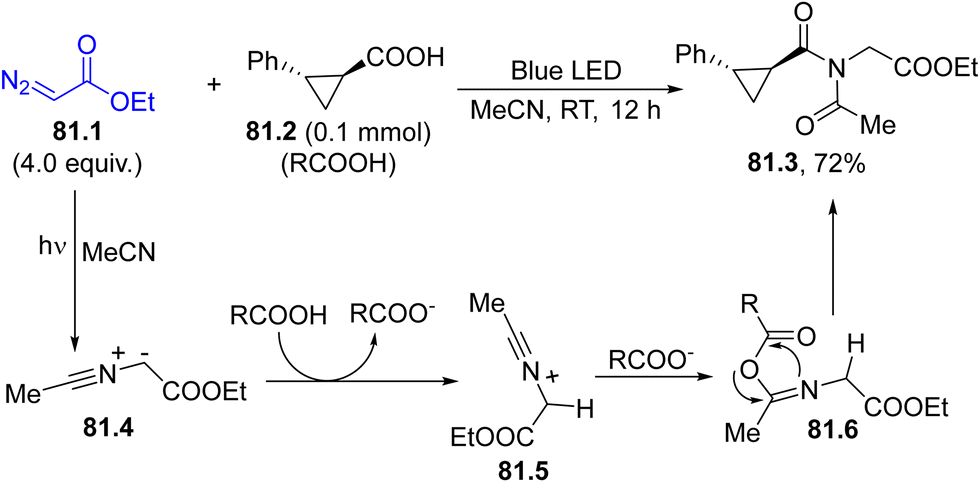 | ||
| Scheme 81 Visible-light synthesis of a diacylglycine ester. | ||
In a related process, the nitrile ylide formed by the addition of a carbene onto MeCN promoted the synthesis of polysubstituted oxazoles in 95% yield, thanks to the presence of a catalytic amount of (iPr)3SiCl as the Lewis acid catalyst.283
A formal enantioselective N–H insertion can be achieved when the carbene is generated in the presence of an excess of DMSO (32 equiv.), a chiral phosphoric acid and an aniline. The carbene was initially trapped by DMSO, forming a C–S bond in an achiral sulfoxonium ylide. Protonation of the ylide by the chiral acid led to a diastereomeric mixture of sulfoxonium ions, which underwent a preferential attack of the amine to form the desired enantioenriched α-aminoester via dynamic kinetic resolution.284
A formal C–N insertion may take place when the carbene adds to the nitrogen atom. The carbene generated from 82.1 was intercepted by allyl amine 82.2, yielding the α,α-disubstituted amino ester 82.3 in a good yield (Scheme 82). The desired compound resulted from a [2,3]-sigmatropic shift occurring on the first-formed ammonium ylide 82.4 intermediate.285
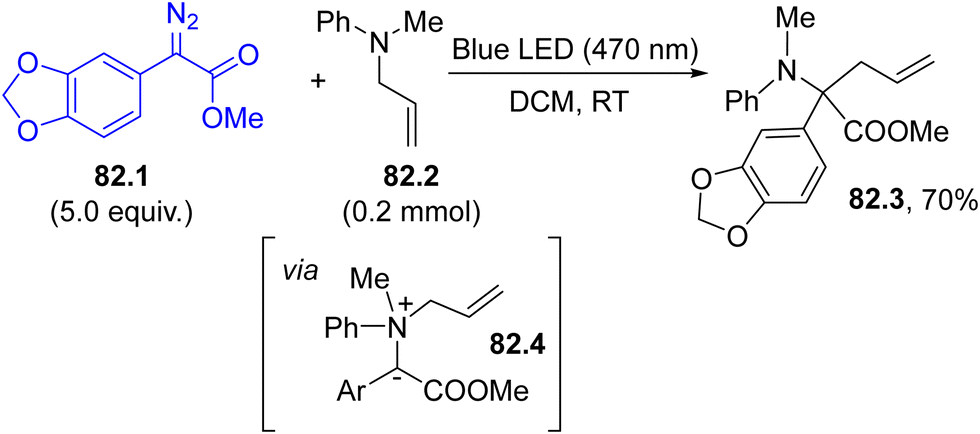 | ||
| Scheme 82 [2,3]-Sigmatropic shift induced by the addition of a photogenerated carbene onto allyl amines. | ||
The photodecomposition of aryl diazoacetates was adopted for the smooth preparation of nitrones. The carbene (obtained from 83.1 under Blue LED irradiation) here coupled with a nitrosoarene (83.2), leading to nitrone 83.3 again without the need for any additives and catalysts (Scheme 83). The protocol developed showed a good broad substrate scope and functional group tolerance for both reacting partners.286
 | ||
| Scheme 83 Photoinduced coupling between a diazocompound and a nitrosoarene. | ||
In rare instances, the formation of a C–N bond was promoted by a sulfur ylide formed upon addition of the carbene onto a disulfide. This step initiated a Cu-catalyzed asymmetric [4+1] cycloaddition with ethynyl benzoxazinones, resulting in the production of several chiral indolines bearing C2-quaternary stereocenters.287
6.4. C–O bond formation
The construction of a C–O bond may be achieved through the insertion of carbenes onto the O–H bond in water or other OH containing derivatives or by reaction with cyclic ethers (e.g., THF). α-Hydroxy and α-alkoxy esters or ketones are the core backbones of a wide range of biologically active compounds including, among the others, cefamandole and paclitaxel. In this frame, a metal-free strategy based on the O–H bond insertion by a photogenerated carbene (in turn obtained from diazoesters 84.1a–e) was described (Scheme 84). The protocol was optimized under both batch and continuous flow conditions. In particular, by using a mesoflow photoreactor, the product 84.2a was isolated on a multigram scale in 91% yield with a residence time of 4 h.288 Cyclic diazoamides have been also employed as photoactivatable substrates in the preparation of α-hydroxy and α-alkoxy amides by using (fluorinated) alcohols and phenols as oxygenated partners.289 Analogously, O-, S- and N-alkylated phenylacetate esters have been prepared in good to satisfactory yields by using 2-pyridones (phenols), 2-mercaptopyridine and anilines, respectively, as the coupling agents.290 | ||
| Scheme 84 Preparation of α-hydroxy esters via photogenerated carbenes. | ||
Two orthogonal approaches for the conversion of azirine-2-carboxylic acids 85.2a–c (Scheme 85) by using diazoesters have been recently reported. Indeed, blue light activation of diazoester 85.1 led to the O–H bond insertion of the generated carbene on 85.2a–c, producing derivatives 85.3a–c. Notably, in the presence of an Au(I) catalyst, the nitrogen atom in the azirine ring was the exclusive reacting site and a 1,3-oxazin-6-one ring was formed.291
 | ||
| Scheme 85 O-Functionalization of azirine-2-carboxylic acids. | ||
A divergent strategy for the coupling of the oxime 86.1 and an aryldiazo acetate 86.2 was developed by simply tuning the nature of the reaction media (Scheme 86). Indeed, in neat DCM, direct O–H insertion occurred, releasing the corresponding oxime ether 86.3. In contrast, when tetrahydrofuran or other oxygen-based heterocycles were used, incorporation of the ring opened form of the solvent into the final product 86.4 took place.292 The mechanism involved the initial formation of the ylide 86.5, followed by the nucleophilic induced ring opening of the five-membered ring and addition onto 86.1. The capability of carbenes to activate cyclic ethers was thoroughly investigated through a dual experimental and computational approach, which confirmed the formation of the oxonium ylide 86.5 as the key step.293,294
 | ||
| Scheme 86 Solvent dependent reactivity of carbenes with oxime 86.1. | ||
In this context, THF was intensively exploited for the development of a wide range of multicomponent processes, for the synthesis, among the others, of thiocyanates 87.2a–c (in the presence of elemental sulfur and trimethylsilyl cyanide, Scheme 87a),295S-alkyl dithiocarbamates,296 organophosphorus derivatives 87.4a–c (under aerated conditions in the presence of phosphine oxides as the coupling partner, Scheme 87b),297 polysubstituted nitrogen-based heterocycles298,299 and 1,3-diketones.298
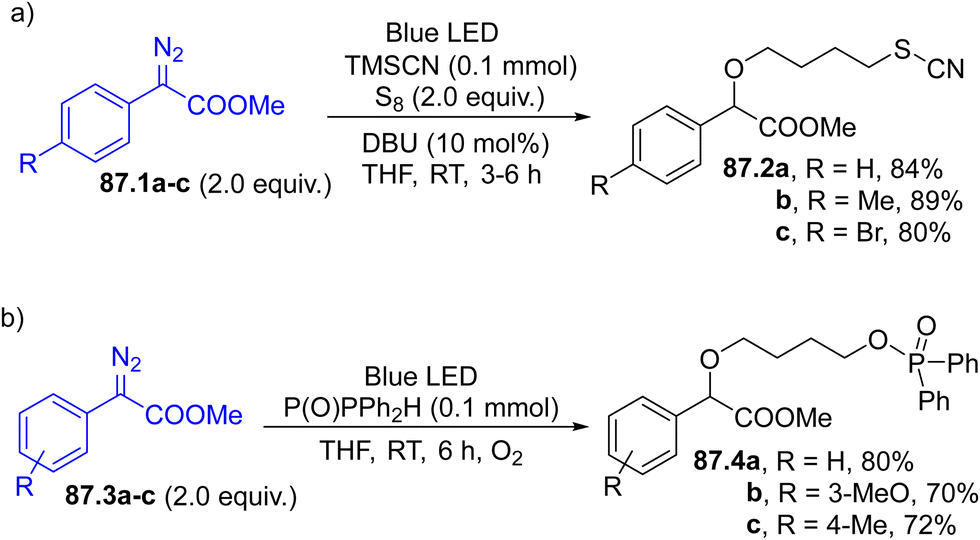 | ||
| Scheme 87 THF as the reaction partner in the carbene promoted C–O bond formation for the preparation of (a) thiocyanates and (b) organophosphorus derivatives. | ||
The formation of an ylide intermediate between a cyclic ether and a carbene was the key step of a ring expansion/contraction process. A versatile synthesis of oxacyclic spirooxindoles (88.3a–c, Scheme 88a) was described to occur from unstrained O-containing heterocycles (e.g. (substituted) tetrahydrofurans 88.2) with 3-diazoindolin-2-ones 88.1a–c. Mechanistic investigation highlighted that the initial formation of carbenes 88.4a–c (path a) and their ensuing addition to ether 88.2 resulted in the formation of ylides 88.5a–c (path b), which in turn underwent ring opening to form zwitterions 88.6a–c (path c), which upon ring closure (path d) afforded the desired spirocompounds.300 A ring expansion was also invoked for the preparation of substituted tetrahydrofurans 88.9a–c (Scheme 88b) or tetrahydrothiophenes starting from the corresponding oxetanes 88.8 or thietanes.301 The protocol was also performed on a gram scale on 3,4-dihydrofurans under continuous flow conditions. In this case, a ring contraction occurred leading to a vinyl oxetane.302 Recently, a carbene was reported to undergo addition to the oxygen atom of ethynylbenziodoxolones. As a result, a visible-light-promoted oxy-alkynylation took place, delivering propargylic esters via an oxonium ylide intermediate.303
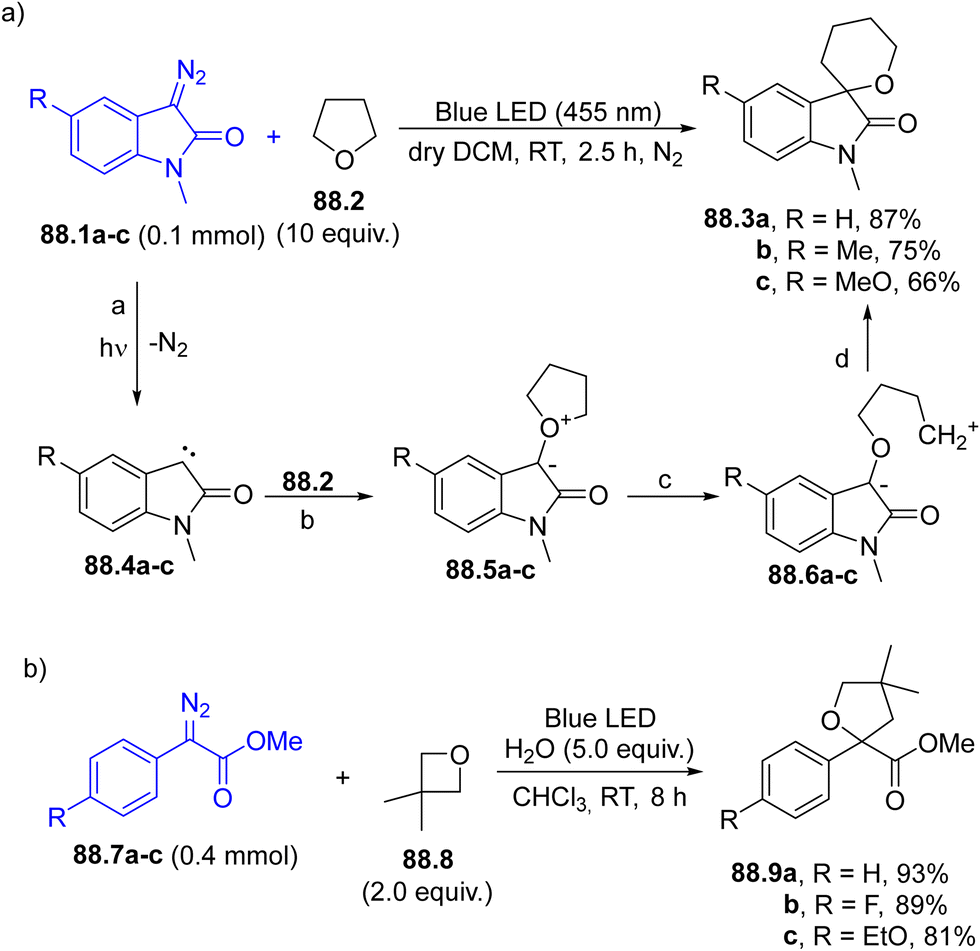 | ||
| Scheme 88 Synthesis of (a) oxacyclic spirooxindoles and (b) tetrahydrofurans via ring enlargement. | ||
An interesting case is the visible-light driven preparation of trisubstituted hydroxylamines (89.4a–d) via multicomponent coupling of an aryldiazo acetate (89.1) a 2-nitrosopyridine (89.2) and β-keto esters 89.3a–d (Scheme 89a). The reaction started with a thermal nitroso aldol reaction between 89.2 and 89.3a–d. The insertion of the carbene onto the O–H bond of the thus formed aldol adduct yielded the desired hydroxylamines 89.4a–d. When switching to THF as the solvent, a four-component reaction occurred in up to quantitative yield.304 A related photochemical three multicomponent carbene transfer reaction is described in Scheme 89b. Here, DBU promoted the deprotonation of the triazolium salt 89.7, releasing a N-heterocyclic carbene (NHC) catalyst, which engaged in a reaction with aldehydes 89.6a–c to form protected hydroxamic acids via a Breslow intermediate. Trapping of these acids with the carbene yielded differently substituted hydroxamic acid esters 89.8a–c in discrete yields. This approach has been successfully exploited in the preparation of modified natural and bioactive products including vitamin E, borneol and anesthetic propofol.305 The carbene formed from aryl diazoacetates was, however, able to attack the oxygen bond in aldehydes. The resulting carbonyl ylides engaged in a [3+2] cycloaddition with substituted maleimides, forming 4,6-dioxo-hexahydro-1H-furo[3,4-c] pyrrole in excellent yields.306
 | ||
| Scheme 89 Synthesis of substituted hydroxamic acid esters starting from (a) β-ketoesters (b) and benzaldehydes. | ||
The photogeneration of carbenes from aryldiazo acetate esters 90.1a–c in the presence of H-phosphine oxide 90.2 resulted in an oxyphosphorylation process that occurred under aerated conditions on a gram scale and in good to satisfactory yields (Scheme 90).307 The mechanism here involved the hydrogen abstraction operated by the carbenes 90.4a–c (paths a,b) from 90.2 and the resulting phosphinoyl radical 90.5 was trapped by dioxygen to form, after the decomposition of the resulting peroxy radical, phosphine-oxy radical 90.6 (path c) which then coupled with intermediates 90.7a–c (path d), furnishing the desired product.
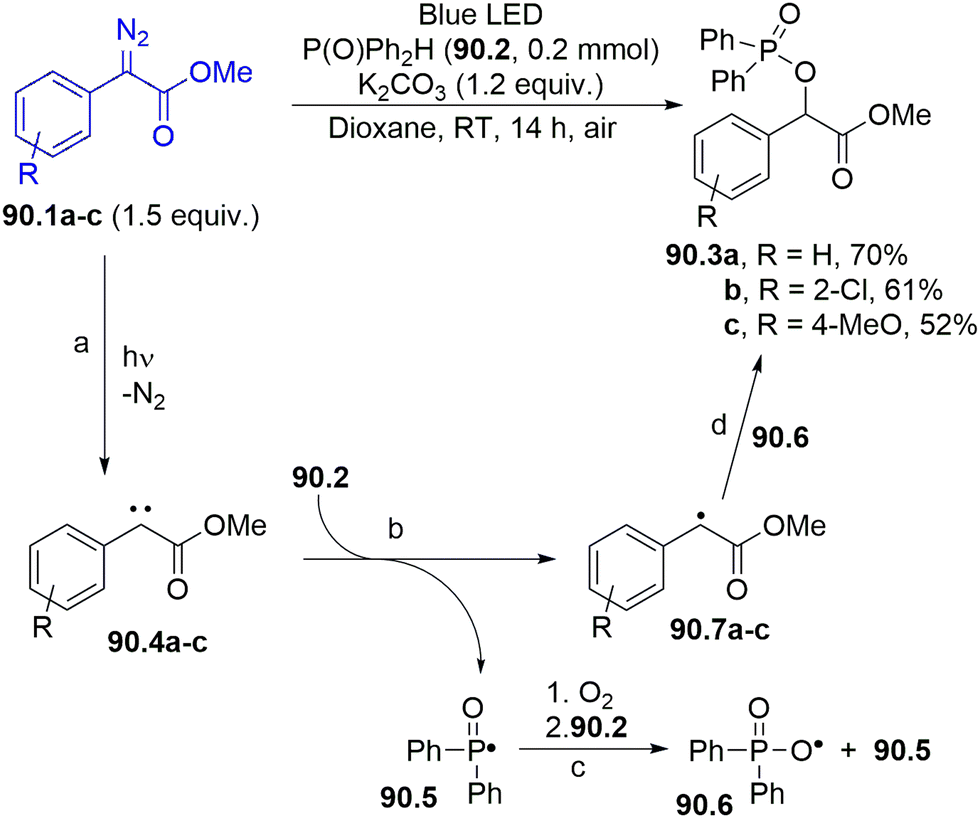 | ||
| Scheme 90 Visible-light mediated oxyphosphorylation of α-diazo esters. | ||
In one instance, the addition of the carbene onto a nitrosoarene led to the synthesis of α-ketoesters via initial addition onto the N atom of the NO group.308
6.5. C–S bond formation
Similar to C–O bond formation, the formation of a C–S bond occurred via a S–H insertion or a reaction deriving from a sulfur ylide obtained by carbene addition onto a sulfur atom. The S–H bond insertion reaction of photogenerated carbenes was exploited in the α-functionalization of esters by aromatic and aliphatic thiols,309 including substituted cysteines310 (91.2, an example in Scheme 91).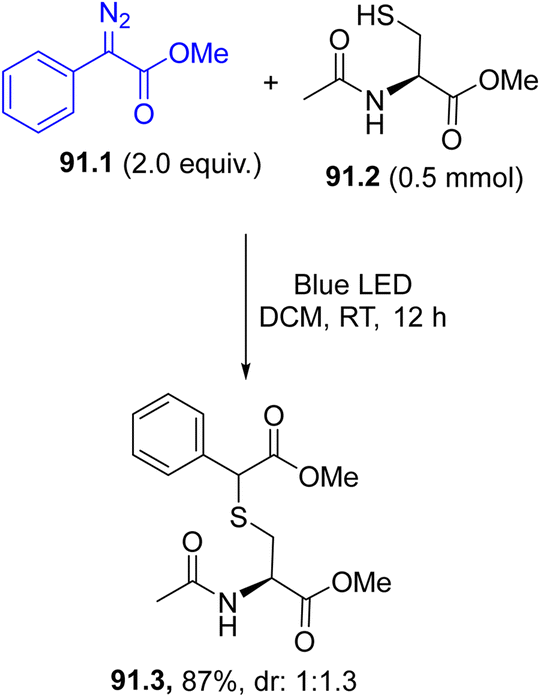 | ||
| Scheme 91 Visible-light construction of a C–S bond via a carbene S–H insertion. | ||
By adopting an approach analogous to those described in Section 6.4, S-alkyl phosphorothioates (92.2a–c, Scheme 92a) were obtained via a three-component coupling of α-diazoesters 92.1a–c, elemental sulfur and H-phosphonates in the presence of DBU as the additive.311 Similarly, a multicomponent reaction involving aryldiazoesters 92.3a–c, carbon disulfide and secondary amines was exploited for the preparation of alkyl dithiocarbamates 92.4a–c (Scheme 92b).312
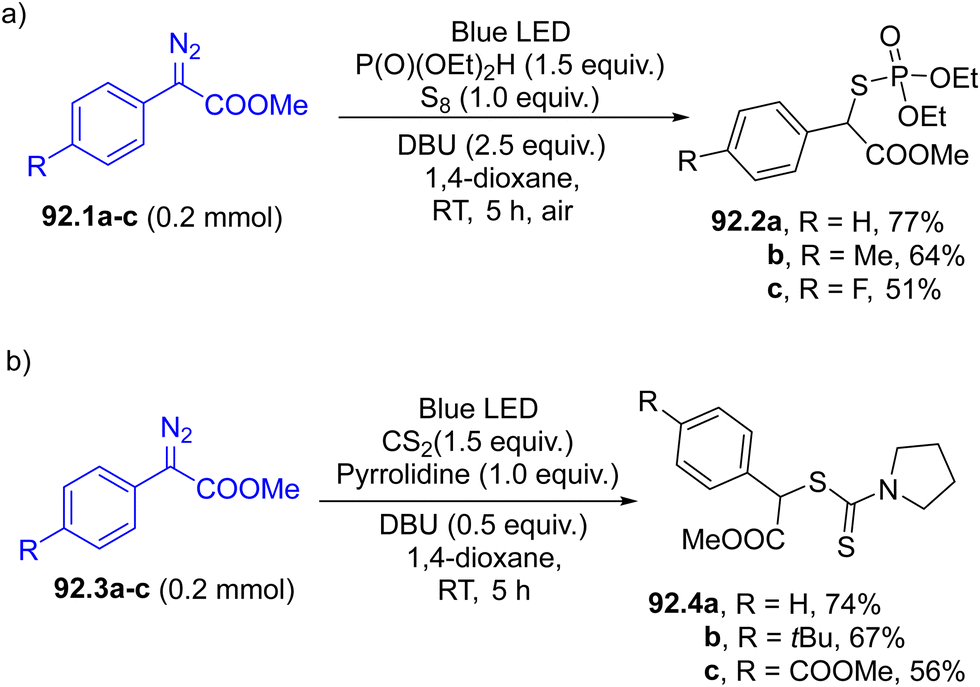 | ||
| Scheme 92 Synthesis of (a) S-alkyl phosphorothioates and (b) alkyl dithiocarbamates. | ||
A C–S bond may result from an initial addition of the carbene on the sulfur atom in sulfides, yielding an S-ylide, which in turn may react with a reaction partner or undergo rearrangement. An exemplary case belonging to the first class was the synthesis of a set of chiral indolines bearing C2-quaternary stereocenters with good enantio- and diastereo-selectivity. These compounds were formed via copper-catalyzed asymmetric [4+1] cycloadditions of ethynylbenzoxazinones with sulfur ylides; the sulfur ylides, in turn, were obtained via coupling between a photogenerated carbene and a sulfide.303
However, the rearrangement of the ylide is the most common case. As an example, upon irradiation of diazoesters 93.1a–c with visible-light and subsequent generation of singlet carbenes 93.3a–c (Scheme 93, path a) in the presence of allylsulfide 93.2, the formation of 3,3-difluoroallylated sulfonium ylides 93.4a–c (path b) was observed. The latter ylides then underwent a [2,3] sigmatropic shift (a Doyle-Kirmse reaction), forming gem-difluoroallyl esters 93.5a–c (path c).313 The same approach was recently applied for the synthesis of 4-allyl-4-(arylthio)-1,4-dihydroisoquinolin-3-ones.314 Notably, when 93.2 was replaced with propargylic sulfides, functionalized allenes were formed.315 In the case of N-sulfenyl phthalimide as the sulfonylated partner, a [1,2] sigmatropic rearrangement took place instead, simultaneously formimg a C–N and a C–S bond.316
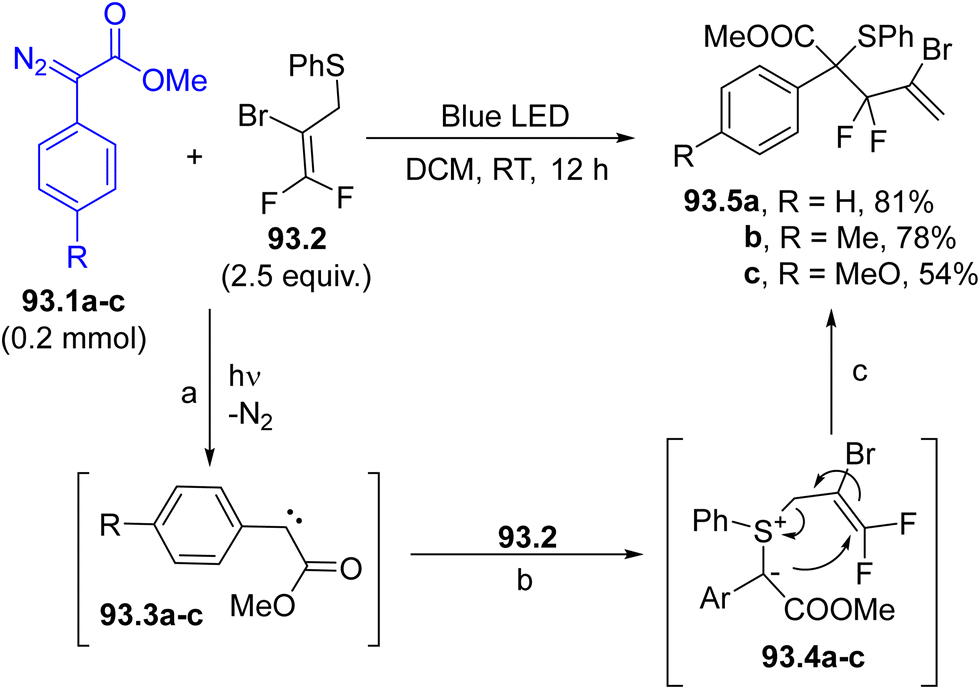 | ||
| Scheme 93 gem-Difluoroallyl esters from a photoinduced Doyle–Kirmse reaction. | ||
Photogenerated carbenes are also reactive towards CS bonds. In fact, upon irradiation of an α-diazo 1,3-diketone (94.1, Scheme 94) with visible-light in the presence of β-ketothioamides 94.2a–c yielded thiazoline derivatives 94.3a–cvia the intermediacy of the N,S-acetals 94.4a–c. The protocol was also performed on a gram scale.317
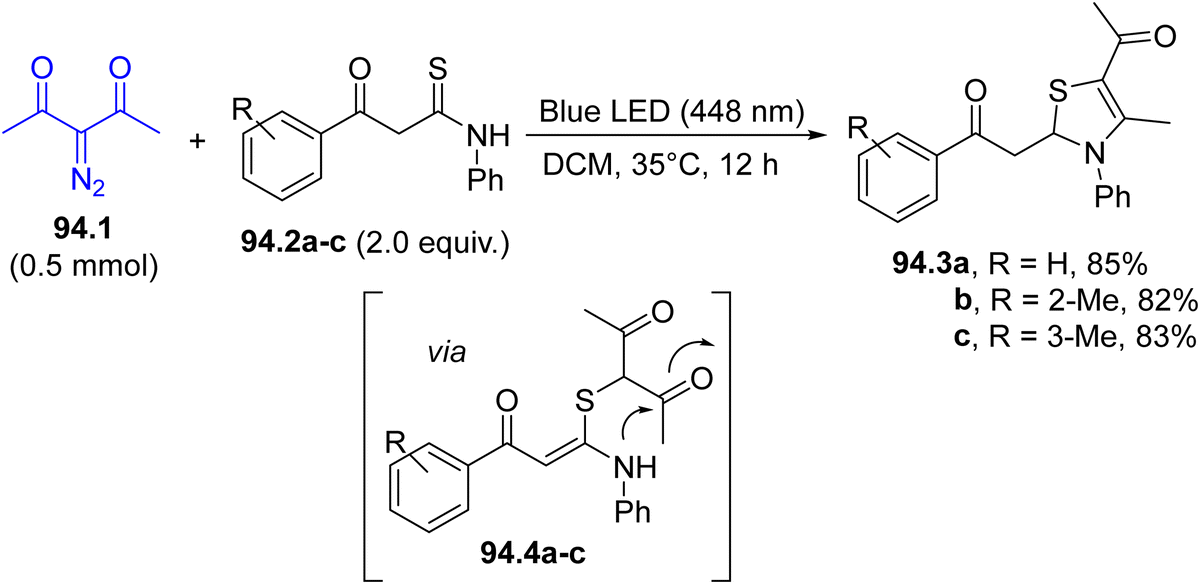 | ||
| Scheme 94 Visible-light promoted preparation of thiazolines. | ||
The visible-light-driven [1+5] annulation of phosphoryl diazomethylarenes (95.1a–d, Scheme 95) and pyridinium 1,4-thiolate 95.2 has been employed for the preparation, in good yields and excellent diasteroselectivity, of a wide range of trifunctionalized dialkyl 1-phosphoryl-1,9a-dihydropyrido[2,1-c][1,4]thiazine-3,4-dicarboxylates 95.3a–d.318 The heterocyclic product was obtained through the addition of the carbene onto the thiolate anion, resulting in zwitterions 95.4a–d where the pyridinium nucleus underwent nucleophilic attack on the CN bond.
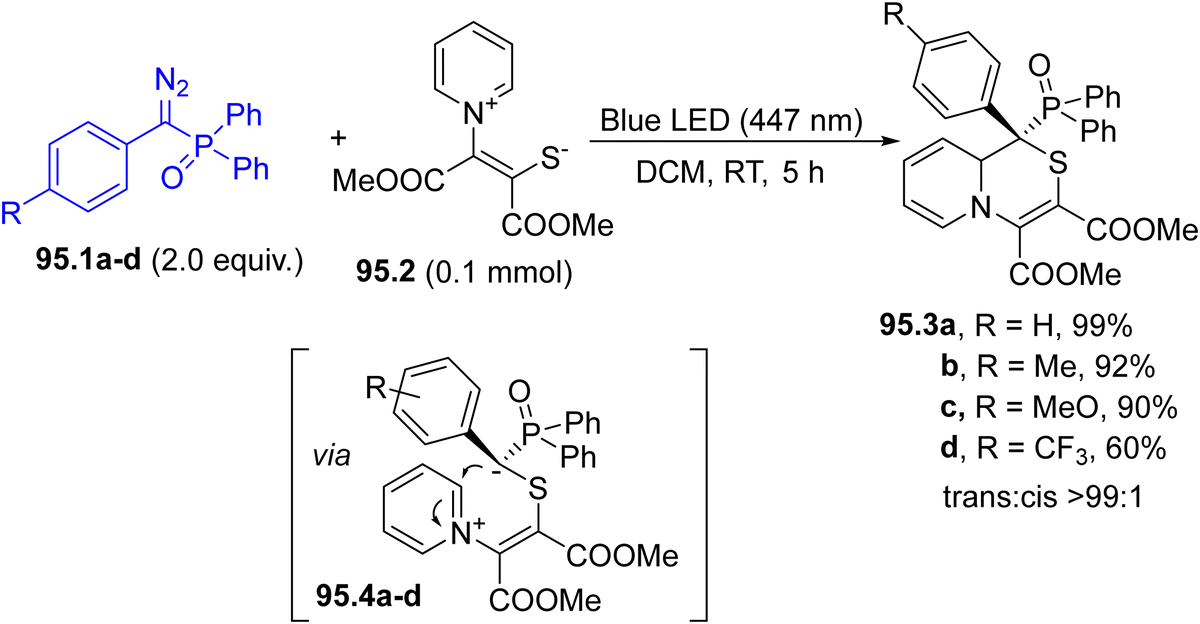 | ||
| Scheme 95 Dihydropyrido[2,1-c][1,4]thiazine derivatives via photogenerated carbenes. | ||
A particular case is the efficient visible-light mediated approach for the trifluoromethylsulfonylation of diazo compounds (96.1a–e, Scheme 96). Here the photogenerated carbene reacted with an intermediate formed between Mn(acac)3 (herein employed as the catalyst) and the rather unstable trifluoromethylsulfonyl radical formed through the Mn catalyzed oxidation of CF3SO2Na to yield 96.3a–e. Reduction to an anion followed by protonation enabled the attainment of the desired sulfonated products 96.2a–e, even on a gram scale.319
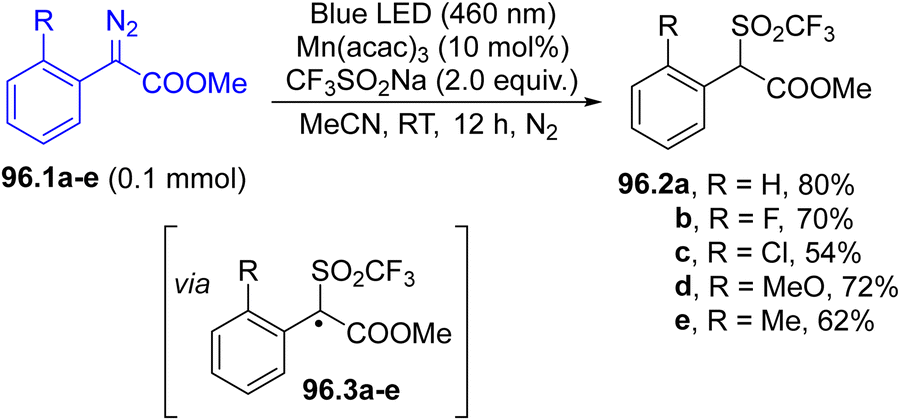 | ||
| Scheme 96 Visible-light mediated trifluoromethylsulfonylation of diazo compounds. | ||
6.6. Other C–X bond formation
In rare instances, the carbene formed from the photolysis of a coloured compound led to the formation of other C–X bonds. Very few examples described the trapping of a carbene intermediate by a boron containing derivative. Singlet siloxycarbenes arising from the irradiation of acylsilanes afforded α-alkoxyorganoboronate esters via insertion reactions within the B–H bond in HBpin derivatives.232 A B–H insertion took place likewise in the synthesis of 97.3, starting from aryldiazoacetate 97.1, and NHC-borane 97.2 (Scheme 97). However, when using N-heterocyclic carbene (NHC)-borane complexes, a site-selective cyclopropanation of the double bond of the heterocyclic ring occurred.320 | ||
| Scheme 97 B–H Insertion in NHC-boranes. | ||
A rare case of the formation of a C–Si bond was observed through the insertion of carbenes into Si–H bonds in trialkylsilanes, resulting in the preparation of α-trialkylsilyl esters 98.2a–c (Scheme 98).321
 | ||
| Scheme 98 Visible-light mediated synthesis of α-trialkylsilyl esters. | ||
More recently, acylsilanes (e.g., 99.1, Scheme 99) were employed in the preparation of α-hydroxyphosphorus oxides 99.3a–c in satisfactory yields by using H-phosphorus oxides 99.2a–c as the coupling partner. The suggested mechanism again highlighted the role of a photogenerated siloxycarbene 99.4 as the key intermediate.322
 | ||
| Scheme 99 C–P bond formation in the synthesis of α-hydroxyphosphorus oxides. | ||
6.7. Via ketenes
When coloured α-diazo ketones are used in place of the corresponding esters, a new chemistry appears due to the conversion of the photogenerated α-ketocarbenes (upon nitrogen loss) into electrophilic ketenes via Wolff rearrangement.323 One of the possible pathways for the ketenes (e.g., 100.3) is the nucleophile addition, as exemplified in Scheme 100. Thus, differently substituted 1H-indene-3-carboxylates (100.2a–d) were produced in discrete to good yields via visible-light mediated Wolff rearrangement of 1-diazonaphthalen-2(1H)-one 100.1 in the presence of different alcohols as nucleophilic additives.324 | ||
| Scheme 100 Alcohols as nucleophiles in the synthesis of 1H-indene-3-carboxylates. | ||
Apart from alcohols, even phenols (e.g., 101.2) may be used to obtain esters with the help of a benzotetramisole-type chiral catalyst such as (S)-101.3, acting as the nucleophilic catalyst (Scheme 101). Thus, the ketene formed through the visible-light irradiation of α-diazoketone 101.1 was trapped by (S)-101.3, forming ammonium enolate 101.4, which upon protonation and nucleophilic attack of the resulting phenate yielded α,α-disubstituted carboxylic ester 101.5 with a good enantiomeric ratio, which on subsequent hydrolysis yielded (R)-ibuprofen 101.6 in 90% yield.325
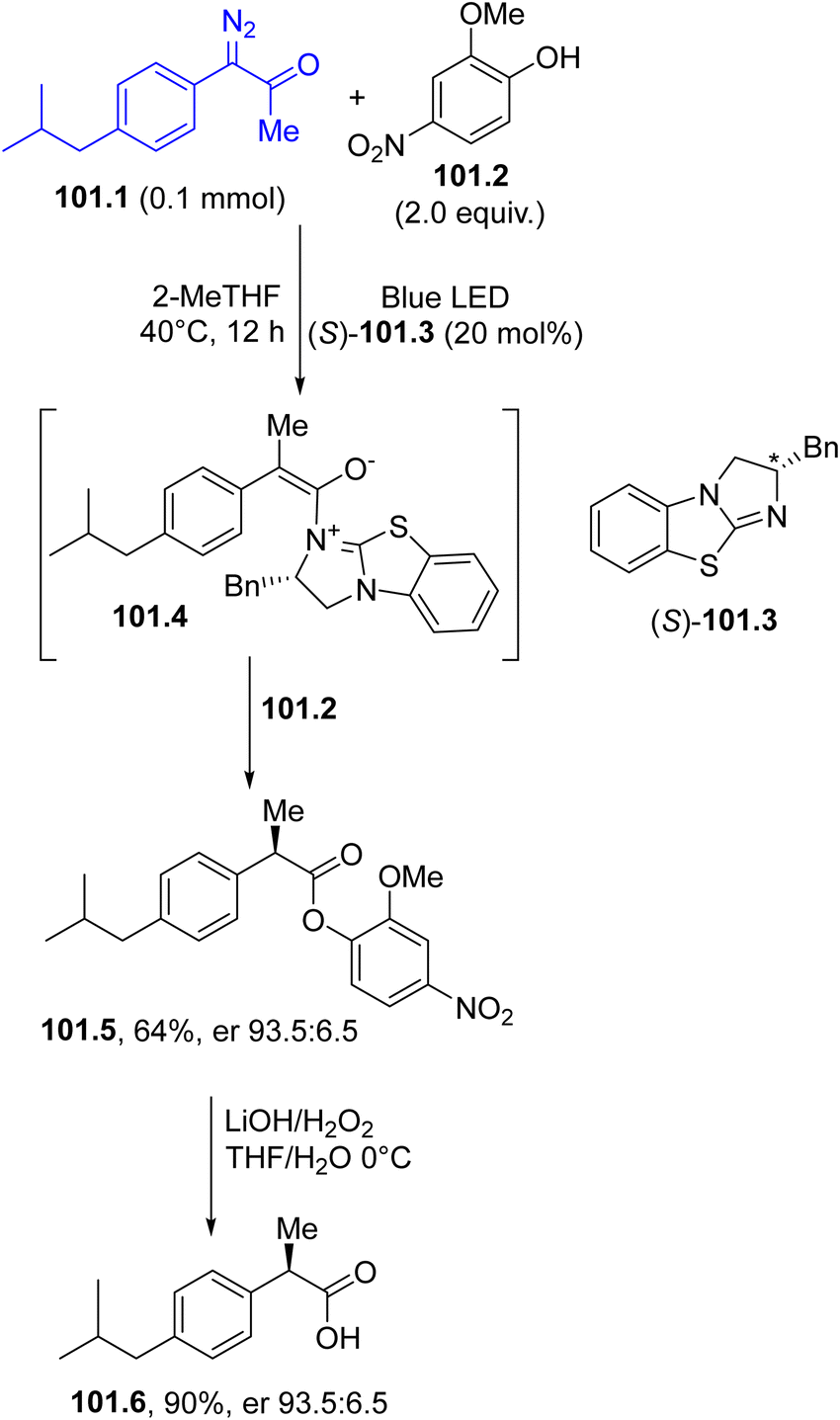 | ||
| Scheme 101 Photochemical synthesis of (R)-ibuprofen. | ||
A similar approach was exploited in the enantioselective preparation of α-chlorinated carboxylic acid esters from α-diazoketones by reaction with N-chloro succinimide (NCS) as the electrophilic chlorine source mediated by an isothiourea-based chiral catalyst.326 The nucleophile can be a primary amine and the presence of a chiral phosphoric acid in the double role of accelerating the ketene capture by amines and promoting an enantioselective proton-transfer led to the formation of an enantioenriched chiral amide.327
1,3-Dicarbonyl sulfoxonium ylides 102.3a–c were prepared via purple light irradiation of azoketone 102.1 in the presence of sulfoxonium ylides 102.2a–c as the nucleophiles (Scheme 102).328,329 The process relied on the nucleophilic addition of the ylide onto the photogenerated ketene to give intermediates 102.4a–c that ultimately led to the formation of compounds 102.3a–c upon proton transfer. Notably, the process has been also optimized under continuous flow conditions, allowing for a decrease in the reaction time (tr = 28 min) and an improvement of the overall yields.328
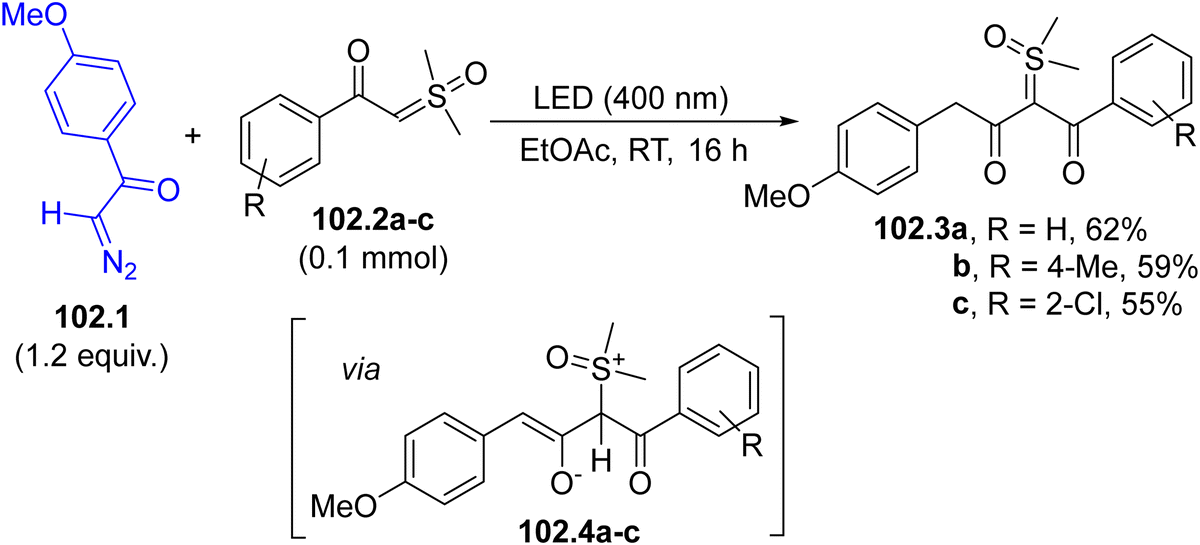 | ||
| Scheme 102 Synthesis of 1,3-dicarbonyl sulfoxonium ylides via photogenerated ketenes. | ||
Again, an ammonium enolate intermediate was the key compound for the synthesis of 3,4-dihydrobenzofuro[3,2-b]pyridin-2(1H)-ones 103.4a–c (Scheme 103).330 These heterocycles were obtained via [4+2] annulation of a photogenerated ketene 103.5 with the aurone derived α,β-unsaturated imines 103.2a–c in the presence of a chiral isothiourea 103.3 as the catalyst.330 In detail, ketene 103.5 (from path a) coupled with the chiral catalyst 103.3 to form ammonium enolate 103.6 (path b), which in turn acted as a nucleophile towards the Michael acceptors 103.2a–c (path c). The resulting intermediates 103.7a–c then underwent intramolecular amidation, yielding the desired products 103.4a–c. An analogous [4+2] cyclization was employed in the preparation of substituted benzothiazolopyrimidines from 2-benzothiazolimines and α-diazo ketones.331
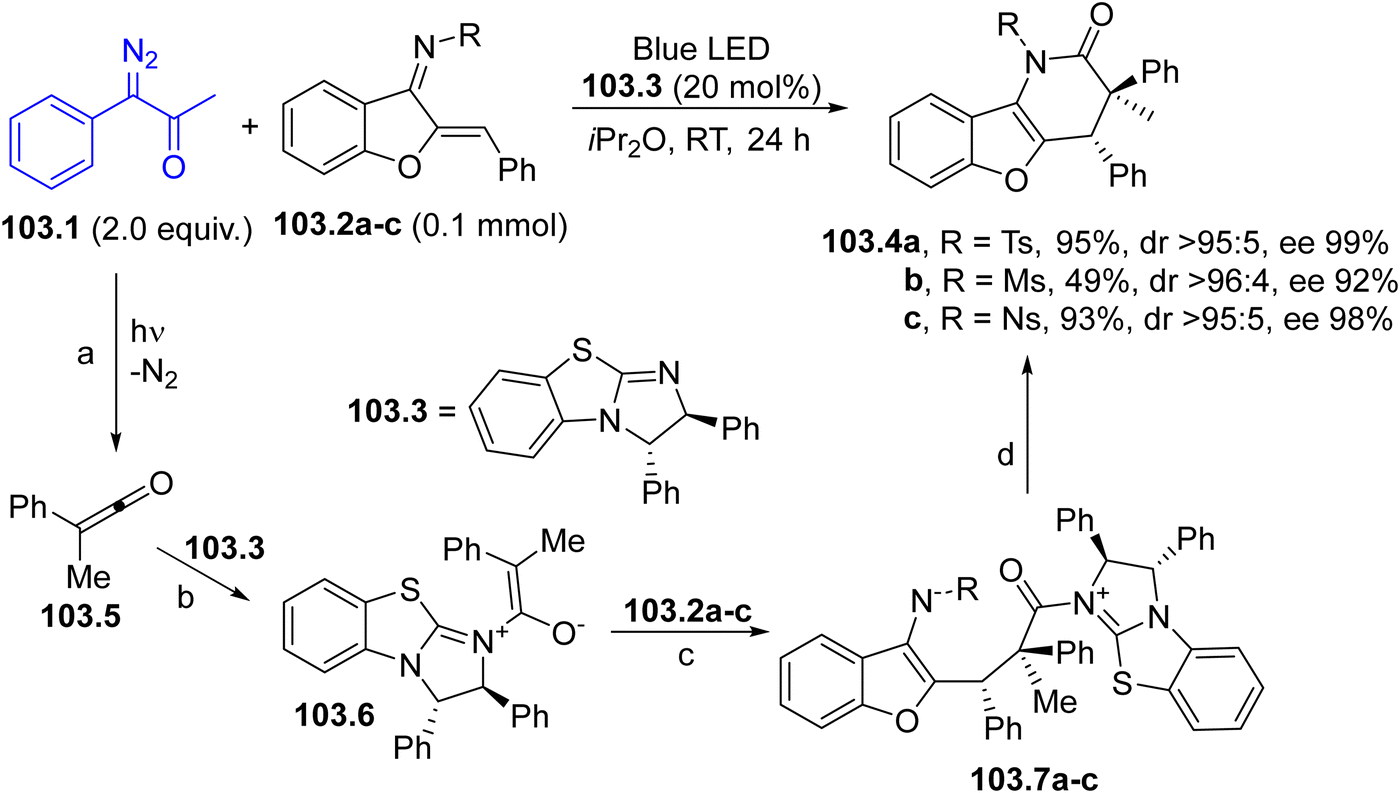 | ||
| Scheme 103 Enantioselective preparation of 4-dihydrobenzofuro[3,2-b]pyridin-2(1H)-ones. | ||
Moreover, a chiral NHC may function as an organocatalyst for the stereoselective construction of a six-membered ring between 3-alkylenyloxindole 104.2 and α-diazoketone 104.1 under visible-light irradiation (Scheme 104). Thus, triazolium salt 104.3 may release the corresponding NHC under basic conditions and the consecutive addition of the ketene and 104.2 enabled the enantio and stereoselective preparation of tetrahydropyrano[2,3-b]indoles (e.g., 104.4) bearing an all-carbon quaternary stereocenter.332
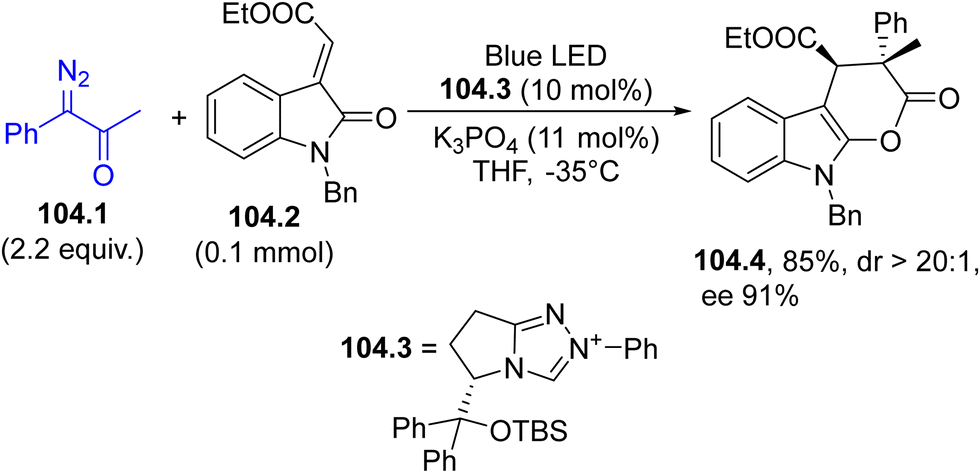 | ||
| Scheme 104 NHC catalyzed preparation of tetrahydropyrano[2,3-b]indoles. | ||
In a similar fashion, the reaction of ketene with sultam-fused dihydropyridinones has been performed via dual light driven/NHC catalyzed asymmetric [4+2] annulation of saccharine-derived azadienes and α-diazoketones.333
Photogenerated ketenes have also been efficiently employed as a partner in cycloaddition reactions.
Scheme 105 illustrates an example where this approach was included in a multicomponent process. Thus, the ketene 3-component Staudinger reaction (K-3CSR) for the synthesis of β-lactams 105.4a–c was devised starting from α-diazoketones 105.1a–c, a primary amine 105.2 and an aldehyde 105.3. The first step of the reaction (the in situ formation of the imine intermediate) proceeded in dark, whereas the generation of the ketene (via Wolff rearrangement of 105.1a–c) occurred under visible-light irradiation.334 Analogously, a sequential Wolff rearrangement of diazoketones and Staudinger cycloaddition of the photogenerated ketenes onto pyrazolone ketimines led to the formation of spiro-pyrazolone-β-lactams in more than 90% yield in several cases.335
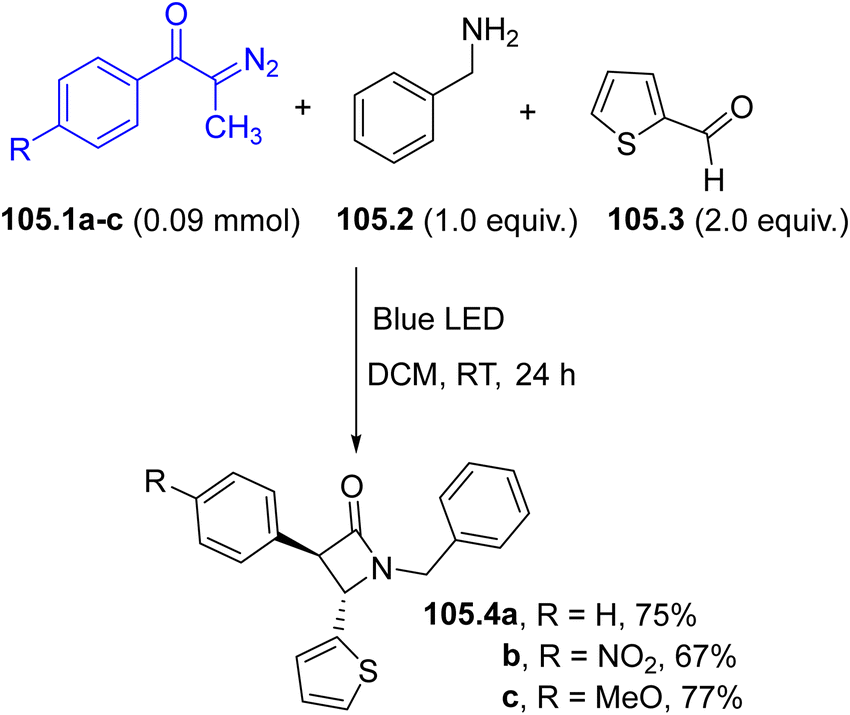 | ||
| Scheme 105 Visible-light promoted ketene 3-component Staudinger reaction. | ||
A powerful [4+2] annulation strategy was adopted for the construction of dihydroquinolinone derivatives. The reaction took place through visible-light irradiation of a mixture of an α-diazoketone and ortho-amino Morita–Baylis–Hillman (MBH) carbonate in the presence of DMAP as the Lewis base catalyst. An initial SN2′ reaction promoted by the addition of the base onto the unsaturated system of the MHB-carbonate was followed by ketene addition and an intramolecular ring-closure.336
Alternative kinds of cycloaddition reactions may be promoted by Pd catalysis. The Pd-catalyzed [3+2] cycloaddition of ketenes with vinyl cyclopropanes 106.2 in the presence of a hybrid P,S-ligand 106.3 was found to yield a wide range of substituted tetrahydrofurans 106.4a–cvia a tandem C–C/C–O bond formation (Scheme 106). The role of Pd(0) here was to convert the vinyl cyclopropane into a reactive ring opened Pd-containing 1,3-dipolar intermediate.337 Similarly an enantioselective [8+2] cycloaddition was carried out between a ketene and a Pd-containing 1,8-dipole (in turn generated from a vinyl carbamate derivative) to afford a 10-membered nitrogen-based heterocycle.338
 | ||
| Scheme 106 Visible-light mediated, Pd-catalyzed preparation of tetrahydrofurans. | ||
7. Reactions via other intermediates
7.1. Biradicals
Typical reactions belonging to this class are Norrish-type I or II reactions. Thiones underwent an intramolecular hydrogen atom transfer reaction when excited with visible-light.52 As shown in Scheme 107, irradiation of β-substituted arylalkyl thione 107.1 in benzene triggered a Norrish-type II reaction forming the 1,3-biradical 107.2, which upon radical recombination afforded the cyclopropyl thiol 107.3 in a good yield.339,340 | ||
| Scheme 107 Intramolecular hydrogen transfer in thioketones. | ||
α-Diketones are well-known substrates for visible-light-induced photochemical reactions. As an example, the irradiation of syn-9,10-epoxy-1,4-dihydro-1,4-dipropyl-1,4-ethanonaphthalene-2,3-dione (syn-108.1) resulted in the efficient formation of 1,4-di-n-propylnaphthalene (108.5, Scheme 108). In the suggested mechanism, a Norrish-type I homolytic cleavage led to the formation of biradical 108.2. This biradical, in turn, upon losing carbon monoxide, led to the formation of intermediate 108.3, which upon radical recombination and fast carbon dioxide loss from lactone 108.4, produced naphthalene 108.5 in more than 90% yield.341
 | ||
| Scheme 108 Norrish-type I cleavage in α-diketones. | ||
A visible-light triggered Norrish-type II intramolecular hydrogen abstraction reaction on 1,2-diketones has also been reported. As a matter of fact, the exposure of compound 109.1 to a daylight lamp prompted the hydrogen abstraction from one of the two carbonyls groups, targeting the hydrogen located adjacent to the oxygen atom of the ring (Scheme 109). The so obtained biradical 109.2 fragmentated, releasing Z photoenol 109.3 which upon heating underwent an intramolecular aldol reaction affording the final product 109.4.342,343 This approach was a simple route to achieve the synthesis of densely functionalized cyclopentitols starting from pyranoses exhibiting a surprisingly diastereoselectivity.
 | ||
| Scheme 109 Synthesis of functionalized cyclopentitols. | ||
When the structure of the starting carbohydrate containing a 1,2-diketone moiety was converted to 1-glycosyl-2,3-butanodiones, the occurrence of the Norrish-type II reaction led to a biradical that cyclized, accessing chiral 1-hydroxy-1-methyl-5-oxaspiro[3.5]nonan-2-ones (bearing the 1-hydroxy-1-methyl-2-cyclobutanone moiety).344 A Norrish–Yang cyclization reaction was instead proposed as the strategy to prepare a potent inhibitor of the 20S proteasome, (+)-lactacystin, starting from a tetrahydro-3H,5H-pyrrolo[1,2-c]oxazol-5-one.345 The photochemically induced cyclization of 1,2-ketones for the preparation of functionalized 2-hydroxycyclobutanones was likewise performed with blue LED radiation under flow conditions.346
The photoreactivity of 1,2-diketones has also been exploited for the release of smaller chemical entities, which can be used in situ for other chemical transformations. For instance, the visible-light exposure of 2-methyl-1,6-dihydro-3a,6-ethanoisoindole-3,8,9(2H)-trione 110.1 resulted in the liberation of two equivalents of carbon monoxide along with the formation of lactam 110.2 (Scheme 110a). Thus, the CO liberated in the process was successfully combined with palladium catalysis for the coupling of non-activated alkyl iodides 110.3 with amines 110.4, yielding amides 110.5 (Scheme 110b).347
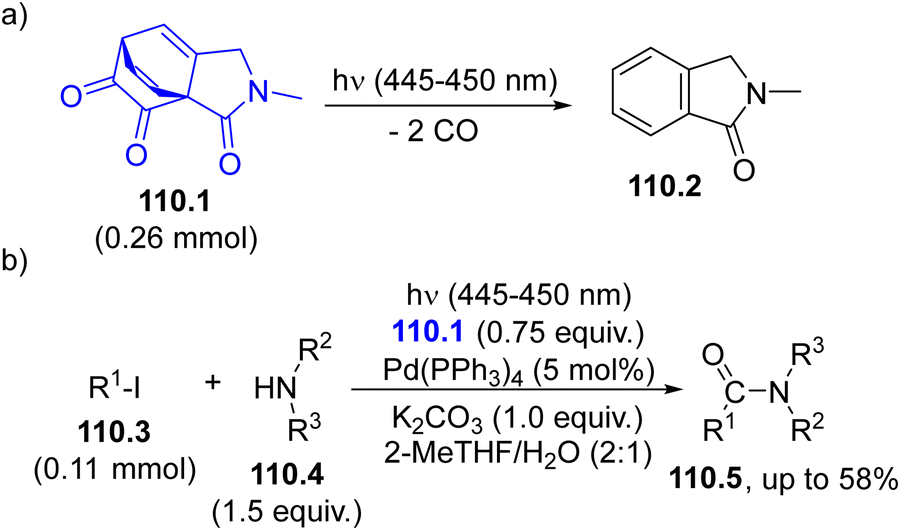 | ||
| Scheme 110 Visible-light fragmentation of diketone 110.1 for (a) the carbon monoxide release to be used in (b) the synthesis of amides. | ||
α-Ketoamides (see, for instance, 1-phenyl-2-(piperidin-1-yl)ethane-1,2-dione 111.1 in Scheme 111) upon photolysis with blue light underwent a Norrish–Yang type II reaction, giving access to α-hydroxy-β-lactam 111.3via biradical 111.2.348 The reaction was performed on a film of the starting compound 111.1 applied to the glass surface of a 20 mL vial, under solvent-free conditions.
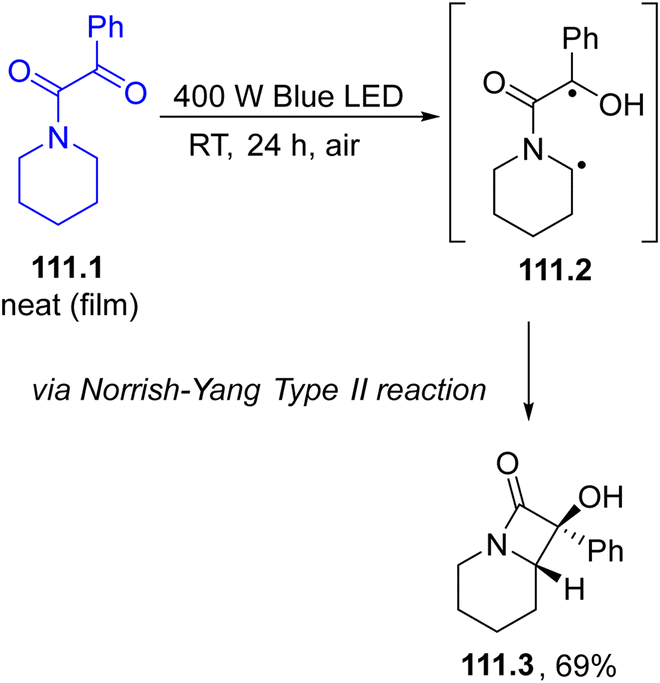 | ||
| Scheme 111 Norrish–Yang type II reaction of α-ketoamides. | ||
Recently, a peculiar aspect of the pyrazolo[1,2-a]pyrazolones 112.1 photoreactivity has been highlighted (Scheme 112a). Under exposure to visible-light in neat DCM, these compounds experience the homolytic cleavage of the C5–N4 and/or C1–N8 bonds, furnishing substituted pirazoles 112.2 or diazepines 112.3, respectively. Remarkably, a selective photochemical ring-opening process could be achieved using different additives. For instance, when pyrazolo[1,2-a]pyrazoles were irradiated in the presence of diethyl bromomalonate and 2,6-lutidine yielded after C7–N8 cleavage N-acryloyl-substituted pyrazoles 112.4 (Scheme 112b). On the other hand, shifting to other nucleophilic species (e.g., alcohols and amines), induced the photochemical C5–N4 photocleavage, resulting in the formation of functionalized pyrazoles 112.5 (Scheme 112b).349
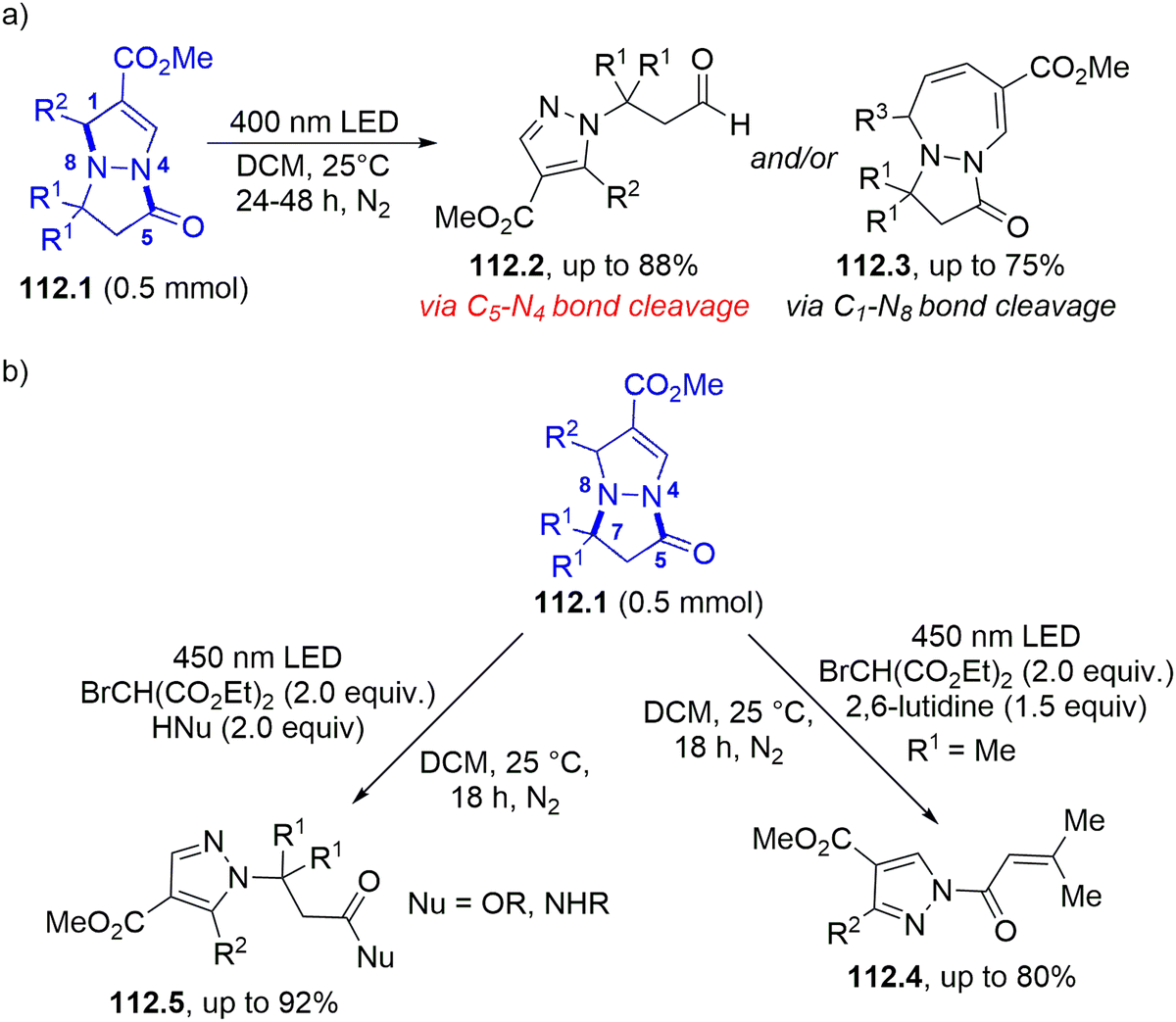 | ||
| Scheme 112 Photochemical reaction of pyrazolo[1,2-a]pyrazolones. | ||
The latter example deals with the use of a nitroarene as oxygen transfer. Excitation of 113.1 populates the triplet nπ* state, allowing it to readily add to an electron-rich CC bond in styrenes 113.2a–c, yielding a biradical intermediate that smoothly cyclized to the short-lived 1,3,2-dioxazolidine intermediates 113.3a–c that underwent fragmentation to the corresponding carbonyl products 113.4a–c (Scheme 113). This nonstereospecific radical cycloaddition of nitroarenes onto an alkene was the key for a mild alternative oxidative cleavage protocol, such as ozonolysis.350 A related oxidative cleavage made use of 3,5-dinitrobenzotrifluoride as the oxidant.351
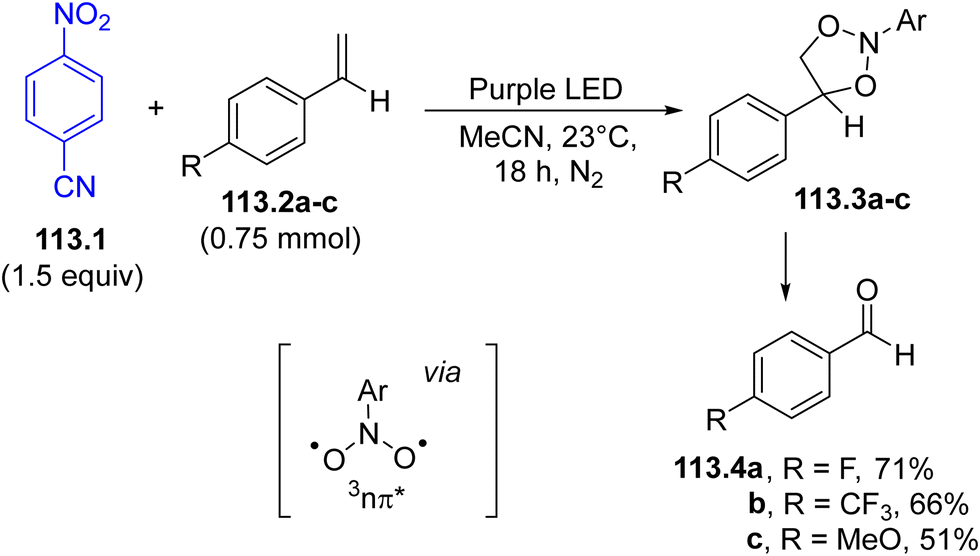 | ||
| Scheme 113 Oxidative cleavage of alkenes promoted by excited nitroarenes. | ||
7.2. Radical ions and ions
Cyanoarenes were known for having a good reactivity towards electron donors352 under visible-light irradiation promoting a photoaddition/photosubstitution on the aromatic ring.353 For example, the reaction between 114.1 and the α-silyl amine 114.2 was fueled by an initial electron transfer; the subsequent loss of the trimethylsilyl cation from the radical cation 114.2˙+ followed by radical–radical anion recombination yielded dinitrile 114.3 (Scheme 114). In some cases, the substitution occurred on the ipso position upon cyanide anion loss.354 | ||
| Scheme 114 Visible-light functionalization of cyanoarenes. | ||
Visible-light was successfully applied in the arylation of substituted arenaz-2-(2-nitro)-propanes 115.1. These compounds in aqueous HCl are able to absorb purple light, promoting the nitrogen loss and releasing a singlet aryl cation 115.2 which was immediately trapped by a variety of nucleophiles NuH (Scheme 115). The cation 115.2 was stabilized by the in situ photochemically generated counter ion 115.3, avoiding any intersystem crossing or single electron transfer processes. This approach was a good strategy for the formation of C(sp2)–X bonds, allowing for the synthesis of compounds 115.4.355
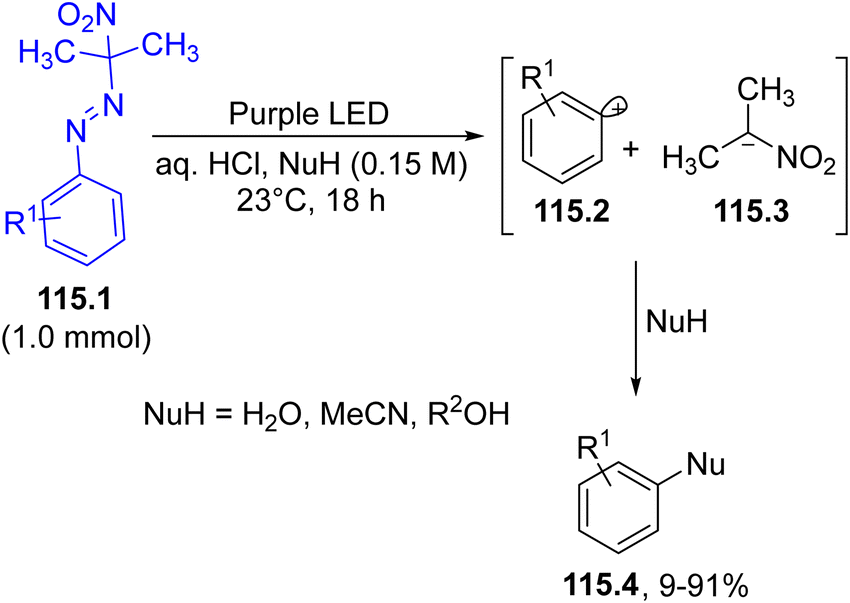 | ||
| Scheme 115 Photochemically induced synthesis of C(sp2)–X bonds. | ||
8. Conclusions and outlook
Although most organic compounds are not able to absorb in the visible-light region, the use of an expensive high-energy UV-photons source,13 has been mainly avoided by utilizing photocatalytic strategies that employ low-energy demanding LED lamps covering the entire visible-light region (Scheme 1a). Despite its fast development, this strategy (still the main used) is often hindered by the cost of the (metal-based) photocatalyst used, and a rather limited number of approaches for the activation of the starting substrates.In this context, the conversion of a photoreactive coloured compound into the desired product is the preferred option for exploiting visible photons in synthesis, thus behaving as the ideal promoting reagents, and several classes of organic molecules can be photochemically activated by visible-light-photons without the need for promoters or catalysts. These compounds can absorb coloured radiation related to a specific transition (π → π*, n → π* or n → σ*) due to a given chromophore (in 1,2-diketones, diazo compounds, dichalcogenides, etc.). However, the application of these compounds in synthetic strategies remains quite limited, and, apart from their use in carbene generation from diazo compounds,37 there has been little advancement in the last 10–15 years. This demonstrated that there is still a hard work to do for the satisfying exploitation of the photoconversion of known classes or coloured organic compounds following the strategy depicted in Scheme 1g.
As an example, nitroaromatics (apart those having benzylic hydrogens in the ortho position) were always considered photochemically unreactive and unimportant in synthesis due to the short lifetime of their excited states.356 However, these arenes are recently becoming interesting substrates for the photogeneration of nitrenes by tuning the right reaction conditions.224,225 Moreover, due to the recent development of visible LEDs technology it is possible to carry out reactions on coloured compounds, previously promoted by multichromatic sources which mostly emit in the UV region. The easy accessibility to these sources allowed for the discovery of new processes on known chromophores.
Along with this traditional and re-discovered approach, alternative strategies may be adopted for the in situ formation of the coloured species or the installation of a new devised photoreactive chromophore by a functional group interconversion on colourless derivatives. The latter case belongs to the dyedauxiliary groups strategy35 (Scheme 1f) as in the cases of arylazo sulfones, dihydropyridines and Barton esters. The use of such compounds in organic synthesis can take advantage of their visible-light photoactivity since they generate reactive intermediates without the need for harsh conditions (transition metal chemistry, high temperatures, strong bases, etc.). In doing so, these kinds of processes become appealing from the industrial point of view due to the low operational cost, leaving open the possibility of using known dyedauxiliary groups for the release of new intermediates (e.g., carbon or heteroatom-based radicals). Alternatively, the development of of new dyedauxiliary groups may open up fascinating new routes for the photogeneration of reactive intermediates, although it would require a chemical step to install the photoreactive chromophore. However, this strategy, together with the largely unexplored in situ chromophore activation (Scheme 1b and c) or the nucleophilic activation (Scheme 1e) could represent a significant advancement in the near future, paving the way for a new era for visible photons based on a photocatalyst-free chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from UniPV and MUR through the program ‘Departments of Excellence’ (2023–2027). SP is grateful to the Italian Ministry for Universities and Research, PRIN-PNRR, “Xilonite” project P2022HSF3R for financial support. MF acknowledges support from the project PRIN PNRR “LIGHT CAT” P2022RHMCM supported by the European Commission – NextGeneration EU program – M4C2. LN is grateful to the support of the IDEX Paris-Saclay, “ADI 2022” ANR-11-IDEX-0003-02 for the doctoral fellowships. LDT acknowledges support from the European Union’s Life Programme under grant agreement No 101074164 (CROSS-LIFE – CROtonic acid from Sewage Sludge).References
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS.
- M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed.
- M. Di Filippo, C. Bracken and M. Baumann, Molecules, 2020, 25, 356 CrossRef CAS PubMed.
- S. Protti, M. Fagnoni and A. Albini, in Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue, John Wiley & Sons, Chichester, 2nd edn, 2018, pp. 373–406 Search PubMed.
- A. Albini and M. Fagnoni, ChemSusChem, 2008, 1, 63–66 CrossRef CAS PubMed.
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1–6 RSC.
- N. Hoffmann, Photochem. Photobiol. Sci., 2012, 11, 1613–1641 CrossRef CAS.
- A. Albini and M. Fagnoni, in New methodologies and Techniques for a Sustainable Organic Chemistry, ed. A. Mordini and F. Faigl, Springer Science + Business Media B.V., 2008, pp. 279–293 Search PubMed.
- A. Albini and M. Fagnoni, in Green Chemical Reactions, ed. P. Tundo and V. Esposito, Springer Science + Business Media B.V., 2008, pp. 173–189 Search PubMed.
- S. Protti, S. Manzini, M. Fagnoni and A. Albini, in Eco-friendly synthesis of fine chemicals, RSC green chemistry book series, ed. R. Ballini, Royal society of chemistry, 2009, pp. 80–111 Search PubMed.
- A. Albini and L. Germani, Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH Verlag, Weinheim, 2010, pp. 1–24 Search PubMed.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- Chemical Photocatalysis, ed. B. Koenig, De Gruyter, Berlin, Germany, 2013 Search PubMed.
- Visible Light Photocatalysis in Organic Chemistry, ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley-VCH, Weinheim, Germany, 2018 Search PubMed.
- Photoorganocatalysis in organic synthesis, ed. M. Fagnoni, P. Protti and D. Ravelli, World Scientific Publishing Europe, Ltd, Singapore, 2019 Search PubMed.
- Green Photocatalysts. Environmental Chemistry for a Sustainable World, ed. M. Naushad, S. Rajendran and E. Lichtfouse, Springer Nature Switzerland AG, 2020, vol. 34 Search PubMed.
- R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766–897 RSC.
- A. Albini and M. Fagnoni, Photochemically-generated intermediates in synthesis, John Wiley & Sons, Hoboken, 2013, pp. 1–40 Search PubMed.
- S. Protti, D. Ravelli and M. Fagnoni, in Enabling Tools and Techniques for Organic Synthesis: A Practical Guide to Experimentation, Automation, and Computation, ed. S. G. Newman, John Wiley & Sons, Hoboken, NJ, 2023, pp. 37–72 Search PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338–14349 CrossRef CAS PubMed.
- C. Brenninger and T. Bach, Top. Catal., 2018, 61, 623–629 CrossRef CAS PubMed.
- C. G. S. Lima, T. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- Y.-Q. Yuan, S. Majumder, M.-H. Yang and S.-R. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS.
- H. F. Piedra, C. Valdés and M. Plaza, Chem. Sci., 2023, 14, 5545–5568 RSC.
- B. Saxena, R. I. Patel and A. Sharma, Adv. Synth. Catal., 2023, 365, 1538–1564 CrossRef CAS.
- A. K. Wortman and C. R. J. Stephenson, Chem, 2023, 9, 2390–2415 CAS.
- B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–138 CrossRef CAS PubMed.
- D. Qiu, C. Lian, J. Mao, M. Fagnoni and S. Protti, J. Org. Chem., 2020, 85, 12813–12822 CrossRef CAS PubMed.
- R. Chawla, A. K. Singh and P. K. Dutta, Org. Biomol. Chem., 2024, 22, 869–893 RSC.
- Y. Guo, C. Empel, C. Pei, I. Atodiresei, T. Fallon and R. M. Koenigs, Org. Lett., 2020, 22, 5126–5130 CrossRef CAS PubMed.
- D. Beaudoin and J. D. Wuest, Chem. Rev., 2016, 116, 258–286 CrossRef CAS.
- T. Doba, T. Ichikawa and H. Yoshida, Bull. Chem. Soc. Jpn., 1977, 50, 3158–3163 CrossRef CAS.
- H. M. D. Bandarab and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- C. Raviola and D. Ravelli, Synlett, 2019, 803–808 CAS.
- S. Kamijo, K. Kamijo, K. Maruoka and T. Murafuji, Org. Lett., 2016, 18, 6516–6519 CrossRef CAS.
- M. B. Rubin, Photochemistry and Organic Synthesis, Topics in Current Chemistry, Springer, Berlin, Heidelberg, 1985, vol. 129 Search PubMed.
- C. Huang, M. Zheng, J. Xu and Y. Zhang, Molecules, 2013, 18, 2942–2966 CrossRef CAS.
- X.-Y. Wang, Y.-Q. He, Y. Zhou, L. Lu, X.-R. Song, Z.-Z. Zhou, W.-F. Tian and Q. Xiao, Org. Lett., 2023, 25, 3847–3852 CrossRef CAS.
- P. Franceschi, S. Cuadros, G. Goti and L. Dell’Amico, Angew. Chem., Int. Ed., 2023, 62, e202217210 CrossRef CAS PubMed.
- V. Dixit, A. Sharma, A. Jangid and N. Jain, Adv. Synth. Catal., 2023, 365, 892–899 CrossRef CAS.
- A. A. Fadeev and M. Kotora, Org. Biomol. Chem., 2023, 21, 6174–6179 RSC.
- S. J. Sarma and P. B. Jones, J. Org. Chem., 2010, 75, 3806–3813 CrossRef CAS PubMed.
- T. Yoshihara, M. Yamaji, T. Itoh, J. Nishimura, H. Shizuka and H. S. Tobita, J. Photochem. Photobiol., A, 2001, 140, 7–13 CrossRef CAS.
- J. D. Coyle, Tetrahedron, 1985, 41, 5393–5425 CrossRef CAS.
- J. Xu, Beilstein J. Org. Chem., 2020, 16, 1357–1410 CrossRef CAS PubMed.
- M. Mella, M. Fagnoni, M. Freccero, E. Fasani and A. Albini, Chem. Soc. Rev., 1998, 27, 81–89 RSC.
- A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS PubMed.
- S. Dong, A. Ong and C. Chi, J. Photochem. Photobiol., C, 2019, 38, 27–46 CrossRef CAS.
- V. Brega, Y. Yan and S. W. Thomas, III, Org. Biomol. Chem., 2020, 18, 9191–9209 RSC.
- D. E. Marschner, P. W. Kamm, H. Frisch, A.-N. Unterreiner and C. Barner-Kowollik, Chem. Commun., 2020, 56, 14043–14046 RSC.
- D. L. Priebbenow, J. Org. Chem., 2019, 84, 11813–11822 CrossRef CAS PubMed.
- A. C. S. Page, S. O. Scholz, K. N. Keenan, J. N. Spradlin, B. P. Belcher, S. M. Brittain, J. A. Tallarico, J. M. McKenna, M. Schirle, D. K. Nomura and F. D. Toste, Chem. Sci., 2022, 13, 3851–3856 RSC.
- D. Maiti, R. Das and S. Sen, J. Org. Chem., 2021, 86, 2522–2533 CrossRef CAS PubMed.
- R. D. C. Gallo, G. Cariello, T. A. C. Goulart and I. D. Jurberg, Chem. Commun., 2023, 59, 7346–7360 RSC.
- S. Protti, D. Ravelli and M. Fagnoni, Trends Chem., 2022, 4, 305–317 CrossRef CAS.
- E. Fasani, A. Albini and M. Mella, Tetrahedron, 2008, 64, 3190–3196 CrossRef CAS.
- E. Fasani, M. Fagnoni, D. Dondi and A. Albini, J. Org. Chem., 2006, 71, 2037–2045 CrossRef CAS PubMed.
- P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC.
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed.
- L. Nicchio, J. Médard, P. Decorse, S. Gam-Derouich, A. Chevillot-Biraud, Y. Luo, C. Mangeney, A. Berisha, F. Averseng, M. Fagnoni, S. Protti and J. Pinson, Chem. – Eur. J., 2023, 29, e202301006 CrossRef CAS PubMed.
- S. Kanti Bera, R. Bhanja and P. Mal, Synthesis, 2023, 1467–1486 CrossRef.
- V. A. Schmidt, R. K. Quinn, A. T. Brusoe and E. J. Alexanian, J. Am. Chem. Soc., 2014, 136, 14389–14392 CrossRef CAS PubMed.
- I. H. Shah, S. Kumar, J. Kumar, S. Raheem, M. A. Rizvi and B. A. Shah, ChemPhotoChem, 2022, 6, e202100231 CrossRef CAS.
- R. Narobe and B. Koenig, Org. Chem. Front., 2023, 10, 1577–1586 RSC.
- K. Tsuchii, Y. Tsuboi, S.-I. Kawaguchi, J. Takahashi, N. Sonoda, A. Nomoto and A. Ogawa, J. Org. Chem., 2007, 72, 415–423 CrossRef CAS PubMed.
- A. Ogawa, K. Yokoyama, H. Yokoyama, R. Obayashi, N. Kambe and N. Sonoda, J. Chem. Soc., Chem. Commun., 1991, 1748–1750 RSC.
- S. Protti and M. Fagnoni, ACS Org. Inorg. Au, 2022, 2, 455–463 CrossRef CAS PubMed.
- P. Klan, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef CAS PubMed.
- M. Fagnoni, in Sustainable Organic Synthesis: Tools and Strategies, ed. S. Protti, A. Palmieri, The Royal Society of Chemistry, 2022, ch. 6, pp. 150–180 Search PubMed.
- T. Nevesely, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650–2694 CrossRef CAS PubMed.
- M. E. I. Khan, L. Di Terlizzi, S. Protti and A. Palmieri, Eur. J. Org. Chem., 2022, e202200635 CrossRef CAS.
- J. Sui, M. Li, Z. Zhang, Y. Liang, T. Wang and Z. Zhang, Asian J. Org. Chem., 2023, 12, e202200701 CrossRef CAS.
- J. Xu, N. Liu, H. Lv, C. He, Z. Liu, X. Shen, F. Cheng and B. Fan, Green Chem., 2020, 22, 2739–2743 RSC.
- A. Arunachalampillai, P. Chandrappa, A. Cherney, R. Crockett, J. Doerfler, G. Johnson, V. C. Kommuri, A. Kyad, J. McManus, J. Murray, T. Myren, N. F. Nathel, I. Ndukwe, A. Ortiz, M. Reed, H. Rui, M. V. S. Elipe, J. Tedrow, S. Wells, S. Yacoob and K. Yamamoto, Org. Lett., 2023, 25, 5856–5861 CrossRef CAS PubMed.
- M. Sicignano, R. I. Rodríguez and J. Alemán, Eur. J. Org. Chem., 2021, 3303–3321 CrossRef CAS PubMed.
- W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC.
- E. H. Southgate, J. Pospech, J. Fu, D. R. Holycross and D. Sarlah, Nat. Chem., 2016, 8, 922–928 CrossRef CAS PubMed.
- K. Ikeda, R. Kojima, K. Kawai, T. Murakami, T. Kikuchi, M. Kojima, T. Yoshino and S. Matsunaga, J. Am. Chem. Soc., 2023, 145, 9326–9333 CrossRef CAS PubMed.
- C.-F. Zhu, J. Zhang, Y.-L. Zhu, W.-J. Hao, S.-J. Tu, D.-C. Wang and B. Jiang, Org. Chem. Front., 2021, 8, 1952–1958 RSC.
- X. Song, J. Gu, E. Zhang, Y. Jiang, M. Xin, Y. Meng, A. S. C. Chan and Y. Zou, ACS Sustainable Chem. Eng., 2022, 10, 16399–16407 CrossRef.
- Q.-B. Zhang, Y. Yang, S. Zhang and Q. Liu, Adv. Synth. Catal., 2023, 365, 3556–3571 CrossRef CAS.
- M. D’Auria, Photochem. Photobiol. Sci., 2019, 18, 2297–2362 CrossRef PubMed.
- K. A. Rykaczewski and C. S. Schindler, Org. Lett., 2020, 22, 6516–6519 CrossRef CAS PubMed.
- J. Zheng, X. Dong and T. P. Yoon, Org. Lett., 2020, 22, 6520–6525 CrossRef CAS PubMed.
- R. Tinelli, D. Ravelli, A. Basso, S. C. Tarantino and L. Capaldo, Photochem. Photobiol. Sci., 2022, 21, 695–703 CrossRef CAS PubMed.
- X. Xu, F. Yang, X. Zhang, Y. Gao and W. Su, Asian J. Org. Chem., 2023, 12, e202300069 CrossRef CAS.
- D. Zhang, C. Wu, H. Zhou, Y. Ma and Y. Zhu, Asian J. Org. Chem., 2022, 11, e202200561 CrossRef CAS.
- A. Sharma, V. Dixit, S. Kumar and N. Jain, Org. Lett., 2021, 23, 3409–3414 CrossRef CAS PubMed.
- Z.-W. Qiu, L. Long, Z.-Q. Zhu, H.-F. Liu, H.-P. Pan, A.-J. Ma, J.-B. Peng, Y.-H. Wang, H. Gao and X.-Z. Zhang, ACS Catal., 2022, 12, 13282–13291 CrossRef CAS.
- V. P. Charpe, A. Ragupathi, A. Sagadevan, Y.-S. Ho, M.-J. Cheng and K. C. Hwang, Chem. – Eur. J., 2023, 29, e202300110 CrossRef CAS PubMed.
- S.-Z. Zhang, S.-S. Zhang, J.-L. Li, S. Shen, X.-L. Yang and X. Niu, J. Org. Chem., 2023, 88, 9094–9104 CrossRef CAS PubMed.
- J. Woo, A. H. Christian, S. A. Burgess, Y. Jiang, U. F. Mansoor and M. D. Levin, Science, 2022, 376, 527–532 CrossRef CAS PubMed.
- J. Woo, C. Stein, A. H. Christian and M. D. Levin, Nature, 2023, 623, 77–82 CrossRef CAS PubMed.
- M. F. Saraiva, M. R. C. Couri, M. Le Hyaric and M. V. de Almeida, Tetrahedron, 2009, 65, 3563–3572 CrossRef CAS.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- D. H. R. Barton, B. Garcia, H. Togo and S. Z. Zard, Tetrahedron Lett., 1986, 27, 1327–1330 CrossRef CAS.
- D. H. R. Barton, J. Cs Jaszberenyi and E. A. Theodorakis, Tetrahedron Lett., 1991, 32, 321–3324 Search PubMed.
- D. H. R. Barton, J. Cs Jaszberenyi and E. A. Theodorakis, Tetrahedron, 1992, 48, 2613–2626 CrossRef CAS.
- X. Chen, X. Luo, K. Wang, F. Liang and P. Wang, Synlett, 2021, 733–737 CAS.
- L. Buzzetti, A. Prieto, S. R. Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 15039–15043 CrossRef CAS PubMed.
- J. Liu, W. Zhang, X. Tao, Q. Wang, X. Wang, Y. Pan, J. Ma, L. Yan and Y. Wang, Org. Lett., 2023, 25, 3083–3088 CrossRef CAS PubMed.
- Y. Sato, K. Nakamura, Y. Sumida, D. Hashizume, T. Hosoya and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 9938–9943 CrossRef CAS PubMed.
- Y. Miyamoto, Y. Sumida and H. Ohmiya, Org. Lett., 2021, 23, 5865–5870 CrossRef CAS PubMed.
- Y. Sato, Y. Goto, K. Nakamura, Y. Miyamoto, Y. Sumida and H. Ohmiya, ACS Catal., 2021, 11, 12886–12892 CrossRef CAS.
- S. G. E. Amos, D. Cavalli, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 23827–23834 CrossRef CAS PubMed.
- J. He and S. P. Cook, Chem. Sci., 2023, 14, 9476–9481 RSC.
- P. Girard, N. Guillot, W. B. Motherwell, R. S. Hay-Motherwell and P. Potier, Tetrahedron, 1999, 55, 3573–3584 CrossRef CAS.
- D. S. Masterson and N. A. Porter, Org. Lett., 2002, 4, 4253–4256 CrossRef CAS PubMed.
- T. Huang, P.-F. Yuan, K. Dong, Y.-Y. Zong, C. Liu, R.-H. Wang, X.-L. Jin and Q. Liu, Org. Chem. Front., 2023, 10, 4559–4564 RSC.
- D. H. R. Barton and J. Zhu, J. Am. Chem. Soc., 1993, 115, 2071–2072 CrossRef CAS.
- D. H. R. Barton, S. D. Géro, P. Holliday, B. Quielet-Sire and S. Z. Zard, Tetrahedron, 1998, 54, 6751–6756 CrossRef CAS.
- D. H. R. Barton, B. Lather, B. Misterkiewicz and S. Z. Zard, Tetrahedron, 1988, 44, 1153–1158 CrossRef CAS.
- M. E. Attardi and M. Taddei, Tetrahedron Lett., 2001, 42, 3519–3522 CrossRef CAS.
- D. H. R. Barton and M. Ramesh, J. Am. Chem. Soc., 1990, 112, 891–892 CrossRef CAS.
- D. Zheng, S. Ploeger, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 8547–8551 CrossRef PubMed.
- K. Li, X. Zhang, J. Chen, Y. Gao, C. Yang, K. Zhang, Y. Zhou and B. Fan, Org. Lett., 2019, 21, 9914–9918 CrossRef CAS PubMed.
- Y. Yamamoto, E. Kuroyanagi, H. Suzuki and T. Yasui, Adv. Synth. Catal., 2021, 363, 4932–4940 CrossRef CAS.
- C. Pan, C. Yang, K. Li, K. Zhang, Y. Zhu, S. Wu, Y. Zhou and B. Fan, Org. Lett., 2021, 23, 7188–7193 CrossRef CAS PubMed.
- P. Capurro, R. Valentina, P. Lova, C. Lambruschini, S. Protti and A. Basso, ACS Omega, 2022, 7, 48564–48571 CrossRef CAS PubMed.
- A. Marotta, H. Fang, C. E. Adams, K. S. Marcus, C. G. Daniliuc and J. J. Molloy, Angew. Chem., Int. Ed., 2023, 62, e202307540 CrossRef CAS PubMed.
- P. Huang, Z. Yan, J. Ling, P. Li, J. Wang, J. Li, B. Sun and C. Jin, Green Chem., 2023, 25, 3989–3994 RSC.
- F. Zhang, Z. Wei, W. Wu, N. Liu, X. Li, L. Zou, K. Wang, J. Xu and B. Fan, Org. Biomol. Chem., 2023, 21, 719–723 RSC.
- Y.-B. Wang, F. Chen, M. Li, Q. Bu, Z. Du, J. Liu, B. Dai and N. Liu, Green Chem., 2023, 25, 1191–1200 RSC.
- I. I. Roslan, H. Zhang, K.-H. Ng, S. Jaenicke and G.-K. Chuah, Adv. Synth. Catal., 2021, 363, 1007–1013 CrossRef CAS.
- H. Wang, Y. Cheng and S. Yu, Sci. China: Chem., 2016, 59, 195–198 CrossRef CAS.
- L. Chen, P. Ma, B. Yang, X. Zhao, X. Huang and J. Zhang, Chem. Commun., 2021, 57, 1030–1033 RSC.
- Á. M. Pálvölgyi, F. Ehrschwendtner, M. Schnürch and K. Bica-Schröder, Org. Biomol. Chem., 2022, 20, 7245–7249 RSC.
- F.-L. Zeng, K.-C. Xie, Y.-T. Liu, H. Wang, P.-C. Yin, L.-B. Qu, X.-L. Chen and B. Yu, Green Chem., 2022, 24, 1732–1737 RSC.
- P. Jiang, L. Liu, J. Tan and H. Du, Org. Biomol. Chem., 2021, 19, 4487–4491 RSC.
- X. Shan, X. Wang, E. Chen, J. Liu, K. Lu and X. Zhao, J. Org. Chem., 2023, 88, 319–328 CrossRef CAS PubMed.
- J. Yang, M. Song, H. Zhou, G. Wang, B. Ma, Y. Qi and C. Huo, Org. Lett., 2020, 22, 8407–8412 CrossRef CAS PubMed.
- W. Shi, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2022, 9, 6513–6519 RSC.
- N. Kamigata and M. Kobayashi, Sulfur Rep., 1982, 2, 87–134 CrossRef CAS.
- S. Crespi, S. Protti and M. Fagnoni, J. Org. Chem., 2016, 81, 9612–9621 CrossRef CAS PubMed.
- H. O. Abdulla, A. A. Amin, C. Raviola, T. Opatz, S. Protti and M. Fagnoni, Eur. J. Org. Chem., 2020, 1448–1452 CrossRef CAS.
- P. E. da Silva Júnior, H. I. M. Amin, A. M. Nauth, F. da Silva Emery, S. Protti and T. Opatz, ChemPhotoChem, 2018, 2, 878–883 CrossRef.
- J. Xu, H. Zhang, J. Zhao, Z. Ni, P. Zhang, B.-F. Shi and W. Li, Org. Chem. Front., 2020, 7, 4031–4042 RSC.
- H. Jung, J. Lee and D. Kim, Bull. Korean Chem. Soc., 2018, 39, 1003–1006 CrossRef CAS.
- C. Sauer, Y. Liu, A. De Nisi, S. Protti, M. Fagnoni and M. Bandini, ChemCatChem, 2017, 9, 4456–4459 CrossRef CAS.
- L. Di Terlizzi, S. Scaringi, C. Raviola, R. Pedrazzani, M. Bandini, M. Fagnoni and S. Protti, J. Org. Chem., 2022, 87, 4863–4872 CrossRef CAS PubMed.
- L. Di Terlizzi, I. Cola, C. Raviola, M. Fagnoni and S. Protti, ACS Org. Inorg. Au, 2021, 1, 68–71 CrossRef PubMed.
- A. Dossena, S. Sampaolesi, A. Palmieri, S. Protti and M. Fagnoni, J. Org. Chem., 2017, 82, 10687–10692 CrossRef CAS PubMed.
- M. Malacarne, S. Protti and M. Fagnoni, Adv. Synth. Catal., 2017, 359, 3826–3830 CrossRef CAS.
- Y. Xu, X. Yang and H. Fang, J. Org. Chem., 2018, 83, 12831–12837 CrossRef CAS PubMed.
- C. Lian, G. Yue, J. Mao, D. Liu, Y. Ding, Z. Liu, D. Qiu, X. Zhao, K. Lu, M. Fagnoni and S. Protti, Org. Lett., 2019, 21, 5187–5191 CrossRef CAS PubMed.
- D. Qiu, C. Lian, J. Mao, Y. Ding, Z. Liu, L. Wei, M. Fagnoni and S. Protti, Adv. Synth. Catal., 2019, 361, 5239–5244 CrossRef CAS.
- J. Liu, M. Tian, Y. Li, X. Shan, A. Li, K. Lu, M. Fagnoni, S. Protti and X. Zhao, Eur. J. Org. Chem., 2020, 7358–7367 CrossRef.
- A. Li, Y. Li, J. Liu, J. Chen, K. Lu, D. Qiu, M. Fagnoni, S. Protti and X. Zhao, J. Org. Chem., 2021, 86, 1292–1299 CrossRef CAS PubMed.
- H. I. M. Amin, C. Raviola, A. A. Amin, B. Mannucci, S. Protti and M. Fagnoni, Molecules, 2019, 24, 2164 CrossRef PubMed.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, Boca Raton, FL, 2003 Search PubMed.
- J. H. Kuhlmann, J. H. Dickoff and O. García Mancheño, Chem. – Eur. J., 2023, 29, e202203347 CrossRef CAS PubMed.
- H. Sheng, B. B. Zhang, Q. Liu, Z. S. Yang, Z. X. Wang and X. Y. Chen, Sci. China: Chem., 2022, 65, 2494–2499 CrossRef CAS.
- J. L. Esker and M. Newcomb, J. Org. Chem., 1994, 59, 2779–2786 CrossRef CAS.
- Z. Zhang, J. Feng, C. Yang, H. Cui, W. Harrison, D. Zhong, B. Wang and H. Zhao, Nat. Catal., 2023, 6, 687–694 CrossRef CAS PubMed.
- C. G. Na and E. J. Alexanian, Angew. Chem., Int. Ed., 2018, 57, 13106–13109 CrossRef CAS PubMed.
- L. Song, L. Zhang, S. Luo and J.-P. Cheng, Chem. – Eur. J., 2014, 20, 14231–14234 CrossRef CAS PubMed.
- C. Wang, D. Ma, Y. Tu and C. Bolm, Org. Lett., 2020, 22, 8937–8940 CrossRef CAS PubMed.
- D. V. Patil, T. Si, H. Y. Kim and K. Oh, Org. Lett., 2021, 23, 3105–3109 CrossRef CAS PubMed.
- H. O. Abdulla, S. Scaringi, A. A. Amin, M. Mella, S. Protti and M. Fagnoni, Adv. Synth. Catal., 2020, 362, 2150–2154 CrossRef.
- H. Okamura, M. Iida, Y. Kaneyama and F. Nagatsugi, Org. Lett., 2023, 25, 466–470 CrossRef CAS PubMed.
- E. J. McClain, A. K. Wortman and C. R. J. Stephenson, Chem. Sci., 2022, 13, 12158–12163 RSC.
- Z. Wu and D. A. Pratt, Nat. Rev. Chem., 2023, 7, 573–589 CrossRef PubMed.
- J. Feng, Y. Zhang, X. Wang, J. Liu, V. Benazzi, K. Lu, X. Zhao and S. Protti, Adv. Synth. Catal., 2023, 365, 3413–3431 CrossRef CAS.
- T. Ma, M. Bian, X. Lin, Z. Yang, X. Yang, J. Duan, N. Zhu, C. Liu, Z. Fang and K. Guo, ChemPhotoChem, 2022, 6, e202200208 CrossRef CAS.
- L. Li, Y. Liu, J. Li, Q. Chen, P. Zhang, J. Shen and J. Wu, Adv. Synth. Catal., 2023, 365, 3112–3117 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8, 583272 CrossRef CAS PubMed.
- N. Wang, H. Wang, L.-X. Wan, X.-H. Li, X.-L. Zhou, J.-H. Li, S. De Jonghe, D. Schols, J.-B. Xu and F. Gao, Org. Lett., 2023, 25, 597–602 CrossRef CAS PubMed.
- E. Hao, B. Lu, Y. Liu, T. Yang, H. Yan, X. Ding, Y. Jin and L. Shi, Org. Lett., 2023, 25, 5094–5099 CrossRef CAS PubMed.
- W. L. Czaplyski, C. G. Na and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 13854–13857 CrossRef CAS PubMed.
- Q. Liu, F. Liu, H. Yue, X. Zhao, J. Li and W. Wei, Adv. Synth. Catal., 2019, 361, 5277–5282 CrossRef CAS.
- Y. Lv, Q. Liu, F. Liu, H. Yue, J. Li and W. Wei, Tetrahedron Lett., 2020, 61, 151335 CrossRef CAS.
- R. Chawla, S. Jaiswal, P. K. Dutta and L. D. S. Yadav, Tetrahedron Lett., 2020, 61, 151898 CrossRef CAS.
- R. Chawla, S. Jaiswal, P. K. Dutta and L. D. S. Yadav, Org. Biomol. Chem., 2021, 19, 6487–6492 RSC.
- L. Di Terlizzi, L. Nicchio, C. Callegari, S. Scaringi, L. Neuville, M. Fagnoni, S. Protti and G. Masson, Org. Lett., 2023, 25, 9047–9052 CrossRef CAS PubMed.
- Q. Liu, Y. Lv, R. Liu, X. Zhao, J. Wang and W. Wei, Chinese Chem. Lett., 2021, 32, 136–139 CrossRef CAS.
- L. Di Terlizzi, A. Martinelli, D. Merli, S. Protti and M. Fagnoni, J. Org. Chem., 2023, 88, 6313–6321 CrossRef CAS PubMed.
- L. Di Terlizzi, E. M. Galathri, S. Protti, C. G. Kokotos and M. Fagnoni, ChemSusChem, 2023, 16, e202201998 CrossRef CAS PubMed.
- E. M. Galathri, L. Di Terlizzi, M. Fagnoni, S. Protti and C. G. Kokotos, Org. Biomol. Chem., 2023, 21, 365–369 RSC.
- W. Li and L. Zhou, Green Chem., 2021, 23, 6652–6658 RSC.
- D. Zhao, K. Sun, M. Tian, B. Yan, W. Li, Q. Sun, G. Zheng and Q. Zhang, Org. Chem. Front., 2023, 10, 3313–3320 RSC.
- M. R. Mutra, Y.-T. Chen and J.-J. Wang, Adv. Synth. Catal., 2023, 365, 1012–1019 CrossRef.
- Z.-P. Ye, J.-S. Yang, S.-J. Yang, M. Guo, C.-P. Yuan, Y.-Q. Ye, H.-B. Chen, H.-Y. Xiang, K. Chen and H. Yang, Org. Lett., 2023, 25, 7062–7066 CrossRef CAS PubMed.
- L. Zhuo, S. Xie, H. Wang and H. Zhu, Eur. J. Org. Chem., 2021, 3398–3402 CrossRef CAS.
- E. S. Conner, K. E. Crocker, R. G. Fernando, F. R. Fronczek, G. G. Stanley and J. R. Ragains, Org. Lett., 2013, 15, 5558–5561 CrossRef CAS PubMed.
- G.-Q. Liu, W. Yi, P.-F. Wang, J. Liu, M. Ma, D.-Y. Hao, L. Ming and Y. Ling, Green Chem., 2021, 23, 1840–1846 RSC.
- G.-Q. Liu, C.-F. Zhou, Y.-Q. Zhang, W. Yi, P.-F. Wang, J. Liu and Y. Ling, Green Chem., 2021, 23, 9968–9973 RSC.
- H.-Y. Liu, J.-R. Zhang, G.-B. Huang, Y.-H. Zhou, Y.-Y. Chen and Y.-L. Xu, Adv. Synth. Catal., 2021, 363, 1656–1661 CrossRef CAS.
- L. Lu, X. Zhao, W. Dessie, X. Xia, X. Duan, J. He, R. Wang, Y. Liu and C. Wu, Org. Biomol. Chem., 2022, 20, 1754–1758 RSC.
- S.-Y. Tian, J.-J. Ai, J.-H. Han, W. Rao, S.-S. Shen, D. Sheng and S.-Y. Wang, J. Org. Chem., 2023, 88, 828–837 CrossRef CAS PubMed.
- K. P. Sujith and A. Lee, Eur. J. Org. Chem., 2023, e202300257 Search PubMed.
- Z. Zhang, P. Tan, S. Wang, H. Wang, L. Xie, Y. Chen, L. Han, S. Yang and K. Sun, Org. Lett., 2023, 25, 4208–4213 CrossRef CAS PubMed.
- X.-L. Ma, Q. Wang, X.-Y. Feng, Z.-Y. Mo, Y.-M. Pan, Y.-Y. Chen, M. Xin and Y.-L. Xu, Green Chem., 2019, 21, 3547–3551 RSC.
- H. Chen, R. Ding, H. Tang, Y. Pan, Y. Xu and Y. Chen, Chem. – Asian J., 2019, 14, 3264–3268 CrossRef CAS PubMed.
- X. J. Zhou, H.-Y. Liu, Z.-Y. Mo, X.-L. Ma, Y.-Y. Chen, H.-T. Tang, Y.-M. Pan and Y.-L. Xu, Chem. – Asian J., 2020, 15, 1536–1539 CrossRef CAS PubMed.
- S. R. Sahoo, B. Das, D. Sarkar, F. Henkel and H. Reuter, Eur. J. Org. Chem., 2020, 891–896 CrossRef CAS.
- Z. Chen, X. Zheng, S.-F. Zhou and X. Cui, Org. Biomol. Chem., 2022, 20, 5779–5783 RSC.
- F. Pan, H. Xie, W. Xie, Y.-C. Wang, J.-B. Liu and G. Qiu, Synthesis, 2022, 3105–3113 CrossRef CAS.
- M. Roy, R. Jamatia, A. Samanta, K. Mohar and D. Srimani, Org. Lett., 2022, 24, 8180–8185 CrossRef CAS PubMed.
- I. D. Lemir, W. D. Castro-Godoy, A. A. Heredia, L. C. Schmidt and J. E. Argüello, RSC Adv., 2019, 9, 22685–22694 RSC.
- A. Ogawa, K. Yokoyama, R. Obayashi, L.-B. Han, N. Kambe and N. Sonoda, Tetrahedron, 1993, 49, 1177–1188 CrossRef CAS.
- T. Mitamura, Y. Tsuboi, K. Iwata, K. Tsuchii, A. Nomoto, M. Sonoda and A. Ogawa, Tetrahedron Lett., 2007, 48, 5953–5957 CrossRef CAS.
- N. Li, Y. Wang, S. Gu, C. Hu, Q. Yang, Z. Jin, W.-T. Ouyang, J. Qiao and W.-M. He, Org. Biomol. Chem., 2023, 21, 370–374 RSC.
- L. Man, R. C. B. Copley and A. L. Handlon, Org. Biomol. Chem., 2019, 17, 6566–6569 RSC.
- S. Borra, L. Borkotoky, U. D. Newar, A. Kalwar, B. Das and R. A. Maurya, Org. Biomol. Chem., 2019, 17, 5971–5981 RSC.
- S. Borra, L. Borkotoky, U. D. Newar, B. Das and R. A. Maurya, Adv. Synth. Catal., 2020, 362, 3364–3368 CrossRef CAS.
- J. Liu, N. Liu, Q. Yang and L. Wang, Org. Chem. Front., 2021, 8, 5296–5302 RSC.
- S. C. Patel and N. Z. Burns, J. Am. Chem. Soc., 2022, 144, 17797–17802 CrossRef CAS PubMed.
- T. J. Pearson, R. Shimazumi, J. L. Driscoll, B. D. Dherange, D.-I. Park and M. D. Levin, Science, 2023, 381, 1474–1479 CrossRef CAS PubMed.
- B. Li, A. Ruffoni and D. Leonori, Angew. Chem., Int. Ed., 2023, 62, e202310540 CrossRef CAS PubMed.
- X. Tian, L. Song and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2020, 59, 12342–12346 CrossRef CAS PubMed.
- I. D. Jurberg, R. A. Nome, S. Crespi, T. D. Z. Atvars and B. König, Adv. Synth. Catal., 2022, 364, 4061–4068 CrossRef CAS.
- Y. Guo, C. Pei and R. M. Koenigs, Nat. Commun., 2022, 13, 86 CrossRef CAS PubMed.
- Y. Guo, C. Pei, C. Empel, S. Jana and R. M. Koenigs, ChemPhotoChem, 2022, 6, e202100293 CrossRef CAS.
- P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Chem, 2023, 9, 3401–3414 CAS.
- R. Sánchez-Bento, B. Roure, J. Llaveria, A. Ruffoni and D. Leonori, Chem, 2023, 9, 3685–3695 Search PubMed.
- E. Matador, M. J. Tilby, I. Saridakis, M. Pedron, D. Tomczak, J. Llaveria, I. Atodiresei, P. Merino, A. Ruffoni and D. Leonori, J. Am. Chem. Soc., 2023, 145, 27810–27820 CrossRef CAS PubMed.
- Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC.
- S. Jana, Y. Guo and R. M. Koenigs, Chem. – Eur. J., 2021, 27, 1270–1281 CrossRef CAS PubMed.
- C. Empel and R. M. Koenigs, Synlett, 2019, 1929–1934 CAS.
- D. L. Priebbenow, Adv. Synth. Catal., 2020, 362, 1927–1946 CrossRef CAS.
- S. Jana, C. Pei, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2021, 60, 13271–13279 CrossRef CAS PubMed.
- J.-H. Ye, L. Quach, T. Paulisch and F. Glorius, J. Am. Chem. Soc., 2019, 141, 16227–16231 CrossRef CAS PubMed.
- J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS.
- C. Empel, C. Pei and R. M. Koenigs, Chem. Commun., 2022, 58, 2788–2798 RSC.
- W.-F. Zuo, Q. Liu, X. Xie, Q. Pang, W. Li, C. Peng, X. Li and B. Han, Org. Chem. Front., 2023, 10, 4474–4487 RSC.
- N. R. Candeias and C. A. M. Afonso, Curr. Org. Chem., 2009, 13, 763–787 CrossRef CAS.
- L. W. Ciszewski, K. Rybicka-Jasińska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432–448 RSC.
- W. P. Hong, H. N. Lim and I. Shin, Org. Chem. Front., 2023, 10, 819–836 RSC.
- C. Pei, C. Empel and R. M. Koenigs, Org. Lett., 2023, 25, 169–173 CrossRef CAS PubMed.
- Y. Zhang, G. Zhou, X. Gong, Z. Guo, X. Qi and X. Shen, Angew. Chem., Int. Ed., 2022, 61, e202202175 CrossRef CAS PubMed.
- A. Bunyamin, C. Hua, A. Polyzos and D. L. Priebbenow, Chem. Sci., 2022, 13, 3273–3280 RSC.
- E. Yamaguchi, H. Inagawa and A. Itoh, Photochem. Photobiol. Sci., 2022, 21, 813–818 CrossRef CAS PubMed.
- X. Zhang, C. Du, H. Zhang, X.-C. Li, Y.-L. Wang, J.-L. Niu and M.-P. Song, Synthesis, 2019, 889–898 Search PubMed.
- J. Chauhan, M. K. Ravva, L. Gremaud and S. Sen, Org. Lett., 2020, 22, 4537–4541 CrossRef CAS PubMed.
- Y. Guo, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2019, 21, 8814–8818 CrossRef CAS PubMed.
- S. Zhao, X.-X. Chen, N. Gao, M. Qian and X. Chen, J. Org. Chem., 2021, 86, 7131–7140 CrossRef CAS PubMed.
- R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203–1207 CrossRef CAS PubMed.
- F. He and R. M. Koenigs, Chem. Commun., 2019, 55, 4881–4884 RSC.
- N. Tanbouza, V. Carreras and T. Ollevier, Org. Lett., 2021, 23, 5420–5424 CrossRef CAS PubMed.
- X. Cheng, B.-G. Cai, H. Mao, J. Lu, L. Li, K. Wang and J. Xuan, Org. Lett., 2021, 23, 4109–4114 CrossRef CAS PubMed.
- I. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC.
- J. V. Santiago, K. Orłowska, M. Ociepa and D. Gryko, Org. Lett., 2023, 25, 6267–6271 CrossRef CAS PubMed.
- A. R. S. Mondal, B. Ghorai and D. Prasad Hari, Org. Lett., 2023, 25, 4974–4979 CrossRef PubMed.
- B. Zhao, H. Li, F. Jiang, J.-P. Wan, K. Cheng and Y. Liu, J. Org. Chem., 2023, 88, 640–646 CrossRef CAS PubMed.
- L. Zheng, X. Guo, Y.-C. Li, Y. Wu, X.-S. Xue and P. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216373 CrossRef CAS PubMed.
- R. D. C. Gallo, M. Duarte, A. F. da Silva, C. Y. Okada, Jr., V. M. Deflon and I. D. Jurberg, Org. Lett., 2021, 23, 8916–8920 CrossRef CAS PubMed.
- S. Li, C. Zhang, G. Pan, L. Yang, Z. Su, X. Feng and X. Liu, ACS Catal., 2023, 13, 4656–4666 CrossRef CAS.
- H. Zhang, Z. Wang, Z. Wang, Y. Chu, S. Wang and X.-P. Hui, ACS Catal., 2022, 12, 5510–5516 CrossRef CAS.
- W. Li, Y. Yang, Z. Tang, X. Yu, J. Lin and Y. Jin, J. Org. Chem., 2022, 87, 13352–13362 CrossRef CAS PubMed.
- J. R. Vale, R. F. Gomes, C. A. M. Afonso and N. R. Candeias, J. Org. Chem., 2022, 87, 8910–8920 CrossRef CAS PubMed.
- C. Ye, B.-G. Cai, J. Lu, X. Cheng, L. Li, Z.-W. Pan and J. Xuan, J. Org. Chem., 2021, 86, 1012–1022 CrossRef CAS PubMed.
- S. Muthusamy and C. Ramesh, J. Org. Chem., 2023, 88, 5609–5621 CrossRef CAS PubMed.
- Y.-R. Zhao, L. Li, G.-Y. Xu and J. Xuan, Adv. Synth. Catal., 2022, 364, 506–511 CrossRef CAS.
- D. Maiti, R. Das, T. Prabakar and S. Sen, Green Chem., 2022, 24, 3001–3008 RSC.
- T. Xiao, M. Mei, Y. He and L. Zhou, Chem. Commun., 2018, 54, 8865–8868 RSC.
- K. Ishida, F. Tobita and H. Kusama, Chem. – Eur. J., 2018, 24, 543–546 CrossRef CAS PubMed.
- L. Ma, Y. Yu, L. Xin, L. Zhu, J. Xia, P. Ou and X. Huang, Adv. Synth. Catal., 2021, 363, 2573–2577 CrossRef CAS.
- D. L. Priebbenow, R. L. Pilkington, K. N. Hearn and A. Polyzos, Org. Lett., 2021, 23, 2783–2789 CrossRef CAS PubMed.
- R. Vaggu, N. Thadem, M. Rajesh, R. Gree and S. Das, Org. Lett., 2023, 25, 2594–2599 CrossRef CAS PubMed.
- Z. Fan, Y. Yi, S. Chen and C. Xi, Org. Lett., 2021, 23, 2303–2307 CrossRef CAS PubMed.
- Y. Ueda, Y. Masuda, T. Iwai, K. Imaeda, H. Takeuchi, K. Ueno, M. Gao, J.-Y. Hasegawa and M. Sawamura, J. Am. Chem. Soc., 2022, 144, 2218–2224 CrossRef CAS PubMed.
- P. Becker, D. L. Priebbenow, H.-J. Zhang, R. Pirwerdjan and C. Bolm, J. Org. Chem., 2014, 79, 814–817 CrossRef CAS PubMed.
- H.-J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F.-F. Pan and C. Bolm, Adv. Synth. Catal., 2012, 354, 2157–2161 CrossRef CAS.
- Y. Hussain, C. Empel, R. M. Koenigs and P. Chauhan, Angew. Chem., Int. Ed., 2023, 62, e202309184 CrossRef CAS PubMed.
- A. F. da Silva, M. A. S. Afonso, R. A. Cormanich and I. D. Jurberg, Chem. – Eur. J., 2020, 26, 5648–5653 CrossRef CAS PubMed.
- M. L. Stivanin, A. A. G. Fernandes, A. F. da Silva, C. Y. Okada Jr and I. D. Jurberg, Adv. Synth. Catal., 2020, 362, 1106–1111 CrossRef CAS.
- C. Stuckhardt, M. Wissing and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 18605–18611 CrossRef CAS PubMed.
- J. Reimler and A. Studer, Chem. – Eur. J., 2021, 27, 15392–15395 CrossRef CAS PubMed.
- C. Empel, F. W. Patureau and R. M. Koenigs, J. Org. Chem., 2019, 84, 11316–11322 CrossRef CAS PubMed.
- S. Rath, B. Mohanty and S. Sen, J. Org. Chem., 2023, 88, 1036–1048 CrossRef CAS PubMed.
- K. Zhu, M. Cao, G. Zhao, J. Zhao and P. Li, Org. Lett., 2022, 24, 5855–5859 CrossRef CAS PubMed.
- B.-G. Cai, W.-Z. Yao, L. Li and J. Xuan, Org. Lett., 2022, 24, 6647–6652 CrossRef CAS PubMed.
- J. Bai, D. Qi, Z. Song, B. Li, L. Guo, C. Yang and W. Xia, Org. Biomol. Chem., 2023, 21, 5511–5515 RSC.
- W. Guo, Y. Zhou, H. Xie, X. Yue, F. Jiang, H. Huang, Z. Han and J. Sun, Chem. Sci., 2023, 14, 843–848 RSC.
- F. Li, F. He and R. M. Koenigs, Synthesis, 2019, 4348–4358 CAS.
- B.-G. Cai, L. Li, G.-Y. Xu, W.-J. Xiao and J. Xuan, Photochem. Photobiol. Sci., 2021, 20, 823–829 CrossRef CAS PubMed.
- B.-L. Qu, B. Shi, L. He, J.-W. Shi, W.-J. Xiao and L.-Q. Lu, Org. Chem. Front., 2023, 10, 3498–3503 RSC.
- J. Bai, D. Qi, Z. Song, B. Li, L. Guo, C. Yang and W. Xia, Synlett, 2022, 2048–2052 CAS.
- C. Empel, D. Verspeek, S. Jana and R. M. Koenigs, Adv. Synth. Catal., 2020, 362, 4716–4722 CrossRef CAS.
- J. Yang, G. Wang, H. Zhou, Z. Li, B. Ma, M. Song, R. Sun and C. Huo, Org. Biomol. Chem., 2021, 19, 394–398 RSC.
- G. D. Titov, G. I. Antonychev, M. S. Novikov, A. F. Khlebnikov, E. V. Rogacheva, L. A. Kraeva and N. V. Rostovskii, Org. Lett., 2023, 25, 2707–2712 CrossRef CAS PubMed.
- M. L. Stivanin, M. Duarte, L. P. M. O. Leão, F. A. Saito and I. D. Jurberg, J. Org. Chem., 2021, 86, 17528–17532 CrossRef CAS PubMed.
- Y.-N. Wang, X. Wang, S.-J. Li and Y. Lan, Org. Chem. Front., 2022, 9, 1247–1253 RSC.
- M. L. Stivanin, R. D. C. Gallo, J. P. M. Spadeto, R. A. Cormanich and I. D. Jurberg, Org. Chem. Front., 2022, 9, 1321–1326 RSC.
- Z. Wang, R. Liu, C. Qu, X.-E. Zhao, Y. Lv, H. Yue and W. Wei, Org. Chem. Front., 2022, 9, 3565–3570 RSC.
- Y. Lv, R. Liu, H. Ding, W. Wei, X. Zhao and L. He, Org. Chem. Front., 2022, 9, 3486–3492 RSC.
- C. Qu, J. Hao, H. Ding, Y. lv, X.-E. Zhao, X. Zhao and W. Wei, J. Org. Chem., 2022, 87, 12921–12931 CrossRef CAS PubMed.
- H. Ding, Z. Wang, C. Qu, Y. Lv, X. Zhao and W. Wei, Org. Chem. Front., 2022, 9, 5530–5535 RSC.
- K. Zhu, X. Zhou, Y. Ren, L. Dong, G. Zhao, J. Zhao and P. Li, Chem. Commun., 2023, 59, 631–634 RSC.
- D. Qi, J. Bai, Z. Song, B. Li, C. Yang, L. Guo and W. Xia, Org. Lett., 2023, 25, 506–511 CrossRef CAS PubMed.
- S. Jana, Z. Yang, C. Pei, X. Xu and R. M. Koenigs, Chem. Sci., 2019, 10, 10129–10134 RSC.
- D. Qi, J. Bai, H. Zhang, B. Li, Z. Song, N. Ma, L. Guo, L. Song and W. Xia, Green Chem., 2022, 24, 5046–5051 RSC.
- M. Cao, Y. Ren, R. Zhang, H. Xu, P. Cheng, H. Xu, Y. Xu and P. Li, Org. Lett., 2023, 25, 6300–6304 CrossRef CAS PubMed.
- B.-G. Cai, Q. Li, Q. Zhang, L. Li and J. Xuan, Org. Chem. Front., 2021, 8, 5982–5987 RSC.
- B.-G. Cai, Q. Li, C. Empel, L. Li, R. M. Koenigs and J. Xuan, ACS Catal., 2022, 12, 11129–11136 CrossRef CAS.
- H. Khan, S. Guha, M. Baruah, S. Yadav, S. Maheshwari, S. Sainani, D. Maiti and S. Sen, Chem. – Asian J., 2023, 18, e202300420 CrossRef CAS PubMed.
- H. Zhou, G. Wang, C. Wang and J. Yang, Org. Lett., 2022, 24, 1530–1535 CrossRef CAS PubMed.
- S. Roy, G. Kumar and I. Chatterjee, Org. Lett., 2021, 23, 6709–6713 CrossRef CAS PubMed.
- J. Yang, G. Wang, S. Chen, B. Ma, H. Zhou, M. Song, C. Liu and C. Huo, Org. Biomol. Chem., 2020, 18, 9494–9498 RSC.
- L.-Z. Qin, X. Yuan, Y.-S. Cui, Q. Sun, X. Duan, K.-Q. Zhuang, L. Chen, J.-K. Qiu and K. Guo, Adv. Synth. Catal., 2020, 362, 5093–5104 CrossRef CAS.
- C. Qu, R. Liu, Z. Wang, Y. Lv, H. Yue and W. Wei, Green Chem., 2022, 24, 4915–4920 RSC.
- Y. Lv, R. Liu, H. Ding, W. Wei, X. Zhao and L. He, Org. Chem. Front., 2022, 9, 3486–3492 RSC.
- J. Yang, J. Wang, H. Huang, G. Qin, Y. Jiang and T. Xiao, Org. Lett., 2019, 21, 2654–2657 CrossRef CAS PubMed.
- J. Xie, M. Suleman, Z. Wang, X. Mao, B. Mao, J. Fan, P. Lu and Y. Wang, Org. Biomol. Chem., 2021, 19, 6341–6345 RSC.
- K. Orłowska, K. Rybicka-Jasińska, P. Krajewski and D. Gryko, Org. Lett., 2020, 22, 1018–1021 CrossRef PubMed.
- Z. Yang, Y. Guo and R. M. Koenigs, Chem. – Eur. J., 2019, 25, 6703–6706 CrossRef CAS PubMed.
- M. Arbaz Ansari, D. Yadav and M. S. Singh, Chem. – Eur. J., 2020, 26, 8083–8089 CrossRef PubMed.
- S. Sun, Y. Wei and J. Xu, Org. Lett., 2022, 24, 6024–6030 CrossRef CAS PubMed.
- J. Bai, S. Li, D. Qi, Z. Song, B. Li, L. Guo, L. Song and W. Xia, Org. Lett., 2023, 25, 2410–2414 CrossRef CAS PubMed.
- Z.-L. Chen, C. Empel, K. Wang, P.-P. Wu, B.-G. Cai, L. Li, R. M. Koenigs and J. Xuan, Org. Lett., 2022, 24, 2232–2237 CrossRef CAS PubMed.
- F. He, F. Li and R. M. Koenigs, J. Org. Chem., 2020, 85, 1240–1246 CrossRef CAS PubMed.
- X. Mo, Y. Xie, L. Wei, X. Gu, M. Zhang, X. Zhang and J. Jiang, Org. Lett., 2023, 25, 2338–2343 CrossRef CAS PubMed.
- W. Kirmse, Eur. J. Org. Chem., 2002, 2193–2256 CrossRef CAS.
- X. Yue, Y. Zhou, Y. Zhang, T. Meng, Y. Zhao and W. Guo, Chem. Commun., 2023, 59, 6363–6366 RSC.
- J. Meng, W.-W. Ding and Z.-Y. Han, Org. Lett., 2019, 21, 9801–9805 CrossRef CAS PubMed.
- D. Weinzierl, M. Piringer, P. Zebrowski, L. Stockhammer and M. Waser, Org. Lett., 2023, 25, 3126–3130 CrossRef CAS PubMed.
- J.-B. Pan, Z.-C. Yang, X.-G. Zhang, M.-L. Li and Q.-L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202308122 CrossRef CAS PubMed.
- R. Echemendia, K. T. de Oliveira and A. C. B. Burtoloso, Org. Lett., 2022, 24, 6386–6390 CrossRef CAS PubMed.
- Y. Dong, Y. Tian, Z. Zhang and T. Wang, Adv. Synth. Catal., 2022, 364, 4026–4030 CrossRef CAS.
- T. Fan, Z.-J. Zhang, Y.-C. Zhang and J. Song, Org. Lett., 2019, 21, 7897–7901 CrossRef CAS PubMed.
- J.-M. Wang, Y.-X. Chen, C.-S. Yao and K. Zhang, Asian J. Org. Chem., 2022, 11, e202200238 CrossRef CAS.
- C. Wang, Z. Wang, J. Yang, S.-H. Shi and X.-P. Hui, Org. Lett., 2020, 22, 4440–4443 CrossRef CAS PubMed.
- Y. Chen, B. Shi, H. Yin, Y. Liu, C. Yu, K. Zhang, T. Li and C. Yao, Org. Chem. Front., 2022, 9, 5191–5196 RSC.
- F. Minuto, C. Lambruschini and A. Basso, Eur. J. Org. Chem., 2021, 3270–3273 CrossRef CAS.
- J. Tang, Z.-H. Yan, G. Zhan, Q.-Q. Yang, Y.-Y. Cheng, X. Li and W. Huang, Org. Chem. Front., 2022, 9, 4341–4346 RSC.
- J. Zhou, C. Chen, Q. Pang, W.-F. Zuo, X. Li, G. Zhan, Q.-Q. Yang and B. Han, Org. Chem. Front., 2023, 10, 1034–1041 RSC.
- J. Liu, M.-M. Li, B.-L. Qu, L.-Q. Lu and W.-J. Xiao, Chem. Commun., 2019, 55, 2031–2034 RSC.
- Q.-L. Zhang, Q. Xiong, M.-M. Li, W. Xiong, B. Shi, Y. Lan, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 14096–14100 CrossRef CAS PubMed.
- A. Couture, J. Gdmez and P. de Mayo, J. Org. Chem., 1981, 46, 2010–2016 CrossRef CAS.
- A. Couture, M. Hoshino and P. de Mayo, J. Chem. Soc., Chem. Commun., 1976, 131–132 RSC.
- C. C. Liao, H. S. Lin, T. H. Hseu, C. P. Tang and J. L. Wang, J. Am. Chem. Soc., 1982, 104, 292–294 CrossRef CAS.
- D. Alvarez-Dorta, E. León, A. R. Kennedy, C. Riesco-Fagundo and E. Suárez, Angew. Chem., Int. Ed., 2008, 47, 8917–8919 CrossRef CAS PubMed.
- D. Alvarez-Dorta, E. Leon, A. R. Kennedy, A. Martin, I. Perez-Martin, C. Riesco-Fagundo and E. Suarez, Chem. – Eur. J., 2014, 20, 2663–2671 CrossRef CAS PubMed.
- A. J. Herrera, M. Rondón and E. Suárez, J. Org. Chem., 2008, 73, 3384–3391 CrossRef CAS PubMed.
- S. Yoshioka, M. Nagatomo and M. Inoue, Org. Lett., 2015, 17, 90–93 CrossRef CAS PubMed.
- F. Secci, S. Porcu, A. Luridiana, A. Frongia and P. C. Ricci, Org. Biomol. Chem., 2020, 18, 3684–3689 RSC.
- L. K. De La Cruz, N. Bauer, A. Cachuela, W. S. Tam, R. Tripathi, X. Yang and B. Wang, Org. Lett., 2022, 24, 4902–4907 CrossRef CAS PubMed.
- J. S. Ham, B. Park, M. Son, J. B. Roque, J. Jurczyk, C. S. Yeung, M.-H. Baik and R. Sarpong, J. Am. Chem. Soc., 2020, 142, 13041–13050 CrossRef CAS PubMed.
- N. Petek, H. Brodnik, U. Grošelj, J. Svete, F. Požgan and B. Štefane, Org. Lett., 2021, 23, 5294–5298 CrossRef CAS PubMed.
- D. E. Wise, E. S. Gogarnoiu, A. D. Duke, J. M. Paolillo, T. L. Vacala, W. A. Hussain and M. Parasram, J. Am. Chem. Soc., 2022, 144, 15437–15442 CrossRef CAS PubMed.
- A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 610, 81–86 CrossRef CAS PubMed.
- I. R. Gould, D. Ege, J. E. Moser and S. Farid, J. Am. Chem. Soc., 1990, 112, 4290–4301 CrossRef CAS.
- Y. T. Jeon, C.-P. Lee and P. S. Mariano, J. Am. Chem. Soc., 1991, 113, 8847–8863 CrossRef CAS.
- E. Hasegawa, M. A. Brumfield, P. S. Mariano and U.-C. Yoon, J. Org. Chem., 1988, 53, 5435–5442 CrossRef CAS.
- D. V. Patil, K. Ramesh, H. Y. Kim and K. Oh, Org. Lett., 2023, 25, 7204–7208 CrossRef CAS PubMed.
- M. Takezaki, N. Hirota and M. Terazima, J. Phys. Chem. A, 1997, 101, 3443–3448 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |