
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zn-based batteries for sustainable energy storage: strategies and mechanisms
Lei
Tang†
a,
Haojia
Peng†
a,
Jiarui
Kang
a,
Han
Chen
a,
Mingyue
Zhang
a,
Yan
Liu
c,
Dong Ha
Kim
 *b,
Yijiang
Liu
*d and
Zhiqun
Lin
*ab
*b,
Yijiang
Liu
*d and
Zhiqun
Lin
*ab
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore, 117585, Singapore. E-mail: z.lin@nus.edu.sg
bDepartment of Chemistry and Nano Science, Ewha Womans University, 52 Ewhayeodae-gil, Seodaemun-gu, Seoul 03760, Republic of Korea. E-mail: dhkim@ewha.ac.kr
cInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore
dCollege of Chemistry, Key Lab of Environment-Friendly Chemistry and Application in Ministry of Education, Xiangtan University, Xiangtan 411105, Hunan Province, P. R. China. E-mail: liuyijiang84@xtu.edu.cn
First published on 10th April 2024
Abstract
Batteries play a pivotal role in various electrochemical energy storage systems, functioning as essential components to enhance energy utilization efficiency and expedite the realization of energy and environmental sustainability. Zn-based batteries have attracted increasing attention as a promising alternative to lithium-ion batteries owing to their cost effectiveness, enhanced intrinsic safety, and favorable electrochemical performance. In this context, substantial endeavors have been dedicated to crafting and advancing high-performance Zn-based batteries. However, some challenges, including limited discharging capacity, low operating voltage, low energy density, short cycle life, and complicated energy storage mechanism, need to be addressed in order to render large-scale practical applications. In this review, we comprehensively present recent advances in designing high-performance Zn-based batteries and in elucidating energy storage mechanisms. First, various redox mechanisms in Zn-based batteries are systematically summarized, including insertion-type, conversion-type, coordination-type, and catalysis-type mechanisms. Subsequently, the design strategies aiming at enhancing the electrochemical performance of Zn-based batteries are underscored, focusing on several aspects, including output voltage, capacity, energy density, and cycle life. Finally, challenges and future prospects of Zn-based batteries are discussed.
 Lei Tang | Lei Tang received his bachelor's degree in Bioengineering from Sichuan University in 2014, and his master's degree in Chemical Engineering and Technology from Tianjin University in 2017. He received his PhD in Physical Chemistry from the University of Chinese Academy of Sciences in 2022, under the supervision of Prof. Dehui Deng. He is currently a postdoctoral research associate in the Department of Chemical and Biomolecular Engineering at National University of Singapore under the supervision of Prof. Zhiqun Lin. His current research focuses on energy electrocatalysis and Zn-based batteries. |
 Haojia Peng | Haojia Peng received his BS degree in Material Science and Engineering from Beijing Institute of Technology in 2023. He is currently working toward his master's degree in Energy Systems in the Department of Chemical and Biomolecular Engineering at National University of Singapore. His research interests include electrocatalysis for alkaline HER and cathode materials for lithium-ion batteries. |
 Jiarui Kang | Jiarui Kang is a PhD student in the Department of Chemical and Biomolecular Engineering at National University of Singapore. She conducts her research under the mentorship of Professor Zhiqun Lin. Her scholarly pursuits are twofold: one project focuses on the development of polymer hydrogel electrolytes, while the other examines the anode interface in zinc-ion batteries. Each project aims to expand the frontiers of knowledge in energy storage technology. |
 Han Chen | Han Chen is currently a PhD student in the Department of Chemical and Biomolecular Engineering at National University of Singapore. His research mainly focuses on the rational design of advanced electrolytes for high energy density lithium–metal batteries. |
 Mingyue Zhang | Mingyue Zhang received his BS, master's, and PhD degrees in Materials Science and Engineering from the Pennsylvania State University, University of Pennsylvania, and Georgia Institute of Technology in 2016, 2018, and 2022 respectively. He is now a research fellow in the Department of Chemical and Biomolecular Engineering at National University of Singapore. His research interests include non-linear polymers, chiral polymers, functional nanocrystals, self-assembly, and nanotechnologies for energy related applications. |
 Yan Liu | Dr Yan Liu is a principal scientist at the Institute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), Singapore. Her research interests include the development of multifunctional materials for thermal catalysis, electrocatalysis, and sonocatalysis for CO2 utilization and green hydrogen production. |
 Dong Ha Kim | Dr Dong Ha Kim is a professor of the Department of Chemistry and Nanoscience at Ewha Womans University. He received his PhD degree from the Department of Fiber and Polymer Science of Seoul National University (2000). He conducted postdoctoral research at the University of Massachusetts at Amherst and the Max Planck Institute for Polymer Research. In 2006, he joined the faculty of the Division of Nanoscience at Ewha Womans University. His research focuses on the development of hybrid nanostructures for energy, catalysis, memory, display, and theragnosis. Currently, he is a Fellow of Royal Society of Chemistry and Associate Editor of Nanoscale. |
 Yijiang Liu | Dr Yijiang Liu is a professor in the College of Chemistry at Xiangtan University, China. She received her MS degree from Xiangtan University in 2009 and PhD from the Institute of Chemistry, Chinese Academy of Sciences, in 2015. She was a visiting scholar at Georgia Institute of Technology during 2017–2018. Her research work focuses on the design and preparation of advanced functional materials: porous carbon materials for efficient and durable electrocatalysts in energy conversion and storage devices; the crafting of highly stable perovskite nanocrystals by using functional polymers as nanoreactors; and Janus materials for concurrent catalysis and emulsification. |
 Zhiqun Lin | Dr Zhiqun Lin is a Professor in the Department of Chemical and Biomolecular Engineering at the National University of Singapore. His research interests include electrocatalysis, batteries, solar energy conversion, photocatalysis, semiconductor organic–inorganic nanocomposites, multifunctional nanocrystals, conjugated polymers, block copolymers, hierarchical structure formation and assembly, and surface and interfacial properties. |
1. Introduction
The increasing use of fossil fuels and growing environmental concerns have placed a great demand for efficient and sustainable energy storage systems capable of harnessing energy from renewable and clean sources such as wind, solar, and tidal power.1–9 This surge in demand has played a major role in driving the widespread adoption of electrification and rechargeable batteries. Lithium-ion batteries have prominently featured in various secondary battery applications across industries, including communication (mobile phones), portable electronic devices (smartwatches), and transportation (automotive vehicles), owing to their high energy density, well-established manufacturing technology, and low weight.10–15 Between 2020 and 2021, the worldwide demand for batteries amounted to approximately 960 GW h, with battery production reaching a range of 7 to 10 million tons (Fig. 1a).16 The anticipated growth in market demand over the upcoming years will continue to hinge on batteries, with projections indicating a surpassing of 3200 GW h by 2030 (Fig. 1a). The escalating demand cannot be solely addressed by Li-ion batteries, given the projected depletion of the world's lithium resources within the span of a century. The diminishing availability of economically extractable lithium reserves has prompted increasing concerns regarding global energy security, emphasizing the critical urgency for battery technologies rooted in abundant earth metals. Furthermore, the utilization of Li-ion batteries for large-scale energy storages faces impediments due to their elevated costs, as well as apprehensions related to safety and environmental considerations.17 Therefore, the urgency to explore alternative battery systems beyond lithium-ion batteries is paramount in order to effectively meet the rising requirements for affordable, safe, and high-performance energy storage systems. | ||
| Fig. 1 Motivation to study Zn-based batteries. (a) Projected battery demand (GW h) for portable electronic devices, electric vehicles, and energy storage systems spanning the period 2020 to 2030. (b) Redox potential and specific capacity of various metals. (c) Energy density and price of various batteries. (d) Comparison of Zn-based batteries and Li-based batteries from multiple metrics. | ||
Battery chemistries employing abundant elements like sodium, potassium, zinc, magnesium, aluminum, and calcium as the anode have attracted considerable attention as alternative options.18–31 Within the realm of “beyond Li-ion” battery technologies, rechargeable Zn-based batteries show considerable promise for large-scale energy storage applications due to distinct advantages associated with metallic zinc. These advantages encompass a high theoretical capacity (820 mA h g−1 or 5854 mA h cm−3) as illustrated in Fig. 1b, a lower cost of zinc (∼US$2 kg−1) compared to alkali metals (e.g., lithium ∼US$19 kg−1; potassium ∼US$13 kg−1; sodium ∼US$3 kg−1), stability and reversibility in aqueous electrolytes, non-toxicity, and ease of material processing and battery fabrication.32–36 In addition, the low anodic potential (−0.763 V vs. standard hydrogen electrode) and the two-electron transfer during the battery operation contribute to elevated energy densities.37–43 Additional benefits of Zn-based batteries include the affordability, safety, and high ionic conductivity (up to 1 S cm−1) of commonly used aqueous electrolytes, in contrast to flammable organic electrolytes employed in lithium-ion batteries (∼1–10 mS cm−1).37,44–48 The projected cost of a Zn-based battery utilizing a Zn anode and a MnO2 cathode is anticipated to be less than $100 per kW h, representing approximately half the cost of contemporary lithium-ion batteries (∼$200 per kW h) (Fig. 1c).49 Notably, the anticipated cost of Zn–air batteries, estimated at approximately $100 per kW h and possessing significantly higher energy density than Li-ion batteries, is markedly lower than that of Li-ion batteries. Given these advantageous attributes (Fig. 1d), Zn-based batteries demonstrate significant potential for practical applications.
Historically, metal zinc served as the initial anode material for batteries, specifically in the Volta Pile, dating back to 1799.50 Currently, zinc finds extensive application in diverse battery technologies, including the Zn-ion battery, Zn–air battery, Zn–CO2 battery, Zn-based flow battery, and Zn-based flexible battery. Compared to Li-based batteries, Zn-based batteries have distinct advantages in price and safety, yet their performance needs to be further improved to ensure large-scale practical applications (Fig. 1d).21,51–59 For battery performance, energy density and cycle life are the two most important evaluation indicators. Essentially, energy density is derived from the product of a battery's specific capacity and voltage (energy density = capacity × voltage).60 Consequently, four primary design strategies are employed to attain high-performance Zn-based batteries, focusing on advancements in high capacity, increased voltage, enhanced energy density, and prolonged cycle life. Design strategies for achieving high capacity in Zn-based batteries center primarily on cathode materials and electrodes,61–63 and the design strategies for enhancing voltage in Zn-based batteries primarily involve the optimization of the electrolyte to facilitate high-potential reactions.57,64,65 Particularly, exploring innovative battery configurations and optimizing the energy storage mechanism stand out as crucial strategies for achieving high energy density in Zn-based batteries.66–69 In addition, the design strategies for long-cycle-life Zn-based batteries focus mainly on the Zn anode and its interface with the electrolyte.70–78 Influenced by these strategies, Zn-based batteries have achieved notable advancements, wherein the metrics of output voltage, capacity, and energy density attain levels of 2.92 V, 2000 mA h g−1, and 2372 W h kg−1, respectively (as determined by the mass of active materials), as depicted in Table 1. Simultaneously, Zn-based batteries display a prolonged cycle life, surpassing 1000 hours.
| Types of Zn-based batteries | Cathode material | Electrolyte | Capacity (mA h g−1)/working voltage (V) | Energy density (W h kg−1)/cycle life (h or cycles) | Mechanism | Ref. |
|---|---|---|---|---|---|---|
| Zn-ion battery | V6O13/carbon cloth | 3 M ZnSO4 | 520/0.98 | 511/1000 cycles | Insertion-type mechanisms | 191 |
| MnVO | 3 M Zn(CF3SO3)2 | 610.2/0.77 | 469/4000 cycles | 192 | ||
| La–Ca co-doped MnO2 | 1 M ZnSO4 + 0.4 M MnSO4 | 297/1.35 | 401/200 cycles | 215 | ||
| MnO2 | 1 M (NH4)2SO4 + 0.1 M MnSO4 | 365/1.35 | 493/4000 cycles | 234 | ||
| CoFe(CN)6 | 4 M Zn(CF3SO3)2 | 173.4/1.75 | 303/2200 cycles | 66 | ||
| LiVPO4F | 21 M LiTFSI + 2 M Zn(OTf)2 | 140/1.65 | 230/600 cycles | 242 | ||
| Co0.247V2O5 | 20 M LiTFSI + 1 M Zn (TFSI)2 | 432/1.15 | 497/7500 cycles | 67 | ||
| α-MnO2 | 1 M Al(CF3SO3)3 + 1 M Zn(CF3SO3)2 | 265/1.5 | 395/1000 cycles | 245 | ||
| CuHCF | WIG-NaCl/ZnSO4/SA | 260/1.7 | 440/450 cycles | 116 | ||
| S@C | Gelatin/CuSO4//gelatin/ZnSO4 | 2063/1.15 | 2372/100 cycles | Conversion-type mechanisms | 204 | |
| S/KB | 0.5 M CuSO4//0.5 M Na2SO4//0.5 M ZnSO4 | 2000/1.15 | 2300/100 cycles | 201 | ||
| S@CNTs-50 | 1 M Zn(CH3COO)2 + 0.05 wt% I2 | 1105/0.455 | 502/50 cycles | 203 | ||
| PbO2 | 4 M H2SO4 + 0.5 M K2SO4//6 M KOH + 0.8 M Zn(CH3COO)2 | 86/2.92 | 252.4/250 cycles | 64 | ||
| SeS5.76@3D-NPCF | 3 M ZnSO4 + 0.1 wt% I2 | 1222/0.71 | 867.6/500 cycles | 205 | ||
| Se/CMK-3 | 2 M ZnTFSI/PEG/water | 611/1.01 | 751/500 cycles | 206 | ||
| PAC/I2 | ZnCl2 + LiCl + CH3CN | 594/1.26 | 750/6000 cycles | 236 | ||
| Co3O4 | 2 M ZnSO4 + 0.2 M CoSO4 | 200/1.8 | 361/5000 cycles | 241 | ||
| MnO2 | 1 M MnSO4 + 1 M ZnSO4 + 0.1 M H2SO4 | 570/1.95 | 1100/1800 cycles | 246 | ||
| MnO2 | 0.5 M Mn(Ac)2 + 0.5 M Zn(Ac)2 + 2 M KCl | 616/1.58 | 973/400 cycles | 253 | ||
| BQ-NCC | 3 M ZnSO4 | 489/1.1 | 538/500 cycles | Coordination-type mechanisms | 233 | |
| C4Q | 3 M Zn(CF3SO3)2 | 335/1 | 335/1000 cycles | 147 | ||
| HAQ-COF | 2 M ZnSO4 | 344/0.84 | 289/10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles 000 cycles |
199 | ||
| Zn–air battery | F–Ni27Fe10-LDH(charge cathode)//FeNC(discharge cathode) | 6 M KOH + 0.2 M Zn(Ac)2 | 786.6/1.2 | 959.6/5220 h | Catalysis-type mechanisms | 456 |
| HESA | 6 M KOH + 0.2M Zn(Ac)2 |
779/0.96 | 748/200 h | 411 | ||
| NPMC foam | 6 M KOH | 735/1.14 | 835/240 h | 386 | ||
| XC72R | 1 M Zn(OTf)2 | 684/1 | 684/1600 h | 502 | ||
| Pt/C + Co3O4 | Gel electrolyte | 742/1.3 | 964/200 h | 513 | ||
| Pt/C + IrO2 | ZnSO4 + LiTFSI//H2SO4 + LiTFSI | 802.3/1.5 | 1243.6/2000 h | 481 | ||
| Fe3C-NG/Ni foam | 6 M KOH + 0.2M Zn(Ac)2 |
810/0.87 | 706.4/1000 h | 492 | ||
| NiCo2S4/N-CNT | 6 M KOH + 0.2 ZnCl2 | 431.1/1.29 | 554.6/150 cycles | 504 | ||
| Zn–CO2 battery | CHF | The ionic liquid | 340/0.85 | 288.3/192 h | 518 | |
| Zn–C2H2 battery | Cu dendrites | 1 M KOH | 786/0.27 | 213.8/40 h | 521 | |
| Zn–NO2− battery | C/Co3O4 | 1 M KOH//0.5 M K2SO4 + 50 mM NO2− | 764.3/0.67 | 512.1/24 h | 519 |
For Zn-based batteries, beyond the pursuit of high-performance batteries, understanding energy storage mechanisms and exploring new reaction mechanisms have also emerged as the key research areas.52,79–83 The reaction mechanism of Zn-based batteries diverges from the established energy storage chemistry reliant on Li/Na ions (including intercalation, conversion, and alloying reactions), remaining enigmatic and subject to frequent debate. Using the Zn–MnO2 battery as an example, three distinct energy storage mechanisms have been proposed, encompassing (1) Zn2+ insertion/extraction reaction, (2) H+/Zn2+ co-insertion reaction, and (3) conversion reaction. To deepen our comprehension of the reaction mechanism in Zn-based batteries and advance the design of higher-performance counterparts, ongoing efforts involve the use of diverse electrochemical methods, advanced characterization techniques (both in situ and ex situ), and theoretical calculations. Since battery performance is affected by multiple factors such as the electrolyte, electrodes, and the energy storage mechanism, more efforts are required to reveal the reaction mechanism underlying the overall battery device.
This review surveys the latest efforts in designing high-performance Zn-based batteries and elucidating energy storage mechanisms (Fig. 2). We will discuss the four types of redox mechanisms (i.e., insertion-type, conversion-type, coordination-type and catalysis-type mechanisms), highlight the design strategies employed in enhancing the performance of Zn-based batteries while concurrently presenting challenges encountered and providing insights into the future outlook of these batteries. It is imperative to acknowledge that the references discussed in this review are not meant to be exhaustive; instead, they serve as initial literature for more in-depth studies. Additionally, this review leverages valuable insights from numerous outstanding reviews on the Zn-ion battery, Zn–air battery, and flexible battery, enhancing our understanding of reaction mechanisms and design strategies for high-performance Zn-based batteries.45,84–99 Furthermore, our discussion extends beyond the scope of Zn-based batteries, as the strategies and mechanisms delineated herein are applicable to a wider array of battery systems.
 | ||
| Fig. 2 Overview of energy storage mechanisms and design strategies employed in Zn-based battery systems. | ||
2. Mechanisms of Zn-based batteries
The energy storage mechanism of Zn-based batteries is presently in its nascent research phase, contributing to controversy and misunderstanding in comprehending the reaction process and mechanism. In addition, the intricate nature of battery devices adds complexity to the exploration and comprehension of the reaction mechanism in Zn-based batteries. Various configurations of Zn-based battery devices share a common structure, predominantly featuring Zn anode, electrolyte, and cathode. Irrespective of their classification, the anodes in all Zn-based batteries function based on the Zn2+/Zn redox couple. Nevertheless, diverse electrochemistry can be achieved on the cathode side owing to variations in electrolyte and cathode materials. Therefore, in this review, we mainly discuss various redox mechanisms on the cathode side. In this section, four types of redox mechanisms based on the different electrochemical behaviors of the cathode will be introduced: (1) insertion-type mechanisms, (2) conversion-type mechanisms, (3) coordination-type mechanisms, and (4) catalysis-type mechanisms. Among the four reaction mechanisms, the insertion-type mechanism is predominantly observed in Zn-ion batteries featuring inorganic materials (such as Mn-based oxide or V-based oxide) as the cathode electrode, while the conversion-type mechanism is primarily evident in Zn-ion batteries and Zn-based redox flow batteries. In particular, the coordination-type mechanism mainly appears in Zn-ion batteries with organic materials as the cathode electrode (Zn–organic batteries). In addition, the catalysis-type mechanism is predominantly present in Zn-based batteries wherein the cathode relies on a catalyst to facilitate redox reactions, exemplified by Zn–air batteries and Zn–CO2 batteries.2.1. Insertion-type mechanisms
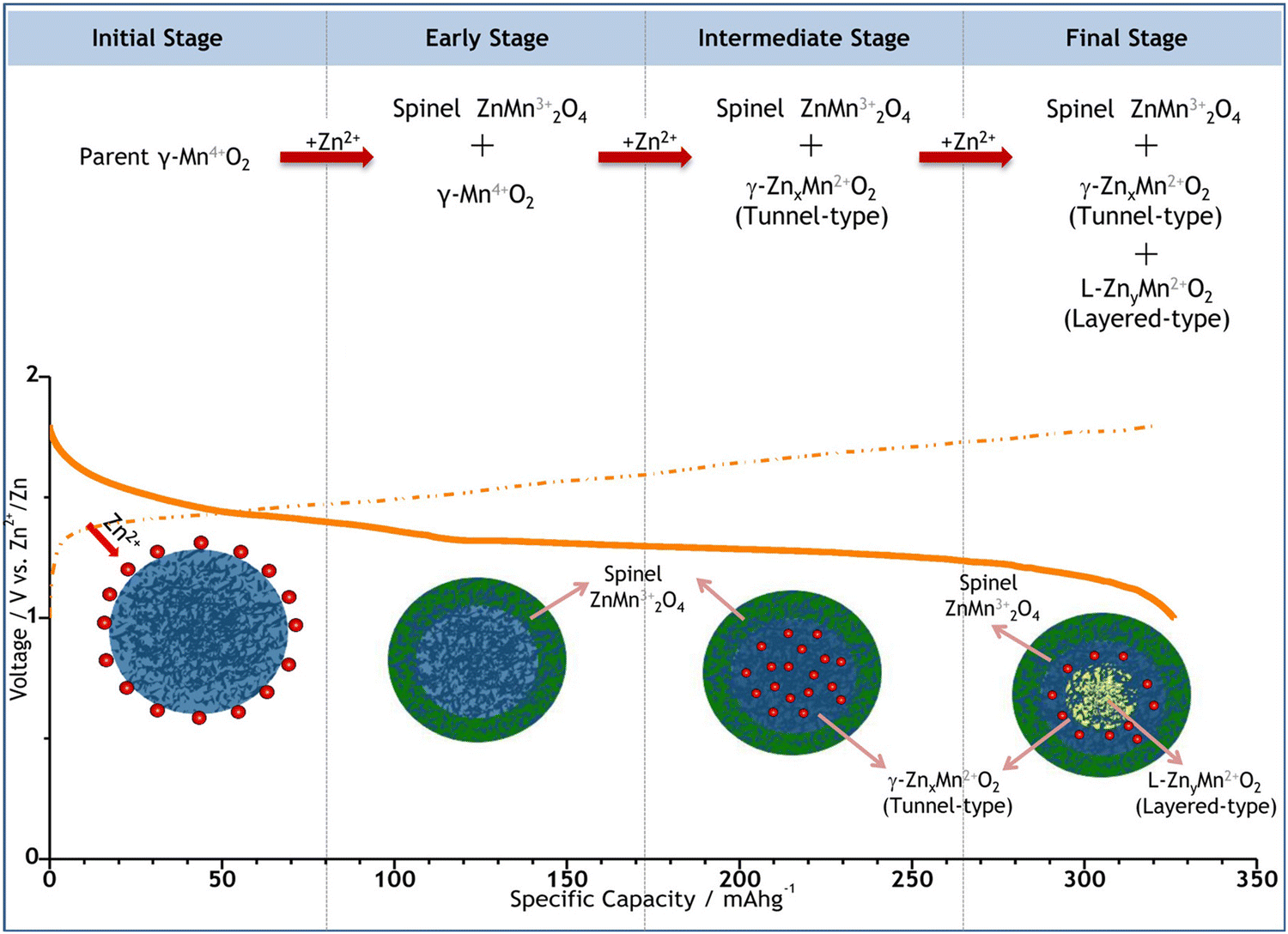 | ||
| Fig. 3 Schematic depiction of the electrochemically induced multi-step phase transition and the corresponding Zn2+ insertion process in the γ-MnO2 cathode. Reproduced with permission from ref. 103. Copyright 2015 American Chemical Society. | ||
Apart from the aforementioned Mn-based active materials, the exploration of electroactive cathode host materials for reversible zinc ion insertion/extraction has extended to include other substances with layered or tunnel-type structures, such as V-based oxides.84,88,104–107 Illustratively, Mai et al. employed layered Na0.33V2O5 nanowires as cathode materials for Zn-ion batteries, utilizing X-ray photoelectron spectroscopy (XPS), XRD, scanning electron microscopy (SEM), and transmission electron microscopy (TEM), to unveil the reversible insertion/extraction mechanism of zinc ions in cathode materials.108 The schematic diagram in Fig. 4 depicts the complete charge and discharge process. During discharge, a segment of the Na0.33V2O5 nanowire undergoes a phase change, transforming into a novel ZnxNa0.33V2O5 phase. This structural transformation is due to the crystal distortion caused by the insertion of Zn2+. In contrast, during the charging phase, the new phase formed during the discharge process gradually dissipates owing to the progressive deintercalation of Zn2+, underscoring the high reversibility of the phase change and structural alteration in Na0.33V2O5.
 | ||
| Fig. 4 Schematic depiction of the Zn2+ insertion mechanism in the Na0.33V2O5 cathode. Reproduced with permission from ref. 108. Copyright 2018 John Wiley & Sons. | ||
 | ||
| Fig. 5 Co-insertion mechanism of H+ and Zn2+ ions. (a) GITT curve depicting the behavior of Zn/MnO2 batteries. (b) Galvanostatic charge–discharge curves of Zn/MnO2 batteries in an organic Zn-based electrolyte, with and without H2O addition. (c) Discharge curve of the MnO2 cathode in a MnSO4 solution, with or without ZnSO4 as the electrolyte. (d) XRD patterns depicting the MnO2 cathode post-discharge at 1.0 V and 1.3 V. Reproduced with permission from ref. 111. Copyright 2017 American Chemical Society. | ||
The co-insertion reaction mechanism involving Zn2+ and H2O has garnered considerable attention and scrutiny within various Zn-ion battery systems. Notably, Naza and her colleagues recently conducted an investigation into the co-insertion mechanism of H2O and Zn ions in Zn/V2O5 batteries (Fig. 6).112 After a series of electrochemical characterization and structural characterization (XRD, TEM and XPS), the authors revealed the specific evolution process of Zn2+ and H2O insertion and extraction in layered V2O5. Throughout the discharge process, as Zn2+ and H2O were gradually inserted, the initially layered V2O5 underwent a transformation into the layered ZnxV2O5·nH2O phase, incorporating water. Concurrently, the interlayer spacing expanded from the original 4.4 Å to 13.4 Å, owing to the co-insertion of H2O molecules and hydrated Zn2+ ions. In the subsequent charging process, the newly formed phase from the discharge gradually dissipated as the hydrated Zn ions detached from the lattice of the cathode material. Because the crystal structure of the cathode host material usually has charge repulsion with Zn2+ ions, the co-insertion of water can effectively slow down this charge repulsion. When only organic electrolytes were used without water, the battery exhibited very limited capacity, demonstrating that water co-insertion can reduce charge repulsion.
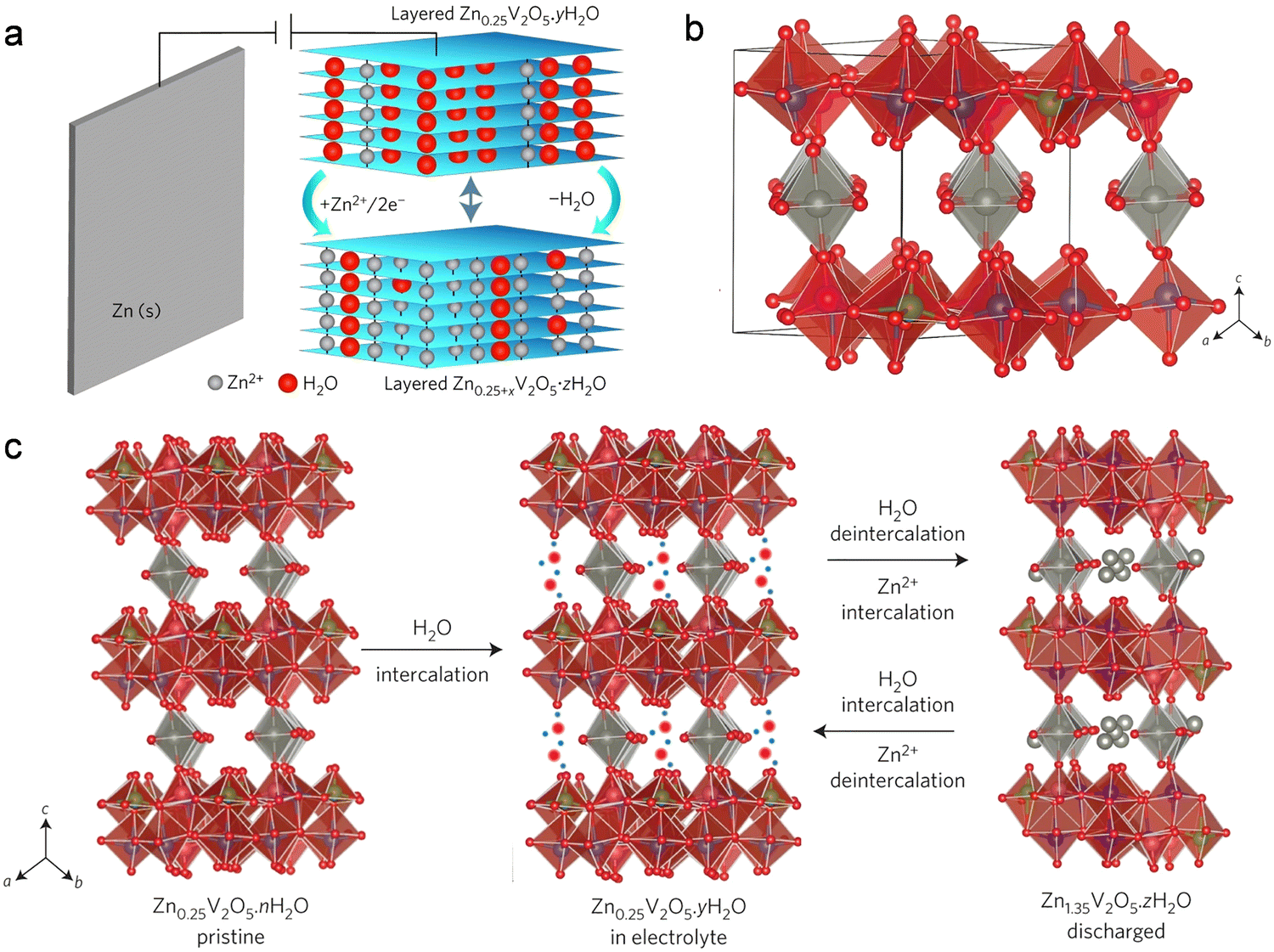 | ||
| Fig. 6 Co-insertion mechanism of H2O and Zn2+. (a) Schematic representation illustrating the co-insertion of H2O and Zn2+ during discharge. (b) Crystal structure depiction of Zn0.25V2O5. (c) Schematic diagram delineating the reversible co-insertion/extraction process of H2O and Zn ions in Zn0.25V2O5·nH2O. Reproduced with permission from ref. 112. Copyright 2016 Springer Science & Business Media. | ||
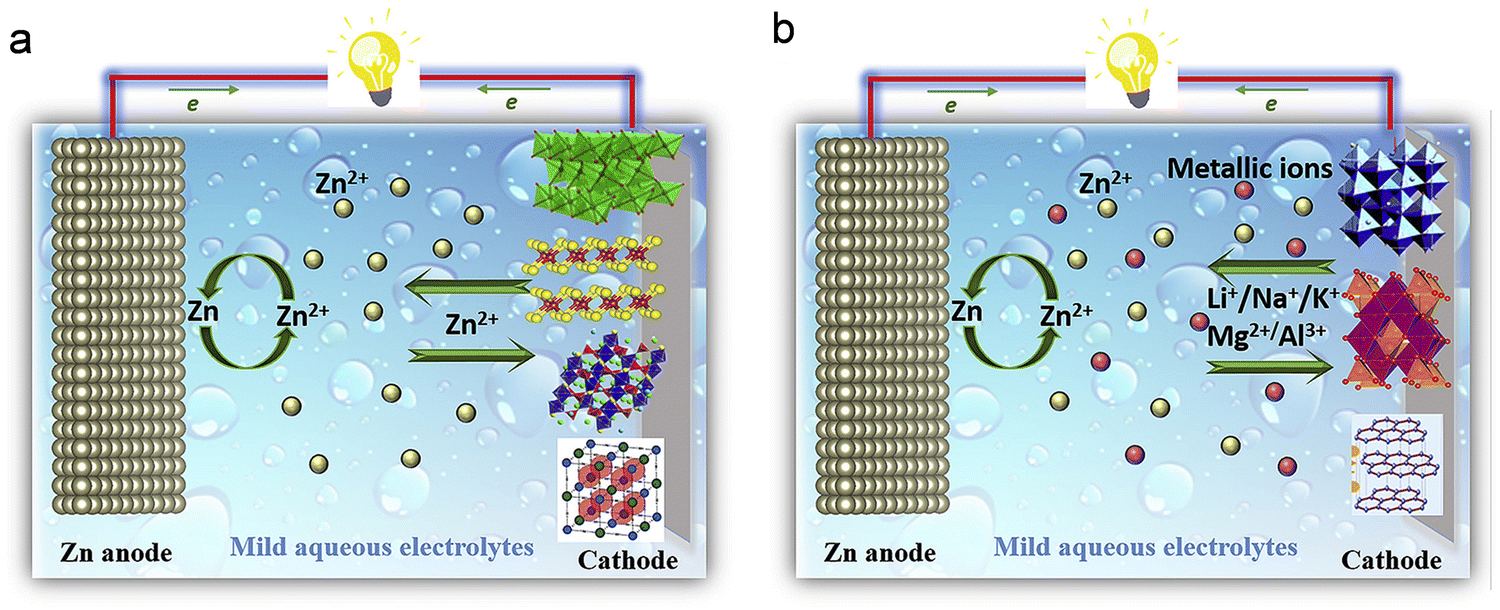 | ||
| Fig. 7 Comparing the operational principles of Zn-ion batteries. (a) Zn-ion battery (Zn2+ insertion/extraction mechanism). (b) Hybrid Zn-ion battery (Li/Na/K/Mg/Al insertion/extraction mechanism). Reproduced with permission from ref. 114. Copyright 2019 Elsevier. | ||
For a more comprehensive comprehension of the hybrid insertion mechanism, the LiMn2O4//Zn battery serves as an illustrative example to delineate the cathode and anode evolution throughout the charge and discharge processes.122 The electrolyte for this battery comprises a blended aqueous solution containing LiCl and ZnCl2. By using XRD to monitor the evolution of spinel LiMn2O4 and Zn anodes during battery charge and discharge, the hybrid insertion–extraction mechanism is revealed. Concerning spinel LiMn2O4, as the charging process unfolds, the characteristic peaks associated with the (111), (311), and (400) planes of spinel LiMn2O4 progressively shift to higher angles. This shift indicates both the desorption of Li+ ions and the formation of the Mn2O4 phase during charging.123 Conversely, throughout the discharge process, these characteristic peaks exhibit a gradual shift towards lower angles, ultimately returning to their original positions. This pattern reflects the reversible insertion/extraction process of Li ions in the LiMn2O4 material. For zinc anodes, XRD results also confirmed the reversible deposition/dissolution process of zinc during charge and discharge. Preceding the charging process, only the characteristic peaks of the carrier are discernible in the XRD spectrum. Post-charging, the emergence of characteristic peaks corresponding to (002), (010), and (011) of Zn in the XRD spectrum substantiates the deposition of zinc ions from the mixed electrolyte onto the carrier. In this hybrid insertion–extraction mechanism, the cathode experiences an intercalation/deintercalation process involving alkali metal ions, while the anode undergoes a reversible Zn deposition/dissolution process.
2.2. Conversion-type mechanisms
 | ||
| Fig. 8 Conversion mechanism based on protonation/deprotonation. (a) Schematic diagram of phase evolution during discharge. Reproduced with permission from ref. 124. Copyright 2019 Springer Science & Business Media. (b) The XRD pattern of α-MnO2 in its fully discharged state unequivocally validates the generation of MnOOH. Reproduced with permission from ref. 125. Copyright 2016 Springer Science & Business Media. | ||
Beyond the charge/discharge processes observed in mild Zn-ion batteries, governed by the deprotonation/protonation mechanism, certain alkaline Zn-ion batteries (e.g., Ni–Zn, Co–Zn batteries) also follow the deprotonation/protonation mechanism. Due to this mechanism, these alkaline Zn-ion batteries typically achieve higher operating voltages, approximately in the range of 1.7–1.8 V, in comparison to other aqueous rechargeable batteries. In alkaline electrolytes, materials such as Co or Ni-based oxides/hydroxides, sulfides, and phosphides interact with OH− under voltage-driven conditions.126 Charge storage is achieved through deprotonation/protonation conversion reactions during charge and discharge. Illustrating the deprotonation/protonation mechanism of Ni or Co-based hydroxides, the process can be succinctly represented by the equation A(OH)2 + OH− ↔ AOOH + H2O + e−, where A denotes metal cations like Co or Ni.
Lu et al. employed the deprotonation/protonation reaction mechanism to fabricate a high-performance Zn-ion battery, utilizing 2D nickel cobaltite (NiCo2O4) nanosheets endowed with abundant O defects as the active material.127 In the charging process, NiCo2O4 nanosheets undergo an oxidation reaction with OH−, transforming into NiOOH and CoO2. Simultaneously, [Zn(OH)4]2− in the electrolyte is also reduced to Zn. Conversely, in the discharge process, both CoO2 and NiOOH are reduced to lower valence oxides, and Zn undergoes oxidation with the hydroxide anion in the electrolytes, resulting in the formation of [Zn(OH)4]2−. This deprotonation/protonation reaction mechanism is succinctly represented by the following equation:
| Cathode: NiCo2O4 + H2O + OH− ↔ 2CoOOH + NiOOH + e− | (1) |
| CoOOH + OH− ↔ H2O + CoO2 + e− | (2) |
| Anode: Zn(OH)42− + 2e− ↔ 4OH− + Zn | (3) |
| 3Zn(OH)42− + 2NiCo2O4 ↔ 2NiOOH + 4CoO2 + 6OH− + 2H2O + 3Zn | (4) |
In addition to transition metal hydroxides, nickel- or cobalt-based sulfides and phosphides are also used in alkaline Zn-ion batteries, and their charge and discharge processes are also based on the deprotonation/protonation conversion mechanism. Concurrently, batteries employing cathode materials based on nickel or cobalt sulfides and phosphides exhibit enhanced performance owing to their conductivity and more suitable electronic structure in comparison to transition metal oxides.128 Transition metal sulfides and phosphides participate in reversible redox reactions with OH− in aqueous electrolytes during charge and discharge processes, facilitating charge storage. Exemplifying this with transition metal sulfides, the reaction mechanism in alkaline Zn-ion batteries is succinctly represented by the equation AS + OH− ↔ ASOH + e−, where A denotes the metal cation.
| Zn + I3− ↔ Zn2+ + 3I−, E = 1.299 V. | (5) |
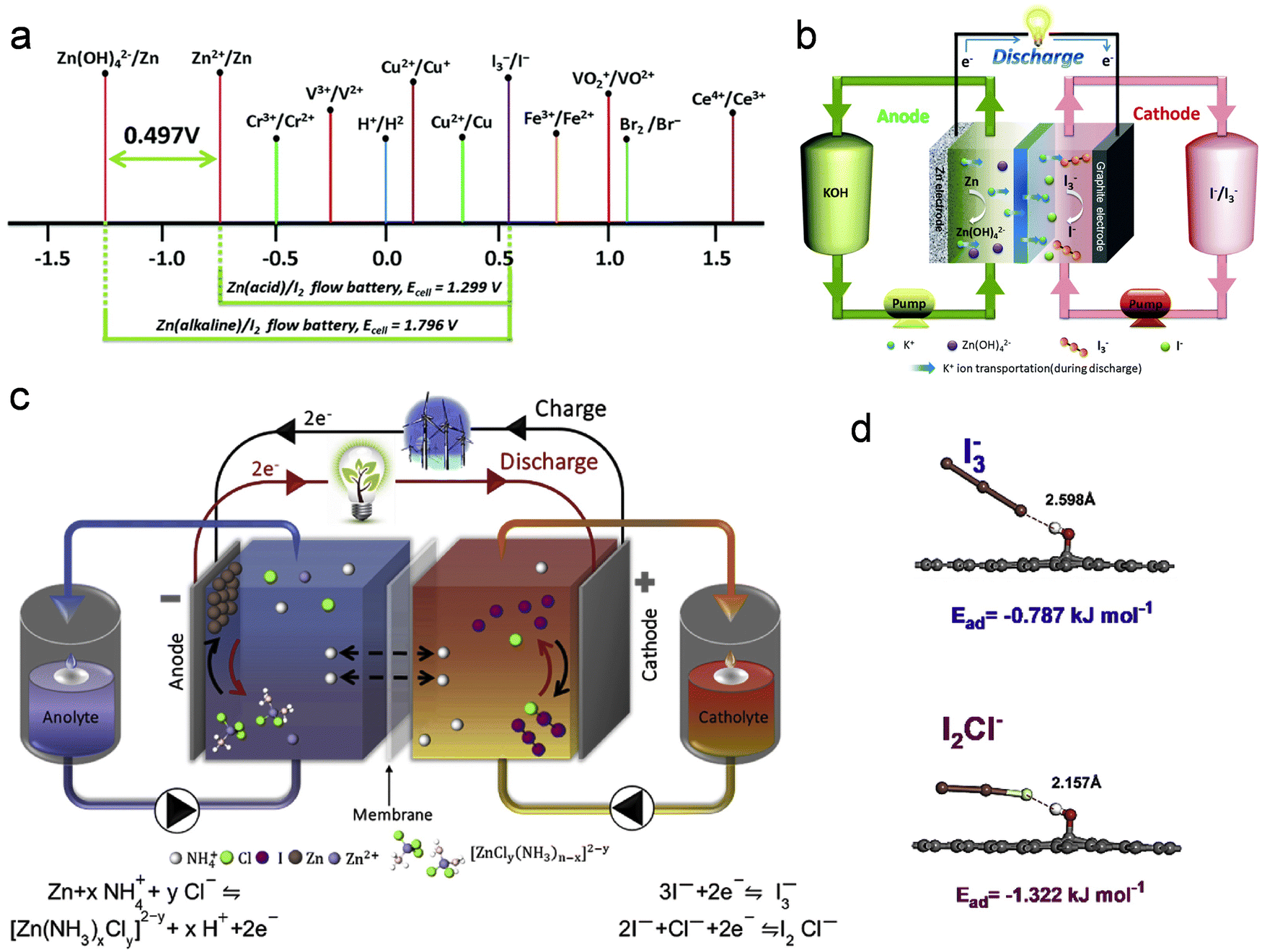 | ||
| Fig. 9 Zn–I2 battery based on the redox-couple conversion mechanism. (a) Standard redox potentials for various candidate redox couples. (b) Illustration outlining the operational mechanism during the discharge phase of an alkaline Zn–I2 redox flow battery. Reproduced with permission from ref. 136. Copyright 2018 Royal Society of Chemistry. (c) Illustrative depiction elucidating the operational principles of a Zn–I2 redox flow battery utilizing an electrolyte containing NH4Cl. (d) DFT optimized molecular structure, and anions on the cathode surface. Reproduced with permission from ref. 137. Copyright 2020 Elsevier. | ||
From the results of the above equation, it can be seen that the theoretical electrode voltage of the traditional Zn–I2 flow battery is only 1.299 V. Therefore, it is necessary to innovate on the basis of the traditional Zn–I2 flow battery to increase its voltage. In this context, the electrolyte decoupling strategy emerges as an effective approach to elevate the voltage of Zn–I2 flow batteries. A case in point is the design by Chen and colleagues, who implemented a hybrid alkaline Zn–I2 redox flow battery by transitioning the anolyte from acidic to alkaline conditions (Fig. 9b).136 This hybrid Zn–I2 flow battery exhibits an elevated electrode voltage, marked by a potential enhancement of 0.497 V compared to the conventional Zn–I2 flow batteries. The redox mechanism of this hybrid system can be succinctly captured by the following equation:
| Zn + 4OH− + I3− ↔ 3I− + Zn(OH)42−, E = 1.796 V | (6) |
In Zn–I2 flow batteries, the challenges of zinc dendrite formation and the precipitation of insoluble iodine (I2) are primary contributors to their compromised cycle stability and low energy efficiency. These issues significantly impede the large-scale practical applications of Zn–I2 flow batteries. Recently, Chen et al. successfully addressed both challenges by incorporating ammonium chloride (NH4Cl) into the electrolyte of Zn–I2 flow batteries (Fig. 9c).137 The addition of NH4Cl serves a dual purpose in enhancing Zn–I2 flow battery performance. Firstly, it effectively modulates the chemical composition of the electrolyte, suppressing both the formation of Zn dendrites and the generation of insoluble iodine. Secondly, NH4Cl not only accelerates the reaction kinetics at the cathode and anode but also involves Cl− in the formation of I2Cl, contributing to additional capacity release (Fig. 9d). This mechanism-guided design proves instrumental in effectively ameliorating Zn–I2 flow battery performance.
2.3. Coordination-type mechanisms
Coordination-type mechanisms are primarily characteristic of Zn–organic batteries. In recent years, investigations have revealed that numerous organic compounds, including caliquinone, p-tetrachlorobenzoquinone, and tetrachloro-1, among others, serve as effective cathode materials in reversible Zn-ion batteries.90,138–146 Through coordination with Zn2+, the organic compounds undergo alterations in their charge states during the redox processes. This evolution in the charge state of the electroactive group serves as a valuable indicator for elucidating the redox mechanism in Zn–organic batteries. To pinpoint the specific sites within organic compounds that engage in coordination with Zn2+, Chen and collaborators conducted a meticulous analysis, determining the active sites by comparing electrostatic potentials (ESP) across various locations within organic compounds (Fig. 10a–e). ESP, a widely employed metric in organic chemistry, proves instrumental in assessing electrophilic or nucleophilic reactions.147 Because the cathode will involve the absorption of Zn2+ during battery discharge, sites with more negative ESP in organic compounds are beneficial to the discharge reaction. By comparing the ESP of each site in the calix[4]quinone (C4Q) molecule, it can be seen that the carbonyl groups on the molecule and below the molecule show a lower ESP than the bilateral carbonyl groups, which indicates that the carbonyl groups on the molecule and below the molecule are more conducive to the adsorption of Zn2+. | ||
| Fig. 10 Inferring the active site and structural evolution of quinone cathodes. (a)–(e) The ESP-mapped van der Waals surface of diverse organic molecules. (f) Optimized molecular structure of C4Q before and after Zn2+ absorption. Reproduced with permission from ref. 147. Copyright 2018 American Association for the Advancement of Science. | ||
In contrast to their inorganic counterparts, organic cathode materials often exhibit intrinsically low crystallinity, posing challenges for accurate characterization through conventional methods such as XRD and TEM. Nevertheless, these organic cathode materials boast an abundance of oxygen- or nitrogen-containing functional groups. Consequently, the discernment of the reaction mechanism in Zn–organic batteries during redox processes is achievable through Raman spectroscopy, ultraviolet-visible spectroscopy, and attenuated total reflection Fourier transform infrared spectroscopy. For instance, Chen and colleagues employed various in situ spectroscopic techniques to investigate the energy storage mechanism of C4Q organic molecules during charging and discharging. As the discharge progresses, the two carbonyl groups positioned atop the C4Q molecule coordinate with a single Zn2+ ion, accepting two electrons. Simultaneously, the four carbonyl groups located at the base of the C4Q molecule coordinate with the remaining two Zn2+ ions. During the subsequent charging phase, C4Q undergoes oxidation, resulting in the release of the stored electron and zinc ion during the reverse discharging process (Fig. 10f). The coordination evolution between C4Q and Zn2+ throughout the charge and discharge phases can be succinctly expressed by the following equation:
| C4Q + 3Zn2+ ↔ Zn3C4Q | (7) |
2.4. Catalysis-type mechanisms
The catalysis-type mechanism denotes the operational process in Zn-based batteries, wherein a catalyst is essential for facilitating the completion of the redox reaction at the cathode. This mechanism is observed in various Zn-based battery configurations, encompassing Zn–air batteries and Zn–CO2 batteries. Extensive research has been undertaken to elucidate the mechanism of the oxygen reduction reaction (ORR) during discharging and the oxygen evolution reaction (OER) during charging in Zn–air batteries. To augment the overall performance of Zn–air batteries, numerous advanced catalysts, including high-performance bifunctional electrocatalysts, have been devised based on both OER and ORR mechanisms. In contrast to the swift progress observed in Zn–air batteries, the development of Zn–CO2 batteries, an emerging category of Zn-based batteries, is at the beginning of its exploration phase. The catalytic conversion of carbon dioxide in Zn–CO2 batteries is notably intricate compared to that of oxygen, rendering the mechanism more complex. Beyond the incorporation of the catalytic conversion mechanisms of oxygen and carbon dioxide into Zn-based batteries, various other catalytic conversion mechanisms have been introduced. Examples include the catalytic conversion of acetylene (Zn–acetylene battery) and the catalytic conversion of nitrate (Zn–nitrate battery). This section focuses predominantly on elucidating the mechanisms of Zn–air batteries and Zn–CO2 batteries. | ||
| Fig. 11 Illustration depicting the fundamental structure and reaction mechanism of Zn–air batteries. Reproduced with permission from ref. 155. Copyright 2020 Springer Science & Business Media. | ||
During the discharging and charging process of the Zn–air batteries, O2 in the atmosphere penetrates into the gas diffusion layer with a porous structure of the air cathode, as depicted in the schematic diagram. Subsequently, the ORR occurs on the catalyst layer of the cathode. Concurrently, hydroxide ions, produced as a byproduct at the air cathode, gradually migrate towards the zinc anode, culminating in the completion of the Zn–air battery discharge reaction. The charging process of Zn–air batteries is a reversible counterpart of the discharge phase. The overarching electrochemical reaction mechanism for Zn–air batteries is succinctly expressed by the following equation:
| Anode: Zn + 4OH− ↔ Zn(OH)42− + 2e− | (8) |
| Zn(OH)42− ↔ H2O + ZnO + 2OH− | (9) |
| Cathode: 2H2O + O2 + 4e− ↔ 4OH− | (10) |
| Overall reaction: 2Zn + O2 → 2ZnO | (11) |
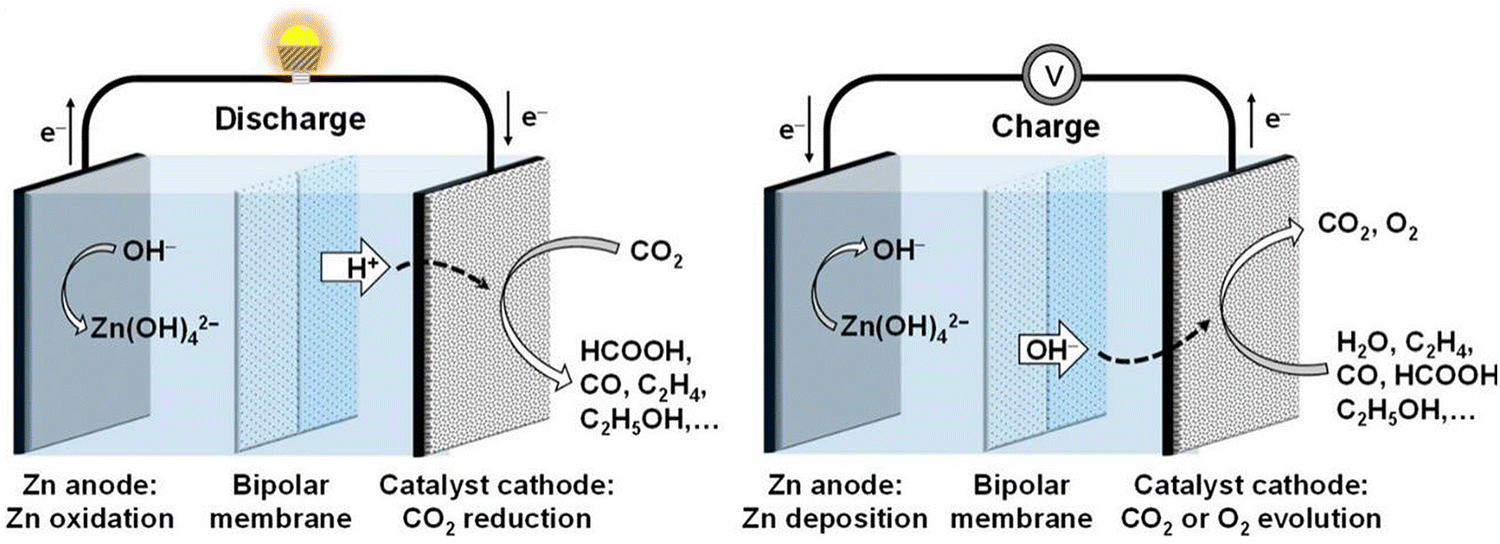 | ||
| Fig. 12 Illustration depicting the discharge and charging processes of an aqueous Zn–CO2 battery. Reproduced with permission from ref. 166. Copyright 2022 John Wiley & Sons. | ||
Cathode:
| CO2 + 2e− + H+ → CO + H2O, E0 = −0.11 V | (12) |
| CO2 + 2e− + 2H+ → HCOOH, E0 = −0.20 V | (13) |
| 2CO2 + 12e− + 12H+ → 4H2O + C2H4, E0 = −0.07 V | (14) |
| 2CO2 + 12e− + 12H+ → 3H2O + C2H5OH, E0 = 0.08 V | (15) |
Anode:
| Zn + 2OH− → H2O + ZnO +2e−, E0 = −1.26 V | (16) |
Based on the standard electrode potential, the spontaneous discharge of aqueous Zn–CO2 batteries is thermodynamically viable. However, the efficiency of these batteries is hindered by the low electrocatalytic efficiency in CO2 conversion, attributed to the chemical inertness of CO2 molecules and the competition for hydrogen evolution reaction (HER) in aqueous electrolytes. In recent years, researchers have innovatively developed efficient CO2 conversion electrocatalysts, encompassing heteroatom-doped carbon, single-atom electrocatalysts, and metal catalysts. Aqueous Zn–CO2 batteries employing these electrocatalysts have demonstrated successful reduction of CO2 to CO, HCOOH, C2H5OH, and CH4.176,177 However, in these Zn–CO2 battery systems, the charging process is still based on the water oxidation process (OER).174,178 In addition, Wang and his colleagues accomplished a reversible conversion of CO2 to formic acid (HCOOH) in an aqueous Zn–CO2 battery, employing bifunctional Pd as a cathode catalyst.179 As the discharge process proceeds, CO2 is converted into HCOOH on the Pd catalyst. For the charging reaction, the reversible process of converting HCOOH into CO2 will occur at the Pd cathode (CO2 + 2e− + 2H+ ↔ HCOOH).
3. Design strategies for high-performance Zn-ion batteries
The Zn-ion battery is the earliest studied and most representative type of battery in Zn-based batteries. Capitalizing on attributes such as high safety, ample reserves, low price, and substantial energy density, the rechargeable Zn-ion battery has garnered considerable attention as an efficient energy storage solution. Nevertheless, when compared to conventional Li-based rechargeable batteries, the performance of Zn-ion batteries falls short of the target due to constraints like a narrow electrochemical stable potential window, limited discharge capacity, intricate side reactions, and low operating voltage. Addressing these challenges and striving for an ideal Zn-ion battery characterized by heightened reversibility and energy density, diverse strategies have been employed to optimize the electrochemistry, cathode materials, the Zn anode, and the electrolyte. The ensuing sections will delve into these design strategies, with a particular focus on enhancing electrochemical performance concerning capacity, working voltage, energy density, and cycle life.3.1. Design strategies for high-capacity Zn-ion batteries
 | ||
| Fig. 13 Theoretical capacity. (a) Theoretical capacity calculation of electrode materials. (b) Factors affecting high theoretical capacity. (c) Comparing the theoretical capacities of various cathode materials. | ||
In comparison to inorganic compounds, the coordination-type organic compounds have lower theoretical capacity due to the substantial proportion of inert carbon forming the skeleton (Fig. 13c).90,196–198 Fortunately, since the carbon skeleton in coordination organic compounds has flexible and adjustable characteristics, it is very possible to introduce active coordination groups on the carbon skeleton in coordination-type organic compounds. In addition to being easy to introduce active groups, coordination-type organic compounds also have the advantage of low mass, so they are regarded as ideal candidates for electrode materials possessing high theoretical capacities.90 As an illustration, enhancement of activity and augmentation of active coordination sites can be achieved by introducing redox-active quinone groups into 1,4,5,8,9,12-hexaazatriphenylene-based covalent organic frameworks (HAQ-COFs), leading to an increased capacity of 344 mA h g−1.199 Furthermore, the hierarchical arrangement of poly(para-aminophenol, pAP) and poly(1,5-naphthalene-diamine, 1,5-NAPD) on nanoporous carbon is able to enhance the full activation of active coordination centers (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/CO) for Zn2+.200 Conversely, while certain coordination organic compounds, exemplified by benzoquinone (BQ) small molecules, demonstrate high theoretical capacities, the cathode incorporating these organic small molecules is deficient in adequate covalent structure, thereby compromising battery stability. Therefore, while pursuing enhanced capacity in coordination-type organic compounds, it becomes imperative to prioritize the stability of the underlying structure. The target is to attain a harmonious equilibrium between electrochemical performance (capacity and cycling stability) and structure (active groups and carbon skeleton).
N/CO) for Zn2+.200 Conversely, while certain coordination organic compounds, exemplified by benzoquinone (BQ) small molecules, demonstrate high theoretical capacities, the cathode incorporating these organic small molecules is deficient in adequate covalent structure, thereby compromising battery stability. Therefore, while pursuing enhanced capacity in coordination-type organic compounds, it becomes imperative to prioritize the stability of the underlying structure. The target is to attain a harmonious equilibrium between electrochemical performance (capacity and cycling stability) and structure (active groups and carbon skeleton).
Unlike cathode materials of insertion or coordination types, conversion-type cathode materials hold a distinguished status in the realm of high-theoretical-capacity categories. A notable illustration of this is the prevalent utilization of sulfur as the electrode material in Zn-ion batteries, due to its engagement in diverse redox reaction pairs and ultrahigh theoretical capacity,201–204 including S/Cu2S (approximately 3350 mA h g−1) and S/ZnS (approximately 1675 mA h g−1) (Fig. 13c). Nevertheless, inherent challenges within Zn//S batteries, such as irreversible by-products, sluggish kinetics, and specific pH specifications, constrain both the practical electrode potential and the capacity of aqueous Zn//S batteries. To handle these issues, many methods have been proposed. The incorporation of the electrolyte additive I2 has demonstrated effectiveness, as I2 acts as a medium for Zn2+ while concurrently reducing reaction barriers. This property proves advantageous for achieving high energy efficiency and low-voltage hysteresis in Zn–S batteries.203 Moreover, the utilization of an infinite solid solution between sulfur (S) and selenium (Se), denoted as S–Se, serves to optimize the band structure, electronic structure as well as activation energy of S. Consequently, this leads to a significant improvement in both reactivity and conductivity.205 Beyond S, the proposal to utilize Se as an electrode material for Zn-ion batteries holds promise, offering a notable capacity of 611 mA h g−1.206 Despite the intrinsic advantages of elemental S and Se concerning the electron transfer number and mass, a persistent challenge lies in their low output voltage range (0.455–1.01 V). A feasible solution involves pairing them with high-voltage redox reactions, exemplified by the S/Cu2S system, which operates at a working voltage of 1.15 V.
 | ||
| Fig. 14 Schematic diagram of the adverse effects of various factors (strong repulsive effect, insufficient active sites, and low conductivity) on the theoretical capacity of cathode materials. | ||
In Zn-ion batteries based on the traditional insertion-type mechanism, a noteworthy challenge arises from the intense electrostatic repulsion between the Zn2+ ion and the host materials. This phenomenon results in suboptimal utilization of the active material and diminished battery capacity.207 To address this challenge, an effective approach involves the preliminary insertion of certain ions182,208–211 or molecules107,212–214 into layered cathode materials. This process serves to mitigate electrostatic interactions and widen the spacing between layers, consequently enhancing the storage capacity of Zn-ion batteries (425–550 mA h g−1). It is noteworthy, however, that the introduction of inactive ingredients during insertion may lead to a reduction in valency and ratio of active ingredients within the cathode material. Additionally, semiconductor oxides face a challenge in their limited electron transfer capabilities, undermining the actual capacity. Broadly speaking, the enhancement of electrical conductivity can be attained by tuning the band gap of materials through heteroatom doping. As an illustration, the co-doping scheme involving the introduction of La and Ca into MnO2, yielding Ca/La co-doped MnO2, has demonstrated reduced charge transfer resistance (29.8 vs. 198.1 Ω) and increased capacity (297 vs. 199 mA h g−1) in comparison to pure MnO2.215 In addition, defect engineering stands out as an additional effective strategy for enhancing actual capacity. The introduction of defects proves effective in diminishing electrostatic repulsion and elevating carrier concentration.106,190,216–220 On the other hand, it is inevitable to accurately control and characterize these defects, while it can often be a complex and challenging endeavor.
Carbonaceous materials and lightweight carbon-based materials, like graphene, MXene, carbon nanotubes, and carbon fiber, are extensively employed as supports for active materials. Their utilization is driven by their substantial active surface area and high conductivity, facilitating enhanced contact between the cathode material and ions in the electrolyte and improving charge transfer rates.221–225 As a consequence, there is a notable augmentation in the capacity of the cathode.226 Beyond the intrinsic attributes of the active material that influence actual capacity, extrinsic factors also contribute to determining the practical capacity of the electrode materials. Frequently, binders obstruct a segment of the effective active area of the cathode material, impeding direct contact between active materials and charge carriers.227–231 Carbon substrates featuring surface functional groups obviate the necessity for binders, facilitating adhesive-free immobilization of the cathode material and maximizing the exposure area of active materials. A case in point is the substantial enhancement in the actual capacity of the Co-Mn3O4 material, achieved by combining it with a carbon nanosheet array – from 220 mA h g−1 to 362 mA h g−1.232 Likewise, the incorporation of N-doped carbon cloth effectively secures benzoquinone, preventing its dissolution and thereby stabilizing its capacity.233
 | ||
| Fig. 15 Mechanism innovation in design strategies. (a) Illustration portraying the H+/NH4+ co-insertion mechanism. (b) Alterations in local geometry and charge induced by the co-insertion of H+ and NH4+ into MnO2. Reproduced with permission from ref. 234. Copyright 2021 John Wiley & Sons. (c) Schematic diagram of direct conversion reaction. (d) Energy changes based on the two processes of the Zn2+ migration mechanism and the direct conversion mechanism. Reproduced with permission from ref. 237. Copyright 2021 John Wiley & Sons. | ||
Compared with the insertion-type mechanism and the coordination-type mechanism, the conversion-type mechanism has some unique advantages such as high selectivity towards charge carriers and strong practical versatility. This suggests the potential for employing various redox reaction systems in Zn-ion batteries. When designing high-capacity energy storage mechanisms, it is essential to consider electron transfer numbers and the mass of active materials. Consequently, it is viable to pursue continuous transformation reactions involving large numbers of transferred electrons and exploit them for high-capacity energy storage mechanisms. For example, a unique continuous conversion reaction mechanism centered on continuous I+/I2/I− conversion reaction, which involves a four-electron transfer process, has been finely tailored.135,235 This innovation has elevated Zn–I batteries to a remarkable capacity level of 594 mA h g−1.236 Furthermore, because the conversion reaction mechanism has the advantage of versatility, a variety of cathode materials with conversion-active functional groups can adapt to the conversion-type mechanism, thereby realizing high-capacity Zn-ion batteries. Recently, the conversion mechanisms based on Cu–CuI have been explored, and other types of Cu-based materials (Cu2O, Cu3N, Cu2S, and so on) have shown high electrochemical capacity with their unique structures through this conversion-type mechanism (Fig. 15c and d).237 Although the chemical process based on the conversion-type mechanism appears to be a relatively simple process, in addition to reaction stability, carrier compatibility with the electrolyte, anode, and additional electrolyte mass must also be considered.
The paramount objective in realizing high-energy-density Zn-ion batteries is the design of batteries with elevated capacity. Through the introduction and advancement of diverse design strategies, a considerable number of Zn-ion batteries have made substantial strides in enhancing their capacity,238 and the capacity of some Zn-ion batteries can even reach 2000 mA h g−1. Achieving high-capacity performance alone is insufficient due to the inherent drawback of low working voltages (0.4–1.3 V). This limitation stems from the high ratio of the Gibbs free energy change (ΔG) to the number of electron transfers in the cathodic redox reaction (Fig. 16a).
 | ||
| Fig. 16 Design strategies for high-voltage Zn-ion batteries. (a) Design principles for high potential reactions. (b) Various strategies for expanding the electrochemical stability potential window. | ||
3.2. Design strategies for high-voltage Zn-ion batteries
Currently, while high-capacity Zn-ion batteries find extensive application, only a restricted subset of high-voltage Zn-ion batteries have attained a relatively high energy density, ranging from 200 to 440 W h kg−1. This is attributed to their narrow electrochemical stability potential window and inherently low theoretical capacity. It is evident that numerous challenges persist in achieving groundbreaking advancements in both high-capacity and high-voltage Zn-ion batteries due to their intrinsic limitations. Therefore, dedicated efforts are required to explore strategies that enable the simultaneous achievement of high-voltage and high-capacity Zn-ion batteries.
3.3. Design strategies for high-energy-density Zn-ion batteries
 | ||
| Fig. 17 Optimizing the conversion reaction mechanism. (a) Performance comparison of various Zn-based batteries. Reproduced with permission from ref. 246. Copyright 2019 John Wiley & Sons. (b) Charging and discharging curves of Zn//Te batteries and schematic diagram of their six-electron reversible transfer chemistry. Reproduced with permission from ref. 264. Copyright 2023 American Chemical Society. | ||
Cathode:
| Mn2+ + 2H2O ↔ 4H+ + MnO2 + 2e− E0 = 1.228 V vs. SHE | (17) |
Anode:
| Zn2+ + 2e− ↔ Zn E0 = −0.763 V vs. SHE | (18) |
Overall:
| Zn2+ + 2H2O + Mn2+ ↔ Zn + MnO2 + 4H+ E = 1.991 V vs. Zn/Zn2+ | (19) |
While the MnO2 dissolution/deposition process within the MnO2/Mn2+ conversion mechanism opens up new possibilities for high-energy-density Zn-ion batteries, it introduces the complexity of a liquid–solid phase reaction. To navigate this complexity, effective design strategies for the liquid–solid phase interface become crucial.247 Examining the MnO2 dissolution/deposition process during charge and discharge reveals two elementary reactions: MnO2 to Mn3+ and Mn3+ to Mn2+. Consequently, the diffusion and aggregation of Mn2+, Mn3+, and H+ at the liquid–solid interface significantly impact the discharging voltage and platform of the Zn//MnO2 battery. It is noteworthy that during discharge, the ion insertion mechanism may prevail over the conversion mechanism. The reversible MnO2 deposition/dissolution capability is intricately tied to the specific surface area of the current collector and the number of redox active sites. To optimize performance, precautions should be taken to avert the excessive generation of a MnO2 layer on the cathode surface. This is essential, as an overly thick MnO2 layer can compromise cathode conductivity, leading to diminished reaction reversibility and sluggish reaction kinetics. At the same time, the use of strong acidic electrolytes will also cause the side reaction of HER on the zinc anode.248 For each of the factors mentioned above, it will adversely affect the output voltage, coulombic efficiency, and energy efficiency of Zn–MnO2 batteries. To circumvent the challenges associated with MnO2 dissolution/deposition in Zn–MnO2 batteries following the conversion mechanism, researchers typically adopt a specific battery configuration. However, this approach inevitably leads to increased volume and mass of the battery. Subsequent to the pivotal findings in this domain, numerous researchers have proposed innovative strategies. These include designing decoupled electrolytes and incorporating liquid flow devices, all aligned with the conversion mechanism, to address the aforementioned issues. These strategic designs have successfully elevated the output voltage of Zn//MnO2 batteries beyond 2.0 V, achieving an extraordinary energy density ranging from 600 to 1700 W h kg−1.249,250
While optimizing the conversion mechanism in a strong acidic electrolyte can improve the energy density of Zn–MnO2 batteries, it comes with a trade-off. The strong acidic electrolyte has the drawback of corroding the electrode and inducing the side reaction of HER on the zinc anode. This underscores the importance of balancing electrolyte conditions to ensure both performance enhancement and prevention of undesired side reactions. In this regard, optimizing the conversion mechanism in a mild, less corrosive electrolyte can both achieve high energy density and increase battery durability. Conversely, employing a low-acidity electrolyte introduces its own set of challenges. The reduced acidity can also lead to low Coulombic efficiency in the battery due to the competing side reaction between OER and MnO2 deposition during charging. In order to inhibit the occurrence of OER, the Mn2+ concentration in the electrolyte should be adjusted to a range that is conducive to the reversible MnO2/Mn2+ reaction.251 Following this, the researchers implemented a cathode composed of a Mn2+-rich multivalent manganese oxide layer supported on carbon cloth. The presence of oxygen vacancies in this cathode material serves to diminish local oxygen mobility and restrain the onset of OER. Simultaneously, the abundance of Mn2+ enhances the dynamics of electron transfer.252 In addition, the nature of anions in the electrolytes contributes to influencing the electrochemical behavior of the Zn//MnO2 battery utilizing the conversion mechanism. In a mild electrolyte containing CH3COO−, the coordination of CH3COO− facilitates the direct conversion of Mn2+ into MnO2 during the charging process, bypassing the intermediate state of Mn3+. This also greatly improves the reversibility of the battery.253,254 However, the use of CH3COO− will reduce the output voltage of the battery, so an appropriate CH3COO− concentration needs to be used. In addition, adding redox mediators such as KI to the electrolyte can promote the dissolution of residual MnO2 in the electrolyte,255 thereby promoting the output performance of Zn-based batteries. Hence, the use of redox mediators also provides a new direction for optimizing the conversion mechanism.
Additionally, beyond the mentioned MnO2 cathodes relying on the conversion mechanism, diverse conversion cathodes have emerged and been investigated for Zn-ion batteries in recent years, encompassing halogens (Cl2, Br2, and I2) and chalcogens (S, Se, and Te).205,206,256–260 Te, one of the chalcogen elements, is used as an example to further illustrate the optimization strategy of the conversion mechanism. Given Te's plentiful valence state transitions spanning from −2 to +4 and its substantial intrinsic conductivity of 2 × 10−2 S m−1, a Zn-ion battery employing Te as the cathode boasts an exceedingly high theoretical capacity of 1692 mA h g−1 (under six electron transfers) and the prospect of a high electrode potential.261 Yet, the present Zn–Te battery, with Te as the cathode material, only demonstrates a modest output voltage of 0.59 V.262 This low output voltage is caused by the fact that the conversion reaction of Te is limited to the redox of Te2−/Te0 rather than the expected positive valence state transition from Te0 to Ten+, which also limits the progression of the Zn–Te battery toward high output voltage and high energy density.239,263 Recently, Zhi and his colleagues successfully activated the required reversible Te2−/Te0/Te2+/Te4+ redox behavior and the transfer of up to six electrons by using high-concentration Cl-containing electrolytes (Fig. 17b).264 This Zn–Te battery has three flat discharge platforms (at 1.24, 0.77 and 0.51 V respectively), and its total capacity can reach 802.7 mA h g−1. In addition, a modified ionic liquid was used as the electrolyte, which improved the stability of Te2+/Te4+ and enhanced the cycle stability, thereby achieving an impressive energy density of 542 W h kgTe−1.
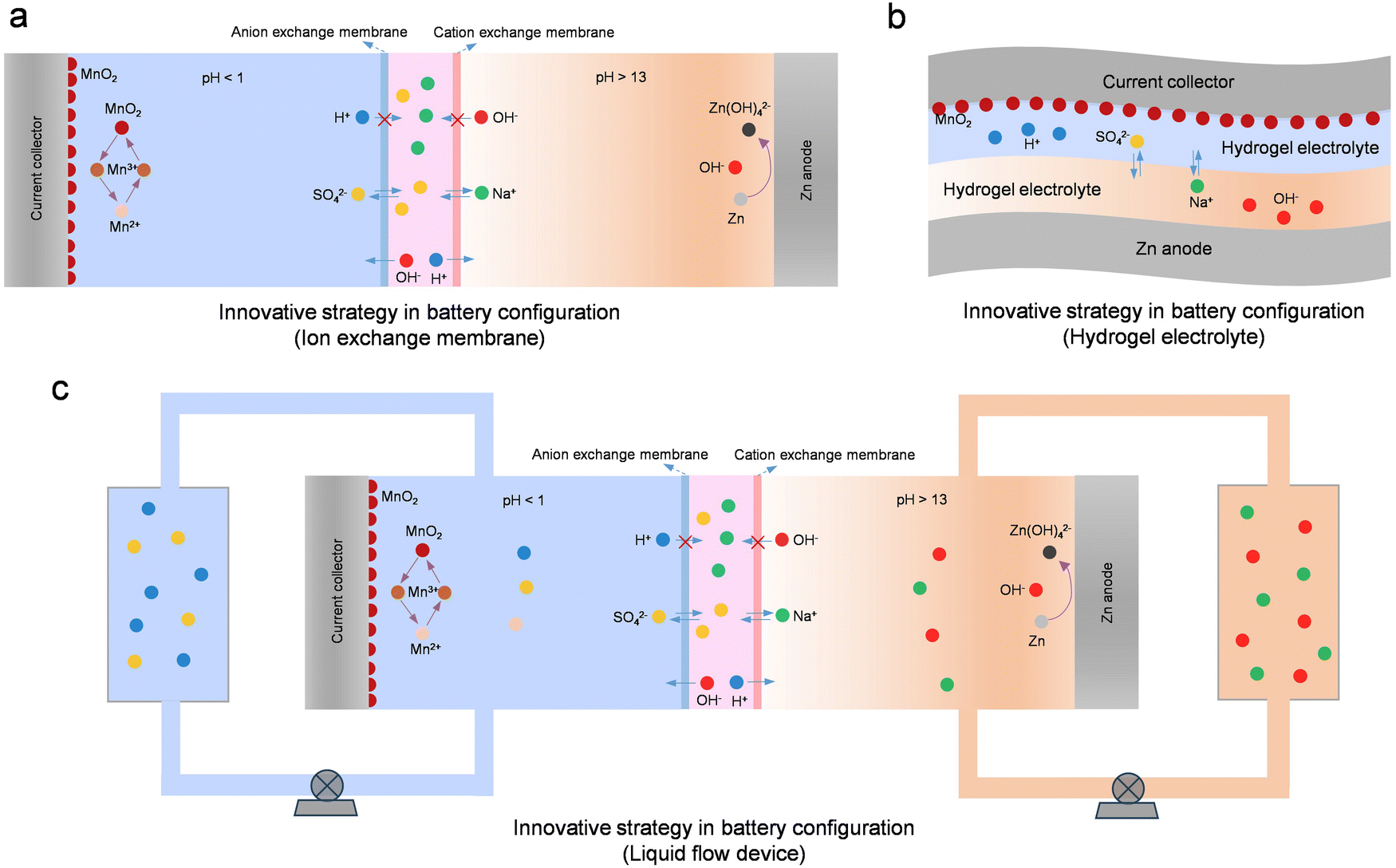 | ||
| Fig. 18 Innovative strategies in battery configuration. (a) Decoupled battery based on the ion exchange membrane. (b) Hydrogel-based decoupled battery. (c) Decoupled batteries using liquid flow devices. | ||
While ion exchange membranes effectively decouple the electrolyte in Zn-ion batteries, their high cost significantly limits their widespread application. The use of low-cost hydrogels instead of ion exchange membranes offers a promising approach to decoupling electrolytes (Fig. 18b). Because the water content in the hydrogel is relatively low, the ion mobility inside it is relatively low. Therefore, the zinc anode and cathode can be effectively separated even without the use of the ion exchange membrane.267 At the same time, because hydrogel materials have the advantages of mechanical flexibility and good electrochemical stability, Zn-ion batteries using hydrogels as the electrolyte have a great practical application value, such as being used as flexible and wearable batteries.267,268 Moreover, employing a decoupled hydrogel electrolyte can effectively augment the operating voltage and energy density of battery devices. For instance, in a Zn//MnO2 battery utilizing hydrogels as the electrolyte, its operating voltage can reach 2.4 V, and the energy density can reach an impressive 1700 W h kg−1.249 Yet, when utilizing a hydrogel electrolyte, addressing challenges like compromised battery cycle stability and diminished voltage efficiency arising from the crossover of cathode and anode ions is imperative.
While ion exchange membranes and hydrogel electrolytes can effectively decouple battery devices, ion crossover between the anode and the cathode poses a persistent challenge. Throughout the battery's charging and discharging processes, water in the electrolyte decomposes into OH− and H+. Driven by the electric field, H+ passes through the cation exchange membrane, neutralizing the alkaline electrolyte, while OH− moves through the anion exchange membrane, neutralizing the strongly acidic electrolyte. Therefore, strategies are needed to further address the ion crossover problem. In this regard, integrating the respective advantages of flow batteries and decoupled batteries provides a very promising strategy to avoid ion crossover (Fig. 18c). In recent years, various high-energy-density Zn-ion batteries based on this integration strategy have been proposed, such as Zn//Br2, Zn//MnO2, Zn//V, Zn//Fe, Zn//Ce, and others.269–277 The introduction of liquid flow devices provides new possibilities for realizing high-energy-density Zn-ion batteries.
3.4. Design strategies for long-cycle-life Zn-ion batteries
 | ||
| Fig. 19 Modification of the zinc anode. (a) Summary of undesirable reactions on Zn anodes. (b) Illustration depicting the introduction of additives on the Zn anode. (c) Schematic diagram of surface modification of the Zn anode. (d) Schematic diagram illustrating the structural design applied to the Zn anode. | ||
Surface modification of electrodes emerges as a very effective strategy for enhancing the cycling stability of the Zn anode (Fig. 19c). This approach enhances the accessibility of the zinc anode bulk and surface, mitigates the occurrence of side reactions, and augments the reversibility of the zinc anode.71,291–306 In recent years, researchers have formulated diverse protective layers, encompassing organic materials, metals, and metal compounds, to improve the longevity of the Zn-ion battery by modifying zinc anodes. Notably, Kang and co-workers used a CaCO3 coating with porous structure as a protective layer for the zinc anode to achieve uniform zinc stripping/plating.307 This approach proves effective in suppressing zinc dendrite growth, preventing short circuits, and ultimately enhancing the cycle stability of Zn-ion batteries. The zinc anode coated with a porous nano-CaCO3 layer was then assembled into a Zn–MnO2 battery. This coated zinc anode exhibited remarkable performance in capacity retention, delivering 177 mA h g−1 even after 1000 cycles at a current of 1 A g−1. In stark contrast, a battery assembled with a pure Zn anode under identical conditions provided a lower capacity of 124 mA h g−1. In addition, Mai et al. used atomic layer deposition technology (ALD) to successfully coat an ultra-thin TiO2 layer on the zinc anode.308 The amorphous TiO2 coating with high conductivity can serve as a protective layer for the zinc anodes to suppress irreversible by-products and side reactions (HER), thus promoting the cycle stability of the Zn-ion battery. The zinc electrode with a TiO2 coating was incorporated into a Zn–MnO2 battery, exhibiting exceptional cycle stability. It retained 85% of its capacity even after undergoing 1000 cycles at a current density of 3 mA cm−2.
The design of the structure of zinc anodes plays a crucial role in achieving reversibility as it directly influences the generation of zinc dendrites, the overall structural changes of the electrodes, and the internal resistance levels (Fig. 19d).309,310 For instance, introducing a porous structure to achieve zinc anodes with high specific surface area is beneficial to improving the contact between the Zn electrode and the electrolyte, thereby reducing ion transmission distances and significantly improving zinc utilization. Moreover, a zinc anode with high surface area minimizes the deposition overpotential of Zn2+, effectively restraining the generation of Zn dendrites and the generation of the by-product. Various configurations of zinc anodes with substantial specific surface areas, including sheets, fibers, ribbons, spheres, and foams, have been documented.311 Taking a cue from this, Rolison and colleagues crafted a three-dimensional, sponge-like zinc anode characterized by a highly porous structure, aiming to enhance the utilization of zinc anodes.312 The three-dimensional porous structure, featuring a high specific surface area, facilitates a more uniform distribution of charge during battery charging and discharging. This design proves effective in inhibiting the growth of zinc dendrites. In addition, this three-dimensional porous structure can limit the deposition of Zn2+ inside the pore channels, which further reduces the generation of zinc dendrites during charge and discharge. Leveraging its distinctive three-dimensional framework, the sponge-like zinc anode exhibits a dendrite-free performance even under high-rate cycling conditions, reaching up to 188 mA h g−1. Building upon this foundation, Rolison and collaborators subsequently refined this innovative three-dimensional porous sponge-like zinc anode, culminating in the development of a nickel-3D zinc rechargeable battery with high safety and high energy density.313 In battery performance tests, the Ni-3D Zn battery was able to provide high specific energy competitive with commercial lithium-ion batteries. In addition, this Ni-3D Zn battery can achieve ultra-long cycle life and therefore has a great practical value.
Moreover, the introduction of additives into the electrolyte is another avenue to enhance the cycle stability of Zn-ion batteries. For instance, the inclusion of MnSO4 in the ZnSO4 electrolyte serves as an effective measure to prevent the cathode from dissolving in the Zn–MnO2 battery.125 Furthermore, incorporating additives proves instrumental in addressing issues related to Zn anodes that impact battery cycle stability, including the generation of Zn dendrites, the occurrence of HER, and the formation of by-products.286,342–346 Various types of additives (such as dimethyl sulfoxide,347 polyacrylamide,348 arginine,349 polyethylene oxide,350 urea,351 lignin sulfate,352etc.) have been reported to be feasible for improving the cycle stability of the battery. For example, Hu and colleagues added nontoxic xylitol as an additive to the traditional ZnSO4 electrolyte to achieve dendrite-free stripping/plating of zinc metal anodes.344 According to experimental tests and theoretical simulations, the introduction of a small amount of xylitol (100 mM) effectively regulates the interaction between Zn2+ ions, H2O molecules, and SO42− ions through the rearrangement of hydrogen bonds. Concurrently, xylitol molecules exhibit a preference for adsorption on the zinc surface, creating a shielding buffer layer that delays sedimentation and hinders the planar diffusion of Zn2+ ions. For batteries containing xylitol molecules, their cycle stability is significantly better than the cycle stability of batteries that do not use additives. How to stabilize the pH environment between the electrode and the electrolytes while suppressing dendrite growth and side reactions is also a key point in achieving the high cycle stability of Zn-ion batteries. In a recent study, Parkin and collaborators introduced a multifunctional additive, ammonium dihydrogen phosphate, which not only suppressed the generation of Zn dendrites and side reactions but also maintained a stable pH environment (Fig. 20).353 Electrochemical investigations, coupled with theoretical calculations, reveal that NH4+ in the additive exhibits a preference for adsorption on the zinc electrode surface, creating a “shielding effect” that diminishes contact between the zinc electrode surface and H2O. Additionally, the interaction between (H2PO4)− and NH4+ contributes to pH stabilization at the electrode–electrolyte interface. This multifunctional additive facilitates highly reversible zinc plating/stripping behavior in Zn//Zn and Zn//Cu cells.
 | ||
| Fig. 20 Optimization of electrolytes using additives. (a) Schematic diagram of the zinc deposition process in a 1 M ZnSO4 electrolyte. (b) Cycle performance of Zn//Zn symmetrical batteries in 1 M ZnSO4 electrolytes. (c) Schematic diagram of the zinc deposition process in 1 M ZnSO4 and additive electrolyte. (d) Cycle performance of Zn//Zn symmetrical batteries in 1 M ZnSO4 and additive electrolyte. Reproduced with permission from ref. 353. Copyright 2023 John Wiley & Sons. | ||
000 cycles at a substantial current density of 10 A g−1.354 In addition to using chain polymers as carriers, small organic molecules with redox active centers can also be grafted onto covalent organic framework materials (COFs). Utilizing COFs as carriers endows small organic molecules with the advantageous attributes of high specific surface area and exceptional stability inherent to COFs. This enhancement facilitates the swift transport of ions during charge and discharge processes, contributing to an extended cycle life.355–360 As an illustration, Kurungot and colleagues employed a hydroquinone-linked β-ketoenamine COF as zinc ion anchors in Zn–organic batteries.361 Owing to the high porosity and stable structure of the COF, the battery maintains an impressive 95% of its initial capacity after enduring 1000 cycles at a substantial current density of 3750 mA g−1.
4. Design strategies for high-performance Zn–air batteries
Zn–air batteries have garnered widespread research interest due to their remarkable theoretical energy density (1086 W h kg−1), intrinsic safety associated with aqueous electrolytes and stable zinc anode, cost-effectiveness of abundant Zn ore (20 times less than Li), and the straightforward preparation, storage, and environmentally friendly nature of Zn.47,98,362–374 Significantly, Zn–air batteries offer the potential for synergy with other energy storage systems, providing complementary advantages. In terms of commercial viability, Zn–air batteries are widely regarded as holding substantial promise, garnering extensive attention over the past decade. Despite the escalating reports on Zn–air batteries, challenges persist, including the sluggish kinetics and instability of the air electrode, limited Coulombic efficiency of the anode, and the generation of dendrites. These issues contribute to limited discharge capacity, low energy density, and shortened cycle life. To address these challenges and achieve Zn–air batteries with high performance, diverse design strategies have been proposed and developed. The subsequent sections will delve into various approaches aimed at enhancing the capacity, output voltage, energy density, and cycle life of Zn–air batteries.4.1. Design strategies for high-capacity Zn–air batteries
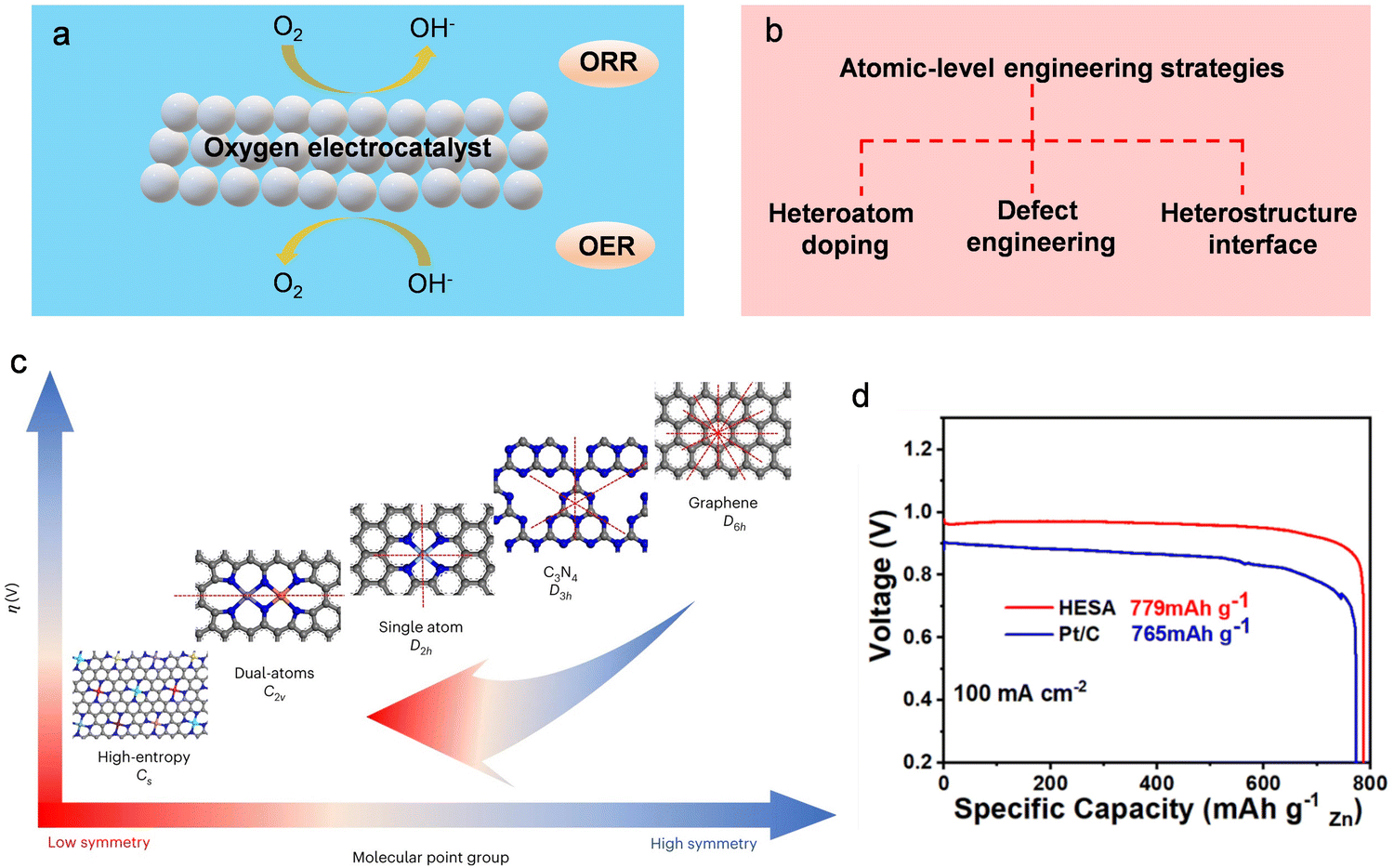 | ||
| Fig. 21 Tuning the local electronic structure and surface/interface properties of oxygen electrocatalysts through atomic-scale modulation engineering. (a) The schematic delineates the operational principles governing the oxygen electrocatalyst at the three-phase interface. (b) Schematic diagram of the content of atomic-scale modulation engineering. (c) Relationship between symmetry of atomic modulation and electrocatalytic performance. (d) The specific capacity normalized with the consumed zinc for HESA and Pt/C (20%) at 100 mA cm−2. Reproduced with permission from ref. 411. Copyright 2023 Springer Science & Business Media. | ||
High-performance oxygen electrocatalysts with bifunctional properties (ORR/OER) play a pivotal role in facilitating the practical application of Zn–air batteries.376 The oxygen electrocatalytic reaction typically takes place at the three-phase interface involving gas (O2), solid (catalyst), and liquid (electrolyte). The intrinsic activity of this reaction is intricately linked to the electronic structure on the surface of the solid electrocatalyst. Consequently, optimizing the adsorption/desorption behavior of the reaction intermediate, reducing the energy barrier of the reaction, and expediting OER and ORR kinetics necessitate precise tuning of the electronic structure. This underscores the significance of atomic-level modulation engineering.377–385 Currently, numerous reports have surfaced regarding the application of atomic-level engineering strategies to improve the OER and ORR kinetics in electrocatalysts.151,386–404 This approach aims to augment the capacity of Zn–air batteries. Various atomic-level engineering strategies, such as heteroatom doping, defect engineering, and hybrid structure construction, are considered as effective methods to promote oxygen electrocatalysis (Fig. 21b). Heteroatom doping involves the incorporation of heteroatom elements characterized by distinct electronegativity and atomic size in the crystalline structure of the foundational electrocatalyst. This process serves to finely tune the catalyst's inherent electronic configuration, consequently influencing its catalytic performance.405–410 As an illustration, high-entropy single-atom-doped carbon catalysts exhibit exceptional catalytic performance in both OER and ORR, surpassing the capabilities of Pt/C and RuO2 catalysts.411 Their utilization in Zn–air batteries yields significantly increased specific capacity compared to a benchmark RuO2 and Pt/C electrocatalyst (779 mA h gZn−1versus 765 mA h gZn−1, respectively) (refer to Fig. 21c and d). Concurrently, defect engineering stands as a widely acknowledged method for adjusting the electronic structure of host materials.219,372,412–417 Surface defects play a pivotal role in the modulation of electron density in proximity to the Fermi level, facilitating enhanced orbital hybridization. This effect yields a surplus of unsaturated coordination sites and dangling bonds. Moreover, at the heterostructure interface, there is room for efficient adsorption of electron-donating and electron-withdrawing groups, consequently promoting rapid electrocatalytic rates characterized by high reaction kinetics.418 Crucially, the fabrication of heterostructures stands as a rational strategy for attaining bifunctional properties in catalysts.419
Strategies involving atomic-scale engineering for oxygen electrocatalysts exhibit notable enhancements in OER/ORR catalytic performance,420,421 mitigating the concentration polarization issue in Zn–air batteries. Nonetheless, the exclusive reliance on atomic engineering to modify the surface/interface of electrocatalysts falls short of fully optimizing the actual capacity of Zn–air batteries. This discrepancy arises from the variance between the operational conditions of the rotating disk electrode (RDE) and the authentic working environment of air electrodes in Zn–air batteries. In RDE tests, the swift rotation of the RDE minimizes mass transfer resistance, thereby alleviating concentration polarization and maximizing the intrinsic catalytic activity of the bifunctional electrocatalyst. In contrast, the static conditions prevalent in the air electrodes of Zn–air batteries highlight reactant diffusion resistance as a pivotal factor influencing mass transfer efficiency.422 Despite the development of advanced OER/ORR electrocatalysts, enhancing the practical capacity of Zn–air batteries for real-world applications remains imperative. Therefore, in tandem with the atomic-scale design of oxygen electrocatalysts, a concurrent focus on optimizing the electrode structure is essential to enhance mass transfer efficiency,423 ultimately realizing the goal of augmenting the actual capacity of the battery. Typically, air cathodes are fabricated through drop-coating or spray-coating processes, establishing a constrained two-dimensional interface between the gas diffusion layer and the electrocatalyst layer. However, the inadequacy in leveraging the active site and maintaining stable interfacial contacts renders the air cathode prone to inactive reactivity, leading to diminished battery capacity. To address this issue, Yu et al. advocated the use of asymmetric air electrodes, allowing the catalytic process to also take place inside the gas diffusion layer.424 The asymmetric configuration of the air cathode facilitates gas diffusion and electrolyte permeation within this 3D conductive framework. This interconnected 3D framework structure markedly amplifies the active site density and expedites mass transfer. Consequently, the Zn–air battery featuring an asymmetric air cathode demonstrates an elevated capacity (800 mA h gZn−1) compared to its symmetric cathode counterpart (749 mA h gZn−1).
An additional significant challenge encountered in two-electrode Zn–air batteries arises from the zinc anode. The formation of zinc dendrites and the occurrence of HER on the Zn electrode surface during the charging process contribute to diminished Coulombic efficiency and short circuits.309,451 Moreover, elevated discharge depths of the metallic zinc plate per cycle can induce uneven charging/discharging, resulting in shape alterations and irreversible damage of the Zn electrode throughout repeated cycles.452 Recent research efforts predominantly center on developing protective layers for zinc anodes, aiming to impede dendrite growth and mitigate hydrogen evolution.453,454 Nevertheless, these studies conducted each cycle at a low depth of discharge to extend cycling stability, a practice that fails to meet the capacity demands essential for the anode in practical Zn–air batteries.455 Therefore, only after solving the problems of anode and cathode simultaneously, Zn–air batteries can have high discharge capacity, and then it is possible to meet the requirements of actual commercialization. To address the challenges inherent in traditional two-electrode Zn–air batteries, a three-electrode Zn–air battery was devised. This configuration includes a hydrophilic charging cathode, a hydrophobic discharge cathode, and a Zn-free anode (Fig. 22).456 This innovative three-electrode Zn–air battery capitalizes on the decoupling of the cathode, facilitating rapid OER and ORR kinetics. The separation of cathodes allows the OER and ORR processes to unfold on distinct cathodes, preventing the oxidation of the ORR electrocatalyst. Simultaneously, a Zn-free anode, comprising tin-plated copper foam, is employed. This anode induces growth along the (002)Zn plane, effectively curbing hydrogen evolution and mitigating zinc corrosion. Consequently, the novel three-electrode Zn–air battery demonstrates an impressive high capacity of 800 mA h cm−2 per cycle and a minimal voltage gap between charge and discharge platforms, measuring at 0.66 V.
 | ||
| Fig. 22 Working principle and performance of three-electrode Zn–air batteries. (a) Illustration delineating the operational principle of a three-electrode Zn–air battery. Its overall structure includes three parts: a hydrophilic charging cathode, a hydrophobic discharge cathode, and a Zn-free anode. During charging, the anode is linked to the hydrophilic charging cathode, while in discharge the anode is connected to the hydrophobic discharging cathode. (b) Anode design that inhibits hydrogen evolution and produces dendrite-free (002)Zn growth. (c) High-resolution electron microscope image of (002)Zn. (d) Comparison of capacity and stability between three-electrode Zn–air battery and other Zn–air battery systems. Reproduced with permission from ref. 456. Copyright 2023 John Wiley & Sons. | ||
4.2. Design strategies for high-voltage Zn–air batteries
 | ||
| Fig. 23 Design strategies for high-voltage Zn–air batteries. (a) Schematic diagram of how a decoupled Zn–air battery works during discharge. (b) Illustration depicting the charging process of a decoupled Zn–air battery. (c) Rate performance of decoupled Zn–air batteries. Reproduced with permission from ref. 461. Copyright 2016 John Wiley & Sons. (d) Illustration delineating the operational principle of a light-involved Zn–air battery. (e) Discharging/charging curves at different current densities. Reproduced with permission from ref. 466. Copyright 2020 John Wiley & Sons. | ||
4.3. Design strategies for high-energy-density Zn–air batteries
 | ||
| Fig. 24 Design strategies for high-energy-density Zn–air batteries. (a) A visual representation illustrating the operational principles of conventional Zn–air batteries. (b) A visual depiction outlining the operational principles of hybrid Zn–Ni/air batteries. (c) Working mechanism of hybrid Zn–Ni/air batteries. Reproduced with permission from ref. 467. Copyright 2019 Royal Society of Chemistry. (d) Schematic diagram of the design principles and working principle of decoupled Zn–air batteries. (e) Discharge and charging curves of decoupled Zn–air batteries at different current densities. (f) Comparing the discharge capacity curves of the zinc anode in decoupled Zn–air batteries and traditional Zn–air batteries at different currents. Reproduced with permission from ref. 481. Copyright 2022 Elsevier Inc. | ||
Despite the effective decoupling of the alkaline electrolyte in Zn–air batteries achieved through the use of an ion exchange membrane, the substantial cost associated with this membrane has significantly constrained its large-scale application. At this point, low-cost hydrogel electrolytes are viewed as a promising alternative.482 Hydrogel electrolytes, owing to their comparatively low water content, impede ion migration and prevent the crossover of ions in decoupled hydrogel electrolytes. This characteristic effectively mitigates the mixing of anode and cathode electrolytes without the need for additional membranes. In addition, using hydrogel electrolytes to replace alkaline water electrolytes can form flexible and stretchable Zn–air batteries, which have broad practical application prospects in wearable devices, robots, etc. These types of zinc–air batteries that use hydrogels as the electrolyte are called quasi-solid-state Zn–air batteries. These quasi-solid-state Zn–air batteries mainly have two configurations: sandwich type and cable type.156,157,483 In sandwich-type quasi-solid-state Zn–air batteries, hydrogel electrolytes are positioned between the air electrode and the zinc electrode. This configuration ensures stable performance output even when subjected to various bending or twisting conditions. Conversely, cable-type quasi-solid-state Zn–air batteries embrace a coil design, positioning the zinc electrode at the center and completely surrounded by the hydrogel electrolyte. The air electrode, featuring the catalyst layer, is encased around the outer periphery. Presently, quasi-solid-state Zn–air batteries demonstrate remarkable resilience to intricate mechanical forces and deformations. Additionally, their output performance can be customized by interconnecting battery packs in either series or parallel configurations. Despite substantial enhancements in the output performance of quasi-solid-state Zn–air batteries in recent years, the semi-open form of the air electrode poses a challenge, potentially resulting in the depletion of H2O within the hydrogel electrolyte. The advent of all-solid-state electrolytes emerges as an appealing solution to address these issues. All-solid-state systems can deliver high energy density to the power supply in the form of bipolar stacked electrodes and provide higher volumetric energy density by reducing dead space. As an illustration, Lee and colleagues showcased a practical Zn–air pouch battery featuring a cathode composed of (101) faceted copper phosphorus sulfide [CPS(101)], coupled with an antifreeze chitosan biocellulose serving as the superionic conductor electrolyte.484 The employed CPS(101) demonstrates exceptional activity and stability, surpassing 30000 cycles for reversible ORR and OER. Its performance outshines the performance of Pt/C and RuO2 catalysts. Simultaneously, the hydroxide superionic conductor, comprising polymerized chitosan biocellulose, exhibits noteworthy electrical conductivity (86.7 mS cm−1 at 25 °C) along with robust mechanical and chemical stability. The resulting commercially viable all-solid-state Zn–air batteries, at 1 A h scale, achieve an ultrahigh battery-level energy density of 460 W h kgcell−1 and 1389 W h l−1.
4.4. Design strategies for long-cycle-life Zn–air batteries
By structurally designing the electrode, the zinc anode with a continuous pore network architecture can be developed, thereby enhancing the stable phase change between ZnO and Zn throughout the charge and discharge cycles. Simultaneously, the three-dimensional framework structure, characterized by its high specific surface area, elevated electronic conductivity, and fully exposed zinc affinity sites, proves advantageous for the even deposition of zinc and mitigates the risk of uncontrolled zinc dendrite proliferation.486,487 Moreover, the utilization of zinc foam featuring a three-dimensional pore structure proves effective in mitigating passivation issues, thereby enhancing the accessibility of zinc to 90%. Compared with the traditional zinc powder, the introduction of continuous nanoporous structures into zinc electrodes has been proven to be able to effectively stabilize the electrochemical transformation between metallic Zn and ZnO, thereby exhibiting excellent battery cycle stability. For example, Chen's team synthesized a Zn electrode with a bicontinuous nanoporous structure.488 This nanoporous zinc can stabilize the phase transition of zinc during charge and discharge while retaining a continuous network of conductive pores. Conversely, in the initial charge and discharge cycles, the zinc powder lacking a nanoporous structure exhibited a notable alteration in surface morphology. Subsequently, the nanoporous zinc anode was assembled into a Zn–air coin cell, and the electrochemical performance test results showed that the Zn–air battery with a nanoporous anode achieved enhanced charge–discharge behavior and high energy efficiency after 80 cycles.
Modifications at the anode interface, including the incorporation of additives and coatings, represent another effective strategy for enhancing the reversibility of zinc anodes.489–491 The introduction of small amounts of additives into the zinc anode brings about a substantial enhancement in its electrochemical reversibility, resulting in the enhanced long-term cycling performance of the Zn–air battery. Specifically, incorporating carbon-based materials with expansive specific surface areas into zinc anodes offers ample zinc-philic sites for Zn nucleation. This approach helps prevent the formation of Zn dendrites, ensuring the uniform deposition of Zn2+ on the electrode. For example, Yang et al. introduced graphene into a zinc anode and prepared a Zn/graphene composite electrode.492 This Zn/graphene composite anode can effectively prevent Zn corrosion in harsh electrolytes. Simultaneously, Zn–air batteries employing Zn/graphene composites as anodes exhibit lower overpotential and more consistent electrochemical behavior compared to Zn–air batteries utilizing pure zinc anodes over extended charge and discharge cycles. This outcome underscores the positive impact of introducing graphene additives in mitigating the self-corrosion and dendrite generation of Zn anodes. In addition, Qian and colleagues improved the reversibility of zinc during charge and discharge by coating a chemical buffer layer on the zinc metal anode.493 Its chemical buffer layer is constructed by uniformly dispersing ZnO nanorods in the graphene skeleton. For the buffer layer, the adsorption affinity of graphene to zinc ions and the role of ZnO nanorods as uniformly deposited crystal nuclei cooperate with each other to achieve the purpose of reducing passivation and dissolution at the zinc anode interface, thus achieving the reversible precipitation–dissolution behavior of zinc. The integration of a zinc anode with a buffer layer into a Zn–air battery resulted in robust stability over 100 cycles, characterized by minimal voltage fluctuations. As the deposition time of Zn2+ increases, the zinc anode featuring a buffer layer maintains a relatively flat electrode surface, in stark contrast to the evident dendrite formation observed on the surface of the zinc electrode lacking a buffer layer. Consequently, the Zn anode with a buffer layer demonstrated an impressive long-term cycling performance, lasting for 450 hours in an alkaline Zn–air battery. Overall, this chemical buffering strategy provides new ideas and strategies for developing Zn–air batteries with long cycle life.
Recently, the exploration of high-concentration Zn salts, such as ZnCl2, Zn(ClO4)2, Zn(CF3SO3)2, Zn(Ac)2, Zn(OTF)2, Zn(TFSI)2, and ZnSO4, as non-alkaline electrolytes is emerging as a promising avenue to enhance the cycling stability of Zn–air batteries.496–501 Zn–air batteries utilizing highly concentrated non-alkaline electrolytes demonstrate superior reversibility compared to conventional alkaline Zn–air batteries, primarily attributed to the reduction of by-reactions. For example, Wang and his colleagues achieved an efficient and stable operation of Zn–air batteries using a high-concentration Zn(OTF)2 aqueous solution as the electrolyte.502 In particular, owing to the use of the high-concentration aqueous electrolyte, the traditional four-electron process in alkaline Zn–air batteries is transformed into a novel two-electron Zn–O2/ZnO2 electrochemical process. Under the influence of the electric field, hydrophobic OTF anions tend to establish a Zn2+-rich/H2O-poor inner Helmholtz layer (IHL) on the surface of the air electrode (Fig. 25a). The Zn2+-rich/H2O-poor environment, on the one hand, diminishes the opportunities for H2O molecules to participate in electrode reactions. On the other hand, this environment also provides better contact with Zn2+ for the intermediate species generated by the redox reaction of the oxygen cathode. Theoretical calculations and kinetic simulation results indicate that the formation of superoxide species takes place from the weak interaction between OTF anions and Zn2+ in the high concentration electrolyte (Zn(OTF)2). This interaction promotes the subsequent highly reversible ZnO2 chemical reaction. Consequently, a two-electron oxygen reduction mechanism is realized in the high-concentration aqueous electrolyte instead of the four-electron oxygen reduction mechanism in alkaline electrolytes. Consequently, this non-alkaline Zn–air battery with the Zn(OTF)2 electrolyte achieved stable operation for 1600 hours at a current density of 0.1 mA cm−2 and 160 hours at a current density of 1 mA cm−2 in ambient air (Fig. 25b). Moreover, Wang and colleagues engineered a H2O-proof and Zn2+-conducting SEI layer by elevating the content of the Zn(OTf)2 electrolyte and incorporating trimethylethyl ammonium trifluoromethanesulfonate (Me3EtNOTF) as an additive (Fig. 25c).503 The development of this SEI layer notably enhances the electrochemical output performance of the Zn-based battery. The assembled Zn–air battery demonstrates a high energy density of 325 W h kg−1 and maintains robust cycling stability over 300 cycles.
 | ||
| Fig. 25 Electrolyte optimization strategy. (a) Illustration outlining the reaction process of the double capacitor layer on the cathode surface in a Zn(OTf)2 electrolyte. (b) Long-term cycling stability of Zn–air batteries at different current densities. Reproduced with permission from ref. 502. Copyright 2021 American Association for the Advancement of Science. (c) Schematic illustration of in situ generation of the ZnF2-rich SEI layer. Reproduced with permission from ref. 503. Copyright 2021 Springer Science & Business Media. | ||
Substituting traditional alkaline aqueous electrolytes with hydrogel electrolytes proves to be an effective strategy in improving the long cycle life of Zn–air batteries.505,506 Unfortunately, to date, the development of high-performance hydrogel electrolytes has severely lagged behind the development of bifunctional catalysts.507 Conventional polyvinyl alcohol (PVA)-based hydrogel electrolytes encounter challenges such as low ionic conductivity, limited stretchability, and inadequate water retention capacity.508 In order to obtain hydrogel electrolytes with better performance, researchers began to try to modify PVA. For example, combining PVA with additives with high hydrophilicity and/or alkali resistance (such as guar hydroxypropyltrimonium chloride,509 quaternary ammonium510) can greatly improve the ionic conductivity and stability of PVA. In addition, if other polymers with excellent water retention capabilities, such as polyacrylic acid (PAA)511 and polyacrylamide (PAM),512 are combined with PVA, it can also improve the water retention capacity of PVA. As an illustration, Zhong and colleagues synthesized alkaline gel polymer electrolytes via multiple cross-linking reactions involving polyacrylic acid (PAA), polyvinyl alcohol (PVA), graphene oxide, alkali, and potassium iodide (KI) (Fig. 26a).513 The developed hydrogel electrolyte surpasses conventional PVA gel electrolytes in terms of mechanical strength, ionic conductivity, and H2O retention capacity. Moreover, the incorporation of iodide ions alters the conventional oxygen evolution pathway on the air electrode, resulting in a thermodynamically more favorable route. With this optimized alkaline gel electrolyte, the flexible Zn–air battery achieves an extremely low charging potential of 1.69 V and maintains long-term cycling stability for 200 hours (Fig. 26b and c). Considering that the stability of a battery is determined by multiple factors, subsequent research should also invest more efforts in electrolyte design and battery configuration while developing high-performance electrocatalysts.
 | ||
| Fig. 26 Hydrogel electrolytes for long-cycle-life Zn–air batteries. (a) Diagram outlining the process of hydrogel electrolyte preparation and the corresponding configuration of the Zn–air battery. (b) Galvanostatic discharge and charge curves at 2 mA cm−2. (c) Examining the disparities in cycle life and charging potential between sandwich-type KI-PVAA-GO-based Zn–air batteries and certain previously documented flexible Zn–air batteries. Reproduced with permission from ref. 513. Copyright 2020 John Wiley & Sons. | ||
5. Design strategies for Zn–CO2 batteries and other Zn-based batteries
Since the industrial revolution, the levels of CO2 in the atmosphere have continued to rise, far exceeding the upper limit recommended by environmentalists (350 ppm).176 At the same time, CO2, as the main greenhouse gas and acidic gas, is closely related to the environment and ecology. High concentrations of CO2 in the atmosphere will change the global climate and increase the acidity of the ocean, thereby causing damage to the marine ecosystem. But from another perspective, if there are suitable environmentally friendly technologies that can realize the conversion and utilization of CO2, CO2 will be the most abundant and cheapest C1 raw material in nature. It is noteworthy that Takechi et al. have recently ascertained that the incorporation of CO2 positively influences the energy density of Li-air batteries,514 since the presence of CO2 contributes to the rapid consumption of superoxide anion radicals and slows down the filling of Li2CO3 in the cathode. This research result shows that it is possible to design metal–CO2 batteries and use them as a platform to convert CO2.164 In metal–CO2 batteries, the typical operational mechanism involves the oxidation of the metal at the anode. Subsequently, the generated electrons are conveyed through an external circuit to the cathode, where they participate in the reduction of CO2. Compared with commonly used CO2 conversion and utilization technologies, metal–CO2 battery technology is unique in that it combines CO2 conversion with energy generation, rather than pure energy input.Amidst the array of presently documented metal–CO2 batteries, Zn–CO2 batteries stand out as a promising environmentally efficient option due to their avoidance of organic electrolytes. As an emerging Zn-based battery, there are currently few reports on Zn–CO2 batteries. Because of the chemical inertness of CO2 molecules, current design strategies for Zn–CO2 batteries mainly focus on the design of efficient electrocatalysts.515–517 As an illustration, Hou and colleagues devised a gas diffusion strategy to craft nitrogen-doped porous carbon polyhedra featuring minute quantities of isolated iron atoms.178 The optimized electrocatalyst, featuring Fe–N3 sites, demonstrates outstanding performance in CO2 reduction, achieving a CO faradaic efficiency of 96% at −0.5 V, coupled with remarkable stability. Its CO2-to-CO conversion efficiency surpasses that of nearly all previously documented non-noble metal single-atom electrocatalysts relying on nitrogen-doped carbon. This exceptional performance can be ascribed to the enhanced intrinsic activity and the heightened exposure of active sites. The augmented exposure of catalytic sites results primarily from size adjustments designed to amplify the electrochemical surface area. Simultaneously, the heightened intrinsic activity stems predominantly from nitrogen species doping, inducing alterations in the electronic structure. Subsequently, the synthesized material was integrated into a Zn–CO2 battery as a cathode catalyst, yielding robust battery performance characterized by a notable power density of 526 mW cm−2 and an open circuit voltage of 0.727 V. Additionally, Wang et al. introduced and successfully implemented a reversible Zn–CO2 battery, capitalizing on the reversible conversion between carbon dioxide and liquid HCOOH facilitated by a bifunctional Pd cathode (Fig. 27).179 Leveraging their abundant edge and pore structure, the three-dimensional porous Pd nanosheets, upon preparation, manifest a highly electrochemically active surface. This characteristic facilitates selective CO2 reduction during discharging and HCOOH oxidation at low overpotential. When Pd nanosheets are assembled into Zn–CO2 batteries as cathode catalysts, the charging voltage is only 1 V, the cycle durability can exceed 100 cycles, and the energy efficiency can be as high as 81.2%.
 | ||
| Fig. 27 Zn–CO2 batteries. (a) Illustration outlining the prospective application of the Zn–CO2 battery in future energy storage. (b) Visual representation elucidating the operational principles of a reversible aqueous Zn–CO2 battery. (c) Charge and discharge polarization curve. (d) Charge and discharge cycle curve under 0.56 mA cm−2 constant current. Reproduced with permission from ref. 179. Copyright 2018 John Wiley & Sons. | ||
Apart from the emerging Zn–CO2 batteries,518 recent years have witnessed the proposal and development of various other Zn-based batteries, such as the Zn–acetylene battery, Zn–nitrate battery, and several hybrid battery configurations. For these emerging Zn-based batteries, current design strategies mainly focus on the design of cathode electrocatalysts and innovation of battery configurations.519,520 Illustratively, Zhang et al. employed copper dendrites, known for their superior catalytic performance in acetylene semi-hydrogenation, as the cathode catalyst to establish a novel Zn–C2H2 battery. This innovative design integrates zinc oxidation at the anode with alkyne semi-hydrogenation at the cathode.521 This new type of battery can simultaneously achieve selective alkyne reduction and power generation. The engineered Zn–C2H2 battery demonstrates significant open circuit potential and elevated energy density throughout its charge and discharge cycles. Moreover, in the stable discharge phase, the Zn–C2H2 battery achieves exceptional faradaic efficiency and ethylene productivity, yielding polymer-grade ethylene raw materials. Consequently, the advancement of functional Zn–C2H2 batteries not only introduces innovative concepts and applications for next-generation batteries but also presents a promising avenue for energy-efficient and cost-effective alkyne removal technology in olefin purification. Furthermore, some high-performance hybrid batteries have also been proposed and developed through the design of electrolytes. As an illustration, Xia and collaborators devised Zn–air/iodide hybrid batteries, employing an innovative dual-network structured hydrogel as the electrolyte.522 The dual-network structure in the electrolyte imparts enhanced mechanical strength and heightened ionic conductivity to the material. Notably, the incorporation of iodine species brings about more favorable cathode kinetics in the iodide/iodate redox process compared to oxygen electrocatalysis. Simultaneously, iodine species influence the solvation structure of Zn2+, contributing to improved interface stability. Consequently, the assembled hybrid battery demonstrates an outstanding long cycle stability of 110 hours and a commendable energy efficiency of 80%. While these emerging Zn-based batteries are in the nascent stages of research, further endeavors are imperative to explore diverse strategies aimed at achieving optimal battery performance.
6. Conclusions and outlook
Exploring the energy storage mechanisms of batteries and enhancing the performance of diverse battery types remain a prevalent theme in the realm of energy storage. The pursuit of these aims is intricately tied to both human life and societal advancement. Zn-based batteries, recognized for their low cost, environmental sustainability, inherent safety, and substantial theoretical energy density, emerge as an ideal candidate for the next generation of secure battery technologies. This review first systematically summarizes various energy storage mechanisms existing in Zn-based batteries, including insertion-type, conversion-type, coordination-type, and catalysis-type mechanisms. Furthermore, the design strategies aiming at enhancing their electrochemical performance in terms of output voltage, specific capacity, energy density, and cycle life are introduced. In addition to these achievements discussed in this review (Table 1), there are still many issues that need to be solved in order to render high-performance Zn-based batteries. Because of the complexity of battery processes and devices, more efforts need to be invested to deeply understand the reaction mechanism of Zn-based batteries. Concurrently, it is necessary to realize that the excellent performance of Zn-based batteries stems from the collective enhancement of each constituent rather than isolated improvements in individual components. The following points represent unsolved and important areas in Zn-based batteries that require further research and exploration (Fig. 28): | ||
| Fig. 28 Direction for future development of Zn-based batteries. | ||
(1) Further understanding of energy storage mechanisms
While the insertion-type, conversion-type, coordination-type, and catalysis-type mechanisms collectively encompass the spectrum of redox reactions occurring at the cathode in Zn-based batteries, it is noteworthy that high-performance variants often exhibit a concurrent presence of multiple reaction mechanisms, rather than being exclusively governed by a singular type of redox reaction. Simultaneously, the intricate nature of the constituents within Zn-based batteries contributes to heightened challenges in comprehending the underlying reaction mechanisms. To glean insights into the factors influencing the performance of Zn-based batteries and to facilitate the design of high-performance variants, it is imperative to dedicate increased efforts towards the exploration and comprehension of the associated reaction mechanisms. In this regard, the utilization of diverse advanced characterization techniques and theoretical calculations opens avenues for enhanced comprehension of reaction mechanisms.Despite recent advances in the performance of Zn-based batteries, the mechanisms leading to electrode material deactivation and battery failure processes remain elusive, posing a constraint on further evolution of Zn-based battery technology. The seamless functionality of Zn-based batteries is intricately linked to the stability of the zinc anode/electrolyte/cathode interface. Consequently, substantial efforts are warranted to elucidate the intricacies of the charge and discharge processes occurring at this interface. Specifically, the employment of diverse in situ characterization techniques, such as in situ XRD, in situ Raman, in situ XAFS, and in situ TEM, is crucial to monitor dynamic alterations at the electrolyte/electrode interface. Such effort facilitates a comprehensive understanding of the battery failure process. Furthermore, a profound comprehension of Zn-based battery failures serves as a valuable reference for the design of high-performance Zn-based batteries.
(2) Exploring new reaction mechanisms to achieve high performance
Zn-ion batteries inherently benefit from the natural capacity advantage offered by sulfur in conversion-type materials. However, the proposed sulfur redox reactions thus far exhibit limited valence changes, resulting in a constrained electrode potential. To enhance energy density, a promising approach involves devising a novel conversion reaction mechanism that facilitates a broader valence change range for sulfur in redox processes. Achieving an expanded valence change range for sulfur in redox reactions necessitates the minimization of side reactions, such as the HER and OER. Consequently, the implementation of a high-concentration electrolyte, specifically a water-in-salt configuration, emerges as an effective strategy to circumvent side reactions. This approach successfully eliminates undesired reactions, thereby widening the valence change range for sulfur in the redox process.In the realm of alkaline Zn–air batteries, the well-established 4e−/O2 chemistry is a cornerstone. Nevertheless, the incorporation of functional additives possesses the potential to induce alterations in the Zn2+ solvation sheath structure, hydrogen bonding configuration, or the reaction pathway within the alkaline electrolyte. By incorporating high-concentration additives, the conventional four-electron process undergoes a transformation into a two-electron Zn–oxygen/Zn peroxide chemistry, exemplifying a notable alteration in reaction dynamics. These changes, in turn, yield unexpected performance enhancements, effectively overcoming inherent bottlenecks. Concurrently, the adoption of a non-alkaline electrolyte not only circumvents the constraints associated with an alkaline electrolyte but also ensures excellent reversibility. The commendable performance observed in non-alkaline Zn–air batteries signifies a promising avenue for the design of sustainable batteries capable of prolonged and stable operation. The anticipation for innovative electrolytes and novel reaction mechanisms is high, yet their development poses a significant challenge.
(3) Developing Zn-based batteries with high energy density
High energy density in Zn-based batteries necessitates the concurrent optimization of both capacity and voltage, as energy density is inherently determined by the product of these two factors. Cathode materials, including Prussian blue materials, polyanionic compounds, and Co-based oxides, demonstrate distinctive high-voltage features, yet their theoretical capacity remains unsatisfactory. Consequently, a critical focus lies in enhancing the actual capacity of cathode materials with high voltage characteristics. Various strategies, including introduction of multispecies redox reactions and adjustment of ion concentrations in the cathode material, can be employed to reduce the proportion of inactive components and increase the electron transfer number. This, in turn, enhances the intrinsic capacity of redox-active materials. Conversely, addressing the intrinsic low electrode potential of high-capacity cathode materials remains a persistent challenge. While maintaining stable operation of the Zn anode, the introduction of various charge carriers, such as Al3+, Li+, Na+, etc., offers the potential to establish a higher output voltage platform. It is essential, however, to ensure the compatibility of introduced charge carriers with the structure of the cathode material.Beyond the investigation of cathode materials exhibiting simultaneous high capacity and voltage, exploring innovative battery configurations stands out as a highly promising avenue for achieving elevated energy density in zinc-based batteries. Currently, the predominant method for decoupling electrolytes involves the utilization of ion exchange membranes. This design approach facilitates a substantial increase in the potential difference between anode and cathode, expanding the stable electrochemical window of the electrolyte and, consequently, realizing high-energy-density Zn-based batteries. However, it is imperative to acknowledge the associated cost escalation, primarily due to the expense of ion exchange membranes commonly employed for decoupling. Future investigations should prioritize the development of more economical ion exchange membranes. Additionally, exploring the feasibility of cost-effective gel electrolytes as substitutes for ion exchange membranes holds promise as a crucial avenue for further research. Furthermore, the integration of flow devices presents an additional avenue for enhancing the energy density of Zn-based batteries.
(4) Designing Zn-based batteries with ultra-long cycle life
The growing demand for practical applications underscores the increasing need for Zn-based batteries characterized by ultra-long cycle life. Given the intimate connection between reversibility at the zinc anode and the long-term cycling stability of Zn-based batteries, an effective strategy to prolong cycle life involves the careful adjustment of the zinc anode and its interface with the electrolyte. Through the modification and structural design of the zinc anode, it is possible to mitigate the formation of zinc dendrites, prevent the occurrence of the side reaction (HER), and restrict the production of irreversible by-products. Moreover, the introduction of additives into the electrolyte facilitates the effective modulation of the interface between the zinc anode and the electrolyte, thereby enabling the reversible dissolution and deposition of zinc.The electrolyte also plays a crucial role in influencing the cycle stability of Zn-based batteries. Currently, the decoupling of electrolytes stands out as a highly effective approach to enhance the cycle stability of Zn-based batteries. This method involves isolating the cathode and anode in distinct electrolyte environments, effectively mitigating the occurrence of side reactions and preserving the stability of the electrode structure. The two predominant methods for decoupling electrolytes are through the use of ion exchange membranes and hydrogel electrolytes. In the case of ion exchange membranes, the high cost necessitates further research and development focused on achieving more cost-effective alternatives. Regarding gel electrolytes, considerations should extend to the water retention effect and compatibility with both positive and negative electrodes.
(5) Developing flexible and wearable Zn-based batteries with high performance
The pursuit of flexible, wearable, and multifunctional Zn-based batteries with innovative features, including self-protection, self-healing, stretchability, photoelectric detection, shape memory, and thermal response, represents a highly promising direction. This endeavor is poised to potentially shape the next generation of electronic products. A primary challenge is manifested in the development of solid or quasi-solid electrolytes that concurrently exhibit robust mechanical strength, high ionic conductivity, and multifunctionality. Addressing this challenge requires not only the optimization of device structures but also the incorporation of smart designs. These considerations aim to improve compatibility between the multifunctional electrolyte and the intelligent material, as well as other parts within battery devices. The overarching goal is to realize multifunctional devices without compromising electrochemical performance significantly.(6) Developing high-performance Zn-based batteries for extreme-condition applications
While Zn-based batteries have found extensive applications in the consumable electronics market, their performance experiences significant degradation under extreme conditions, including polar expeditions, ocean exploration, and high-altitude drone operations. As such, the imperative lies in the development of Zn-based batteries characterized by heightened performance in challenging environmental conditions, encompassing low and high temperatures, elevated pressure, humidity, and radiation exposure. Extreme conditions frequently influence the electrochemical behavior of specific components within battery devices. For instance, low temperature may result in diminished ion transport rates, while elevated temperatures can induce undesirable side reactions. Therefore, in subsequent research, it is important to design corresponding electrolytes and electrode structures for specific extreme conditions.(7) From laboratory research to industry implementation
Zn-based batteries hold considerable promise for sustainable advancements in future energy and environmental initiatives. Despite extensive research and development efforts in laboratory settings, the transition from theoretical concepts to practical implementation poses substantial challenges. It is crucial to acknowledge that testing conditions in the laboratory substantially differ from those encountered in practical applications. Therefore, careful consideration of performance variations arising from disparate testing conditions becomes imperative. Notably, the path forward requires a holistic approach, emphasizing the synergistic effects among battery components to enhance overall Zn-based battery performance, rather than isolated improvements in individual elements. Furthermore, the broad adoption of Zn-based batteries calls for a thoughtful equilibrium between cost considerations and the pursuit of performance enhancement.Overall, the prospect of employing a singular strategy as a panacea for addressing the myriad challenges inherent in Zn-based batteries appears improbable. Clearly, the key to realizing large-scale applications entails the judicious integration of diverse strategies. Nanotechnology, by introducing innovative avenues, holds promise in mitigating these multifaceted issues. Concurrently, the utilization of advanced characterization techniques contributes significantly by furnishing additional insights crucial for informed material design. Given the active research landscape surrounding Zn-based batteries, concerted endeavors in mechanism exploration, strategic design, and battery engineering are vital to render their viability feasible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NUS Startup Grant, the National Natural Science Foundation of China (52173207), the Science Fund for Distinguished Young Scholars of Hunan Province of China (2023JJ10040), and the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2020R 1A 2C 3003958).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- L. Tang, X. G. Meng, D. H. Deng and X. H. Bao, Adv. Mater., 2019, 31, 1901996 CrossRef CAS PubMed.
- O. Schmidt, S. Melchior, A. Hawkes and I. Staffell, Joule, 2019, 3, 81–100 CrossRef.
- O. Schmidt, A. Hawkes, A. Gambhir and I. Staffell, Nat. Energy, 2017, 2, 17110 CrossRef.
- R. P. Fang, S. Y. Zhao, Z. H. Sun, W. Wang, H. M. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef PubMed.
- D. H. Liu, Z. Y. Bai, M. Li, A. P. Yu, D. Luo, W. W. Liu, L. Yang, J. Lu, K. Amine and Z. W. Chen, Chem. Soc. Rev., 2020, 49, 5407–5445 RSC.
- J. X. Zheng, M. S. Kim, Z. Y. Tu, S. Choudhury, T. Tang and L. A. Archer, Chem. Soc. Rev., 2020, 49, 2701–2750 RSC.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- D. C. Lin, Y. Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- Z. P. Cano, D. Banham, S. Y. Ye, A. Hintennach, J. Lu, M. Fowler and Z. W. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- Y. C. Ding, P. W. Cai and Z. H. Wen, Chem. Soc. Rev., 2021, 50, 1495–1511 RSC.
- Y. Zhao, Y. Q. Kang, J. Wozny, J. Lu, H. Du, C. L. Li, T. Li, F. Y. Kang, N. Tavajohi and B. H. Li, Nat. Rev. Mater., 2023, 8, 623–634 CrossRef CAS.
- C. J. Xu, B. H. Li, H. D. Du and F. Y. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- Y. L. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- Z. X. Liu, Y. Huang, Y. Huang, Q. Yang, X. L. Li, Z. D. Huang and C. Y. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 RSC.
- B. Y. Tang, L. T. Shan, S. Q. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- D. L. Chao, W. H. Zhou, F. X. Xie, C. Ye, H. Li, M. Jaroniec and S. Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- Y. M. Sui and X. L. Ji, Chem. Rev., 2021, 121, 6654–6695 CrossRef CAS PubMed.
- Y. Y. Liu, X. Lu, F. L. Lai, T. X. Liu, P. R. Shearing, I. P. Parkin, G. J. He and D. J. L. Brett, Joule, 2021, 5, 2845–2903 CrossRef CAS.
- W. C. Du, E. H. X. Ang, Y. Yang, Y. F. Zhang, M. H. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- Z. Hou, Y. Gao, H. Tan and B. A. Zhang, Nat. Commun., 2021, 12, 3083 CrossRef CAS PubMed.
- L. J. Kong, M. Liu, H. Huang, Y. H. Xu and X. H. Bu, Adv. Energy Mater., 2022, 12, 2100172 CrossRef CAS.
- T. Xue and H. J. Fan, J. Energy Chem., 2021, 54, 194–201 CrossRef CAS.
- J. X. Zheng and L. A. Archer, Sci. Adv., 2021, 7, eabe0219 CrossRef CAS PubMed.
- X. M. Xu, F. Y. Xiong, J. S. Meng, X. P. Wang, C. J. Niu, Q. Y. An and L. Q. Mai, Adv. Funct. Mater., 2020, 30, 1904398 CrossRef CAS.
- N. Zhang, F. Y. Cheng, Y. C. Liu, Q. Zhao, K. X. Lei, C. C. Chen, X. S. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- G. Z. Fang, J. Zhou, A. Q. Pan and S. Q. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- N. Zhang, X. Y. Chen, M. Yu, Z. Q. Niu, F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y. K. Sun and S. T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- N. Zhang, F. Y. Cheng, J. X. Liu, L. B. Wang, X. H. Long, X. S. Liu, F. J. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef PubMed.
- Y. C. Shi, Y. Chen, L. Shi, K. Wang, B. Wang, L. Li, Y. M. Ma, Y. H. Li, Z. H. Sun, W. Ali and S. J. Ding, Small, 2020, 16, 2000730 CrossRef CAS PubMed.
- M. Song, H. Tan, D. L. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- H. Jia, Z. Q. Wang, B. Tawiah, Y. D. Wang, C. Y. Chan, B. Fei and F. Pan, Nano Energy, 2020, 70, 104523 CrossRef CAS.
- P. Yu, Y. X. Zeng, H. Z. Zhang, M. H. Yu, Y. X. Tong and X. Lu, Small, 2019, 15, 1804760 CrossRef PubMed.
- F. Wan, Z. M. Hao, S. Wang, Y. X. Ni, J. C. Zhu, Z. W. Tie, S. S. Bi, Z. Q. Niu and J. Chen, Adv. Mater., 2021, 33, 2102701 CrossRef CAS PubMed.
- P. He, Y. L. Quan, X. Xu, M. Y. Yan, W. Yang, Q. Y. An, L. He and L. Q. Mai, Small, 2017, 13, 1702551 CrossRef PubMed.
- B. Li, X. T. Zhang, T. L. Wang, Z. X. He, B. A. Lu, S. Q. Liang and J. Zhou, Nano-Micro Lett., 2022, 14, 6 CrossRef CAS PubMed.
- X. L. Xu, Y. Chen, D. Zheng, P. C. Ruan, Y. H. Cai, X. J. Dai, X. X. Niu, C. J. Pei, W. H. Shi, W. X. Liu, F. F. Wu, Z. Y. Pan, H. Li and X. H. Cao, Small, 2021, 17, 2101901 CrossRef CAS PubMed.
- C. Xia, J. Guo, Y. J. Lei, H. F. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed.
- X. X. Jia, C. F. Liu, Z. G. Neale, J. H. Yang and G. Z. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- V. Verma, S. Kumar, W. Manalastas and M. Srinivasan, ACS Energy Lett., 2021, 6, 1773–1785 CrossRef CAS.
- Y. P. Deng, R. L. Liang, G. P. Jiang, Y. Jiang, A. P. Yu and Z. W. Chen, ACS Energy Lett., 2020, 5, 1665–1675 CrossRef CAS.
- W. H. Yang, Y. Yang, H. J. Yang and H. S. Zhou, ACS Energy Lett., 2022, 7, 2515–2530 CrossRef CAS.
- R. F. Service, Science, 2021, 372, 890–891 CrossRef CAS PubMed.
- A. R. Mainar, L. C. Colmenares, J. A. Blázquez and I. Urdampilleta, Int. J. Energy Res., 2018, 42, 903–918 CrossRef.
- D. L. Chao and S. Z. Qiao, Joule, 2020, 4, 1846–1851 CrossRef.
- X. Wang, Z. C. Y. Zhang, B. J. Xi, W. H. Chen, Y. X. Jia, J. K. Feng and S. L. Xiong, ACS Nano, 2021, 15, 9244–9272 CrossRef CAS PubMed.
- Y. L. Zhao, Y. H. Zhu and X. B. Zhang, InfoMat, 2020, 2, 237–260 CrossRef CAS.
- Y. Fang, X. S. Xie, B. Y. Zhang, Y. Z. Chai, B. A. Lu, M. K. Liu, J. Zhou and S. Q. Liang, Adv. Funct. Mater., 2022, 32, 2109671 CrossRef CAS.
- J. Fu, R. L. Liang, G. H. Liu, A. P. Yu, Z. Y. Bai, L. Yang and Z. W. Chen, Adv. Mater., 2019, 31, 1805230 CrossRef PubMed.
- J. H. Song, K. Xu, N. Liu, D. Reed and X. L. Li, Mater. Today, 2021, 45, 191–212 CrossRef CAS.
- J. P. Yan, E. H. Ang, Y. Yang, Y. F. Zhang, M. H. Ye, W. C. Du and C. C. Li, Adv. Funct. Mater., 2021, 31, 2010213 CrossRef CAS.
- B. F. Cui, X. P. Han and W. B. Hu, Small Struct., 2021, 2, 2000128 CrossRef CAS.
- Y. X. Zeng, D. Y. Luan and X. W. Lou, Chem, 2023, 9, 1118–1146 CAS.
- Y. Lu, Y. Y. Lu, Z. Q. Niu and J. Chen, Adv. Energy Mater., 2018, 8, 1702469 CrossRef.
- X. Wang, Y. G. Li, S. Wang, F. Zhou, P. Das, C. L. Sun, S. H. Zheng and Z. S. Wu, Adv. Energy Mater., 2020, 10, 2000081 CrossRef CAS.
- B. Yong, D. T. Ma, Y. Y. Wang, H. W. Mi, C. X. He and P. X. Zhang, Adv. Energy Mater., 2020, 10, 2002354 CrossRef CAS.
- Q. L. Li, Q. C. Zhang, C. L. Liu, Z. Y. Zhou, C. W. Li, B. He, P. Man, X. N. Wang and Y. G. Yao, J. Mater. Chem. A, 2019, 7, 12997–13006 RSC.
- Y. P. Xu, P. W. Cai, K. Chen, Y. C. Ding, L. Chen, W. F. Chen and Z. H. Wen, Angew. Chem., Int. Ed., 2020, 59, 23593–23597 CrossRef CAS PubMed.
- Y. H. Zhu, Y. F. Cui, Z. L. Xie, Z. B. Zhuang, G. Huang and X. B. Zhang, Nat. Rev. Chem., 2022, 6, 505–517 CrossRef CAS PubMed.
- L. T. Ma, S. M. Chen, C. B. Long, X. L. Li, Y. W. Zhao, Z. X. Liu, Z. D. Huang, B. B. Dong, J. A. Zapien and C. Y. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- L. T. Ma, N. Li, C. B. Long, B. B. Dong, D. L. Fang, Z. X. Liu, Y. W. Zhao, X. L. Li, J. Fan, S. M. Chen, S. J. Zhang and C. Y. Zhi, Adv. Funct. Mater., 2019, 29, 1906142 CrossRef CAS.
- C. Zhong, B. Liu, J. Ding, X. R. Liu, Y. W. Zhong, Y. Li, C. B. Sun, X. P. Han, Y. D. Deng, N. Q. Zhao and W. B. Hu, Nat. Energy, 2020, 5, 440–449 CrossRef CAS.
- D. L. Chao, C. Ye, F. X. Xie, W. H. Zhou, Q. H. Zhang, Q. F. Gu, K. Davey, L. Gu and S. Z. Qiao, Adv. Mater., 2020, 32, 2001894 CrossRef CAS PubMed.
- C. Han, W. J. Li, H. K. Liu, S. X. Dou and J. Z. Wang, Nano Energy, 2020, 74, 104880 CrossRef CAS.
- Q. Zhang, J. Y. Luan, Y. G. Tang, X. B. Ji and H. Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- Z. H. Yi, G. Y. Chen, F. Hou, L. Q. Wang and J. Liang, Adv. Energy Mater., 2021, 11, 2003065 CrossRef CAS.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. S. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- X. Z. Yang, C. Li, Z. T. Sun, S. Yang, Z. X. Shi, R. Huang, B. Z. Liu, S. Li, Y. H. Wu, M. L. Wang, Y. W. Su, S. X. Dou and J. Y. Sun, Adv. Mater., 2021, 33, 2105951 CrossRef CAS PubMed.
- Z. Wang, J. H. Huang, Z. W. Guo, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- P. Xue, C. Guo, L. Li, H. P. Li, D. Luo, L. C. Tan and Z. W. Chen, Adv. Mater., 2022, 34, 2110047 CrossRef CAS PubMed.
- P. Sun, L. Ma, W. H. Zhou, M. J. Qiu, Z. L. Wang, D. L. Chao and W. J. Mai, Angew. Chem., Int. Ed., 2021, 60, 18247–18255 CrossRef CAS PubMed.
- F. W. Ming, Y. P. Zhu, G. Huang, A. H. Emwas, H. F. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS PubMed.
- H. Chen, C. L. Dai, F. Y. Xiao, Q. J. Yang, S. N. Cai, M. W. Xu, H. J. Fan and S. J. Bao, Adv. Mater., 2022, 34, 2109092 CrossRef CAS PubMed.
- F. X. Xie, H. Li, X. S. Wang, X. Zhi, D. L. Chao, K. Davey and S. Z. Qiao, Adv. Energy Mater., 2021, 11, 2003419 CrossRef CAS.
- F. Wang, W. Sun, Z. Shadike, E. Y. Hu, X. Ji, T. Gao, X. Q. Yang, K. Xu and C. S. Wang, Angew. Chem., Int. Ed., 2018, 57, 11978–11981 CrossRef CAS PubMed.
- D. Kundu, S. H. Vajargah, L. W. Wan, B. Adams, D. Prendergast and L. F. Nazar, Energy Environ. Sci., 2018, 11, 881–892 RSC.
- P. Li, Y. Q. Wang, Q. Xiong, Y. Hou, S. Yang, H. L. Cui, J. X. Zhu, X. L. Li, Y. B. Wang, R. Zhang, S. C. Zhang, X. Q. Wang, X. Jin, S. C. Bai and C. Y. Zhi, Angew. Chem., Int. Ed., 2023, 62, e202303292 CrossRef CAS PubMed.
- F. Wan and Z. Q. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS PubMed.
- Q. Yang, Q. Li, Z. X. Liu, D. H. Wang, Y. Guo, X. L. Li, Y. C. Tang, H. F. Li, B. B. Dong and C. Y. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed.
- M. Li, Z. L. Li, X. P. Wang, J. S. Meng, X. Liu, B. K. Wu, C. H. Han and L. Q. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC.
- S. Guo, L. P. Qin, T. S. Zhang, M. Zhou, J. Zhou, G. Z. Fang and S. Q. Liang, Energy Storage Mater., 2021, 34, 545–562 CrossRef.
- S. Liu, L. Kang, J. M. Kim, Y. T. Chun, J. Zhang and S. C. Jun, Adv. Energy Mater., 2020, 10, 2000477 CrossRef CAS.
- V. Mathew, B. Sambandam, S. Kim, S. Kim, S. Park, S. Lee, M. H. Alfaruqi, V. Soundharrajan, S. Islam, D. Y. Putro, J. Y. Hwang, Y. K. Sun and J. Kim, ACS Energy Lett., 2020, 5, 2376–2400 CrossRef CAS.
- Z. W. Tie and Z. Q. Niu, Angew. Chem., Int. Ed., 2020, 59, 21293–21303 CrossRef CAS PubMed.
- H. Zhang, X. Liu, H. H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598–616 CrossRef CAS PubMed.
- T. Xiong, Y. X. Zhang, W. S. V. Lee and J. M. Xue, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS.
- H. Y. Liu, J. G. Wang, Z. Y. You, C. G. Wei, F. Y. Kang and B. Q. Wei, Mater. Today, 2021, 42, 73–98 CrossRef CAS.
- Q. C. Wang, S. Kaushik, X. Xiao and Q. Xu, Chem. Soc. Rev., 2023, 52, 6139–6190 RSC.
- J. Yi, P. C. Liang, X. Y. Liu, K. Wu, Y. Y. Liu, Y. G. Wang, Y. Y. Xia and J. J. Zhang, Energy Environ. Sci., 2018, 11, 3075–3095 RSC.
- T. P. Zhou, N. Zhang, C. Z. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1132–1153 RSC.
- Y. G. Li and H. J. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- P. Tan, B. Chen, H. R. Xu, H. C. Zhang, W. Z. Cai, M. Ni, M. L. Liu and Z. P. Shao, Energy Environ. Sci., 2017, 10, 2056–2080 RSC.
- Z. Z. Yuan, Y. B. Yin, C. X. Xie, H. M. Zhang, Y. Yao and X. F. Li, Adv. Mater., 2019, 31, 1902025 CrossRef CAS PubMed.
- H. F. Li, L. T. Ma, C. P. Han, Z. F. Wang, Z. X. Liu, Z. J. Tang and C. Y. Zhi, Nano Energy, 2019, 62, 550–587 CrossRef CAS.
- J. H. Huang, Z. Wang, M. Y. Hou, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- P. He, M. Y. Yan, G. B. Zhang, R. M. Sun, L. N. Chen, Q. Y. An and L. Q. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- M. H. Alfaruqi, V. Mathew, J. Gim, S. Kim, J. Song, J. P. Baboo, S. H. Choi and J. Kim, Chem. Mater., 2015, 27, 3609–3620 CrossRef CAS.
- Y. Y. Lu, T. Y. Zhu, W. van den Bergh, M. Stefik and K. Huang, Angew. Chem., Int. Ed., 2020, 59, 17004–17011 CrossRef CAS PubMed.
- S. Z. Deng, Z. S. Yuan, Z. W. Tie, C. D. Wang, L. Song and Z. Q. Niu, Angew. Chem., Int. Ed., 2020, 59, 22002–22006 CrossRef CAS PubMed.
- J. Cao, D. D. Zhang, Y. L. Yue, X. Wang, T. Pakornchote, T. Bovornratanaraks, X. Y. Zhang, Z. S. Wu and J. Q. Qin, Nano Energy, 2021, 84, 105876 CrossRef CAS.
- S. C. Liu, H. Zhu, B. H. Zhang, G. Li, H. K. Zhu, Y. Ren, H. B. Geng, Y. Yang, Q. Liu and C. C. Li, Adv. Mater., 2020, 32, 2001113 CrossRef CAS PubMed.
- P. He, G. B. Zhang, X. B. Liao, M. Y. Yan, X. Xu, Q. Y. An, J. Liu and L. Q. Mai, Adv. Energy Mater., 2018, 8, 1702463 CrossRef.
- Y. F. Yuan, R. Sharpe, K. He, C. H. Li, M. T. Saray, T. C. Liu, W. T. Yao, M. Cheng, H. L. Jin, S. Wang, K. Amine, R. Shahbazian-Yassar, M. S. Islam and J. Lu, Nat. Sustain., 2022, 5, 890–898 CrossRef.
- S. M. Chen, Y. Ying, L. T. Ma, D. M. Zhu, H. T. Huang, L. Song and C. Y. Zhi, Nat. Commun., 2023, 14, 2925 CrossRef CAS PubMed.
- W. Sun, F. Wang, S. Y. Hou, C. Y. Yang, X. L. Fan, Z. H. Ma, T. Gao, F. D. Han, R. Z. Hu, M. Zhu and C. S. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- Z. G. Yao, Q. P. Wu, K. Y. Chen, J. J. Liu and C. L. Li, Energy Environ. Sci., 2020, 13, 3149–3163 RSC.
- X. H. Zeng, J. N. Hao, Z. J. Wang, J. F. Mao and Z. P. Guo, Energy Storage Mater., 2019, 20, 410–437 CrossRef.
- L. P. Wang, P. F. Wang, T. S. Wang, Y. X. Yin, Y. G. Guo and C. R. Wang, J. Power Sources, 2017, 355, 18–22 CrossRef.
- W. D. Pan, Y. F. Wang, X. L. Zhao, Y. Zhao, X. H. Liu, J. Xuan, H. Z. Wang and D. Y. C. Leung, Adv. Funct. Mater., 2021, 31, 2008783 CrossRef CAS.
- J. N. Hao, J. Long, B. Li, X. L. Li, S. L. Zhang, F. H. Yang, X. H. Zeng, Z. H. Yang, W. K. Pang and Z. P. Guo, Adv. Funct. Mater., 2019, 29, 1903605 CrossRef.
- H. Z. Li, C. J. Firby and A. Y. Elezzabi, Joule, 2019, 3, 2268–2278 CrossRef CAS.
- P. Hu, T. Zhu, X. P. Wang, X. F. Zhou, X. J. Wei, X. H. Yao, W. Luo, C. W. Shi, K. A. Owusu, L. Zhou and L. Q. Mai, Nano Energy, 2019, 58, 492–498 CrossRef CAS.
- F. Wan, L. L. Zhang, X. Dai, X. Y. Wang, Z. Q. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed.
- J. L. Liu, C. H. Xu, Z. Chen, S. B. Ni and Z. X. Shen, Green Energy Environ., 2018, 3, 20–41 CrossRef.
- J. Yan, J. Wang, H. Liu, Z. Bakenov, D. Gosselink and P. Chen, J. Power Sources, 2012, 216, 222–226 CrossRef CAS.
- K. W. Nam, W. S. Yoon, H. Shin, K. Y. Chung, S. Choi and X. Q. Yang, J. Power Sources, 2009, 192, 652–659 CrossRef CAS.
- Y. F. Huang, J. Mou, W. B. Liu, X. L. Wang, L. B. Dong, F. Y. Kang and C. J. Xu, Nano-Micro Lett., 2019, 11, 49 CrossRef CAS PubMed.
- H. L. Pan, Y. Y. Shao, P. F. Yan, Y. W. Cheng, K. S. Han, Z. M. Nie, C. M. Wang, J. H. Yang, X. L. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- T. Wang, H. C. Chen, F. Yu, X. S. Zhao and H. X. Wang, Energy Storage Mater., 2019, 16, 545–573 CrossRef.
- Y. X. Zeng, Z. Z. Lai, Y. Han, H. Z. Zhang, S. L. Xie and X. H. Lu, Adv. Mater., 2018, 30, 1802396 CrossRef PubMed.
- P. Hu, T. S. Wang, J. W. Zhao, C. J. Zhang, J. Ma, H. P. Du, X. G. Wang and G. L. Cui, ACS Appl. Mater. Interfaces, 2015, 7, 26396–26399 CrossRef CAS PubMed.
- L. Y. Zhang, R. Z. Feng, W. Wang and G. H. Yip, Nat. Rev. Chem., 2022, 6, 524–543 CrossRef PubMed.
- C. K. Zhang, L. Y. Zhang, Y. Ding, X. L. Guo and G. H. Yu, ACS Energy Lett., 2018, 3, 2875–2883 CrossRef CAS.
- J. A. Luo, B. Hu, M. W. Hu, Y. Zhao and T. L. Liu, ACS Energy Lett., 2019, 4, 2220–2240 CrossRef CAS.
- L. T. Shan, Y. R. Wang, S. Q. Liang, B. Y. Tang, Y. Q. Yang, Z. Q. Wang, B. A. Lu and J. Zhou, InfoMat, 2021, 3, 1028–1036 CrossRef CAS.
- B. P. Gindt, D. G. Abebe, Z. J. Tang, M. B. Lindsey, J. Chen, R. A. Elgammal, T. A. Zawodzinski and T. Fujiwara, J. Mater. Chem. A, 2016, 4, 4288–4295 RSC.
- L. Hu, L. Gao, M. T. Di, X. B. Jiang, X. M. Wu, X. M. Yan, X. F. Li and G. H. He, Energy Storage Mater., 2021, 34, 648–668 CrossRef.
- D. Lin and Y. Li, Adv. Mater., 2022, 34, 2108856 CrossRef CAS PubMed.
- J. Zhang, G. P. Jiang, P. Xu, A. G. Kashkooli, M. Mousavi, A. P. Yu and Z. W. Chen, Energy Environ. Sci., 2018, 11, 2010–2015 RSC.
- M. Mousavi, G. P. Jiang, J. Zhang, A. G. Kashkooli, H. Z. Dou, C. J. Silva, Z. P. Cano, Y. Niu, A. P. Yu and Z. W. Chen, Energy Storage Mater., 2020, 32, 465–476 CrossRef.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- Y. Lu, Q. Zhang, L. Li, Z. Q. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS.
- K. W. Nam, H. Kim, Y. Beldjoudi, T. W. Kwon, D. J. Kim and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 2541–2548 CrossRef CAS PubMed.
- H. Y. Shi, Y. J. Ye, K. Liu, Y. Song and X. Q. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 CrossRef CAS PubMed.
- Z. W. Tie, L. J. Liu, S. Z. Deng, D. B. Zhao and Z. Q. Niu, Angew. Chem., Int. Ed., 2020, 59, 4920–4924 CrossRef CAS PubMed.
- N. Wang, X. L. Dong, B. L. Wang, Z. W. Guo, Z. Wang, R. H. Wang, X. Qiu and Y. G. Wang, Angew. Chem., Int. Ed., 2020, 59, 14577–14583 CrossRef CAS PubMed.
- Y. R. Wang, C. X. Wang, Z. G. Ni, Y. M. Gu, B. L. Wang, Z. W. Guo, Z. Wang, D. Bin, J. Ma and Y. G. Wang, Adv. Mater., 2020, 32 Search PubMed.
- H. Z. Zhang, Y. B. Fang, F. Yang, X. Q. Liu and X. H. Lu, Energy Environ. Sci., 2020, 13, 2515–2523 RSC.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. C. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- Q. Zhao, W. W. Huang, Z. Q. Luo, L. J. Liu, Y. Lu, Y. X. Li, L. Li, J. Y. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- J. Park, M. Park, G. Nam, J. S. Lee and J. Cho, Adv. Mater., 2015, 27, 1396–1401 CrossRef CAS PubMed.
- Y. G. Li, M. Gong, Y. Y. Liang, J. Feng, J. E. Kim, H. L. Wang, G. S. Hong, B. Zhang and H. J. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- J. Pan, Y. Y. Xu, H. Yang, Z. H. Dong, H. F. Liu and B. Y. Xia, Adv. Sci., 2018, 5, 1700691 CrossRef PubMed.
- H. F. Wang, C. Tang and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1803329 CrossRef.
- P. Yu, L. Wang, F. F. Sun, Y. Xie, X. Liu, J. Y. Ma, X. W. Wang, C. G. Tian, J. H. Li and H. G. Fu, Adv. Mater., 2019, 31, 1901666 CrossRef PubMed.
- X. P. Han, X. F. Ling, Y. Wang, T. Y. Ma, C. Zhong, W. B. Hu and Y. D. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed.
- H. F. Wang, C. Tang, B. Wang, B. Q. Li and Q. Zhang, Adv. Mater., 2017, 29, 1702327 CrossRef PubMed.
- H. S. Shang, X. Y. Zhou, J. C. Dong, A. Li, X. Zhao, Q. H. Liu, Y. Lin, J. J. Pei, Z. Li, Z. L. Jiang, D. N. Zhou, L. R. Zheng, Y. Wang, J. Zhou, Z. K. Yang, R. Cao, R. Sarangi, T. T. Sun, X. Yang, X. S. Zheng, W. S. Yan, Z. B. Zhuang, J. Li, W. X. Chen, D. S. Wang, J. T. Zhang and Y. D. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- P. Gu, M. B. Zheng, Q. X. Zhao, X. Xiao, H. G. Xue and H. Pang, J. Mater. Chem. A, 2017, 5, 7651–7666 RSC.
- J. Fu, Z. P. Cano, M. G. Park, A. P. Yu, M. Fowler and Z. W. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef PubMed.
- Z. Zhang, X. Huang, Z. Chen, J. J. Zhu, B. Endrödi, C. Janáky and D. H. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302789 CrossRef CAS PubMed.
- L. Tang, S. B. Zhang, Q. L. Wu, X. R. Wang, H. Wu and Z. Y. Jiang, J. Mater. Chem. A, 2018, 6, 2964–2973 RSC.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- S. I. Seneviratne, M. G. Donat, A. J. Pitman, R. Knutti and R. L. Wilby, Nature, 2016, 529, 477–483 CrossRef CAS PubMed.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- Z. J. Xie, X. Zhang, Z. Zhang and Z. Zhou, Adv. Mater., 2017, 29, 1605891 CrossRef PubMed.
- F. Wang, Y. Li, X. H. Xia, W. Cai, Q. G. Chen and M. H. Chen, Adv. Energy Mater., 2021, 11, 2100667 CrossRef CAS.
- M. J. Wu, G. X. Zhang, H. M. Yang, X. H. Liu, M. Dubois, M. A. Gauthier and S. H. Sun, InfoMat, 2022, 4, e12265 CrossRef CAS.
- J. F. Xie and Y. B. Wang, Acc. Chem. Res., 2019, 52, 1721–1729 CrossRef CAS PubMed.
- Y. Qiao, J. Yi, S. C. Wu, Y. Liu, S. X. Yang, P. He and H. S. Zhou, Joule, 2017, 1, 359–370 CrossRef CAS.
- C. Yang, K. K. Guo, D. W. Yuan, J. L. Cheng and B. Wang, J. Am. Chem. Soc., 2020, 142, 6983–6990 CrossRef CAS PubMed.
- Y. Xiao, F. Du, C. G. Hu, Y. Ding, Z. L. Wang, A. Roy and L. M. Dai, ACS Energy Lett., 2020, 5, 916–921 CrossRef CAS.
- L. Song, C. G. Hu, Y. Xiao, J. P. He, Y. Lin, J. W. Connell and L. M. Dai, Nano Energy, 2020, 71, 104595 CrossRef CAS.
- B. Liu, Y. L. Sun, L. Y. Liu, J. T. Chen, B. J. Yang, S. Xu and X. B. Yan, Energy Environ. Sci., 2019, 12, 887–922 RSC.
- J. F. Xie, Z. Zhou and Y. B. Wang, Adv. Funct. Mater., 2020, 30, 1908285 CrossRef CAS.
- S. S. Gao, Y. F. Liu, Z. Y. Xie, Y. Qiu, L. C. Zhuo, Y. J. Qin, J. Q. Ren, S. S. Zhang, G. Z. Hu, J. Luo and X. J. Liu, Small Methods, 2021, 5, 2001039 CrossRef CAS PubMed.
- W. T. Bi, C. Z. Wu and Y. Xie, ACS Energy Lett., 2018, 3, 624–633 CrossRef CAS.
- K. Wang, Y. Y. Wu, X. B. Cao, L. Gu and J. Hu, Adv. Funct. Mater., 2020, 30, 1908965 CrossRef CAS.
- J. Gu, C. S. Hsu, L. C. Bai, H. M. Chen and X. L. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- T. T. Wang, X. H. Sang, W. Z. Zheng, B. Yang, S. Y. Yao, C. J. Lei, Z. J. Li, Q. G. He, J. G. Lu, L. C. Lei, L. M. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed.
- J. F. Xie, X. Y. Wang, J. Q. Lv, Y. Y. Huang, M. X. Wu, Y. B. Wang and J. N. Yao, Angew. Chem., Int. Ed., 2018, 57, 16996–17001 CrossRef CAS PubMed.
- Y. Q. Yang, Y. Tang, G. Z. Fang, L. T. Shan, J. S. Guo, W. Y. Zhang, C. Wang, L. B. Wang, J. Zhou and S. Q. Liang, Energy Environ. Sci., 2018, 11, 3157–3162 RSC.
- X. M. Ma, X. X. Cao, M. L. Yao, L. T. Shan, X. D. Shi, G. Z. Fang, A. Q. Pan, B. A. Lu, J. Zhou and S. Q. Liang, Adv. Mater., 2022, 34, 2105452 CrossRef CAS PubMed.
- Q. Zong, Q. Q. Wang, C. F. Liu, D. W. Tao, J. Y. Wang, J. J. Zhang, H. W. Du, J. F. Chen, Q. L. Zhang and G. Z. Cao, ACS Nano, 2022, 16, 4588–4598 CrossRef CAS PubMed.
- X. Zhang, J. P. Hu, N. Fu, W. B. Zhou, B. Liu, Q. Deng and X. W. Wu, InfoMat, 2022, 4, e12306 CrossRef CAS.
- X. Guo, J. Zhou, C. L. Bai, X. K. Li, G. Z. Fang and S. Q. Liang, Mater. Today Energy, 2020, 16, 100396 CrossRef.
- C. F. Liu, Z. Neale, J. Q. Zheng, X. X. Jia, J. J. Huang, M. Y. Yan, M. Tian, M. S. Wang, J. H. Yang and G. Z. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC.
- F. W. Ming, H. F. Liang, Y. J. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2018, 3, 2602–2609 CrossRef CAS.
- D. Selvakumaran, A. Q. Pan, S. Q. Liang and G. Z. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- B. Y. Tang, J. Zhou, G. Z. Fang, F. Liu, C. Y. Zhu, C. Wang, A. Q. Pan and S. Q. Liang, J. Mater. Chem. A, 2019, 7, 940–945 RSC.
- Y. Q. Yang, Y. Tang, S. Q. Liang, Z. X. Wu, G. Z. Fang, X. X. Cao, C. Wang, T. Q. Lin, A. Q. Pan and J. Zhou, Nano Energy, 2019, 61, 617–625 CrossRef CAS.
- C. Y. Zhu, G. Z. Fang, S. Q. Liang, Z. X. Chen, Z. Q. Wang, J. Y. Ma, H. Wang, B. Y. Tang, X. S. Zheng and J. Zhou, Energy Storage Mater., 2020, 24, 394–401 CrossRef.
- P. G. He, J. H. Liu, X. D. Zhao, Z. P. Ding, P. Gao and L. Z. Fan, J. Mater. Chem. A, 2020, 8, 10370–10376 RSC.
- S. Q. Wei, S. M. Chen, X. Z. Su, Z. H. Qi, C. D. Wang, B. B. Ganguli, P. J. Zhang, K. F. Zhu, Y. Y. Cao, Q. He, D. F. Cao, X. Guo, W. Wen, X. J. Wu, P. M. Ajayan and L. Song, Energy Environ. Sci., 2021, 14, 3954–3964 RSC.
- J. W. Ding, Z. G. Du, B. Li, L. Z. Wang, S. W. Wang, Y. J. Gong and S. B. Yang, Adv. Mater., 2019, 31, 1904369 CrossRef CAS PubMed.
- H. Luo, B. Wang, F. Wang, J. Yang, F. D. Wu, Y. Ning, Y. Zhou, D. L. Wang, H. K. Liu and S. X. Dou, ACS Nano, 2020, 14, 7328–7337 CrossRef CAS PubMed.
- J. W. Ding, H. Y. Zheng, H. G. Gao, Q. N. Liu, Z. Hu, L. F. Han, S. W. Wang, S. D. Wu, S. M. Fang and S. L. Chou, Adv. Energy Mater., 2021, 11, 2100973 CrossRef CAS.
- J. Cui, Z. W. Guo, J. Yi, X. Y. Liu, K. Wu, P. C. Liang, Q. Li, Y. Y. Liu, Y. G. Wang, Y. Y. Xia and J. J. Zhang, ChemSusChem, 2020, 13, 2160–2185 CrossRef CAS PubMed.
- Z. H. Li, J. Tan, C. Y. Gao, Y. Wang, Y. G. Wang, M. X. Ye and J. F. Shen, Energy Environ. Sci., 2023, 16, 2398–2431 RSC.
- Z. R. Lin, H. Y. Shi, L. Lin, X. P. Yang, W. L. Wu and X. Q. Sun, Nat. Commun., 2021, 12, 4424 CrossRef CAS PubMed.
- W. X. Wang, V. S. Kale, Z. Cao, Y. J. Lei, S. Kandambeth, G. D. Zou, Y. P. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 CrossRef CAS PubMed.
- Y. Zhao, Y. X. Huang, F. Wu, R. J. Chen and L. Li, Adv. Mater., 2021, 33, 2106469 CrossRef CAS PubMed.
- X. Y. Wu, A. Markir, L. Ma, Y. K. Xu, H. Jiang, D. P. Leonard, W. Shin, T. P. Wu, J. Lu and X. L. Ji, Angew. Chem., Int. Ed., 2019, 58, 12640–12645 CrossRef CAS PubMed.
- C. L. Dai, L. Y. Hu, X. T. Jin, H. Chen, X. Q. Zhang, S. H. Zhang, L. Song, H. Y. Ma, M. W. Xu, Y. Zhao, Z. P. Zhang, H. H. Cheng and L. T. Qu, Adv. Mater., 2021, 33, 2105480 CrossRef CAS PubMed.
- W. Li, K. L. Wang and K. Jiang, Adv. Sci., 2020, 7, 2000761 CrossRef CAS PubMed.
- C. L. Dai, X. T. Jin, H. Y. Ma, L. Y. Hu, G. Q. Sun, H. Chen, Q. J. Yang, M. W. Xu, Q. W. Liu, Y. K. Xiao, X. Q. Zhang, H. S. Yang, Q. Guo, Z. P. Zhang and L. T. Qu, Adv. Energy Mater., 2021, 11, 2003982 CrossRef CAS.
- W. Li, Y. S. Ma, P. Li, X. Y. Jing, K. Jiang and D. H. Wang, Adv. Funct. Mater., 2021, 31, 2101237 CrossRef CAS.
- Z. Chen, F. N. Mo, T. R. Wang, Q. Yang, Z. D. Huang, D. H. Wang, G. J. Liang, A. Chen, Q. Li, Y. Guo, X. L. Li, J. Fan and C. Y. Zhi, Energy Environ. Sci., 2021, 14, 2441–2450 RSC.
- J. Shin, D. S. Choi, H. J. Lee, Y. Jung and J. W. Choi, Adv. Energy Mater., 2019, 9, 1900083 CrossRef.
- N. Zhang, M. Jia, Y. Dong, Y. Y. Wang, J. Z. Xu, Y. C. Liu, L. F. Jiao and F. Y. Cheng, Adv. Funct. Mater., 2019, 29, 1807331 CrossRef.
- D. Bin, Y. Liu, B. B. Yang, J. H. Huang, X. L. Dong, X. Zhang, Y. G. Wang and Y. Y. Xia, ACS Appl. Mater. Interfaces, 2019, 11, 20796–20803 CrossRef CAS PubMed.
- T. J. Sun, Q. S. Nian, S. B. Zheng, X. M. Yuan and Z. L. Tao, J. Power Sources, 2020, 478, 228758 CrossRef CAS.
- Y. X. Zeng, X. F. Lu, S. L. Zhang, D. Y. Luan, S. Li and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 22189–22194 CrossRef CAS PubMed.
- D. Bin, W. C. Huo, Y. B. Yuan, J. H. Huang, Y. Liu, Y. X. Zhang, F. Dong, Y. G. Wang and Y. Y. Xia, Chem, 2020, 6, 968–984 CAS.
- H. L. Peng, C. H. Liu, N. N. Wang, C. G. Wang, D. D. Wang, Y. L. Li, B. Chen, J. Yang and Y. T. Qian, Energy Environ. Sci., 2022, 15, 1682–1693 RSC.
- T. T. Lv, Y. Y. Liu, H. Wang, S. Y. Yang, C. S. Liu and H. Pang, Chem. Eng. J., 2021, 411, 128533 CrossRef CAS.
- M. S. Zhang, W. X. Wu, J. W. Luo, H. Z. Zhang, J. Liu, X. Q. Liu, Y. Y. Yang and X. H. Lu, J. Mater. Chem. A, 2020, 8, 11642–11648 RSC.
- T. Xiong, Z. G. Yu, H. J. Wu, Y. H. Du, Q. D. Xie, J. S. Chen, Y. W. Zhang, S. J. Pennycook, W. S. V. Lee and J. M. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- Z. Q. Li, Y. K. Ren, L. Mo, C. F. Liu, K. Hsu, Y. C. Ding, X. X. Zhang, X. L. Li, L. H. Hu, D. H. Ji and G. Z. Cao, ACS Nano, 2020, 14, 5581–5589 CrossRef CAS PubMed.
- M. M. Han, J. W. Huang, S. Q. Liang, L. T. Shan, X. S. Xie, Z. Y. Yi, Y. R. Wang, S. Guo and J. Zhou, Iscience, 2020, 23, 100797 CrossRef CAS PubMed.
- Y. Q. Zhang, L. Tao, C. Xie, D. D. Wang, Y. Q. Zou, R. Chen, Y. Y. Wang, C. K. Jia and S. Y. Wang, Adv. Mater., 2020, 32, 1905923 CrossRef CAS PubMed.
- Z. C. Y. Zhang, B. J. Xi, X. Wang, X. J. Ma, W. H. Chen, J. K. Feng and S. L. Xiong, Adv. Funct. Mater., 2021, 31, 2103070 CrossRef CAS.
- X. L. Li, Z. D. Huang, C. E. Shuck, G. J. Liang, Y. Gogotsi and C. Y. Zhi, Nat. Rev. Chem., 2022, 6, 389–404 CrossRef PubMed.
- W. Y. Du, L. Miao, Z. Y. Song, X. W. Zheng, Y. K. Lv, D. Z. Zhu, L. H. Gan and M. X. Liu, J. Power Sources, 2022, 536, 231512 CrossRef CAS.
- N. Zhang, J. C. Wang, Y. F. Guo, P. F. Wang, Y. R. Zhu and T. F. Yi, Coord. Chem. Rev., 2023, 479, 215009 CrossRef CAS.
- Y. Z. Wu, M. C. Wang, Y. Tao, K. Zhang, M. L. Cai, Y. Ding, X. P. Liu, T. Hayat, A. Alsaedi and S. Y. Dai, Adv. Funct. Mater., 2020, 30, 1907120 CrossRef CAS.
- S. Z. Deng, Z. W. Tie, F. Yue, H. M. Cao, M. J. Yao and Z. Q. Niu, Angew. Chem., Int. Ed., 2022, 61, e202115877 CrossRef CAS PubMed.
- X. D. Zhu, Z. Y. Cao, W. J. Wang, H. J. Li, J. C. Dong, S. P. Gao, D. X. Xu, L. Li, J. F. Shen and M. X. Ye, ACS Nano, 2021, 15, 2971–2983 CrossRef CAS PubMed.
- Y. X. Zeng, X. Y. Zhang, Y. Meng, M. H. Yu, J. N. Yi, Y. Q. Wu, X. H. Lu and Y. X. Tong, Adv. Mater., 2017, 29, 1700274 CrossRef PubMed.
- Y. Q. Fu, Q. L. Wei, G. X. Zhang, X. M. Wang, J. H. Zhang, Y. F. Hu, D. N. Wang, L. C. Zuin, T. Zhou, Y. C. Wu and S. H. Sun, Adv. Energy Mater., 2018, 8, 1801445 CrossRef.
- B. K. Wu, G. B. Zhang, M. Y. Yan, T. F. Xiong, P. He, L. He, X. Xu and L. Q. Mai, Small, 2018, 14, 1703850 CrossRef PubMed.
- J. J. Wang, J. G. Wang, H. Y. Liu, Z. Y. You, Z. Li, F. Y. Kang and B. Q. Wei, Adv. Funct. Mater., 2021, 31, 2007397 CrossRef CAS.
- Q. Ni, B. Kim, C. A. Wu and K. Kang, Adv. Mater., 2022, 34, 2108206 CrossRef CAS PubMed.
- J. Ji, H. Z. Wan, B. Zhang, C. Wang, Y. Gan, Q. Y. Tan, N. Z. Wang, J. Yao, Z. H. Zheng, P. Liang, J. Zhang, H. B. Wang, L. Tao, Y. Wang, D. L. Chao and H. Wang, Adv. Energy Mater., 2021, 11, 2003203 CrossRef CAS.
- Z. Q. Luo, S. L. Zheng, S. Zhao, X. Jiao, Z. S. Gong, F. S. Cai, Y. Q. Duan, F. J. Li and Z. H. Yuan, J. Mater. Chem. A, 2021, 9, 6131–6138 RSC.
- S. Wang, Z. S. Yuan, X. Zhang, S. S. Bi, Z. Zhou, J. L. Tian, Q. C. Zhang and Z. Q. Niu, Angew. Chem., Int. Ed., 2021, 60, 7056–7060 CrossRef CAS PubMed.
- Y. Q. Yang, S. Q. Liang, B. A. Lu and J. Zhou, Energy Environ. Sci., 2022, 15, 1192–1200 RSC.
- Y. P. Zou, T. T. Liu, Q. J. Du, Y. Y. Li, H. B. Yi, X. Zhou, Z. X. Li, L. J. Gao, L. Zhang and X. Liang, Nat. Commun., 2021, 12, 170 CrossRef CAS PubMed.
- J. N. Hao, L. B. Yuan, B. Johannessen, Y. L. Zhu, Y. Jiao, C. Ye, F. X. Xie and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 25114–25121 CrossRef CAS PubMed.
- H. Yang, T. S. Zhang, D. Chen, Y. C. Tan, W. H. Zhou, L. Li, W. Li, G. S. Li, W. Han, H. J. Fan and D. L. Chao, Adv. Mater., 2023, 35, 2300053 CrossRef CAS PubMed.
- Z. Chen, H. L. Cui, Y. Hou, X. Q. Wang, X. Jin, A. Chen, Q. Yang, D. H. Wang, Z. D. Huang and C. Y. Zhi, Chem, 2022, 8, 2204–2216 CAS.
- L. Y. Zhang, L. Chen, X. F. Zhou and Z. P. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- L. T. Ma, S. M. Chen, H. F. Li, Z. H. Ruan, Z. J. Tang, Z. X. Liu, Z. F. Wang, Y. Huang, Z. X. Pei, J. A. Zapien and C. Y. Zhi, Energy Environ. Sci., 2018, 11, 2521–2530 RSC.
- Z. X. Liu, Q. Yang, D. H. Wang, G. J. Liang, Y. H. Zhu, F. N. Mo, Z. D. Huang, X. L. Li, L. T. Ma, T. C. Tang, Z. G. Lu and C. Y. Zhi, Adv. Energy Mater., 2019, 9, 1902473 CrossRef CAS.
- Z. G. Hou, X. Q. Zhang, X. N. Li, Y. C. Zhu, J. W. Liang and Y. T. Qian, J. Mater. Chem. A, 2017, 5, 730–738 RSC.
- F. Wan, Y. Zhang, L. L. Zhang, D. B. Liu, C. D. Wang, L. Song, Z. Q. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 7062–7067 CrossRef CAS PubMed.
- N. Li, G. Q. Li, C. J. Li, H. C. Yang, G. W. Qin, X. D. Sun, F. Li and H. M. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 13790–13796 CrossRef CAS PubMed.
- D. L. Chao, W. H. Zhou, C. Ye, Q. H. Zhang, Y. G. Chen, L. Gu, K. Davey and S. Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS PubMed.
- X. S. Xie, H. W. Fu, Y. Fang, B. A. Lu, J. Zhou and S. Q. Liang, Adv. Energy Mater., 2022, 12, 2102393 CrossRef CAS.
- C. Liu, X. W. Chi, Q. Han and Y. Liu, Adv. Energy Mater., 2020, 10, 1903589 CrossRef CAS.
- P. C. Ruan, S. Q. Liang, B. G. Lu, H. J. Fan and J. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202200598 CrossRef CAS PubMed.
- P. C. Ruan, X. L. Xu, D. Zheng, X. H. Chen, X. Y. Yin, S. Q. Liang, X. W. Wu, W. H. Shi, X. H. Cao and J. Zhou, ChemSusChem, 2022, 15, e202201118 CrossRef CAS PubMed.
- X. F. Shen, X. N. Wang, Y. R. Zhou, Y. H. Shi, L. M. Zhao, H. H. Jin, J. T. Di and Q. W. Li, Adv. Funct. Mater., 2021, 31, 2101579 CrossRef CAS.
- L. X. Dai, Y. Wang, L. Sun, Y. Ding, Y. Q. Yao, L. D. Yao, N. E. Drewett, W. Zhang, J. Tang and W. T. Zheng, Adv. Sci., 2021, 8, 2004995 CrossRef CAS PubMed.
- C. X. Xie, T. Y. Li, C. Z. Deng, Y. Song, H. M. Zhang and X. F. Li, Energy Environ. Sci., 2020, 13, 135–143 RSC.
- G. D. Li, W. Chen, H. Zhang, Y. J. Gong, F. F. Shi, J. Y. Wang, R. F. Zhang, G. X. Chen, Y. Jin, T. Wu, Z. Y. Tang and Y. Cui, Adv. Energy Mater., 2020, 10, 1902085 CrossRef CAS.
- J. F. Lei, Y. X. Yao, Z. Y. Wang and Y. C. Lu, Energy Environ. Sci., 2021, 14, 4418–4426 RSC.
- Z. Chen, C. Li, Q. Yang, D. H. Wang, X. L. Li, Z. D. Huang, G. J. Liang, A. Chen and C. Y. Zhi, Adv. Mater., 2021, 33, 2105426 CrossRef CAS PubMed.
- Z. Chen, Q. Yang, F. N. Mo, N. Li, G. J. Liang, X. L. Li, Z. D. Huang, D. H. Wang, W. C. Huang, J. Fan and C. Y. Zhi, Adv. Mater., 2020, 32, 2001469 CrossRef CAS PubMed.
- X. L. Li, N. Li, Z. D. Huang, Z. Chen, Y. W. Zhao, G. J. Liang, Q. Yang, M. Li, Q. Huang, B. B. Dong, J. Fan and C. Y. Zhi, ACS Nano, 2021, 15, 1718–1726 CrossRef CAS PubMed.
- X. L. Li, Y. L. Wang, Z. Chen, P. Li, G. J. Liang, Z. D. Huang, Q. Yang, A. Chen, H. L. Cui, B. B. Dong, H. Y. He and C. Y. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202113576 CrossRef CAS PubMed.
- X. Y. Wu, Y. K. Xu, C. Zhang, D. P. Leonard, A. Markir, J. Lu and X. L. Ji, J. Am. Chem. Soc., 2019, 141, 6338–6344 CrossRef CAS PubMed.
- Z. Chen, Y. W. Zhao, F. N. Mo, Z. D. Huang, X. L. Li, D. H. Wang, G. J. Liang, Q. Yang, A. Chen, Q. Li, L. T. Ma, Y. Guo and C. Y. Zhi, Small Struct., 2020, 1, 2000005 CrossRef.
- J. Ibers, Nat. Chem., 2009, 1, 508 CrossRef CAS PubMed.
- X. F. Zhang, S. Q. Jiao, J. G. Tu, W. L. Song, X. Xiao, S. J. Li, M. Y. Wang, H. P. Lei, D. H. Tian, H. S. Chen and D. N. Fang, Energy Environ. Sci., 2019, 12, 1918–1927 RSC.
- Z. Chen, S. N. Wang, Z. Q. Wei, Y. Q. Wang, Z. X. Wu, Y. Hou, J. X. Zhu, Y. B. Wang, G. J. Liang, Z. D. Huang, A. Chen, D. H. Wang and C. Y. Zhi, J. Am. Chem. Soc., 2023, 145, 20521–20529 CrossRef CAS PubMed.
- J. H. Huang, Y. H. Xie, L. Yan, B. L. Wang, T. Y. Kong, X. L. Dong, Y. G. Wang and Y. Y. Xia, Energy Environ. Sci., 2021, 14, 883–889 RSC.
- Z. X. Zhu, T. L. Jiang, M. Ali, Y. H. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610–16751 CrossRef CAS PubMed.
- H. M. Tang, Y. Yin, Y. Huang, J. W. Wang, L. X. Liu, Z. Qu, H. Zhang, Y. Li, M. S. Zhu and O. G. Schmidt, ACS Energy Lett., 2021, 6, 1859–1868 CrossRef CAS.
- G. G. Yadav, D. Turney, J. C. Huang, X. Wei and S. Banerjee, ACS Energy Lett., 2019, 4, 2144–2146 CrossRef CAS.
- S. Gu, K. Gong, E. Z. Yan and Y. S. Yan, Energy Environ. Sci., 2014, 7, 2986–2998 RSC.
- K. Gong, X. Y. Ma, K. M. Conforti, K. J. Kuttler, J. B. Grunewald, K. L. Yeager, M. Z. Bazant, S. Gu and Y. S. Yan, Energy Environ. Sci., 2015, 8, 2941–2945 RSC.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS PubMed.
- C. X. Xie, H. M. Zhang, W. B. Xu, W. Wang and X. F. Li, Angew. Chem., Int. Ed., 2018, 57, 11171–11176 CrossRef CAS PubMed.
- D. G. Kwabi, Y. L. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS PubMed.
- S. N. Wang, Z. Y. Wang, Y. B. Yin, T. Y. Li, N. N. Chang, F. T. Fan, H. M. Zhang and X. F. Li, Energy Environ. Sci., 2021, 14, 4077–4084 RSC.
- Z. M. Zhao, X. H. Liu, M. Q. Zhang, L. Y. Zhang, C. K. Zhang, X. F. Li and G. H. Yu, Chem. Soc. Rev., 2023, 52, 6031–6074 RSC.
- D. L. Yu, L. P. Zhi, F. F. Zhang, Y. Song, Q. Wang, Z. Z. Yuan and X. F. Li, Adv. Mater., 2023, 35, 2209390 CrossRef CAS PubMed.
- J. Wu, C. G. Yuan, T. Y. Li, Z. Z. Yuan, H. M. Zhang and X. F. Li, J. Am. Chem. Soc., 2021, 143, 13135–13144 CrossRef CAS PubMed.
- S. Q. Jiao, J. M. Fu, M. Z. Wu, T. Hua and H. B. Hu, ACS Nano, 2022, 16, 1013–1024 CrossRef CAS PubMed.
- L. T. Ma, S. M. Chen, N. Li, Z. X. Liu, Z. J. Tang, J. A. Zapien, S. M. Chen, J. Fan and C. Y. Zhi, Adv. Mater., 2020, 32, 1908121 CrossRef CAS PubMed.
- F. Wang, O. Borodin, T. Gao, X. L. Fan, W. Sun, F. D. Han, A. Faraone, J. A. Dura, K. Xu and C. S. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- D. Yuan, J. Zhao, H. Ren, Y. Q. Chen, R. Chua, E. T. J. Jie, Y. Cai, E. Edison, W. Manalastas, M. W. Wong and M. Srinivasan, Angew. Chem., Int. Ed., 2021, 60, 7213–7219 CrossRef CAS PubMed.
- Z. D. Zhao, R. Wang, C. X. Peng, W. J. Chen, T. Q. Wu, B. Hu, W. J. Weng, Y. Yao, J. X. Zeng, Z. H. Chen, P. Y. Liu, Y. C. Liu, G. S. Li, J. Guo, H. B. Lu and Z. P. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- Q. Zhang, Y. L. Ma, Y. Lu, X. Z. Zhou, L. Lin, L. Li, Z. H. Yan, Q. Zhao, K. Zhang and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 23357–23364 CrossRef CAS PubMed.
- L. B. Yuan, J. N. Hao, C. C. Kao, C. Wu, H. K. Liu, S. X. Dou and S. Z. Qiao, Energy Environ. Sci., 2021, 14, 5669–5689 RSC.
- D. E. Turney, J. W. Gallaway, G. G. Yadav, R. Ramirez, M. Nyce, S. Banerjee, Y. C. K. Chen-Wiegart, J. Wang, M. J. D'Ambrose, S. Kolhekar, J. C. Huang and X. Wei, Chem. Mater., 2017, 29, 4819–4832 CrossRef CAS.
- Z. F. Yang, C. Hu, Q. Zhang, T. Q. Wu, C. L. Xie, H. Wang, Y. G. Tang, X. B. Ji and H. Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202308017 CrossRef CAS PubMed.
- X. H. Zheng, T. Ahmad and W. Chen, Energy Storage Mater., 2021, 39, 365–394 CrossRef.
- Y. M. Zhang, J. D. Howe, S. Ben-Yoseph, Y. T. Wu and N. Liu, ACS Energy Lett., 2021, 6, 404–412 CrossRef CAS.
- F. Moser, F. Fourgeot, R. Rouget, O. Crosnier and T. Brousse, Electrochim. Acta, 2013, 109, 110–116 CrossRef CAS.
- H. F. Li, C. J. Xu, C. P. Han, Y. Y. Chen, C. G. Wei, B. H. Li and F. Y. Kang, J. Electrochem. Soc., 2015, 162, A1439–A1444 CrossRef CAS.
- L. S. Cao, D. Li, T. Deng, Q. Li and C. S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19292–19296 CrossRef CAS PubMed.
- X. Wang, J. P. Meng, X. G. Lin, Y. D. Yang, S. Zhou, Y. P. Wang and A. Q. Pan, Adv. Funct. Mater., 2021, 31, 2106114 CrossRef CAS.
- J. L. Yang, J. Li, J. W. Zhao, K. Liu, P. H. Yang and H. J. Fan, Adv. Mater., 2022, 34, 2202382 CrossRef CAS PubMed.
- Y. Yang, C. Y. Liu, Z. H. Lv, H. Yang, Y. F. Zhang, M. H. Ye, L. B. Chen, J. B. Zhao and C. C. Li, Adv. Mater., 2021, 33, 2007388 CrossRef CAS PubMed.
- M. H. Yu, N. Chandrasekhar, R. K. M. Raghupathy, K. H. Ly, H. Z. Zhang, E. Dmitrieva, C. L. Liang, X. H. Lu, T. D. Kühne, H. Mirhosseini, I. M. Weidinger and X. L. Feng, J. Am. Chem. Soc., 2020, 142, 19570–19578 CrossRef CAS PubMed.
- M. S. Zhu, J. P. Hu, Q. Q. Lu, H. Y. Dong, D. D. Karnaushenko, C. Becker, D. Karnaushenko, Y. Li, H. M. Tang, Z. Qu, J. Ge and O. G. Schmidt, Adv. Mater., 2021, 33, 2007497 CrossRef CAS PubMed.
- P. Chen, X. H. Yuan, Y. B. Xia, Y. Zhang, L. J. Fu, L. L. Liu, N. F. Yu, Q. H. Huang, B. Wang, X. W. Hu, Y. P. Wu and T. van Ree, Adv. Sci., 2021, 8, 2100309 CrossRef CAS PubMed.
- Y. H. Cui, Q. H. Zhao, X. J. Wu, X. Chen, J. L. Yang, Y. T. Wang, R. Z. Qin, S. X. Ding, Y. L. Song, J. W. Wu, K. Yang, Z. J. Wang, Z. W. Mei, Z. B. Song, H. Wu, Z. Y. Jiang, G. Y. Qian, L. Y. Yang and F. Pan, Angew. Chem., Int. Ed., 2020, 59, 16594–16601 CrossRef CAS PubMed.
- Z. K. Guo, L. S. Fan, C. Y. Zhao, A. S. Chen, N. N. Liu, Y. Zhang and N. Q. Zhang, Adv. Mater., 2022, 34, 2105133 CrossRef CAS PubMed.
- H. B. He, H. Tong, X. Y. Song, X. P. Song and J. Liu, J. Mater. Chem. A, 2020, 8, 7836–7846 RSC.
- L. Hong, X. M. Wu, L. Y. Wang, M. Zhong, P. Y. Zhang, L. S. Jiang, W. Huang, Y. L. Wang, K. X. Wang and J. S. Chen, ACS Nano, 2022, 16, 6906–6915 CrossRef CAS PubMed.
- C. Huang, X. Zhao, S. Liu, Y. S. Hao, Q. L. Tang, A. P. Hu, Z. X. Liu and X. H. Chen, Adv. Mater., 2021, 33, 2100445 CrossRef CAS PubMed.
- Q. Yang, Y. Guo, B. X. Yan, C. D. Wang, Z. X. Liu, Z. D. Huang, Y. K. Wang, Y. R. Li, H. F. Li, L. Song, J. Fan and C. Y. Zhi, Adv. Mater., 2020, 32, 2001755 CrossRef CAS PubMed.
- Q. Zhang, J. Y. Luan, X. B. Huang, Q. Wang, D. Sun, Y. G. Tang, X. B. Ji and H. Y. Wang, Nat. Commun., 2020, 11, 3961 CrossRef CAS PubMed.
- J. H. Zhou, M. Xie, F. Wu, Y. Mei, Y. T. Hao, R. L. Huang, G. L. Wei, A. N. Liu, L. Li and R. J. Chen, Adv. Mater., 2021, 33, 2101649 CrossRef CAS PubMed.
- R. T. Guo, X. Liu, F. J. Xia, Y. L. Jiang, H. Z. Zhang, M. Huang, C. J. Niu, J. S. Wu, Y. Zhao, X. P. Wang, C. H. Han and L. Q. Mai, Adv. Mater., 2022, 34, 2202188 CrossRef CAS PubMed.
- L. T. Kang, M. W. Cui, F. Y. Jiang, Y. F. Gao, H. J. Luo, J. J. Liu, W. Liang and C. Y. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- K. N. Zhao, C. X. Wang, Y. H. Yu, M. Y. Yan, Q. L. Wei, P. He, Y. F. Dong, Z. Y. Zhang, X. D. Wang and L. Q. Mai, Adv. Mater. Interfaces, 2018, 5, 1800848 CrossRef.
- H. Yu, Y. X. Zeng, N. W. Li, D. Y. Luan, L. Yu and X. W. Lou, Sci. Adv., 2022, 8, eabm5766 CrossRef CAS PubMed.
- S. B. Wang, Q. Ran, R. Q. Yao, H. Shi, Z. Wen, M. Zhao, X. Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef CAS PubMed.
- W. J. Lu, C. X. Xie, H. M. Zhang and X. F. Li, ChemSusChem, 2018, 11, 3996–4006 CrossRef CAS PubMed.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117–1124 RSC.
- J. F. Parker, C. N. Chervin, I. R. Pala, M. Machler, M. F. Burz, J. W. Long and D. R. Rolison, Science, 2017, 356, 414–417 CrossRef PubMed.
- J. N. Hao, L. B. Yuan, C. Ye, D. L. Chao, K. Davey, Z. P. Guo and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- A. Naveed, H. J. Yang, J. Yang, Y. N. Nuli and J. L. Wang, Angew. Chem., Int. Ed., 2019, 58, 2760–2764 CrossRef CAS PubMed.
- X. H. Zeng, J. F. Mao, J. N. Hao, J. T. Liu, S. L. Liu, Z. J. Wang, Y. Y. Wang, S. L. Zhang, T. Zheng, J. W. Liu, P. H. Rao and Z. P. Guo, Adv. Mater., 2021, 33, 2007416 CrossRef CAS PubMed.
- C. Li, A. Shyamsunder, A. G. Hoane, D. M. Long, C. Y. Kwok, P. G. Kotula, K. R. Zavadil, A. A. Gewirth and L. F. Nazar, Joule, 2022, 6, 1103–1120 CrossRef CAS.
- H. Y. Liu, J. G. Wang, W. Hua, L. B. Ren, H. H. Sun, Z. D. Hou, Y. Huyan, Y. J. Cao, C. G. Wei and F. Y. Kang, Energy Environ. Sci., 2022, 15, 1872–1881 RSC.
- G. Q. Ma, L. C. Miao, Y. Dong, W. T. Yuan, X. Y. Nie, S. L. Di, Y. Y. Wang, L. B. Wang and N. Zhang, Energy Storage Mater., 2022, 47, 203–210 CrossRef.
- L. C. Miao, R. H. Wang, S. L. Di, Z. F. Qian, L. Zhang, W. L. Xin, M. Y. Liu, Z. Q. Zhu, S. Q. Chu, Y. Du and N. Zhang, ACS Nano, 2022, 16, 9667–9678 CrossRef CAS PubMed.
- X. H. Zeng, K. X. Xie, S. L. Liu, S. L. Zhang, J. N. Hao, J. T. Liu, W. K. Pang, J. W. Liu, P. H. Rao, Q. H. Wang, J. F. Mao and Z. P. Guo, Energy Environ. Sci., 2021, 14, 5947–5957 RSC.
- S. W. Huang, L. Hou, T. Y. Li, Y. C. Jiao and P. Y. Wu, Adv. Mater., 2022, 34, 2110140 CrossRef CAS PubMed.
- T. C. Li, Y. Lim, X. L. Li, S. Z. Luo, C. J. Lin, D. L. Fang, S. W. Xia, Y. Wang and H. Y. Yang, Adv. Energy Mater., 2022, 12, 2103231 CrossRef CAS.
- S. L. Liu, J. F. Mao, W. K. Pang, J. Vongsvivut, X. H. Zeng, L. Thomsen, Y. Y. Wang, J. W. Liu, D. Li and Z. P. Guo, Adv. Funct. Mater., 2021, 31, 2104281 CrossRef CAS.
- S. L. Liu, R. Z. Zhang, J. F. Mao, Y. L. Zhao, Q. Cai and Z. P. Guo, Sci. Adv., 2022, 8, eabn5097 CrossRef CAS PubMed.
- Y. Q. Lv, Y. Xiao, L. T. Ma, C. Y. Zhi and S. M. Chen, Adv. Mater., 2022, 34, 2106409 CrossRef CAS PubMed.
- X. Y. Nie, L. C. Miao, W. T. Yuan, G. Q. Ma, S. L. Di, Y. Y. Wang, S. G. Shen and N. Zhang, Adv. Funct. Mater., 2022, 32, 2203905 CrossRef CAS.
- P. J. Wang, S. Q. Liang, C. Chen, X. S. Xie, J. W. Chen, Z. H. Liu, Y. Tang, B. A. Lu and J. Zhou, Adv. Mater., 2022, 34, 2202733 CrossRef CAS PubMed.
- Y. Y. Wang, Z. J. Wang, F. H. Yang, S. L. Liu, S. L. Zhang, J. F. Mao and Z. P. Guo, Small, 2022, 18, 2107033 CrossRef CAS PubMed.
- H. J. Yang, Y. Qiao, Z. Chang, H. Deng, X. Y. Zhu, R. J. Zhu, Z. T. Xiong, P. He and H. S. Zhou, Adv. Mater., 2021, 33, 2102415 CrossRef CAS PubMed.
- M. Yang, J. C. Zhu, S. S. Bi, R. Wang and Z. Q. Niu, Adv. Mater., 2022, 34, 2201744 CrossRef CAS PubMed.
- W. N. Xu, K. N. Zhao, W. C. Huo, Y. Z. Wang, G. Yao, X. Gu, H. W. Cheng, L. Q. Mai, C. G. Hu and X. D. Wang, Nano Energy, 2019, 62, 275–281 CrossRef CAS.
- Y. P. Zhu, J. Yin, X. L. Zheng, A. H. Emwas, Y. J. Lei, O. F. Mohammed, Y. Cui and H. N. Alshareef, Energy Environ. Sci., 2021, 14, 4463–4473 RSC.
- Y. Tang, C. X. Liu, H. R. Zhu, X. S. Xie, J. W. Gao, C. B. Deng, M. M. Han, S. Q. Liang and J. Zhou, Energy Storage Mater., 2020, 27, 109–116 CrossRef.
- H. J. Yang, Z. Chang, Y. Qiao, H. Deng, X. W. Mu, P. He and H. S. Zhou, Angew. Chem., Int. Ed., 2020, 59, 9377–9381 CrossRef CAS PubMed.
- H. Y. Lu, J. S. Hu, L. T. Wang, J. Z. Li, X. Ma, Z. C. Zhu, H. Q. Li, Y. J. Zhao, Y. J. Li, J. X. Zhao and B. G. Xu, Adv. Funct. Mater., 2022, 32, 2112540 CrossRef CAS.
- G. J. Liang, Z. J. Tang, B. Han, J. X. Zhu, A. Chen, Q. Li, Z. Chen, Z. D. Huang, X. L. Li, Q. Yang and C. Y. Zhi, Adv. Mater., 2023, 35, 2210051 CrossRef CAS PubMed.
- H. Jiang, L. T. Tang, Y. K. Fu, S. T. Wang, S. K. Sandstrom, A. M. Scida, G. X. Li, D. Hoang, J. Hong, N. C. Chiu, K. C. Stylianou, W. F. Stickle, D. H. Wang, J. Li, P. A. Greaney, C. Fang and X. L. Ji, Nat. Sustain., 2023, 6, 806–815 CrossRef.
- Z. Liu, G. Pulletikurthi and F. Endres, ACS Appl. Mater. Interfaces, 2016, 8, 12158–12164 CrossRef CAS PubMed.
- G. Kasiri, R. Trócoli, A. B. Hashemi and F. La Mantia, Electrochim. Acta, 2016, 222, 74–83 CrossRef CAS.
- B. Sambandam, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, J. Mater. Chem. A, 2018, 6, 3850–3856 RSC.
- D. S. Liu, Z. Y. Zhang, Y. F. Zhang, M. H. Ye, S. Huang, S. Z. You, Z. J. Du, J. F. He, Z. P. Wen, Y. C. Tang, X. Q. Liu and C. C. Li, Angew. Chem., Int. Ed., 2023, 62, e202215385 CrossRef CAS PubMed.
- W. Zhang and G. J. He, Angew. Chem., Int. Ed., 2023, 62, e202218466 CrossRef CAS PubMed.
- H. F. Wang, W. Q. Ye, B. W. Yin, K. X. Wang, M. S. Riaz, B. B. Xie, Y. J. Zhong and Y. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218872 CrossRef CAS PubMed.
- M. J. Qiu, P. Sun, Y. Wang, L. Ma, C. Y. Zhi and W. J. Mai, Angew. Chem., Int. Ed., 2022, 61, e202210979 CrossRef CAS PubMed.
- Y. Wang, T. R. Wang, S. Y. Bu, J. X. Zhu, Y. B. Wang, R. Zhang, H. Hong, W. J. Zhang, J. Fan and C. Y. Zhi, Nat. Commun., 2023, 14, 1828 CrossRef CAS PubMed.
- L. S. Cao, D. Li, E. Y. Hu, J. J. Xu, T. Deng, L. Ma, Y. Wang, X. Q. Yang and C. S. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed.
- X. M. Zeng, X. J. Meng, W. Jiang, M. Ling, L. J. Yan and C. D. Liang, Electrochim. Acta, 2021, 378, 138106 CrossRef CAS.
- Z. H. Chen, H. Z. Chen, Y. C. Che, L. Cheng, H. Zhang, J. Chen, F. Y. Xie, N. Wang, Y. S. Jin and H. Meng, ACS Sustainable Chem. Eng., 2021, 9, 6855–6863 CrossRef CAS.
- M. D. Yan, C. L. Xu, Y. Sun, H. L. Pan and H. Li, Nano Energy, 2021, 82, 105739 CrossRef CAS.
- Z. G. Hou, M. F. Dong, Y. L. Xiong, X. Q. Zhang, H. S. Ao, M. K. Liu, Y. C. Zhu and Y. T. Qian, Small, 2020, 16, 2001228 CrossRef CAS PubMed.
- W. J. Zhou, M. F. Chen, Q. H. Tian, J. Z. Chen, X. W. Xu, X. Han and J. L. Xu, J. Colloid Interface Sci., 2021, 601, 486–494 CrossRef CAS PubMed.
- W. Zhang, Y. H. Dai, R. W. Chen, Z. M. Xu, J. W. Li, W. Zong, H. X. Li, Z. Li, Z. Y. Zhang, J. X. Zhu, F. Guo, X. Gao, Z. J. Du, J. T. Chen, T. L. Wang, G. J. He and I. P. Parkin, Angew. Chem., Int. Ed., 2023, 62, e202212695 CrossRef CAS PubMed.
- Y. Zhao, Y. N. Wang, Z. M. Zhao, J. W. Zhao, T. Xin, N. Wang and J. Z. Liu, Energy Storage Mater., 2020, 28, 64–72 CrossRef.
- Z. Q. Luo, L. J. Liu, J. X. Ning, K. X. Lei, Y. Lu, F. J. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed.
- S. Gu, S. F. Wu, L. J. Cao, M. C. Li, N. Qin, J. Zhu, Z. Q. Wang, Y. Z. Li, Z. Q. Li, J. J. Chen and Z. G. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- X. J. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- D. G. Wang, T. J. Qiu, W. H. Guo, Z. B. Liang, H. Tabassum, D. G. Xia and R. Q. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC.
- L. J. Kong, M. Zhong, W. Shuang, Y. H. Xu and X. H. Bu, Chem. Soc. Rev., 2020, 49, 2378–2407 RSC.
- X. X. Luo, W. H. Li, H. J. Liang, H. X. Zhang, K. D. Du, X. T. Wang, X. F. Liu, J. P. Zhang and X. L. Wu, Angew. Chem., Int. Ed., 2022, 61, e202117661 CrossRef CAS PubMed.
- M. A. Khayum, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 RSC.
- B. J. Hopkins, C. N. Chervin, J. W. Long, D. R. Rolison and J. F. Parker, ACS Energy Lett., 2020, 5, 3405–3408 CrossRef CAS.
- K. W. Leong, Y. F. Wang, M. Ni, W. D. Pan, S. J. Luo and D. Y. C. Leung, Renewable Sustainable Energy Rev., 2022, 154, 111771 CrossRef CAS.
- L. T. Ma, S. M. Chen, Z. X. Pei, Y. Huang, G. J. Liang, F. N. Mo, Q. Yang, J. Su, Y. H. Gao, J. A. Zapien and C. Y. Zhi, ACS Nano, 2018, 12, 1949–1958 CrossRef CAS PubMed.
- B. Q. Li, S. Y. Zhang, B. Wang, Z. J. Xia, C. Tang and Q. Zhang, Energy Environ. Sci., 2018, 11, 1723–1729 RSC.
- W. J. Zang, A. Sumboja, Y. Y. Ma, H. Zhang, Y. Wu, S. S. Wu, H. J. Wu, Z. L. Liu, C. Guan, J. Wang and S. J. Pennycook, ACS Catal., 2018, 8, 8961–8969 CrossRef CAS.
- Y. Y. Guo, P. F. Yuan, J. N. Zhang, Y. F. Hu, I. S. Amiinu, X. Wang, J. G. Zhou, H. C. Xia, Z. B. Song, Q. Xu and S. C. Mu, ACS Nano, 2018, 12, 1894–1901 CrossRef CAS PubMed.
- C. Guan, A. Sumboja, H. J. Wu, W. N. Ren, X. M. Liu, H. Zhang, Z. L. Liu, C. W. Cheng, S. J. Pennycook and J. Wang, Adv. Mater., 2017, 29, 1704117 CrossRef PubMed.
- Y. Y. Guo, P. F. Yuan, J. A. Zhang, H. C. Xia, F. Y. Cheng, M. F. Zhou, J. Li, Y. Y. Qiao, S. C. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- D. X. Ji, L. Fan, L. L. Li, S. J. Peng, D. S. Yu, J. N. Song, S. Ramakrishna and S. J. Guo, Adv. Mater., 2019, 31, 1808267 CrossRef PubMed.
- X. F. Lu, Y. Chen, S. B. Wang, S. Y. Gao and X. W. Lou, Adv. Mater., 2019, 31, 1902339 CrossRef PubMed.
- C. Tang, B. Wang, H. F. Wang and Q. Zhang, Adv. Mater., 2017, 29, 1703185 CrossRef PubMed.
- B. Q. Li, C. X. Zhao, S. M. Chen, J. N. Liu, X. Chen, L. Song and Q. Zhang, Adv. Mater., 2019, 31, 1900592 CrossRef PubMed.
- Y. B. Li, C. Zhong, J. Liu, X. Q. Zeng, S. X. Qu, X. Han, Y. P. Deng, W. B. Hu and J. Lu, Adv. Mater., 2018, 30, 1703657 CrossRef PubMed.
- C. X. Zhao, J. N. Liu, B. Q. Li, D. Ren, X. Chen, J. Yu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2003619 CrossRef CAS.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, B. Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- D. H. Deng, K. S. Novoselov, Q. Fu, N. F. Zheng, Z. Q. Tian and X. H. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- L. Tang, M. H. Xia, S. Y. Cao, X. Bo, S. B. Zhang, Y. L. Zhang, X. Liu, L. Z. Zhang, L. Yu and D. H. Deng, Nano Energy, 2022, 101, 107562 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. Y. Chen, H. Li, C. Fan, Q. W. Meng, Y. W. Tang, X. Y. Qiu, G. T. Fu and T. Y. Ma, Adv. Mater., 2020, 32, 2003134 CrossRef CAS PubMed.
- C. H. Zhou, X. Chen, S. Liu, Y. Han, H. B. Meng, Q. Y. Jiang, S. M. Zhao, F. Wei, J. Sun, T. Tan and R. F. Zhang, J. Am. Chem. Soc., 2022, 144, 2694–2704 CrossRef CAS PubMed.
- Y. S. Zhao, J. W. Wan, H. Y. Yao, L. J. Zhang, K. F. Lin, L. Wang, N. L. Yang, D. B. Liu, L. Song, J. Zhu, L. Gu, L. Liu, H. J. Zhao, Y. L. Li and D. Wang, Nat. Chem., 2018, 10, 924–931 CrossRef CAS PubMed.
- C. G. Hu, R. Paul, Q. B. Dai and L. M. Dai, Chem. Soc. Rev., 2021, 50, 11785 RSC.
- Y. Jiang, Y. P. Deng, R. L. Liang, J. Fu, R. Gao, D. Luo, Z. Y. Bai, Y. F. Hu, A. P. Yu and Z. W. Chen, Nat. Commun., 2020, 11, 5858 CrossRef CAS PubMed.
- Y. Jiang, Y. P. Deng, R. L. Liang, N. Chen, G. King, A. P. Yu and Z. W. Chen, J. Am. Chem. Soc., 2022, 144, 4783–4791 CrossRef CAS PubMed.
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- J. Yin, Y. X. Li, F. Lv, Q. H. Fan, Y. Q. Zhao, Q. L. Zhang, W. Wang, F. Y. Cheng, P. X. Xi and S. J. Guo, ACS Nano, 2017, 11, 2275–2283 CrossRef CAS PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- Q. C. Wang, Y. J. Ji, Y. P. Lei, Y. B. Wang, Y. D. Wang, Y. Y. Li and S. Y. Wang, ACS Energy Lett., 2018, 3, 1183 CrossRef CAS.
- Y. Tong, P. Z. Chen, T. P. Zhou, K. Xu, W. S. Chu, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 7121–7125 CrossRef CAS PubMed.
- M. C. Luo, Z. L. Zhao, Y. L. Zhang, Y. J. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. N. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. J. Guo, Nature, 2019, 574, 81 CrossRef CAS PubMed.
- X. Liu, L. Wang, P. Yu, C. G. Tian, F. F. Sun, J. Y. Ma, W. Li and H. G. Fu, Angew. Chem., Int. Ed., 2018, 57, 16166–16170 CrossRef CAS PubMed.
- Q. Liu, Y. B. Wang, L. M. Dai and J. N. Yao, Adv. Mater., 2016, 28, 3000–3006 CrossRef CAS PubMed.
- Y. J. Chen, S. F. Ji, S. Zhao, W. X. Chen, J. C. Dong, W. C. Cheong, R. A. Shen, X. D. Wen, L. R. Zheng, A. I. Rykov, S. C. Cai, H. L. Tang, Z. B. Zhuang, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- J. Yin, Y. X. Li, F. Lv, M. Lu, K. Sun, W. Wang, L. Wang, F. Y. Cheng, Y. F. Li, P. X. Xi and S. J. Guo, Adv. Mater., 2017, 29, 1704681 CrossRef PubMed.
- J. Zhang, Q. X. Zhou, Y. W. Tang, L. Zhang and Y. G. Li, Chem. Sci., 2019, 10, 8924–8929 RSC.
- H. F. Wang and Q. Xu, Matter, 2019, 1, 565–595 CrossRef CAS.
- S. S. Ren, X. D. Duan, S. Liang, M. D. Zhang and H. G. Zheng, J. Mater. Chem. A, 2020, 8, 6144–6182 RSC.
- S. S. Li, X. G. Hao, A. Abudula and G. Q. Guan, J. Mater. Chem. A, 2019, 7, 18674–18707 RSC.
- Z. F. Huang, J. J. Song, S. Dou, X. G. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 CrossRef.
- G. T. Fu, Y. Wang, Y. W. Tang, K. Zhou, J. B. Goodenough and J. M. Lee, ACS Mater. Lett., 2019, 1, 123–131 CrossRef CAS.
- Y. Y. Huang, Y. Q. Wang, C. Tang, J. Wang, Q. Zhang, Y. B. Wang and J. T. Zhang, Adv. Mater., 2019, 31, 1803800 CrossRef PubMed.
- L. Yan, Z. Y. Xu, W. K. Hu, J. Q. Ning, Y. J. Zhong and Y. Hu, Nano Energy, 2021, 82, 105710 CrossRef CAS.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- Z. Z. Liang, N. N. Kong, C. X. Yang, W. Zhang, H. Q. Zheng, H. P. Lin and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 12759–12764 CrossRef CAS PubMed.
- H. Yang, S. Gao, D. W. Rao and X. H. Yan, Energy Storage Mater., 2022, 46, 553–562 CrossRef.
- T. T. Cui, Y. P. Wang, T. Ye, J. Wu, Z. Q. Chen, J. Li, Y. P. Lei, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CrossRef CAS PubMed.
- Y. J. Li, L. Cui, P. F. Da, K. W. Qiu, W. J. Qin, W. B. Hu, X. W. Du, K. Davey, T. Ling and S. Z. Qiao, Adv. Mater., 2018, 30, 1804653 CrossRef PubMed.
- G. G. Yang, J. W. Zhu, P. F. Yuan, Y. F. Hu, G. Qu, B. A. Lu, X. Y. Xue, H. B. Yin, W. Z. Cheng, J. Q. Cheng, W. J. Xu, J. Li, J. S. Hu, S. C. Mu and J. N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- H. J. Qiu, P. Du, K. L. Hu, J. J. Gao, H. L. Li, P. Liu, T. Ina, K. Ohara, Y. Ito and M. W. Chen, Adv. Mater., 2019, 31, 1900843 CrossRef PubMed.
- X. Lei, Q. Y. Tang, Y. P. Zheng, P. Kidkhunthod, X. L. Zhou, B. F. Ji and Y. B. Tang, Nat. Sustainable, 2023, 6, 816–826 CrossRef.
- D. F. Yan, Y. X. Li, J. Huo, R. Chen, L. M. Dai and S. Y. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- C. Xie, D. F. Yan, W. Chen, Y. Q. Zou, R. Chen, S. Q. Zang, Y. Y. Wang, X. D. Yao and S. Y. Wang, Mater. Today, 2019, 31, 47–68 CrossRef CAS.
- X. C. Yan, Y. Jia and X. D. Yao, Chem. Soc. Rev., 2018, 47, 7628–7658 RSC.
- H. J. Huang, A. M. Huang, D. Liu, W. T. Han, C. H. Kuo, H. Y. Chen, L. L. Li, H. Pan and S. J. Peng, Adv. Mater., 2023, 35, 2303109 CrossRef CAS PubMed.
- H. Tian, A. L. Song, P. Zhang, K. A. Sun, J. J. Wang, B. Sun, Q. H. Fan, G. J. Shao, C. Chen, H. Liu, Y. D. Li and G. X. Wang, Adv. Mater., 2023, 35, 2210714 CrossRef CAS PubMed.
- F. Liu and Z. X. Fan, Chem. Soc. Rev., 2023, 52, 1723–1772 RSC.
- M. H. Luo, W. P. Sun, B. B. Xu, H. G. Pan and Y. Z. Jiang, Adv. Energy Mater., 2021, 11, 2002762 CrossRef CAS.
- Y. Yan, S. Liang, X. Wang, M. Y. Zhang, S. M. Hao, X. Cui, Z. W. Li and Z. Q. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110036118 CrossRef CAS PubMed.
- M. Jiao, Q. Zhang, C. Ye, Z. Liu, X. Zhong, J. Wang, C. Li, L. Dai, G. Zhou and H. M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202202119 CrossRef CAS PubMed.
- M. Jiao, Q. Zhang, C. Ye, R. Gao, L. Dai, G. Zhou and H. M. Cheng, ACS Nano, 2022, 16, 13223–13231 CrossRef CAS PubMed.
- Z. H. Liu, Y. Du, R. H. Yu, M. B. Zheng, R. Hu, J. S. Wu, Y. Y. Xia, Z. C. Zhuang and D. S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202212653 CrossRef CAS PubMed.
- W. Z. Li, L. K. Cheng, X. Y. Chen, Y. F. Liu, Y. Liu, Q. J. Liu and Y. Huang, Nano Energy, 2023, 106, 108039 CrossRef CAS.
- J. Yu, B. Q. Li, C. X. Zhao, J. N. Liu and Q. Zhang, Adv. Mater., 2020, 32, 1908488 CrossRef CAS PubMed.
- C. X. Zhao, J. N. Liu, J. Wang, C. D. Wang, X. Guo, X. Y. Li, X. Chen, L. Song, B. Q. Li and Q. Zhang, Sci. Adv., 2022, 8, eabn5091 CrossRef CAS PubMed.
- J. Wang, C. X. Zhao, J. N. Liu, Y. W. Song, J. Q. Huang and B. Q. Li, Nano Energy, 2022, 104, 107927 CrossRef CAS.
- P. Z. Chen, T. P. Zhou, L. L. Xing, K. Xu, Y. Tong, H. Xie, L. D. Zhang, W. S. Yan, W. S. Chu, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- X. N. Li, L. H. Liu, X. Y. Ren, J. J. Gao, Y. Q. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef CAS PubMed.
- J. Wang, H. H. Wu, D. F. Gao, S. Miao, G. X. Wang and X. H. Bao, Nano Energy, 2015, 13, 387–396 CrossRef CAS.
- Y. L. Zhao, L. Xu, L. Q. Mai, C. H. Han, Q. Y. An, X. Xu, X. Liu and Q. J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19569–19574 CrossRef CAS PubMed.
- R. Cepitis, N. Kongi, J. Rossmeisl and V. Ivanistsev, ACS Energy Lett., 2023, 8, 1330–1335 CrossRef CAS PubMed.
- Q. Liu, L. Wang and H. G. Fu, J. Mater. Chem. A, 2023, 11, 4400–4427 RSC.
- P. Zhang, K. Chen, J. Y. Li, M. M. Wang, M. Li, Y. Q. Liu and Y. Pan, Adv. Mater., 2023, 35, 2303243 CrossRef CAS PubMed.
- Y. Gao, B. Z. Liu and D. S. Wang, Adv. Mater., 2023, 35, 2209654 CrossRef CAS PubMed.
- Z. J. Li, S. Q. Ji, C. Wang, H. X. Liu, L. P. Leng, L. Du, J. C. Gao, M. Qiao, J. H. Horton and Y. Wang, Adv. Mater., 2023, 35, 2300905 CrossRef CAS PubMed.
- B. Chen, X. W. Zhong, G. M. Zhou, N. Q. Zhao and H. M. Cheng, Adv. Mater., 2022, 34, 2105812 CrossRef CAS PubMed.
- G. Yasin, S. Ali, S. Ibraheem, A. Kumar, M. Tabish, M. A. Mushtaq, S. Ajmal, M. Arif, M. A. Khan, A. Saad, L. Qiao and W. Zhao, ACS Catal., 2023, 13, 2313–2325 CrossRef CAS.
- B. Y. Lu, B. Chen, D. S. Wang, C. Li, R. H. Gao, Y. Q. Liu, R. Mao, J. L. Yang and G. M. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216933120 CrossRef CAS PubMed.
- R. C. Iwata, L. N. Zhang, K. L. Wilke, S. Gong, M. F. He, B. M. Gallant and E. N. Wang, Joule, 2021, 5, 887–900 CrossRef CAS.
- L. Tang, L. Yu, C. Ma, Y. Song, Y. C. Tu, Y. L. Zhang, X. Bo and D. H. Deng, J. Mater. Chem. A, 2022, 10, 6242–6250 RSC.
- L. K. Gao, X. Cui, C. D. Sewell, J. Li and Z. Q. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- L. K. Gao, X. Cui, Z. W. Wang, C. D. Sewell, Z. L. Li, S. Liang, M. Y. Zhang, J. Li, Y. J. Hu and Z. Q. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023421118 CrossRef CAS PubMed.
- T. Wang, Y. He, Y. J. Liu, F. J. Guo, X. F. Li, H. B. A. Chen, H. M. Li and Z. Q. Lin, Nano Energy, 2021, 79, 105487 CrossRef CAS.
- J. T. Fan, M. Chen, Z. L. Zhao, Z. Zhang, S. Y. Ye, S. Y. Xu, H. J. Wang and H. Li, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- W. D. Xue, Q. X. Zhou, X. Cui, J. W. Zhang, S. J. Zuo, F. Mo, J. W. Jiang, X. Y. Zhu and Z. Q. Lin, Angew. Chem., Int. Ed., 2023, 62, e2023075 Search PubMed.
- R. Ma, X. Cui, X. X. Xu, Y. L. Wang, G. Q. Xiang, L. K. Gao, Z. Q. Lin and Y. K. Yang, Nano Energy, 2023, 108, 108179 CrossRef CAS.
- B. F. Ji, J. L. Gou, Y. P. Zheng, X. H. Pu, Y. H. Wang, P. Kidkhunthod and Y. B. Tang, Adv. Mater., 2023, 35, 2300381 CrossRef CAS PubMed.
- B. F. Ji, J. L. Gou, Y. P. Zheng, X. L. Zhou, P. Kidkhunthod, Y. H. Wang, Q. Y. Tang and Y. B. Tang, Adv. Mater., 2022, 34, 2202714 CrossRef CAS PubMed.
- T. Meng, P. P. Sun, F. Yang, J. Zhu, B. G. Mao, L. R. Zheng and M. H. Cao, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 2214089119 CrossRef PubMed.
- Z. J. Li, S. Q. Ji, C. Xu, L. P. Leng, H. X. Liu, J. H. Horton, L. Du, J. C. Gao, C. He, X. Y. Qi, Q. Xu and J. F. Zhu, Adv. Mater., 2023, 35, 2209644 CrossRef CAS PubMed.
- W. J. Lu, C. K. Zhang, H. M. Zhang and X. F. Li, ACS Energy Lett., 2021, 6, 2765–2785 CrossRef CAS.
- C. Y. Yang, J. L. Xia, C. Y. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan, E. Thimsen, J. J. Xu, W. T. Song, E. Y. Hu, X. Ji, S. Y. Hou, X. Y. Zhang, M. S. Ding, S. Hwang, D. Su, Y. Ren, X. Q. Yang, H. Wang, O. Borodin and C. S. Wang, Nat. Sustain., 2023, 6, 325–335 CrossRef.
- Y. X. Zeng, P. X. Sun, Z. H. Pei, Q. Jin, X. T. Zhang, L. Yu and X. W. Lou, Adv. Mater., 2022, 34, 2200342 CrossRef CAS PubMed.
- Y. H. Zou, X. Z. Yang, L. Shen, Y. W. Su, Z. Y. Chen, X. Gao, J. Zhou and J. Y. Sun, Energy Environ. Sci., 2022, 15, 5017–5038 RSC.
- J. F. Parker, J. S. Ko, D. R. Rolison and J. W. Long, Joule, 2018, 2, 2519–2527 CrossRef CAS.
- X. W. Zhong, Y. F. Shao, B. Chen, C. Li, J. Z. Sheng, X. Xiao, B. M. Xu, J. Li, H. M. Cheng and G. M. Zhou, Adv. Mater., 2023, 35, 2301952 CrossRef CAS PubMed.
- J. S. Lee, S. T. Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- D. U. Lee, J. Y. Choi, K. Feng, H. W. Park and Z. W. Chen, Adv. Energy Mater., 2014, 4, 1301389 CrossRef.
- T. Zhang, N. Imanishi, Y. Shimonishi, A. Hirano, Y. Takeda, O. Yamamoto and N. Sammes, Chem. Commun., 2010, 46, 1661–1663 RSC.
- L. J. Li, Y. Z. Fu and A. Manthiram, Electrochem. Commun., 2014, 47, 67–70 CrossRef CAS.
- L. J. Li and A. Manthiram, Adv. Energy Mater., 2016, 6, 1502054 CrossRef.
- L. J. Li, S. H. Chai, S. Dai and A. Manthiram, Energy Environ. Sci., 2014, 7, 2630–2636 RSC.
- J. X. Li, K. Zhang, B. J. Wang and H. S. Peng, Angew. Chem., Int. Ed., 2022, 61, e202213026 CrossRef CAS PubMed.
- J. Q. Lv, S. C. Abbas, Y. Y. Huang, Q. Liu, M. X. Wu, Y. B. Wang and L. M. Dai, Nano Energy, 2018, 43, 130–137 CrossRef CAS.
- X. R. Liu, Y. F. Yuan, J. Liu, B. Liu, X. Chen, J. Ding, X. P. Han, Y. D. Deng, C. Zhong and W. B. Hu, Nat. Commun., 2019, 10, 4767 CrossRef PubMed.
- D. F. Du, S. Zhao, Z. Zhu, F. J. Li and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 18140–18144 CrossRef CAS PubMed.
- W. X. Shang, W. T. Yu, P. Tan, B. Chen, Z. Wu, H. R. Xu and M. Ni, J. Mater. Chem. A, 2019, 7, 15564–15574 RSC.
- X. W. Wang, F. X. Wang, L. Y. Wang, M. X. Li, Y. F. Wang, B. W. Chen, Y. S. Zhu, L. J. Fu, L. S. Zha, L. X. Zhang, Y. P. Wu and W. Huang, Adv. Mater., 2016, 28, 4904–4911 CrossRef CAS PubMed.
- L. T. Ma, S. M. Chen, Z. X. Pei, H. F. Li, Z. F. Wang, Z. X. Liu, Z. J. Tang, J. A. Zapien and C. Y. Zhi, ACS Nano, 2018, 12, 8597–8605 CrossRef CAS PubMed.
- D. U. Lee, J. Fu, M. G. Park, H. Liu, A. G. Kashkooli and Z. W. Chen, Nano Lett., 2016, 16, 1794–1802 CrossRef CAS PubMed.
- C. C. Chang, Y. C. Lee, H. J. Liao, Y. T. Kao, J. Y. An and D. Y. Wang, ACS Sustainable Chem. Eng., 2019, 7, 2860–2866 CrossRef CAS.
- B. Li, X. M. Ge, F. W. T. Goh, T. S. A. Hor, D. S. Geng, G. J. Du, Z. L. Liu, J. Zhang, X. G. Liu and Y. Zong, Nanoscale, 2015, 7, 1830–1838 RSC.
- P. Tan, B. Chen, H. R. Xu, W. Z. Cai, W. He and M. Ni, Electrochim. Acta, 2018, 283, 1028–1036 CrossRef CAS.
- P. Tan, B. Chen, H. R. Xu, W. Z. Cai, W. He and M. Ni, Appl. Catal., B, 2019, 241, 104–112 CrossRef CAS.
- K. L. Wang, P. C. Pei, Z. Ma, H. C. Chen, H. C. Xu, D. F. Chen and X. Z. Wang, J. Mater. Chem. A, 2015, 3, 22648–22655 RSC.
- J. F. Parker, E. S. Nelson, M. D. Wattendorf, C. N. Chervin, J. W. Long and D. R. Rolison, ACS Appl. Mater. Interfaces, 2014, 6, 19471–19476 CrossRef CAS PubMed.
- X. C. Chen, Z. Zhou, H. E. Karahan, Q. Shao, L. Wei and Y. Chen, Small, 2018, 14, 1801929 CrossRef PubMed.
- J. X. Zheng, D. C. Bock, T. Tang, Q. Zhao, J. F. Yin, K. R. Tallman, G. Wheeler, X. T. Liu, Y. Deng, S. Jin, A. C. Marschilok, E. S. Takeuchi, K. J. Takeuchi and L. A. Archer, Nat. Energy, 2021, 6, 398 CrossRef CAS.
- J. N. Hao, X. L. Li, X. H. Zeng, D. Li, J. F. Mao and Z. P. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- L. An, Z. Y. Zhang, J. R. Feng, F. Lv, Y. X. Li, R. Wang, M. Lu, R. B. Gupta, P. X. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed.
- Y. F. Cui, Y. H. Zhu, J. Y. Du, Y. L. Zhang, K. Li, W. Q. Liu, G. Huang and X. B. Zhang, Joule, 2022, 6, 1617–1631 CrossRef CAS.
- M. L. Jiao, L. X. Dai, H. R. Ren, M. T. Zhang, X. Xiao, B. R. Wang, J. L. Yang, B. L. Liu, G. M. Zhou and H. M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202301114 CrossRef CAS PubMed.
- J. Park, M. Park, G. Nam, J. S. Lee and J. Cho, Adv. Mater., 2015, 27, 1396 CrossRef CAS PubMed.
- S. S. Shinde, J. Y. Jung, N. K. Wagh, C. H. Lee, D. H. Kim, S. H. Kim, S. U. Lee and J. H. Lee, Nat. Energy, 2021, 6, 592–604 CrossRef CAS.
- J. Yin, Y. Z. Wang, Y. P. Zhu, J. J. Jin, C. L. Chen, Y. Y. Yuan, Z. Bayhan, N. Salah, N. A. Alhebshi, W. L. Zhang, U. Schwingenschlogl and H. N. Alshareef, Nano Energy, 2022, 99, 107331 CrossRef CAS.
- P. Xiong, Y. Zhang, J. R. Zhang, S. Ha Baek, L. X. Zeng, Y. Yao and H. S. Park, Energychem, 2022, 4, 100076 CrossRef CAS.
- S. X. Qu, B. Liu, X. Y. Fan, X. R. Liu, J. Liu, J. Ding, X. P. Han, Y. D. Deng, W. B. Hu and C. Zhong, ChemistrySelect, 2020, 5, 8305–8310 CrossRef CAS.
- L. Y. Li, Y. C. A. Tsang, D. W. Xiao, G. Y. Zhu, C. Y. Zhi and Q. Chen, Nat. Commun., 2022, 13, 2870 CrossRef CAS PubMed.
- D. Yang, J. S. Li, C. P. Liu, W. Xing and J. B. Zhu, J. Energy Chem., 2023, 82, 122–138 CrossRef CAS.
- W. X. Shang, W. T. Yu, Y. F. Liu, R. X. Li, Y. W. Dai, C. Cheng, P. Tan and M. Ni, Energy Storage Mater., 2020, 31, 44–57 CrossRef.
- L. P. Wang, N. W. Li, T. S. Wang, Y. X. Yin, Y. G. Guo and C. R. Wang, Electrochim. Acta, 2017, 244, 172–177 CrossRef CAS.
- X. X. Yang, X. C. Zheng, H. X. Li, B. C. Luo, Y. K. He, Y. Yao, H. H. Zhou, Z. H. Yan, Y. F. Kuang and Z. Y. Huang, Adv. Funct. Mater., 2022, 32, 2200397 CrossRef CAS.
- W. Sun, M. M. Ma, M. G. Zhu, K. L. Xu, T. Xu, Y. C. Zhu and Y. T. Qian, Small, 2022, 18, 2106604 CrossRef CAS PubMed.
- P. Thakur, K. Alam, A. Roy, C. Downing, V. Nicolosi, P. Sen and T. N. Narayanan, ACS Appl. Mater. Interfaces, 2021, 13, 33112–33122 CrossRef CAS PubMed.
- D. J. Dong, T. R. Wang, Y. Sun, J. Fan and Y. C. Lu, Nat. Sustain., 2023, 6, 1474–1484 CrossRef.
- Y. Yamada, J. H. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- N. K. Wagh, D. H. Kim, S. H. Kim, S. S. Shinde and J. H. Lee, ACS Nano, 2021, 15, 14683–14696 CrossRef CAS PubMed.
- W. Sun, V. Küpers, F. Wang, P. Bieker and M. Winter, Angew. Chem., Int. Ed., 2022, 61, e202207353 CrossRef CAS PubMed.
- C. Wang, Z. X. Pei, Q. Q. Meng, C. M. Zhang, X. Sui, Z. W. Yuan, S. J. Wang and Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 990–997 CrossRef CAS PubMed.
- Y. A. Zhang, D. S. Wu, F. L. Huang, Y. B. Cai, Y. G. Li, H. Z. Ke, P. F. Lv and Q. F. Wei, Adv. Funct. Mater., 2022, 32, 2203204 CrossRef CAS.
- C. Y. Chen, K. Matsumoto, K. Kubota, R. Hagiwara and Q. Xu, Adv. Energy Mater., 2019, 9, 1900196 CrossRef.
- W. Sun, F. Wang, B. Zhang, M. Y. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. S. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- L. S. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Y. Yang, L. Chen, J. Vatamanu, E. Y. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Y. Hou, K. Gaskell, J. T. Fourkas, X. Q. Yang, K. Xu, O. Borodin and C. S. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
- X. P. Han, X. Y. Wu, C. Zhong, Y. D. Deng, N. Q. Zhao and W. B. Hu, Nano Energy, 2017, 31, 541–550 CrossRef CAS.
- X. Zhong, Z. Zheng, J. Xu, X. Xiao, C. Sun, M. Zhang, J. Ma, B. Xu, K. Yu, X. Zhang, H. M. Cheng and G. Zhou, Adv. Mater., 2023, 35, 2209980 CrossRef CAS PubMed.
- M. Jiao, L. Dai, H. R. Ren, M. Zhang, X. Xiao, B. Wang, J. Yang, B. Liu, G. Zhou and H. M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202301114 CrossRef CAS PubMed.
- X. Y. Fan, H. Z. Wang, X. R. Liu, J. Liu, N. Q. Zhao, C. Zhong, W. B. Hu and J. Lu, Adv. Mater., 2023, 35, 2209290 CrossRef CAS PubMed.
- Y. Huang, M. Zhong, F. K. Shi, X. Y. Liu, Z. J. Tang, Y. K. Wang, Y. Huang, H. Q. Hou, X. M. Xie and C. Y. Zhi, Angew. Chem., Int. Ed., 2017, 56, 9141–9145 CrossRef CAS PubMed.
- M. Wang, N. N. Xu, J. Fu, Y. Y. Liu and J. L. Qiao, J. Mater. Chem. A, 2019, 7, 11257–11264 RSC.
- C. Lin, S. S. Shinde, X. P. Li, D. H. Kim, N. W. Li, Y. Sun, X. K. Song, H. J. Zhang, C. H. Lee, S. U. Lee and J. H. Lee, ChemSusChem, 2018, 11, 3215–3224 CrossRef CAS PubMed.
- T. N. T. Tran, H. J. Chung and D. G. Ivey, Electrochim. Acta, 2019, 327, 135021 CrossRef CAS.
- H. F. Li, C. P. Han, Y. Huang, Y. Huang, M. S. Zhu, Z. X. Pei, Q. Xue, Z. F. Wang, Z. X. Liu, Z. J. Tang, Y. K. Wang, F. Y. Kang, B. H. Li and C. Y. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC.
- Z. S. Song, J. Ding, B. Liu, X. R. Liu, X. P. Han, Y. D. Deng, W. B. Hu and C. Zhong, Adv. Mater., 2020, 32, 1908127 CrossRef CAS PubMed.
- K. Takechi, T. Shiga and T. Asaoka, Chem. Commun., 2011, 47, 3463–3465 RSC.
- X. Y. Wang, J. F. Xie, M. A. Ghausi, J. Q. Lv, Y. Y. Huang, M. X. Wu, Y. B. Wang and J. N. Yao, Adv. Mater., 2019, 31, 1807807 CrossRef PubMed.
- Z. P. Zeng, L. Y. Gan, H. B. Yang, X. Z. Su, J. J. Gao, W. Liu, H. Matsumoto, J. Gong, J. M. Zhang, W. Z. Cai, Z. Y. Zhang, Y. B. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed.
- L. L. Han, X. Y. Peng, H. T. Wang, P. F. Ou, Y. Y. Mi, C. W. Pao, J. G. Zhou, J. Wang, X. J. Liu, W. F. Pong, J. Song, Z. Lin, J. Luo and H. L. L. Xin, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2207326119 CrossRef CAS PubMed.
- W. Wang, Y. Wu, X. Cao, L. Gu and J. Hu, Adv. Funct. Mater., 2020, 30, 1908965 CrossRef.
- R. Zhang, S. C. Zhang, Y. Guo, C. Li, J. H. Liu, Z. D. Huang, Y. W. Zhao, Y. Y. Li and C. Y. Zhi, Energy Environ. Sci., 2022, 15, 3024–3032 RSC.
- Z. L. Li, S. L. Ning, J. C. Xu, J. M. Zhu, Z. X. Yuan, Y. L. Wu, J. Chen, F. Y. Xie, Y. S. Jin, N. Wang, H. Meng and S. H. Sun, Energy Environ. Sci., 2022, 15, 5300–5312 RSC.
- S. Y. An, Z. P. Liu, J. Bu, J. Lin, Y. Yao, C. Yan, W. Tian and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202116370 CrossRef CAS PubMed.
- Q. Q. Liu, C. F. Xia, C. H. He, W. Guo, Z. P. Wu, Z. Li, Q. Zhao and B. Y. Xia, Angew. Chem., Int. Ed., 2022, 61, e202210567 CrossRef CAS PubMed.
Footnote |
| † L Tang and H Peng contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |