
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-driven nitrogen fixation routes for green ammonia production
Laura
Collado
 *,
Alejandro H.
Pizarro
,
Mariam
Barawi
,
Miguel
García-Tecedor
,
Marta
Liras
and
Víctor A.
de la Peña O'Shea
*
*,
Alejandro H.
Pizarro
,
Mariam
Barawi
,
Miguel
García-Tecedor
,
Marta
Liras
and
Víctor A.
de la Peña O'Shea
*
Photoactivated Processes Unit, IMDEA Energy Institute, Móstoles, Madrid 28935, Spain. E-mail: laura.collado@imdea.org; victor.delapenya@imdea.org
First published on 10th October 2024
Abstract
The global goal for decarbonization of the energy sector and the chemical industry could become a reality by a massive increase in renewable-based technologies. For this clean energy transition, the versatile green ammonia may play a key role in the future as a fossil-free fertilizer, long-term energy storage medium, chemical feedstock, and clean burning fuel for transportation and decentralized power generation. The high energy-intensive industrial ammonia production has triggered researchers to look for a step change in new synthetic approaches powered by renewable energies. This review provides a comprehensive comparison of light-mediated N2 fixation technologies for green ammonia production, including photocatalytic, photoelectrocatalytic, PV-electrocatalytic and photothermocatalytic routes. Since these approaches are still at laboratory scale, we examine the most recent developments and discuss the open challenges for future improvements. Last, we offer a technoeconomic comparison of current and emerging ammonia production technologies, highlighting costs, barriers, recommendations, and potential opportunities for the real development of the next generation of green ammonia solutions.
 Laura Collado | Laura Collado is a Senior Assistant Researcher at IMDEA Energy in charge of the photo(thermo)catalytic research line at the Photoactivated Processes Unit. She received her PhD in Chemical and Environmental Engineering from Rey Juan Carlos University in 2015, working on solar fuels production by artificial photosynthesis. She performed a PhD stay at Imperial College London (UK) to develop charge dynamics studies. In 2016, she joined Heriot-Watt University (UK) as a postdoctoral researcher. She obtained a GotEnergyTalent Marie-Skłodowska-Curie-cofund (2018) and a Juan de la Cierva-Incorporación 2020 fellowships. Shortly after, she obtained a JIN-type R + D + i project (Principal Investigator) at IMDEA Energy. Her scientific background covers light-driven catalytic processes, synthesis of multifunctional catalysts, advanced characterization and mechanistic studies. |
 Alejandro H. Pizarro | Alejandro Herrero is a postdoctoral researcher at the Photoactivated Process Unit at IMDEA Energy. He received his PhD in Chemical Engineering from Universidad Autónoma de Madrid (UAM) in 2013, working on the catalytic hydrotreatment of chlorinated and nitrogenated compounds in water. In 2014 he moved to Abengoa Research S.L. (Sevilla) for more than two years as a postdoctoral researcher, working on drinking water treatments for the removal of nitrate and disinfection by-products. He also worked on nitrate removal at Université du Poitiers in 2012 and at the UAM in 2017–2018. Since 2022, he was focused on photo(thermo)catalytic production of hydrogen and ammonia at IMDEA Energy. |
 Mariam Barawi | Mariam Barawi is a Senior Assistant researcher in charge of the (photo)electrochemical cells line in Photoactivated Process Unit. She obtained her PhD in 2015 at Universidad Autónoma de Madrid, investigating photoelectrochemical solar energy conversion. Thereafter she started her postdoctoral period at Istituto Italiano di Tecnologia investigating nanocrystals for Smart Windows. In 2017, she joined IMDEA Energy and has been awarded the three more prestigious Spanish Science Ministry grants (Juan de la Cierva-Formación 2017, Juan de la Cierva-Incorporación 2020 and Ramon Y Cajal 2023). Mariam is co-author of more than 50 scientific publications (>2500 citations) and has participated in more than 20 research projects, being Principal Investigator in three of them. |
 Miguel García-Tecedor | Miguel García Tecedor (PhD in Physics 2017, Universidad Complutense de Madrid) is a Senior Assistant Researcher at IMDEA Energy. His PhD focused on the growth and characterization of semiconducting nanostructures and their application in optoelectronics and energy. In 2015, he joined the Institute for Energy Technology (Norway) to work on organic–inorganic composites for silicon solar cells’ passivation. In 2017, he joined the Institute of Advanced Materials at Universitat Jaume I as Research Scientist to investigate (photo)electrocatalytic water splitting and CO2 reduction. In 2021, Miguel joined IMDEA Energy to work on photo(electro)catalytic approaches towards wastewater oxidation, CO2 reduction and N2 fixation. Miguel is co-author of 43 scientific publications and he has participated in 14 research projects, being principal investigator in two. |
 Marta Liras | Marta Liras is a Senior Researcher at the Photoactivated Processes Unit from IMDEA Energy since 2022. She received her PhD in organic chemistry in 2003 from Universidad Complutense de Madrid (UCM) for her work at the Organic Chemistry Institute (IQOG-CSIC) and Science and Technology of Polymers Institute (ICTP-CSIC). In 2008 she joined Marta Liras the J.C. Scaiano research group at the University of Ottawa as a postdoctoral researcher. She was awarded with Juan de la Cierva (2004), JAE-Doc (2013) and Ramon y Cajal (2016) grants. She leads the Specialized Photochemistry Group of Spanish Royal Society of Chemistry GRUFO-RSEQ. Her interests are focused on polymer design for energy applications. |
 Víctor A. de la Peña O'Shea | Víctor A. de la Peña O’Shea is the head of the Photoactivated Processes Unit at IMDEA Energy in Madrid (Spain) from 2015. He obtained a PhD in Physical-Chemistry from the University Autonoma de Madrid and Catalysis and Petrochemistry Institute (ICP-CSIC). His research interest is focused on different aspects of Energy production and storage technologies covering a wide range of experience on: (1) design and synthesis of multifunctional hybrid materials; (2) advanced in-operando characterization combined with theoretical calculations; and (3) construction of automatized reactors form lab to pilot plant scale, covering wide range of application from heterogeneous catalysis and solar chemistry technologies. |
1. Introduction
1.1. Ammonia: the key to a greener future
A transformative solution to the ever-growing energy crisis and global warming is to replace the current fossil feedstock with renewable and carbon-free energy sources. A fundamental pillar of this energy transition relies on the development of affordable, safe and sustainable technologies to store and transport renewables, which would contribute to alleviate the fossil fuels dependency and balance the mismatch between renewable energy supply and demand at the power grid. Storing cheap renewable electricity into chemical bonds (i.e. chemical energy storage) opens the door to a long-term energy storage, which would drive the transition towards carbon-free fuels and commodity chemicals.1,2 Among renewables, solar energy is by far the largest exploitable energy resource on earth (4.3 × 1020 J h−1).3,4 In fact, the total sum of recoverable energy from all renewable and non-renewable reserves only covers 1% of the solar energy arriving to the earth (173![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TW h per hour5), which is almost 10000 times higher than the global primary energy consumption of the world (178899 TW h year−1 in 2022).6 However, the challenge lies in the development of sustainable and cheap technologies able to harvest and storage solar energy as high-energy fuels and high-value chemicals, which could be used in a clean and decentralized manner.2 A strong candidate to pursue the decarbonization of the energy sector is green ammonia (NH3), which due to its versatility (i.e. existence of N and H elements in its structure) could confront two of the most pressing global challenges today: food and energy security in the context of a low-carbon economy.7 Basically, ammonia is a multipurpose chemical due to its composition. N atoms provide the molecule an agrochemical value, while H atoms (ca. 17.6 wt%) enables the cracking of ammonia to hydrogen (H2), which can be used in many sectors such as transportation, electricity generation and energy storage.1,8
000 TW h per hour5), which is almost 10000 times higher than the global primary energy consumption of the world (178899 TW h year−1 in 2022).6 However, the challenge lies in the development of sustainable and cheap technologies able to harvest and storage solar energy as high-energy fuels and high-value chemicals, which could be used in a clean and decentralized manner.2 A strong candidate to pursue the decarbonization of the energy sector is green ammonia (NH3), which due to its versatility (i.e. existence of N and H elements in its structure) could confront two of the most pressing global challenges today: food and energy security in the context of a low-carbon economy.7 Basically, ammonia is a multipurpose chemical due to its composition. N atoms provide the molecule an agrochemical value, while H atoms (ca. 17.6 wt%) enables the cracking of ammonia to hydrogen (H2), which can be used in many sectors such as transportation, electricity generation and energy storage.1,8
In nature, ammonia is produced by a group of diazotrophic microorganisms that transform atmospheric or aqueous N2 into NH3 at mild conditions (<40 °C, atmospheric pressure) with a turnover frequency (TOF) of 40–120 min−1.9,10 This biological N2 fixation (N2 + 6H+ + 6e− → 2NH3) is restricted to especially cyanobacteria (e.g. Anabaena, Azotobacter) containing a metalloenzyme nitrogenase with a FeMo cofactor, which is able to convert 50–150 kg air per hectare.11–14 However, the natural synthesis of ammonia is not sufficient to confront the current global demand. In fact, NH3 is the second most produced chemical worldwide, after sulphuric acid, with approximately 183 Mtons of annual production,15 and it is one of the largest-volume industrial chemicals in terms of energy use and carbon footprint.16 Around 80% of the current NH3 production is used as a key component of mineral fertilizers,17 while the rest is mostly used as industrial refrigerant and chemical feedstock in the production of polyimides, nitric acid, nylon, pharmaceuticals, dyes, explosive materials, cleaning solutions, etc.18–20
Industrial NH3 production is carried out by the traditional thermochemical Haber–Bosch (H–B) process, following the procedure first developed in the beginning of the 20th century. The H–B process requires high-purity (99.99%) H2 and N2 gas streams, and uses Fe- or Ru-based catalysts, typically promoted with K2O, CaO, SiO2, and Al2O3. Besides, it requires high temperature and pressure (ca. 350–550 °C, 20–40 MPa) to enable fast kinetics and to shift the reaction equilibrium towards NH3 yield (1/2N2(g) + 3/2H2(g) ↔ NH3).16,18,21
Although the formation of ammonia is exothermic (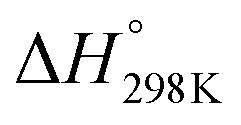 = −45.9 kJ mol−1,
= −45.9 kJ mol−1, 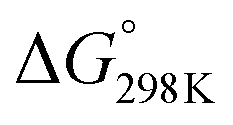 = −16.4 kJ mol−1, Keq = 750) and favoured under low temperatures, the high chemical inertness of the N2 molecule requires high temperatures to break the N
= −16.4 kJ mol−1, Keq = 750) and favoured under low temperatures, the high chemical inertness of the N2 molecule requires high temperatures to break the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond at sufficient rates.16,22 In fact, equilibrium calculations show that more than 99% of ammonia decomposes to N2 and H2 at temperatures higher than 400 °C at 0.1 MPa. Therefore, NH3 production via H–B is operated under high pressures (20–40 MPa) to shift the equilibrium to the right (eqn (1)), reaching ∼15% N2 conversion to NH3 in a single reaction, or ∼97% yield when unreacted N2 and H2 are recirculated to the reactor.19,22 About 96% of the supplied hydrogen is derived from fossil fuels, mainly from steam reforming of methane (72%) from natural gas or coal, and from partial oxidation of coal (26%). The remaining 4% is generated from coal or natural gas electrical generation.18,20 Consequently, H–B is one of the largest energy consumer and greenhouse gas emitter processes. Indeed, it accounts with 3–5% of global annual natural gas consumption and 1–3% of the global electrical energy, resulting in an overall energy usage of more than 30 GJ tNH3−1.20,23,24 Moreover, the strict operational conditions are responsible for about 1.8% of the global CO2 emissions per year, which represent 2.16 kg of carbon dioxide equivalent (CO2eq) for each kg of NH3 produced.18 These CO2 emissions are nearly twice intensive as those of crude steel production and four times those of cement industry.25 Besides, H–B also generates vast amounts of NOx that need to be removed via selective non-catalytic reduction, increasing the costs of operation.18 It should be noted that this process also requires large-scale NH3 plants that increase the capital expenditure and introduce geographical limitations for the construction of production facilities.21 Apart from this, the availability of feedstock and energy supply (i.e. low-cost natural gas and abundant coal reserves) limits the locations where ammonia can be produced. To date, China is the largest NH3 manufacturer (ca. 30% of production and 45% of associated CO2 emissions), followed by United States, European Union, Russia, India and Middle East (ca. 8–10% production each). Important energy savings have been achieved over the past 20th century by shifting away from coal gasification to more efficient natural gas-based production, and implementing large centrifugal compressors, better process control and maintenance, better use of waste heat, and catalyst improvements.26 However, efficiency gains have almost reached the theoretical minimum energy intensity,25 thus making clear the critical need to develop alternative routes for a renewable, efficient and affordable ammonia synthesis. Over the last few years, intensive research activity is currently devoted to achieve the decarbonization of ammonia synthesis, preferably under similar mild conditions as the biological process. In 1921, renewable ammonia started to be produced from hydropower, but only one commercial plant is still operational to date.27 At present, the annual production of renewable ammonia (<0.02 Mt) represents 0.01% of the global ammonia production. Various demonstration plants are today under operation, based on solar and wind energy coupled with electrolyzers for H2 production.27
N bond at sufficient rates.16,22 In fact, equilibrium calculations show that more than 99% of ammonia decomposes to N2 and H2 at temperatures higher than 400 °C at 0.1 MPa. Therefore, NH3 production via H–B is operated under high pressures (20–40 MPa) to shift the equilibrium to the right (eqn (1)), reaching ∼15% N2 conversion to NH3 in a single reaction, or ∼97% yield when unreacted N2 and H2 are recirculated to the reactor.19,22 About 96% of the supplied hydrogen is derived from fossil fuels, mainly from steam reforming of methane (72%) from natural gas or coal, and from partial oxidation of coal (26%). The remaining 4% is generated from coal or natural gas electrical generation.18,20 Consequently, H–B is one of the largest energy consumer and greenhouse gas emitter processes. Indeed, it accounts with 3–5% of global annual natural gas consumption and 1–3% of the global electrical energy, resulting in an overall energy usage of more than 30 GJ tNH3−1.20,23,24 Moreover, the strict operational conditions are responsible for about 1.8% of the global CO2 emissions per year, which represent 2.16 kg of carbon dioxide equivalent (CO2eq) for each kg of NH3 produced.18 These CO2 emissions are nearly twice intensive as those of crude steel production and four times those of cement industry.25 Besides, H–B also generates vast amounts of NOx that need to be removed via selective non-catalytic reduction, increasing the costs of operation.18 It should be noted that this process also requires large-scale NH3 plants that increase the capital expenditure and introduce geographical limitations for the construction of production facilities.21 Apart from this, the availability of feedstock and energy supply (i.e. low-cost natural gas and abundant coal reserves) limits the locations where ammonia can be produced. To date, China is the largest NH3 manufacturer (ca. 30% of production and 45% of associated CO2 emissions), followed by United States, European Union, Russia, India and Middle East (ca. 8–10% production each). Important energy savings have been achieved over the past 20th century by shifting away from coal gasification to more efficient natural gas-based production, and implementing large centrifugal compressors, better process control and maintenance, better use of waste heat, and catalyst improvements.26 However, efficiency gains have almost reached the theoretical minimum energy intensity,25 thus making clear the critical need to develop alternative routes for a renewable, efficient and affordable ammonia synthesis. Over the last few years, intensive research activity is currently devoted to achieve the decarbonization of ammonia synthesis, preferably under similar mild conditions as the biological process. In 1921, renewable ammonia started to be produced from hydropower, but only one commercial plant is still operational to date.27 At present, the annual production of renewable ammonia (<0.02 Mt) represents 0.01% of the global ammonia production. Various demonstration plants are today under operation, based on solar and wind energy coupled with electrolyzers for H2 production.27
In this context, important research efforts are currently focused on finding efficient and environmental-friendly ammonia production routes, fully powered by renewable energy and operated at mild conditions. The so-called artificial N2 fixation or nitrogen reduction reaction (NRR) is a promising candidate for the next generation of ammonia production technologies, which is included within the so-called emerging power-to-X (P2X) technologies. In brief, NRR targets the catalytic conversion of natural feedstocks (N2/air, H2O) into ammonia under milder conditions than the conventional H–B process, powered by renewable energies.28,29 When NRR is powered by solar energy, this process can be carried out by different light-driven routes such as photocatalysis, (photo)electrocatalysis, and photothermal catalysis.
A close look to the state-of-the-art literature reveals an increasing publication rate over the past 10 years (including research articles and patents), which has been especially noticeable over the last 6 years (Fig. 1). This trend highlights the big potential of NRR technology to contribute to the decarbonization of the energy system and the fertilizers industry, but also other pressing sectors such as transportation or chemical industry. However, the production rates and selectivity of the current catalytic systems still fail to reach industrial interest. Therefore, research efforts still need to be directed on the development of efficient catalysts and cost-effective solutions to take a leap forward in performance for practical applications.
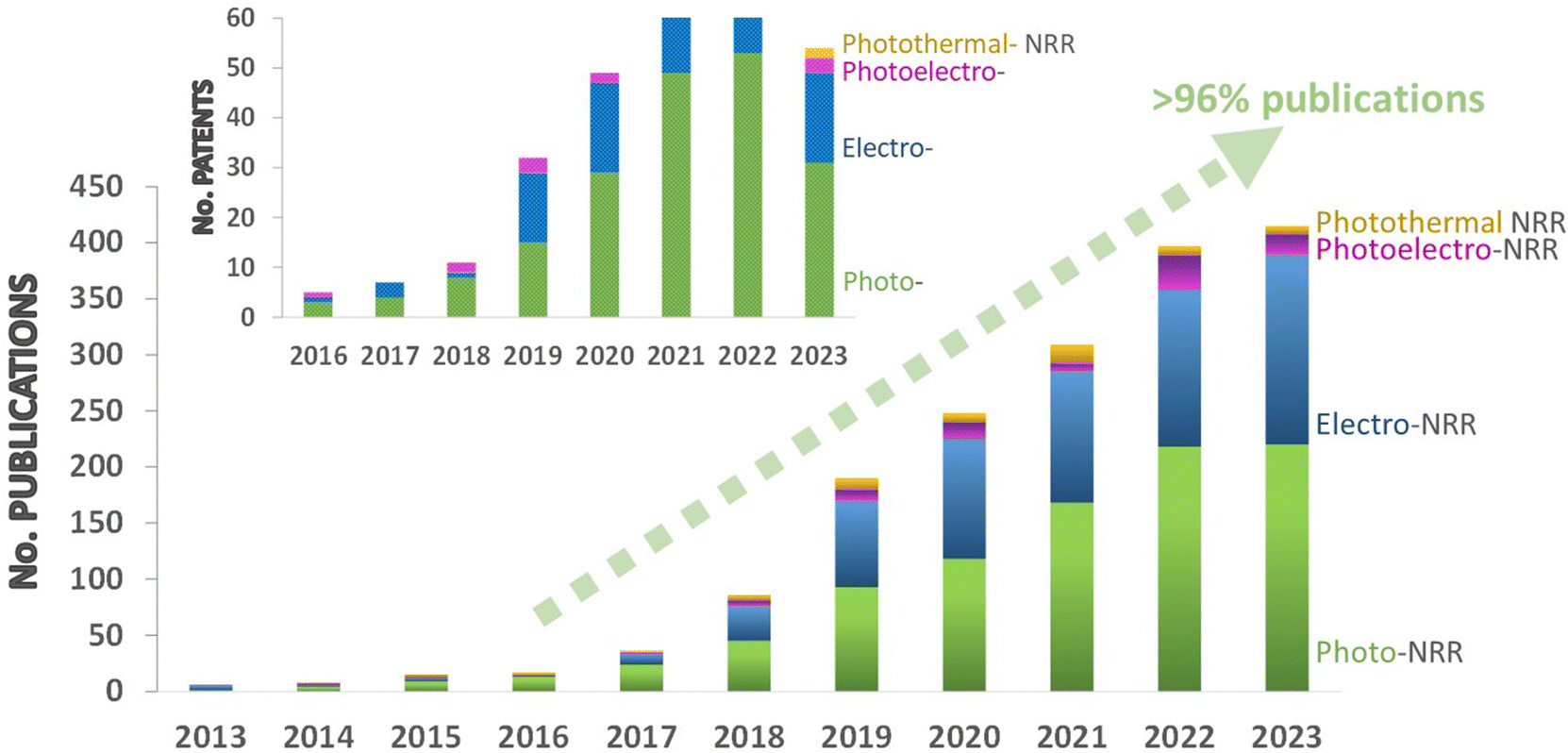 | ||
| Fig. 1 Histogram showing the increasing trend in the number of publications in the NRR field over the past 10 years. Papers are classified by technologies: photocatalytic-NRR (green), electrocatalytic NRR (blue), photoelectrocatalytic NRR (pink), and photothermocatalytic NRR (yellow). Data collected from the main scientific journal databases (∑entries = 1526 in the period January 2013–December 2023). Inset shows a histogram of the numbers of NRR patents per year and per NRR technology. Data collected from Google Patents (∑patents = 325, 2013–2023). | ||
1.2. Scope of this review
Our first goal in this review was to provide a comprehensive comparison of green ammonia production technologies by light-mediated catalytic routes. To do so, here we revise the current status of each technology, highlighting the major advantages and challenges that must to be addressed. Further, and with the aim of progressing on green ammonia technologies, we provide a didactic overview of the fundamentals and key aspects controlling the NRR process, including possible strategies to improve the performance, intuitive guidelines for the development of photoactive materials, and an assessment of experimental methodologies and recommendations for a reliable analysis. Going beyond fundamental research, we also overview the role of ammonia in the future energy scenario, including a technoeconomic comparison of current and emerging NRR technologies for a foreseen practical application, highlighting costs, barriers, opportunities, and recommendations for improvement.2. Fundamentals and challenges of nitrogen activation
Molecular nitrogen (N2) is considered the most stable known diatomic molecule in nature. It contains an extremely stable NN triple bond and a non-bonding pair of electrons (σ* and π*) on each atom (Fig. 2). The molecular orbital configuration of N2 leads to a large energy gap (10.82 eV) between the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively), which hinders the electron injection. In particular, the N2 molecule can be activated by either accepting σ electrons from N2via empty d-orbitals, or donating electrons from partially occupied d-orbitals to the π* antibonding orbitals of N2.
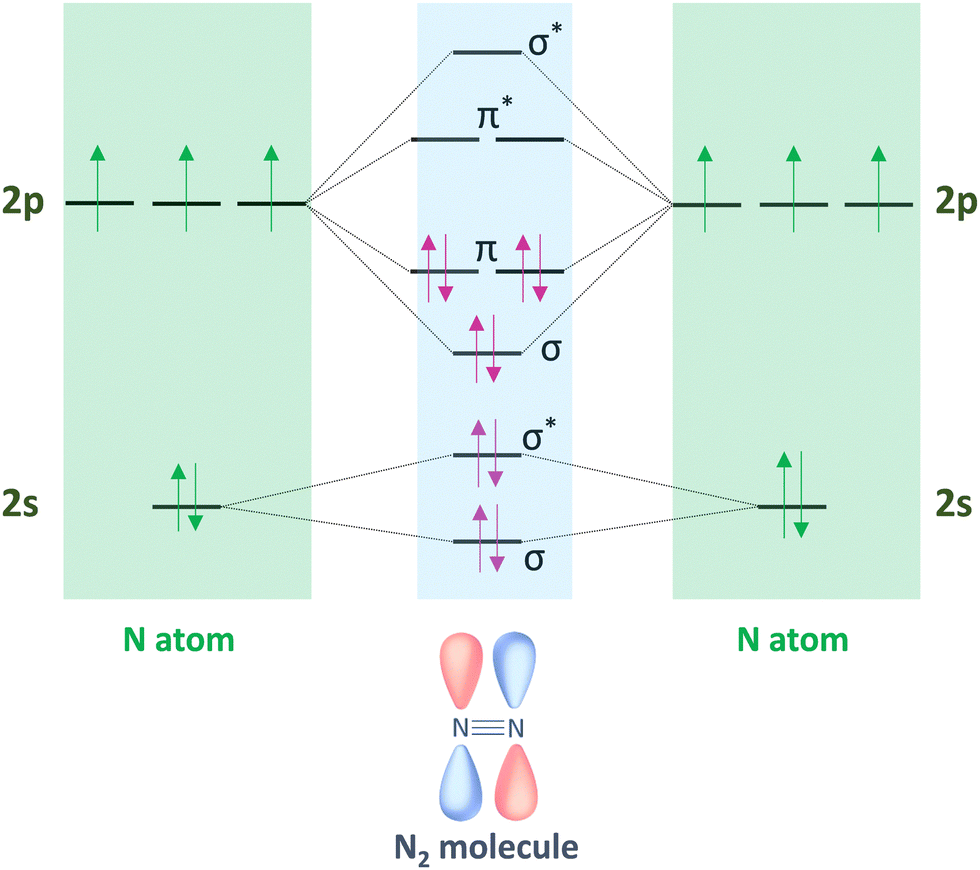 | ||
| Fig. 2 Hybridization of the s–p atomic orbitals in N2 molecule by linear combination of N atoms sharing three pairs of electrons from 2σ and π molecular orbitals. | ||
This configuration provides the N2 molecule a very short bond length of 109.8 pm and high bond strength of 941 kJ mol−1.30–32 However, this high dissociation barrier cannot fully explain the chemical inertness of N2 if compared to other similarly triple-bonded molecules, such as acetylene (HCCH), which has a comparable dissociation energy (ca. 962 kJ mol−1) but substantial more reactivity than N2.33 The difference arises from the high first-bond cleavage energy of N2 (410 kJ mol−1) that almost doubles that of acetylene's (222 kJ mol−1), and thus hinders NN dissociation.22,33 Besides, the N2 molecule lacks a permanent dipole and therefore shows a high ionization potential (15.84 eV), a negative electron affinity (−1.90 eV), and a low proton affinity (493.8 kJ mol−1), which is lower than acetylene's (641.4 kJ mol−1). Besides, its large energy gap (10.82 eV) complicates the electron transfer from the catalyst surface to the N2 molecule. All these factors strength the chemical inertness of N2 and its difficulty for the direct protonation under ambient conditions, despite being perfectly accessible from a thermodynamic point of view31,34,35 (eqn (1)):
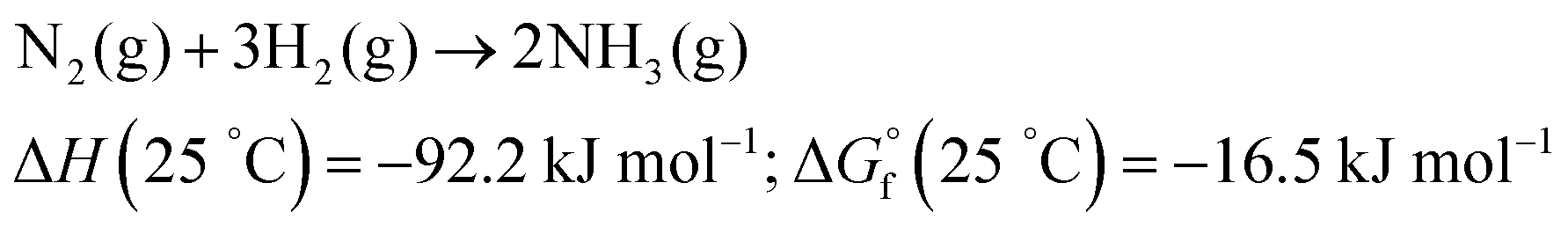 | (1) |
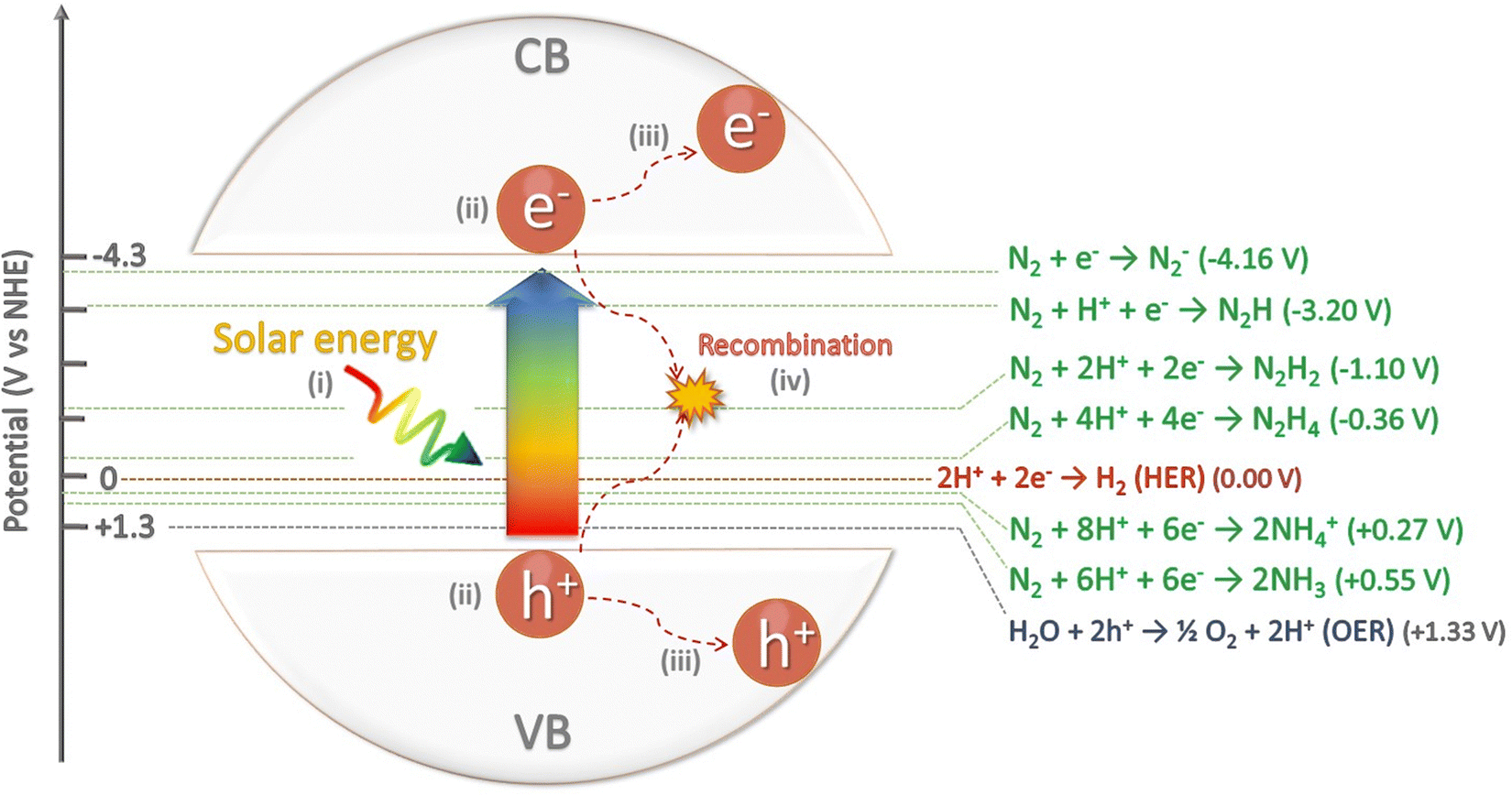 | ||
| Fig. 3 Schematic energy diagram for NRR, HER and OER, including their corresponding redox potentials (V vs. NHE). Additionally, the scheme depicts (i) the absorption of light by the semiconductor, (ii) the subsequent photogeneration of electron/hole pairs, (iii) the migration of the carriers to the surface of the semiconductor, (iv) recombination of carriers. | ||
In general, the catalytic NRR to NH3 requires: (i) chemisorption of N2 and hydrogen atoms on the catalyst active sites; (ii) activation and cleavage of the N2 molecule; (iii) reductive addition of hydrogen atoms to form ammonia (NH3) or ammonium (NH4+) depending on the pH media; and (iv) desorption of NH3/NH4+ from the catalyst surface. The equilibrium between gas-phase ammonia [NH3(g)], dissolved ammonia [NH3(aq)], and ammonium ion [NH4+(aq)] strongly depends on the pH of the reaction media (eqn (2)), mostly existing as NH4+ in acidic solutions.1,37 Under these conditions, protons are highly available and adsorb more easily on the surface of catalysts than N2 molecules. Consequently, most surface active sites are occupied by hydrogen atoms that consume available electrons to drive the competing HER, and consequently lowering both NRR yield and selectivity.38
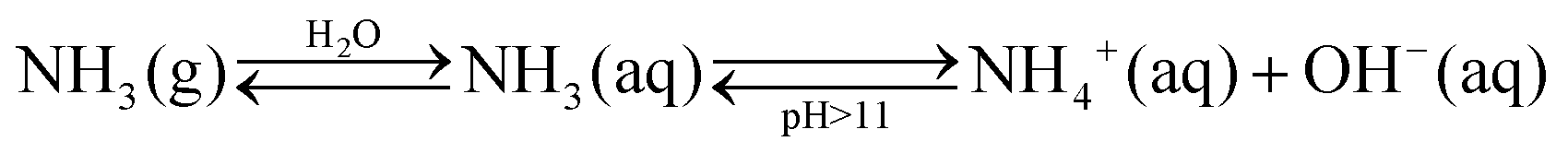 | (2) |
Nevertheless, the undesired HER is dominant in most photo(electro)chemical systems, in which electrons and protons preferentially evolves H2 instead of reducing N2 to ammonia, which is one of the major bottleneck of the NRR.22,38 Besides, the low solubility and low diffusion rates of N2 to active sites impose additional kinetic limitations in aqueous reaction media.31
Apart from H2 evolution, the product distribution of NRR may contain other reduction products such as hydrazine (N2H4) and diazene (N2H2), which are singly-bonded and doubly-bonded partially hydrogenated molecules, respectively; or ammonia oxidation products such as nitrates (NO3−) and nitrites (NO2−). We note that diazene has never been spectroscopically observed in solution due to either its high reactivity and spontaneous disproportionation to N2 and H2 (preferably in acidic media), or its rapid conversion to N2H4.22,40
2.1. Natural N2 fixation and global nitrogen cycle
The nitrogen cycle is one of the most critical biogeochemical cycles on Earth. It manages the fifth most abundant element in our solar system, which is also an essential bioelement present in nucleic acids and proteins.1,41 Unreactive atmospheric N2 can be fixed in nature mainly via (i) lightning strikes, or through (ii) diazotrophic microorganisms containing the enzyme nitrogenase. (i) Lightning strikes dissociate and ionize molecular N2 and O2 into free radicals as result of the instantaneous increase of temperature (as high as 30000 K) and local air pressure. Free radicals combine to generate stable molecules such as nitric oxide (NO), nitrogen dioxide (NO2), N2, ozone (O3) and O2. Nitrogen oxides may further react with atmospheric water to form nitric acid (HNO3), which may act as a soil fertilizer on the earth's surface via reduction of NO3− to NO2− and NH4+ by humic substances.42 (ii) The second route consists on the biological N2 fixation into terrestrial and marine ecosystems by nitrogen fixing microorganisms (bacteria and archaea). The fixed nitrogen is subsequently converted into a diverse range of N-containing compounds (i.e. ammonia, amino acids, nucleic acids, amines, N-heterocycles, nitrates, nitrites, etc.), and finally returned to the atmosphere as molecular nitrogen through microbial denitrification in soils, and fresh/marine waters.1,41–43 These compounds are synthesized through nitrogenase enzymes under ambient conditions and without H2. Nitrogenase (Fig. 4a) is a two-component system composed of two metalloproteins working in tandem: an electron-donating iron protein (Fe4S4 cluster) and a catalytic active site metallocofactor (MoFe protein).12,23,42 In brief, the iron protein (also called dinitrogenase reductase or component II) acts as a strong reducing cluster, hydrolyzing adenosine 5′ triphosphate (ATP) and donating the generated electrons to the MoFe protein (also called dinitrogenase or component I), which finally reduces N2 into NH3.12,23,42,44 The MoFe protein contains two metal clusters (Fig. 4a): (i) the iron–molybdenum cofactor (FeMo-co), which provides the active site for nitrogen binding and subsequent reduction, and (ii) the P-cluster [Fe8S7] that transfers the electrons from the Fe protein to FeMo-co.45 Thus, the FeMo-co is the key element for the N2 fixation mechanism.46,47 Three classes of nitrogenase are known, which differ in the heteroatom present in the catalytic active site (Mo, V or Fe). Among them, the Mo-dependent nitrogenase is the best-studied and the most chemoselective one.22,44,47
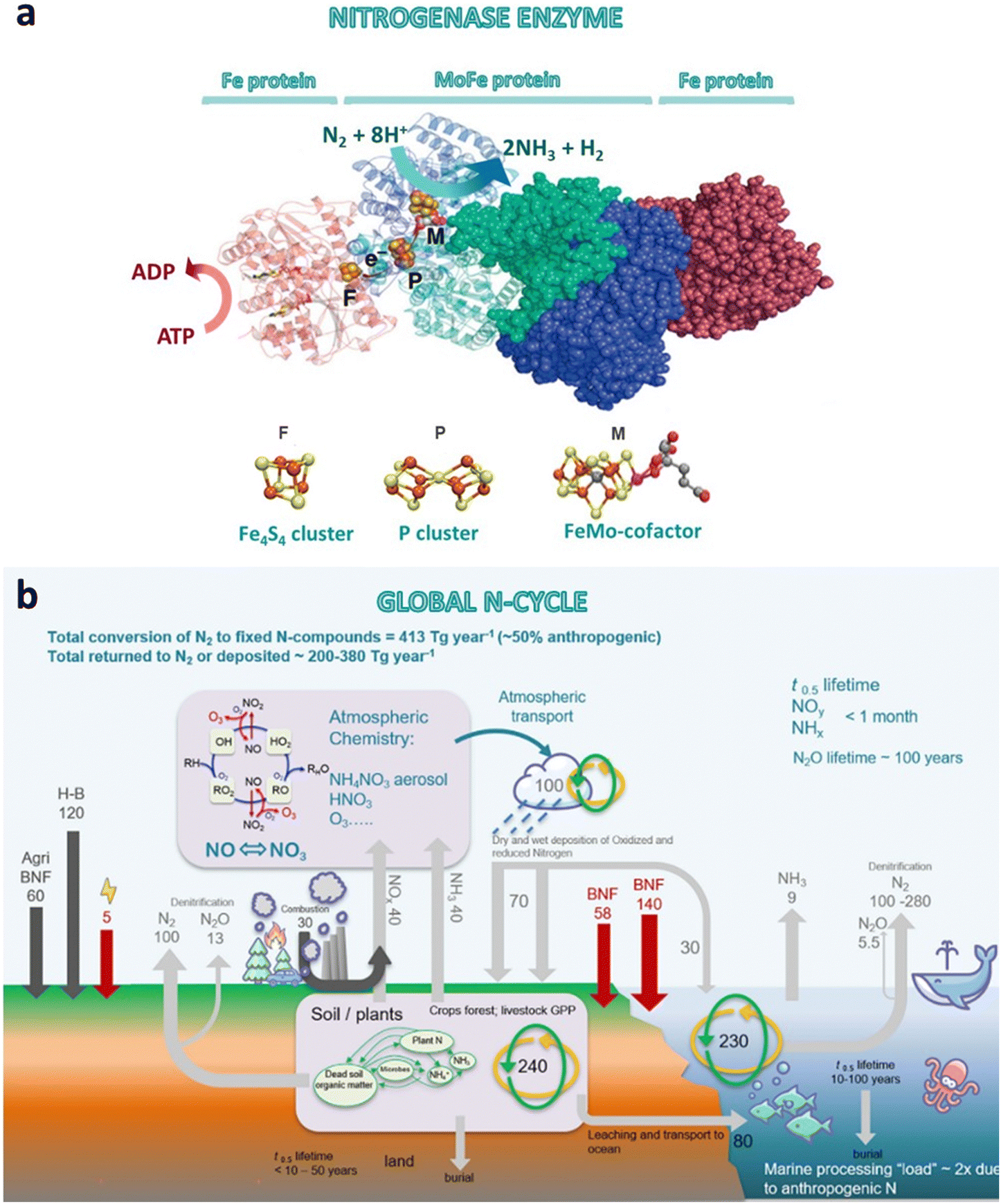 | ||
| Fig. 4 (a) Structure of the nitrogenase enzyme complex, including the MoFe and Fe protein components with the three metalloclusters (Fe4S4 cluster, P cluster and FeMo-cofactor). The Figure also shows the ATP hydrolysis by the Fe protein, the electron transfer to the catalytic site, and the biological nitrogen fixation to ammonia (adapted from ref. 48). (b) Global nitrogen cycle and fluxes in Tg per year (BNF, biological nitrogen fixation; H–B, Haber–Bosch nitrogen fixation; GPP, gross primary productivity). Reproduced with permission from MacFarlane et al.49 Copyright 2020 Elsevier Inc. | ||
For NRR, two electrons are consumed by the MoFe cofactor to bind N2 to Mo, together with a displacement of hydrogen following the catalytic cycle shown in Fig. 4b.23 Then, the structure of nitrogenase limits the access to electrons in order to avoid the competing HER, but the overall NRR entails 8 electron transfer reaction and one H2 molecule release (eqn (3)). Among nitrogenases, the Mo-dependent one produces two molecules of NH3 per H2 (eqn (3)) at a faster rate and more efficiently (requiring less ATP per mol of N2) than the others, which produce more H2 than NH3.22,44,47,50
| N2 + 8e− + 16ATP + 8H+ → 2NH3 + H2 + 16ADP + 16Pi | (3) |
In general, about 16ATP or 26–30 GJ tNH3−1 are required to fix N2 into ammonia under ambient conditions. This corresponds to a substantial energy expense of 244 kJ to reduce one N2 molecule into NH3, which results in a maximum overall efficiency of the enzyme nitrogenase of ca. 10–15% (150–225 GJ tNH3−1).23,51–53
On the other hand, N2 fixation can also occur via oxidative pathway with O2 or H2O to produce NOx (x = 1, 2, 3 corresponding to oxidation states of +2, +3 and +5, respectively). NOx are crucial feedstocks for the fertilizer and pharmaceutical industries, and are mainly produced via thermocatalytic ammonia oxidation. Therefore, finding a renewable nitrogen oxidation reaction (NOR) route to produce nitrate (eqn (4)) is also an appealing approach, alternative to the conventional synthesis of nitrate from NH3 oxidation.1,54 For practical NRR applications, NOR is thermodynamically more favourable than the competing side oxygen evolution reaction (OER, 1.23 V vs. RHE) and therefore, coupling NOR with NRR in a single device is emerging as a disruptive approach for the production of fertilizers directly from N2 and even from air.55
| N2(g) + 6H2O → 2NO3−(aq) + 12H+ + 10e− E0 = 1.076 V vs. RHE | (4) |
Looking globally, the natural N2 fixation provides 413 Tg y−1 (Tg = 1012 g) of reactive nitrogen (i.e. NH3, NH4+, NO, NO2, HNO3, N2O, etc.) to terrestrial and marine ecosystems, of which human activities contribute for half (210 Tg year−1).43,56 The development of intensive agriculture and new high-yielding crops over the past century is largely responsible for this anthropogenic value, which has drastically altered the nitrogen cycle. Indeed, nitrogen fertilizers frequently generate leaching of NO3− in drainage waters that end up in seas and open oceans (40–70 Tg year−1), increasing the amount of reactive nitrogen to be processed in water systems. Fertilizer leakages also contribute to trace emissions of NH3, which together with nitrogen oxides (NOx) emissions from combustion processes, contribute to 100 Tg year−1 to the atmosphere. Additional N2 and N2O emissions are emitted back to the atmosphere from denitrification in marine sediments. In turn, reactive nitrogen in the atmosphere may generate secondary pollutants, such as ozone and other photochemical oxidants and aerosols, mainly ammonium, nitrates and sulfates.43 The human intervention in the nitrogen cycle has been key to securing food security, but is also having a negative impact on climate, biodiversity and human health. As a few examples, N2O is a potent greenhouse gas able to trap heat about 200 times more effectively than the well-known CO2; over-enriching of reactive nitrogen in aquatic ecosystems can lead to eutrophication and loss of biodiversity; while atmospheric aerosols can cause serious health issues such as respiratory illness, cancer, or cardiac diseases.57 These effects highlight the importance of finding solutions to remediate the human disruption of the nitrogen cycle, which has been identified as one of the 14 Grand Challenges for Engineering in the 21st century.57 Most negative effects would be more easily mitigated by cutting reactive nitrogen emissions to the atmosphere, since the lifetime of N-compounds range between a few hours (NH3 and HNO3) to few days-week (aerosols); one exception is N2O with an atmospheric lifetime of approximately 100 years. On the other hand, removal of reactive nitrogen from oceans and terrestrial ecosystems would be slower, needing periods longer than a few decades for the later.43 Bearing this in mind, possible technological solutions suggested by the National Academy of Engineering57 include the recycling of organic waste (e.g. converting manure into pelletized organic fertilizers), and capturing and valorization of derived-greenhouse gases (CH4, N2O) to reduce atmospheric emissions from soils and water systems. Besides, engineering solutions could improve the efficiency of fertilizer application, reducing leaking into water bodies, runoff and erosion.
In this context, finding sustainable routes to produce valuable nitrogen-containing fuels and chemicals (e.g. ammonia, nitric acid, hydrazine, etc.) is also key to restore the nitrogen cycle and to promote an environmental-friendly nitrogen economy.1,41 Indeed, the U.S. Department of Energy recently concluded that there is a lack of sustainable ammonia synthesis technologies and catalysts with enough activity, selectivity, and scalability to fulfil industrial requirements.58 Research is currently in progress to adapt the enzymatic processes underlying the natural nitrogen cycle to artificial N2 fixation routes, which can produce renewable-sourced ammonia in an efficient and affordable manner as discussed in detail in the following sections.
2.2. Artificial N2 fixation
As mentioned above, the ultimate goal of artificial N2 fixation is to mimic the chemistry behind the nitrogenases to photoactive catalytic systems, ultimately looking for a potential scaling-up for industrial application. However, NH3 yields are still very low to fulfil the industrial demand, mainly due to the inertness of the N2 molecule, and the easiness of the competitor HER. Therefore, this technology has become an on-going hot research topic for the next generation of sustainable ammonia production. On this basis, this review addresses the main light-driven catalytic routes (i.e. photocatalytic, photoelectocatalytic, PV-electrocatalytic and photothermal catalytic NRR) in Section 5. We note that some of these processes have recently diversified into alternative catalytic routes such as magnetic field enhanced photocatalysis,59 sono-photocatalysis,60,61 piezo-photocatalysis,62,63 plasma-catalysis,64–66 plasma-electrocatalysis,67–70 plasma photoelectrocatalysis,71 plasma-UV,72 mechanocatalysis,73,74 light-driven chemical looping,75 and even semi-artificial photosynthesis routes based on (photo)bioelectrocatalysis,76–78 which are out of the scope of this review.3. NRR mechanism
As shown in Section 2.1, N2 fixation to ammonia occurs in nature via a multi-proton and electron transfer process, with the hydrolysis of 16 adenosine triphosphate (ATP) molecules per molecule of N2. From a mechanistic point of view, the NRR process involves four main steps: (i) N2 adsorption on the catalytic active sites; (ii) NN bond cleavage by either proton transfer from a proton donor or by electron injection from an external circuit; (iii) hydrogenation through proton-coupled-electron transfer (PCET) processes; and (iv) formation and desorption of NH3.38 The overall hydrogenation of N2 to ammonia is a 6 electron-proton transfer process, in which the first hydrogenation (*N2 + H+ + e− → *N2H) is commonly regarded as the rate-limiting step (where * indicates a surface site).38 Subsequent protonation to NH3/NH4+ occurs via two possible intermediates: hydrazine (N2H4) and diazene (N2H2),79 which can be both hydrogenated more easily than N2 due to their higher proton affinities (853.2 kJ mol−1 and 803 kJ mol−1 for hydrazine and diazene, respectively, vs. 493.8 kJ mol−1 for N2).22
The reaction mechanism for heterogeneous NRR can be divided into dissociative or associative, depending on the adsorption and hydrogenation modes of nitrogen on the catalyst surface (Fig. 5). In the dissociative pathway, the triple NN bond is first cleaved on the catalyst surface and then, the protons and electrons are added to the adsorbed N atoms to form NH3.38,80 However, it is extremely difficult to directly break the NN bond due to its high bond strength (941 kJ mol−1). This explains why the industrial Haber–Bosch process, which follows the dissociative pathway, requires harsh reaction conditions.81 In general, the dissociative pathway is favored over catalytic surfaces with highly negative N2 adsorption energies, such as over early transition metals (e.g. Sc, Y, Ti, Zr).
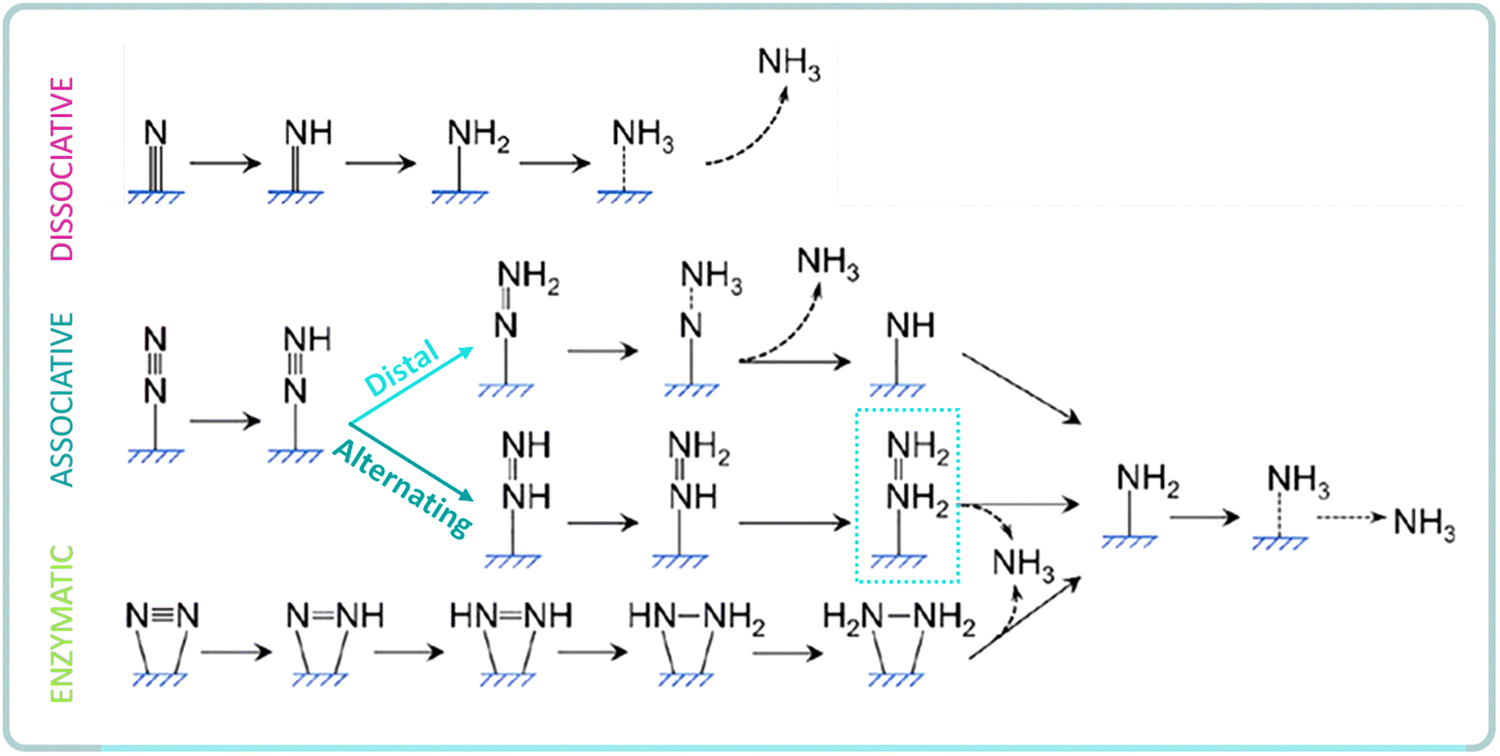 | ||
| Fig. 5 Schematics of possible pathways for NRR to ammonia on heterogeneous catalysts. Adapted from ref. 32. | ||
In the associative pathway, the hydrogenation of N2 proceed before the NN triple bond is broken, leaving one N atom adsorbed on the catalyst surface for further hydrogenation to NH3. Addition of hydrogen can occur following distal or alternating pathways, depending if the two N atoms are hydrogenated alternatively or if the terminal N atom is hydrogenated preferentially.38,80 The possible formation of N2H4 by-product (alternating) is a key distinction between these two pathways. Further, the alternating pathway can also start with a side-on N2 adsorption, known as the enzymatic pathway.81
Moreover, a special pathway for N2 reduction is the Mars–van Krevelen (MvK) mechanism, mostly prevalent for metal nitrides.82–84 In this case, ammonia is produced by the reduction of the surface nitrogen atoms of the catalyst, creating N vacancies that are further replenished by N2 molecules. Thus, the MvK mechanism is thermodynamically more favorable than the dissociative and associative pathways, because the formation of the first NH3 molecule does not involve the breaking down of the triple bond in the N2 molecule (i.e. it is formed from the N site on the nitride surface), which significantly lowers the energy barrier for NH3 formation.85,86 However, the catalysts following this MvK mechanism can easily deactivate if the N vacancy is filled with a different atom or if the adsorbed N2 is not activated.84
An additional surface-hydrogenation mechanism was recently suggested by Ling et al.87 for catalysts with weak N2 binding strength, such as noble-metal catalysts. This approach considers that H+ reduction is the first step rather than N2 adsorption, and thus N2 can be activated and reduced into *N2H2, and further reduced into NH3 spontaneously.
Among all mechanistic pathways, biological N2 fixation is believed to be associative by coordination of N2 to the metal centre in the FeMo nitrogenase. However, the addition of hydrogen via distal or associative pathways is still under investigation.53 In the case of artificial NRR, the dissociative pathway is highly uncommon for catalysts working at room temperature, which generally follow an associative pathway due to the lower energy input required to break the NN bond.8,38,42,53 However, the NRR mechanism still suffer from uncertainties and this limits the performance enhancement. Relevant insights into the natural N2 fixation process have revealed the nitrogenase structure and functioning (i.e. crystallographic structure of Fe and MoFe proteins), the specific role of the metal clusters (FeMo-co and P-cluster, see Section 2.1), or the existence of different types of nitrogenases.45 However, structural or fundamental studies have not provided a full picture of the catalytic mechanism under real operating conditions, which is essential to guide a rational design of catalysts for selective NRR. Unclear key factors include the clarification of the specific catalytic sites for activation and reduction steps, the timescale of charge carriers and catalytic events, the changes on the physicochemical and optoelectronic properties of catalysts under illumination and/or heating, and the associated changes on their structure–reactivity. The combination of in situ characterization, photophysical measurements and theoretical calculations offers a powerful understanding approach to bridge the gap between mechanistic understanding and catalysis.
3.1. Advanced characterization to understand the NRR mechanism
With appropriate experimental design, in situ and operando spectroscopic studies can reveal the chemical structure, electronic states of active sites, and reaction intermediates during the catalysis in real time, providing insights into their dynamic evolution. This type of analysis also enables the identification of the deactivation modes of a catalyst. Besides, in situ or operando characterization can provide a deeper understanding of the catalyst defects, which is a very important feature in photo(thermo)catalysis. In particular, some techniques such as X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR) and X-ray absorption spectroscopy (XAS) can discern the type and concentration of defects, allowing real-time monitoring of the status of defects and their evolution during the NRR.88 In this work, we revise some of the most interesting works that use advanced spectroscopic techniques during reaction to shed some light into the understanding of the light-driven NRR mechanism.In situ XAS can provide information about the electronic properties and oxidation state changes during the reaction. Hou et al.89 examined the electronic properties of W in a WO3-based catalyst during the photocatalytic NRR by using in situ X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS). The W L3 edge variation of the absorption edge position was found to be almost negligible when the catalyst was only immersed in water under N2 atmosphere, while it changed to a lower energy under light irradiation, suggesting that W was partially reduced to generate oxygen vacancies during the reaction. Furthermore, the coordination number of the W–O shell in the catalyst decreased from 5.4 to 4.4 during the photocatalytic NRR, which coincided with the variation of the electronic structure of W but also with the formation of NH3 on the surface of catalysts due to the generation of OVs. This study demonstrates the power of XAS to identify the in situ formation of active sites, such as OVs, and to determine their relevance during catalytic reactions for the understanding of the reaction mechanism.
X-ray absorption technique can be performed using hard or soft radiation, depending on the target elements to analyze. When dealing with light elements, it is necessary to move to in situ soft X-ray absorption spectroscopy (sXAS). While hard XAS provides bulk information, sXAS allows a penetration depth of around few nanometers in total electron yield (TEY) mode, thus enabling a more accurate tracking of slight variations on the catalyst-reactant interface.90 In this regard, in situ synchrotron radiation soft XAS (in situ sXAS) was used to probe the variation of the electronic structure of a model catalyst (i.e. Ni single-atom and CeO2 co-modified reduced graphene oxide), as well as to monitor the photogenerated electron flow during photocatalytic NRR.91In situ sXAS directly revealed the migration of photoexcited electrons from the light-absorbing unit of CeO2 to the Ni active sites atoms via the electron-transfer bridge of reduced graphene oxide, and then flew toward adsorbed N2 molecules, providing a direct evidence for the photogenerated electron flow.
Regarding a more surface characterization, some works in literature report quasi-in situ XPS analyses, while full in situ or operando analysis are very scarce. Quasi in situ XPS can monitor the formation of defects at the atomic level during the catalytic reaction, as well as to identify their chemical state.92 For instance, Hou et al.89 tracked the chemical and defect structure of WO3 during N2 photofixation by monitoring the O 1s XPS spectra before and after exposing the catalyst to reactants and illumination. Zhang et al.93 proved changes in the chemical state and the electron structure of a PdCu/TiO2/Si photocathode during the Li-mediated PEC NRR by using operando synchrotron XPS. By monitoring the photocathode surface under dark and illumination conditions, they observed the generation and separation of photogenerated electrons and their migration from the nanostructured n+p-Si optical absorber to the alloyed PdCu nanoparticles. The resulting electron-rich Pd and Cu sites may serve as a Lewis base, facilitating the adsorption and reduction of Li+ to Li on the PdCu/TiO2/Si photocathode surface. A very recent work by Zeng et al.94 reports a true in situ XPS analysis to elucidate the NRR mechanism. In particular, they used this technique to analyze the charge transfer between a heterojunction composed by δ-Bi2O3 and BWO catalysts. In situ XPS revealed that red-shift of the binding energies of W 4f, Bi 4f and O 1s under illumination, compared to the analyses in dark conditions. The shift of binding energies demonstrated that the photogenerated electrons flowed from the conduction band of BWO to the valence band of δ-Bi2O3 under illumination, also suggesting a Z-scheme charge transfer pathway within the heterojunction. Another very recent work by Cheng et al.95 also used in situ XPS to show the light-induced electron transfer from ZnIn2S4 to MXene quantum dots during photocatalytic NRR.
Other techniques such as in situ EPR spectroscopy allow to analyse the attenuation and generation of defects during N2 photoreduction. According to the EPR principles, unpaired free electrons in atomic or molecular orbitals will be excited to higher energy levels upon the application of an external magnetic field, thus generating a characteristic signal. For instance, in situ EPR experiments under constant N2 flow recently revealed the role of defects and electron-rich Cuδ+ in promoting the N2 photoreduction performance of zinc aluminium layered double hydroxide (ZnAl-LDH) nanosheets.96 The authors found that the 0.5%Cu–ZnAl-LDH sample exhibited a stronger EPR signal, Cu2+ (3d9), than the CuZnAl-LDH containing much more copper (as Cu2+). This implied that the generation of oxygen defects increased the number of free electrons in the Cu sites of 0.5%Cu–ZnAl-LDH. Furthermore, the EPR signal intensity of Cu2+ for 0.5%Cu–ZnAl-LDH decreased under UV-vis irradiation, demonstrating the transfer of photogenerated electrons to the Cu centers.
In surface chemistry, operando infrared spectroscopy can generally identify adsorbed intermediates or products, while operando Raman spectroscopy is able to identify other N2-containing reaction intermediates.86 Fang et al.97 recently used in situ Fourier transform infrared (FTIR) spectroscopy to investigate the photocatalytic N2 reduction to ammonia in a BiOBr/MXene-Ti3C2 composite as catalyst. In situ FTIR showed the formation of NxHy intermediates by a continuous protonation process of N2 molecules. Wang et al.98 used in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) to investigate the photocatalytic NRR mechanism over a Fe-doped TiO2 S-scheme anatase/rutile homojunction. Their experiments revealed the appearance of ammonium and NRR intermediates absorption bands upon illumination, namely –N2H, –N2H2, –NH, and –NH4+ centered at 1661, 1585, 1500, and 1319 cm−1, respectively. Besides, they did not find any band associated to the formation of –N2H4, leading them to conclude that the photocatalytic NRR mechanism followed a distal pathway. In other work, Ajmal et al.99 used in situ DRIFT spectra to study the chemical adsorption of N2 at the surface of an electron-deficient boron-doped carbon nitride catalyst. In situ DRIFT and density functional theory (DFT) calculations proved that B–N sites were catalytic active centers for N2 chemisorption by electron pair acceptance (σ-donation), while the presence of electron-withdrawing boron in the carbon nitride inhibited HER and promoted N2 adsorption through electron back-donation to N2.
Surface-enhanced Raman spectroscopy (SERS) can be used to identify key intermediate species for nitrogen fixation at the catalyst-reactant interface, providing mechanistic insights into NRR mechanisms and leading to the design of more efficient catalysts. Nazemi et al.1 used operando SERS to identify NRR intermediates at the electrode–electrolyte interface over plasmonic transition metal nanoparticle hybrids. They found that hydrazine is consumed as an intermediate of the NRR, following an overall associative reaction mechanism. We also highlight that the recent advances in microscopic techniques may also enable to image nanoscale defects and their associated changes during NRR. In situ scanning electron microscopy (SEM) and transmission electron microscopy (TEM) provide a unique possibility to obtain real-time data on the morphological and microstructural evolution of defective catalyst surfaces at the atomic level,100,101 as well as atomic disorder and local rearrangement under heating conditions.102
4. Strategies to boost NRR
The performance of NRR catalysts is mostly limited by the sluggish activation of the inert N2, the competing HER, and some technical difficulties associated with the reaction systems. Different strategies are presented below to control the main reaction limitations of the NRR.4.1. Overcoming the main thermodynamic and kinetic limitations
From a thermodynamic point of view, the activation and reduction of the N2 molecule is constrained by its short bond length (109.76 pm), the high dissociation energy (941 kJ mol−1) of the NN bond, the high ionization potential (15.85 eV) for N2 adsorption on the catalyst surface, the endothermic first proton addition to N2 (ΔH0 = 37.6 kJ mol−1), and its low electron affinity (−1.903 eV).81 On the kinetics point of view, NRR is mainly impeded by the large energy gap (10.82 eV) between the HOMO and LUMO of N2 (i.e. electron transfer process limitation), the lack of a dipole moment and its low polarizability.81,82 Besides, finding catalysts with fast NRR kinetics is difficult since their catalytic activity strongly depends on the adsorption energy of the different surface reaction intermediates (Fig. 6a).103 For instance, transition metals that bind nitrogen too weakly (right-side of the volcano plot, e.g. Cu, Ag) are limited by N2 activation (N2 adsorption as N2H*), whereas strong-binding metals (left-side of the volcano plot, e.g. Ru, Mo) are limited by hydrogenation due to a slow N–H formation, either through protonation of NH* to form NH2* or by removal of NH2* as NH3.103–105 Metals located at the top of the volcano plot (e.g. Ru, Fe, CoMo alloy) are expected to be highly active for NRR, since they have an intermediate nitrogen binding. However, these metals have a higher adsorption ability towards H atoms and exhibit a significant activation barrier for both N2 dissociation and N–H bond formation. For that reason, the Haber–Bosch process is conducted at high temperature to reach acceptable reaction rates.104,105 Unfortunately, no metal exists with an intermediate N* binding and a low N2 dissociation energy (bottom middle of volcano plot).105 Therefore, active site engineering is key to lower the activation barriers and achieve a preferential adsorption of nitrogen rather than hydrogen. Widely studied strategies include heteroatom doping, vacancies construction, co-catalyst loading, morphology control, etc. (more details in Section 4.4).
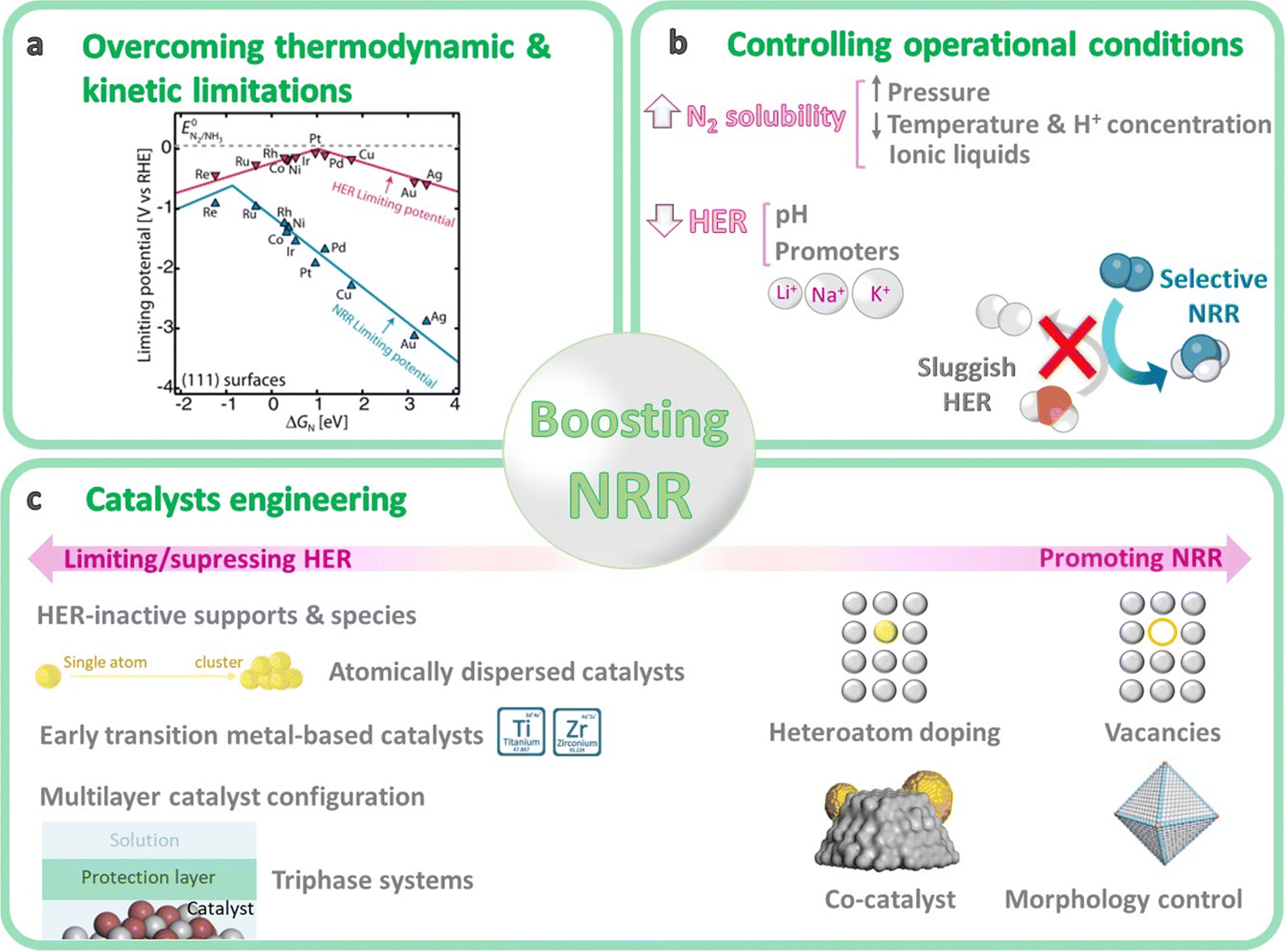 | ||
| Fig. 6 General overview of the main strategies to boost NRR performance, in terms of reaction engineering (a) and (b) and catalyst engineering (c). Volcano plot in Fig. 6a reprinted from ref. 104 Copyright 2017 The American Association for the Advancement of Science. | ||
4.2. Limiting the competing hydrogen evolution reaction (HER)
The competitor hydrogen evolution reaction (HER, 2H+ +2e− → H2) is a two-electron transfer reaction with one catalytic intermediate (H*, where * indicates a surface site), in which the overall reaction rate is limited by the adsorption of hydrogen (Volmer step), its binding strength to the catalyst surface, and the final desorption of the H2 molecule (Heyrovsky–Tafel steps) (eqn (5)–(7)).104| Volmer step: H+ + e− + * → H* | (5) |
| Heyrovsky step: H* + H+ + e− → H2 +* | (6) |
| Tafel step: 2H* → H2 +2* | (7) |
Thus, both NRR and HER involve proton-coupled electron transfer (PCET) reactions. The difference between them is that the HER is dependent on the electron–proton concentration in the reaction medium (i.e. first-order kinetics: HER α [H+]1 [e−]1), while NRR is independent (HER α [H+]0 [e−]0).38,82,106 Thus, an effective strategy to improve the selectivity to NH3 consists on limiting the accessibility of protons and electrons to the catalyst surface. On the one hand, restricting the accessibility of protons is important because H atoms adsorb more easily than nitrogen.107 This can be achieved by either modifying the operational conditions (e.g. pH and reaction media, details in Section 4.3), or designing specific catalyst configurations (details in Section 4.4.3). On the other hand, limiting the electron accessibility during NRR can help to control HER, and this can be also achieved through catalyst engineering (details in Section 4.4.3).
4.3. Tuning the operational conditions
The rational control of the operational conditions during NRR is crucial to promote N2 activation and limit the competing HER (Fig. 6b). The poor solubility of N2 in the aqueous medium (about 2 vol%) is one of the most crucial factors responsible for the low NH3 production rates. The low solubility of N2 implies mass transfer limitations. Lowering the reaction temperature (room temperature and slightly below) can enhance N2 solubility, and boost NRR selectivity due to the inhibition of the H2 production at some extent, which is favored at higher temperatures (>40 °C).38 However, low temperatures also restrict N2 diffusion, which could be overcome by increasing the reaction pressure. Increasing the operating pressure helps to shift the chemical equilibrium towards NRR, by suppressing HER that is predominant under ambient pressure conditions. However, higher pressures may also add technological complexity and cost.38,88 Moreover, N2 solubility can be improved by increasing the N2 flow rate, or using gas-diffusion electrodes (GDEs) in (photo)electrochemical flow cell reactors to increase the local N2 concentration. Further, another efficient strategy to improve N2 solubility is the use of aprotic ionic liquids to solvate N2 gas in the reaction solution.82 This approach can solubilize 20 times more N2 than aqueous solutions, and limits the proton reduction side reactions due to the lower water content.10 This growing research field has shown interesting improvements, some related to a better interaction of N2 with the polar part of the ionic liquids (e.g. 1-butyl-3-methylimidazolium tetrafluoroborate, [bmim][BF4];108 1-alkyl-3-methylimidazolium (Cnmim, n = 2,4,6) tris(pentafluoroethyl)-trifluorophosphate, [eFAP]109), or a better solvation of N2 in the presence of highly fluorinated anions.110On the other hand, changes on the reaction media may shift the chemical equilibrium towards NRR, suppressing the competing HER. For instance, controlling the pH medium is a simple way to limit the concentration of protons available for the NRR process. For aqueous solutions (the most common medium for ambient NRR), the pH determines whether H2O (basic medium) or H3O+ (acidic medium) are the proton donors during NRR (Fig. 7). Neutral solutions or electrolytes (e.g. phosphate-buffered saline, PBS), with intrinsically limited proton availability, are the most suitable reaction media for NRR.82 Under this conditions, H2 generation is constrained because of a restricted Volmer step (Fig. 7). This involves a first high energy barrier for water dissociation, which does not exist in acidic medium,38 thus retarding HER and favoring NRR.
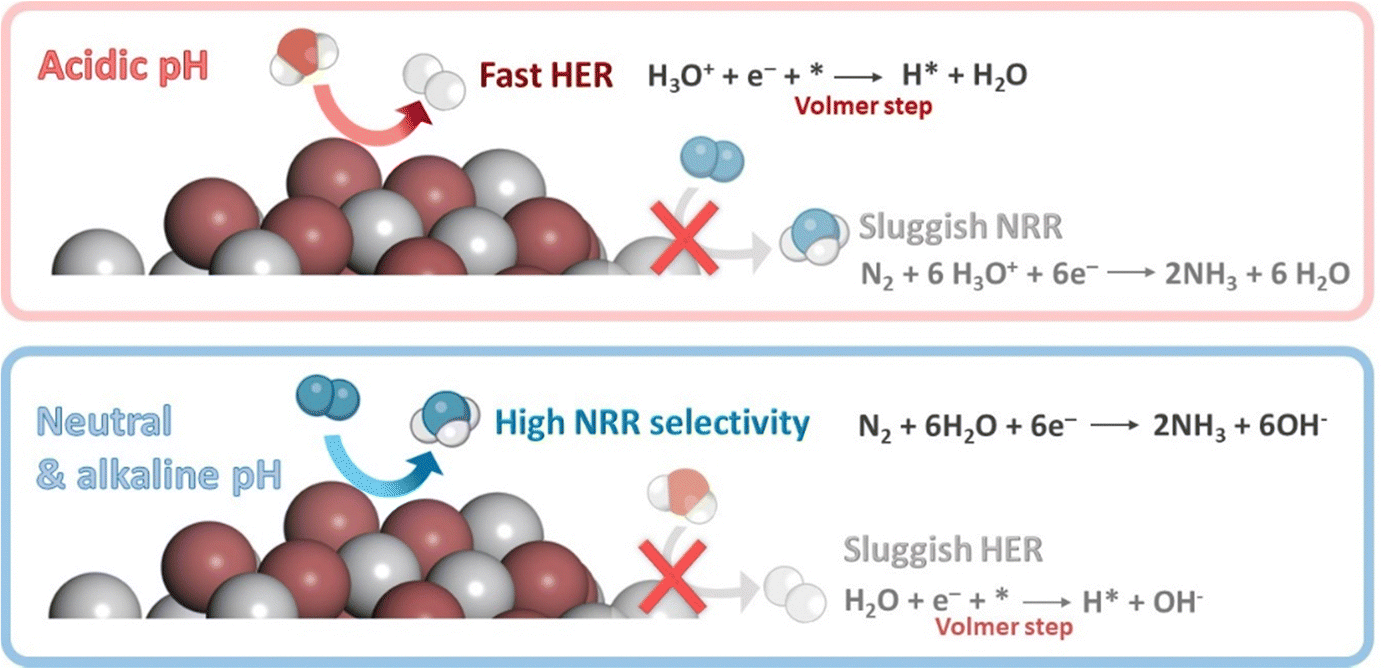 | ||
| Fig. 7 Effect on pH on the competition between the HER and NRR under ambient conditions. | ||
Another effective change for decreasing the availability of protons consists on adding alkali metal cations (e.g. Li+, Na+, K+) to the reaction medium. These ions tend to form solvation shells and hence steric effects, which remarkably restrict the transfer rate of H2O molecules to the catalyst surface, thus suppressing HER. In general, the smaller the cation, the easier is the formation of the dehydrated cation layer on the surface of the catalyst.38,82 Thus, the smallest Li+ ions are preferred for non-aqueous electrolytes, although commercial Li salts need to be pre-treated to remove NOx impurities (see Section 6.3). However, high concentrations (>1.0 mol L−1) of bigger cations such as K+ prevent HER side reactions, and may facilitate the adsorption and activation of N2 by creating electronic and electric-field effects that polarize nitrogen molecules.10,111
4.4. Rational catalysts engineering
Previous sections reflect that the challenging NRR usually impose several limitations to the conventional catalysts, such as high energy barriers for N2 adsorption and activation, slow electron transfer kinetics to N2, and unwanted side reactions (hydrogen evolution, ammonia oxidation, nitrogen oxidation). From the viewpoint of material design, an efficient light-driven NRR catalyst should possess suitable surface properties and composition to facilitate the strong binding with N-adatoms rather than H-adatoms, well-defined nanostructure (e.g. nanoparticles), high density of active sites (i.e. high surface area and porosity), suitable band alignment, high conductivity to facilitate electron transfer, and long durability.112Regarding composition, catalytic active sites need to have strong interactions with the N2 molecule to achieve an effective photochemical activation. Transition metals (e.g. Fe, Mo, V, Ti, Al) can weaken or break the strong triple NN bond by donating electrons from their atomic d orbitals into the antibonding π* orbitals of N2. In contrast, main-group elements (e.g. B, C, Bi, S) lack accessible d orbitals but have abundant valence electrons, so they activate N2 by p-electron backdonation into the unoccupied antibonding orbitals of N2.32,81,113 Besides, some main group non-metals, such as C, B, P, S and F can construct abundant defect sites for N2 adsorption, either through heteroatom doping or vacancy engineering.81
This section summarizes the main catalyst engineering strategies to boost NRR performance, which are mainly based on structure modulation and interface engineering (Fig. 6c). These strategies can improve the adsorption and activation of N2 on the catalytic active sites, tailor the band structure of the semiconductors, and enhance the charge transfer processes. Widely studied approaches include morphology control (plane/corner/edge sites, pores, etc.), crystal regulation (amorphous layers, lattice strain, etc.), heteroatom doping, vacancies construction (oxygen, nitrogen and metal vacancies), co-catalyst loading, etc. In addition, other engineering strategies focus on restricting or even suppressing the HER activity, mainly using inactive HER supports, early transition metals with stronger nitrogen binding ability, atomically dispersed catalysts or multilevel configurations (Fig. 6c and Fig. 8).
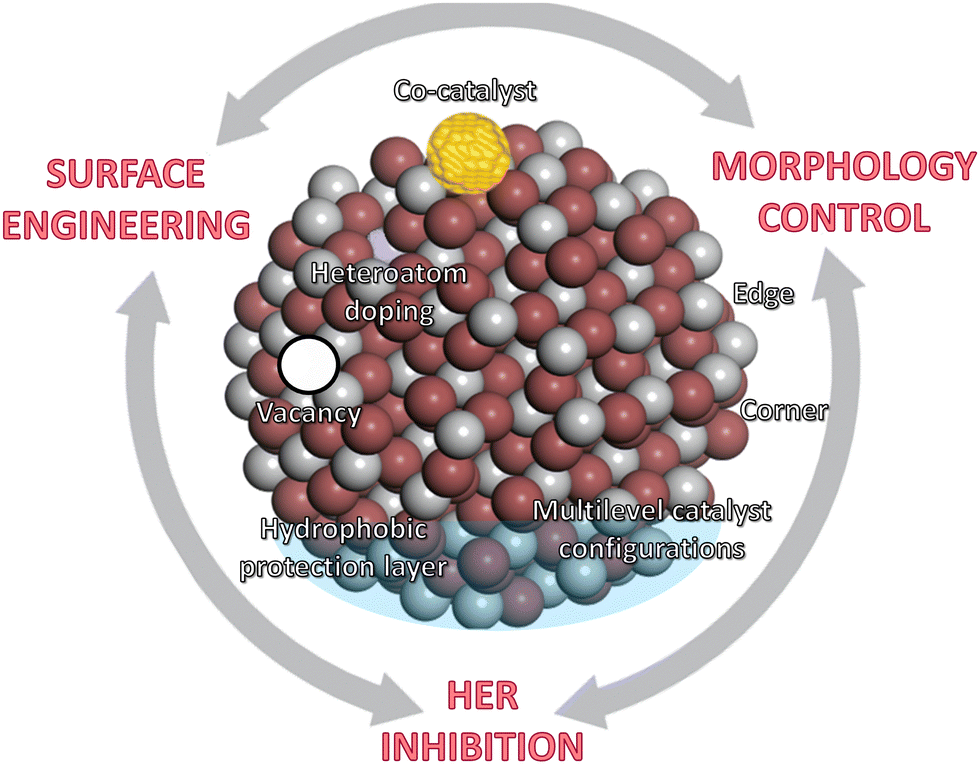 | ||
| Fig. 8 Catalyst engineering strategies to boost NRR performance, mainly based on surface modification and functionalization, morphology control, and synthetic strategies for suppressing HER. | ||
4.4.1.1. Vacancy engineering. Defect-rich materials (with anion/cation vacancies) generally exhibit remarkably higher NRR performance than their pristine counterparts, due to the introduction of new active sites, the modulation of their energy-band structure, and the associated improvement of surface reaction kinetics.31
Anion vacancies (oxygen, nitrogen and sulfur) can serve as electron traps, promoting the separation of photogenerated carriers in the parent semiconductor, and lowering the reaction barriers through the generation of electron-rich Lewis-base sites for N2 activation.31,116 Among them, oxygen vacancies (OVs) are the most popular anion defects in transition-metal oxides due to their low formation energy.116 They can be easily synthesized by different methods, including thermal annealing, heterogeneous ion doping, wet chemical reduction, and high-energy particle (such as electrons or Ar+ ions) bombardment.81 Among advantages, OVs expose extra coordinatively unsaturated sites for the adsorption of the inert N2 molecule, and these sites increase in concentration as the thickness of photo(electro)catalysts approaches atomic scales (i.e. ≈1 nm).117 Besides, OVs facilitate the activation of NN bond by promoting the electron-donating ability of adjacent metal atoms to the π* antibonding orbitals of N2 molecule.81 Besides, OVs may narrow the band gap by up-lifting the valence band maximum and conduction band minimum.118 This effect results in an extended visible light absorption range, and better separation of photogenerated charges.116,119 However, high amounts of OVs can excessively distort the crystal structure of the metal oxides, causing electronic delocalization and creating recombination centers of the photogenerated electron–hole pairs.119,120
For instance, Hirakawa et al.121 reported the creation of a large number of surface oxygen vacancies in a commercial rutile TiO2 sample (JRC-TIO-6), in which surface Ti3+ species efficiently produced NH3 from N2 and water under ambient conditions with a solar-to-chemical conversion (SCC) efficiency 0.02%. Zhao et al.117 demonstrated that ultrathin CuCr-layered double hydroxides (LDHs) nanosheets containing abundant OVs were able to reduce N2 at wavelengths up to 500 nm. The incorporation of Cu(II) ions introduced structural distortions and compressive strain, creating a high concentration of OVs that improved N2 adsorption and photoinduced charge transport in the nanosheets. As result, CuCr-LDH achieved a NH3 evolution rate of ≈7.1 μmol L−1 under monochromatic illumination at 500 nm (quantum yield (QY) ≈0.10%). However, the concentration of defects could not be fully controlled because it was dependent to the morphology control (i.e. minimum LDH thickness synthetically achievable of 2.4 nm). Later on, Zhao et al.122 were able to control the concentration of OVs in ultrathin TiO2 nanosheets by doping with copper ions. The introduction of Cu as a dopant (6 mol% Cu) in TiO2 introduced substantial compressive strain and created additional OVs, which extended the photoresponse up to 700 nm wavelength. Ammonia evolution rates of 78.9 μmol h−1 g−1 were achieved under solar irradiation, while full visible light illumination led to NH3 production rates of 1.54 μmol h−1 g−1 (600 nm) and 0.72 μmol h−1 g−1 (700 nm), with corresponding QY of 0.08% and 0.05%, respectively.
The beneficial effect of strain-induced OV defects was recently demonstrated by Li et al.123 from distortion-corrected elemental electron energy loss spectroscopy (EELS) mapping images and spectra, and DFT calculations. These authors constructed a semiconductor/plasmonic heterostructure composed by cerium oxide nanosheets with abundant strain-OV defects, on which they anchored Au hollow nanomushrooms. They found a large formation of Ce3+ and OVs at the interface, induced by interfacial strain, which promoted the activation of NN bonds and facilitated the adsorption/desorption of N intermediates. Besides, they found that the hot electrons generated by plasmonic Au were efficiently transferred into the cerium oxide through the heterostructure interface, which also inhibited their reverse movement by the Schottky barrier, thus facilitating an efficient electron–hole separation. As result, the cerium oxide/Au heterostructure photocatalyst delivered a NH3 production rate of 215.14 μmol h−1 g−1 and a solar-to-chemical conversion (SCC) efficiency of 0.1% under simulated AM 1.5G solar illumination.
Yang et al.124 reported a photocatalytic system based on Au nanocrystals anchored on ultrathin TiO2 nanosheets with oxygen vacancies. Here, OVs act as active sites for N2 chemisorption and activation, while N2 is reduced to NH3 by the hot electrons generated from plasmon excitation of Au nanocrystals. Au/TiO2-OV (1.5 wt% Au) reached a NH3 yield of 130.5 μmol h−1 g−1 with an apparent quantum efficiency (AQE) of 0.82% at 550 nm. Sun et al.125 presented a non-noble-metal photocatalyst based on sulfur vacancy-rich oxygen-doped 1T-MoS2 nanosheets. The authors associated the high content of sulfur vacancies with a better N2 adsorption and activation, enhanced light absorption, and better charge separation and transfer, which led to an excellent photocatalytic performance of 8220.83 μmol L−1 h−1 g−1 (AQE = 4.4%) under simulated solar light illumination.
Nitrogen vacancies (NVs) may also facilitate N2 adsorption and activation, since they provide an electron-deficient environment due to their similar structure and size with N atoms in the N2 molecule. Besides, they are an ideal active site for NRR owing to their relatively weak HER activity.81 Nevertheless, we note that NVs have found to be unstable for electrochemical NRR, leading to deactivation or false-positive results.81 For instance, Guo et al.126 embedded plasmonic Au nanoparticles in the mesopores of nitrogen-deficient hollow carbon nitride spheres. The NVs served as N2 chemisorption and activation sites, while the interfacial plasmon-induced charge separation led to a visible-light driven ammonia production of 783.4 μmol h−1 g−1, with an apparent quantum yield of 0.64% (at 550 nm) and a solar-to-ammonia (STA) conversion efficiency of 0.032% in pure water and under simulated AM1.5 G sunlight. Liang et al.127 also explored the creation of nitrogen vacancies in a boron-doped graphitic carbon nitride, finding that nitrogen defects improved N2 adsorption, visible light absorption, and charge carrier separation efficiency. Their optimized catalyst achieved a NH3 conversion rate of 435.28 μmol h−1 g−1 under visible light illumination.
On the other hand, the construction of cation vacancies is a less explored field for NRR, and most examples include electrocatalysts. However, this is also an interesting approach for light-driven NRR applications. The introduction of these local charge defects in transition metal oxides can change their surface electronic structure, create new active sites, and improve the charge transfer processes. The typical preparation methods of cation vacancies include chemical synthesis, plasma/chemical etching, thermal annealing in a reducing atmosphere, ion irradiation, heterovalent doping, and stripping.115 Ding et al.128 investigated the effect of titanium vacancies (VTi) on the photocatalytic NRR performance of an undoped anatase TiO2, which was changed from n-type to p-type with VTi. They found that cation vacancies lead to local charge defects that enhance carrier separation and transport, while trapped electrons can activate and reduce N2 to NH3. This material achieved a N2 fixation performance in air, water and UV-visible illumination of 47.13 μmol h−1 g−1, which increased to 64.82 μmol h−1 g−1 under N2 atmosphere.
4.4.1.2 Heteroatom doping. Heteroatom doping is an effective approach to improve N2 activation and hydrogenation, as result of the modification of the chemical composition of the catalysts, their electrical properties, and the creation of structural distortions that increase the concentration of OVs.31,129 Specifically, electron-deficient dopants induce defect levels that may promote conductivity, provide coordination active sites and even suppress HER by limiting proton adsorption (Lewis acid H+).129 Zhang et al.130 reported a photocatalytic NH3 yield of 195.5 μmol h−1 g−1 (STA of 0.028%), from pure water and AM 1.5G light irradiation, using Mo-doped W18O49 (1 mol% Mo) ultrathin nanowires. This study reveals that the defect states created upon Mo doping polarize the chemisorbed N2 molecules, facilitating the dissociation of the N
N bond through proton coupling; and elevate the defect-band center toward the Fermi level increasing the driving force towards NRR. Li et al.131 studied the beneficial effect of a non-metal dopant (boron) on the photocatalytic NRR performance of the carbon nitride g-C3N5. The doped B-C3N5 showed a NH3 production rate of 421.18 μmol h−1 g−1, which was 1.72 times as high as that of g-C3N5. The authors inferred that B sites (B–O–H), acting as Lewis acid, played an excellent role in adsorption and protonation of nitrogen, while O–H sites act as Brønsted acid and would protonate the activated nitrogen. Xie and co-workers132 explored the photocatalytic NRR performance of a single Cu atom-modified carbon nitride, achieving 186 μmol h−1 g−1 under visible illumination and in the presence of ethanol as electron scavenger, with a quantum efficiency (1.01%) at 420 nm monochromatic light.
On the other hand, NRR performance can be improved with electron-rich dopants (e.g. Fe, Mn, Ni, Co). Bo et al.133 reported a 5.3-times higher NH3 production of Fe-doped TiO2 nanofibers than pristine TiO2 (12.1 vs. 64.2 μmol h−1 g−1, respectively). Besides, they combined in situ characterizations with first-principles simulations to demonstrate that Fe dopants modulate the electronic structure nearby oxygen vacancies, facilitating the adsorption and polarization of N2, and even altering the hydrogenation of N2 from the associative alternating pathway to a more favorable associative distal pathway. Li et al.134 also demonstrated that doping with metal ions with exposed d orbitals, such as Fe3+, promotes the generation of OVs that are found to be the main active centers for photocatalytic NRR. The authors created OVs in Fe-doped Bi2MoO6 by alcohothermal synthesis, showing that metal–alcohol (e.g. Fe3+–ethylene glycol) coordination promotes the reduction of Bi3+ to Bi0 to generate OVs, which in turn promotes charge separation and photocatalytic activity. They obtained a 16-fold enhancement of photoactivity, compared to bulk Bi2MoO6, with an external quantum yield (EQY) of 2.8% at 420 nm. Regarding Bi-based semiconductors, Yang et al.135 demonstrated the regulation of the electronic structure of Bi2MoO6 by Co doping, which led to the creation of dual active sites (i.e. Bi and Co sites) that achieved 7.2 higher NH3 production (95.5 μmol h−1 g−1) than bare Bi2MoO6.
4.4.1.3 Co-catalysts. Surface functionalization of semiconductors with co-catalysts, such as noble metal or non-metal nanoparticles, is another strategy to boost NRR. In particular, co-catalysts can be used to trap and separate the photogenerated charge carriers, act as active sites for the adsorption and activation of the N2 molecule, and decrease the NRR overpotential.136
Regarding noble metal co-catalysts, Ranjit et al.137 concluded that the photocatalytic NH3 yield was highly dependent on the nature of noble metals (Ru, Rh, Pt, Pd) deposited on TiO2. In particular, they found higher productions and selectivity to NH3 by successively increasing the metal–hydrogen bond strength (Ru > Rh > Pd > Pt), due to the stabilization of H adatoms (H*) that effectively suppress HER. Qiu et al.138 anchored plasmonic Au nanocrystals on Mo doped W18O49 nanowires, reaching a photocatalytic NH3 production rate of 399.24 μmol h−1 g−1 and a AQE of 0.611% at 540 nm. The synergistic effect created between Mo doping and Au nanocrystals not only extended the visible light absorption range, but also decreased the desorption energy of NH3, which accelerated the regeneration of the active sites and further improved NRR.
Bu et al.111 studied the combined effect of plasmonic sensitization and the addition of alkali metal cations (Li+, Na+, and K+) on the overall improvement of the photocatalytic NRR. Plasmonic Au/P25 achieved a NH3 yield and selectivity of 85 μmol h−1 g−1 and 75% under UV-visible illumination, which increased to 0.43 mmol h−1 g−1 and 94.5% (AQE of 0.62% at 550 nm) when promoted by K+. The authors concluded that the addition of the alkali promoter chemically facilitated the activation and reduction of N2, while the attachment of Au nanocrystals sensitized the photocatalyst with hot electrons to improve nitrogen fixation.
Liu et al.139 studied different transition metal (Cu, Fe Ni) modified TiO2 nanosheets founding a better NRR performance and stability than pristine TiO2. Under the simulated sunlight illumination, the catalyst Cu/TiO2-1.0% reached a NH4+ generation rate of 6.78 mmol h−1 g−1 using glycerol as the sacrificial agent, which was 6-times higher than that of TiO2. This better performance was ascribed to the enlarged specific surface area and visible absorption range, as well as to a lower radiative recombination and more efficient charge transfer after loading copper. Ray et al.140 reported a robust nickel phosphide (NixPy) co-catalyst coupled to ZnO nanorods, which achieved a NH4+ production rate of 2304 μmol h−1 g−1 without using any organic scavenger, which was 3.3 times higher than that of the neat ZnO sample. The NixPy co-catalyst increased the visible light absorption ability of ZnO, accelerated the transfer of photoexcitons, and subsequently facilitated the NRR.
2-Dimensional (2D) co-catalysts has been also proposed to facilitate charge separation and promote N2 chemisorption and activation. For instance, Liao et al.141 increased by 5 times the NH3 production of bare TiO2 P25 by using a Ti3C2 MXene as a co-catalyst, achieving 10.74 μmol h−1 g−1 under full spectrum light irradiation and using water as the proton source. They inferred that Ti3C2 MXene co-catalysts favored the separation of photogenerated carriers by storing the photogenerated electrons in P25, and by promoting the chemisorption and activation of N2 molecules. Similarly, Shen et al.142 reported the use of a 2D black phosphorus nanosheets (BPNSs) co-catalyst coupled to CdS to achieve a rapid charge separation and transfer within the heterojunction. The authors obtained a 3.73-fold higher NH3 production (359.51 μmol h−1 g−1) than that of bare CdS, in the presence of methanol as the sacrificial reagent and under visible light illumination (λ > 420 nm).
In addition, morphological control can be employed as a strategy for broadening the light absorption range of semiconductors, aiming at full spectrum utilization. In particular, tailoring the particle size of the catalysts can change their band gap energy through quantum sizing effects. For instance, Sun et al.148 reported the synthesis of bismuth monoxide (BiO) quantum dots (size of 2–5 nm), which delivered an ammonia synthesis rate of 1226 μmol h−1 g−1 in water and under simulated solar illumination, which did not show obvious deactivation even after 120 h. Later on, Liang et al.149 proposed a heterojunction between BiO quantum dots and nitrogen defective ultrathin carbon nitride. This system comprised low-valent Bi2+ and nitrogen vacancies, which enhanced the optical absorption ability of the heterojunction, improve charge separation, and led to a NH3 production rate of 576.11 μmol h−1 g−1 under visible light illumination (AQE = 0.53% at 400 nm).
4.4.3.1. Single-atom catalysts. Atomically dispersed catalysts have recently emerged as an appealing strategy to boost N2 activation and regulate the catalytic pathways (i.e. increase NRR selectivity and inhibit HER).150 This approach affords highly exposed metal centers in which d orbitals of transition metals are hybridized with p orbitals of main group elements, leading to a partial electron transfer from the metal atom to the support material.150 The so-called single-atom catalysts (SACs) are generally used for electrocatalytic applications, including e-NRR. However, their use in light-driven NRR is scarce to date and mostly based on theoretical studies.151,152 Reported experimental works are based on single metal atoms (Mo, Cu) immobilized on a carbon nitride (C3N4) photocatalyst. Li and co-workers153 synthesized a Mo-C3N4 forming two-coordinated MoN2 species, which effectively adsorbed N2 and weaken the N
N bond. These authors achieved a visible-light driven NH3 evolution rate of 50.9 μmol g−1 h−1 in pure water, and 830 μmol h−1 g−1 using ethanol as electron scavenger with a quantum efficiency of 0.70% at 400 nm. Liu et al.154 studied the incorporation of La single atoms in an oxygen-deficient MoO3−x, founding a strong N2 activation ability due to the back-donation of electrons from La atoms to 2π* molecular orbitals of adsorbed N2. This single atom La photocatalyst delivers an impressive NH3 production rate of 209.0 μmol h−1 g−1 under visible illumination, without any sacrificial agent. Li et al.155 successfully anchored single Pt atoms at the –N3 sites of stable and ultrathin covalent triazine framework (CTF) nanosheets, aiming to achieve a better separation of photogenerated carriers. They obtained an averaged NH4+ production rate of 171.4 μmol h−1 g−1 (AQE = 1.4% at 420 nm) under visible light irradiation and in the absence of any sacrificial agent.
4.4.3.2. Multilevel catalyst configurations. An additional approach consists on building multilevel catalyst configurations, which may help to limit the electron accessibility during the NRR to control HER. However, it should be noted that an excessive low electron transfer to the active sites can conduce to low NRR efficiencies. Examples of this type of configuration include support–semimetal catalyst (e.g. Sn, Bi), support–conductive polymer catalyst (e.g. polyaniline (PAN), polyimide, polypyrrole), and support–insulator–catalyst (see Fig. 9).38
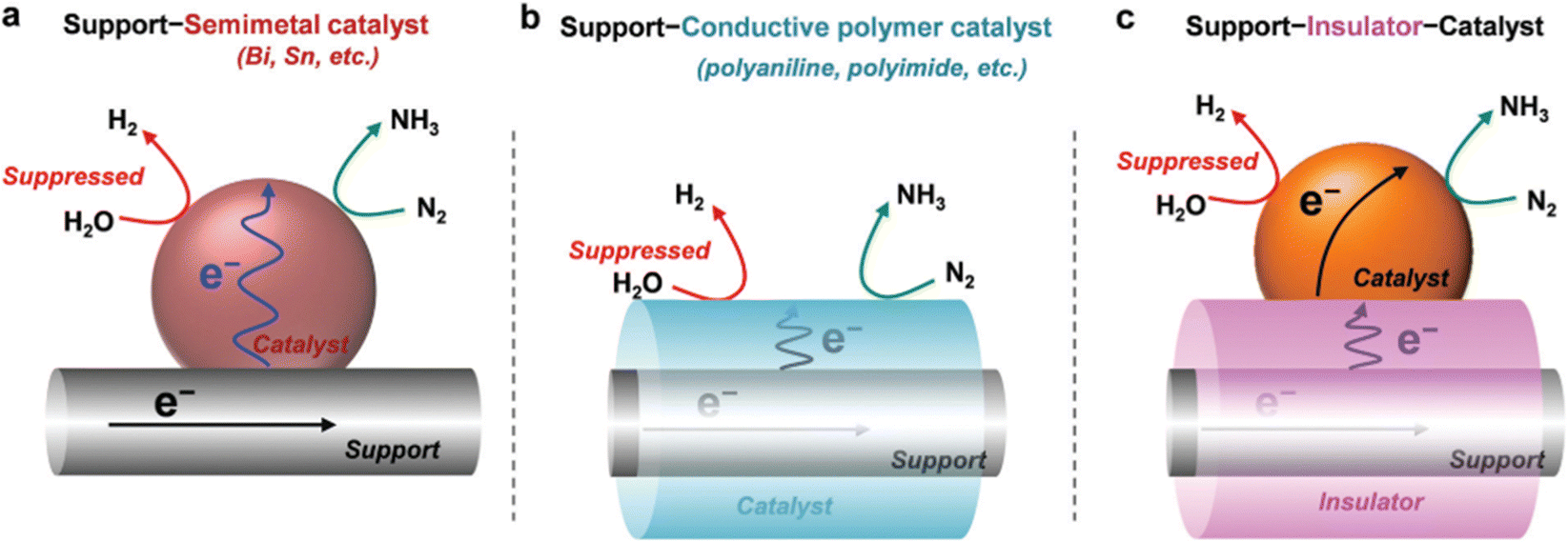 | ||
| Fig. 9 Multilevel catalyst configurations for limiting the electron accessibility in NRR, including (a) support–semimetal catalyst; (b) support–conducive polymer catalyst; and (c) support–insulator catalyst. Reprinted with permission from ref. 38 Copyright 2021, Royal Society of Chemistry. | ||
4.4.3.3. Hydrophobic protection layer. Since N2 molecules have poor solubility and diffusion coefficient in water, the photo(electro)catalytic ammonia production rates can be limited by diffusion processes in conventional liquid–solid diphase systems.42 Gas pre-saturation can improve the reaction efficiency but the slow gas diffusion to the catalytic centers cannot be increased, and this inevitably results in low reaction kinetics.156 An efficient strategy to circumvent these solubility and diffusion limitations consists on building a hydrophobic protection coating on the catalytic active sites, creating a gas–solid–liquid triphasic system. This configuration is based on the cooperative interaction between micro-/nanocomposites and low surface energy materials. Here, catalyst nanoparticles are located at the gas–liquid interface through surface tension regulation, and reactant gases (N2 or air) are continuously fed to the system. The creation of such gas–solid–liquid triphase limits the coverage of H2O molecules on the catalyst surface, because this configuration forms gas pockets trapped inside the structure when they are immersed in water. Thus, N2 molecules can be readily delivered to the catalytic sites from the gas phase, reaching much higher local nitrogen concentrations and eliminating the gas diffusion limitation of conventional diphase systems.42,156 Besides, this approach is an efficient strategy to lower HER kinetics and thus favor NRR selectivity. In addition, triphase systems holds a great economic potential because they also restrict the possible oxidation of ammonia, thus allowing the direct use of air instead of pure nitrogen.156
Some successful examples of hydrophobic layers reported for light-driven NRR include alkanethiols, porous poly(tetrafluoroethylene) (PTFE) frameworks,157 Poly(N-isopropylacrylamide) (PNIPAm),158 polydimethylsiloxane (PDMS),159 carbon cloth,160 or zeolitic imidazolate frameworks (ZIF)85 with hydrophobic cavities that block water and favor N2 permittivity.
5. Ammonia production technologies
5.1. Thermocatalytic-NRR
The thermocatalytic synthesis of ammonia from its elements is considered as one of the greatest discoveries of the 20th century, providing enough fertilizer supply to sustain the growing world population.161 Ammonia is produced nowadays in large plants (1000 to 1500 t day−1)20via the H–B ammonia synthesis loop (Fig. 10a), which is also known as the industrial process that made bread from air.162,163 At present, conventional H–B plants produce ammonia using natural gas (50%), oil (31%) or coal (19%) as fossil feedstock.164 Ammonia is produced by shifting the reaction equilibrium (1/2N2 + 3/2H2 ↔ NH3) of a high purity N2/H2 gas mixture towards the formation of ideally 22.7 mol% NH3 (relative to stoichiometric conversion),165 under high temperature and pressure (ca. 350–550 °C, 20–40 MPa) and in the presence of an alkali-promoted Fe catalyst (either magnetite or wüstite).16,18 This historical technology is able to convert N2 and H2 to NH3 in a single reaction with around ∼15% efficiency, and requires the recirculation of unreacted N2 and H2 to achieve overall NH3 conversion of ca. 97%.16,22 | ||
| Fig. 10 (a) Simplified diagram of Haber–Bosch ammonia synthesis. Reprinted with permission from ref. 26 (b) Volcano plot for ammonia synthesis on late transition metals, showing the ammonia production rate as a function of the nitrogen adsorption energy. The point denoted as ‘‘CoMo’’ represents a mixed site containing both Co and Mo. Reprinted with permission from ref. 166 Copyright 2001 ACS. | ||
Over the past years, alternative catalysts have emerged for low-temperature ammonia synthesis such as electrides, hydrides, oxides and amides, among others. Electrides are very unique crystals with cavity-trapped electrons, which can act as electron-donating materials. Some examples studied for ammonia synthesis include Ru/C12A7:e−,175–177 Ru/Ca2N:e−,178 LaRuSi,179 Ru/BaO/CaH2,180 Ru/LaScSi.181 An outstanding NH3 production rate of 2.7 mmol h−1 g−1 was obtained by Kitano and co-workers177 using Ru/C12A7:e− (360 °C, 0.3 MPa). They used the high electron density of electrides and their reversible hydrogen storage–release ability to avoid the well-known hydrogen poisoning of Ru. A year later, these authors developed a best performing Ru/Ca2N:e− electride178 able to work even at 200 °C, which delivered a maximum NH3 production rate of 3386 mmol h−1 g−1 (at 340 °C, 0.1 MPa) comparable to the reference Ru(2%)–Cs/MgO. Under similar reaction conditions, these authors also obtained outstanding ammonia production rates (ca. 4002 mmol h−1 g−1) exploring the partial conversion of a pure hydride (Ru/CaH2) into an electride (Ru/CaH2−x(e−)x), due to the easy desorption of hydrogen (>200 °C) from CaH2 surface in presence of a Ru catalyst.178
In the case of hydrides, these materials are attractive candidates for ammonia production due to their strong reducing character and their reversible H2 storage capacity, as they are used as electron/proton donors. Some examples of the most active metallic hydrides reported for thermocatalytic NH3 production include LiH,182,183 VH,184 binary hydrides (BaH2,180,185 TiH2,186 CaH2,178,180 ZrH2187), mixed-anion hydrides (Ca2NH, Li4RuH6188), nitride-hydrides (Mn4N,189 BaCrHN190), perovskite and lantanide oxyhydrides (Ru/BaCeO3−xNyHz, BaTiO2−xHx),191–194 hydride-based electrides.195 Interestingly, hydrides have achieved NH3 synthesis in the range of 50–100 μmol h−1 g−1 using mild temperatures such as 50–150 °C, and using catalysts based on Co–BaH2, CaFH hydride, Fe–BaH2–BaO/CaH2 and BaCr nitride-hydride (BaCrHN).190,196,197
Other interesting materials include metal oxides (e.g. oxygen vacancy-rich CeO2)198–200 or amide-based catalysts, which have shown a strong electron donation from Ca(NH2)2 to Ru metallic phase, achieving NH3 productions at temperatures below 200 °C during 700 h.201–206
000 to 40000 mL g−1 h−1, and can achieve ammonia productions higher than 100 mmolNH3 h−1 g−1.171,173,207 The catalytic converters are resistant to carbonization171 and steel embrittlement due to hydrogen diffusion.208,209 However, new steel alloys were introduced to prevent the nitridation of iron when it is in contact with ammonia at high temperatures.171 The centrifugal compressors, which replace reciprocating compressors,208 allow to reach the required operation pressures (150–250 bar) in the synthesis loop, as well as to recycle the gas stream to reach more than 20% of ammonia in each pass.171 Today, industrial ammonia plants have an integrated design to improve energy recovery and efficiency. Relevant technological improvements include: revamping or modernization of existing converters; use of bigger converters with more efficient designs; development of innovative radial-flow ammonia converters with a very low pressure drop to increase ammonia yield; use of lower pressure systems; and implementation of energy saving designs.210,211
5.2. Photocatalytic-NRR
| Photocatalyst | T (°C) | Phase | Light source | Reagents | Hole scavenger | NH3 production ratea (mmol h−1 g−1) | STA (%) | AQE,bξc (%) | Year | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Photocatalytic activity determined from experimental data from each reference. b Apparent quantum efficiency. c Photonic efficiency. d Quantum dots. e Triethanolamine. f CL: carbonized loofah sponge. g POF: porous organic framework. h NC: nitrogen defects and cyano groups. | ||||||||||
| Porous graphdiyne with Co QDd | 20 | Liquid | 300 W Xe lamp | N2, Na2SO4 (0.1 M) | — | 26.5 | — | 8.72b (500 nm) | 2021 | 214 |
| Bi defective K/g-C3N4 | 5 | Liquid | 300 W Xe lamp | Air, H2O | Methanol | 23.5 | — | — | 2022 | 215 |
| g-C3N4/CoFe2O4 | Room | Liquid | 250 W Xe lamp | N2, H2O | — | 14.7 | — | — | 2023 | 216 |
| Bi5O7Br | Room | Liquid | 300 W Xe lamp | N2, H2O | — | 12.7 | — | — | 2020 | 217 |
| Ni-ZrO2/Bi2O3 | Room | Liquid | Sunlight (65500 lux) | N2, H2O | TEOAe | 9.7 | — | — | 2023 | 218 |
| Porous few-layer g-C3N4 | Room | Liquid | 500 W Xe lamp (AM 1.5G) | N2, H2O | Methanol | 8.2 | — | — | 2020 | 146 |
| Bi2Te3/BiOCl | Room | Liquid | UV and Vis | N2, H2O | Methanol | 7.9 | — | — | 2018 | 219 |
| Cu/TiO2 | Room | Liquid | 300 W Xe lamp (AM 1.5G) | N2, H2O | Glycerol | 6.8 | — | — | 2020 | 139 |
| S-doped g-C3N4 | Room | Liquid | 500 W Xe lamp | N2, H2O | Methanol | 6.0 | — | — | 2018 | 220 |
| FeS2-FeP-CeO2 | 25 | Liquid | 250 W Hg lamp | N2, H2O | Methanol | 5.6 | — | — | 2019 | 221 |
| UiO-66(−NH2)/CuInS2 | Room | Liquid | 250 W Xe lamp | N2, H2O | Methanol | 4.0 | — | — | 2023 | 222 |
| Ru/HxMoO3-y | Room | Gas | 300 W Xe lamp (420 nm filter) | N2, H2 | — | 4.0 | — | 6.0b (650 nm) | 2022 | 223 |
| In(OH)3/C3N4 | 25 | Liquid | 300 W Xe lamp (420 nm filter) | N2, H2O | TEOAe | 3.8 | — | — | 2020 | 224 |
| KOH treated g-C3N4 | 25 | Liquid | 300 W Xe lamp | N2, H2O | Methanol | 3.6 | — | 21.5b (420 nm) | 2018 | 225 |
| Ag-Bi2S3/MnOS | Room | Liquid | 300 W tungstate lamp | N2, H2O | Methanol | 3 | — | — | 2023 | 226 |
| Au-Os/Cs2O | 60 | Gas | Solar simulator | N2, H2 | — | 2.7 | — | — | 2015 | 227 |
| CL@BiOBr-OV/Auf | Room | Liquid | 300 W Xe lamp | N2, H2O | — | 2.6 | — | — | 2022 | 228 |
| Black phosphorus | Room | Liquid | LED lamp (420 nm) | N2, H2O | Na2SO3 and Na2S·9H2O | 2.4 | — | — | 2020 | 144 |
| MOF-74@C3N4 | Room | Liquid | 300 W Xe lamp | N2, H2O | Methanol | 2.3 | — | — | 2020 | 229 |
| TiO2/SrTiO3/g-C3N4 | Room | Liquid | 300 W Xe lamp | N2, H2O | Methanol | 2.2 | — | 3.03b (365 nm) | 2020 | 230 |
| Bi5O7I | 20 | Liquid | 300 W Xe lamp | N2, H2 | Methanol | 2.2 | — | 5.1b (365 nm) | 2016 | 231 |
| ZnInCuS QDd/BiOI | Room | Liquid | 250 W Hg lamp (420 nm filter) | N2, H2O | — | 2.1 | 0.24 | — | 2023 | 232 |
| g-C3N4 | Room | Liquid | 300 W Xe lamp (AM 1.5G filter) | N2, H2O | Methanol | 1.9 | — | 7.79b (370 nm) | 2023 | 233 |
| BiMnO/BiOIBr | Room | Liquid | Solar simulator | N2, H2O | Methanol | 1.8 | — | 3.6b (420 nm) | 2023 | 234 |
| MPc-POFg | Room | Liquid | 300 W Xe lamp | N2, H2O | Methanol | 1.8 | — | 0.5b (400 nm) | 2023 | 235 |
| Graphdiyne@Janus magnetite | Room | Liquid | 300 W Xe lamp | N2, Na2SO4 (0.1 M) | — | 1.8 | — | — | 2021 | 236 |
| ZnCuInS–BiOI | Room | Liquid | 250 W Hg lamp | N2, H2O | — | 1.6 | 0.36 | — | 2023 | 232 |
| NC-g-C3N4h | Room | Liquid | 300 W Xe lamp (AM1.5) | N2, H2O | Methanol | 1.6 | — | — | 2019 | 237 |
| Zr/MOFs | Room | Liquid | 150 W Xe lamp (420 nm filter) | N2, H2O | — | 1.5 | — | — | 2023 | 238 |
| Bi@BiOBr | Room | Liquid | 300 W Xe lamp | N2, H2O | — | 1.4 | — | — | 2021 | 239 |
| TiO2/BiOBr | Room | Liquid | 300 W Xe lamp | N2, H2O | Methanol | 1.4 | 2021 | 240 | ||
| WS2@TiO2 | Room | Liquid | Simulated sunlight (AM1.5) | N2, H2O | Na2SO3 (0.01 M) | 1.4 | — | — | 2020 | 241 |
| Bi5O7Br | Room | Liquid | 300 W Xe lamp (400 nm filter) | N2, H2O | — | 1.4 | — | 2.3b (420 nm) | 2017 | 242 |
| H-Bi2MoO6 | Room | Liquid | 300 W Xe lamp | Air, H2O | — | 1.3 | — | 2.4b (420 nm) | 2016 | 243 |
| Ru–Pd/g-C3N4 | Room | Liquid | 300 W Xe lamp | N2, H2O | Ethanol | 1.3 | — | 6c | 2021 | 244 |
| BiO quantum dots | 25 | Liquid | 500 W Xe lamp | N2, H2O | — | 1.2 | — | — | 2017 | 148 |
| CeCO3OH/g-C3N4/CeO2 | Room | Liquid | 500 W Xe lamp | Air, H2O | — | 1.2 | 2019 | 245 | ||
| CeCO3OH | Room | Liquid | 300 W Xe lamp | N2, H2O | — | 1.1 | — | 0.98b (380 nm) | 2022 | 246 |
| TiO2/Au/K+ | Room | Liquid | Xe lamp (400nm filter) |
N2, H2O | Methanol | 1.0 | — | 0.93b (550nm) |
2021 | 247 |
| TiO2/MIL-88A(Fe)/g-C3N4 | 25 | Liquid | 300 W Xe lamp | N2, H2O | Methanol | 1.0 | — | — | 2023 | 248 |
| Fe-doped BiOCl | Room | Liquid | 300 W Xe lamp | N2, H2O | — | 1.0 | — | 1.8b (420 nm) | 2019 | 249 |
The most active NRR photocatalyst reported to date is based on stable porous graphdiyne (GDY) decorated with Co quantum dots,214 which achieved a maximum NH3 production rate of 26.5 mmol h−1 g−1 and a AQE of 8.7% without any sacrificial agent (see Fig. 11 and Table 1). This high catalytic performance was ascribed to the rapid conversion of valence states of Co species (Co2+/Co3+) together with the GDY. Recently, a similar heterojunction based on magnetite/GDY236 showed a remarkable NH3 production of 1.8 mmol h−1 g−1 without sacrificial agent. These authors demonstrated that the coordination environment and valence state of the Fe atoms in magnetite can be also modified by incorporating GDY, resulting in a high-performance heterojunction.
 | ||
| Fig. 11 (a) Schematics of photocatalytic NRR on porous graphdiyne decorated with Co quantum dots; (b) NH3 production rates in different solutions and (c) in independent experiments; (d) 15N2/14N2 isotopic labeling tests under (red) and without (blue) irradiation. Reprinted with permission from ref. 214 Copyright 2021 Elsevier. (e) Photocatalytic NH3 production with different g-C3N4 samples; (f) 1H NMR spectra for nanostructured g-C3N4. Reprinted with permission from ref. 215 Copyright 2022 Elsevier. | ||
An extraordinary NH3 production rate of 23.5 mmol h−1 g−1 was reported by Liu et al.215 using g-C3N4 nanorod arrays modified with cyano groups (–CN) and K centers. Interestingly, this work used air as N2 source and reported an operational temperature of 5 °C. This good performance was explained by the coexistence of both sites: –CN groups with electron donation effect for N2 activation, and K centers that act as electron trapping sites, promoting charge separation. Zhang et al.216 combined a 3D porous g-C3N4 with NVs and CoFe2O4 to create a Z-scheme heterojunction that achieved a NH3 production rate of 14.7 mmol h−1 g−1. This catalytic system presented wider light absorption, faster electron transfer rates and stronger N2 adsorption and activation than the g-C3N4 counterpart. Fan et al.224 supported OV-rich In(OH)3 on g-C3N4, obtaining a OD/2D heterojunction with many chemisorption sites for N2, efficient electron–hole separation and thus prolonged carriers lifetime.
In the case of bismuth oxyhalides (BiOX, X = Cl, Br, I), the formation of OVs with abundant localized electrons on the surface play a critical role for NRR, mainly for N2 adsorption and activation.217–239 These materials have been combined with other inorganic semiconductors within binary heterojunctions to boost performance.219–240
Of special interest is the more recently emerged sunlight-driven nitrogen fixation from air. The big challenge of this approach relays on the competition between N2 and O2 for photogenerated electrons, since O2 is more easily reduced than N2 due to its lone pair electrons and this suppresses NH3 formation.250 Hirakawa et al.121 demonstrated that air bubbling suppresses NH3 formation over a commercial TiO2 sample with a large number of surface Ti3+ species. Despite this, the sustainability benefits of aerobic NRR is encouraging the development of research works on this field using a wide variety of materials, such as bismuth molybdate,243 Fe–WO3,251 Ti/Mo composite,252 CoF2O4,253 Co3O4@Schiff,254 and binary255–257 or ternary258 heterojunctions. In this aerobic route, both NRR and NOR may coexist (Fig. 12). Zhao et al.256 reported that the reaction mechanism towards ammonia follows an alternative redox pathway, in which N2 and O2 can be trapped at OVs, generating nitric oxide (*NO) as active intermediate that is further photoreduced to ammonia. DFT calculations supported this point by founding a lower free energy barriers for N2 activation (*N2 → *N–NO, −0.08 eV) and and rate-limiting step (*NO → *NHO, 1.23 eV) than traditional NRR (*N2 → *N–NH, 1.48 eV and H2N–NH2 → *NH2, 1.29 eV, respectively).
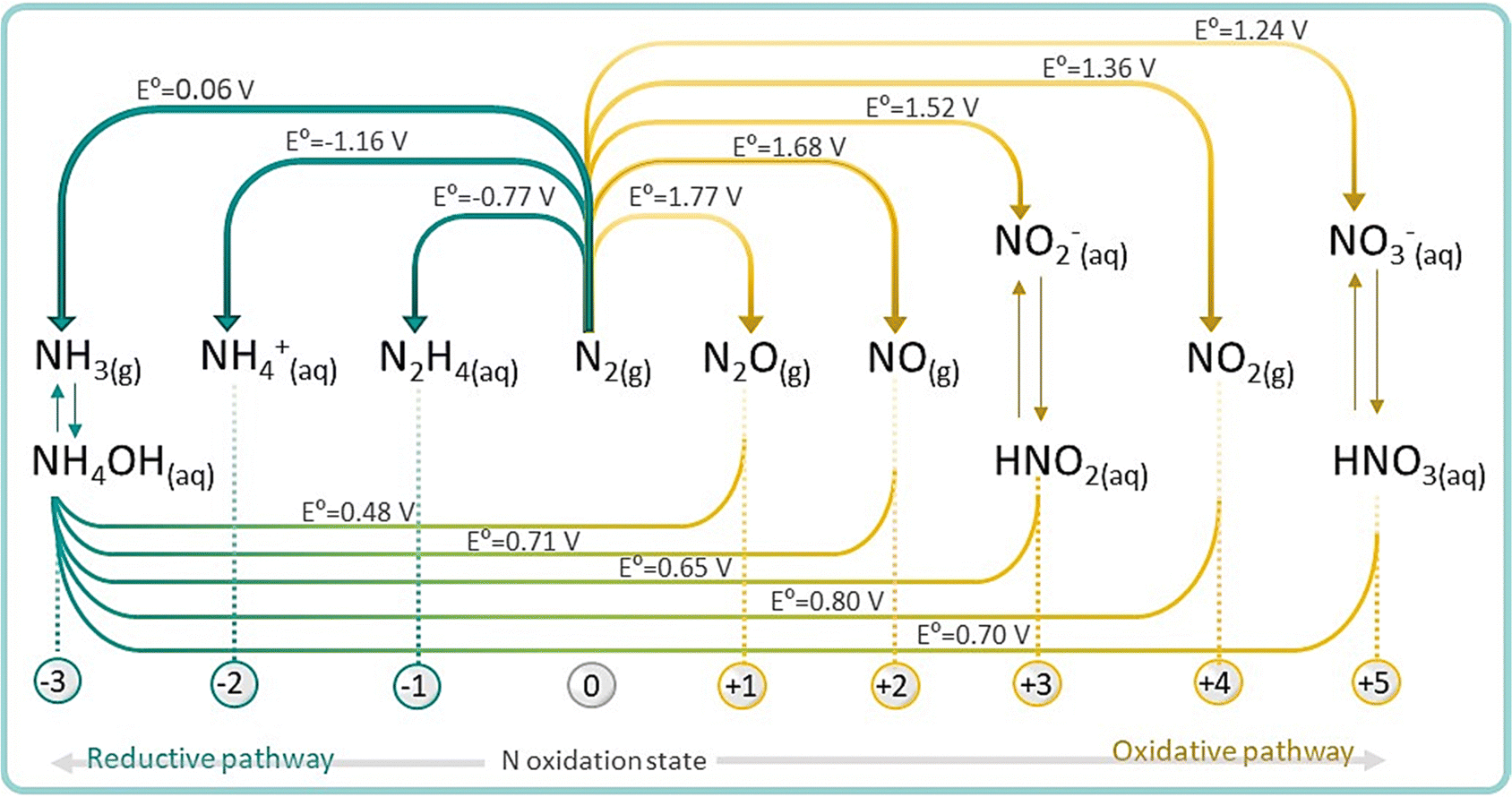 | ||
| Fig. 12 Possible reductive and oxidative pathways (left- and right-side figure, respectively) in N2 photofixation. Redox potentials (E°) are referred to the normal hydrogen electrode (NHE). Adapted from ref. 259. | ||
A rational configuration of photocatalytic reaction systems holds the potential to improving ammonia yield and selectivity. Next-generation photoreactors should enable the spatial separation of ammonia and oxidative species to avoid back reactions, for instance by using products separation membranes. This spatial separation would also maximize the utilization of photogenerated carriers towards target products, that is the production of ammonia using photoelectrons, and the synthesis of nitrates consuming photoholes. This approach would further increase the added value of the photocatalytic process if nitrogen fixation products are even enriched by forming ammonium nitrate or urea.
5.3. Photoelectrocatalytic- and (PV)-electrocatalytic- NRR
The use of the light to reduce the necessary potential of a certain electrocatalytic reaction has already been extensively employed among the scientific community.260,261 As previously occurred with other interesting chemical reactions, there are two main strategies to achieve this purpose: (i) the direct use of photoelectrocatalytic materials (i.e. semiconductors able to generate electron–hole pairs and also drive the desired reaction262,263), or (ii) coupling of a photovoltaic cell, which generates the carriers, with an electrocatalytic material that drives the chemical reaction.264–266 Aiming to ease the readers comprehension, these two ammonia production technologies will be described separately in the following sections.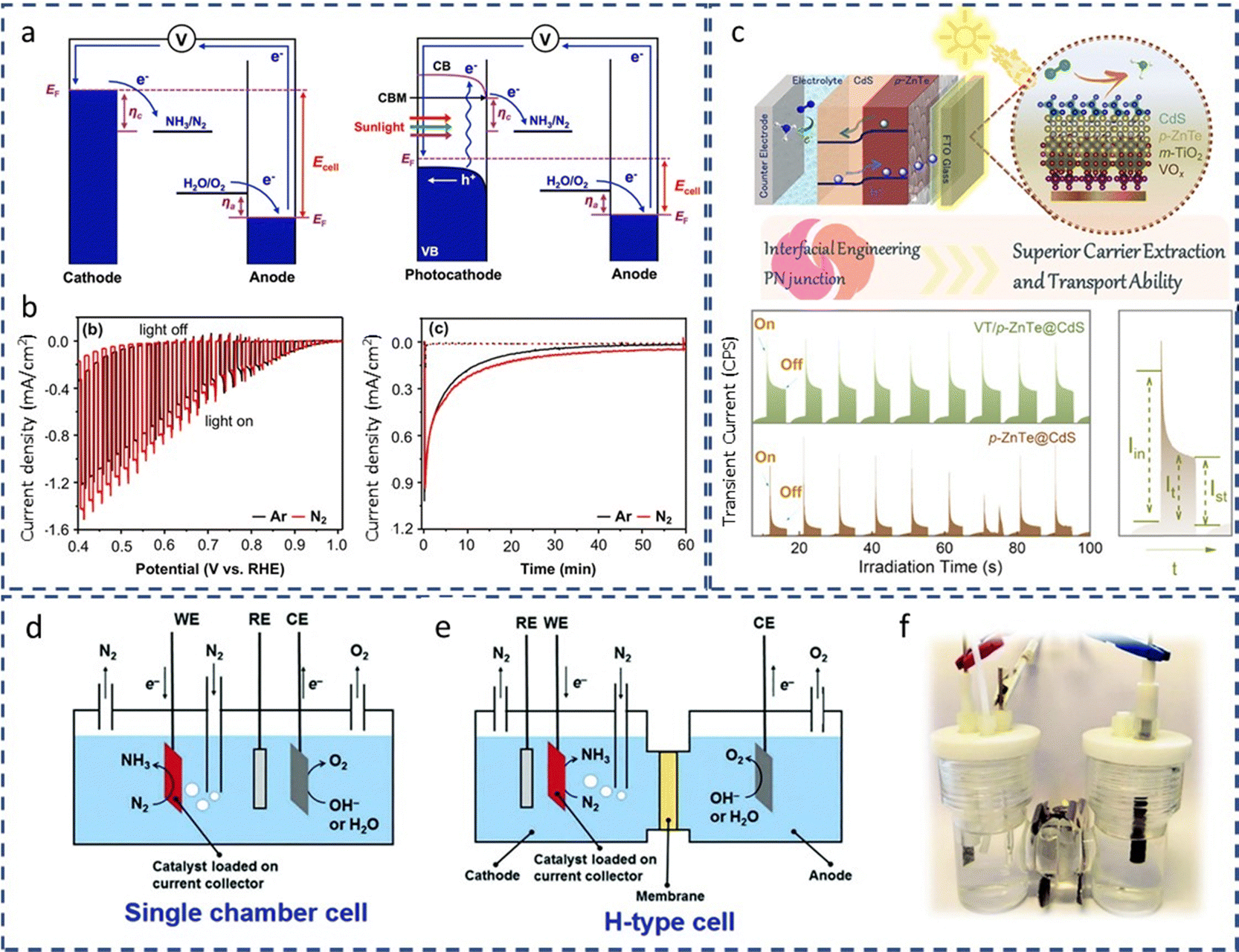 | ||
| Fig. 13 (a) Comparison of electrochemical (left) and photoelectrochemical NRR (right), depicting the beneficial effect of using a photocathode to decrease the overall cell voltage needed for NRR. (b) J–V plot (5 mV s−1) and J–t plot (at 0.6 V vs. RHE) of a CuO measured under Ar (black) and 15N2 (red) in 0.1 M KOH solutions under AM 1.5G, 100 mW cm−2. The dark currents in the J–t plot are shown as dotted lines. Reproduced with permission from Jang et al.267 Copyright 2020 American Chemical Society. (c) Scheme of a solar-electrolytic cell, transient photocurrent and dynamics for VT/p-ZnTe@CdS and p-ZnTe@CdS photocathodes. Reproduced with permission from Jia et al.268 Copyright 2023 American Chemical Society. Schematic representation of (d) a single-chamber and (e) an H-type cell with a proton exchange membrane. (f) Photograph showing an H-type cell, highlighting the working electrode (WE), reference electrode (RE), counter electrode (CE), and membrane. Reproduced with permission from Pang et al.269 Copyright 2021 Royal Society of Chemistry. | ||
Since the first photoelectrochemical cell (PEC) publication in 1972,262 its applications have evolved from water splitting270 to other fields such as fuel production through CO2 conversion,271 reduction of organic compounds and the degradation of contaminants.271 However, the use of PEC cells in the field of nitrogen reduction reaction (NRR) is still quite limited.272 Photoelectrochemical N2 reduction involves three principal stages: (i) charge carriers photogeneration, (ii) separation of charge carriers to the active sites, and (iii) N2 molecules absorption and reduction reaction, very similar to the photocatalytic process. However, in this case, photogenerated electrons and holes separate to different reaction sites, unless charge recombination. Electrons migrate to the surface of the photocathode and holes goes to the (photo)anode. Thus, electrons drive the N2 reduction on the active sites of the photocathode (N2 + 6H+ + 6e− → 2NH3, E0 = −0.148 V vs. RHE), while holes drive the oxidation evolution reaction (OER) on the (photo)anode (2H2O + 4h+ → 4H+ + O2, E0 = 1.23 V vs. RHE). Despite its promising features, PEC-NRR is still at a development stage since the catalytic performance is not yet suitable for practical applications. Typically, PEC-NRR occurs on the surface of p-type materials (photocathodes) with intrinsic structural defects, which may lead to poor visible-light absorption, high exciton recombination, and serious light corrosion. Over the last years, PEC-NRR investigations have focused on developing high-performance photoelectrode materials, which include inorganic semiconductors with suitable band gaps, metallic modifications, organometallic frameworks with tunable structures, and single atoms with notable active sites (see Table 2). Metal oxides have been thoroughly explored as potential photo(electro)catalysts for various catalytic reactions in the energy and environmental sectors. These materials provide benefits such as high catalytic activity, cost-effectiveness, and straightforward preparation. Especially, p-type semiconductors have been tested as photocathodes for PEC-NRR due to their suitable band structures and efficient carrier transport.
| Photoelectrode material | Electrolyte | Light source | Max. photocurrent density (mA cm−2) | NH3 production rate | Year | Ref. |
|---|---|---|---|---|---|---|
| a COF/GO: Covalent organic framework/graphite oxide. b PANI-ASSM: Polyaniline-coated anodized stainless-steel mesh. c LDH: Layered double hydroxide. d PANI: Polyaniline. e NV: Nitrogen vacancy. f PTFE: poly(tetrafluoroethylene). g TS: Ti layer on Si surface. h OVs: oxygen vacancies. | ||||||
| COF/GOa | 0.5 M Na2SO4 | 300 W Xe lamp, 200 mW cm−2 | −0.3 mA at −0.7 V RHE | 108.20 μg h−1 mg−1 | 2024 | 273 |
| Ti–WO3/SrWO4 | 0.05 M H2SO4 | AM 1.5G, 100 mW cm−2 | −2.05 mA at −0.6 V RHE | 11.17 μg h−1 cm−2 | 2024 | 274 |
| Vo–TiO2/Ag/TiO2 | 0.2 M Na2SO4 | 300 W Xe lamp | −0.5 mA at −0.4 V RHE | 51.20 g h−1 cm−2 | 2023 | 275 |
| PdCu/TiO2/Si | 1 M LiClO4 | AM 1.5G, 100 mW cm−2 | −4 mA at 0 V vs. Li | 43.09 μg h−1 cm−2 | 2023 | 93 |
| Mo2C/GaN/InGaN | 0.05 M H2SO4 | 100 W Xe lamp (AM 1.5G) | −1.4 mA at −0.8 V RHE | 7.93 μg h−1 cm−2 | 2023 | 276 |
| VOx/m-TiO2/p-ZnTe@CdS | 0.05 M H2SO4 | Xe lamp, 200 mW cm−2 | −4 mA at −0.7 V vs. RHE | 6.23 μg h−1 cm−2 | 2023 | 268 |
| PANI-ASSMb/CdS-Co3S4 | H2SO4 | 40 W blue LED | −22 mA at 0.5 V | 12.33 μg mol−1 h−1 | 2022 | 277 |
| CoV-LDHc | 0.1 M Na2SO4 | AM 1.5G, 100 mW cm−2 | −1 mA at −0.4 V RHE | 8.97 × 10−7 mol h−1 cm−2 | 2022 | 278 |
| Mo-doped WO3@CdS | 0.5 M H2SO4 + 5% H2O2 | 300 W Xe lamp | −0.05 mA at −0.3 V vs. RHE | 38.99 μg h−1 mg−1 | 2022 | 279 |
| Bi2S3 quantum dots on MoS2 | 0.1 M Na2SO4 | 300 W Xe lamp | −1.7 mA at −0.6 V vs. RHE | 18.50 μg h−1 mg−1 | 2022 | 280 |
| Metal–organic iron terephthalate structure (MOF-235) | 0.1 M Na2SO4 | Hg lamp | −20 mA at −0.7 V vs. Ag/AgCl | 0.72 μg h−1 cm2 | 2022 | 281 |
| BiVO4/PANId | 0.1 M Li2SO4 | 300 W Xe lamp | −0.05 mA at −0.35 RHE | 0.93 μg h−1 cm−2 | 2021 | 282 |
| p-type BiVO4 | 0.1 M Li2SO4 | 300 W Xe lamp | −0.2 mA at −0.2 V RHE | 1.6 × 10−8 mol h−1 cm−2 | 2021 | 283 |
| Cu2S–In2S3 | 0.2 M K2SO4 | Xe lamp (100 mW cm−2) | −3 mA at −0.9 V vs. RHE | 23.67 mg h−1 cm−2 | 2021 | 284 |
| Au/SiO2/Si | 0.05 M potassium phosphate buffer | AM 1.5G, 100 mW cm−2 | −0.05 mA at 0.2 V vs. RHE | 22.0 μg h−1 cm−2 | 2021 | 285 |
| MoSe2@g-C3N4 | 0.1 M KOH | 300 W Xe lamp | −12 mA at −0.5 V vs. RHE | 7.72 μmol h−1 cm−2 | 2021 | 286 |
| CuO and Cu2O | 0.1 M KOH | 300 W Xe lamp | −1.5 mA at −0.4 V vs. RHE | 5.3 μg h−1 cm−2 (CuO) | 2020 | 267 |
| 7.2 μg h−1 cm−2 (Cu2O) | ||||||
| W18O49 | 0.5 M Na2SO4 | Xe lamp (1 sun) | −150 mA at −1.8 V Ag/AgCl | 9.82 μg h−1 cm−2 | 2020 | 287 |
| Black phosphorus nanosheets | 0.1 M HCl | Xe lamp | −10 mA at −0.5 V vs. RHE | 102.4 μg h−1 g−2 | 2020 | 288 |
| NVe-g-C3N5/BiOBr | 0.05 M HCl + 0.05 M Na2SO4 | Xe lamp (solar simulator) | −1.2 mA at −0.2 V | 29.4 μg h−1 mg−1 | 2020 | 289 |
| Au/PTFEf/TSg | 0.05 M H2SO4 + 0.05 M Na2SO3 | AM 1.5G, 100 mW cm−2 | — | ≈18.9 μg cm−2 h−1 | 2019 | 157 |
| MoS2@TiO2 | 0.1 M Na2SO4 | 300 W Xe lamp | −4 mA at −0.6 V vs. RHE | 1.42 × 10−6 mol h−1 cm−2 | 2019 | 290 |
| BiOI-OVsh | N2-saturated H2O | 100 mW cm−2 | 0.4 V vs. RHE | 140 nmol h−1 cm−2 | 2019 | 291 |
Fig. 13a shows a recent study from Jang et al.267 who explored the feasibility of PEC-NRR using CuO and Cu2O photocathodes, as well as its associated reaction mechanism. The authors observed that both systems exhibited substantial faradaic efficiencies for NH3 production (17% and 20%, respectively) with minimal H2 production. Interestingly, the external bias (0.6 V vs. RHE for CuO and 0.4 V vs. RHE for Cu2O) were less negative than the thermodynamic reduction potential for NRR (0.092 V vs. RHE at pH = 13), demonstrating the potential of the photogenerated electrons used for NRR depend on the conduction band minimum of the photocathode. However, they found cathodic photocorrosion of CuO (see Fig. 13b), and pointed out the need for a protection layer. In literature, the surface of Cu2O photocathodes has been coated with different systems to improve the NH3 yield and stability, for instance using Cu metal–organic frameworks (MOF) to build Cu-MOF/Cu2O292 or Ag-doped Ni MOF heterojunctions.292
Very recently, He et al.273 tested a novel donor-site-acceptor system based on a covalent organic framework (COF) and a graphite oxide. They found that the active boron sites, embedded between the acceptor and the donor units, effectively activated chemisorbed N2 with a continuous electron flow after photoexcitation. This system achieved a faradaic efficiency around 91.6% and 108.2 μg h−1 mgcat−1 (at −0.4 V versus RHE) in several cycles, reaching the target value set by the U.S. Department of Energy (90%).
Regarding other active metals for PEC-NRR, Mo is a very interesting candidate since natural nitrogenase contains Mo active sites. Based on DFT calculations, Nørskov and co-workers293 studied the prospect of electrochemical reduction of N2 to NH3 under ambient conditions, and anticipated that Mo provides many additional active sites for N2 reduction. On this basis, several Mo-based catalysts have been developed for (photo)electrochemical N2 fixation, and are actually the subject of a recent bibliographic review.129 We highlight the work by Ye et al.290 who developed a functional MoS2@TiO2 structure as a photocathode for PEC-NRR under ambient conditions. They achieved a NH3 yield rate of 1.42 × 10−6 mol h−1 cm−2 and a faradaic efficiency of 65.52%, which was a remarkable result at that time. This performance was due to an efficient interfacial charge transport and the synergistic effects between the MoS2 and TiO2 counterparts. Another recent example286 published in 2021 reported the use of MoSe2@g-C3N4 as photocathodes, which demonstrated to be inherently active, selective and robust catalysts for PEC-NRR. These cathodes exhibited good NH3 yield rate (7.72 μmol h−1 cm−2) and FE (28.91%) at −0.3 V versus RHE at room temperature and pressure. This performance was attributed to the synergistic effects and electronic coupling between MoSe2 and g-C3N4 and the improvement in the light harvesting capacity and charge separation efficiency, which led to slow recombination rates and longer lifetime of photogenerated electron–hole pairs.
Other strategy to improve PEC-NRR performance is the construction of a PN junction to accelerate the photogeneration of charge carriers and improve their spatial separation.289 In this line, we highlight the contribution of Jia et al.268 who developed a photocathode composed by a PN junction (p-ZnTe@CdS) and a VOx/TiO2 interlayer. This heterostructured photocathode achieved an AQE of 0.6% and a N2 to NH3 conversion performance of 6.23 μg h−1 cm−2 (Fig. 13c). Very recently, Wang et al.274 prepared a Ti–WO3/SrWO4 heterojunction by simple hydrothermal and spin-coating approaches and use it as photocathode. They found out that this catalyst inhibited the photogenerated carrier's complexation. Ti acted as the N2 adsorption site, promoting nitrogen activation and thereby improving the carrier transport efficiency and NH3 synthesis performance. At −0.5 V vs. RHE, the ammonia production reached 11.17 μg h−1 cm−2, which surpassed that of SrWO4 by four times. Moreover, Ti–WO3/SrWO4 exhibited high stability and N-gas sensitivity during NRR, showing its suitability for photoelectrocatalytic ammonia synthesis.
On the other hand, vanadium-based materials are promising catalysts for N2 fixation, as vanadium nitrogenase acts as an important component of the N2 cycle in nature. In this transition metal, the unoccupied 3d orbital of V facilitates the adsorption of N2 and serves as a bridge to allow the transfer of photoexcited electrons from the catalyst to the N2 molecules. BiVO4 is a very suitable matrix for V localization due to its good physicochemical properties, low cost, and very high photostability. However, BiVO4 is generally considered as an n-type semiconductor and therefore, it is frequently used as a photoanode in PEC systems but not for NRR. In contrast, Bai et al.283 reported for the first time the use of a p-BiVO4 photocathode for PEC-NRR under ambient conditions. Interestingly, density functional theory (DFT) calculations revealed that V sites worked as essential active centres for N2 adsorption/activation. They obtained a NH3 evolution rate of 11.6 × 10−2 μmol h−1 cm−2 and a faradaic efficiency of 16.2% at 0.1 V versus RHE in 0.1 M Li2SO4 solution. Bai et al.282 incorporated PANI (polyaniline) on the BiVO4 surface to create a Z-scheme heterojunction, which accelerated the separation and transfer of photogenerated carriers leading to a NH3 yield rate of 0.93 μg h−1 cm−2 and FE = 26.43%.
Interestingly, in the two previous studies, NO3− was used as the nitrogen source for PV-electrocatalytic ammonia production. The reasons behind, are that nitrates present unique advantages as nitrogen sources for electrocatalytic NH3 production. First, nitrate is widely present in the nature, being a habitual pollutant of water resources, accumulating over time due to agriculture and industrial activity.296,297 Then, producing ammonia from nitrates, is both a renewable energy source and a pollution mitigation strategy. And secondly, the bond energy of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (204 kJ mol−1) is around 4 times weaker than the triple NN bond, making this bond easily activated at lower energies.298
O (204 kJ mol−1) is around 4 times weaker than the triple NN bond, making this bond easily activated at lower energies.298
In non-solid single-PEC cell systems, both oxidation and reduction reactions take place in the same cell. As a result, re-oxidation of NH3 products on the anode is likely to be detrimental to the NH3 performance. Therefore, as in other PEC reactions, separating the cathode and anode into two divided chambers is an effective way to correct these deficiencies and increase the NRR performance. This type of reactor is known as double-chamber cell or H-cell in the literature. Generally, H-cells are in the form of consist on an H-shaped apparatus separated by a proton exchange membrane (PEM) or an anion exchange membrane (AEM). During the electrochemical reaction, the protons or hydroxide ions produced at the anode move through the membrane to the cathode to participate in the NRR under mild catalytic conditions. On the other hand, most of the NH3 produced (NH4+ in aqueous solutions) cannot cross these membranes, thus avoiding the re-oxidation and the loss of efficiency of single-chamber cells. That is why it is essential to select suitable membranes that minimize the NH3/NH4+ crossover, allowing the total concentration of NH3 in both chambers’ electrolytes, as a portion of the products can be still transferred through the membrane to the anodic chamber. Furthermore, this anti-crossing effect allows the use of two different electrolytes. This additional degree of freedom can lead to even more optimized reaction conditions by electrolytes combinations. For instance, Kim et al.301 reported a bio-electrolyte system using LiCl/ethylenediamine (EDA) as catholyte and H2SO4 solution as anolyte. Regarding membranes, they have a fundamental role in the performance of such cells. The most commonly used cation exchange membrane (CEM) so far in H-cell testing is Nafion 212, which leads to a significant NH4+ crossover, limiting its suitability. Alternative membranes have been examined, including AEM and porous polypropylene (PP) membranes; the AEMs showed minimal NH4+ crossing, while the PP porous membrane exhibited high NH4+ crossing. The AEM PiperION-A80 is particularly promising as it shows negligible NH4+ crossover in acidic and neutral electrolytes, making it suitable for experiments with H-cell electrolyzers, generating NH3 under these conditions. However, it is permeable to NH3 in basic electrolytes (pH 13), thus restricting its selective suitability depending on the electrolyte environment.302
5.4. Photothermocatalytic-NRR
The photothermal NRR route is based on the synergistic combination of the photo- and thermo-chemical contributions of sunlight (i.e. light and heat). This approach allows for a more effective harvesting of the solar spectrum through the potential absorption of photons in the UV-visible-infrared range.303,304 Light absorption at catalytic active sites (i.e. absorber) causes an increase of the local temperature at the nanoscale, which has the potential to render higher reaction rates. The local heating of the absorber can be maximized by using supports with low thermal conductivity (e.g. strontium titanate, silicon, etc.), due to their slower heat dissipation losses.305 This section summarizes the most relevant advances on the photothermocatalytic N2 reduction to ammonia (Table 3), covering active catalysts and reactor configurations.| Photothermal catalyst | T (°C) | Light source | Reagents | NH3 production ratea (mmol h−1 g−1) | STA | AQE,bξc | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Photocatalytic activity determined from experimental data from each reference. b Apparent quantum efficiency. c Photonic efficiency. d Value obtained under 6 bar pressure. | ||||||||
| Ru–Cs/ZrO2 | 350 | 300 W Xe lamp (1 Sun) | N2, H2 | 5.1 | — | 39%b | 2023 | 306 |
| Ru–Cs/MgO | 333 | Blue LED light | N2, H2 | 4.5 | — | — | 2019 | 307 |
| Ru/C | 380 | Concentrated sunlight | N2, H2 | 3.5 (0.1 MPa) | — | — | 2023 | 308 |
| CsRu@SrTiO3 | 360 | 300 W Xe lamp | N2, H2 | 3.5 | — | — | 2022 | 305 |
| Fe–MoS2 | 270 | 70 W tungsten lamp | N2, H2O | 2.1–17d (6 bar) | 0.24–0.18% (solar furnace) | 37.1%b (432 nm) | 2021 | 309 |
| Pt-Ptn-TiN | 280 | Xe lamp (6 Sun) | N2, H2 | 0.5 | — | 0.03%b (465 nm) | 2023 | 310 |
| Ni/TiO2 | 400 | 300 W Xe lamp | N2, H2 | 0.2 | — | — | 2021 | 311 |
| K/Ru/TiO2−xHx | 360 | 300 W Xe lamp | N2, H2 | 0.1 | — | — | 2018 | 312 |
| TiO2−xHy/Fe | 495 | Xe lamp (102 Sun) | N2, H2 | — | 3.9 × 10−4 | — | 2019 | 39 |
In 2019, Li et al.307 reported a photothermal plasmonic system based on a cesium-promoted ruthenium NPs supported on magnesium oxide (Ru–Cs/MgO). The authors found that the creation of light-induced controlled thermal gradients in an illuminated packed catalyst bed, by photothermal effect, improved NH3 reaction rates without external heating and working under atmospheric pressure. They demonstrated that photothermal heating created a negative thermal gradient in the catalyst bed, in which a hot top region accelerated nitrogen cleavage, while generated NH3 was moved to the cold bottom region of the catalyst bed due to thermophoretic forces, thus preventing NH3 decomposition (Fig. 14a). The authors defined an equivalent catalyst temperature assuming the formation of a linear temperature gradient from the top to the bottom of the isothermal catalyst bed. They obtained a NH3 reaction rate of 858 μmol h−1 g−1 under concentrated blue LED illumination (455 nm, 4.7 W cm−2) as sole energy source at an equivalent catalyst temperature of 261 °C. They obtained a NH3 production of 1.5 mmol h−1 g−1 under dark thermal conditions (333 °C by supplying external heating), while the production rate increased up to 4.5 mmol h−1 g−1 by combining LED illumination and external heating, due to the creation of a strong negative thermal gradient. Mao et al.39 also explored a dual-temperature-zone ammonia synthesis, using TiO2−xHy/Fe nanocomposites upon solar illumination (10.2 W cm−2). The authors spatially separated a local hot zone (Fe) to dissociate N2via photogenerated hot electrons, and a local cold zone (TiO2−xHy) for hydrogenation of spilled-over N from Fe. The plasmonic local heating effect of Fe raised the catalyst surface temperature up to 495 °C, with a local temperature gradient of 137 °C between hot and cold zones. Under these conditions, the authors reported ηSTA values of 3.9 × 10−4% and 9.2 × 10−3% at 1 atm and 10 atm, respectively.
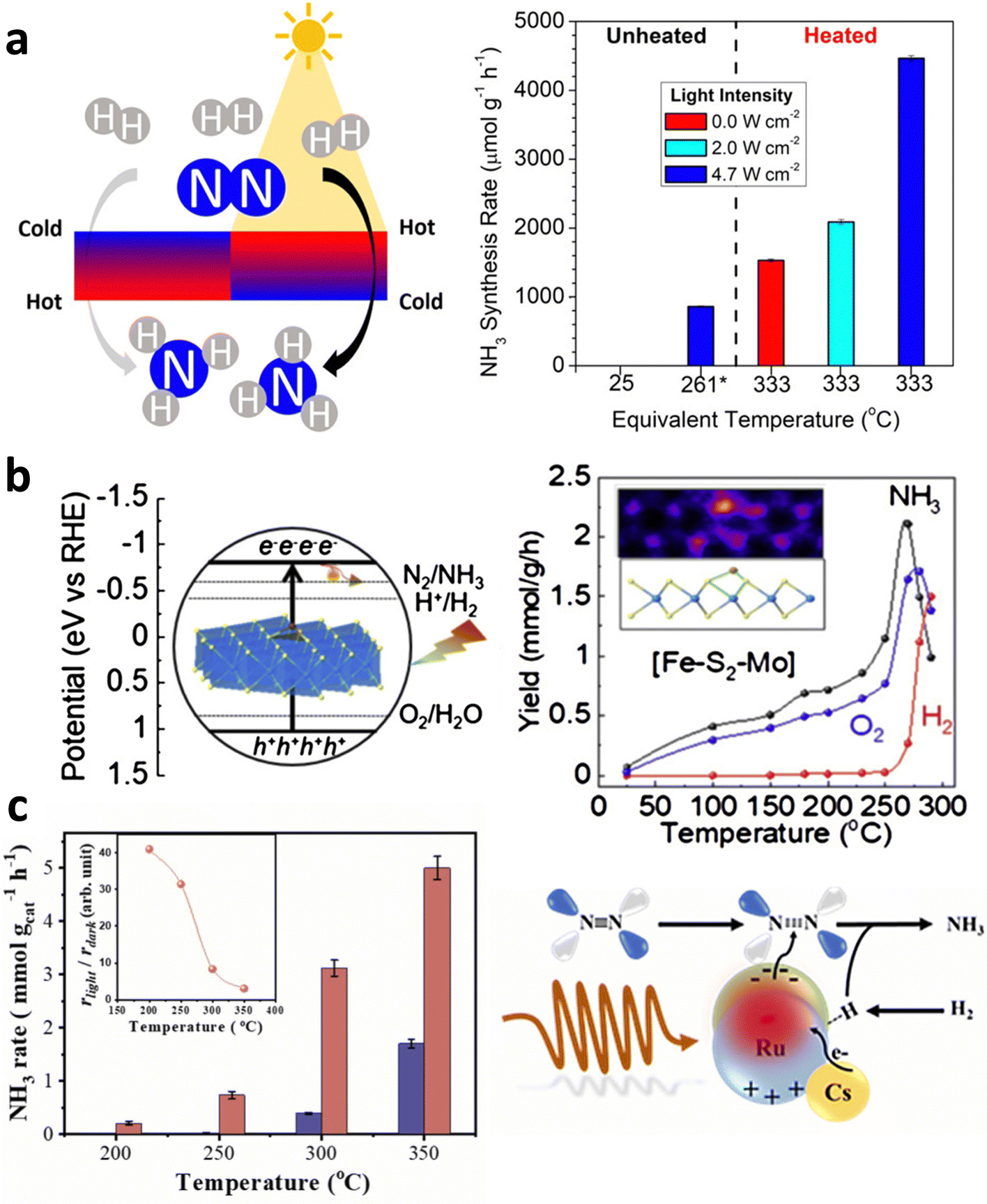 | ||
| Fig. 14 (a) Thermal gradients created and controlled by photothermal heating of Ru–Cs/MgO catalyst, and associated plasmonic photothermal NH3 production rates under dark and illuminated conditions (blue LED). Reprinted from ref. 307 Copyright 2019 ACS. (b) Schematic energy diagram of Fe–MoS2, and production rates of NH3, H2, and O2 as a function of temperature. Dark experiments are shown as empty symbols. Reprinted from ref. 309 Copyright 2021 Elsevier. (c) Temperature dependence of NH3 production rates with CsRu@ZrO2 under dark (blue) and light illumination (red). Inset: Performance enhancement under illumination. Reprinted from ref. 306 Copyright 2023 Elsevier. | ||
Zheng et al.309 prepared a Fe–MoS2 catalyst mimicking the FeMoCo structure by locating Fe on a single molecular layer of MoS2. These authors achieved an ammonia production rate of 2.1 mmol h−1 g−1 with water and UV illumination (70 W tungsten lamp), reaching a remarkable STA of 0.24% at 270 °C. They observed an increase in ammonia selectivity at 180 °C, whereas it dramatically decreased at temperatures over 250 °C due to the competition with the HER. Further, Fe–MoS2 was tested in a solar furnace to mimic the solar illumination, reaching reaction rates of 17 mmol h−1 g−1 at 6 bar (Fig. 14b).
In 2022, Wang et al.311 reported the photothermal hydrogenation of N2 over Ni/TiO2 catalysts, achieving a NH3 production rate of 0.2 mmol h−1 g−1 at 400 °C and under illumination with a 300 W solar simulator. The authors found that the oxygen vacancies on TiO2 photocatalytically activated N2 and trapped photoelectrons, while Ni atoms thermocatalytically dissociated H2 and hosted the holes, finally resulting in the photothermal N2 hydrogenation to ammonia. Recently, Peng et al.305 reported the hydrogenation of N2 to NH3 under visible-NIR illumination using Cs-decorated strontium titanate-supported Ru nanoparticles. They achieved 3345 μmol h−1 g−1 at 350 °C and near 1 Sun power illumination, with a slight decay of activity during a continuous 120 h reaction. They found that NIR illumination was more efficient than visible-light wavelengths to activate N2, due to a greater generation of hot electrons and local heating at Ru NPs. In contrast, UV light did not contribute to NH3 production. Besides, they inferred that the partially reduced Cs promoter transferred electrons to Ru sites, contributing to the formation of oxy-azide-related species that were sequentially hydrogenated to NH3. Very recently, these authors reported a N2 hydrogenation catalyst based on highly dispersed Cs-decorated Ru sub-nanometric clusters (ratio Ru/Cs = 6) supported on ZrO2 NPs (Fig. 14c).306 This material achieved 1.6 mmolNH3 h−1 g−1 in the dark (350 °C, 0.1 MPa), which increased up to 5.1 mmol h−1 g−1 under 1 sun power illumination with a remarkable extended stability for 100 h irradiation under continuous flow. They found the role of Cs species in increasing the basicity of the ZrO2 support, and donating electron density to the adjacent Ru sites that favored a pre-activation of the adsorbed N2. Also in the last year, Bian et al.308 published a strategy for determining the actual reaction temperature in photothermal catalysts, identifying and independently quantifying the contributions of hot electrons and local heating effects on photothermal catalysis on the basis of Le Chatelier's principle. They studied the photothermal ammonia synthesis over a carbon-supported Ru catalyst, in which they found that hot electrons generated by Ru reduced the activation energy of N2 fixation with respect to thermal catalysis (54.9 vs. 126.0 kJmol−1), and simultaneously protected the carbon support from methanation. This effect provided the catalyst with high stability during an operation time of 1000 h, while under thermal conditions the material deactivated before 150 h on stream. They estimated a hot electron contribution of 73.6% under their experimental conditions (350 °C, 5.3 W cm−2).
:N2 ratio of 70%:30% or a mixture of N2 and H2O, at a mildly elevated temperature (<450 °C).
In photothermal reactors, heat can be generated via (i) light irradiation, (ii) external heating or (iii) coupling both illumination and external heating. (i) In the absence of external heating, thermal energy is generated by the conversion of incident photons to heat (phonons) over the catalyst surface. In this case, heat generated from light can completely replace the external heating source or reduce the temperature required for the reaction.304 In general, the heating effect provided by a light source may vary from 110–160 °C depending on different factors, such as the type of light source, light intensity, or distance to the catalyst surface.313 (ii) In contrast, heat can be supplied externally by using electrical furnaces, heating ribbons and heating plates. External heating is usually employed either when the catalyst cannot generate enough heat by light absorption, or to provide a constant heating for the progression of the chemical reaction. However, in this case, NRR reaction may proceed mostly via thermal catalysis and therefore, the process should be referred as “photo-assisted”.313 Lastly, the outlet product stream is generally directed to acid traps (e.g. 0.05 M H2SO4, 0.1 M HCl) for subsequent analysis by colorimetry or ionic chromatography.
6. NRR analysis
6.1. Detection methods
Ammonia is a small, polar molecule with basic character (i.e. it accepts protons from water) and a high solubility in aqueous phase (482 g L−1 at 24 °C),22,314 compared to the gases that can be found in the reaction media (e.g. N2, O2 and H2), which are less soluble (specially H2) and easily determined by gas chromatography (GC).22 In contrast, ammonia quantification is usually performed in aqueous solution by different techniques. In liquid phase, ammonia can exist as ammonium ion (NH4+) or un-ionized ammonia (NH3) depending on the pH and the temperature of the reaction media. In general, the fraction of NH3 increases as the pH or temperature of the solution increases, and the equilibrium fully shifts to gas-phase ammonia for pH higher than 11 (eqn (2), Section 2).22,37 Therefore, alkaline aqueous solutions may contain gaseous ammonia both in the head space and in the liquid phase as dissolved gas. This complicates the storage of the samples for quantitative analysis, which is preferred to be done immediately after sampling.37 The current methods for the detection and quantification of ammonia include colorimetric assays, ion chromatography (IC), fluorescence, ion-selective electrodes, 15N-labeled N2, 1H nuclear magnetic resonance (1H NMR) spectroscopy, and Fourier-transform infrared spectroscopy.30,315At a glance, colorimetric assays are the most commonly employed methods due to their low cost and easy application.30 However, the method should be carefully chosen depending on the particular reaction conditions. For instance, Nessler's reagent method works well in both alkaline and acidic solutions, the indophenol blue method is suitable in alkaline media, whereas IC works best with acidic solutions. Besides, certain sacrificial agents are not compatible with the cation chromatography columns, which can limit the application of IC.31 Experimental data also reveal that Nessler's reagent method and IC are preferred for aqueous ammonia quantification over a wide concentration range (0–8 mg L−1), whereas the indophenol blue method may overestimate the ammonia concentrations above 500 μg L−1.316 Further details for each method are given in the following subsections.
6.1.1.1. Nessler's reagent method. Nessler's reagent consists of mercury(II) iodide and potassium iodide (K2HgI4) and an alkaline solution of sodium hydroxide (NaOH) or potassium hydroxide (KOH). Iodide and mercury ions react with ammonia under alkaline conditions to produce a reddish-brown complex, which exhibits strong absorbance at 420 nm (eqn (8)).
| 2[HgI4]2− + NH3 + 3OH− → Hg2ONH2I↓ + 7I− + 2H2O | (8) |
This absorbance is directly proportional to the NH4+ concentration in the absence of interferants, such as metal cations (except sodium and potassium), hydrazine, carbonyl compounds, etc.31,316 The high alkalinity of the Nessler's reagent can cause the precipitation of metal hydroxides, creating turbidity that interferes with colorimetric analyses. Zinc sulfate and sodium hydroxide solutions can be added to clarify turbid samples.22 Besides, Rochelle salt (KNaC4H4O6 × 4H2O) is often added during the analysis to minimize possible interferences from other ions (Fe3+, Co2+, Ni2+, Cr3+, Ag+, S2−, etc.) in solution and to prevent the formation of cloudy samples.31,316 Ammonia is quantified colorimetrically at wavelengths between 380 and 550 nm.
It should be noted that mercury ions in Nessler's reagent are toxic and thus the reagent should be handled and disposed carefully; Nessler's reagent solution must be prepared using ultrapure water; the lifetime of Nessler's reagent is relatively short (around three weeks); the quantification of NH3 should be performed with a reaction time from 10 to 30 min to avoid degradation and erroneous results.316
6.1.1.2. Indophenol blue method. The indophenol blue method is based on the Berthelot reaction (eqn (13)), and involves the reaction of ammonia with phenol and hypochlorite under alkaline conditions to generate a blue-colored indophenol product. Blue colored indophenol is formed from the reaction of ammonia with hypochlorite and phenol in an alkaline medium, following consecutive steps. First, ammonia and hypochlorite react at pH 9.7–11.5 to give monochloramine, which then reacts with phenol to give quinone chloramine that further reacts with phenol to form yellow indophenol. Then, the indophenol dissociates in an alkaline medium to give the blue color (eqn (9)). Sodium nitroprusside is used as a catalyst to intensify the color change in indophenol reaction, and citrate buffer is used to stabilize the pH of the reaction solution and avoid interferences with magnesium and calcium precipitates. Indophenol can be quantitatively determined by colorimetry between 630 and 650 nm.22,31,316
 | (9) |
This method is suitable for the detection of low-concentration ammonia ranging from 0–0.6 mg NH3–N L−1, and slightly interfered by other species such as organic nitrogen compounds, nitrites or nitrates. Its major drawback is the time-consuming sample preparation.22,37,317
6.1.1.3. Salicylate method. This is a modification of the indophenol blue method in which phenol is substituted for sodium salicylate to prevent the formation of o-chlorophenol. Therefore, this method is much safer and more stable than the indophenol blue method. However, it is less sensitive, needing much higher concentration of salicylate for ammonia detection, and reagents are more expensive. In this method ammonia and hypochlorite react to give monochloramine, which forms 5-aminosalicylate with salicylate (eqn (10)). This method gives colors changing from light yellow (excess reagent) to green and then to blue with increasing ammonia concentration. Colorimetric quantification is performed at 640 nm.22 Some metal ions (e.g. Cu2+) can interfere with the analysis, although it can be solved using masking agents.116
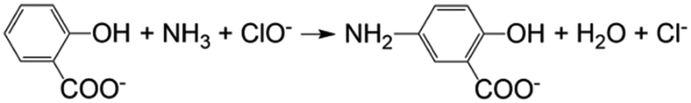 | (10) |
Ammonia can also be detected by fluorescence. This method is based on the reaction of NH3 with o-phthaldialdehyde and sulfite, which produces a strongly fluorescent compound that exhibits maximum excitation and emission wavelengths at 362.5 and 423.0 nm, respectively. This fluorescence is then correlated to the concentration of ammonia in sample solutions, reaching a detection limit up to 1 nmol L−1. Measurement interferences can arise from amines and amino acids in the sample solution.22 On the other hand, in situ FTIR provides useful information about changes in the IR bands during the NRR, such as –H–N–H bending, –NH2 wagging and N–N stretching of adsorbed N2Hy species.22,116
Apart from ammonia, we also note that the product distribution in N2 fixation can include oxidation products such as NOx, most frequently nitrates, or partial hydrogenated intermediates such as diazene (N2H2) and hydrazine (N2H4). Diazene is a highly reactive molecule that has never been spectroscopically detected in solution due to its rapid conversion to N2H4 or disproportionation to N2.22 In contrast, hydrazine is a common by-product that can be determined by the method invented by Watt and Chrisp.320 Briefly, a mixture of para-(dimethylamino)benzaldehyde, dilute hydrochloric acid and ethanol is used as colour reagent in solutions containing hydrazine, which can be spectrophotometrically detected at 455 nm.
In the case of the analysis of oxidation products, NOx (NO2− and NO3−) can be determined by gas or ionic chromatography, although they are rarely reported as by-products in N2 fixation.30,321,322 Additional NOx detection methods include the reduction of nitrate by the cadmium reduction method, which is based on the heterogeneous reduction of NO3− with a copperized cadmium column followed by the spectrophotometric detection of NO2− formed.323 Besides, the formation of nitrites can be analyzed by the Griess assay324 in which nitrite reacts with sufanilic acid to produce a red–violet colored azo dye (λmax ≈ 540 nm). This is a simple and effective technique for the detection of nitrites in several matrices for concentrations in the range of 0.02 and 2 μM.325 An additionally recent spectrophotometric method for nitrite detection is based on the reduction of nitrates with UV lamps, which avoids the use of cadmium.326
6.2. Performance evaluation
The activity of NRR catalysts can be determined by means of: (i) production rate; (ii) catalyst stability; and (iii) performance metrics (e.g. apparent quantum yield (AQY), faradaic efficiency, solar to ammonia yield).The ammonia production rate represents the amount of ammonia (mol of NH3 or NH4+) produced per unit mass/area of catalyst per unit time, and it is intended to monitor the effectiveness of the catalysts. It can be expressed as μmol h−1 g−1 for gas- and liquid-phase reactions, μM h−1 for liquid-phase reactions, and μmol h−1 cm−2 for (photo)electrochemical cells.
The apparent quantum yield (AQY) or apparent quantum efficiency (AQE) represents the ratio between the number of electrons participating in the NRR to the total number of incident photons (eqn (11)).
 | (11) |
The AQY/AQE should be adopted as a generalized reported parameter to allow the comparison of data acquired under different experimental conditions, such as light source, catalyst loading, reaction time, and illumination area. In general, most materials present low photons to ammonia conversion efficiencies, showing AQY lower than 2.5%,12 except for a KOH-treated carbon nitride with an outstanding AQE of 21.5%.225
The faradaic efficiency (ηF, %) measures the selectivity of the (photo)electrochemical NH3 production, and refers to the ratio of the current used for N2 reduction to the total current passed through the circuit (eqn (12)).
| ηF (%) = (xF × n × V)/(M × Q) | (12) |
485 C mol−1); n is the measured concentration of NH3 or NH4+ (μg mL−1); V is the volume of the electrolyte (mL); M is the relative molecular mass of NH3 (M = 17 g mol−1); and Q is the total charge passed through the electrodes.22,327
The solar-to-ammonia (STA) conversion efficiency is calculated from eqn (13):
 | (13) |
STA efficiencies higher than 0.1% have been reported for graphitic carbon nitride catalysts328 and ZnCuInS–BiOI heterojunctions232 without the use of sacrificial agents, while higher STA of 0.24% has been achieved using a photothermal catalyst (Fe–MoS2) working at 270 °C.309 Substantial room for improvement exists in order to progress in NRR, which efficiencies are still one order of magnitude lower than that of the photocatalytic water splitting.12 In this regard, estimations foresee that STA higher than 0.1% need to be reached to compete with Haber–Bosch process for the production of ammonia as a nitrogen fertilizer, whereas STA higher than 20% would be needed for its potential application as a fuel.30
The STA efficiency of the (photo)electrochemical processes is not directly measured but can be estimated based on the electrical energy conversion efficiency329 (eqn (14)):
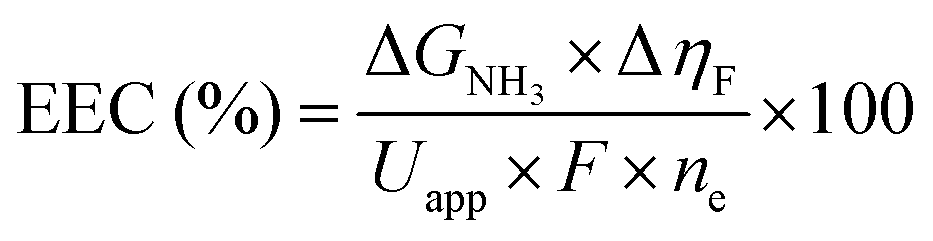 | (14) |
6.3. Control experiments and interferences
For the evaluation of the NRR performance, one should bear in mind that previous quantification methods are prone to interferences and false-positives at low ammonia yields, which is the case of most reported photoactivities (ranging from nano- to micromolar concentrations). On the one hand, colorimetric methods provide in theory reliable results for simple solutions of ammonia in water.316 However, certain reaction conditions (e.g. changes in solution pH) or the presence of interferants (e.g. scavengers, N-containing electrolytes and surface capping agents, solvents, impurity of water, N-ligands) may overestimate the concentration of ammonia due to the formation of colored complexes in the spectrophotometric assays.30,316 In particular, Nessler's reagent method may lead to false-positives in aqueous solutions containing organic scavengers (e.g. methanol, ethanol, isopropanol, triethanolamine, etc.) due to the formation of oxidized carbonyl compounds (formaldehyde, acetaldehyde, and acetone) that severely affect the color development. The presence of some metal ions (Fe2+, Ni2+, Ru3+, In3+) in acidic solutions can also interfere with ammonia detection due to the formation of colored complexes. Additional interferences may arise from nanoparticulated precipitates formed in old Nessler solutions.Further inconsistencies may arise when using certain capping agents (e.g. oleyl amine, thioacetamide, hexamethylenetetramine) because they easily decompose into ammonia and other products under light irradiation. Nafion, frequently used in N2 (photo)electroreduction experiments (as separating membrane or electrolyte), may accumulate and release NH4+ through ion exchange with acid groups. N-containing photo(electro)catalysts are also susceptible to decomposition or contain potential ammonia or amine surface groups (e.g. NHx species on the surface of g-C3N4) that may lead to inaccurate ammonia yields. Accordingly, a thorough washing step with ultrapure water is critical to guarantee the removal of ammonia or amine groups adsorbed on the catalyst surface.31 In this regard, control experiments using isotope-labelled 15N2 are essential for N-containing materials, in order to verify that ammonia yields originate from gaseous N2.31,330 However, we should note that isotope-labelled 15N2 gas usually contains ammonia and 15NOx impurities that may lead to experimental artefacts at low ammonia yields.315 These impurities are generated as by-products during the fabrication of 15N2via the CuO-catalyzed oxidation of 15NH3, and can reach 15NOx concentrations as high as 0.1 mol% with respect to 15N2.331 Andersen et al.330 estimated that bubbling 15N2 gas with a purity of 99.13% for 28 h at a rate of 10 mL min−1 would potentially generate 3600 μg h−1 of N-containing impurities, thus overestimating the final ammonia yield. This adventitious contamination in the gas stream can be trapped using acidic aqueous solutions or appropriate adsorbents (see good practices in Section 6.3.1).315 Additional interferences may arise during the sample preparation and handling due to the ubiquitous character of ammonia. Indeed, NH3 is highly soluble in water and easily adsorbs on a wide range of surfaces, including common laboratory materials (e.g. glassware).314 This can potentially overestimate the quantification and compromise a reproducible analysis. Another source of contamination may come from ambient air, which contains a non-negligible amount of ammonia (ranging from 0.05–250 ppm), which can affect unclosed reaction systems.315 In fact, gaseous ammonia has a short ambient lifetime of 12 h to 5 days because of its rapid deposition on surfaces, whereas aerosolized ammonium can last in the atmosphere up to 5–10 days. Therefore, gaseous ammonia always provides a background level of contamination either from gas phase dissolution in open reaction systems or solid surface desorption.314 An additional contamination may originate from the electrolytes in (photo)electrochemical experiments. Commercial electrolytes such as lithium salts (e.g. Li2CO3, Li2SO4, LiClO4) generally contain trace amounts of NO3− and NO2− that can lead to false positives.38 As a general rule, inorganic salts should be subjected to a high-temperature annealing (e.g. 800 °C for 4 h under Ar atmosphere332) before use to eliminate possible NOx impurities.
On the other hand, N-impurities on the catalyst surface (NOx, NH4+) can be removed by pre-reduction of the materials before experiments.103 The reaction system should be cleaned between uses by boiling in ultra-pure water and subsequent drying in oven.103 The use of catalysts precursors that contain nitrogen (e.g. urea, amino compounds or nitrate/nitrite salts) usually lead to residual N contents that can lead to the formation of ammonia under illumination or reductive conditions.334 Some active catalysts, especially those based in amino and nitride groups, present nitrogen in its composition, making necessary to determine the nitrogen balance, by comparing the N produced in reaction and the N content from the catalyst (e.g. by elemental analysis). A especial care should be taken with some synthesis methods that can promote the incorporation of N species on the catalyst surface,335 and which can react giving false positives.
Then, catalytic experiments should start with background measurements of NH3/NH4+ and NOx in control tests. For instance, experiments under inert atmosphere (e.g. Argon/Helium) should be carried out under the same experimental conditions as the NRR tests, in order to quantify any possible amounts of adventitious ammonia or NOx coming from the catalyst surface or the reaction system. Photoelectrocatalytic experiments would require control tests under argon, with and without potential applied, and control tests under N2 at open circuit potential over the same duration. In all cases, ammonia yields should be considered reliable if substantially exceed those obtained in control tests.315,330,331
Detection of N-contamination should be addressed with improved gas-scrubbing or through appropriate catalyst pre-treatment. Ammonia production should be confirmed by more than one quantification technique, always including precise detection methods such as IC or 1H NMR. In this regard, a recent comparative study315 on ammonia quantification methodologies found that almost all testing methods showed poor reproducible results for ammonia concentrations below 0.2 ppm. Indeed, they found detection errors of 20.5% with Nessler's reagent method and 71.2% with IC for the analysis of a standard NH3 solution (0.1 ppm), thus raising strong concerns about the reliable quantification of low ammonia concentrations. Therefore, the use of 15N isotopically labeled control experiments is highly recommended to verify the origin of products and exclude false-positives. The amount of 15NH3 produced in the experiments should agree with the amount of 14NH3 produced in the control tests with 14N2 under equivalent conditions, keeping a 1:1 ratio.330 Moreover, the formation of 15NH3 can be further cross-checked by using a combination of 15N2 and 1H NMR experiments. However, we note that this approach requires a complete protonation of ammonia in order to observe a clear ammonium triplet for 14N2 and doublet for 15N2, which is not always well controlled. Besides, one should bear in mind that ammonium protons rapidly exchange in reaction solution, and consequently, 1H NMR measurements can suffer from interferences if the deuterated solvent contains any labile deuterium, thus leading to uncontrolled levels of hydrogen/deuterium exchange. For that reason, the use of more stable solvents such as DMSO-d6 and CDCl3 is preferred.331
6.4. Proposed standardization of experimental devices and parameters
NRR field is growing rapidly, as reflected by the increasing number of publications of the different NRR technologies every year (Fig. 1). However, reported ammonia productions are strongly affected by false positives and experimental artifacts that overestimate the catalyst performance. The reason behind these issues is the lack of standards for benchmarking performance. A general agreement on standardized protocols and performance metrics would greatly facilitate the comparison of reported data, essential to progress on the development of this technology. In fact, the creation of a global network of research institutions sharing NRR testing could facilitate the development of meaningful techno-economic life cycle analysis (LCA) and socio-economic studies, which are crucial for profitability analyses. Besides, considering the growing rate of this research field, the prompt adoption of a common set of operating conditions would avoid to reach a high volume of overestimated and non-comparable data, as happened before for CO2 reduction reaction.336 Reference NRR experiments could be performed using the agreed set of operation conditions and commercial catalysts for a reliable global comparison. These experiments also call for benchmark reactors, due to the common use of home-made lab-scale reactors that limit a rapid and accurate comparison of experimental results. Finally, reported data should always include efficiency metrics (such as AQY, STA efficiency and/or faradaic efficiency) to guarantee reproducibility and to pursue good practices for a real development of the technology. Standardization of experimental procedures and performance metrics could be in line with the successful qualification test protocols for photovoltaics pioneered at the National Renewable Energy Laboratory (NREL). This would clearly impact on a high data reliability for technology transfer, hopefully paving the way to the development of NRR demonstrators, pilot plants and ultimately solar ammonia refineries. Fig. 15 depicts a flow chart protocol with the abovementioned suggested experimental practices.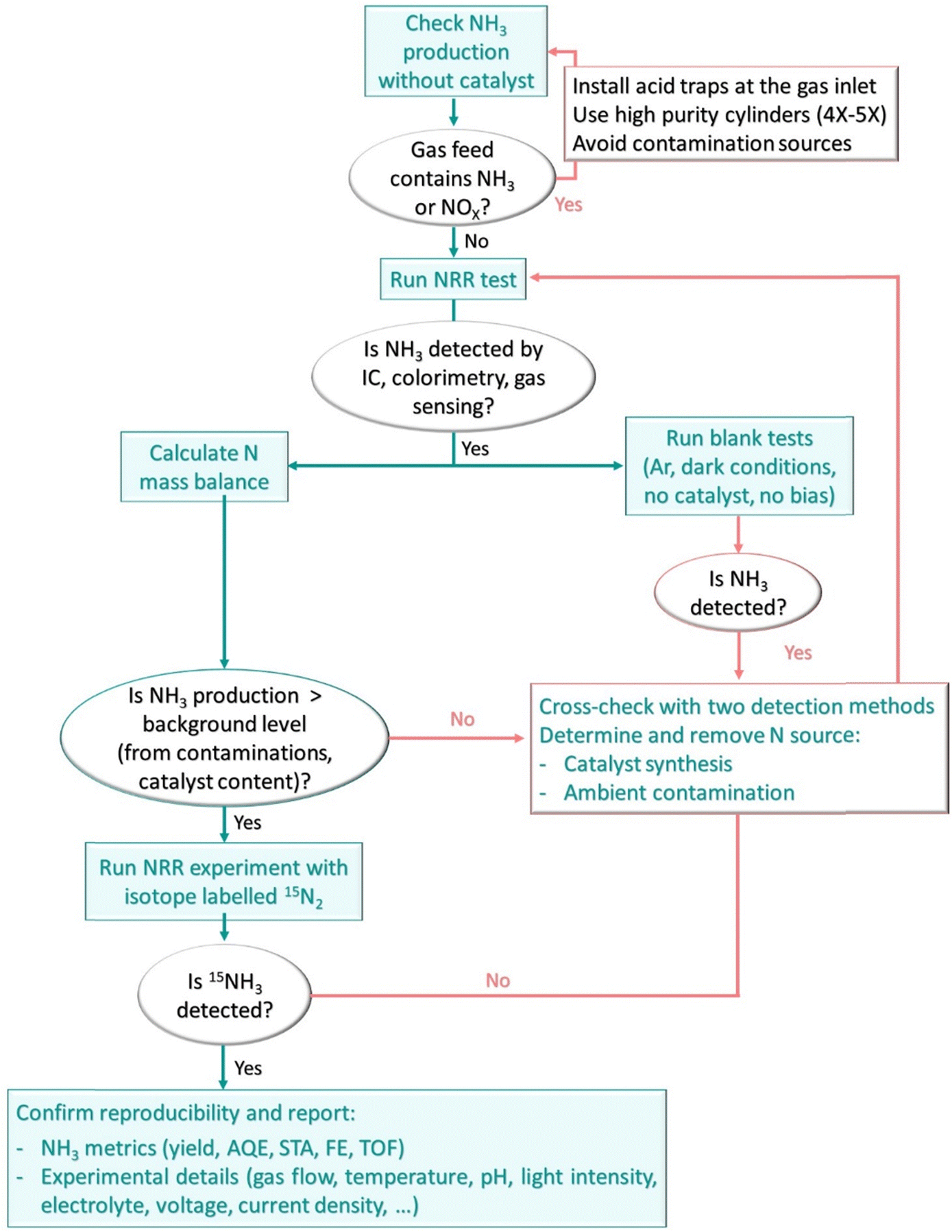 | ||
| Fig. 15 Suggested protocol for benchmarking light-driven NRR experiments. | ||
7. Uses of ammonia and future opportunities
7.1. Versatile properties of ammonia for wide-range applications
Ammonia is a colourless gas with a sharp and penetrating odour. The NH3 molecule has a trigonal pyramidal shape with three hydrogen atoms and an unshared pair of electrons attached to the nitrogen atom. It is a polar molecule with strong intermolecular hydrogen bonding. Its boiling point is 239.8 K, its freezing point 195.5 K and its density 0.73 kg m−3. Ammonia has an auto-ignition temperature of 930 K (compared to methane, 859 K) under atmospheric conditions, an octane number of ∼130 and a high heat of vaporization (1371 kJ kg−1 compared to ∼271 kJ kg−1 of gasoline). The combustion of ammonia is challenging due to its low reactivity, although it can release energy yielding only water and nitrogen as by-products (4NH3 + 3O2 → 2N2 + 6H2O + heat), with a stoichiometric air fuel ratio (AFR) of 6.06 by weight.18 Due to its low reactivity, the hazards derived from accidental combustion or explosions are low compared to other fuels. In this regard, anhydrous ammonia is non-flammable while ammonia vapour in air is flammable and may explode when ignited. In the event of leaks, ammonia is very soluble in water and may be corrosive due to a rapid increase in pH to 11.6. Besides, ammonia spills generally form a dense aerosol cloud after evaporation, which rapidly dilutes since it is lighter than air. Dry, warm and windy weather diffuses ammonia to the atmosphere faster than humid, cold and low wind conditions.18 For that reason, guidelines recommend to include a refuge point upwind from the point of storage/use of ammonia in large scale facilities.18Regarding health hazards, ammonia is toxic for humans and most invertebrates. The degree of toxicity mainly depends on the concentration and the time of exposure. However, it is not carcinogenic and can be easily detected by odour at very low concentrations (5–25 ppm). At low concentrations in air (50 ppm to 100 ppm), ammonia may irritate tissue surfaces such as eyes, respiratory system and skin, while the inhalation of higher concentrations rapidly produces suffocation and may cause respiratory burn injuries or even death. Human exposure limits range between 25 and 50 ppm, with severe health consequences for concentrations above 300 ppm.18,337 Main properties are summarized below in Table 4.
| a Compared to 859 K for methane under atmospheric conditions. b Compared to ∼271 kJ kg−1 for gasoline. c Compared to 81 for water at 298.2 K. d Compared to 120 MJ kg−1 for hydrogen. | |
|---|---|
| CAS number | 7664-41-7 |
| Physical state | Liquid |
| Color | Colorless |
| Odor threshold | 5–25 ppm |
| Molar weight | 17.03 g mol−1 |
| pKa | 9.25 |
| Molecular structure | Symmetrical pyramid, N at apex |
|
|
| H–N–H bond angle | 106.67° |
| N–H length | 1.1024 Å |
| Dipole moment | 1.47 debye |
| Boiling point | 239.8 K |
| Melting point | 195 K |
| Freezing point | 195.5 K |
| Critical temperature | 406.4 K |
| Auto-ignition temperature | 930 Ka |
| Density | 0.73 kg m−3 |
| Vapor pressure | 10 atm (at 298.7 K) |
| Thermal conductivity (gas) | 2.45 × 10−4 J cm−1 s−1 K−1 (at 299.7 K) |
| Refractive index (liq) | 1.325 (at 289.5 K) |
| Dielectric constant (liq) | 16.9 (at 298 K) |
| Octane number | ∼130 |
| Heat of vaporization (at 1 bar) | 1.37 MJ kgb |
| Latent heat of evaporation | 1.37 kJ g−1 |
| Dielectric constant | 22 (at 239.2 K)c |
| Ionization potential | 10.2 eV |
| Energy content | 18.8 MJ kg−1 (LHV)d |
| Ideal gas properties (298 K, 1 atm) | |
| Specific heat (Cp) | 35.65 J K−1 mol−1 |
| Standard entropy | 192.77 J K−1 mol−1 |
| Standard enthalpy of formation | −45.89 kJ mol−1 |
| Free energy of formation | −16.37 kJ mol−1 |
| Equilibrium constant (logKf) |
2.87 |
| Standard enthalpy of formation (gas at 298 K) | −46.22 kJ mol−1 |
| Standard entropy of evaporation | 87.75 J K−1 mol−1 |
| Standard entropy of fusion | 28.93 J K−1 mol−1 |
| Latent heat of fusion | 0.33 kJ g−1 |
| Heat of solution in water | 2.18 kJ gNH3(gas)−1 |
| Solubility in water | 51–53 gNH3(gas)/100 mL |
| Flammability limits of ammonia in air | 16–25% |
| Flammability limits of ammonia in oxygen | 15–79% |
| Auto ignition temperature | 924 K |
| Hazard statements | H221-Flammable gas |
| H280-Contains gas under pressure; may explode if heated | |
| H314-Causes severe skin burns | |
| H318-Serious eye damage irritation | |
| H331-Toxic if inhaled | |
| H400, H411-Very toxic to the aquatic environment with long lasting effects | |
7.2. Current and potential end-uses of renewable ammonia
Nowadays, around 85% of the overall ammonia production is destined to the fertilizer industry (i.e. urea, ammonium nitrate, ammonium sulphate, diammonium phosphate (DAP) or dry ammonia). Around 15% production is used as synthesis platform of multitude of everyday products (chemicals, plastics, textiles, pharmaceuticals, refrigerants, abatement of nitrogen oxides (NOx), etc.). For instance, the textile industry uses ammonia for softening cotton and for the production of synthetic fibers (such as nylon and rayon). Ammonia is also used for the production of explosives, such as ammonium nitrate (NH4NO3) or antibacterial drugs.339 Its unique properties and wide application (Fig. 16) explain why it is considered one of the seven basic chemicals, alongside ethylene, propylene, methanol and BTX aromatics (benzene, toluene and xylene).27 More recently, ammonia produced from renewables (<1%) has risen attention as an attractive candidate to replace fossil fuels in relevant sectors such as electric power generation and maritime transport, as well as for the storage and delivery of hydrogen in the move towards a low-carbon economy. The key advantage of NH3 is that it contains 40% more hydrogen (17.75 wt%) than methanol (12.6 wt%), and can be produced from renewable hydrogen and nitrogen from air, without any carbon-species for the synthesis.340 Thus, ammonia has the potential to be a major contributor to the clean energy transition in the context of “Power-to-X” technologies, in which sustainable fuels are synthesized from excess power. For instance, in countries with excess power from non-intermittent low-carbon energy sources (e.g. nuclear), ammonia could provide stable exports of energy to other markets; while in countries with intermittent energy resources (e.g. renewables), ammonia could store the excess generation, dealing with the seasonal variability and balancing the energy system.339 Besides, ammonia could offer decentralized sources of green energy to remote locations with difficult access to energy supply. Below are listed the most promising applications of this carbon-free energy vector.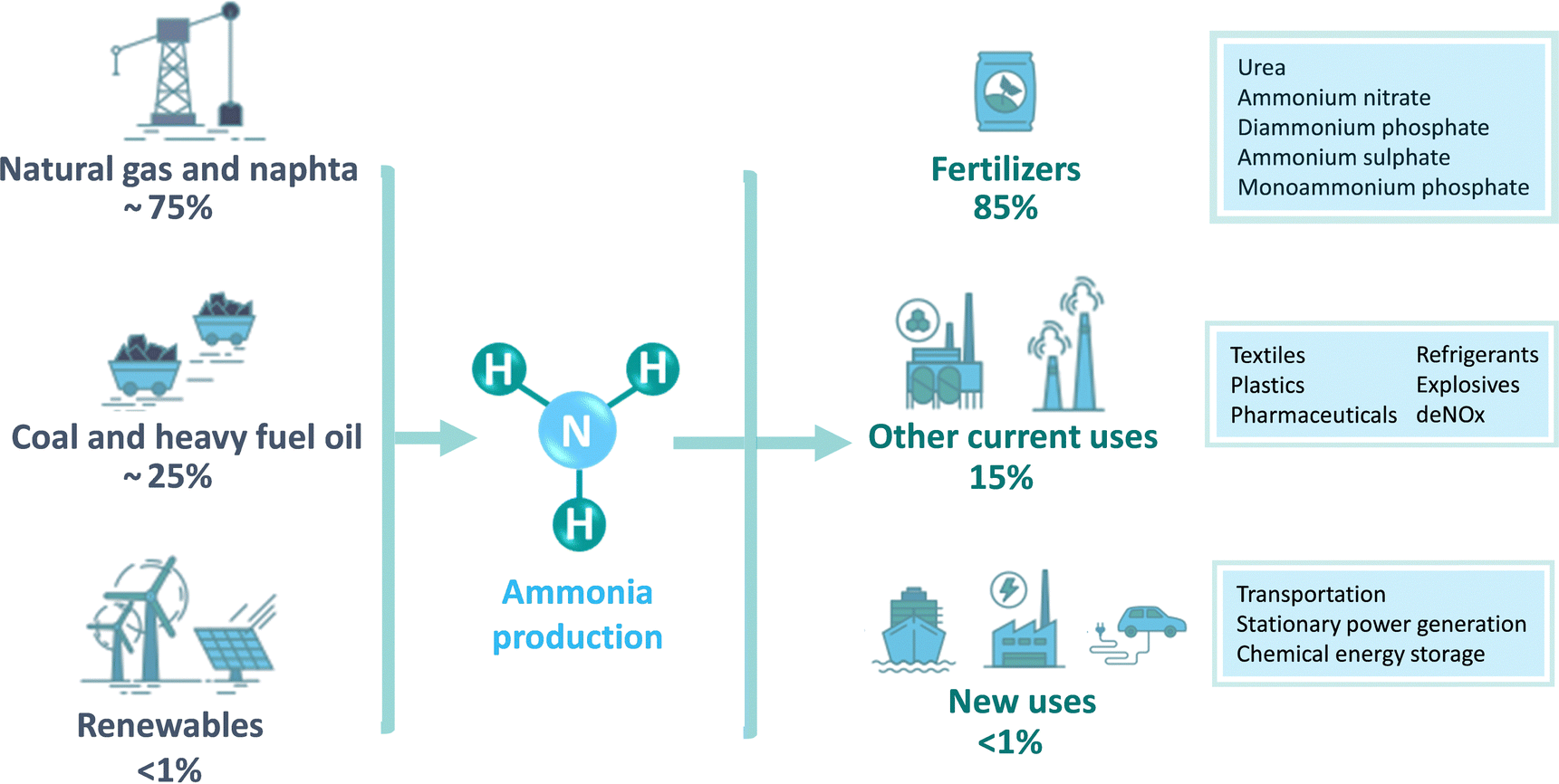 | ||
| Fig. 16 Current and potential uses of ammonia. Adapted from ref. 27. | ||
| Technology | Energy density (W h L−1) | Maximum storage capacity (GW h) | Maximum storage duration | Advantages | Disadvantages | ||
|---|---|---|---|---|---|---|---|
| Electrical | Supercapacitors | 2–10 | 0.0001 | Seconds–minutes | Easy storage and distribution high round-trip efficiency (90–94%) | Low energy density | |
| Low storage capacity | |||||||
| High costs per installed density | |||||||
| Electrochemical | Li-ion batteries | 200–350 | 0.5 (for stationary module systems) | Hours–days | Medium energy density high efficiency (95%) long lifetime transportable | High cost | |
| Complex management systems | |||||||
| Mechanical | Pumped hydro | 0.3–1.5 | 3 (potentially 14) | Days–months | Long storage time | Low energy density | |
| Medium efficiency (75–82%) | Limited storage (constrained by geography) and distribution large-scale units for economically feasible applications | ||||||
| Chemical | Natural gas | 10.1 6200 (LNG) | 1260 (underground caverns) | Weeks–months | High energy density | Expensive electrolysers | |
| Hydrogen | 2400 (liquid) | 0.13 (tanks) | Large storage capacity | Low efficiency | |||
| 124–167 (underground caverns) | Long storage time | Natural gas (30–35%) | |||||
| Ammonia | Steam reforming | 3194–4325 (liquid) | 0.25 (cooling tanks) | Low energy installation costs | Hydrogen (30–60%) | ||
| 300 (underground caverns) | Ammonia-steam reforming (20–25%) | ||||||
| Ammonia-electrolysis (40–72%) | |||||||
| High power installation costs | |||||||
The current technology for maritime propulsion consists on the two-stroke engine, which could be adapted to use ammonia as a fuel, with an energy efficiency of ca. 45–50% on a lower heating value (LHV) basis.27 MAN Energy Solutions (Research Center Copenhagen) is currently thoroughly testing a two-stroke engine for large-scale container ships, operating on 100% ammonia fuel by 2025.343 Moreover, the technology group Wärtsilä is leading a powerful consortium of shipping stakeholders to develop demonstrators for two-stroke and 10 MW four-stroke marine engines running on ammonia fuel.344 Four-stroke engines for marine applications are believed to be able to reach energy efficiencies around 50% on LHV basis.27 Further developments for maritime propulsion are also focused on ammonia-fed solid oxide fuel cells (SOFC) with potentially higher energy efficiency (ca. 55–60% on LHV basis).27 A few examples of ammonia-fuelled vehicles have been achieved so far, such as the implementation of an ammonia-fed engine in the Toyota GT86-R Marangoni sports car led by Bigas International;345 or the development of an ammonia-gasoline dual fuel prototype vehicle in the South Korean Institute for Energy Research (KIER), which uses a fuel ratio of 70% ammonia to 30% gasoline to power a spark ignition engine.345 The application of ammonia in internal combustion engines greatly benefits from the absence of SOx or particulate emissions, and their limited N2O/NOx emissions in comparison to heavy oils.
7.3. Decarbonization scenarios
Fossil-based ammonia production routes emit around 2.0 tCO2 tNH3−1 on average. About two-third of the CO2 emissions come from the generation of hydrogen through hydrocarbon reforming, while a third originates from fuel combustion for ammonia synthesis (about 7.2–9.0 GJ tNH3−1).23 The significant carbon footprint of conventional routes has triggered researchers to look for an urgent decarbonization of the process, based on three key criteria: energy efficiency, scalability/modularity, and CO2-free emissions.314 As a result, three possible scenarios with five overlapping technology generations have emerged, in which synthetic ammonia is classified by colors depending on the production route as: brown, grey, blue, turquoise or green ammonia (Fig. 17).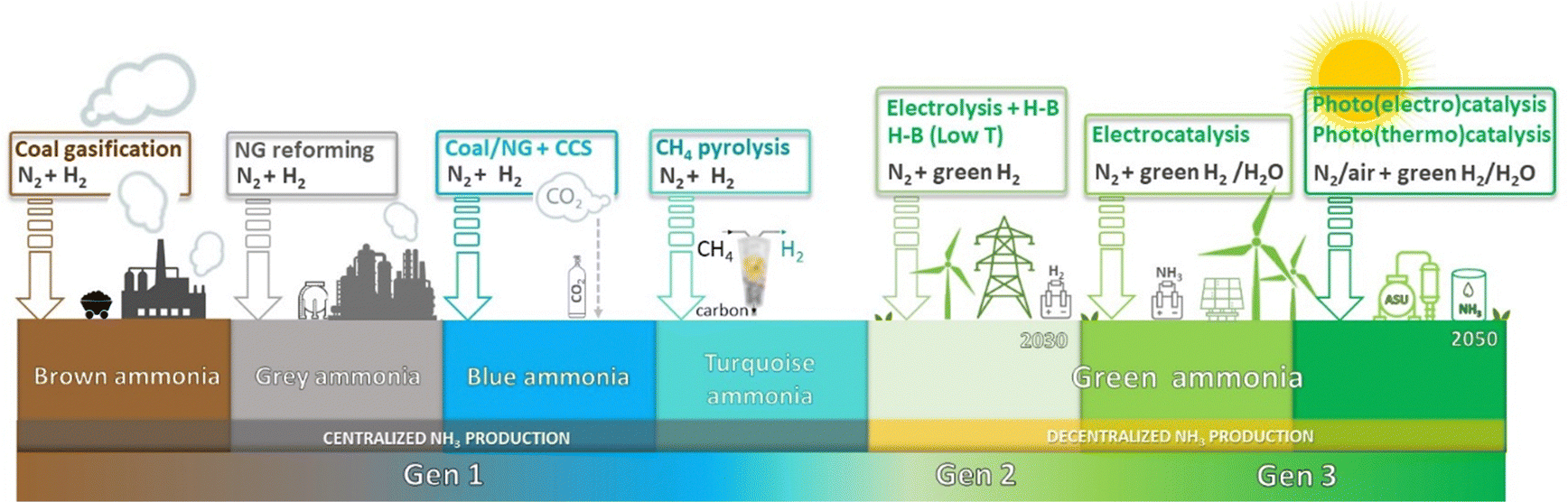 | ||
| Fig. 17 Classification of synthetic ammonia scenarios based on the energy source and production technology. | ||
Ammonia produced from fossil fuels is labelled as brown when hydrogen supply comes from coal gasification, or grey when it comes from natural gas reforming. Both categories correspond to conventional synthetic routes and emit between 2.5–3.8 tCO2 tNH3−1 and 1.6 tCO2 tNH3−1 for brown and grey ammonia, respectively.24,207,349,350 Blue ammonia uses the same feedstock as brown and grey ammonia, but includes CO2 sequestration, while turquoise ammonia uses pyrolysis to convert methane into pure carbon and hydrogen, which is reacted with nitrogen to make NH3. This first approach, also known as Generation 1 (Gen 1) scenario, would avoid more than 90% CO2 emissions generated by conventional routes, and would even bring the net carbon impact of the ammonia production to zero by supplying green hydrogen to the H–B process. Generation 2 (Gen 2) refers to ammonia produced in a modified small-scale H–B process using renewable sourced hydrogen from water electrolysis; and finally, Generation 3 (Gen 3) refers to the production of green ammonia by alternative synthetic routes using renewable electricity (electrochemical) or sunlight (photochemical), without the participation of the H–B process.350,351 The term “green” implies that the hydrogen is generated from water electrolysis without releasing carbon emissions on combustion and the nitrogen is extracted from air through an air separation unit, using renewable electricity (e.g. biomass, solar, wind, hydro, geothermal), meanwhile the reaction is driven at ambient temperature and pressure.24,207,349,350
Synthetic ammonia based on the previous approaches is currently going from pure academic research to pilot-scale plants. Some of the most relevant advances include: shipments of blue ammonia (40 tonnes) for power generation from Saudi Arabia to Japan in 2020, where ammonia imports for power generation are expected to reach 0.5–1 Mt year−1 in 2025, 3–5 Mt year−1 in 2030 and 30 Mt year−1 in 2050;27 industrial developments to produce green ammonia via water electrolysis (Proton Ventures, Morris, Hy2gen, ACME group, NFUEL®, Fertiberia, Stamicarbon, Topsoe, Yara, BASF, Kapsom, Casale, Thyssenkrupp, Siemens, etc.);26,27 Pilot plant testing of ammonia fueled SOFCs (1 kW class power) by IHI cooperation;207 A feasibility study on a 900 MW hydro-based renewable power plant (Sarawak, Malaysia) conducted by South Korea's Samsung Engineering for the production of green hydrogen (7000 t year−1) and green/blue ammonia (>600000 t year−1), amongst others, expected to end before 2027;352 another pre-feasibility study of a hydrogen project for blue ammonia production (1 million t year−1) in the Northern Territory (Australia), completed in 2022;353 as well as near future blue/green ammonia plants, which include a 20 million m3-per-day blue hydrogen and ammonia production plant in Louisiana (operations expected to start in 2026),354 and a green ammonia production facility (100000–200000 t year−1) in Jordan, which will include a 530 MW solar farm, an energy storage facility, electrolyzers of unspecified capacity, and a water desalination plant.355
All these initiatives highlight the unique opportunities offered by ammonia to achieve a global zero carbon transition, within the concept of Power-to-X (P2X) technologies, based on long-term renewable energy storage into carbon-neutral synthetic fuels, which can then be used in other sectors or stored until needed.
7.4. Recent advances towards a solar ammonia refinery
Current decarbonization plans are putting the focus on the development of green ammonia production technologies at sufficient industrial-scale for the new green economic model. The possibility of producing ammonia from just N2/air, H2O and sunlight is a great challenge for our solar fuels’ community. On this pathway towards a solar ammonia refinery, there are currently two main lines of thinking: (i) the one that supports the storage of renewable electricity as chemical energy only when there is an excess of electricity that otherwise would be wasted; and (ii) the one that believes on a future solar-to-chemicals (S2C) industry, even if it entails long-term research and developments.16 In the second approach, Centi and Perathoner recently suggested the possibility of fabricating artificial tree-like devices to produce fuels and chemicals from N2, CO2 and H2O at the consumer level, which would act as small solar fuels industries (Fig. 18).356 | ||
| Fig. 18 Schematic representation of an artificial-tree for the local generation of solar fuels and chemicals. Reproduced with permission from Centi et al.356 | ||
These artificial trees would consist on the combination of “leaves”, representing photoelectrocatalytic devices, along with the “branches and trunk” that would act as distribution/collection elements. This in situ solar-based chemical industries would have greater adaptability to different applications, increased resilience, very low carbon footprint, lower time-to-market, and reduced economic costs and environmental impact, mainly associated with avoided transportation. In a nearer-term scenario, technological developments are working on turning the solar ammonia refinery in a real concept. At present, great efforts are directed at replacing or re-configuring H–B plants with CO2 capture as a short-term solution, while mid-to-long-term scenarios foresee a large-scale green ammonia production from water, air and solar energy, which could be extended to the production of fertilizer derivatives. The latter option is still constrained by the cost of electricity/energy and the current low ammonia yields (mmol h−1 g−1 range).357 Both factors impose limitations for the industrial scale up of the technology, which needs for a breakthrough in the areas of materials science, reactor/cell engineering, and the use of artificial intelligence and robotization. Following this concept, over 20 renewable and low-carbon ammonia European projects have been announced to start in 2030, gathering partnerships between different ammonia and fertilizer manufacturers and hydrogen suppliers, such as Yara, Fertiberia, Iberdrola, Hyperion, Siemens, Orsted, etc.358 The world's first green ammonia plant was developed by Kapsom, a global leader in sustainable energy solutions, in 2020. The facility is located in North-eastern India and uses solar power to produce more than 1500 t year−1 of green ammonia.359 The same year, the Australian Renewable Energy Agency (ARENA) supported a feasibility study for the installation of a solar refinery in the Pilbara region of Western Australia.360 The so-called Yuri Renewable Hydrogen to Ammonia Project is led by Yara Pilbara Fertilisers Pty Ltd (Yara), an important wide word fertilizer company, and ENGIE, a global leader in low-carbon energy and services. The project started in 2022 with the support of ARENA, ENGINE and Mitsui & Co. Ltd (Mitsui), and is expected to finish in 2028. This would be one of the world's first industrial-scale green ammonia plants using off-grid intermittent renewable H2via electrolysis. In particular, this project will develop a 10 MW electrolyzer powered by 18 MW of solar PV, and will be supported by an 8 MW battery energy storage system. In 2022, the Spanish fertilizer producer Fertiberia inaugurated the first green ammonia plant in Spain (Puertollano), within the Green H2F project,361 in partnership with Iberdrola and with the technical support of Spain's National Hydrogen Center. This plant (Fig. 19) includes a 100 MW PV facility that powers a 20 PEM MW electrolyzer with a capacity of 3000 tH2 year−1 (i.e. 360 kgH2 h−1). The solar intermittency is addressed by battery electrochemical storage (5 MW/20 MW h lithium-ion battery) and hydrogen storage (6000 kg capacity), which is enough to feed the H–B plant during low solar irradiation hours. The surplus of electricity generation is used to refill the storage systems or is exported to the grid.358 Moreover, a second phase with up to 800 MW of additional capacity is already under development through 2027.361
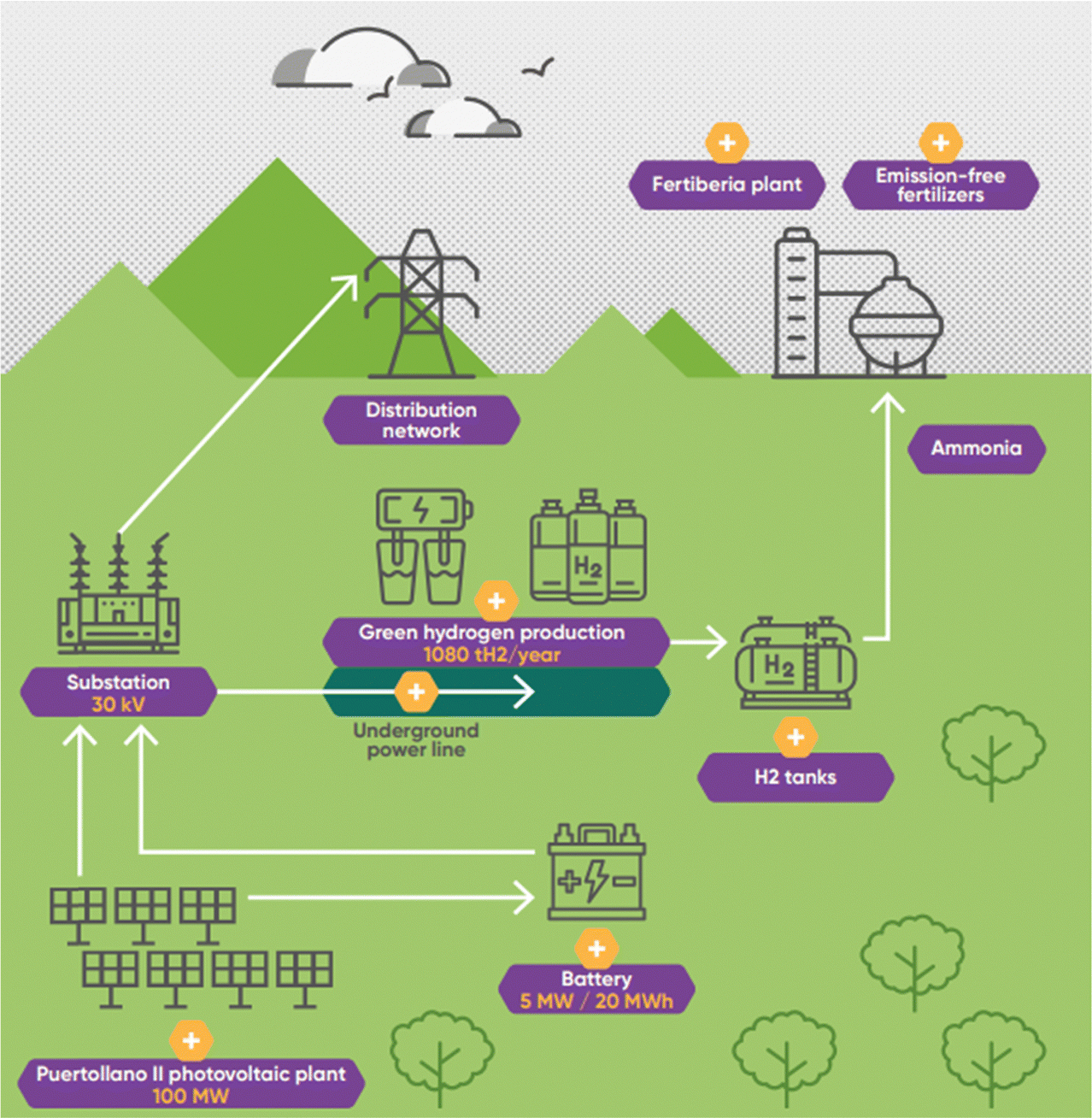 | ||
| Fig. 19 Schematics of Puertollano ammonia plant. Source: Iberdrola. | ||
Very recently, Fertiberia has joined Cepsa as a strategic partner to develop a 1-GW electrolysis project in Palos de la Frontera (Spain), as part of the Andalusian green hydrogen valley.362 Green hydrogen production will start in 2026, and it will be consumed by Cepsa and Fertiberia for the manufacture of advanced biofuels, ammonia, AdBlue and sustainable crop nutrition solutions. Another very recent project (2023) is the first green ammonia plant developed by Kapsom in Colombia.363 This project has an annual NH3 output of 2000 t year−1, and will significantly reduce CO2 emissions during the production process if compared to the traditional coal-based or natural gas-based ammonia synthesis (6400 tCO2 and 3600 tCO2, respectively). Additional forthcoming projects for green ammonia production are summarized in Fig. 20.
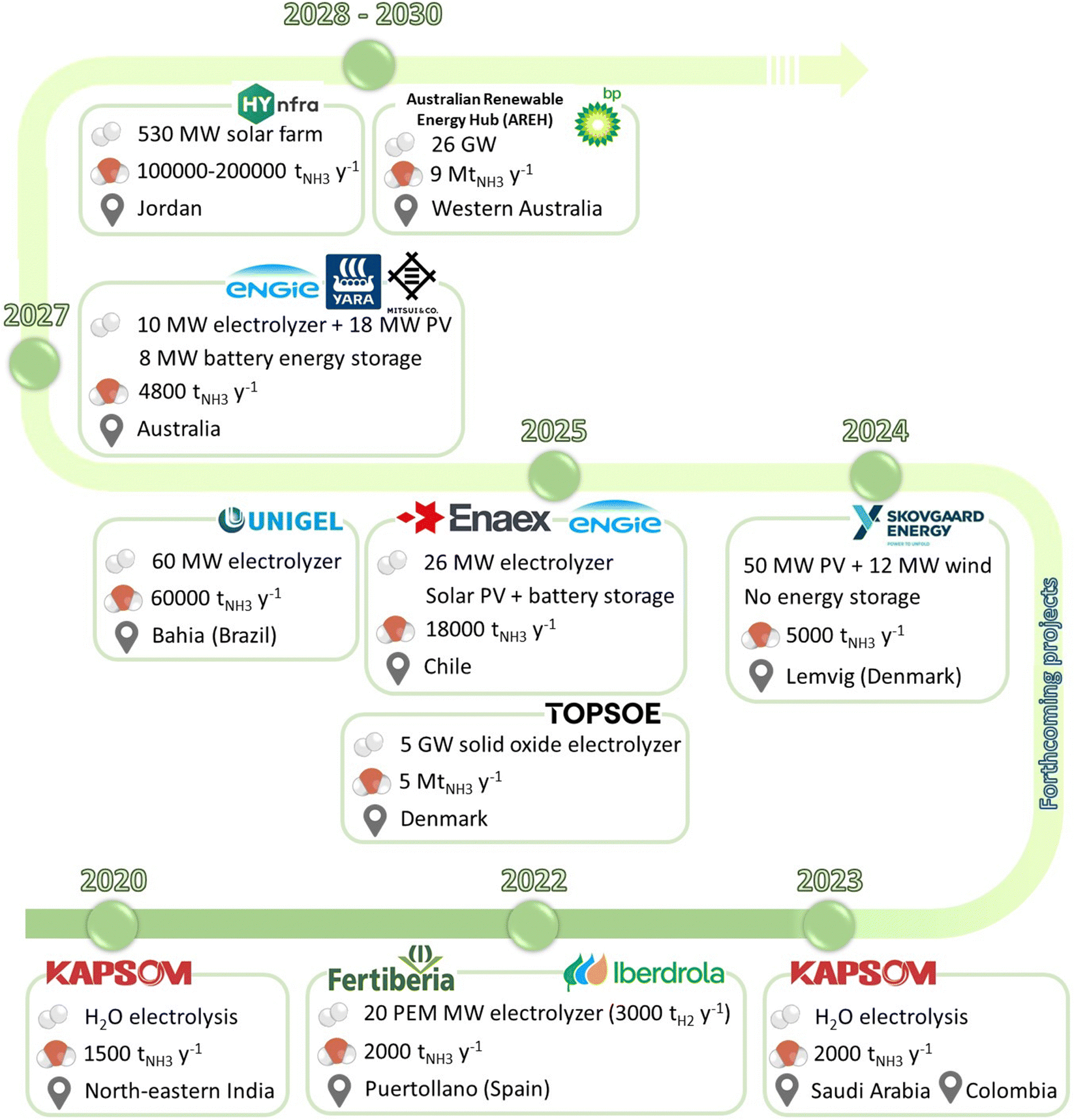 | ||
| Fig. 20 Timeline of operational and forthcoming green ammonia projects worldwide. | ||
8 Techno-economic comparison of ammonia production technologies
8.1. Ammonia production in conventional Haber–Bosch plants
The earth's population growth increases at a constant ratio, demanding for a continuous production of fertilizers alongside an increasing energy and electricity consumption (Fig. 21a). About 80% of the total ammonia production via Haber–Bosch (H–B) process is converted into nitrogen-rich fertilizers (Fig. 21b). Among them, ammonium nitrate is mainly used in the European Union, urea is more frequently used in China and Bharat (India), while USA commonly injects ammonia directly into soil (10–20 cm below surface to prevent losses by volatilization).103,162,364,365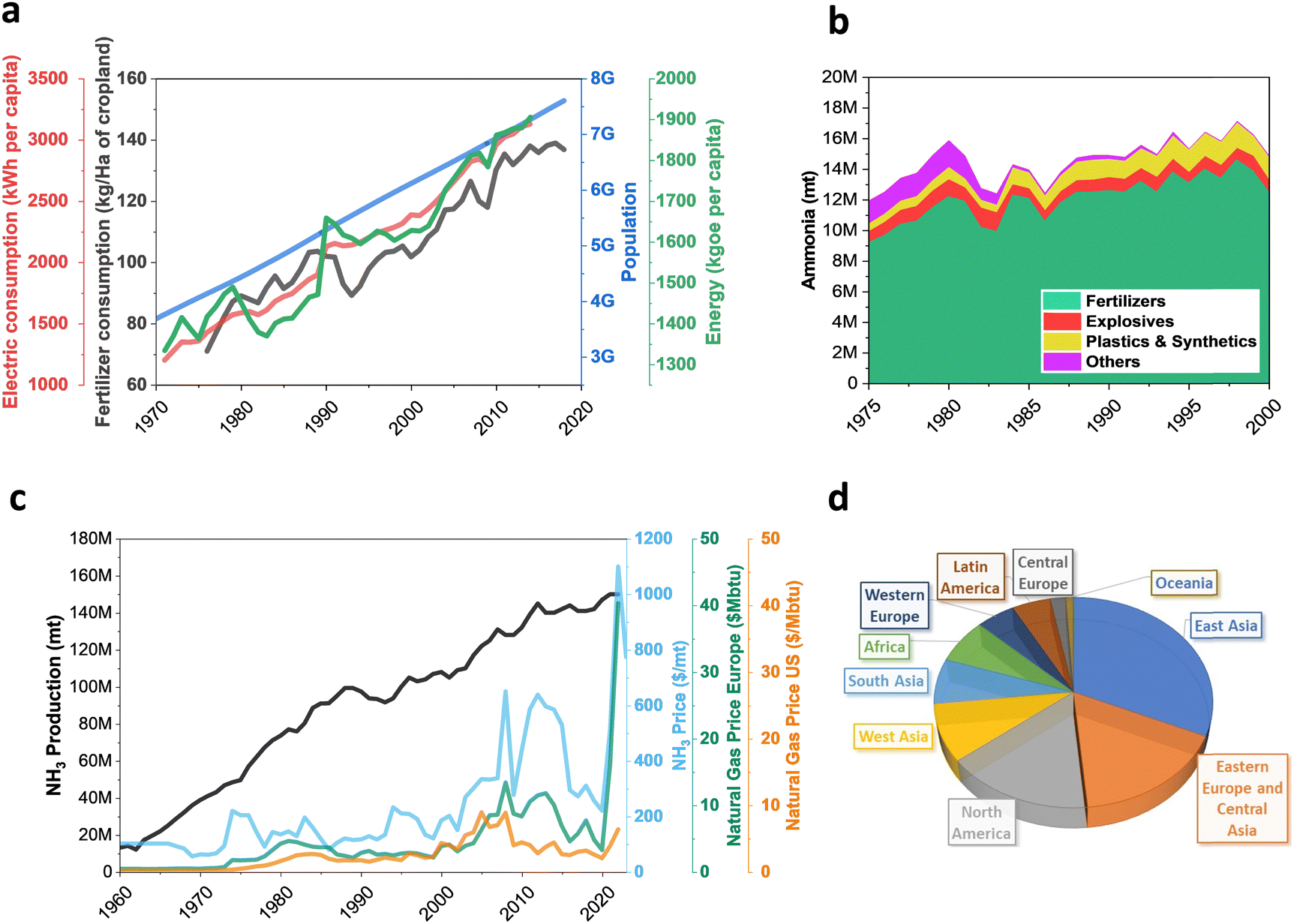 | ||
| Fig. 21 (a) Temporal evolution of world population (blue line), fertilizer consumption (grey) and electricity/energy consumption (red/green) in the period 1970–2020 (data extracted from the World Bank366); (b) end uses of ammonia (USGS);367 (c) evolution of the global ammonia production (USGS368,369), and price evolution of ammonia (USGS) and natural gas in USA and Europe (World Bank370); (d) distribution of the global fertilizer production capacity in 2021 by region.371 | ||
The energy-intensive H–B process has experienced significant technological improvements over the past 20th century, reducing the energy consumption from 40–100 GJ tNH3−1 to 28–32 GJ tNH3−1 in actual single-train ammonia plants207,211 with the best available techniques implemented. Today, H–B technology consumes around 1% of the world energy production372 to reach ammonia generation capacities of more than 3000 t d−1. The highest energy consumption in current ammonia production plants comes from natural gas reformers and CO2 removal systems. However, steam methane reforming (SMR) (72%) is nowadays considered the best available technique (BAT) for ammonia synthesis, given its higher energy efficiency and lower carbon emissions compared to others such as coal gasification (22%, 56–64 GJ tNH3−1), naphta (1%) or partial oxidation of heavy oil (4%). Coal gasification and heavy oil partial oxidation consumes much more energy (1.3 and 1.7 times more energy, respectively)19 and release more CO2 (ca. 6 tCO2 tNH3−1) than methane reforming (ca. 1.6 tCO2 tNH3−1).208,373,374 As a result, the ammonia production industry emits 500 Mt year−1, which represents 1% of the global CO2 emissions.372 Coupling H–B plants with carbon capture technologies (CCS) can considerably reduce the amount of CO2 emissions to 0.2–0.4 t of CO2 per t of NH3.375,376 Additional recent improvements include the development of energy recovery systems with more efficient designs, new catalysts, adsorbent/absorbent enhanced H–B processes, new converter designs, and electrochemical H–B process.377
Regarding costs, the global ammonia production has been historically influenced by the evolution of the natural gas price (Fig. 21c), suffering important fluctuations during economic/energy crisis periods, especially in importing regions such as the European Union. In particular, the recent disruptions in the global economy as a consequence of the 2020–2021 pandemic, and the rise in natural gas prices as a consequence of the Russia–Ukraine conflict led to a circumstantial increase in ammonia prices, which varied from less than $300 tNH3−1 to more than $1600 tNH3−1 in 2022.378 This price rising was worsened by the increase in transportation costs, among other factors, which disrupted the supply chains. The fertilizer prices have fallen 71% in 2023 due to a decrease in the natural gas prices and the adaptation of markets to the new geopolitical scenario.379 Despite that, ammonia prices are still high compared to historical standards, and the risk of appreciation remains constant following the same instability previously observed during the oil (1973) and financial (2008) crisis. Today, ammonia production is one of the most important markets worldwide, accounting for $80 billion per year.29 The current production system is based on a centralized generation mainly ruled by the biggest fertilizer manufacturers (China, India, Russia and USA) (Fig. 21d). These countries produce cheaper ammonia due to the lower costs in natural gas, achieving ammonia production costs around ∼$160 tNH3−1 in large H–B plants (>2000 tNH3 d−1), although costs can be doubled small plants (<100 tNH3 d−1).380,381 In contrast, ammonia producers in other countries are unable to produce at less than $500 tNH3.382 We note that the production costs of ammonia in large H–B plants may further increase due to storage and transportation, eventually reaching market prices around $600 tNH3−1.383 However, the long-distance transportation of ammonia by pipeline is more efficient than that of H2 and natural gas, making the transportation and storage of ammonia much more economic.384 Transportation of anhydrous ammonia by pipeline is economically viable for connecting ammonia plants with fertilizer production facilities located in market areas. In contrast, shipping is adequate for large volumes and exportations with good terminal costs and infrastructure. Transport by rail is suitable for moderate ammonia productions, while transport by truck is relatively expensive although flexible to reach isolated areas at short distances.385 In general, the main factor determining the selection of the transportation media is the distance to the agricultural land. For instance, the costs of transportation reported for a specific case varied from $0.0153, $0.0161, $0.0215 to $0.0365 t−1 km−1 for pipelines, ships, rail tankers and trucks, respectively.329 Additional costs may arise from the storage of pressurized/refrigerated anhydrous ammonia for its stational selling (75%) during the planting season.386 Besides, the total ammonia cost may considerably increase considering the social costs as externality.380
8.2. Green ammonia with H–B synthesis loop
The traditional H–B synthesis loop could enable the production of green ammonia by (i) fully electrifying the process with renewable energy, (ii) decoupling H2 production by methane steam reforming, or (iii) using renewable hydrogen as feedstock.164 In this regard, H2 can be obtained from other fossil-free sources, for instance, through biomass gasification (syngas), biomass reforming (biogas from anaerobic digestion), or renewable water electrolysis.387 Indeed, early ammonia production plants in 1920's already integrated H2 produced by electrolysis in regions with hydroelectric power generation. Afterwards, the lower cost of natural gas made methane reforming more competitive, thus outcompeting other H2 production processes.15 Nowadays, decarbonization scenarios estimate that green hydrogen will contribute to more than half of the total ammonia production in 2050 (>550 Mt year−1).27,388 In particular, Saygin et al.374 estimate an increase in 1100 GW electrolyzer capacity and 2300 GW of renewable generation capacity to supply 5500 TW h year−1 of electricity (from the actual 29000389–30000 TW h390).
Table 6 collects relevant data from reported technoeconomic studies on ammonia production using H2 from biomass conversion.
| H2 source | Plant capacity (tpd or t year−1*) | CO2 emissions (kgCO2 kgNH3−1) | NH3 cost** ($ t−1) | Ref. |
|---|---|---|---|---|
| Biomass pyrolysis and gasification | 14–384 (pyrolysis) | — | 570–1369 (pyrolysis) | 384 |
| 10–1022 (gasification) | 488–1519 (gasification) | |||
| Biomass gasification | 1200 | 0.5–0.88 | 386 | 397 |
| Biomass, biogas, wind | 2000–28000* |
— | 680–2300 | 398 |
| Biomass gasification | 700 | — | 750–1200 | 399 |
| Biomass gasification | 1187 | 0.3 | 968 | 400 |
| Biomass gasification | 65–200 | 0.59–0.84 | 1153 (65 t) | 396 |
| 740 (1200 t) |
Large-scale green ammonia production with electrolysis requires around 30–36 GJ t−1 of electricity,207,401 although the potential to reduce these values is still high.164 Around 80–90% of this energy consumption is used for H2 production (50–55 kW h kgH2−1), while the remaining energy is used for air separation, gas compression and synthesis loop.374 Cryogenic air separation at large scale (> 600 tN2 day−1) also accounts for approximately 25% total cost of a whole H–B plant, ranging between $0.03–0.05 kgN2−1.250 The CO2 emissions derived from E/H–B are very low, between 0.12 to 1.3 tCO2 tNH3−1, which is far below other technologies such as SMR or coal gasification.164 Life cycle analyses of conventional and renewable ammonia production routes showed that obtaining N2 from cryogenic distillation and H2 from electrolysis reduce GHG emissions by 91% compared to conventional SMR plants.402 However, the most challenging drawbacks of this approach are the current cost and the intermittency of renewable electricity, which requires the dynamic operation of electrolyzers and H–B loops.375 For instance, power-to-ammonia plants in North Europe become competitive at gas prices of €70 MW h−1 and CO2 emissions prices of €200 tCO2−1.403 Thus, a decrease in the costs of renewable electricity would substantially make green ammonia synthesis more economically feasible.404 At present, H2O electrolyzers are commercially available at a scale larger than 100 MW power per stack.17,349,405,406 Among several electrolyzer's designs, proton exchange membrane (PEM) electrolyzers are the most competitive with H2 production costs at $5–23 kg−1. However, the average costs for SMR are around $1.28 kg−1.375,393,407 The costs associated to the electrolysis unit remains expensive ($1 M per MW), but foreseen technological improvements would reduce costs by half in the mid-term.408,409 These technological improvements will also need to address current engineering limitations related to the durability of high-temperature electrolyzers (700–900 °C), powered by renewable energy and thus working in intermittent conditions.15 Other costs associated with the nitrogen separation unit (membranes, Pressure Swing Adsorption, or distillation) should be addressed depending on the production capacity and the required purity.410
Further, it should be noted that ammonia production via E/H–B strongly depends on the levelized cost of electricity (LCOE) (0.04–0.19 € per kW h411,412). In this regard, economic analyses showed the advantage of using Ru-based catalysts in small scale ammonia plants (< 100 tNH3 d−1), where the cost associated with reactors and heat exchangers are dominant compared to the catalyst's cost.413,414
Table 7 shows that average green ammonia prices double or triple those of grey ammonia,375,415 due to the large capital costs of renewable power (LCOE) and electrolyzers.17,349 In this regard, Salmon and Bañares-Alcántara349 estimated that 50% reduction in the electrolyzer's capital costs could reduce ammonia costs to $400 t−1, and this could be achieved by combining wind and solar energy as the power supply.
| H2 source | Plant capacity (tpd or t yeara) | CO2 emissions (kgCO2 kgNH3−1) | NH3 costb ($ t−1) | Ref. |
|---|---|---|---|---|
| a tNH3 year−1. b If not specified ($ tNH3−1). | ||||
| Wind/solar | 7–490 (solar) | — | 660–2342 (wind) | 384 |
| 5–263 (wind) | 830–5951 (solar) | |||
| Wind | 48 | — | 655–720 | 416 |
| Wind | 1202.55 | 0.97 | 742 | 417 |
| Offshore wind | 300 | — | 1566 | 418 |
| Solar/wind, biogas, anaerobic digestion | 0.219 | 0–3.82 | — | 387 |
| Wind | 109.6 | — | 526–861 (2023) | 419 |
| 431–931 (2030) | ||||
| N.A. | — | — | 713–1457 | 420 |
| Offshore wind | 300 | 0.09–0.478 | 583–1224 | 421 |
| Wind | 1000 | — | 273 | 404 |
| Wind/solar PV | 300 | — | 1350–1380 € t−1 | 422 |
| Wind/solar PV/hydro | — | — | 431–528 € t−1 | 403 |
| Hydroelectric | 200 | — | 335–380 € t−1 | 423 |
| Wind/solar | 83220a |
588 £ (2025–2030) | 424 | |
| Wind | 0.18 | — | — | 425 |
| Wind, solar | 35000a |
— | < 500 | 401 |
| Renewable | 20000a |
— | 798 | 426 |
| Solar | 1840 | — | 450–718 | 427 |
| Renewable (2040) | 91–2000 | — | 515–613 | 380 |
| Wind/solar PV | 0.3–2.33 | — | 391–644 | 428 |
| Wind (floating offshore ammonia) | 300 | — | 1574–1724 | 418 |
| Steam turbine | 50000a |
— | 374–670 | 415 |
| Renewable | 11.6 | 754–815 € t−1 | 429 | |
| Wind, solar | 1320–1728 | — | 842 € t−1 | 430 |
In the current geopolitical context, high gas prices and low capital costs of electrolyzers could improve the competitiveness of green ammonia.374 In the meantime, blue ammonia is foreseen as a viable near-future option to replace conventional ammonia production under present-day conditions (i.e. 2024), as long as natural gas leakage rates are maintained low. However, this transition scenario will need to face some other important aspects such as the cost of H2 storage solutions (e.g. underground storage), the energy losses in the DC/AC/DC electricity conversions and other processes involved, safety of H2 transportation and related costs, use of feedstock buffers (i.e. oversized electrolyzers for H2 storage), etc.17,431
The technoeconomic study of Mayer et al.432 concluded that the implementation of green ammonia would require further developments to become an efficient alternative, needing to achieve higher efficiencies for electrolyzers and PV panels (80% and 70%, respectively). In contrast, Cesaro et al.412 made an optimistic estimate of green ammonia price, ranging from $771 t−1 in 2020 to less than $400 t−1 in 2040. Nayak-Luke and Bañares-Alcántara17 studied the viability of producing islanded green ammonia in 534 locations in 70 countries. The study found that the current islanded ammonia production with renewable energies (wind and solar) is not competitive with conventional fossil fuel plants, founding the production of H2 (electrolyzer CAPEX and OPEX) as the most significant cost components. However, predictions by 2030 foresee an achievable islanded green levelized cost of ammonia (LCOA) below $350 t−1 in the most favorable locations. We note that the LCOA depends on the cost of air separation, the capital cost per unit area of reactor ($ m−2), and the STA conversion efficiency.250 Sagel et al.433 concluded that combined wind and ammonia energy storage system could be competitive with fossil-based alternatives coupled to carbon capture and storage (CCS) in some locations, such as small island developing states (SIDS), although ammonia imports are still the most feasible option at market prices above $500 tNH3−1.434 In good agreement with previous works, Fasihi et al.435 estimated the cost of green ammonia with hybrid wind/PV energy production at the most favorable sites of the world, which varied between 440–630 € t−1 in 2020, and were predicted to decrease to 345–420, 300–330 and 260–290 € t−1 in 2030, 2040 and 2050, respectively. The limitation of this hybrid technology, based on solar panels and wind turbines, is their intermittency and the intense land occupation, which could be solved with off-shore energy generation.436
The TRL of electric H–B varies depends on the type of technology, with examples being alkaline (8–9), PEM (6–9) or solid oxide (3–6) electrolysis.164,437 Several projects have been started and announced during the last years with ammonia production capacities ranging from 1 to 1200000 t year−1. Some companies are developing or announcing green ammonia projects349,438 like Yara, First Ammonia, BASF, Siemens Energy, Fertiberia, Proton Ventures, Morris, Hy2gen, ACME group, NFUEL®,439 Stamicarbon, Yara, Kapsom, Casale, Thyssenkrupp, Hygenco, Ammpower, SIP, Haldor Topsoe, CF Industries, Engie & Enaex, AREH, Origin Energy, Origin Energy, NEOM, ACME, Ökowind, EE GmbH, Starfire Energy, ReMo Energy, Greenfield Nitrogen, North Ammonia or nium.
8.3. Green ammonia produced by electrocatalysis
The electrocatalytic production of ammonia is considered the next step towards a low carbon economy transition. Briefly, this technology generates H2 from water electrolysis, and produces ammonia via electrocatalytic N2 reduction. Three systems have been proposed already: (i) one-step direct electrosynthesis of NH3 in water,440 (ii) two-steps electrosynthesis of H2 and NH3,375,440 and (iii) Li-mediated synthesis of NH3 by Li nitridation.441 Depending on the reaction temperature, the electrocatalytic ammonia production can be conducted in high temperature solid state reactors (400–750 °C), molten salts and composite membrane reactors (100–500 °C), composite membranes and liquid electrolytes, and solid state electrolyte reactors (ambient-100 °C).376,442 In general, solid-state synthesis at high temperatures increases the ammonia production rates, although it is restricted by the thermodynamic equilibrium of ammonia formation and the degradation of the materials. On the other hand, the synthesis of ammonia using molten salts usually reaches faradaic efficiencies (FE) below 10% and ammonia formation rates of 10−8 mol s−1 cm−1.443 Liquid electrolyte reactors, which use water as the proton source, usually reach FE below 1% and similar ammonia production rates. In contrast, FE as high as 50–60% can be obtained at low temperatures with ammonia formation rates in the order of 10−9 mol s−1 cm−1.444–448 Advised values for a potential commercialization of electrocatalytic ammonia are FE of at least 50%, and NH3 yield higher than 10−6 mol s−1 cm−2.449 Regarding the economic viability, a substantial decrease in costs is foreseen when plants are operated at low electricity prices. However, it should be considered that LCOA for electrochemical ammonia synthesis plants is more sensitive to variations in operation factors (LCOE, FE and the current density) than capital costs (electrochemical reactors and electrodes).440,450Hochman et al.380 affirmed that direct electrochemical nitrogen reduction plants could achieve more competitive costs than E/HB technology, with higher FE and overpotential, reaching production costs between $400–500 t−1 by 2040. Other studies even predict that electrocatalytic NH3 synthesis could be feasible in a fully decentralized ammonia production system at low production scales (<10 t d−1), in locations where natural gas H–B plants or E/H–B plants are not competitive.380,440 In such locations, a production of 0.03 tNH3 d−1 (10950 kg year−1), in a 100 ha farm that requires 100 kgN ha−1 year−1 (10000 kg year−1), could be obtained at prices below $600 t−1 under certain predictions (particular LCOE values, FE and current densities).329,440 Data collected in Table 8 show that electrocatalytic ammonia synthesis could be competitive for low productions in isolate locations, or if the energy input is reduced to 6 MW h tNH3−1 with electricity prices below $0.025 kW−1.1
The TRL of direct electrocatalytic ammonia synthesis ranges between 1–5, depending on the operational conditions.164,452 Some examples of companies currently developing direct electrocatalytic ammonia production are Nitricity, Jupiter ionics, Nitrofix solutions, Liquium, Plasmaleap, Nitrocapt, Atmonia or Nitrofix.
Fig. 22 depicts the historic prices trend for ammonia produced from fossil fuels in the US during the last 60 years, spiking during the periods of crisis, especially during 2020–2023 when prices reached more than $1100 t−1. The graphic also introduces the production cost ranges for E/H–B, biomass/H–B and electrosynthesis of green ammonia estimated in all the articles reviewed previously. Nowadays these technologies can compete with traditional H–B when the prices of fossil fuels rise. The estimations for 2040 predicted in recent works are in the range of $300–600 t−1 for electric H–B and electrosynthesis.380,412,435
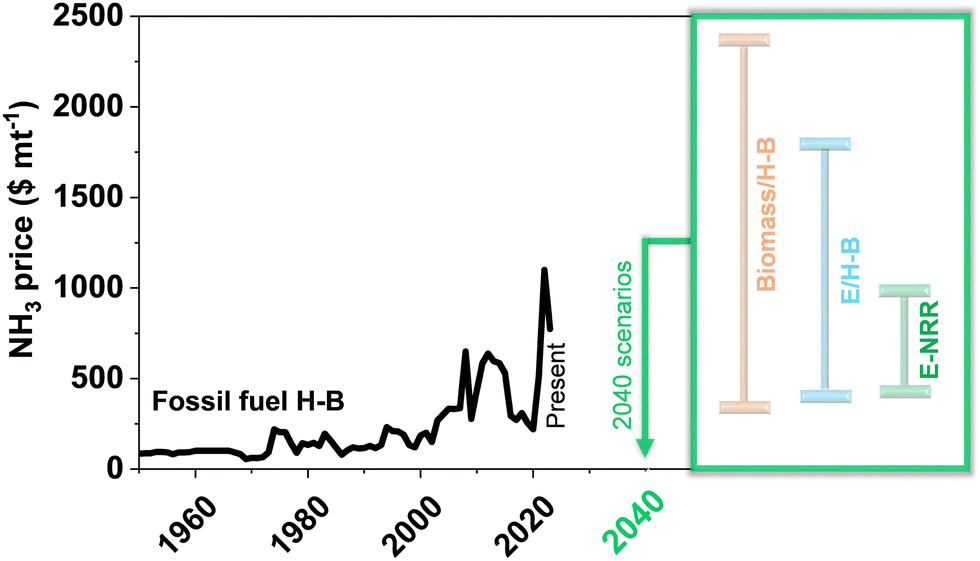 | ||
| Fig. 22 Historical evolution of ammonia H–B market prices in US (black line),367–369 and estimation of costs intervals for green ammonia production in three different 2040 scenarios: biomass H–B, electrified H–B (E/H–B), and electrocatalytic NRR (E-NRR). Estimations by 2040 are based on data from ref. 380,412,435 | ||
8.4. Green ammonia produced via photo(electro)catalysis
There is a lack of technoeconomic studies centered on photo(electro)catalytic ammonia synthesis. However, some works highlight the potential of combining photo(electro)catalytic H2 production with PEC-based electrocatalytic NRR.450,453 At present, a solar to fuel efficiency of 18% can be obtained using the best solar cells available in the market.454 Exergoeconomic analyses in large-scale plants calculate hydrogen and ammonia costs of $3.24 kg−1 and $0.84 kg−1, respectively.4508.5. Green ammonia produced by direct solar photo(thermal)catalysis
One of the main drawbacks of grid-connected technologies for ammonia synthesis is their strong dependency on the LCOE. An interesting alternative to avoid this vulnerability is the direct synthesis of green ammonia by solar photocatalysis. This technology, developed in aqueous or gas-phase, offers the opportunity to completely decentralize the production of fertilizers in remote locations, reducing the dependency on grid-based electricity. Besides, this approach reduces the vulnerability to energy crisis events and brings economic savings, since ammonia can be produced locally at the consumer point avoiding transportation costs. This would also imply more efficient fertilization processes in terms of time, effort and energy consumption. Moreover, aqueous-phase photocatalytic NRR can also reuse side-products from agricultural and industrial activities as sacrificial agents (e.g. bioethanol or glycerol), valorizing wasted compounds in the same process, reducing the total organic carbon from aqueous effluents, reducing the management cost of residues, and promoting a more rational use of nitrogen following the precision farming concept.455A recent noteworthy approach is the so-called “aerobic” photocatalytic nitrogen fixation, in which NRR is conducted in aqueous phase using air as the N2 source. This alternative technology avoids the need for air separation (indispensable for H–B, electrocatalytic NRR, or gas-phase photo(thermo)catalytic NRR using H2:N2) considerably reducing the operation costs, which usually represent around 70% of the total expenses. However, it should be noted that the presence of oxygen may corrode the catalyst surface, limit the solubility of N2, or react with surface NH species, negatively impacting on the catalyst performance.121,250 The use of metals with limited oxygen adsorption (e.g. Co, Mo or V) may help to overcome this drawback.456
From a practical point of view, the key parameter for the application of direct solar ammonia synthesis relies on the amount of ammonia that can be produced per unit area, since it directly affects to the land requirements. Likewise, the occupied area by photoreactors strongly depends on the performance of the catalysts. Best performing NRR photocatalysts have reached ammonia production rates close to 30 mmol h−1 g−1,214 and solar to ammonia efficiencies of 0.3%.232 However, most photocatalysts usually deliver STA below 0.1%, which is the reference value required for practical application.333,457 This target value imposes serious limitations to photocatalytic NRR to compete with H–B process for centralized ammonia production. However, this may become a competitive technology for off-grid distributed production of solar fertilizers.329 As a reference, the current nitrogen requirements of crop fields vary between 20–200 kgN ha−1 year−1,458–461 even reaching values as high as 400 kgN ha−1 year−1 in some countries (e.g. China).462 Such variations depend on the type of crop, soil, or geography.463 Considering average fertilizer demands of 50–200 kgN ha−1 year−1, some works in literature estimate area requirements for solar capture lower than 10% in regions with high solar flux and using photocatalysts with STA of 0.1%.329 Comer et al.329 estimated 1% arable land area for solar capture, corresponding to 100 m2 ha−1 (equivalent to 6 typical solar panels per hectare), assuming 50 kgN ha−1 year−1. Other estimations foresee farmland requirements of 66 m2 g−1 for photothermal yields of 2.1 mmol h−1 g−1.309
Regarding costs, technoeconomic analyses464 indicate the viability of ammonia production, especially via aerobic NRR, at competitive costs using different photoreactor configurations, such as slurry ($5 m−2), fixed panel array ($20 m−2) and solar concentrator array ($100 m−2) reactors, and predict achievable production costs under $600 tNH3−1 assuming STA efficiencies of 1% in slurry reactors (see Table 9). On-going and future research on highly active photocatalysts, with high STA efficiencies and enough tolerance to the presence of dissolved oxygen, could facilitate the implementation of aerobic photocatalytic NRR for green ammonia production.
| N2 supply | Reactor | Plant capacity (tpd) | NH3 cost ($ t−1) |
|---|---|---|---|
| Air (no separation) | Slurry | <1 t d−1 | 30 (STA 10%) |
| 600 (STA 1%) | |||
| Panel | 120 (STA 10%) | ||
| 600 (STA 2.5%) | |||
| Concentrated array | 580 (STA 10%) | ||
| N2 (separation by membrane) | Slurry | <1 t d−1 | 110 (STA 10%) |
| Panel | 200 (STA 10%) | ||
| Concentrated array | 660 (STA 10%) |
8.6. Implementation challenges and policies for green ammonia technologies
The technoeconomic studies summarized in this section have shown the potential role of green ammonia in the transition to a sustainable circular economy in the chemical industry, transport and energy-related sectors. Estimations based on the existing degree of technological readiness of ammonia production (Table 10) agree on its key role for a decarbonized future scenario, in which public perception will be determinant for a successful transition.| Ammonia production method | TRL |
|---|---|
| E/H–B | 5–9 |
| Modified E/H–B (R&D: lower temperature catalysts) | 1–4 |
| Modified E/H–B (R&D: lower pressure operation) | 1–5 |
| Chemical looping | 1–4 |
| Electrochemical production | 1–3 |
| Light-driven catalytic production | 1 |
However, there is a lack of technoeconomic studies and life cycle analyses of electrocatalytic and photo(electro)catalytic NRR technologies for green ammonia production, which should be developed in the next years to push the real development and implementation of these sustainable technologies. In the meantime, the International Renewable Energy Agency (IRENA) recommends that the initial target application for green ammonia should be the chemical and fertilizer industries,27 specially to gain leverage in the retrofitting of existing ammonia plants. To do so, the implementation of renewable ammonia should be accompanied by an adequate regulatory framework, policies and incentives. Indeed, policy uncertainties are associated with high weighted average capital costs (WACC) and thus high levelized costs, which could be decreased through technology demonstrations. Policies should be also oriented to induce demand, and to overcome the main existing barriers for green ammonia development:27
Implementation of appropriate taxation policies. Policies may ensure an equitable tax treatment and a long-term guaranteed price for green ammonia. IRENA recommends that those taxes should be based on energy content rather than volume (e.g. USD per kilowatt-hour [kW h] instead of USD per litre).
Near-term efforts should focus on green ammonia production via retrofitted existing technologies (e.g. E/H–B or combinations of technologies) rather than emerging routes. In this regard, light-driven technologies (i.e. photo(electro)catalytic, PV-electrocatalytic or photothermal NRR) may find a future niche market in remote locations with high electricity or transportation costs, where these alternative technologies could provide both decentralized production and energy independency. However, the short-term implementation plan of these technologies is still unclear because its dependency on different key issues, such as the volatile price of electricity, the implementation cost of new advanced technologies, their energy requirements, the associated land occupancy (e.g. 0.4 ha t−1 for average 1 tNH3 d−1 production in E/H–B plants465), transport limitations and geopolitical aspects.
Implementation of regulations on CO2 emissions. CO2 penalties could help to decarbonize ammonia production using the current infrastructure, which would aid to bridge the gap between fossil-based ammonia with unmitigated emissions and green ammonia production.
De-risking early investment projects. Governments may de-risk part of the CAPEX side of the investment of green ammonia plants through grants, loans, and loan guarantees. The OPEX side of the investments may be de-risked with contracts for difference, procurement contracts, off-take guarantees, etc. While the CAPEX for green ammonia is higher than fossil-based ammonia plants, its OPEX can be substantially lower (low operating expenses), making green ammonia competitive in the market as a commodity chemical and a renewable fuel.
9. Summary and outlook
Light-driven green ammonia production has been intensively studied over the past 10 years, as a potential solution to contribute to the decarbonization of the energy system and the fertilizer industry, as well as other pressing sectors such as transportation or chemical industry. This approach also allows for a long-term energy storage that would enable the distribution of excess energy, mitigating both seasonal variability and unpredictable vulnerabilities of the actual centralized production model (e.g. price rising during crisis periods). Besides, they can contribute to reach a total decentralized production of solar fertilizers in next-generation farms. Despite the great potential of NRR technologies to achieve a decentralized ammonia production, they are still at an early stage of development at laboratory scale. At present, best reported light-driven ammonia production rates are close to 30 mmol h−1 g−1, with STA efficiencies of 0.3%. However, most photo(electro)catalysts usually deliver STA lower than 0.1%, which is below the reference values required for practical applications (i.e. STA > 0.1% or STA > 20% for its potential application as a fertilizer or fuel, respectively). Thus, the main technological challenge consists on developing stable and high-performing catalysts, which are able to operate with the lowest energy consumption. Catalyst engineering and processes development should also focus their efforts on improving cost competitiveness (e.g. avoiding precious metals or complex installations, or using air instead of pure N2 to avoid separation costs). Besides, the production of ammonia via light-driven NRR technologies confronts other challenges different from the technological ones, such as the land use requirements for solar capture, the intermittency of solar irradiation, the access to water reservoirs for H2 production, or the fluctuation of electricity prices. On this basis, on-going research efforts can make possible to find efficient catalysts and cost-effective technological solutions to push the real implementation of light-driven NRR systems. Besides, the development of technoeconomic studies and life cycle analyses would help to evaluate the technological feasibility and economic viability of these processes, reinforcing the future of green ammonia production technologies for a net-zero carbon society.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the National Projects ARMONIA (PID2020-119125RJ-I00), NovaCO2 (PID2020- 118593RB-C22), 2D-Photo2Change (TED2021-129999A- C33), SOLARCHEM5.0 (TED2021-130173B-C41), SOL-Future (PLEC2021- 0079069), and SolarCPP-Bat (CNS2022-135380), funded by both MCIN/AEI/10.13039/501100011033 and NextGenerationEU/PRTR. Additional funding has been received from FET Proactive program project Hysolchem, and ERC-PoC2022 Demonia (Ref. 101069268). This work received also support from “La Caixa” foundation fellowship (ID 100010434, fellowship code LCF/BQ/PR23/11980046).References
- M. Nazemi and M. A. El-Sayed, Acc. Chem. Res., 2021, 54, 4294–4304 CrossRef CAS PubMed.
- J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- J. Su and L. Vayssieres, ACS Energy Lett., 2016, 1, 121–135 CrossRef CAS.
- N. S. Lewis, G. Crabtree, A. J. Nozik, M. R. Wasielewski and P. Alivisatos, Basic Energy Sci. Work. Sol. Energy Util., 2005, 1–260 Search PubMed.
- D. L. C. Chandler, 2011.
- Global Primary Energy.
- M. Ravi and J. W. Makepeace, Chem. Sci., 2022, 13, 890–908 RSC.
- J. Chen, W. Zhang, H. Li, W. Li and D. Zhao, SusMat, 2021, 1, 174–193 CrossRef CAS.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3011 CrossRef CAS PubMed.
- S. L. Meng, X. B. Li, C. H. Tung and L. Z. Wu, Chemistry, 2021, 7, 1431–1450 CrossRef CAS.
- H. Wang, L. Wang, Q. Wang, S. Ye, W. Sun, Y. Shao, Z. Jiang, Q. Qiao, Y. Zhu, P. Song, D. Li, L. He, X. Zhang, J. Yuan, T. Wu and G. A. Ozin, Angew. Chem., Int. Ed., 2018, 57, 12360–12364 CrossRef CAS PubMed.
- D. Ziegenbalg, J. Zander and R. Marschall, ChemPhotoChem, 2021, 5, 792–807 CrossRef CAS.
- X. Chen, N. Li, Z. Kong, W. J. Ong and X. Zhao, Mater. Horizons, 2018, 5, 9–27 RSC.
- A. Braun, D. K. Bora, L. Lauterbach, E. Lettau, H. Wang, S. P. Cramer, F. Yang and J. Guo, Catal. Today, 2022, 387, 186–196 CrossRef CAS PubMed.
- K. H. R. Rouwenhorst, A. S. Travis and L. Lefferts, Sustainable Chem., 2022, 3, 149–171 CrossRef CAS.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- R. M. Nayak-Luke and R. Bañares-Alcántara, Energy Environ. Sci., 2020, 13, 2957–2966 RSC.
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef.
- A. E. Yüzbaşıoğlu, C. Avşar and A. O. Gezerman, Curr. Res. Green Sustainable Chem., 2022, 5, 100307 CrossRef.
- S. Ghavam, M. Vahdati, I. A. G. Wilson and P. Styring, Front. Energy Res., 2021, 9, 1–19 Search PubMed.
- Z. Yan, M. Ji, J. Xia and H. Zhu, Adv. Energy Mater., 2020, 10, 1–35 Search PubMed.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C. P. Chen, M. R. Smith and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Techno-Economic Challenges of Green Ammonia as an Energy Vector, Elsevier Inc., 2021, pp. 41–83 Search PubMed.
- The Royal Society, Ammonia: zero-carbon fertiliser, fuel and energy store. Policy Briefing, 2020.
- International Energy Agency (IEA), Ammonia Technology Roadmap, 2021.
- L. Collado, A. Herrero and V. A. de la Peña O’Shea, Powerfuels – Status & Prospects, Springer, 2024, p. Under editing (edition ID 447710) Search PubMed.
- IRENA (International Renewable Energy Agency), Innovation Outlook: Renewable Ammonia, 2022.
- H. Ali, M. Masar, A. C. Guler, M. Urbanek, M. Machovsky and I. Kuritka, Nanoscale Adv., 2021, 3, 6358–6372 RSC.
- R. Daiyan, I. Macgill and R. Amal, ACS Energy Lett., 2020, 5, 3843–3847 CrossRef CAS.
- Y. Zhao, Y. Miao, C. Zhou and T. Zhang, Mol. Catal., 2022, 518, 112107 CrossRef CAS.
- S. Zhang, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, EnergyChem, 2019, 1, 100013 CrossRef.
- X. Hui, L. Wang, Z. Yao, L. Hao and Z. Sun, Front. Chem., 2022, 10, 1–26 Search PubMed.
- H. P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- Y. Q. Le, J. Gu and W. Q. Tian, Chem. Commun., 2014, 50, 13319–13322 RSC.
- S. Singh, A. K. Mohammed, A. A. AlHammadi, D. Shetty and K. Polychronopoulou, Int. J. Hydrogen Energy, 2023, 48, 34700–34739 CrossRef CAS.
- J. John, D. K. Lee and U. Sim, Nano Convergence, 2019, 6, 1–16 CrossRef PubMed.
- R. Zaffaroni, D. Ripepi, J. Middelkoop and F. M. Mulder, ACS Energy Lett., 2020, 5, 3773–3777 CrossRef CAS.
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 RSC.
- C. Mao, H. Li, H. Gu, J. Wang, Y. Zou, G. Qi, J. Xu, F. Deng, W. Shen, J. Li, S. Liu, J. Zhao and L. Zhang, Chemistry, 2019, 5, 2702–2717 CrossRef CAS.
- H. Kisch, Eur. J. Inorg. Chem., 2020, 1376–1382 CrossRef CAS.
- N. Gruber and J. N. Galloway, Nature, 2008, 451, 293–296 CrossRef CAS PubMed.
- R. Shi, X. Zhang, G. I. N. Waterhouse, Y. Zhao and T. Zhang, Adv. Energy Mater., 2020, 10, 1–10 Search PubMed.
- D. Fowler, M. Coyle, U. Skiba, M. A. Sutton, J. N. Cape, S. Reis, L. J. Sheppard, A. Jenkins, B. Grizzetti, J. N. Galloway, P. Vitousek, A. Leach, A. F. Bouwman, K. Butterbach-Bahl, F. Dentener, D. Stevenson, M. Amann and M. Voss, Philos. Trans. R. Soc. B Biol. Sci., 2013, 368, 20130164 CrossRef PubMed.
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Annu. Rev. Biochem., 2009, 78, 701–722 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- D. Mallamace, G. Papanikolaou, S. Perathoner, G. Centi and P. Lanzafame, Int. J. Mol. Sci., 2021, 22, 1–18 Search PubMed.
- R. R. Eady, Chem. Rev., 1996, 96, 3013–3030 CrossRef CAS PubMed.
- G. E. D. Oldroyd and R. Dixon, Curr. Opin. Biotechnol, 2014, 26, 19–24 CrossRef CAS PubMed.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- H. R. Rucker and B. Kaçar, Trends Microbiol., 2023, 1–11 Search PubMed.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef CAS PubMed.
- J. Norskov and J. Chen, Sustainable Ammonia Synthesis. Exploring the scientific challenges associated with discovering alternative, sustainable processes for ammonia production, 2016.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- H. Wan, A. Bagger and J. Rossmeisl, J. Phys. Chem. Lett., 2022, 13, 8928–8934 CrossRef CAS PubMed.
- G. Centi, S. Perathoner, C. Genovese and R. Arrigo, Chem. Commun., 2023, 59, 3005–3023 RSC.
- J. N. Galloway, Environ. Pollut., 1998, 102, 15–24 CrossRef CAS.
- NAE Grand Challenges for Engineering. National Academy of Engineering., www.engineeringchallenges.org/challenges.aspx.
- J. Baltrusaitis, ACS Sustainable Chem. Eng., 2017, 5, 9527 CrossRef CAS.
- Y. Feng, Z. zhao, T. Wang, J. Li, M. Xu, H. Jiao, C. Wang, H. Li and M. Feng, Ceram. Int., 2022, 48, 20062–20069 CrossRef CAS.
- Z. Ding, M. Sun, W. Liu, W. Sun, X. Meng and Y. Zheng, Sep. Purif. Technol., 2021, 276, 119287 CrossRef CAS.
- H. Maimaitizi, A. Abulizi, T. Zhang, K. Okitsu and J. Jie Zhu, Ultrason. Sonochem., 2020, 63, 104956 CrossRef CAS PubMed.
- X. Ning, D. Jia, S. Li, M. F. Khan and A. Hao, Ceram. Int., 2023, 49, 21658–21666 CrossRef CAS.
- L. Chen, J. Wang, X. Li, J. Zhang, C. Zhao, X. Hu, H. Lin, L. Zhao, Y. Wu and Y. He, Green Energy Environ., 2023, 8, 1630–1643 CrossRef CAS.
- J. Feng, P. Ning, K. Li, X. Sun, C. Wang, L. Jia and M. Fan, ACS Sustainable Chem. Eng., 2023, 11, 804–814 CrossRef CAS.
- I. Muzammil, Y.-N. Kim, H. Kang, D. K. Dinh, S. Choi, C. Jung, Y.-H. Song, E. Kim, J. M. Kim and D. H. Lee, ACS Energy Lett., 2021, 6, 3004–3010 CrossRef CAS.
- X. Li, Y. Jiao, Y. Cui, C. Dai, P. Ren, C. Song and X. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 52498–52507 CrossRef CAS PubMed.
- W. Li, S. Zhang, J. Ding, J. Liu, Z. Wang, H. Zhang, J. Ding, L. Chen and C. Liang, ACS Sustainable Chem. Eng., 2023, 11, 1168–1177 CrossRef CAS.
- Y. Cui, H. Yang, C. Dai, P. Ren, C. Song and X. Ma, Ind. Eng. Chem. Res., 2022, 61, 4816–4823 CrossRef CAS.
- R. K. Sharma, H. Patel, U. Mushtaq, V. Kyriakou, G. Zafeiropoulos, F. Peeters, S. Welzel, M. C. M. van de Sanden and M. N. Tsampas, ACS Energy Lett., 2021, 6, 313–319 CrossRef CAS.
- J. Sun, D. Alam, R. Daiyan, H. Masood, T. Zhang, R. Zhou, P. J. Cullen, E. C. Lovell, A. (Rouhollah) Jalili and R. Amal, Energy Environ. Sci., 2021, 14, 865–872 RSC.
- P. Lamichhane, B. Chandra Adhikari, L. N. Nguyen, R. Paneru, B. Ghimire, S. Mumtaz, J. Sup Lim, Y. June and E. Ha Choi DOI:10.1088/1361.
- D. Zhou, R. Zhou, R. Zhou, B. Liu, T. Zhang, Y. Xian, P. J. Cullen, X. Lu and K. (Ken) Ostrikov, Chem. Eng. J., 2021, 421, 129544 CrossRef CAS.
- J. A. DeWitt, E. V. Phillips, K. L. Hebisch, A. W. Tricker and C. Sievers, Faraday Discuss., 2023, 243, 65–76 RSC.
- A. W. Tricker, K. L. Hebisch, M. Buchmann, Y.-H. Liu, M. Rose, E. Stavitski, A. J. Medford, M. C. Hatzell and C. Sievers, ACS Energy Lett., 2020, 5, 3362–3367 CrossRef CAS.
- D. F. Swearer, N. R. Knowles, H. O. Everitt and N. J. Halas, ACS Energy Lett., 2019, 4, 1505–1512 CrossRef CAS.
- E. M. Gaffney, M. Grattieri, K. Beaver, J. Pham, C. McCartney and S. D. Minteer, Electrochim. Acta, 2013, 337, 135731 CrossRef PubMed.
- M. Grattieri, K. Beaver, E. M. Gaffney, F. Dong and S. D. Minteer, Chem. Commun., 2020, 56, 8553–8568 RSC.
- R. Yadav, P. Chiranjeevi, S. Yadav, R. Singh and S. A. Patil, J. CO2 Util., 2022, 60, 101997 CrossRef CAS.
- X. Xue, R. Chen, C. Yan, P. Zhao, Y. Hu, W. Zhang, S. Yang and Z. Jin, Nano Res., 2019, 12, 1229–1249 CrossRef CAS.
- B. Puértolas, M. Comesaña-Hermo, L. V. Besteiro, M. Vázquez-González and M. A. Correa-Duarte, Adv. Energy Mater., 2022, 12, 2103909 CrossRef.
- L. Li, C. Tang, H. Jin, K. Davey and S. Z. Qiao, Chemistry, 2021, 7, 3232–3255 CrossRef CAS.
- A. Biswas, S. Bhardwaj, T. Boruah and R. S. Dey, Mater. Adv., 2022, 3, 5207–5233 RSC.
- H. Xu, K. Ithisuphalap, Y. Li, S. Mukherjee, J. Lattimer, G. Soloveichik and G. Wu, Nano Energy, 2020, 69, 104469 CrossRef CAS.
- Q. Qin and M. Oschatz, ChemElectroChem, 2020, 7, 878–889 CrossRef CAS.
- X. Yan, D. Liu, H. Cao, F. Hou, J. Liang and S. X. Dou, Small Methods, 2019, 3, 1800501 CrossRef.
- Z. Qiao, D. Johnson and A. Djire, Cell Rep. Phys. Sci., 2021, 2, 100438 CrossRef CAS.
- C. Ling, Y. Zhang, Q. Li, X. Bai, L. Shi and J. Wang, J. Am. Chem. Soc., 2019, 141, 18264–18270 CrossRef CAS PubMed.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chemistry, 2021, 7, 1708–1754 CrossRef CAS.
- T. Hou, Y. Xiao, P. Cui, Y. Huang, X. Tan, X. Zheng, Y. Zou, C. Liu, W. Zhu, S. Liang and L. Wang, Adv. Energy Mater., 2019, 9, 1–8 Search PubMed.
- J. Timoshenko and B. R. Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- Q. Wang, Y. Xiao, S. Yang, Y. Zhang, L. Wu, H. Pan, D. Rao, T. Chen, Z. Sun, G. Wang, J. Zhu, J. Zeng, S. Wei and X. Zheng, Nano Lett., 2022, 22, 10216–10223 CrossRef CAS PubMed.
- H. Shen, M. Yang, L. Hao, J. Wang, J. Strunk and Z. Sun, Nano Res., 2022, 15, 2773–2809 CrossRef CAS.
- X. Zhang, Y. Lyu, H. Zhou, J. Zheng, A. Huang, J. Ding, C. Xie, R. De Marco, N. Tsud, V. Kalinovych, S. P. Jiang, L. Dai and S. Wang, Adv. Mater., 2023, 35, 1–9 Search PubMed.
- T. Zeng, Y. Hu, Z. Yuan, H. He, X. Zhao, X. Zhao, X. Jian, H. Zhang and X. Gao, J. Alloys Compd., 2023, 975, 172851 CrossRef.
- X. Cheng, R. Guan, Z. Wu, Y. Sun, W. Che and Q. Shang, InfoMat, 2024, 6, 1–15 CrossRef.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2020, 10, 1–10 Search PubMed.
- Y. Fang, Y. Cao, B. Tan and Q. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 42624–42634 CrossRef CAS.
- Y. Wang, Z. Li, H. Zhu, X. Xu, Z. Zeng, X. Liu, H. Wang, M. Gong, X. Liu and Y. Wang, Chem. Eng. J., 2024, 484, 149583 CrossRef CAS.
- S. Ajmal, A. Rasheed, N. Q. Tran, X. Shao, Y. Hwang, V. Q. Bui, Y. D. Kim, J. Kim and H. Lee, Appl. Catal., B, 2023, 321, 122070 CrossRef CAS.
- S. Li, Z. Yao, J. Zheng, M. Fu, J. Cen, S. Hwang, H. Jin, A. Orlov, L. Gu, S. Wang, Z. Chen and D. Su, Angew. Chem., Int. Ed., 2020, 59, 22092–22099 CrossRef CAS PubMed.
- S. Kondo, T. Mitsuma, N. Shibata and Y. Ikuhara, Sci. Adv., 2016, 2, 1–8 Search PubMed.
- O. Kwon, Y. I. Kim, K. Kim, J. C. Kim, J. H. Lee, S. S. Park, J. W. Han, Y. M. Kim, G. Kim and H. Y. Jeong, Nano Lett., 2020, 20, 8353–8359 CrossRef CAS PubMed.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Prim., 2021, 1, 56 CrossRef CAS.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706–709 CrossRef CAS.
- Y. Cheng, X. Xu, M. Wang, C. Deng, Y. Sun, C. Yan and T. Qian, Adv. Funct. Mater., 2023, 33, 20232302332 Search PubMed.
- J. Jacquemin, M. F. Costa Gomes, P. Husson and V. Majer, J. Chem. Thermodyn., 2006, 38, 490–502 CrossRef CAS.
- D. Almantariotis, S. Stevanovic, O. Fandiño, A. S. Pensado, A. A. H. Padua, J.-Y. Coxam and M. F. C. Gomes, J. Phys. Chem. B, 2012, 116, 7728–7738 CrossRef CAS PubMed.
- C. S. M. Kang, X. Zhang and D. R. MacFarlane, J. Phys. Chem. C, 2018, 122, 24550–24558 CrossRef CAS.
- T. A. Bu, Y. C. Hao, W. Y. Gao, X. Su, L. W. Chen, N. Zhang and A. X. Yin, Nanoscale, 2019, 11, 10072–10079 RSC.
- C. Guo, J. Ran, A. Vasileff and S. Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- D. L. J. Broere and P. L. Holland, Science, 2018, 359, 871 CrossRef CAS PubMed.
- R. Shi, Y. Zhao, G. I. N. Waterhouse, S. Zhang and T. Zhang, ACS Catal., 2019, 9, 9739–9750 CrossRef CAS.
- M. Yang, J. Liu, H. Xu, Y. Pei, C. Jiang, D. He and X. Xiao, ChemPhysMater, 2022, 1, 155–182 CrossRef.
- D. Yan, H. Li, C. Chen, Y. Zou and S. Wang, Small Methods, 2019, 3, 1800331 CrossRef.
- Y. Zhao, Y. Zhao, G. I. N. Waterhouse, L. Zheng, X. Cao, F. Teng, L. Z. Wu, C. H. Tung, D. O’Hare and T. Zhang, Adv. Mater., 2017, 29, 1–10 Search PubMed.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS PubMed.
- W. Zhou and H. Fu, Inorg. Chem. Front., 2018, 5, 1240–1254 RSC.
- Z. Zafar, S. Yi, J. Li, C. Li, Y. Zhu, A. Zada, W. Yao, Z. Liu and X. Yue, Energy Environ. Mater., 2022, 5, 68–114 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1–9 Search PubMed.
- H. Li, J. Zhang, X. Deng, Y. Wang, G. Meng, R. Liu, J. Huang, M. Tu, C. Xu, Y. Peng, B. Wang and Y. Hou, Angew. Chem., Int. Ed., 2024, 63, 1–11 Search PubMed.
- J. Yang, Y. Guo, R. Jiang, F. Qin, H. Zhang, W. Lu, J. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS PubMed.
- B. Sun, Z. Liang, Y. Qian, X. Xu, Y. Han and J. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7257–7269 CrossRef CAS PubMed.
- Y. Guo, J. Yang, D. Wu, H. Bai, Z. Yang, J. Wang and B. Yang, J. Mater. Chem. A, 2020, 8, 16218–16231 RSC.
- C. Liang, H. Y. Niu, H. Guo, C. G. Niu, D. W. Huang, Y. Y. Yang, H. Y. Liu, B. Bin Shao and H. P. Feng, Chem. Eng. J., 2020, 396, 125395 CrossRef CAS.
- W. Ding, X. Li, S. Su, Z. Liu, Y. Cao, L. Meng, S. Yuan, W. Wei and M. Luo, Nanoscale, 2023, 15, 4014–4021 RSC.
- M. Arif, M. Babar, U. Azhar, M. Sagir, M. Bilal Tahir, M. Asim Mushtaq, G. Yasin, M. Mubashir, J. Wei Roy Chong, K. Shiong Khoo and P. Loke Show, Chem. Eng. J., 2023, 451, 138320 CrossRef CAS.
- N. Zhang, A. Jalil, D. Wu, S. Chen, Y. Liu, C. Gao, W. Ye, Z. Qi, H. Ju, C. Wang, X. Wu, L. Song, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 9434–9443 CrossRef CAS PubMed.
- K. Li, W. Cai, Z. Zhang, H. Xie, Q. Zhong and H. Qu, Chem. Eng. J., 2022, 435, 135017 CrossRef CAS.
- P. Huang, W. Liu, Z. He, C. Xiao, T. Yao, Y. Zou, C. Wang, Z. Qi, W. Tong, B. Pan, S. Wei and Y. Xie, Sci. China: Chem., 2018, 61, 1187–1196 CrossRef CAS.
- Y. Bo, H. Wang, Y. Lin, T. Yang, R. Ye, Y. Li, C. Hu, P. Du, Y. Hu, Z. Liu, R. Long, C. Gao, B. Ye, L. Song, X. Wu and Y. Xiong, Angew. Chem., 2021, 133, 16221–16228 CrossRef.
- H. Li, G. Wang, Q. Deng, W. Hu and W. Hou, Appl. Catal., B, 2024, 344, 123652 CrossRef CAS.
- C. Yang, Y. Zhang, F. Yue, R. Du, T. Ma, Y. Bian, R. Li, L. Guo, D. Wang and F. Fu, Appl. Catal., B, 2023, 338, 123057 CrossRef CAS.
- L. Wang, Y. Xia and J. Yu, Chemistry, 2021, 7, 1983–1985 CrossRef CAS.
- K. T. Ranjit, T. K. Varadarajan and B. Viswanathan, J. Photochem. Photobiol., A, 1996, 96, 181–185 CrossRef CAS.
- P. Qiu, C. Huang, G. Dong, F. Chen, F. Zhao, Y. Yu, X. Liu, Z. Li and Y. Wang, J. Mater. Chem. A, 2021, 9, 14459–14465 RSC.
- Y. Liu, Z. Yu, S. Guo, L. Yao, R. Sun, X. Huang and W. Zhao, New J. Chem., 2020, 44, 19924–19932 RSC.
- A. Ray, S. Sultana, S. P. Tripathy and K. Parida, ACS Sustainable Chem. Eng., 2021, 9, 6305–6317 CrossRef CAS.
- Y. Liao, J. Qian, G. Xie, Q. Han, W. Dang, Y. Wang, L. Lv, S. Zhao, L. Luo, W. Zhang, H. Y. Jiang and J. Tang, Appl. Catal., B, 2020, 273, 119054 CrossRef CAS.
- Z. K. Shen, Y. J. Yuan, P. Wang, W. Bai, L. Pei, S. Wu, Z. T. Yu and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 17343–17352 CrossRef CAS PubMed.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- S. Bian, M. Wen, J. Wang, N. Yang, P. K. Chu and X. F. Yu, J. Phys. Chem. Lett., 2020, 11, 1052–1058 CrossRef CAS PubMed.
- D. Cui, S. Wang, X. Yang, L. Xu and F. Li, Small, 2024, 20, 2306229 CrossRef CAS PubMed.
- Y. Xue, X. Kong, Y. Guo, Z. Liang, H. Cui and J. Tian, J. Mater., 2020, 6, 128–137 Search PubMed.
- L. Li, Y. Wang, S. Vanka, X. Mu, Z. Mi and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 8701–8705 CrossRef CAS PubMed.
- S. Sun, Q. An, W. Wang, L. Zhang and J. Liu, J. Mater. Chem. A, 2017, 5, 201–209 RSC.
- C. Liang, H. Y. Niu, H. Guo, C. G. Niu, Y. Y. Yang, H. Y. Liu, W. W. Tang and H. P. Feng, Chem. Eng. J., 2021, 406, 126868 CrossRef CAS.
- S. M. Stratton, S. Zhang and M. M. Montemore, Surf. Sci. Rep., 2023, 78, 100597 CrossRef CAS.
- S. Dutta and S. K. Pati, Catal. Today, 2023, 424, 113804 CrossRef CAS.
- X. Lv, W. Wei, F. Li, B. Huang and Y. Dai, Nano Lett., 2019, 19, 6391–6399 CrossRef CAS PubMed.
- X. W. Guo, S. M. Chen, H. J. Wang, Z. M. Zhang, H. Lin, L. Song and T. B. Lu, J. Mater. Chem. A, 2019, 7, 19831–19837 RSC.
- X. Liu, Y. Luo, C. Ling, Y. Shi, G. Zhan, H. Li, H. Gu, K. Wei, F. Guo, Z. Ai and L. Zhang, Appl. Catal., B, 2022, 301, 120766 CrossRef CAS.
- J. Li, P. Liu, Y. Tang, H. Huang, H. Cui, D. Mei and C. Zhong, ACS Catal., 2020, 10, 2431–2442 CrossRef CAS.
- L. Chen and X. Feng, Chem. Sci., 2020, 11, 3124–3131 RSC.
- J. Zheng, Y. Lyu, M. Qiao, R. Wang, Y. Zhou, H. Li, C. Chen, Y. Li, H. Zhou, S. P. Jiang and S. Wang, Chemistry, 2019, 5, 617–633 CrossRef CAS.
- C. Lee, H. Kim and Y. J. Jang, ACS Appl. Energy Mater., 2022, 5, 11018–11024 CrossRef CAS.
- X. Wang, X. Shi, S. Yin, P. She, J. Zheng, Y. Song and H. Sun, J. Mater. Chem. A, 2023, 11, 9976–9988 RSC.
- Z. Lu, S. E. Saji, J. Langley, Y. Lin, Z. Xie, K. Yang, L. Bao, Y. Sun, S. Zhang, Y. H. Ng, L. Song, N. Cox and Z. Yin, Appl. Catal., B, 2021, 294, 120240 CrossRef CAS.
- S. Vaclav, Nature, 1999, 400, 415 CrossRef.
- V. Smil, 2000.
- Y. Liu, S. Zhou, J. Li, Y. Wang, G. Jiang, Z. Zhao, B. Liu, X. Gong, A. Duan, J. Liu, Y. Wei and L. Zhang, Appl. Catal., B, 2015, 168–169, 125–131 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- R. Michalsky, B. J. Parman, V. Amanor-Boadu and P. H. Pfromm, Energy, 2012, 42, 251–260 CrossRef CAS.
- C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS PubMed.
- G. Ertl, M. Weiss and S. B. Lee, Chem. Phys. Lett., 2013, 589, 18–20 CrossRef CAS.
- A. Ozaki, Acc. Chem. Res., 1981, 14, 16–21 CrossRef CAS.
- F. Rosowski, A. Hornung, O. Hinrichsen, D. Herein, M. Muhler and G. Ertl, Appl. Catal., A, 1997, 151, 443–460 CrossRef CAS.
- K. Aika, H. Hori and A. Ozaki, J. Catal., 1972, 27, 424–431 CrossRef CAS.
- M. Appl, Ammonia: Principles and Industrial Practice, Wiley Online Books, 1999, pp. 65–176 Search PubMed.
- N. Saadatjou, A. Jafari and S. Sahebdelfar, Chem. Eng. Commun., 2015, 202, 420–448 CrossRef CAS.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainable Res., 2021, 2, 2000043 CrossRef CAS.
- Z. Yang, W. Guo, J. Lin and D. Liao, Chin. J. Catal., 2006, 27, 378–380 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- M. Hara, M. Kitano and H. Hosono, ACS Catal., 2017, 7, 2313–2324 CrossRef CAS.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- M. Kitano, Y. Inoue, H. Ishikawa, K. Yamagata, T. Nakao, T. Tada, S. Matsuishi, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2016, 7, 4036–4043 RSC.
- J. Li, J. Wu, H. Wang, Y. Lu, T. Ye, M. Sasase, X. Wu, M. Kitano, T. Inoshita and H. Hosono, Chem. Sci., 2019, 10, 5712–5718 RSC.
- M. Hattori, T. Mori, T. Arai, Y. Inoue, M. Sasase, T. Tada, M. Kitano, T. Yokoyama, M. Hara and H. Hosono, ACS Catal., 2018, 8, 10977–10984 CrossRef CAS.
- J. Wu, Y. Gong, T. Inoshita, D. C. Fredrickson, J. Wang, Y. Lu, M. Kitano and H. Hosono, Adv. Mater., 2017, 29, 1700924 CrossRef PubMed.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- W. Gao, S. Feng, H. Yan, Q. Wang, H. Xie, L. Jiang, W. Zhang, Y. Guan, H. Wu, H. Cao, J. Guo and P. Chen, Chem. Commun., 2021, 57, 8576–8579 RSC.
- Y. Cao, A. Saito, Y. Kobayashi, H. Ubukata, Y. Tang and H. Kageyama, ChemCatChem, 2021, 13, 191–195 CrossRef CAS.
- W. Gao, P. Wang, J. Guo, F. Chang, T. He, Q. Wang, G. Wu and P. Chen, ACS Catal., 2017, 7, 3654–3661 CrossRef CAS.
- Y. Kobayashi, Y. Tang, T. Kageyama, H. Yamashita, N. Masuda, S. Hosokawa and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18240–18246 CrossRef CAS PubMed.
- L. Li, T. Zhang, J. Cai, H. Cai, J. Ni, B. Lin, J. Lin, X. Wang, L. Zheng, C.-T. Au and L. Jiang, J. Catal., 2020, 389, 218–228 CrossRef CAS.
- Q. Wang, J. Pan, J. Guo, H. Anton Hansen, H. Xie, L. Jiang, L. Hua, H. Li, Y. Guan, P. Wang, W. Gao, L. Liu, H. Cao, Z. Xiong, T. Vegge and P. Chen, Ternary ruthenium complex hydrides for ammonia synthesis Hydrogen content Search PubMed.
- F. Chang, Y. Guan, X. Chang, J. Guo, P. Wang, W. Gao, G. Wu, J. Zheng, X. Li and P. Chen, J. Am. Chem. Soc., 2018, 140, 14799–14806 CrossRef CAS PubMed.
- Y. Guan, W. Zhang, Q. Wang, C. Weidenthaler, A. Wu, W. Gao, Q. Pei, H. Yan, J. Cui, H. Wu, S. Feng, R. Wang, H. Cao, X. Ju, L. Liu, T. He, J. Guo and P. Chen, Chem. Catal., 2021, 1, 1042–1054 CrossRef CAS.
- Y. Tang, Y. Kobayashi, N. Masuda, Y. Uchida, H. Okamoto, T. Kageyama, S. Hosokawa, F. Loyer, K. Mitsuhara, K. Yamanaka, Y. Tamenori, C. Tassel, T. Yamamoto, T. Tanaka and H. Kageyama, Adv. Energy Mater., 2018, 8, 1801772 CrossRef.
- M. Kitano, J. Kujirai, K. Ogasawara, S. Matsuishi, T. Tada, H. Abe, Y. Niwa and H. Hosono, J. Am. Chem. Soc., 2019, 141, 20344–20353 CrossRef CAS PubMed.
- H. Yamashita, T. Broux, Y. Kobayashi, F. Takeiri, H. Ubukata, T. Zhu, M. A. Hayward, K. Fujii, M. Yashima, K. Shitara, A. Kuwabara, T. Murakami and H. Kageyama, J. Am. Chem. Soc., 2018, 140, 11170–11173 CrossRef CAS PubMed.
- K. Ooya, J. Li, K. Fukui, S. Iimura, T. Nakao, K. Ogasawara, M. Sasase, H. Abe, Y. Niwa, M. Kitano and H. Hosono, Adv. Energy Mater., 2021, 11, 2003723 CrossRef CAS.
- H. Mizoguchi, M. Okunaka, M. Kitano, S. Matsuishi, T. Yokoyama and H. Hosono, Inorg. Chem., 2016, 55, 8833–8838 CrossRef CAS PubMed.
- M. Hattori, S. Iijima, T. Nakao, H. Hosono and M. Hara, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- H. Michikazu, H. Masashi, O. Natsuo and K. Hiyori, Nat. Portf, 2022, 1–15 Search PubMed.
- Y. Ogura, K. Sato, S. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230–2237 RSC.
- Y. Ogura, K. Tsujimaru, K. Sato, S. Miyahara, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustainable Chem. Eng., 2018, 6, 17258–17266 CrossRef CAS.
- K. Sato, S. Miyahara, Y. Ogura, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustainable Chem. Eng., 2020, 8, 2726–2734 CrossRef CAS.
- Y. Inoue, M. Kitano, K. Kishida, H. Abe, Y. Niwa, M. Sasase, Y. Fujita, H. Ishikawa, T. Yokoyama, M. Hara and H. Hosono, ACS Catal., 2016, 6, 7577–7584 CrossRef CAS.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Angew. Chem., Int. Ed., 2018, 57, 2648–2652 CrossRef CAS PubMed.
- H. Yamamoto, H. Miyaoka, S. Hino, H. Nakanishi, T. Ichikawa and Y. Kojima, Int. J. Hydrogen Energy, 2009, 34, 9760–9764 CrossRef CAS.
- P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS PubMed.
- Y. Kojima, Y. Kawai and N. Ohba, J. Power Sources, 2006, 159, 81–87 CrossRef CAS.
- W. Gao, J. Guo, P. Wang, Q. Wang, F. Chang, Q. Pei, W. Zhang, L. Liu and P. Chen, Nat. Energy, 2018, 3, 1067–1075 CrossRef CAS.
- K. H. R. Rouwenhorst, A. G. J. Van der Ham, G. Mul and S. R. A. Kersten, Renew. Sustainable Energy Rev., 2019, 114, 109339 CrossRef CAS.
- A. S. Travis, Casale: The first 100 years, Casale S.A., 2022 Search PubMed.
- A. S. Travis, Nitrogen Capture: The Growth of an International Industry (1900–1940), Springer International Publishing, Cham (Switzerland), 2018, pp. 93–127 Search PubMed.
- A. S. Travis, Nitrogen Capture: The Growth of an International Industry (1900–1940), Springer International Publishing, Cham (Switzerland), 2018, pp. 347–360 Search PubMed.
- V. Pattabathula and J. Richardson, Chem. Eng. Prog., 2016, 112, 69–75 Search PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- G. N. Schrauzer, in Energy Efficiency and Renewable Energy Through Nanotechnology, ed. L. Zang, Springer London, London, 2011, pp. 601–623 Search PubMed.
- Y. Liu, Y. Xue, L. Hui, H. Yu, Y. Fang, F. He and Y. Li, Nano Energy, 2021, 89, 106333 CrossRef CAS.
- G. Liu, Z. Tang, X. Gu, N. Li, H. Lv, Y. Huang, Y. Zeng, M. Yuan, Q. Meng, Y. Zhou and C. Wang, Appl. Catal., B, 2022, 317, 121752 CrossRef CAS.
- S. Zhang, X. Rong, T. Sun, P. Gao, J. Liu, X. Qiu, X. Zhou and Z. Wu, Diam. Relat. Mater., 2023, 138, 110167 CrossRef CAS.
- P. Li, Z. Zhou, Q. Wang, M. Guo, S. Chen, J. Low, R. Long, W. Liu, P. Ding, Y. Wu and Y. Xiong, J. Am. Chem. Soc., 2020, 142, 12430–12439 CrossRef CAS PubMed.
- A. R. S. C. Lazuli, R. Thapa and B. Neppolian, Catal. Today, 2023, 420, 114034 CrossRef.
- X. Rong, Y. Mao, J. Xu, X. Zhang, L. Zhang, X. Zhou, F. Qiu and Z. Wu, Catal. Commun., 2018, 116, 16–19 CrossRef CAS.
- S. Cao, B. Fan, Y. Feng, H. Chen, F. Jiang and X. Wang, Chem. Eng. J., 2018, 353, 147–156 CrossRef CAS.
- S. Sultana, S. Mansingh and K. M. Parida, J. Mater. Chem. A, 2019, 7, 9145–9153 RSC.
- R. Bariki, S. K. Pradhan, S. Panda, S. K. Nayak, A. R. Pati and B. G. Mishra, Langmuir, 2023, 39, 7707–7722 CrossRef CAS PubMed.
- H. Yin, Z. Chen, Y. Peng, S. Xiong, Y. Li, H. Yamashita and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202114242 CrossRef CAS PubMed.
- J. Fan, M. Zuo, Z. Ding, Z. Zhao, J. Liu and B. Sun, Chem. Eng. J., 2020, 396, 125263 CrossRef CAS.
- X. Li, X. Sun, L. Zhang, S. Sun and W. Wang, J. Mater. Chem. A, 2018, 6, 3005–3011 RSC.
- M. H. Urgesa, G. S. Wolde and D.-H. Kuo, Chem. Eng. J., 2023, 464, 142717 CrossRef CAS.
- H. Zeng, S. Terazono and T. Tanuma, Catal. Commun., 2015, 59, 40–44 CrossRef CAS.
- X. Wang, B. Wang, S. Yin, M. Xu, L. Yang and H. Sun, J. Cleaner Prod., 2022, 360, 132162 CrossRef CAS.
- Z. Ding, S. Wang, X. Chang, D. H. Wang and T. Zhang, RSC Adv., 2020, 10, 26246–26255 RSC.
- R. Tao, X. Li, X. Li, C. Shao and Y. Liu, Nanoscale, 2020, 12, 8320–8329 RSC.
- Y. Bai, L. Ye, T. Chen, L. Wang, X. Shi, X. Zhang and D. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 27661–27668 CrossRef CAS PubMed.
- D. Prusty, S. Mansingh and K. M. Parida, Catal. Sci. Technol., 2023, 13, 1311–1324 RSC.
- Y. Xue, C. Ma, Q. Yang, X. Wang, S. An, X. Zhang and J. Tian, Chem. Eng. J., 2023, 457, 141146 CrossRef CAS.
- M. H. Urgesa, G. S. Wolde and D.-H. Kuo, J. Alloys Compd., 2023, 947, 169589 CrossRef CAS.
- S. Liu, M. Wang, H. Ji, L. Zhang, J. Ni, N. Li, T. Qian, C. Yan and J. Lu, Adv. Mater., 2023, 35, 2211730 CrossRef CAS PubMed.
- Y. Fang, Y. Xue, L. Hui, H. Yu and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3170–3174 CrossRef CAS PubMed.
- Y. Xue, Y. Guo, Z. Liang, H. Cui and J. Tian, J. Colloid Interface Sci., 2019, 556, 206–213 CrossRef CAS PubMed.
- B. Bagherpour and S. Dehghanpour, J. Solid State Chem., 2023, 324, 124079 CrossRef CAS.
- M. Lan, N. Zheng, X. Dong, H. Ma and X. Zhang, Colloids Surf., A, 2021, 623, 126744 CrossRef CAS.
- J. Wang, Y. Fang, W. Zhang, X. Yu, L. Wang and Y. Zhang, Appl. Surf. Sci., 2021, 567, 150623 CrossRef CAS.
- L. Shi, Z. Li, L. Ju, A. Carrasco-Pena, N. Orlovskaya, H. Zhou and Y. Yang, J. Mater. Chem. A, 2020, 8, 1059–1065 RSC.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1–7 Search PubMed.
- Y. Hao, X. Dong, S. Zhai, H. Ma, X. Wang and X. Zhang, Chem. – Eur. J., 2016, 22, 18722–18728 CrossRef CAS PubMed.
- I. F. de Sá, P. H. P. R. Carvalho, H. A. Centurion, R. V. Gonçalves and J. D. Scholten, ACS Sustainable Chem. Eng., 2021, 9, 8721–8730 CrossRef.
- X. Feng, H. Chen, F. Jiang and X. Wang, Catal. Sci. Technol., 2019, 9, 2849–2857 RSC.
- H. Zhang, Y. Chen, L. Bao and Y. J. Yuan, J. Catal., 2022, 412, 1–9 CrossRef CAS.
- T.-A. Bu, Y.-C. Hao, W.-Y. Gao, X. Su, L.-W. Chen, N. Zhang and A.-X. Yin, Nanoscale, 2019, 11, 10072–10079 RSC.
- Q. Ding, X. Zou, J. Ke, Y. Dong, Y. Cui and H. Ma, J. Colloid Interface Sci., 2023, 649, 148–158 CrossRef CAS PubMed.
- N. Zhang, L. Li, Q. Shao, T. Zhu, X. Huang and X. Xiao, ACS Appl. Energy Mater., 2019, 2, 8394–8398 CrossRef CAS.
- Y. H. Liu, C. A. Fernández, S. A. Varanasi, N. N. Bui, L. Song and M. C. Hatzell, ACS Energy Lett., 2022, 7, 24–29 CrossRef CAS.
- Y. Shen, J. Shou, L. Chen, W. Han, L. Zhang, Y. Chen, X. Tu, S. Zhang, Q. Sun, Y. Chang and H. Zheng, Appl. Catal., A, 2022, 643, 118739 CrossRef CAS.
- L. Chen, J. Shou, Y. Chen, W. Han, X. Tu, L. Zhang, Q. Sun, J. Cao, Y. Chang and H. Zheng, Chem. Eng. J., 2023, 451, 138592 CrossRef CAS.
- Y. Shen, L. Chen, L. Zhang, W. Han, M. Jiang and H. Zheng, Mol. Catal., 2022, 518, 112091 CrossRef CAS.
- Y. Shen, L. Chen, L. Zhang, W. Han, Y. Chang and H. Zheng, Mol. Catal., 2022, 524, 112338 CrossRef CAS.
- K. Pournemati, A. Habibi-Yangjeh and A. Khataee, J. Colloid Interface Sci., 2023, 641, 1000–1013 CrossRef CAS PubMed.
- W. Zhao, J. Qin, W. Teng, J. Mu, C. Chen, J. Ke, J. C. Huang, B. Liu and S. Wang, Appl. Catal., B, 2022, 305, 121046 CrossRef CAS.
- S. Rostami, R. Tayebee and B. Mahdavi, RSC Adv., 2023, 13, 31303–31313 RSC.
- T. Wang, J. Liu, P. Wu, C. Feng, D. Wang, H. Hu and G. Xue, J. Mater. Chem. A, 2020, 8, 16590–16598 RSC.
- D. R. MacFarlane, A. N. Simonov, T. M. Vu, S. Johnston and L. M. Azofra, Faraday Discuss., 2023, 243, 557–570 RSC.
- A. J. Bard, Science, 1980, 207, 139–144 CrossRef CAS PubMed.
- M. Bellardita, V. Loddo, F. Parrino and L. Palmisano, ChemPhotoChem, 2021, 5, 767–791 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- D. R. Strongin and G. A. Somorjai, Surface Science and Catalytic Study of the Effects of Aluminum Oxide and Potassium on the Ammonia Synthesis Over Iron Single-Crystal Surfaces, Catalytic Ammonia Synthesis: Fundamentals and Practice, Springer US, 1991, ch. 4, pp. 133–177 Search PubMed.
- J. Brillet, J. H. Yum, M. Cornuz, T. Hisatomi, R. Solarska, J. Augustynski, M. Graetzel and K. Sivula, Nat. Photonics, 2012, 6, 824–828 CrossRef CAS.
- Y. Shi, T. Y. Hsieh, M. A. Hoque, W. Cambarau, S. Narbey, C. Gimbert-Surinãch, E. Palomares, M. Lanza and A. Llobet, ACS Appl. Mater. Interfaces, 2020, 12, 55856–55864 CrossRef CAS PubMed.
- D. Cardenas-Morcoso, M. García-Tecedor, T. Merdzhanova, V. Smirnov, F. Finger, B. Kaiser, W. Jaegermann and S. Gimenez, Mater. Adv., 2020, 1, 1202–1211 RSC.
- Y. J. Jang, A. E. Lindberg, M. A. Lumley and K. Choi, ACS Energy Lett., 2020, 5, 1834–1839 CrossRef CAS.
- Y. Jia, J. Gao, Z. Xiao, Z. Tian, Y. Xia and C. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 26111–26119 CrossRef CAS PubMed.
- Y. Pang, C. Su, G. Jia, L. Xu and Z. Shao, Chem. Soc. Rev., 2021, 50, 12744–12787 RSC.
- Y. Zhao, Z. Niu, J. Zhao, L. Xue, X. Fu and J. Long, Recent Advancements in Photoelectrochemical Water Splitting for Hydrogen Production, Springer Nature Singapore, 2023, vol. 6 Search PubMed.
- W. Zhang, Z. Jin and Z. Chen, Adv. Sci., 2022, 9, 1–30 Search PubMed.
- A. Das, K. Panigrahi and P. Howli, Catal. Today, 2023, 423, 113979 CrossRef CAS.
- Y. He, M. Wang, L. Zhang, Q. Cheng, S. Liu, X. Sun, Y. Jiang, T. Qian and C. Yan, Adv. Funct. Mater., 2024, 2315548 CrossRef CAS.
- K. Chen, X. Xu, Q. Mei, J. Huang, G. Yang and Q. Wang, Appl. Catal., B, 2024, 341, 123299 CrossRef CAS.
- S. Lin, J. B. Ma, J. J. Fu, L. Sun, H. Zhang, J. Cheng and J. F. Li, J. Phys. Chem. C, 2023, 127, 1345–1354 CrossRef CAS.
- P. Gnanasekar, K. Peramaiah, H. Zhang, M. K. Eswaran, R. R. Pradhan, U. Schwingenschlögl, T. K. Ng, Q. Gan, J. Kulandaivel, K.-W. Huang and B. S. Ooi, ACS Appl. Energy Mater., 2023, 6, 10784–10793 CrossRef CAS.
- R. Karimi, F. Yousefi, M. Ghaedi, K. Dashtian and G. Yasin, J. Environ. Chem. Eng., 2022, 10, 108549 CrossRef CAS.
- W. Ye, C. He, M. A. Mushtaq, K. Lin and X. Xing, Eur. J. Inorg. Chem., 2022, e202200325 CrossRef CAS.
- M. A. Mushtaq, A. Kumar, G. Yasin, M. Arif, M. Tabish, S. Ibraheem, X. Cai, W. Ye, X. Fang, A. Saad, J. Zhao, S. Ji and D. Yan, Appl. Catal., B, 2022, 317, 121711 CrossRef CAS.
- N. Gao, H. Yang, D. Dong, D. Dou, Y. Liu, W. Zhou, F. Gao, C. Nan, Z. Liang and D. Yang, J. Colloid Interface Sci., 2022, 611, 294–305 CrossRef CAS PubMed.
- L. P. Camargo, P. R. C. da Silva, A. Batagin-Neto, V. Klobukoski, M. Vidotti and L. H. Dall’Antonia, Appl. Mater. Today, 2022, 28, 101540 CrossRef.
- Y. Bai, H. Bai, Z. Fang, X. Li, W. Fan and W. Shi, Chem. Commun., 2021, 57, 10568–10571 RSC.
- Y. Bai, J. Lu, H. Bai, Z. Fang, F. Wang, Y. Liu, D. Sun, B. Luo, W. Fan and W. Shi, Chem. Eng. J., 2021, 414, 128773 CrossRef CAS.
- K. Bi, Y. Wang, D. M. Zhao, J. Z. Wang, D. Bao and M. M. Shi, J. Mater. Chem. A, 2021, 9, 10497–10507 RSC.
- J. Zheng, Y. Lyu, J. P. Veder, B. Johannessen, R. Wang, R. De Marco, A. Huang, S. P. Jiang and S. Wang, J. Phys. Chem. C, 2021, 125, 23041–23049 CrossRef CAS.
- M. A. Mushtaq, M. Arif, X. Fang, G. Yasin, W. Ye, M. Basharat, B. Zhou, S. Yang, S. Ji and D. Yan, J. Mater. Chem. A, 2021, 9, 2742–2753 RSC.
- M.-H. Vu, C.-C. Nguyen and T.-O. Do, ACS Sustainable Chem. Eng., 2020, 8, 12321–12330 CrossRef CAS PubMed.
- D. Liu, J. Wang, S. Bian, Q. Liu, Y. Gao, X. Wang, P. K. Chu and X. F. Yu, Adv. Funct. Mater., 2020, 30, 1–7 Search PubMed.
- M. Li, Q. Lu, M. Liu, P. Yin, C. Wu, H. Li, Y. Zhang and S. Yao, ACS Appl. Mater. Interfaces, 2020, 12, 38266–38274 CrossRef CAS PubMed.
- W. Ye, M. Arif, X. Fang, M. A. Mushtaq, X. Chen and D. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 28809–28817 CrossRef CAS PubMed.
- Y. Bai, H. Bai, K. Qu, F. Wang, P. Guan, D. Xu, W. Fan and W. Shi, Chem. Eng. J., 2019, 362, 349–356 CrossRef CAS.
- Y. Liu, H. Bai, Q. Zhang, Y. Bai, X. Pang, F. Wang, Y. Yang, J. Ding, W. Fan and W. Shi, Chem. Eng. J., 2021, 413, 127453 CrossRef CAS.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- D. V. J. Li, Y. Zhang, C. Liu, L. Zheng, E. Petit, K. Qi, Y. Zhang, H. Wu, W. Wang, A. Tiberj, X. Wang, M. Chhowalla, L. Lajaunie and R. Yu, Adv. Funct. Mater., 2021, 32, 2108316 CrossRef.
- K. Y. C. Lim, H. Roh, E. H. Kim, H. Kim, T. Park and D. Lee, Small, 2023, 2304274 CrossRef PubMed.
- S. K. Singh, R. K. Rai and D. Tyagi, Eur. J. Inorg. Chem., 2017, 2450–2456 Search PubMed.
- S. Xu, D. C. Ashley, H. Y. Kwon, G. R. Ware, C. H. Chen, Y. Losovyj, X. Gao, E. Jakubikova and J. M. Smith, Chem. Sci., 2018, 9, 4950–4958 RSC.
- A. Stirling, I. Pápai, J. Mink and D. R. Salahub, J. Chem. Phys., 1994, 100, 2910–2923 CrossRef CAS.
- E. E. Van Tamelen and B. Akermark, J. Am. Chem. Soc., 1968, 90, 4492–4493 CrossRef CAS.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- K. Kim, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F1523 CrossRef CAS.
- L. M. Wilder, K. Wyatt, C. A. Skangos, W. E. Klein, M. R. Parimuha, J. L. Katsirubas, J. L. Young and E. M. Miller, ACS Appl. Energy Mater., 2024, 7, 536–545 CrossRef CAS PubMed.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC.
- M. Gao, T. Zhang and G. W. Ho, Nano Res., 2022, 15, 9985–10005 CrossRef CAS.
- Y. Peng, J. Albero, A. Franconetti, P. Concepción and H. García, ACS Catal., 2022, 12, 4938–4946 CrossRef CAS PubMed.
- Y. Peng, A. Melillo, R. Shi, A. Forneli, A. Franconetti, J. Albero and H. García, Appl. Catal., B, 2023, 339, 123143 CrossRef CAS.
- X. Li, X. Zhang, H. O. Everitt and J. Liu, Nano Lett., 2019, 19, 1706–1711 CrossRef CAS PubMed.
- X. Bian, Y. Zhao, G. I. N. Waterhouse, Y. Miao, C. Zhou, L. Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, 1–8 Search PubMed.
- J. Zheng, L. Lu, K. Lebedev, S. Wu, P. Zhao, I. J. McPherson, T.-S. Wu, R. Kato, Y. Li, P.-L. Ho, G. Li, L. Bai, J. Sun, D. Prabhakaran, R. A. Taylor, Y.-L. Soo, K. Suenaga and S. C. E. Tsang, Chem. Catal., 2021, 1, 162–182 CrossRef CAS.
- C. Mao, J. Wang, Y. Zou, Y. Shi, C. J. Viasus, J. Y. Y. Loh, M. Xia, S. Ji, M. Li, H. Shang, M. Ghoussoub, Y. F. Xu, J. Ye, Z. Li, N. P. Kherani, L. Zheng, Y. Liu, L. Zhang and G. A. Ozin, J. Am. Chem. Soc., 2023, 145, 13134–13146 CrossRef CAS PubMed.
- S. Wang, W. Yu, S. Xu, K. Han and F. Wang, ACS Sustainable Chem. Eng., 2022, 10, 115–123 CrossRef CAS.
- C. Mao, L. Yu, J. Li, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 224, 612–620 CrossRef CAS.
- W. K. Fan and M. Tahir, Chem. Eng. J., 2022, 427, 131617 Search PubMed.
- L. F. Greenlee, J. N. Renner and S. L. Foster, ACS Catal., 2018, 8, 7820–7827 CrossRef CAS.
- Y. Zhao, F. Wu, Y. Miao, C. Zhou, N. Xu, R. Shi, L. Wu, J. Tang and T. Zhang, Angew. Chem., 2021, 133, 21896–21899 CrossRef.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- M. Li, H. Huang, J. Low, C. Gao, R. Long and Y. Xiong, Small Methods, 2018, 1800388 Search PubMed.
- K. Wang, D. Smith and Y. Zheng, Carbon Resour. Convers., 2018, 1, 2–31 CrossRef.
- M. Kolen, W. A. Smith and F. M. Mulder, ACS Omega, 2021, 6, 5698–5704 CrossRef CAS PubMed.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- S. J. Yuan, J. J. Chen, Z. Q. Lin, W. W. Li, G. P. Sheng and H. Q. Yu, Nat. Commun., 2013, 4, 2249 CrossRef PubMed.
- Y. Shiraishi, M. Hashimoto, K. Chishiro, K. Moriyama, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2020, 142, 7574–7583 CrossRef CAS PubMed.
- C. Gal, W. Frenzel and J. Möller, Microchim. Acta, 2004, 146, 155–164 CrossRef CAS.
- P. Griess, Ber. Dtsch. Chem. Ges., 1879, 12, 426–428 CrossRef.
- M. J. Moorcroft, J. Davis and R. G. Compton, Talanta, 2001, 54, 785–803 CrossRef CAS PubMed.
- A. Machado, G. Marshall, A. A. Bordalo and R. B. R. Mesquita, Anal. Methods, 2017, 9, 1876–1884 RSC.
- G. F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L. X. Ding and H. Wang, Small Methods, 2019, 3, 1–20 Search PubMed.
- Y. Shiraishi, S. Shiota, Y. Kofuji, M. Hashimoto, K. Chishiro, H. Hirakawa, S. Tanaka, S. Ichikawa and T. Hirai, ACS Appl. Energy Mater., 2018, 1, 4169–4177 CrossRef CAS.
- B. M. Comer, P. Fuentes, C. O. Dimkpa, Y. H. Liu, C. A. Fernandez, P. Arora, M. Realff, U. Singh, M. C. Hatzell and A. J. Medford, Joule, 2019, 3, 1578–1605 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- J. Choi, B. H. R. Suryanto, D. Wang, H. Du, R. Y. Hodgetts, F. M. F. Vallana, D. R. Macfarlane and A. N. Simonov, Nat. Commun., 2020, 11(5546), 1–10 Search PubMed.
- L. Li, C. Tang, D. Yao, Y. Zheng and S. Z. Qiao, ACS Energy Lett., 2019, 4, 2111–2116 CrossRef CAS.
- P. W. Huang and M. C. Hatzell, Nat. Commun., 2022, 13, 1–7 Search PubMed.
- E. Truszkiewicz, W. Raróg-Pilecka, K. Schmidt-Szałowski, S. Jodzis, E. Wilczkowska, D. Łomot, Z. Kaszkur, Z. Karpiński and Z. Kowalczyk, J. Catal., 2009, 265, 181–190 CrossRef CAS.
- L.-M. Kwedi-Nsah and T. Kobayashi, Ultrason. Sonochem., 2020, 66, 105051 CrossRef CAS PubMed.
- L. Collado, P. Reñones, J. Fermoso, F. Fresno, L. Garrido, V. Pérez-Dieste, C. Escudero, M. D. Hernández-Alonso, J. M. Coronado, D. P. Serrano and V. A. de la Peña O’Shea, Appl. Catal., B, 2022, 303, 120931 CrossRef CAS.
- G. Chehade and I. Dincer, Fuel, 2021, 299, 120845 CrossRef CAS.
- D. R. Strongin and G. A. Somorjai, Surface Science and Catalytic Study of the Effects of Aluminum Oxide and Potassium on the Ammonia Synthesis Over Iron Single-Crystal Surfaces, in Catalytic Ammonia Synthesis: Fundamentals and Practice, ed. J. R. Jennings, Springer US, 1991, ch. 4, pp. 133–177 Search PubMed.
- A. Patonia, Ammonia as a storage solution for future decarbonized energy systems, 2020 Search PubMed.
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustainable Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. De Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Energy Fuels, 2021, 35, 6964–7029 CrossRef CAS.
- J. W. Makepeace, T. J. Wood, H. M. A. Hunter, M. O. Jones and W. I. F. David, Chem. Sci., 2015, 6, 3805–3815 RSC.
- M. E. Solutions, Unlocking ammonia's potential for shipping, https://www.man-es.com/discover/two-stroke-ammonia-engine.
- Wärtsilä, AMMONIA 2-4 project, https://www.ammonia2-4.eu/.
- A. E. Association, Ammonia fueled sports car: Marangoni Toyota GT86 Eco Explorer, https://www.ammoniaenergy.org/articles/ammonia-fueled-sports-car-marangoni-toyota-gt86-eco-explorer/ Search PubMed.
- International Energy Agency, Net Zero by 2050: A Roadmap for the Global Energy Sector, 2021.
- JERA, JERA and IHI to Start a Demonstration Project Related to Ammonia Co-firing at a Large-Scale Commercial Coal-Fired Power Plant, https://www.jera.co.jp/en/news/information/20210524_677.
- N. Morlanés, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, Chem. Eng. J., 2021, 408, 127310 CrossRef.
- N. Salmon and R. Bañares-Alcántara, Sustainable Energy Fuels, 2021, 5, 2814–2839 RSC.
- K. Smart, Johnson Matthey Technol. Rev., 2021, 230–244 Search PubMed.
- L. Ye, R. Nayak-Luke, R. Bañares-Alcántara and E. Tsang, Chemistry, 2017, 3, 712–714 CrossRef CAS.
- A. E. Association, H2biscus: green ammonia in Malaysia, https://www.ammoniaenergy.org/articles/h2biscus-green-ammonia-in-malaysia/.
- Ammonia Energy Association, Blue ammonia in the Northern Territory & Wyoming, https://www.ammoniaenergy.org/articles/blue-ammonia-in-the-northern-territory-wyoming/.
- Ammonia Energy Association, $4.5 billion blue ammonia project in Louisiana, https://www.ammoniaenergy.org/articles/4-5-billion-blue-ammonia-project-in-louisiana/.
- Ammonia Energy Association, “Jordan Green Ammonia”: a new ammonia partnership in the Middle East, https://www.ammoniaenergy.org/articles/jordan-green-ammonia-a-new-ammonia-partnership-in-the-middle-east/.
- G. Centi and S. Perathoner, Green Chem., 2023, 26, 15–41 RSC.
- O. A. Ojelade, S. F. Zaman and B. J. Ni, J. Environ. Manage., 2023, 342, 118348 CrossRef PubMed.
- Clean Ammonia in the Future Energy System. Hydrogen Europe., 2023.
- Kapsom, The World's First Green Ammonia Plant Was Made By Kapsom., https://www.kapsom.com/the-worlds-first-green-ammonia-plant-was-made-by-kapsom/#:˜:text=Green ammonia produced from renewable,to a renewable energy drive.
- ENGIE-YARA, Renewable Hydrogen and Ammonia Deployment in Pilbara. YURI Phase 0: Feasibility study public report, 2020.
- H2F project. Green hydrogen, green ammonia and green fertiliser plant, https://www.fertiberia.com/en/amoniacoverde/h2f-project/ Search PubMed.
- CEPSA, Cepsa and Fertiberia form a strategic alliance to boost green hydrogen production and decarbonize industry in Huelva., https://www.cepsa.com/en/press/cepsa-and-fertiberia-will-produce-green-hydrogen Search PubMed.
- Kapsom, The First 2,000 TPA Green Ammonia Pilot Project in South America, https://www.kapsom.com/the-first- 2000-tpa-green-ammonia-pilot-project-in-south-america/ (http://-tpa-green-ammonia-pilot-project-in-south-america/) Search PubMed.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- S. A. Nosherwani and R. C. Neto, J. Energy Storage, 2021, 34, 102201 CrossRef.
- The World Bank indicators.
- USGS, historical-statistics-mineral-and-material-commodities. Nitrogen Search PubMed.
- Nitrogen (fixed) ammonia statistics.
- USGS.
- W. Bank, Worls Bank Commodity Prices.
- Statista, Distribution of agricultural fertilizer production capacity worldwide in 2021, by region.
- R. F. Service, Science, 2018, 361, 120–123 CrossRef CAS PubMed.
- OECD/IEA, Tracking industrial energy efficiency and CO2 emissions, Paris, 2007.
- D. Saygin, H. Blanco, F. Boshell, J. Cordonnier, K. Rouwenhorst, P. Lathwal and D. Gielen, Sustainability, 2023, 15 Search PubMed.
- M. Wang, M. A. Khan, I. Mohsin, J. Wicks, A. H. Ip, K. Z. Sumon, C. T. Dinh, E. H. Sargent, I. D. Gates and M. G. Kibria, Energy Environ. Sci., 2021, 14, 2535–2548 RSC.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Ammonia Production Technologies, Techno-Economic Challenges of Green Ammonia as an Energy Vector, Elsevier Inc., 2021, ch. 4, pp. 41–83 Search PubMed.
- V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou and M. Stoukides, Joule, 2020, 4, 142–158 CrossRef CAS.
- A. Raghuveer and Tejasvi Wilczewski, Today in Energy, 2022 Search PubMed.
- S. Canel Soria and S. Soh, Analysis: Global ammonia prices fall 50% on year, sparking concerns over future low-carbon market, https://cilive.com/commodities/energy-transition/thought-leadership/082323-conflive-analysis-global-ammonia-prices-fall.
- G. Hochman, A. S. Goldman, F. A. Felder, J. M. Mayer, A. J. M. Miller, P. L. Holland, L. A. Goldman, P. Manocha, Z. Song and S. Aleti, ACS Sustainable Chem. Eng., 2020, 8, 8938–8948 CrossRef CAS.
- A. Makhlouf, T. Serradj and H. Cheniti, Environ. Impact Assess. Rev., 2015, 50, 35–41 CrossRef.
- N. Morlanés, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, Chem. Eng. J., 2021, 408, 127310 CrossRef.
- G. Schnitkey, Farmdoc Dly.
- J. Bartels, PhD thesis, Iowa State University, 2008.
- UNIDO & IFCD, Fertilizer Manual, Kluwer Academic Publisher, 1998 Search PubMed.
- T. A. Czuppon, S. A. Knez and J. M. Rovner, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. C. Ley, Ammonia, Wiley, 5th edn, 2000 Search PubMed.
- D. Frattini, G. Cinti, G. Bidini, U. Desideri, R. Cioffi and E. Jannelli, Renewable Energy, 2016, 99, 472–482 CrossRef CAS.
- D. Saygin and D. Gielen, Energies, 2021, 14 Search PubMed.
- Our World in Data, Electricity production by source, World Search PubMed.
- Enerdata, World Energy & Climate Statistics-Yearbook 2022.
- A. Faaij, Mitig. Adapt. Strateg. Glob. Chang., 2006, 11, 343–375 CrossRef.
- D. B. Pal, A. Singh and A. Bhatnagar, Int. J. Hydrogen Energy, 2022, 47, 1461–1480 CrossRef CAS.
- B. Parkinson, P. Balcombe, J. F. Speirs, A. D. Hawkes and K. Hellgardt, Energy Environ. Sci., 2019, 12, 19–40 RSC.
- S. G. Nnabuife, J. Ugbeh-Johnson, N. E. Okeke and C. Ogbonnaya, Carbon Capture Sci. Technol., 2022, 3, 100042 CrossRef CAS.
- J. R. Bartels, M. B. Pate and N. K. Olson, Int. J. Hydrogen Energy, 2010, 35, 8371–8384 CrossRef CAS.
- P. Arora, A. F. A. Hoadley, S. M. Mahajani and A. Ganesh, Ind. Eng. Chem. Res., 2016, 55, 6422–6434 CrossRef CAS.
- P. Gilbert, S. Alexander, P. Thornley and J. Brammer, J. Cleaner Prod., 2014, 64, 581–589 CrossRef CAS.
- P. Tunå, C. Hulteberg and S. Ahlgren, Environ. Prog. Sustainable Energy, 2014, 33, 1290–1297 CrossRef.
- J. Andersson and J. Lundgren, Appl. Energy, 2014, 130, 484–490 CrossRef CAS.
- L. Tock, F. Maréchal and M. Perrenoud, Can. J. Chem. Eng., 2015, 93, 356–362 CrossRef CAS.
- J. Armijo and C. Philibert, Int. J. Hydrogen Energy, 2020, 45, 1541–1558 CrossRef CAS.
- X. Liu, A. Elgowainy and M. Wang, Green Chem., 2020, 22, 5751–5761 RSC.
- J. Ikäheimo, J. Kiviluoma, R. Weiss and H. Holttinen, Int. J. Hydrogen Energy, 2018, 43, 17295–17308 CrossRef.
- P. H. Pfromm, J. Renew. Sustainable Energy, 2017, 9, 34702 CrossRef.
- H2view, Plug Power reveals record production of 122MW PEM electrolyser stacks in Q1.
- Y. Ishimoto, M. Voldsund, P. Nekså, S. Roussanaly, D. Berstad and S. O. Gardarsdottir, Int. J. Hydrogen Energy, 2020, 45, 32865–32883 CrossRef CAS.
- M. H. Hasan, T. M. I. Mahlia, M. Mofijur, I. M. Rizwanul Fattah, F. Handayani, H. C. Ong and A. S. Silitonga, A comprehensive review on the recent development of ammonia as a renewable energy carrier, Energies, 2021, 14, 3732 CrossRef CAS.
- A. H. Reksten, M. S. Thomassen, S. Møller-Holst and K. Sundseth, Int. J. Hydrogen Energy, 2022, 47, 38106–38113 CrossRef CAS.
- G. Bristowe and A. Smallbone, Hydrogen, 2021, 2, 273–300 CrossRef CAS.
- A. Sánchez and M. Martín, Sustainable Prod. Consum., 2018, 16, 176–192 CrossRef.
- C. Kost, S. Shammugam, V. Jülch, H.-T. Nguyen and T. Schlegl, Levelized cost of electricity, Renewable Energy Technol., 2018 Search PubMed.
- Z. Cesaro, M. Ives, R. Nayak-Luke, M. Mason and R. Bañares-Alcántara, Appl. Energy, 2021, 282, 116009 CrossRef CAS.
- K. H. R. Rouwenhorst, A. G. J. Van der Ham and L. Lefferts, Int. J. Hydrogen Energy, 2021, 46, 21566–21579 CrossRef CAS.
- M. Yoshida, T. Ogawa, Y. Imamura and K. N. Ishihara, Int. J. Hydrogen Energy, 2021, 46, 28840–28854 CrossRef CAS.
- H. Zhang, L. Wang, J. Van herle, F. Maréchal and U. Desideri, Appl. Energy, 2020, 259, 114135 CrossRef CAS.
- R. Bañares-Alcántara, G. Dericks, M. Fiaschetti, G. Philipp, L. Joaquín Masa, E. Tsang, Y. Aidong, Y. Lin and Z. Shangyi, Analysis of Islanded Ammonia-based Energy Storage Systems, Oxford, 2015 Search PubMed.
- M. Matzen, M. Alhajji and Y. Demirel, J. Adv. Chem. Eng., 2015, 5, 3 Search PubMed.
- V. Parmar, J. Manwell and J. McGowan, J. Phys. Conf. Ser., 2020, 1452, 012015 CrossRef CAS.
- ISPT, Power to Ammonia: Feasibility study for the value chains and business cases to produce CO2-free ammonia suitable for various market applications (REF TESI115001), 2017.
- G. Wang, A. Mitsos and W. Marquardt, AIChE J., 2017, 63, 1620–1637 CrossRef CAS.
- E. R. Morgan, J. F. Manwell and J. G. McGowan, ACS Sustainable Chem. Eng., 2017, 5, 9554–9567 CrossRef CAS.
- A. Sánchez and M. Martín, J. Clean. Prod., 2018, 178, 325–342 CrossRef.
- M. Rivarolo, G. Riveros-Godoy, L. Magistri and A. F. Massardo, ChemEngineering, 2019, 3 Search PubMed.
- R. Nayak-Luke, R. Bañares-Alcántara and I. Wilkinson, Ind. Eng. Chem. Res., 2018, 57, 14607–14616 CrossRef CAS.
- A. Allman, M. J. Palys and P. Daoutidis, AIChE J., 2019, 65, e16434 CrossRef.
- B. Lin, T. Wiesner and M. Malmali, ACS Sustainable Chem. Eng., 2020, 8, 15517–15531 CrossRef CAS.
- O. Osman, S. Sgouridis and A. Sleptchenko, J. Cleaner Prod., 2020, 271, 121627 CrossRef CAS.
- M. J. Palys and P. Daoutidis, Comput. Chem. Eng., 2020, 136, 106785 CrossRef CAS.
- C. Quintero-Masselski, J.-F. Portha and L. Falk, Chem. Eng. Res. Des., 2022, 177, 826–842 CrossRef CAS.
- N. Campion, H. Nami, P. R. Swisher, P. Vang Hendriksen and M. Münster, Renewable Sustainable Energy Rev., 2023, 173, 113057 CrossRef CAS.
- O. Elishav, D. R. Lewin, G. E. Shter and G. S. Grader, Appl. Energy, 2017, 185, 183–188 CrossRef CAS.
- P. Mayer, A. Ramirez, G. Pezzella, B. Winter, S. M. Sarathy, J. Gascon and A. Bardow, iScience, 2023, 26, 107389 CrossRef CAS PubMed.
- V. N. Sagel, K. H. R. Rouwenhorst and J. A. Faria, Renewable Sustainable Energy Rev., 2022, 161, 112381 CrossRef CAS.
- V. N. Sagel, K. H. R. Rouwenhorst and J. A. Faria, Energies, 2022, 15 Search PubMed.
- M. Fasihi, R. Weiss, J. Savolainen and C. Breyer, Appl. Energy, 2021, 294, 116170 CrossRef CAS.
- N. Salmon and R. Bañares-Alcántara, J. Clean. Prod., 2022, 367, 133045 CrossRef CAS.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Ullmann's Encycl. Ind. Chem., 2020, 1–20 Search PubMed.
- I. C. Ten Have, Feeding and fueling a greener future: everything you need to know about green ammonia Search PubMed.
- A. Patil, L. Laumans and H. Vrijenhoef, Procedia Eng., 2014, 83, 322–327 CrossRef CAS.
- C. A. Fernandez and M. C. Hatzell, J. Electrochem. Soc., 2020, 167, 143504 CrossRef CAS.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- F. B. Juangsa, A. R. Irhamna and M. Aziz, Int. J. Hydrogen Energy, 2021, 46, 14455–14477 CrossRef CAS.
- B. Yang, W. Ding, H. Zhang and S. Zhang, Energy Environ. Sci., 2021, 14, 672–687 RSC.
- Z.-H. Xue, S.-N. Zhang, Y.-X. Lin, H. Su, G.-Y. Zhai, J.-T. Han, Q.-Y. Yu, X.-H. Li, M. Antonietti and J.-S. Chen, J. Am. Chem. Soc., 2019, 141, 14976–14980 CrossRef CAS PubMed.
- H. He, Q. Q. Zhu, Y. Yan, H. W. Zhang, Z. Y. Han, H. Sun, J. Chen, C. P. Li, Z. Zhang and M. Du, Appl. Catal., B, 2022, 302, 120840 CrossRef CAS.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 341 CrossRef CAS PubMed.
- Y. Liu, Q. Li, X. Guo, X. Kong, J. Ke, M. Chi, Q. Li, Z. Geng and J. Zeng, Adv. Mater., 2020, 32, 1907690 CrossRef CAS PubMed.
- Y. Liu, L. Huang, X. Zhu, Y. Fang and S. Dong, Nanoscale, 2020, 12, 1811–1816 RSC.
- M. A. Mushtaq, M. Arif, G. Yasin, M. Tabish, A. Kumar, S. Ibraheem, W. Ye, S. Ajmal, J. Zhao, P. Li, J. Liu, A. Saad, X. Fang, X. Cai, S. Ji and D. Yan, Renewable Sustainable Energy Rev., 2023, 176, 113197 CrossRef CAS.
- Y. Bicer and I. Dincer, Int. J. Hydrogen Energy, 2019, 44, 18875–18890 CrossRef CAS.
- J. R. Gomez, J. Baca and F. Garzon, Int. J. Hydrogen Energy, 2020, 45, 721–737 CrossRef CAS.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- Y. Bicer and I. Dincer, ACS Sustainable Chem. Eng., 2017, 5, 8035–8043 CrossRef CAS.
- N. C. Kani, A. Prajapati and M. R. Singh, ACS ES&T Eng., 2022, 2, 1080–1087 Search PubMed.
- J. H. Guo, X. J. Liu, Y. Zhang, J. L. Shen, W. X. Han, W. F. Zhang, P. Christie, K. W. T. Goulding, P. M. Vitousek and F. S. Zhang, Science, 2010, 327, 1008–1010 CrossRef CAS PubMed.
- Y. Wei, W. Jiang, Y. Liu, X. Bai, D. Hao and B. J. Ni, Nanoscale, 2022, 14, 2990–2997 RSC.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- S. M. Swarbreck, M. Wang, Y. Wang, D. Kindred, R. Sylvester-Bradley, W. Shi, Varinderpal-Singh, A. R. Bentley and H. Griffiths, Trends Plant Sci., 2019, 24, 892–904 CrossRef CAS PubMed.
- Fertilizer consumption (kilograms per hectare of arable land)—World, 2021.
- R. Einarsson, A. Sanz-Cobena, E. Aguilera, G. Billen, J. Garnier, H. J. M. van Grinsven and L. Lassaletta, Sci. Data, 2021, 8, 288 CrossRef CAS PubMed.
- W. Adalibieke, X. Cui, H. Cai, L. You and F. Zhou, Sci. Data, 2023, 10, 617 CrossRef PubMed.
- World Bank, Fertilizer consumption (kilograms per hectare of arable land)—China, 2021.
- K. Smart, Johnson Matthey Technol. Rev., 2022, 66, 230–244 CrossRef CAS.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- J. Osorio-Tejada, N. N. Tran and V. Hessel, Sci. Total Environ, 2022, 826, 154162 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |