
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpin states of metal centers in electrocatalysis
Yuwei
Zhang†
a,
Qian
Wu†
 a,
Justin Zhu Yeow
Seow
ab,
Yingjie
Jia
c,
Xiao
Ren
*c and
Zhichuan J.
Xu
*ab
a,
Justin Zhu Yeow
Seow
ab,
Yingjie
Jia
c,
Xiao
Ren
*c and
Zhichuan J.
Xu
*ab
aSchool of Materials Science and Engineering, Nanyang Technological University, 639798, Singapore. E-mail: xuzc@ntu.edu.sg
bEnergy Research Institute@NTU (ERI@N), Interdisciplinary Graduate Programme, Nanyang Technological University, 639798, Singapore
cBeijing National Laboratory for Molecular Engineering, College of Chemistry and Molecular Engineering, Peking University, 100871, China. E-mail: renxiao_@pku.edu.cn
First published on 15th July 2024
Abstract
Understanding the electronic structure of active sites is crucial in efficient catalyst design. The spin state, spin configurations of d-electrons, has been frequently discussed recently. However, its systematic depiction in electrocatalysis is lacking. In this tutorial review, a comprehensive interpretation of the spin state of metal centers in electrocatalysts and its role in electrocatalysis is provided. This review starts with the basics of spin states, including molecular field theory, crystal field theory, and ligand field theory. It further introduces the differences in low spin, intermediate spin, and high spin, and intrinsic factors affecting the spin state. Popular characterization techniques and modeling approaches that can reveal the spin state, such as X-ray absorption microscopy, electron spin resonance spectroscopy, Mössbauer spectroscopy, and density functional theory (DFT) calculations, are introduced as well with examples from the literature. The examples include the most recent progress in tuning the spin state of metal centers for various reactions, e.g., the oxygen evolution reaction, oxygen reduction reaction, hydrogen evolution reaction, carbon dioxide reduction reaction, nitrogen reduction reaction, nitrate reduction reaction, and urea oxidation reaction. Challenges and potential implications for future research related to the spin state are discussed at the end.
 Yuwei Zhang | Yuwei Zhang is currently a PhD candidate in Professor Zhichuan J. Xu's group at the School of Materials Science and Engineering, Nanyang Technological University, Singapore. Her research focuses on electrocatalyst design for clean energy conversion. |
 Qian Wu | Qian Wu received her PhD from Shandong University. She is currently a Research Fellow at Nanyang Technological University working with Professor Zhichuan J. Xu. Her current research focuses on revealing the intrinsic mechanism of electrocatalytic activity, and engineering novel electrocatalysts for clean energy conversion. |
 Zhichuan J. Xu | Zhichuan J. Xu is a President's Chair Professor in the School of Materials Science and Engineering, Nanyang Technological University and a Fellow of the Academy of Engineering, Singapore. He serves as the director of the Centre of Excellence in Maritime Energy & Sustainable Development (MESD) and the director of the Centre of Advanced Catalysis Science and Technology. His major research interests lie in the fields of catalysis and related materials. |
Key learning points(1) Role of spin states in electronic structures for electrocatalytic reactions. (2) Fundamental principles of the spin state in the electronic structure of common first-row transition metals. (3) Extrinsic factors affecting the spin states and the regulating strategies. (4) Characterization techniques including experimental approaches and theoretic modeling methods to determine the spin state. (5) Challenges and future research directions related to spin states in first-row transition metals for electrocatalysis. |
1. Introduction
The current energy supply intensely relies on fossil fuel combustion, which leads to massive emission of greenhouse gases, posing a threat to the global ecosystem.1 Natural disasters and extreme weather events occur frequently, thereby endangering people's lives and impeding sustainable social development. Additionally, fossil fuels are vital raw materials for value-added chemical products, such as plastics, rubber, and lubricants. Therefore, finding ways to reduce the overall societal reliance on fossil fuels through energy source diversification is crucial to ensuring sustainable growth. Electrocatalytic reactions are involved in many renewable energy conversion and storage processes. For example, the production of green hydrogen is through water electrolysis powered by renewable energy sources.For electrocatalysts, diverse element compositions and different atomic coordinates are challenges in high-efficiency catalyst design. To overcome these challenges, studying the intrinsic properties of electrocatalysts is significant for understanding electrocatalyst behavior towards reactions. Great efforts2–4 have been made to reveal the relationship between the electrocatalyst structure and electrocatalysis performance at the electronic level, as well as the interaction between the electrocatalyst surface and reactants. Extensive attention has been paid to the interaction between reactant molecule orbitals and electron orbitals of atoms on the catalyst surface. The d band center theory proposed by Hammer and Norskov4 explains that the adsorption energy could be controlled by the antibonding state and orbital overlap integrals of the interacting atoms. Another descriptor eg occupancy,5 proposed by the Yang group, describes the near-unity occupancy of the eg orbital of surface transition metal ions and high covalency as optimized parameters to enhance the intrinsic activity of catalysts. The success of the relationship between the electronic structure and electrocatalyst performance proves the necessity of understanding at the electronic level and calls for researchers to delve into more fundamental levels.
In the past decade, the spin states of 3d transition metal centers in catalysts have garnered considerable attention. As a regulatory factor at the electronic level, the role of spin states is highlighted in electrocatalysts based on Cr,6 Mn,7 Fe,8–10 Co,11–19 and Ni.20,21 The involved electrocatalytic reactions include the oxygen evolution reaction (OER),11–19 oxygen reduction reaction (ORR),8–10,22–26 hydrogen evolution reaction (HER),27–30 carbon dioxide reduction reaction (CO2RR),31,32 nitrogen reduction reaction (NRR),6,33,34 nitrate reduction reaction (NO3−RR),35,36 and urea oxidation reaction (UOR).21 The role of spin states in central metals reflects the ability of metal sites to interact with intermediates and can be revealed in the electrocatalytic performance. However, attention toward the key role of the spin state in electrocatalysis is deficient. A systematic spin state depiction in electrocatalysis is lacking.
In this review, we outline the role of the spin state of metal centers in electrocatalysis, aiming to provide a tutorial for researchers in spin-state-related electrocatalysis. This review focusing on the spin state covers the origin, approaches, applications, and perspectives (Fig. 1). The spin state nature is illustrated with three classic principles, crystal field theory, molecular orbital theory, and ligand field theory. Approaches to characterizing the spin state through experimental techniques and theoretical modeling are described and the strengths of each approach are discussed. The specific role of spin states in the electrocatalytic reactions is analyzed, covering the OER, ORR, HER, CO2RR, CORR, NRR, NO3−RR and UOR. Finally, we propose the challenges and opportunities in spin-state-related electrocatalysis. Through this review, we emphasize the vital role of the spin state in electrocatalysis research and encourage researchers in the electrocatalysis field to take the spin state into consideration.
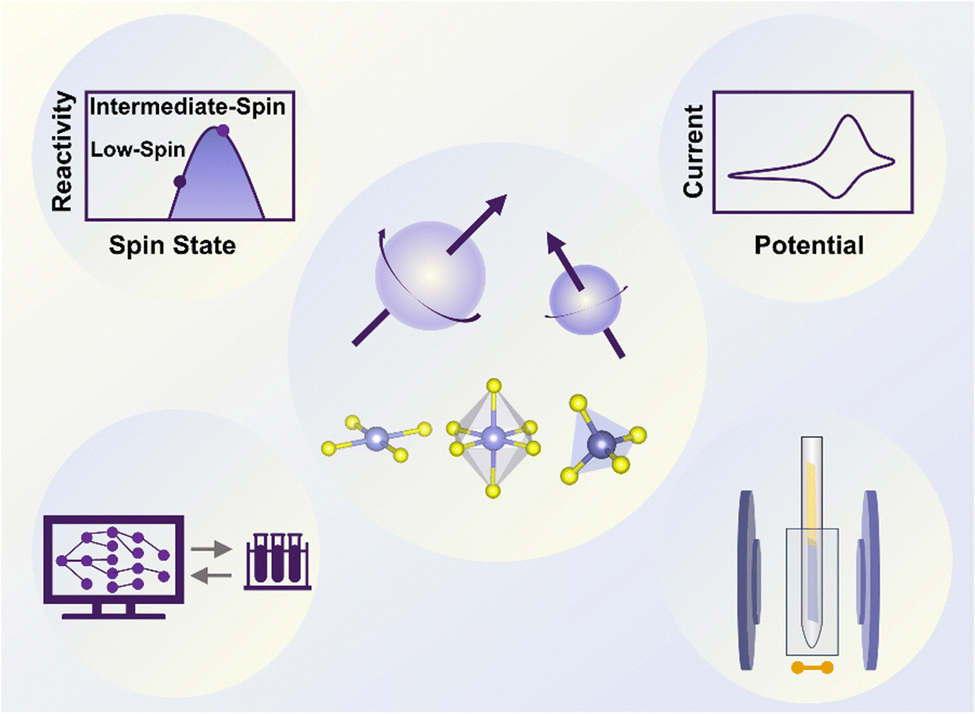 | ||
| Fig. 1 Overview of the study of spin states in electrocatalysis from aspects of theory, experiment, modeling, and characterization. In the center, three typical configurations of central metals are depicted. The figure in the top left shows the possible relationship between the electrocatalyst reactivity and the spin state of the central metal. In the top right figure, cyclic voltammetry, a commonly used electrochemical technique, is depicted as an example for evaluating the performance of electrocatalysts. In the bottom left, computational calculations used to predict the properties of electrocatalysts, such as the spin state of the metal centers, and assist the experiment, are depicted. The figure in the bottom right shows one typical spin-state characterization technique, electron spin resonance, serving to identify the spin state of the central metal. | ||
2. Basic principles
2.1 Molecular orbital theory (MOT), crystal field theory (CFT), and ligand field theory (LFT)
Spin37 refers to the angular momentum carried by elementary particles, and the spin state refers to the spin configuration of the d electrons in transition metals (TMs). In general, they are two different concepts. This article focuses on the spin state and its relationship with electrocatalysis. To understand the spin state, three main models, MOT,38 CFT,39 and LFT,40 are required. Among these three models, MOT is the basis for chemical bonding theory, while LFT is an application of MOT to TM compounds and complexes. In CFT, the spin states of TM atoms or ions are explained in terms of energy.MOT is a chemical bonding hypothesis that was created in the early 20th century by Friedrich Hund and Robert Mulliken to explain the composition and characteristics of various molecules. Valence bond theory makes it hard to fully explain the electronic properties of molecules, such as those in resonance-stabilized compounds that have two or more equivalent bonds with a non-integral bond order with a strength that is between those of single bonds and double bonds. Molecular orbitals can be classified as bonding molecular orbitals, antibonding molecular orbitals, and non-bonding molecular orbitals. A bonding molecular orbital always has a lower energy than its parent orbital, while an antibonding molecular orbital always has a higher energy. Since electrons have a higher tendency to occupy molecular orbitals with a lower orbital energy, when the energies of the interacting atomic orbitals are similar, they can combine most effectively to form molecular bonding orbitals.
According to CFT, the attraction between the positively charged metal cation and the negatively charged non-bonding electron of the ligand causes TM–ligand interaction. In this interaction, the alike charges between the electrons in the TM d-orbitals and the non-bonding electrons in the ligand repel each other. As a result, within the TM cation, electrons in the d-orbitals closest to the ligand possess higher energy compared to those in other farther d-orbitals. The directional nature of this TM–ligand interaction causes the splitting of the energy level of the d-orbital electrons, resulting in a loss of degeneracy. The characteristics of metal ions, such as their oxidation state, the arrangement of the ligands surrounding the metal ions, and the coordination number of the metal ions, have an impact on the manner in which their energy levels split. The energy level difference between the higher-energy and lower-energy d-orbitals increases with the strength of the ligand in the TM–ligand interaction. Due to the separation of the d-orbital energy levels, the electrons in the d-orbital of the central TM atom will be rearranged to preferentially occupy the lower energy levels, reducing the total energy of the system, and stabilizing the complex.
LFT explains the bonding in the coordination complexes and orbital configuration of TM-based complexes based on MOT. Five n d, one (n + 1) s, and three (n + 1) p orbitals make up the nine valence atomic orbitals of a TM ion possessing the right amount of energy to chemically interact with ligands to form bonds. Although the complex geometry has a significant impact on the results of LFT analysis, most explanations start by describing octahedral complexes. Hence, LFT is a combination of CFT and the then-emerging MOT.
2.2 Low spin, intermediate spin, and high spin
Spin states can be classified into low-spin (LS), intermediate-spin (IS), and high-spin (HS) (Fig. 2), which are determined by the crystal field splitting energy and spin pairing energy.39 Generally, electrons fill up each of the lower-energy orbitals with two electrons of opposite spins before filling up orbitals at higher energy levels using the Aufbau principle when the crystal field splitting energy of the system is higher than the pairing energy (i.e., the energy required for two electrons to occupy the same orbital). Conversely, if the pairing energy is higher than the crystal field energy of a system, electrons will initially fill all orbitals with similar energy levels (e.g., eg and t2g levels of d-orbitals) with one electron each before pairing as dictated by Hund's rule. In this case, once all orbitals at the lower energy level are singly occupied, the next electron will singly occupy the orbital at the higher energy level until the energy required to occupy the next empty orbital becomes higher than the pairing energy. Since the system tends to lower its total energy, a system with high crystal field splitting energy relative to the pairing energy will tend to be LS, whereas one with low crystal field splitting energy tends to be HS.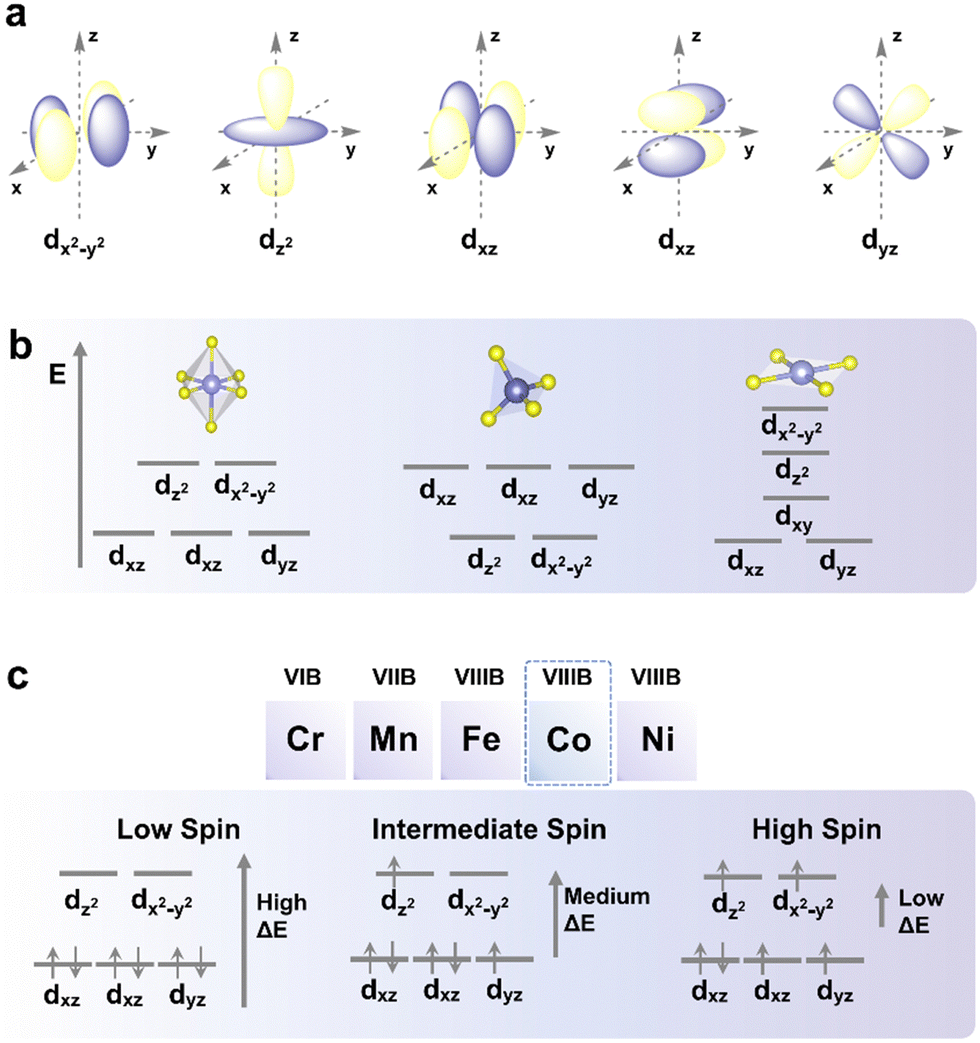 | ||
| Fig. 2 Illustrations of the spin state. (a) Spatial illustration of the five d-orbitals, (b) crystal field splitting of d-orbitals in a transition metal cation when ligands interacted with the cation in octahedral, tetrahedral, and square planar molecular geometries, and (c) elements reported in the previous research involving spin states, as well as d-orbital energy levels and possible spin states (low spin state, intermediate spin state, and high spin state) of Co(III) when interacting with ligands in octahedral molecular geometry as an example to illustrate the effect of d-orbital energy level splitting on the spin state of transition metal cations. | ||
2.3 Intrinsic factors affecting the spin states
Rearrangement of d-electron orbitals may lead to changes in the relative value between crystal field splitting energy and spin pairing energy, inducing a spin state change. Orbital rearrangement results from coordination structure distortion in central metals. Several methods are proposed to alter the metal coordination structure and change the spin states. Lattice orientation conversion,41 size control,42 and crystallinity manipulation43 are confirmed to adjust the coordination structure of central metals. The polyhedra of central metals are changed and d-electron orbitals rearrange, leading to the change in spin states. Apart from a coordination change in the overall crystal structure, in the local structure around central metals, heteroatom substitution,19 and defect induction44 can tune the neighboring coordination environment of central metals and change spin states. Replacing the ligands31 and neighboring atoms19 connected with central metals could also affect metals’ coordination structure. Hybridization between metal d orbitals and ligand orbitals changes, and spin states change. For detailed discussions on strategies to regulate spin states in oxygen electrocatalysis, such as defect induction, heteroatom substitution, and size modulation, we refer readers elsewhere.453. Approaches for spin state characterization and modeling
Identifying spin states is an essential step in spin state-related electrocatalysis studies. Experimental techniques including XAS, ESR, Mössbauer spectroscopy, and temperature-dependent magnetization are commonly used in the literature. Spin-polarized density functional theory is a typical modeling method to investigate the spin state.3.1 X-Ray absorption spectroscopy (XAS)
In XAS, electrons from the 1s or 2p orbitals can be excited to wavelengths of approximately 1 Å with high-energy X-ray excitation at synchrotron facilities. The XAS results provide comprehensive information on the chemical environment and the electronic structure of the investigated atoms (Fig. 3a).46 During the measurement, the measured transmitted X-ray intensity, I, is related to the incident X-ray intensity, I0 through the equation: I = I0e−μx, where μ is the incident-photon-energy-dependent X-ray absorption coefficient and x represents sample thickness. A sharp increase in X-ray absorption (typically represented by μ or normalized absorbance), known as X-ray absorption edges, will emerge at certain X-ray photon energy characteristics of the investigated element in the sample. This situation occurs when the energy of the X-ray photon is equal to the energy required to ionize the inner-shell electrons of the investigated atoms. The X-ray intensity variation before and after the interaction with the sample will be analyzed to provide details on the chemical environment around the constituent atoms. The X-ray absorption near-edge structure spectroscopy (XANES) ranges from 30 eV to 50 eV above the absorption edge of the element, while the extended X-ray absorption fine structure (EXAFS) ranges from 50 eV to 1000 eV above the absorption edge. Through the use of Fourier transformation (FT) or wavelet transformation (WT) analysis, EXAFS is effective in identifying the coordination number and chemical bond length related to the investigated element. In further detail, the distinctive peaks corresponding to metal–metal or metal–nonmetal bonding may be used to identify the bonding mode of doped heteroatoms after translating the EXAFS signals into R space using FT.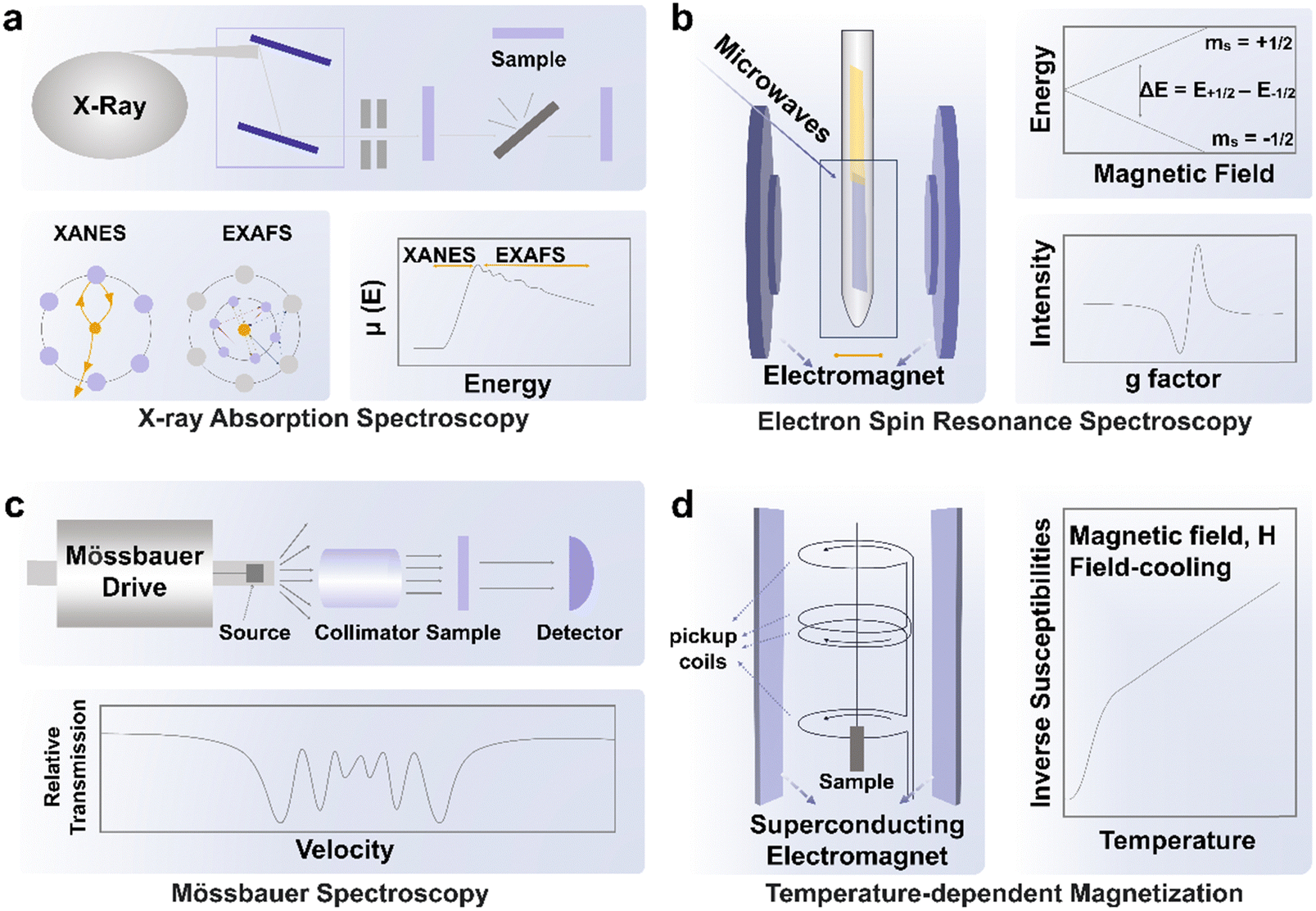 | ||
| Fig. 3 Advanced characterization techniques for verification of spin state during electrocatalysis, including (a) XAS, (b) ESR, (c) Mössbauer spectroscopy, and (d) temperature-dependent magnetization. | ||
Using in situ/operando near edge X-ray absorption fine structure (NEXAFS) spectroscopy, Hyung-Suk Oh's group19 found that the reconstructed Fe–CoOOH phase transitioned from the LS state to an IS state during the OER. It was clear that the cobalt oxide nanoparticles (CPs) after the OER (CP–O) included LS Co(II) (t62ge1g) and Co(III) (t62ge0g) species because open circuit voltage (OCV) conditions allowed CP–O to show Co(II) and Co(III) peaks with filled t2g orbitals. The Co L-edge peak of CP–O was moved to higher energy when the potential was raised to 1.43 V, and the t2g peak (O K-edge spectrum) of CoO was visible. The mixed species of Co(II), Co(III), and some converted Co(IV) (CoO2) species are shown by the wide, displaced Co L-edge peak. The results gained from this spectrum are consistent with those from the in situ/operando Raman measurements. The CoO2 produced during the OER was predicted to have an HS (t32ge2g) state based on the t2g peak of CP–O. Under OER conditions, cobalt oxide nanoparticles after Fe and S treatment (CP–FeSO) likewise demonstrated a higher spin state relative to that at OCV, although the change was slightly different. A significant percentage of the Co(III) state (Fe–CoOOH) of the CP–FeSO catalyst under OER conditions was shown by the Co L-edge of CP–FeSO at 1.43 V, which almost maintained the location of the major peaks with decreasing signals of Co(II) species. A modest t2g peak of CP–FeSO was found despite the presence of a significant amount of Co(III) species, indicating that Fe–CoOOH in CP–FeSO underwent a transition from the LS state (t62ge0g) to an IS state (t52ge1g) during the OER.
In XAS, information on a single element can be gathered independently even in small amounts. Surface and bulk information of first-row transition metals in bulk materials can be identified using soft (lower energy X-rays) XAS. However, the limitation of XAS lies in that the results are obtained as an average value of all absorbed atoms. For metal centers with more than one type of coordination, the coordination of the catalytic reaction center is difficult to be quantified due to the diverse structure of the active site.
3.2 Electron spin resonance (ESR) spectroscopy
ESR is based on the Zeeman effect, where electron energy levels of degenerate electronic states would split in the presence of an external magnetic field due to the difference in work done to align the magnetic moment of each electron (with varying spin) to the external field (Fig. 3b).47 For an unpaired free electron, the energy difference between the two electronic states after the split in the presence of a magnetic field is defined by ΔE = geμBB0, where ge is known as the free electron g-factor with a value of approximately 2.002, μB is the Bohr magneton and B0 is the applied external magnetic field. In this case, resonance can occur when the probe microwave with energy that matches ΔE interacts with the unpaired free electron in an external magnetic field B0, causing its transition between the two energy states (and subsequent absorption or emission of microwaves) and consequently, the reduction in the transmitted intensity of the probe microwave through the instrument. While the instrument measures the absorbance of the probe microwave, the absorbance signal is usually presented as the first derivative of the measured quantity. Hence, the g-factor would be estimated from the applied magnetic field strength B0 at which the absorbance peak (i.e., usually at the middle of the ESR pattern when the first derivative signal is zero), and when it is similar to ge, it belongs to free unpaired electrons. The environment of the unpaired electron would influence the effective magnetic field around the electron. However, since the alteration in the effective magnetic field could not be measured, the equation used has been modified into ΔE = (ge + Δg)μBB0, where the estimated quantity Δg can be used as the fingerprint of the analyzed molecule, providing sample information on the atomic, molecular, and chemical structures of the analyzed substance. Both Δg and the analysis of hyperfine interactions could be used to determine if the spin signals produced by magnetic moments of unpaired electrons are from either those trapped in vacancies, those delocalized within multiple nuclei, or those localized to a single nucleus. Additionally, by using a low testing temperature, the ESR signal may be enhanced by reducing thermal disturbance, which is convenient for some tests that require low temperature. It is worth noting that room temperature for detecting transition metal ions is not always suitable because some transition metal ions (e.g. Co(III)) have short relaxation times.47 Relaxation time is the time to characterize the return of an excited spin system to the ground state. Short relaxation times broaden the spectral shape and may be beyond detection. Cryogenic temperatures (below 77 K) are supposed to be used as the detection temperature. Wu et al.48 utilized low-temperature EPR to validate the higher ratio of IS/HS Co(III) in Bi0.1Sr0.9CoO3−δ and Bi0.05Sr0.95CoO3−δ than those in SrCoO3−δ. The resonant intensity at g = 2.306, which corresponds to the peak of IS/HS Co(III), increased in Bi-doped samples.ESR stands out for its high sensitivity toward transition metal ions, even at low concentrations (in μM levels). The technique's applicability to diverse material types (e.g. powders, solutions) and its non-destructive nature are also its strengths. Its noticeable limitation is associated with the magnetism of species, which can restrict its application to paramagnetic species with unpaired electrons. When considering the reuse of materials post-testing for specific reactions, attention is required for materials that may be adversely affected by the magnetic field.
3.3 Mössbauer spectroscopy
For Fe-based catalysts, 57Fe Mössbauer spectroscopy is regarded as the most convenient technique to identify the spin states. The absorption of γ-rays by an atomic nucleus results in transitions between the ground state and the first excited spin state (Fig. 3c).49 The interaction between the electromagnetic moment of the nuclear charge and the electromagnetic field produced by electrons could be studied through Mössbauer spectroscopy. The flexible method of Mössbauer spectroscopy can be used to offer highly accurate details on the chemical, structural, magnetic, and time-dependent characteristics of a material. Numerous energy-level transitions occur in the nuclei of atoms, frequently accompanied by the emission or absorption of gamma rays. These energy levels are affected by their electromagnetic and electrical surroundings, which could split or transform them. Resonance fluorescence is used to monitor these variations in energy levels since it can provide details on the immediate surroundings of an atom within a system. In Frédéric Jaouen's work,50 the change in the HS Fe(II) moiety and IS/LS Fe(II) moiety is traced during potentiostatic operation, where the spin state of Fe(II) is identified using in situ57Fe Mössbauer spectroscopy. It is shown that the HS Fe(II) moiety irreversibly changes from a ferric HS state to a ferrous HS state while the IS or LS Fe(II) moiety is stable after 50 h potentiostatic operation. The advantages of Mössbauer spectroscopy for spin state studies are robust anti-interference capabilities. The strong anti-interference ability arises from resonance absorption of specific atomic nuclei. For the tested samples, this method is applicable to diverse sample types, including powders, thin films, and bulk materials. Importantly, Mössbauer spectroscopy does not cause damage to the sample during testing. However, only 57Fe and 119Sn have been extensively utilized in Mössbauer spectroscopy. Consequently, in the context of spin state studies, Mössbauer spectroscopy is predominantly employed for Fe-based electrocatalysts.3.4 Temperature-dependent magnetization
Temperature-dependent magnetization (M–T) measurements (Fig. 3d) are conducted at a chosen magnetic field under field-cooling procedures to unveil the spin state of metal ions in ferromagnetic materials, which is commonly carried out on a superconducting quantum design (SQUID) magnetometer.51 Ferromagnetic materials will lose their permanent magnetic properties when heated above the Curie temperature (TC). Over their TC, ferromagnetic materials become paramagnetic. In the paramagnetic region, the susceptibilities derived from the magnetizations obey the paramagnetic Curie–Weiss law: χ = C/(T − TC), where C is the Curie constant and TC is the Curie–Weiss temperature. After obtaining the value of C, the effective magnetic moment μeff could be calculated through the equation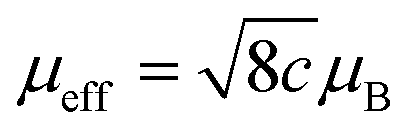 , where μB is Bohr magneton (a constant, 9.274 × 10−24 J T−1). The spin state of metal ions could be derived using the equation μeff = gμBSeff, where the g is the Lande factor and Seff is the effective spin quantum number. Take Co(III) as an example, Seff of HS Co(III) (SHS) is 2 and Seff of LS Co(III) (SLS) is 0. Duan et al. utilized M–T measurement with a magnetic field of 100 Oe under field-cooling procedures to reveal the spin state of Co(III) in LaCo1−xFexO3 (x = 0.00, 0.10, 0.25, 0.50, 0.75, 1.00).52 Zhou et al. obtained the spin state of Co(III) in a series of LaCoO3 with different particle sizes through M–T measurement with a magnetic field of 1 kOe under field-cooling procedures.42 In both studies, a mixture of HS and LS Co(III) is considered as the spin state of Co(III) in LaCo1−xFexO3 and volume fractions of HS and LS Co(III) are calculated through the relationship,
, where μB is Bohr magneton (a constant, 9.274 × 10−24 J T−1). The spin state of metal ions could be derived using the equation μeff = gμBSeff, where the g is the Lande factor and Seff is the effective spin quantum number. Take Co(III) as an example, Seff of HS Co(III) (SHS) is 2 and Seff of LS Co(III) (SLS) is 0. Duan et al. utilized M–T measurement with a magnetic field of 100 Oe under field-cooling procedures to reveal the spin state of Co(III) in LaCo1−xFexO3 (x = 0.00, 0.10, 0.25, 0.50, 0.75, 1.00).52 Zhou et al. obtained the spin state of Co(III) in a series of LaCoO3 with different particle sizes through M–T measurement with a magnetic field of 1 kOe under field-cooling procedures.42 In both studies, a mixture of HS and LS Co(III) is considered as the spin state of Co(III) in LaCo1−xFexO3 and volume fractions of HS and LS Co(III) are calculated through the relationship, 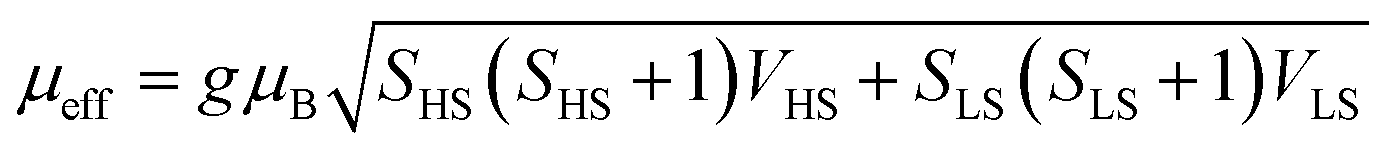 , where VHS and VLS are the volume fractions for HS and LS Co(III), respectively. The advantage of M–T measurements for spin state identification is that it is easy to process data following the Curie–Weiss law. However, for samples with high TC (over 800 K) or non-ferromagnetic materials, the Curie constant C might not be obtained easily and other techniques for spin state characterization should be considered.
, where VHS and VLS are the volume fractions for HS and LS Co(III), respectively. The advantage of M–T measurements for spin state identification is that it is easy to process data following the Curie–Weiss law. However, for samples with high TC (over 800 K) or non-ferromagnetic materials, the Curie constant C might not be obtained easily and other techniques for spin state characterization should be considered.
3.5 Modeling and calculations
Theoretical modeling and calculations aid in understanding and predicting the structures and facilitate further investigation into the underlying material mechanisms and reaction processes. They can include material properties such as crystal symmetry, element composition, and crystal lattice parameters that can be fine-tuned in modeling to reveal intrinsic electronic properties. More detailed discussions on affecting factors can be found in section 2.3 – Intrinsic factors affecting the spin states.Nria López's team53 analyzed the potential for SCO in two separate cobalt hexacyanoferrate model systems at their resting states to explain the OER properties using DFT calculations (Fig. 4a and b). They demonstrate that the overpotential may be greatly reduced by 0.7 V with the occurrence of SCO during the OER, resulting in an overpotential of about 0.3 V, which is consistent with the experimentally determined value (Fig. 4c). It was concluded that the cobalt hexacyanoferrate favors the SCO-assisted pathway as the main reaction pathway under the external potential above 1.5 V versus SHE. To explore why adding 25% Fe boosts the OER performance of NiOOH, He et al. performed DFT calculations and found that the LS state of Fe(III) in NiOOH renders surface Fe sites highly active for the OER.54 When the Fe content exceeds 25%, a spin state transition occurs in Fe(III) from LS to HS, which explains the drop of NiFeOOH OER activity with the increasing Fe content.
 | ||
| Fig. 4 (a) Catalytic OER cycle, which is divided into four proton-coupled electron-transfer (PCET) steps (OER 1–4), where *Co denotes the active site and □ denotes the vacancy. (b) SCO between LS *Co–OH and IS *Co–OH via the minimum energy crossing points, for which the SCO energy ΔGSCO is required. (c) Reaction free energies and spin densities involved in OER 2. (a)–(c) are reproduced from ref. 53 with permission from American Chemical Society, Copyright 2022. (d) Structure and dynamics of the N–FeN4C10 moiety in the ORR with optimized geometries. Reproduced from ref. 55 with permission from Elsevier, Copyright 2020. | ||
Considering the critical role of *O2− and *OH− intermediates during the ORR process, Liu's team55 employed DFT analyses to investigate the potential-relevant structural and electrical evolutions of the active single-Fe-atom moieties. To provide a theoretical understanding of the structural and dynamic evolutions of atomically scattered Fe moieties during the ORR, quantum chemical investigations were carried out. The whole structural and dynamic cycles of the N–FeN4C10 (D3 symmetry) moiety in the ORR are displayed (Fig. 4d). As demonstrated, *OH− adsorption causes the central Fe atom to partly shift toward the N4-plane (the symmetry of N–FeN4C10 approach D3), while the Fe–N bond is slightly shortened. The formation of the O2-FeIIN5 intermediate results in the electronic configuration conversion of the Fe(II) from HS to LS, which is consistent with the increased ligand field, and further reduces the length of the Fe–N bond. The central Fe atom is completely moved into the N4-plane. The energy barrier for the dynamic cycle of the N–FeN4C10 moiety in the ORR is greatly lowered by the SCO of Fe(II) in the D3 symmetry upon the creation of the *O2− intermediate.
4. Examples of spin state studies in electrocatalysis
In this section, the influence of spin state on popular electrocatalytic reactions is discussed. To date, most studies involving the spin state of metal centers are about oxygen electrocatalysis (OER11–19 and ORR8–10,22–26). A trend is also merging to other reactions such as the HER,27–30 CO2RR,31,32 NRR,6,33,34 NO3−RR,35,36 and UOR.214.1 Oxygen evolution reaction
The OER is the key reaction and main bottleneck in water electrolysis. Perovskite cobaltite (e.g., LaCoO3, LCO) and spinel cobaltite (e.g. Co3O4) have been extensively studied for catalyzing the OER. Co(III) in octahedra favors LS (t62ge0g) due to a high crystal field splitting energy (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1)56 relative to the spin pairing energy (11600 cm−1).57 The σ bonding between eg orbitals and oxygen-containing adsorbates influences the reaction rate and rate-determining steps (RDS) of the OER. Shao-Horn's group reported that the half-filled eg orbitals in octahedral Co(III) have optimal interaction with the adsorbed intermediates, resulting in the best OER performance.5 In octahedral LS Co(III), no eg orbital is occupied. Thus, tuning the spin states of Co(III) to a higher spin state than the LS state could be regarded as a strategy to enhance OER activity.
000 cm−1)56 relative to the spin pairing energy (11600 cm−1).57 The σ bonding between eg orbitals and oxygen-containing adsorbates influences the reaction rate and rate-determining steps (RDS) of the OER. Shao-Horn's group reported that the half-filled eg orbitals in octahedral Co(III) have optimal interaction with the adsorbed intermediates, resulting in the best OER performance.5 In octahedral LS Co(III), no eg orbital is occupied. Thus, tuning the spin states of Co(III) to a higher spin state than the LS state could be regarded as a strategy to enhance OER activity.
For LCO, Qian et al. reported that the OER performance could be promoted through doping Ce and tuning the spin state of Co(III) ions from LS to IS.11 Zhou et al. utilized crystal facet engineering and a Sr doping strategy to regulate the spin states of Co(III) ions from LS LCO to IS (Sr-doped LCO) and achieved a higher OER activity.12 Doping of anions, sulfur13 or fluorine,14 in LCO accompanied with O vacancy introduction has been reported to enhance the OER performance through tuning the spin state of octahedral Co(III) ions from LS to IS. Zhou et al. decreased the LCO particle size to tune the ratio of HS and LS Co(III) states to around 1.5 for optimal OER activity.42 They claimed that while only HS Co(III) ions in LCO contribute to the OER activity, an excessive proportion of HS Co(III) ions with too few LS Co(III) ions lowers the charge transfer ability. For Co3O4, Ramsundar et al. optimized the content of HS Co(III) ions in Zn0.8Co2.2O4 by substituting 80% tetrahedral Co(II) ions with Zn(II) ions to lower the overpotential.17 Zhang et al. modified CoO6 octahedra with SO42− and boosted an LS–IS transition of Co in deformed Co3O4–SO42−.18 Meanwhile, the shifted 3d band center of Co in Co3O4–SO42− led to a less-filled antibonding orbital and stronger chemical interaction with oxygen atoms and adsorbed intermediates than that on Co3O4 and surface Co sites.
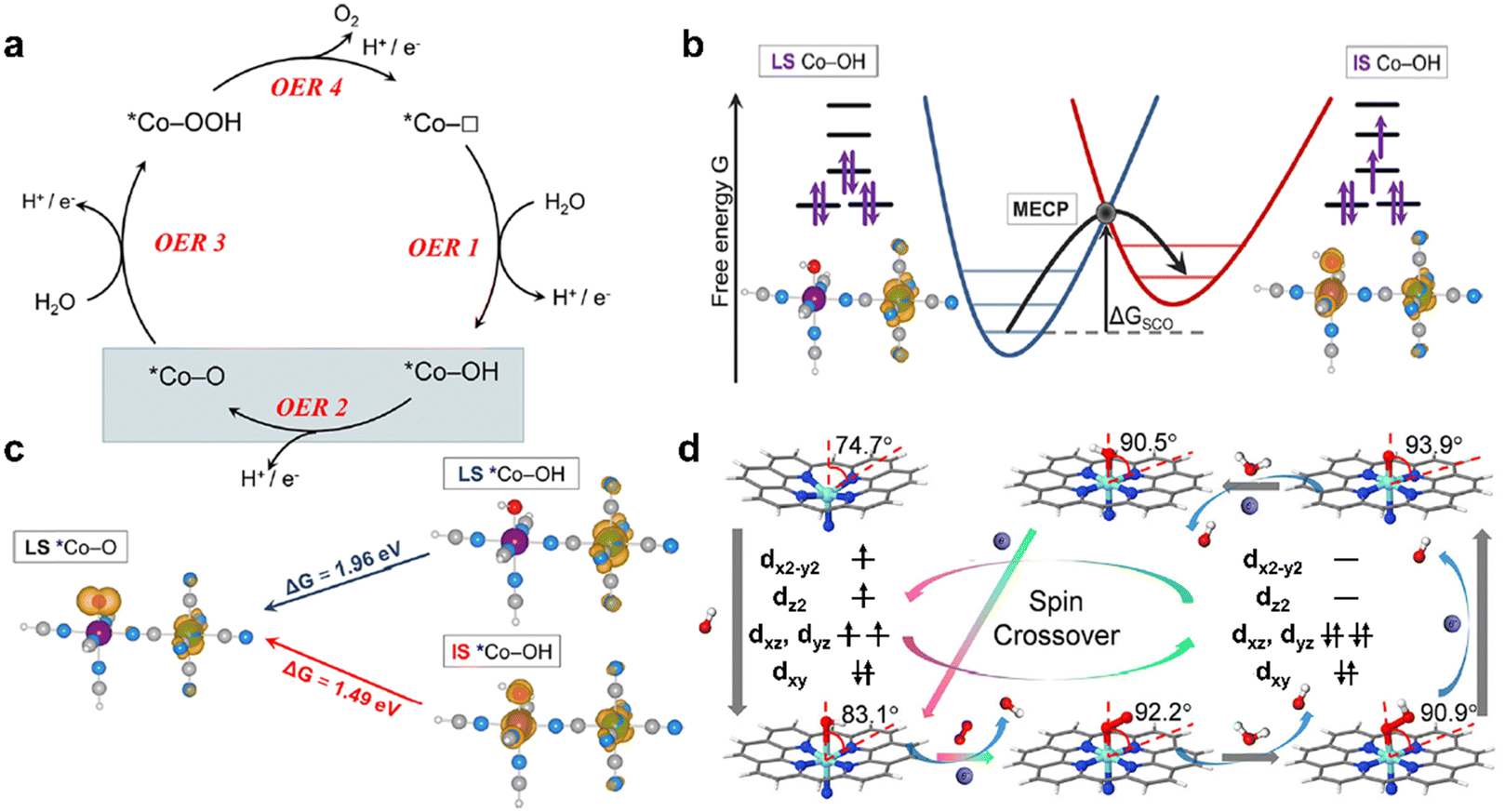
Notably, the surface of some unstable electrocatalysts may undergo reconstruction during the OER and the reconstructed surface provides the real active site instead of the original phase. Probing the intrinsic properties (such as spin state) in the reconstructed layer (CoOxHy) is critical for the OER. Lee et al. fabricated Fe-doped CoOxHy with sulfur treatment and tuned the spin state of Co(III) ions from LS in CoOxHy to IS in Fe–CoOOH.19 The doped Fe in Fe–CoOOH could hinder the conversion of IS Co(III) ions to HS Co(IV) ions under OER conditions, which corresponds to high and stable OER activity. In CF–O@1.4 V (Fig. 5a), superoxo intermediates (OO–) are stabilized on high-spin Co(IV), and slow dioxygen release becomes the RDS, resulting in low OER activity. HS Co(IV) (t32ge2g) with an eg orbital occupancy of about 2, which is higher than the optimal value of 1.2. In CF–FeSO@1.4 V (Fig. 5b), Fe–CoOOH is stable with IS Co(III) and the adsorption energy of the OER intermediates is optimal, leading to high OER catalytic activity. With Fe substitution, IS Co(III) possesses optimal eg orbital occupancy with a value of 1. The reconstructed layer acts as the active electrocatalyst. The kinetics of reconstruction may be affected by the original phase. For NiFe-based electrocatalysts, electron transfer occurs between Fe sites and adsorbed OH− during reconstruction. To optimize the reconstruction process, Du et al. triggered the spin state of Fe from LS to HS in Fe5Ni4S8(FNS) by modifying the FNS surface with anionic surfactant.58 HS Fe with a low d-band center promotes OH− accumulation and accelerates electron transfer to enhance reconstruction kinetics. More studies about the intrinsic activities (such as spin states) of (pre)catalysts and reconstructed layers are expected to deepen the fundamental understanding of OER electrocatalysts.
 | ||
| Fig. 5 Illustration of the role of spin state during the OER. In the left part with no frame, spin states under open-circuit voltage (OCV) for (a) cobalt foam (CF–O)@OCV and (b) cobalt foam after sulfur and iron treatment (CF–FeSO)@OCV. Within the dotted frames, the OER conditions, the chemical structure at active sites, and Co 3d–O 2p orbital coupling diagram under OER conditions are illustrated for (a) CF–O@1.4 V and (b) CF–FeSO@1.4 V. Dotted arrows in light purple and blue colors indicate the spin state before the OER and arrows in dark purple and blue represent the converted spin state during the OER. (a) and (b) are reproduced from ref. 19 with permission from Macmillan Publishers Limited, Copyright 2022. | ||
4.2 Oxygen reduction reaction
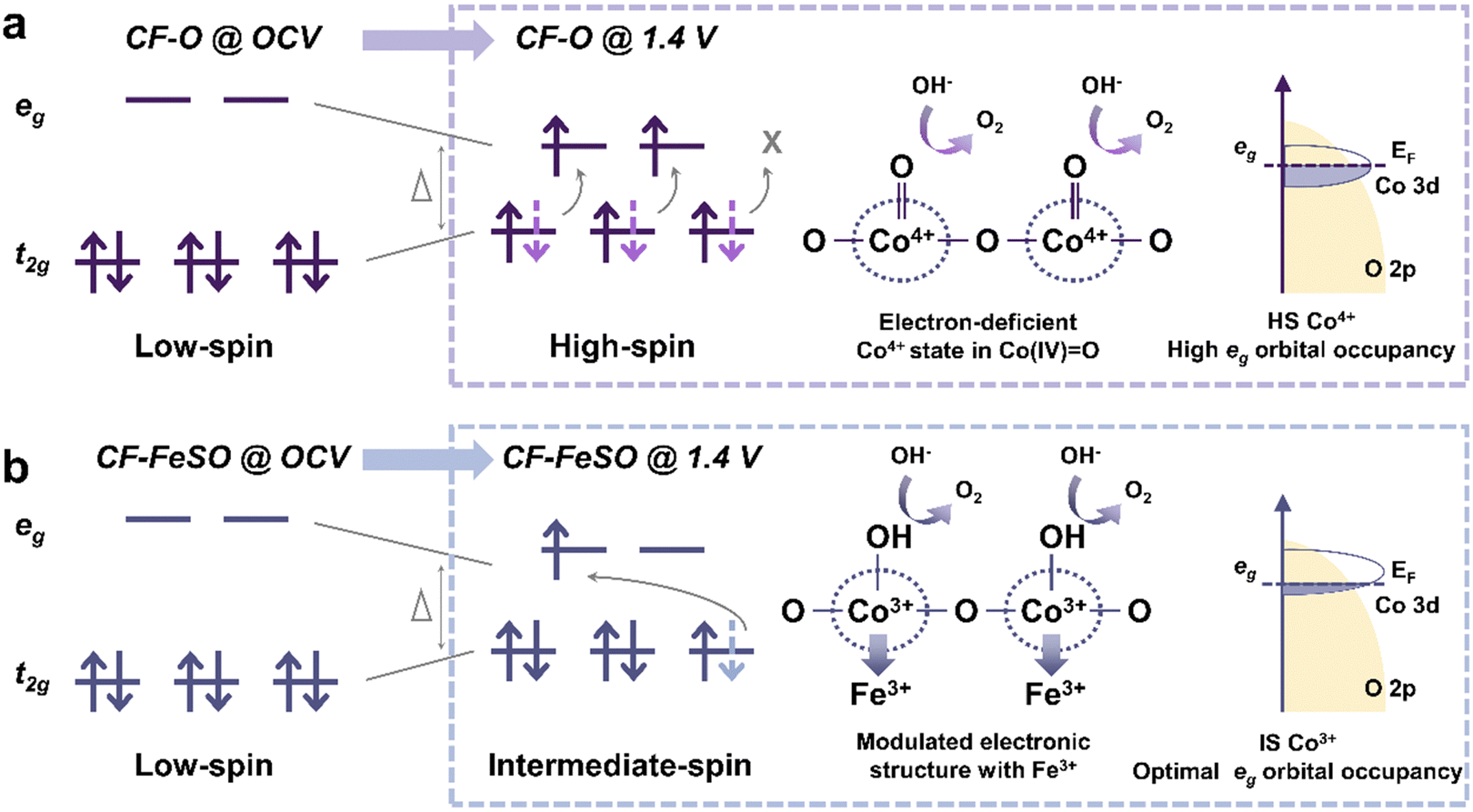
The ORR is the cathode reaction in fuel cells and metal–air batteries. Some transition metal oxides have been found with high ORR activity. An effort has been made to explain the observed activity from the aspect of their electronic structure and coordination environment. Suntivich et al. proposed that ORR activity is closely related to the σ*-orbital (eg) filling and covalency strength between oxygen and transition-metal B sites of perovskite.22 The rate limiting step (RLS) is determined by the σ*-orbital filling and metal-oxide covalency strength. When the eg filling of perovskite is close to 1, electrocatalysts exhibit excellent ORR activity. The eg filling is determined by the spin state of metal centers and thus tuning the spin state is possible to regulate ORR performance. Mu et al. found that octahedral LS Co(II) ions rather than HS Co(II) ions in Co2VO4 possess better ORR activities, where eg filling of LS Co(II) is 1.23 This HS–LS transition (Fig. 6a) in octahedral Co(II) ions originates from the higher crystal field splitting energy in LS Co(II) ions than in HS Co(II) ions, induced by Co–O octahedron contraction. The RLS barrier (Fig. 6b) in Co2VO4 with octahedral LS Co(II) ions (0.59 eV) is lower than that with HS Co(II) ions (0.94 eV), contributing to better ORR performance.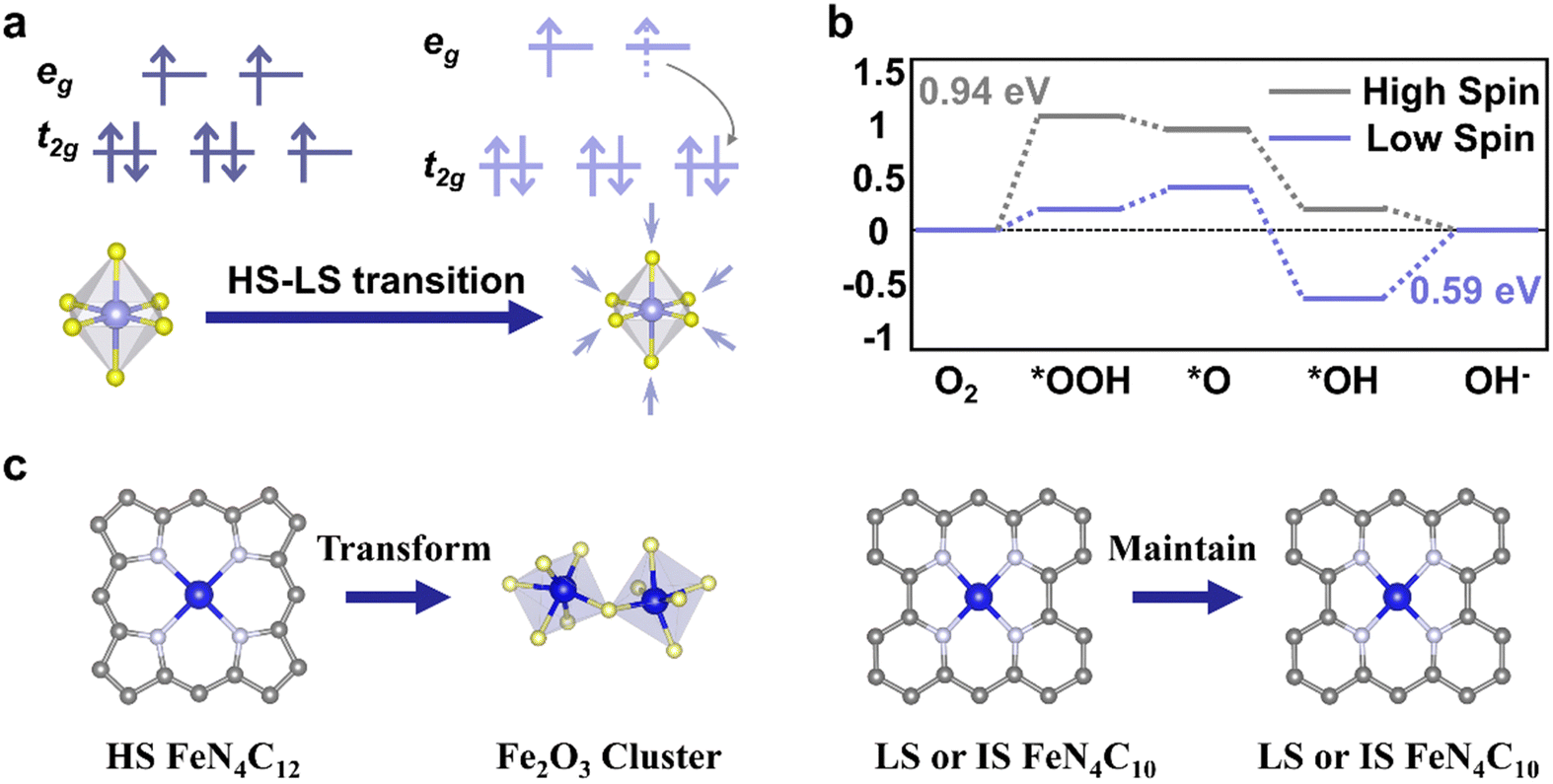 | ||
| Fig. 6 Relationship between spin state regulation and ORR performance tuning. (a) Electronic configurations of octahedral Co(II) in HS and LS states. (b) Calculated ORR free energy diagram at 1.23 V vs. RHE on HS and LS Oh-site Co(II). (a) and (b) are reproduced from ref. 23 with permission from Wiley-VCH, Copyright 2020. (c) Structural change of HS FeN4C12 in the III/II oxidation state and LS or IS Fe(II)N4C10. Reproduced from ref. 9 with permission from Macmillan Publishers Limited, Copyright 2020. | ||
Another type of transition metal-based catalyst is iron–nitrogen–carbon (Fe–N–C).26,44,59 The diversity of FeNx moieties challenges the identification of active sites in Fe–N–C electrocatalysts. Cai et al. synthesized single Fe(II)N4 species and found that such LS Fe(II)N4 species are highly active for the ORR.8 Chen et al. reported that ORR performance could be optimized through sulfur-doped Fe–N–C, which provides more LS Fe(III) sites than pristine Fe–N–C and promotes the OH* desorption process.60 Li et al. proposed that Fe–N–C catalysts initially comprise two distinct FeNx sites and both contribute to the ORR activity. However, only the LS or IS Fe(II)N4C10 moiety remained after a 50 h test, while another site identified as an HS FeN4C12 moiety in the III/II oxidation state transformed into iron oxides during the test (Fig. 6c).9 Such insights indicate that regulating the spin state of moieties is a way to improve the durability of Fe–N–C catalysts. To obtain an optimal spin state of Fe in FeN4 species, Chen et al. decorated Ti3C2 MXene substrate with sulfur terminals to tune the spin state of Fe(II) from IS to HS.24 An upshifted d-band center enables Fe(II) on FeN4 to bind oxygen with an end-on adsorption mode, regulating oxygen-containing intermediate adsorption and the corresponding ORR kinetics. Li et al. synthesized HS Fe(II) centers in Fe–N–C using iron phthalocyanine (FePc) and Ti3C2Tx Mxene rich in hydroxyl and fluorine terminals, yielding better dioxygen adsorption and reduction than IS Fe(II) centers.10 Wang et al. utilized SeO2 to modify FeN4, showing that the distorted square-planar coordination of FeN4 could boost ORR activity.25 Yang et al. proposed that the ORR preferentially takes place on IS Fe(III) sites in FeN4 where the one electron occupying the eg orbital readily penetrates the antibonding π-orbital of oxygen.26 They fabricated dual-metal atom-dispersed Fe,Mn/N–C catalysts, in which the atomically dispersed Mn–N moieties could induce the LS–IS transition of Fe(III) sites in FeN4, thus improving ORR performance.
4.3 Other reactions
Besides the OER and ORR, the spin state effect has been studied for other electrochemical reactions. In the case of the HER, H atom adsorption and O–H bond cleavage are closely related to the electronic structure of metal sites and therefore the spin state plays a role here. Liu et al. doped Mn (t52ge0g) into CoSe2 (Co, t62ge1g) ultrathin nanosheets and introduced a subtle crystallographic distortion for additional edge sites.27 Compared to pristine CoSe2, the adsorption of H atom onto Mn-doped CoSe2 is weaker and the activation barrier of H–H bond formation is lower. Similarly, Xia et al. reported that the high HER performance of Ni0.33Co0.67Se2 is attributed to the spin state difference between Ni(t62ge2g) and Co(t62ge1g).28 The additional active sites and distorted lattice induced by doped Ni in CoSe2 contribute to high HER activity. Zhang et al. found that partially substituting Se with S in CoSe2 could trigger an LS–HS transition in octahedral Co.29 Compared to CoSe2, CoSe1.03S0.97 has a larger Se–Se/S bond distance and smaller Se–Se/S bonding–antibonding splitting, which facilitated the electrons transfer from t2g to eg orbital (Fig. 7a). On the other hand, the spin state of Co in CoSe1.03S0.97, which exhibits the best HER performance among a batch of S-doped marcasite CoSe2 with different unpaired d electron numbers (Fig. 7b), was modulated to higher spin states, thus leading to improved HER performance. Pei et al. also found that a higher spin state of Co in the NiCo2S4/ReS2 interface could contribute to facile water dissociation and superior HER activity.30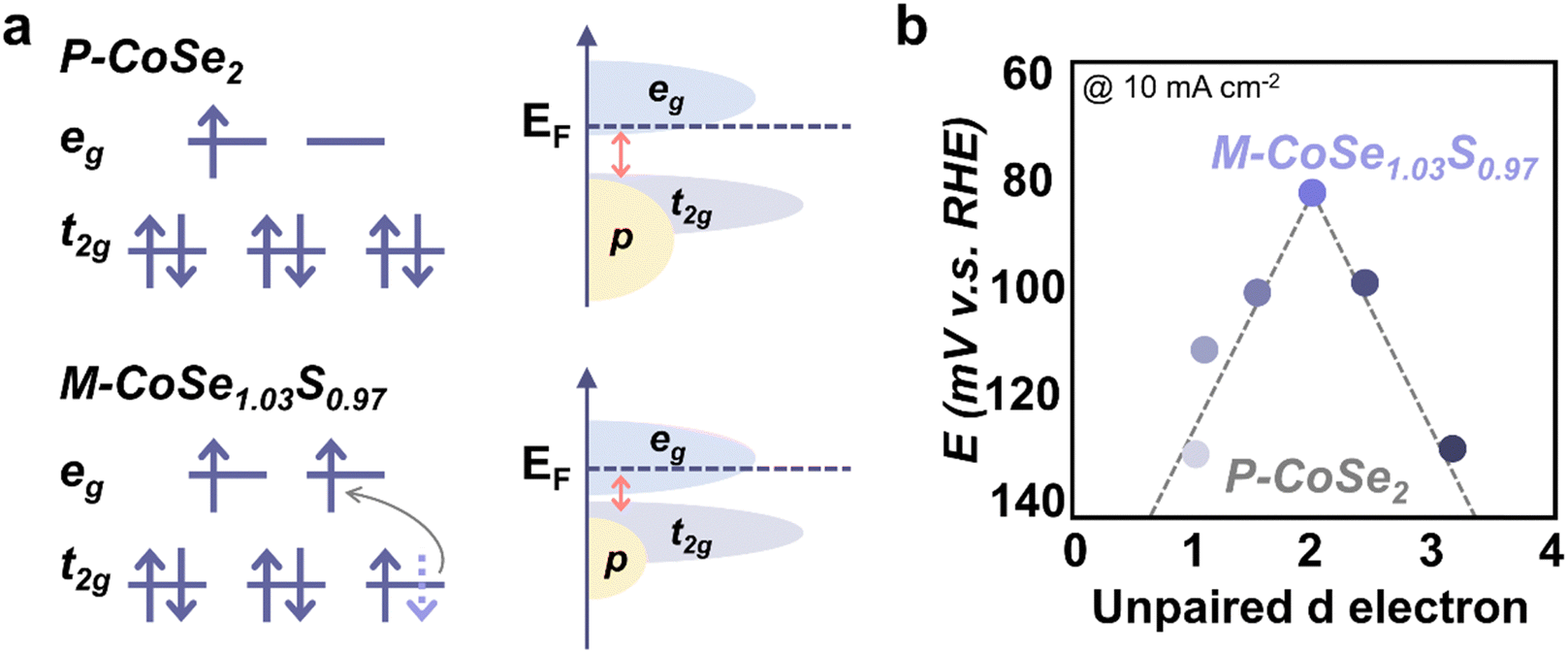 | ||
| Fig. 7 (a) Schematic illustration of the Co(II) spin state and energy band diagrams of pyrite CoSe2 (P-CoSe2) and marcasite CoSe1.03S0.97 (M-CoSe1.03S0.97). (b) Volcano relationship between the HER activity and the number of unpaired d electrons for S-doped CoSe2 (M-CoSe2−xSx, where 0 < x < 1). M-CoSe1.03S0.97 with an unpaired d electron number of 2.03 is at the top of the volcano. Reproduced from ref. 29 with permission from American Association for the Advancement of Science, Copyright 2023. | ||
In the studies of the CO2RR, there are mentions of the influence of the spin state on performance. For example, Wang et al. reported that HS Ni(II) phthalocyanine (NiPc) gives a higher CO faradaic efficiency than LS NiPc.32 The HS Ni center possesses more unpaired 3d electrons than the LS Ni center, and hence, exhibits an improved *COOH adsorption capacity and accelerated conversion of adsorbed CO2 to *COOH. On the other hand, Saha et al. reported that the selectivity of products can be affected by the spin state of metal centers as well. They found the spin state of Fe–COOH determining the product selectivity between CO and HCOOH.31 They compared the spin state of Fe(II)–COOH and CO2RR pathways on iron porphyrin (FeEs4) and Fe–chlorin. Fe(II)–COOH of FeEs4 displaying the LS state favors selective 2e−/2H+ electrocatalytic reduction of CO2 to CO, while Fe(II)–COOH of the Fe–chlorin complex displaying the HS state favors generating HCOOH as the only product. For Co-based electrocatalysts, Kong et al.61 detected the spin state of Co(II) on Co–salophen-X, (X represents Cl, Br, and I) and determined that the high ratio of HS Co(II) in Co–salophen–Br facilitates CO2 activation. On Co–salophen–Br with a high ratio of HS Co(II), electron transfer from Co(II) 3d orbitals (3dz2 and 3dx2–y2) to the CO2 unoccupied π* orbital is promoted and the faradaic efficiency of CO2–CO reduction is boosted. Besides in the CO2RR, HS Co(II) in single-Co-atom catalysts is also indicated as the atomic center for high selectivity of CH3OH in CO reduction.62 Electron backdonation via the dxy/dyz − 2π* bond could be improved and C–O bonding in *CO could be weakened on the HS Co(II) center. Compared with the LS Co(II) atomic center, the CORR RDS on HS Co(II) is methanol desorption instead of *CO/*CHxO hydrogenation and the RDS energy carrier is decreased from 0.75 to 0.43 eV.
As for the N2 reduction reaction (NRR), challenges lie in the adsorption and activation of N2 on the catalytic sites as well as the breaking of the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond. In this regard, Fang et al. revealed that tuning the spin state of the transition metal in single-atom-catalysts (to increase the magnetic moment) deviates the scaling relationships between key N-containing intermediates (*N2H/*N2 and *NH2/*N2), providing the active site with a balanced N-affinity critical for efficient eNRR.6 Wang et al. unveiled that HS Fe(II) species were regulated to IS Fe(II) species in Fe, Mo-co-coordinated polyphthalocyanine (FeMo-PPc).33 The IS state with empty d orbitals and unpaired d electrons facilitated the overlap of Fe 3d and N 2p orbitals, resulting in efficient activation of the NN triple bond. In contrast, Song et al. tuned the spin state of Fe(III) ions to HS via F modification in TiO2 nanoparticles, where HS Fe(III) ions facilitated the backdonation of Fe 3d electrons to N 1πg* orbital.34 In both cases, the activation of N2 molecule is promoted and the limiting potential of the NRR is reduced.
N triple bond. In this regard, Fang et al. revealed that tuning the spin state of the transition metal in single-atom-catalysts (to increase the magnetic moment) deviates the scaling relationships between key N-containing intermediates (*N2H/*N2 and *NH2/*N2), providing the active site with a balanced N-affinity critical for efficient eNRR.6 Wang et al. unveiled that HS Fe(II) species were regulated to IS Fe(II) species in Fe, Mo-co-coordinated polyphthalocyanine (FeMo-PPc).33 The IS state with empty d orbitals and unpaired d electrons facilitated the overlap of Fe 3d and N 2p orbitals, resulting in efficient activation of the NN triple bond. In contrast, Song et al. tuned the spin state of Fe(III) ions to HS via F modification in TiO2 nanoparticles, where HS Fe(III) ions facilitated the backdonation of Fe 3d electrons to N 1πg* orbital.34 In both cases, the activation of N2 molecule is promoted and the limiting potential of the NRR is reduced.
Another popular reaction for N-cycles, the electrochemical reduction of nitrate to ammonia, has been studied as well for the effect of the spin state of metal centers. Dai et al. proposed that increasing the HS Fe(II) content in Fe1–Ti pairs boosted the ammonia yield through the electrochemical NO3RR.63 They introduced oxygen vacancies into the Fe-anchored monolithic Ti electrode to create more HS Fe(II) and yielded ammonia five times higher than those with less HS Fe(II), which could be attributed to boosted NO3− deoxygenation and *NO hydrogenation on HS Fe(II) in Fe1–Ti pairs. Sun et al. reported that the Co spin state in the Cu–Co pair on rock-salt high-entropy oxides influences nitrate reduction.35 The HS Co ion of the Cu–Co pair in Mg0.2Co0.2Ni0.2Cu0.2Zn0.2O facilitates ammonia production, while Li incorporation into the oxide lowers the spin state of Co and weakens the Cu–Co synergetic effects. In the coupling of nitrate reduction with carbon dioxide reduction for urea synthesis, Wang et al. demonstrated that FeOOH with LS Fe doubled the urea yield rate compared to that with IS Fe.36 Electron pair transfer from the occupied orbitals of *NO intermediate to the empty 3d orbitals of Fe is promoted by LS Fe, lowing the energy barrier to improve urea generation.
A recent study also reports an example of spin state influence in the urea electrooxidation reaction (UOR). The reaction may proceed through a PCET dehydrogenation mechanism and direct chemical coupling reaction pathway. A near-unity eg orbital occupancy of active sites is reported as an optimal condition for key intermediate adsorption. Li et al. replaced octahedral Co(III) ions with Ni(III) ions in NiCo2O4 and found that the increase of LS octahedral Ni(III) ionic content improved the urea oxidation rate.21 Specifically, LS octahedral Ni(III) ions in NixCo3xO4 facilitated the adsorption of two N atoms in CO(NH2)2 on the octahedral Ni(III) active sites, thus improving the UOR activity.
5. Challenges and opportunities
In a nutshell, the spin state of first-row TM-based catalysts could be marked as one of the fundamental features and plays a significant role in their catalysis of electrochemical reactions. While we opine that the spin state is a pivotal feature in electrocatalysis, the development of the electrocatalysis field is still in its infancy with many potentially impactful research directions. Therefore, to assist researchers to navigate through the challenges and make the best use of scientific opportunities, we suggest some propositions for this topic (Fig. 8). | ||
| Fig. 8 An outlook on future spin-state-related research development from the aspects of advanced techniques, modern theoretical calculation, and the possible effects of spin states on electrocatalysts. | ||
5.1 Distinguish the spin state of central metals in the active phase/surface from the bulk of electrocatalysts
Depending on the reaction conditions, the phase and/or surface of electrocatalysts may be changed during the reaction. Hence, more effort is needed to study the change in the spin state during the reactions and their influence on the performance over a long-time scale. In the context of the OER catalyzed by first-row TM oxide catalysts, it has been widely observed that the surface reconstruction of a (pre-)catalyst generates an active phase/surface that is different from the pristine state of the (pre-)catalyst. Most attention has been paid to the chemical composition and electronic structure of the reconstructed surface phase. However, the possible spin state change due to this kind of phase change has not been fully investigated. The surface reconstruction may not be a phenomenon only for OER catalysts. It has been found in several reactions, such as the ORR,64 HER,65 CO2RR,66–69 NRR,70 and NO3RR.66,71 If the spin state of metal centers changes in these reactions, how the change affects the reaction performance will need more studies to be revealed.5.2 Tracking of spin state evolution at active sites during electrocatalysis
The evolution of the spin state of active sites (within the persisted surface phase) during electrocatalysis has not been thoroughly studied. Some studies have shown how the change or preservation of the spin state influences electrocatalysis utilizing ex situ characterization approaches. In a process involving multiple electron transfer, the spin state of active sites may change across different reaction steps, which might have an impact on intermediate adsorption or desorption. Moreover, some TM-based catalysts have exhibited reversible surface reconstruction during electrocatalysis, generating temporary active sites that catalyze those reactions that cannot be observed once the conditions to generate them (e.g., applied potential) have been removed. Hence, the use of in situ/operando characterization approaches is essential to provide time-evolution data on the spin state of metal centers, which will provide important information for correlating the spin state change with the thermodynamics and kinetics of each stage in the reaction.5.3 Inclusion of the spin state in predictive theoretical calculations
Theoretical calculation methods, such as DFT and molecular dynamics, are useful tools for forecasting experimental results, especially for data that cannot be obtained directly from experiments. One such data is the spin state of catalysts as theoretical calculations can help to overcome challenges arising from the operational complexity of the characterization of the spin state. Recently, Fang et al. precisely modulated the spin state of Co(III) in LaCo1−xFexO3 (x = 0, 1/9, 2/9……8/9, 1) through adjusting the second coordination spheres (SCS) of Co6−y–[Co]–Fey (y = 0–6).72 The Co–O binding energy of Co–O6 is affected by SCS and the corresponding spin state is regulated. The highest OER activity of LaCo7/9Fe2/9O3 with IS Co(III) among LaCo1−xFexO3 is predicted in DFT calculations and confirmed in experiments. A similar result was reported previously by Duan et al. that LaCo0.9Fe0.1O3 with a mixture of HS and LS Co(III) gave the best OER activity.52 Thanks to the comparably easier access to theoretical calculation, the impacts of many complicated or extreme experimental conditions on spin states could be simulated. Various external forces, such as thermal field, magnetic field, and temperature conditions, can be introduced to predict their impact on the electrocatalytic performance. Besides, benefitting from the vigorous development of calculation techniques, the combination of material simulation and machine learning acts as an extremely modern and sharp tool to predict the material performance and screen catalyst candidates, reducing the tremendous manpower and material resources that would have been needed for unaided scientific exploration. Following the results acquired from the predictive calculation, the experiment might be executed in a relatively efficient manner.5.4 Difference between the spin states in bulk and surface phases
Generally, the properties of a material surface may be very different from the bulk. Since catalytic reactions mostly occur on the surface, it is intuitive that the surface properties need to be studied precisely. As discussed, several cations have varied spin states in different conditions, including their position relative to the surface. During the characterization of spin states, the difference between spin states in the bulk and on the surface should be emphasized. While the reactants and intermediates are adsorbed or desorbed on the surface, the electron transport in the bulk may also influence the catalytic performance. As such, in choosing characterization techniques, their probe areas and depth need to be taken into consideration. Moreover, the interaction between surface and bulk should be considered especially since a spin pinning effect has been previously observed at the interface between the bulk phase and the reconstructed surface.73 Therefore, it is worth noting that a systematic discussion on the catalytic behavior with distinction between spin states at the surface and in the bulk is necessary for a complete discussion on the role of spin states in an electrocatalyst.5.5 Increase in electrical conductivity of catalysts due to changes in the spin states
It is commonly understood that the electrical conductivity of catalysts contributes to the electron transport between the electrocatalytic surface and the current collector interfaces and is regarded as one of the factors in their catalytic performance. As previously reported,23,74 the optimization of spin states could affect the electrical conductivity and thus improve the catalytic performance. The improved conductivity of catalysts brought by spin state optimization validates the significant role of spin states in electrocatalysis. Therefore, it is also interesting to determine the spin states when the electrical conductivity of electrocatalysts changes. Furthermore, in electrical conductivity studies, the effects of spin states and their regulation in the optimization of electrical conductivity should be considered. Finally, it should be noted that only the reactions limited by the conductivity may show a remarkable influence on the electrocatalysis performance in this case. For the reactions limited by the reaction kinetics at the active site, the conductivity in principle won’t have a significant influence.5.6 Relationship between working conditions, spin state, and electrocatalysis
Working conditions such as pH,75 temperature,76,77 pressure,77 and operating potential78 may affect the spin states of electrocatalysts. The metal coordination and orbital overlapping between metals and ligands can be changed through varying these conditions, resulting in a spin state change in metal centers. The metal centers in optimal spin states can give d-orbital fillings to achieve the desired interactions with intermediates. It is worth noting that, in general, an external magnetic field cannot change the spin state except under some extreme conditions.79 For example, it is reported that a pulsed magnetic field of up to 67 T can only induce 14% LS–IS transition of Co(III) in LaCoO3.80 While it has been reported that an HS–LS transition of Co(III) in Co0.8Mn0.2-MOF is triggered by a high-frequency alternating magnetic field in the range of 0.1 T for 20 min, the change is attributed to the magnetic heating generated by alternative magnetic stimulation.81 The working conditions may affect the spin states of metal centers in electrocatalysts. However, insufficient attention has been paid to the spin state change with those employed working conditions in electrocatalysis. Investigating the relationship between working conditions, the spin state of metal centers, and electrocatalytic performance will facilitate the development of fundamental understandings as well as electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Agency for Science, Technology and Research (A*STAR) MTC Individual Research Grants (IRG) M22K2c0078 and the Singapore Ministry of Education Tier 1 Grant (RG78/22). This research was also supported by the National Nature Science Foundation of China (Grant No. 52202202).Notes and references
- F. Johnsson, J. Kjärstad and J. Rootzén, Clim. Policy, 2018, 19, 258–274 CrossRef.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- J. K. Norskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- C. Fang and W. An, Nano Res., 2021, 14, 4211–4219 CrossRef CAS.
- J. Kim, X. Yin, K. C. Tsao, S. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS PubMed.
- H. Cai, B. Chen, X. Zhang, Y. Deng, D. Xiao, D. Ma and C. Shi, Nano Res., 2020, 14, 122–130 CrossRef.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2020, 4, 10–19 CrossRef.
- Z. Li, Z. Zhuang, F. Lv, H. Zhu, L. Zhou, M. Luo, J. Zhu, Z. Lang, S. Feng, W. Chen, L. Mai and S. Guo, Adv. Mater., 2018, 30, e1803220 CrossRef PubMed.
- J. Qian, T. Wang, Z. Zhang, Y. Liu, J. Li and D. Gao, Nano Energy, 2020, 74, 104948 CrossRef CAS.
- Y.-N. Zhou, F.-G. Wang, Y.-N. Zhen, J. Nan, B. Dong and Y.-M. Chai, Sci. China Mater., 2022, 65, 2665–2674 CrossRef CAS.
- J. Ran, T. Wang, J. Zhang, Y. Liu, C. Xu, S. Xi and D. Gao, Chem. Mater., 2020, 32, 3439–3446 CrossRef CAS.
- J. Ran, L. Wang, M. Si, X. Liang and D. Gao, Small, 2023, 19, 2206367 CrossRef CAS PubMed.
- C. N. R. Rao, O. M. Parkash and P. Ganguly, J. Solid State Chem., 1975, 15, 186–192 CrossRef CAS.
- D. V. Karpinsky, I. O. Troyanchuk, K. Bärner, H. Szymczak and M. Tovar, J. Phys.: Condens. Matter, 2005, 17, 7219–7226 CrossRef CAS.
- R. M. Ramsundar, V. K. Pillai and P. A. Joy, Phys. Chem. Chem. Phys., 2018, 20, 29452–29461 RSC.
- R. Zhang, L. Wang, L. Pan, Z. Chen, W. Jia, X. Zhang and J.-J. Zou, Appl. Catal., B, 2020, 277, 119237 CrossRef CAS.
- W. H. Lee, M. H. Han, Y. J. Ko, B. K. Min, K. H. Chae and H. S. Oh, Nat. Commun., 2022, 13, 605 CrossRef CAS PubMed.
- Z. Wang, S. Shen, Z. Lin, W. Tao, Q. Zhang, F. Meng, L. Gu and W. Zhong, Adv. Funct. Mater., 2022, 32, 2112832 CrossRef CAS.
- Y. Li, H. Jiang, Z. Cui, S. Zhu, Z. Li, S. Wu, L. Ma, X. Han and Y. Liang, J. Phys. Chem. C, 2021, 125, 9190–9199 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- C. Mu, J. Mao, J. Guo, Q. Guo, Z. Li, W. Qin, Z. Hu, K. Davey, T. Ling and S. Z. Qiao, Adv. Mater., 2020, 32, e1907168 CrossRef PubMed.
- S. Chen, X. Liang, S. Hu, X. Li, G. Zhang, S. Wang, L. Ma, C. L. Wu, C. Zhi and J. A. Zapien, Micro Nano Lett., 2023, 15, 47 CrossRef CAS PubMed.
- R. Wang, L. Zhang, J. Shan, Y. Yang, J.-F. Lee, T.-Y. Chen, J. Mao, Y. Zhao, L. Yang, Z. Hu and T. Ling, Adv. Sci., 2022, 9, e2203917–e2203917 CrossRef PubMed.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- Y. Liu, X. Hua, C. Xiao, T. Zhou, P. Huang, Z. Guo, B. Pan and Y. Xie, J. Am. Chem. Soc., 2016, 138, 5087–5092 CrossRef CAS PubMed.
- C. Xia, H. Liang, J. Zhu, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2017, 7, 1602089 CrossRef.
- X.-L. Zhang, P.-C. Yu, X.-Z. Su, S.-J. Hu, L. Shi, Y.-H. Wang, P.-P. Yang, F.-Y. Gao, Z.-Z. Wu, L.-P. Chi, Y.-R. Zheng and M.-R. Gao, Sci. Adv., 2023, 9, eadh2885 CrossRef CAS PubMed.
- C. Pei, M. C. Kim, Y. Li, C. Xia, J. Kim, W. So, X. Yu, H. S. Park and J. K. Kim, Adv. Funct. Mater., 2023, 33, 2210072 CrossRef CAS.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- X. Wang, Y. Fu, D. Tranca, K. Jiang, J. Zhu, J. Zhang, S. Han, C. Ke, C. Lu and X. Zhuang, ACS Appl. Energy Mater., 2021, 4, 2891–2898 CrossRef CAS.
- Y. Wang, W. Cheng, P. Yuan, G. Yang, S. Mu, J. Liang, H. Xia, K. Guo, M. Liu, S. Zhao, G. Qu, B. A. Lu, Y. Hu, J. Hu and J. N. Zhang, Adv. Sci., 2021, 8, e2102915 CrossRef PubMed.
- G. Song, R. Gao, Z. Zhao, Y. Zhang, H. Tan, H. Li, D. Wang, Z. Sun and M. Feng, Appl. Catal., B, 2022, 301, 120809 CrossRef CAS.
- S. Sun, C. Dai, P. Zhao, S. Xi, Y. Ren, H. R. Tan, P. C. Lim, M. Lin, C. Diao, D. Zhang, C. Wu, A. Yu, J. C. J. Koh, W. Y. Lieu, D. H. L. Seng, L. Sun, Y. Li, T. L. Tan, J. Zhang, Z. J. Xu and Z. W. Seh, Nat. Commun., 2024, 15, 260 CrossRef CAS PubMed.
- C. Wang, C. Yu, B. Qian, Y. Ren, L. Wang, Y. Xie, X. Tan, X. He and J. Qiu, Small, 2023, 2307349 Search PubMed.
- R. G. J. Fraser and H. M. Macdonald, Proc. R. Soc. London, Ser. A, 1927, 114, 212–221 CAS.
- A. Streitwieser and H. W. Salzberg, J. Electrochem. Soc., 1962, 109, 116Cb CrossRef.
- R. G. Burns, Mineralogical Applications of Crystal Field Theory, Cambridge University Press, Cambridge, 2 edn, 1993, pp. 7–43 Search PubMed.
- J. Griffith and L. Orgel, Quarterly Reviews, Chemical Society, 1957, vol. 11, pp. 381–393 Search PubMed.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Chem, 2017, 3, 812–821 CAS.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 CrossRef CAS PubMed.
- Y.-Z. Jin, Z. Li, J.-Q. Wang, R. Li, Z.-Q. Li, H. Liu, J. Mao, C.-K. Dong, J. Yang, S.-Z. Qiao and X.-W. Du, Adv. Energy Mater., 2018, 8, 1703469 CrossRef.
- W. Cheng, P. Yuan, Z. Lv, Y. Guo, Y. Qiao, X. Xue, X. Liu, W. Bai, K. Wang, Q. Xu and J. Zhang, Appl. Catal., B, 2020, 260, 118198 CrossRef CAS.
- Z. Zhang, P. Ma, L. Luo, X. Ding, S. Zhou and J. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202216837 CrossRef CAS PubMed.
- F. de Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS PubMed.
- S. A. Bonke, T. Risse, A. Schnegg and A. Brückner, Nat. Rev. Methods Primers, 2021, 1, 33 CrossRef CAS.
- C. Wu, X. Wang, Y. Tang, H. Zhong, X. Zhang, A. Zou, J. Zhu, C. Diao, S. Xi, J. Xue and J. Wu, Angew. Chem., Int. Ed., 2023, 62, e202218599 CrossRef CAS PubMed.
- T. E. Cranshaw, J. Phys. E: Sci. Instrum., 1974, 7, 497 CrossRef CAS.
- M. T. Sougrati, V. Goellner, A. K. Schuppert, L. Stievano and F. Jaouen, Catal. Today, 2016, 262, 110–120 CrossRef CAS.
- Y.-A. Soh and G. Aeppli, J. Appl. Phys., 1999, 85, 4607–4609 CrossRef CAS.
- Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou, G. Zhang, H. Yang, Y. Du and Z. J. Xu, Chem. Mater., 2017, 29, 10534–10541 CrossRef CAS.
- F. S. Hegner, J. R. Galan-Mascaros and N. Lopez, J. Phys. Chem. Lett., 2022, 13, 4104–4110 CrossRef CAS PubMed.
- Z.-D. He, R. Tesch, M. J. Eslamibidgoli, M. H. Eikerling and P. M. Kowalski, Nat. Commun., 2023, 14, 3498 CrossRef CAS PubMed.
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T.-S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS.
- W. L. Roth, J. Phys. Chem. Solids, 1964, 25, 1–10 CrossRef CAS.
- S. Ohnishi and S. Sugano, J. Phys. C: Solid State Phys., 1981, 14, 39 CrossRef CAS.
- Z. Du, Z. Meng, X. Gong, Z. Hao, X. Li, H. Sun, X. Hu, S. Yu and H. Tian, Angew. Chem., 2023, e202317022 Search PubMed.
- M. Xiao, J. Zhu, L. Ma, Z. Jin, J. Ge, X. Deng, Y. Hou, Q. He, J. Li, Q. Jia, S. Mukerjee, R. Yang, Z. Jiang, D. Su, C. Liu and W. Xing, ACS Catal., 2018, 8, 2824–2832 CrossRef CAS.
- Z. Chen, H. Niu, J. Ding, H. Liu, P. H. Chen, Y. H. Lu, Y. R. Lu, W. Zuo, L. Han, Y. Guo, S. F. Hung and Y. Zhai, Angew. Chem., Int. Ed., 2021, 60, 25404–25410 CrossRef CAS PubMed.
- X. Kong, J. Ke, Z. Wang, Y. Liu, Y. Wang, W. Zhou, Z. Yang, W. Yan, Z. Geng and J. Zeng, Appl. Catal., B, 2021, 290, 120067 CrossRef CAS.
- J. Ding, Z. Wei, F. Li, J. Zhang, Q. Zhang, J. Zhou, W. Wang, Y. Liu, Z. Zhang and X. Su, Nat. Commun., 2023, 14, 6550 CrossRef CAS PubMed.
- J. Dai, Y. Tong, L. Zhao, Z. Hu, C.-T. Chen, C.-Y. Kuo, G. Zhan, J. Wang, X. Zou, Q. Zheng, W. Hou, R. Wang, K. Wang, R. Zhao, X.-K. Gu, Y. Yao and L. Zhang, Nat. Commun., 2024, 15, 88 CrossRef PubMed.
- Q. Li, D. Zhang, J. Wu, S. Dai, H. Liu, M. Lu, R. Cui, W. Liang, D. Wang and P. Xi, Adv. Mater., 2023, 2309266 Search PubMed.
- S. Banerjee, A. Kakekhani, R. B. Wexler and A. M. Rappe, ACS Catal., 2023, 13, 4611–4621 CrossRef CAS.
- J. Vavra, G. P. L. Ramona, F. Dattila, A. Kormányos, T. Priamushko, P. P. Albertini, A. Loiudice, S. Cherevko, N. Lopéz and R. Buonsanti, Nat. Catal., 2024, 7, 89–97 CrossRef CAS.
- S. Liang, J. Xiao, T. Zhang, Y. Zheng, Q. Wang and B. Liu, Angew. Chem., 2023, 135, e202310740 CrossRef.
- Y. Jia, Y. Ding, T. Song, Y. Xu, Y. Li, L. Duan, F. Li, L. Sun and K. Fan, Adv. Sci., 2023, 10, 2303726 CrossRef CAS PubMed.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- Y. Ling, Q. Feng, H. Xie, X. Zheng, X. Chen, Z. Zou, A. Liu, J. Tang, Y. Li and Q. Wang, ACS Sustainable Chem. Eng., 2023, 11, 12345–12354 CrossRef CAS.
- L. Qiao, D. Liu, A. Zhu, J. Feng, P. Zhou, C. Liu, K. W. Ng and H. Pan, Appl. Catal., B, 2024, 340, 123219 CrossRef CAS.
- Z. Fang, M. Chai, Y. Lv, W. Zhao, X. Hou and Y. Hou, Adv. Funct. Mater., 2024, 2315039 CrossRef CAS.
- T. Wu, X. Ren, Y. Sun, S. Sun, G. Xian, G. G. Scherer, A. C. Fisher, D. Mandler, J. W. Ager, A. Grimaud, J. Wang, C. Shen, H. Yang, J. Gracia, H.-J. Gao and Z. J. Xu, Nat. Commun., 2021, 12, 3634 CrossRef CAS PubMed.
- X. Zhao, H. Zhang, Y. Yan, J. Cao, X. Li, S. Zhou, Z. Peng and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 328–332 CrossRef CAS PubMed.
- S. Kim and H. Kim, Catal. Today, 2017, 295, 119–124 CrossRef CAS.
- M. Ya, Y. Wang, J. Wang, G. Gao, X. Zhao, Y. Wei, Z. Geng, G. Li and L. Li, ACS Sustainable Chem. Eng., 2023, 11, 15451–15459 CrossRef CAS.
- G. Vankó, J.-P. Rueff, A. Mattila, Z. Németh and A. Shukla, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 024424 CrossRef.
- L. Ma, S.-F. Hung, L. Zhang, W. Cai, H. B. Yang, H. M. Chen and B. Liu, Ind. Eng. Chem. Res., 2018, 57, 1441–1445 CrossRef CAS.
- S. Luo, K. Elouarzaki and Z. J. Xu, Angew. Chem., Int. Ed., 2022, 61, e202203564 CrossRef CAS PubMed.
- K. Sato, A. Matsuo, K. Kindo, Y. Kobayashi and K. Asai, J. Phys. Soc. Jpn., 2009, 78, 093702 CrossRef.
- G. Zhou, P. Wang, H. Li, B. Hu, Y. Sun, R. Huang and L. Liu, Nat. Commun., 2021, 12, 4827 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |