
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
5-Hydroxymethylcytosine: the many faces of the sixth base of mammalian DNA
Edita
Kriukienė
 ,
Miglė
Tomkuvienė
and
Saulius
Klimašauskas
*
,
Miglė
Tomkuvienė
and
Saulius
Klimašauskas
*
Institute of Biotechnology, Life Sciences Center, Vilnius University, Saulėtekio al. 7, LT-10257 Vilnius, Lithuania. E-mail: saulius.klimasauskas@bti.vu.lt
First published on 11th January 2024
Abstract
Epigenetic phenomena play a central role in cell regulatory processes and are important factors for understanding complex human disease. One of the best understood epigenetic mechanisms is DNA methylation. In the mammalian genome, cytosines (C) in CpG dinucleotides were long known to undergo methylation at the 5-position of the pyrimidine ring (mC). Later it was found that mC can be oxidized to 5-hydroxymethylcytosine (hmC) or even further to 5-formylcytosine (fC) and to 5-carboxylcytosine (caC) by the action of 2-oxoglutarate-dependent dioxygenases of the TET family. These findings unveiled a long elusive mechanism of active DNA demethylation and bolstered a wave of studies in the area of epigenetic regulation in mammals. This review is dedicated to critical assessment of recent data on biochemical and chemical aspects of the formation and conversion of hmC in DNA, analytical techniques used for detection and mapping of this nucleobase in mammalian genomes as well as epigenetic roles of hmC in DNA replication, transcription, cell differentiation and human disease.
 Miglė Tomkuvienė, Saulius Klimašauskas and Edita Kriukienė | Edita Kriukienė received her PhD in Biochemistry in 2007 from the Institute of Biotechnology in Vilnius, Lithuania for her work on biochemical characterization of bacterial restriction-modification enzymes in the lab of Arvydas Lubys. Then she joined the group of Saulius Klimašauskas at Vilnius University, Lithuania as a postdoctoral researcher and later as a group leader and Research Professor. Her work is concerned with genome-wide studies of epigenetic DNA modifications and chromatin structure in development and disease. Miglė Tomkuvienė received her PhD in Biochemistry in 2013 from the Institute of Biotechnology, Vilnius University, Lithuania, under supervision of Saulius Klimašauskas and in collaboration with Béatrice Clouet-d’Orval at Paul Sabatier University, Toulouse, France. She continued in the Klimašauskas group as a researcher and then senior researcher, where her research focuses on natural and synthetic DNA modifications and rare genetic syndromes associated with epigenetic dysregulation. Saulius Klimašauskas, FRSC graduated Organic Chemistry at Vilnius University, and then worked with Arvydas Janulaitis at the Institute of Applied Enzymology Fermentas to receive his PhD in Bioorganic Chemistry in 1987. Then he worked at Cold Spring Harbor Laboratory as a postdoctoral scientist in Structural and Molecular Biology with Richard J. Roberts. After starting his own group in 1995, he grew through the ranks to become a Distinguished Professor of Vilnius University in 2017. His long standing research interests include mechanistic studies and molecular engineering of AdoMet-dependent methyltransferases and epigenetic mechanisms involving biological modification of DNA and RNA. |
Key learning points(1) Mechanistic understanding of the genesis and conversions of 5-hydroxymethylcytosine in the context of DNA metabolism.(2) Strengths and limitations of different methodologies used to study the occurrence and distribution of 5-hydroxymethylcytosine in mammalian genomes. (3) Major regulatory roles that this and related epigenetic modifications play in DNA repair, replication, transcription and cell differentiation. (4) Potential utility of 5-hydroxymethylcytosine as a disease marker and therapeutic agent. |
1. Introduction
The key function of the genome is the storage, replication and transmission of the encoded genetic information. A meaningful and timely reading of the genome consisting of billions of recurring G:C or A:T base pairs in different types of cells is possible by using a sort of (epi)genetic “bookmarking” system – a mechanism that allows living organisms to maintain or reprogram the identity of each cell during development or in response to environmental cues. A key player in this process is DNA methylation which occurs by enzymatic transfer of a methyl group from the ubiquitous cofactor S-adenosyl-L-methionine (SAM) onto specific targets in DNA. Three major products of DNA methyltransferases are N6-methyladenine, N4-methylcytosine or 5-methylcytosine (mC). Notably, the biologically installed methyl groups do not alter the pairing specificity of the target nucleobases preserving the original genetic content of the genome. Their exposure in the major groove of the DNA helix (Fig. 1) enables readability of these “steric” groups by specialized cellular proteins, enzymes or large multicomponent complexes. These features make such modified bases well suited to serve as epigenetic marks for biological signaling that operates as an additional regulatory layer “above” the genome. All three types of DNA methylation are found in microorganisms and occur sequence-specifically. In vertebrate animals, the dominant methylation product is mC (Fig. 2a); the mC methylation occurs in sequence-specific (predominantly but not exclusively at CpG dinucleotides) and in a locus-specific manner.1,2 DNA methylation levels vary dramatically during development, but in somatic tissues the majority (70–80%) of CpGs are methylated except those localized in so-called CpG islands (genomic regions highly enriched in CpG sites).3,4 Traditionally, mCs, when localized at CpG islands, are important transcriptional silencers of gene promoters. Three major types of DNA methyltransferases are active on mammalian genomes. Initial methylation patterns are thought to be established by so-called de novo DNA methyltransferases Dnmt3a and Dnmt3b, whereas preservation of the CpG methylation marks across cell divisions is carried out by the maintenance methyltransferase Dnmt1. Underlying its importance, mC is often called the fifth base of DNA.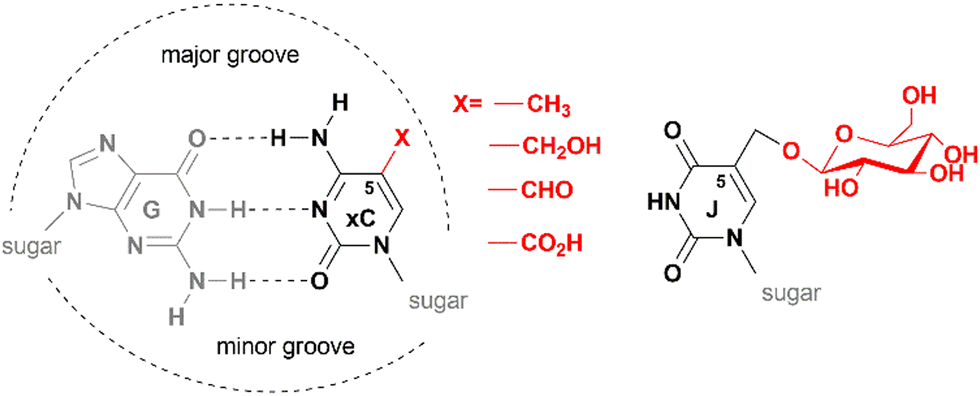 | ||
| Fig. 1 Base pairing and epigenetic modifications in DNA of eukaryotic organisms. Biological cytosine-5 modifications (xC, x = methyl, hydroxymethyl, formyl of carboxyl) point to the major groove of the B-helical DNA and do not interfere with G:C base pairing interactions (left). Structure of base-J (5-(β-D-glucosyl)oxymethyluracil) present in DNA of flagellated protozoa (right). | ||
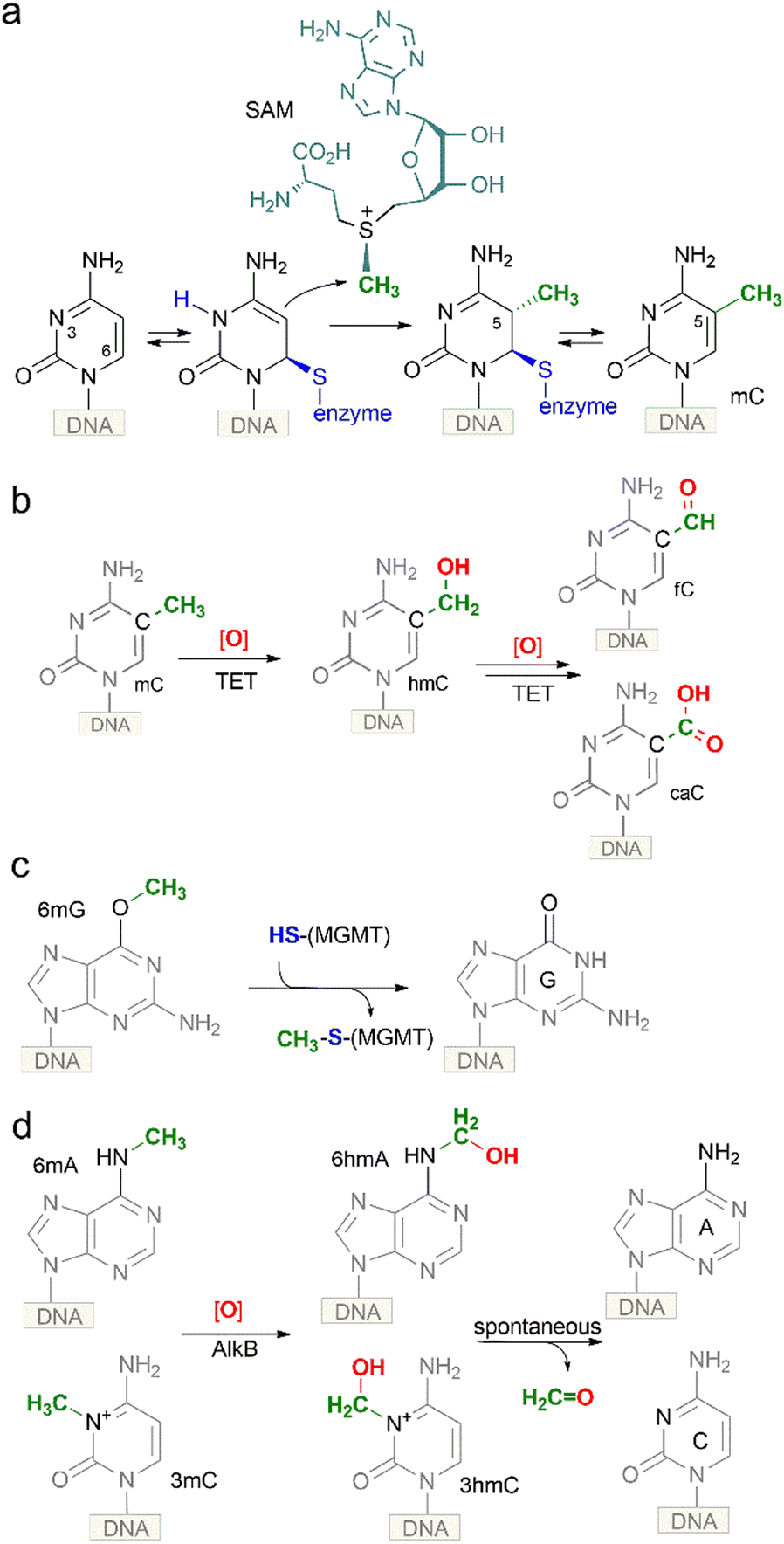 | ||
| Fig. 2 Chemical strategies for biological methylation and demethylation of DNA nucleobases at O, N and C atoms. (a), methylation of cytosine by C5-MTases via an SN2 transfer of a sulfonium-bound methyl group onto a covalently activated target cytosine residue; (b) oxidation of mC in DNA by mammalian TET dioxygenases yields chemically stable hmC, which is further oxidized to fC or caC to be removed by dedicated glycosylases; (c) a product of DNA alkylation damage, O6-methylguanine, is reverted to guanine directly via an SN2 transfer of the O6-methyl group onto a cysteine residue of a methylguanine DNA methyltransferase (MGMT) protein; (d) oxidation of representative N-alkylated nucleobases (N3-methylcytosine, N6-methyladenine) in DNA by the AlkB family of dioxygenases yields corresponding N-hydroxymethyl derivatives, which then undergo spontaneous hydrolytic release of formaldehyde to directly generate the unmodified base. | ||
2. Occurrence of 5-hydroxymethylated nucleobases in DNA
The presence of hmC in DNA was first observed in certain bacteriophages. In T-even phages, 5-hydroxymethylated cytosine is incorporated into the genome during DNA synthesis and is subsequently modified by phage α- and β-glucosyltransferases creating highly glucosylated DNA containing 5-glucosyloxymethylcytosine (glc-hmC) residues. Such DNA becomes resistant to cleavage by host restriction endonucleases. A similar modified base-J (5-(β-D-glucosyl) oxymethyluracil) (Fig. 1) is present in DNA of flagellated protozoa (which include parasites Trypanosoma brucei and Leishmania sp.) and closely related unicellular alga.5 It replaces up to 1% of thymines and is mostly found in telomeric repeats. It is produced by the oxidation of thymine residues in DNA by the JBP1/JBP2 2-oxoglutarate-dependent dioxygenases, followed by enzymatic glycosylation of the produced hmU. Base-J was demonstrated to be essential for transcription termination at the ends of the polycistronic gene clusters that are a hallmark of Leishmania and related trypanosomatids.6,7The presence of hmC in genomic DNA extracted from the brain of adult mice, rats and frogs was first reported in the early seventies.8 However, no confirmation of this finding was obtained in other labs for a period of almost 40 years.9 In 2009, two groups independently re-confirmed the existence of hmC in mouse brain cells and mouse embryonic stem cells (ESC).10,11 By using thin layer chromatography Kriaučionis and Heinz found that hmC constitutes 0.6% of total nucleotides in Purkinje cells and ∼0.2% in granule cells.10 Based on knowledge of the above-mentioned trypanosome proteins JBP1 and JBP2, Rao and co-workers identified their mammalian counterparts, the ten-eleven-translocation (TET) proteins TET1, TET2 and TET3 as potential modifiers of mC to hmC (Fig. 2b).11 The three homologs contain the common features of 2-oxoglutarate (2OG)- and Fe(II)-dependent dioxygenases. Two years later, the groups of Zhang and Xu have demonstrated that the TET enzymes are capable of further converting mC and hmC to 5-formylcytosine (fC) and 5-carboxylcytosine (caC) in vitro and in vivo,12,13 and, independently, the presence of low levels of genomic fC has been identified in mouse ESCs by the Carell group.14 Altogether this evidence and subsequent studies convincingly demonstrated that hmC is indeed an endogenous biological component of mammalian genomic DNA (also named the sixth base of DNA) assuming the role of a key intermediate in the long searched pathway of active DNA demethylation.
Besides the well-established TET-mediated pathway, one possible source of hmC could be direct hydroxymethylation of cytosine in DNA. This chemical precedent comes from in vitro experiments with representative SAM-dependent C5-MTases, which unexpectedly showed that these enzymes can catalyze covalent addition of formaldehyde to the C5-position of their target cytosine residues in DNA yielding hmC.15 Formaldehyde is an essential metabolite naturally present at ∼0.1 mM concentrations in mammalian cells, tissues and fluids, although the concentration can vary depending on the cell type and physiological conditions.16 However, the significance of this chemo-enzymatic route has not been confirmed in vivo.
Another potential way of hmC arrival in DNA is through the nucleotide salvage pathway (NSP) (Fig. 3). In addition to de novo synthesis, cells recycle nucleotides arising from the breakdown of DNA (DNA repair, apoptosis etc.) or from nutrient uptake. Akin to deoxycytidine, modified cytidines could enter deoxynucleotide pools via an enzymatic phosphorylation cascade which involves deoxycytidine kinase (DCK) to produce a monophosphate, followed by conversion into a diphosphate by cytidine monophosphate kinases (CMPK), and phosphorylation into a triphosphate by a family of nucleoside diphosphate kinases. However, a key barrier to the formation of 5-modified dCTPs and entrance of the modified cytidines into DNA is a high selectivity of CMPK for unmodified dCMP leading to exclusion of nucleotides carrying C5-modifications. Nevertheless, along with mC, hmC is readily deaminated to hmU at the level of dNMP (or dN depending on the cell type)17–19 to produce hmU 5′-mononucleotide. Since thymidylate kinase (DTYMK) is promiscuous with respect to 5-modifications, hmdUMP could be phosphorylated and then incorporated into DNA,19 where hmU then be targeted for excision by SMUG1 or TDG. To avoid cytotoxic complications associated with the appearance of hmU, cells deploy a dedicated enzyme – 2′-deoxynucleoside 5′-monophosphate N-glycosidase (DNPH1 also known as RCL), which degrades hmdUMP eliminating it from the nucleotide pool.17 These two main barriers of the NSP guard the mammalian DNA from sporadic incorporation of 5-hydroxymethylated pyrimidines, and thereby preventing their potential interference with the regulatory mechanisms mediated by the mC-derived epigenetic modifications. This safeguarding system is often weakened in fast proliferating cancer cells, which confers them susceptible to treatment with modified cytidines as potential therapeutic agents.
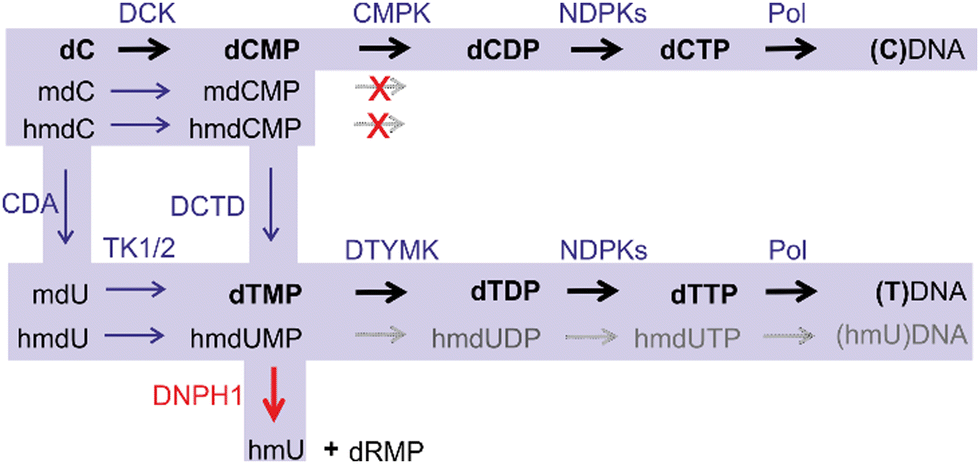 | ||
| Fig. 3 Nucleotide salvage pathway of dC and modified 2′-deoxycytidines (mdC and hmdC). DCK, deoxycytidine kinase; CMPK, cytidine monophosphate kinases; NDPKs, nucleoside-diphosphate kinases; DNPH1, 2′-deoxynucleoside 5′-monophosphate N-glycosidase; CDA, cytidine deaminase; TK1/2, thymidine kinase 1 and 2; DTYMK, deoxythymidylate kinase; DCDT, deoxycytidylate deaminase; Pol, DNA polymerase. Action of CMPK and DNPH1 guards against incorporation of hmC and hmU into DNA. | ||
3. Reversal of genomic cytosine-5 methylation
Loss of mC in genomic DNA can occur through a passive or an active DNA demethylation pathway. In the first scenario, the methylation marks are diluted from DNA in a replication-dependent manner in the absence of the methylation maintenance activity on the daughter strand. This mechanism has been well proven to operate in many cases, when DNMT1 is downregulated or excluded.20,21 Alternatively, mC can be “actively” converted back to unmodified C in the framework of the same DNA strand independently of DNA replication. The existence of active DNA demethylation has long been known in plants. In Arabidopsis thaliana, a group of DNA glycosylases named Demeter excise mC by cleaving the N-glycosydic bond, resulting in an abasic site on the DNA strand.22 Then, AP lyases and AP endonucleases form a single nucleotide gap that is subsequently filled by action of DNA polymerases and ligases. However, no such glycosylases have been found to act directly on mC in mammals. The reality of active demethylation in vertebrates had long remained elusive, but it all suddenly became clear with the discovery of hmC in DNA.3.1 Chemistry of DNA demethylation
As mentioned above, biological methylation is performed by DNA MTases which catalyze the SN2 transfer of sulfonium-bound methyl group from the cofactor SAM to defined positions on DNA (Fig. 2a). Although all reactions, in theory, are reversible, only one example of direct DNA demethylation via an SN2 reaction is known to date. O6-Methylguanine, a product of DNA alkylation damage by exogenous or endogenous compounds, is reverted to guanine directly via transfer of the O6-methyl group onto a cysteine residue of methylguanine DNA methyltransferase (MGMT) protein (Fig. 2c). This reaction requires stoichiometric amounts of the protein, which is a costly burden for the cell, perhaps justified by the rarity and severity (altered base-pairing) of the lesion. This seems to illustrate that a direct SN2 demethylation is only instrumental for O-bound methyl groups, and perhaps it is chemically unfeasible in the case of N- or C-methylation due to the high thermodynamic stability of the N–CH3 and C–CH3 bonds. Indeed, in all other reported demethylation cases, a multi-step mechanism based on radical redox chemistry is deployed. The latter reactions are carried out by a large family of 2OG-dependent dioxygenases. For N-alkylated nucleobases (N3-methylcytosine, N6-methyladenine), an enzymatic oxidation by the AlkB family of oxygenases leads to corresponding N-hydroxymethyl derivatives (Fig. 2d), which then undergo spontaneous hydrolytic release of formaldehyde from the nitrogen to directly generate the original unmodified base. Demethylation of biological N-methyl marks in histones occurs through a similar route. However, it turns out that hydroxymethyl groups produced at the C5-position of cytosine are chemically quite stable and long-lived under physiological conditions. In this case, additional cycles of enzymatic hydroxylation are required to produce modified bases that “appear” like DNA lesions and can be further processed by the repair machinery to ultimately yield unmodified cytosine (Fig. 2b).As mentioned above, the TET proteins were identified in mammals as the modifiers of mC to hmC11 and were also shown to be capable of oxidizing the formed hmC to fC and caC (collectively ox-mC).12,13 He et al. demonstrated that mC and hmC are almost fully (90%) converted to caC by TET1 and TET2 without appearance of fC, whereas Ito et al. report that fC accumulates relative to caC.12,13 Subsequent enzymatic and structural studies showed that TET2 can yield fC and caC by iteratively acting in a single encounter with mC-containing DNA, without release of the hmC intermediate; but once released, hmC is a less favorable substrate than is mC.23,24 Altogether, only a fraction of hmC is further oxidized to fC/caC, making hmC a rather stable cytosine modification. Altogether, these results suggest that the efficiency and the final product of the oxidation steps performed by TET proteins depend on various conditions which are not fully understood yet.
3.2 Modulators of TET activity
Not surprisingly, the enzymatic activity of the TET proteins can be affected by the availability of the small molecules that are involved in its catalytic reaction: oxygen (O2), 2-oxoglutarate (2OG) and Fe(II) (Fig. 4). The activity of TET enzymes often, but not always,25,26 is reduced in hypoxic environment due to oxygen shortage,27 which is one of the key elements in cancer development and potential treatment targets (reviewed in28). Similarly, exogenous 2OG increases TET activity and the hmC/mC ratio in cultured mouse embryos,29 in different tissues of adult mice30 and significantly improves anti-cancer treatment through epigenetic modulation.31 Intracellular levels of 2OG are thought to direct the timing of ESC differentiation.32 On the other hand, certain metabolic components such as N-oxalylglycine (NOG)33–35 and 2-hydroxyglutarate (2HG)36,37 can act as potent inhibitors of 2OG-dependent oxygenases. Similarly, supplementation with Fe(II) in cell culture results in elevated levels of hmC.38 Iron-deficient diets during neurodevelopment result in decreased TET activity and reduced global hydroxymethylation in rat brain.39 Apart from the reaction substrates, it was found that vitamin C enhances the abundance of ox-mC in various cell cultures, from iPSCs and ESCs to cancer cell lines and also activates histone demethylation by Jhdm.40 Although it was originally prescribed a role in maintaining iron in the reduced form Fe(II), no other reducing agents were found to exert similar stimulation of the oxygenase activity. Biochemical studies showed direct interaction of this compound with the catalytic domain of TET2.41 Curiously, vitamin C in its hydrated form can itself serve as a substrate in an oxidative mC modification catalyzed by an algal TET homolog, CMD1 (Fig. 4).42 The latter finding, highlighting a structural similarity between 2OG and ascorbate hydrate (both contain the 2-oxocarboxylate moiety), may point at some other mechanisms of ascorbate-induced TET stimulation apart from keeping iron in the reduced state; for example, if TETs could directly utilize ascorbate for the generation of the active oxygen species in lieu of 2OG, the reaction would then yield 5-carbon L-lyxonic and/or L-xylonic acid (stereoisomers at C4), which were long ago identified as metabolites of vitamin C.43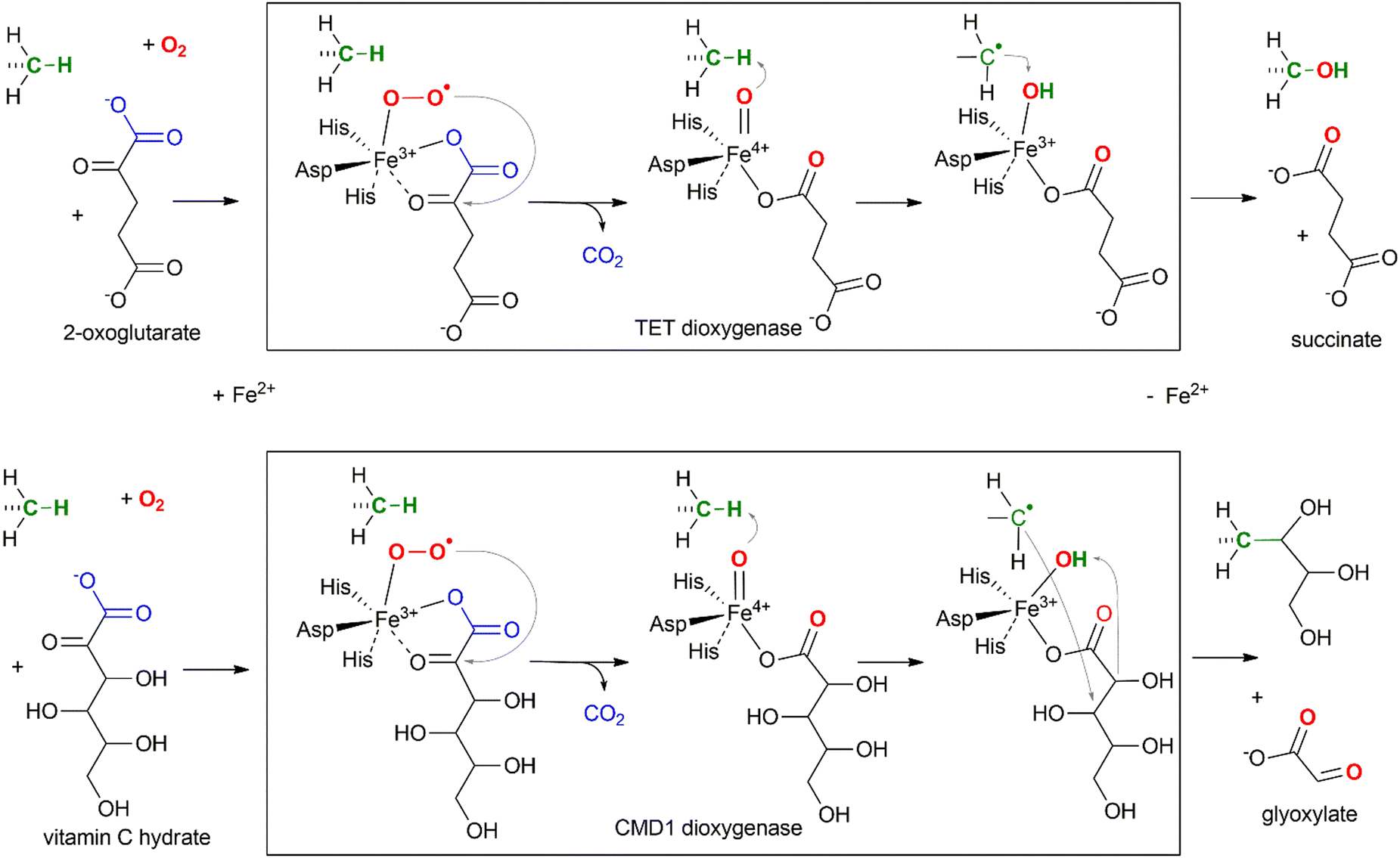 | ||
| Fig. 4 Mechanism of TET-directed hydroxylation (upper) and CMD1-directed glycerylation (lower) of 5-methyl groups of mC in DNA. | ||
3.3. Processing of ox-mC modifications
Further down the pathway, thymine-DNA glycosylase (TDG) whose primary function was thought to repair T-G mismatches produced in DNA upon sporadic deamination of mC, was found to directly excise fC and caC, while leaving mC and hmC untouched.13,44 The produced abasic sites in DNA are then repaired via the enzymatic cascade of the base excision repair (BER) whereby the nucleotides are replaced by newly incorporated unmodified nucleotides. Based on these observations, a pathway for active demethylation involving iterative mC oxidation by TET proteins coupled with TDG-mediated BER has been proposed (Fig. 5). A major caveat to this mechanism was that double-strand DNA breaks could be generated at densely methylated loci or if demethylation occurred on both strands of a CG/CG site. However, these concerns were alleviated by productive sequential demethylation of symmetrically methylated CGs and thereby avoiding DNA double-strand break formation in a reconstituted TET–TDG–BER system in vitro.45 It is now well established that both BER and nucleotide excision repair (NER) pathways play a central role in active DNA demethylation in the mouse zygote.46,47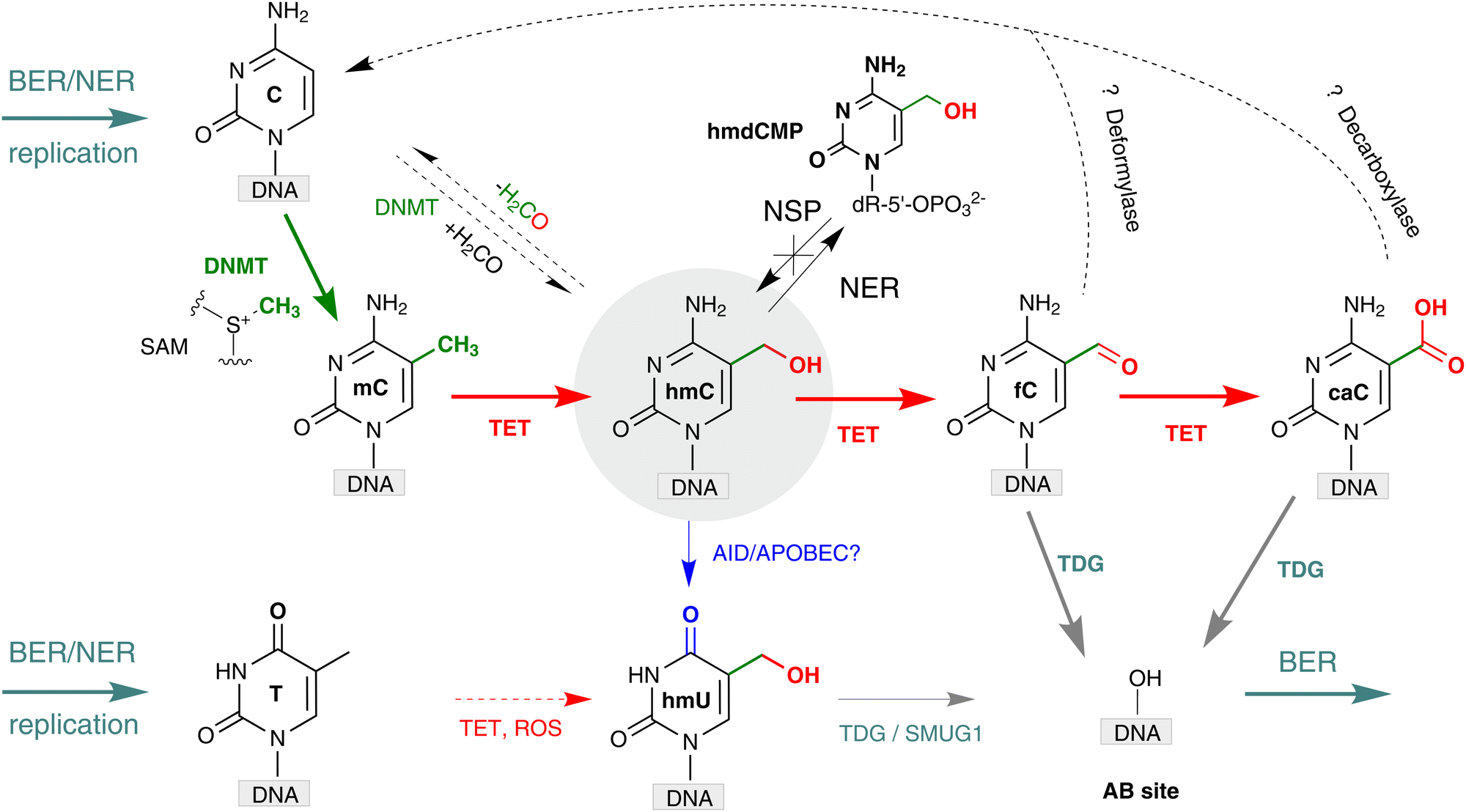 | ||
| Fig. 5 Formation and transformations of hmC in mammalian DNA. Cytosine (C) is converted to 5-methylcytosine (mC) by action of endogenous DNA MTases of the DNMT family (green pathway). Several mechanisms for DNA demethylation, in which 5-methylcytosine (mC) is converted back to C, have been proposed. Red arrows represent oxidation-based pathways performed by TET dioxygenases yielding 5-hydroxymethylcytosine (hmC), 5-formylcytosine (fC) and 5-carboxycytosine (caC), and may also produce minor amounts of 5-hydroxymethyluracil (hmU). Blue arrow shows deamination-based pathways where hmC is deaminated to hmU by AID/APOBEC or other deaminases. Grey arrows denote base excision repair (BER) pathways initiated by TDG, and SMUG1 glycosylases, and DNA replication. Black dashed arrows denote the hydroxymethylation and dehydroxymethylation reactions performed by cytosine-5 methyltransferases in vitro and putative deformylation and decarboxylation of fC and caC, respectively, yielding unmodified C. BER, base excision repair; NER, nucleotide excision repair; NSP, nucleotide salvage pathway. | ||
An alternative pathway for hmC processing was proposed based on the ability of some DNA glycosylases to excise hmU, which might occur via deamination of hmC (Fig. 5).48 Notably, some DNA glycosylases, single-stranded monofunctional uracil-DNA glycosylase 1 (SMUG1) and MBD4 have no significant activity for excision of caC or hmC,13 but they efficiently remove hmU.49,50 TDG was also shown to exhibit excision activity against hmU-G mispairs in dsDNA.49 This pathway relies on the assumption that AID/APOBEC deaminases (or some other unknown deaminase) can effectively deaminate hmC in duplex DNA in vivo, although in vitro studies indicated a strong preference of these enzymes for unmodified cytosines located in single stranded DNA. Some studies have suggested that hmU likely does not result from the deamination of hmC, as most hmU in DNA originates from the TET-catalyzed oxidation of thymine.51 Therefore, the deamination-mediated pathway still requires strong biochemical support.52,53
Yet another way, which lately gained substantial support, is based on direct conversion of hmC, fC or caC to C via a C–C bond cleavage, and thus altogether avoiding the troubles related to the generation of abasic sites. For example, it was shown that active demethylation in certain cases can occur in the absence of TDG. An early hint was provided by DNA C5-MTases, which are not only capable of coupling of formaldehyde to cytosine, but can also promote sequence-specific conversion of hmC15,54 or caC55 to unmodified C in vitro. The MTase-directed reaction proceeds via a covalent intermediate at C6 (Fig. 6a) resembling the thiol-mediated56 or bisulfite-mediated decarboxylation of caU and deformylation of fC (Fig. 6).57 These chemical precedents suggest that certain DNMTs or some dedicated enzymes may in principle perform the removal of the oxidized groups (5-hydroxymethyl, 5-formyl or 5-carboxyl) to give unmodified cytosine residues.14,53 To this end, deformylation and decarboxylation activity has recently been reported in mammalian cell extracts and in vivo.58–60 It seems that this scenario is operational in certain cases, however, no enzyme or other cellular component directly responsible for these reactions has yet been identified.
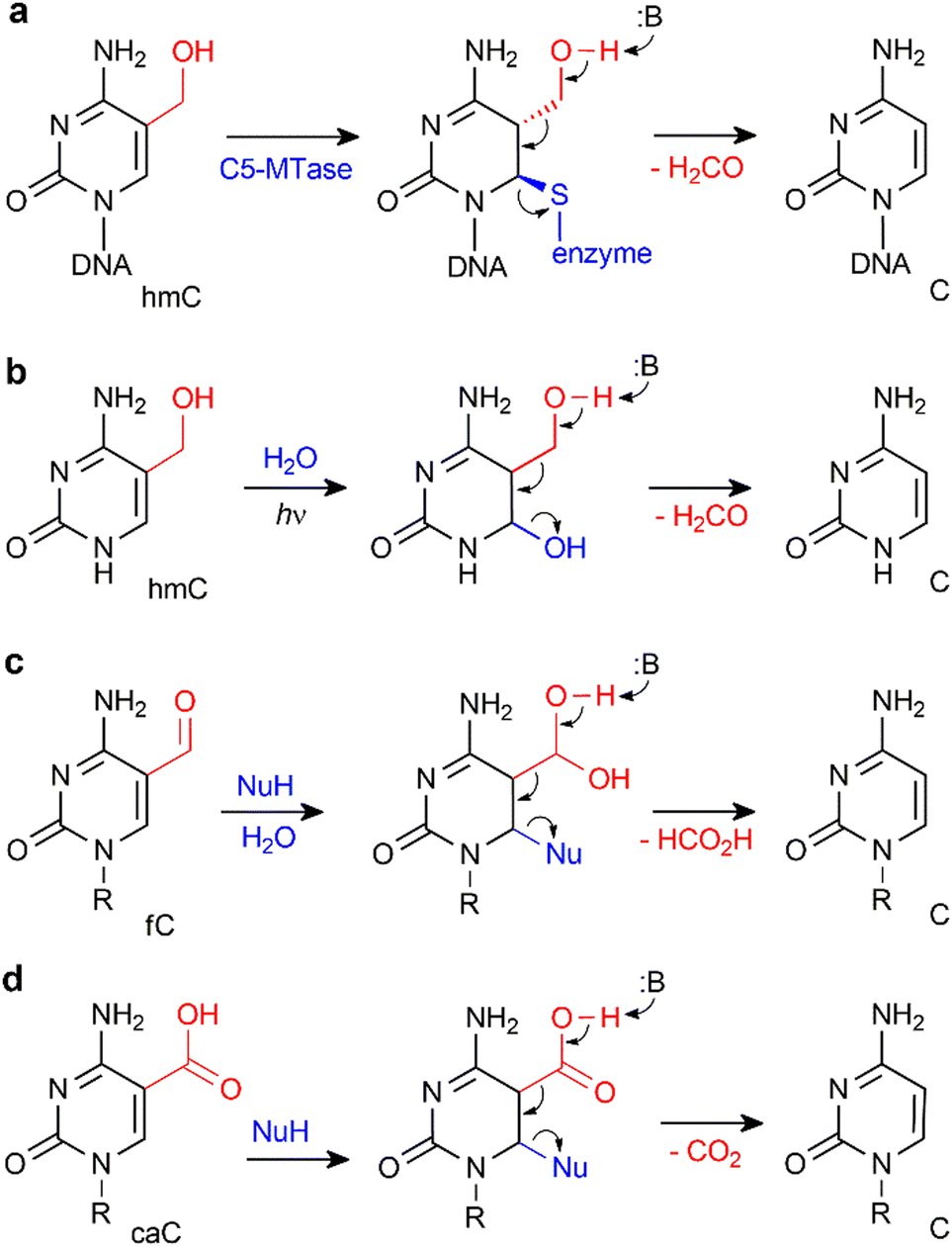 | ||
| Fig. 6 Removal of 5-hydroxymethyl or 5-carboxyl groups is facilitated by transient nucleophilic addition at C6 of the pyrimidine ring. (a) DNA cytosine-5 methyl-transferase directed conversion of hmC to C residues in DNA. (b) Light-induced dehydroxymethylation of cytosine.61 (c) and (d) Nucleophile-promoted deformylation of fC and decarboxylation of caC, respectively. | ||
4. Production and analysis of hmC in DNA in vitro
4.1. Production of hmC in DNA in vitro
Production of DNA substrates containing hmC residues is required for studies of chemo-enzymatic transformations of this modified nucleotide as well as for the development and validation of new analytical techniques. Chemical synthesis of hmC containing oligodeoxyribonucleotides was first proposed in 1997,62 which used phosphoramidite precursors with reversible 2-cyanoethyl O-derivatization of the 5-hydroxymethyl group along with the canonical N-benzoylation of the exocyclic amine. More recently, an alternative strategy based on N,O-carbamoyl bridging of both exocyclic groups afforded improved yields and purity of the target oligomers.63,64 Both types of phosphoramidites are commercially available and used by DNA synthesis services.Akin to the cellular reactions, hmC-modified DNA can in principle be produced from methylated DNA in vitro by oxidation of mC residues using TET oxygenases but the reaction should be firmly controlled to avoid further oxidation products. This has been demonstrated by directed engineering of the catalytic processivity of human TET2 oxygenase65 and further by developing a series of reactions permitting interconversion of mC and its oxi-forms for analytical applications (see below). The positions of produced hmC residues would then be determined by those of the original mCs. Direct sequence-specific installation of hmC in DNA is possible using an atypical chemo-enzymatic reaction of DNA-C5 methyltransferases,15,66 which catalyze a reversible coupling of formaldehyde to their target cytosine residues in vitro. The reaction is highly specific for the methyltransferase target sites, although the efficiency of hydroxymethylation may vary for each enzyme used. Random replacement of C with hmC in DNA in vitro67 or in vivo68 is achieved by supplying the 2′-deoxynucleoside-5′-triphosphate (hmdCTP) in DNA polymerase-dependent strand extension reactions including PCR.
4.2. Detection and quantification of hmC
Several techniques have been developed or adapted for detection and quantitation of hmC in DNA. Thin layer chromatography of radiolabeled nucleotides had been previously widely used for analysis of RNA and DNA modifications including hmC.10–12 Despite the high sensitivity, its use is more and more limited due to hazards associated with handling and disposing of radioactive materials in standard laboratories.The gold standard for analysis of global nucleoside composition of unlabeled DNA samples is liquid chromatography coupled with UV or MS detection. A higher selectivity and sensitivity can be achieved with modern MS/MS detectors, and reliable quantitation of the nucleosides is possible using synthetic stable-isotope labeled internal standards.69,70 These methods are well suited for quantitation of global hmC, fC and caC levels in genomic DNA, however they require specialized equipment. A high-throughput version has been developed using direct-injection MS, whereby DNA hydrolyzates are mass-analyzed without chromatographic fractionation of nucleosides.71
Due to the rarity of fC and caC and sometimes hmC, special care is required during sample processing72 and base-specific derivatization could be used to increase the sensitivity (by MS or optical detection) and enable enrichment of the target fragments from bulk genomic DNA.73,74 A particularly useful analytical modification of hmC is its enzymatic glucosylation to a much bulkier and distinctive residue, 5-glucosyloxy-methylcytosine, using T4 β-glucosyltransferase (BGT) and the uridine-5′-diphospho-D-glucose (UDP-glc) cofactor or its analogs (see below). The BGT reaction is robust and highly selective for the 5-hydroxymethyl group of hmC (or hmU). Such treatment can be used to attach tritium labeled glucose moieties from UDP-[3H]glucose to DNA permitting direct quantification of hmC by scintillation counting.75
New hmC detection methods that involve fluorescence strategies such as fluorescence resonance energy transfer,76,77 fluorescent tags introduced by enzymatic78–80 and chemical81 functionalization have been developed in recent years. These methods offer high sensitivity, specificity and are usually amplification-free. Their combination with different amplification techniques, rolling circle amplification,82 loop-mediated isothermal amplification,83 and isothermal exponential amplification84 accomplishes hmC detection even more sensitively and in a simple and time-saving manner. Furthermore, the unique MTase-catalyzed reaction to replace the hydroxyl- group of hmC by sulfhydryl group from cysteamine15 was combined with fluorescence85 or electrochemiluminescence86,87 and photoelectrochemical methods88,89 to create sensitive biosensors for detection of hmC in tissues.
5. Technologies to map hmC in genomic DNA
Numerous studies showed that although all mammalian tissues contain similar levels of mC (4–6% from C),2,90 hmC amounts are tissue dependent and vary from 0.03% to 1.2%.75,91 The most abundant hmC was detected in mouse Purkinje neurons, the brain parts responsible for cognitive functions (0.6–1.3% hmC/C) and embryonic stem cells (up to 0.5%), while much lower amounts are present in other tissues (0.03–0.3% hmC/C).10–12,92 hmC levels increase with age93 and are strongly depleted in tumor samples, suggesting the utility of hmC as a prognostic biomarker (see below). Other forms of ox-mC, fC and caC, are present at 100–1000-fold lower levels and vary from 0.00002 to 0.002% for fC, whereas caC amounts do not exceed 0.0003% from C.12,94,95The sparsity of ox-mC in genomes poses challenges in developing a universal method that would be sensitive and specific and most importantly, would inform on the cytosine modification status of each CpG. A myriad of methods has been developed over the years after the discovery of hmC to investigate its genomic profiles, which can be divided into enrichment-based methods and single nucleotide resolution approaches (Fig. 7). The methods considerably differ in genomic coverage, experimental and analysis strategies and cost.
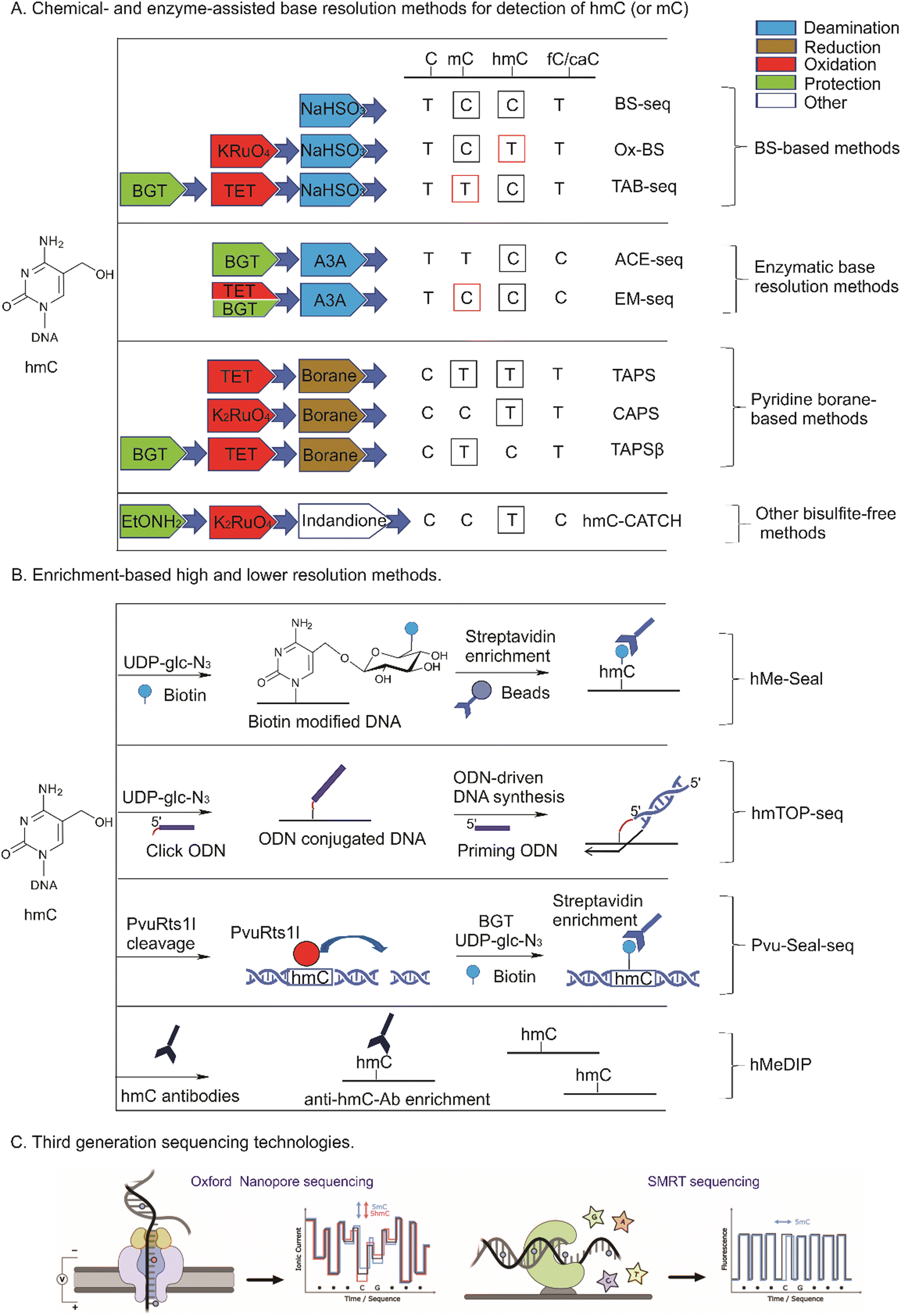 | ||
| Fig. 7 Analytical approaches for whole-genome mapping of hmC and associated DNA cytosine modifications. Bases read in one track are squared in black; bases read by subtraction of a related signal are squared in red. ODN, oligodeoxyribo-nucleotide. | ||
5.1. Affinity enrichment-based methods
Methods based on affinity enrichment rely on selective binding of short hmC-containing DNA fragments (usually 200–500 bp) to hmC-specific antibodies, other hmC-binding proteins or derivatization of hmC with reporter groups permitting their physical extraction from the rest of DNA for analysis using quantitative PCR, DNA microarrays or sequencing. The length of fragmented genomic DNA defines the resolution limit of all enrichment-based methods. Akin to methylated DNA immunoprecipitation (MeDIP), antibodies have been raised against hmC96,97 for immunoprecipitation of hmC DNA and its subsequent sequencing by the hMeDIP (hydroxymethylated-DNA immunoprecipitation) approach (Fig. 7b). However, the results from different studies show considerable variation even in the analysis of the same genomes, perhaps due to typical shortcoming of antibody-based pull-down approaches, such as possible cross-reactivity with methylated and unmodified cytosines, as these bases bear very few chemical differences for discrimination. Very recently the reliability of the current DIP protocols has been put into question, as differences were shown to arise due to the intrinsic affinity of IgG antibodies for short unmodified DNA repeats.98The development of covalent hmC modification significantly advanced the sensitivity and specificity of enrichment-based strategies. To discriminate between mC and hmC, many of such approaches involve a highly selective enzymatic glycosylation of hmC using BGT to a much bulkier and distinctive residue glc-hmC. The most widely used technique is hMe-Seal in which BGT catalyzes hmC derivatization by transferring an azide-modified glucose from the chemically modified cofactor analogue UDP-glc-azide.99 A biotin is subsequently attached through click chemistry and used for selective streptavidin–biotin pull down of hmC-containing DNA and sequencing (Fig. 7b). Using this technique, a myriad of studies to map hmC genome-wide in various cell lines, tissues and cfDNA have been performed.100,101 Its further advancement, Nano-hmC-Seal, demonstrated hmC mapping capabilities of ultra-low quantities of genomic DNA.102
An alternative chemo-enzymatic derivatization of hmC that requires no UDP-glc-azide cofactor has been proposed by Liutkevičiūtė et al.103 It was found that M.SssI (and a few other DNA C5-MTases) can catalyse sequence-specific replacement of 5-hydroxyl groups in hmCpG sites with alkylthio moieties carrying functional amino groups. The attached aliphatic amino group permits chemical ligation of NHS-biotin reagents for selective enrichment and analysis of hmC containing DNA.
5.2. Tethered oligonucleotide-primed sequencing
To push the resolution limits from 200–500 bp offered by the enrichment-based methods to a single nucleotide, an approach called TOP-seq (Tethered Oligonucleotide-Primed sequencing) has been proposed (Fig. 7b). The salient feature of the approach is that a DNA oligonucleotide is covalently tethered instead of a biotin moiety to enable non-homologous priming of the DNA polymerase strand synthesis right at the covalently tagged CpG genomic site.104 In its further extension, hmTOP-seq, a DNA oligonucleotide is tethered through copper(I)-promoted azide–alkyne click chemistry on BGT azide-derivatized glycosylated hmC residues, which then serves as a primer for generating amplicons in which the starting sequence marks a precise position of the original hmC in the genome.66 Pilot hmTOP-seq studies demonstrated wide applicability of the method for high-resolution and cost-efficient profiling of hmC in mammalian genomic samples and cfDNA.26,66,105 Independently, a similar approach has been proposed that exploited copper-free click chemistry to attach a DNA hairpin to hmC for sequence readout around (20 bp window) the modified cytosines;106 in this case, a much bulkier tethering moiety is produced which seems to negatively affect the precision and efficiency of the polymerase priming as compared to the copper(I)-promoted tagging.665.3. Chemical conversion-based base resolution methods
For many years the gold standard method for mC profiling genome-wide at single C resolution was bisulfite sequencing (BS-seq). The differential reactivity of mC and C in the presence of bisulfite, forms the basis for the method: mC is very stable to bisulfite-promoted deamination and is subsequently read as normal C whereas unmodified C is read as T. With the discovery of ox-mCs, the informativity of standard bisulfite treatment appeared insufficient as the bisulfite attack at the 5-hydroxymethyl group yields a hydrolytically stable 5-sulfonylmethylcytosine (smC, also called cytosine 5-methylenesulfonate) and cannot be discriminated from mC (Fig. 7a and 8). Moreover, the products of further hmC oxidation, caC and fC, are interpreted as unmodified C (reads in the T lane). The indistinguishable behavior of mC and hmC or caC and fC in BS is most likely the reason why ox-mC were long missed in DNA modification analysis. Therefore, as the standard bisulfite chemistry provides only an aggregated mC + hmC signal versus unmodified cytosine (plus very rare fC and caC), this evoked the demand for specific pre-treatment approaches to detect hmC. Two most widely used methods are based on enzymatic or chemical oxidation of hmC prior to standard BS (Fig. 7a and 8). The TET-assisted bisulfite sequencing, TAB-seq,107 is a chemoenzymatic approach that exploits the protection of hmC by glycosylation and subsequent oxidation of mC to caC by the mouse TET1 enzyme. Following bisulfite sequencing, hmC is read as C, while all other cytosine forms are interpreted as unmodified C (reads in the T lane). In another method called the oxidative bisulfite sequencing, OxBS,108 hmC is oxidized by potassium perruthenate (KRuO4) into fC and then is read as T, whereas mC remains as C. The absolute amount and genomic positions of hmC in OxBS are determined by the subtraction of the hmC track from the standard BS signal and thus, necessitates parallel processing of two samples. The subtraction-based analysis strategy to discriminate between hmC and mC is also required in TAB-seq (the TAB-seq signal is subtracted from BS) which considerably increases the analysis cost, as sequencing depth for each cytosine must be considerably high to detect low-level hmCs in both approaches.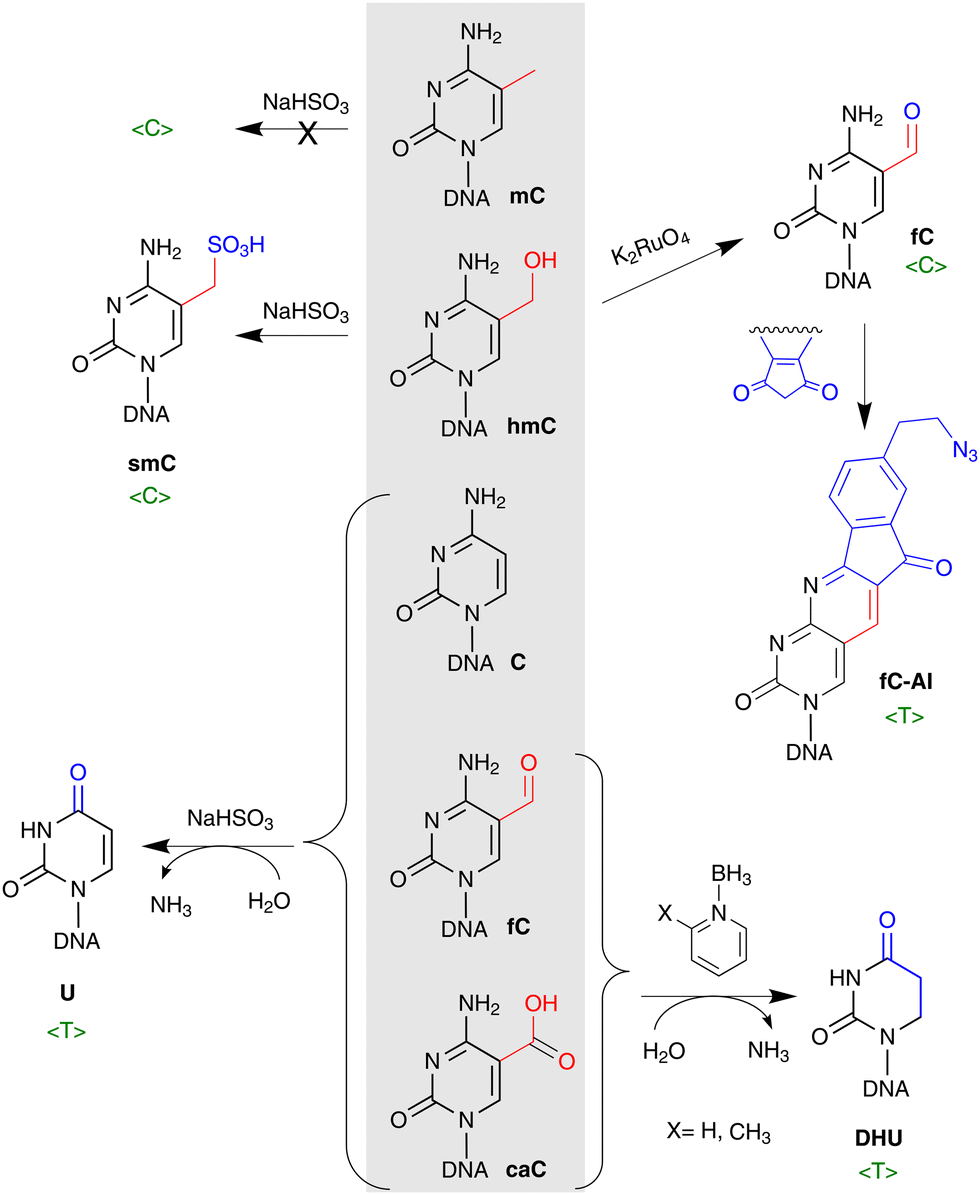 | ||
| Fig. 8 Chemical conversions of C5-modified cytosines for their analysis by DNA sequencing. Upon treatment of DNA with bisulfite (leftward reactions), hmC forms a hydrolytically stable 5-sulfonylmethylcytosine (smC) and cannot be discriminated from unreactive mC (both read in lane C, 〈C〉), whereas caC and fC are deaminated and interpreted as unmodified C (read as T). hmC can be oxidized by potassium perruthenate (K2RuO4) into fC and sequenced directly (read as C) or further derivatized using an azido derivative of 1,3-indandione (fC-AI) for biotin pull down sequencing (read as T). In pyridine-borane treatment-based methods, mC and hmC are first converted to caC and then caC is reduced to dihydrouracil (DHU) and is read as T, whereas C remains unaffected during the pyridine-borane workout (bottom right). | ||
On the downside, bisulfite treatment leads to extensive degradation of input DNA109 while deamination of all unmodified cytosines causes analytical challenges due to reduced sequence complexity. This sparked the appearance of alternative bisulfite-free single base analysis methods (see Fig. 7a and 8). Such novel chemical strategy is used in a chemoenzymatic bisulfite-free method, termed TET-assisted pyridine borane sequencing, TAPS.110 The method involves TET proteins to convert mC and hmC into caC and then, caC is reduced to dihydrouracil by pyridine borane (PB) and is sequenced as T. The important advancements of pyridine-borane chemistry is the direct readout of modified bases preserving unmodified C intact and less destructive nature that allows improved sequence quality, mapping rate, and coverage compared to BS.110 Although the first protocol of TAPS provided information on the aggregated mC + hmC signal, the recently introduced optimizations to replace the TET-mediated oxidation by the treatment with potassium ruthenate (K2RuO4) and the protection of hmC with glycosylation prior to PB (or picolyl borane (pic-borane)) conversion enabled an independent mapping of hmC and mC in the CAPS and TAPSβ approaches, respectively.111 Another novel bisulfite-free chemical strategy to detect hmC, termed hmC-CATCH, is based on the selective oxidation of hmC to fC with K2RuO4, and subsequent labeling of newly generated fC using derivatives of 1,3-indandione that results in the transformation of hmC into T in sequencing, while endogenous fC is blocked before the oxidation reaction (Fig. 7a).112 hmC-CATCH causes minimal DNA degradation, and offers single base hmC analysis with limited amounts of input DNA as shown in application of this method to analysis of cfDNA.
5.4. Enzyme-assisted base resolution methods
Milder treatment of DNA compared to BS provides advantages in the ACE-seq (APOBEC-coupled epigenetic sequencing) and EM-seq (Enzymatic Methyl-Seq) methods which both employ hydrolytic deamination of C into U using the DNA deaminases from the AID/APOBEC family (Fig. 7a).113,114 The ACE-seq approach provides direct sequencing of hmC, whereas EM-seq detects the common mC + hmC signal and demands for the ACE-seq acquired signal subtraction to localize mC. Although both APOBEC deamination-based methods contain a non-destructive feature, they still may suffer from the low complexity of base contents after C deamination, and thus, analysis of low DNA amounts may be complicated. A similar set of enzymatic nucleotide conversions is used in a recently proposed sequencing platform that can derive the four genetic bases and two epigenetic states of cytosine (mC and hmC) avoiding lane subtraction. This is achieved by linking and intricate processing of both strands of each genomic fragment in one experimental workflow.115Restriction endonuclease cleavage has been long employed in DNA modification analysis as a simple to perform and cost-efficient strategy. Owing to the strict sequence specificity of restriction endonucleases, such methods offer base-resolution DNA modification profiling. However, since their target sites are longer than two nucleotides (typically 4–6 bp), the approach can query only a small subset of all genomic CpG sites. Several methods for hmC analysis have been developed that combine restriction enzyme digestion with enzymatic glucosylation of hmC. The first of them adapted for genome-wide analysis of hmC employs the differential sensitivity of the HpaII and MspI restriction endonucleases to cytosine modification within their target sequence CCGG. BGT-directed glycosylation makes ChmCGG sites resistant to MspI cleavage, which can be enriched and analyzed using qPCR,116 microarrays or next-generation sequencing.117,118
More recent example strategies include Aba-seq approach119 and Pvu-Seal-seq.120 In Aba-seq, the AbaSI restriction endonuclease cleaves at a narrow range of distances away from the recognized glycosylated hmC and then, the cleaved fragments are pulled down and sequenced. The PvuRts1l restriction endonuclease recognizes hydroxymethylated target sites and cleaves 11–12 bp downstream in Pvu-Seal-seq (see Fig. 7b). The PvuRts1l-cleavage is further combined with the enrichment of hydroxymethylated target sites by covalent labeling of hmC with BGT and UDP-azido-glucose for specific separation of modified DNA as described for the hmC-selective chemical labeling approach hMe-Seal (see above). Although all restriction endonuclease-based methods cannot determine the absolute amounts of hmC genome-wide, their inherent feature is sensitivity to detect lowly hydroxymethylated target sites without requirement for deep sequencing necessary in BS-based approaches.
5.5. Single-molecule long read biophysical detection
Several approaches permit single-molecule profiling of epigenetic modifications in native stretches of DNA. One such method is optical mapping of hmC in native chromosomal DNA. This method uses a two-step labeling of hmC with a fluorophore, followed by stretching the DNA strands in nano-channels for fluorescence image analysis. Physical positioning of the fluorophores along the linear DNA strand generates epigenetic profiles of long DNA molecules at the sub-megabase scale,102 which can be interpreted alone or along with a genetic (sequence-specific) fluorescence profile. The mapping resolution of 1 kb was determined by the detecting microscope technology and physical fluctuations of the DNA strands in the nano channel, which can reach 10 nm or just 20 bp as demonstrated in pilot experiments using DNA stretching on solid surfaces and dSTORM imaging.121Third-generation sequencing technologies generally avoid DNA derivatization and DNA amplification whereby native stretches of DNA are sequenced directly. In nanopore sequencing, commercially offered by Oxford Nanopore Technologies (ONT), sequencing is achieved by reading ionic current profiles as a nucleic acid strand passes through a nanopore. Single-molecule real-time (SMRT) sequencing, offered by Pacific Biosciences, tracks the incorporation of fluorescently labeled dNTPs by a DNA polymerase immobilized in a zero-mode waveguide (Fig. 7C). In this case base modifications present on a template strand alter the kinetics of nucleotide incorporation (dNTP arrival and residence times) permitting their identification in multiple sequencing rounds. In both cases, DNA modifications result in subtle and context-dependent deviations of the signal measured. Therefore, it comes to extensive signal processing and machine learning using in vitro prepared training DNA substrates, to decipher the presence and identity of the modified bases.122 Although a proof of principle for the discrimination among the five cytosine variants in certain CpG contexts by their nanopore signal signatures has been demonstrated,123 selective signal enhancement for improved SMRT detection of mC and hmC can be achieved by their enzymatic conversion to caC124 and glc-hmC,125 respectively. Both ONT and PacBio have integrated their tools into their default sequencing software. For native DNA, currently detection is restricted to CpG sites and is only available for mC and hmC (ONT Remora) or mC (PacBio). Further dynamic developments in third-generation sequencing seem about to deliver the benefits of single molecule sensitivity, reliable discrimination of native cytosine modifications, long reads and high throughput automation.
6. Structure and interactions of hmC-DNA
6.1. DNA structure
hmC induces no significant structural alterations of the B- or A-helical DNA as observed by X-crystallography or NMR (Fig. 9).126–130 There is a general tendency for reduced twist and increased roll angles when C is substituted by mC or hmC, probably due to a steric hindrance of the cytosine 5-substituent.129 hmC results in a 0.8 Å (4.5%) widening of the major groove at the site of modification,127 similarly to mC or other C5 modifications,131 reviewed in.132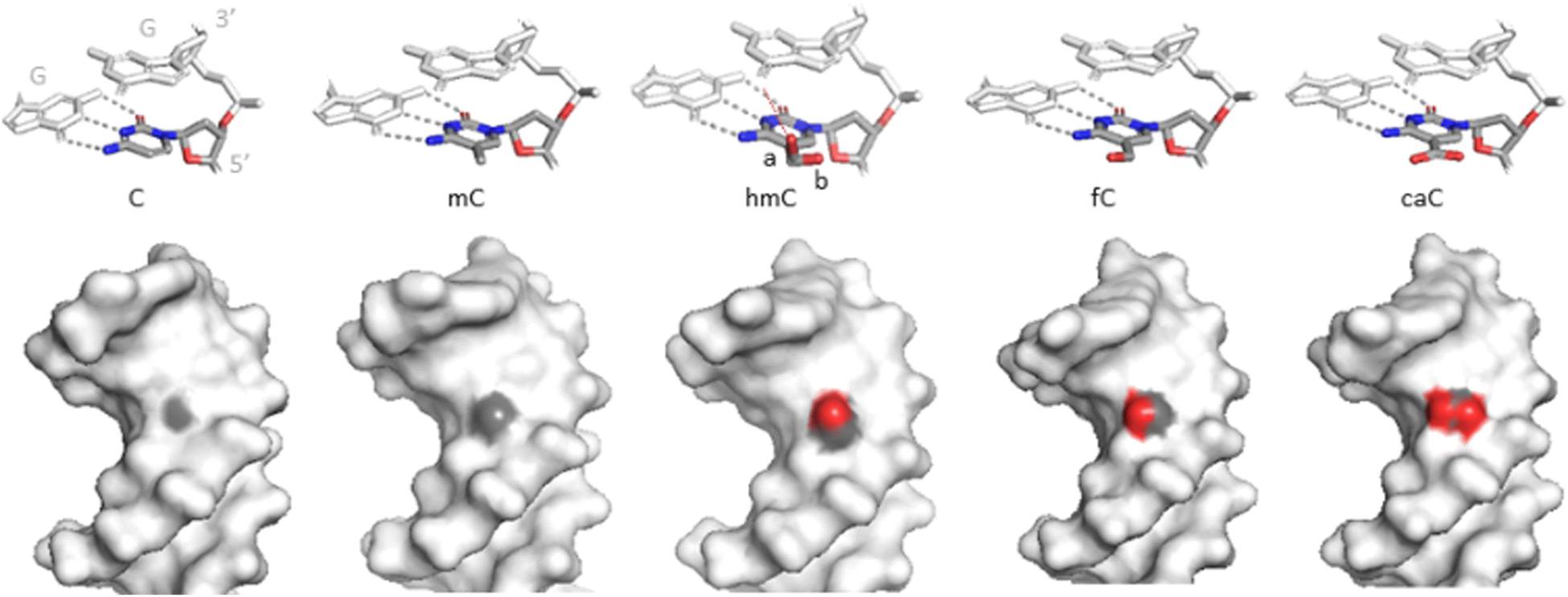 | ||
| Fig. 9 Structures of the C5-modified cytosines in DNA. Upper, stick representations of C and the 5-modified cytosines together with base-paired (grey dashes) and 3′-neighbouring (CpG) guanine residues. hmC is found in two major conformations: a (70–80% occupancy) and b (20–30%). Bottom, spacefill representations of the cytosine variants in the major groove of DNA. Light grey – Dickerson-Drew dodecamer. Grey – carbons, red – oxygens at position C5 of the (modified) cytosine no. 9 (PDB ID 3u2n, 4glg, 4i9v, 4pwm, 4qc7). | ||
The hydroxymethyl moiety extends into the major groove (Fig. 9), and the main orientation of the hydroxyl group (70–80% occupation) points towards the 3′ neighboring base. An alternative conformation (20–30%) has the hydroxyl interacting with a backbone phosphodiester oxygen via a bridging water molecule.127,128,130 NMR experiments showed no evidence for increased imino tautomerization of hmC consistent with Watson–Crick base pairing. The dynamic opening rates of the modified base pair is similar to that of C or mC and do not correlate with the excision preferences of the oxidized mC forms by TDG.44,130
Notably, further oxidation of the hydroxymethyl group to a formyl or carboxyl group substantially alters the electronic properties of the nucleobase (polarity, charge distribution, stability of the N-glycosidic bond),44,133 which has been shown to affect the global structural properties of DNA to different degrees depending on experimental contexts.127,134–136 As for hmC, there is no unanimous agreement on whether it makes DNA duplex more flexible or more stiff, more stable or more melting-prone as compared to unmodified and methylated DNA. UV melting temperature, circularisation with ligation or FRET, nucleosome assembly/disassembly, molecular force assays, molecular dynamics simulations, etc. – the in vitro approaches give conflicting results among independent studies or even in the same study among different DNA sequences or the same sequence with different hmC modification densities. In general, hmC is considered to reverse the impacts of methylation on DNA physical properties. Numerous studies have shown that while mC increases the duplex melting temperature, hmC brings it back closer to the one observed with C.68,126,127,129
A single-molecule mechanical force study showed that the mechanical stability of DNA (resistance to zipper-mode strand separation) increases with the number of hmC bases.137 Different approaches have been taken to evaluate the effects of epigenetic DNA modifications on the DNA flexibility with anticipation of finding a direct mechanistic connection to nucleosome binding and gene expression regulation. However, DNA circularisation with either ligation or FRET, nucleosome assembly or disassembly, and different molecular dynamics simulations have led to contrasting conclusions.129,136 Finally, there can be differences between effects of solitary hmC modifications and their cooperative impact in larger clusters.129,131
Among non-canonical DNA structures, G-quadruplexes (G4) have emerged as a significant player in regulation of gene expression, replication and other genome metabolic processes. G4s are shown to form not only in telomeric regions but also throughout the genome in promoters, replication start sites,138 therefore partly overlapping with methylation sites.139 While some studies find no marked changes in the formation or stability of G4 structures after incorporation of hmC,140 others report more complex results,138 and infer their significance from the perspective of structural impact on promoter G4s formation in senescent cells.
6.2. Protein–DNA interactions
Clearly, the thermodynamic and structural effects of the natural cytosine modifications on DNA are quite weak and diverse to account for their biological effects on physical grounds only. Rather the dominant belief is that the regulatory roles of the cytosine modifications are largely exerted at the level of protein-DNA interactions.141 To this end, compared to a methyl group, a hydroxymethyl group is slightly larger, more polar and can serve as a donor or acceptor of a H-bond. However, for the most part dedicated hmC “readers” have remained elusive as a large fraction of hmC binders would also bind mC.86,142,143 For example, methyl-CpG-binding protein 2 (MeCP2) can bind mC and hmC,144,145 although the methyl-CpG binding domain protein 1 (MBD1) can specifically bind to mC but not to ox-mCs.146 Altogether, hmC appears incapable of interacting with a number of proteins that recognize mC, suggesting that hmC is an anti-binding modification that serves to exclude mC readers.147 Nonetheless, there are proteins that bind mC and hmC using distinct interaction modes. A particular thermodynamic signature of binding hmC-DNA, compared to methylated or unmethylated DNA, has been shown for MeCP2; mutations affecting the hmCpG – MeCP2 interactions are associated with the phenotype of the Rett syndrome.148 UHRF2 has been shown to use a base-flipping mechanism, first discovered in a DNA MTase,149,150 for exclusive recognition and enhanced binding of hmC-DNA.149Altering the target specificity or de novo engineering artificial hmC binders for biotechnological applications have been demonstrated for TALEs151 and MeCp2.152 Notably, plant glycosylases DME and ROS1, which serve to remove mC in plants, have also been shown to excise hmC in vitro. As no hmC was detected in Arabidopsis thaliana, this activity may simply reflect a promiscuous substrate specificity of this family of proteins, which could be applied for epigenetic editing.22
Another important class of proteins are the mammalian CpG-specific DNMTs. In particular, DNMT1 which is largely involved in replication of the methylation patterns on the daughter strand, shows a strong preference (80-fold on average) for hemimethylated CpG sites as compared to unmethylated sites. Several studies using different model DNA substrates and variants of the protein pointed towards the notion that hemihydroxymethylated CpG sites (hmCG/CG) are poor substrates (similar to unmodified sites) and thus hmC marks would be passively erased in the progeny.50,153 However, in more recent studies, two groups reported that the DNMT1 methylation preferences in vitro gradually decline in the row mC > hmC > fC≈C,154 and the mC/hmC preference is on average only 13-fold while the hmC/C maintenance preference is still ∼7-fold.155 These findings suggest that hemi-hydroxymethylated sites could be partially maintained (appear as attenuated hemimethylated sites) and thus passive DNA demethylation by hmC generation may not not be as efficient as previously thought. As the DNMT1 activity is strongly regulated by many external factors and features on target DNA, the role of hmC in the loss of methylation patterns during DNMT1-directed methylation maintenance in vivo requires further attention.
As discussed above, in line with its distinctive chemical properties (chemically active aldehyde group), fC is a target for numerous specific binders.142 To this end, fC has been shown to form a reversible covalent bond (Schiff base) to lysine and arginine residues of bound histones in vitro and in vivo, thereby potentially acting as a modulator of nucleosome organization and gene expression.156–158
7. Biological roles of hmC in mammals
The discovered active and passive mC removal provided evidence that DNA methylation is a bi-directional and dynamically regulated process; methylation and demethylation events occur at different times and genomic regions under different developmental programs, and environmental cues. After more than a decade of hmC studies, its biological implications are beginning to emerge and are still under extensive debate. Although hmC is generated from its precursor mC in the active TET-mediated demethylation pathway, hmC appears to persist in many cell types,159 arguing against hmC as being solely a demethylation intermediate.7.1. hmC role in replication, repair and recombination
An intimate interaction of hmC and DNA repair stems from the involvement of BER/NER pathways in the TET-induced DNA demethylation (Fig. 5). TET activity leading to enhanced levels of hmC also generates more fC and caC, which in turn activate BER. This idea is consistent with a decreased mutational frequencies in hmC rich regions, in contrast to mC, which marks hotspots of genomic CpG depletion due to mC → T mutations.160 But, notably, hmC, as opposed to related nucleobases hmU, U, fC and caC, is not a substrate for the base excision machinery, and thus can accumulate to substantial levels in genomic DNA.Genomic profiling studies detected elevated hmC levels at the sites of DNA damage161 and at the stalled replication forks.162 The presence of hmC was also found at DNA double strand breaks during meiotic recombination of gametes.163,164 It was suggested that the conversion of mC to hmC may prevent binding of factors with affinity for mC.163 However, experiments in which the TET activity is altered to vary the levels of hmC, do not make it clear if the generated hmC is directly involved in recruiting the DNA repair components to the problematic loci, or is just a passive by-stander produced along with the actively excised fC and caC.
Replication origins in mammals, which are not clearly marked by DNA methylation, specific histone modification or consensus sequences, have been shown to be enriched for hmC.165 hmC appears to direct the assembly of the pre-replication complex in G1 and M phases, however, prior or during origin firing, hmC has to be removed and then again installed in newly replicated DNA. Therefore, elevated hmC levels delay the progression through the G1 phase, reducing cell division. These observations point at direct involvement of hmC in cell cycle regulation and may explain why the non-dividing neuron populations show the highest hmC content, whereas rapidly dividing neural progenitors are almost devoid of hmC.165
On the other hand, it was proposed that, vice versa, the genomic hmC content may in fact depend on the duration of the cell cycle.159 Metabolic isotope labeling of cytosine derivatives in DNA of mammalian cells and tissues, showed that, in contrast to DNA methylation, which occurs during or immediately after replication, hmC forms slowly over a 30 h period following DNA synthesis. This delayed hmC appearance thus can explain why rapidly diving cells would contain less hmC, in line with the observed inverse correlation between the global levels of hmC and the rate of cell proliferation.
7.2. hmC involvement in transcription
Rausch et al. have measured mC and hmC effects on the transcription and replication of the whole genome in live bacterial, yeast and mammalian cells. They conclude that while mC stabilizes the DNA helix and reduces the DNA helicase and RNA/DNA polymerase speed, hmC reverts the duplex stabilizing and genome metabolic effects to the level of unmodified cytosine.68 In biochemical assays, the presence of hmC at the ICV promoter strongly inhibited transcription, while its presence within gene bodies had almost no effect on transcription.166Different hmC profiling studies have found its preferential distribution at gene bodies of active genes, 3’UTRs, active or poised enhancers, promoters of development-associated genes, and around transcription factor binding sites.99,107,167,168 Involvement of hmC in epigenetic regulation is already indicated by the differential accumulation of hmC and fC/caC across the genome. For example, in mouse ESC, >90% hmCG positions do not correspond to those of fC and caC, and only ∼19% fC/caC sites overlap with hmC.169 Moreover, fC enrichment is stronger in active enhancers which usually show lower DNA methylation as compared to hmC-containing loci.170 Therefore, hmC may mark less active chromatin loci than caC and fC (the suggested order is caC > fC > hmC).171 The positioning of hmC at exon-intron boundaries and its abundance in constitutive exons compared to alternatively spliced exons raised an idea of its role in alternative splicing.117 It was demonstrated that the major genome insulator and multifunctional regulator methyl-sensitive protein CTCF binds to intragenic hmC in alternative exons and is associated with alternative exon inclusion.172 Interestingly, hmC levels were found to be higher on the sense strand.173,174 Indeed, the association of gene body hmC with gene expression is widely attested, although its influence is bi-directional: for example, it promotes gene expression in ESC and many other cell types, but negatively influences transcription in neuronal progenitors.173,175,176 In contrast, highly expressed genes usually show hmC depletion at promoters in many cell types.173
The role of hmC and other ox-mC as independent epigenetic regulators is indirectly supported by the existence of partially different sets of their binders, which represent mainly transcriptional regulators.86,142,143 For example, MeCP2 can bind mC and hmC, which alters chromatin structure and facilitates gene expression in neural cells.144,145 In contrast, MBD1 can specifically bind to mC but not to ox-mCs.146 mC-bound MBD1 can recruit the histone methyltransferase SETDB1 and thus, promotes H3K9 methylation and gene repression.146 In addition, hmC distribution at gene bodies modulates the deposition of H3K36me3 marks which indicate active transcription177 thereby collectively altering the configuration of chromatin for turning on or off gene activity in heterochromatic and/or euchromatic regions.176
7.3. hmC in cell differentiation and reprogramming
Some degree of controversy exists in defining the role of TET proteins and hmC in stem cell pluripotency. In mouse ESC, the depletion of highly expressed TET1 or TET2, or both did not affect pluripotency or development though it decreases hmC levels and induces transcriptional changes.178–181 However, the TET1/TET2 deficiency may delay transcriptional changes during differentiation, or induce the commitment of ESCs towards specific lineages.97,180 Moreover, the triple-mutant (TET1, TET2, and TET3) mESCs were found to be viable and pluripotent though showed depletion of hmC, hypermethylation of promoters and impaired differentiation potential,182 supporting the essential role of the active DNA demethylation in differentiation.It seems that a major role of TET-mediated oxidation in ESC is the maintenance of the demethylated state of the regulatory regions, particularly enhancers and promoters,183 which are the primary targets in reprogramming. In ESC, the high-level hmC marks silent bivalent promoters, which contain both repressive and activating histone marks H3K27me3 and H3K4me3, respectively, and typically are activated on differentiation.53 It seems that TET and DNMT proteins compete with each other to regulate the methylation status of enhancers and promoters;184,185 TET1 can exclude DNMT3A1 from promoters where TET1 preferentially binds in ESC.186 Moreover, in tight cooperation with transcription factors, the TET proteins mediate the stepwise oxidation of mC and regulate gene expression of developmental genes.187 For example, TET1 and TET2 interact with the pluripotency factor Nanog to enhance reprogramming efficiency,188 or with various heterochromatin- associated proteins, such as HDAC histone deacetylases, to remodel chromatin and regulate transcription.97,189
Other studies demonstrate that TET proteins and ox-mCs are important for telomere length maintenance, which is important in sustaining pluripotency, self-renewal and genomic stability. For example, the double TET1/TET2 mutant results in elevated amounts of DNMT3b and the mC/hmC ratio, which leads to telomere shortening.183,190
Interestingly, mutational engineering of the TET2 activity toward the preferential production of hmC permitted to dissect the biological significance of hmC versus the other ox-mCs during reprogramming of induced pluripotent cells (iPSC).191 The increased generation of hmC itself appeared insufficient to drive the epigenetic changes, and the formation of fC/caC was necessary to promote rapid changes at reprogramming loci. In addition, during differentiation of pluripotent stem cell into neurons, glial cells and hepatocytes, the increased caC amounts were detected in promoters of cell-specific genes, thus, it is thought that caC-pathway might exists as a general epigenetic rewiring mechanism for establishing cell lineages.192,193
In mouse zygotic reprogramming, most of hmC is generated from de novo mC, indicating a specific hmC role in the early embryo development.194 Generally, the epigenetic role of hmC becomes apparent in the context of cell differentiation. During erythropoiesis, a decrease in global hmC level was observed, but hmC at certain genomic loci remains highly enriched despite multiple rounds of DNA replication.195 In asymmetrically dividing stem cells, higher hmC has been shown to identify immortal DNA strand chromosomes.196 In neuronal cells, hmC is depleted from TSS, regardless of gene expression level or CpG content.145,174 It was suggested that in early development, brain-specific enhancers initially become hydroxymethylated, but later, both hydroxymethylation and demethylation stages at enhancers are differentially regulated in a cell-type specific manner. Overall, these data point to the requirement of properly functioning TET-mediated DNA demethylation for differentiation, reprogramming and cell fate decisions.
7.4. hmC as a diagnostic and prognostic marker
The balance between proper DNA methylation and demethylation maintains the DNA methylation landscape of healthy cells, which becomes heavily disturbed in cancers. It has been well documented that global levels of hmC are significantly reduced in many cancer types proposing the potential clinical relevance of hmC. One might think that in such intensively dividing cells as cancer cells the decreased hmC might be related to the hmC removal through passive DNA demethylation. However, in the healthy developing cells, hmC at certain genomic loci remains highly enriched despite multiple rounds of DNA replication and decreased global hmC levels.195 In many cancers genetic alterations of TETs are absent and thus the basis for global loss of hmC is not always clear. Some tumors display increased activity of isocitrate dehydrogenase (due to gain-of-function mutations in IDH1 or IDH2), which leads to production of 2HG, an oncometabolite and inhibitor of the TET function.36,37 In addition, the decreased glucose/glutamine levels in nutrient-deprived conditions of a tumor may impact the intracellular 2OG/succinate ratio which downregulates TET and evoke hmC loss even in the absence of the genetic TET and IDH mutations.197 Other factors, such as the shortage of Fe(II), ascorbate, or oxygen also may attenuate the activity of TETs (see above). The supplementation of cancer cells with ascorbic acid may restore hmC levels and inhibit cancer growth and migration.198,199 Generally, the mechanisms for hmC loss in cancers are likely more global than just defined by abberant functioning of TET or IDH enzymes. Interestingly, it was shown that simple re-installation of hmC and TET enzymes may reduce the tumor growth and aggressiveness.200 The correlation between decreased hmC and tumor aggressiveness has been suggested by multiple studies10,201 raising the idea that hmC level could be used to predict the metastasis, recurrence, and prognosis. There were also attempts to predict cancer types or their stages from hmC amounts or tissue-specific distribution of hmC in cfDNA.101,202–204 Besides widely attested links between hmC and cancer, the clinical relevance of tissue-specific hmC monitoring was demonstrated for other complex diseases, for example in non-invasive diagnostics of fetal Down syndrome, Alzheimer disease, and Type 2 diabetes.105,205,206In addition to its value as a disease marker, hmC may find some therapeutic value. As mentioned above, cells deploy a two-tier safeguarding system to avoid sporadic incorporation of the modified nucleosides in DNA via the nucleotide salvage pathway (Fig. 3). However, this system is more promiscuous in fast proliferating cancer cells, which confers them more susceptible to treatment with the modified cytidines and thus opens new ways for potential therapeutic options in controling cancer.17,18
8. Conclusions
From biological standpoint, the generated hmC presents an epigenetic state of cytosine in which the proper 5-methyl mark is no longer present, but an exocyclic group on the C5-position is still present precluding its remethylation. How is this chemically stable epigenetic state (functionally demethylated) of cytosine resolved? Notably, hmC is not recognized by some of the methyl-CpG binding domain proteins, such as transcriptional repressors MBD1 and MBD250,207 suggesting that conversion of mC to hmC may attenuate the gene silencing effect of mC. In the presence of maintenance MTases such as Dnmt1, hmC patterns would be partially replicated into mC on the daughter strand (passively reduced). It is reasonable to assume that, although being produced from a primary epigenetic mark, mC, hmC may play its unique regulatory role as a secondary epigenetic mark; in many cases though it may behave as an attenuated (partially muted, demoted) methyl group in DNA.Several other factors to consider are as follows. The distribution of hmC is not uniform across the genome and is different from that of the other modified cytosines (see above). The abundance of hmC is lower than that of mC, but in certain cell types and genetic loci, is quite substantial to persist as a causative epigenetic mark. A tight safeguarding system precludes sporadic incorporation of hmC nucleosides in mammalian DNA via NSP (Fig. 3). All this indicates that the occurrence or absence of hmC itself (rather than loss of mC) in certain loci and certain times has functional importance. There are several well-established examples of direct hmC interactions with the cellular machinery leading to defined consequences for the cell. Therefore, it seems fair to conclude that besides its well-established role as an intermediate in the demethylation pathway, hmC is exploited to play additional epigenetic roles as a biological marker and/or controller of biological processes in the mammalian cell.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank our colleagues at the Department of Biological DNA Modification for their valuable suggestions and support. This work was supported by the University Excellence Initiative Programme of the Ministry of Education, Science and Sports of Lithuania (Measure No. 12-001-01-01-01).References
- R. Lister, E. A. Mukamel, J. R. Nery, M. Urich, C. A. Puddifoot, N. D. Johnson, J. Lucero, Y. Huang, A. J. Dwork, M. D. Schultz, M. Yu, J. Tonti-Filippini, H. Heyn, S. Hu, J. C. Wu, A. Rao, M. Esteller, C. He, F. G. Haghighi, T. J. Sejnowski, M. M. Behrens and J. R. Ecker, Science, 2013, 341, 1237905 CrossRef PubMed.
- J. Arand, D. Spieler, T. Karius, M. R. Branco, D. Meilinger, A. Meissner, T. Jenuwein, G. L. Xu, H. Leonhardt, V. Wolf and J. Walter, PLoS Genet., 2012, 8, 1002750 CrossRef PubMed.
- M. M. Suzuki and A. Bird, Nat. Rev. Genet., 2008, 9, 465–476 CrossRef CAS PubMed.
- Z. D. Smith and A. Meissner, Nat. Rev. Genet., 2013, 14, 204–220 CrossRef CAS PubMed.
- P. Borst and R. Sabatini, Annu. Rev. Microbiol., 2008, 62, 235–251 CrossRef CAS PubMed.
- B. C. Jensen, I. Q. Phan, J. R. McDonald, A. Sur, M. A. Gillespie, J. A. Ranish, M. Parsons and P. J. Myler, mSphere, 2021, 6, 204 CrossRef PubMed.
- H. G. A. M. Van Luenen, C. Farris, S. Jan, P. A. Genest, P. Tripathi, A. Velds, R. M. Kerkhoven, M. Nieuwland, A. Haydock, G. Ramasamy, S. Vainio, T. Heidebrecht, A. Perrakis, L. Pagie, B. Van Steensel, P. J. Myler and P. Borst, Cell, 2012, 150, 909–921 CrossRef CAS PubMed.
- N. W. Penn, R. Suwalski, C. O’Riley, K. Bojanowski and R. Yura, Biochem. J., 1972, 126, 781–790 CrossRef CAS PubMed.
- R. M. Kothari and V. Shankar, J. Mol. Evol., 1976, 7, 325–329 CrossRef CAS.
- S. Kriaučionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef PubMed.
- M. Tahiliani, K. P. Koh, Y. H. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 1979, 324, 930–935 CrossRef PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 1979, 333, 1300–1303 CrossRef PubMed.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Y. Tang, J. P. Ding, Y. Y. Jia, Z. C. Chen, L. Li, Y. Sun, X. X. Li, Q. Dai, C. X. Song, K. L. Zhang, C. He and G. L. Xu, Science, 1979, 333, 1303–1307 CrossRef PubMed.
- T. Pfaffeneder, B. Hackner, M. Truss, M. Munzel, M. Muller, C. A. Deiml, C. Hagemeier and T. Carell, Angew. Chem. Int. Ed., 2011, 50, 7008–7012 Search PubMed.
- Z. Liutkevičiūtė, G. Lukinavičius, V. Masevičius, D. Daujotytė and S. Klimašauskas, Nat. Chem. Biol., 2009, 5, 400–402 CrossRef PubMed.
- H. Heck and M. Casanova, Regul. Toxicol. Pharmacol., 2004, 40, 92–106 CrossRef CAS PubMed.
- K. Fugger, I. Bajrami, M. Silva, D. Santos, S. J. Young, S. Kunzelmann, G. Kelly, G. Hewitt, H. Patel, R. Goldstone, T. Carell, S. J. Boulton, J. Macrae, I. A. Taylor and S. C. West, Science, 1979, 372, 156–165 CrossRef PubMed.
- M. Zauri, G. Berridge, M. L. Thézénas, K. M. Pugh, R. Goldin, B. M. Kessler and S. Kriaucionis, Nature, 2015, 524, 114–118 CrossRef CAS PubMed.
- M. J. Peña-Gómez, P. Moreno-Gordillo, M. Narmontė, C. B. García-Calderón, A. Rukšėnaitė, S. Klimašauskas and I. V. Rosado, Cell Death Dis., 2022, 13, 503 CrossRef PubMed.
- F. von Meyenn, M. Iurlaro, E. Habibi, N. Q. Liu, A. Salehzadeh-Yazdi, F. Santos, E. Petrini, I. Milagre, M. Yu, Z. Xie, L. I. Kroeze, T. B. Nesterova, J. H. Jansen, H. Xie, C. He, W. Reik and H. G. Stunnenberg, Mol. Cell, 2016, 62, 848–861 CrossRef CAS.
- F. Spada, S. Schiffers, A. Kirchner, Y. Zhang, G. Arista, O. Kosmatchev, E. Korytiakova, R. Rahimoff, C. Ebert and T. Carell, Nat. Chem. Biol., 2020, 16, 1411–1419 CrossRef CAS PubMed.
- H. Jang, H. Shin, B. F. Eichman and J. H. Huh, Biochem. Biophys. Res. Commun., 2014, 446, 1067–1072 CrossRef CAS PubMed.
- L. Hu, J. Lu, J. Cheng, Q. Rao, Z. Li, H. Hou, Z. Lou, L. Zhang, W. Li, W. Gong, M. Liu, C. Sun, X. Yin, J. Li, X. Tan, P. Wang, Y. Wang, D. Fang, Q. Cui, P. Yang, C. He, H. Jiang, C. Luo and Y. Xu, Nature, 2015, 527, 118–122 CrossRef CAS PubMed.
- D. J. Crawford, M. Y. Liu, C. S. Nabel, X. J. Cao, B. A. Garcia and R. M. Kohli, J. Am. Chem. Soc., 2016, 138, 730–733 CrossRef CAS PubMed.
- C. J. Mariani, A. Vasanthakumar, J. Madzo, A. Yesilkanal, T. Bhagat, Y. Yu, S. Bhattacharyya, R. H. Wenger, S. L. Cohn, J. Nanduri, A. Verma, N. R. Prabhakar and L. A. Godley, Cell Rep., 2014, 7, 1343–1352 CrossRef CAS PubMed.
- M. Narmontė, P. Gibas, K. Daniūnaitė, J. Gordevičius and E. Kriukienė, Front. Cell Dev. Biol., 2021, 9, 727353 CrossRef PubMed.
- B. Thienpont, J. Steinbacher, H. Zhao, F. D’Anna, A. Kuchnio, A. Ploumakis, B. Ghesquière, L. Van Dyck, B. Boeckx, L. Schoonjans, E. Hermans, F. Amant, V. N. Kristensen, K. P. Koh, M. Mazzone, M. L. Coleman, T. Carell, P. Carmeliet and D. Lambrechts, Nature, 2016, 537, 63–68 CrossRef CAS PubMed.
- R. Matuleviciute, P. P. Cunha, R. S. Johnson and I. P. Foskolou, FEBS J., 2021, 288, 7143–7161 CrossRef CAS PubMed.
- Z. Zhang, C. He, L. Zhang, T. Zhu, D. Lv, G. Li, Y. Song, J. Wang, H. Wu, P. Ji and G. Liu, Reproduction, 2019, 158, 123–133 Search PubMed.
- H. Yang, H. Lin, H. Xu, L. Zhang, L. Cheng, B. Wen, J. Shou, K. Guan, Y. Xiong and D. Ye, Cell Res., 2014, 24, 1017–1020 CrossRef CAS PubMed.
- N. Liu, J. Zhang, M. Yan, L. Chen, J. Wu, Q. Tao, B. Yan, X. Chen and C. Peng, Cell Death Dis., 2023, 14, 170 CrossRef CAS PubMed.
- I. Y. Hwang, S. Kwak, S. Lee, H. Kim, S. E. Lee, J. H. Kim, Y. A. Kim, Y. K. Jeon, D. H. Chung, X. Jin, S. Park, H. Jang, E. J. Cho and H. D. Youn, Cell Metab., 2016, 24, 494–501 CrossRef CAS PubMed.
- N. R. Rose, M. A. Mc Donough, O. N. F. King, A. Kawamura and C. J. Schofield, Chem. Soc. Rev., 2011, 40, 4364–4397 RSC.
- W. Xu, H. Yang, Y. Liu, Y. Yang, P. Wang, S. H. Kim, S. Ito, C. Yang, P. Wang, M. T. Xiao, L. X. Liu, W. Q. Jiang, J. Liu, J. Y. Zhang, B. Wang, S. Frye, Y. Zhang, Y. H. Xu, Q. Y. Lei, K. L. Guan, S. M. Zhao and Y. Xiong, Cancer Cell, 2011, 19, 17–30 CrossRef CAS PubMed.
- M. Xiao, H. Yang, W. Xu, S. Ma, H. Lin, H. Zhu, L. Liu, Y. Liu, C. Yang, Y. Xu, S. Zhao, D. Ye, Y. Xiong and K. L. Guan, Genes Dev., 2012, 26, 1326–1338 CrossRef CAS.
- P. S. Ward, J. Patel, D. R. Wise, O. Abdel-Wahab, B. D. Bennett, H. A. Coller, J. R. Cross, V. R. Fantin, C. V. Hedvat, A. E. Perl, J. D. Rabinowitz, M. Carroll, S. M. Su, K. A. Sharp, R. L. Levine and C. B. Thompson, Cancer Cell, 2010, 17, 225–234 CrossRef CAS PubMed.
- R. A. Cairns and T. W. Mak, Cancer Discovery, 2013, 3, 730–741 CrossRef CAS PubMed.
- B. Zhao, Y. Yang, X. Wang, Z. Chong, R. Yin, S. H. Song, C. Zhao, C. Li, H. Huang, B. F. Sun, D. Wu, K. X. Jin, M. Song, B. Z. Zhu, G. Jiang, J. M. Rendtlew Danielsen, G. L. Xu, Y. G. Yang and H. Wang, Nucleic Acids Res., 2014, 42, 1593–1605 CrossRef CAS PubMed.
- A. Barks, M. M. Beeson, T. C. Hallstrom, M. K. Georgieff and P. V. Tran, Dev. Neurosci., 2022, 44, 80–90 CrossRef CAS PubMed.
- K. Blaschke, K. T. Ebata, M. M. Karimi, J. A. Zepeda-Martínez, P. Goyal, S. Mahapatra, A. Tam, D. J. Laird, M. Hirst, A. Rao, M. C. Lorincz and M. Ramalho-Santos, Nature, 2013, 500, 222–226 CrossRef CAS PubMed.
- R. Yin, S. Q. Mao, B. Zhao, Z. Chong, Y. Yang, C. Zhao, D. Zhang, H. Huang, J. Gao, Z. Li, Y. Jiao, C. Li, S. Liu, D. Wu, W. Gu, Y. G. Yang, G. L. Xu and H. Wang, J. Am. Chem. Soc., 2013, 135, 10396–10403 CrossRef CAS PubMed.
- J. H. Xue, G. D. Chen, F. Hao, H. Chen, Z. Fang, F. F. Chen, B. Pang, Q. L. Yang, X. Wei, Q. Q. Fan, C. Xin, J. Zhao, X. Deng, B. A. Wang, X. J. Zhang, Y. Chu, H. Tang, H. Yin, W. Ma, L. Chen, J. Ding, E. Weinhold, R. M. Kohli, W. Liu, Z. J. Zhu, K. Huang, H. Tang and G. L. Xu, Nature, 2019, 569, 581–585 CrossRef CAS PubMed.
- J. Kanfer, G. Ashwell and J. J. Burns, J. Biol. Chem., 1960, 235, 2518–2521 CrossRef CAS PubMed.
- A. Maiti and A. C. Drohat, J. Biol. Chem., 2011, 286, 35334–35338 CrossRef CAS PubMed.
- A. R. Weber, C. Krawczyk, A. B. Robertson, A. Kusnierczyk, C. B. Vågbø, D. Schuermann, A. Klungland and P. Schar, Nat. Commun., 2016, 7, 10806 CrossRef CAS PubMed.
- P. Hajkova, S. J. Jeffries, C. Lee, N. Miller, S. P. Jackson and M. A. Surani, Science, 2010, 329, 78–82 CrossRef CAS PubMed.
- M. Wossidlo, T. Nakamura, K. Lepikhov, C. J. Marques, V. Zakhartchenko, M. Boiani, J. Arand, T. Nakano, W. Reik and J. Walter, Nat. Commun., 2011, 2, 241 CrossRef PubMed.
- J. U. Guo, Y. J. Su, C. Zhong, G. L. Ming and H. J. Song, Cell Cycle, 2011, 10, 2662–2668 CrossRef CAS PubMed.
- S. Cortellino, J. F. Xu, M. Sannai, R. Moore, E. Caretti, A. Cigliano, M. Le Coz, K. Devarajan, A. Wessels, D. Soprano, L. K. Abramowitz, M. S. Bartolomei, F. Rambow, M. R. Bassi, T. Bruno, M. Fanciulli, C. Renner, A. J. Klein-Szanto, Y. Matsumoto, D. Kobi, I. Davidson, C. Alberti, L. Larue and A. Bellacosa, Cell, 2011, 146, 67–79 CrossRef CAS PubMed.
- H. Hashimoto, Y. Liu, A. K. Upadhyay, Y. Chang, S. B. Howerton, P. M. Vertino, X. Zhang and X. Cheng, Nucleic Acids Res., 2012, 40, 4841–4849 CrossRef CAS PubMed.
- T. Pfaffeneder, F. Spada, M. Wagner, C. Brandmayr, S. K. Laube, D. Eisen, M. Truss, J. Steinbacher, B. Hackner, O. Kotljarova, D. Schuermann, S. Michalakis, O. Kosmatchev, S. Schiesser, B. Steigenberger, N. Raddaoui, G. Kashiwazaki, U. Müller, C. G. Spruijt, M. Vermeulen, H. Leonhardt, P. Schär, M. Müller and T. Carell, Nat. Chem. Biol., 2014, 10, 574–581 CrossRef CAS PubMed.
- N. Bhutani, D. M. Burns and H. M. Blau, Cell, 2011, 146, 866–872 CrossRef CAS PubMed.
- X. Wu and Y. Zhang, Nat. Rev. Genet., 2017, 18, 517–534 CrossRef CAS PubMed.
- C. C. Chen, K. Y. Wang and C. K. J. Shen, J. Biol. Chem., 2012, 287, 33116–33121 CrossRef CAS PubMed.
- Z. Liutkevičiūtė, E. Kriukienė, J. Ličytė, M. Rudytè, G. Urbanavičiūtė and S. Klimašauskas, J. Am. Chem. Soc., 2014, 136, 5884–5887 CrossRef PubMed.
- S. Schiesser, B. Hackner, T. Pfaffeneder, M. Müller, C. Hagemeier, M. Truss and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 6516–6520 CrossRef CAS PubMed.
- K. Isono, K. Asahi and S. Suzuki, J. Am. Chem. Soc., 1969, 91, 7490–7505 CrossRef CAS PubMed.
- Y. Feng, J. J. Chen, N. Bin Xie, J. H. Ding, X. J. You, W. B. Tao, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 11322–11329 RSC.
- K. Iwan, R. Rahimoff, A. Kirchner, F. Spada, A. S. Schröder, O. Kosmatchev, S. Ferizaj, J. Steinbacher, E. Parsa, M. Müller and T. Carell, Nat. Chem. Biol., 2018, 14, 72–78 CrossRef CAS PubMed.
- E. Kamińska, E. Korytiaková, A. Reichl, M. Müller and T. Carell, Angew. Chem., Int. Ed., 2021, 60, 23207–23211 CrossRef PubMed.
- E. Privat and L. C. Sowers, Chem. Res. Toxicol., 1996, 9, 745–750 Search PubMed.
- S. Tardy-Planechaud, J. Fujimoto, S. S. Lin and L. C. Sowers, Nucleic Acids Res., 1997, 25, 553–559 CrossRef CAS PubMed.
- M. Munzel, D. Globisch, C. Trindler and T. Carell, Org. Lett., 2010, 12, 5671–5673 CrossRef CAS PubMed.
- Q. Dai and C. A. He, Org. Lett., 2011, 13, 3446–3449 CrossRef CAS PubMed.
- M. Y. Liu, H. Torabifard, D. J. Crawford, J. E. DeNizio, X. J. Cao, B. A. Garcia, G. A. Cisneros and R. M. Kohli, Nat. Chem. Biol., 2017, 13, 181–187 CrossRef CAS PubMed.
- P. Gibas, M. Narmontė, Z. Staševskij, J. Gordevičius, S. Klimašauskas and E. Kriukienė, PLoS Biol., 2020, 18, 3000684 CrossRef PubMed.
- A. B. Robertson, J. A. Dahl, C. B. Vagbo, P. Tripathi, H. E. Krokan and A. Klungland, Nucleic Acids Res., 2011, 39, 55 CrossRef PubMed.
- C. Rausch, P. Zhang, C. S. Casas-Delucchi, J. L. Daiß, C. Engel, G. Coster, F. D. Hastert, P. Weber and M. Cristina Cardoso, Nucleic Acids Res., 2021, 49, 12870–12894 CrossRef CAS PubMed.
- M. Munzel, D. Globisch, T. Bruckl, M. Wagner, V. Welzmiller, S. Michalakis, M. Muller, M. Biel and T. Carell, Angew. Chem., Int. Ed., 2010, 49, 5375–5377 CrossRef PubMed.
- F. R. Traube, S. Schiffers, K. Iwan, S. Kellner, F. Spada, M. Müller and T. Carell, Nat. Protoc., 2019, 14, 283–312 CrossRef CAS PubMed.
- Y. Sun, S. Stransky, J. Aguilan, S. Koul, S. J. Garforth, M. Brenowitz and S. Sidoli, Anal. Chim. Acta, 2021, 1180, 338880 CrossRef CAS PubMed.
- F. Yuan, Y. Bi, J. Y. Zhang, Y. L. Zhou, X. X. Zhang and C. X. Song, RSC Adv., 2019, 9, 29010–29014 RSC.
- J. Dietzsch, D. Feineis and C. Höbartner, FEBS Lett., 2018, 592, 2032–2047 CrossRef CAS PubMed.
- J. Ličytė, K. Kvederavičiūtė, A. Rukšėnaitė, E. Godliauskaitė, P. Gibas, V. Tomkutė, G. Petraitytė, V. Masevičius, S. Klimašauskas and E. Kriukienė, Open Biol., 2022, 12, 210302 CrossRef PubMed.
- A. Szwagierczak, S. Bultmann, C. S. Schmidt, F. Spada and H. Leonhardt, Nucleic Acids Res., 2010, 38, 181 CrossRef PubMed.
- T. Wang, C. E. Loo and R. M. Kohli, Mol. Metab., 2022, 57, 101314 CrossRef CAS PubMed.
- T. Hong, T. Wang, P. Guo, X. Xing, F. Ding, Y. Chen, J. Wu, J. Ma, F. Wu and X. Zhou, Anal. Chem., 2013, 85, 10797–10802 CrossRef CAS PubMed.
- L. Cui, J. Hu, M. Wang, C. C. Li and C. Y. Zhang, Anal. Chem., 2019, 91, 1232–1236 CrossRef CAS PubMed.
- S. Avraham, L. Schütz, L. Käver, A. Dankers, S. Margalit, Y. Michaeli, S. Zirkin, D. Torchinsky, N. Gilat, O. Bahr, G. Nifker, M. Koren-Michowitz and E. Weinhold, ChemBioChem, 2023, 30, e202300400 CrossRef PubMed.
- T. Shahal, N. Gilat, Y. Michaeli, O. Redy-Keisar, D. Shabat and Y. Ebenstein, Anal. Chem., 2014, 86, 8231–8237 CrossRef CAS PubMed.
- K. Chen, M. Juhasz, F. Gularek, E. Weinhold, Y. Tian, U. F. Keyser and N. A. W. Bell, Nano Lett., 2017, 17, 5199–5205 CrossRef CAS PubMed.
- Z. Y. Wang, M. Wang, Y. Zhang and C. Y. Zhang, Chem. Commun., 2018, 54, 8602–8605 RSC.
- Z. Zhang, T. He, Y. Qi, Y. Dai, K. Lao and X. Gou, RSC Adv., 2022, 12, 19885–19889 RSC.
- M. S. Reid, X. C. Le and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 11856–11866 CrossRef CAS PubMed.
- Y. An, Y. Li, X. Chen, L. Han, X. Xin, H. Nie, X. Zhang and C. Li, Sens. Actuators, B, 2023, 393, 134315 CrossRef CAS.
- L. Bai, G. Yang, Z. Qin, J. Lyu, Y. Wang, J. Feng, M. Liu, T. Gong, X. Li, Z. Li, J. Li, J. Qin, W. Yang and C. Ding, Adv. Sci., 2021, 8, 2101426 CrossRef CAS PubMed.
- C. Sui, Q. Wang, Y. Zhou, D. Zhang, H. Yin and S. Ai, Talanta, 2020, 219, 121211 CrossRef CAS PubMed.
- Z. Yang, Y. Shi, W. Liao, H. Yin and S. Ai, Sens. Actuators, B, 2016, 223, 621–625 CrossRef CAS.
- Y. Zhou, H. Yin, C. Sui, Y. Wang and S. Ai, Chem. Eng. J., 2019, 357, 94–102 CrossRef CAS.
- D. Globisch, M. Münzel, M. Müller, S. Michalakis, M. Wagner, S. Koch, T. Brückl, M. Biel and T. Carell, PLoS One, 2010, 5, 15367 CrossRef PubMed.
- C. E. Nestor, R. Ottaviano, J. Reddington, D. Sproul, D. Reinhardt, D. Dunican, E. Katz, J. M. Dixon, D. J. Harrison and R. R. Meehan, Genome Res., 2012, 22, 467–477 CrossRef CAS PubMed.
- M. Wagner, J. Steinbacher, T. F. J. Kraus, S. Michalakis, B. Hackner, T. Pfaffeneder, A. Perera, M. Müller, A. Giese, H. A. Kretzschmar and T. Carell, Angew. Chem., Int. Ed., 2015, 54, 12511–12514 CrossRef CAS PubMed.
- Y. Xu, L. Zhong, H. Wei, Y. Li, J. Xie, L. Xie, X. Chen, X. Guo, P. Yin, S. Li, J. Zeng, X. J. Li and L. Lin, Front. Aging Neurosci., 2022, 14, 934224 CrossRef CAS PubMed.
- M. Bachman, S. Uribe-Lewis, X. Yang, H. E. Burgess, M. Iurlaro, W. Reik, A. Murrell and S. Balasubramanian, Nat. Chem. Biol., 2015, 11, 555–557 CrossRef CAS PubMed.
- Y. Tang, S. J. Zheng, C. B. Qi, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2015, 87, 3445–3452 CrossRef CAS PubMed.
- S. G. Jin, X. Wu, A. X. Li and G. P. Pfeifer, Nucleic Acids Res., 2011, 39, 5015 CrossRef CAS PubMed.
- G. Ficz, M. R. Branco, S. Seisenberger, F. Santos, F. Krueger, T. A. Hore, C. J. Marques, S. Andrews and W. Reik, Nature, 2011, 473, 398–402 CrossRef CAS PubMed.
- A. Lentini, C. Lagerwall, S. Vikingsson, H. K. Mjoseng, K. Douvlataniotis, H. Vogt, H. Green, R. R. Meehan, M. Benson and C. E. Nestor, Nat. Methods, 2018, 15, 499–504 CrossRef CAS PubMed.
- C. X. Song, K. E. Szulwach, Y. Fu, Q. Dai, C. Yi, X. Li, Y. Li, C. H. Chen, W. Zhang, X. Jian, J. Wang, L. Zhang, T. J. Looney, B. Zhang, L. A. Godley, L. M. Hicks, B. T. Lahn, P. Jin and C. He, Nat. Biotechnol., 2011, 29, 68–75 CrossRef CAS PubMed.
- C. X. Song, S. Yin, L. Ma, A. Wheeler, Y. Chen, Y. Zhang, B. Liu, J. Xiong, W. Zhang, J. Hu, Z. Zhou, B. Dong, Z. Tian, S. S. Jeffrey, M. S. Chua, S. So, W. Li, Y. Wei, J. Diao, D. Xie and S. R. Quake, Cell Res., 2017, 27, 1231–1242 CrossRef CAS PubMed.
- W. Li, X. Zhang, X. Lu, L. You, Y. Song, Z. Luo, J. Zhang, J. Nie, W. Zheng, D. Xu, Y. Wang, Y. Dong, S. Yu, J. Hong, J. Shi, H. Hao, F. Luo, L. Hua, P. Wang, X. Qian, F. Yuan, L. Wei, M. Cui, T. Zhang, Q. Liao, M. Dai, Z. Liu, G. Chen, K. Meckel, S. Adhikari, G. Jia, M. B. Bissonnette, X. Zhang, Y. Zhao, W. Zhang, C. He and J. Liu, Cell Res., 2017, 27, 1243–1257 CrossRef CAS PubMed.
- D. Han, X. Lu, A. H. Shih, J. Nie, Q. You, M. M. Xu, A. M. Melnick, R. L. Levine and C. He, Mol. Cell, 2016, 63, 711–719 CrossRef CAS PubMed.
- Z. Liutkevičiūtė, E. Kriukienė, I. Grigaitytė, V. Masevičius and S. Klimašauskas, Angew. Chem., Int. Ed., 2011, 50, 2090–2093 CrossRef PubMed.
- Z. Staševskij, P. Gibas, J. Gordevičius, E. Kriukienė and S. Klimašauskas, Mol. Cell, 2017, 65, 554–564 CrossRef PubMed.
- J. Gordevičius, M. Narmontė, P. Gibas, K. Kvederavičiūtė, V. Tomkutė, P. Paluoja, K. Krjutškov, A. Salumets and E. Kriukienė, Clin. Epigenet., 2020, 12, 153 CrossRef PubMed.
- L. Hu, Y. Liu, S. Han, L. Yang, X. Cui, Y. Gao, Q. Dai, X. Lu, X. Kou, Y. Zhao, W. Sheng, S. Gao, X. He and C. He, J. Am. Chem. Soc., 2019, 141, 8694–8697 CrossRef CAS PubMed.
- M. Yu, G. C. Hon, K. E. Szulwach, C. X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen, B. Park, J. H. Min, P. Jin, B. Ren and C. He, Cell, 2012, 149, 1368–1380 CrossRef CAS PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 1979, 336, 934–937 CrossRef PubMed.
- K. Tanaka and A. Okamoto, Bioorg. Med. Chem. Lett., 2007, 17, 1912–1915 CrossRef CAS PubMed.
- Y. Liu, P. Siejka-Zielińska, G. Velikova, Y. Bi, F. Yuan, M. Tomkova, C. Bai, L. Chen, B. Schuster-Böckler and C. X. Song, Nat. Biotechnol., 2019, 37, 424–429 CrossRef CAS PubMed.
- Y. Liu, Z. Hu, J. Cheng, P. Siejka-Zielińska, J. Chen, M. Inoue, A. A. Ahmed and C. X. Song, Nat. Commun., 2021, 12, 618 CrossRef PubMed.
- H. Zeng, B. He, B. Xia, D. Bai, X. Lu, J. Cai, L. Chen, A. Zhou, C. Zhu, H. Meng, Y. Gao, H. Guo, C. He, Q. Dai and C. Yi, J. Am. Chem. Soc., 2018, 140, 13190–13194 CrossRef CAS PubMed.
- E. K. Schutsky, J. E. Denizio, P. Hu, M. Y. Liu, C. S. Nabel, E. B. Fabyanic, Y. Hwang, F. D. Bushman, H. Wu and R. M. Kohli, Nat. Biotechnol., 2018, 36, 1083–1090 CrossRef CAS PubMed.
- R. Vaisvila, V. K. C. Ponnaluri, Z. Sun, B. W. Langhorst, L. Saleh, S. Guan, N. Dai, M. A. Campbell, B. S. Sexton, K. Marks, M. Samaranayake, J. C. Samuelson, H. E. Church, E. Tamanaha, I. R. Corrêa, S. Pradhan, E. T. Dimalanta, T. C. Evans, L. Williams and T. B. Davis, Genome Res., 2021, 31, 1280–1289 CrossRef PubMed.
- J. Füllgrabe, W. S. Gosal, P. Creed, S. Liu, C. K. Lumby, D. J. Morley, T. W. B. Ost, A. J. Vilella, S. Yu, H. Bignell, P. Burns, T. Charlesworth, B. Fu, H. Fordham, N. J. Harding, O. Gandelman, P. Golder, C. Hodson, M. Li, M. Lila, Y. Liu, J. Mason, J. Mellad, J. M. Monahan, O. Nentwich, A. Palmer, M. Steward, M. Taipale, A. Vandomme, R. S. San-Bento, A. Singhal, J. Vivian, N. Wójtowicz, N. Williams, N. J. Walker, N. C. H. Wong, G. N. Yalloway, J. D. Holbrook and S. Balasubramanian, Nat. Biotechnol., 2023, 41, 1457–1464 CrossRef PubMed.
- T. Davis and R. Vaišvila, J. Vis. Exp., 2011, 1, 2661 Search PubMed.
- T. Khare, S. Pai, K. Koncevičius, M. Pal, E. Kriukienė, Z. Liutkevičiūtė, M. Irimia, P. Jia, C. Ptak, M. Xia, R. Tice, M. Tochigi, S. Moréra, A. Nazarians, D. Belsham, A. H. C. Wong, B. J. Blencowe, S. C. Wang, P. Kapranov, R. Kustra, V. Labrie, S. Klimašauskas and A. Petronis, Nat. Struct. Mol. Biol., 2012, 19, 1037–1044 CrossRef CAS PubMed.
- N. Plongthongkum, D. H. Diep and K. Zhang, Nat. Rev. Genet., 2014, 15, 647–661 CrossRef CAS PubMed.
- Z. Sun, J. Terragni, J. G. Borgaro, Y. Liu, L. Yu, S. Guan, H. Wang, D. Sun, X. Cheng, Z. Zhu, S. Pradhan and Y. Zheng, Cell Rep., 2013, 3, 567–576 CrossRef CAS PubMed.
- Z. Sun, N. Dai, J. G. Borgaro, A. Quimby, D. Sun, I. R. Corrêa, Y. Zheng, Z. Zhu and S. Guan, Mol. Cell, 2015, 57, 750–761 CrossRef CAS PubMed.
- R. K. Neely, P. Dedecker, J. Hotta, G. Urbanavičiūtė, S. Klimašauskas and J. Hofkens, Chem. Sci., 2010, 1, 453–460 RSC.
- S. L. Amarasinghe, M. E. Ritchie and Q. Gouil, Gigascience, 2021, 10, 1–7 CrossRef PubMed.
- Z. L. Wescoe, J. Schreiber and M. Akeson, J. Am. Chem. Soc., 2014, 136, 16582–16587 CrossRef CAS PubMed.
- T. A. Clark, X. Lu, K. Luong, Q. Dai, M. Boitano, S. W. Turner, C. He and J. Korlach, BMC Biol., 2013, 11, 4 CrossRef CAS PubMed.
- C.-X. Song, T. A. Clark, X.-Y. Lu, A. Kislyuk, Q. Dai, S. W. Turner, C. He and J. Korlach, Nat. Methods, 2012, 9, 75–77 CrossRef CAS PubMed.
- D. Renciuk, O. Blacque, M. Vorlickova and B. Spingler, Nucleic Acids Res., 2013, 41, 9891–9900 CrossRef CAS PubMed.
- L. Lercher, M. A. McDonough, A. H. El-Sagheer, A. Thalhammer, S. Kriaucionis, T. Brown and C. J. Schofield, Chem. Commun., 2014, 50, 1794–1796 RSC.
- M. W. Szulik, P. S. Pallan, B. Nocek, M. Voehler, S. Banerjee, S. Brooks, A. Joachimiak, M. Egli, B. F. Eichman and M. P. Stone, Biochemistry, 2015, 54, 1294–1305 CrossRef CAS PubMed.
- F. Battistini, P. D. Dans, M. Terrazas, C. L. Castellazzi, M. Labrador, N. Villegas, I. Brun-Heath, C. González and M. Orozco, PLoS Comput. Biol., 2021, 17, 1009547 CrossRef PubMed.
- R. Wang, S. V. Ranganathan, V. A. Valsangkar, S. M. Magliocco, F. Shen, A. Chen and J. Sheng, Chem. Commun., 2015, 51, 16389–16392 RSC.
- M. Tomkuvienė, M. Meier, D. Ikasalaitė, J. Wildenauer, V. Kairys, S. Klimašauskas and L. Manelytė, Nucleic Acids Res., 2022, 50, 6549–6561 CrossRef PubMed.
- S. Li, Y. Peng and A. R. Panchenko, Curr. Opin. Struct. Biol., 2022, 75, 102430 CrossRef CAS PubMed.
- M. T. Bennett, M. T. Rodgers, A. S. Hebert, L. E. Ruslander, L. Eisele and A. C. Drohat, J. Am. Chem. Soc., 2006, 128, 12510–12519 CrossRef CAS PubMed.
- E. A. Raiber, P. Murat, D. Y. Chirgadze, D. Beraldi, B. F. Luisi and S. Balasubramanian, Nat. Struct. Mol. Biol., 2015, 22, 44–49 CrossRef CAS PubMed.
- J. S. Hardwick, D. Ptchelkine, A. H. El-Sagheer, I. Tear, D. Singleton, S. E. V. Phillips, A. N. Lane and T. Brown, Nat. Struct. Mol. Biol., 2017, 24, 544–552 CrossRef CAS PubMed.
- T. T. M. Ngo, J. Yoo, Q. Dai, Q. Zhang, C. He, A. Aksimentiev and T. Ha, Nat. Commun., 2016, 7, 10813 CrossRef CAS PubMed.
- P. M. D. Severin, X. Zou, K. Schulten and H. E. Gaub, Biophys. J., 2013, 104, 208–215 CrossRef CAS PubMed.
- S. Matsumoto, H. Tateishi-Karimata, T. Ohyama and N. Sugimoto, RSC Adv., 2021, 11, 37205–37217 RSC.
- A. J. Stevens, L. de Jong and M. A. Kennedy, Int. J. Mol. Sci., 2022, 23, 2407 CrossRef CAS PubMed.
- R. K. Morgan, M. M. Molnar, H. Batra, B. Summerford, R. M. Wadkins and T. A. Brooks, J. Nucleic Acids, 2018, 2018, 9281286 CrossRef PubMed.
- G. L. Xu and M. Bochtler, Nat. Chem. Biol., 2020, 16, 1160–1169 CrossRef CAS PubMed.
- M. Iurlaro, G. Ficz, D. Oxley, E. A. Raiber, M. Bachman, M. J. Booth, S. Andrews, S. Balasubramanian and W. Reik, Genome Biol., 2013, 14, 119 CrossRef PubMed.
- C. G. Spruijt, F. Gnerlich, A. H. Smits, T. Pfaffeneder, P. W. T. C. Jansen, C. Bauer, M. Münzel, M. Wagner, M. Müller, F. Khan, H. C. Eberl, A. Mensinga, A. B. Brinkman, K. Lephikov, U. Müller, J. Walter, R. Boelens, H. Van Ingen, H. Leonhardt, T. Carell and M. Vermeulen, Cell, 2013, 152, 1146–1159 CrossRef CAS PubMed.
- C. Frauer, T. Hoffmann, S. Bultmann, V. Casa, M. C. Cardoso, I. Antes and H. Leonhardt, PLoS One, 2011, 6, 21306 CrossRef PubMed.
- M. Mellén, P. Ayata, S. Dewell, S. Kriaucionis and N. Heintz, Cell, 2012, 151, 1417–1430 CrossRef PubMed.
- A. Marco, T. Kisliouk, T. Tabachnik, A. Weller and N. Meiri, Diabetes, 2016, 65, 2258–2267 CrossRef CAS PubMed.
- G. P. Pfeifer, P. E. Szabó and J. Song, J. Mol. Biol., 2020, 432, 1718–1730 CrossRef CAS PubMed.
- D. Ortega-Alarcon, R. Claveria-Gimeno, S. Vega, O. C. Jorge-Torres, M. Esteller, O. Abian and A. Velazquez-Campoy, Int. J. Biol. Macromol., 2023, 232, 123373 CrossRef CAS PubMed.
- T. Zhou, J. Xiong, M. Wang, N. Yang, J. Wong, B. Zhu and R. M. Xu, Mol. Cell, 2014, 54, 879–886 CrossRef CAS PubMed.
- S. Klimasauskas, S. Kumar, R. J. Roberts and X. Cheng, Cell, 1994, 76, 357–369 CrossRef CAS PubMed.
- L. Liu, Y. Zhang, M. Liu, W. Wei, C. Yi and J. Peng, J. Mol. Biol., 2020, 432, 1035–1047 CrossRef CAS PubMed.
- B. C. Buchmuller, J. Dröden, H. Singh, S. Palei, M. Drescher, R. Linser and D. Summerer, J. Am. Chem. Soc., 2022, 144, 2987–2993 CrossRef CAS PubMed.
- V. Valinluck and L. C. Sowers, Cancer Res., 2007, 67, 946–950 CrossRef CAS PubMed.
- C. L. Seiler, J. Fernandez, Z. Koerperich, M. P. Andersen, D. Kotandeniya, M. E. Nguyen, Y. Y. Sham and N. Y. Tretyakova, Biochemistry, 2018, 57, 6061–6069 CrossRef CAS PubMed.
- S. Adam, V. Klingel, N. E. Radde, P. Bashtrykov and A. Jeltsch, Nucleic Acids Res., 2023, 51, 6622–6633 CrossRef CAS PubMed.
- E. A. Raiber, G. Portella, S. Martínez Cuesta, R. Hardisty, P. Murat, Z. Li, M. Iurlaro, W. Dean, J. Spindel, D. Beraldi, Z. Liu, M. A. Dawson, W. Reik and S. Balasubramanian, Nat. Chem., 2018, 10, 1258–1266 CrossRef CAS PubMed.
- S. Ji, H. Shao, Q. Han, C. L. Seiler and N. Y. Tretyakova, Angew. Chem., 2017, 129, 14318–14322 CrossRef.
- F. Li, Y. Zhang, J. Bai, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 10617–10620 CrossRef CAS PubMed.
- M. Bachman, S. Uribe-Lewis, X. Yang, M. Williams, A. Murrell and S. Balasubramanian, Nat. Chem., 2014, 6, 1049–1055 CrossRef CAS PubMed.
- M. Tomkova, M. Mcclellan, S. Kriaucionis and B. Schuster-Boeckler, eLife, 2016, 5, 17082 CrossRef PubMed.
- G. R. Kafer, X. Li, T. Horii, I. Suetake, S. Tajima, I. Hatada and P. M. Carlton, Cell Rep., 2016, 14, 1283–1292 CrossRef CAS PubMed.
- S. S. Kharat, X. Ding, D. Swaminathan, A. Suresh, M. Singh, S. K. Sengodan, S. Burkett, H. Marks, C. Pamala, Y. He, S. D. Fox, E. C. Buehler, K. Muegge, S. E. Martin and S. K. Sharan, Sci. Signal, 2020, 13, 8091 CrossRef PubMed.
- Y. Imai, M. Biot, J. A. J. Clément, M. Teragaki, S. Urbach, T. Robert, F. Baudat, C. Grey and B. de Massy, eLife, 2020, 9, 1–33 CrossRef PubMed.
- K. Brick, F. Pratto and R. Daniel Camerini-Otero, Genes Dev., 2020, 34, 731–732 CrossRef CAS PubMed.
- T. Prikrylova, J. Robertson, F. Ferrucci, D. Konorska, H. Aanes, A. Manaf, B. Zhang, C. B. Vågbø, A. Kuśnierczyk, K. M. Gilljam, C. Løvkvam-Køster, M. Otterlei, J. A. Dahl, J. Enserink, A. Klungland and A. B. Robertson, Sci. Rep., 2019, 9, 11065 CrossRef PubMed.
- J. Robertson, A. B. Robertson and A. Klungland, Biochem. Biophys. Res. Commun., 2011, 411, 40–43 CrossRef CAS PubMed.
- W. A. Pastor, U. J. Pape, Y. Huang, H. R. Henderson, R. Lister, M. Ko, E. M. McLoughlin, Y. Brudno, S. Mahapatra, P. Kapranov, M. Tahiliani, G. Q. Daley, X. S. Liu, J. R. Ecker, P. M. Milos, S. Agarwal and A. Rao, Nature, 2011, 473, 394–397 CrossRef CAS PubMed.
- H. Wu, A. C. D’Alessio, S. Ito, Z. Wang, K. Cui, K. Zhao, Y. E. Sun and Y. Zhang, Genes Dev., 2011, 25, 679–684 CrossRef CAS PubMed.
- H. Wu, X. Wu, L. Shen and Y. Zhang, Nat. Biotechnol., 2014, 32, 1231–1240 CrossRef CAS PubMed.
- B. Xia, D. Han, X. Lu, Z. Sun, A. Zhou, Q. Yin, H. Zeng, M. Liu, X. Jiang, W. Xie, C. He and C. Yi, Nat. Methods, 2015, 12, 1047–1050 CrossRef CAS PubMed.
- X. Lu, D. Han, B. S. Zhao, C. X. Song, L. S. Zhang, L. C. Doré and C. He, Cell Res., 2015, 25, 386–389 CrossRef CAS PubMed.
- R. J. Marina, D. Sturgill, M. A. Bailly, M. Thenoz, G. Varma, M. F. Prigge, K. K. Nanan, S. Shukla, N. Haque and S. Oberdoerffer, EMBO J., 2016, 35, 335–355 CrossRef CAS PubMed.
- J. Yang, N. Bashkenova, R. Zang, X. Huang and J. Wang, Development, 2020, 147, 183129 CrossRef PubMed.
- L. Wen, X. Li, L. Yan, Y. Tan, R. Li, Y. Zhao, Y. Wang, J. Xie, Y. Zhang, C. Song, M. Yu, X. Liu, P. Zhu, X. Li, Y. Hou, H. Guo, X. Wu, C. He, R. Li, F. Tang and J. Qiao, Genome. Biol., 2014, 15, 49 CrossRef PubMed.
- L. Tan, L. Xiong, W. Xu, F. Wu, N. Huang, Y. Xu, L. Kong, L. Zheng, L. Schwartz, Y. Shi and Y. G. Shi, Nucleic Acids Res., 2013, 41, 84 CrossRef PubMed.
- D. Q. Shi, I. Ali, J. Tang and W. C. Yang, Front. Genet., 2017, 8, 100 CrossRef PubMed.
- A. Klungland and A. B. Robertson, Free Radic. Biol. Med., 2017, 107, 62–68 CrossRef CAS PubMed.
- M. M. Dawlaty, K. Ganz, B. E. Powell, Y. C. Hu, S. Markoulaki, A. W. Cheng, Q. Gao, J. Kim, S. W. Choi, D. C. Page and R. Jaenisch, Cell Stem Cell, 2011, 9, 166–175 CrossRef CAS PubMed.
- M. Ko, H. S. Bandukwala, J. An, E. D. Lamperti, E. C. Thompson, R. Hastie, A. Tsangaratou, K. Rajewsky, S. B. Koralov and A. Rao, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14566–14571 CrossRef CAS PubMed.
- K. P. Koh, A. Yabuuchi, S. Rao, Y. Huang, K. Cunniff, J. Nardone, A. Laiho, M. Tahiliani, C. A. Sommer, G. Mostoslavsky, R. Lahesmaa, S. H. Orkin, S. J. Rodig, G. Q. Daley and A. Rao, Cell Stem Cell, 2011, 8, 200–213 CrossRef CAS PubMed.
- W. Li and M. Liu, J. Nucleic Acids, 2011, 2011, 870726 Search PubMed.
- M. M. Dawlaty, A. Breiling, T. Le, M. I. Barrasa, G. Raddatz, Q. Gao, B. E. Powell, A. W. Cheng, K. F. Faull, F. Lyko and R. Jaenisch, Dev. Cell, 2014, 29, 102–111 CrossRef CAS PubMed.
- F. Lu, Y. Liu, L. Jiang, S. Yamaguchi and Y. Zhang, Genes Dev., 2014, 28, 2103–2119 CrossRef CAS PubMed.
- N. Verma, H. Pan, L. C. Doré, A. Shukla, Q. V. Li, B. Pelham-Webb, V. Teijeiro, F. González, A. Krivtsov, C. J. Chang, E. P. Papapetrou, C. He, O. Elemento and D. Huangfu, Nat. Genet., 2018, 50, 83–95 CrossRef CAS PubMed.
- J. Charlton, E. J. Jung, A. L. Mattei, N. Bailly, J. Liao, E. J. Martin, P. Giesselmann, B. Brändl, E. K. Stamenova, F. J. Müller, E. Kiskinis, A. Gnirke, Z. D. Smith and A. Meissner, Nat. Genet., 2020, 52, 819–827 CrossRef CAS PubMed.
- T. Gu, X. Lin, S. M. Cullen, M. Luo, M. Jeong, M. Estecio, J. Shen, S. Hardikar, D. Sun, J. Su, D. Rux, A. Guzman, M. Lee, L. S. Qi, J. J. Chen, M. Kyba, Y. Huang, T. Chen, W. Li and M. A. Goodell, Genome Biol., 2018, 19, 88 CrossRef PubMed.
- J. Xiong, Z. Zhang, J. Chen, H. Huang, Y. Xu, X. Ding, Y. Zheng, R. Nishinakamura, G. L. Xu, H. Wang, S. Chen, S. Gao and B. Zhu, Mol. Cell, 2016, 64, 913–925 CrossRef CAS PubMed.
- Y. Costa, J. Ding, T. W. Theunissen, F. Faiola, T. A. Hore, P. V. Shliaha, M. Fidalgo, A. Saunders, M. Lawrence, S. Dietmann, S. Das, D. N. Levasseur, Z. Li, M. Xu, W. Reik, J. C. R. Silva and J. Wang, Nature, 2013, 495, 370–374 CrossRef CAS PubMed.
- K. Williams, J. Christensen, M. T. Pedersen, J. V. Johansen, P. A. C. Cloos, J. Rappsilber and K. Helin, Nature, 2011, 473, 343–472 CrossRef CAS PubMed.
- J. Yang, R. Guo, H. Wang, X. Ye, Z. Zhou, J. Dan, H. Wang, P. Gong, W. Deng, Y. Yin, S. Q. Mao, L. Wang, J. Ding, J. Li, D. L. Keefe, M. M. Dawlaty, J. Wang, G. L. Xu and L. Liu, Cell Rep., 2016, 15, 1809–1821 CrossRef CAS PubMed.
- B. A. Caldwell, M. Y. Liu, R. D. Prasasya, T. Wang, J. E. DeNizio, N. A. Leu, N. Y. A. Amoh, C. Krapp, Y. Lan, E. J. Shields, R. Bonasio, C. J. Lengner, R. M. Kohli and M. S. Bartolomei, Mol. Cell, 2021, 81, 859–869 CrossRef CAS PubMed.
- L. M. Wheldon, A. Abakir, Z. Ferjentsik, T. Dudnakova, S. Strohbuecker, D. Christie, N. Dai, S. Guan, J. M. Foster, I. R. Corrêa, M. Loose, J. E. Dixon, V. Sottile, A. D. Johnson and A. Ruzov, Cell Rep., 2014, 7, 1353–1361 CrossRef CAS PubMed.
- L. C. Lewis, P. C. K. Lo, J. M. Foster, N. Dai, I. R. Corrêa, P. M. Durczak, G. Duncan, A. Ramsawhook, G. P. Aithal, C. Denning, N. R. F. Hannan and A. Ruzov, Epigenetics, 2017, 12, 277–286 CrossRef PubMed.
- R. Amouroux, B. Nashun, K. Shirane, S. Nakagawa, P. W. S. Hill, Z. D’Souza, M. Nakayama, M. Matsuda, A. Turp, E. Ndjetehe, V. Encheva, N. R. Kudo, H. Koseki, H. Sasaki and P. Hajkova, Nat. Cell Biol., 2016, 18, 225–233 CrossRef CAS PubMed.
- J. Madzo, H. Liu, A. Rodriguez, A. Vasanthakumar, S. Sundaravel, D. B. D. Caces, T. J. Looney, L. Zhang, J. B. Lepore, T. Macrae, R. Duszynski, A. H. Shih, C. X. Song, M. Yu, Y. Yu, R. Grossman, B. Raumann, A. Verma, C. He, R. L. Levine, D. Lavelle, B. T. Lahn, A. Wickrema and L. A. Godley, Cell Rep., 2014, 6, 231–244 CrossRef CAS PubMed.
- Y. H. Huh, J. Cohen and J. L. Sherley, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16862–16867 CrossRef CAS PubMed.
- B. W. Carey, L. W. S. Finley, J. R. Cross, C. D. Allis and C. B. Thompson, Nature, 2015, 518, 413–416 CrossRef CAS PubMed.
- D. Peng, G. Ge, Y. Gong, Y. Zhan, S. He, B. Guan, Y. Li, Z. Xu, H. Hao, Z. He, G. Xiong, C. Zhang, Y. Shi, Y. Zhou, W. Ci, X. Li and L. Zhou, Clin. Epigenet., 2018, 10, 94 CrossRef PubMed.
- C. B. Gustafson, C. Yang, K. M. Dickson, H. Shao, D. Van Booven, J. W. Harbour, Z. J. Liu and G. Wang, Clin. Epigenet., 2015, 7, 51 CrossRef PubMed.
- C. G. Lian, Y. Xu, C. Ceol, F. Wu, A. Larson, K. Dresser, W. Xu, L. Tan, Y. Hu, Q. Zhan, C. W. Lee, D. Hu, B. Q. Lian, S. Kleffel, Y. Yang, J. Neiswender, A. J. Khorasani, R. Fang, C. Lezcano, L. M. Duncan, R. A. Scolyer, J. F. Thompson, H. Kakavand, Y. Houvras, L. I. Zon, M. C. Mihm, U. B. Kaiser, T. Schatton, B. A. Woda, G. F. Murphy and Y. G. Shi, Cell, 2012, 150, 1135–1146 CrossRef CAS PubMed.
- Y. Liao, J. Gu, Y. Wu, X. Long, D. Ge, J. Xu and J. Ding, Oncol. Lett., 2016, 11, 3753–3760 CrossRef CAS PubMed.
- J. Cai, L. Chen, Z. Zhang, X. Zhang, X. Lu, W. Liu, G. Shi, Y. Ge, P. Gao, Y. Yang, A. Ke, L. Xiao, R. Dong, Y. Zhu, X. Yang, J. Wang, T. Zhu, D. Yang, X. Huang, C. Sui, S. Qiu, F. Shen, H. Sun, W. Zhou, J. Zhou, J. Nie, C. Zeng, E. K. Stroup, X. Zhang, B. C. H. Chiu, W. Y. Lau, C. He, H. Wang, W. Zhang and J. Fan, Gut, 2019, 68, 2195–2205 CrossRef CAS PubMed.
- G. D. Guler, Y. Ning, C. J. Ku, T. Phillips, E. McCarthy, C. K. Ellison, A. Bergamaschi, F. Collin, P. Lloyd, A. Scott, M. Antoine, W. Wang, K. Chau, A. Ashworth, S. R. Quake and S. Levy, Nat. Commun., 2020, 11, 5270 CrossRef CAS PubMed.
- H. Y. Chen, W. L. Zhang, L. Zhang, P. Yang, F. Li, Z. R. Yang, J. Wang, M. Pang, Y. Hong, C. Yan, W. Li, J. Liu, N. Xu, L. Chen, X. B. Xiao, Y. Qin, X. H. He, H. Liu, H. C. Zhu, C. He, J. Lin and H. M. Jing, Clin. Epigenet., 2021, 13, 33 CrossRef CAS PubMed.
- L. Chen, Q. Shen, S. Xu, H. Yu, S. Pei, Y. Zhang, X. He, Q. Wang and D. Li, J. Alzheimer's Dis., 2022, 85, 573–585 Search PubMed.
- Y. Yang, C. Zeng, X. Lu, Y. Song, J. Nie, R. Ran, Z. Zhang, C. He, W. Zhang and S. M. Liu, Clin. Chem., 2019, 65, 1414–1425 CrossRef CAS PubMed.
- S. G. Jin, S. Kadam and G. P. Pfeifer, Nucleic Acids Res., 2010, 38, 125 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |