
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding deNOx mechanisms in transition metal exchanged zeolites
Jamal
Abdul Nasir
 *a,
Andrew M.
Beale
ab and
C. Richard A.
Catlow
*abc
*a,
Andrew M.
Beale
ab and
C. Richard A.
Catlow
*abc
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: c.r.a.catlow@ucl.ac.uk
bUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, R92 Harwell, Oxfordshire OX11 0FA, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK
First published on 23rd October 2024
Abstract
Transition-metal-containing zeolites have wide-ranging applications in several catalytic processes including the selective catalytic reduction (SCR) of NOx species. To understand how transition metal ions (TMIs) can effect NOx reduction chemistry, both structural and mechanistic aspects at the atomic level are needed. In this review, we discuss the coordination chemistry of TMIs and their mobility within the zeolite framework, the reactivity of active sites, and the mechanisms and intermediates in the NH3-SCR reaction. We emphasise the key relationship between TMI coordination and structure and mechanism and discuss approaches to enhancing catalytic activity via structural modifications.
 Jamal Abdul Nasir | Jamal Abdul Nasir received his MSc and MPhil degree in Inorganic Chemistry from Quaid-i-Azam University Pakistan in 2014 and 2016 respectively. He joined the same institute as a PhD student in September 2016. He then started a 2nd PhD in computational chemistry under Prof. Richard Catlow's group at UCL and is now a postdoctoral research fellow at UCL. He has a strong interest in the both experimental and computational chemistry e.g. MM, DFT and hybrid QM/MM methodologies. He is currently working on Quantum Computing applications for Catalysis; and on Machine learning Interatomic Potentials for inorganic-based materials. |
 Andrew M. Beale | Andrew (Andy) Beale was awarded a BSc from the University of Sussex followed by a PhD at the Royal Institution of Great Britain. He then worked as a Postdoctoral researcher, VENI Research Fellow and Assistant Professor in the Department of Inorganic Chemistry and Catalysis, at Utrecht University in the Netherlands. He returned to the UK in 2013 as an EPSRC Early career fellow. He is currently a Professor of Inorganic Chemistry at UCL Group Leader at the Research Complex@Harwell and Chief Scientific Officer of Finden Ltd. Current research interests concern the study of functional materials with in situ and operando methods. |
 C. Richard A. Catlow | C. Richard A. Catlow obtained his doctorate from the University of Oxford and subsequently held academic appointments at University College London, Keele University, the Daresbury Laboratory, and the Royal Institution. He was a Vice-President and Foreign Secretary of the Royal Society from 2016–2021. He currently has a joint professorial position with UCL and Cardiff University. His research interests are in the field of the chemistry of complex materials, including those for applications in catalytic and energy applications, investigated by a combination of computational and experimental techniques. He has published over 1200 research articles in this field. |
Introduction
The development of catalysts for removing nitrogen oxides (NOx) from the emissions of power plants and internal combustion engines is amongst the most intensively studied fields of environmental catalysis. Zeolites containing transition metal ions (TMIs) are effective for these key catalytic processes, but despite extensive study, there remain uncertainties concerning structural and mechanistic aspects. This review will discuss the status of the field, emphasising the complementary nature of experiments with computationally based theory and with a strong focus on the ammonia-assisted reduction reaction.As will be discussed in greater detail below, zeolites are tecto-aluminosilicate nano-porous materials, which possess exceptional physicochemical properties that are useful in a wide range of applications including ion-exchange, gas separation, and catalysis;1 an analogous class of nano-porous alumino-phosphate (ALPO) and related materials can also show useful catalytic functionality. To modify their adsorption properties and to incorporate new functionality into zeolites, ion-exchange with transition metal ions (TMI) is a widely used strategy.2,3 TMIs, which may also be introduced into framework sites through isomorphous substitution or annealing, play a key role in zeolite catalytic chemistry because of their variable oxidation and coordination states, which are essential in many catalytic reactions. The study of their properties is facilitated by characterisation techniques such as diffraction and spectroscopy including X-ray absorption fine structure (XAFS), electron paramagnetic resonance (EPR), UV-visible, and mid-infrared (MIR) spectroscopy. Computational techniques can also be effectively applied to model structural and mechanistic properties of TMI centres in zeolites.
TMI-zeolites are highly active in a variety of key redox reactions including hydroxylation of benzene4 and methane5 to phenol and methanol, respectively. The use of TMI-containing zeolites for the remediation of NOx from combustion engines has attracted particular attention and is the focus of this review. The selective catalytic reduction (SCR) of NOx to N2 in the presence of ammonia plays an important role in mitigating the emission of NOx from power plants and combustion engines.6–8 According to the California Air Resources Board (CARB) Emission Factors 2021 inventory model, heavy-duty diesel vehicles (HDDVs) were expected to account for about 70% of on-road mobile source NOx emissions in 2021.9 Industrial processes commonly generate flue gases at relatively low temperatures, often around 300 °C. Consequently, SCR catalysts must exhibit activity within a temperature range of 100–300 °C. Whereas the V2O5–WO3(MoO3)/TiO2 (VTW) catalyst is a well-known and efficient choice for NH3-SCR in medium-temperature (100–400 °C) processes,10 it does possess some inherent limitations including a relatively narrow and high working temperature window (functioning optimally within the range of 300–500 °C), and reduced selectivity for converting NOx to N2 in the high-temperature range.11,12 SCR on stationary emissions (i.e. nitric acid plants) often operates at stable temperatures of 300–400 °C and uses a variety of reductants, including H2 and hydrocarbons. With mobile emissions, the catalyst temperature varies with the drive cycle and hence the advantage of NH3-SCR is that it can work at lower temperatures (onset of activity is ∼130–150 °C with metal-containing zeolites) [178]. VTW catalysts show the same sorts of limitations as Cu-zeolites with ammonia combustion being the main problem at high temperatures. The main reason VTW are not more widely used, outside Europe, concerns the limitations with using V5+ containing formulations in the US as it is classified as a carcinogen. Various transition metal-based zeolite catalysts have been extensively explored for the low-temperature NH3-SCR reaction owing to their redox properties, cost-effectiveness, and high hydrothermal stability.13–15
The NH3-SCR reaction either involves oxygen (standard SCR) (eqn (1) or NO2 (Fast SCR) (eqn (2) to generate nitrogen and water from NH3 and NO.16 In addition, there is the NO-activation pathway (eqn (3), where the rate does not depend on the concentration of NO2 in contrast to Fast SCR.17 Several other reactions can occur in this mixture of NOx, NH3, O2, and H2O, including the oxidation of ammonia or the formation of N2O. These side reactions influence the selectivity to N2, and therefore, can affect the overall catalytic performance.
| 4NO + 4NH3 + O2 → 4N2 + 6H2O | (1) |
| NO + 2NH3 + NO2 → 4N2 + 3H2O | (2) |
| 3NO + 2NH3 + O2 → 2N2 + 3H2O + NO2 | (3) |
Following the breakthrough discovery of Cu/ZSM-5 as an effective catalyst for nitrogen oxide (NOx) removal in 1986 by Iwamoto et al.,18 several zeolite-based catalysts have undergone thorough investigation in the field of NH3-SCR. This interest stems, as noted, mainly from their broad operational temperature range (200–600 °C), elevated catalytic efficiency and selectivity towards N2, as well as good hydrothermal stability.19 The stable and ordered framework structure of zeolite catalysts, along with their high specific surface area, not only contributes to their exceptional hydrothermal stability but also facilitates the effective dispersion of active metals. This enhanced dispersion enables the diffusion of both reacting and stable product species, thereby enhancing the overall reactivity. Additionally, the concentration and distribution of active metals in zeolites can be controlled by adjusting the aluminium content (in aluminosilicate zeolites) and matching it with the desired metal loadings, which may be achieved through the electrostatic stabilisation effects,20,21 leading to improved hydrothermal stability and catalytic activity of the catalysts in NH3-SCR.
In this review, we first discuss the industrial importance and applications of these catalysts which we follow by discussing the relevant basics of zeolite science and then describe the key aspects of TMI chemistry: their distribution and location within the pores of the zeolite framework, the possible coordination to the lattice, and especially the formation of amine and metal oxo-complexes. We review in detail the structural complexity of TMI-based-zeolites and their reactivity in activated materials in NH3-SCR. We describe the state-of-the-art characterisation practices employed to study the structural and chemical identity of metals in the zeolite framework. We then focus on the role of TMIs in the NH3-SCR reaction, where we provide a summary of the mobility and restructuring of TMI-active sites and their ultimate effect on the NH3-SCR reactivity. We discuss the chemistry of the intermediate species and their formation. Finally, we briefly consider the stability of TMI-zeolites, in particular the ageing impact of materials in NH3-SCR activity, and the potentially interesting possibilities of designing ideal zeolite-based heterogeneous catalysts for NH3-SCR. We conclude by considering future perspectives.
NH3-SCR process and its industrial importance
SCR technology is primarily employed to reduce NOx emissions, which contribute to air pollution, and smog formation, and have adverse health effects. NH3-SCR, in particular, has gained widespread industrial importance due to its efficiency, versatility, and environmental benefits.22 One of the key motives for the practical significance of the NH3-SCR is its ability to reduce effectively NOx emissions across a wide range of industrial sectors. Power plants, chemical manufacturing plants, refineries, and other industrial facilities are major sources of NOx emissions,23 and more than 1000 SCR plants have been implemented in the United States alone to mitigate the emissions of NOx from industrial boilers, steel mills and process heaters.24 Mobile sources have seen a rapid upsurge in the utilisation of SCR systems in diesel vehicles, owing to the inherent inadequacy of three-way catalysts (TWC) in tackling the reduction of NOx emissions in lean-burning gasoline and diesel engines.25 TWCs, designed for gasoline engines, struggle with the higher NOx emissions characteristic of the higher temperatures encountered in diesel combustion. SCR systems offer a more effective solution by injecting urea into the exhaust stream to convert NOx into harmless nitrogen and water vapour, thus meeting emissions regulations and improving air quality.26Diesel engines are widely used in worldwide road transport owing to their efficiency and durability. European studies show that 40% of atmospheric NOx comes from road transport, with 75% attributed to diesel engines.27,28 Stringent measures since 1993 have reduced NOx emissions by approximately 95% from heavy-duty diesel engines.28 Catalytic materials are now employed to reduce further emissions in line with EU legislation. Upcoming Euro VII standards will require optimising new catalytic technologies, crucial for the automotive industry.29 By implementing NH3-SCR technology, these industries can achieve further reductions in NOx emissions, thereby complying with these new environmental regulations and improving air quality. Furthermore, the NH3-SCR process offers advantages in terms of its efficiency and cost-effectiveness.30,31 The use of a catalytic converter ensures high conversion rates of NOx, even at low temperatures, making it appropriate for a wide range of industrial applications.
The NH3-SCR process also offers flexibility in terms of integration into existing infrastructure without requiring major modifications, making it an attractive option for industries seeking to retrofit their facilities for emissions reduction. For example, it typically requires the addition of a urea dosing unit, the urea decomposing to yield 2 moles of NH3 and one mole of CO2.32 Additionally, the technology can be customised to suit specific industrial needs and accommodate variations in operating conditions, including gas composition and temperature. This adaptability enables the NH3-SCR process to be implemented in several industries.
Zeolites: fundamentals and challenges
Zeolites are crystalline alumino-silicate or silica nanoporous materials with large specific surface areas (typically 100's of m2 g−1); their framework architectures lead to porosity in the form of channels and or cages of molecular dimensions in 1, 2 and 3D. They have adjustable acidity and basicity and are widely used in industrial applications owing to their excellent catalytic activity and product selectivities.33,34 The materials occur naturally as minerals, but there is also an extensive range of synthetic materials.35 Their production, topology, and behaviour raise fascinating basic questions, and their extensive industrial usage has stimulated both the improvement of current zeolitic materials and the exploration of new ones with novel functionalities.More than 200 zeolite topologies have been reported so far. The topology of the channel/cage system is crucially important as are the dimensions of the rings which control access to the cages.36 Different rings are symbolised with the notation n-MR (8-MR, 6-MR, and 4-MRs where n signifies the number of tetrahedral (T) atoms); but zeolites are flexible frameworks and molecules whose size might be considered to preclude their passage through a window may be able to “squeeze” through at higher temperatures. Indeed, the actual window diameter is a dynamic property owing to the breathing motion of the framework.37
A key, basic feature of zeolite chemistry is the isomorphous replacement of Si4+ with Al3+ which has two consequences: first, as the distance between Al–O bonds is longer than Si–O bonds, the tetrahedrally coordinated Al3+ with four lattice oxygen influences the shape and free diameter of the rings within the framework. Second, the incorporation of Al3+ creates a locally negative charge, which must be charge-neutralised by the introduction of cationic species which can include protons, as discussed further below. These cations are exchangeable with the other cations including TMI. The location of these exchanged cations depends on several aspects including the Si/Al ratio, the charge and concentration of the TMI, the preparation method, and the activation conditions.38
Related to zeolites are zeotypes: other classes of nano-porous materials possessing often similar but sometimes unique frameworks of which the most extensive are AlPOs (aluminophosphates) and SAPOs (silicoaluminophosphate) which offer distinctive characteristics and properties.39 MgAlPOs, synthesised using aluminium and phosphorus precursors, exhibit acid-catalytic activity due to the presence of framework magnesium. SAPOs, which incorporate silicon, aluminium, and phosphorus, have the potential for enhanced structural flexibility, allowing for a broader range of pore sizes and shapes depending on their specific composition and synthesis conditions.40,41 The range of nano-porous materials has also in recent decades been greatly extended by the development of nano-porous materials including metal–organic frameworks (MOFs) which have been very extensively studied,42 for example, ZIF-8,43 MOF-74,44 Cu-BDC (HKUST-1),45 but they generally have larger pores than zeolites.
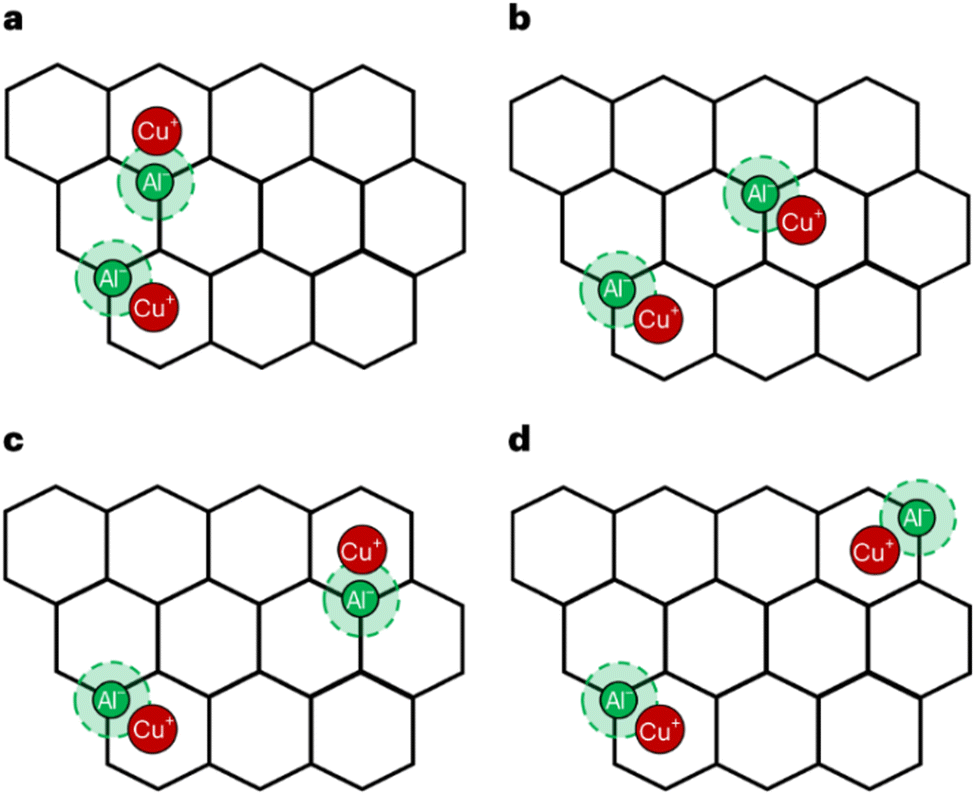
The distribution of Al3+ over tetrahedral sites has been widely studied and debated. Al3+ tetrahedra are not located in direct neighbouring sites in the framework (no Al–O–Al linkage) according to Lowenstein's rule,46,47 which appears to be widely obeyed in zeolites. Computational investigations have shown that small clusters of zeolites have lower energy for “Lowensteinian” aluminium distributions,46 suggesting that the origin of the rule may be in the energetics of the small clusters from which zeolites nucleate. The rule results in an upper limit of isomorphous substitution of Si by Al, of 1. There are further rules to describe the distribution of Al3+: Dempsey's rule proposes that the Al3+ substitution follows, as far as possible, an evenly dispersed Al distribution.48 However, deviations from Dempsey's rule occur in synthetic zeolites.49 The experimental analysis shows that Al3+ substituents may be in close proximity rather than in a dispersed distribution, but Lowenstein's rule still holds50 and the possible aluminium configurations in different ring structures with 5 > Si/Al > 1 for the 4-, 5-, 6-, 8-, and 10-MR of zeolites are shown in Fig. 1.51 However, Fletcher et al.47 predict deviations from Löwenstein's rule in both protonated and sodium-containing forms of the zeolite, SSZ-13 which could lead to new catalytic routes and materials, enhancing existing zeolite catalysts. Additional rules include that of Takaishi and Kato52 which states that within 5-membered ring (5-MR) containing zeolites only one Al3+ substitution is allowed.
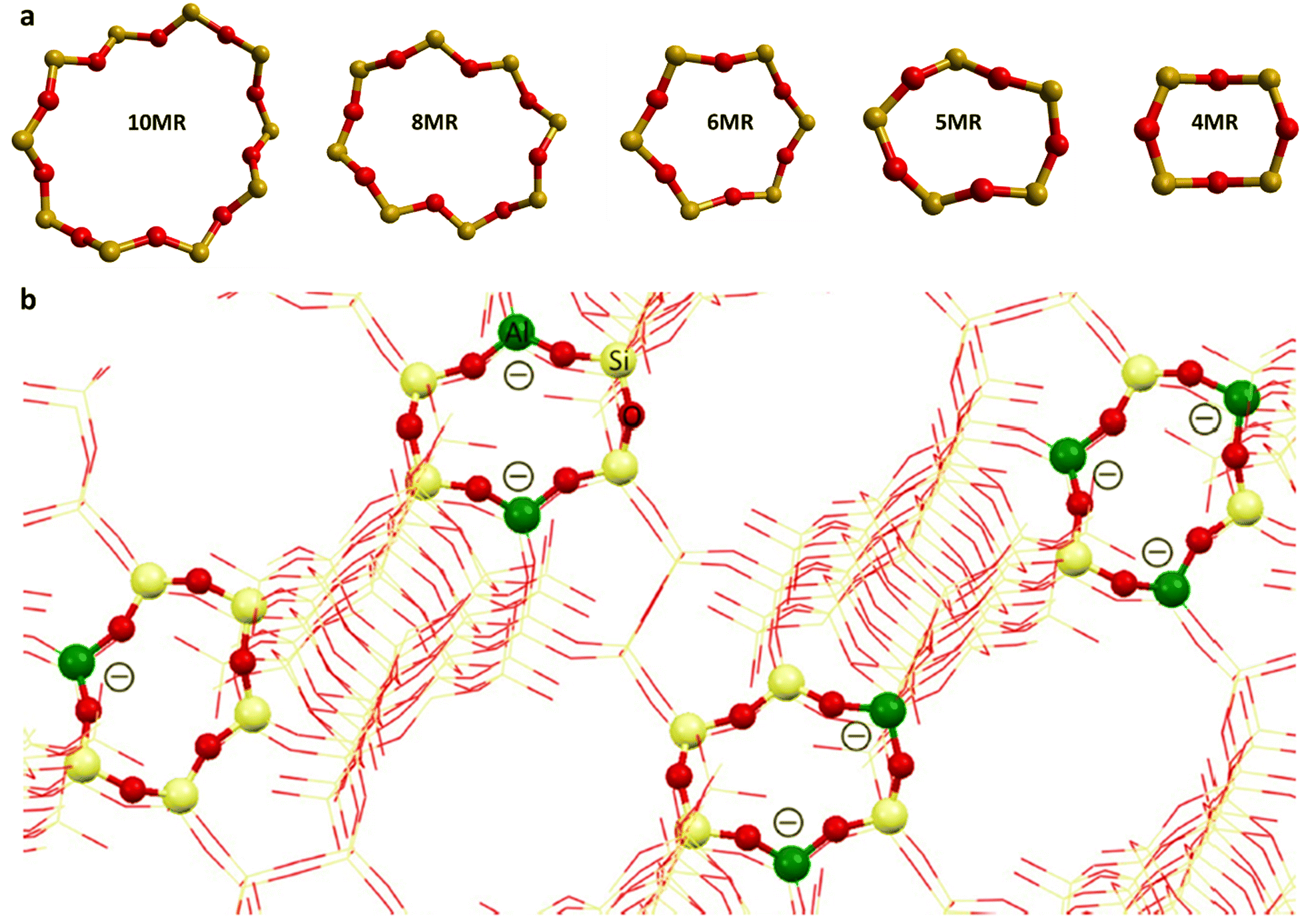 | ||
| Fig. 1 (a) The representative rings (10-MR, 8-MR, 6-MR, 5-MR, and 4-MR) of CHA and ZSM-5 zeolites, and (b) examples of allowed Al distributions in β-6-MR sites, Al distributions in zeolites allow for one, two, or three Al T-sites as per qualitative rules. Colour code: O, red; Si, grey; Al, green. Reproduced from ref. 51 with permission from American Chemical Society, copyright 2018. | ||
Several experimental findings indicate that the Al distribution in zeolites is non-random and does not adhere to straightforward statistical principles.53–55 Daniel et al. determined quantitatively the spatial distribution of Al using atomic-scale analysis (e.g. Atom Probe Tomography).55 They observed a very non-random distribution of Al within highly crystalline zeolites. Upon steaming, the non-random, Al distribution involves clustering. This pronounced clustering appears to occur at the regions near diffusion barriers supposed to separate the zeolite subunits and hence, behave as a route for the transportation of Al towards the outer surface of zeolites. In addition, Li et al.56 reported a steady transition of Al towards the crystal surface during crystallization. Sometimes, Si enrichment of the crystal surface is also detected.57 The incorporation of high-aluminium-containing precursor species to existing crystals, especially when occurring in the late synthesis stages, leads to an uneven Al distribution within zeolite ZSM-5 crystals, observed using focused ion beam (FIB), energy dispersive X-ray spectroscopy (EDX), and scanning transmission electron microscopy (STEM).56 FIB milling enabled the analysis of precise middle sections of crystals, facilitating the quantification of Si/Al ratios in different crystal regions.
The siting and distribution of Al are also closely coupled with the location of charge compensating cations. The Si/Al ratio and the distribution of Al in the zeolite framework are key factors in influencing catalytic reactions, and the nature and siting of the charge-compensating cation have a major impact on zeolite catalytic activity. Zeolites as a host facilitate high levels of cation dispersion across multiple sites and environments. Differences in coordination surrounding the cations in extra framework sites may result in cation-zeolite combinations with distinctive acid–base characteristics and can, therefore, affect redox chemistry.58,59 The Si/Al ratio and the Al location can influence the nuclearity of the TMI species and in some circumstances, it has been observed that binuclear species similar to those seen in enzymes i.e. methane monooxygenase (MMO)60 have been identified.
Brønsted acid sites are created when protons balance the negative charge in aluminosilicate zeolites,61 with the protons binding to the lone pairs of the bridging oxygen atoms. The phenomenon of Brønsted acidity is normally used to describe both acid density and acid strength. While acid density is relatively straightforward, acid strength needs more extensive discussion.62 Intrinsic acid strength is determined by the proton affinity at acid sites, which is affected by charge dispersion within the zeolite framework and correlates with the local electric field. The Brønsted acid sites of different zeolites have significantly different strengths63 which are mainly linked to the ring structures that make up the zeolite rather than the bulk topology. In addition, the acid strength shows a dependency also on the location of the Al3+ in particular topologies.64 Both Brønsted acid sites and TMI play a significant role in SCR catalysis. TMI exhibit the necessary redox functionality and the Brønsted sites seem to be important in terms of NH3 storage/activation.65,66
Lewis acid Al3+ in zeolites is more versatile,67 but understanding Lewis acid structures remains uncertain due to diverse potential sites arising from aluminium's structural versatility68,69 with Al being identified with 3, 4, 5 and 6 coordination.70 Lewis acidic aluminium can be produced via post-synthesis procedures including steaming and calcination, forming extra-framework aluminium species71,72 which could be considered as a catalytic site for NH3-SCR reaction as reported by Lezcano-Gonzalez et al.73 Furthermore, Lewis acidic Al3+ can also be an inherent characteristic of the zeolite framework, and there are therefore, several proposals for Lewis acid site structures, originating from the framework, framework-associated, and extra-framework aluminium,74–76 and identification of Lewis acid structures and properties will remain challenging.77,78
The influence of the Al distribution within zeolite frameworks has a significant effect on the structure, properties, and catalytic activity of TMIs e.g., Cu, Fe, and Co ion species.79,80 The local negative framework charge, determined by isolated Al substitutions or pairs, critically influences the structure and activity of metal ion species. Dědeček et al.81 investigated how Al, either paired as Al–O–(Si–O)2–Al sequences within a single ring or isolated as Al–O–(Si–O)n>2–Al sequences across separate rings, affect the performance of metal ion species in SCR-NOx. They selected BEA, MFI, and FER structures, each with a similar overall composition but different Al distributions. The concentrations of Al pairs and isolated Al atoms were determined using 29Si MAS NMR and visible spectra analysis of dehydrated Co–zeolites with maximally exchanged Co2+ ions. The structure and reactivity of the metal sites were characterised using UV-Vis spectroscopy, FTIR, and TPR-H2. Co2+, Cu2+, and Fe2+ ions are stabilised by Al pairs, while Co-oxo and Cu-oxo or Cu+ species are associated with isolated Al atoms. Cu+ ions near isolated Al atoms in Cu-MFI zeolites show the highest activity in NO decomposition. Krishna et al.82 studied the influence of Al on NH3-SCR of NOx activity and showed that higher Al densities enhance the mobility and reactivity of Cu ions, crucial for the formation of active binuclear Cu2+ complexes necessary for NOx reduction. As the Al density increases, the rate of Cu+ oxidation rises significantly, improving SCR performance, particularly in low-oxygen conditions. However, Cu2+ reduction rates exhibit a non-linear relationship with Al density, peaking at intermediate levels, suggesting a balance between active site availability and potential inhibition by co-cations like NH4+. These findings highlight the importance of optimizing the Al distribution in CHA zeolites to maximize NOx conversion efficiency. More details on zeolites in deNOx catalysis including Al distribution effects on metal ion catalysis in zeolites, can be found in other reviews.12,13,83,84
Following this introductory account of zeolite's structural and chemical properties, in the next section, we will focus on the NH3-SCR of NOx reaction on metal-exchanged zeolite.
NH3-SCR of NOx in zeolites
In the “NH3-SCR” reaction, NOx gases, (mainly NO) are treated in the presence of O2 and NH3 to generate N2 and H2O.85 The primary exhaust gases emitted from industrial and diesel engines consist predominantly of NO (>90%), instead of NO2, hence the reaction of NH3-SCR is generally recognized as the “Standard SCR”.86 As noted earlier, in the 1980s, Iwamoto et al.87 showed that Cu-exchanged zeolites had high activity and stability for deNOx catalysis.84 Since then, zeolites have been widely investigated for this key catalytic reaction.88,89 The term de-NOx is often used for the remediation of both NO and NOx, present in exhaust gases. Considering the availability and activity, a small number of TMIs (Cu, Fe, Ag,) have been developed as viable catalysts for NH3-SCR unlike other TMIs (Mn, Cr, Co, Zn, Ni, La).90–98 Ion-exchanged zeolites including Fe or Cu-exchanged ZSM-5,99 SSZ-13,100 SAPO-34,101 FAU,102 Beta,103 USY,104 and MOR,105 offer the potential for NH3-SCR applications with broad temperature windows (150–450 °C) and unique active sites. However, challenges remain, including hydrothermal stability, SO2 tolerance, and hydrocarbon coking.13,106 It is important to note that the Si/Al for NH3-SCR catalysts is generally >15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) since this represents a balance in terms of enabling a sufficient number of metal active sites and hydrothermal stability.85,107
since this represents a balance in terms of enabling a sufficient number of metal active sites and hydrothermal stability.85,107
Despite the efficacy of TMI-exchanged zeolites, for stationary NH3-SCR, the preferred catalyst is still vanadium oxide-based catalysts owing to their low cost, lower oxidation activity for the conversion of SO2 to SO3, and excellent thermal stability.108 However, mobile applications have highly dynamic reaction conditions, with the gas flow rate of the lean-burned exhaust gases, and varying temperature demands. They, therefore, require catalysts that will not only be able to show high activity at low temperatures (<300 °C) but also have better hydrothermal stability at high temperatures (>500–600 °C).86,109 Several zeolites including mordenite (MOR), zeolite Y (FAU), ZSM-5 (MFI), and BETA (BEA) have received significant interest owing to their applications in catalysis and stability under different temperature conditions.
Some important and widely studied zeolite structures in NH3-SCR of NOx are shown in Fig. 2. Medium-pore size zeolites including ZSM-5 and mordenite were thought to be more active than larger pore sizes (e.g. BETA, USY, and Y); and, in recent years most focus has been on smaller pore size zeolites mainly with the CHA topology.28,110 Although many other TMIs, as mentioned earlier, have also been explored, to date, Cu and Fe have been the main focus of research.
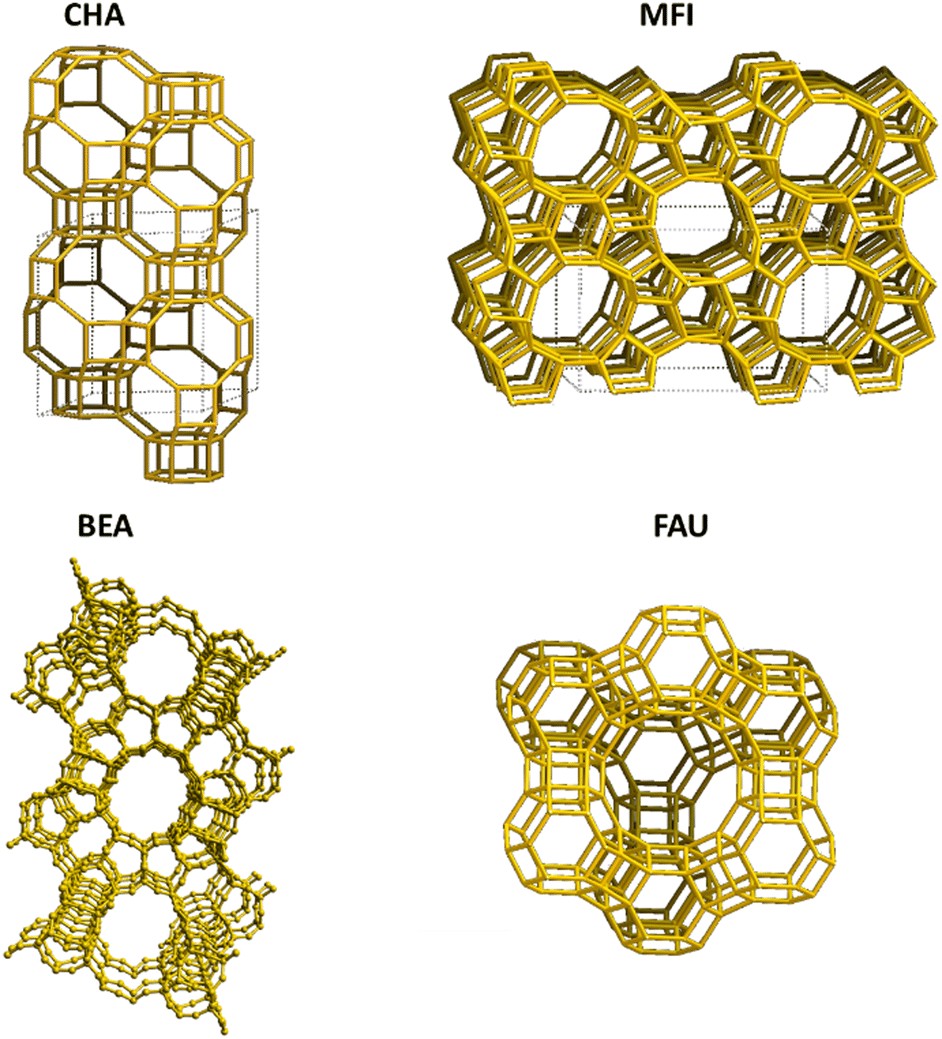 | ||
| Fig. 2 The important zeolite framework structures used in NH3-SCR reaction. | ||
Several studies111,112 consistently demonstrate that zeolites containing Cu or Fe display enhanced efficiency in NH3-SCR compared to catalysts based on other transition metals. This superiority stems from the effective activation of NO over a wide temperature spectrum. The Cu-exchanged zeolites are optimized for SCR within the range of 150–300 °C. Beyond 300 °C, undesirable side reactions such as ammonia oxidation become pronounced over Cu-based zeolites, leading to a decline in NOx conversion.113,114 In contrast, zeolites containing Fe require higher reaction temperatures.
Several highly effective catalysts based on Cu or Fe-exchanged zeolites for NH3-SCR have been recognized including Cu/SSZ-13, Cu/LTA, Fe/Beta, Cu/SAPO-34, and Fe/SSZ-13, which, with fine-tuning of metal content and zeolite carrier types, demonstrate promising industrial applications; and as noted, the small pore Cu/SSZ-13 (SSZ indicating a small-pore, siliceous framework zeolites), which has been effectively deployed in the exhaust deNOx of diesel vehicles.110,115 The zeolite, SSZ-13 (CHA) is an exception because of its unique structure and relatively small pore size (0.37 × 0.42 nm).116 NH3-SCR is thought to be effective in SSZ-13 firstly because of the metal speciation, secondly, as the small pore restricts poisons such as carbon-based molecules and thirdly, the small pore zeolites have high hydrothermal stability. In addition, its catalytic efficacy is assisted by the presence of double 6-MRs serving as preferred Cu sites and the inherent stability of its framework structure.117–119 Indeed, Cu-zeolites were found to provide the highest performance at lower temperatures (i.e., <200 °C) whereas Fe exhibited the maximum activity above ∼350 °C.120
In MFI the sites which contribute to the catalytic reactions are the 10-MR rings with a diameter of ∼0.55 nm (e.g. in CHA it is 0.38 nm) where the TMI is located either in straight or zigzag channels so that a smaller pore size makes it difficult for the reacting molecules to access the active sites. Noting the importance of the pore size, the same approach can be made for other zeolites. The advantage of larger pore sizes is that they not only allow rapid diffusion of reactants to gain access to the active sites but also make the desorption process more favourable.61 On the other hand, larger pore zeolites allow a greater number of poisons to enter the pores. Moreover, it is often considered that zeolites, by confinement effects (transition state/product shape), can positively influence product distribution, which will be less effective in large pore zeolites.
Several theoretical and experimental investigations have been performed to elucidate the Standard SCR mechanism. These mechanisms are increasingly well understood, with both mononuclear and dynamic binuclear Cu species identified as the primary active sites.111,121 NH3-bonded to the zeolite was investigated by Giordanino et al.122 using NH3-temperature programmed desorption (TPD). They identified three types of adsorbed ammonia species, which can participate in SCR reactions. At low temperatures (T < 400 °C), NH3 linked to the Cu-site appears to be more stable, whereas, at temperature (T > 400 °C), NH3 preferentially bonded to Brønsted sites. In addition, NH3 can bind with ammonium ions giving rise to NH4+·nNH3 associations. Lezcano-Gonzalez et al.73 identified a fourth species, namely ammonia species coordinating to extra-framework Al and that these played a positive role in promoting NOx reduction.
High-temperature SCR activity is an important aspect, particularly owing to the significant heating of NOx emissions during engine acceleration. Above 450 °C, a decrease in NOx conversion is noted on the Cu-SSZ-13 catalyst, ascribed to the strong oxidizing nature of the Cu species, which facilitates the oxidation of NH3 by oxygen.123–125 To address this issue, various transition metal-modified Cu zeolite catalysts have been widely investigated, including Fe–Cu/SZM-5,124 CeOx–Cu-SAPO-34,126 MnCe/Cu-SSZ-13,127 which demonstrated excellent high-temperature SCR catalytic activity, providing potential solutions to enhance the performance and overcome the limitations of Cu-SSZ-13 catalysts in high-temperature conditions. On the other hand, cold start conditions pose challenges for catalytic converters, where lower temperatures hinder catalyst performance until optimal operating temperatures are reached.
The concentration of NO2 present in the gas mixture can affect the performance of Fe-ZSM-5, and therefore, oxidation of NO to NO2 is considered the rate-limiting step in NH3-SCR.120,128,129 It is also found that Fe-based catalysts are more prone to the NH3 inhibition effect.130,131 The experimental data strongly indicate that an excess of NH3, whether in the gas phase or on the catalyst surface, inhibits NO conversion at low temperatures,132 which suggests an optimal NH3 surface concentration for effective conversion. Further comparison between Cu and Fe zeolites showed that Cu is less sensitive to NO2133,134 and is, therefore, under standard SCR conditions expected to exhibit better activity than Fe at a lower temperature. Recently, the investigation of Fe and Cu has moved one step further combining both metal ions, using monoliths made up of different sequential compositions.135
For, Fe-exchanged zeolites, the Fe in pentasil ring zeolites is notable for its stability for NOx conversion in the presence of a high content of water.136 Fe-ZSM-5 is also active in the SCR of NOx with olefins137 or paraffins (C3–C4)138 but in the presence of methane, it is not so efficient.135 Kucherov et al. also tested the Fe-Beta catalyst with the introduction of different Cu loadings128,139 to test their impact on the catalytic activity of NOx reduction. Catalysts with Cu content above 1.25 wt% showed decreased high-temperature activity, indicating the critical role of Cu loading.
Even though, both Cu and Fe are widely investigated systems for NH3-SCR, other TM metals including Ce and Mn have also been investigated owing to their redox properties.140–144 Ce-modified Cu/SSZ-13 increases the reducibility of the catalyst and increases the NH3 and NOx adsorption capacity, which in turn improves the NH3-SCR activity.145 Furthermore, the presence of Ce promotes the formation of nitrate species, which play an important role as prominent intermediates at low temperatures.145,146 Ce doping has been found to boost the stability of active Cu ions and avoid the dealumination of the framework during hydrothermal ageing which is a promising strategy to enhance the performance and stability of Cu-SSZ-13 catalysts for NH3-SCR applications.145
Mn-doped zeolites have displayed excellent activity as explored by Nam et al.143,144 They developed Mn(20 wt%)/ZSM-5 and Mn(20 wt%)–Fe(10 wt%)/ZSM-5, and tested them for the NH3-SCR de-NOx reaction. The presence of 10 wt% of Fe boosts the catalytic activity impressively and surpasses the mono counterpart Fe(10)/ZSM-5 and Mn(20)-/ZSM-5 under low-temperature treatment (below 250 °C). Furthermore, it achieved 80% conversion of NO into N2 at 160 °C while the Cu/ZSM-5 and Cu/zeolites-based commercial catalysts convert only 40% and 30% at 160 °C (NO/NH3 = 500/500 ppm; O2 = 5 vol%). Although at high-temperature treatment (above 350 °C) the deNOx ability of Mn-doped zeolite was comparatively lower than that of Cu and Co-doped zeolites, the increased content of Mn enhances the hydrothermal stability.147
In addition, the N2O decomposition over TMI-exchanged zeolites has been also widely studied.148–151 Like NO, N2O is also an environmentally undesired product in NH3-SCR, and its conversion is a possible route for the generation of N2. The decomposition of N2O on a single Fe site and binuclear O bridged Fe site both in the presence and absence of NO and water has been investigated by Heyden et al.152 Their findings indicate that NO-assisted N2O decomposition occurs more favourably when compared to NH3-SCR owing to the absence of H2O poisoning effect on the active sites, as opposed to NO. In line with this finding, Bruggemann et al.149 deemed it a crucial aspect of the SCR pathways in exhaust streams abundant in NO2. It is interesting that N2O is sometimes observed as a by-product of the SCR reaction and that there has been some investigation into the decomposition of this using zeolite-based catalysts.153–155 Often though N2O decomposition is performed in the absence of ammonia and oxygen.
Detailed characterisation of TMIs is essential for an understanding of zeolite-based catalytic processes. Next, we will discuss the mode of attachment and coordination of TMI in zeolites. We will focus on the TMI distribution and location within the zeolite framework and their role in de-NOx catalysis. We then turn to the key aspect of the activation of O2 and oxo complexes of TMI in zeolites and their role in NH3-SCR catalysis.
TMI location, speciation and coordination state
The location and distribution of TMIs in zeolite are crucial as they directly impact the zeolite's catalytic abilities, influencing its overall reactivity and performance in NH3-SCR of NOx reaction. The role of TMI speciation, in particular, their formation and transformation in the zeolite framework, the activation procedure, and initial chemical composition are of particular interest.156–158Cu-based zeolitic systems
Early diffraction data from the mineral chabazite identified only one cationic ion-exchange site in the CHA topology although for Cu-SSZ-13 X-ray powder diffraction data (XRPD) has shown that there are two crystallographically distinct sites in the 6-MR and the 8-MR that Cu2+ occupies when CHA is dehydrated and measurements are made at room temperature.159 When fully hydrated (i.e. forming a hexa-aqua complex) this amalgamates to one site in the centre of the cage.159,160 It has been proposed that Cu in both sites after dehydration is off-centre and in the 8-MR is hydroxylated (Cu–OH) as determined by IR spectroscopy and computational methods whilst the location of Cu2+ in the 6-MR and its properties show a dependency on whether they are close to 1 or 2 Al3+ species.161 Operando spectroscopy and scattering data have shown that Cu is typically in the 2+ oxidation state at temperatures above 200 °C.119 A similar finding was reported by Uzunova et al.61 who performed extensive calculations using DFT with periodic models.The proposal for isolated Cu2+ species in Cu-CHA during NH3-SCR originated from the work of Fickel et al.162 and Korhonen et al.163 who proposed a formation of single Cu2+ ions within the plane of the 6-MR of Cu-SSZ-13 during dehydration using XRD & X-ray absorption (XAS) spectroscopy. Work followed by Deka et al.164 using XAFS/XRD showed that mononuclear Cu2+ ions are seen under operando NH3-SCR conditions, largely reside in the plane of the 6-MR of SSZ-13 at temperatures above 473 K. They also performed PXRD analysis on Cu-SAPO-34 and correlated the excellent SCR activity to the active sites of isolated Cu2+ species located in the 6-MR of Cu-SAPO-34.165 Utilizing XAFS/XRD, the study provides insights into the local copper environment under operando NH3-SCR conditions. Confirming the presence of mononuclear Cu2+ species in Cu-SSZ-13, the study emphasizes the significance of the local copper environment for the catalyst's catalytic activity. Recent studies suggested that CHA possesses several active sites for both Cu+ and Cu2+ ions as discussed before.85,119,156,166
McEwen and coworkers167 examined Cu-SSZ-13 and Cu-SAPO-34 performing periodic DFT calculations confirming that it is the 6-MR of CHA that hosts the isolated Cu+ species. This finding is similar to that reported in ref. 168. In addition, Göltl et al.169 introduced a new theoretical method that integrates DFT calculations and statistical approaches to predict the placement of Cu2+ ions within Cu-CHA, adjusting the exchange level of Cu. Furthermore, they show the allocation of the cations amongst the various active sites for a range of exchange levels (ELs) or proportion of charge balancing cations. EL represents the proportion of occupied Cu2+ sites in the catalyst. At an EL of 1.0, there is a single Cu ion for every two Al ions. When the EL is set at 0.2, configurations (a), (b), and (f) depicted in Fig. 3a will be filled. At this low EL, configurations (a) and (b) are already fully occupied, while configuration (f) rises with increasing EL values. Site (c) is only occupied just before reaching full exchange. Fig. 3b illustrates the graphical representation of active site distribution across EL values ranging from 0.2 to 1.0. By integrating this information with experimental findings provided by Kwak et al.,170 it becomes possible to discern configurations (a) and/or (b) as the active sites involved in the SCR of NOx reduction.
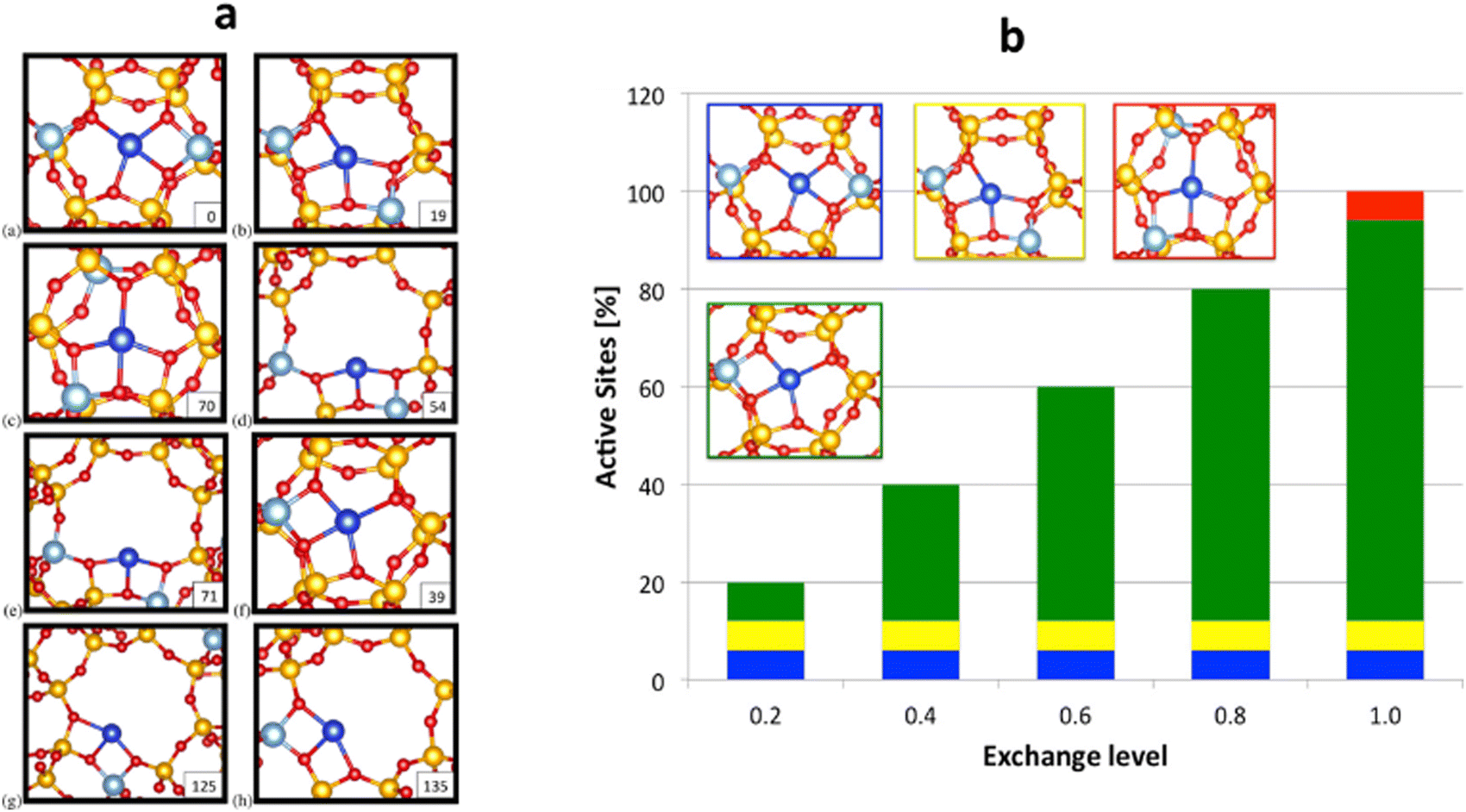 | ||
| Fig. 3 (a) shows eight distinct cation locations for Cu2+ in SSZ-13. Each Cu cation, along with a proton, must be linked with two Al ions. For example, for sites (a)–(e), both Al ions are situated within the same unit cell, whereas sites (f)–(h) represent scenarios with significant Al separation. Si is depicted in yellow, Al in silver, O in red, and cations in blue. The stability energy (ΔECu(II)exc [kJ mol−1]) of these sites is presented in the lower right-hand corner, and (b) displays the distribution of metal ions within Cu2+-SSZ-13 at various ELs, as determined by total energy calculations. (a) (blue), (b) (yellow), and (f) (green) are the most stable sites occupied at an EL of 0.2. As EL increases, the occupancy of site (f) also increases, and site (c) is only occupied at the highest EL. These findings are then compared with experimental data (see ref. 170) showing the stable sites whereas (a) and (b) are the active centres. Reproduced from ref. 169 with permission from American Chemical Society, copyright 2013. | ||
Low-temperature (below 250 °C) studies claimed Cu dimer formation in Cu-SSZ-13.85,111,171,172 For example, through a combination of several experimental modelling techniques such as XAS, steady-state and transient kinetic measurements, and first-principles calculations through DFT and ab initio molecular dynamics (AIMD), Paolucci and coworkers85 have found that at low-temperature, mobilized Cu+ ions can move through zeolite 'windows' and generate transient ion pairs. These ion pairs are actively involved in an oxygen (O2)-mediated Cu+ → Cu2+ redox step crucial to the SCR process. The emergence of these dynamic multinuclear sites, originating from mobilized single atoms, is a distinct and important feature of NH3-SCR zeolite catalysis.
Lomachenko et al.,173 studied the dynamic behaviour of the Cu-SSZ-13 catalyst in NH3-assisted SCR of NOx across different temperatures. They employed advanced operando X-ray spectroscopies, including XAS and X-ray emission spectroscopy (XES). The investigation uncovered two distinctive regimes dictating the atomic-scale conduct of Cu active sites. At lower temperatures, up to around 200 °C, a balanced distribution of both Cu+ and Cu2+ active sites is observed, with mobile ammonia-solvated Cu-species predominating. However, with an increase in temperature, specifically from 250 °C onwards, there is a notable shift, showing the prevalence of framework-coordinated Cu2+ sites. These findings suggest that the framework-coordinated Cu2+ sites probably represent the active sites for high-temperature SCR which helps to provide a detailed overview of the temperature-dependent evolution of Cu active sites within the Cu-CHA zeolite catalyst.
Izquierdo et al.174 and Morpurgo and coworkers175 employed DFT- our own N-layered integrated molecular orbital and molecular mechanics (ONIOM) and cluster methods to calculate the isolated and the dimer species of Cu in Cu-ZSM-5. Their study showed that both species can catalyse NO decomposition but the process has a higher activation energy for a dimeric Cu+ species.176
The nuclearity of ion-exchanged Cu is influenced by the structural and compositional characteristics of the zeolite host. Paolucci and coworkers demonstrate differences in Cu species near 1 or 2 Al in SSZ-13's 6-MR and the significance of binuclear species as active sites.177 The nature of these binuclear species, specifically whether they are bridged by one or two oxygen ions as shown in Fig. 4, is a crucial aspect influencing their catalytic behaviour and electronic structure. Understanding this configuration aids in enhancing heterogeneous catalyst design and elucidating catalytic mechanisms.
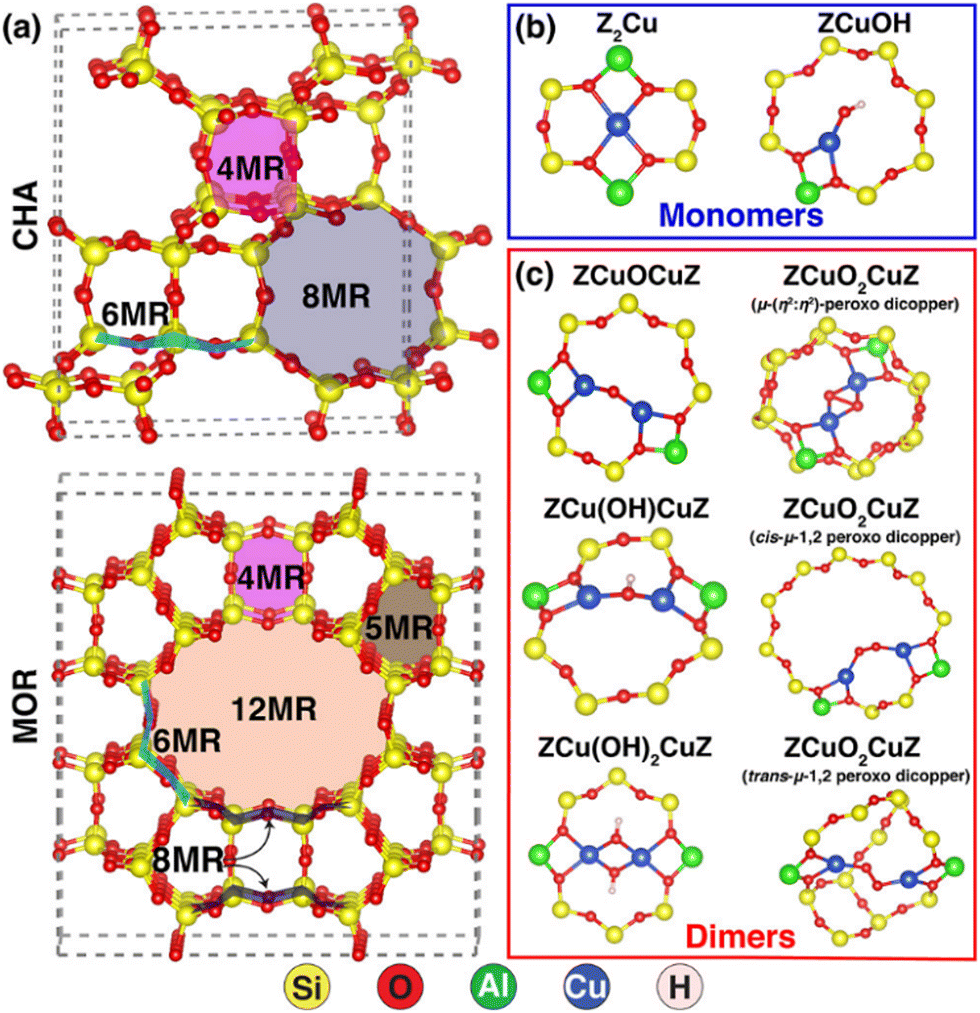 | ||
| Fig. 4 Depicts various ring structures and additional framework Cu ions in zeolites, showcasing the CHA framework (top) and the MOR framework (bottom), along with Cu-monomer species and Cu-dimer species. Reproduced from ref. 177 with permission from American Chemical Society, copyright 2024. | ||
Pierloot et al.178 investigated the local environment of Cu2+ in zeolites Y (FAU), A (LTA), and ZK-4 (LTA) and provided insight into coordination environments in 6-MR. They showed that Cu is coordinated with the four framework oxygen ions. For the low Si/Al ratio, the distance of Cu to the four neighbouring oxygen ions was reported as 1.9, 2, and 2.2 Å for the first two, third, and fourth respectively. However, with increasing Si/Al ratio the interactions of Cu with neighbouring oxygen also changed with three oxygen ions at ∼1.95–2.0 Å and with the fourth one at ∼2.4 Å. Their calculations were based on geometry optimisations on non-rigid frameworks, and in experimental studies using EXAFS, three and not four Cu–O bonds are seen. The fourth oxygen is likely to be closer to 2.8 Å, where the Al/Si contributions lie. A single-crystal study conducted on Cu–Y also supported this observation showing that Cu (at site II, see Fig. 5) coordinated with three oxygens at 1.85–2.0 Å and the fourth one at 2.5 Å.179 The resulting geometry of Cu, in this case, corresponds to the distorted square-planar Cu2+ located at one side of the 6-MR. In FAU, the TMI preferably resides in the hexagonal prisms after dehydration. At higher dosages, TMI occupies the most accessible sites (sites II and III) as shown in Fig. 5. In this case, sites I and II are important as both belong to 6-MR. The nonequivalent oxygen atoms are labeled as 1 through 4. This study does not provide evidence for any specific ordering of silicon and aluminum ions at the tetrahedral sites, though it is anticipated that Loewenstein's46,47 and Dempsey's48 rules would still apply.
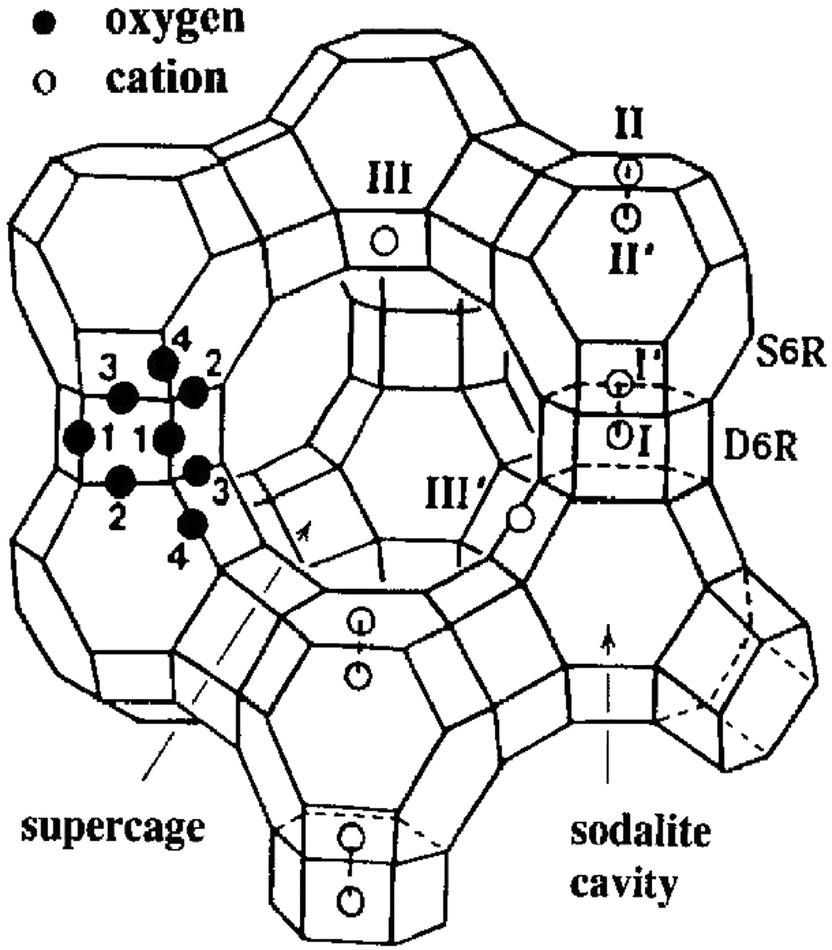 | ||
| Fig. 5 Stylized drawing of the framework structure of zeolite Y. Near the centre of each line segment is an oxygen atom. Extraframework cation positions are labelled with Roman numerals. Reproduced from ref. 179 with permission from American Chemical Society, copyright 2012. | ||
UV/Vis., and IR spectroscopies can be employed to gain further insight into the structural features of cations, hydroxyl groups and other extra framework species under operando or in situ conditions.180 The presence of isolated Cu2+ species in Cu-SSZ-13 during NH3-SCR has been demonstrated by Korhonen and coworkers employing the UV/Vis technique, alongside XAFS.163 Mid-IR vibrational frequencies to detect adsorbed molecules (e.g. CO, NO, and NH3) are used to confirm the chemical environment of cation sites in zeolites.181–183 Kwak et al.184 utilized H2-temperature-programmed reduction (TPR) and FTIR techniques to characterize the Cu ion composition within Cu-CHA across various ion exchange levels (Fig. 6a). They collected findings from both H2-TPR and FT-IR data to determine the position of Cu2+ ions under different ion ELs; their data suggest that Cu2+ ions located in 6-MR of CHA at the low exchange level and occupy the large cage when the level of exchange increases. Further, DRIFTS is extensively used to explore the reaction mechanism.185,186 Wang et al.187 performed DRIFTS and showed that under low-temperature NH3-SCR NH4NO3 intermediates are first generated followed by their subsequent reduction by NO.
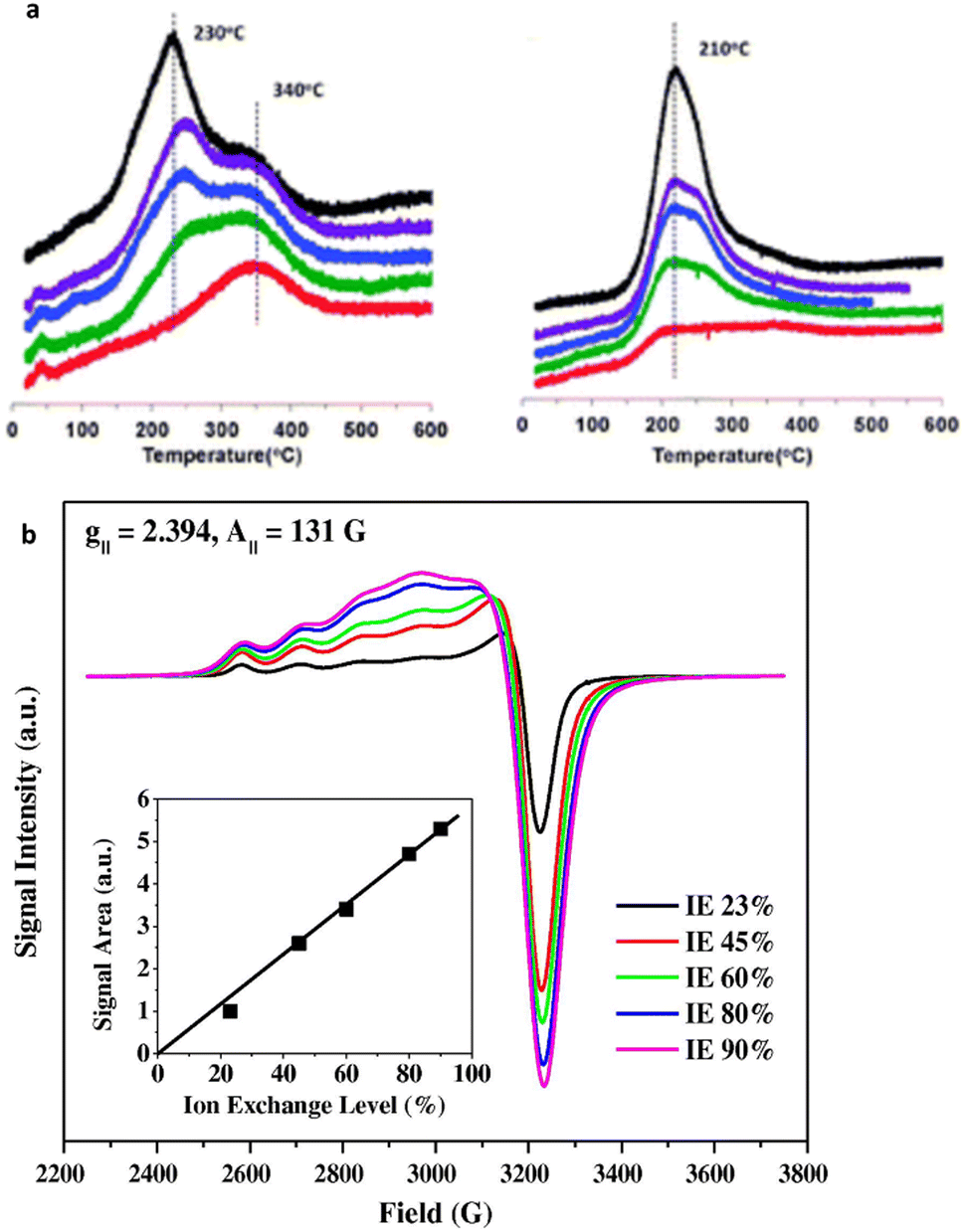 | ||
| Fig. 6 Characterization techniques for the isolated Cu2+ active sites (a) the consumption profiles of hydrogen (H2) during hydrogen temperature-programmed reduction (H2-TPR) on Cu-SSZ-13 are depicted. Reproduced from ref. 184 with permission from The Royal Society of Chemistry, copyright 2012, and (a) hydrated Cu-SSZ-13 EPR spectra at 155 K. The colours indicating different Cu ion exchange levels are as follows: red (20%), green (40%), blue (60%), purple (80%), and black (100%), arranged from bottom to top. Reproduced from ref. 188 with permission from The Royal Society of Chemistry, copyright 2012. | ||
EPR is a useful technique for investigating the cation environment and can determine the concentration, location, oxidation state, symmetry, and coordination number of TMI.180,189,190 Gao et al. reported a detailed study quantifying the isolated Cu2+ ions employing the EPR technique.189,191 EPR also offers insights into TMI magnetic properties and catalytic potential. The Cu2+ ion ESR exhibits an axially symmetrical and split signal characterized by parameters of the parallel component, where gII = 2.33 and AII = 140—160 G.103,192 It is evident from Fig. 6b that increasing Cu loadings results in increasing signal intensity alongside the broadening of the EPR features. This broadening is attributed to distance-dependent dipole–dipole interactions among Cu2+ ions. The line width observed in the IE 23% sample closely resembles that of a sample with low Cu loading (approximately 2%), where dipolar interactions are negligible, suggesting that even within the IE 23% sample, Cu–Cu distances are sufficiently large to weaken dipolar interactions, thereby preventing significant line broadening. Following the in situ EPR analysis, Yu et al.193 identified the chemical state of Cu species under the SCR operating conditions using Cu-SAPO-34 as a catalyst. They showed using EPR that during NH3-SCR catalysis, Cu2+ ions are converted to Cu+ when exposed to NH3, and then re-oxidised to Cu2+ when the adsorbed NH3 is consumed by the purging NOx environment. TPD and TPR are also helpful techniques. For example, NH3-TPD can determine the poisoning of acidity by metals including Pt, Zn, and the effect of hydrothermal ageing on acidity.186,194
Previous findings have demonstrated that NH3-SCR involves the reduction of Cu2+ to Cu+.111,180,195 Subsequently, Cu+ is re-oxidised by O2 (or NO + O2) to regenerate Cu2+ and complete the cycle. If a catalyst containing a transition metal, apart from the widely recognized Fe- and Cu-based catalysts, demonstrates similar characteristics, it could be assessed as a possible alternative catalyst for NH3-SCR.90,196,197 The existence of multiple sites of Cu may arise from the location of Cu-species depending strongly on the physio-chemical environment, the composition of the sample, and the Si/Al or Cu/Al ratio in the framework.
We now discuss active sites and metal speciation to understand the detailed chemistry of the TMI in zeolite under SCR conditions. Some studies have reported that there are at least two types of Cu species in CHA zeolites, and the presence of such species within the cage depends on the Cu loading; some reports suggested that the Cu2+/Cu+ occurs mainly at lower loading (i.e. 0.5–1 wt%), whereas CuO occupies the CHA framework at higher loading (i.e. 2–3 wt%),185,198,199 Cu speciation in Cu-CHA depends on temperature, gas composition, catalyst activation, and composition. Therefore, the structure, coordination, and positions of active Cu sites in SCR catalysts may differ in different catalytic reaction conditions and are likely to change during SCR of NOx reaction.
The activation of zeolites is a key step for both sorption and catalysis because it is necessary to evacuate water and other molecules to make active sites accessible for reactant species. Residual H2O or hydrocarbons (due to incomplete activation) may block the active sites or modify their activity.200 In ion-exchanged zeolites under aqueous conditions, copper is present as a hydrated Cu2+ species. However, upon dehydration, hydrated Cu2+ species undergo progressive reduction to Cu+ species under an inert atmosphere and at increasingly elevated temperatures.157
Several important species generated during SCR reaction play significant roles in defining the reaction pathway. For example, the activation of NO in the presence of oxygen results in the formation of nitrate intermediates which have been detected and characterized using various analytical techniques, including XAS, EPR, and FTIR;168,187 and surface bidentate nitrates have been suggested to play a crucial role in Fast SCR.201 The subsequent reduction of these nitrates by NO to form nitrites is considered the rate-limiting step, whereas the reoxidation of nitrates is fast, even at very low temperatures. However, NH3 acts as an inhibitor in this redox step, probably by obstructing the surface nitrates.202,203
In computational modelling work, discussed in more detail later, we observed that nitrate species tend to be more stable on physisorbed ammonia/water-occupied sites than on bare sites. This suggests that solvents such as ammonia and water could potentially enhance the re-oxidation phase of the NH3-SCR cycle.195,204 This phenomenon is more pronounced in the presence of H2O compared to NH3. In addition, there have been limited studies indicating that NO2 can improve the efficiency of NH3-SCR over Cu-CHA catalytic systems. In contrast, NO2 has been also found to inhibit NO conversion over aluminium-rich Cu-SSZ-13 catalysts owing to the production of NH4NO3, a phenomenon known as the “abnormal fast NH3-SCR reaction”.205 Additionally, previous research demonstrated the reaction between NO and NH4NO3 at Brønsted acid sites over the H-SSZ-13 catalyst.206
Gao et al. addressed the impact of Si/Al ratios on the redox behaviour of Cu-SSZ-13 catalysts and for this reason, they collected the H2 TPR data for a set of samples.107 They analysed the redox mediation of Cu-ions, which can be systematically tuned by changing the composition of the materials. The results showed that an increase in the Si/Al ratio leads to alterations in multiple Cu species, influencing the Cu redox barriers, which in turn, enhances the reaction rates for NO oxidation. Intriguingly, Brønsted acidity was found to be insignificant in influencing this reaction. Moreover, the work demonstrated that residual Brønsted acidity positively influences both low and high-temperature conditions in typical ammonia-assisted SCR. Nevertheless, regarding NH3 oxidation, no definitive pattern emerged, indicating a complex interaction among the Cu ion centre, redox properties, and Brønsted acidity.
The presence of divalent [Cu(H2O)n]2+ complexes and their stabilization is favoured where two framework Al ions are in close proximity. In this case, dehydration may dissociate water molecules into [Cu(H2O)n]+ and H+ (eqn (4)) and the two adjacent frameworks of Al atoms are balanced by these two cationic species, as has been confirmed from the DFT-supported XES and XAS studies performed by Borfecchia et al.207 They showed that [Cu(H2O)n]2+ species can be stabilized at higher temperatures in an oxidative environment; otherwise, as a consequence of extra ligand loss they undergo “self-reduction”. Alternatively, bare Cu2+ cations can be generated due to the dehydration of [Cu(H2O)n]2+ complex (eqn (5)). However, in those sites where there is only one Al in the proximity (1Al Z sites), the aqueous ion exchange leads to the formation of monovalent [Cu(H2O)n(OH)]+ which do not require any water dissociation upon dehydration (eqn (6)). In addition, several Raman vibrational features appeared, after O2 activation on Cu2+, correspond to the O2 activated Cu2+208 species as shown in Fig. 7. [CuO2˙]+ end-on uperoxol species, within the 1000–1200 cm−1 spectral range for Cu–O, with two strong peaks appearing at 1100 and 1155 cm−1. These features signify the presence of this species in equilibrium with the side-on species, as can be seen in Fig. 7b. While the [Cu(trans-μ-1,2-O2)Cu]2+ vibrational feature appeared at 510, 580, and 830 cm−1, species shown in Fig. 7c for a pictorial representation of this complex. These distinctive spectral components represent the spectroscopic fingerprints of these Cu2+ species following O2 activation.
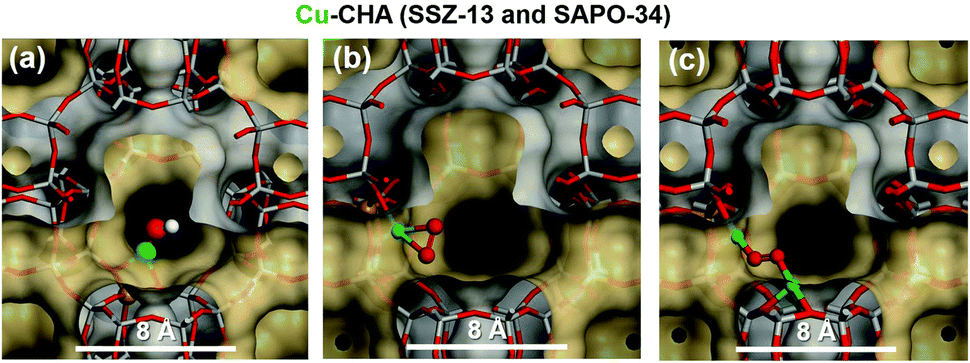 | ||
| Fig. 7 Proposed local sites of the copper species in the cage of the CHA at high temperature and under oxygen treatment (a) [CuOH]+ (b) [CuO2˙]+ superoxide species (c) [Cu(trans-μ-1,2-O2)Cu]2+. Colour code: Si grey, Al gold, Cu green, O red, H, white. Reproduced from ref. 208 with permission from The Royal Society of Chemistry, copyright 2018. | ||
Borfecchia et al.207 also investigated the tri-coordinated [CuOH]+ as a site for O2-activation while the bare bi-coordinated Cu+ ions were found to be the most dominant species in the 8-MR. The presence of dimeric oxo-Cu species appears to be unlikely which shows that monomeric [CuOH]+ species are mostly found in the Cu-CHA, and may play an important role in the SCR mechanism.
| [Cu(H2O)n]2+ → [Cu(H2O)n)2+ + (n − 1)(H2O)↑ → [Cu(OH)]+ + H+ → Cu+ + (OH)↑ + H+ | (4) |
| [Cu(H2O)n]2+ → Cu2+ + n(H2O)↑ | (5) |
| [Cu(H2O)n(OH)]+ → [Cu(OH)]+ + n(H2O)↑ → Cu+ + (OH)↑ | (6) |
From synchrotron-XRD analysis, cation exchange sites were identified for TMI divalent cations such as Cu2+209 and Ni2+210 in zeolites. For Cu, it has been proposed that there may be six sites in Cu-MFI (Cu-ZSM-5) as reported by Mentzen et al.211 Cu-ZSM-5 were synthesized via the reaction of H-MFI samples with 0.1 M Cu2+ acetate solutions. Analysis using the Rietveld method of PXRD profiles of dehydrated Cu-MFI found the presence of multiple independent extra framework Cu centres in Fig. 8. In the compositions H1.04Cu(I)1.19Cu(II)0.51MFI and H1.99Cu(I)2.37Cu(II)0.00MFI, four and five Cu cations were identified, respectively. Within these phases, the Cu1/Cu2/Cu2′/Cu3/Cu3′ populations per unit cell were determined as 0.0/1.07/0.02/0.17/0.45 and 0.18/1.12/0.44/0.20/0.50, respectively. Cu2 was the most prevalent. The shortest distances observed between framework oxygen atoms and copper sites were associated with the Cu1, Cu2, and Cu2′ sites, falling within the range of 1.99–2.32 Å. Additionally, Cu1, Cu3, and Cu3′ sites were found to be situated within secondary 5 and 6-MR channel sections of the MFI framework. In contrast, Cu2 and Cu2′ were close to the 10-MR of the straight channel. Given their immediate accessibility to guest molecules, one or both of these cations, namely Cu2 and Cu2′, emerge as the principal contenders for the catalytically active sites within Cu-MFI materials.
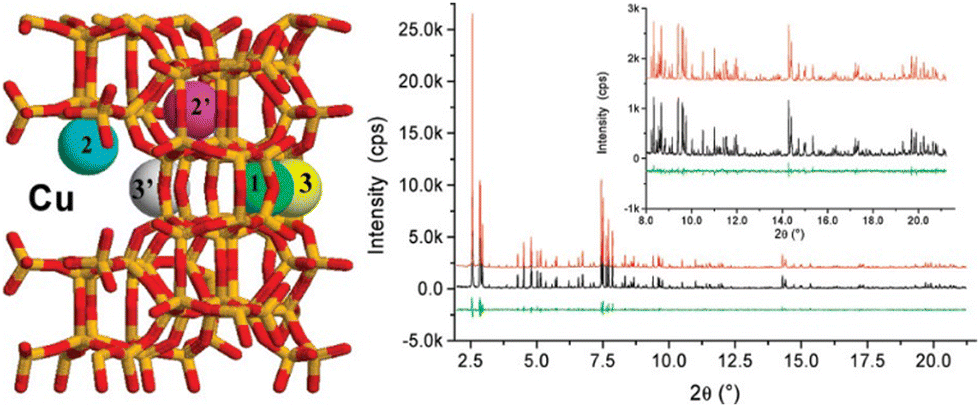 | ||
| Fig. 8 Rietveld refinement plots illustrating the dehydrated H1.99Cu(I)2.37Cu(II)0.00MFI phase at room temperature (25 °C) are presented. The plots include observed profiles (in the middle, shown in black), calculated profiles (above, depicted in red), and difference profiles (below, represented in green). These plots were generated using X-ray synchrotron radiation with a wavelength of λ = 0.50013 Å. The fitting parameters Rp/Rwp/Rexp/RI = 7.3/8.4/5.8/6.2%. The populations of Cu1/Cu2/Cu2′/Cu3/Cu3′ within a unit cell are 0.0/1.07/0.02/0.17/0.45 and 0.18/1.12/0.44/0.20/0.50, respectively. Reproduced from ref. 211 with permission from American Chemical Society, copyright 2007. | ||
Fluorescence spectroscopy of Cu-ZSM-5 suggests that there are two types of Cu species: (i) Cu ions located in close proximity to a single framework Al ion (540 nm emission), and (ii) Cu ions neighbouring two framework Al ions with the 480 nm emission which linked to Cu ions (Cuα sites) (as can be seen in Fig. 11) adjacent to two framework Al ions.212 The 540 nm emission is thought to correspond to Cu ions (Cuβ sites) located in a less densely packed environment near one framework Al ion. However, this site has not yet been identified unequivocally. As discussed earlier the Cu2+ ESR spectrum displays axial symmetry, causing a signal split. The parameters for the parallel component are gII = 2.33 and AII = 140–160 G, denoted as Cuα while for the Cuβ sites, they are gII = 2.27 and AII = 170–180 G, and are associated with a monovalent state of copper. The Cuα sites possess more tightly packed coordination environments, shorter-wavelength emissions, and higher positive charges on the copper cation.
Lobo et al.13,213 conducted nuclear magnetic resonance (NMR) experiments within a Li+-exchanged zeolite Beta to delineate cation exchange sites, drawing insights from interactions (or their absence) with oxygen molecules. Initially, they postulated sites denoted as Cu1, Cu2, and Cu3, located within pentasil or 6-MR of the structure (refer to Fig. 9a). However, their NMR analysis revealed that only a fraction of these sites were accessible to oxygen. Considering the known activity of Cu-Beta in the deNOx reaction, it was inferred that structurally accessible sites must be present. Hence, a revised depiction in Fig. 9b presents Cu cations positioned slightly differently. Cu1 and Cu2 maintain their locations in structurally analogous 6-MR, with Cu1 now facing the 12-MR channel. However, Cu3, close to two framework O ions with a bond distance between Cu and O is 1.3 Å, was relocated to an alternate site within a different 5-MR, facing the 12-MR channel. This adjustment ensures the availability of Cu in the most accessible sites conducive to catalytic processes. Notably, both Cu1 and Cu3 in the revised model occupy positions akin to the planar position within 6-MR of the SSZ-13 structure, suggesting their potential activity in the SCR reaction.
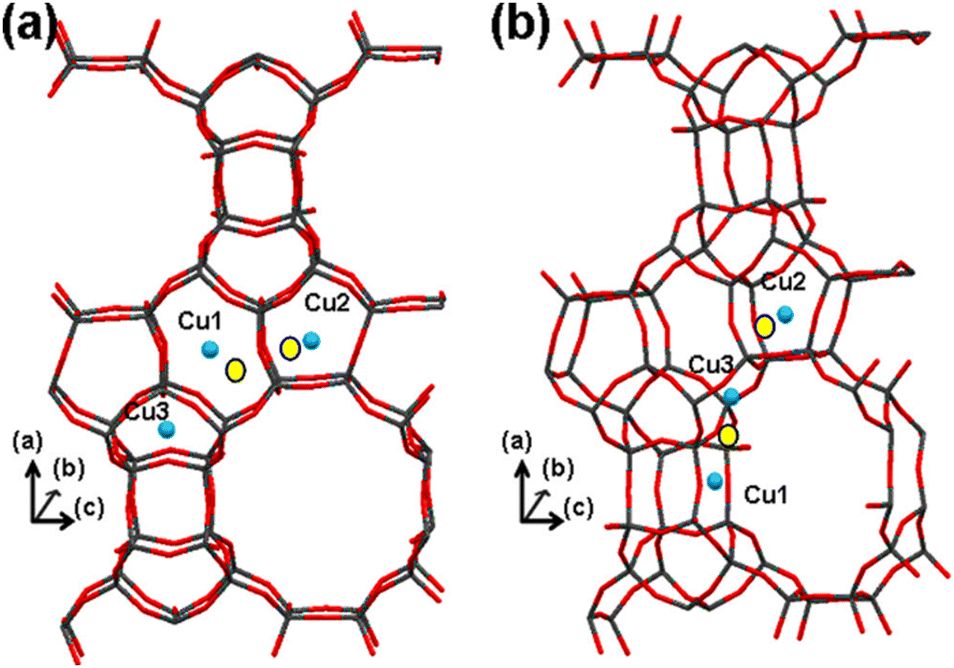 | ||
| Fig. 9 Illustrates the positioning of Cu cations within Beta zeolite. Structure (a) is derived from NMR experiments conducted with Li+ as a cation,213 whereas structure (b) offers an alternative configuration related to structure (a). In the diagram, blue spheres indicate the positions of the Cu cations, and yellow circles represent adjusted positions, allowing for close coordination (approximately 2 Å) with at least three oxygen atoms. Reproduced from ref. 28 with permission from American Chemical Society, copyright 2013. | ||
Fe-based zeolitic systems
Fe speciation tends to be more varied than with other TMIs in zeolites, with techniques including UV/Vis and DRIFTS used to identify the presence of single ions (both Oh and Td in nature), clusters and polymeric species.120,214–217 Generally, it has been shown that all species can perform NH3-SCR but the monomeric species218 are usually active at lower temperatures below 250 °C,218 particularly those species located in the 8-MR of MFI zeolites (often termed the Beta site). Li et al.219 employed ab initio thermodynamic techniques to assess the stability of various Fe complexes within ZSM-5 with Si/Al ratio of 47, and concluded that mononuclear Fe site-local (FeO)+ is unaffected by the local environment, whereas binuclear sites tend to predominantly occupy the larger eight-membered ring (8-MR) γ site in the sinusoidal channel. In the coordination of Fe ions to the framework under low loading (≤1.2 wt% Fe), most of the species are present as a monomer at the ion-exchange positions while at higher loading (2.5–5.1 wt% Fe), oligomers of Fe are formed which mainly reside inside the micro-pores of zeolites.220 Consequently, at high loading, the lower coordination of the TMI to the lattice O occurs which is difficult to verify as they are masked by other Fe–O species at high loading. The increased loading of TMI creates more heterogeneous sites which need extra-lattice ligands such as H2O, and OH. The removal of these ligands under high-temperature pretreatment induces the reduction of TMI.Considering monomeric species (FeO)+ as an active site, various reaction pathways were explored and the energy for several species was described by Li et al.221 and Bruggeman et al.149 However, some experimental studies suggest that in the presence of two closely exchanged Fe species, the dimer of Fe (i.e. bridging Fe–O–Fe dimers) species is readily generated which is evident from several investigations.222–224 These dimer species are extensively studied by DFT and are considered to be active species in the NH3-SCR reaction and for N2O and NO decomposition.150,216,225,226 In addition, site occupancy of the Fe ions in zeolites varies with the sample preparation method, while the loading of the metal, which is only partially determined by the Al content, can also affect the coordination of TMI to the lattice O.
Brandenberger et al.227 utilised a Poisson distribution, a method employed for the prediction of rare but significant events in a fixed interval to compute the fraction of content of several Fe species in zeolites. The results obtained show the correlation between the degree of Fe-exchanged species and the presence of particular active sites. In particular, they modelled the distribution of several Fe species including Fe–O–Fe bridged and higher nuclear Fe-species within Fe-ZSM-5 zeolite. These findings have sufficient accuracy to show the correlation of the degree of Fe-exchanged species with active sites. Furthermore, the calculated probabilities are compared with experimental data obtained from DR-UV/Vis and DRIFTS which offers insights into the proportions of various iron species and helps validate the analytical findings. Consistent with the observation, there is further agreement that more than one site can be present in the zeolite Fig. 10, and the monomeric Fe ions will predominate only for low levels of exchange.227 Moreover, their investigation showed that the formation of the monomer represents the rate-limiting stage in the SCR which could stem from the re-oxidation of Fe species, resulting in the subsequent conversion (oxidation) of NO to NO2. However, the discussion regarding the nature of Fe species (e.g. monomer and dimer) may be an oversimplification since the nature of the monomeric and dimeric species could be more complicated than what is shown in Fig. 8 for example, undercoordinated Fe monomers or else di-oxo bridged dimeric species.
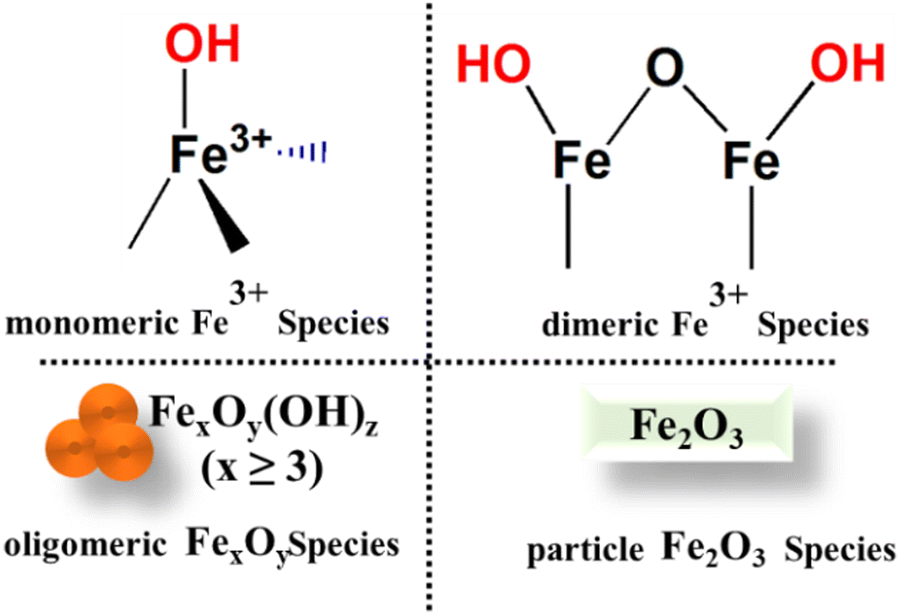 | ||
| Fig. 10 Schematic illustration of four possible Fe species in Fe-zeolites. | ||
We reported that Fe3+ species have demonstrated varying levels of activity under different conditions.228 Isolated octahedral Fe3+ entities hosted on H-ZSM-5 zeolite surfaces have exhibited high reactivity. These octahedral sites displayed a propensity for reduction upon exposure to ammonia under SCR conditions. Conversely, isolated tetrahedral Fe3+ sites demonstrated comparatively lower redox properties, resulting in diminished NO conversion rates. Furthermore, clusters and FexOy particles observed on H-SSZ-13 zeolite surfaces showed subdued SCR performance although this improves at higher temperatures, all of which suggested a critical role of Fe3+ speciation.
The self-reduction of metal ions during the reaction has a major impact on the coordination of the metal to the zeolite framework. Under high temperatures and an inert atmosphere, TMI-Zeolites (Co3+, Fe3+, Cu2+) can undergo auto reduction.180,229,230 The auto reduction of Fe3+ to Fe2+ under He treatment has been characterised by in situ XAS.231,232 The switching of Fe3+ to Fe2+ is attributed to the desorption and dehydration of Fe3+(OH)2 to liberate O2 and form two Fe2+(OH) species.233 It is also argued that bare Fe3+ only coordinates with oxygen and will be unstable in high silica zeolites due to the inadequate negative charge within the zeolite lattice to counterbalance the trivalent ions.234 The characterisation of such trivalent ions especially Fe3+ is challenging. The identification and the chemistry of the Fe ions in an aqueous solution are, of course, well defined from the existence of various mono-, di, and poly- complexes bearing different species including oxygen, water molecules, and OH groups (depending on the reaction media, especially pH).235,236 Fe ions (Fe2+ and Fe3+ ions) can be incorporated into the framework of high silica zeolites; subsequent high-temperature treatment of these ferrisilicates leads to the ejection of Fe from the framework and can be considered another approach for the generation of extra-framework species.234
Other TMI-systems
Besides Cu and Fe, other TMI196,237 within zeolite frameworks are also studied for their coordination, stability, and catalytic properties, employing techniques like DFT, XAS, and IR spectroscopy. Sklenak et al.238 studied the cationic sites of ferrierite exchanged with Co2+ and Cu2+ and the impact of Al placement within six-membered rings on the structure and binding energies of Me2+ ions (Me = Co and Cu) using periodic DFT molecular dynamics. They demonstrated that the binding of both cations to these sites prompts substantial rearrangement of the local structure within the zeolite framework, particularly for the α and β-2 sites, yet minimal for the β-1 site (see Fig. 9).Rice and co-workers239,240 examined several divalent transition metal cations including Zn, Pt, Cu, Ni, and Co in ZSM-5, and investigated the coordination, location, stability, and structural properties using the gradient-corrected DFT technique. They modelled the coordination of these ions at isolated charge-exchange sites and pairs at adjacent exchange sites. The data obtained indicate that Fe2+, Cu2+, Ni2+ and Co2+ are coordinated to 5-MR containing 2Al, whereas Rh2+, Pd2+, Pt2+, Zn2+ and Ru2+ are preferentially coordinated to 6-MR of ZSM-5.
The detailed analysis of Co-doped zeolites by Bellman et al.241 prepared Co-ZSM-5 via wet ion-exchange (IE) with various Co contents (0.32, 0.34, 1.96, 2.44 and 3.19 wt%) and found that not only Co3+ but also Co2+ were present in oxide-like clusters (i.e. Co3O4) and at ion exchange position(s). The UV-Vis spectra show isolated Co2+ species, and the triple band is from both γ-, β-, and α-type Co2+ sites in the zeolite and those dispersed on different oxide supports’ surfaces. Co3+ species, present in the oxide layer oxidized NO and itself reduced to Co2+ which is subsequently reoxidized to Co3+ upon exposure to O2, therefore, establishing an active Co3+/Co2+ redox pair. In contrast, the Co2+ located at the exchange position assists the chemisorption of NO and hence acts as a storage material for NO. The diffuse reflectance spectra of Co2+ ions in the Co–zeolites, including BEA and ZSM-5 indicate the presence of three sites denoted as α, β, and γ.242–244 The positions and the structure of these sites in the local framework of ZSM-5 are shown in Fig. 11.
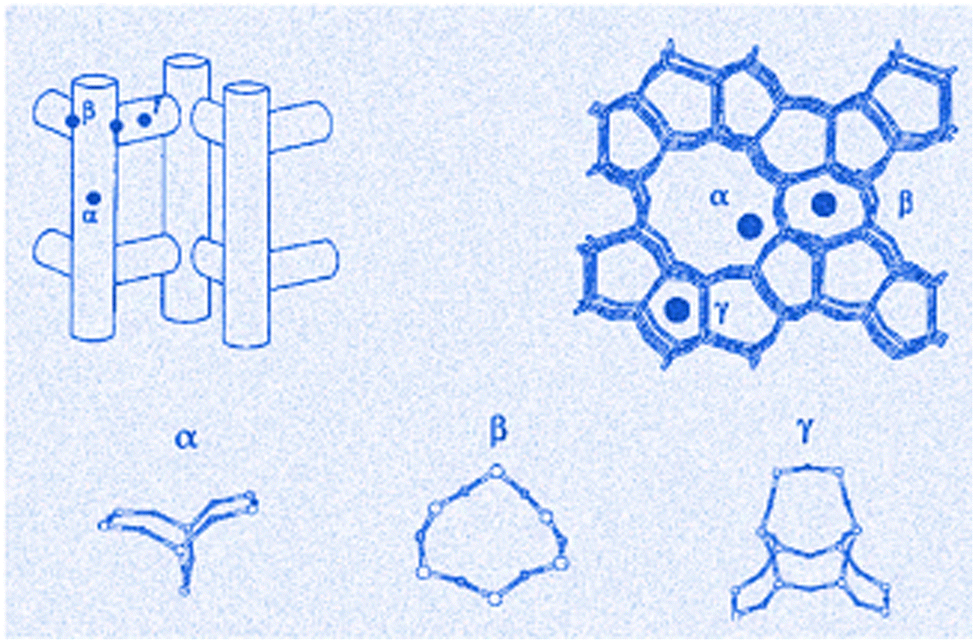 | ||
| Fig. 11 Illustration showing Co siting in ZSM-5 structure. Local framework structures of the a-, b-, and c-sites. Reproduced from ref. 245 with permission from Elsevier, copyright 2000. | ||
Dooryhee et al.246 studied Ni2+ exchanged Y zeolite (Si/Al = 2.25) by X-ray absorption and diffraction at room temperature and suggest that there is a significant distortion in the shape of the d6-MR cavity when occupied by Ni2+ ions, signifying that the framework of the zeolite adjusts in response to the migration of Ni ions during the process of dehydration. There are several reasons (i) first, are the electrostatic contacts between the cations and the framework; and second, TMIs where possible expand their coordination number to gain a stable configuration within the zeolite framework.
Similarly, coordination of TMI to specific exchange sites leads to a strong distortion in the framework of zeolites, as shown by detailed ab initio quantum-chemical analysis.247–249 However, other reports indicate that Ni ions reside within the 6-MR, specifically at a corner position. It has been suggested that XRD, when subject to position averaging, may not have the capability to discern any distortion as reported by Pierloot et al.,178 and indeed the work of Dooryhee et al.246 proposed distortion based on the combination of XRD and EXAFS data.
Smeets and coworkers250 and Vanelderen et al.251 have examined the coordination chemistry of TMI (Cu, Fe, Co) in zeolites in particular, to investigate the coordination chemistry of the activated species and demonstrated their ability to activate O2 and oxo-complexes. They explore two aspects of TMIs (Cu, Co, and Fe) in zeolites: their coordination within the lattice and the presence of activated oxygen species. TMIs tend to occupy 6-MR exchange sites, coordinating with Al tetrahedra at low loading. High loading results in diverse TMI species on the zeolite surface. The activated O2 species are suggested to oxidize (i) NO into NO2 and (ii) reoxidize the TMI to its appropriate oxidation state.85,252
Modelling mobility and restructuring of TMI
One of the key aspects of TMI zeolite catalysts is their location and their migration. According to AIMD and DFT studies, the migration of metal ions is limited by their electrostatic tethering to the zeolite framework.253 The presence of extra-framework cations and coordinating molecules changes the coordination environment of the TMI sites which ultimately enables their migration.Paolucci et al.85 conducted both computational and experimental analyses on coordinated Cu speciation under operando SCR, in situ and ex situ conditions as a function of the catalyst composition. They demonstrated that the number, type, and nature of Cu sites depend on the bulk composition of the zeolites as well as on the environmental conditions. In a notable addition, they demonstrated that the Cu speciation, mobility, and siting are strongly influenced by environmental conditions and are not dependent on the initial cation sites and framework of zeolites. Using molecular dynamics, the mobility of [Cu(NH3)2]+ inside the CHA framework employing ab initio metadynamics was predicted. They calculated the free energy (using Cu–Al coordination distance as a collective variable) (Fig. 12a) in a CHA supercell of 72 sites. The free energy minimum is located at a distance of ∼4.7 Å which increases to a Cu–Al distance of 8 Å when Cu ions move through 8-MR between two cages as can be seen in Fig. 12b. However, the free energy decreases again as the Cu ion enters the adjacent cage (product state) until the distance is greater than 9 Å.
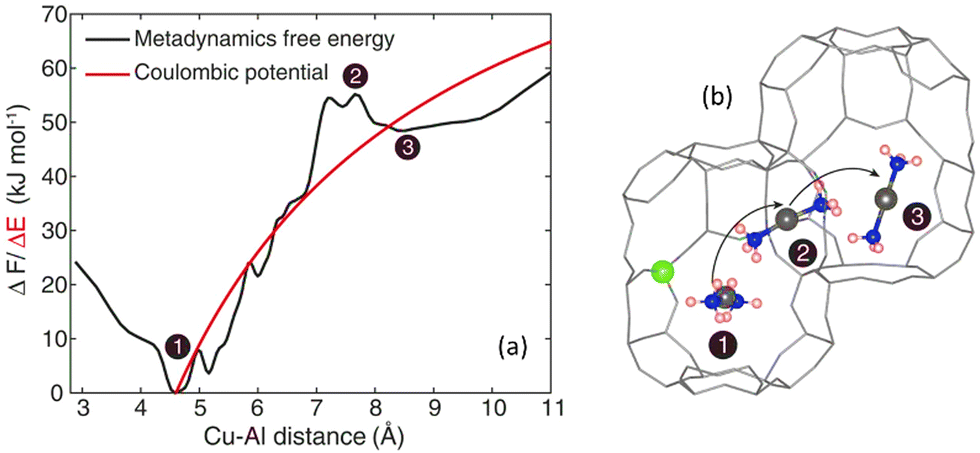 | ||
| Fig. 12 (a) The free energy profile – varies with the Cu–Al distance, involving the mobility of [Cu(NH3)2]+ species and Al ions within the CHA framework to maintain charge balance. The first stage involves the reactant [Cu(NH3)2]+. (2) Transition state [Cu(NH3)2]+ (3) product state [Cu(NH3)2]+. For comparison, the computed point-charge model has also been reported as a red line.16 (b) [Cu(NH3)2]+ configuration. Cu; grey, Al; green N; blue, and H; reddish. Reproduced from ref. 85 with permission from The American Association for the Advancement of Science, copyright 2017. | ||
The window of CHA is sufficiently large for the diffusion of SCR gases and Cu species to lead to a potential interaction between species within and cluster or dimer formation.85 As illustrated previously, the isolated Cu ions are mostly present in the 6-MR and the [Cu–O–Cu]2+ dimer is generated as a result of two proximal Cu species.254,255 This species was thought to be located inside the cage; however, migration of Cu ions can occur which affects the SCR deactivation reaction.
In zeolite Y the sites SI, SII, and SIII for Cu2+, or Cu+ ions are considered to be more predominant locations for the Cu species178,256,257 where site I is located in the hexagonal prisms connecting sodalite cages, site II positioned in front of the 6-MR inside the super-cages, and sites III found in the super-cages near the 12-MR or 4-MR windows.257,258 In accord with the above argument, site SII, located in the 6-MR is considered to be the active site at low temperatures. Deka et al.28 show, that Cu ions move from SI to SII sites under reaction conditions and the gases can reach these Cu ions via the cage. The Cu ions are highly mobile and can easily migrate to the supercages, changing their coordination with the oxygen ions within them. They started in the supercages when the Cu ions were first introduced since the hexa-aqua species will not be stable elsewhere. Similarly, at the SIII site, a dimeric species [Cu–O–Cu] could be formed as a result of interaction with the Cu of the SII site, demonstrating that mobility leads to the generation of multiple species.259
Krishna et al.82 elucidated the complex behaviour of Cu cations within a zeolite framework during the NH3-SCR of NOxFig. 13. They proposed that initially, NH3 coordinates to Cu cations, which remain connected to charge-balancing framework Al centres through two O ions. This dynamic system involves the conversion between mononuclear Cu+ and binuclear Cu2+ sites during the oxidation half-cycle. This conversion is assisted by the movement of NH3-solvated Cu+ complexes within CHA cages and their migration through 8-MR windows into neighbouring cages. Migration into nearby cages is suggested to have lower energy barriers, as shown in Fig. 13a, with an activation energy of 20 kJ mol−1 at 423 K for empty cages and approximately 35 kJ mol−1 when the cage is occupied. Fig. 13b indicates higher free-energy barriers of 60–80 kJ mol−1 at 473 K for migration into CHA cages that are farther from the charge-balancing Al centre. Such migration dynamics affect steady-state SCR rates, which depend on Cu density at two different sites.
 | ||
| Fig. 13 The illustrations display the arrangement of Cu+ cations, which are linked electrostatically to framework Al and labelled from (a) to (d). Additionally, the illustrations depict SSZ-13 lattices with varying degrees of separation between Cu+ pairs. This separation is indicated by the number of required CHA cage migrations: n = 0 (a), 1 (b), 2 (c), or 3 (d). CHA lattice is shown in two dimensions as a hexagonal lattice, where faces correspond to a CHA 8-MR. Furthermore, each SSZ-13 cage is linked to three other SSZ-13 cages via an 8-MR. Reproduced from ref. 82 with permission from Springer Nature, copyright 2023. | ||
The zeolite's compositional and structural properties, particularly the arrangement of framework AlO4 anionic charges, play a crucial role in influencing Cu cation proximity and mobility, affecting SCR rates. The theoretical predictions, including DFT calculations, emphasise the relevance of Cu+(NH3)2 migration over O2 activation in controlling SCR rates. Spatial isolation of Cu+ sites is noted to impede reactivity with O2, highlighting the complexity and sensitivity of SCR processes to zeolite properties.
Nucleophiles including H2O, NH3, and NO2 bind strongly to the cation and can enhance the mobility of the ions at the active sites e.g. Cu(NH3)2. This active site has an effective area of diffusion which significantly restricts its interaction with other active sites within the overlapping area. Kwak et al.198 compared Cu-Beta, Cu-SSZ-13, and Cu-SSZ-5 and concluded that although very few changes can be observed in the Cu species located in the SSZ-13, the Cu-SSZ-5 and Cu-Beta have CuO or Cu aluminate on the surface. The mobility of TMI ions as well as Al can act as an indicator for the activity of the NH3-SCR reaction as shown by in situ impedance- ‘modulus’ spectroscopy) and theoretical studies, the discussion of which is, however, beyond the scope of this review.260
It is argued by Merberger et al.261 that at (high temperatures 400 °C), the ammoniated–Cu complex including (Cu+(NH3)2 and Cu2+(NH3)4) decomposes, and the Cu ion loses an NH3 ligand as well as its mobility. Subsequently, Cu ions become attached to the wall of zeolite and hence lose their ability to form dimers dynamically and activate oxygen. It is, therefore, conceivable that the oxygen-activated NO step described by Janssens et al.17 becomes an appropriate step in NH3-SCR at high temperatures.
Millan et al. conducted extensive studies to investigate the mobility of Cu ions in Cu-SSZ-13 and its implications for the NH3-SCR.262,263 They aimed to elucidate the mobility-dependent SCR reaction and developed a machine-learning interatomic potential capable of correctly reproducing ab initio results, enabling simulations of large supercells and various chemical compositions. Employing a combination of biased and unbiased simulations, the researchers explored the mobility of [Cu(NH3)2]+ in Cu-CHA catalysts. Their findings indicated that Al pairing in 8-MR facilitated local hopping with the increased concentration of NH3, and so enhanced long-range diffusion. Furthermore, AIMD simulations were used to investigate the migration of [Cu(NH3)2]+ under NH3-SCR-NOx reaction conditions. Results indicated that the diffusion of these complexes was hindered by additional NO or NH3 in the initial cavity, while the presence of O2 also impacted the NO and Cu+ interaction. Expanding their analysis to a larger 1 × 2 × 1 unit cell to simulate the catalyst, they examined diffusion towards a third cavity, C (Fig. 14). An activation-free energy barrier of 76 kJ mol−1 was computed for diffusion through the 8-MR window connecting cavities B and C. The significant destabilization observed during the migration of the [Cu(NH3)2]+ to cavities B and C was attributed to the interactions between [Cu(NH3)2]+ and the negatively charged framework surrounding the Al ion residing in the 8-MR connecting cavities A and B, as depicted in Fig. 14.
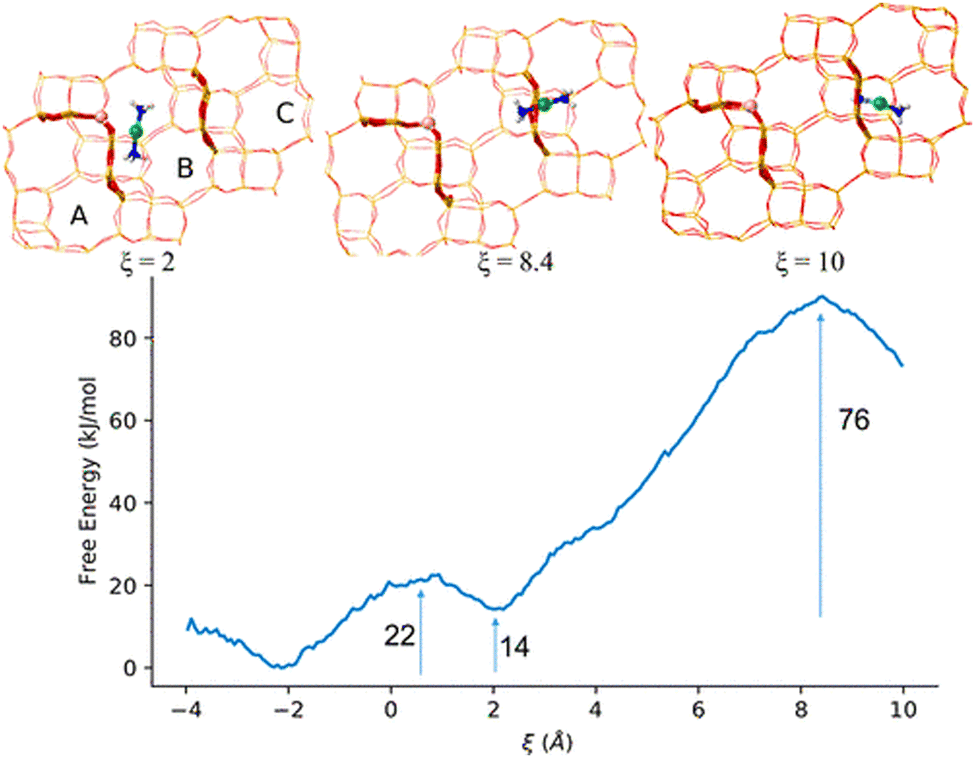 | ||
| Fig. 14 The figure illustrates the diffusion pathway of [Cu(NH3)2]+ through different 8-MR of CHA, along with the corresponding free energy profile at 423 K. The O and Si ions are depicted as red and yellow sticks, while Al, Cu, N, and H atoms are represented as thatch, green, blue, and white balls, respectively. Reproduced from ref. 262 with permission from American Chemical Society, copyright 2021. | ||
Still, more experiments (especially high temperature-driven reactions) are required to understand the detailed chemistry behind the NH3-SCR mechanism over TMI zeolite especially Cu-zeolite. Further modelling studies are also clearly needed.
Reaction mechanism and kinetics of NH3-SCR
In the exploration of NH3-SCR mechanisms, a synergistic relationship between experimental and theoretical techniques has emerged, each offering unique perspectives on the complex processes at play. While experimental techniques provide insights into reactant adsorption and kinetics on catalyst surfaces, theoretical methods, particularly DFT, offer predictive capabilities for reaction pathways and intermediate states. The synergy between these approaches enables a comprehensive understanding of catalytic processes. Theoretical investigations often afford deeper mechanistic insights, in part to their ability to control variables and predict highly transient species that may evade detection by experimental techniques, which can be limited by time resolution and sensitivity. However, since the detection of intermediates via experimental methods is limited to a handful of species, as a result, we focus primarily on the theoretical studies that have yielded very detailed insights into the reaction pathways and intrinsic activity of metal species.Although the role of TMI has been widely explored, the reaction mechanism is still under debate and its investigation is ongoing.170,173,188,264 Generally, the reaction is thought to follow a Langmuir–Hinshelwood (L–H) mechanism: the reactant species (NH3, NOx) simultaneously adsorb on the catalysts and then react to generate H2O and N2. However, ammonia can react with NO via the Eley–Rideal (E–R) route. These reactions lead to the formation of NHx–NOx intermediates, which afterwards decompose into H2O and N2. Experimental techniques, including surface analysis and reaction kinetics measurements, offer insights into reactant adsorption and subsequent reactions on catalyst surfaces. Theoretical methods, especially DFT enable the prediction of reaction pathways and intermediate states. By integrating experimental data with theoretical models, a comprehensive understanding of catalytic processes can be achieved.
Both acidic and redox sites have important roles in the NH3-SCR reaction. The catalyst's redox chemistry controls its activity at low temperatures, while the acidic properties of the catalyst's surface govern its activity at high temperatures (HT).265,266
A catalyst possessing appropriate redox capability can produce active NO2 and NH2, thereby enhancing reaction efficiency via the E–R pathway and so can follow “Fast SCR”. Increased acidity is beneficial for establishing adsorbed NH3/NH4+ species, which are important in generating NH2 or reacting with active nitrites and nitrates. Both redox and acidic sites are necessary for achieving a wide temperature range of activity. A common reaction pathway exists for the SCR process over both zeolite and metal oxide.13 In the E–R pathway, ammonia attaches to acidic sites, resulting in the production of NH3/NH4+, which are subsequently oxidized by high-oxidation redox sites to produce NH2 species. Next, NH3 or NH2 then can directly react with NO, producing NH2NO/NH3NO, which further decomposes into H2O and N2. Following the L–H mechanism, ammonia binds to acidic sites to create NH4+ species, while NO undergoes oxidation to produce active nitrate/bridged bidentate nitrate/monodentate nitrite. These active intermediates then react with NH4+ to produce NH4NO2/NH4NO3, which subsequently decompose into H2O and N2. Concurrently, the high-oxidation redox sites experience reduction to a low-valent and, therefore, re-oxidized by oxygen to complete the redox cycle.
Cu-based zeolitic systems
The possible mechanisms and reaction pathways for NH3-SCR reaction observed in Cu-based zeolitic systems are studied here, shedding light on their catalytic activity and the formation of intermediate species essential for NH3-SCR. Understanding these processes is crucial for optimizing catalytic performance and reducing harmful NOx emissions. As proposed by Janssens et al.17 in the NH3-SCR over Cu-CHA reaction, the formation of Cu-nitrate species plays an important role in completing the Cu redox cycle. They mainly used theoretical investigations employing DFT, supported by experiments to determine this behaviour. They observed that it is possible to split the redox cycle and separate the oxidation step from the reduction step by switching between the NH3 + NO and NO + O2 atmospheres to study the individual half-cycles. The Cu2+–nitrate species formed as a result of the NO + O2 oxidation process is converted to the solvated Cu+–NH3 under NH3 + NO reductive conditions. Cu2+–nitrate species are reformed when switching back to NO + O2 oxidation conditions, closing the catalytic cycle. They proposed a simple mechanism for an NH3-SCR cycle on isolated Cu-ions, in which the NO reacts with the nitrate intermediate to generate gaseous NO2 and Cu-nitrate species. This observation agrees with the experimental data (i.e. detection of nitrate bands using IR) as the transient formation of gaseous NO2 was successfully detected during the SCR reaction.In lower temperature (below 300 °C) processes, a significant puzzle relates to the kinetics of the Cu+ → Cu2+ half-cycle as the Cu concentration decreases which corresponds to a transition from second-order to the first-order dependency of SCR rate on Cu density.264 The reduction in the apparent reaction order is observed for O2 from 0.3 to 0.8 with a decrease in Cu density.85,264 Recently, researchers have investigated these observations using steady-state and transient kinetic measurements, first-principles calculations, and XAS techniques. From the findings of Paolluci et al., the NH3-solvated Cu+ ions can form transient ion pairs while migrating through zeolite windows and activating oxygen.85
Understanding the mechanistic details of the SCR of NOx with NH3 over Cu-exchanged zeolites is crucial for optimising catalyst design and enhancing performance. Operando techniques, especially XAS, (both XANES and EXAFS) and UV-Vis have been significant in providing detailed, real-time insights into redox transformations under reaction conditions.
Pappas et al.267 investigated the nature of Cu ions in H-MOR samples with Si/Al ratios of 11 and 7, and varying Cu loadings, using a range of techniques including XAS, high-energy resolution fluorescence detected (HERFD) XANES coupled with multivariate curve resolution (MCR). MCR is a chemometric technique that deconvolves complex spectroscopic data into pure component spectra. Their study revealed the coexistence of both active and inactive framework-coordinated Cu2+ species after O2 activation, identifying a specific framework-coordinated Cu2+ moiety as the active site for methane activation.
Paolucci et al.85 utilized XAS to monitor the oxidation state changes of Cu in Cu-SSZ-13 catalysts under working conditions, establishing a direct correlation between the catalytic activity and the Cu redox cycle. To investigate the mechanistic origins of changes in kinetic behaviour during standard SCR, they employed XANES spectroscopy to quantify the Cu oxidation state as shown in Fig. 15a which presents the steady-state Cu+ fraction obtained from XANES fitting as a function of Cu density for various Cu-CHA zeolites. As observed, site-isolated Cu2+ ions evolve into a mixture of Cu+ and Cu2+ during SCR at 473 K catalysis, indicating redox cycling between these oxidation states.119 The data reveal an inverse relationship between Cu+ fraction and Cu ion density, contrary to expectations for a single-site catalyst where the oxidation state should depend solely on reaction conditions. At high Cu densities, Cu+ is the minority oxidation state, but it becomes predominant at lower Cu densities.
 | ||
| Fig. 15 (a) Dependence of the Cu+ fraction on Cu ion volumetric density during steady-state standard SCR at 473 K, measured by XANES (details in SM Section S3). The inset shows NH3-solvated Cu+ and Cu2+ species observed and computed at 473 K.119 The coloured arrow is a visual guide; error bars reflect a 5% uncertainty from XANES fitting. Reproduced from ref. 85 with permission from The American Association for the Advancement of Science, copyright 2017, and (b) Operando UV/Vis. DR spectra during an RHC transient at 175 °C (dark blue to dark grey curves) after NH3 saturation (dashed blue line); (c) NO inlet (dashed red line), NO outlet (red symbols), and redox model predictions (red solid line); (d) Cu2+ reduction dynamics from MCR analysis (blue symbols) and model predictions (blue solid line). Reproduced from ref. 268 with permission from John Willey and Sons, copyright 2024. | ||
Negri et al.268 utilised UV/Vis Diffuse Reflectance (DR) spectroscopy in conjunction with a packed bed flow reactor to monitor the kinetics of Cu2+/Cu+ redox dynamics during the NH3-SCR process over Cu-CHA catalysts, as illustrated in Fig. 15b–d. In their detailed analysis, they describe how the spectra in Panel (b) reveal the progressive reduction of Cu2+ to Cu+. Initially, the UV/Vis DR spectra exhibit a broad band centered around 15100 cm−1, characteristic of Cu2+ d–d transitions. As the reaction proceeds, the intensity of this band diminishes, indicating the reduction of Cu2+ to Cu+, which lacks d–d transitions due to its closed-shell d10 configuration. Concurrently, changes in the charge transfer region, particularly around 39700 cm−1, signify the gradual replacement of oxygen ligands by nitrogen from NH3 in the coordination sphere of copper as the reduction occurs. In Panel (c), they present the NO concentration transients at the reactor inlet and outlet, alongside model predictions. The observed decrease in NO concentration at the reactor outlet is in line with the reduction of Cu2+ sites, as NO is consumed in this process. The close match between the experimental NO outlet data (red symbols) and the model predictions (solid red line) underscores the validity of the kinetic model. Panel (d) shows the time-dependent fraction of Cu2+ (Cu2+/CuTOT) derived from Multivariate Curve Resolution (MCR) analysis of the operando UV/Vis data. They highlight that the Cu2+ fraction starts at about 90% and decreases steadily as Cu2+ is reduced to Cu+. The model's fit to these data (solid blue line) supports the proposed second-order dependency of the reduction half-cycle rate on the oxidized Cu fraction, providing strong validation for their kinetic parameters. They emphasize that the consistency between the spectroscopic data and gas-phase measurements not only supports the kinetic model but also demonstrates the effectiveness of their operando spectroscopic approach in capturing the dynamics of the catalyst under reaction conditions.
Pankin et al.269 studied the speciation of Cu and Fe in composite zeolite catalysts under NH3-SCR conditions using operando XAS, providing valuable insights into the catalytic behaviour of these metals in both monometallic and mixed catalysts. For iron, the Fe K-edge XANES and EXAFS spectra show that iron in both the monometallic Fe-MOR and the Cu–Fe-mix catalysts primarily forms small Fe2O3 clusters with a structure similar to hematite (α-Fe2O3). These clusters, probably located on the exterior of the MOR framework, contribute to NH3 oxidation and influence Cu-speciation in the mixed catalyst, particularly at high temperatures. The Fe2O3 clusters enhance NH3 oxidation, reducing the formation of Cu+ species in the Cu–Fe-mix catalyst at higher temperatures. Additionally, a minor fraction of isolated Fe species, possibly monomeric or dimeric Fe3+, may be active at high temperatures during SCR. These findings suggest that the iron sites in both Fe-MOR and Cu–Fe-mix primarily consist of small Fe2O3 clusters that efficiently catalyze NH3 oxidation, with isolated Fe3+ species contributing to the high-temperature SCR activity. Fig. 16a and b, which displays the Fe K-edge XANES spectra, effectively captures these characteristics.
 | ||
| Fig. 16 Cu and Fe speciation in NH3-SCR catalysts (a) normalized Cu K-edge XANES spectra, (b) k2-weighted phase-uncorrected FT of EXAFS spectra at various temperatures under NH3-SCR conditions, comparing the monometallic Cu-CZC (black) and bimetallic Cu–Fe-mix (green) catalysts, and (c) Fe K-edge XANES spectra for the Cu–Fe-mix catalyst at RT, 200 °C, and 500 °C after oxygen activation, compared to bulk α-Fe2O3 (dashed line). The inset shows a magnified view of the background-subtracted pre-edge peak. Reproduced from ref. 269 with permission from The Royal Society of Chemistry, copyright 2024. | ||
The Cu K-edge XANES and EXAFS analyses shown in Fig. 16c reveal significant differences in the behaviour of Cu species in the pure Cu-CZC and the mixed Cu–Fe-mix catalysts under NH3-SCR conditions. In the monometallic Cu-CZC, mobile linear Cu+ di-amino complexes dominate at lower temperatures (below 300 °C), while Cu2+ species become more prominent as the temperature increases, which indicates a dynamic redox process for Cu during the SCR reaction. In contrast, the Cu–Fe-mix catalyst shows a higher abundance of reduced Cu+ species, particularly at lower temperatures (150–300 °C), suggesting that Fe enhances the reduction of Cu sites. This interaction, which is probably facilitated by the migration of NH3-solvated Cu species between different zeolite frameworks, results in improved NOx reduction performance at lower temperatures, making the mixed catalyst more effective across a broader temperature range.
Yu et al.270 demonstrated the NH3-SCR mechanism on Cu-SAPO-34 using in situ EPR and DRIFTs experiments, where, NH3 binds to both hydroxyls and Cu2+ while the NO is oxidized into nitrates or nitrites on Cu2+ sites. The generated nitrates and nitrites then react with NH3 forming ammonium nitrates and N2O intermediates respectively. The subsequent decomposition of these intermediates ultimately produces H2O and N2. Similarly, Wang et al.187 suggested a mechanism for Cu-SAPO-34 in NH3-SCR employing in situ DRIFTS and explored the mechanism on the Lewis acid sites, which involves first the generation of NH4NO3 species and then its reaction with NO to generate H2O and N2. The adsorbed NH3 on Brønsted acid sites migrates to Lewis acid sites, providing more NH3 for the reaction. Owing to the high reactivity of NH4NO3, DRIFTS spectra were unable to detect its presence; however, its involvement in the reaction could not be ruled out. In contrast, Su et al.271 confirmed the consequent formation and decomposition of NH4NO2 species using temperature-programmed surface reaction and in situ DRIFTS which are generated as a result of the sequential reaction between NH3 and the generated NO2. To explore the role of NH3 in the SCR reaction, Giordanino et al.122 investigated three potentially stable adsorption sites for the NH3 molecule via NH3-TPD and IR spectroscopy: the NH3 coordinated to Cu site and as solvated ammonium ions are more stable for T < 400 °C, whereas, at higher temperatures, the Brønsted acid sites with a bound NH3 is found to be more stable.
Chen et al.172 present a comprehensive reaction mechanism elucidating NH3-assisted SCR of NOx over Cu-CHA under low temperatures. The reaction proceeds in a multisite manner, implicating solvated Cu cations by ammonia (Cu+(NH3)2), and Brønsted acid sites. Cu sites play an essential role in oxygen activation and the generation of important intermediates, including HONO and H2NNO, while Brønsted acid sites facilitate the subsequent breakdown of these intermediates into N2 and H2O. Oxygen activation necessitates pairs of Cu(NH3)2 complexes, with the coupling of HONO and H3NNO potentially occurring on single complexes. They presented the free-energy landscape of the reaction at 150 °C, as illustrated in Fig. 17. According to the diagram, O2 undergoes endergonic adsorption over the Cu complex pair, resulting in a loss of 0.44 eV due to changes in rotational and translational entropy. The subsequent adsorption and activation of O2 to structure III has an energy barrier of 0.53 eV. The binding of NO leads to an energy change of −0.24 eV, generating nitrosonium (NO+) species. The measured binding energy of NO is computed as −0.90 eV. Furthermore, the coordination of NH3 molecules from the gas phase to NO+ yields structure IV (intermediate C) forming an N–N bond length of 1.9 Å. This step results in an overall free-energy change of +0.52 eV, with an adsorption energy of −0.11 eV. The transition from III to structure IV incurs a total energy change of +0.28 eV. Upon coordination with NO+, the NH3 molecule activates and extends the N–H bond by 0.03 Å towards oxygen. The transfer of this proton to oxygen, generating an OH− group and a nitrosamide (H2NNO) species (structure V), occurs with a free-energy barrier of 0.07 eV.
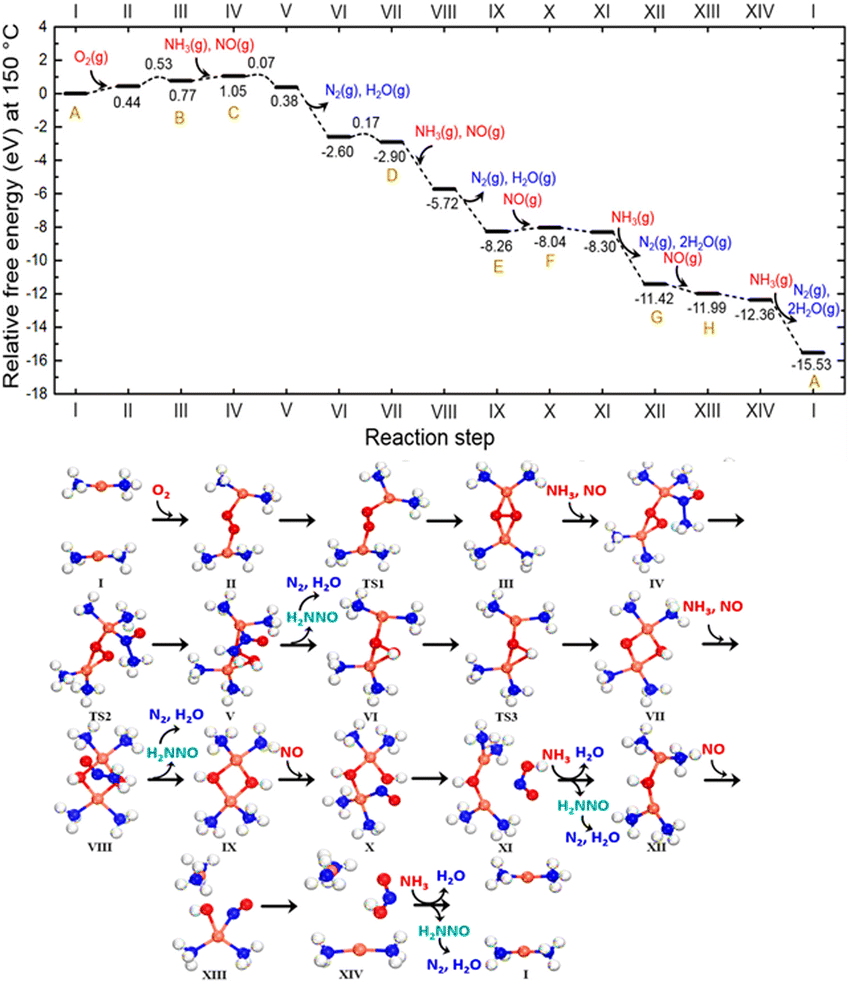 | ||
| Fig. 17 The free-energy landscape of the complete reaction cycle at 150 °C. In the lower panel, the structures of each intermediate are displayed. Reactants, intermediates, and product species are shown in red, green, and blue, respectively. To enhance clarity, the zeolite framework is not illustrated. A to H letters in the energy diagram correspond to the respective species shown in the cycle. Atom colour codes are as follows: Cu (pink), N (blue), O (red), and H (white). Reproduced from ref. 172 with permission from American Chemical Society, copyright 2020. | ||
The breakdown of the H2NNO intermediate on a Brønsted acid site, when released from the Cu+ active site, yields N2 and H2O as products. The desorption of H2NNO from the Cu site results in a minimal free-energy barrier of 0.2 eV (determined through constrained molecular dynamics), leaving behind an O–OH group on the Cu sites (structure VI). The O–OH bond readily splits with an activation energy of 0.17 eV, generating VII. In structure VII, the oxygen atom carries a formal charge of −2, resulting in a multiplicity of doublet (species D). The highly reactive oxygen atom (see structure VII) readily combines with another H2NNO molecule, resulting in structure IX. To this end, both Cu ions now exhibit a +2 oxidation state (E). The Cu2(OH)22+ species commonly observed in enzymatic reactions272,273 have been experimentally determined in crystal structures.274,275 NO binds exothermically to the Cu sites in E leading to the Cu–OH bond, and so generating structure X (species F). The binding of NO+ is accompanied by the reduction of Cu2+ to Cu+.
A HONO intermediate forms and is released from the Cu sites (structure XI) without any energy barrier. Subsequently, HONO then can react with gas-phase ammonia, generating an NH3–HONO complex (ammonium nitrite). This complex decomposes into one N2 molecule and two H2O molecules. Adsorption of an additional NO molecule onto one of the Cu sites in Structure XII (belonging to species G) leads to the second Cu2+ to Cu+ reduction. This adsorption process triggers the breaking of a Cu–OH bond, leading to the generation of two distinct Cu complexes (referred to as Structure XIII and species H and). Subsequently, HONO intermediate emerges on the Cu site hosting OH− and NO+ groups. Furthermore, the HONO species undergoes a reaction with gas-phase NH3, resulting in NH3–HONO formation, which consequently decomposes into one N2 molecule and two H2O molecules, thereby concluding a standard NH3-SCR cycle. According to the free-energy landscape, the activation of O2 represents the highest barrier, with a relative energy of 0.97 eV compared to the gas phase.
Shan et al.206 proposed that the Fast-SCR reaction mechanism over Cu-SSZ-13 involves a stepwise process. In their study, they found that ZCu(II)OH initiates the reaction by adsorbing an NH3 molecule, reaching a coordinatively saturated state as shown in Fig. 18. This adsorbed NH3 then interacts with NO2, forming HNO3 without encountering any discernible energy barrier. The species denoted as B was identified as NH4NO3 adsorbed on Cu sites. Subsequently, gas phase NO then reacts with the adsorbed HNO3, with an associated energy barrier of 0.87 eV. This reaction leads to the generation of adsorbed HNO2 and the simultaneous release of a NO2 molecule (C → D). Furthermore, the adsorbed HNO2 engages in a reaction with the NH3 ligand, producing NH2NO and H2O. Desorption of N2 and H2O molecules occurs, allowing NH3 and NO2 to adsorb at the Cu site, leading to subsequent reactions and the generation of NH2NO and –OH groups. NH2NO decompose into N2 and H2O, and the ZCuOH site undergoes regeneration. The rate-limiting step in their Fast SCR cycle corresponds to the reaction between adsorbed HNO2 and an NH3 ligand over the ZCuOH site, resulting in the production of NH2NO and H2O (1.58eV activation energy).
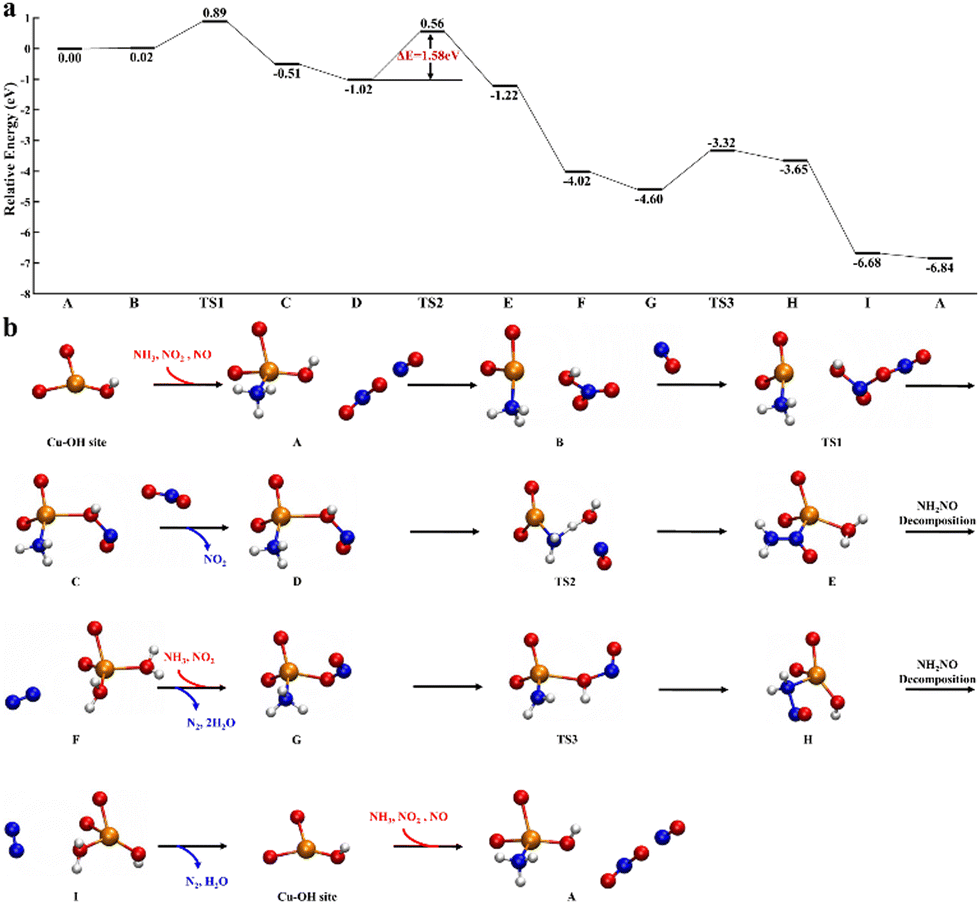 | ||
| Fig. 18 Fast SCR cycle at ZCu2+OH site (a) the free energy profile is illustrated, showcasing the energetics of the reaction, and (b) the optimized geometries of the reactants, products for each elementary step, and the transition states (TSs) are provided in the lower panel. For clarity, atoms of the zeolite framework, excluding the two O atoms linked to the Cu–OH group, are omitted. Reproduced from ref. 206 with permission from Springer Nature, copyright 2022. | ||
Water and NH3 vapours are significant constituents of NOx-containing exhaust gases, and when present they are inevitable and could result in the adsorption on TMI sites, consequently impacting the energetics of intermediate species.276,277 Liu et al.204 explored the impact of water on the NH3-SCR activity employing DFT calculations and using Cu-LTA as a model catalyst and discovered that water exerts a beneficial influence on low-temperature SCR activity, potentially through the formation of solvated [H2O–Cu–NH3]+ species. Similarly, Yu et al.278 conducted catalytic performance testing of Cu-SAPO-34, showing a positive effect of water. They observed that water presence improves the reducibility of Cu2+ species at high temperatures while inhibiting NH3 oxidation. Additionally, Lee et al.279 stated that NH3 does not hinder the generation of nitrate species when water is present in the feed. Furthermore, several studies indicate that Cu-ion mobility is facilitated by physisorbed water and ammonia molecule ligation, resulting in improved low-temperature NH3-SCR activity.85,204,280
Work by Nasir et al.195 and Greenaway et al.281 reported the study of the NH3-SCR reaction mechanism using DFT techniques employing a quantum mechanical/molecular mechanical (QM/MM) technique together with concentration modulation excitation DRIFTS, with a specific focus on understanding solvent effects on Cu-CHA reactivity and the corresponding reaction mechanism respectively.
Examining the NH3-SCR reaction, the potential energy landscape was first studied, without physisorbed solvent molecules as can be seen in Fig. 19 with black lines, showcasing exothermic O2 adsorption, followed by NO adsorption generating Cu–NO3 species (species B). Gaseous NO2 reacts with the Cu+-site, forming bidentate Cu–NO3 intermediates. The reaction of NH3 with nitrite produces an intermediate (species D), and NH3 also reacts with the Cu–HONO, generating an NH3–Cu–HONO species, which subsequently decomposes, leading to the generation of the crucial Cu-nitrosamine species, considered important in the NH3-SCR reaction. Next, the influence of water solvation has been examined (Fig. 19 red lines), as water is a primary product of the NH3-SCR reaction, necessitating consideration of hydrated active sites. Water represents a significant product of the NH3-SCR reaction, demonstrating the importance of considering the hydrated state of the active sites.207 The presence of water enhances the migration of Cu species, indicating that unanchored Cu ions migrate to create active sites, thereby promoting the SCR reaction.281 The computed binding energies of H2O on both Cu+ and Cu2+ sites indicate variations in the NH3-SCR reaction mechanism on solvated Cu-CHA sites. Analysing the impact of water on intermediate species formation, notable differences in the absence and presence of physisorbed water are found. Water promotes the re-oxidation half-cycle, favouring nitrate species formation, while on a water-solvated Cu-CHA site, the generation of Cu-nitrosamine (NH2NO) is less favourable, potentially slowing down the reduction half-cycle of the NH3-SCR. The overall influence of H2O on NH2NO is more important than on nitrate, suggesting a net negative influence on activity. Additionally, previous reports indicate that hydrocarbons or residual H2O can alter or block active sites. Regarding the effect of NH3 solvation (Fig. 19; green lines), previous experimental evidence supports the improvement of NOx reduction with preadsorbed NH3 at temperatures 300 °C or 250 °C.278 However, it was observed that the activity for NO reduction remains low when temperatures fall below 150 °C.278 The adsorption energies of NH3 for Cu+-CHA and Cu2+-CHA sites have also been calculated suggesting that reactive sites occupied by NH3 molecules can affect the mechanism. Investigating the effect of NH3 on intermediate species formation, revealed that Cu bidentate nitrate (species B) formation is more favourable on physisorbed NH3 sites compared to bare sites. Moreover, the formation of Cu-nitrosamine (species G) is relatively less favourable on physisorbed NH3 active sites than on bare sites, indicating a potential deactivation of active sites for the reduction part of the NH3 + NO activation half cycle.
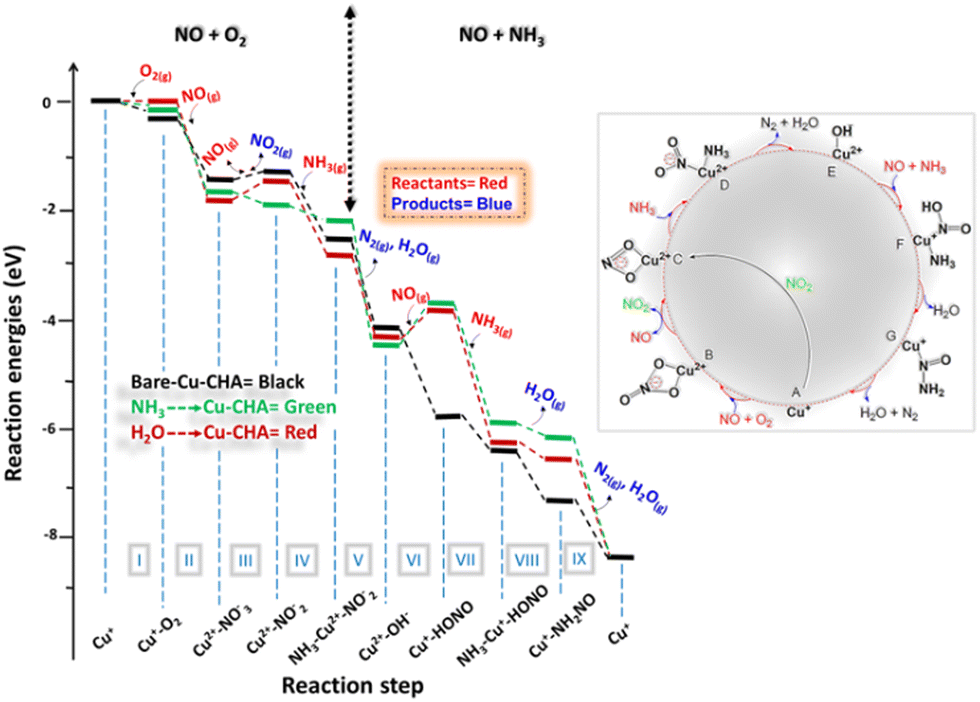 | ||
| Fig. 19 The NH3-SCR reaction on the Cu-CHA site shows energy landscapes: black represents the bare site, green with physisorbed ammonia, and red indicates water-solvated sites. The inset on the right illustrates the NH3-SCR cycle. Reproduced from ref. 195 with permission from American Chemical Society, copyright 2023. | ||
Cheng et al.282 suggested the Cu–Mn/zeolite as a potential candidate for the NH3-assisted SCR reaction using in situ DRIFTS. They found that the formation of both NH3 and NOx on the Cu-zeolite and Cu–Mn/zeolite are similar and suggested that the introduction of Mn only enhanced the concentration of the intermediate. The bidentate nitrate coordinates with the metal ion followed by the desorption of water after the adsorption of NO from the catalyst.
Marberger et al.261 correlated the rate-limiting step to the redox mediation of [Cu(NH3)2]+ to [Cu(NH3)2]+2 using transient and quick-scanning EXAFS methods. They found that the re-oxidation process is inhibited by the interaction of NH3 with the Cu+ complex. The analysis of Marberger et al.261 mainly relies on data from low Cu-loading samples. Although in these studies, the formation and chemistry of the redox mediation of [Cu(NH3)2]+ and [Cu(NH3)2]+2 complexes are well documented, the nature and role of these species in relation to other reaction intermediates are less clear.
Fe-based zeolitic systems
Previous studies134,283–286 have found several differences between Cu and Fe/zeolite SCR catalysts. Fe/zeolites exhibit low standard SCR activities below 250 °C due to strong NH3 inhibition effects. Contrary to this, Cu/zeolites can maintain high activity even around ∼175 °C. Cu/zeolites also demonstrate less sensitivity to NO2/NOx ratios in the reactant feed. The variance in NOx conversion between standard (NO2/NOx = 0) and fast (NO2/NOx = 0.5) SCR conditions is typically minimal for Cu/zeolite catalysts. While for Fe/zeolites, a NO2/NOx = 0.5 ratio is preferred for low-temperature reactivity. Furthermore, Fe/zeolites tend to be less selective in N2O formation, particularly at NO2/NOx ≤ 0.5.The NH3-SCR pathway on Fe-based zeolitic systems including ZSM-5 and MOR has been widely investigated. Through experiments and calculations, several steps have been studied including important steps such as NO oxidation and NH2NO generation, aiming to understand catalytic pathways. Li et al. reported a mechanism investigated through DFT calculations for the whole NH3-SCR process for extra framework Fe and Brønsted acid sites of the ZSM-5 zeolite.221,287 They proposed the catalytic decomposition of NO over the H-form of the zeolite (using a cluster model with 5-T sites), and found the NO oxidation step as the rate-limiting step (with an energy barrier of 15.6 kcal mol−1) at 373 K. Subsequently, the NH2NO species decomposes to H2O and N2via a hydrogen transfer mechanism. This process can be catalytically driven by the Z-[FeO]+ species over Fe-exchanged zeolite. Here, NO reacts with NH3 on the FeO+ to generate NH2NO and [FeOH]+. The activation barrier is reduced by 3.0 kcal mol−1 due to the FeO+ species.
Yasumara and colleagues7 explored the reduction half cycle in MOR by investigating the elementary steps involved in Fe3+ reduction using a mixture of NO + NH3. They utilized operando IR measurements to track intermediates and gas-phase products, considering the instability of surface intermediates during catalysis. Upon exposure to NO, they identified NO+ species with vibrational signatures at 2190 cm−1 (ref. 288 and 289) and Fe2+–NO with 1880 cm−1,290 accompanied by the detection of adsorbed NO2 and H2O (1620 cm−1) in the gas phase. They also observed the depletion of Fe3+–OH species which appeared at 3682 cm−1 (ref. 291) and Brønsted acid sites at 3638 cm−1.292 Their investigation extended to examining the Fe cationic species reactivity in zeolites towards HONO generation through transition state calculations employing a T5 (where T indicates tetrahedral site) cluster model as demonstrated in Fig. 20a. The T4 site in the MOR framework293 is recognized as a replacement site for Al among the T sites. The stability of mononuclear iron species in SSZ-13 zeolite containing isolated Al has also been investigated by Schneider group.294 According to their calculations, the phase diagram for Fe species in the presence of H2O revealed that [Fe3+(OH−)2]+ species are the most stable at temperatures below 600 °C. Hence, in their study, [Fe3+(OH−)2]+ was used as the initial structure for the active iron species. Through theoretical calculations, they considered singlet, triplet, and quintet spin multiplicities, with quintet states exhibiting the lowest energy.295,296 The energy diagram of the quintet state illustrated in Fig. 20b shows the formation of HONO species, facilitated by NO molecules attaching to an OH group of [Fe3+(OH−)2]+ with an energy barrier of 29.2 kJ mol−1. Subsequently, the generated HONO molecule desorbed with 76.3 kJ mol−1 of desorption energy. These findings implied the facile formation of HONO species, with the high desorption energy of NH3 from Brønsted acid sites (145–153 kJ mol−1)293 indicating the migration of generated HONO intermediates into Brønsted acid sites for N2 generation.
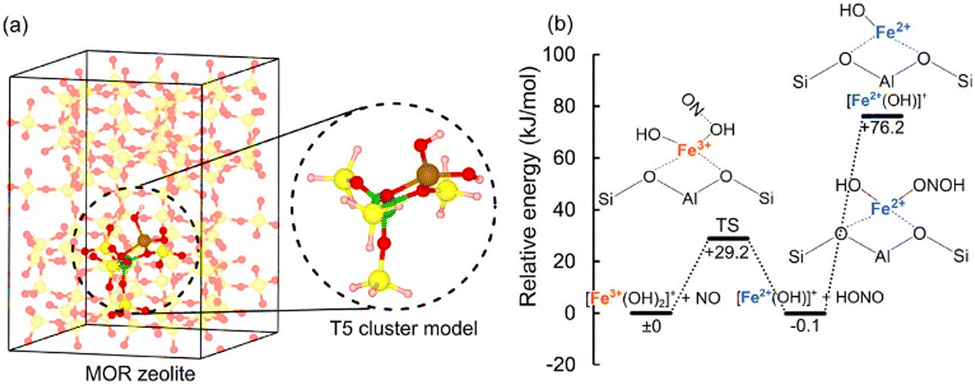 | ||
| Fig. 20 (a) Illustrates the MOR zeolite framework and the T5 cluster model used in the study, and (b) the energy diagram depicts the formation of HONO from [Fe3+(OH−)2]+ and adsorbed NO within MOR zeolites. Brown represents Fe, yellow for Si, green for Al, red for O, and white for H, providing a visual reference for molecular structures and energy transitions. Reproduced from ref. 7 with permission from American Chemical Society, copyright 2022. | ||
Bruggemann et al. also performed a series of tests to probe the NH3-SCR mechanism on ZSM-5 containing 5T atoms using DFT.297 First, they compared the NO oxidation step and the conversion of trans–cis N2O3 as an intermediate to NH2NO on Brønsted acid sites of ZSM-5. They subsequently calculated the energy barrier for the formation of nitrosamine (NH2NO) in the gas phase and over the ZSM-5 zeolite using DFT. Their results showed that the energy barrier in the case of the zeolite of 20 kcal mol−1 is more compared to the gas phase (0.16 kcal mol−1). Further, they extended the system and investigated Fe-zeolites using the cluster model approach.149,298 The catalytical cycle in this case only produces two species: nitramide (NH2NO2) and NH2NO, and consider Fe as a mononuclear species Z-[FeO]+, as mentioned earlier. In addition, several species were found on Fe sites, including FeO2N, FeONO, and FeO2NO which are considered the most prominent species in the NO oxidation cycle based on the calculated Gibbs free and activation energies which are calculated by gradient corrected DFT.
Recently, Rudolph and Jacob compared the mechanism of the SCR of NOx over Fe and Cu-doped zeolites.299 The pathway starts with the neutral models [Z-Fe3+(OH)2] and [Z-Cu2+(OH)] and proceeds via two pathways. In the first NH3 is adsorbed to generate the [Z-M(OH)2(NH3)] intermediate. This step is quite exothermic and requires a subsequent shifting of the proton from NH3 to the OH− resulting in the formation of [Z-Fe3+(OH)(H2O)(NH3)] and then release water molecules leading to [Z-Fe3+(OH)(NH2)] with overall endothermic energy of 82 kJ mol−1 in the case of Fe while the same steps for Cu need only 56 kJ mol−1. Some previous reports proposed this proton transfer (in the case of Cu) to the zeolite framework rather than to the OH− ligands85,300,301 which requires 114 kJ mol−1 energy. The intermediate [Z-Fe3+(OH)(NH2)] interacts with NO in two different ways: first, NO coordinates with this intermediate to generate [Z-Fe2+(OH)(NH2)(NO)] and reduce the Fe3+ to Fe2+ followed by rearrangement that leads to the formation of [Z-Fe2+(OH)(NONH2)].
Other TMI-based zeolitic systems
Employing operando IR experiments, Yasumura et al.302 compared the transient reactions of pre-adsorbed NH3 with NO and NO + O2. Prior to the experiment, the IR disk of pre-oxidised Cr2.3β5 – beta zeolite (Si/Al = 5) with 2.3 wt% chromium. Cr2.3β5 was subjected to 0.1% NH3 for 1200 seconds at 200 °C, resulting in the observation of B–NH3 (1450 cm−1) and L–NH3 (1628 cm−1) species. Upon exposure to NO, more than half of the B–NH3 species were consumed within 2000 seconds. Concurrently, the generation of N2 was detected by mass spectrometry (MS). During TPR of pre-oxidised Cr2.3β5 with NO, in situ IR and gas-phase analysis showed the presence of NO+ and NO3− species below 200 °C.303–305 NO+ decreased while NO3− initially increased (up to 70 °C) before declining. NO2 evolved in the low-temperature range (35–300 °C), peaking around 130 °C. NO reduction Cr6+/Cr5+ led to Cr3+ species formation on Cr2.3β5, with NO2 possibly originating from surface intermediates: NO → NO+ → NO3− → NO2.When the same sample was treated with NO + O2 gas flow, the complete consumption of B–NH3 occurred within 2000 seconds, accompanied by a higher total production of N2 compared to the NO case. The consumption of B–NH3 under NO + O2 atmosphere was attributed to the persistent reduction/oxidation cycles (pseudo-steady-state SCR). The enhanced reactivity of B–NH3 in the presence of NO + O2 compared to NO has also been reported for V2O5-based catalysts and Cu-zeolites.306 The reaction mechanism that follows steady-state NH3-SCR as shown in Fig. 21, does not follow the NH3 adsorption species. However, the reactions between NOx and preadsorbed NH3 play a crucial role during transient diesel deNOx conditions. IR investigations on the NH3-SCR mechanism have focused on the reaction between adsorbed NO + O2 and NH3. However, some reports306 considered this approach unsuitable for analysing the NH3-SCR process over Cu-CHA under realistic conditions. Particularly, for the species, such as Cr3+, which are formed through the reduction of Cr species by NH3 and adsorbed NO. These reduced species can be re-oxidised by O2, thereby ensuring a continuous formation of N2 through the ongoing redox cycles of the reaction between the reduced high-valent Cr species and OCH (pseudo-steady-state SCR).
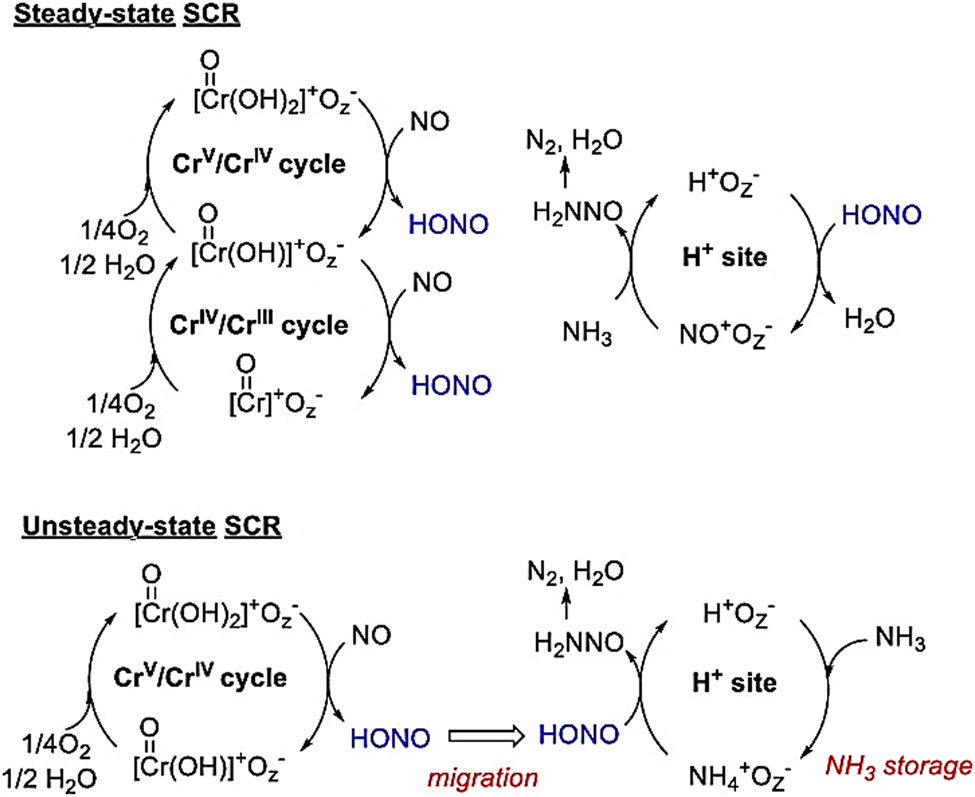 | ||
| Fig. 21 Mechanism of NH3-SCR Catalyzed with Cr-zeolite catalysts in the steady-state (top) and unsteady-state (bottom) in which the mobile HONO intermediate migrates and reacts with the adsorbed NH3. Reproduced from ref. 17 with permission from American Chemical Society, copyright 2015. | ||
Deactivation mechanisms
TMI stability
SCR catalysts for mobile applications must meet demanding criteria, including stable operation across a wide range of temperatures (from low engine start temperatures ∼150 °C to high hydrocarbon contaminant burn-off temperatures > 500 °C), long lifespans, and resistance to poisoning.307 Cu-ZSM-5 was initially introduced for NH3-SCR in 198687 but falls short of meeting lifespan conditions owing to deactivation in tailpipe conditions.90,308,309 Research indicates that zeolite catalysts with larger pores are more susceptible to the influence of hydrocarbon (HC) species, as demonstrated by several studies.310–312 For instance, medium-pore zeolites like Cu-ZSM-5 and large-pore zeolites such as Cu-Beta tend to deactivate more readily compared to small-pore zeolites like Cu-SSZ-13.313 This difference arises because the small-pore structure of Cu-SSZ-13 restricts the diffusion of HC species into the zeolites effectively. The unique small-pore configuration of Cu-SSZ-13, with channels approximately 3.8 Å in diameter, minimizes the impact of long-chain hydrocarbons on its performance, as it restricts the entry of these molecules into the zeolite pores.313Generally, small-pore zeolites containing metal ions (Fe, Cu, Co, Mn) are hydrothermally more stable than medium or large-pore zeolites.314,315 The hydrothermal stability of Cu-CHA has been extensively studied recently and is considered a highly important criterion for effective NOx-SCR316–319 catalysts. Despite its activity, the evolution of Cu from an active to inactive form during NOx-SCR is seriously problematic.320–322 Lee et al.323 studied the NH3-SCR performance and hydrothermal stability of Cu-SSZ-13, Cu-SSZ-5, Cu-Beta, and Cu-UZM-35 after treating at 750 °C, and in the order as follows; Cu-SSZ-13> Cu-UZM-35> Cu-SSZ-5> Cu-Beta. A similar investigation was described by Kwak et al.324
Ryu et al.325 studied the Cu-exchanged LTA zeolite (high silica polymorph) and demonstrated that LTA is a new contender to substitute for Cu-SSZ-13. They tested the Cu-LTA zeolite after hydrothermal ageing at a high temperature (900 °C) under which commercial SSZ-13 deactivates. Also, they found that the local environment of Cu2+ in LTA is different from that in SSZ-13. Cu2+ is present at the centre of the 6-MR and not only acts as a catalytically active site but also as a suppressor of dealumination.
To examine the role of Brønsted acid site (BAS) strength after undergoing hydrothermal ageing, Wu et al.326 performed operando electron EPR analysis on different Cu-exchanged zeolites. To ensure the catalyst's structural integrity while inducing dealumination, an ageing temperature of 850 °C was chosen. This temperature effectively removes Al from the supports without causing complete structural damage. Upon comparing the levels of isolated Cu2+ ions in the hydrothermally aged catalysts, referred to as “HTA850,” with the fresh catalysts, it becomes apparent that ageing leads to a partial reduction in isolated Cu2+ ions due to the generation of EPR-silent Cu species like Cu-aluminates and CuxOy clusters. The most significant reduction in isolated Cu2+ ions was observed in Cu/LTA, followed by Cu/CHA-b (Si/Al ratio = 10), Cu/CHA-a (Si/Al ratio = 17), and Cu/AEI.
Fig. 22a and b depict a comparison of the 27Al magic angle spinning (MAS)-NMR spectra between the HTA850 and fresh catalysts. In the fresh catalysts, there is a noticeable 27Al signal at approximately 50 ppm, indicating the presence of framework tetrahedral Al (AlTd) ions with a signal that appeared around 0 ppm which can be attributed to extra-framework octahedral Al (AlOh) species. Conversely, for HTA850 catalysts, all spectra exhibit an additional broad 27Al signal at around 30 ppm, indicating the presence of penta-coordinated Al (AlP) species. The order Cu/LTA > Cu/CHA-a > Cu/AEI > Cu/CHA-b in terms of hydrothermal stability suggests that hydrothermal stability relies on both the composition of the zeolite supports and the framework topology.
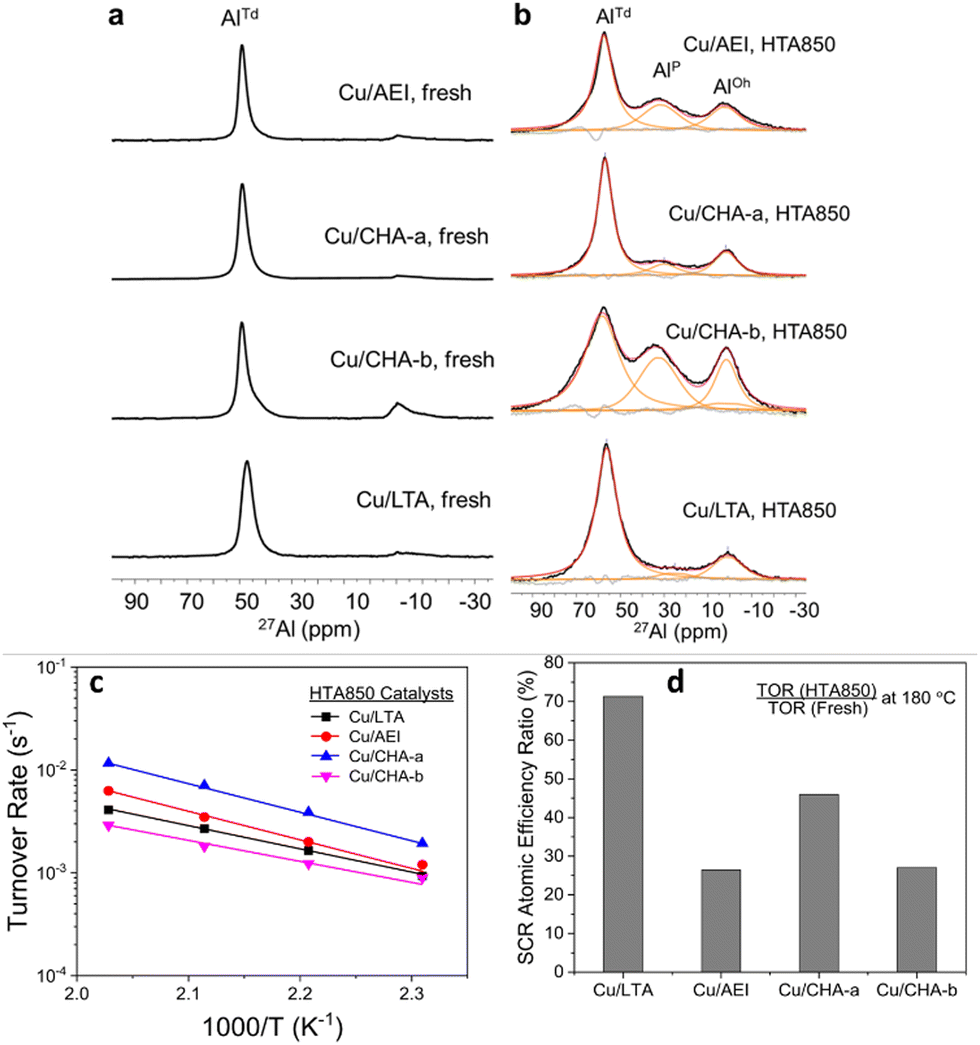 | ||
| Fig. 22 (a) Spectra of fresh (a) and hydrothermally aged (b) Cu-zeolites. Arrange the turnover rates (TORs) as Arrhenius plots (c), and illustrate turnover rate (TOR) ratios at 180 °C between freshly prepared catalysts and (d) those subjected to hydrothermal ageing. Reproduced from ref. 326 with permission from Springer Nature, copyright 2023. | ||
Additionally, Fig. 22c illustrates turnover rates derived from the first-order kinetic analysis of light-off SCR data. When comparing the effectiveness of Cu sites in aged Cu/LTA with those in aged Cu/AEI and Cu/CHA, a notable alignment is observed, indicating the significant role of zeolite Brønsted acid sites in regulating rates. By removing these sites through dealumination, their kinetic effect diminishes. The examination of turnover rates (TOR) values for the fresh and HTA850 catalysts at 180°C are shown in Fig. 20d, which shows that the aged catalysts maintain an atomic efficiency order: Cu/LTA > Cu/CHA-a > Cu/AEI > Cu/CHA-b, further supporting the importance of Brønsted acid sites in kinetics. Noteworthy is the migration of Cu+ ions initially located within the sterically distant sod cages of Cu/LTA to the 6-MRs at 750–900°C. This migration, coupled with oxidation to Cu2+ ions, contributes to the increase in low-temperature NH3-SCR activity and Cu atomic efficiency observed in aged Cu/LTA as reported by Nak et al.327 Thus, the zeolite cage topology emerges as a critical factor suggesting the sustained high Cu atomic efficiency in aged Cu/LTA.
Furthermore, Wu et al.326 conducted an operando EPR study on fresh catalysts across a temperature range of 50 to 350 °C, specifically examining temperatures of 50, 150, 225, and 350 °C. At 50 °C, which is below the SCR light-off temperature, Cu ions are found exclusively in the +2 oxidation state and are solvated by NH3. This occurs due to ligand displacement from H2O to NH3, which binds more strongly to Cu2+ ions. The study reveals that not all Cu2+ cations exhibit the same level of mobility. Cu2+ ions with restricted mobility show anisotropic properties (g|| = 2.25, A|| = 170 G, high-field signals around 3300 G), whereas those with unrestricted mobility are isotropic (gave = 2.18, high-field signals around 3260 G). The Cu2+ ions with restricted mobility likely have both NH3 and zeolite lattice oxygen ligands, while the freely mobile ions are associated with Cu2+(NH3)4 and Cu2+(OH)(NH3)3 complexes. NH3-solvated Cu2+ ions in Cu/LTA show greater mobility compared to those in Cu/AEI and Cu/CHA, resembling the mobility of H2O-solvated Cu2+ ions. At 50°C, the formation of a mobile [Cu2+(NH3)3(NO3)]+ complex, as demonstrated by Negri et al.,328 is unlikely under steady-state conditions since Cu2+–(NOx) y-type species only form when the catalyst is not fully covered by NH3, as noted by Marberger et al.261 Even if small amounts of [Cu2+(NH3)3(NO3)]+ complex form, its signal would be overwhelmed by the stronger signals of [Cu2+(NH3)4] and [Cu2+(OH)(NH3)3].
The hydrothermal ageing of the Cu–CHA catalyst resulted in a notable decline in NH3 oxidation at high temperatures.329 The TPD profiles were primarily resolved into three peaks, centred at 317 °C, 456 °C, and 526 °C, respectively. Specifically, the intensity of the peak at 317 °C increased with hydrothermal ageing, while the intensities of the peaks at 456 °C and 526 °C witnessed a decrease and, therefore, a reduction in the availability of active sites for NH3 oxidation. While the study did not explicitly address the effects of ageing on standard SCR activities, the decrease in NH3 oxidation suggests a potential indirect influence on the overall SCR activity of the Cu–CHA catalyst.
As noted, Cu-based zeolites have generally been considered to be the most efficient contender in the SCR of NOx. However, zeolites e.g. ERI, AEI, CHA, and AFX have also been explored with Fe ions as an active site.330 Some comparison regarding reaction kinetics for both Fe and Cu was made by Gao and co-workers.331 Fresh Fe-zeolites are comparatively less active than Cu-zeolites under wet conditions in SSZ-13 as shown in Fig. 23.332 However, on comparing the fresh and aged versions of both catalysts, the Fe-version retained more activity than the Cu-version after ageing at high temperatures (above 500 °C).
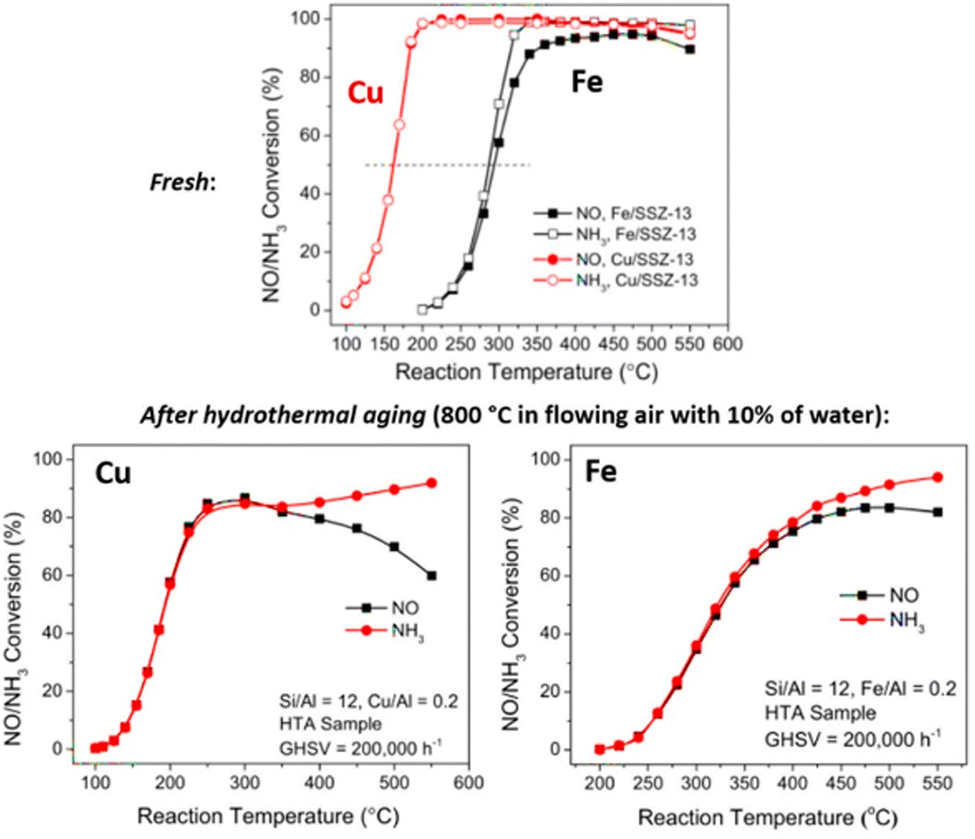 | ||
| Fig. 23 A comparison between Cu-SSZ-13 and Fe-SSZ-13 was conducted under standard wet SCR conditions, both before (fresh) and after hydrothermal ageing. Details of the conditions are provided in the inset. Reproduced from ref. 332 with permission from American Chemical Society, copyright 2018. | ||
In another report, they also studied the effect of Fe on high silica LTA at a hydrothermal ageing temperature of 900 °C,333 and investigated the stability and hydrothermal activity of LTA and compared with that of commercial SSZ-13. The hydrothermal stability of both catalysts was found to decrease at 900 °C; however, the extent of decrease in the case of Fe-LTA was smaller as compared to Fe-SSZ-13. Furthermore, the activity profile of Fe-LTA was much better than that of Fe-SSZ-13. The iron species, including Fe3+ and small oligomeric species such as FexOy, present in Fe-LTA, exhibit greater stability compared to those in Fe-SSZ-13 owing to variations in the environment surrounding the Fe sites within the framework.
Ruggeri et al. examined the temperature dependence of hydrothermal ageing treatment and its influence on the duration of the Fe-zeolite catalyst.334 They carefully examined the NH3 adsorption, oxidation, and its reaction with NOx. The deactivation was very fast and the primary effect of hydrothermal ageing on the SCR reaction was the oxidation of NH3.
The inclusion of Fe into small-pore zeolites leads to a hydrothermally stable NH3-SCR catalyst.330 However, conventional iron exchanged methodologies appear to be difficult as the size of the Fe3+ aqueous (hexahydrate) ions (9 Å) is bigger than the typical pore size of the zeolite (~ 3.8 Å)335 which is also the case for the Cu hexahydrate. Martin et al.336 investigated the nature of Fe-doped zeolites (prepared by the one-pot method) using different characterization techniques. They discovered that the material (Fe-zeolite) synthesised through a one-pot procedure exhibited better NO conversion and hydrothermal stability (due to the improved dispersion of Fe ions) as compared to the post-synthetic ion-exchange method. In addition, the same group prepared Fe-Beta zeolites using two different reaction media: (i) alkaline conditions and (ii) a fluoride medium for synthesizing iron-containing zeolites. A comprehensive investigation of the direct preparation of the Fe-containing material was conducted to gain insights into how the physicochemical conditions of the synthesized catalyst affect the SCR-NOx conversion and the hydrothermal stability under rigorous conditions.337 The Fe–Beta zeolite, synthesized without alkali cations at a Si/Al ratio of approximately 13, exhibited high NO conversion (>90%) but suffered severe deactivation at high temperatures in steam. Conversely, Fe–CHA zeolites, prepared under alkaline conditions with the same Si/Al ratio using N,N,N-trimethyladamantylammonium as a template, demonstrated superior catalytic activity and high hydrothermal stability, particularly after selective removal of sodium cations.
Manganese-based ZSM-5 catalysts have been the subject of detailed investigation. Lou et al.338 explored the impact of calcination on the performance of Mn-ZSM-5 catalyst activity for NH3-SCR. They observed that at lower temperatures (<500 °C), Mn3O4 and amorphous MnO2 were predominant, while at higher temperatures (600 °C), Mn2O3 formed, which negatively affected the activity. The catalyst treated at 300 °C exhibited excellent performance which can be attributed to the presence of an abundance of amorphous MnO2, and Mn3O4. Additionally, Zr-doped Mn-ZSM-5 catalysts achieved over 90% NOx conversion at elevated temperatures, owing to enhanced Mn dispersion, increased Mn species on ZSM-5 surfaces, and the interaction between Mn and Zr.13
Sue et al.199 conducted a comparative analysis of SSZ-13 (Si/Al = 10) and SAPO-34 (Si/P/Al = 0.19:0.63:1) during hydrothermal ageing at 800 °C, noting that SAPO-34 exhibits greater stability compared to SSZ-13. The findings indicated dealumination within the SSZ-13 zeolite, resulting in loss of crystallinity and a significant reduction in Brønsted acid site concentration. However, during hydrothermal ageing, both samples experience desilication (in SAPO-34) and dealumination (in SSZ-13).
Catalyst poisons
Catalyst deactivation due to poisoning is a significant concern in industrial catalytic processes including NH3-SCR of NOx reduction, as it can lead to reduced efficiency and increased operational costs. Sulfur (S) poisoning is widely recognized and thoroughly investigated, with substantial literature detailing its mechanisms and effects.339–343 However, other potential poisons, such as phosphorus and zinc, also merit consideration.8,344,345 These elements, often introduced through contaminants in feedstocks or additives in lubricants, may influence catalyst performance and longevity. Although their impact is sometimes debated, understanding the breadth of catalyst poisons is crucial for developing more resilient catalytic systems. Sulfur oxides present in the exhaust gas can deactivate both metal oxide and metal cation-exchanged zeolite catalysts utilized in NH3–SCR. Enhancing chemical poisoning tolerance, especially against SO2, P, and alkali metals, is crucial for catalyst development. Despite thermal treatment's promise in recovering S-poisoned zeolites, steady-state SO2 poisoning remains problematic due to ammonium sulphate and copper bisulphite formation. Incorporating sacrificial metals shows potential for enhancing SO2 resistance.The deposition of (NH4)2SO4/NH4HSO4 and metal sulfate can also block and impair active sites.346 While (NH4)2SO4/NH4HSO4 deposition may undergo decomposition under specific conditions, the primary concern remains the irreversible chemical poisoning of active sites.
The Cu-CHA catalyst is susceptible to SO2 poisoning, leading to the formation of compounds like (NH4)2SO4 and CuSO4, as well as the chemisorption of SO2 on isolated Cu2+ ion sites.347 The main cause of Cu2+ deactivation is proposed to be the blockage caused by (NH4)2SO4 on the Cu2+ sites.348 Cu-CHA is known to contain significant quantities of ZCuOH, making it prone to SO2 adsorption, resulting in severe poisoning through the reaction between ZCuOH species and SO2.349 Moreover, the external environment has also an obvious effect. For example, in dry conditions, weakly bound SO2 and copper sulfate are observed, while in humid environments, there is a higher content of copper sulfate.350
(NH4)2SO4 formation is not considered the ‘death knell’ of the SCR catalyst however as it decomposes completely, restoring catalyst activity after treatment at 500 °C. However, the activity of Cu-CHA cannot fully return to its original level due to the high stability of the resultant copper sulfate. In the absence of oxygen, catalyst regeneration to overcome sulfur poisoning proves effective as it avoids the formation of stable sulfates and occurs at lower temperatures. However, in the presence of oxygen, ammonium sulfate converts into stable metal sulfates, posing an obstacle.351
In the case of deactivated Cu-ZSM-5 retaining crystallinity, there is a loss of framework Al due to their migration through the 10-MRs, coupled with copper mobility and sintering. However, Cu-SSZ-13 remains unaffected under the same conditions.186,308,309 Nevertheless, there are substantial gaps in recognizing the precise deactivation process. The reason might be due to the small size of deactivating species that renders them difficult to determine by experimental techniques, such as X-ray diffraction, while the presence of paramagnetic copper can disrupt locally sensitive methods like NMR. Hence, reconstructing the distribution of single ions in three dimensions is essential for a comprehensive understanding of deactivation processes.
Phosphorus poisoning in SCR catalysts, caused by engine oils like zinc dialkyldithiophosphate (ZDDP), is commonly considered irreversible and affects catalytic activity through physical and chemical mechanisms.352,353 Studies on Cu-exchanged CHA catalysts345,354,355 reveal phosphorus’ dual role as a promoter and inhibitor, impacting both high- and low-temperature selectivity/conversion and hydrothermal stability. Phosphorus affects Cu ions differently, with P2O5 and PO3−/PO43− responsible for poisoning Z2Cu and ZCuOH, respectively.354 Recent research356 suggests that controlled phosphate addition can preserve zeolite frameworks and enhance SCR activity. Low phosphorus content may improve SCR performance.352 While phosphorus has minimal impact on Cu/FER catalysts, its co-addition with sodium significantly deactivates them.353
Addressing the challenge of maintaining catalyst activity in alkaline dust-containing conditions prevalent in industrial exhaust is essential. Such conditions, loaded with compounds including calcium, potassium, and sodium, can poison SCR catalysts, particularly V-based ones.357–359 Mitigation strategies include increasing active phase content, periodic washing of poisoned catalysts, and designing optimized catalysts.360
Understanding NH3-SCR mechanisms from experiments and theory
To construct a more unified understanding of the NH3-SCR mechanisms, it is essential to integrate insights from both experimental and theoretical studies, highlighting areas of agreement and pinpointing discrepancies that necessitate further investigation. The reviewed literature presents a complex picture of the SCR process, with multiple mechanisms proposed, particularly concerning the roles of Cu and Fe ions in zeolite frameworks. A key point of contention lies in the comparison between the L–H and E–R mechanisms. The NH3-SCR reaction is often described as following the L–H mechanism, where both NH3 and NOx adsorb onto the catalyst surface before reacting. However, an alternative E–R mechanism suggests that pre-adsorbed NH3 can react directly with gas-phase NO.The coexistence of these mechanisms and their relative contributions under different conditions (e.g., temperature, gas composition) remain areas of active research. Theoretical studies support the occurrence of both mechanisms, but the experimental evidence is sometimes conflicting, particularly regarding the dominance of one pathway over the other under specific reaction conditions. In Cu-based zeolites, the redox cycling between Cu+ and Cu2+ is critical for SCR activity. Experimental evidence, including XANES and operando UV-Vis spectroscopy, confirms the dynamic nature of Cu species under reaction conditions, with the Cu+ to Cu2+ transition being rate-limiting at lower temperatures. However, discrepancies arise regarding the exact nature of intermediate species, such as the role of Cu–nitrate complexes and whether these intermediates are essential or merely spectators in the reaction. Some studies emphasize the importance of these complexes in the SCR cycle, while others argue that their role may be overstated, pointing to a need for more targeted experiments to clarify these points
The behavior of Fe-based systems in NH3-SCR also presents unique challenges. The reaction mechanisms in Fe-zeolites differ significantly from those in Cu-zeolites, with Fe species showing a preference for higher temperature operations and different sensitivities to NO2/NOx ratios. Theoretical models suggest that the presence of Fe3+ and FeO+ species is crucial, yet the exact mechanistic details, such as the formation and decomposition of intermediates like NH2NO, remain under debate. The complexity of Fe species' behaviour, particularly in mixed Fe–Cu systems, adds another layer of uncertainty, with experimental data sometimes contradicting theoretical predictions.
Water and sulfur poisoning are additional factors that complicate the NH3-SCR reaction. Water generally enhances low-temperature SCR activity by facilitating Cu ion mobility, while sulfur tends to poison active sites, leading to catalyst deactivation. The literature presents a unified understanding of the detrimental effects of sulfur, yet there are gaps as to how different poisoning mechanisms affect various zeolite frameworks and active sites. For example, the regeneration of sulfur-poisoned catalysts under different conditions remains an area where experimental results are inconsistent, indicating a need for more systematic studies.
Machine learning in catalysis
The integration of machine learning (ML) techniques into the field of catalysis, particularly for the NH3-SCR, is assisting in the optimisation of catalytic processes. Martini et al.361 utilise in situ XAS combined with wavelet transform (WT) analysis and ML-assisted fitting to investigate the effects of zeolite composition on the structure and reactivity of Cu-CHA catalysts. This approach enables a detailed assessment of the local coordination environment of copper ions during the catalytic cycle. The Si/Al ratio significantly influences the formation and stability of dicopper(II) complexes during the oxidation phase. Al-rich zeolites (low Si/Al ratio) promote a planar coordination motif with longer Cu–Cu distances, enhancing NOx conversion efficiency at low temperatures (200 °C). During the NH3-SCR process, Cu ions dynamically switch between framework-coordinated and mobile configurations.The Cu density and its distribution within the zeolite framework are critical for effective O2 activation, which is essential for the oxidation of Cu+ to Cu2+. Fig. 24 illustrates the in situ Cu K-edge XANES for various Cu-CHA catalysts collected at 200 °C after pretreatment in O2, at the end of the reduction step, during and after the oxidation step. The Pi charts included show the Cu+/Cu2+ percentages evaluated by XANES linear combination fitting (LCF) at the end of the oxidation step for each catalyst. This figure depicts the influence of zeolite composition on the structure of the Cu species during the NH3-SCR process, underscoring the role of the Si/Al ratio in dictating the catalytic efficiency.
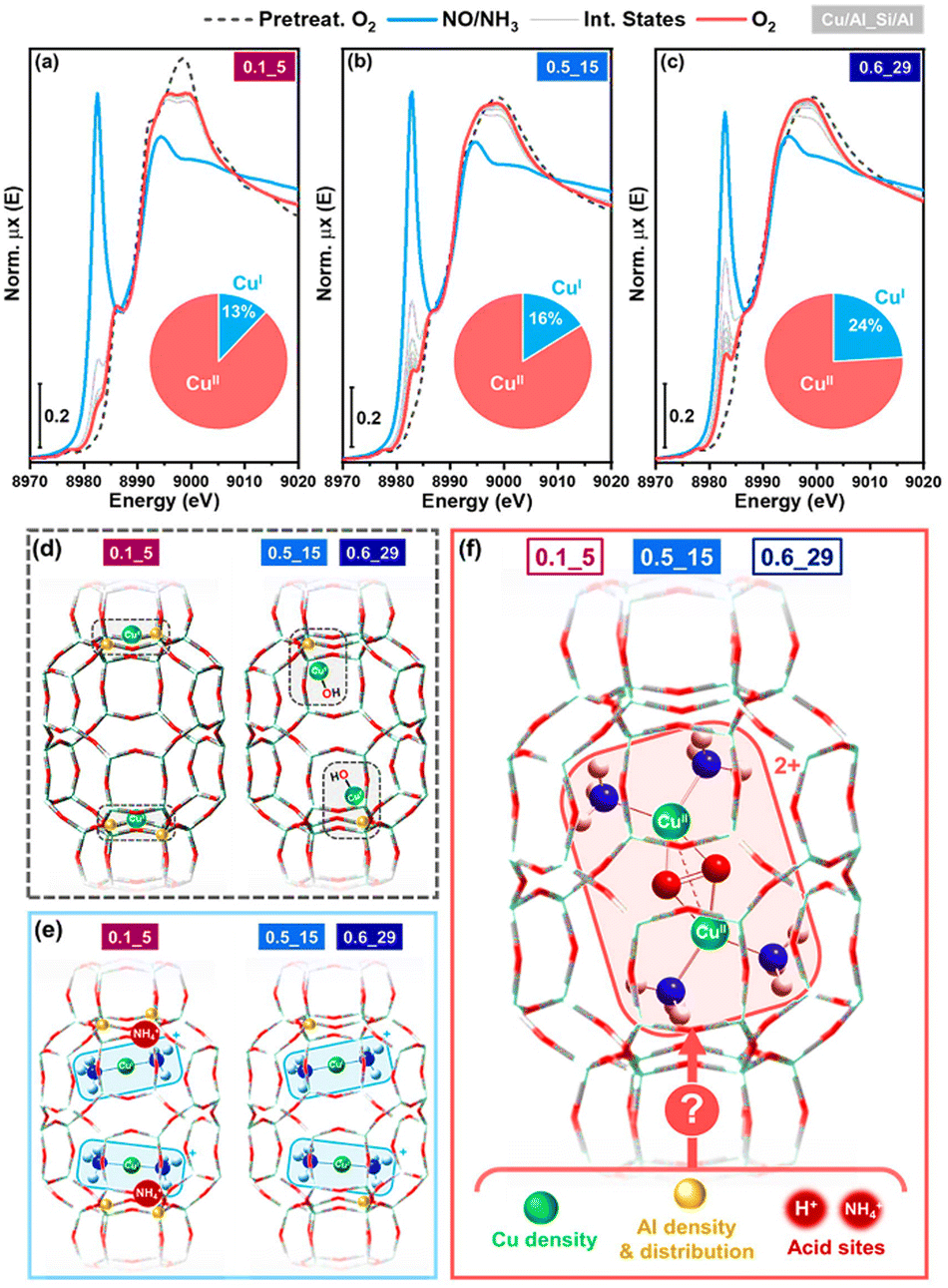 | ||
| Fig. 24 In situ Cu K-edge XANES spectra for Cu-CHA catalysts: (a) 0.1_5, (b) 0.5_15, and (c) 0.6_29. These spectra were collected at 200 °C following pretreatment in O2, after the reduction step, and after the oxidation step. The accompanying pie charts represent the relative percentages of Cu+ and Cu2+ species, as determined by XANES linear combination fitting (LCF) at the end of the oxidation step for each catalyst. Under the experimental conditions used, Cu+ and Cu2+ correspond to the complexes [Cu(NH3)2]+ and [Cu2(NH3)4O2]2+, respectively. Panels (d)–(f) depict schematic representations of the predominant Cu species at each stage, highlighting the impact of catalyst composition and raising questions about the factors that influence the coordination environment in [Cu2(NH3)4O2]2+ complexes, particularly in part (f). Reproduced from ref. 361 with permission from American Chemical Society, copyright 2022. | ||
Conclusions and outlook
This review has highlighted the complexity of the chemistry of transition metal exchanged zeolites used in the burgeoning field of NH3-assisted SCR of NOx reactions. Developing a comprehensive picture of the structure and mechanism in TMI exchanged zeolites is an essential step in understanding these catalytic reactions if it is to become possible to design the next generation of catalysts with improved performance metrics. To this end the identification of the most active TMI species, differentiating between the various monomer, dimer, and oligomers and their properties is clearly necessary although will be difficult to control synthetically.Simulations reveal the dynamic nature of metal-ion active species at the molecular level, with the diffusion of the Cu ions being determined by the electrostatic binding to the zeolite framework, which shows how the zeolite framework controls the movement of cationic species. Modelling studies, including the use of QM/MM techniques, can also give detailed information on mechanisms. QM/MM modelling of TMI zeolites for SCR is promising, offering detailed insights into catalytic mechanisms. In addition, quantum computing holds the potential to screen more pathways and consider realistic scenarios including temperature and solvent effects, advancing catalyst optimization for SCR. Conventional spectroscopic techniques including in situ and operando DRIFTS, XANES, EPR, and DR-UV-vis, can provide direct evidence regarding, for example, NH3 solvation on isolated metal sites by detecting the change in the metal coordination environment, but currently lack the resolution or sensitivity to unravel detailed mechanistic steps. However, modern interrogation and analysis methods utilising modulated excitation (ME) or MCR have become increasingly popular and available ‘seeing’ more of the transient metal ion species present at various stages of reaction.
MCR is useful in catalytic studies where multiple transient species coexist. However, ME improves transient species detection by modulating external parameters and analysing system responses with phase-sensitive detection. For example, the ME technique can detect changes in metal ion coordination during NH3 solvation, offering detailed information on solvation effects in catalysis. The combination of experiment and theory appears to be critical for detailed mechanistic insight.
The main obstacle for zeolite-based SCR reactions is their durability under hydrothermal conditions, although metal ions containing small-pore zeolites are consistently more hydrothermally stable than medium or large-pore materials. Alas, there are few small pore zeolites and none whose performance matches that of CHA, AEI and LTA for NH3-SCR. The salient structural characteristics of these active topologies comprise 6-MR and 8-MR leading to large cages and hence future zeolite topology synthesis strategies could consider using synthesis conditions (i.e. templates and Si/Al > 15) known to favour the formation of small pore topologies. However, the design of reaction-specific zeolites is still a long way from being a reality and even if new topologies could be obtained there is little assurance that they will fulfil all the diverse operation requirements i.e. exhibiting high activity at low temperatures with stability at high (hydrothermal) temperatures. It is interesting to consider the need for further evolution and understanding of zeolite-based NH3-SCR catalysts considering the transition away from liquid fuels particularly in Europe by 2035. Here we note that less developed countries will find it harder to decarbonise and the use of liquid fuels is expected to continue as the population grows and the quality of life improves hence there remains a need to continue researching this field. Furthermore, static NOx emissions from e.g. nitric acid plants will also continue to grow in order to support population growth again requiring mitigation using SCR technologies. Heavy-duty vehicles requiring more torque than what electrical powertrains typically provide are also being investigated to run off hydrogen internal combustion engines (H2-ICE) which will also require NOx emission mitigation.
A detailed understanding of the structures and dynamics of transition metal cationic species through the combination of experimental and computational modelling will be essential in understanding TMI as catalytic sites in zeolites and in gaining further mechanistic insight into the NH3-SCR reaction.
Author contributions
The manuscript was drafted by JAN and was reviewed, edited and modified by CRAC and AB.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the UK Catalysis Hub funded by EPSRC grants EP/R026939/1, and EP/R026815/1. We would like to acknowledge the financial support for Jamal Abdul Nasir from the Project Management Unit (PMU) Higher Education Department of KPK, Pakistan. We are grateful to Dr Alexey A. Sokol for several useful discussions on zeolite science.Notes and references
- Y. Li and J. Yu, Nat. Rev. Mater., 2021, 6, 1156–1174 CrossRef CAS.
- A. Nicula, D. Stamires and J. Turkevich, J. Chem. Phys., 1965, 42, 3684–3692 CrossRef CAS.
- J. Turkevich, Y. Ono and J. Soria, J. Catal., 1972, 25, 44–54 CrossRef CAS.
- R. Hamada, Y. Shibata, S. Nishiyama and S. Tsuruya, Phys. Chem. Chem. Phys., 2003, 5, 956–965 RSC.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef CAS PubMed.
- S. Yasumura, Y. Qian, T. Kato, S. Mine, T. Toyao, Z. Maeno and K.-I. Shimizu, ACS Catal., 2022, 12, 9983–9993 CrossRef CAS.
- A. Guo, K. Xie, H. Lei, V. Rizzotto, L. Chen, M. Fu, P. Chen, Y. Peng, D. Ye and U. Simon, Environ. Sci. Technol., 2021, 55, 12619–12629 CrossRef CAS PubMed.
- Y. Tan, S. Yoon, C. R. Ruehl, J. Herner, P. Henderick, T. Montes, J. Latt, A. Lee, E. Florea and S. Lemieux, Environ. Sci. Technol., 2021, 55, 13657–13665 CrossRef CAS PubMed.
- L. Huang, Y. Zeng, Z. Chang, Y. Zong, H. Wang, S. Zhang and Y. Yu, Mol. Catal., 2021, 506, 111565 CrossRef CAS.
- A. M. Beale, I. Lezcano-Gonzalez, T. Maunula and R. G. Palgrave, Catal., Struct. React., 2015, 1, 25–34 Search PubMed.
- Y. Xin, Q. Li and Z. Zhang, ChemCatChem, 2018, 10, 29–41 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J.-Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- W.-B. Zhang, J.-L. Chen, G. Li, W. Zheng, G.-H. Wang, S.-K. Zheng and X.-Q. Wu, J. Fuel Chem. Technol., 2021, 49, 1294–1315 CrossRef CAS.
- J. Chen, W. Huang, S. Bao, W. Zhang, T. Liang, S. Zheng, L. Yi, L. Guo and X. Wu, RSC Adv., 2022, 12, 27746–27765 RSC.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- T. V. Janssens, H. Falsig, L. F. Lundegaard, P. N. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko and C. Lamberti, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- M. Iwamoto, H. Furukawa and S. Kagawa, Studies in Surface Science and Catalysis, Elsevier, 1986, vol. 28, pp. 943–949 Search PubMed.
- S. Mohan, P. Dinesha and S. Kumar, Chem. Eng. J., 2020, 384, 123253 CrossRef CAS.
- J. Cheng, S. Han, Q. Ye, S. Cheng, T. Kang and H. Dai, Res. Chem. Intermed., 2019, 45, 2023–2044 CrossRef CAS.
- J. Wang, D. Fan, T. Yu, J. Wang, T. Hao, X. Hu, M. Shen and W. Li, J. Catal., 2015, 322, 84–90 CrossRef CAS.
- M. Kang, E. D. Park, J. M. Kim and J. E. Yie, Appl. Catal., A, 2007, 327, 261–269 CrossRef CAS.
- B. Ye, B. Jeong, M.-J. Lee, T. H. Kim, S.-S. Park, J. Jung, S. Lee and H.-D. Kim, Nano Convergence, 2022, 9, 51 CrossRef CAS PubMed.
- J. L. Sorrels, D. D. Randall, K. S. Schaffner and C. R. Fry, EPA air pollution control cost manual, 2019, p. 7 Search PubMed.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- M. K. A. Wardana and O. Lim, Catalysts, 2022, 13, 67 CrossRef.
- F. Klingstedt, K. Arve, K. Eränen and D. Y. Murzin, Acc. Chem. Res., 2006, 39, 273–282 CrossRef CAS PubMed.
- U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, ACS Catal., 2013, 3, 413–427 CrossRef CAS.
- N. Sandhu, X. Yu and M. Zheng, Int. J. Automot. Technol., 2024, 4 Search PubMed.
- P. C. Ogugua, E. Wang, Z. Jinyang, Q. Wang and H. Su, Environ. Sci. Pollut. Res., 2023, 30, 84972–84998 CrossRef CAS PubMed.
- Y. Sun, Y. Shan, W. Ding, J. Du, Z. Liu, X. Shi, M. Ren, Y. Wang, X. Wang and Y. Yu, Catal. Today, 2024, 433, 114690 CrossRef CAS.
- D. Peitz, A. Bernhard and O. Kröcher, Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, Springer, 2014, pp. 485–506 Search PubMed.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- Q. Sun, N. Wang and J. Yu, Adv. Mater., 2021, 33, 2104442 CrossRef CAS PubMed.
- C. Baerlocher and L. McCusker, Google Scholar, there is no corresponding record for this reference, 2021.
- B. E. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2017, 118, 2718–2768 CrossRef PubMed.
- M. Deem, J. Newsam and J. Creighton, J. Am. Chem. Soc., 1992, 114, 7198–7207 CrossRef CAS.
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- G. Yang, Y. Wei, S. Xu, J. Chen, J. Li, Z. Liu, J. Yu and R. Xu, J. Phys. Chem. C, 2013, 117, 8214–8222 CrossRef CAS.
- Q. Sun, Z. Xie and J. Yu, Natl. Sci. Rev., 2018, 5, 542–558 CrossRef CAS.
- Q. Wang and D. Astruc, Chem. Rev., 2019, 120, 1438–1511 CrossRef PubMed.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- C. Catlow, A. George and C. Freeman, Chem. Commun., 1996, 1311–1312 RSC.
- C.-S. Yang, J. M. Mora-Fonz and C. R. A. Catlow, J. Phys. Chem. C, 2011, 115, 24102–24114 CrossRef CAS.
- T. Takaishi, M. Kato and K. Itabashi, J. Phys. Chem., 1994, 98, 5742–5743 CrossRef CAS.
- M. Sato, K. Maeda and K. Hirasawa, Studies in Surface Science and Catalysis, Elsevier, 1994, vol. 84, pp. 589–596 Search PubMed.
- J. Dědeček, Z. Sobalík and B. Wichterlová, Catal. Rev., 2012, 54, 135–223 CrossRef.
- B. E. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2017, 118, 2718–2768 CrossRef PubMed.
- T. Takaishi, M. Kato and K. Itabashi, Zeolites, 1995, 15, 21–32 CrossRef CAS.
- O. H. Han, C. S. Kim and S. B. Hong, Angew. Chem., Int. Ed., 2002, 41, 469–472 CrossRef CAS PubMed.
- V. Gábová, J. Dědeček and J. Čejka, Chem. Commun., 2003, 1196–1197 RSC.
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanović, L. Kovarik, B. W. Arey, J. A. Lercher, S. R. Bare and B. M. Weckhuysen, Nat. Commun., 2015, 6, 7589 CrossRef PubMed.
- T. Li, F. Krumeich, M. Chen, Z. Ma and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2020, 22, 734–739 RSC.
- N. Omori, A. Candeo, S. Mosca, I. Lezcano-Gonzalez, I. K. Robinson, L. Li, A. G. Greenaway, P. Collier and A. M. Beale, Angew. Chem., Int. Ed., 2021, 60, 5125–5131 CrossRef CAS PubMed.
- J. Armor, Microporous Mesoporous Mater., 1998, 22, 451–456 CrossRef CAS.
- W. Li, Y. Chai, G. Wu and L. Li, J. Phys. Chem. Lett., 2022, 13, 11419–11429 CrossRef CAS PubMed.
- C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. Ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen and E. E. Stangland, Chem. – Eur. J., 2012, 18, 15735–15745 CrossRef CAS PubMed.
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Acc. Chem. Res., 2021, 54, 2894–2904 CrossRef CAS PubMed.
- J. F. Haw, Phys. Chem. Chem. Phys., 2002, 4, 5431–5441 RSC.
- C. Chizallet, C. Bouchy, K. Larmier and G. Pirngruber, Chem. Rev., 2023, 123, 6107–6196 CrossRef CAS PubMed.
- M. Yabushita, R. Osuga and A. Muramatsu, CrystEngComm, 2021, 23, 6226–6233 RSC.
- Z. Gao, J. Pihl, T. LaClair and B. Fricke, Chem. Eng. J., 2021, 406, 127120 CrossRef CAS.
- J. Gong, K. Narayanaswamy and C. J. Rutland, Ind. Eng. Chem. Res., 2016, 55, 5874–5884 CrossRef CAS.
- W. Haag, R. Lago and P. Weisz, Nature, 1984, 309, 589–591 CrossRef CAS.
- J. Brus, L. Kobera, W. Schoefberger, M. Urbanová, P. Klein, P. Sazama, E. Tabor, S. Sklenak, A. V. Fishchuk and J. Dědeček, Angew. Chem., Int. Ed., 2015, 54, 541–545 CrossRef CAS PubMed.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, J. Phys. Chem. C, 2019, 123, 15139–15144 CrossRef CAS.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS PubMed.
- Z. Yu, A. Zheng, Q. Wang, L. Chen, J. Xu, J. P. Amoureux and F. Deng, Angew. Chem., 2010, 122, 8839–8843 CrossRef.
- J. Klinowski, C. A. Fyfe and G. C. Gobbi, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 3003–3019 RSC.
- I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale, Phys. Chem. Chem. Phys., 2014, 16, 1639–1650 RSC.
- G. Woolery, G. Kuehl, H. Timken, A. Chester and J. Vartuli, Zeolites, 1997, 19, 288–296 CrossRef CAS.
- B. Gil, S. I. Zones, S.-J. Hwang, M. Bejblová and J. Čejka, J. Phys. Chem. C, 2008, 112, 2997–3007 CrossRef CAS.
- P. A. Jacobs and H. K. Beyer, J. Phys. Chem., 1979, 83, 1174–1177 CrossRef CAS.
- E. Derouane, J. C. Vedrine, R. R. Pinto, P. Borges, L. Costa, M. Lemos, F. Lemos and F. R. Ribeiro, Catal. Rev., 2013, 55, 454–515 CrossRef CAS.
- T. K. Phung and G. Busca, Appl. Catal., A, 2015, 504, 151–157 CrossRef CAS.
- A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2014, 136, 8296–8306 CrossRef CAS PubMed.
- P. Sazama, E. Tabor, P. Klein, B. Wichterlova, S. Sklenak, L. Mokrzycki, V. Pashkkova, M. Ogura and J. Dedecek, J. Catal., 2016, 333, 102–114 CrossRef CAS.
- J. Dědeček, L. Čapek, P. Sazama, Z. Sobalik and B. Wichterlova, Appl. Catal., A, 2011, 391, 244–253 CrossRef.
- S. H. Krishna, A. Goswami, Y. Wang, C. B. Jones, D. P. Dean, J. T. Miller, W. F. Schneider and R. Gounder, Nat. Catal., 2023, 6, 276–285 CrossRef CAS.
- M. S. Kumar, M. Alphin, P. S. Kumar and S. Raja, Fuel, 2023, 334, 126828 CrossRef.
- Y. Shan, J. Du, Y. Zhang, W. Shan, X. Shi, Y. Yu, R. Zhang, X. Meng, F.-S. Xiao and H. He, Natl. Sci. Rev., 2021, 8, nwab010 CrossRef CAS PubMed.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets and J. T. Miller, Science, 2017, 357, 898–903 CrossRef CAS PubMed.
- M. Koebel, M. Elsener and M. Kleemann, Catal. Today, 2000, 59, 335–345 CrossRef CAS.
- M. Iwamoto, H. Furukawa, Y. Mine, F. Uemura, S.-I. Mikuriya and S. Kagawa, J. Chem. Soc., Chem. Commun., 1986, 1272–1273 RSC.
- D. Peitz, A. Bernhard and O. Kröcher, Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, 2014, 485–506 Search PubMed.
- G. Centi and S. Perathoner, Appl. Catal., A, 1995, 132, 179–259 CrossRef CAS.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev., 2008, 50, 492–531 CrossRef CAS.
- K. Rahkamaa-Tolonen, T. Maunula, M. Lomma, M. Huuhtanen and R. L. Keiski, Catal. Today, 2005, 100, 217–222 CrossRef CAS.
- R. Long, R. Yang and K. Zammit, J. Air Waste Manage. Assoc., 2000, 50, 436–442 CrossRef CAS PubMed.
- G. Ramis, L. Yi, G. Busca, M. Turco, E. Kotur and R. J. Willey, J. Catal., 1995, 157, 523–535 CrossRef CAS.
- A. Salker and W. Weisweiler, Indian J. Chem., Sect. A, 2004, 43, 1167–1171 Search PubMed.
- A. Salker and W. Weisweiler, Appl. Catal., A, 2000, 203, 221–229 CrossRef CAS.
- A. Salker, B. Maurer and W. Weisweiler, Chem. Ing. Tech., 1998, 70, 566–570 CrossRef CAS.
- A. Garrido Pedrosa, M. Souza, A. Silva, D. Melo and A. Araujo, J. Therm. Anal. Calorim., 2006, 84, 503–509 CrossRef CAS.
- W. Van Kooten, J. Kaptein, C. Van den Bleek and H. Calis, Catal. Lett., 1999, 63, 227–231 CrossRef CAS.
- X. Shi, F. Liu, L. Xie, W. Shan and H. He, Environ. Sci. Technol., 2013, 47, 3293–3298 CrossRef CAS PubMed.
- T. Zhang, J. Li, J. Liu, D. Wang, Z. Zhao, K. Cheng and J. Li, AIChE J., 2015, 61, 3825–3837 CrossRef CAS.
- M. Xu, J. Wang, T. Yu, J. Wang and M. Shen, Appl. Catal., B, 2018, 220, 161–170 CrossRef CAS.
- J. H. Kwak, J. Szanyi and C. H. Peden, Catal. Today, 2004, 89, 135–141 CrossRef CAS.
- N. Wilken, K. Wijayanti, K. Kamasamudram, N. W. Currier, R. Vedaiyan, A. Yezerets and L. Olsson, Appl. Catal., B, 2012, 111, 58–66 Search PubMed.
- M. Al-Sabawi and H. de Lasa, AIChE J., 2009, 55, 1538–1558 CrossRef CAS.
- X. Zhang, Q. Shen, C. He, C. Ma, J. Cheng, L. Li and Z. Hao, ACS Catal., 2012, 2, 512–520 CrossRef CAS.
- M. Bian, K. Liu, D. Zheng, X. Han, X. Yang, Y. Fang, C. Liu, J. Zhao, Y. Zhang and X. Yang, Chem. Eng. J., 2024, 481, 148563 CrossRef CAS.
- F. Gao, N. M. Washton, Y. Wang, M. Kollár, J. Szanyi and C. H. Peden, J. Catal., 2015, 331, 25–38 CrossRef CAS.
- J.-K. Lai and I. E. Wachs, ACS Catal., 2018, 8, 6537–6551 CrossRef CAS.
- P. L. Gabrielsson, Top. Catal., 2004, 28, 177–184 CrossRef CAS.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. Peden, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- J. Szanyi, F. Gao, J. H. Kwak, M. Kollár, Y. Wang and C. H. Peden, Phys. Chem. Chem. Phys., 2016, 18, 10473–10485 RSC.
- H. Zhan, S. Li, G. Yin, E. Hu and Z. Huang, Combust. Flame, 2024, 263, 113129 CrossRef CAS.
- M. Jabłońska, M. E. Potter and A. M. Beale, ChemCatChem, 2024, 16, e202301214 CrossRef.
- F. Göltl, R. E. Bulo, J. R. Hafner and P. Sautet, J. Phys. Chem. Lett., 2013, 4, 2244–2249 CrossRef.
- M. Lee, S. Hong, D. Kim, E. Kim, K. Lim, J. C. Jung, H. Richter, J.-H. Moon, N. Choi and J. Nam, ACS Appl. Mater. Interfaces, 2019, 11, 3946–3960 CrossRef CAS PubMed.
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef CAS PubMed.
- J. Wang, H. Zhao, G. Haller and Y. Li, Appl. Catal., B, 2017, 202, 346–354 CrossRef CAS.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass and J. T. Miller, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- Q. Liu, C. Bian, S. Ming, L. Guo, S. Zhang, L. Pang, P. Liu, Z. Chen and T. Li, Appl. Catal., A, 2020, 607, 117865 CrossRef CAS.
- L. N. Wilcox, S. H. Krishna, C. B. Jones and R. Gounder, Catal. Sci. Technol., 2021, 11, 7932–7942 RSC.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- S. J. Schmieg, S. H. Oh, C. H. Kim, D. B. Brown, J. H. Lee, C. H. Peden and D. H. Kim, Catal. Today, 2012, 184, 252–261 CrossRef CAS.
- L. Kovarik, N. M. Washton, R. Kukkadapu, A. Devaraj, A. Wang, Y. Wang, J. Szanyi, C. H. Peden and F. Gao, ACS Catal., 2017, 7, 2458–2470 CrossRef CAS.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Chem. Eng. J., 2013, 225, 323–330 CrossRef CAS.
- X. Dong, J. Wang, H. Zhao and Y. Li, Catal. Today, 2015, 258, 28–34 CrossRef CAS.
- Q. Liu, Z. Fu, L. Ma, H. Niu, C. Liu, J. Li and Z. Zhang, Appl. Catal., A, 2017, 547, 146–154 CrossRef CAS.
- M. Devadas, O. Kröcher, M. Elsener, A. Wokaun, N. Söger, M. Pfeifer, Y. Demel and L. Mussmann, Appl. Catal., B, 2006, 67, 187–196 CrossRef CAS.
- M. Schwidder, S. Heikens, A. De Toni, S. Geisler, M. Berndt, A. Brückner and W. Grünert, J. Catal., 2008, 259, 96–103 CrossRef CAS.
- J.-Y. Luo, X. Hou, P. Wijayakoon, S. J. Schmieg, W. Li and W. S. Epling, Appl. Catal., B, 2011, 102, 110–119 CrossRef CAS.
- M. Devadas, O. Kröcher, M. Elsener, A. Wokaun, G. Mitrikas, N. Söger, M. Pfeifer, Y. Demel and L. Mussmann, Catal. Today, 2007, 119, 137–144 CrossRef CAS.
- I. Nova, M. Colombo, E. Tronconi, V. Schmeisser and M. Weibel, SAE Int. J. Engines, 2011, 4, 1822–1838 CrossRef.
- A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, Top. Catal., 2009, 52, 1837 CrossRef CAS.
- M. Colombo, I. Nova and E. Tronconi, Catal. Today, 2010, 151, 223–230 CrossRef CAS.
- H.-Y. Chen, X. Wang and W. M. Sachtler, Appl. Catal., A, 2000, 194, 159–168 CrossRef.
- B. Wichtelová, Z. Sobalík and J. Dědeček, Appl. Catal., B, 2003, 41, 97–114 CrossRef.
- H.-Y. Chen and W. M. Sachtler, Catal. Today, 1998, 42, 73–83 CrossRef CAS.
- T. V. Voskoboinikov, H.-Y. Chen and W. M. Sachtler, Appl. Catal., B, 1998, 19, 279–287 CrossRef CAS.
- A. V. Kucherov, D. E. Doronkin, A. Y. Stakheev, A. L. Kustov and M. Grill, J. Mol. Catal. A: Chem., 2010, 325, 73–78 CrossRef CAS.
- X. Lu, Y. Zheng, Y. Zhang, H. Qiu and H. Zou, Micro Nano Lett., 2015, 10, 666–669 CrossRef CAS.
- F. Gao, X. Tang, H. Yi, S. Zhao, C. Li, J. Li, Y. Shi and X. Meng, Catalysts, 2017, 7, 199 CrossRef.
- J. Chen, Y. Zheng, W. Cai, H. Zou, W. Zheng, Y. Zhang, X. Chen and B. Fu, Micro Nano Lett., 2017, 12, 6–10 CrossRef CAS.
- Y. J. Kim, H. J. Kwon, I. Heo, I.-S. Nam, B. K. Cho, J. W. Choung, M.-S. Cha and G. K. Yeo, Appl. Catal., B, 2012, 126, 9–21 CrossRef CAS.
- J. W. Choung, I.-S. Nam, H. J. Kwon, Y.-J. Kim, D.-H. Kang and M.-S. Cha, US Pat., 8048393, 2011.
- Y. Shen, T. Li, J. Yang, A. Wang, L. Wang, W. Zhan, Y. Guo and Y. Guo, Chem. Eng. J., 2023, 473, 145275 CrossRef CAS.
- B. Guan, H. Jiang, X. Peng, Y. Wei, Z. Liu, T. Chen, H. Lin and Z. Huang, Appl. Catal., A, 2021, 617, 118110 CrossRef CAS.
- E.-Y. Choi, I.-S. Nam and Y. G. Kim, J. Catal., 1996, 161, 597–604 CrossRef CAS.
- F. Lin, T. Andana, Y. Wu, J. Szanyi, Y. Wang and F. Gao, J. Catal., 2021, 401, 70–80 CrossRef CAS.
- T. Bruggemann and F. Keil, J. Phys. Chem. C, 2011, 115, 23854–23870 CrossRef.
- H. Guesmi, D. Berthomieu, B. Bromley, B. Coq and L. Kiwi-Minsker, Phys. Chem. Chem. Phys., 2010, 12, 2873–2878 RSC.
- M.-L. Tsai, R. G. Hadt, P. Vanelderen, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 3522–3529 CrossRef CAS PubMed.
- A. Heyden, N. Hansen, A. T. Bell and F. J. Keil, J. Phys. Chem. B, 2006, 110, 17096–17114 CrossRef CAS PubMed.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- J. M. Jeong, J. H. Park, J. H. Baek, R. H. Hwang, S. G. Jeon and K. B. Yi, Korean J. Chem. Eng., 2017, 34, 81–86 CrossRef CAS.
- L. Alves, L. I. Holz, C. Fernandes, P. Ribeirinha, D. Mendes, D. P. Fagg and A. Mendes, Renewable Sustainable Energy Rev., 2022, 155, 111916 CrossRef CAS.
- C. W. Andersen, E. Borfecchia, M. Bremholm, M. R. V. Jørgensen, P. N. R. Vennestrøm, C. Lamberti, L. F. Lundegaard and B. B. Iversen, Angew. Chem., Int. Ed., 2017, 56, 10367–10372 CrossRef CAS PubMed.
- F. Llabrés, I. Xamena, P. Fisicaro, G. Berlier, A. Zecchina, G. T. Palomino, C. Prestipino, S. Bordiga, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2003, 107, 7036–7044 CrossRef.
- W. J. Mortier, Compilation of extra framework sites in zeolites, Butterworth Scientific Limited on behalf of the Structure Commission of the International Zeolite Association, 1982 Search PubMed.
- C. W. Andersen, M. Bremholm, P. N. R. Vennestrøm, A. B. Blichfeld, L. F. Lundegaard and B. B. Iversen, IUCrJ, 2014, 1, 382–386 CrossRef CAS PubMed.
- A. M. Beale, I. Lezcano-Gonzalez, W. Slawinksi and D. Wragg, Chem. Commun., 2016, 52, 6170–6173 RSC.
- R. Zhang, J.-S. McEwen, M. R. Kollár, F. Gao, Y. Wang, J. Szanyi and C. H. Peden, ACS Catal., 2014, 4, 4093–4105 CrossRef CAS.
- D. W. Fickel and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Chem. Commun., 2011, 47, 800–802 RSC.
- U. Deka, A. Juhin, E. A. Eilertsen, H. Emerich, M. A. Green, S. T. Korhonen, B. M. Weckhuysen and A. M. Beale, J. Phys. Chem. C, 2012, 116, 4809–4818 CrossRef CAS.
- U. Deka, I. Lezcano-Gonzalez, S. J. Warrender, A. L. Picone, P. A. Wright, B. M. Weckhuysen and A. M. Beale, Microporous Mesoporous Mater., 2013, 166, 144–152 CrossRef CAS.
- J. M. Fedeyko, B. Chen and H.-Y. Chen, Catal. Today, 2010, 151, 231–236 CrossRef CAS.
- J.-S. McEwen, T. Anggara, W. Schneider, V. Kispersky, J. Miller, W. Delgass and F. Ribeiro, Catal. Today, 2012, 184, 129–144 CrossRef CAS.
- T. V. Janssens, H. Falsig, L. F. Lundegaard, P. N. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko and C. Lamberti, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- F. Göltl, R. E. Bulo, J. R. Hafner and P. Sautet, J. Phys. Chem. Lett., 2013, 4, 2244–2249 CrossRef.
- J. H. Kwak, D. Tran, J. Szanyi, C. H. Peden and J. H. Lee, Catal. Lett., 2012, 142, 295–301 CrossRef CAS.
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. Janssens, M. Cutini, S. Bordiga and G. Berlier, J. Am. Chem. Soc., 2020, 142, 15884–15896 CrossRef CAS PubMed.
- L. Chen, T. V. Janssens, P. N. Vennestrøm, J. Jansson, M. Skoglundh and H. Gronbeck, ACS Catal., 2020, 10, 5646–5656 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- R. Izquierdo, L. J. Rodríguez, R. Añez and A. Sierraalta, J. Mol. Catal. A: Chem., 2011, 348, 55–62 CrossRef CAS.
- S. Morpurgo, G. Moretti and M. Bossa, J. Mol. Catal. A: Chem., 2012, 358, 134–144 CrossRef CAS.
- Y. Mao, H. F. Wang and P. Hu, Int. J. Quantum Chem., 2015, 115, 618–630 CrossRef CAS.
- A. Wijerathne, A. Sawyer, R. Daya and C. Paolucci, JACS Au, 2024, 4, 197–215 CrossRef CAS PubMed.
- K. Pierloot, A. Delabie, M. H. Groothaert and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 2001, 3, 2174–2183 RSC.
- S. M. Seo, W. T. Lim and K. Seff, J. Phys. Chem. C, 2012, 116, 963–974 CrossRef CAS.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- R. Martínez-Franco, M. Moliner, P. Concepcion, J. R. Thogersen and A. Corma, J. Catal., 2014, 314, 73–82 CrossRef.
- Z. Nour, H. Petitjean and D. Berthomieu, J. Phys. Chem. C, 2010, 114, 17802–17811 CrossRef CAS.
- J. Guan, Y. Lu, K. Sen, J. Abdul Nasir, A. W. Desmoutier, Q. Hou, X. Zhang, A. J. Logsdail, G. Dutta and A. M. Beale, Philos. Trans. R. Soc., A, 2023, 381, 20220234 CrossRef CAS PubMed.
- J. H. Kwak, H. Zhu, J. H. Lee, C. H. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758–4760 RSC.
- D. Wang, L. Zhang, J. Li, K. Kamasamudram and W. S. Epling, Catal. Today, 2014, 231, 64–74 CrossRef CAS.
- D. Wang, Y. Jangjou, Y. Liu, M. K. Sharma, J. Luo, J. Li, K. Kamasamudram and W. S. Epling, Appl. Catal., B, 2015, 165, 438–445 CrossRef CAS.
- D. Wang, L. Zhang, K. Kamasamudram and W. S. Epling, ACS Catal., 2013, 3, 871–881 CrossRef CAS.
- F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. Peden, J. Catal., 2013, 300, 20–29 CrossRef CAS.
- F. Gao, E. D. Walter, N. M. Washton, J. Szanyi and C. H. Peden, Appl. Catal., B, 2015, 162, 501–514 CrossRef CAS.
- A. Godiksen, F. N. Stappen, P. N. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS.
- F. Gao, E. D. Walter, N. M. Washton, J. Szanyi and C. H. Peden, ACS Catal., 2013, 3, 2083–2093 CrossRef CAS.
- W. Gruenert, N. W. Hayes, R. W. Joyner, E. S. Shpiro, M. R. H. Siddiqui and G. N. Baeva, J. Phys. Chem., 1994, 98, 10832–10846 CrossRef.
- T. Yu, J. Wang, Y. Huang, M. Shen, W. Li and J. Wang, ChemCatChem, 2014, 6, 2074–2083 CrossRef CAS.
- I. Lezcano-Gonzalez, U. Deka, H. Van Der Bij, P. Paalanen, B. Arstad, B. Weckhuysen and A. M. Beale, Appl. Catal., B, 2014, 154, 339–349 CrossRef.
- J. Abdul Nasir, J. Guan, T. W. Keal, A. W. Desmoutier, Y. Lu, A. M. Beale, C. R. A. Catlow and A. A. Sokol, J. Am. Chem. Soc., 2022, 145, 247–259 CrossRef PubMed.
- A. Penkova and K. Hadjiivanov, Catal. Commun., 2003, 4, 485–491 CrossRef CAS.
- A. Goswami, S. H. Krishna, R. Gounder and W. F. Schneider, ACS Catal., 2024, 14, 8376–8388 CrossRef CAS PubMed.
- J. H. Kwak, D. Tran, S. D. Burton, J. Szanyi, J. H. Lee and C. H. Peden, J. Catal., 2012, 287, 203–209 CrossRef CAS.
- W. Su, Z. Li, Y. Peng and J. Li, Phys. Chem. Chem. Phys., 2015, 17, 29142–29149 RSC.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC.
- R. P. Vélez, U. Bentrup, W. Grünert and A. Brückner, Top. Catal., 2017, 60, 1641–1652 CrossRef.
- A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, J. Catal., 2008, 256, 312–322 CrossRef CAS.
- A. Grossale, I. Nova and E. Tronconi, J. Catal., 2009, 265, 141–147 CrossRef CAS.
- Y. Liu, W. Xue, S. Seo, X. Tan, D. Mei, C.-J. Liu, I.-S. Nam and S. B. Hong, Appl. Catal., B, 2021, 294, 120244 CrossRef CAS.
- L. Xie, F. Liu, K. Liu, X. Shi and H. He, Catal. Sci. Technol., 2014, 4, 1104–1110 RSC.
- Y. Shan, G. He, J. Du, Y. Sun, Z. Liu, Y. Fu, F. Liu, X. Shi, Y. Yu and H. He, Nat. Commun., 2022, 13, 4606 CrossRef CAS PubMed.
- E. Borfecchia, K. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563 RSC.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- M. P. Attfield, S. J. Weigel and A. K. Cheetham, J. Catal., 1997, 170, 227–235 CrossRef CAS.
- M. C. Dalconi, G. Cruciani, A. Alberti, P. Ciambelli and M. Rapacciuolo, Microporous Mesoporous Mater., 2000, 39, 423–430 CrossRef CAS.
- B. F. Mentzen and G. Bergeret, J. Phys. Chem. C, 2007, 111, 12512–12516 CrossRef CAS.
- B. Wichterlova, J. Dědeček, Z. Sobalık, A. Vondrova and K. Klier, J. Catal., 1997, 169, 194–202 CrossRef CAS.
- R. J. Accardi and R. F. Lobo, Microporous Mesoporous Mater., 2000, 40, 25–34 CrossRef CAS.
- F. Gao, J. Szanyi, Y. Wang, B. Schwenzer, M. Kollár and C. H. Peden, Top. Catal., 2016, 59, 882–886 CrossRef CAS.
- M. L. Bols, H. M. Rhoda, B. E. Snyder, E. I. Solomon, K. Pierloot, R. A. Schoonheydt and B. F. Sels, Dalton Trans., 2020, 49, 14749–14757 RSC.
- F. Gao, Y. Zheng, R. K. Kukkadapu, Y. Wang, E. D. Walter, B. Schwenzer, J. Szanyi and C. H. Peden, ACS Catal., 2016, 6, 2939–2954 CrossRef CAS.
- J. A. Nasir, J. Guan, T. W. Keal, Y. Lu, A. A. Sokol and C. R. A. Catlow, J. Catal., 2024, 115696 CrossRef.
- D. Wierzbicki, A. H. Clark, O. Kröcher, D. Ferri and M. Nachtegaal, J. Phys. Chem. C, 2022, 126, 17510–17519 CrossRef CAS.
- G. Li, E. A. Pidko, R. A. Van Santen, C. Li and E. J. Hensen, J. Phys. Chem. C, 2013, 117, 413–426 CrossRef CAS.
- M. Høj, M. J. Beier, J.-D. Grunwaldt and S. Dahl, Appl. Catal., B, 2009, 93, 166–176 CrossRef.
- J. Li and S. Li, J. Phys. Chem. C, 2008, 112, 16938–16944 CrossRef CAS.
- S. Shwan, J. Jansson, L. Olsson and M. Skoglundh, Appl. Catal., B, 2014, 147, 111–123 CrossRef CAS.
- J. Kim, A. Jentys, S. M. Maier and J. A. Lercher, J. Phys. Chem. C, 2013, 117, 986–993 CrossRef CAS.
- S. Shwan, J. Jansson, J. Korsgren, L. Olsson and M. Skoglundh, Catal. Today, 2012, 197, 24–37 CrossRef CAS.
- N. Hansen, A. Heyden, A. T. Bell and F. J. Keil, J. Phys. Chem. C, 2007, 111, 2092–2101 CrossRef CAS.
- H. Guesmi, D. Berthomieu and L. Kiwi-Minsker, J. Phys. Chem. C, 2008, 112, 20319–20328 CrossRef CAS.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Appl. Catal., A, 2010, 373, 168–175 CrossRef CAS.
- M. Agote-Arán, I. Lezcano-González, A. G. Greenaway, S. Hayama, S. Díaz-Moreno, A. B. Kroner and A. M. Beale, Appl. Catal., A, 2019, 570, 283–291 CrossRef.
- D. Chen, Y. Yan, A. Guo, V. Rizzotto, H. Lei, Z. Qiao, H. Liang, M. Jabłońska, X. Jiang and J. Jiang, Appl. Catal., B, 2023, 322, 122118 CrossRef CAS.
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed.
- A. Zecchina, M. Rivallan, G. Berlier, C. Lamberti and G. Ricchiardi, Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 RSC.
- S. Lai, Y. She, W. Zhan, Y. Guo, Y. Guo, L. Wang and G. Lu, J. Mol. Catal. A: Chem., 2016, 424, 232–240 CrossRef CAS.
- L. J. Lobree, I.-C. Hwang, J. A. Reimer and A. T. Bell, J. Catal., 1999, 186, 242–253 CrossRef CAS.
- B. Wichterlová, Top. Catal., 2004, 28, 131–140 CrossRef.
- C. F. Baes, Jr. and R. E. Mesmer, The Hydrolysis of Cations, (John Wiley and Sons, New York, 1976; second printing, Robert E. Krieger, Malabar, FL, 1986).
- P. Ratnasamy and R. Kumar, Catal. Today, 1991, 9, 329–416 CrossRef.
- M. Mihaylov, A. Penkova, K. Hadjiivanov and M. Daturi, J. Mol. Catal. A: Chem., 2006, 249, 40–46 CrossRef CAS.
- S. Sklenak, P. C. Andrikopoulos, S. R. Whittleton, H. Jirglova, P. Sazama, L. Benco, T. Bucko, J. Hafner and Z. Sobalik, J. Phys. Chem. C, 2013, 117, 3958–3968 CrossRef CAS.
- M. J. Rice, A. K. Chakraborty and A. T. Bell, J. Phys. Chem. B, 2000, 104, 9987–9992 CrossRef CAS.
- M. J. Rice, A. K. Chakraborty and A. T. Bell, J. Catal., 2000, 194, 278–285 CrossRef CAS.
- A. Bellmann, C. Rautenberg, U. Bentrup and A. Brückner, Catalysts, 2020, 10, 123 CrossRef CAS.
- J. Dědeček and B. Wichterlová, J. Phys. Chem. B, 1999, 103, 1462–1476 CrossRef.
- J. Dědeček, D. Kaucký and B. Wichterlová, Microporous Mesoporous Mater., 2000, 35, 483–494 CrossRef.
- J. Dědeček, L. Čapek, D. Kaucký, Z. Sobalık and B. Wichterlova, J. Catal., 2002, 211, 198–207 CrossRef.
- J. Dědeček, D. Kaucký and B. Wichterlová, Microporous Mesoporous Mater., 2000, 35, 483–494 CrossRef.
- E. Dooryhee, C. Catlow, J. Couves, P. Maddox, J. Thomas, G. Greaves, A. Steel and R. Townsend, J. Phys. Chem., 1991, 95, 4514–4521 CrossRef CAS.
- M. H. Groothaert, K. Pierloot, A. Delabie and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 2003, 5, 2135–2144 RSC.
- A. Delabie, K. Pierloot, M. H. Groothaert, R. A. Schoonheydt and L. G. Vanquickenborne, Eur. J. Inorg. Chem., 2002, 515–530 CrossRef CAS.
- K. Pierloot, A. Delabie, A. Verberckmoes and R. Schoonheydt, The Interplay between DFT and Conventional Quantum Chemistry: Transition Metals in Zeolites, in Density Functional Theory: A Bridge Between Chemistry and Physics, ed. P. Geerlings, F. De Proft and W. Langenaeker, VUB University Press, Brussels, 1999, pp. 169–188 Search PubMed.
- P. J. Smeets, J. S. Woertink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed.
- P. Vanelderen, J. Vancauwenbergh, B. F. Sels and R. A. Schoonheydt, Coord. Chem. Rev., 2013, 257, 483–494 CrossRef CAS.
- G. Delahay, S. Kieger, N. Tanchoux, P. Trens and B. Coq, Appl. Catal., B, 2004, 52, 251–257 CrossRef CAS.
- H. Lei, V. Rizzotto, A. Guo, D. Ye, U. Simon and P. Chen, Catalysts, 2021, 11, 52 CrossRef CAS.
- M. K. Neylon, C. L. Marshall and A. J. Kropf, J. Am. Chem. Soc., 2002, 124, 5457–5465 CrossRef CAS PubMed.
- S. C. Larsen, A. Aylor, A. T. Bell and J. A. Reimer, J. Phys. Chem., 1994, 98, 11533–11540 CrossRef CAS.
- S. M. Seo, W. T. Lim and K. Seff, J. Phys. Chem. C, 2011, 116, 963–974 CrossRef.
- S. Bordiga, D. Scarano, G. Spoto, A. Zecchina, C. Lamberti and C. O. Areán, Vib. Spectrosc., 1993, 5, 69–74 CrossRef CAS.
- S. Bordiga, E. Garrone, C. Lamberti, A. Zecchina, C. O. Arean, V. B. Kazansky and L. M. Kustov, J. Chem. Soc., Faraday Trans., 1994, 90, 3367–3372 RSC.
- S. Kieger, G. Delahay, B. Coq and B. Neveu, J. Catal., 1999, 183, 267–280 CrossRef CAS.
- P. Chen, V. Rizzotto, A. Khetan, K. Xie, R. Moos, H. Pitsch, D. Ye and U. Simon, ACS Appl. Mater. Interfaces, 2019, 11, 8097–8105 CrossRef CAS PubMed.
- A. Marberger, A. W. Petrov, P. Steiger, M. Elsener, O. Kröcher, M. Nachtegaal and D. Ferri, Nat. Catal., 2018, 1, 221 CrossRef CAS.
- R. Millan, P. Cnudde, V. Van Speybroeck and M. Boronat, JACS Au, 2021, 1, 1778–1787 CrossRef CAS PubMed.
- R. Millan, E. Bello-Jurado, M. Moliner, M. Boronat and R. Gomez-Bombarelli, ACS Cent. Sci., 2023, 9, 2044–2056 CrossRef CAS PubMed.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- C. Tang, H. Zhang and L. Dong, Catal. Sci. Technol., 2016, 6, 1248–1264 RSC.
- L. Lietti, J. Alemany, P. Forzatti, G. Busca, G. Ramis, E. Giamello and F. Bregani, Catal. Today, 1996, 29, 143–148 CrossRef CAS.
- D. K. Pappas, K. Kvande, M. Kalyva, M. Dyballa, K. A. Lomachenko, B. Arstad, E. Borfecchia, S. Bordiga, U. Olsbye and P. Beato, Catal. Today, 2021, 369, 105–111 CrossRef CAS.
- C. Negri, N. Usberti, G. Contaldo, M. Bracconi, I. Nova, M. Maestri and E. Tronconi, Angew. Chem., Int. Ed., 2024, e202408328 CAS.
- I. A. Pankin, H. I. Hamoud, K. A. Lomachenko, S. B. Rasmussen, A. Martini, P. Bazin, V. Valtchev, M. Daturi, C. Lamberti and S. Bordiga, Catal. Sci. Technol., 2021, 11, 846–860 RSC.
- T. Yu, T. Hao, D. Fan, J. Wang, M. Shen and W. Li, J. Phys. Chem. C, 2014, 118, 6565–6575 CrossRef CAS.
- W. Su, H. Chang, Y. Peng, C. Zhang and J. Li, Environ. Sci. Technol., 2015, 49, 467–473 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1046 CrossRef CAS PubMed.
- H. V. Obias, Y. Lin, N. Murthy, E. Pidcock, E. I. Solomon, M. Ralle, N. J. Blackburn, Y.-M. Neuhold, A. D. Zuberbühler and K. D. Karlin, J. Am. Chem. Soc., 1998, 120, 12960–12961 CrossRef CAS.
- N. Kitajima, K. Fujisawa, C. Fujimoto, Y. Morooka, S. Hashimoto, T. Kitagawa, K. Toriumi, K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 1277–1291 CrossRef CAS.
- N. Akter, X. Chen, J. Parise, J. A. Boscoboinik and T. Kim, Korean J. Chem. Eng., 2018, 35, 89–98 CrossRef CAS.
- M. Wallin, C.-J. Karlsson, M. Skoglundh and A. Palmqvist, J. Catal., 2003, 218, 354–364 CrossRef CAS.
- T. Yu, J. Wang, M. Shen, J. Wang and W. Li, Chem. Eng. J., 2015, 264, 845–855 CrossRef CAS.
- H. Lee, I. Song, S. W. Jeon and D. H. Kim, J. Phys. Chem. Lett., 2021, 12, 3210–3216 CrossRef CAS PubMed.
- Y. Wan, G. Yang, J. Xiang, X. Shen, D. Yang, Y. Chen, V. Rac, V. Rakic and X. Du, Dalton Trans., 2020, 49, 764–773 RSC.
- A. G. Greenaway, A. Marberger, A. Thetford, I. Lezcano-González, M. Agote-Arán, M. Nachtegaal, D. Ferri, O. Kröcher, C. R. A. Catlow and A. M. Beale, Chem. Sci., 2020, 11, 447–455 RSC.
- M. Cheng, B. Jiang, S. Yao, J. Han, S. Zhao, X. Tang, J. Zhang and T. Wang, J. Phys. Chem. C, 2017, 122, 455–464 CrossRef.
- O. Krocher and M. Elsener, Ind. Eng. Chem. Res., 2008, 47, 8588–8593 CrossRef CAS.
- A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, Top. Catal., 2009, 52, 1837–1841 CrossRef CAS.
- P. S. Metkar, M. P. Harold and V. Balakotaiah, Appl. Catal., B, 2012, 111, 67–80 CrossRef.
- P. S. Metkar, M. P. Harold and V. Balakotaiah, Chem. Eng. Sci., 2013, 87, 51–66 CrossRef CAS.
- J. Li and S. Li, Phys. Chem. Chem. Phys., 2007, 9, 3304–3311 RSC.
- S. Li, Y. Zheng, F. Gao, J. Szanyi and W. F. Schneider, ACS Catal., 2017, 7, 5087–5096 CrossRef CAS.
- H. Kubota, C. Liu, T. Toyao, Z. Maeno, M. Ogura, N. Nakazawa, S. Inagaki, Y. Kubota and K.-I. Shimizu, ACS Catal., 2020, 10, 2334–2344 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev., 2000, 42, 71–144 CrossRef CAS.
- M. Ahrens, O. Marie, P. Bazin and M. Daturi, J. Catal., 2010, 271, 1–11 CrossRef CAS.
- J. N. Kondo, R. Nishitani, E. Yoda, T. Yokoi, T. Tatsumi and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 11576–11586 RSC.
- M. Niwa, K. Suzuki, N. Katada, T. Kanougi and T. Atoguchi, J. Phys. Chem. B, 2005, 109, 18749–18757 CrossRef CAS PubMed.
- S. Li, Y. Wang, T. Wu and W. F. Schneider, ACS Catal., 2018, 8, 10119–10130 CrossRef CAS.
- G. Li, E. A. Pidko, R. A. Van Santen, Z. Feng, C. Li and E. J. Hensen, J. Catal., 2011, 284, 194–206 CrossRef CAS.
- Á. Szécsényi, E. Khramenkova, I. Y. Chernyshov, G. Li, J. Gascon and E. A. Pidko, ACS Catal., 2019, 9, 9276–9284 CrossRef.
- T. C. Brüggemann and F. J. Keil, J. Phys. Chem. C, 2008, 112, 17378–17387 CrossRef.
- T. C. Brüggemann and F. J. Keil, J. Phys. Chem. C, 2011, 115, 2114–2133 CrossRef.
- T. Usui, Z. Liu, H. Igarashi, Y. Sasaki, Y. Shiramata, H. Yamada, K. Ohara, T. Kusamoto and T. Wakihara, ACS Omega, 2019, 4, 3653–3659 CrossRef CAS PubMed.
- F. Maugé, P. Gallezot, J.-C. Courcelle, P. Engelhard and J. Grosmangin, Zeolites, 1986, 6, 261–266 CrossRef.
- D. W. Fickel and R. F. Lobo, J. Phys. Chem. C, 2009, 114, 1633–1640 CrossRef.
- S. Yasumura, Y. Qian, T. Toyao, Z. Maeno and K.-I. Shimizu, J. Phys. Chem. C, 2022, 126, 11082–11090 CrossRef CAS.
- J. Szanyi, J. H. Kwak and C. H. Peden, J. Phys. Chem. B, 2004, 108, 3746–3753 CrossRef CAS.
- S. Yasumura, C. Liu, T. Toyao, Z. Maeno and K.-I. Shimizu, J. Phys. Chem. C, 2021, 125, 1913–1922 CrossRef CAS.
- K. Hadjiivanov, J. Saussey, J. Freysz and J. Lavalley, Catal. Lett., 1998, 52, 103–108 CrossRef CAS.
- H. Kubota, T. Toyao, Z. Maeno, Y. Inomata, T. Murayama, N. Nakazawa, S. Inagaki, Y. Kubota and K.-I. Shimizu, ACS Catal., 2021, 11, 11180–11192 CrossRef CAS.
- J. E. Schmidt, R. Oord, W. Guo, J. D. Poplawsky and B. M. Weckhuysen, Nat. Commun., 2017, 8, 1666 CrossRef PubMed.
- K. Kharas, H. Robota and D. Liu, Appl. Catal., B, 1993, 2, 225–237 CrossRef CAS.
- J. Yan, G.-D. Lei, W. Sachtler and H. Kung, J. Catal., 1996, 161, 43–54 CrossRef CAS.
- L. Ma, W. Su, Z. Li, J. Li, L. Fu and J. Hao, Catal. Today, 2015, 245, 16–21 CrossRef CAS.
- A. Wang, Y. Wang, E. D. Walter, N. M. Washton, Y. Guo, G. Lu, C. H. Peden and F. Gao, Catal. Today, 2019, 320, 91–99 CrossRef CAS.
- L. Ma, J. Li, Y. Cheng, C. K. Lambert and L. Fu, Environ. Sci. Technol., 2012, 46, 1747–1754 CrossRef CAS PubMed.
- W. Ding, Y. Sun, J. Liu, S. Xue, X. Han, Z. Yan, Y. Yu, Y. Shan and H. He, Chem. Eng. J., 2024, 149272 CrossRef CAS.
- M. Moliner, C. Martinez and A. Corma, Chem. Mater., 2013, 26, 246–258 CrossRef.
- M. Moliner, C. Franch, E. Palomares, M. Grill and A. Corma, Chem. Commun., 2012, 48, 8264–8266 RSC.
- S. Han, Q. Ye, S. Cheng, T. Kang and H. Dai, Catal. Sci. Technol., 2017, 7, 703–717 RSC.
- T. Usui, Z. Liu, S. Ibe, J. Zhu, C. Anand, H. Igarashi, N. Onaya, Y. Sasaki, Y. Shiramata and T. Kusamoto, ACS Catal., 2018, 8, 9165–9173 CrossRef CAS.
- Y. Wang, X. Shi, Y. Shan, J. Du, K. Liu and H. He, Ind. Eng. Chem. Res., 2020, 59, 6416–6423 CrossRef CAS.
- Y. Shan, J. Du, Y. Yu, W. Shan, X. Shi and H. He, Appl. Catal., B, 2020, 266, 118655 CrossRef CAS.
- Y. J. Kim, J. K. Lee, K. M. Min, S. B. Hong, I.-S. Nam and B. K. Cho, J. Catal., 2014, 311, 447–457 CrossRef CAS.
- J. Song, Y. Wang, E. D. Walter, N. M. Washton, D. Mei, L. Kovarik, M. H. Engelhard, S. Prodinger, Y. Wang and C. H. Peden, ACS Catal., 2017, 7, 8214–8227 CrossRef CAS.
- J. Luo, F. Gao, K. Kamasamudram, N. Currier, C. H. Peden and A. Yezerets, J. Catal., 2017, 348, 291–299 CrossRef CAS.
- J. H. Lee, Y. J. Kim, T. Ryu, P. S. Kim, C. H. Kim and S. B. Hong, Appl. Catal., B, 2017, 200, 428–438 CrossRef CAS.
- J. H. Kwak, D. Tran, S. D. Burton, J. Szanyi, J. H. Lee and C. H. Peden, J. Catal., 2012, 289, 272 CrossRef CAS.
- T. Ryu, N. H. Ahn, S. Seo, J. Cho, H. Kim, D. Jo, G. T. Park, P. S. Kim, C. H. Kim and E. L. Bruce, Angew. Chem., 2017, 129, 3304–3308 CrossRef.
- Y. Wu, W. Zhao, S. H. Ahn, Y. Wang, E. D. Walter, Y. Chen, M. A. Derewinski, N. M. Washton, K. G. Rappé and Y. Wang, Nat. Commun., 2023, 14, 2633 CrossRef CAS PubMed.
- N. H. Ahn, T. Ryu, Y. Kang, H. Kim, J. Shin, I.-S. Nam and S. B. Hong, ACS Catal., 2017, 7, 6781–6785 CrossRef CAS.
- C. Negri, E. Borfecchia, M. Cutini, K. A. Lomachenko, T. V. Janssens, G. Berlier and S. Bordiga, ChemCatChem, 2019, 11, 3828–3838 CrossRef CAS.
- S. E. Bozbag, D. Şanlı, B. Özener, G. Hisar and C. Erkey, Catalysts, 2020, 10, 411 CrossRef CAS.
- F. Gao, M. Kollár, R. K. Kukkadapu, N. M. Washton, Y. Wang, J. Szanyi and C. H. Peden, Appl. Catal., B, 2015, 164, 407–419 CrossRef CAS.
- F. Gao, Y. Wang, M. Kollár, N. M. Washton, J. Szanyi and C. H. Peden, Catal. Today, 2015, 258, 347–358 CrossRef CAS.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS PubMed.
- T. Ryu, Y. Kang, I.-S. Nam and S. B. Hong, React. Chem. Eng., 2019, 4, 1050–1058 RSC.
- M. P. Ruggeri, I. Nova, E. Tronconi, V. Schmeißer and M. Weibel, Chem. Ing. Tech., 2018, 90, 803–815 CrossRef CAS.
- J. Kielland, J. Am. Chem. Soc., 1937, 59, 1675–1678 CrossRef CAS.
- N. Martín, P. N. Vennestrøm, J. R. Thøgersen, M. Moliner and A. Corma, ChemCatChem, 2017, 9, 1754–1757 CrossRef.
- N. Martín, P. N. Vennestrøm, J. R. Thøgersen, M. Moliner and A. Corma, Chem. – Eur. J., 2017, 23, 13404–13414 CrossRef PubMed.
- X. Lou, P. Liu, J. Li, Z. Li and K. He, Appl. Surf. Sci., 2014, 307, 382–387 CrossRef CAS.
- L. Olsson, K. Wijayanti, K. Leistner, A. Kumar, S. Y. Joshi, K. Kamasamudram, N. W. Currier and A. Yezerets, Appl. Catal., B, 2016, 183, 394–406 CrossRef CAS.
- Y. Jangjou, D. Wang, A. Kumar, J. Li and W. S. Epling, ACS Catal., 2016, 6, 6612–6622 CrossRef CAS.
- P. S. Hammershøi, A. D. Jensen and T. V. Janssens, Appl. Catal., B, 2018, 238, 104–110 CrossRef.
- X. Jiang, J. Yang, Y. Liang, H. Zhang, Y. Zhou, R. Shan, Q. Liu, W. Liu and L. Yao, J. Environ. Chem. Eng., 2023, 11, 109822 CrossRef CAS.
- A. Wang and L. Olsson, Chem. Eng. J., 2020, 395, 125048 CrossRef CAS.
- S. Shwan, Metal-Exchanged Zeolites for NH 3-SCR Applications–Activity and Deactivation Studies, Chalmers Tekniska Hogskola, Sweden, 2014 Search PubMed.
- K. Xie, A. Wang, J. Woo, A. Kumar, K. Kamasamudram and L. Olsson, Appl. Catal., B, 2019, 256, 117815 CrossRef CAS.
- L. Xu, C. Wang, H. Chang, Q. Wu, T. Zhang and J. Li, Environ. Sci. Technol., 2018, 52, 7064–7071 CrossRef CAS PubMed.
- J. Liang, Y. Mi, G. Song, H. Peng, Y. Li, R. Yan, W. Liu, Z. Wang, P. Wu and F. Liu, J. Hazard. Mater., 2020, 398, 122986 CrossRef CAS PubMed.
- X. Yong, H. Chen, H. Zhao, M. Wei, Y. Zhao and Y. Li, Chin. J. Chem. Eng., 2022, 46, 184–193 CrossRef CAS.
- X. Auvray, M. Arvanitidou, Å. Högström, J. Jansson, S. Fouladvand and L. Olsson, Emiss. Control Sci. Technol., 2021, 7, 232–246 CrossRef CAS.
- K. Wijayanti, K. Xie, A. Kumar, K. Kamasamudram and L. Olsson, Appl. Catal., B, 2017, 219, 142–154 CrossRef CAS.
- V. Mesilov, S. Dahlin, S. L. Bergman, S. Xi, J. Han, L. Olsson, L. J. Pettersson and S. L. Bernasek, Appl. Catal., B, 2021, 299, 120626 CrossRef CAS.
- Z. Chen, C. Fan, L. Pang, S. Ming, P. Liu and T. Li, Appl. Catal., B, 2018, 237, 116–127 CrossRef CAS.
- M.-L. Tarot, E. E. Iojoiu, V. Lauga, D. Duprez, X. Courtois and F. Can, Appl. Catal., B, 2019, 250, 355–368 CrossRef CAS.
- A. Wang, K. Xie, D. Bernin, A. Kumar, K. Kamasamudram and L. Olsson, Appl. Catal., B, 2020, 269, 118781 CrossRef CAS.
- K. Xie, J. Woo, D. Bernin, A. Kumar, K. Kamasamudram and L. Olsson, Appl. Catal., B, 2019, 241, 205–216 CrossRef CAS.
- H. Zhao, Y. Zhao, M. Liu, X. Li, Y. Ma, X. Yong, H. Chen and Y. Li, Appl. Catal., B, 2019, 252, 230–239 CrossRef CAS.
- P. Kern, M. Klimczak, T. Heinzelmann, M. Lucas and P. Claus, Appl. Catal., B, 2010, 95, 48–56 CrossRef CAS.
- J. Li, Y. Peng, H. Chang, X. Li, J. C. Crittenden and J. Hao, Front. Environ. Sci. Eng., 2016, 10, 413–427 CrossRef CAS.
- J. Due-Hansen, A. L. Kustov, S. B. Rasmussen, R. Fehrmann and C. H. Christensen, Appl. Catal., B, 2006, 66, 161–167 CrossRef CAS.
- J. Zhang, Z. Huang, Y. Du, X. Wu, H. Shen and G. Jing, Environ. Sci. Technol., 2019, 54, 595–603 CrossRef PubMed.
- A. Martini, C. Negri, L. Bugarin, G. Deplano, R. K. Abasabadi, K. A. Lomachenko, T. V. Janssens, S. Bordiga, G. Berlier and E. Borfecchia, J. Phys. Chem. Lett., 2022, 13, 6164–6170 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |