
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water structures on acidic zeolites and their roles in catalysis
Qiang
Liu
 a and
Jeroen A.
van Bokhoven
*ab
a and
Jeroen A.
van Bokhoven
*ab
aInstitute for Chemical and Bioengineering, ETH Zurich, Vladimir Prelog Weg 1, 8093 Zurich, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch
bLaboratory for Catalysis and Sustainable Chemistry, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland
First published on 19th February 2024
Abstract
The local reaction environment of catalytic active sites can be manipulated to modify the kinetics and thermodynamic properties of heterogeneous catalysis. Because of the unique physical–chemical nature of water, heterogeneously catalyzed reactions involving specific interactions between water molecules and active sites on catalysts exhibit distinct outcomes that are different from those performed in the absence of water. Zeolitic materials are being applied with the presence of water for heterogeneous catalytic reactions in the chemical industry and our transition to sustainable energy. Mechanistic investigation and in-depth understanding about the behaviors and the roles of water are essentially required for zeolite chemistry and catalysis. In this review, we focus on the discussions of the nature and structures of water adsorbed/stabilized on Brønsted and Lewis acidic zeolites based on experimental observations as well as theoretical calculation results. The unveiled functions of water structures in determining the catalytic efficacy of zeolite-catalyzed reactions have been overviewed and the strategies frequently developed for enhancing the stabilization of zeolite catalysts are highlighted. Recent advancement will contribute to the development of innovative catalytic reactions and the rationalization of catalytic performances in terms of activity, selectivity and stability with the presence of water vapor or in condensed aqueous phase.
 Qiang Liu | Qiang Liu received his PhD degree of Chemical Engineering from Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences in 2018. He worked as a postdoc student in the research group of Prof. Johannes A. Lercher at Technical University of Munich (TUM) from 2018 to 2020. Then, he came to work with Prof. Jeroen A. van Bokhoven at ETH Zurich and Paul Scherrer Institute as a postdoctoral fellow. His current research interest focuses on the design of advanced nanocatalysts and their related applications in heterogeneous catalysis. |
 Jeroen A. van Bokhoven | Jeroen A. van Bokhoven completed a degree in chemistry at Utrecht University (Netherlands) in 1995 and went on to obtain a PhD in inorganic chemistry and catalysis from the same university in 2000 (with honours). From 1999 until 2002, he was head of the XAS (X-ray absorption spectroscopy) users-support group at Utrecht University. In 2002, he moved to the ETH Zurich, where he worked as a postdoctoral researcher in the group of Professor Prins. In 2006 he obtained an SNF assistant professorship in the Department of Chemistry and Applied Biology. He was the 2008 recipient of the Swiss Chemical Society Werner Prize. Since 2010, Jeroen A. van Bokhoven has a Chair in Heterogeneous Catalysis at the Institute for Chemical and Bioengineering at ETH Zurich and is Head of Laboratory for Catalysis and Sustainable Chemistry at Paul Scherrer Institute. |
1. Introduction
The variations of the local environment near heterogeneous catalyst surface make it possible to manipulate catalytic chemistry.1–3 A catalytic reaction usually involves active sites, reactants, reactive intermediates, products and solvent (if involved). Water acts as either a co-feed reactant, or a co-product of the reaction or the solvent from condensed aqueous phase for a number of surface-catalyzed chemical transformations. The critical role of water in heterogeneous catalytic reactions at the solid–gas and solid–liquid interfaces has recently become an important topic in catalysis research.4–6 Water molecules adsorbed/existed within the microenvironment of active sites can mediate the sorption behavior of reactants and surface intermediates on the surface of heterogeneous catalysts, leading to tailored reaction pathways and distinct catalytic performances.7–10 The nature as well as the structure of active sites on solid catalysts can be altered by the presence of water molecules, thus making differences in the activation energy barrier of the elementary steps for a catalytic reaction.11 On the other hand, the promotional effects of water in terms of the solvation of reaction partners, the influences on the surface chemistry of active sites and the stabilizations of surface intermediates and activated complexes, have been identified as well.12,13 Performing heterogeneous catalytic reactions in aqueous phase appears to be profitable for reducing energy input (low reaction temperature), and achieving high catalytic reaction rate and product selectivity, which are different from those of gas-phase reactions.14,15 It is noteworthy that an aqueous phase environment usually complicates the understanding of the specific interactions of solvent molecules with reactants and active sites, thus bringing challenges to the insights of reaction mechanisms and structure–performance relations.3,16 Apart from these above, the influence of water on the structural and chemical stability of solid catalysts, e.g., crystalline microporous zeolites should be additionally considered for their efficient applications in heterogeneous catalysis.17,18Zeolitic materials, Brønsted- and Lewis-acidic tectosilicates, are built up from SiO4 tetrahedra to feature an ordered distribution of micropores with the molecular dimensions.19,20 Since the report of one of the first synthetic zeolites, aluminosilicate mordenite (MOR),21 a great number of zeolites have been developed and deployed in petrochemical industry, mostly for catalysis, separation and sorption.22–24 The confinement in the zeolite pores is vital to affect the sorption energetics of reacting molecules, facilitate the dispersive interactions of reactive intermediates and transition states within zeolite channel structure.25–27 Besides, the nanoscopic environments in zeolite structures vary with the confined species and the hydrophilic/hydrophobic nature of the micropores, which eventually determine the reaction pathways and thus the reaction rates and catalytic efficiencies.28,29 Zeolites are effective heterogeneous catalysts for catalyzing a wide variety of chemical reactions, particularly performed in the presence of water. Water molecules can be initially stabilized on Brønsted acid sites, framework heteroatom metal sites and structural defects, followed with the formation of hydrated hydronium ion clusters and extended hydrogen-bonded water networks, which differ substantially from the bulk water. Intraporous water structures occluded in the zeolite pores could make distinct differences for heterogeneous catalysis. Understanding about the formation of water structures in zeolite pores and the corresponding functions in catalytic reactions are desirable to gain the fundamental bases for enhancing catalytic reaction rates and efficiencies at the solid–gas and solid–liquid interfaces. Brønsted and Lewis acidic zeolites will be the focus of this Review, given their wide applications in the development of renewable catalytic processes for transportation fuels and valuable chemicals. The roles of water in metal-containing zeolites-catalyzed reactions (e.g., methane oxidation to methanol,30–32 selective catalytic reduction of nitrogen oxides (SCR-NOx),33,34 Wacker oxidation of ethylene,35,36 high-temperature alkane conversion and biomass valorization37) and water effects in the dynamic evolution of metal sites confined in zeolite structures are beyond the scope of the present Review. Detailed progress on the latter respect can be found from a recently published Review paper by Hu et al.38
Recently, the research group of Resasco provided comprehensive reviews of water-mediated heterogeneous catalysis4 and the interactions of water with zeolites.39 Stanciakova et al. reviewed the water-active site interactions in zeolites based mostly on the viewpoint of molecular modeling approaches.18 In addition, Lin et al. briefly summarized the positive effects brought with water molecules on traditional heterogeneous catalytic reactions, such as aqueous-phase reforming, aqueous-phase methane activation and aqueous-phase Fischer–Tropsch synthesis catalyzed by noble metal or transition metal nanoparticle-based materials.5 Nonetheless, it should be pointed out that advances in the nature and structures of microscale intrazeolite water on the surface of both Brønsted and Lewis acidic zeolites, and the consequent various functions of water structures in zeolite catalysis have not been well summarized in previous works. In the present Review, we first showcase the adsorption behaviors of water from the gas and aqueous phase, and the resulted structural properties of water confined in zeolitic materials. After that, we stress the critical roles of water in impacting the heterogeneously zeolite-catalyzed reactions in terms of the chemical thermodynamics, catalytic reactivity and catalytic/structural stability. Considering that the stability of zeolite materials with the presence of water is of great importance, the potential strategies developed to contribute to zeolite stabilization are also described. Advancements in the research outlined above could distinguish this work from the previous reviews and enlarge the research progress of understanding water-active site interactions on a variety of acidic zeolites. Finally, future research perspectives associated with zeolite catalysis involving the specific interactions between zeolite framework and water structures are highlighted.
2. Water structures on acidic zeolite materials
2.1. Brønsted acidic zeolites
For aluminosilicate zeolites, substituting tetrahedrally coordinated Si4+ with typically Al3+ heteroatom results in the formation of Brønsted acid site (BAS), that is, a hydroxyl group is bridged an aluminum and silicon atom in case of protons as charge balancing cation (Si(OH)Al).40 Aluminosilicate zeolites are the most relevant active materials for catalyzing the reactions including hydrocarbon conversions, such as cracking24,40,41 and isomerization,40 and alkylation,11 alcohol dehydration,42,43 ketonization,44,45 and esterification reactions.46 Silicoaluminophosphate (SAPO) molecular sieves such as SAPO-34 is another class of microporous crystalline material, which are widely applied in the methanol-to-olefins (MTO) conversion.22,47,48 The Brønsted acid sites in SAPO molecular sieves are generated by the incorporation of Si into AlPO4 structures with the presence of charge compensation cations, i.e., protons. Zeolite Si(OH)Al groups usually manifest strongly covalent character in the absence of adsorbates in the pores. With the presence of adsorbates inside of zeolite structures, the polarity of OH bond increases due to the acid–base interactions through hydrogen bonding (H-bonding).49 The protonation of substrate molecules of different base strength by the BAS on the surface or pore of zeolites often constitutes as the initial step for solid acid-catalyzed reactions. Water (H2O), as a good proton acceptor, is employed in general to get insights into the fundamental aspects in terms of the nature of zeolite acidity and the protonation ability of the BAS of zeolite.50–53 Intensive research works have pointed to study the interactions of water molecules with the BAS on Brønsted acidic zeolites in the past decades. The adsorption behavior of water molecules and the structural configuration of water clusters on zeolitic Brønsted acid sites will be briefly summarized.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes (one water molecule per BAS) on the acidic site of H-ZSM-5 zeolite (ZSM-5: Zeolite Socony Mobil-5).57 It resulted in the disappearance of the bond of bridging hydroxyl at 3609 cm−1, followed by formation of bonds at ∼2900 cm−1 (A band) and ∼2400 cm−1 (B band) due to the H-bonding of BAS with the water molecule.58,59 For a long time, the structure of single water adsorption on Brønsted acidic zeolites has been debated. Adsorption of a single water molecule on the BAS site of zeolite could create a structure that features a hydrogen-bonded model (Fig. 1b),60,61 a protonated model (Fig. 1c)62 or the hydrogen-bonded model co-existed with the protonated one.51,63,64 FT-IR on H218O adsorption on H-ZSM-5 has provided spectroscopic evidence for the generation of hydrogen-bonded neutral adduct in the process of the adsorption of the single water molecule.65 Car–Parrinello molecular dynamics demonstrate that the water molecule forms a strong H-bond with the OH group of the BAS and a weak H-bond with a second neighboring oxygen atom in the framework of zeolite chabazite (CHA, H-SSZ-13).66 The hydrogen-bonded water-proton complex has been identified as the favorable model by far for the special interaction of a single water with the BAS on Brønsted acidic zeolites.66–68 Upon interacting with a single water, the proton from the BAS cannot be sufficiently stabilized; instead, the adsorbed water molecule is hydrogen-bonded to the Si(OH)Al site as a neutral complex without the proton transference from zeolite framework in the pores.68,69
:1 complexes (one water molecule per BAS) on the acidic site of H-ZSM-5 zeolite (ZSM-5: Zeolite Socony Mobil-5).57 It resulted in the disappearance of the bond of bridging hydroxyl at 3609 cm−1, followed by formation of bonds at ∼2900 cm−1 (A band) and ∼2400 cm−1 (B band) due to the H-bonding of BAS with the water molecule.58,59 For a long time, the structure of single water adsorption on Brønsted acidic zeolites has been debated. Adsorption of a single water molecule on the BAS site of zeolite could create a structure that features a hydrogen-bonded model (Fig. 1b),60,61 a protonated model (Fig. 1c)62 or the hydrogen-bonded model co-existed with the protonated one.51,63,64 FT-IR on H218O adsorption on H-ZSM-5 has provided spectroscopic evidence for the generation of hydrogen-bonded neutral adduct in the process of the adsorption of the single water molecule.65 Car–Parrinello molecular dynamics demonstrate that the water molecule forms a strong H-bond with the OH group of the BAS and a weak H-bond with a second neighboring oxygen atom in the framework of zeolite chabazite (CHA, H-SSZ-13).66 The hydrogen-bonded water-proton complex has been identified as the favorable model by far for the special interaction of a single water with the BAS on Brønsted acidic zeolites.66–68 Upon interacting with a single water, the proton from the BAS cannot be sufficiently stabilized; instead, the adsorbed water molecule is hydrogen-bonded to the Si(OH)Al site as a neutral complex without the proton transference from zeolite framework in the pores.68,69
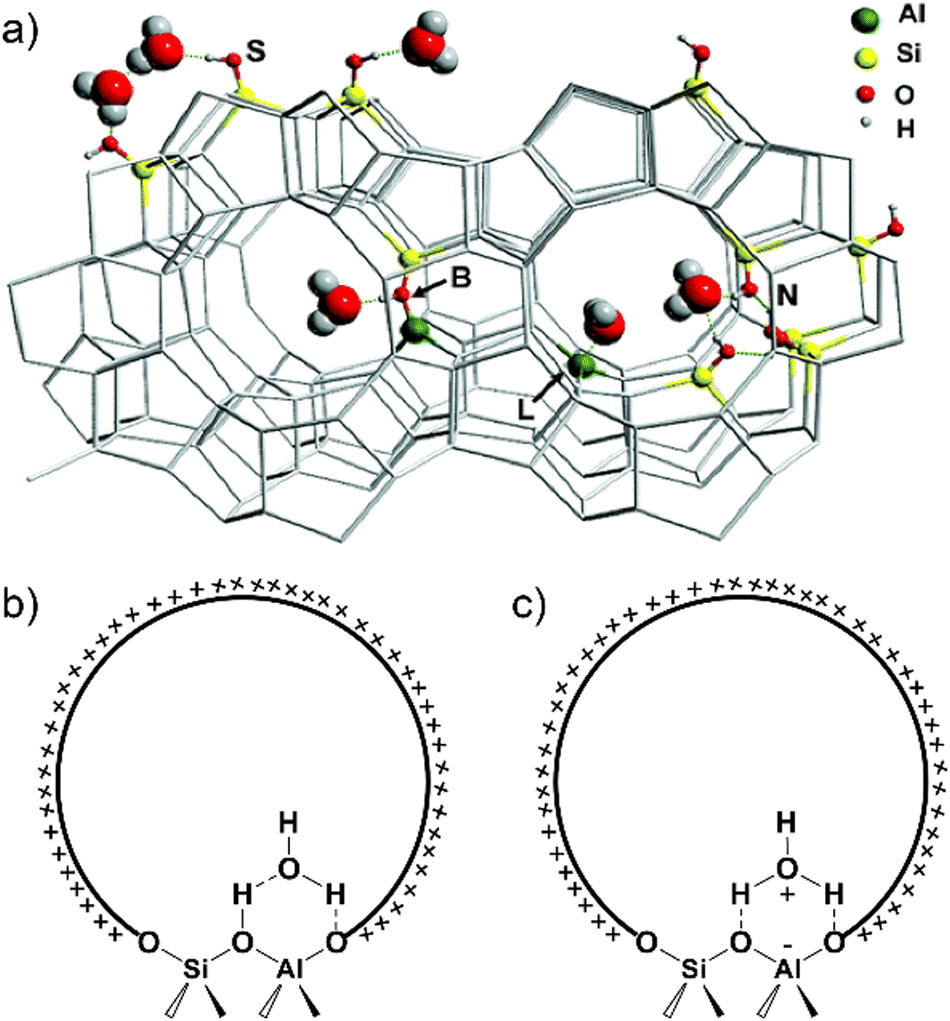 | ||
| Fig. 1 (a) Possible interactions between water molecule and (acidic) sites in H-MFI zeolite cavities. B: Brønsted acid site, L: Lewis acid site, N: silanol nests, S: external surface Si–OH groups. Reproduced with permission from ref. 54. Copyright 2006, American Chemical Society. Structural model of water adsorption in H-form zeolite pore: (b) hydrogen-bonded model; (c) protonated model. Dashed lines represent the formation of hydrogen bonds between water species and zeolite framework. | ||
Increasing the dose of water on Brønsted acidic zeolites could convert the monomeric water complex to dimeric species. The dimeric water was predominately assigned to be stable H5O2+ species (that is the typical “Zundel” cation) by the aid of FT-IR spectra58,70,71 and temperature-programmed desorption analysis.72 Comparatively, NMR and molecular dynamics studies have suggested that the proton involved in the dimeric water structures can be transferred from the framework Si(OH)Al group to the sorbed water molecules,73,74 because water dimer has a higher value of proton affinity (806 kJ mol−1) than the water monomer (694 kJ mol−1).63 Water dimer, as the most stable equilibrium structure based on the PBE (Perdew, Burke and Ernzerhof) function, is able to deprotonate the bridging hydroxyl on the zeolite such as H-SSZ-13.66 The as-formed H5O2+ species comprising a hydronium ion core (H3O+) and the second hydrogen-bonded water are ion-paired with the framework aluminum sites on H-ZSM-5 zeolite.68 Notably, these H5O2+ species show quite low stability according to the observations from density functional theory (DFT) optimization, where the protons undergo dynamic rearrangement between the BAS and H5O2+ in the zeolite pores. In addition to proton affinity, the loading of water and temperature are the critical aspects that determine the degree of the protonation of water structures by the zeolitic BAS.53,63,75 It has been known that water adsorption uptakes in zeolite pores are proportional to the content of framework aluminum atoms in H-form zeolitic materials.54,76–78 Ion-paired hydronium ions with higher protonation degree and larger cluster size can be created upon the stepwise adsorption of three or more water molecules on the framework aluminum sites of Brønsted acidic zeolite (vide infra). Meanwhile, the H-bonding interactions between water clusters and zeolite framework and those within water clusters tend to decrease with increasing temperature, as indicated by the study using reactive force field (ReaxFF) method.75 Moreover, zeolite topology often affects the formation and stabilization of protonated water adducts in the zeolite pore networks; protonated water clusters with more than two water molecules are susceptible to be stabilized in small-pore zeolites.49,66,67,79 Brønsted acidic zeolites with unique water structures adsorbed on their frameworks most probably exhibit differently affected catalytic performance, which will be discussed in section 3 dedicated to catalytic roles of water–zeolite interactions.
:1 and 10:1 ratios of water to aluminum) are not stable and the acidic protons could move back to zeolite framework at elevated temperature.75 A recent study by Wang et al. provided experimental evidence for the nature and stability of hydronium ions formed in the hydrated H-ZSM-5 zeolite channels.88 The generation of a hydronium ion when more than two water molecules interact with the BAS in zeolite is unambiguously indicated by the 9-ppm signal in 1H NMR spectra (Fig. 2a). In the 1H-29Si cross polarization NMR spectra collected with increasing water loading on the surface of acidic zeolite, the mobility of the protonated water cluster starts to be promoted, even with the presence of two water molecules on BAS site. Molecularly-confined water clusters tend to have high proton affinities and facilitate the delocalization of the protons of hydroxyl groups (Si(OH)Al) from the zeolite framework in the form of H+(H2O)n.88 The authors also used periodic DFT-based AIMD at the GGA level of theory to calculate the probability of distance between the oxygen atoms and the center of mass (COM) for the system in H-ZSM-5 and another system of protonated eight water molecules in the gas phase. Calculation results suggested that the zeolite-confined water clusters are chemically different from those in case of gas phase without confinement; in particular, water clusters in the zeolite micropores are more compressed and feature with an enhanced proton mobility. Above established findings have been confirmed by inelastic neutron scattering as well as AIMD (at the GGA level) in the work of Jiménez-Ruiz et al.69
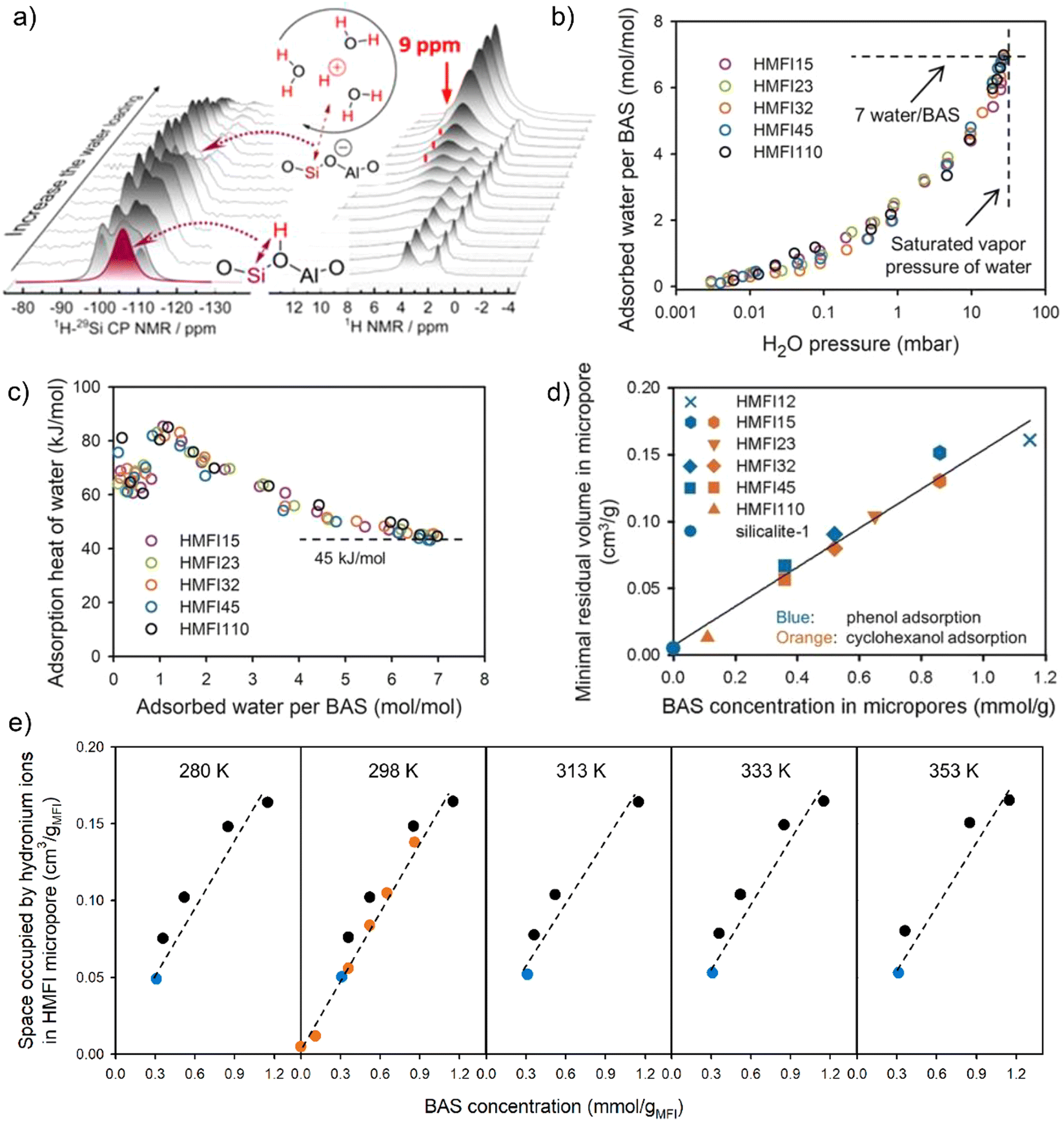 | ||
| Fig. 2 (a) 1H and 1H-29Si CP NMR spectra collected with increasing of water loading on H-MFI zeolite (Si/Al = 15 or 40). Reproduced with permission from ref. 88. Copyright 2019, American Chemical Society. (b) Gas-phase water adsorption isotherms on various H-MFI zeolites (Si/Al = 15, 23, 32, 45, 110) at 298 K, with the value of uptake normalized to the concentration of the BAS. (c) The adsorption heat of water as a function of the number of water per BAS. (d) The micropore space volume occupied by water in zeolite H-MFI at the saturated adsorption of cyclohexanol and phenol in the aqueous phase. Reproduced with permission from ref. 95. Copyright 2019, Wiley-VCH. (e) The space occupied by hydronium ions in zeolite H-MFI micropores as a function of BAS concentration at different temperatures. Reproduced with permission from ref. 99. Copyright 2021, AAAS. | ||
The composition of H+(H2O)n in zeolites can be measured from water vapor adsorption isotherms by the means of thermogravimetric analysis. Both the BAS and silanol groups (i.e., the external silanol and internal defect sites) on zeolite surface serve as the stabilization sites for water molecules. In addition, the density of silanol groups usually relies on the synthetic parameters and the structural properties of resulting zeolitic materials. These thus lead to differences in the quantification of the number of adsorbed water per acid site on Brønsted acidic zeolites.89,90 For example, Chen previously reported a stoichiometric ratio of four water molecules on each framework aluminum from the water adsorption isotherm at a relative pressure P/P0 of 0.6.91 Zechhina et al. obtained an averaged three-to-five interval of water molecules for the H+(H2O)n, by normalizing the total number of water filled in zeolite pores to the number of BAS site.49 Olson et al. identified the tetrahydrate of the proton, H9O4+, as the stable water ionic complexes in H-ZSM-5 with the Si/Al ratios range from 37.5 to 4330.77 A relatively high ratio of five water molecules per the site of framework aluminum on zeolite H-ZSM-5 was observed from the work of Sano et al.92,93 Moreover, Chen et al. found that the size of water clusters formed around each acid site of H-MFI zeolite varied with the density of acid sites.94 Zeolites with a Si/Al ratio of 140 typically showed an adsorption capacity of around eight water molecules per the acid site.
Very recently, Eckstein et al.95 reported a composition of 7 ± 1 water/BAS by subtracting the amount of water adsorbed on the defect sites of H-ZSM-5 zeolite based on the data from Olson and Sano.77,92,93 The authors also researched the gas-phase water adsorption on series of H-MFI zeolites by stepwise dosing water vapor until adsorption saturation (31.6 mbar) at 25 °C. Water adsorption uptake for the hydrophobic silicalite-1 was determined to be one water molecule per unit cell. The BAS on zeolite H-MFI eventually function as the selective adsorption sites for water molecules from the gas phase. An identical adsorption isotherm was obtained by normalizing the amount of adsorbed water to the concentration of the BAS site (Fig. 2b). Hydronium ion cluster comprising of one proton and seven water molecules can be generated at the stage of the saturation of water vapor. The adsorption heat for the first water on BAS is about 65 ± 5 kJ mol−1 (Fig. 2c). With increasing the number of water adsorbed on the acidic sites, water adsorption heat firstly increases to 85 kJ mol−1 and then presents a decreasing trend to 45 kJ mol−1 which is consistent with the water condensation heat (ΔH = 43.5 kJ mol−1) at 30 °C. In the work of Bates et al.,96 the intracrystalline water structures on H-BEA zeolites with 0.11 to 2.0 proton per unit cell were studied by means of the gas-phase adsorption isotherms and in situ IR spectroscopy. At low water partial pressure (P/P0 < 0.2), the generation of (H3O+)(H2O)6±1 clusters on the acidic sites of H-BEA was favored, whereas extended hydrogen-bonded networks started to be formed within the pores at P/P0 > 0.2. In order to precisely quantify the structure of hydronium ions confined in the pores of H-BEA, Kim et al.97 synthesized a wide range of H-BEA materials with varied BAS concentrations and minimized amount of defect sites. In this work, each hydronium ion is measured to be composed of 10 water molecules and one proton (H+(H2O)10). This also contradicts with the result from DFT calculations on H-BEA previously performed by Mei et al.80 Such discrepancy indicates that the composition of hydronium ion clusters is likely dependent on the zeolite topologies. According to Kim et al.,97 the larger size of hydronium ions in H-BEA than those in H-ZSM-5 resulted from the lower entropy loss in larger H-BEA pores.
The composition of water clusters in zeolite pores can also be measured by the saturated adsorption uptake of organic substrate, e.g., cyclohexanol in aqueous phase.95 Assuming the packing density of hydronium ion equals to that of free water, the volume of hydronium ions is the difference between micropore volume of zeolite and volumetric uptake of organic adsorbates. A linear correlation of the volume of hydronium ion with respect to the concentration of BAS on H-ZSM-5 zeolites was reported by Eckstein et al. (Fig. 2d).95 The composition of hydronium ions confined within the H-ZSM-5 micropores is H+(H2O)7±1, which is consistent with that measured from gas-phase water adsorption. As further indicated in the study of Grifoni et al.98 by the means of ab initio metadynamics calculation at the PBE + DFT-D2 level (D2: Grimme's second-generation dispersion corrections), water clusters fabricated around the BAS of H-ZSM-5 bring out limited size because of the decreased stabilization by the inner energy of water molecules that are hydrogen-bonded in the hydrated hydronium ion clusters. By examining a selected group of zeolite frameworks (MFI, CHA, FAU, GIS), the authors demonstrated the minor role of zeolite confinement in determining the structural behavior of water clusters and the adsorbed water molecules are primarily surrounded the BAS without far away diffusion in the zeolite cavities.
Each hydronium ion cluster (H+(H2O)8) has been determined to occupy a volume of (239 ± 15) Å3 in the micropores of zeolite H-MFI with varied Si/Al ratios.95 Based on the diameter of H-MFI zeolite cavities (5.6 × 5.4 Å in diameter for the 10-membered ring channels), a cylindric model was assumed for H+(H2O)8 cluster with a radius of ca. 2.8 Å and a length value of about 10 Å. This implies that the hydrated hydronium ions are tightly confined in zeolite micropores. In addition, the hydronium ions confined inside of the H-MFI pores are stable with respect to their composition at the temperature between 280 and 353 K (Fig. 2e).99 The formation of a stable H+(H2O)8 water cluster has been confirmed by the calculated radial distribution function, where an identical structure of local coordination environment was observed for hydronium ions with eight and 16 H2O molecules loaded on zeolite.88 Additional water molecules are prone to adsorb onto the silanol groups and/or act as the free water outside the zeolite channels. Apart from the above advances, there is a lack of experimental information for the structural and composition of water clusters formed in large pore-sized zeolites (e.g., H-FAU) and other microporous materials. The question arises as to what is the stable composition of water clusters in the presence of adsorbates such as alcohols and bases of various strength that can potentially replace water molecules in the zeolite pores. Moreover, the structure of water clusters remains unclear under realistic reaction conditions, since zeolitic materials are catalytically applied at higher pressure and temperature. It will be thus essential for future studies to evaluate above aspects related to the water structures on zeolite surface.
The molecular structure and H-bonding configuration of water clusters in protonic zeolite was recently studied by Hack et al.,100 enabled by collecting 2D IR spectra and AIMD simulations using DFT at the revPBE/DZVP level of theory. Water clusters with a composition of H+(H2O)8 are simulated to have branched configurations composed of a Y-shaped cluster at the channel intersections and an elongated configuration inside the straight channels (Fig. 3a and b). For a H+(H2O)8 water cluster occluded in H-ZSM-5 zeolite, water molecules those donate two H-bonds (DD), donate one single H-bond while one free O–H bond (SD), and those donate no H-bonds (ND) are considered and quantified from the AIMD simulations. Results indicate that zeolite intersections are capable of accommodating a high degree of connectivity and the water molecules adsorbed in channel intersections possess a slightly larger fraction of SD, whereas those in the straight channel bring out a greater fraction of ND (Fig. 3c). The tight confinement effect in zeolite pores make the fractions of the H-bonding of water molecules in H+(H2O)8 water clusters completely different to those of gas-phase protonated water clusters that manifest the T-shape and cage structures as the stable configurations (Fig. 3). The authors anticipated that these findings could enhance the understanding of zeolite hydration behaviors under hydrous conditions and the mechanistic descriptions of zeolite acid chemistry.
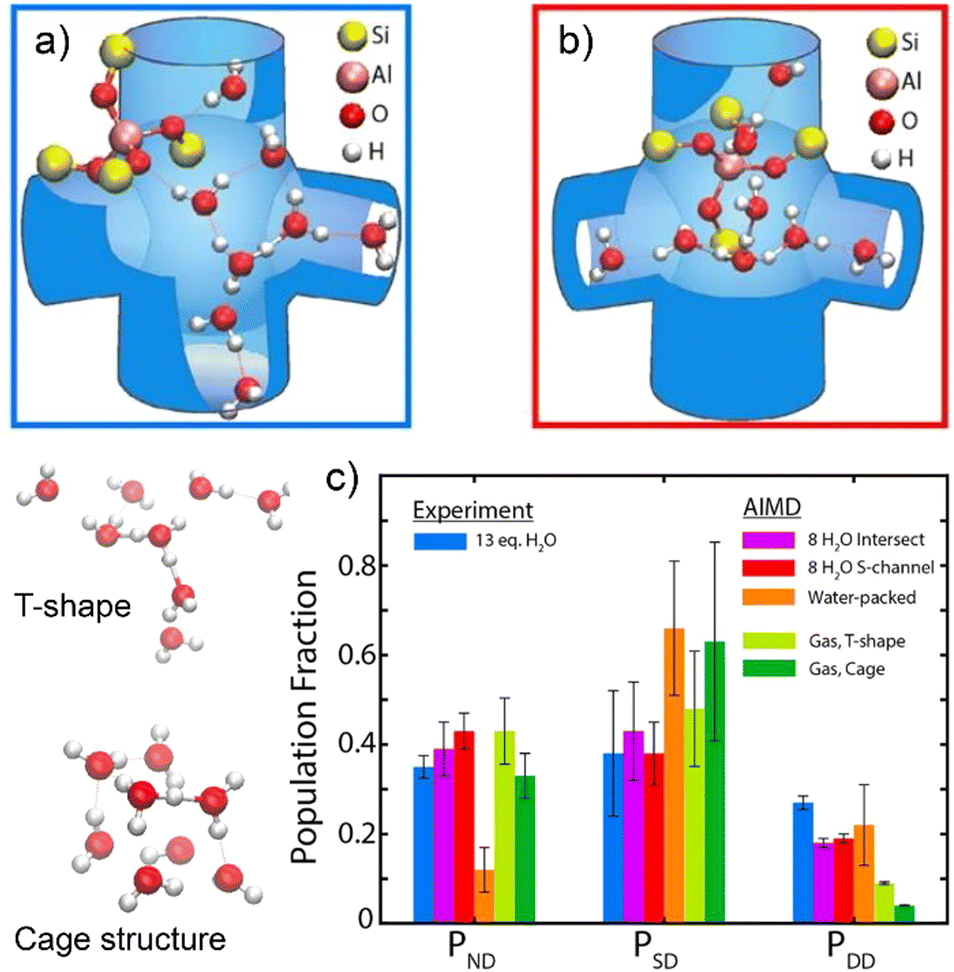 | ||
| Fig. 3 AIMD simulation of the configuration of protonated water cluster in (a) the straight channel and (b) the intersection with zigzag channel of H-ZSM-5. (c) Comparisons of the distributions of ND, SD, and DD water molecules between IR spectra fitting and AIMD simulations. The models of gas-phase water clusters with T-shape and cage configuration are presented on the left side of plot. Reproduced with permission from ref. 100. Copyright 2021, American Chemical Society. | ||
2.2. Lewis acidic zeolites
The structure of the BAS site on aluminosilicate zeolites has been well defined as the bridging Si(OH)Al unit.40 Comparatively, the generation and the structures of Lewis acid sites (LAS) related to the Lewis acidic aluminum have remained not well-understood.101–103 This originates from the dependencies on the zeolite framework, Si/Al ratios, hydrothermal synthetic and postsynthetic procedures (calcination, steaming, acid leaching, and so on), exposure to varied conditions (in the dehydrated or hydrated form) and practical catalytic reaction conditions. In general, the aluminum ions with three-coordinated, tetrahedral, penta-coordinated and octahedral coordination environment are the potential precursor species for the formation of Lewis acidic sites in zeolite structures.102–108 Lewis acidic aluminum sites on the aluminosilicates can be originated from framework, framework-associated or extra-framework species. It calls for detailed structural understandings about these sites by employing various instrumental techniques, e.g., MAS NMR.103–105 In addition, a reversible switch between tetrahedral and octahedral coordination exists for the framework-associated aluminum sites.101,102 Assigning Lewis acid sites to the extra-framework aluminum debris and further correlating to Lewis acidic property of a zeolite do not hold in most studies.101 Exchanging of a portion of framework Si4+ with tetravalent atoms, such as Ti4+, Sn4+ and Hf4+ could result in discrete Lewis acid sites and unique hydrophilic/hydrophobic properties of zeolite.109,110 Lewis acidic zeolites can serve as the active and selective acidic catalysts for alkene epoxidation,111 sugar isomerization,112–115 alcohol dehydration,116,117 Baeyer–Villiger oxidation118 and many other reactions.119,120 In addition to be a solvent for catalytic reactions, water may possess the opportunity of contacting (or binding) with zeolites upon calcination and subsequent exposure to ambient air or under post-synthetic conditions (e.g., ion-exchange treatment). On Lewis acidic zeolites, the adsorption of water mainly occurs on the isolated heteroatom substitutions and silanol groups or framework defects in the pores. Detailed understanding of the interactions of water with certain types of Lewis acidic zeolites is pivotal as well to performing catalytic transformations with optimal efficiency.The composition of water structures stabilized on Lewis acid sites can be determined by gaseous water uptakes.127,128 For example, Bregante et al.128 reported that each framework titanium atom with a structure of Ti(OSi)4 in Lewis acidic Ti-substituted MFI zeolites was able to stabilize about five water molecules at the saturation pressure of water at 293 K. Si-MFI zeolite was measured under a similar condition to subtract the contribution of water adsorption on silanol nest defects. Since the chemical potential of water adsorbed at the titanium site equals that of saturated water in gas phase and even that of water in bulk aqueous phase, the number of water adsorbed on the Ti-MFI zeolite reflects the size of water clusters when engulfed in condensed water. Interestingly, niobium (Nb)- and tantalum (Ta)-substituted MFI zeolites were characterized to contain weak BAS in this work.128 Each framework niobium and tantalum stabilized seven to eight water molecules upon the saturated adsorption of water molecules, showing the similarities with the size of water clusters that are formed on the aluminum-containing Brønsted acidic zeolites (except for H-BEA97) in gas or liquid water phase.88,95,98–100
Difference in the densities of defect sites could cause an obvious variation in the water uptakes inside of the pores. In the work of Cordon et al.,125 at the partial pressure (P/P0) between 0 and 0.2, about 20–25 times higher water uptakes were measured on hydrophilic Ti-Beta zeolites synthesized in a OH− condition than on hydrophobic Ti-Beta zeolites that were prepared in fluorine media. Hydrogen-bonded water networks are prone to form on hydrophilic Ti-Beta zeolites with increasing of water chemical potential, as indicated by the full perturbation of hydroxyl groups from the IR spectra upon water adsorption. Bregante et al.128 reported that the density of surface silanol groups on Lewis acidic MFI zeolite varied with the ratio of hydrofluoric acid (HF) to structure-directing agent (tetrapropylammonium hydroxide, SDA) in the process of hydrothermal synthesis. The MFI zeolite synthesized with a HF/SDA ratio of 1.5 was determined to possess a water uptake 100 times less than that of the analogous material prepared in the OH− condition (i.e., zero HF-to-SDA ratio).
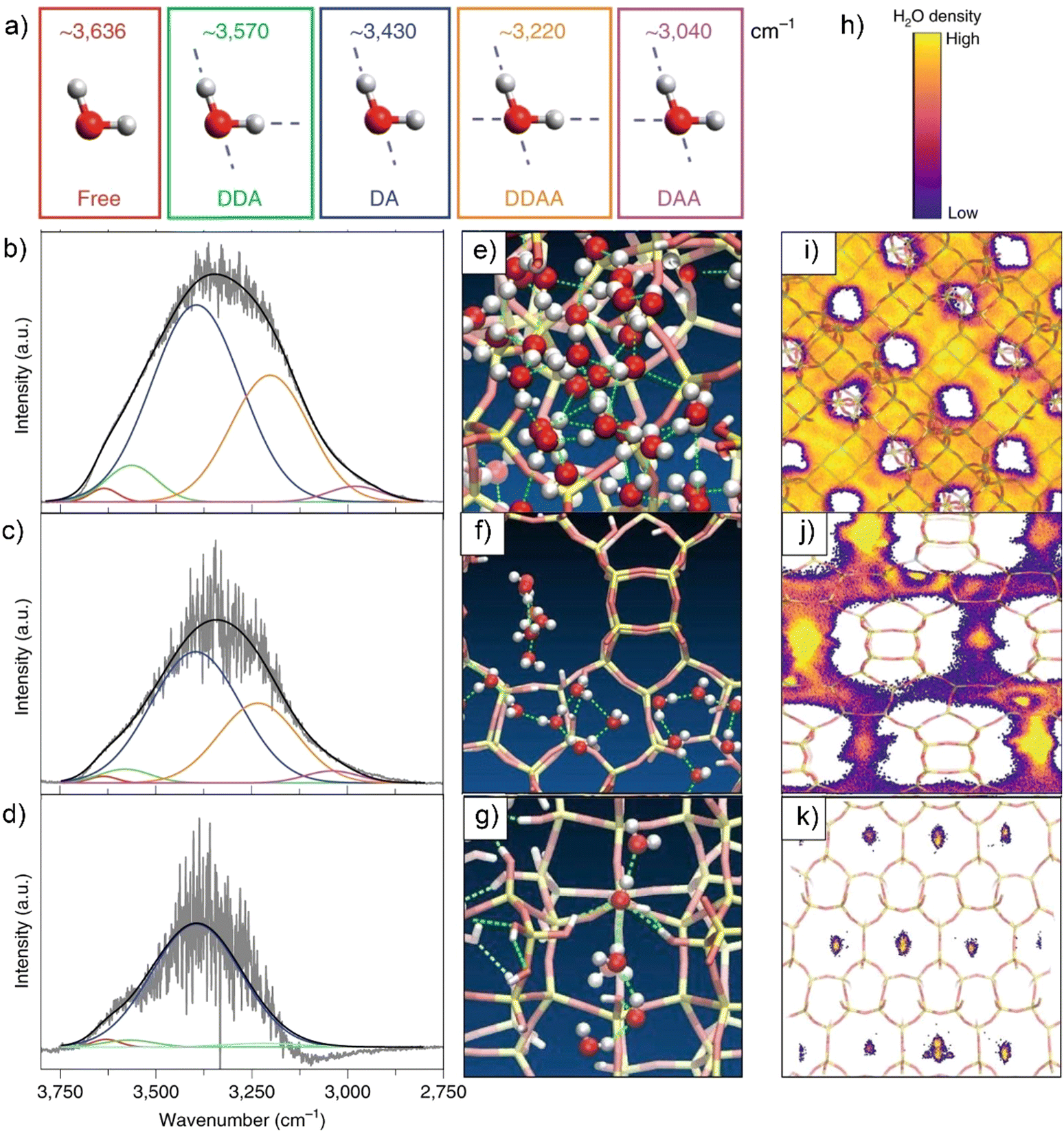 | ||
| Fig. 4 (a) Spectroscopically observed H-bonding configuration of H2O (Details for the abbreviations: “free” means water molecules are lack of hydrogen-bonding (HB); DDA: water donates two and accepts one HB; DA: water donates one and accepts one HB; DDAA: water donates two and accepts two HBs; DAA: water donates one and accepts two HBs). IR spectra of H2O over a ZnSe internal reflection element within the pores of Ti-FAU-OH (b), Ti-BEA-OH (c) and Si-CDO-OH (d). Snapshot of MD simulations of H2O in the pores of Ti-FAU-OH (e), Ti-BEA-OH (f) and Si-CDO-OH (h) constructed with five Si(OH)4 per unit cell. The dashed lines denote as the H-bonding between H2O molecules. Time-averaged spatial distributions of H2O after 1 μs MD simulation projected onto 2D images of Ti-FAU-OH (i), Ti-BEA-OH (j) and Si-CDO-OH (k). (h) The heat map bar for water spatial distributions with low (purple) to high (yellow) densities. The experiments of collecting IR spectra and MD simulations are all conducted at 313 K. Reproduced with permission from ref. 126. Copyright 2021, Springer Nature. | ||
In above work, as calculated from the IR vibrational spectra and MD simulations, the average number of hydrogen bonds decrease with the decreasing of pore dimensions.126 Comparing to Ti-FAU-OH, the hydrophilic Ti-BEA-OH, Ti-MFI-OH and Si-CDO-OH zeolites possess a gradually increased and higher percentage of hydrogen bonds that are formed between water and silanol defects. These lead the authors to state that water molecules in zeolite BEA, MFI, and CDO pores coalesce into one-dimensional oligomers/chains, which are different from that fabricated in the FAU pores (Fig. 4e–k). Of note, forming hydrogen bonds confer the stability to the water structures in larger pores, but these water structures are susceptible to lose stability with decreasing the pore diameter from 1.3 to 0.45 nm. These pore-size-dependent water structures have to reorganize themselves through breaking hydrogen bonds to enable surface catalysis and alter the free energy landscape of catalysis at the solid–liquid interfaces (vide infra).
3. Roles of water structures in zeolite catalysis
3.1. Tailoring the chemical thermodynamics of reactive substrates and intermediates on the surface
Catalytic reactions on acidic zeolites usually involve an initial adsorption of reactant molecules into the pores and a subsequent interaction (e.g., H-bonding or protonation) with the surface acidic sites (e.g., BAS, LAS), before the intrinsic activation step. Zeolite pores can stabilize gaseous reactant molecules through van der Waals forces that depend on the size of pores and the diameter of reactant molecules. In contrast, in liquid-filled pores of zeolites the thermodynamics of liquid-phase reactions can be greatly altered by the solvent molecules (e.g., water). The thermodynamic terms mainly cover the energies of sorption, solvation and interactions of reactants or between reactant and surroundings.16 The adsorption behaviors of reactant molecules on Brønsted and Lewis acidic zeolites from anhydrous and hydrous gas phase and aqueous liquid phase are illustrated in Scheme 1. Typical effects of water structures on the thermodynamics for reactant molecules and surface intermediates on zeolites are discussed as follows.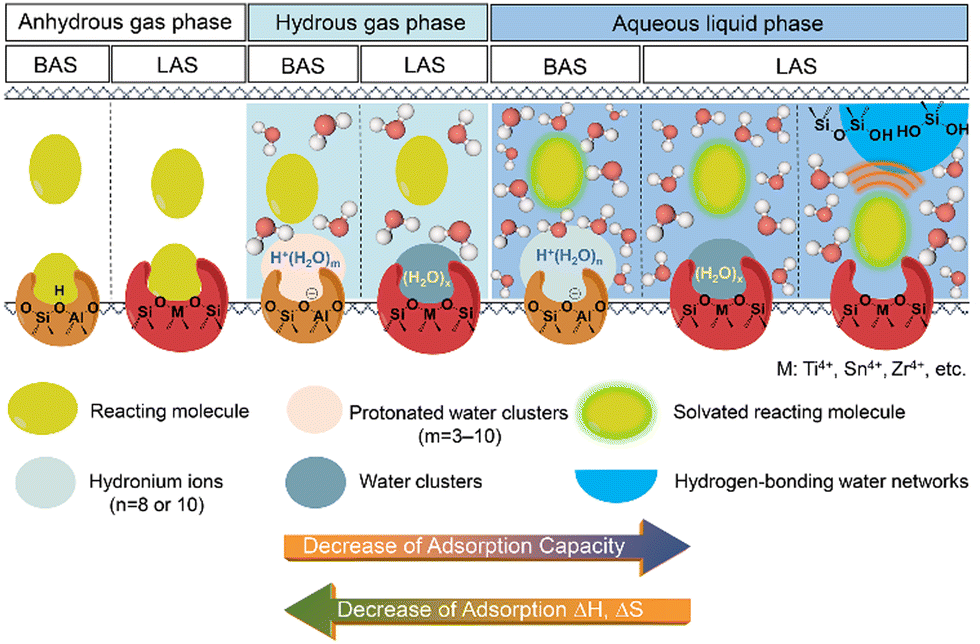 | ||
| Scheme 1 The illustrations of the adsorption of reactant molecule on the BAS and LAS sites of zeolite in anhydrous and hydrous gas phase and aqueous liquid phase. | ||
Competitive adsorption between co-feeding water molecules and coke precursors has been positively applied to depress coke formation in the MTO process.135–137 The consequences in terms of either eliminating catalytic reactivity or quenching the deactivation of zeolites are reliant on the concentration of co-fed water in the reactant stream.135 In addition, for the conversion of methanol with dimethyl ether to produce propylene on H-ZSM-5 zeolite, water in the stream was found to facilitate olefins desorption and ultimately allow for obtaining the desired product distribution.138 The competition-induced desorption by water was also used to explain the achieved high selectivity of linear alkanes from n-hexadecane hydrocracking process on a Pt-containing shape-selective MFI catalyst.139 In this case, water at the MFI acid sites effectively minimized the probability of the secondary cracking and isomerization of the primary cracking products, achieving 80% selectivity of linear cracking products at 80% n-C16 conversion at a low temperature (225–235 °C) and high space times (3500–28000 kg s mol−1). Note that a large portion of water molecules adsorbed on the BAS sites are prone to desorb with decreased equilibrium pressure and at elevated temperatures, which might be accompanied by the diminished concentration of hydrogen-bonded complexes and protonated water clusters in the zeolite pores.50,53,132 That is, water competitive adsorption phenomena may become less pronounced when operating a catalytic reaction under high-temperature conditions.45,133,140 It has been shown to be the case for ethene to aromatics reaction on H-ZSM-5 zeolite when operating the reaction at above 300 °C, where the deterioration effects of water in terms of suppressing the conversion of ethene, the yield of aromatics and hydrogen transfer process, and changing product distributions became relatively insignificant.133
Water molecules also participate in the formation or transformation of reactive intermediates on the surface of acidic zeolites. For example, co-feeding of water was found to shift the equilibrium constant of formaldehyde hydrolysis, which can result in a low concentration of formaldehyde and the reduced initiation and termination rates during the MTO conversion on H-SSZ-13 zeolite.141 Such mechanistic role of water is responsible for the long induction period and high total turnovers observed for MTO with water cofeeds. Not limited to water, note that some reactant molecules such as low-carbon chain alcohols (e.g., methanol, ethanol) can be protonated by the BAS of zeolites.142 Water-reactant-clusters such as (C2H5OH)(H+)(H2O)n cluster have been identified on H-BEA zeolite for the gas-phase ethanol dehydration at a low water pressure (< 10 kPa).96 In addition, water-ethanol dimers (co-existed with the reactive ethanol–ethanol dimers) have been observed on Sn-BEA zeolites and their coverages on the surface depend on the polarity of zeolite pore environment.116 The formation of water-reactant-clusters usually leads to inhibitory effect on zeolite catalysis.
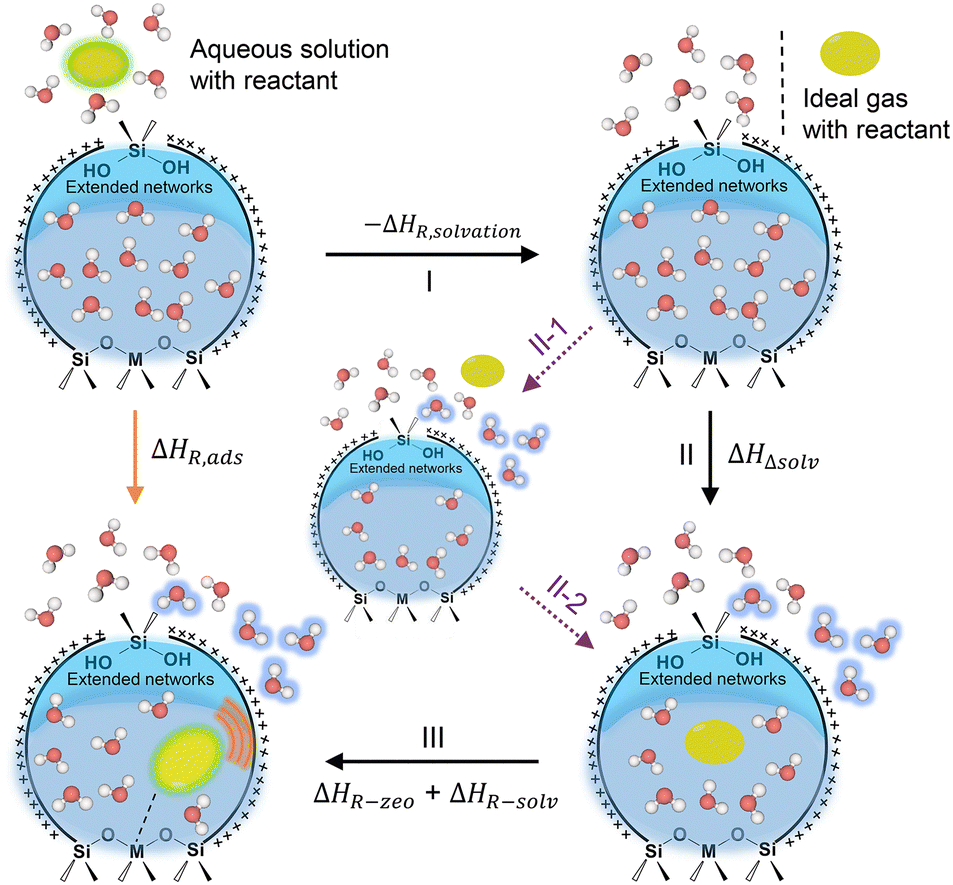 | ||
| Scheme 2 Thermochemical cycle for the deconvoluting enthalpic contributions of ΔHR,ads. Herein, Lewis acidic zeolite pore with heteroatom substitutions (M: Ti, Sn, Zr, etc.) and silanol defect sites on the internal surface is taken as the example. I step: formation of ideal gas reactant molecule from aqueous solution; II step: reactant molecule adsorbs into solvent-filled zeolite pore from gas phase, accompanied by the displacement of inside water molecules (further illustrated by dashed purple arrows in this scheme); III step: adsorbed reactant molecule interacts with water structures and zeolite pores. | ||
As for Brønsted acidic zeolites, hydrated hydronium ions are inclined to occupy the hydrophilic volume space of micropores, showing a strong dependency on the concentration of framework aluminum sites.95Fig. 5a shows that organic molecules preferentially adsorb into the hydrophobic volume space that are preserved to be unoccupied by the hydronium ions in H-MFI channels/cavities. The distance between the center of hydrated hydronium ion and the center of nonpolar domains (dh-n) sharply decreases with the increasing amount of the BAS (that is decreasing of Si/Al ratios) (Fig. 5b). Consequently, the adsorption uptake of organic molecules into the hydrophobic volume from the aqueous phase are decreased.144 On the other hand, zeolite-confined hydronium ions provide a “quasi solid electrolyte” environment and thereby a specific ionic strength in the pores.95 The values of ionic strength determined by normalizing the concentration of BAS to the micropore volume of H-MFI display a proportional relationship to the BAS concentration.95,97 A high ionic strength does in turn dictate the chemical potential of sorbed cyclohexanol and its stabilization state as well. A trend observed for cyclohexanol adsorption on H-MFI illustrates that the chemical potential of cyclohexanol (μ+) increase with the decrease of distance between the center of polar and nonpolar domains (dh-n) (Fig. 5b). This trend indicates the stronger destabilization of cyclohexanol caused by the presence of confined hydronium ions in zeolite pores. The typical exhibition of destabilization effect is the smaller adsorption heat and adsorption constant in an aqueous phase. As a result, the interactions of the organic part of reactant with zeolite are weakened and the corresponding interactions with the protons are favored in the non-ideal system with distinct ionic strength. Recently, different extent of the destabilization effect on reactant molecules was observed by comparative studies on the hydronium ions confined within the pores of H-ZSM-5 and H-BEA zeolites.97 A slight increase in the size of hydronium ions (i.e., from H+(H2O)8 to H+(H2O)10) could cause obvious differences on the calculated excess chemical potentials of the sorbed substrate (i.e., 0.3–2.4 kJ mol−1 for H-ZSM-5 vs. 0.1–1.8 kJ mol−1 for H-BEA).
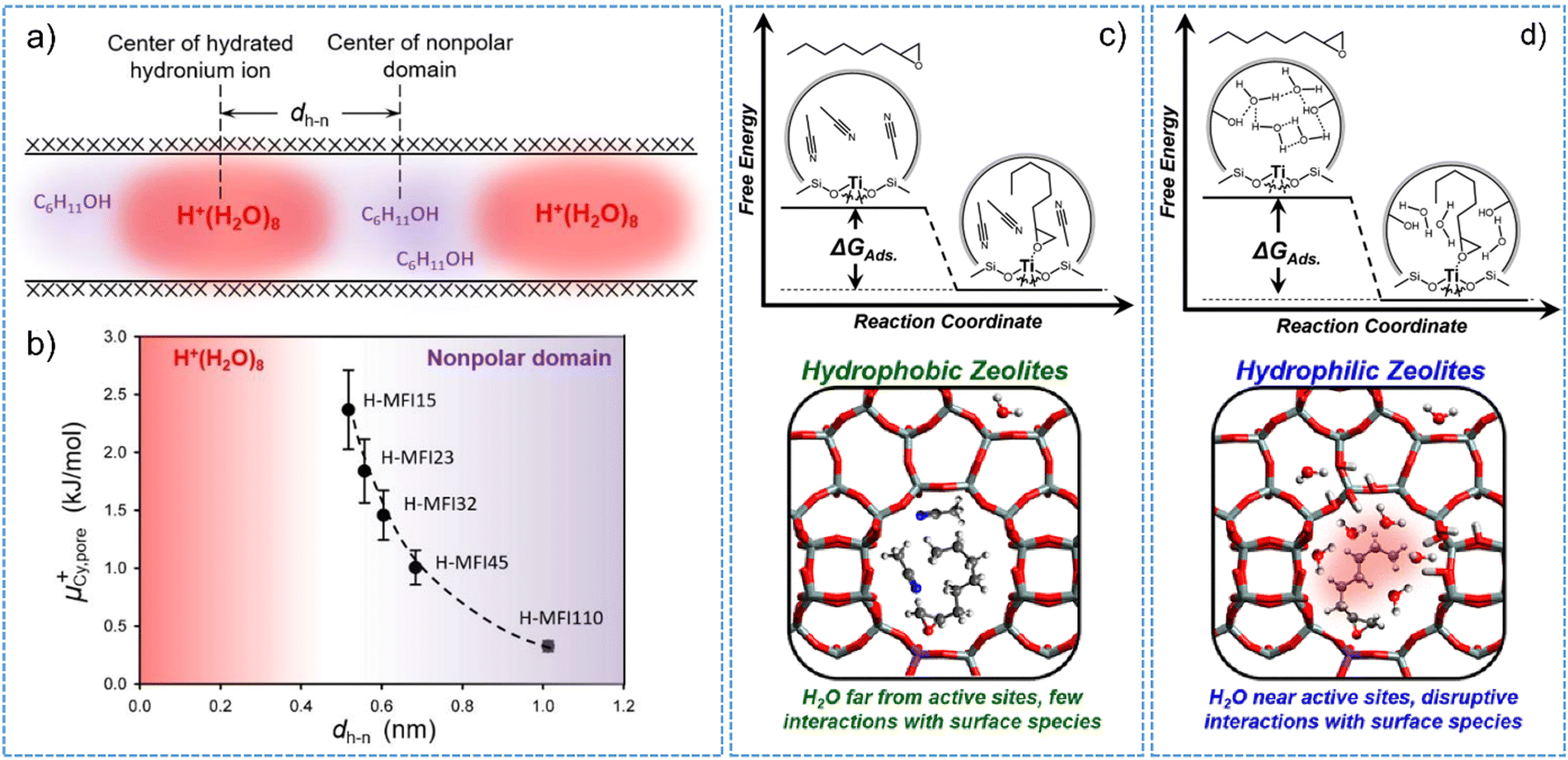 | ||
| Fig. 5 (a) Schematic illustration of hydronium ions and nonpolar domain in H-MFI channels. (b) Chemical potential of cyclohexanol adsorbed in zeolite pores as a function of dh-n. The dh-n is the distance between the center of polar and nonpolar domains. Reproduced with permission from ref. 95. Copyright 2019, Wiley-VCH. Free energy profile (upper) and schematic illustrations (bottom) for the adsorption of C8H16O (1,2-epoxyoctane) in hydrophobic (c) and hydrophilic (d) pores of Ti-BEA. Reproduced with permission from ref. 143. Copyright 2019, American Chemical Society. | ||
The thermodynamics of adsorption in acidic zeolite micropores with the presence of different solvent including water have been studied by employing pyridine, a typical base probe molecule.145,146 Owing to the efficient stabilization effect of water solvation on the protons of BAS, desorption of protonated pyridine from H-ZSM-5 pores shifted to occur at lower temperature, comparing to the system with liquid acetonitrile, n-heptane or to that upon exposure to a vacuum condition.145 The adsorption enthalpy and entropy of pyridine molecules on H-ZSM-5 in the aqueous phase were lower in magnitude than those measured from the gas phase.146 A low enthalpy value was the typical consequence of the intermolecular interactions between adsorbate and water molecules, and the displacement of water molecules occluded in zeolite pores upon the adsorption of substrate molecules from water phase. The less-extreme value of adsorption entropy was associated to the restrictions by zeolite confinement effect on the translational/rotational degrees of freedom. In addition, compared to mesoporous materials (e.g., MCM-41 and SBA-15), the formation of Brønsted acid site-pyridine interactions could be facilitated on H-ZSM-5 and H-BEA zeolite, showing typical Langmuir characters for the measured pyridine adsorption isotherms in water atmosphere.146 These works additionally point to the importance of an elaborate choice of solvent and the potential effects of solvent on the catalytic reactions performed in condensed aqueous phase.
The hydrogen-bonded water clusters on Lewis acidic zeolite can also impact the reactant adsorption thermodynamics and subsequent catalysis processes.110,143 In addition to the dependency of adsorption capability on the zeolite chemical functionality, the formation of stable surface intermediates following the reactant adsorption and molecular interactions with intrazeolite water clusters should be crucially considered.126,130,143 An example for this is the Lewis acidic Ti-BEA zeolite, on which the hydrogen-bonded water molecules stabilized by silanol nests (i.e., [Si(OH)]4) must reorganize to accommodate the surface intermediates that are adsorbed on the active sites (Fig. 5c and d).143 The consequence is that the values of adsorption enthalpy and entropy for C8H16O (1,2-epoxyoctane) were observed to be 19 kJ mol−1 and 75 J mol−1 K−1 more-positive on Ti-BEA with five silanol groups per unit cell than those determined from the defect-free hydrophobic Ti-BEA sample. Such differences are negligible for the Ti-BEA zeolite measured with the absence of water in an acetonitrile solvent. The free energies of adsorption typically reflect the restructuring of water molecules nearby the framework Ti active sites and lean on the specific interactions at the solid–liquid interfaces, which can be described as excess free energy contributions. A linear free energy relationship between the adsorption and transition state for 1-octene epoxidation reaction suggests the crucial role of surface chemistry involving water structures in impacting the stability of surface adsorbed intermediates and altering the chemical thermodynamics at the solid–liquid interfaces.126 More recently, using alkene epoxidation reactions on Ti-BEA zeolites in liquid solvent, Potts et al.130 have investigated the kinetic and thermodynamic consequences of the specific interactions in solvent-filled pores and revealed the origins of these interactions. The requisite reorganization of solvent molecules (e.g., CH3CN, water, or CH3CN–water mixture) inside of the pores of zeolite Ti-BEA allows the adsorption and suitable accommodation of alkene molecules with different carbon chains. The amount of solvent molecules displaced from zeolite pores determines the stability of adsorbed intermediates, thereby influencing rates and selectivity of catalytic reaction.
3.2. Impacting the catalytic reactivity of surface catalysis on zeolite
Ways of tailoring the reactivity of zeolite catalysis by water structures can be diverse, which have been depicted in Scheme 3. Competitive adsorption phenomena exists in both gas phase and aqueous phase for determining the accessibility of reactant molecules to active sites in the pores of zeolites. Water structures on Brønsted and Lewis acidic zeolites can actively participate in the formation of surface intermediates, induce the stabilization/destabilization of reacting intermediates and transition states, thereby modifying the activation Gibbs free energies for a catalytic reaction. In addition, the changes in the nature of active sites and zeolite pore structural properties in the presence of water are also responsible for the structure–performance relations of zeolite catalysis.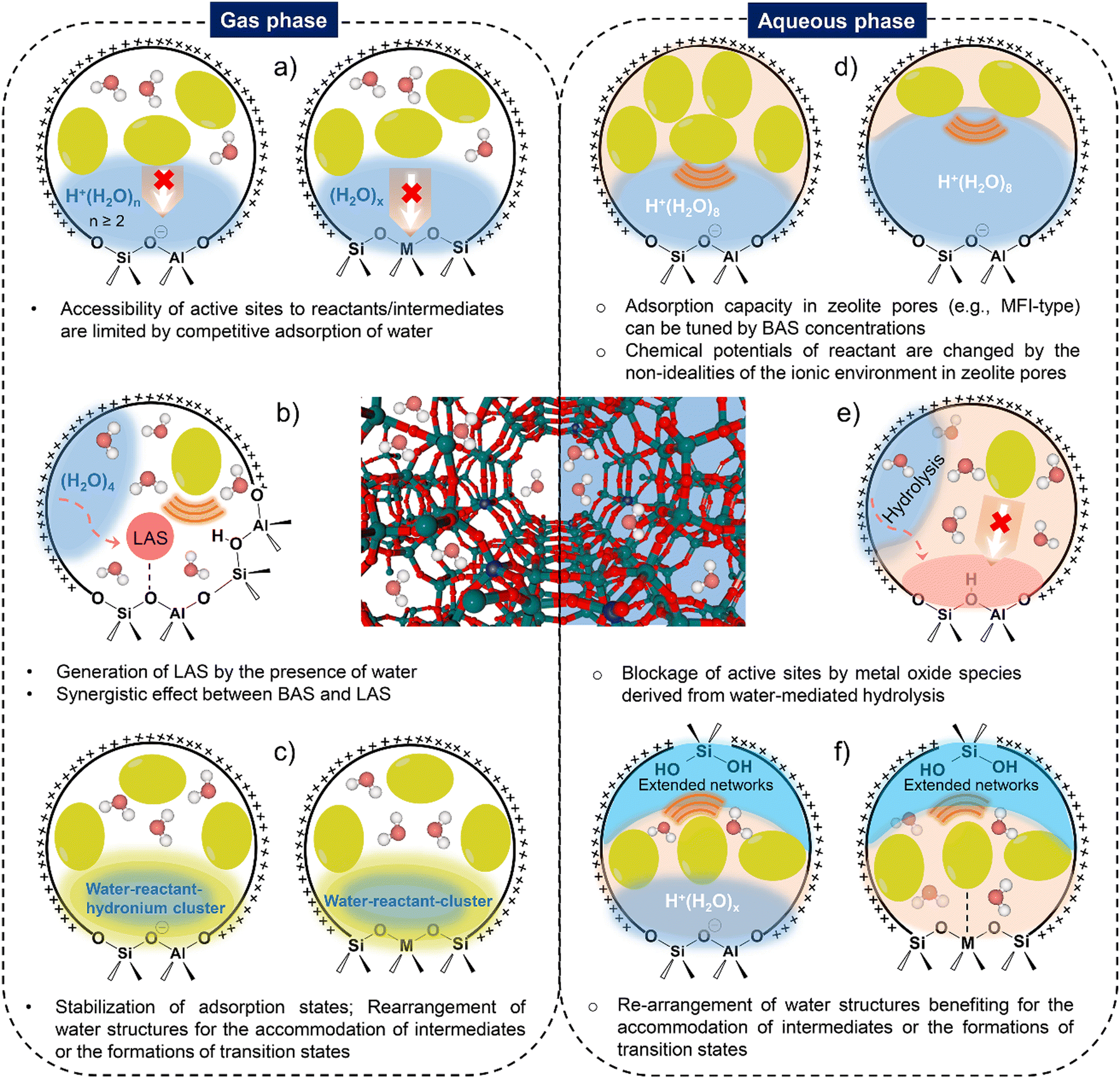 | ||
| Scheme 3 The illustrations of the different ways by water structures for manipulating the zeolite-catalyzed reactions in hydrous gas and in aqueous phase. | ||
Water inhibitions have been intensively observed for gas-phase alcohol dehydration on acidic zeolites (Scheme 3c).96,116,150 For example, at low water pressure (<10 kPa), a −1 order dependence on the water pressure for the ethanol dehydration was measured. It was the consequence of the need to displace one water molecule in the (C2H5OH)(H+)(H2O)n cluster by ethanol on the acid sites of H-BEA.96 Increasing of water pressure led to the formation of (C2H5OH)(H+)(H2O)4–5 with the maximum water–ethanol cluster size and the additional water molecules tended to be hydrogen-bonded in zeolite microporous voids by solvating the water-ethanol clusters. Severe water inhibition with up to −3 of reaction order with respect to the water pressures (10–75 kPa) was ascribed to the more extensive disruption of hydrogen bonds involved at the transition states along the reaction coordinates. Furthermore, in the work of Zhi et al.150 for the catalytic propanol dehydration on H-MFI catalyst, water molecules strongly and preferentially stabilize the ground state of propanol molecules prior to the transition state by forming protonated propanol-water dimer in the zeolite micropores, which results in a high activation barrier and thereby a decrease in the reaction rate. DFT simulations on propanol adsorption on BAS site with the presence of water molecules further suggested that water enhanced propanol adsorption (adsorption energies increased from −151 kJ mol−1 for the case of without water to −163 kJ mol−1 with two water molecules and −188 kJ mol−1 with the presence of four water molecules) through hydrogen bonding.81 The stability of propanol adsorption can be enhanced by forming protonated dimeric propanol and propanol-water complexes. These findings indicate that the formation of water-reactant-clusters on zeolites is detrimental to the catalytic events at the solid–gas interfaces with the presence of gaseous water.
More importantly, the polarity of confining environment of zeolite determines the extent of water inhibition and the catalytic rates for a chemical reaction. Specifically, the coverage of inhibitory ethanol–water dimers was found sensitive to the silanol defects on polar Sn-Beta-OH and non-polar Sn-Beta-F zeolites in the process of gas-phase bimolecular ethanol dehydration.116 The equilibrium constant for the formation of ethanol–water and ethanol–ethanol dimer species on Sn-Beta-OH were determined to be three to four times higher than that in the case of Sn-Beta-F. However, similar kinetic rate constants were obtained for the employed Sn-Beta zeolites, irrespective of whether a highly polar or a less polar framework was involved. It was concluded that ethanol dehydration catalysis responds to the coverage of most abundant reactive intermediate, showing a sensitivity to the H-bonding interactions in zeolite confining nano-environments.116 This work may provide a potential way to circumvent the issue of water inhibition on the catalytic reactivity of zeolite catalysis.
In addition to the inhibitive consequences on zeolite activities, the effect of water on zeolite-catalyzed reaction can become positive. Gešvandtnerová et al.151 observed that water molecules generated from catalytic reaction could mediate proton transfer processes and strongly stabilize the π-complexes of alkenes when performing DFT calculations to explore the monomolecular mechanism of isobutanol conversion to butenes on acidic chabazite zeolite. In another typical work of Chau et al.,152 enhanced rates were observed for cumene dealkylation on H-ZSM-5 zeolites upon water co-feeding. Upon water adsorption, protons from the framework can be delocalized in the form of hydronium ion clusters and the interactions between cumene and hydronium ion clusters were proposed to result in a reaction transition state with more degrees of freedom and thus a less negative activation entropy, comparing to the condition without the presence of water. This work highlights the necessity of exploring and understanding the roles of water on catalytic transformations of different hydrocarbon molecules. Above cases show that the active participation of water in catalytic reactions can be reflected by the formation/stabilization of surface intermediates and transition states, thus influencing the catalytic rates on zeolitic materials.96,116,148–152
Moreover, the positive effect of water on gas-phase catalytic reactions can be resulted from manipulating the diffusivity of reactive compounds and products around the acidic active sites in zeolite pores. Based on solid-state NMR measurements and theoretical simulations, Wang et al.153 have recently demonstrated that water clusters generated on the BAS could promote the diffusion of benzene molecules in zeolite channels and drive their interactions with surface methoxy species (SMS) to obtain highly active SMS-Benzene complexes, eventually improving the methylation reactions of benzene with methanol. This work provides the atomic- and molecular-level insights on the micro-hydrophobic effect induced by water in confined zeolite-catalyzed reactions. In addition, performing DFT simulations to study the reactivity of zeolite-catalyzed alkylphenol dealkylation reaction at high temperatures, Bocus et al.154 found that the competitive adsorption of water molecules with phenol products on the BAS sites could benefit a short residence time of phenol, which further prevents both the poisoning of active sites and catalytic deactivation from being taken place. Although there are some statement in the literatures that the interactions of water molecules with zeolite internal surface could affect the diffusion of reactants/products and further the reaction kinetics,18,39 this effect is relatively unexplored and in-depth studies are still desired.
Similar to the scenario at the solid–gas interfaces, water clusters and/or extended hydrogen-bonded water networks in zeolite-confined micropores dictate the sorption behavior of reacting molecules from aqueous solution and the formation/stabilization of active surface intermediates and transition states (Scheme 3d and f), eventually leading to differences for catalytic performance.114,144 For example, higher turnover rate for phenol alkylation reaction with ethanol in an aqueous phase was measured on H-MFI zeolite that possesses a low amount of hydronium ions (i.e., a high Si/Al ratio) in the micropores.144 The competition between hydronium ion clusters and phenol molecules for the adsorption sites on zeolite imposed no effects for the intrinsic rate constant and product selectivity. It is increase in the adsorption capability of phenol in zeolite H-MFI pores that contributes to the observed enhanced alkylation rates. In analogy to above case, a decreased accessibility of framework Lewis centers to reactant molecules in Sn-Beta and Ti-Beta zeolite pores is responsible for the decrease in the rate of sugar isomerization in aqueous phase.113,155 In the work of zeolite-catalyzed dehydration of polyalcohols and ethylene-vinyl alcohol copolymer in γ-valerolactone (GVL) polar aprotic solvent, the strong adsorption of GVL on framework BASs was found to mitigate the surface coverage of reacting intermediate and cause reduced dehydration rates.156 Interestingly, the addition of water into GVL solvent could tackle the competition of reacting intermediate with GVL molecules for active sites and improve product formation rates. It was achieved by the delocalization of BAS sites from zeolite framework into hydronium ion clusters, the latter serve as mobile active sites. These works also demonstrate the integral role of optimal solvent for catalytic upgrading of biomass compounds and polymer wastes in liquid phase.
There are several scientific advances recently reported from the Lercher group on the topic of acid catalysis by hydronium ion clusters confined in Brønsted acidic zeolites with varied concentrations of BAS. Zeolite confinement effect can synergistically promote the association of reactant molecules with the confined hydronium ions in comparison to those interacted with un-confined hydronium ions in homogeneous acid solution, e.g., phosphoric acid.157–159 Specifically, the positive impacts of confinement effect in zeolite H-MFI on enhancing the reaction rate for aqueous-phase dehydration of cyclohexanol and secondary alkanols (e.g., 3-heptanol and 2-methyl-3-hexanol) are ascribed to the enthalpic stabilization of transition states in the micropores, although less positive activation entropies were determined from the study of kinetic aspects.158,159
It is interesting to note that the hydrated hydronium ions in the micropores of high-crystalline H-MFI zeolite create a unique ionic environment, which eventually brings in non-ideality to a catalytic system.95,99 The turnover frequencies (TOFs) of cyclohexanol dehydration at 423 K as a function of the BAS concentration were compiled into a volcano-shape plot (Fig. 6a), which firstly shows a six-fold increase of TOF values from the BAS concentration of 0.054 to 0.36 mmol g−1 and then a 60% decrease at the BAS concentration of 0.86 mmol g−1.99 The non-ideality triggered by intraporous ionic environment leads to the deviations of activity coefficients from an ideal state, and thereby the excess chemical potentials of both adsorption ground state and transition states are determined. Positive values of the excess chemical potential for the ground state and the negative values of the excess chemical potential for the transition state contribute to the decreases in the activation barriers and, consequently, the increases in aqueous-phase cyclohexanol dehydration TOFs (Fig. 6b and c). Comparatively, in aqueous hydrochloric acid solution the rates for cyclohexanol dehydration increase monotonically with the ionic strength.
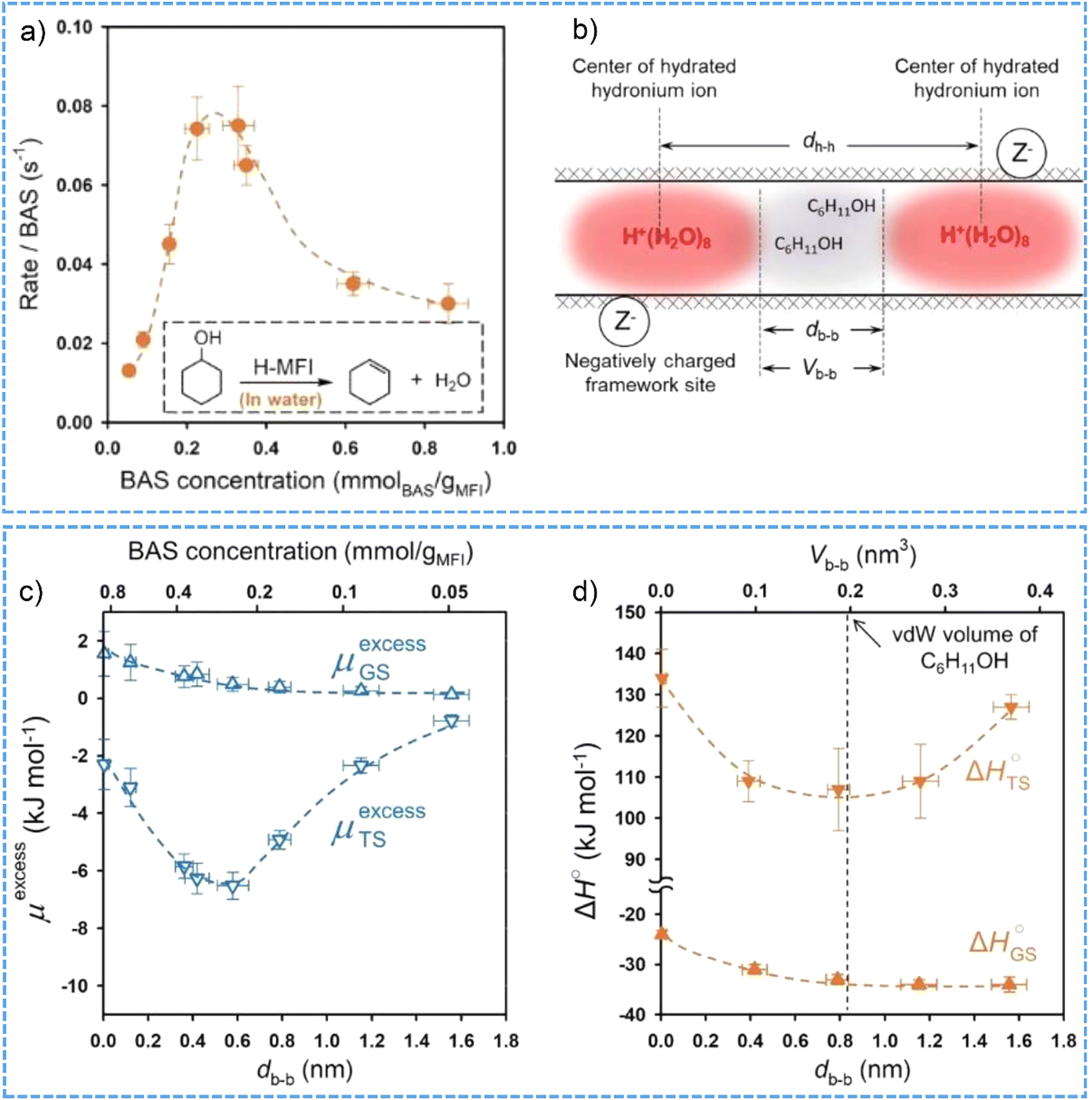 | ||
| Fig. 6 (a) The BAS-normalized reaction rates for aqueous-phase cyclohexanol dehydration on H-MFI zeolites with varied concentration of BAS at 423 K. (b) Schematic illustrations of the mean distance dh-h between two neighboring hydronium ions, the mean distance dh-h and volume Vb-b between the boundaries of hydronium ions in H-MFI pore. The calculated excess chemical potential (c) and enthalpy (d) of the ground (GS) and transition state (TS) as the function of db-b. Reproduced with permission from ref. 99. Copyright 2021, AAAS. | ||
Similar catalytic function (changes of TOF values as a function of the BAS concentrations) was observed for the dehydration of substituted cyclohexanols in aqueous phase catalyzed by varied ionic environment in the H-MFI zeolites.160 However, there were limitations in the rate enhancement for aqueous-phase alcohol dehydration in the ionic environment confined within microporous zeolite. The drop after a maximum position on the volcano plot was likely related to the size exclusion phenomena in the presence of high density of hydronium ions in the molecular-sized pores.99 To be more specific, the adsorption of cyclohexanol molecule in the void space between the boundary of neighboring hydronium ion clusters can be markedly hindered when the void volume is smaller than the van der Waals volume of cyclohexanol (∼0.2 nm3 at 423 K). This thus leads to increased enthalpy of ground state and transition state and, in turn, the decreased TOF values (Fig. 6d). Through measuring the kinetics in the first-order regime to avoid substrate crowding influences, the size exclusion effects have been excluded for the aqueous-phase cyclohexanol dehydration on H-MFI zeolites.161 It was concluded that the rate enhancement at high ionic strength can be partly compensated by the work to separate hydronium ion clusters and rearrange the cation–anion pairs within the zeolite pore network. Therefore, as demonstrated across the whole H-ZSM-5 zeolite crystals, intracrystalline ionic environments lead to a maximum in impact of ionic strength on acid-catalyzed reaction rates.
In analogy to the case of Brønsted acidic zeolites, water structures occluded inside of Lewis acidic zeolite pores alter the reactions of molecules by impacting the excess free energies for the transition states. Cordon et al. reported that, inside of the hydrophobic pores of Ti-Beta-F zeolite, the adsorption of glucose could promote the co-adsorption of water at titanium sites from the solvation sphere of reactant in solution.125 For aqueous-phase glucose isomerization in both zero-order and first-order kinetic regimes, the reaction rate constants were three to twelve times lower on hydrophilic Ti-Beta-OH than those of hydrophobic Ti-Beta-F sample (Fig. 7a), reflected by the entropic destabilization and high Gibbs free energies for the transition state of hydride-shift isomerization on zeolite Ti-Beta-OH with the presence of high density of hydrogen-bonded water networks. The consequences of formation of extended hydrogen-bonded networks of water for sugar isomerization catalysis has also been described for Sn-BEA zeolites in water phase.114,129
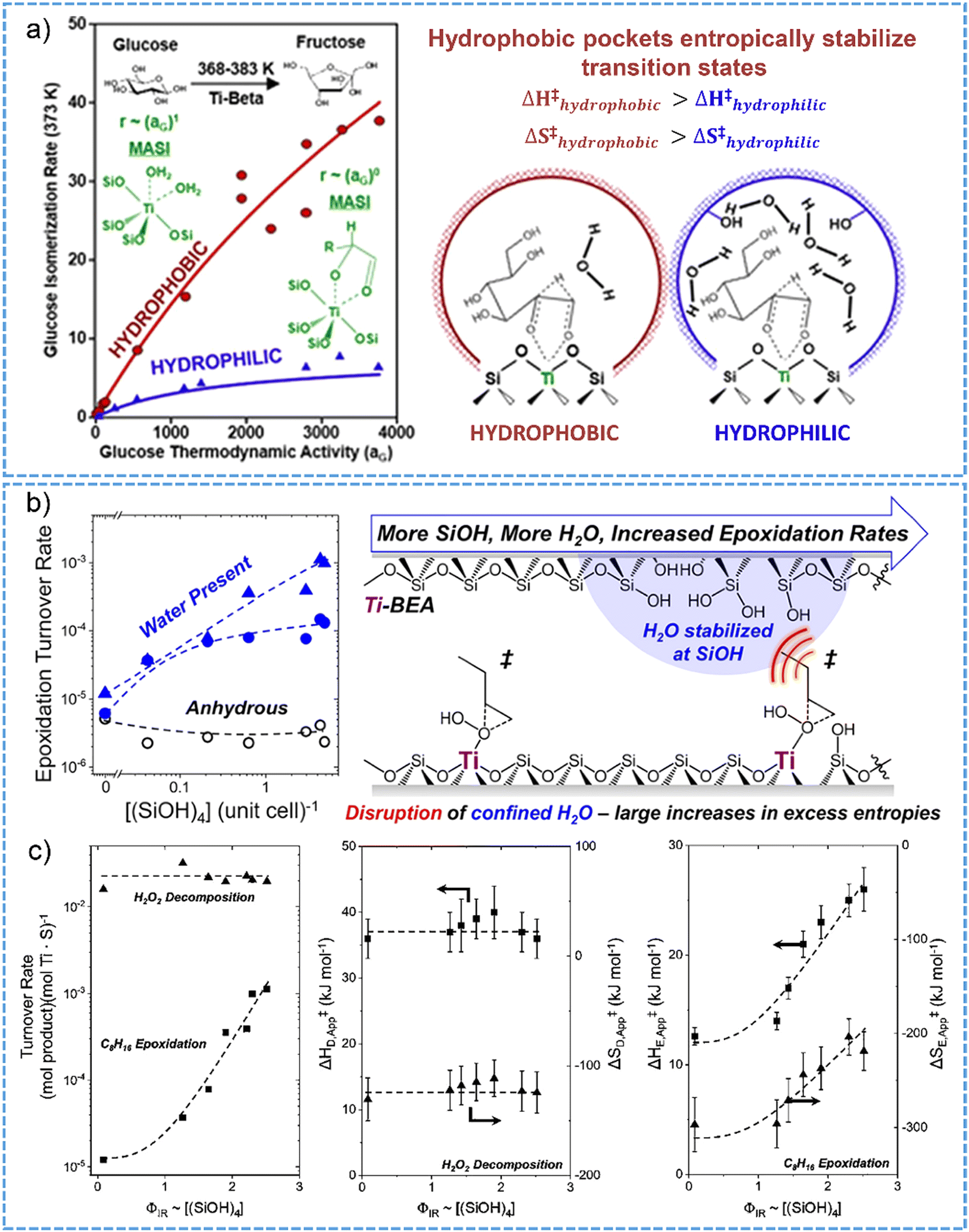 | ||
| Fig. 7 (a) Glucose isomerization on hydrophobic Ti-BEA shows much higher reaction rates than those obtained on hydrophilic counterparts, owing to the entropic stabilization of transition state by hydrophobic pockets. Reproduced with permission from ref. 125. Copyright 2018, American Chemical Society. (b) Epoxidation of 1-octene with H2O2 over hydrophilic Ti-BEA zeolites having certain densities of silanol nests exhibits higher turnover rates in comparison with that catalyzed by hydrophobic T-BEA. (c) The turnover rates for 1-octene epoxidation and H2O2 decomposition at 313 K (left). Apparent activation ethalpies and entropies for H2O2 decomposition (middle) and 1-octene epoxidation (right) as a function of the factions of Si(OH)4 groups. Reproduced with permission from ref. 162. Copyright 2019, American Chemical Society. | ||
Interestingly, the cooperative effects between hydrophilic pores and water structures exist for alkene epoxidation reactions in the presence of Ti-BEA zeolites.130,162 Herein, in hydrophilic pores, the specific interactions between water molecules, reactive intermediates and zeolite surface bring the advantage of enhancing catalytic reaction performance. There are 100-fold larger in the rate and selectivity for 1-octene epoxidation on Ti-BEA zeolite containing five silanol groups per unit cell in comparison with the nearly defect-free Ti-BEA catalysts (Fig. 7b).162 Specifically in this work, the extended hydrogen-bonded networks of water, which are stabilized on the silanol defect sites on hydrophilic Ti-BEA zeolites, benefit the disruptive interactions between water structures and epoxidation transition states in the pores. The rate of 1-octene epoxidation significantly increased with the increasing amount of silanol nests in the series of Ti-BEA zeolites. The rate of H2O2 decomposition did not show such dependence as well as the variations of the apparent activation enthalpies and entropies with the silanol nests concentration (Fig. 7c). Notably, a difference by 12 kJ mol−1 and 93 J mol−1 K−1 for the apparent activation enthalpies and entropies were respectively determined between the most hydrophobic and hydrophilic Ti-BEA zeolite catalysts (Fig. 7c). These results imply that the disruptive interactions with water clusters through the reorganization nearby the titanium sites aids in the stabilization of the transition states in zeolite pores, favorably leading to large-increased gains in the excess entropies and the enhanced catalytic epoxidation rates. Knowledge of the specific interactions with the solvent molecules in zeolite micropores can be applicable for other porous materials to enhance catalytic transformations.130
Water molecules especially those from liquid hot water can also induce the extensive hydrolysis of zeolite framework structures under catalytic conditions, thereby giving rise to the loss of activity. As reported in the work of Vjunov et al.,172 there were 30–40% decrease of micro- and mesopore volume for zeolite H-BEA (Si/Al = 15–75) after aging in water at 160 °C for 48 h. This change in zeolite structures was associated to the pore blocking via framework hydrolysis of Si–O–Si and Si–O–Si–OH groups, and the re-precipitation of silica on crystallite surface. As a result, the accessibility of active BAS sites to cyclohexanol molecules was reduced, leading to a decreased activity for the aqueous-phase alcohol dehydration. The authors stated that the structural and functional stability of zeolites with the presence of intraporous water are important factors determining the activity performance for reactions carried out in liquid hot water.
3.3. Impairing catalytic stability of zeolite by intraporous water structures
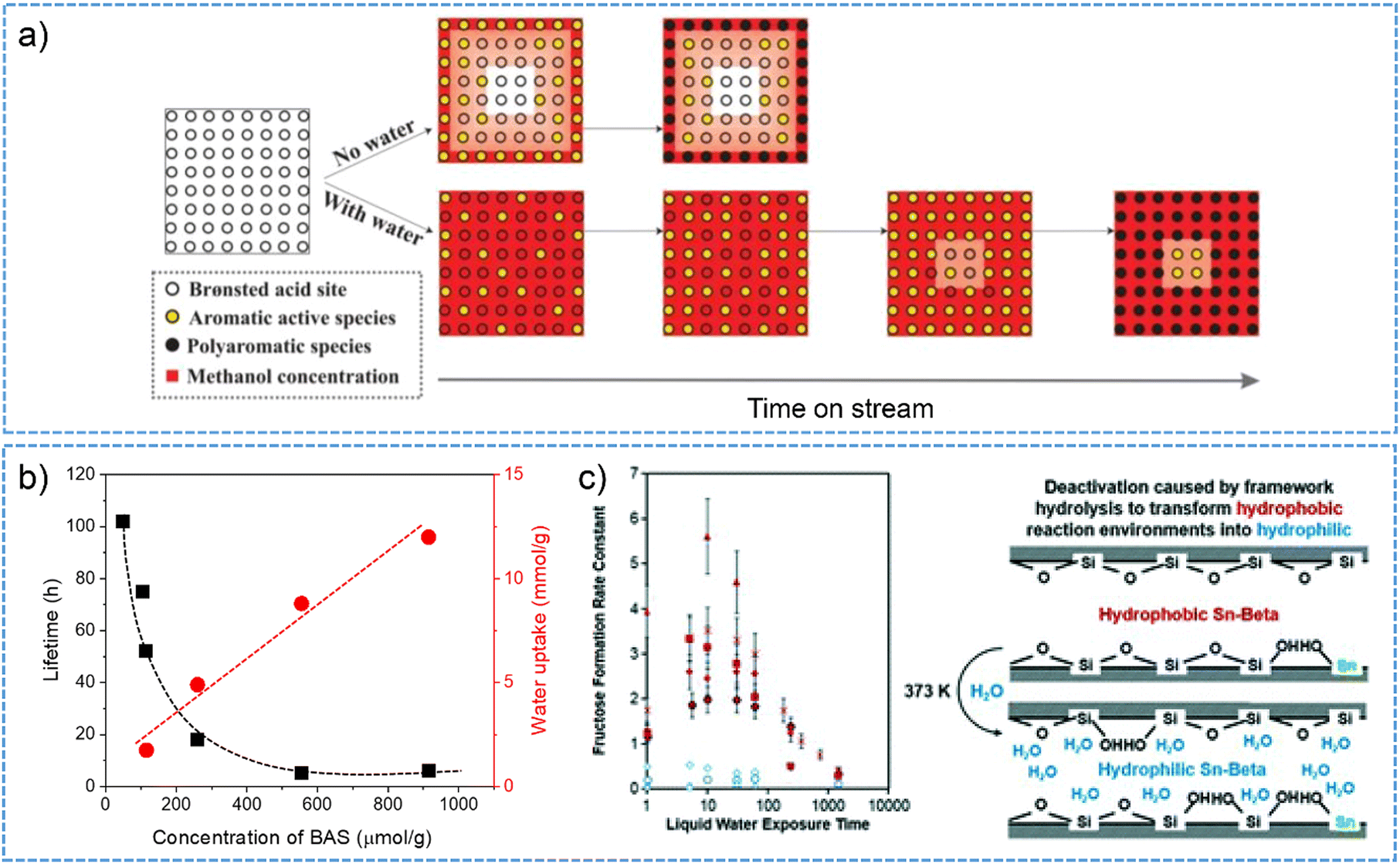 | ||
| Fig. 8 (a) Schematic illustration for water effect on MTO reaction and the formation of aromatic and polyaromatic species. Reproduced with permission from ref. 134. Copyright 2016, American Chemical Society. (b) Zeolite catalytic lifetime and the uptake of water as a function of the concentration of BAS for different H-BEA zeolites. This plot was drown based on the data points from ref. 187. (c) Structural transformation of hydrophobic to hydrophilic micropores of Sn-BEA causes the catalytic deactivation (as a function of exposure time to liquid water) for aqueous-phase glucose isomerization. Reproduced with permission from ref. 193. Copyright 2019, Royal Society of Chemistry. | ||
4. Zeolite stability in the presence of water structures
4.1. Structural and functional stability of zeolitic materials
Exposing to a steam condition at high temperature (above 500 °C) is the method generally adopted on purpose for the preparation of high-performing Brønsted acidic zeolites (e.g., USY and MOR zeolites) and also for the regeneration of spent catalyst materials after the catalytic deactivation, e.g., that in the case of MTO conversion and fluid catalytic cracking.199,200 Heterogeneous catalytic reactions particularly those applied in petrochemical industry often take place at moderate temperatures ranged from 100 to 500 °C. Relating to the competitive adsorption effect, co-feeding water at the solid–gas interfaces is a decent strategy employed to tackle the issue of coke formation in zeolite pore networks134–136 and manipulate the catalytic performance of zeolite materials.133,139 The reversible breaking and formation of T–O–T bonds (i.e., P–O–Al, Si–O–Al) induced by water in the framework of SAPO-34 at moderate-temperature conditions have been revealed by means of 17O NMR spectroscopy.201 The structural dynamic process enables the encapsulation of bulky molecules (e.g., trimethylphosphine (TMP)/pyridine having kinetic diameters larger than the zeolite pore size), which allows the characterization of acidic sites and endows the zeolite with boosted behavior of shape-selective catalysis. On the other hand, it is important for zeolite catalysis to correlate the catalytic properties to the structural changes of zeolite after an exposure in water atmosphere (in gas and liquid phase).202 Upon exposing the aluminum-containing zeolites to water, framework dealumination through the hydrolysis of Si–O–Al bonds takes place to create dislodged or partially dislodged aluminum species that feature as Lewis acid sites.203 The extra-framework aluminum species with the forms like AlO+, Al(OH)2+, Al(OH)3 and Al2O3 can be created by taking the expense of partially coordinated aluminum atoms after a severe steaming treatment on the zeolite.166,204 Many of experimental investigations and theoretical calculations have been conducted for better understandings about the stability of zeolite structures when or after contacting with water molecules.205,206
Zeolite dealumination may render a decrease in the concentration of functional Si(OH)Al, i.e., the BAS105,166,204 and in turn an irreversible activity loss of a given catalytic reaction, for example, catalytic cracking catalyzed by Brønsted acid sites.207 The loss of the BAS is additionally associated with the poisoning effect originating from the extra-framework aluminum species, the latter can be removed by acid-leaching treatment to restore the BAS density to a certain extent.208 Dealumination events may also deteriorate the acid strength of the site209 and the crystal porosity or cause pore collapse of zeolite structure.210–212 Moreover, water even at a small partial pressure in the gas phase (or from the moisture) appears to entangle the identification and quantification of Brønsted and Lewis acidic sites with varied coordination structures on acidic zeolites.102,103 The conversions of part of tetrahedral aluminum atoms in H-BEA, H-MOR, H-Y zeolite frameworks into aluminum species with octahedral coordination have been intensively observed in the presence of water vapor at room temperature.212–214 As measured for zeolite H-MOR, the octahedral aluminum atoms formed in a hydration condition are associated to the framework, correlating to the detected Lewis acid sites by FT-IR spectroscopy in a dehydrated environment.102 Such transformation of framework-associated aluminum enables the switch of Brønsted and Lewis acidity of zeolites and the obtained Lewis acidity can be distinguished with that originated from extra-framework species in zeolites.103 Recently, through the combination of experimental 27Al NMR spectroscopy and biased ab initio molecular dynamics (AIMD), it was revealed that the existence of framework-associated octahedral aluminum can be kinetically feasible and thermodynamically stable on H-MOR and H-CHA zeolites only having BAS sites with a close proximity (low Si/Al ratios) (Fig. 9a and b).215 Moreover, the octahedrally coordinated aluminum species are unstable and they can revert to tetrahedral coordination when the zeolite material is heated to a high temperature (above 383 K),68 or is ion-exchanged to the sodium or ammonium form.103,212,214,216,217 Reversible structural changes are a general property for protonic zeolite materials, which largely depend on the procedures involved in sample pretreatment, as summarized based on our recent investigations (Fig. 9c).103
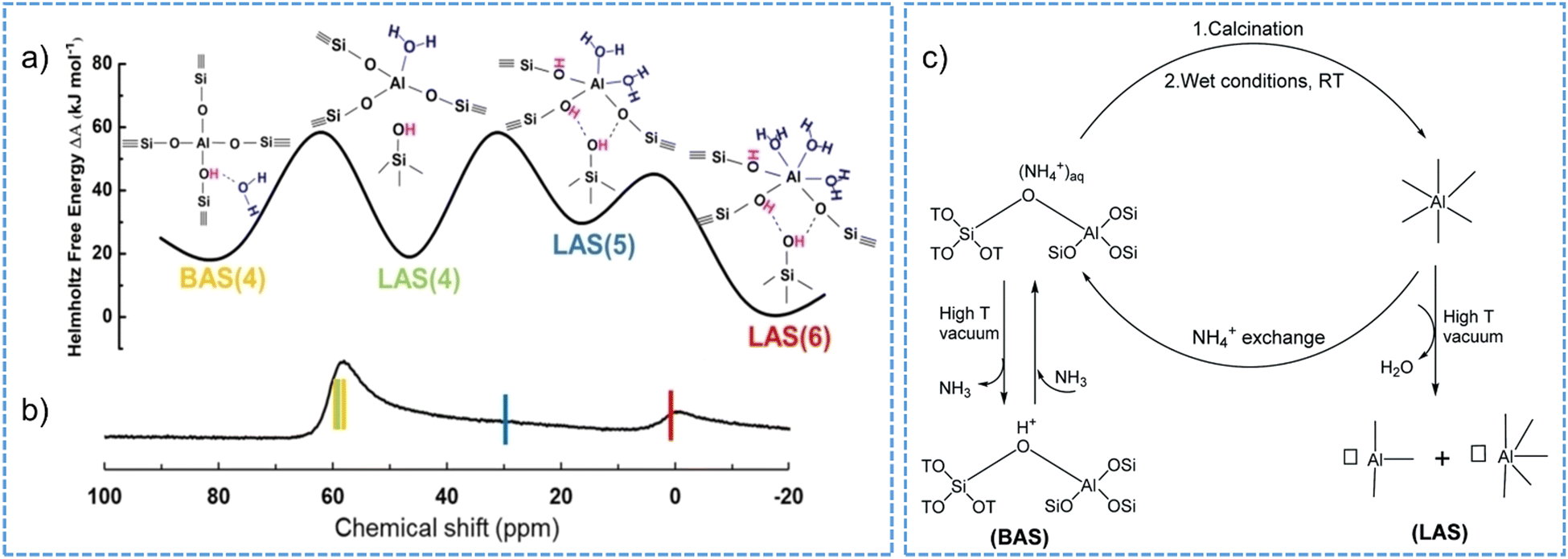 | ||
| Fig. 9 Structural, thermodynamic (a) and NMR (b) characteristics of tetrahedral and octahedral aluminum in CHA zeolite (Si/Al = 3 and 6 water molecules per cage). BAS: Brønsted acid site. LAS: Lewis acid site. The number in the brackets in (a) represents the number of O in the first coordination shell of Al. Reproduced with permission from ref. 215. Copyright 2023, Wiley-VCH. (c) Schematic illustration of the switch between Brønsted and Lewis acidity of zeolite by the transformation of framework-associated aluminum under different synthetic conditions. Reproduced with permission from ref. 103. Copyright 2021, Royal Society of Chemistry. | ||
As for mechanistic understanding of zeolite structural degradation with the presence of water, computational studies have most commonly considered the single-water hydrolysis mechanism, where exists a stepwise interaction of a single water molecule with the zeolite framework.218–220 Water coordinating with an aluminum site is crucial for the breakage of Si–O–Al bonds under the steaming conditions. With respect to the single-water hydrolysis mechanism, the first Al–O(H) bond breaking step is initiated with the adsorption of one water molecule on an aluminum atom in anti-position to the BAS, which leads to the formation of aluminum species with a pentahedral or distorted tetrahedral coordination (Scheme 4).218,220 Subsequent addition of water with up to a number of four enables the successive disconnection of Al–O(H)–Si bonds, thereby giving rise to the generation of dislodged aluminum species (e.g., Al(OH)3H2O) and silanol nest defects. Dissociative water adsorption and proton transfer are included during the adsorption of the first three water molecules, while the adsorption of the fourth water is non-dissociative without the occurrence of proton transfer. There is a high necessity of performing computational modeling under the realistic conditions (i.e., low-pressure water vapor or high-pressure condensed water phase), given that the mechanisms of zeolite dealumination differ with water loadings in the zeolite pores.221 In addition, the regioselectivity of aluminum sites during the process of dealumination are mainly determined by the accessibility of aluminum atoms as well as the reaction conditions in terms of water vapor pressure and temperature.222 Apart from the dealumination behavior, zeolite desilication under the steaming condition requires the dissociated-adsorption of four water molecules for the formation of extra-framework Si species.219
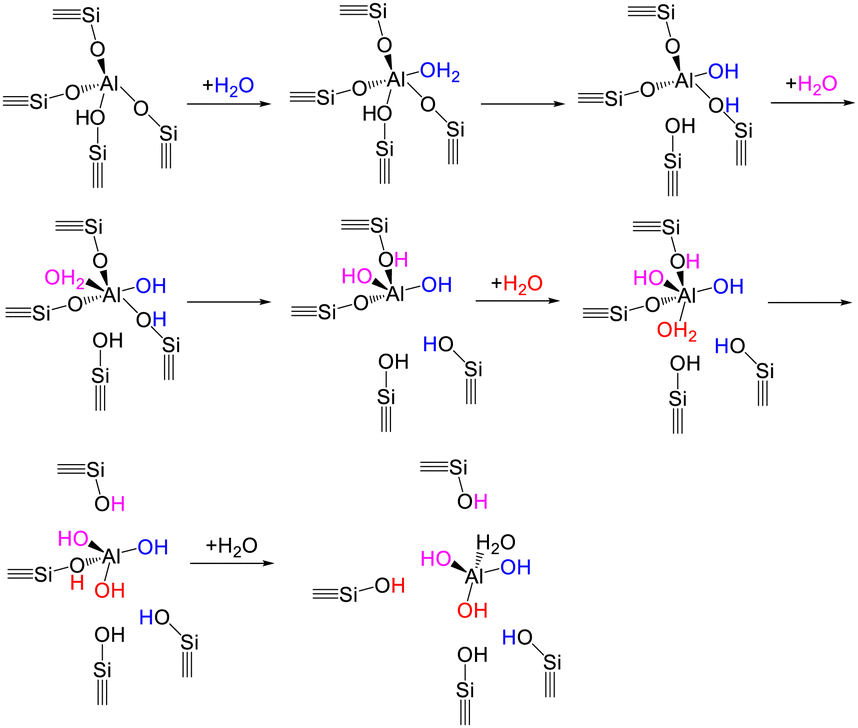 | ||
| Scheme 4 A proposed single-water hydrolysis mechanism for zeolite dealumination process.218,220 | ||
Similar to the behavior in hydrous gas phase, zeolite structures appear to become labile when the pore is subjected to condensed liquid water phase at low-temperature conditions.223,224 By performing AIMD studies on chabazite (CHA) channels fully loaded with water (15 H2O per 36 T site supercell), Heard et al.223 investigated the reversible bond-breakage/hydrolysis of Al–O and Si–O bonds in the hydrated zeolite at room temperature. In analogy to that in Scheme 4, the hydrolysis of Al–O bond involves non-dissociative water adsorption on an aluminum site in the anti-position to the BAS, followed by the inversion of AlO3·H2O tetrahedron (Fig. 10a). The hydrolysis of Si–O bond, which possesses higher free energy barrier than the hydrolysis of Al–O bond, is initiated by the direct interaction of water with silicon through the oxygen atom. Subsequently, a chain of water molecules forms between water within the solvent and the framework oxygen. Proton transfer through water chain to the framework oxygen takes place via the Grotthuss mechanism, giving rise to breaking the Si–O bond together with inverting of the SiO3OH tetrahedron facilitated (Fig. 10a).
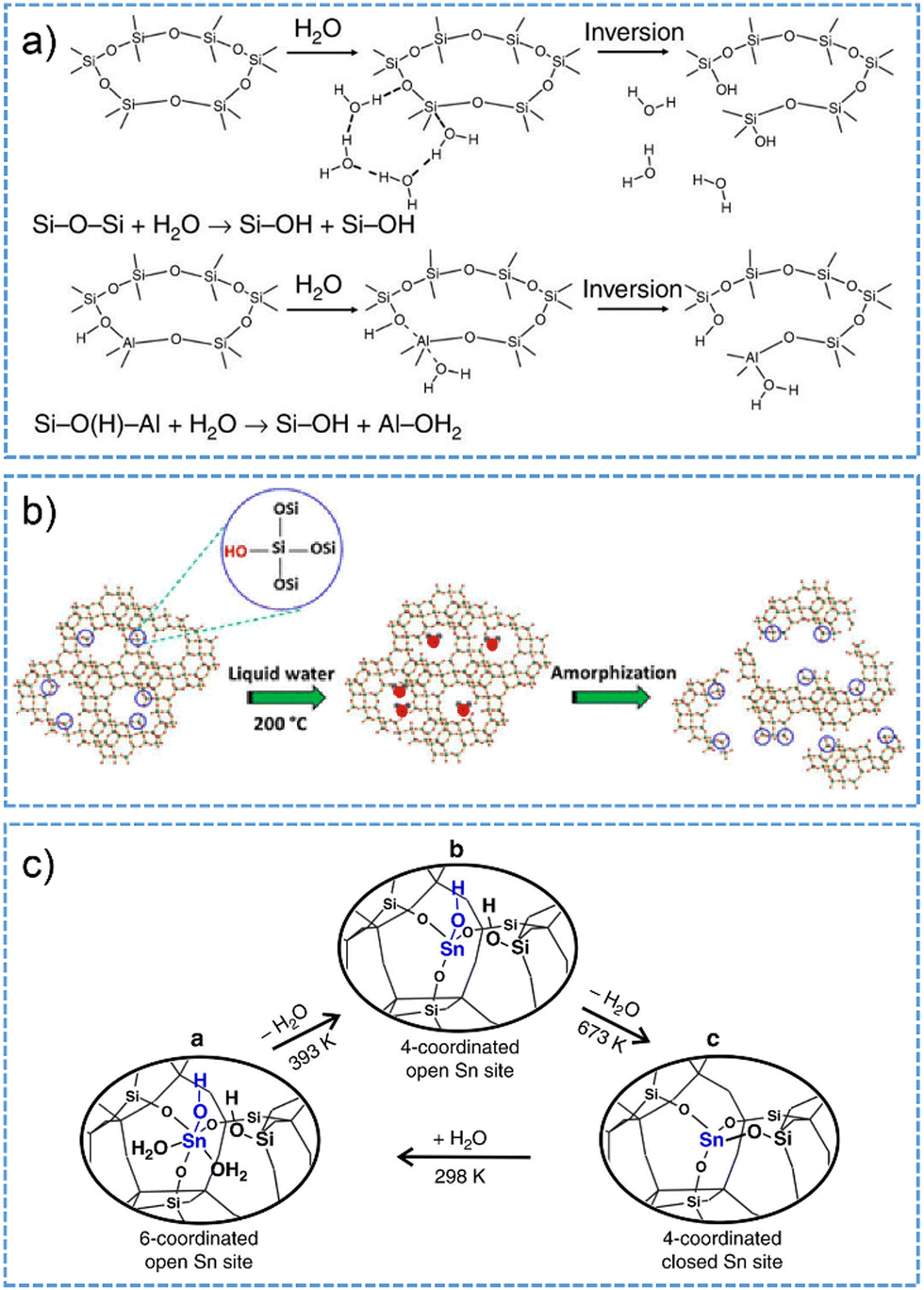 | ||
| Fig. 10 (a) The mechanism proposed for Si–O and Al–O bond breaking reactions on CHA zeolite in water at room temperature. Reproduced with permission from ref. 223. Copyright 2019, Springer Nature. (b) Schematic illustration for the process of zeolite degradation initiated on silanol defect sites in hot liquid water. Reproduced with permission from ref. 185. Copyright 2015, American Chemical Society. (c) The models for the interconversion between open and closed form Sn sites in Sn-BEA zeolite. a: 6-coordinated open form Sn; b: 4-coordinated open form Sn; c: 4-coodrinated closed form Sn. Reproduced with permission from ref. 190. Copyright 2018, Springer Nature. | ||
The hydrothermal stability of zeolitic materials has been studied by most of the research, since the chemical reactions in aqueous phase are often performed at high temperatures.225 Upon short immersion of zeolite Y was shown to induce a fast transformation into amorphous material through the hydrolysis of Si–O–Si bonds by the hydroxyl ions in hot water at 150 and 200 °C.226 Zeolites with dense topologies such as MFI and MOR are more highly resistant toward the degradation in hot water, comparing to the FAU and BEA zeolites with low-density frameworks. Temperature dependence for the degree of zeolite degradation in condensed water phase has been reported for zeolite ZSM-5.227 Besides, the issue concerning zeolite framework degradation tends to become worse with the increased Si/Al ratios228 or with the formed hydrophilic silanol groups in the zeolite pores.226 However, Vjunov et al.184 observed an inverse correlation between the hydrothermal stability and the framework aluminum concentration for zeolite HBEA-12 (Si/Al = 12) and HBEA-75 (Si/Al = 75). Zeolite structural and compositional features such as the distribution of aluminum atoms in lattice, crystal size and the concentration of defects (i.e., silanol nests) were argued to govern the HBEA stability in this case. For HBEA degradation process in hot water, it was proposed to be primarily proceeded by the hydrolysis of siloxane T–O–T groups reside in 4-membered rings, leading to the formation of silanol nests and the collapse of pores. Following this is a subsequent Ostwald–ripening process, where amorphous silica layers are formed on the exterior surface. The amorphous domains in the crystal lattice then undergo the internal decomposition and the final breakdown of zeolite pore structures.
The density of silanol defects, rather than the concentration of BAS, Si–O–Si bonds, framework type and extra-framework aluminum, is the dominant characteristic feature responsible for the disintegration of acidic zeolite frameworks in hot liquid water.185 Silanol defects could facilitate the nucleation of water inside of zeolite pores and thereby promote the hydrolysis of T–O–T groups (Fig. 10b). The loss of structural and functional stability of zeolite can be irreversible in most cases, resulting in a lower degree of zeolite crystallinity and loss of sorption capability. Computational results have shown that the formation of extra-framework silicon is more thermodynamically favorable than the course of zeolite dealumination with the presence of liquid water, which is different to the case when treating zeolite materials under steam. The structure at aluminum sites can therefore be remained to be stable by featuring the tetrahedral coordination without selective removal or substantial redistribution after the treatment in hot liquid water.172,184,226 Despite the above advances, there are also developments in elucidating the nature of defects on zeolite internal and external surfaces by the means of spectroscopic and computational analysis.198,229,230 Providing a critical assessment on the research in zeolite defects is beyond the scope of this section and the reader is referred to the recent excellent reviews from the group of Chizallet.171,198 Since surface defects play important roles in determining the catalytic activities and structural and functional stabilities of zeolitic materials upon contacting with water molecules, comprehensive research efforts in this area are highly desired.
The hydrothermal stability of Lewis acidic zeolite in aqueous phase is, in fact, determined by the defect sites introduced either during the material synthesis process or under the catalytic reaction conditions.193 Typical for zeolite Sn-BEA, the densities of defect sites show a dependency on the framework Sn concentration and they can be hydroxylated to form isolated internal silanol groups when contacting with liquid water molecules.127 In this way, the extensive hydrolysis of Si–O–Sn bonds is initiated followed by an increase in the uptake of water in zeolite micropores, which in turn promotes the process of framework hydrolysis. For the details on the hydrolysis of heteroatoms-substituted zeolites, the readers are referred to an excellent review by Heard et al.232 Current results convince us that the systematic characterization of the metal sites and their overall structures in the presence of water are essential by considering the establishment of structure–performance correlations for Lewis acidic catalysis.
4.2. Synthetic strategies for enhancing zeolite stability
The physicochemical properties of zeolite materials can be tuned generally by modifying the synthetic conditions and through performing post-synthetic treatments. Note again that, the metal-containing zeolite catalysts are not the main focus of this Review, the approach such as substitution of protons with metal cations (such as Na+, Cu+, La3+, etc.)233–235 to strengthen the Al–O bonds and prevent zeolite framework from the attack by water will not be presented in this section. Instead, several typical strategies such as those depicted in Scheme 5 for synthesizing zeolitic catalysts with high structural/catalytic stabilities under steaming conditions or in liquid water phase will be discussed as follows by taking some representative works. | ||
| Scheme 5 Overview of strategies aiming at improving zeolite stability when exposed to steam condition or applied in liquid water phase. (a) Phosphorous impregnation of zeolitic materials. (b) Surface overcoating engineering on the external surface of zeolite with hydrophobic matters (e.g., organosilanes, carbon overlayers, etc.). (c) Healing of internal silanol defect sites by employing organosilanes. (d) Minimizing of defects on zeolite through changing the synthetic conditions from alkaline solution to fluoride media, or synthesizing the zeolite catalysts with high Si/Al ratios. | ||
Of the numerous efforts, the introduction of extra-framework phosphorous (P) species has been proved profitable for stabilization of zeolite catalysts with the presence of water, particularly for applications in hydrocarbon cracking.207,236–239 Wet impregnation of phosphorous precursors (e.g., H3PO4, NH4H2PO4 or (NH4)2HPO4) is the commonly applied post-synthetic method for the incorporation of phosphorous species onto zeolite frameworks.238–240 The dealumination behavior of zeolite upon exposure to a steam condition can be efficiently mitigated by the incorporation of phosphorous species, which thereby aids in obtaining high catalytic performance for the phosphated zeolites.238 For example, zeolite stabilized by phosphorous element has been applied at much higher temperatures for steam catalytic cracking of naphtha, benefiting in the measured high yield and selectivity to ethane and propene.237 Phosphorous species are likely protonated by a certain amount of BAS in zeolite to stabilize the aluminum cations in the zeolitic framework (Scheme 5a),207 while resulting in decreased concentration and strength of the BAS site in most cases.241–243 Optimization of the content of phosphorous during the synthesis of phosphorous-containing zeolite is desirable to allow for avoiding a significant loss of acidity and catalytic activity/selectivity.238,239,243 Nevertheless, the interactions between extra-framework phosphorous species and framework aluminum atoms within the zeolite structures remain to be not well understood. The partially dislodged four-coordinated framework aluminum cations have been proposed to facilitate their interactions with the incorporated phosphate species (P–O–Al interactions) in the ZSM-5 zeolite pores.240,244 Thermal treatment of the zeolite containing P–O–Al interactions at high temperature usually induces the formation of local silico-aluminophosphate interfaces and eventually promotes the hydrothermal stability of phosphated zeolites, as studied by means of multi-spectroscopic technologies including 27Al and 31P MAS NMR and FT-IR.240
Surface overcoating engineering such as the introduction of a hydrophobic barrier layer on the surface of as-synthesized zeolites is expected to be of great interest (Scheme 5b). For example, vapor phase deposition of hydrophobic triphenyl silane could endow BEA zeolite as robust catalyst towards the attack by the steam and thereby achieving high yield of lighter hydrocarbons in steam-assisted catalytic cracking.245 More recently, Wang et al.246 observed that the silane coatings can potentially protect ZSM-5 zeolite under the hydrothermal conditions at ≤300 °C, but these silane coatings provided ineffective protection in the presence of supercritical water (400 °C). Upon the sequence of hydrothermal carbonization with glucose and high-temperature pyrolysis treatment, in this work, the ZSM-5 zeolites were further developed into carbon-coated counterparts to work as the hydrophobic and stable catalysts for dodecane cracking in supercritical water condition.246 Carbon coating layers with an optimal loading of 10 wt% effectively protected the crystallinity of ZSM-5 zeolite and the BAS sites as well, while exerting the influences on the mass diffusion to a lesser extent in comparison with the samples with higher loadings of carbon overlayers.
Functionalization of zeolite external surface with organosilanes have been potentially applied as an efficient post-synthetic tool to address the stability improvement of Brønsted acidic zeolites under liquid-phase catalytic reaction conditions.185,187,247,248 The surface properties of the original zeolite can be replaced with that of introduced organosilanes, eventually becoming hydrophobic. For example, Zapata et al. improved the hydrophobicity of HY zeolite (Si/Al = 30) by performing the silylation treatment on zeolite surface with a silylating agent, e.g., octadecyltrichlorosilane (OTS).247 It was emphasized that the acid sites concentration remained unchanged after the selective introduction of hydrophobic OTS layers on the external surface of HY zeolite. Ethyltrichlorosilane (ETS) has also been applied for the silylation of HY-2.6 (Si/Al = 2.6, CBV 600).185 The stability of functionalized HY zeolites aforementioned were substantially improved by the protection of the internal zeolite pores from contacting with liquid water at 200 °C. In comparison, the parent HY-2.6 zeolite underwent a drastic collapse of the lattice crystal upon a similar treatment in hot water, as indicated by the DRIFT spectra (Fig. 11a). This strategy is flexible, feasible and can be used for the rational design and synthesis of hydrophobic zeolite catalysts by a suitable selection of organosilanes.
 | ||
| Fig. 11 (a) The collected DRIFT spectra and crystallinity losses for steamed HY 2.6 and the corresponding silylated one upon water attack. Reproduced with permission from ref. 185. Copyright 2015, American Chemical Society. (b) Schematic illustration for the silylation of H-BEA with TMS–Cl for the stabilization of internal SiOH nests. Reproduced with permission from ref. 249. Copyright 2016, American Chemical Society. Comparisons of catalytic lifetime as a function of the BAS concentration during the aqueous-phase cyclohexanol dehydration at 170 °C (c) and lattice crystallinity loss in pure water at 170 °C after 48 h (d). These two plots were drawn based on the data points from ref. 186. | ||
In addition to the silylation of the external surface, the direct prevention on Si–O–Si bonds of the hydrolytic attack by water is essential when considering the formation of water with a high rate on the internal space of zeolite crystallites. Prodinger et al. reported an effort related to the improvement of zeolite hydrothermal stability by removing the inner structural defects (Scheme 5c).249 Herein, the internal silanol defects inside of H-BEA were selectively removed by the reaction with trimethylchlorosilane (TMS–Cl). In a flow reactor system, TMS–Cl vapor carried by N2 gas was introduced into H-BEA that were synthesized with certain amounts of Si–OH defect. Upon dosing of TMS–Cl, the Si–OH defect underwent the initial capping by TMS–Cl via a condensation reaction. It leads to the decreased density of Q3 peak at −103 ppm (defect Si(OH)(OSi)3 species) and Q2 peak at −92 ppm (Si(OH)2(OSi)2 species), as observed from the 29Si-CP-MAS-NMR spectra (Fig. 11b). This defect healing process involves the formation of Si–O–Si bonds and the release of methane consequently, as suggested by the identification of primary, secondary and tertiary silylation products with different chemical shifts. Enhanced stability was indicated by the combined characterizations on lattice crystallinity, microporosity and mesoporosity of the silylated BEA zeolites after the treatment in liquid water at 160 °C for 48 h.249 In the following work, silylation-treated BEA with TMS–Cl were reported to show much longer lifetime in aqueous-phase catalytic reactions and less lattice crystallinity loss in pure hot water than their parent defected counterparts (Fig. 11c and d). It was concluded to be the consequences of decreasing the defect densities and reducing the access to water molecules after the treatment of defected BEA zeolite with TMS–Cl.186 This protocol is expected to be applicable for the defect healing of other kinds of zeolite materials to achieve the improvements of hydrothermal stability.
Apart from above approaches, minimizing the concentration of defects and decreasing the amount of hydronium ions in the pores by direct synthesis strategy aid in the improvement of thermal stability of zeolite materials in hot liquid water (Scheme 5d). The utilization of fluoride anions in the synthesis of aluminosilicate and hydrophobic Lewis acidic zeolites is effective in mitigating the formation of defect sites in zeolite pore networks.78,113,231,250 Typically selected as an example, H-BEA zeolites with the Si/Al ratios range from 15 to 230 were synthesized with the presence of fluoride anions and the corresponding defect concentrations were characterized to gradually decrease from 210 to 45 μmol g−1, in contrast to that (380 μmol g−1) of H-BEA-15 sample prepared in an alkaline medium.187 These low-defective H-BEA zeolites exhibited a high resistance toward the disintegration in aqueous phase for Brønsted acid-catalyzed reactions. Synthesizing zeolitic materials with hydrophobic properties by increasing the Si/Al ratios is another effective way to improve their hydrothermal endurance (Scheme 5d). Nevertheless, these strategies cannot be efficiently reliable to attain the defect-free zeolites and further enable a satisfactory structural stability in hot liquid water. On the other hand, aluminum-free materials such as Sn-BEA and Sn-MFI have been widely synthesized as the stable Lewis acidic zeolites for heterogeneously catalyzing the chemical reactions in water.231 In most cases, researchers attribute their unique catalytic performance to the hydrophobic properties of zeolite surface and the suitable Lewis acidity that are originated from the substituted atoms in the zeolitic frameworks.110,115,125,162
5. Summary and outlook
Water as a reactant, by product and solvent could make a difference for heterogeneously catalyzed reactions. Previous studies of zeolite catalysis with the presence of water have highlighted the importance of understanding water structures on acidic zeolites. The interaction of a water molecule with the Brønsted acid site (BAS) on zeolite is initiated with the formation of hydrogen-bonded monomeric complex inside the pores. Increasing the dosages of water in zeolite channels results in the generation of larger-sized water clusters, in which water molecules are protonated by the BAS protons transferred from the framework oxygen. Experimental technologies such as NMR, gas-phase calorimetry, adsorption uptake along with theoretical calculations have verified that the hydrated hydronium ions are composed of eight or ten H2O molecules per the BAS site. Less numbers of water molecules are identified to be stabilized on each of tetravalent framework heteroatom, e.g., tin, titanium for the case of Lewis acidic zeolites. On Brønsted and Lewis acidic zeolite, framework defects (i.e., silanol nests) those are created either during hydrothermal synthesis or under post-synthetic/reaction conditions, endow the zeolitic micropores with hydrophilic property and further promotes water adsorption uptake. In addition to the water clusters strongly interacting with zeolite framework, extended hydrogen-bonded water networks can be subsequently formed with the additional water molecules in the pores. Confinement effect in zeolite leads to the enhanced proton mobility for the intraporous water clusters that are chemically different from those in the gas phase. The character of framework heteroatoms, zeolite topologies and the concentration of framework defects mainly determine the shape and H-bonding configuration of water structures in acidic zeolite pores.Water may play positive or dubious roles in the process of heterogeneous catalysis, which is reliant on different catalytic processes and reaction conditions. Water adsorption on the surface or pores of zeolite brings in competitive adsorption phenomena, from which the adsorption behavior of reactants, intermediates and products are substantially tailored. The consequences can be classified to the following aspects including: (a) the loss of accessibility of active site to the reacting molecules; (b) the tailored nature of active sites after the solvation of acidic BAS protons by water; (c) the modifications on the reactant adsorption kinetics that are responsible for activation barriers; (d) the inhibition of coke formation particularly at the solid–gas interfaces (hence maintaining a good catalytic stability). Water clusters likely participate in the formation of water cluster-reactant complexes/clusters with, e.g., low-carbon chain alcohol molecules, which requires extensive disruption of hydrogen bonds and results in inhibitive role of water in gas-phase alcohol dehydration. In addition, the presence of water structures in the nano-environment of zeolite confined spaces could induce thermodynamic non-idealities to the catalytic reactions. To take as the example, manipulating the highly ionic environment in H-ZSM-5 micropores benefited the determined negative value of excess Gibbs free energies for the transition state, which leads to decreased activation energies and in turn enhanced catalytic turnover rates. Analogously, extended hydrogen-bonded water networks fabricated when exposing in water solvent make differences for the enthalpic or entropic stabilization/destabilization of adsorption and transition states on the Lewis acidic zeolites with unique hydrophilic/hydrophobic characters. Interacting zeolite with water vapor or condensed water phase at high temperature often renders the hydrolysis of framework heteroatoms irreversible, which remains as an important subject of zeolite research. Recent progresses on computational modeling have indicated that not only a single water but also intraporous water clusters in the zeolite channels are responsible for the dynamic evolution of Brønsted acidic zeolite structures. The mechanisms with respect to the dealumination and desilication processes in the presence of water differ significantly from each other. Zeolite degradation when contacting with hot liquid water is typically reflected by the transformation of hydrophobic property into hydrophilic one, the loss of lattice crystallinity as well as the structural porosity. In addition to the dependences of temperature and framework density, the concentration of defect sites in zeolite pores acts as the critical factor determining the degree of structural degradation. The correlations of zeolite lifetime as a function of the concentration of properly confined hydronium ions or the changes in the hydrophobicity of molecularly-sized pores are desirable for acidic chemistry in condensed aqueous phase. Rational design of structurally/catalytically stable zeolitic materials will open up a broader perspective for innovative development of new heterogeneous processes in the aqueous phase. Achieving this has potentially involved the synthesizing low-defect or defect-free zeolites and the hydrophobic-functionalizing zeolite surface through post-synthetic approaches such as the impregnation with phosphorous element and catalyst overcoating engineering with hydrophobic organic (e.g., organosilanes) or inorganic (e.g., carbon) overlayers.
Catalyst structures determine the catalytic performance and the reaction mechanisms. Conventional spectroscopic techniques such as NMR and FTIR spectroscopy have been employed to analyze the initial and final structure of zeolite materials. The obtained catalytic data can be the consequence of many factors originated from the evolution of active sites, i.e., Brønsted and Lewis acidic sites in zeolites during catalytic reactions (even without the presence of water molecules), thus making it challenging to establish precisely the structure–performance relationships.101,171 Performing in situ and operando characterizations to gain the knowledge on the number, location and structure of different type of active sites, particularly the extra-framework aluminum species in the zeolite pores could pave the way for unveiling the underlying reaction mechanisms and designing of stable zeolite materials. First-principles calculations will continue to be the effective approach to study the process dynamics taking place on catalyst surface and help to solve the unanswered open questions about the identification of diverse active sites on both external and internal surface and their synergistic effects (e.g., the synergy of BAS-LAS pairs) on zeolite acid-catalyzed applications.
Future research directions for the susceptibility of zeolites when applied/exposed in catalytic reaction conditions have been provided above, great efforts are also needed to address the water–zeolite interactions and the corresponding roles in zeolite catalysis. Owing to the several advantages of conducting zeolite-catalyzed reactions with water co-feeding or in condensed water phase, a great number of heterogeneous catalytic reactions such as those catalyzed by metal-supported catalysts are being explored increasingly.5,251–254 Diverse important catalytic transformations can be expanded to be performed with the presence of gaseous water or in aqueous phase. In this context, the green-valorization of biosourced compounds and manufacturing of high-value fine chemicals will be expectedly favored. It has been reported for zeolite materials that any exposure to gaseous and liquid water during the zeolite lifetime, i.e., zeolite hydrothermal synthesis, zeolite catalysis, regeneration, etc. can significantly modify the structural properties of active sites and finally manipulate their catalytic performance.18,126,162,186,187,202,223 In addition, the local reaction environment involving water molecules in zeolite pores is varied with the nature of active sites and reacting molecules, zeolite topologies, and reaction conditions, which necessitates the systematical understanding about the promotional roles of water on the catalytic reactions of interest. Catalyst characterization technologies are often conducted in vacuum or in inert gas environments, which cannot provide the direct evidences for the catalytic mechanism of the reactions performed under realistic conditions with the presence of water at high temperatures. Better mechanistic insights from experimental studies as well as computational investigations on the structures of solid–water and solid–gas interfaces are highly desirable. Comparing solid–gas interface characterization, the development of in situ and time-resolved technologies including XAS (both near edge (XANES) and fine structure (EXAFS)), X-ray emission spectroscopy (XES), scanning probe microscopies for the catalytic processes at solid–liquid interfaces are challenging or are still under development.255 A combination of calorimetry, thermogravimetric analysis, attenuated total reflection (ATR)-FTIR and solid state NMR possess the potential to facilitate the quantitative comparison of substrate behaviors and to monitor the reaction progress at the solid–liquid interfaces. Kinetic and thermodynamic insights in addition to the catalyst structural information are also essential for understanding the effects of water structures on determining the activation barriers of each elementary steps and controlling over the reaction rate of a catalytic reaction. Decoupling of thermodynamic terms such as solvation, adsorption and sorbate interactions around the active site has been emphasized for the structure–activity relationships that can be established.145,146,256 There are examples observing differences in the size of hydrated hydronium ions confined in different zeolite topologies and the obvious destabilization effects on sorbed substrates.95,97 There is a pressing need to perform more detailed investigations on the structure of hydronium ion clusters in porous materials and to unveil how the generated nano-environment causes the non-idealities to a catalytic system. Apart from the sorption and reaction steps in the catalytic cycle, diffusion or mass transport of reactive molecules and products may be responsible for the observed catalytic performance in terms of activity, product selectivity and catalytic stability.257–259 At the moment, very few works have addressed the role of water molecules in influencing the diffusivity of reactants, intermediates and products in zeolite pores.153,154 More insights into the effects of water structures in the diffusion behaviors within zeolite pores remain to be provided in the future research.171,260 Moreover, by increasing the model complexity under the realistic catalytic conditions and combining different theoretical methods including density functional theory calculations and molecular dynamics simulations,260 molecular level understandings for the catalytic reactions catalyzed by acidic zeolites with the presence of water can be achieved. Fundamental understanding of water structures and the roles of water molecules in heterogeneous catalysis can benefit the de novo design of highly active and stable zeolite and other solid materials for potential catalytic transformations performed in water vapor or in bulk aqueous phase.
Author contributions
Q. Liu: conceptualization, writing – original draft, writing – review & editing. J. A. van Bokhoven: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
There is no funding or financial support for the present work to declare.References
- J. R. Di Iorio, B. A. Johnson and Y. Román-Leshkov, J. Am. Chem. Soc., 2020, 142, 19379–19392 CrossRef CAS PubMed.
- J. S. Bates and R. Gounder, Chem. Sci., 2021, 12, 4699–4708 RSC.
- D. S. Potts, D. T. Bregante, J. S. Adams, C. Torres and D. W. Flaherty, Chem. Soc. Rev., 2021, 50, 12308–12337 RSC.
- G. Li, B. Wang and D. E. Resasco, ACS Catal., 2020, 10, 1294–1309 CrossRef CAS.
- L. Lin, Y. Ge, H. Zhang, M. Wang, D. Xiao and D. Ma, JACS Au, 2021, 1, 1834–1848 CrossRef CAS PubMed.
- E. Barry, R. Burns, W. Chen, G. X. De Hoe, J. M. M. De Oca, J. J. de Pablo, J. Dombrowski, J. W. Elam, A. M. Felts, G. Galli, J. Hack, Q. He, X. He, E. Hoenig, A. Iscen, B. Kash, H. H. Kung, N. H. C. Lewis, C. Liu, X. Ma, A. Mane, A. B. F. Martinson, K. L. Mulfort, J. Murphy, K. Mølhave, P. Nealey, Y. Qiao, V. Rozyyev, G. C. Schatz, S. J. Sibener, D. Talapin, D. M. Tiede, M. V. Tirrell, A. Tokmakoff, G. A. Voth, Z. Wang, Z. Ye, M. Yesibolati, N. J. Zaluzec and S. B. Darling, Chem. Rev., 2021, 121, 9450–9501 CrossRef CAS PubMed.
- P. Cheung, A. Bhan, G. J. Sunley and E. Iglesia, Angew. Chem., Int. Ed., 2006, 45, 1617–1620 CrossRef CAS PubMed.
- J. F. DeWilde, H. Chiang, D. A. Hickman, C. R. Ho and A. Bhan, ACS Catal., 2013, 3, 798–807 CrossRef CAS.
- C. J. Baranowski, T. Fovanna, M. Roger, M. Signorile, J. McCaig, A. M. Bahmanpour, D. Ferri and O. Kröcher, ACS Catal., 2020, 10, 8106–8119 CrossRef CAS.
- H. Li, D. Guo, N. Ulumuddin, N. R. Jaegers, J. Sun, B. Peng, J.-S. McEwen, J. Hu and Y. Wang, JACS Au, 2021, 1, 1471–1487 CrossRef CAS PubMed.
- Y. Liu, E. Baráth, H. Shi, J. Hu, D. M. Camaioni and J. A. Lercher, Nat. Catal., 2018, 1, 141–147 CrossRef CAS.
- S. K. Desai, V. Pallassana and M. Neurock, J. Phys. Chem. B, 2001, 105, 9171–9182 CrossRef CAS.
- Y. Yoon, R. Rousseau, R. S. Weber, D. Mei and J. A. Lercher, J. Am. Chem. Soc., 2014, 136, 10287–10298 CrossRef CAS PubMed.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- Z. Zhao, R. Bababrik, W. Xue, Y. Li, N. M. Briggs, D.-T. Nguyen, U. Nguyen, S. P. Crossley, S. Wang, B. Wang and D. E. Resasco, Nat. Catal., 2019, 2, 431–436 CrossRef CAS.
- C. Sievers, Y. Noda, L. Qi, E. M. Albuquerque, R. M. Rioux and S. L. Scott, ACS Catal., 2016, 6, 8286–8307 CrossRef CAS.
- A. Martínez-Hernández, G. A. Fuentes and S. A. Gómez, Appl. Catal., B, 2015, 465–474 CrossRef.
- K. Stanciakova and B. M. Weckhuysen, Trends Chem., 2021, 3, 456–468 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- Y. Li and J. Yu, Chem. Rev., 2014, 114, 7268–7316 CrossRef CAS PubMed.
- R. M. Barrer, J. Chem. Soc., 1948, 2158, 2163 Search PubMed.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- M. Moliner, C. Martínez and A. Corma, Chem. Mater., 2014, 26, 246–258 CrossRef CAS.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- D. T. Bregante, N. E. Thornburg, J. M. Notestein and D. W. Flaherty, ACS Catal., 2018, 8, 2995–3010 CrossRef CAS.
- P. Ferri, C. Li, R. Millán, J. Martínez-Triguero, M. Moliner, M. Boronat and A. Corma, Angew. Chem., Int. Ed., 2020, 59, 19708–19715 CrossRef CAS PubMed.
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Acc. Chem. Res., 2021, 54, 2894–2904 CrossRef CAS PubMed.
- H. Kobayashi, H. Yokoyama, B. Feng and A. Fukuoka, Green Chem., 2015, 17, 2732–2735 RSC.
- S. S. Poly, S. M. A. Hakim Siddiki, A. S. Touchy, S. Yasumura, T. Toyao, Z. Maeno and K.-I. Shimizu, J. Catal., 2018, 368, 145–154 CrossRef.
- T. Yumura, Y. Hirose, T. Wakasugi, Y. Kuroda and H. Kobayashi, ACS Catal., 2016, 6, 2487–2495 CrossRef CAS.
- D. Palagin, V. L. Sushkevich and J. A. van Bokhoven, ACS Catal., 2019, 9, 10365–10374 CrossRef CAS.
- V. L. Sushkevich and J. A. van Bokhoven, Catal. Sci. Technol., 2020, 10, 382–390 RSC.
- Y. Liu, W. Xue, S. Seo, X. Tan, D. Mei, C.-J. Liu, I.-S. Nam and S. B. Hong, Appl. Catal., B, 2021, 294, 120244 CrossRef CAS.
- H. Lee, R. J. G. Nuguid, S. W. Jeon, H. S. Kim, K. H. Hwang, O. Kröcher, D. Ferri and D. H. Kim, Chem. Commun., 2022, 58, 6610–6613 RSC.
- J. Imbao, J. A. van Bokhoven, A. Clark and M. Nachtegaal, Nat. Commun., 2020, 11, 1118 CrossRef PubMed.
- J. Imbao, J. A. van Bokhoven and M. Nachtegaal, ACS Catal., 2021, 11, 8684–8691 CrossRef CAS.
- E. B. Clatworthy, S. V. Konnov, F. Dubray, N. Nesterenko, J.-P. Gilson and S. Mintova, Angew. Chem., Int. Ed., 2020, 59, 19414–19432 CrossRef CAS PubMed.
- Z.-P. Hu, J. Han, Y. Wei and Z. Liu, ACS Catal., 2022, 12, 5060–5076 CrossRef CAS.
- D. E. Resasco, S. P. Crossley, B. Wang and J. L. White, Catal. Rev., 2021, 63, 302–362 CrossRef CAS.
- W. O. Haag, R. M. Lago and P. B. Weisz, Nature, 1984, 309, 589–591 CrossRef CAS.
- S. Schallmoser, T. Ikuno, M. F. Wagenhofer, R. Kolvenbach, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, J. Catal., 2014, 316, 93–102 CrossRef CAS.
- D. Liu, A. Bhan, M. Tsapatsis and S. A. Hashimi, ACS Catal., 2011, 1, 7–17 CrossRef CAS.
- A. J. Jones and E. Iglesia, Angew. Chem., Int. Ed., 2014, 53, 12177–12181 CrossRef CAS PubMed.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- A. Gumidyala, T. Sooknoi and S. Crossley, J. Catal., 2016, 340, 76–84 CrossRef CAS.
- M. Milina, S. Mitchell and J. Pérez-Ramírez, Catal. Today, 2014, 235, 176–183 CrossRef CAS.
- M. Yang, D. Fan, Y. Wei, P. Tian and Z. Liu, Adv. Mater., 2019, 31, 1902181 CrossRef CAS PubMed.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- A. Zecchina, F. Geobaldo, G. Spoto, S. Bordiga, G. Ricchiardi, R. Buzzoni and G. Petrini, J. Phys. Chem., 1996, 100, 16584–16599 CrossRef CAS.
- A. Ison and R. J. Gorte, J. Catal., 1984, 89, 150–158 CrossRef CAS.
- L. Smith, A. K. Cheetham, R. E. Morris, L. Marchese, J. M. Thomas, P. A. Wright and J. Chen, Science, 1996, 271, 799–802 CrossRef CAS.
- V. Semmer-Herlédan, L. Heeribout, P. Batamack, C. Dorémieux-Morin, J. Fraissard, A. Gola and E. Benazzi, Micro. Meso. Mater., 2000, 34, 157–169 CrossRef.
- S. Bordiga, L. Regli, C. Lamberti and A. Zecchina, J. Phys. Chem. B, 2005, 109, 7724–7732 CrossRef CAS PubMed.
- V. Bolis and C. Busco, J. Phys. Chem. B, 2006, 110, 14849–14859 CrossRef CAS PubMed.
- L. Heeribout, V. Semmer, P. Batamack, C. Dorémieux-Morin, J. Fraissard and G. Antos, J. Chem. Soc., Faraday Trans., 1, 1995, 91, 3933–3940 RSC.
- L. Heeribout, C. Dorémieux-Morin, J.-P. Nogier, R. Vincent and J. Fraissard, Micro. Meso. Mater., 1998, 24, 101–112 CrossRef CAS.
- A. Jentys, G. Warecka and J. A. Lercher, J. Mol. Catal., 1989, 51, 309–327 CrossRef CAS.
- A. Jentys, G. Warecka, M. Derewinski and J. A. Lercher, J. Phys. Chem., 1989, 93, 4837–4843 CrossRef CAS.
- A. G. Pelmenschikov, J. H. M. C. van Wolput, J. Jaenchen and R. A. van Santen, J. Phys. Chem., 1995, 99, 3612–3617 CrossRef CAS.
- A. G. Pelmenschikov and R. A. van Santen, J. Phys. Chem., 1993, 97, 10678–10680 CrossRef CAS.
- M. J. Rice, A. K. Chakraborty and A. T. Bell, J. Phys. Chem. A, 1998, 102, 7498–7504 CrossRef CAS.
- L. Marchese, J. Chen, P. A. Wright and J. M. Thomas, J. Phys. Chem., 1993, 97, 8109–8112 CrossRef CAS.
- M. Krossner and J. Sauer, J. Phys. Chem., 1996, 100, 6199–6211 CrossRef CAS.
- S. A. Zygmunt, J. Phys. Chem. B, 2001, 105, 3034–3038 CrossRef CAS.
- F. Wakabayashi, J. Phys. Chem., 1996, 100, 1442–1444 CrossRef CAS.
- M. V. Vener, X. Rozanska and J. Sauer, Phys. Chem. Chem. Phys., 2009, 11, 1702–1712 RSC.
- P. Sazama, Z. Tvarůžková, H. Jirglová and Z. Sobalík, Stud. Surf. Sci. Catal., 2008, 174, 821–824 CrossRef.
- A. Vjunov, M. Wang, N. Govind, T. Huthwelker, H. Shi, D. Mei, J. L. Fulton and J. A. Lercher, Chem. Mater., 2017, 29, 9030–9042 CrossRef CAS.
- M. Jiménez-Ruiz, D. S. Gahle, T. Lemishko, S. Valencia, G. Sastre and F. Rey, J. Phys. Chem. C, 2020, 124, 5436–5443 CrossRef.
- A. Zecchina, R. Buzzoni, S. Bordiga, F. Geobaldo, D. Scarano, G. Ricchiardi and G. Spoto, Stud. Surf. Sci. Catal., 1995, 97, 213–222 CrossRef CAS.
- J. N. Kondo, M. Iizuka and K. Domen, Langmuir, 1997, 13, 747–750 CrossRef CAS.
- D. H. Olson, S. A. Zygmunt, M. K. Erhardt, L. A. Curtiss and L. E. Iton, Zeolites, 1997, 18, 347–349 CrossRef CAS.
- Y. Jeanvoine, J. G. Ángyán, G. Kresse and J. Hafner, J. Phys. Chem. B, 1998, 102, 7307–7310 CrossRef CAS.
- P. W. Kletnieks, J. O. Ehresmann, J. B. Nicholas and J. F. Haw, ChemPhysChem, 2006, 7, 114–116 CrossRef CAS PubMed.
- K. L. Joshi, G. Psofogiannakis, A. C. T. van Duin and S. Raman, Phys. Chem. Chem. Phys., 2014, 16, 18433–18441 RSC.
- D. H. Olson, W. O. Haag and R. M. Lago, J. Catal., 1980, 61, 390–396 CrossRef CAS.
- D. H. Olson, W. O. Haag and W. S. Borghard, Micro. Meso. Mater., 2000, 35–36, 435–446 CrossRef CAS.
- K. Zhang, R. P. Lively, J. D. Noel, M. E. Dose, B. A. McCool, R. R. Chance and W. J. Koros, Langmuir, 2012, 28, 8664–8673 CrossRef CAS PubMed.
- S. Jungsuttiwong, J. Limtrakul and T. N. Truong, J. Phys. Chem. B, 2005, 109, 13342–13351 CrossRef CAS PubMed.
- D. Mei and J. A. Lercher, J. Phys. Chem. C, 2019, 123, 25255–25266 CrossRef CAS.
- D. Mei and J. A. Lercher, AIChE J., 2017, 63, 172–184 CrossRef CAS.
- P. Bai, M. Neurock and J. I. Siepmann, J. Phys. Chem. C, 2021, 125, 6090–6098 CrossRef CAS.
- V. Termath, F. Haase, J. Sauer, J. Hutter and M. Parrinello, J. Am. Chem. Soc., 1998, 120, 8512–8516 CrossRef CAS.
- P. Liu and D. Mei, J. Phys. Chem. C, 2020, 124, 22568–22576 CrossRef CAS.
- J. H. Hack, X. Ma, Y. Chen, J. P. Dombrowski, N. H. C. Lewis, C. Li, H. H. Kung, G. A. Voth and A. Tokmakoff, J. Phys. Chem. C, 2023, 127, 16175–16186 CrossRef CAS.
- R. C. Shiery and D. C. Cantu, J. Phys. Chem. C, 2023, 127, 4218–4224 CrossRef CAS.
- C. J. Heard, L. Grajciar and P. Nachtigall, Chem. Sci., 2019, 10, 5705–5711 RSC.
- M. Wang, N. R. Jaegers, M.-S. Lee, C. Wan, J. Z. Hu, H. Shi, D. Mei, S. D. Burton, D. M. Camaioni, O. Y. Gutiérrez, V.-A. Glezakou, R. Rousseau, Y. Wang and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 3444–3455 CrossRef CAS PubMed.
- T. Humplik, R. Raj, S. C. Maroo, T. Laoui and E. N. Wang, Langmuir, 2014, 30, 6446–6453 CrossRef CAS PubMed.
- Z. Li, D. Dittmann, C. Rieg, M. Benz and M. Dyballa, Catal. Sci. Technol., 2022, 12, 5189–5202 RSC.
- N. Y. Chen, J. Phys. Chem., 1976, 80, 60–64 CrossRef CAS.
- T. Sano, N. Yamashita, Y. Iwami, K. Takeda and Y. Kawakami, Zeolites, 1996, 16, 258–264 CrossRef CAS.
- T. Sano, T. Kasuno, K. Takeda, S. Arazaki and Y. Kawakami, Stud. Surf. Sci. Catal., 1997, 105, 1771–1778 CrossRef.
- K. Chen, J. Kelsey, J. L. White, L. Zhang and D. Resasco, ACS Catal., 2015, 5, 7480–7487 CrossRef CAS.
- S. Eckstein, P. H. Hintermeier, R. Zhao, E. Baráth, H. Shi, Y. Liu and J. A. Lercher, Angew. Chem., Int. Ed., 2019, 58, 3450–3455 CrossRef CAS PubMed.
- J. S. Bates, B. C. Bukowski, J. Greeley and R. Gounder, Chem. Sci., 2020, 11, 7102–7122 RSC.
- S. Kim, N. R. Jaegers, W. Hu, J. Z. Hu, F. Chen, Q. Liu, D. M. Camaioni, M. A. Derewinski, O. Y. Gutiérrez, Y. Liu and J. A. Lercher, J. Phys. Chem. C, 2023, 127, 23390–23399 CrossRef CAS.
- E. Grifoni, G. M. Piccini, J. A. Lercher, V.-A. Glezakou, R. Rousseau and M. Parrinello, Nat. Commun., 2021, 12, 2630 CrossRef CAS PubMed.
- N. Pfriem, P. H. Hintermeier, S. Eckstein, S. Kim, Q. Liu, H. Shi, L. Milakovic, Y. Liu, G. L. Haller, E. Baráth, Y. Liu and J. A. Lercher, Science, 2021, 372, 952–957 CrossRef CAS PubMed.
- J. H. Hack, J. P. Dombrowski, X. Ma, Y. Chen, N. H. C. Lewis, W. B. Carpenter, C. Li, G. A. Voth, H. H. Kung and A. Tokmakoff, J. Am. Chem. Soc., 2021, 143, 10203–10213 CrossRef CAS PubMed.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS PubMed.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, J. Phys. Chem. C, 2019, 123, 15139–15144 CrossRef CAS.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Chem. Sci., 2021, 12, 4094–4103 RSC.
- A. V. Yakimov, M. Ravi, R. Verel, V. L. Sushkevich, J. A. van Bokhoven and C. Copéret, J. Am. Chem. Soc., 2022, 144, 10377–10385 CrossRef CAS PubMed.
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S.-B. Liu and A. Zheng, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS PubMed.
- K. Chen, Z. Gan, S. Horstmeier and J. L. White, J. Am. Chem. Soc., 2021, 143, 6669–6680 CrossRef CAS PubMed.
- M. Hu, C. Wang, Y. Chu, Q. Wang, S. Li, J. Xu and F. Deng, Angew. Chem., Int. Ed., 2022, 61, e202207400 CrossRef CAS PubMed.
- Z. Wang, D. Xiao, K. Chen, C. Lou, L. Liang, S. Xu and G. Hou, ACS Catal., 2023, 13, 4960–4970 CrossRef CAS.
- S. Shetty, B. S. Kulkarni, D. G. Kanhere, A. Goursot and S. Pal, J. Phys. Chem. B, 2008, 112, 2573–2579 CrossRef CAS PubMed.
- R. Gounder and M. E. Davis, AIChE J., 2013, 59, 3349–3358 CrossRef CAS.
- N. A. Grosso-Giordano, A. S. Hoffman, A. Boubnov, D. W. Small, S. R. Bare, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2019, 141, 7090–7106 CrossRef CAS PubMed.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724–1728 CrossRef CAS.
- R. Gounder and M. E. Davis, J. Catal., 2013, 308, 176–188 CrossRef CAS.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- L. Botti, S. A. Kondrat, R. Navar, D. Padovan, J. S. Martinez-Espin, S. Meier and C. Hammond, Angew. Chem., Int. Ed., 2020, 59, 20017–20023 CrossRef CAS PubMed.
- J. S. Bates and R. Gounder, J. Catal., 2018, 365, 213–226 CrossRef CAS.
- J. S. Bates, B. C. Bukowski, J. W. Harris, J. Greeley and R. Gounder, ACS Catal., 2019, 9, 6146–6168 CrossRef CAS.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS PubMed.
- J. D. Lewis, S. V. de Vyver and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2015, 54, 9835–9838 CrossRef CAS PubMed.
- J. J. Pacheco and M. E. Davis, Proc. Nat. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- T. Blasco, M. A. Camblor, A. Corma, P. Esteve, J. M. Guil, A. Martínez, J. A. Perdigón-Melón and S. Valencia, J. Phys. Chem. B, 1998, 102, 75–88 CrossRef CAS.
- B. C. Bukowski, J. S. Bates, R. Gounder and J. Greeley, Angew. Chem., Int. Ed., 2019, 58, 16422–16426 CrossRef CAS PubMed.
- E. Gallo, F. Bonino, J. C. Swarbrick, T. Petrenko, A. Piovano, S. Bordiga, D. Gianolio, E. Groppo, F. Neese, C. Lamberti and P. Glatzel, ChemPhysChem, 2013, 14, 79–83 CrossRef CAS PubMed.
- P. Wolf, M. Valla, A. J. Rossini, A. Comas-Vives, F. Núñez-Zarur, B. Malaman, A. Lesage, L. Emsley, C. Copéret and I. Hermans, Angew. Chem., Int. Ed., 2014, 53, 10179–10183 CrossRef CAS PubMed.
- M. J. Cordon, J. W. Harris, J. C. Vega-Vila, J. S. Bates, S. Kaur, M. Gupta, M. E. Witzke, E. C. Wegener, J. T. Miller, D. W. Flaherty, D. D. Hibbitts and R. Gounder, J. Am. Chem. Soc., 2018, 140, 14244–14266 CrossRef CAS PubMed.
- D. T. Bregante, M. C. Chan, J. Z. Tan, E. Z. Ayla, C. P. Nicholas, D. Shukla and D. W. Flaherty, Nat. Catal., 2021, 4, 797–808 CrossRef CAS.
- T. D. Courtney, C.-C. Chang, R. J. Gorte, R. F. Lobo, W. Fan and V. Nikolakis, Micro. Meso. Mater., 2015, 210, 69–76 CrossRef CAS.
- D. T. Bregante, D. S. Potts, O. Kwon, E. Z. Ayla, J. Z. Tan and D. W. Flaherty, Chem. Mater., 2020, 32, 7425–7437 CrossRef CAS.
- J. C. Vega-Vila and R. Gounder, ACS Catal., 2020, 10, 12197–12211 CrossRef CAS.
- D. S. Potts, V. S. Jeyaraj, O. Kwon, R. Ghosh, A. V. Mironenko and D. W. Flaherty, ACS Catal., 2022, 12, 13372–13393 CrossRef CAS.
- A. G. Panov and J. J. Fripiat, Catal. Lett., 1999, 57, 25–32 CrossRef CAS.
- V. Bolis, J. C. Vedrine, J. P. Van de Berg, J. P. Wolthuizen and E. G. Derouane, J. Chem. Soc., Faraday Trans., 1, 1980, 76, 1606–1616 RSC.
- H. Wang, Y. Hou, W. Sun, Q. Hu, H. Xiong, T. Wang, B. Yan and W. Qian, ACS Catal., 2020, 10, 5288–5298 CrossRef CAS.
- K. De Wispelaere, C. S. Wondergem, B. Ensing, K. Hemelsoet, E. J. Meijer, B. M. Weckhuysen, V. V. Speybroeck and J. Ruiz-Martinez, ACS Catal., 2016, 6, 1991–2002 CrossRef CAS.
- J. Valecillos, G. Elordi, A. T. Aguayo and P. Castaño, Catal. Sci. Technol., 2021, 11, 1269–1281 RSC.
- X. Zhao, J. Li, P. Tian, L. Wang, X. Li, S. Lin, X. Guo and Z. Liu, ACS Catal., 2019, 9, 3017–3025 CrossRef CAS.
- C. Wang, L. Yang, M. Gao, X. Shao, W. Dai, G. Wu, N. Guan, Z. Xu, M. Ye and L. Li, J. Am. Chem. Soc., 2022, 144, 21408–21416 CrossRef CAS PubMed.
- J. Gil-Coba, S. C. Marie-Rose and J.-M. Lavoie, Catal. Lett., 2016, 146, 2534–2542 CrossRef CAS.
- R. Brosius, P. J. Kooyman and J. C. Q. Fletcher, ACS Catal., 2016, 6, 7710–7715 CrossRef CAS.
- A. Corma, O. Marie and F. J. Ortega, J. Catal., 2004, 222, 338–347 CrossRef CAS.
- P. Bollini, T. T. Chen, M. Neurock and A. Bhan, Catal. Sci. Technol., 2019, 9, 4374–4383 RSC.
- S. A. F. Nastase, P. Cnudde, L. Vanduyfhuys, K. De Wispelaere, V. Van Speybroeck, C. R. A. Catlow and A. J. Logsdail, ACS Catal., 2020, 10, 8904–8915 CrossRef CAS PubMed.
- D. T. Bregante and D. W. Flaherty, ACS Catal., 2019, 9, 10951–10962 CrossRef CAS.
- S. Eckstein, P. H. Hintermeier, M. V. Olarte, Y. Liu, E. Baráth and J. A. Lercher, J. Catal., 2017, 352, 329–336 CrossRef CAS.
- N. S. Gould and B. Xu, ACS Catal., 2018, 8, 8699–8708 CrossRef CAS.
- N. S. Gould, S. Li, H. J. Cho, H. Landfield, S. Caratzoulas, D. Vlachos, P. Bai and B. Xu, Nat. Commun., 2020, 11, 1060 CrossRef CAS PubMed.
- K. Chen, J. Damron, C. Pearson, D. Resasco, L. Zhang and J. L. White, ACS Catal., 2014, 4, 3039–3044 CrossRef CAS.
- K. Chen, A. Gumidyala, M. Abdolrhamani, C. Villines, S. Crossley and J. L. White, J. Catal., 2017, 351, 130–135 CrossRef CAS.
- M. Bocus, L. Vanduyfhuys, F. De Proft, B. M. Weckhuysen and V. Van Speybroeck, JACS Au, 2022, 2, 502–514 CrossRef CAS PubMed.
- Y. Zhi, H. Shi, L. Mu, Y. Liu, D. Mei, D. M. Camaioni and J. A. Lercher, J. Am. Chem. Soc., 2015, 137, 15781–15794 CrossRef CAS PubMed.
- M. Gešvandtnerová, T. Bučko, P. Raybaud and C. Chizallet, J. Catal., 2022, 413, 786–802 CrossRef.
- H. K. Chau, H. D. Mai, A. Gumidyala, T. N. Pham, D.-P. Bui, A. D. D’Amico, I. Alalq, D. T. Glatzhofer, J. L. White and S. P. Crossley, ACS Catal., 2023, 13, 9158–9170 CrossRef CAS.
- C. Wang, Y. Chu, D. Xiong, H. Wang, M. Hu, Q. Wang, J. Xu and F. Deng, Angew. Chem., Int. Ed., 2024, 63, e202313974 CrossRef CAS PubMed.
- M. Bocus and V. Van Speybroeck, ACS Catal., 2022, 12, 14227–14242 CrossRef CAS.
- W. N. P. van der Graaff, C. H. L. Tempelman, G. Li, B. Mezari, N. Kosinov, E. A. Pidko and E. J. M. Hensen, ChemSusChem, 2016, 9, 3145–3149 CrossRef CAS PubMed.
- H. K. Chau, Q. P. Nguyen, A. C. Jerdy, D.-P. Bui, L. L. Lobban, B. Wang and S. P. Crossley, ACS Catal., 2023, 13, 1503–1512 CrossRef CAS.
- Y. Liu, A. Vjunov, H. Shi, S. Eckstein, D. M. Camaioni, D. Mei, E. Baráth and J. A. Lercher, Nat. Commun., 2017, 8, 14113 CrossRef PubMed.
- H. Shi, S. Eckstein, A. Vjunov, D. M. Camaioni and J. A. Lercher, Nat. Commun., 2017, 8, 15442 CrossRef PubMed.
- M. Shetty, H. Wang, F. Chen, N. Jaegers, Y. Liu, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, Angew. Chem., Int. Ed., 2021, 60, 2304–2311 CrossRef CAS PubMed.
- L. Milaković, P. H. Hintermeier, Y. Liu, E. Baráth and J. A. Lercher, Angew. Chem., Int. Ed., 2021, 60, 24806–24810 CrossRef PubMed.
- Q. Liu, N. Pfriem, G. Cheng, E. Baráth, Y. Liu and J. A. Lercher, Angew. Chem., Int. Ed., 2023, 62, e202208693 CrossRef CAS PubMed.
- D. T. Bregante, A. M. Johnson, A. Y. Patel, E. Z. Ayla, M. J. Cordon, B. C. Bukowski, J. Greeley, R. Gounder and D. W. Flaherty, J. Am. Chem. Soc., 2019, 141, 7302–7319 CrossRef CAS PubMed.
- Y. Chu, X. Yi, C. Li, X. Sun and A. Zheng, Chem. Sci., 2018, 9, 6470–6479 RSC.
- K. Chen, M. Abdolrahmani, S. Horstmeier, T. N. Pham, V. T. Nguyen, M. Zeets, B. Wang, S. Crossley and J. L. White, ACS Catal., 2019, 9, 6124–6136 CrossRef CAS.
- Y. Zhang, R. Zhao, M. Sanchez-Sanchez, G. L. Haller, J. Hu, R. Bermejo-Deval, Y. Liu and J. A. Lercher, J. Catal., 2019, 370, 424–433 CrossRef CAS.
- T. N. Pham, V. Nguyen, B. Wang, J. L. White and S. Crossley, ACS Catal., 2021, 11, 6982–6994 CrossRef CAS.
- S. Li, A. Zheng, Y. Su, H. Zhang, L. Chen, J. Yang, C. Ye and F. Deng, J. Am. Chem. Soc., 2007, 129, 11161–11171 CrossRef CAS PubMed.
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 7024–7033 CrossRef CAS.
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 344, 570–577 CrossRef CAS.
- M. Niwa, K. Suzuki, N. Morishita, G. Sastre, K. Okumura and N. Katada, Catal. Sci. Technol., 2013, 3, 1919–1927 RSC.
- C. Chizallet, C. Bouchy, K. Larmier and G. Pirngruber, Chem. Rev., 2023, 123, 6107–6196 CrossRef CAS PubMed.
- A. Vjunov, M. A. Derewinski, J. L. Fulton, D. M. Camaioni and J. A. Lercher, J. Am. Chem. Soc., 2015, 137, 10374–10382 CrossRef CAS PubMed.
- J. T. C. Wennmacher, S. Mahmoudi, P. Rzepka, S. S. Lee, T. Gruene, V. Paunović and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2022, 61, e202205413 CrossRef CAS PubMed.
- Y. Wei, C. Yuan, J. Li, S. Xu, Y. Zhou, J. Chen, Q. Wang, L. Xu, Y. Qi, Q. Zhang and Z. Liu, ChemSusChem, 2012, 5, 906–912 CrossRef CAS PubMed.
- Y. Nakasaka, J.-i Nishimura, T. Tago and T. Masuda, Chem. Eng. J., 2015, 278, 159–165 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459–8467 CrossRef CAS PubMed.
- D. Kiani, S. Sourav, Y. Tang, J. Baltrusaitis and I. E. Wachs, Chem. Soc. Rev., 2021, 50, 1251–1268 RSC.
- M. Luo, Y. Fu, B. Hu, D. Wang, B. Wang and G. Mao, Appl. Catal., A, 2019, 570, 209–217 CrossRef CAS.
- X. Wu and R. G. Anthony, Appl. Catal., A, 2001, 218, 241–250 CrossRef CAS.
- H. An, F. Zhang, Z. Guan, X. Liu, F. Fan and C. Li, ACS Catal., 2018, 8, 9207–9215 CrossRef CAS.
- L. Yang, C. Wang, L. Zhang, W. Dai, Y. Chu, J. Xu, G. Wu, M. Gao, W. Liu, Z. Xu, P. Wang, N. Guan, M. Dyballa, M. Ye, F. Deng, W. Fan and L. Li, Nat. Commun., 2021, 12, 4661 CrossRef CAS PubMed.
- E. Catizzone, M. Migliori, A. Purita and G. Giordano, J. Energy Chem., 2019, 30, 162–169 CrossRef.
- M. Çaǧlayan, A. L. Paioni, B. Dereli, G. Shterk, I. Hita, E. Abou-Hamad, A. Pustovarenko, A.-H. Emwas, A. Dikhtiarenko, P. Castaño, L. Cavallo, M. Baldus, A. D. Chowdhury and J. Gascon, ACS Catal., 2021, 11, 11671–11684 CrossRef.
- A. Vjunov, J. L. Fulton, D. M. Camaioni, J. Z. Hu, S. D. Burton, I. Arslan and J. A. Lercher, Chem. Mater., 2015, 27, 3533–3545 CrossRef CAS.
- L. Zhang, K. Chen, B. Chen, J. L. White and D. E. Resasco, J. Am. Chem. Soc., 2015, 137, 11810–11819 CrossRef CAS PubMed.
- S. Prodinger, H. Shi, S. Eckstein, J. Z. Hu, M. V. Olarte, D. M. Camaioni, M. A. Derewinski and J. A. Lercher, Chem. Mater., 2017, 29, 7255–7262 CrossRef CAS.
- S. Prodinger, H. Shi, H. Wang, M. A. Derewinski and J. A. Lercher, Appl. Catal., B, 2018, 237, 996–1002 CrossRef CAS.
- M. Boronat, P. Concepción, A. Corma, M. Renz and S. Valencia, J. Catal., 2005, 234, 111–118 CrossRef CAS.
- V. L. Sushkevich, P. A. Kots, Y. G. Kolyagin, A. V. Yakimov, A. V. Marikutsa and I. I. Ivanova, J. Phys. Chem. C, 2019, 123, 5540–5548 CrossRef CAS.
- G. Qi, Q. Wang, J. Xu, Q. Wu, C. Wang, X. Zhao, X. Meng, F. Xiao and F. Deng, Commun. Chem., 2018, 1, 22 CrossRef.
- G. Li, E. A. Pidko and E. J. M. Hensen, Catal. Sci. Technol., 2014, 4, 2241–2250 RSC.
- D. Padovan, L. Botti and C. Hammond, ACS Catal., 2018, 8, 7131–7140 CrossRef CAS.
- M. J. Cordon, J. N. Hall, J. W. Harris, J. S. Bates, S.-J. Hwang and R. Gounder, Catal. Sci. Technol., 2019, 9, 1654–1668 RSC.
- L. Treps, A. Gomez, T. de Bruin and C. Chizallet, ACS Catal., 2020, 10, 3297–3312 CrossRef CAS.
- J. Rey, P. Raybaud and C. Chizallet, ChemCatChem, 2017, 9, 2176–2185 CrossRef CAS.
- L. Treps, C. Demaret, D. Wisser, B. Harbuzaru, A. Méthivier, E. Guillon, D. V. Benedis, A. Gomez, T. de Bruin, M. Rivallan, L. Catita, A. Lesage and C. Chizallet, J. Phys. Chem. C, 2021, 125, 2163–2181 CrossRef CAS.
- T. Jarrin, T. de Bruin and C. Chizallet, ChemCatChem, 2023, 15, e202201302 CrossRef CAS.
- C. Chizallet, ACS Catal., 2020, 10, 5579–5601 CrossRef CAS.
- G. Agostini, C. Lamberti, L. Palin, M. Milanesio, N. Danilina, B. Xu, M. Janousch and J. A. van Bokhoven, J. Am. Chem. Soc., 2010, 132, 667–678 CrossRef CAS PubMed.
- J. Zhou, M. Gao, J. Zhang, W. Liu, T. Zhang, H. Li, Z. Xu, M. Ye and Z. Liu, Nat. Commun., 2021, 12, 17 CrossRef CAS PubMed.
- T. Sun, S. Xu, D. Xiao, Z. Liu, G. Li, A. Zheng, W. Liu, Z. Xu, Y. Cao, Q. Guo, N. Wang, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 20672–20681 CrossRef CAS PubMed.
- A. Zornes, N. B. Abdul Rahman, O. R. Das, L. A. Gomez, S. Crossley, D. E. Resasco and J. L. White, J. Am. Chem. Soc., 2024, 146, 1132–1143 CrossRef CAS PubMed.
- J. A. van Bokhoven, D. C. Koningsberger, P. Kunkeler, H. van Bekkum and A. P. M. Kentgens, J. Am. Chem. Soc., 2000, 122, 12842–12847 CrossRef CAS.
- M. Signorile, F. Bonino, A. Damin and S. Bordiga, J. Phys. Chem. C, 2016, 120, 18088–18092 CrossRef CAS.
- B. Fan, D. Zhu, L. Wang, S. Xu, Y. Wei and Z. Liu, Inorg. Chem. Front., 2022, 9, 3609–3618 RSC.
- X. Bai, J. Zhang, C. Liu, S. Xu, Y. Wei and Z. Liu, Micro. Meso. Mater., 2023, 354, 112555 CrossRef CAS.
- T. Blasco, A. Corma and J. Martínez-Triguero, J. Catal., 2006, 237, 267–277 CrossRef CAS.
- S. M. C. Menezes, V. L. Camorim, Y. L. Lam, R. A. S. San Gil, A. Bailly and J. P. Amoureux, Appl. Catal., A, 2001, 207, 367–377 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Prieto and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 5042–5048 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, J. Catal., 2013, 307, 194–203 CrossRef CAS.
- L. R. Aramburo, E. de Smit, B. Arstad, M. M. van Schooneveld, L. Sommer, A. Juhin, T. Yokosawa, H. W. Zandbergen, U. Olsbye, F. M. F. de Groot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2012, 51, 3616–3619 CrossRef CAS PubMed.
- B. Xu, F. Rotunno, S. Bordiga, R. Prins and J. A. van Bokhoven, J. Catal., 2006, 241, 66–73 CrossRef CAS.
- J. A. van Bokhoven, A. M. J. van der Eerden and D. C. Koningsberger, Stud. Surf. Sci. Catal., 2002, 142, 1885–1890 CrossRef.
- J. A. van Bokhoven, A. M. J. van der Eerden and D. C. Koningsberger, J. Am. Chem. Soc., 2003, 125, 7435–7442 CrossRef CAS PubMed.
- M. Jin, M. Ravi, C. Lei, C. J. Heard, F. Brivio, Z. Tošner, L. Grajciar, J. A. van Bokhoven and P. Nachtigall, Angew. Chem., Int. Ed., 2023, 62, e202306183 CrossRef PubMed.
- A. Omegna, J. A. van Bokhoven and R. Prins, J. Phys. Chem. B, 2003, 107, 8854–8860 CrossRef CAS.
- Y. Ma, J. Ding, L. Yang, X. Wu, Y. Gao, R. Ran and D. Weng, J. Phys. Chem. C, 2023, 127, 16598–16606 CrossRef CAS.
- M.-C. Silaghi, C. Chizallet, J. Sauer and P. Raybaud, J. Catal., 2016, 339, 242–255 CrossRef CAS.
- S. Malola, S. Svelle, F. L. Bleken and O. Swang, Angew. Chem., Int. Ed., 2012, 51, 652–655 CrossRef CAS PubMed.
- M.-C. Silaghi, C. Chizallet, E. Petracovschi, T. Kerber, J. Sauer and P. Raybaud, ACS Catal., 2015, 5, 11–15 CrossRef CAS.
- M. Nielsen, A. Hafreager, R. Y. Brogaard, K. De Wispelaere, H. Falsig, P. Beato, V. V. Speybroeck and S. Svelle, Catal. Sci. Technol., 2019, 9, 3721–3725 RSC.
- K. Stanciakova, B. Ensing, F. Göltl, R. E. Bulo and B. M. Weckhuysen, ACS Catal., 2019, 9, 5119–5135 CrossRef CAS.
- C. J. Heard, L. Grajciar, C. M. Rice, S. M. Pugh, P. Nachtigall, S. E. Ashbrook and R. E. Morris, Nat. Commun., 2019, 10, 4690 CrossRef PubMed.
- S. M. Pugh, P. A. Wright, D. J. Law, N. Thompson and S. E. Ashbrook, J. Am. Chem. Soc., 2020, 142, 900–906 CrossRef CAS PubMed.
- F. Lin, M. Xu, K. K. Ramasamy, Z. Li, J. L. Klinger, J. A. Schaidle and H. Wang, ACS Catal., 2022, 12, 13555–13599 CrossRef CAS.
- R. M. Ravenelle, F. Schüβler, A. D’Amico, N. Danilina, J. A. van Bokhoven, J. A. Lercher, C. W. Jones and C. Sievers, J. Phys. Chem. C, 2010, 114, 19582–19595 CrossRef CAS.
- A. R. Maag, G. A. Tompsett, J. Tam, C. A. Ang, G. Azimi, A. D. Carl, X. Huang, L. J. Smith, R. L. Grimm, J. Q. Bond and M. T. Timko, Phys. Chem. Chem. Phys., 2019, 21, 17880–17892 RSC.
- T. Ennaert, J. Geboers, E. Gobechiya, C. M. Courtin, M. Kurttepeli, K. Houthoofd, C. E. A. Kirschhock, P. C. M. M. Magusin, S. Bals, P. A. Jacobs and B. F. Sels, ACS Catal., 2015, 5, 754–768 CrossRef CAS.
- C. E. Hernandez-Tamargo, A. Roldan and N. H. de Leeuw, J. Solid State Chem., 2016, 237, 192–203 CrossRef CAS.
- F. Dubray, E. Dib, I. Medeiros-Costa, C. Aquino, D. Minoux, S. van Daele, N. Nesterenko, J.-P. Gilson and S. Mintova, Inorg. Chem. Front., 2022, 9, 1125–1133 RSC.
- G. M. Lari, P. Y. Dapsens, D. Scholz, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2016, 18, 1249–1260 RSC.
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Adv. Mater., 2020, 32, 2003264 CrossRef CAS PubMed.
- J. N. Louwen, S. Simko, K. Stanciakova, R. E. Bulo, B. M. Weckhuysen and E. T. C. Vogt, J. Phys. Chem. C, 2020, 124, 4626–4636 CrossRef CAS.
- J. Sun, H. Fang, P. I. Ravikovitch and D. S. Sholl, J. Phys. Chem. C, 2020, 124, 668–676 CrossRef CAS.
- R. C. Shiery, S. J. McElhany and D. C. Cantu, J. Phys. Chem. C, 2021, 125, 13649–13657 CrossRef CAS.
- G. Caeiro, P. Magnoux, J. M. Lopes, F. R. Ribeiro, S. M. C. Menezes, A. F. Costa and H. S. Cerqueira, Appl. Catal., B, 2006, 314, 160–171 CrossRef CAS.
- A. Corma, J. Mengual and P. J. Miguel, Appl. Catal., A, 2012, 421–422, 121–134 CrossRef CAS.
- Y. Kakiuchi, T. Tanigawa, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, Appl. Catal., A, 2019, 575, 204–213 CrossRef CAS.
- R. Simancas, A. Chokkalingam, S. P. Elangovan, Z. Liu, T. Sano, K. Iyoki, T. Wakihara and T. Okubo, Chem. Sci., 2021, 12, 7677 RSC.
- H. E. Van der Bij and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2014, 16, 9892–9903 RSC.
- H. Vinek, G. Rumplmayr and J. A. Lercher, J. Catal., 1989, 115, 291–300 CrossRef CAS.
- G. Lischke, R. Eckelt, H.-G. Jerschkewitz, B. Parlitz, E. Schreier, W. Storek, B. Zibrowius and G. Öhlmann, J. Catal., 1991, 132, 229–243 CrossRef CAS.
- H. E. Van der Bij and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7406–7428 RSC.
- J. N. Louwen, L. van Eijck, C. Vogt and E. T. C. Vogt, Chem. Mater., 2020, 32, 9390–9403 CrossRef CAS.
- U. Khalil, O. Muraza, H. Kondoh, G. Watanabe, Y. Nakasaka, A. Al-Amer and T. Masuda, Fuel, 2006, 168, 61–67 CrossRef.
- Y. Wang, P. Guerra, A. Zaker, A. R. Maag, G. A. Tompsett, L. J. Smith, X. Huang, J. Q. Bond and M. T. Timko, ACS Catal., 2020, 10, 6623–6634 CrossRef CAS.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570–8578 CrossRef CAS PubMed.
- P. A. Zapata, Y. Huang, M. A. Gonzalez-Borja and D. E. Resasco, J. Catal., 2013, 308, 82–97 CrossRef CAS.
- S. Prodinger, M. A. Derewinski, A. Vjunov, S. D. Burton, I. Arslan and J. A. Lercher, J. Am. Chem. Soc., 2016, 138, 4408–4415 CrossRef CAS PubMed.
- M. A. Camblor, A. Corma and S. Valencia, J. Mater. Chem., 1998, 8, 2137–2145 RSC.
- J. Saavedra, H. A. Doan, C. J. Pursell, L. C. Grabow and B. D. Chandler, Science, 2014, 345, 1599–1602 CrossRef CAS PubMed.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- Z. Liu, E. Huang, I. Orozco, W. Liao, R. M. Palomino, N. Rui, T. Duchoň, S. Nemšák, D. C. Grinter, M. Mahapatra, P. Liu, J. A. Rodriguez and S. D. Senanayake, Science, 2020, 368, 513–517 CrossRef CAS PubMed.
- K. Xu, Y. Chen, H. Yang, Y. Gan, L. Wu, L. Tan, Y. Dai and Y. Tang, Appl. Catal., B, 2024, 341, 123244 CrossRef CAS.
- H. Shi, J. A. Lercher and X.-Y. Yu, Catal. Sci. Technol., 2015, 5, 3035–3060 RSC.
- N. S. Gould and B. Xu, Chem. Sci., 2018, 9, 281–287 RSC.
- G. Noh, Z. Shi, S. I. Zones and E. Iglesia, J. Catal., 2018, 368, 389–410 CrossRef CAS.
- V. Shostak, E. Redekop and U. Olsbye, Catal. Today, 2023, 417, 113785 CrossRef CAS.
- P. Cnudde, R. Demuynck, S. Vandenbrande, M. Waroquier, G. Sastre and V. Van Speybroeck, J. Am. Chem. Soc., 2020, 142, 6007–6017 CrossRef CAS PubMed.
- V. Van Speybroeck, M. Bocus, P. Cnudde and L. Vanduyfhuys, ACS Catal., 2023, 13, 11455–11493 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |