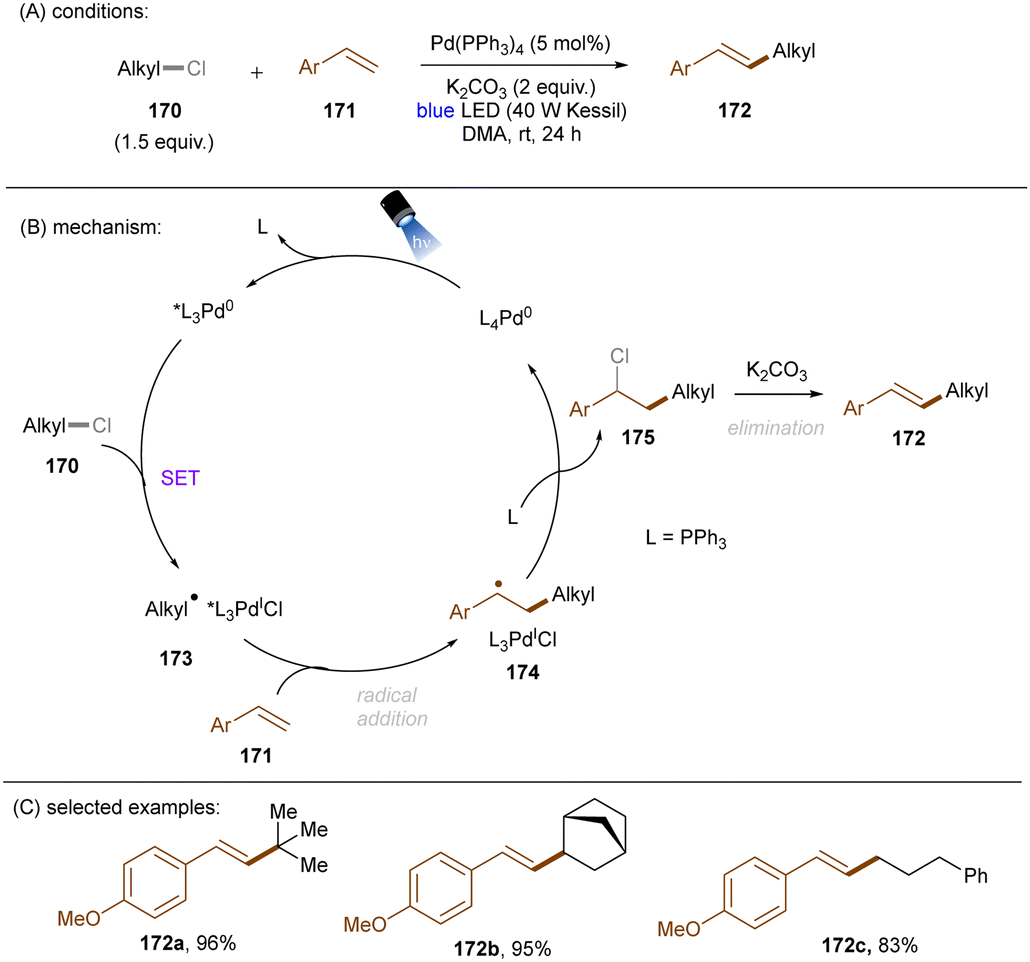
DOI:
10.1039/D2CS00581F
(Review Article)
Chem. Soc. Rev., 2024,
53, 263-316
Photo- and electro-chemical strategies for the activations of strong chemical bonds
Received
31st August 2023
First published on 7th December 2023
Abstract
The employment of light and/or electricity – alternatively to conventional thermal energy – unlocks new reactivity paradigms as tools for chemical substrate activations. This leads to the development of new synthetic reactions and a vast expansion of chemical spaces. This review summarizes recent developments in photo- and/or electrochemical activation strategies for the functionalization of strong bonds – particularly carbon–heteroatom (C–X) bonds – via: (1) direct photoexcitation by high energy UV light; (2) activation via photoredox catalysis under irradiation with relatively lower energy UVA or blue light; (3) electrochemical reduction; (4) combination of photocatalysis and electrochemistry. Based on the types of the targeted C–X bonds, various transformations ranging from hydrodefunctionalization to cross-coupling are covered with detailed discussions of their reaction mechanisms.

Xianhai Tian
| Xianhai Tian was born in Xinyang, P. R. China. He received his PhD in 2019 under the supervision of Prof. A. Stephen K. Hashmi at the University of Heidelberg, Germany, where his doctoral studies investigated gold carbene and nitrene chemistry. In 2020, he joined the group of Dr Joshua P. Barham for postdoctoral research on synthetic photocatalysis and photoelectrochemistry. |

Yuliang Liu
| Yuliang Liu received her PhD in 2017 under the supervision of Prof. Yifeng Wang at Shandong University, P. R. China, where she specialized in photocatalysis. She then joined the group of Prof. Wei Lu at Southern University of Science and Technology for postdoctoral research on photophysics and photochemistry. In 2019, she moved to Nanyang Technological University (NTU) Singapore to pursue postdoctoral research on synthetic photochemistry in the group of Prof. Shunsuke Chiba. Since 2023, she has been working as a senior scientist at Songshan Lake Materials Laboratory, P. R. China. |

Shahboz Yakubov
| Shahboz Yakubov was born in Istaravshan, Tajikistan. He received his MSc in 2020 as part of the Elite Network of Bavaria's SynCat program at the University of Regensburg, Germany. During this time, he worked in the group of Dr Joshua P. Barham where his Master thesis project investigated photocatalytic C–H fluorinations. Currently, he is a PhD student in the group of Dr Joshua P. Barham investigating synthetic photochemistry and flow chemistry. |

Jonathan Schütte
| Jonathan Schütte was born in Westerstede, Germany. He received his MSc in 2023 of the Elite Network of Bavaria's SynCat program at the University of Regensburg, Germany. During this time, he worked in the group of Dr Joshua P. Barham where his Master thesis project investigated catalyst-free and photocatalytic nitrogen centered radical generation under light irradiation. Currently, he is a PhD student in the group of Dr Joshua P. Barham investigating synthetic photocatalysis and photoelectrochemistry. |

Shunsuke Chiba
| Shunsuke Chiba received his PhD in 2006 under the supervision of Prof. Koichi Narasaka at the University of Tokyo. In 2007, he embarked on his independent career as the faculty of Nanyang Technological University (NTU) Singapore, where he is currently Professor of Chemistry. His research group focuses on methodology development in the area of synthetic chemistry and catalysis. In particular, his group has investigated the use of hydride transfer and single electron transfer in organic synthesis. |

Joshua P. Barham
| Joshua P. Barham was born in Watford, UK. He received his industry-based PhD in 2017 under the supervision of Prof. John A. Murphy and Dr Matthew P. John at the University of Strathclyde and GSK, UK. His postdoctoral studies with Prof. Yasuo Norikane and Prof. Yoshitaka Hamashima at AIST and the University of Shizuoka, Japan, specialized in flow chemistry and photoredox catalysis. Since 2019, his independent group has investigated photo-, electro-, photoelectro- and continuous flow organic synthesis at the University of Regensburg, supported by a Sofja Kovalevskaja Award. In 2022, he was awarded an ERC Starting Grant. |
1. Introduction
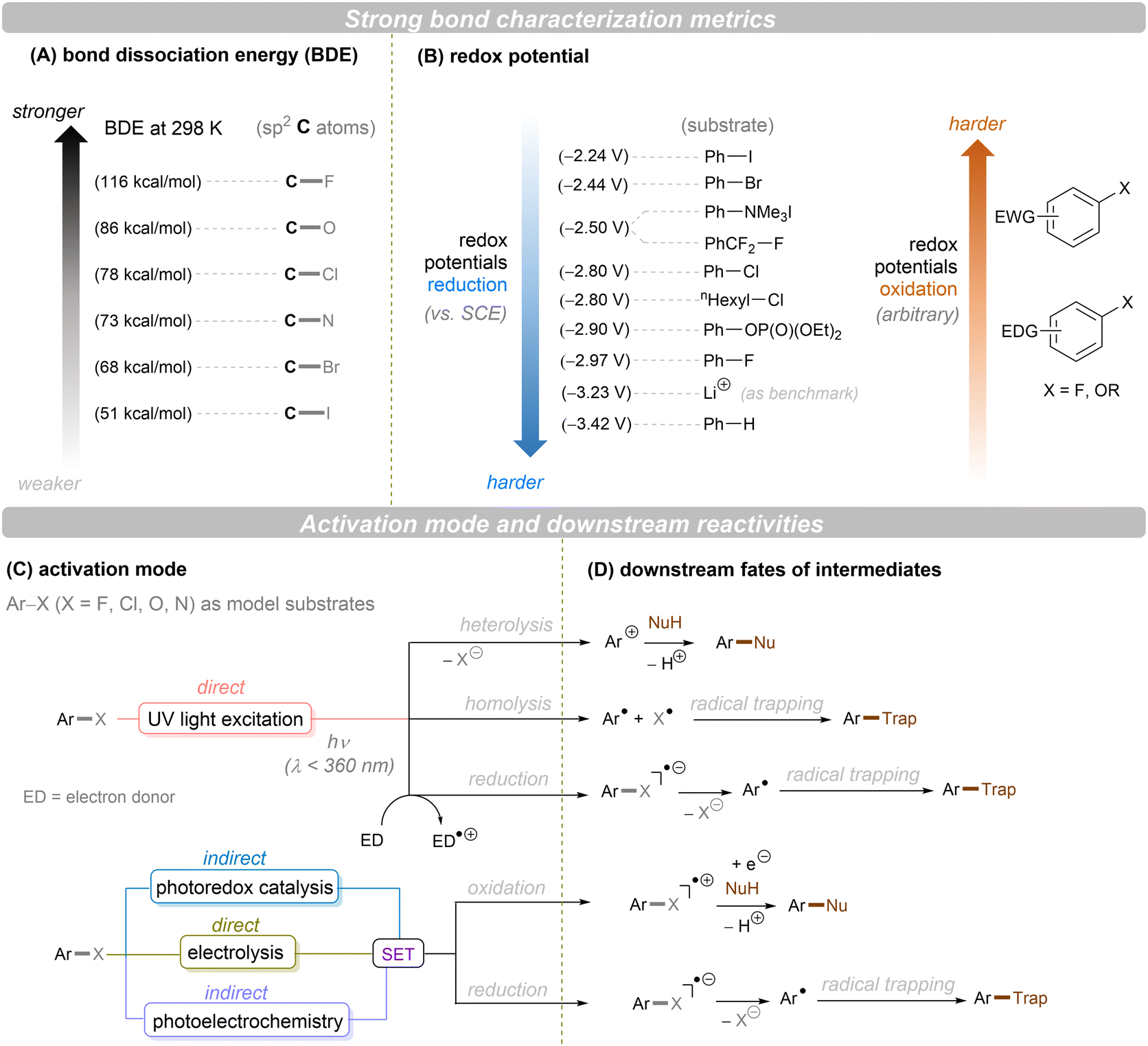
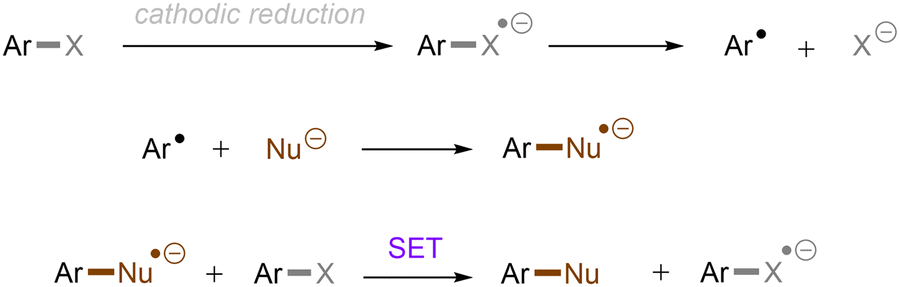
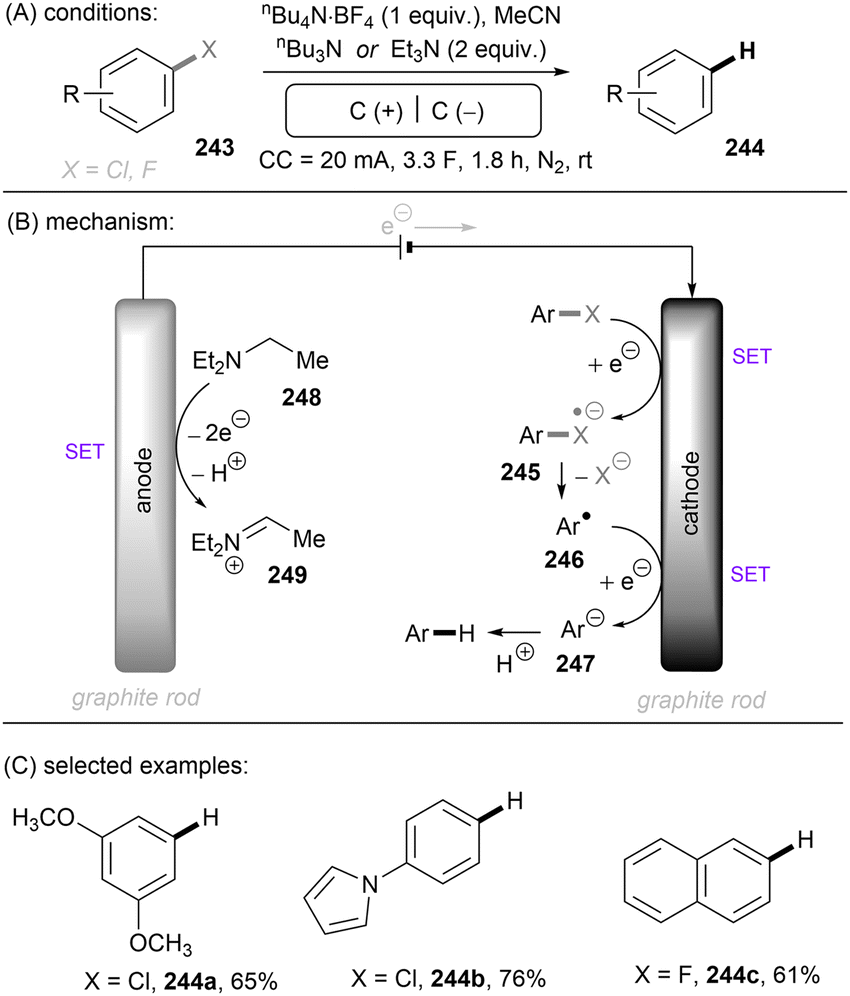
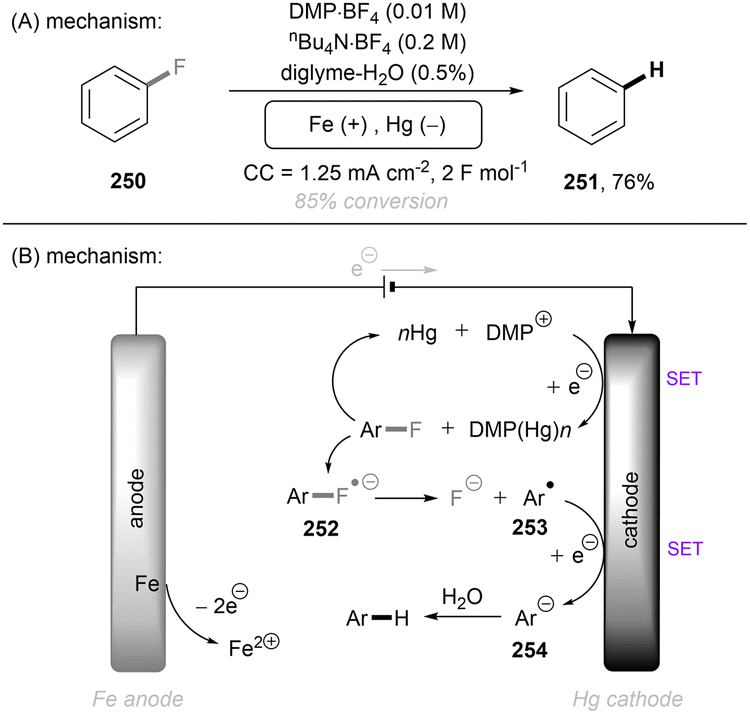
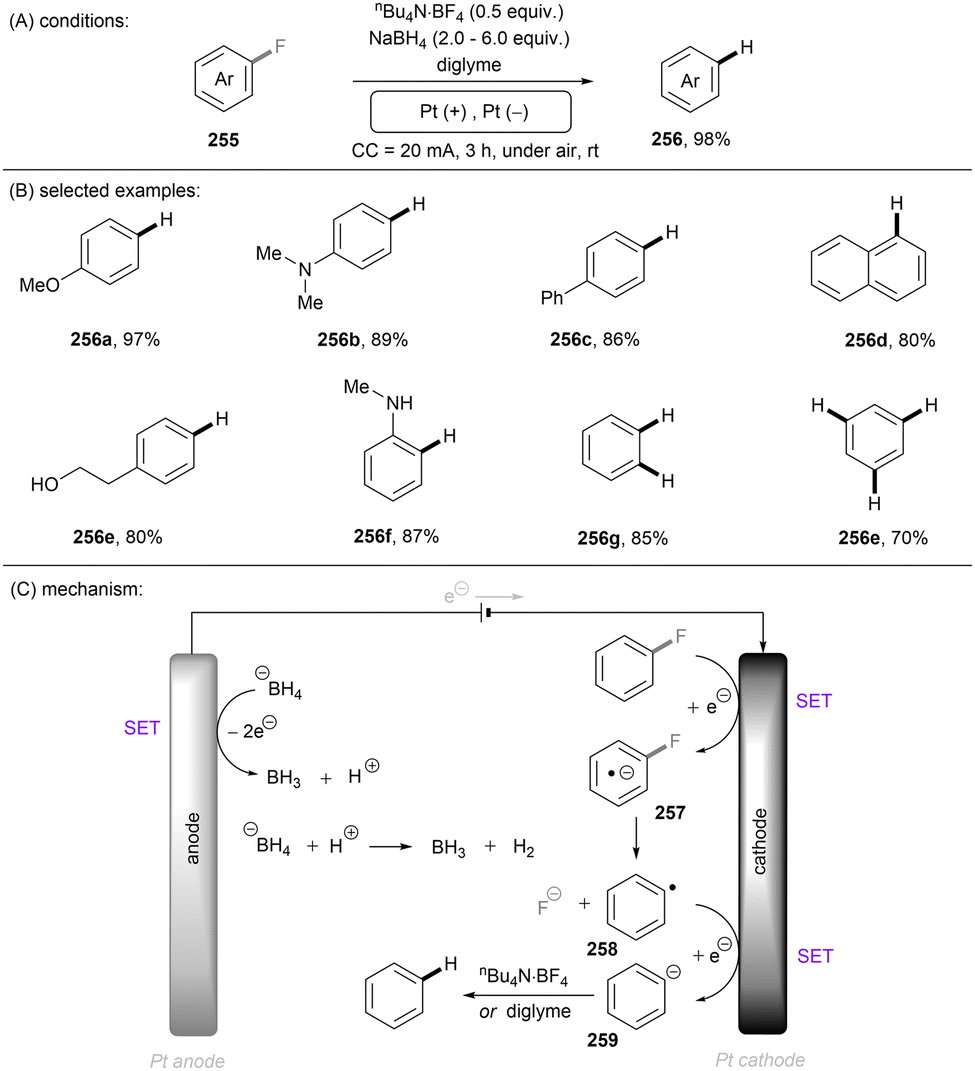
Carbon–heteroatom (C–X) bonds (X = F, Cl, O, N, etc.) are omnipresent as key components in commodity chemicals, biologically-active natural products and drug molecules. The transformations of such C–X bonds to other functional groups expands chemical space for the development of new functional molecules. Taking aryl (pseudo)halide as an example, while aryl bromides and iodides are easier to functionalize, their comparatively poor availability in nature hinders their synthetic utility. In contrast, aryl chlorides are regarded as more ideal substrates to activate for late-stage modifications as they are ubiquitous in nature and comprise over two thirds of commercially available aryl halides. It is especially important to use biomass and persistent chemical waste organic chlorides (like polychlorinated biphenyls) as the feedstock chemicals for drug/agrochemical synthesis. However, the high chemical and redox stabilities of aryl chlorides render them stubborn (Fig. 1A and B). The bond dissociation energy (BDE) of a C(sp2)–Cl bond is generally higher than 72 kcal mol−1, contributing to its decreased reactivity compared to C(sp2)–Br (BDE = 68 kcal mol−1) and C(sp2)–I (BDE = 51 kcal mol−1) bonds (Fig. 1A). Furthermore, these aryl halide substrates have high redox potentials for reduction (Fig. 1B). Substituted benzenes such as fluorobenzene, chlorobenzene, phenyl phosphate etc. generally have deeper negative reduction potentials than bromobenzene (Ph–Br, Ered = −2.44 V) and iodobenzene (Ph–I, Ered = −2.24 V), making them more difficult to reduce via single electron transfer (SET). Therefore, the formidable challenge still remains to realize downstream functionalization of inert Ar–X bonds.
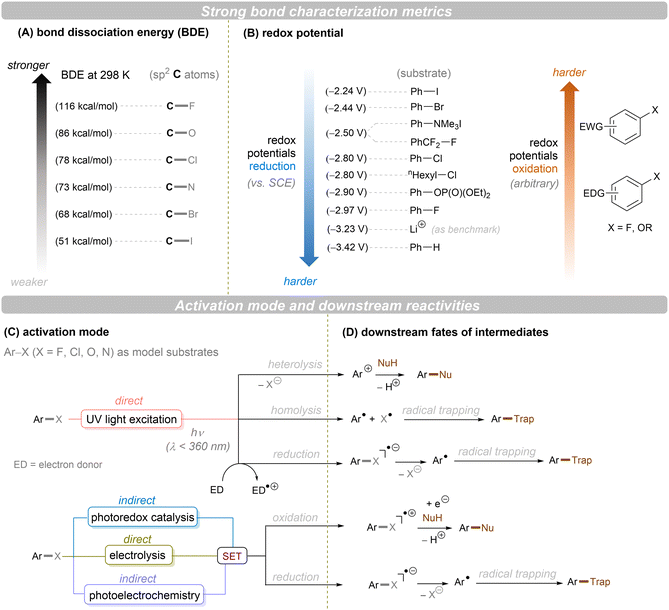 |
| | Fig. 1 Bond types and activation modes mentioned in this review. | |
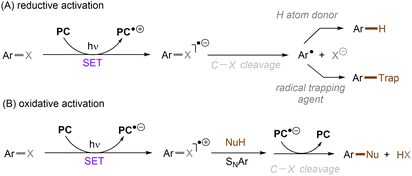
As conventional reductants, alkali metals in liquid ammonia are frequently used for the reductive activation of inert organic compounds.1 However, the functional group tolerance is compromised under these harsh dissolving metal conditions. More benign methods towards challenging activations have been developed, leveraging the platforms of photo- and electrochemistry either directly or indirectly with mediators (Fig. 1C), and these strategies are the theme of this review. UV-light excitation of aryl (pseudo)halides represents a straightforward activation mode.2 The excited arenes undergo either (i) heterolysis, to afford aryl cation intermediates susceptible to nucleophilic attack, or (ii) homolysis, to form aryl radicals that can be trapped by other partners. Furthermore, the excited aryl (pseudo)halides can be reductively or oxidatively quenched by external redox mediators though SET. The reductive quenching followed by a mesolytic C(sp2)–X bond cleavage generates aryl radicals, which can be trapped inter- or intramolecularly. Arene radical cations generated by oxidative quenching (not shown) further undergo a nucleophilic aromatic substitution (SNAr). Although efficient, this strategy requires the use of UV light. Such high-energy light may cause unselective excitations of multiple reaction components, which encourages undesired side reactions. Synthetic chemists have therefore sought to realize C–X bond activations under milder conditions using longer-wavelength visible light. In the last two decades, photoredox catalysis (PRC) has attracted considerable attention.3–8 The development of visible light-absorbing photocatalysts (PCs) has allowed to drive desired synthetic reactions by visible photon energy. Apart from direct halogen-atom-transfer (XAT),9 SET is central to PRC strategies in the activations of organic molecules. While single photonic PRC with neutral PCs as strongly reducing photoreductants (Ered = −2.0–−2.4 V) has enabled the activation of diverse organic molecules, substrates with highly positive redox potentials for oxidation (Eox > +2.0 V) or a highly negative potentials for reduction (Ered < −2.5 V) remain intact. New strategies, such as consecutive photoinduced electron-transfer (conPET)10 – which can generate potent radical ionic photoreductants or other highly reducing species such as solvated electrons and CO2˙− – are able to activate these reductively inert substrates. Generally, such strategies require a plethora of exogenous redox mediators such as trialkylamines and SF6. In a complementary fashion, synthetic organic electrochemistry (SOE) guarantees super redox potentials omitting super stoichiometric amounts of external redox mediators.11–21 However, the applied constant current or voltage may cause over-reductions/-oxidations. Moreover, synthetic chemists have merged PRC and SOE to create a powerful, flexible and fundamentally sustainable tool for the activation of inert chemical bonds – synthetic molecular photoelectrochemistry (PEC).22–26 The combination of advantages of both techniques addresses the aforementioned challenging issues.
This review mainly focuses on photo- and/or electrochemical activations of strong C–X bonds in unactivated organic compounds, particularly (not always) by electron transfer. As a minor theme, reductions of simple unsaturated systems (π-carbon bonds) are also covered. A comprehensive overview of diverse activation strategies including direct UV-light excitation, PRC and/or SOE enabled activations through SET will be presented. In terms of the chemistry, active catalyst species including excited neutral, ionic and radical species as well as other in situ generated species such as CO2˙−, solvated electrons and electron-donor–acceptor (EDA) complexes will be summarized. Reaction types range from hydrodefunctionalizations to cross-couplings. Where available, synthetic applications towards the synthesis of pharmaceutically-relevant compounds and scalabilities in batch or flow will be highlighted. The direct XAT strategy has been well-reviewed elsewhere9 and will not be involved herein. Our review is limited to those activations of inert bonds proposed to proceed by electron transfer, not triplet–triplet energy transfer. Comparisons of different strategies as well as current challenges and future perspectives will be outlined.
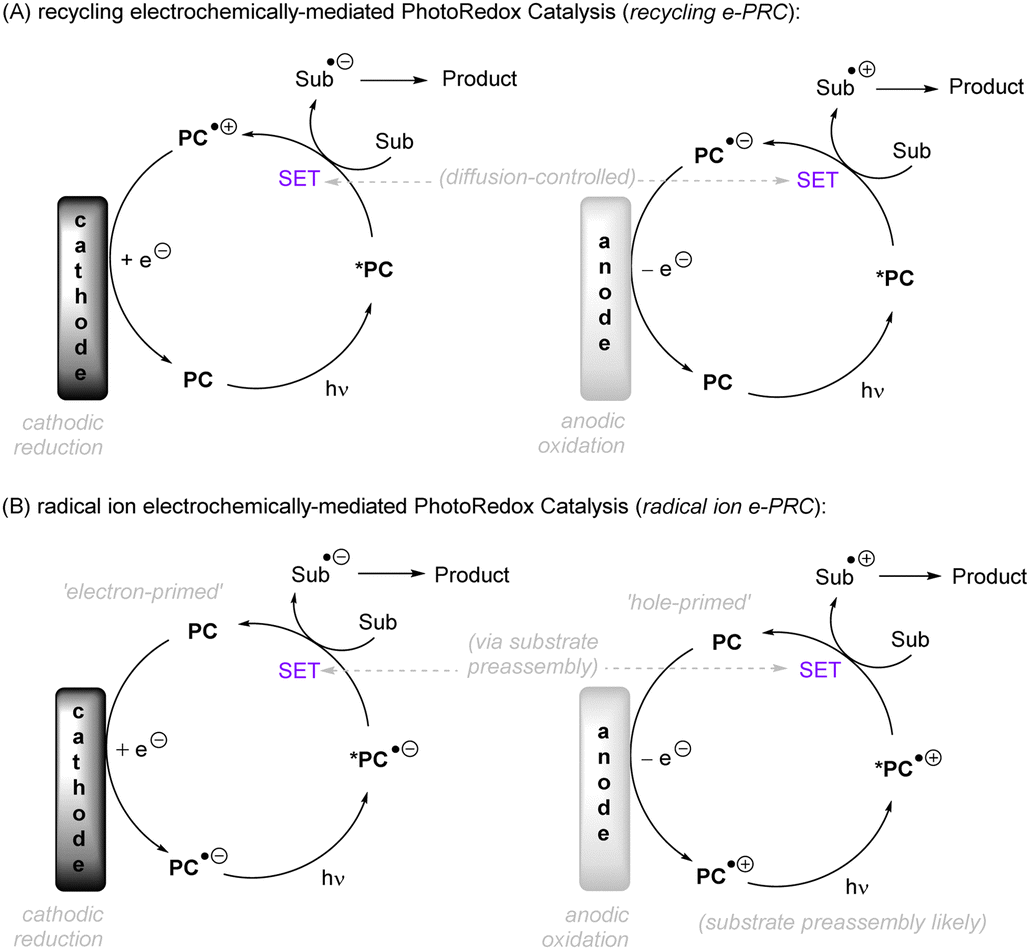
In terms of the composition, we consider the ‘intimacy’ of interaction with molecules (Fig. 1C). Firstly, we discuss direct UV photoexcitation activations of substrates, as the most intimate photochemical manner of interacting with molecules, that provides the highest energy input but also highest risk of side reactions. Secondly, we explore near-UV/visible light activations of substrates indirectly via a photoredox catalyst, as an indirect and more selective method but one which is limited in redox power. Within this section, conPET is covered as a strategy to break through redox limitations. Thirdly, direct electrolysis activations are presented as an intimate (heterogeneous) and flexible manifold for interacting with substrates without redox limits, yet one which suffers from selectivity issues at high applied currents/potentials. Finally, PEC is presented as an indirect platform (rivaling or even exceeding the redox achievements of conPET) that can boast extreme redox potentials without the compromise of selectivity.
Note: unless otherwise specified, all redox potentials given are vs. SCE. All catalyst structures that are drawn represent those proposed by the authors as the active species, even if subsequent studies found transformation of the catalyst to other active species (typically shown in grey). All light sources and temperatures are specified as done in the literature concerned, as far as possible including wavelength and input power and – at the least – ‘colour’, if no specifics were found. For electrochemical reactions, Faradays passed are given as far as possible. Our review's focus is on the activation step and the downstream chemical transformations (Fig. 1D), rather than the detailed photophysical transformations of the catalyst. Therefore, while the nature of the excited state photocatalyst (singlet/doublet/triplet) is specified in some cases, where it is not specified clearly the readers are referred to the reports in question.
2. UV-driven, non-catalytic variants
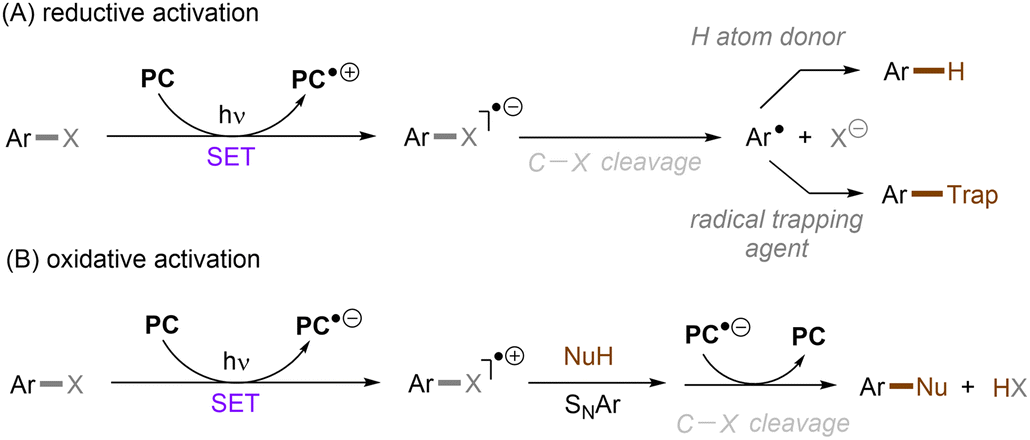
Aromatic rings can be activated by excitation with an ultraviolet (UV) photon, resulting typically in a π → π* locally excited (LE) state electronic transition. In the case of arenes having Ar–X bond(s), their UV photoexcitation can result in Ar–X bond scission in either a homolytic or heterolytic manner.27 The pathway of Ar–X bond cleavage is dictated by the intrinsic nature of the arene substrate's photoexcited state as well as the reaction conditions. Besides Ar–X bond cleavage, photoexcited arenes can also undergo single-electron-transfer (SET) via either oxidative or reductive quenching to form arene radical cations (Ar–X)˙+ or radical anions (Ar–X)˙−, respectively. The resulting radical cations (Ar–X)˙+ could be trapped by suitable nucleophiles to provide the cross-coupling products, whereas the radical anions (Ar–X)˙− may undergo mesolytic Ar–X bond cleavage to generate highly reactive aryl radicals (Ar˙), which can be trapped in various radical arylation reactions.
2.1. Ar–X activation via photoinduced heterolysis
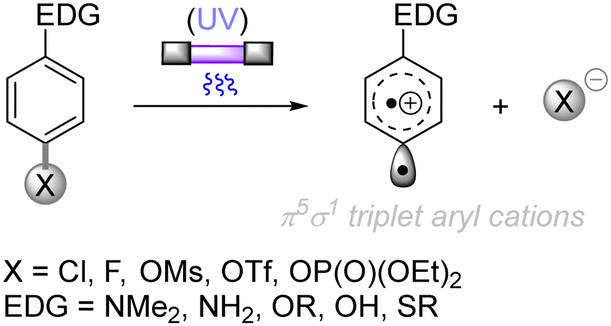
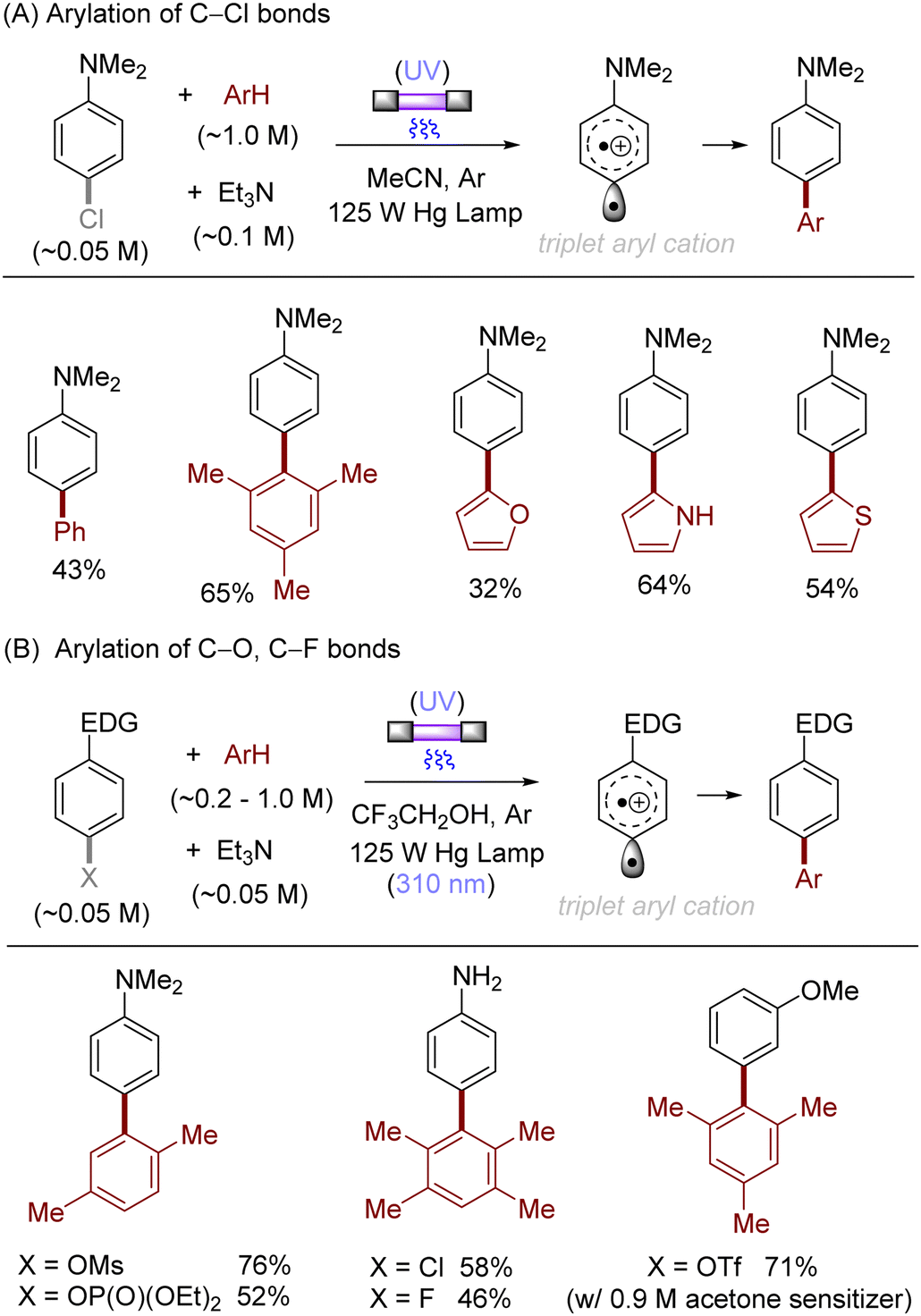
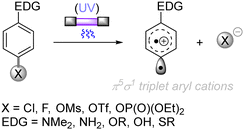
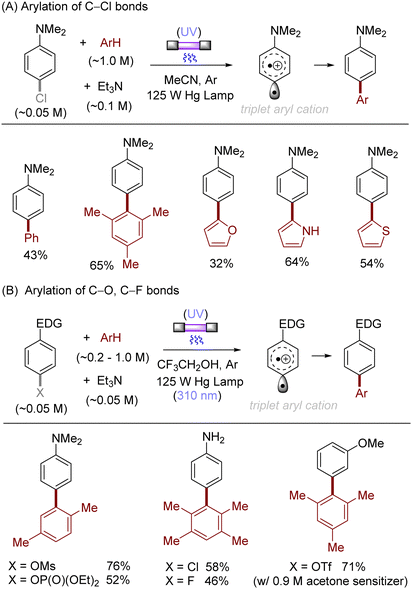
The difficulty in the functionalization of arenes having Ar–X bonds (X = Cl, F, O etc.) is primarily due to the high Ar–X bond dissociation energy (BDE of Ph–Cl = 93.7 kcal mol−1, Ph–F = 125.6 kcal mol−1 and Ph–OAc = 102.9 kcal mol−1). The team of Albini and Fagnoni reported how UV light photoexcitation of Ar–X compounds having an electron-donating group (EDG) in polar aprotic solvents results in Ar–X bond heterolysis to form a triplet biradical of the aryl cation. Its π5σ1 structure is stabilized by the EDGs (Scheme 1).28,29 Amino groups,30 alkoxy/hydroxy groups31 and alkylthio groups32 can serve as EDGs for the photoinduced generation of triplet aryl cations. Albini, Fagnoni and co-workers moreover demonstrated how the triplet aryl cations could be trapped with different kinds of π nucleophiles.30,33,34 For example, benzene, mesitylene and heteroarenes such as furan, thiophene, pyrrole and their derivatives were successfully employed to functionalize the triplet aryl cation derived from 4-chloro-N,N-dimethylaniline, providing the corresponding biaryl motifs (Scheme 2A). The method was extended to the functionalization of aryl fluorides as well as aryl mesylates, triflates, and phosphates (Scheme 2B).35 Carbonate or trialkylamine bases were typically employed to quench generated acid.
 |
| | Scheme 1 Photoinduced generation of the triplet aryl cation. | |
 |
| | Scheme 2 Photoinduced arylation of aryl (pseudo)halides via triplet aryl cations. Carbonate bases were oftentimes employed instead of Et3N, see literature for details. | |
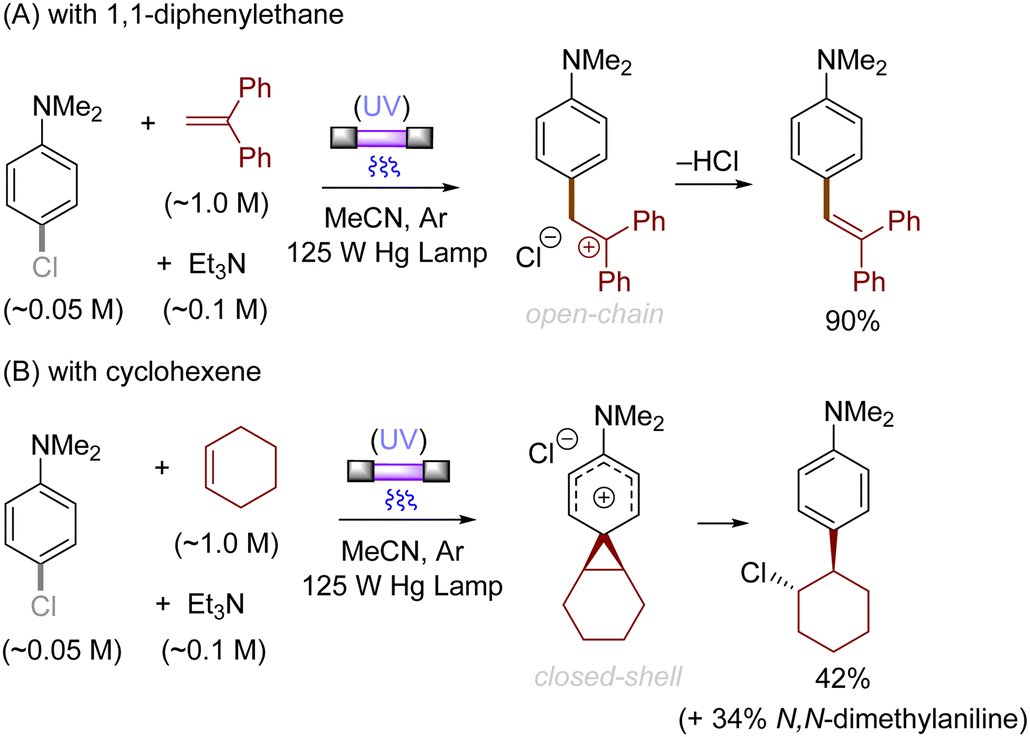
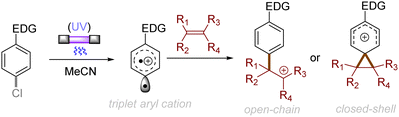
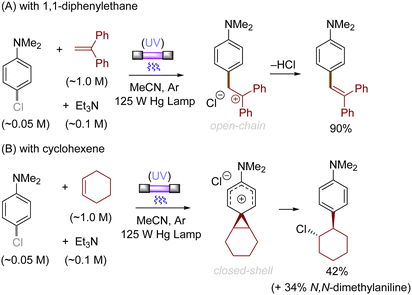
Alkenes could also be employed to intercept the triplet aryl cations. Mechanistically, the process could afford either open-chain carbocations or closed-shell phenonium cations, that could be further functionalized (Scheme 3).29 For example, their reactions with diphenylethylene preferentially affords open-chain benzyl cations, which would undergo deprotonation to deliver the triarylethylene derivatives (Scheme 4A). From the reactions with alkyl-substituted alkenes such as 1-hexene and cyclohexene, closed-shell phenonium ion intermediates could be generated and their downstream functionalization with concomitant σ-nucleophiles derived from the halide leaving groups, solvents, and additives resulted overall in a difunctionalization of alkene nucleophiles (Scheme 4B).29,36
 |
| | Scheme 3 Interception of photogenerated triplet aryl cations by alkenes. | |
 |
| | Scheme 4 Photoinduced coupling reactions between aryl chlorides and alkenes. | |
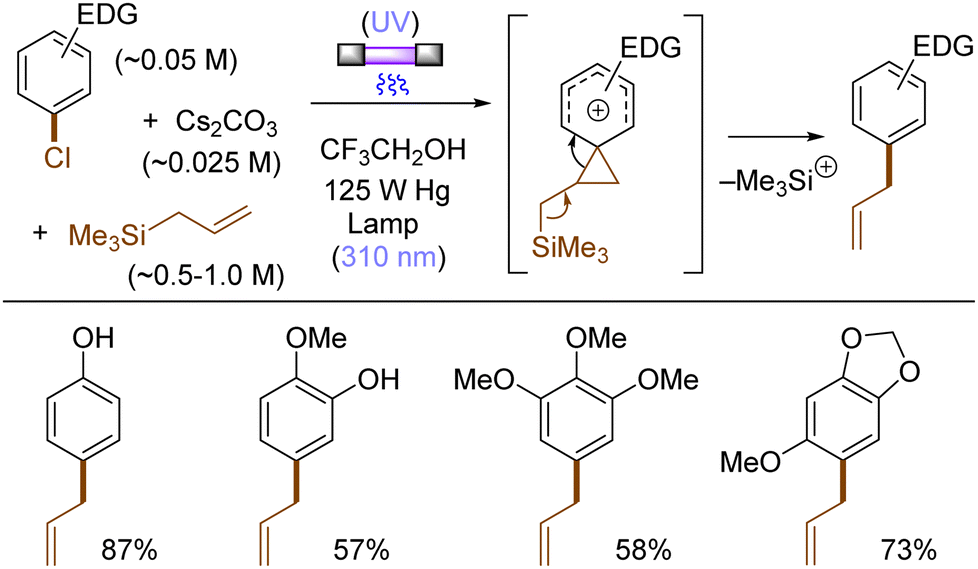
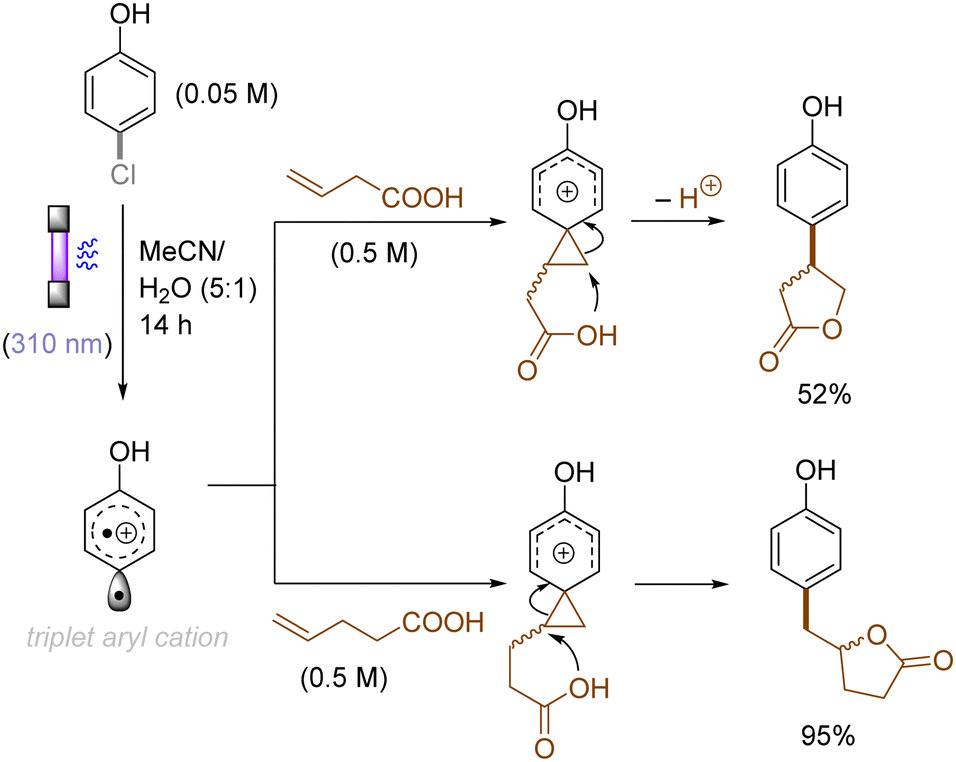
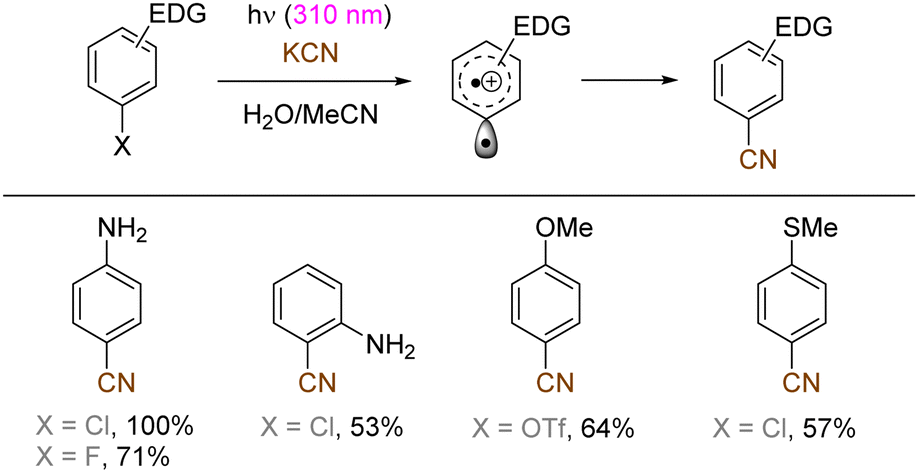
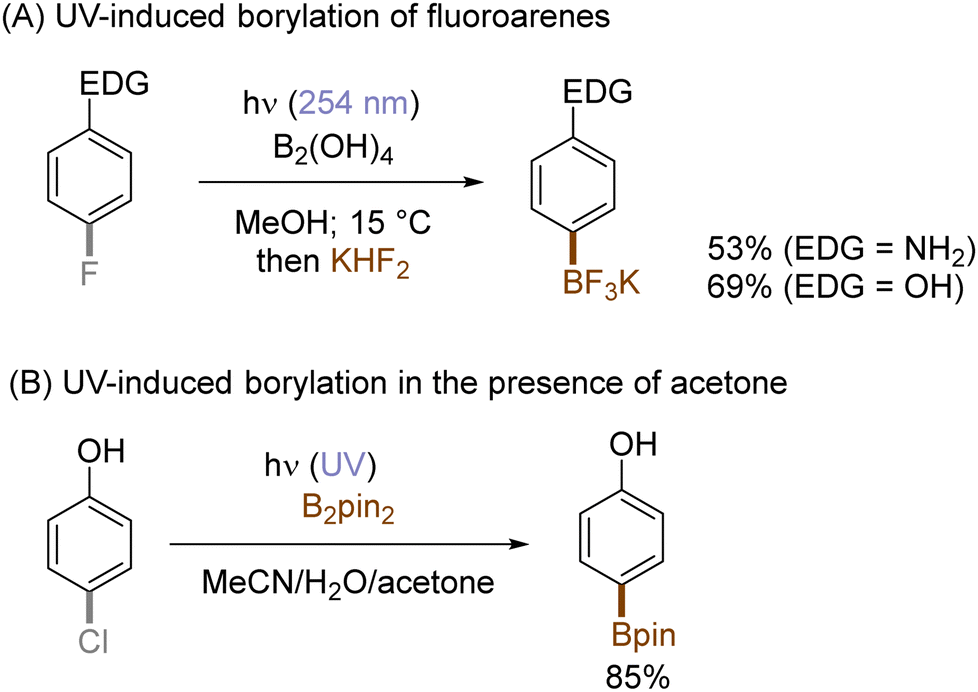
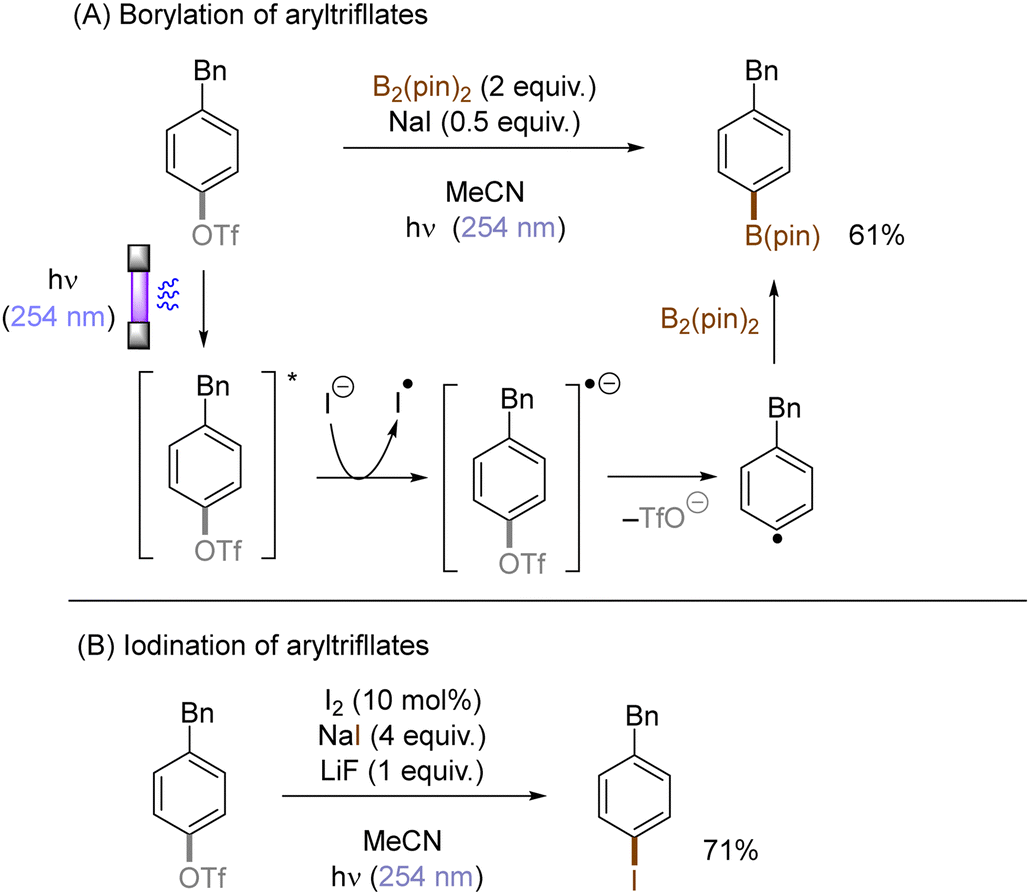
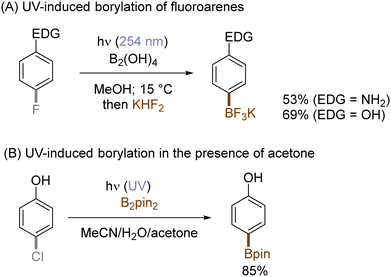
The use of allyltrimethylsilane as a nucleophile induced allylation via elimination of trimethylsilylcation from the closed-shell phenonium cation intermediate (Scheme 5).37 When alkenoic acids were employed as nucleophiles, intramolecular lactonization of the closed-shell phenonium ion intermediates took place to afford lactones (Scheme 6).29,38 The length of the carbon tether of the alkenoic acids governs the mode of lactonization. When the UV light photoinduced Ar–X bond heterolysis of electron-rich aryl (pseudo)halides was undertaken in non-nucleophilic trifluoroethanol as a solvent, terminal alkynes or internal silyl alkynes could be employed as nucleophiles to trap the generated triplet aryl cations. Subsequent deprotonation or desilylation from the closed-shell phenonium ion intermediates liberates arylalkynes (Scheme 7).39 Among σ-nucleophiles, a cyanide is capable of intercepting the triplet aryl cations to afford electron-rich benzonitriles (Scheme 8).40 Larionov developed a protocol for photoinduced borylation of electron-rich aryl fluoride with tetrahydroxydiborone under irradiation with UV light (254 nm) in a polar protic solvent (Scheme 9A).41 On the other hand, Li developed another protocol for borylation of electron-rich aryl fluorides/chlorides/mesylates/phosphates in the presence of acetone as a triplet state UV photosensitizer (Scheme 9B).42
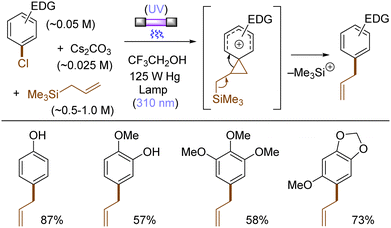 |
| | Scheme 5 Allylation of the photogenerated triplet aryl cations. | |
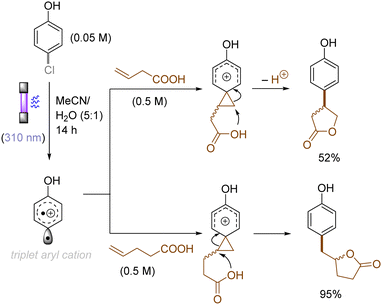 |
| | Scheme 6 Photogenerated triplet aryl cation reactions with alkenoic acids: lactonization. | |
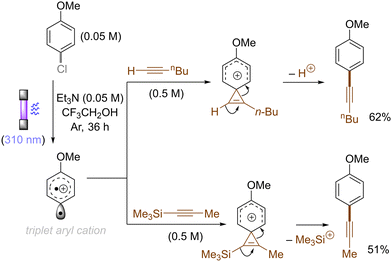 |
| | Scheme 7 Alkynylation of the photogenerated triplet aryl cations. | |
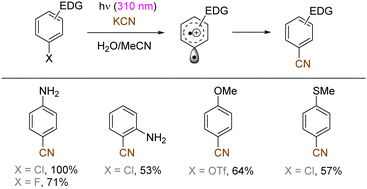 |
| | Scheme 8 Cyanation of the photogenerated triplet aryl cations. | |
 |
| | Scheme 9 Borylation of the photogenerated triplet aryl cation. | |
2.2. Ar–X activation via homolysis
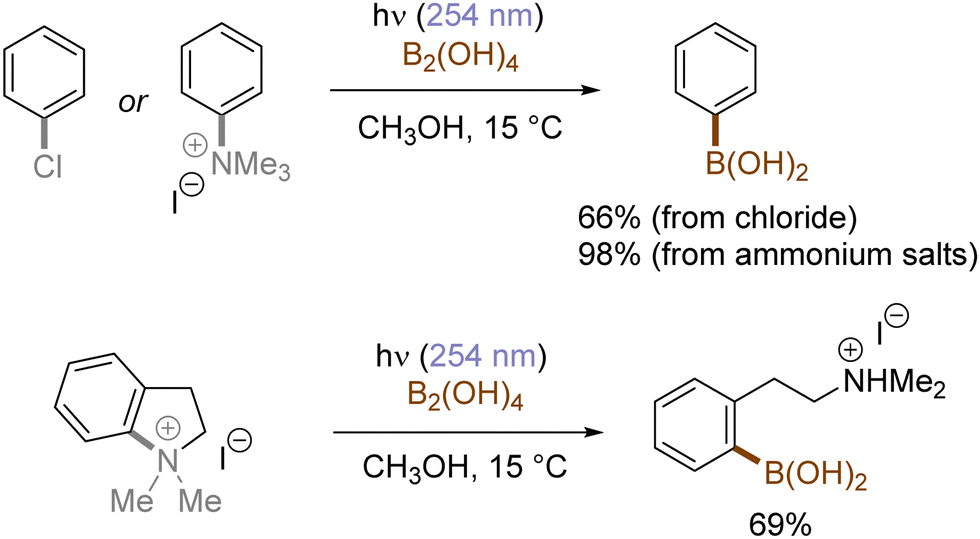
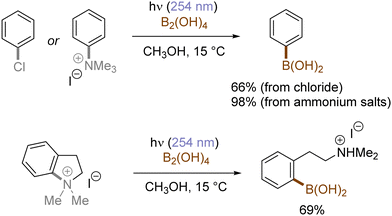
In contrast with the heterolysis of Ar–X bonds being typically observed when photoexcited aryl (pseudo)halides have EDGs, Ar–X bond homolysis usually occurs when aryl (pseudo)halides without EDGs are photoexcited.43 The resulting aryl radicals undergo downstream radical processes under various reaction settings. For example, the Larionov group developed a concise protocol for borylation of aryl (pseudo)halides with tetrahydroxydiboron under irradiation with UV light (Scheme 10). Chloroarenes and quaternary arylammonium salts serve as effective precursors for the borylation upon their photoinduced homolysis.41,44
 |
| | Scheme 10 Photoinduced borylation of aryl chlorides and quaternary arylammonium salts. | |
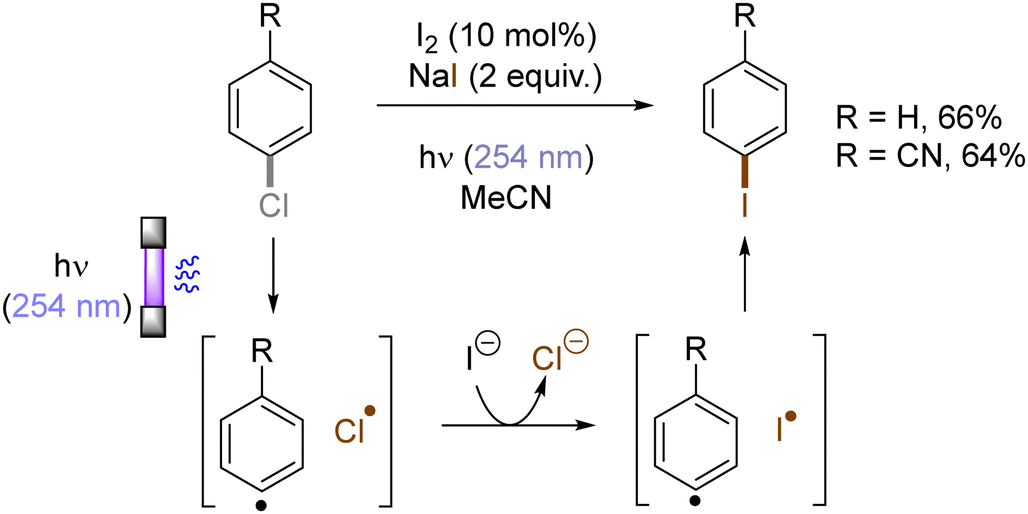
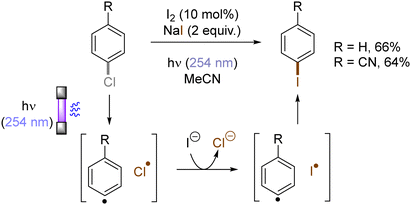
Halogen radicals generated through photoinduced homolysis of aryl halides can serve as SET oxidants. The Li group developed photoinduced iodination of aryl bromides or chlorides in the presence of sodium iodide (NaI, 2 equiv.) and molecular iodine (10 mol%) under irradiation with UV light in acetonitrile, providing more photochemically-susceptible aryl iodides (Scheme 11). A bromine/chlorine radical oxidizes an iodide ion to an iodine radical [E0(I˙/I−) = +0.99 V vs. SCE, E0(Br˙/Br−) = +1.48 V; E0(Cl˙/Cl−) = +1.85 V vs. (SCE)],45 which is intercepted by the simultaneously formed aryl radical. The presence of molecular iodine is helpful to prevent hydrodehalogenation via hydrogen atom transfer (HAT) to aryl radicals from the solvent.46
 |
| | Scheme 11 Photoinduced iodination of the aryl chlorides. | |
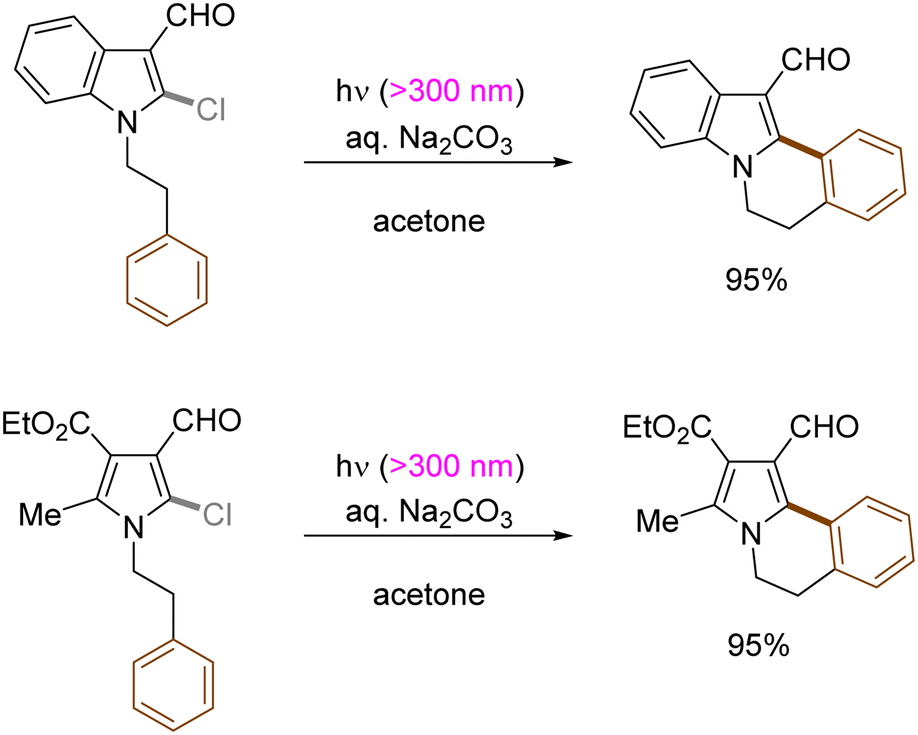
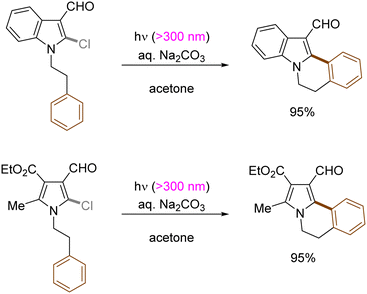
Generation of indolyl/pyrrolyl radicals via photoinduced homolysis of their precursor C(sp2)–chlorides followed by their radical cyclization to an arylalkane tether on the nitrogen was demonstrated by Zhang (Scheme 12).47 The processes were found to be more efficient when conducted in acetone, indicating that photoexcited triplet state of acetone may initiate the generation of photoexcited arenes via a triplet–triplet sensitization.
 |
| | Scheme 12 Construction of indole[2,1-α]isoquinoline derivatives by photoinduced C(sp2)–X bond cleavage. | |
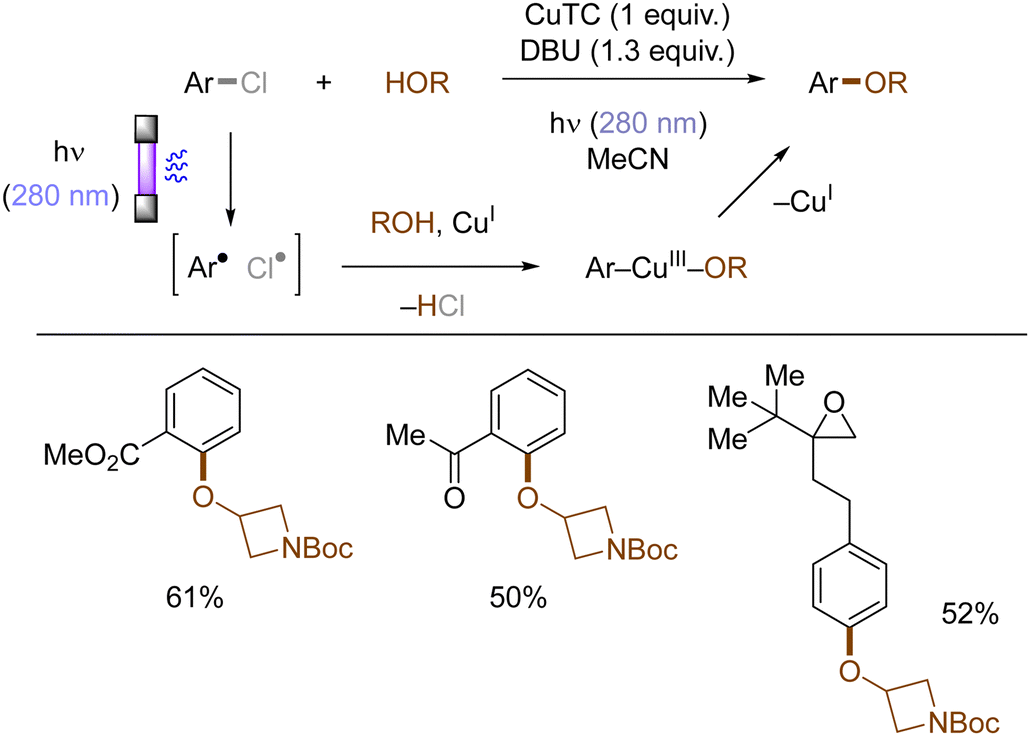
Copper-catalyzed Ullman-type reactions are well-known and attractive approaches to construct aryl ethers from aryl halides and alcohols. However, harsh reaction conditions such as high reaction temperatures are oftentimes required due to high energy barrier for the oxidative addition of aryl halides to copper complexes, which especially hinders the use of less reactive (yet more abundant) aryl chlorides. Ritter reported that photoexcited triplet states of aryl halides facilitates their copper-mediated transformations to aryl ethers, using various aryl halides including aryl chlorides and alcohols without tailored external ligands at ambient conditions (rt) (Scheme 13).48 Upon photoinduced homolysis of Ar–Cl bond, the resulting aryl radical and chlorine radical undergo successive SET oxidative addition to a Cu(I) complex. Ligand exchange between a halide and an alcohol substrate results in the formation of an aryl Cu(III) alkoxide, from which the aryl ether is reductively eliminated as the final product.
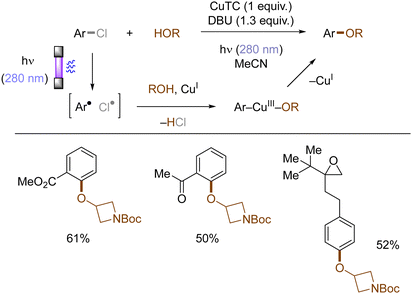 |
| | Scheme 13 Photoinduced Cu(I)-mediated etherification of aryl chlorides. | |
2.3. Ar–X activation via SET to, or from, photoexcited aryl (pseudo)halides
Photoexcited states commonly exhibit enhanced reactivity over their ground states, either as electron donors or electron acceptors. This feature can be adopted in photoinduced functionalization of inert aryl (pseudo)halides. For example, Li developed a concise protocol for photoinduced functional group interconversions of aryl triflates into arylboronates or aryl iodides in the presence of sodium iodide (NaI). SET reduction of the photoexcited aryl triflate by an iodide ion induced mesolysis of the aryl triflate radical anion's C(sp2)–O bond to form aryl radicals with elimination of sodium triflate (NaOTf) (Scheme 14). The resulting aryl radicals were efficiently trapped by iodine or B2(pin)2.49 In a related manner, the same group reported a UV-light promoted cross-coupling of aryl fluorides with diethyl phosphite in the presence of NaH. The reaction proceeded through a reductive quenching of photoexcited fluoroarenes by in situ generated diethyl phosphite anion followed by a radical-radical coupling.50
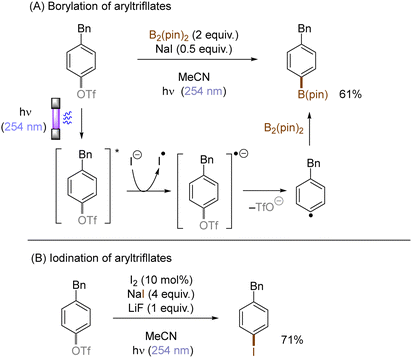 |
| | Scheme 14 Functionalization of the aryl triflates through reduction quenching. | |
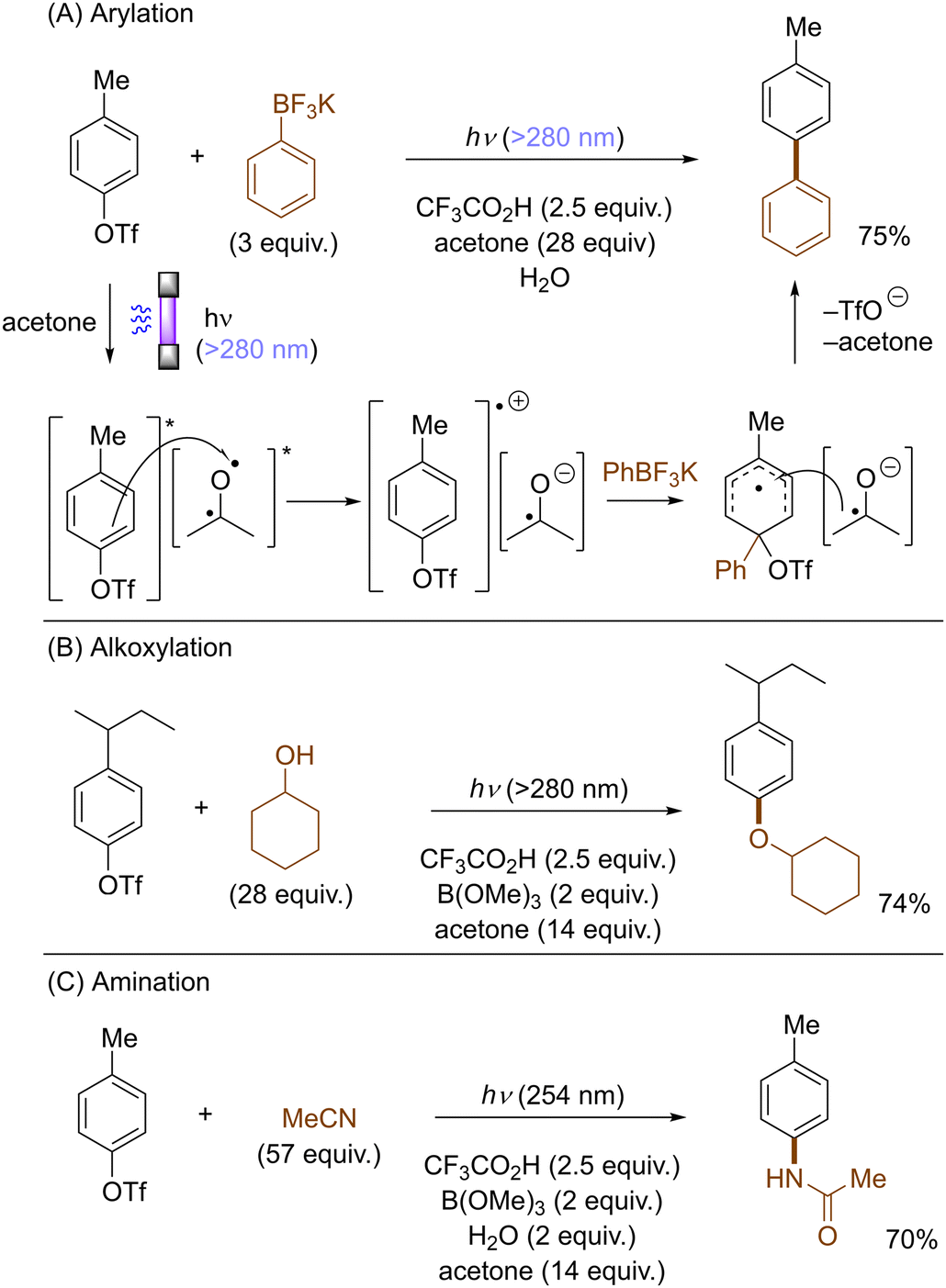
Li also disclosed photoinduced transition-metal free cross-coupling of aryltriflates with potassium aryltrifluoroborates in the presence of acetone (Scheme 15A).51 In this case, photoexcited aryltriflates serve as SET donors to simultaneously photoexcited acetone to generate the corresponding arene radical cations. Their interception by aryltrifluoroborates and rearomatization via SET reduction from the ketyl radical anion and elimination of triflate furnishes the biaryl products. The method also engaged aliphatic alcohols and nitriles as the nucleophile, offering an expedient access to aryl ethers and N-arylamides, respectively (Scheme 15B and C).
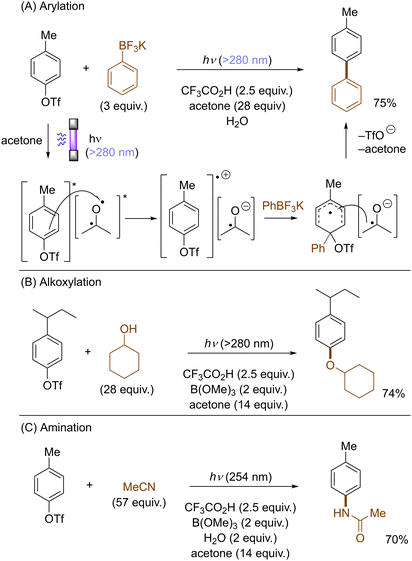 |
| | Scheme 15 Photoinduced functionalizations of aryl triflates through SET oxidation. | |
2.4. Ar–X activation via single-electron-transfer for the selective reduction of arenes
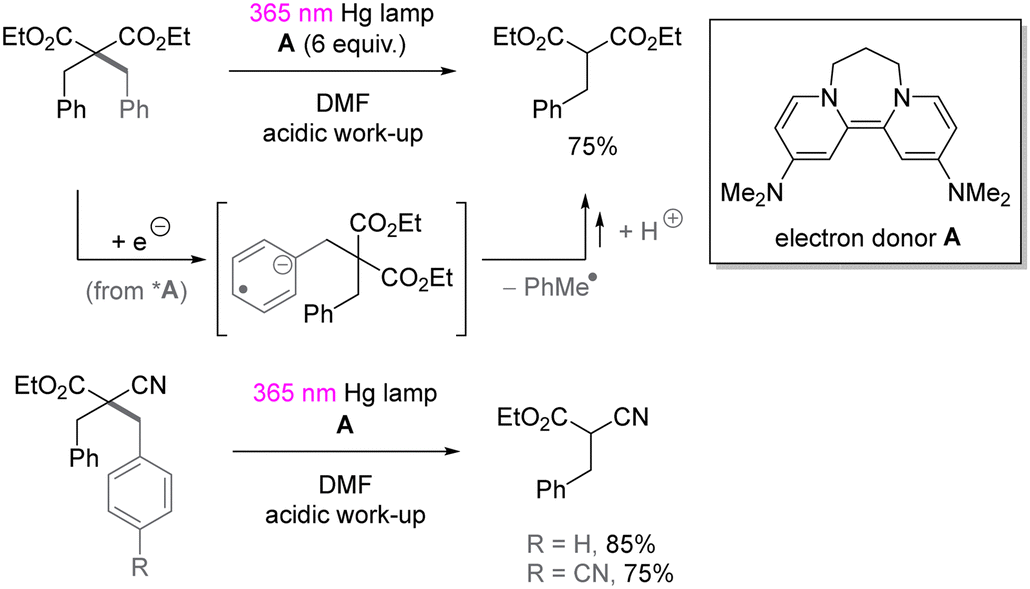
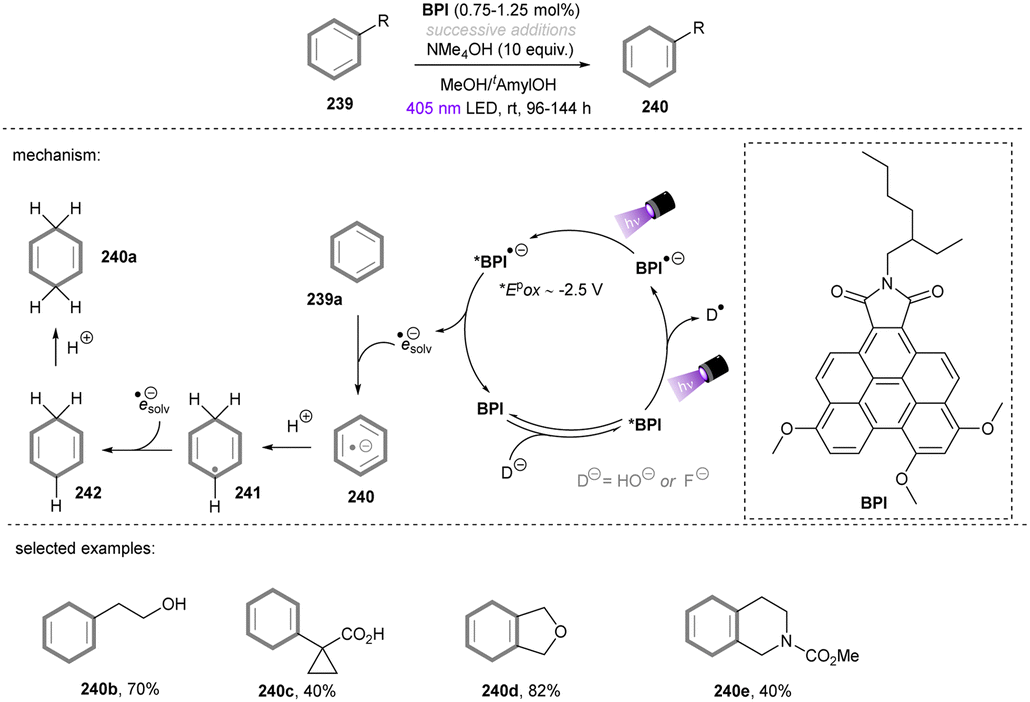
UV-light induced SET reduction is not confined to the functionalization of aryl halides. The Murphy group reported selective reduction of less activated arenes in the presence of alkoxycarbonyl moiety,52 even though the reduction potential of benzene is −3.66 V vs. SCE53 and more negative than the reduction potential of aliphatic esters (ca. −3.0 V vs. SCE).54 The selectivity benefits from donor–acceptor π-stacking interaction between photoexcited organic electron donor A and benzene. As shown in Scheme 16, the selective reduction of benzyl substituted malonate ester as well as cyanoacetate provided the corresponding benzyl-C bond cleavage products in good yields.
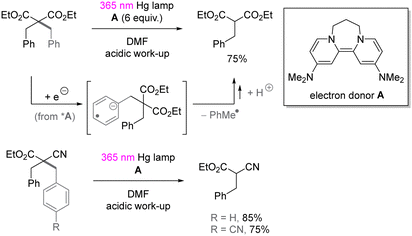 |
| | Scheme 16 Reduction of benzyl malonate and cyanoacetate by organic electron donor. | |
3. Photoredox catalysis
While UV photoactivation provides a powerful platform for the activation of inert substrates for strong bond cleavages, the use of deep UV (<300 nm) light (the cost/lack of selectivity/Norrish-type cleavages etc. resulting from direct photoexcitation, safety profile of reactions with deep UV light <300 nm) is prohibitive and does not align well with the principles of green chemistry. Compared to pressurized Hg bulbs with several hundred watts of power, the use of more energy-efficient longer-wavelength light emitting diodes (LEDs) is desirable and safer. However, visible light photons often do not possess enough energy for the direct activation of such unactivated target substrates mentioned in the introduction. The use of a photocatalyst allows one to bathochromically shift the excitation wavelengths to near-UV or visible light as a neat compromise, still engaging relatively unactivated substrates yet with improved energy efficiency.
This section will focus on photoredox catalysed activation of strong C–X bonds or inert π-systems. Highly reducing or oxidizing species generated through single photonic or multiphotonic paradigms will be summarized. Except for photo-absorbing catalyst variants, also included are other in situ generated redox active species such as electron donor–acceptor complexes, solvated electrons and CO2˙−. Unactivated substrate classes include electronically different aryl or alkyl chlorides/fluorides, ethers, esters, simple arenes and olefins. Due to the enormous body of literature on organic C–Cl reduction, those catalytic systems which cannot engage unactivated organic chlorides – such as unsubstituted chlorobenzene and electron-rich 4-chloroanisole – are generally not reviewed. Reductions of difluoroolefins and polyfluoroarenes55–58 (hexafluorobenzene, pentafluorobenzonitrile etc.) will not be presented herein because radical addition/elimination mechanisms cannot be completely ruled out.
3.1. Single-photon processes
3.1.1. Ar–X (X = F, Cl, O) bond cleavage.
The activation of simple unactivated arenes is highly challenging due to their elusive redox potentials (Ered < −2 V, Eox > 2 V vs. SCE) which exceed the redox potential window of conventional noble metal-based PCs such as Ru(bpy)3(PF6)2 (*E1/2 = −0.81 V vs. SCE) and fac-Ir(ppy)3 (*E1/2 = −1.73 V vs. SCE). Organic photoreductants, generally (but not always) based on an electron-rich anilinic, phenolic or multiply N-substituted olefinic cores, have attracted attention in recent years as alternatives. Among these, catalysts based on a triarylamine moiety such as phenothiazines (e.g. N-phenyl phenothiazine ‘PTH’; *E1/2 = −2.10 V vs. SCE59) and phenoxazines (e.g. those reported by the Miyake group; *E1/2 = −1.70 V vs. SCE60) have emerged as cheaper alternatives to transition metal-based photoredox catalysts, boasting even more potent reducing excited states that allowed their applications to a diverse set of PRC transformations. However, their photocatalytic uses are still limited to the activations of organic bromides, iodides and highly activated aryl chlorides. This challenging problem has been addressed in pace with the development of new PCs and activating strategies. This section will focus on the activation of inert organic compounds through one-photon PRC. Taking Ar–X (X = F, Cl, O) as an example, it can either be reduced or oxidized by the photoexcited PC (Scheme 17). Ar–X radical anion generated by SET reduction affords an aryl radical through C–X mesolysis. Ar–X radical cation generated by SET oxidation, by contrast, undergoes a nucleophilic aromatic substitution (SNAr) to deliver a C–X functionalized compound.
 |
| | Scheme 17 General, divergent mechanisms for single photonic photoredox catalyzed activations of Ar–X bonds, depending on the mode of SET activation. | |
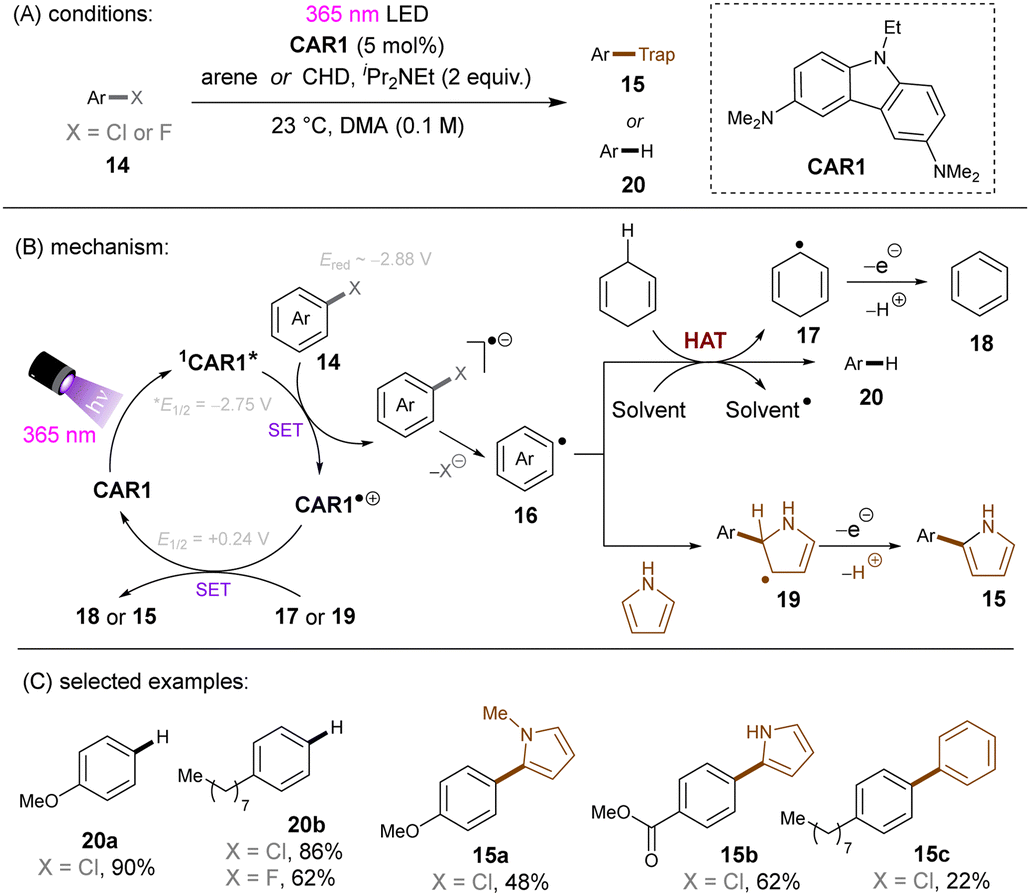
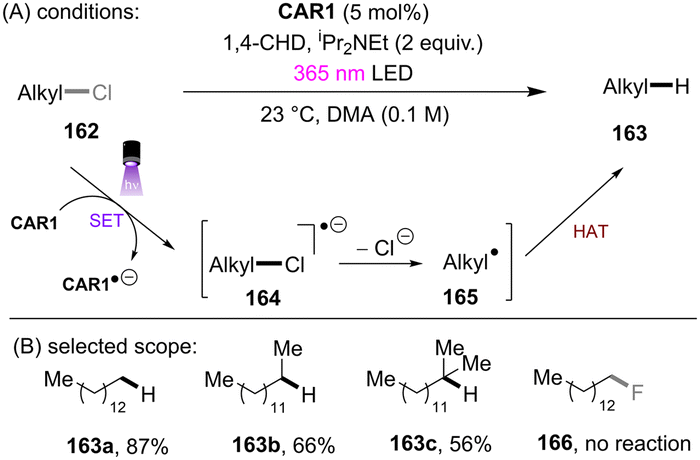
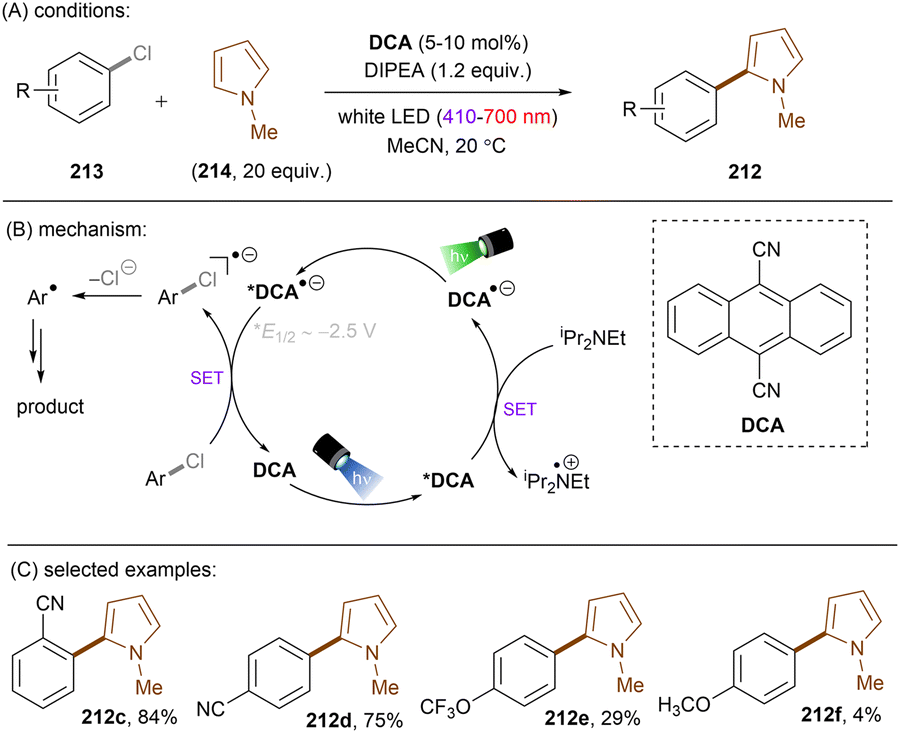
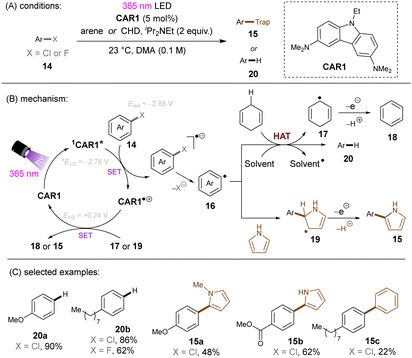
Matsubara and co-workers reported reductive functionalization of electronically different aryl chlorides under near-UV light.61 After a detailed examination of different carbazole-type catalysts by a combination of DFT and cyclic voltammetry (CV) analyses, the authors concluded that CAR1 (Scheme 18) was an ideal organophotocatalyst (orgPC) for reduction of organohalides. Its excited state redox potential was calculated to be E1/2(*CAR1/CAR1˙+) = −2.75 V vs. SCE, which is comparable to the reduction potential of chlorobenzene (Ered = −2.88 V vs. SCE). With CAR1 as the photocatalyst, hydrodechlorination of aryl chlorides with either EWGs or even EDGs proceeded smoothly in good to excellent yields. Furthermore, there was even one example for the reduction of an aryl fluoride, albeit only for hydrodefluorination. Instead of hydrodechlorination, it was also possible to couple aryl chlorides with (hetero)arenes. Electron-poor aryl chlorides worked best with moderate yields, whereas electron-rich aryl chlorides gave poor to moderate yields. The authors proposed a mechanism whereby CAR1 is photoexcited and SET-reduces aryl chloride 14 to give, after loss of chloride, aryl radical 16. Aryl radical 16 can either (i) add to a (hetero)arene to give radical 19, where upon subsequent SET oxidation and deprotonation give product 15, or (ii) undergo HAT with 1,4-cyclohexadiene (CHD) or solvent to furnish product 20. Ground state CAR1 is regenerated by electron transfer from either 17 or 19.
 |
| | Scheme 18 Carbazole-based (CAR) orgPC as a potent photoreductant for reductive activation of aryl chlorides and fluorides. | |
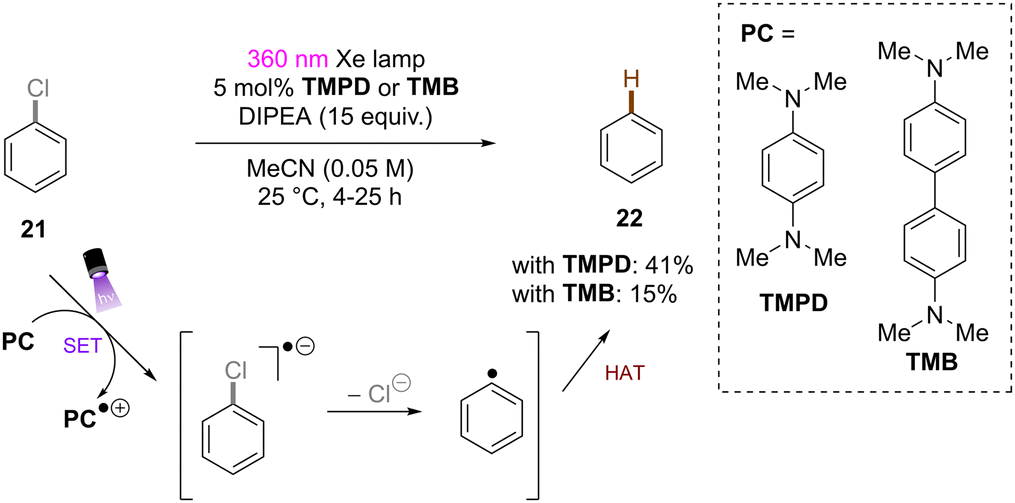
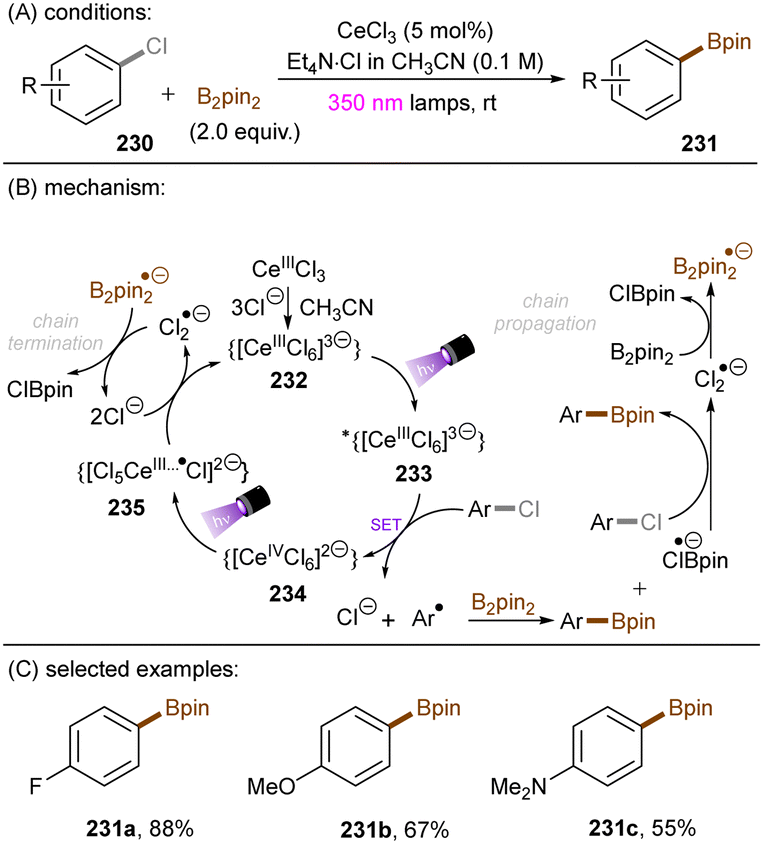
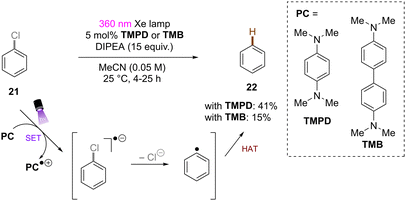
Song and co-workers reported reduction of chlorobenzene based on simpler diamine sensitizers as electron-transfer catalysts (Scheme 19).62N,N,N′,N′-Tetramethyl-p-phenyl-enediamine (TMPD) or N,N,N′,N′-tetramethyl-benzimide (TMB) show S0–S1 absorption up to 380 nm and are strongly reducing excited singlet states. With deep excited state potentials for reduction *Eox(*TMPD/TMPD˙+) = −3.56 V vs. SCE and Eox(*TMB/TMB˙+) = −3.48 V vs. SCE both catalysts are strongly reducing and can therefore be used for the hydrodehalogenation of chlorobenzene despite low process efficiency and high loadings of sacrificial amine electron donors.
 |
| | Scheme 19 Hydrodechlorinatiation of chlorobenzene using TMPD or TMB as an orgPC. | |
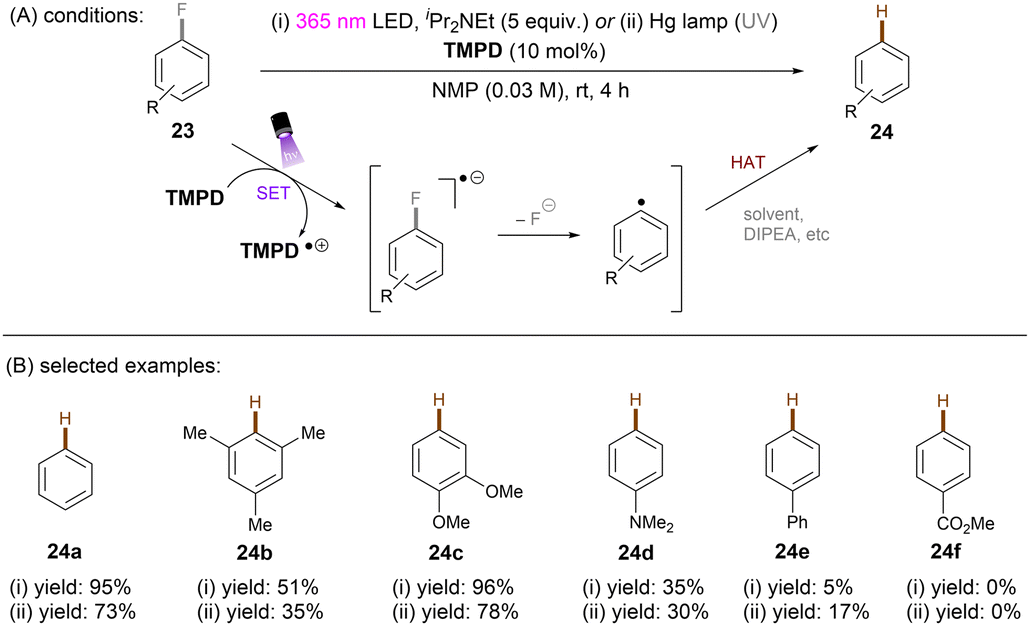
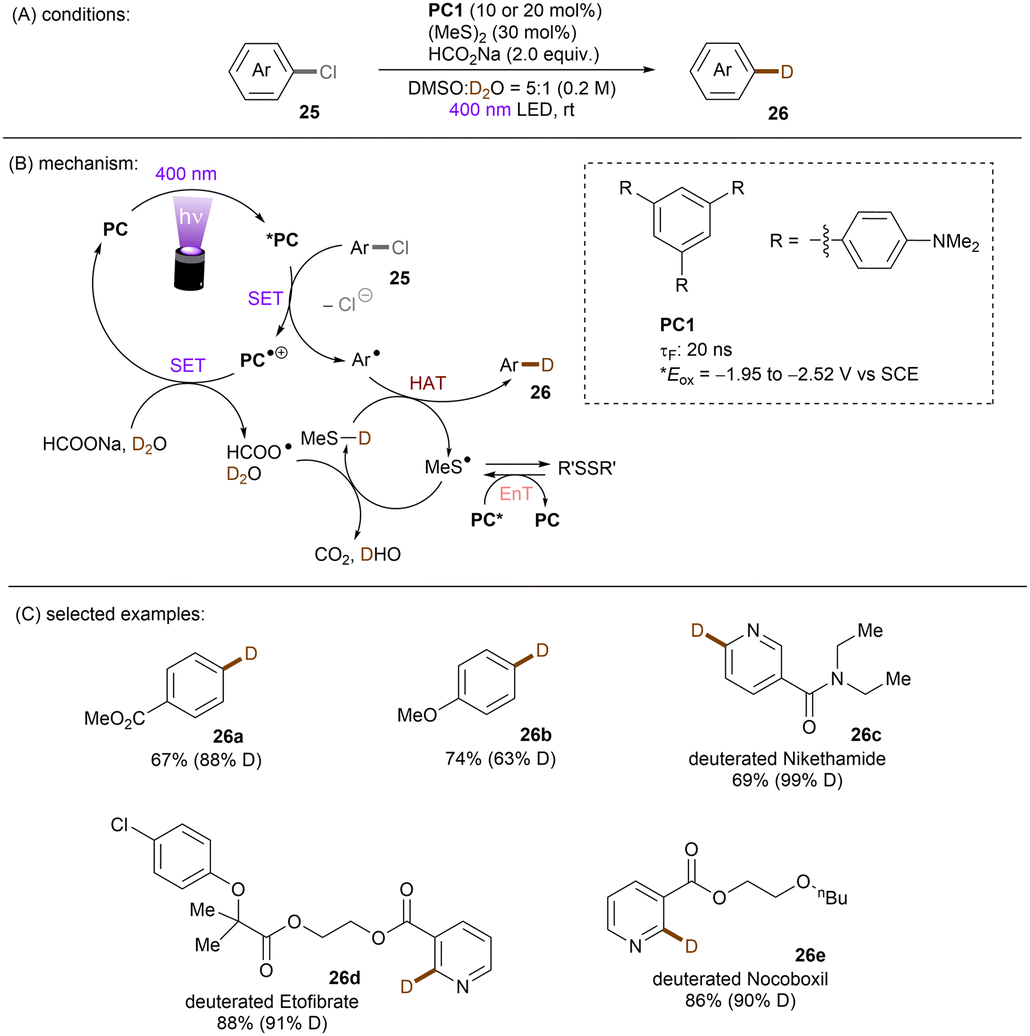
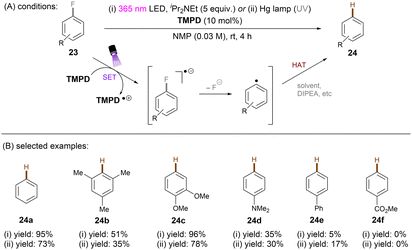
One of the challenging reactions is the activation of C–F bonds in aryl fluorides that are of high thermodynamic stability and kinetic inertness due to the small size (rF = 1.47 Å) and electronegativity of the fluorine atom.63,64 Simultaneous to Song's work on chlorobenzene reduction,62TMPD was employed by Iwasawa and co-workers to effect a hydrodefluorination of monofluoroarenes (Scheme 20).65 Reduction of inert fluorobenzene (Epred = −2.97 V vs. SCE) and methoxy substituted fluoroarenes worked in excellent yields. The sterically-demanding substrate 2,4,6-trimethylfluorobenzene worked in moderate yield. However, phenyl substituted substrates gave poor yields and monofluoroarenes with an electron withdrawing group gave no product. Therefore, elimination of fluoride from electron-deficient monofluoroarene radical anions might be slower than back electron transfer (BET). In the same year, Gong and co-workers published an impressive method for deuterodehalogenation of aryl chlorides using a trianiline-based orgPC, PC1, under irradiation of 400 nm light.66 (Scheme 21). This new catalyst has a long excited state lifetime of 10 ns (presumably in its singlet state) and is strongly reducing E1/2(*PC1/PC1˙+) = −1.95 to −2.52 V vs. SCE, making it a suitable candidate for cleavage of the strong C(sp2)–Cl or C(sp3)–Cl bonds (Ered = −1.8 to −3.0 V). With (MeS)2 and HCO2Na as additives the authors were able to achieve deuterodehalogenation for electron-rich chloroarenes. More importantly, this catalytic system was compatible with a wide variety of electron-poor (hetero)aryl chlorides, representing an important advantage over the aforementioned TMPD catalyst.65 Especially, 4-chloroanisole was compatible despite a moderate deuterium incorporation of 63%. Moreover, selective monodeuteration of polychlorinated products could be achieved while longer reaction times and heating did not lead to polydeuteration. Furthermore, this method offers a powerful tool for the synthesis of deuterated drugs. For example, Etofibrate bearing a deuterium at the ortho-position of the pyridine ring was easily synthesized through a selective pyridinyl C–Cl bond deuteration with the aryl C–Cl bond remaining intact. The authors proposed dual roles for the PC; which serves as both (i) a potent photoreductant for SET reduction of organic chlorides and (ii) an energy transfer catalyst for homolysis of dimethyl disulfide to yield the thiyl radical MeS˙, which leads to deuterium atom transfer in the presence of HCOONa and D2O.
 |
| | Scheme 20 Reduction of aryl fluorides with TMPD as PC under UV-light irradiation reaction conditions. | |
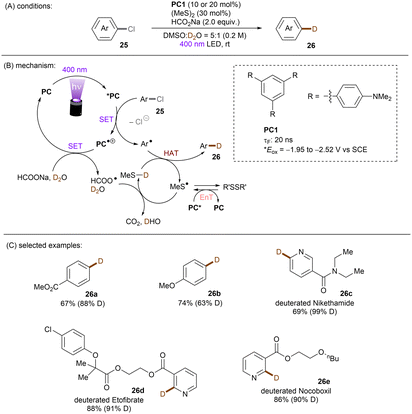 |
| | Scheme 21 Deutero-dehalogenation of organic chlorides using amine-based orgPCs. | |
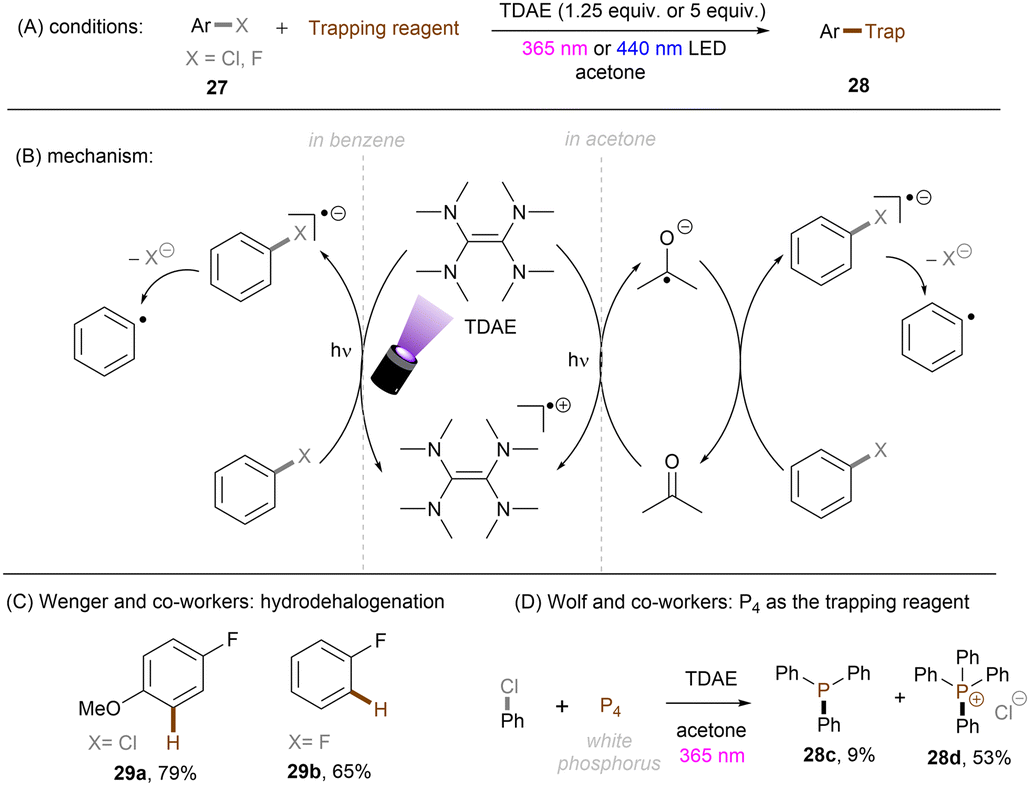
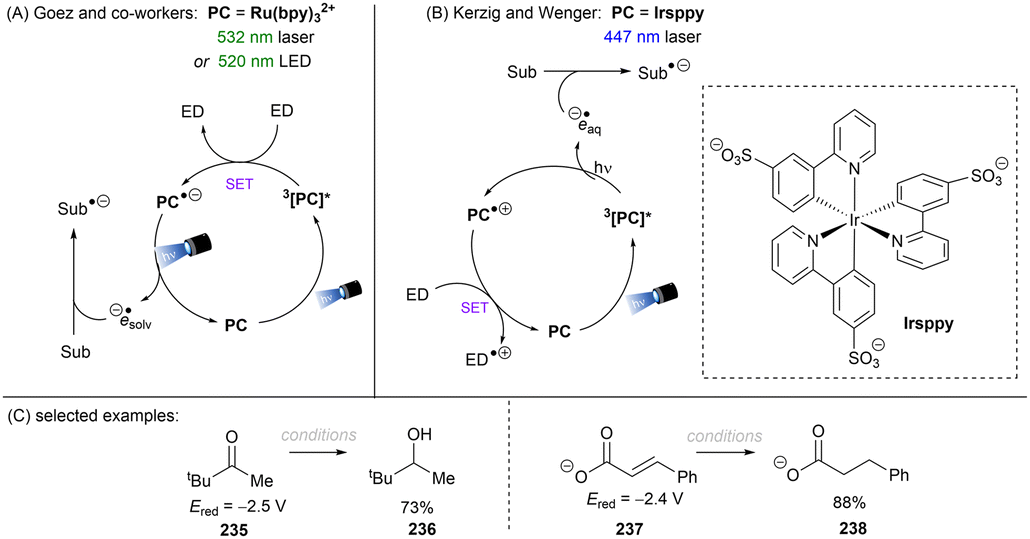
The Wenger group demonstrated the ability of a tetramine, tetrakis(dimethylamino)ethylene (TDAE), to reduce organic halides including aryl fluorides and alkyl chlorides under visible light irradiation (Scheme 22A).67 Stoichiometric amounts of TDAE were used, functioning both as a photosensitizer and electron donor for this reaction. Attempts to use a catalytic amount of TDAE in the presence of other external reductants were unsuccessful as the in situ generated TDAE˙+ cannot be reduced by these reductants. It was found that substrates were reduced by the excited state *TDAE (Epox = −3.4 V vs. SCE) in benzene. In contrast, the reaction mechanism varies when acetone is used as the solvent. The authors claimed that *TDAE reduces acetone (Ered = −2.84 V vs. SCE) rather than the substrates owing to the difference in concentrations. The generated acetone radical anion is a potent enough reductant to engage Ar–X (X = Cl, F). More recently, the Wolf group reported on the photochemical synthesis of arylphosphines and phosphonium salts by leveraging the same aryl chloride activation strategy (Scheme 22D).68 The phenyl radical generated from SET reduction of chlorobenzene by the in situ formed acetone radical anion was intercepted by white phosphorus P4 to form triphenyl phosphine 28c and tetraphenylphosphonium 28d in 62% combined yield. In comparison, other solvents like acetonitrile, benzene or chlorobenzene all afforded diminished yields, demonstrating the important role of acetone as an electron shuttle.
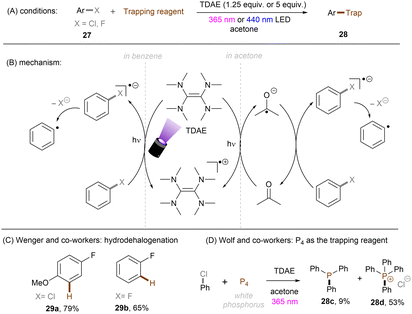 |
| | Scheme 22 Photoreduction of organic halides by in situ generated acetone radical anion. | |
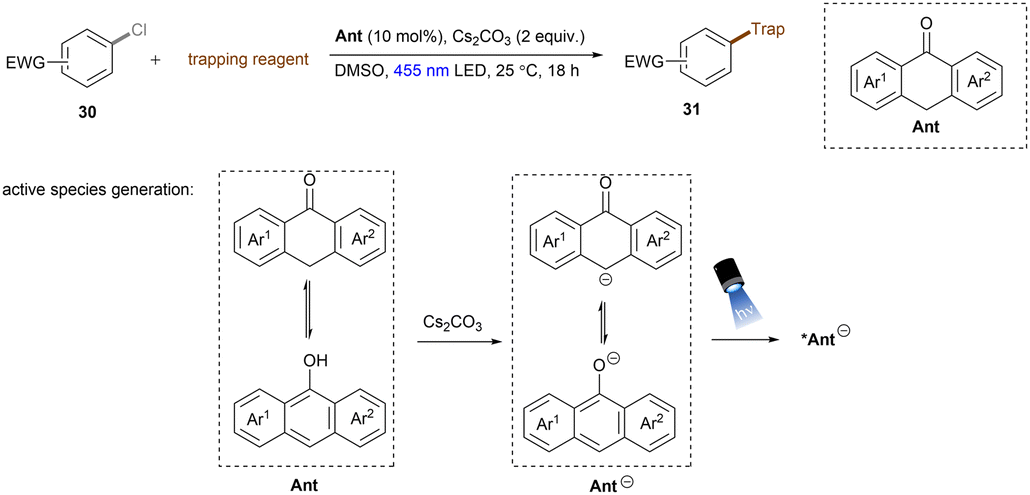
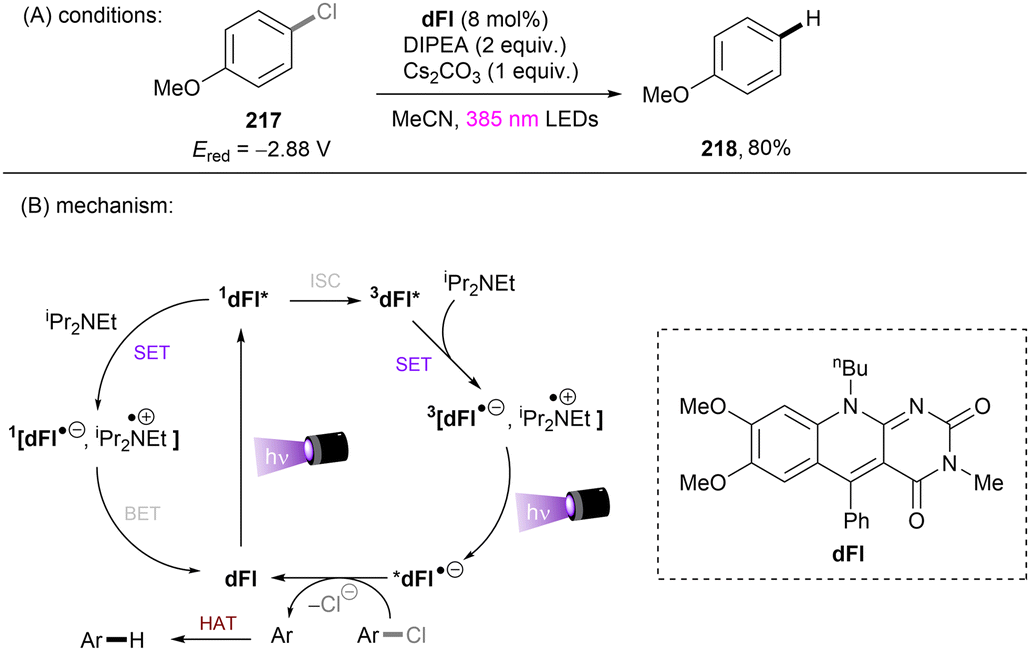
Anionic species are also used as PCs in organic transformations.69 Importantly, they are expected to have higher reducing ability than their corresponding neutral species and pack sufficient redox power for the activations of strong bonds in inert substrates under visible light irradiation. The strategy was extended to electron-poor aryl chloride activations by König and co-workers (Scheme 23).70 The active species, 9-anthrolate, is generated by deprotonation of corresponding anthracenones and is primed for photoexcitation. However, unactivated aryl chlorides remained untouched under these conditions.
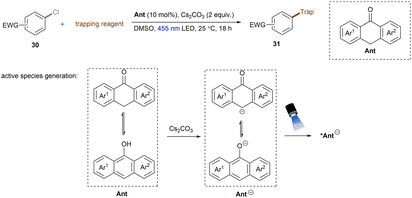 |
| | Scheme 23 Photoexcited anthrolate anion for the reduction of activated aryl chlorides. | |
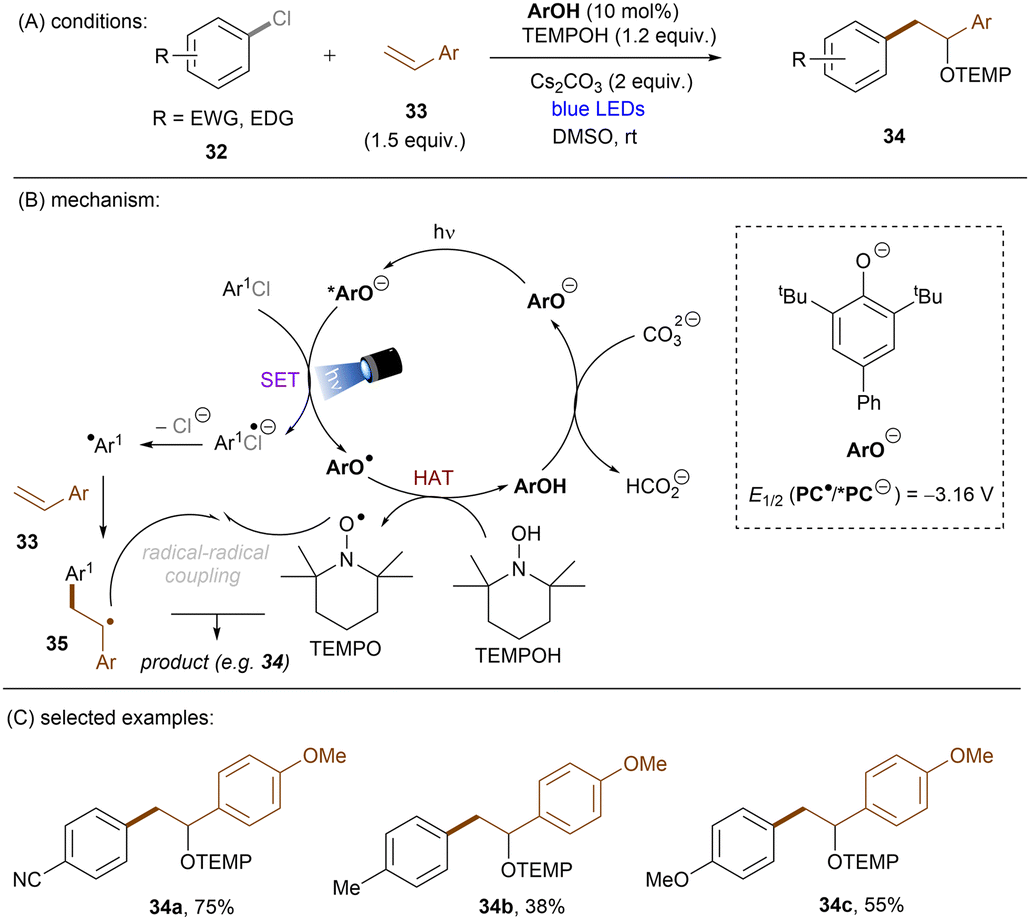
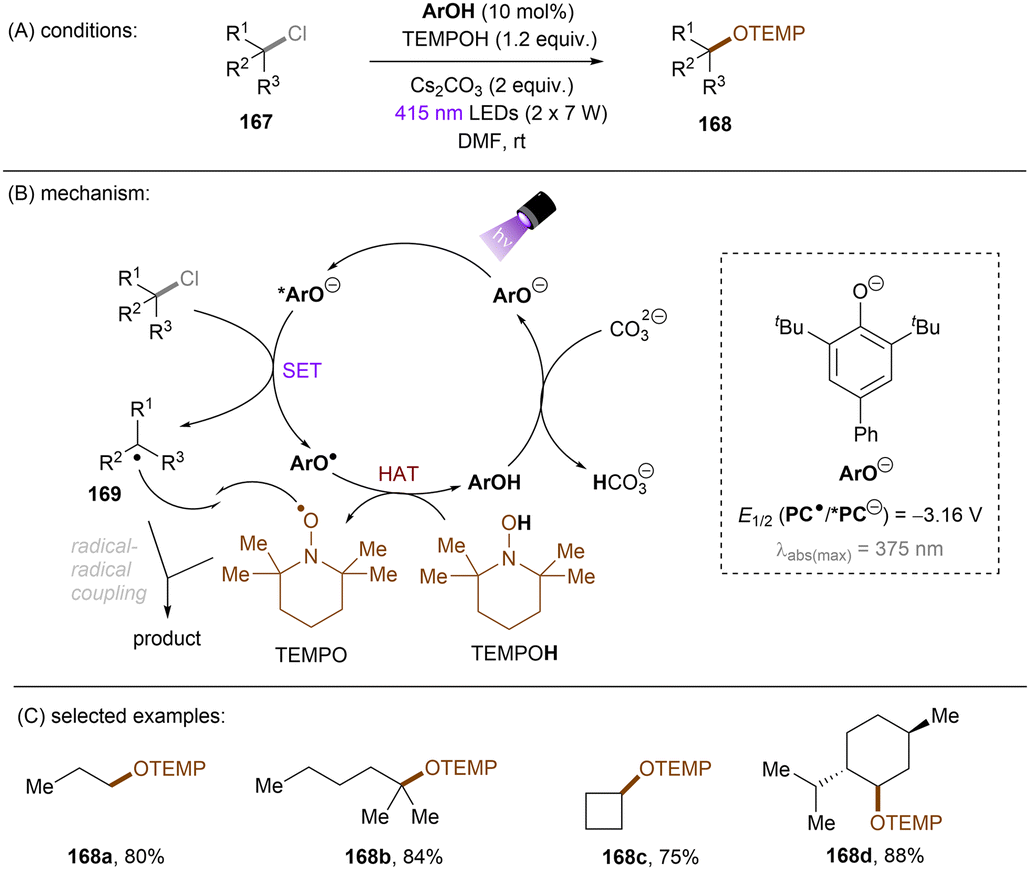
This challenging issue was taken on by Xia and co-workers who reported phenolate anions as visible light excitable species to achieve super reducing power. Inspired by their previous work on the ability of stoichiometric methyl 4-hydroxycinnamate to reduce activated aryl chlorides through deprotonation and subsequent photoexcitation,71 the authors screened a series of substituted phenols in order to find a general catalyst for the activations of more challenging substrates such as electron-rich aryl chlorides. 2,6-Di-tert-butyl-4-phenylphenol (ArOH) was found to be optimal for reducing aryl chlorides under visible light irradiation in the presence of Cs2CO3 (Scheme 24).72 Spectroscopic investigations were conducted to understand the reaction mechanism. While the neutral ArOH catalyst does not absorb visible light (>400 nm), the addition of Cs2CO3 generated a coloured species which showed the same visible light absorption as ArO−. Its spectra was not changed upon addition of aryl halide, this (i) clearly rules out the direct reduction of aryl chlorides by ground state ArO− and (ii) suggests against the formation of an EDA complex. Photoexcited *ArO− was calculated to have a deep redox potential for reduction of −3.16 V vs. SCE (E1/2(ArO˙/*ArO−)) and thus it is able to reduce chlorobenzene (Ered = −2.78 V) ultimately to its aryl radical, which is supported by further Stern–Volmer quenching experiments. The formed ArO˙ is capable to abstract a hydrogen atom from TEMPOH (BDEO–H = 67 kcal mol−1) to regenerate ArOH (BDEO–H = 75 kcal mol−1) catalyst and form a free radical, TEMPO. Meanwhile, the aryl radical generated by the SET reduction step adds to styrene and the adduct is then trapped by TEMPO to afford the final product. Although previous studies have shown how cation–π interactions of alkali metal cations (especially K+ and Cs+) with π-systems/aryl halides can influence reactivity,73–76 suggesting Cs2CO3 may not be innocent as only a base, Xia and co-workers’ reaction also worked with organic bases (DBU).
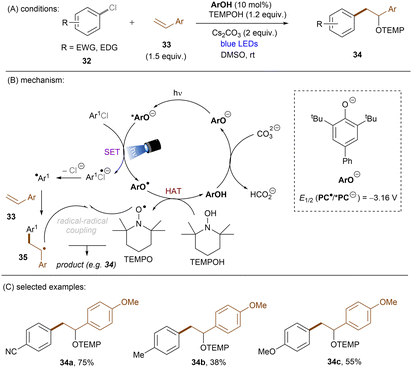 |
| | Scheme 24 Photoexcited phenolate for the reduction of unactivated aryl chlorides. | |
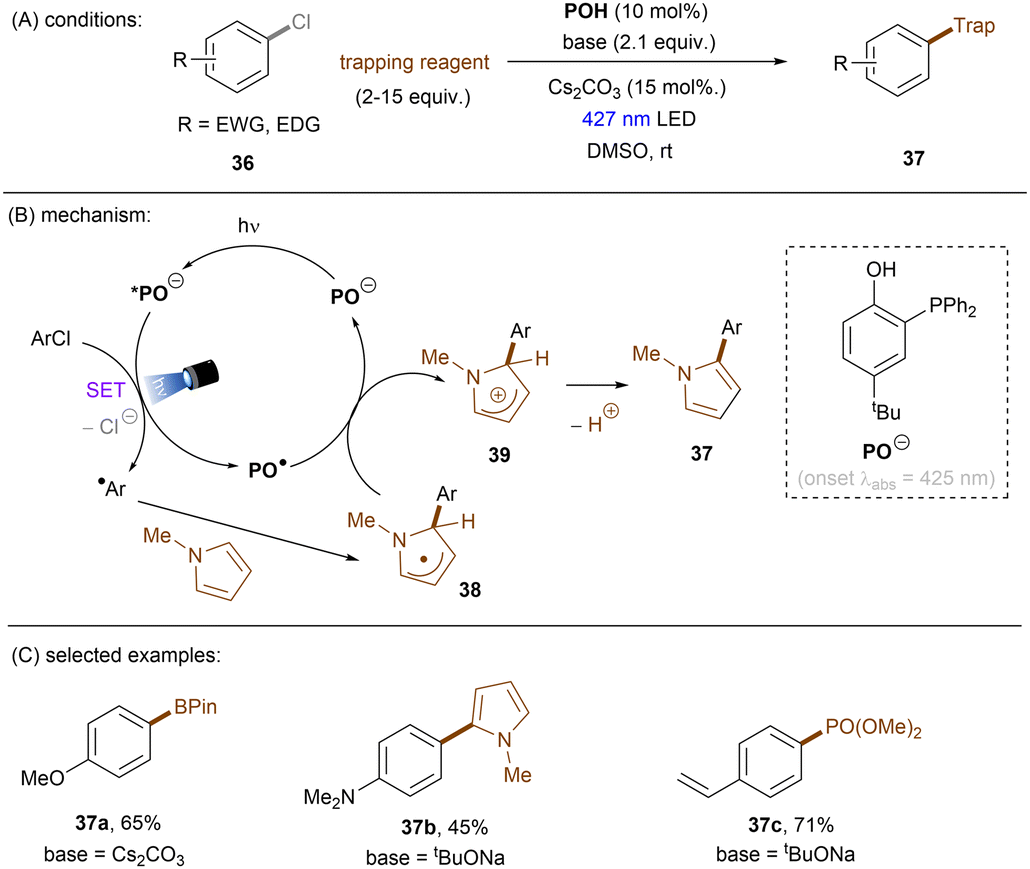
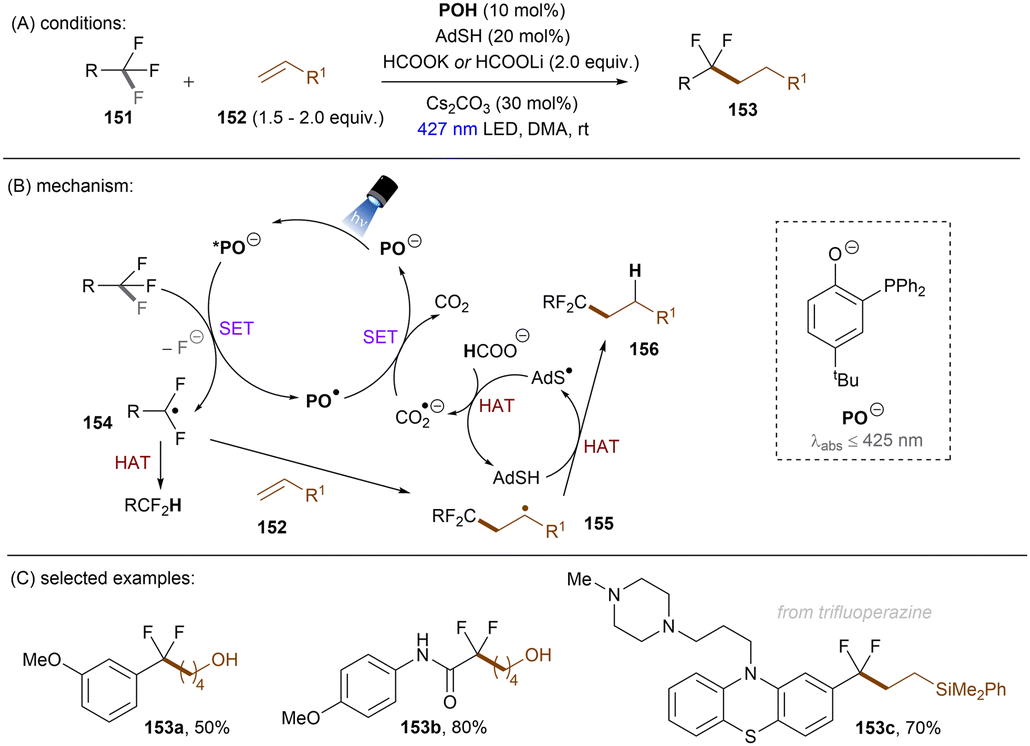
Significant progress has been made towards engaging unactivated aryl chlorides for TEMPO-free radical coupling reactions. Very recently, Guan, Shang and co-workers developed a new phenol catalyst POH encompassing an ortho-diphenylphosphanyl substituent for the activation of aryl chlorides 36 (Scheme 25).77 The generated aryl radicals coupled with B2Pin2, pyrrole and trimethylphosphite instead of TEMPO. Replacing the ortho-PPh2 group with a PCy2 group resulted in a dramatic decrease in product yield, suggesting the importance of the PPh2 moiety where the LUMO of POH is located, as indicated by DFT studies. While the neutral catalyst does not absorb visible light, its anionic species absorbs blue light (427 nm). The reaction exhibited a wide reaction scope in terms of both aryl chlorides 36 and trapping agents. Especially, aryl chlorides bearing strong EDGs such as OMe and NMe2 are amenable to the standard photocatalytic conditions, affording the coupling products (37a, 37b) in good yields.
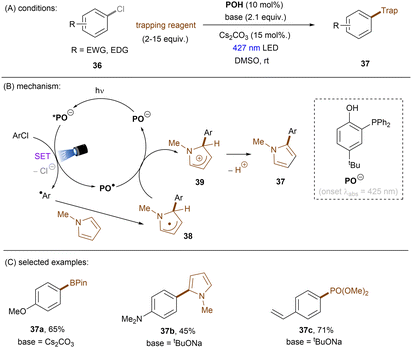 |
| | Scheme 25 Photoexcited deprotonated phosphanyl phenol (POH) for the reduction of unactivated aryl chlorides. | |
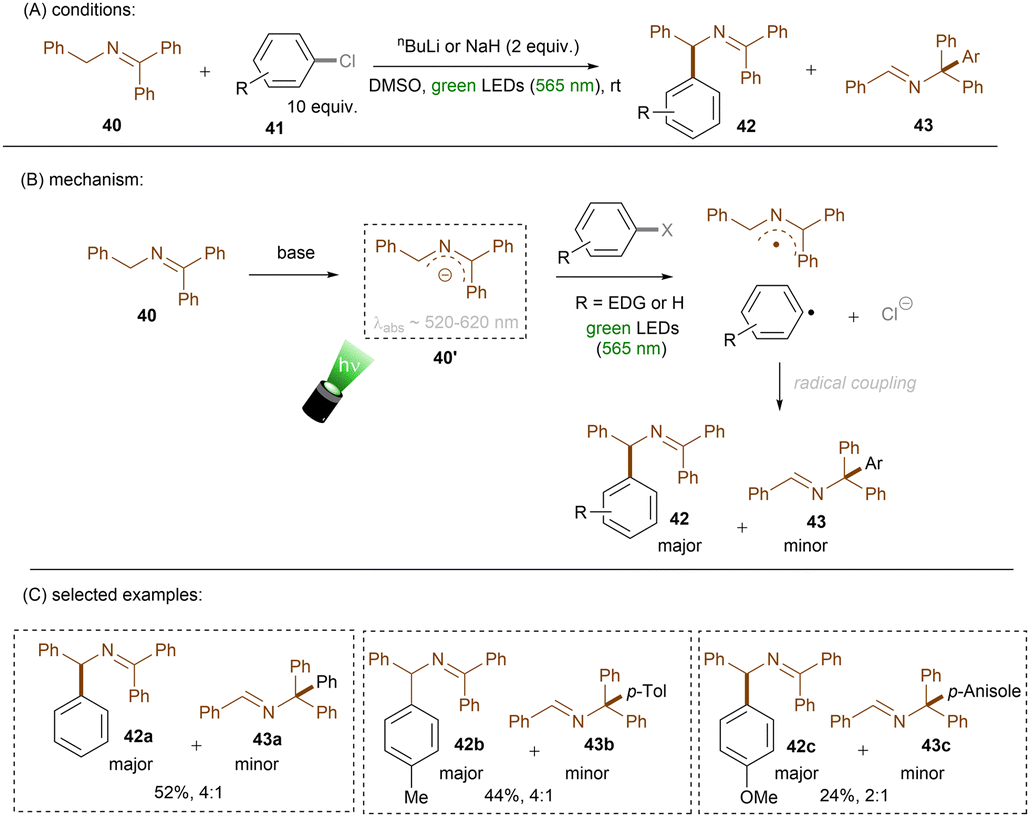
Instead of using an external or in situ generated anion catalyst, Poznik, Chruma and co-workers prepared 2-azaallyl anion 40′ as super electron donor by treating N-benzyl imine 40 with nBuLi (Scheme 26).78 Previously, super electron donors formed in situ from strong bases were mainly leveraged for strongly reductive transformations (i) in the ground state under high temperatures or (ii) in the excited state with UV light.52,79–81 Here, mild, visible green light was able to access a photoexcited species that could achieve SET reduction of an unactivated aryl chloride to form an aryl radical and a 2-azaallyl radical. These two radicals couple to form N-diphenylmethyl imine 42 as a major product along with the generation of minor N-triphenylmethyl imine 43. More electron-rich aryl chlorides afforded lower combined yields of the two products. Despite the drawback that this reaction requires strong bases such as nBuLi and NaH, it provides a facile method for direct aminoalkylations of aryl halides. Although the authors did not mention the role of a dimsyl anion, which would certainly form in the presence of such strong bases,82 Rossi and co-workers earlier reported that photoexcited dimsyl anion was only reductive enough to engage aryl iodides and activated aryl bromides in reduction.83
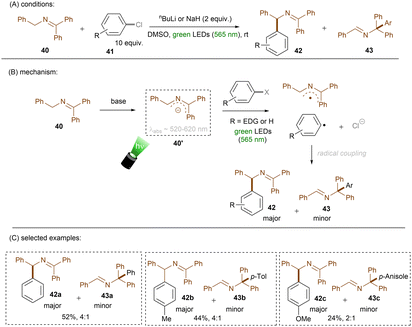 |
| | Scheme 26 Photoexcited 2-azaallyl anion for the reduction of electron-rich aryl chlorides. | |
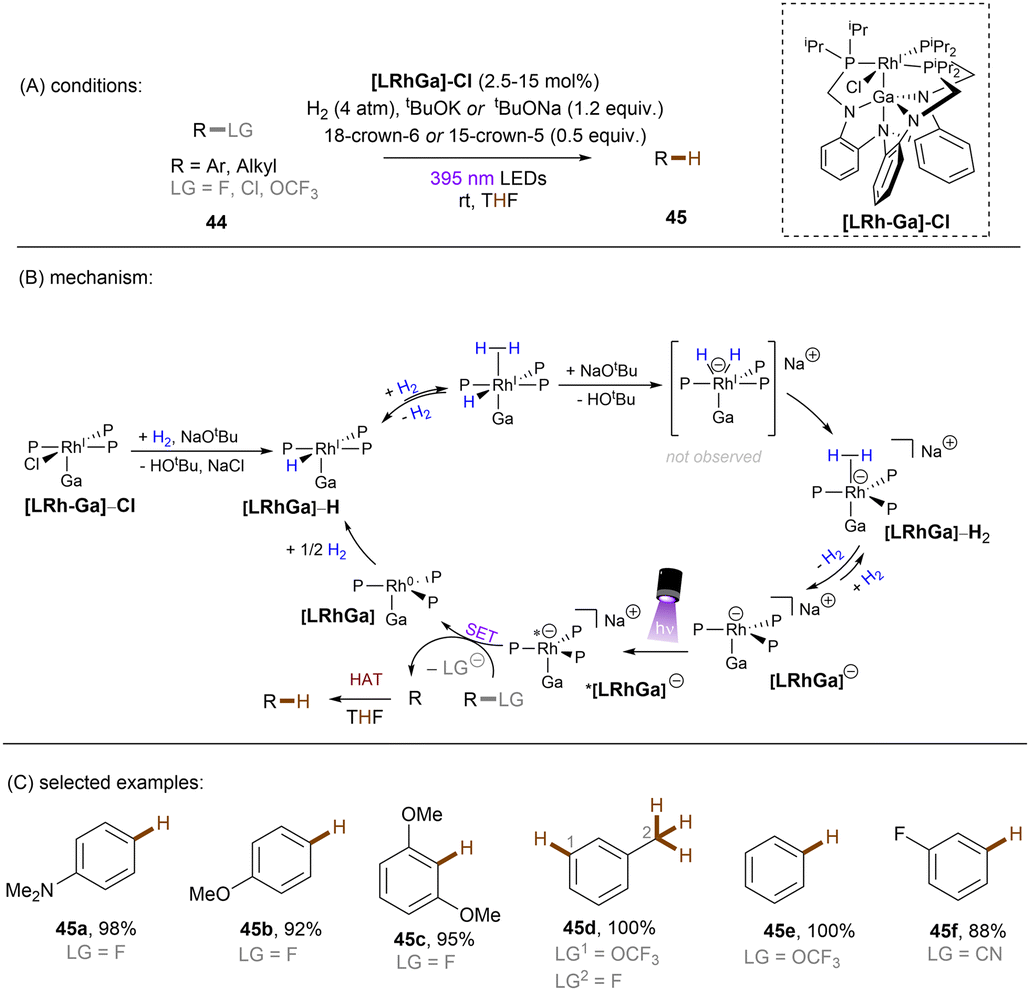
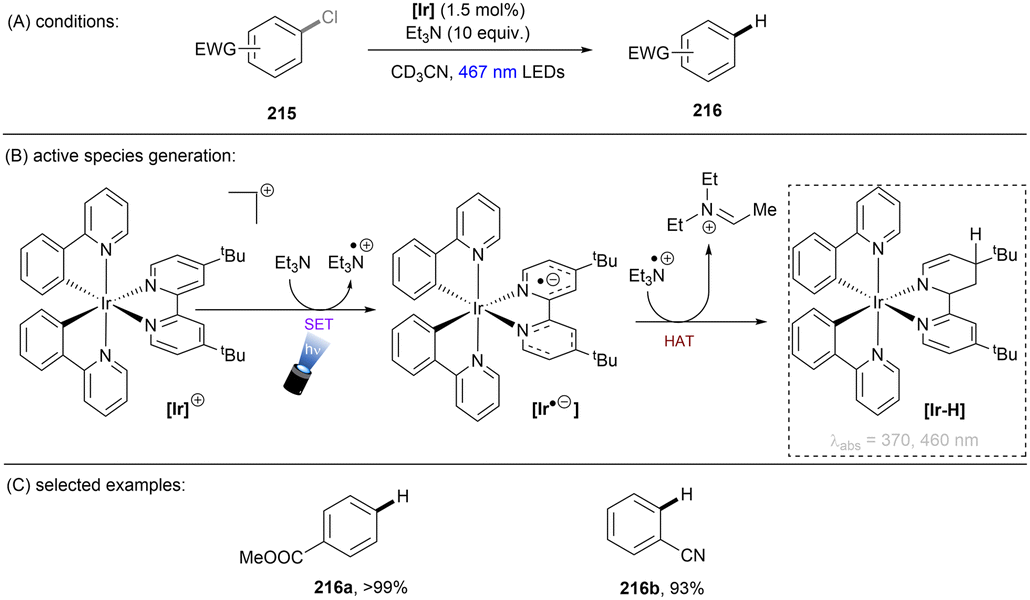
There has also been a report where a metal-centered ionic species has proved to be a potent photoreductant. The Lu group recently achieved photoredox catalysed activations of strong C(sp2)–F/C(sp3)–F, C(sp3)–Cl and C(sp2)–O bonds using a [Rh–Ga] complex catalyst with H2 as the terminal reductant (Scheme 27).84 The photoactive species [LRhGa]− is generated by two consecutive bindings by H2 and a subsequent deprotonation by tBuO−, prior to its photoexcitation. *[LRhGa]− is then a potent photoreductant for SET reduction of inert substrates such as fluorobenzene. The generated phenyl radical abstracts a hydrogen atom from THF solvent to form benzene due to the favourable difference in BDEs. This catalytic system is able to offer extreme reduction potentials and it is particularly powerful for the reduction of unactivated alkyl chlorides and electron-rich fluoroarenes including dimethylamino and methoxy substituted ones. Aryl C–OCF3 and C–CN bonds are readily cleavable under the standard conditions. However, over-reduction is a potential drawback of this method, with 1-fluoro-3-(trifluoromethyl)benzene undergoing full reduction to toluene in 100% yield. Selective activation of a certain functional group in an arene substrate that contains multiple reductively-labile groups would be problematic via this method.
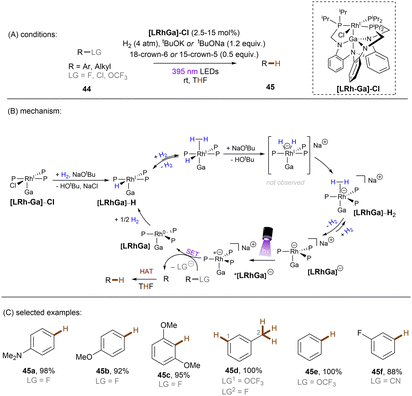 |
| | Scheme 27 Photoexcited [LRhGa]− as the active species for the activation of reductively challenging substrates. | |
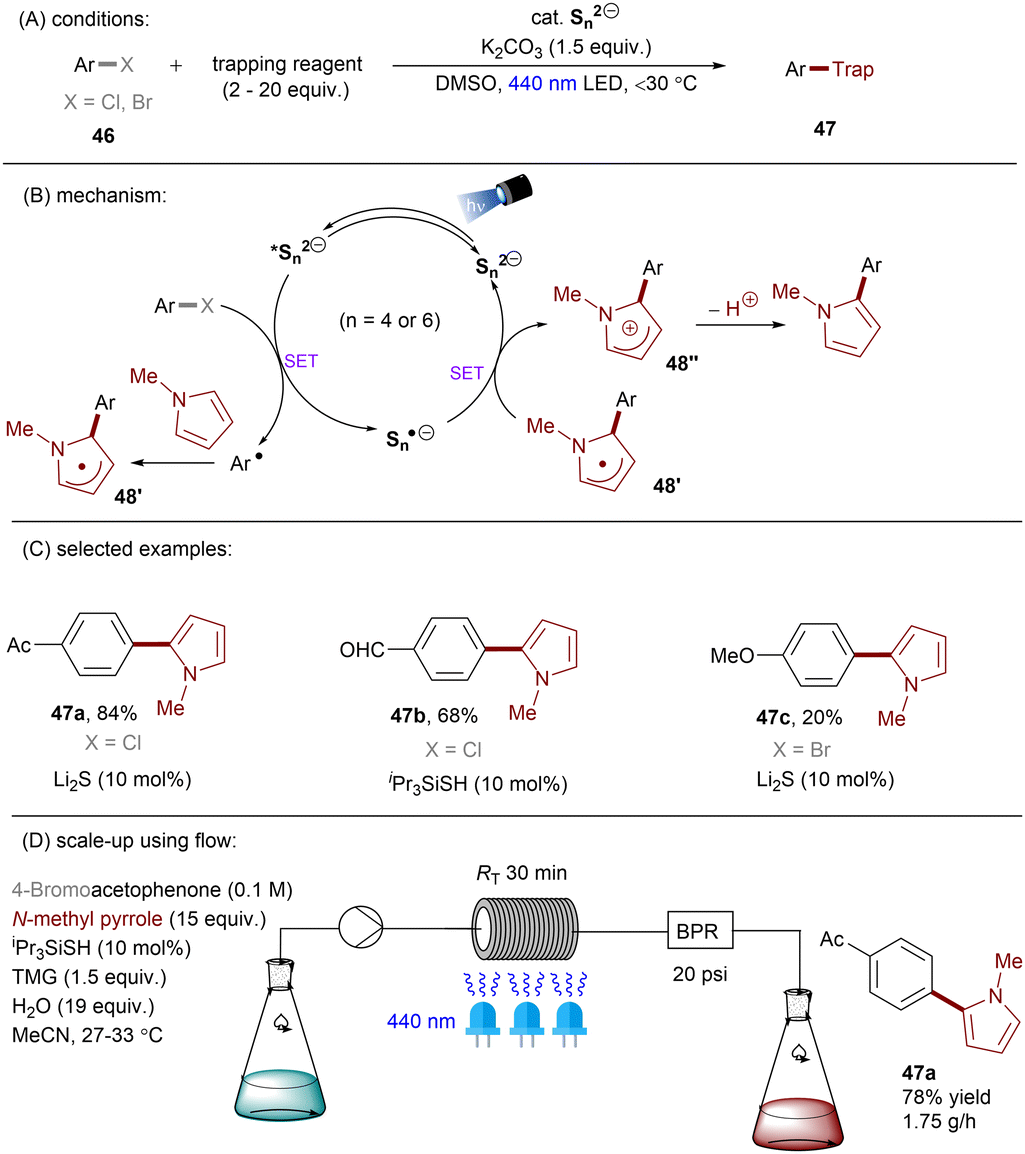
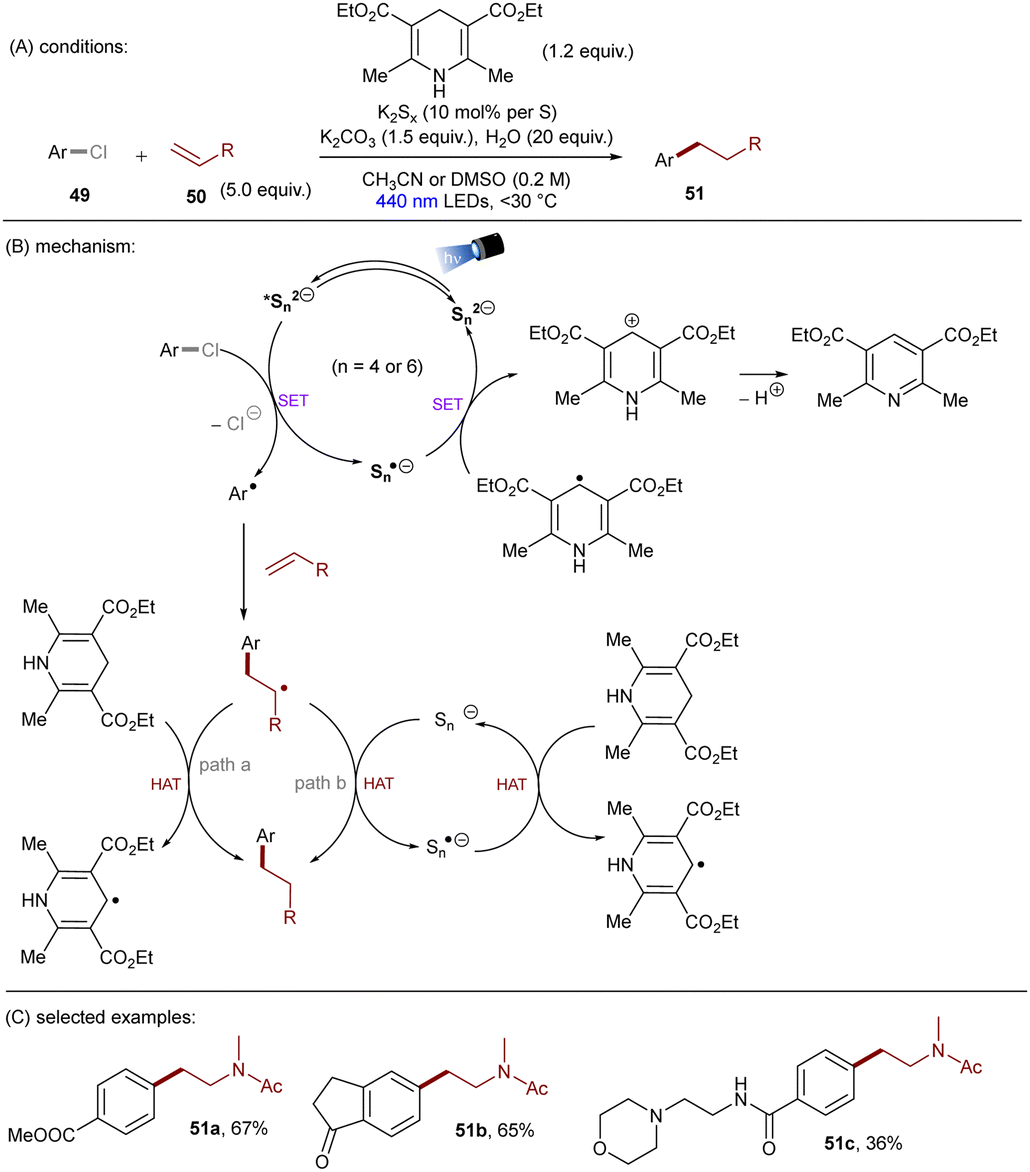
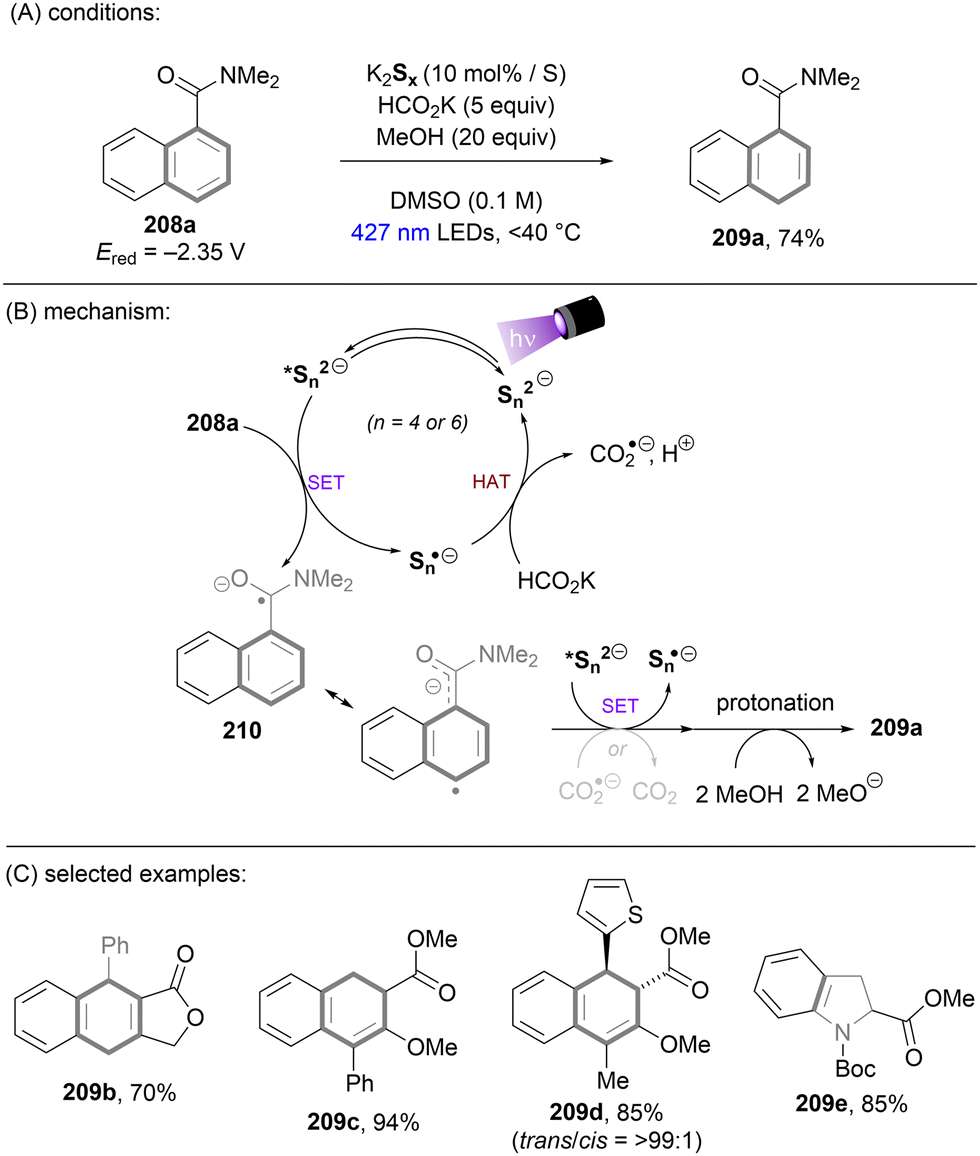
Dianionic species have also been employed to achieve high photoreducing power for the reduction of inert compounds through SET. Polysulfide anions have found applications in alkali metal–sulfur batteries. The photophysical and redox properties of these anionic species have been investigated in detail.85–87 The UV-vis spectrum of K2Sx in DMSO manifested the presence of UV-light-absorbing S32− and visible-light-absorbing S42− and S62−. They are known to have redox potentials Ered(S4˙−/S42−) = −0.85 V vs. SCE and Ered(S6˙−/S62−) = −0.4 V vs. SCE. These distinct light-absorbing species could potentially comprise a new catalytic system where S42− and S62− act as photo-reductants. The Chiba group reported the activation of aryl halides 46 using a catalytic amount of inexpensive K2Sx (Scheme 28).88 Photoexcited dianions *Sn˙2− (n = 4 or 6) are the active species that reduce aryl halides to aryl radicals which then add to N-methyl pyrrole. The concomitantly formed Sn˙− oxidizes the adduct radical to form aromatized coupling products and Sn2−. Besides K2Sx, other pre-catalysts such as Li2S and iPr3SiSH were also identified as precursors of Sn2−. A broad substrate scope of aryl bromides was tolerated, including electron-rich 4-bromoanisole. While aryl chlorides bearing an EWG at their arene ring were coupled in high yields, more electron-rich aryl chlorides were unreactive. The coupling of 4-bromoacetophenone with N-methyl pyrrole was successfully scaled up in a continuous flow reactor, providing the desired product in 78% yield with a productivity of 1.75 g h−1. The same group further described an extension of this strategy to anti-Markovnikov hydroarylation of alkenes (Scheme 29).89 Alkenes were employed to trap aryl radicals formed by *Sn2−-enabled SET reduction of aryl halides. The generated secondary or tertiary alkyl radical intermediates abstract a hydrogen atom from either the Hantzsch ester or HSn− to deliver anti-Markovnikov hydroarylated products. The reaction scope in terms of aryl chlorides was comparable to that of their previous work: only electron-deficient ones were suitable substrates.88
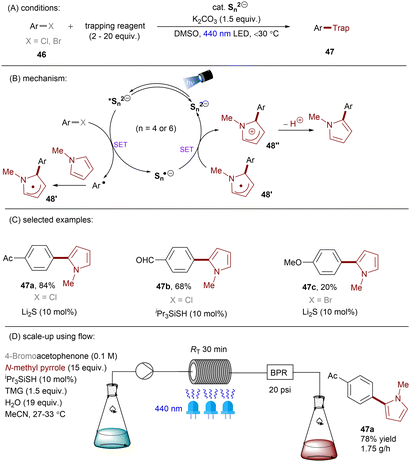 |
| | Scheme 28 Reductive C(sp2)–halogen activations using polysulfide anion photocatalysts. | |
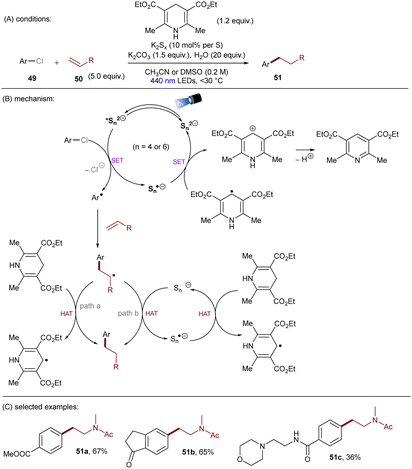 |
| | Scheme 29 Anti-Markovnikov hydroarylation by using polysulfide anion photocatalysts. | |
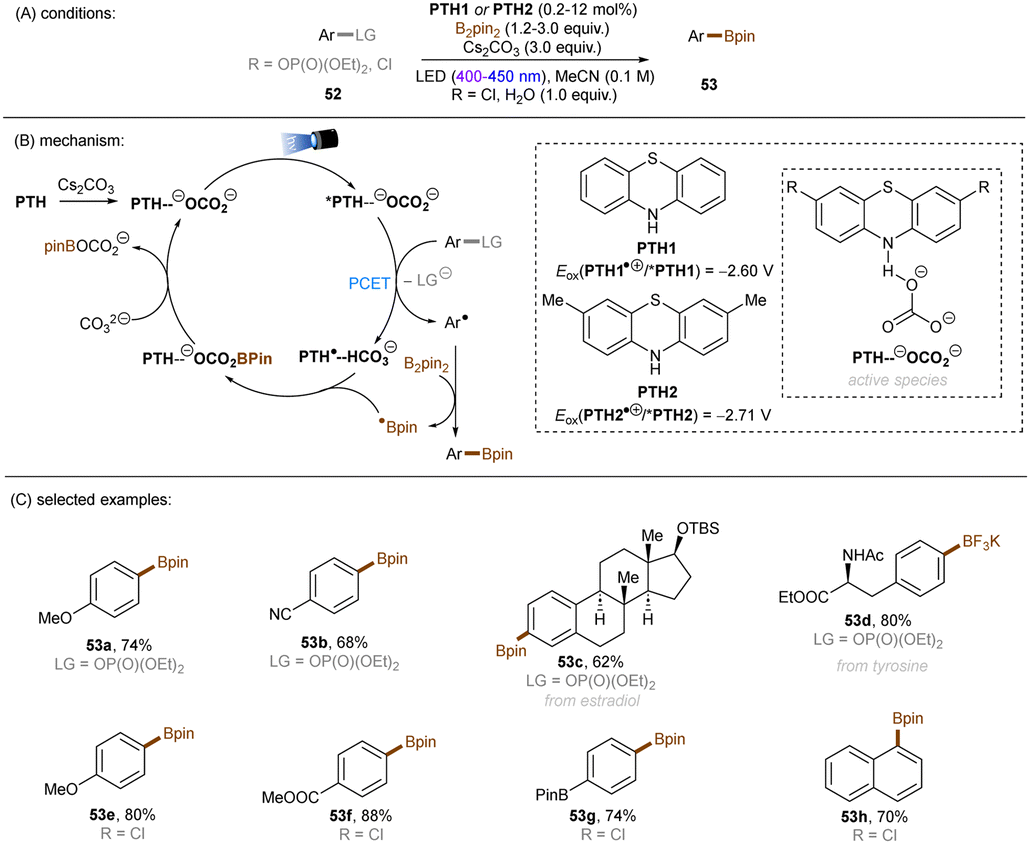
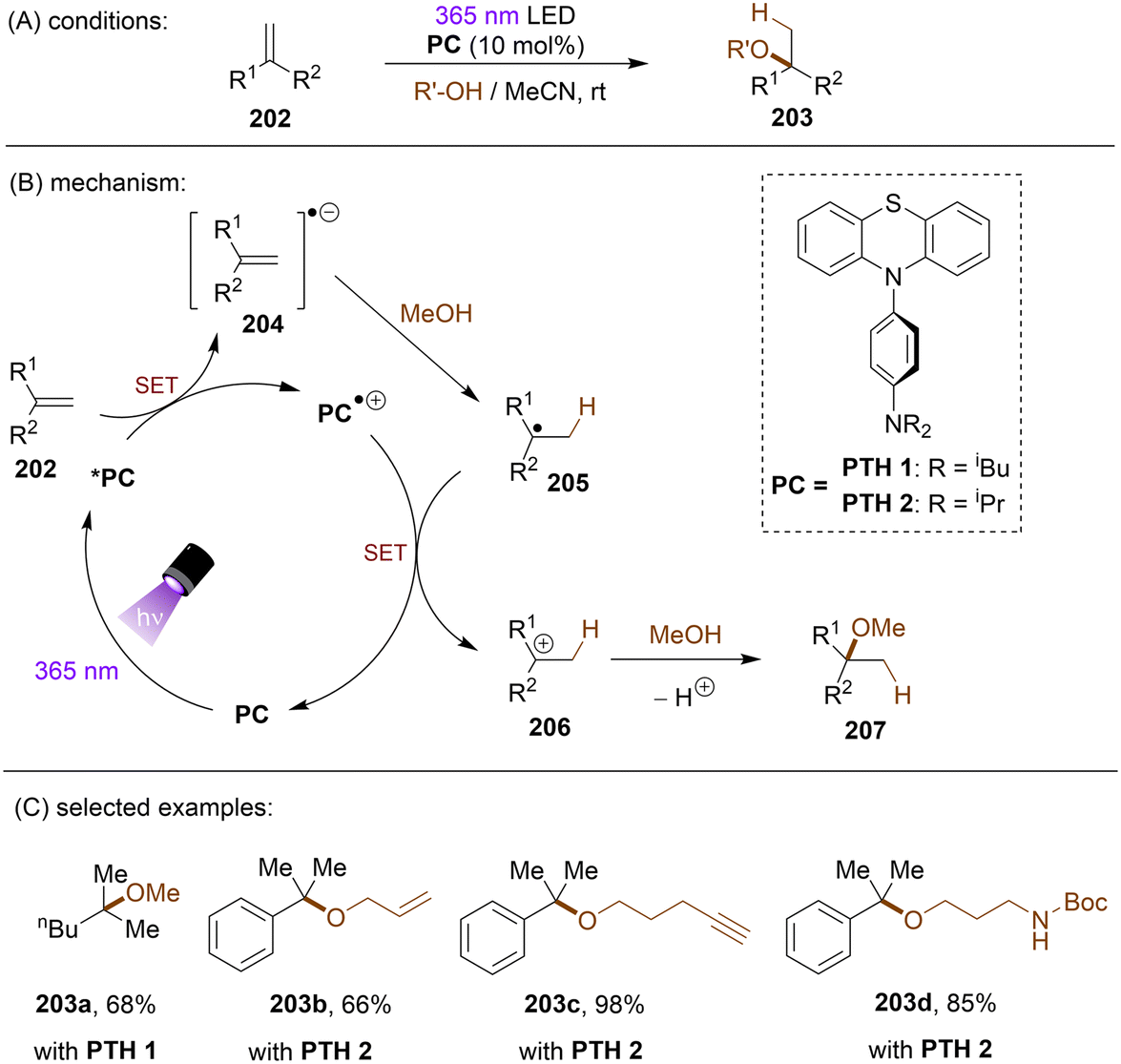
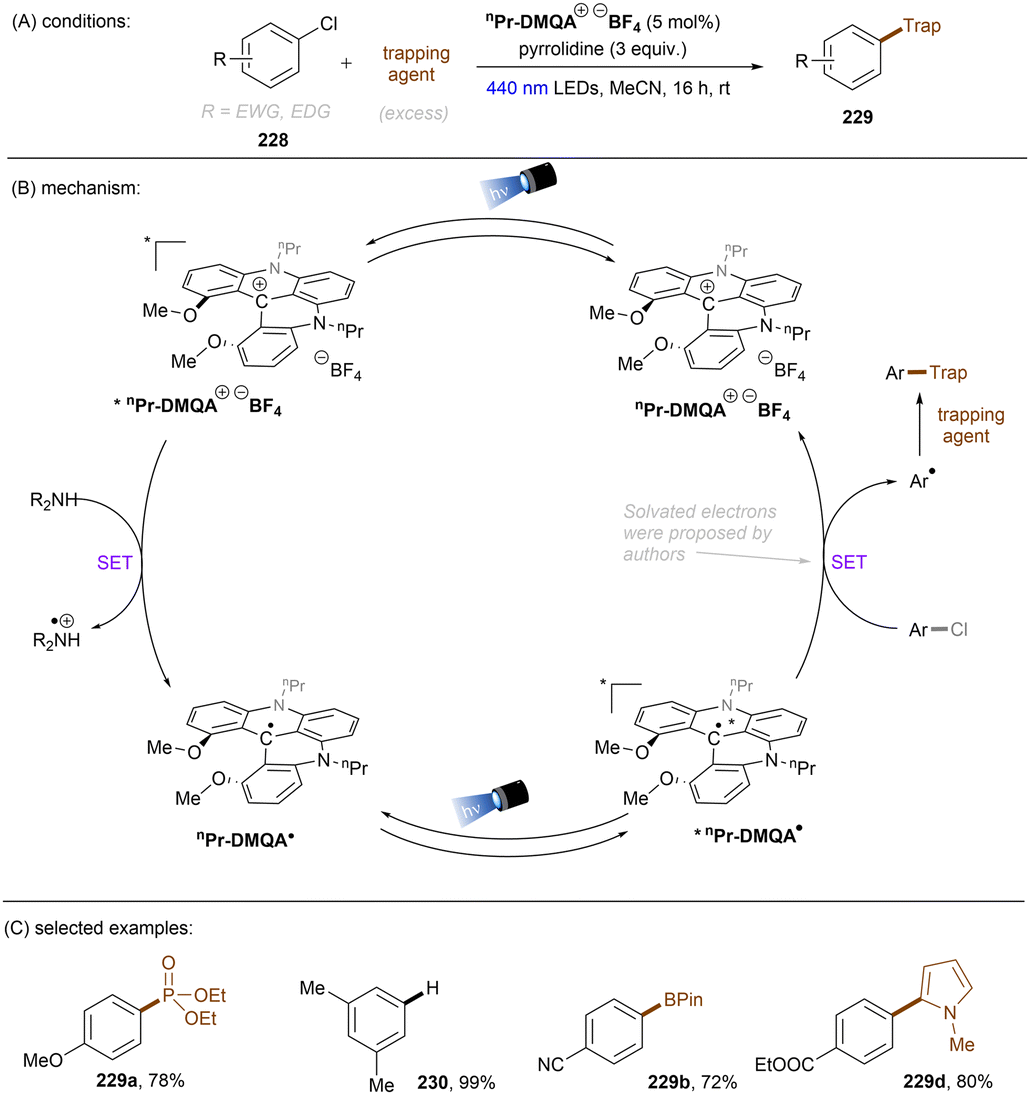
There have also been reports on the reduction of inert aryl halides through in situ formation of an electron donor–acceptor complex. The Larionov group developed a highly efficient method for the borylation of aryl halides and aryl phosphates.90N-unprotected phenothiazine derivatives were employed as strongly photoreducing PCs (Scheme 30). Interestingly, in terms of redox potentials, all the excited neutral PTH catalysts are insufficiently reducing to activate electron-rich aryl chlorides and phosphates. Replacing unprotected phenothiazine with N-Me or N-Phenyl substituted phenothiazines resulted in substantially decreased product yields, demonstrating the importance of the N–H bond in the PTH catalysts. Investigations by NMR and UV-vis ruled out direct SET from the excited state PTH anion – generated by deprotonation and a subsequent photoexcitation – to the arene substrates. Instead, an interaction of this N–H bond with the added carbonate base was found to form a visible light absorbing complex which is capable of donating an electron to reductively challenging substrates upon photoirradiation. Taken together, these results support a proton-coupled electron transfer (PCET) mechanism. For more activated substrates such as aryl chlorides bearing an electron-withdrawing groups at their arene ring, direct SET induced by the excited neutral PTH catalyst cannot be excluded. The authors gained more insights into the mechanism by computational studies. It was suggested that SET reduction of substrates to aryl radicals by singlet excited state of PTH–−OCO2− is thermodynamically favoured, and the further interception of these radicals by B2Pin2 both have a low activation barrier. B2Pin2 herein acts as both a borylation reactant and a mediator for phase transfer.
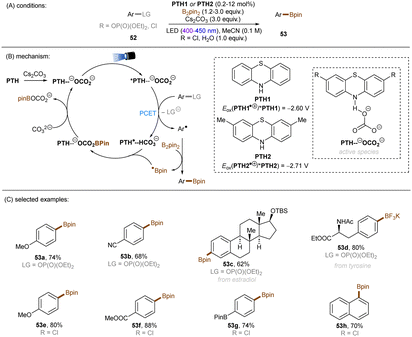 |
| | Scheme 30 Reduction of aryl chlorides and aryl phosphates enabled by proton-coupled electron transfer with a PTH photocatalyst. | |
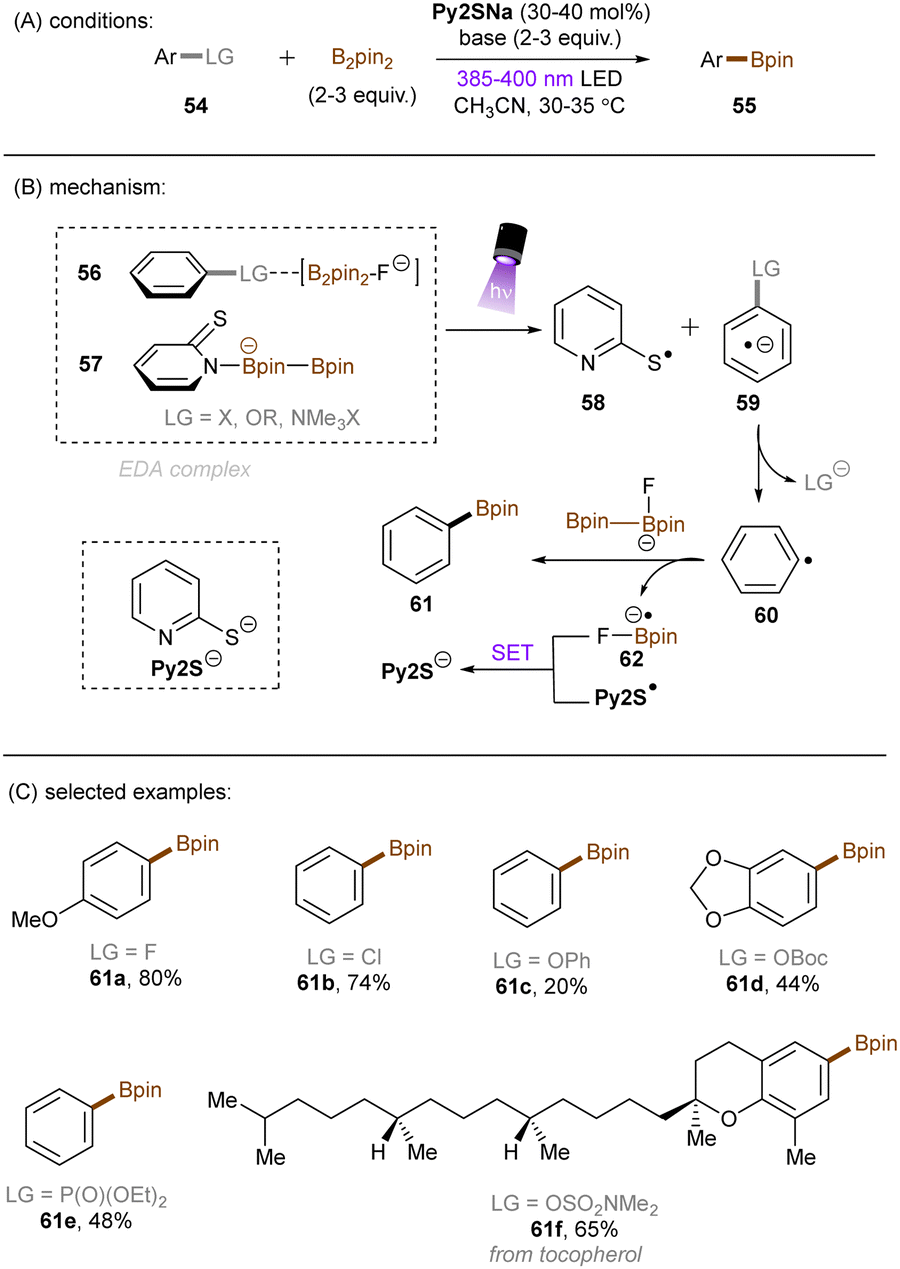
König and co-workers recently developed an extremely reducing system to effect borylations of strong C(sp2)–F, C(sp2)–Cl and C(sp2)–O bonds in substituted arenes through the formation of an EDA complex between the boryl-fluoride-activated substrates 56 and the B2Pin2-interacted ground state thiolate catalyst 57 (Scheme 31).91 NMR and UV-vis spectroscopic measurements supported the formation of such a complex. Subsequent photoirradiation facilitated SET from the thiolate to the arene substrate. The reducing power of this catalytic system was demonstrated by the borylation of diverse and inert aryl fluorides, aryl chlorides, aryl carbonates, aryl carbamates and diaryl ethers. Issues of over-reductions – such as carbonyl reduction – arose when attempting to utilize highly electron-deficient arenes as substrates, and this resulted in low to moderate product yields. This could be due to incompatible electronics for EDA complex formation at the π-system, the specificity of which being a general limitation of EDA photochemistry. Or, it could be due to crossing over into the Marcus inverted region of electron transfer and thus a dramatically decreased rate of SET.
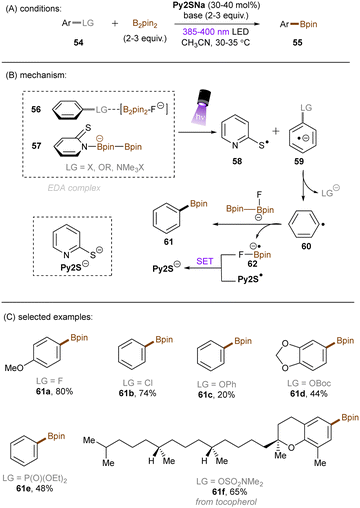 |
| | Scheme 31 Activation of strong C(sp2)–F, C(sp2)–Cl and C(sp2)–O bonds via an EDA complex. | |
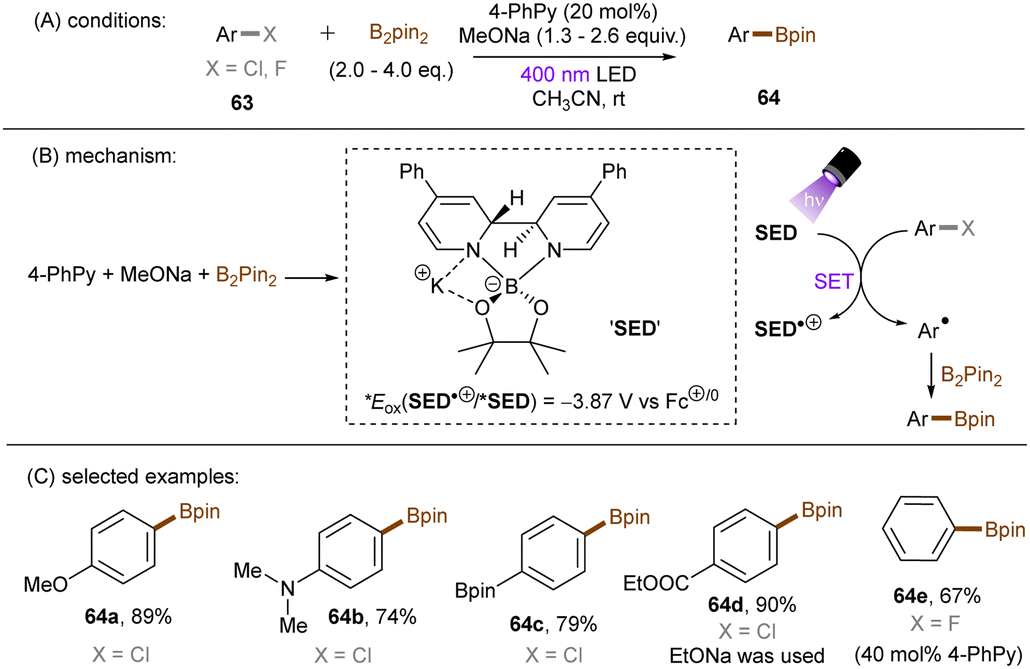
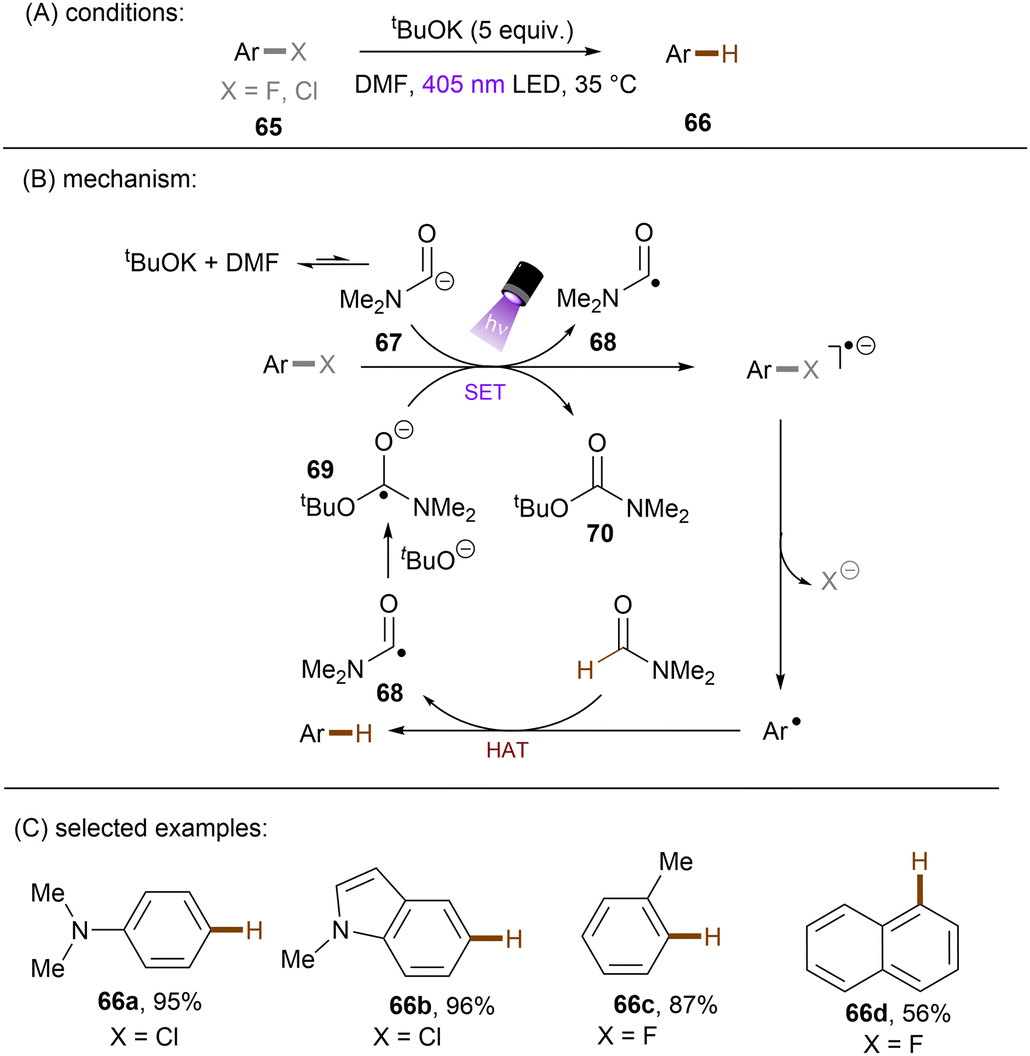
Generating photoactivated super electron donors (SEDs) from simple reactants the also appeared to be a facile method for the activation of reductively challenging compounds. In 2017, the Jiao group reported a visible light induced borylation of aryl halides with B2Pin2, in the presence of KOMe and catalytic amount of 4-phenyl pyridine (4-PhPy).92 While no component absorbs visible light, a complex formed by mixing 4-PhPy, KONa and B2Pin2 was identified to be a visible-light-absorbing SED. Its photoexcited state was calculated to possess a strong reducing ability (*Eox(SED˙+/*SED) = −3.87 V vs. Fc+/0).93,94 Recently, such strategy has been applied to the borylation of aryl chlorides and fluorides (Scheme 32).94 It was observed that the reaction between stoichiometric SED·DME complex and para-chloroanisole in THF under irradiation with 400 nm light afforded anisole in 82% yield, clearly demonstrating that the photoexcited SED is capable of reducing electron-rich chloroarenes. The catalytic reaction also worked efficiently, indicating the efficient in situ generation of SED. Under the catalytic reaction conditions, diverse aryl chlorides including methoxy- and dimethylamino-substituted ones (63a-b) were borylated in high yields. On a gram scale, the reaction proceeded smoothly without significant loss of efficiency. The reducing power of this catalytic system was further demonstrated by engaging aryl fluoride as a competent reaction partner. A potential drawback of this method is the use of sodium methoxide. Although methoxide is inexpensive and readily available, it may encourage side reactions, such as hydrolysis of substrates containing susceptible functional groups. This was observed for the ethyl benzoate substrate and so ethoxide was used as the base. Given Li's report on t-BuOK mediated dehalogenation of bromo-/iodoarenes in DMSO,95 Qu and Kang developed a related catalyst-free photochemical reduction of less activated aryl chlorides and fluorides using t-BuOK as the only additive to the solution of aryl halides in DMF. (Scheme 33).96 The authors claimed that the reaction is initiated by deprotonation of DMF's aldehydic proton by tert-butoxide.82,97 The deprotonated DMF (carbamoyl) anion 67 reduces aryl halides through SET upon irradiation with visible light. The aryl radical, generated by fragmentation of the haloarene radical anion then abstract a hydrogen atom from DMF to deliver a hydrodehalogenated product and DMF radical 68. Radical 68 undergoes a nucleophilic attack by tBuO− to form a carbamate radical anion 69 which is also able to furnish aryl halide reduction by SET. This radical chain process has enabled the activation of a broad range of aryl C–F and C–Cl bonds. Given the strong reducing power of this system, surprisingly, smaller aromatics reacted more efficiently than more easily reducible larger π-systems. This could possibly be due to the influence of entering the Marcus inverted region for SET.
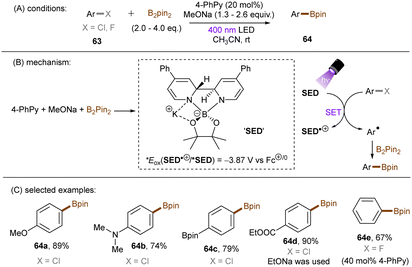 |
| | Scheme 32 Forming SED complex for photoreduction of aryl chlorides and fluorides. | |
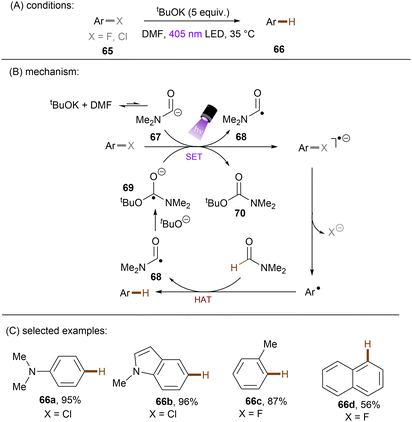 |
| | Scheme 33 Photochemical hydrodehalogenation of aryl chlorides and fluorides using t-BuOK in DMF to form a photoactive electron donor. | |
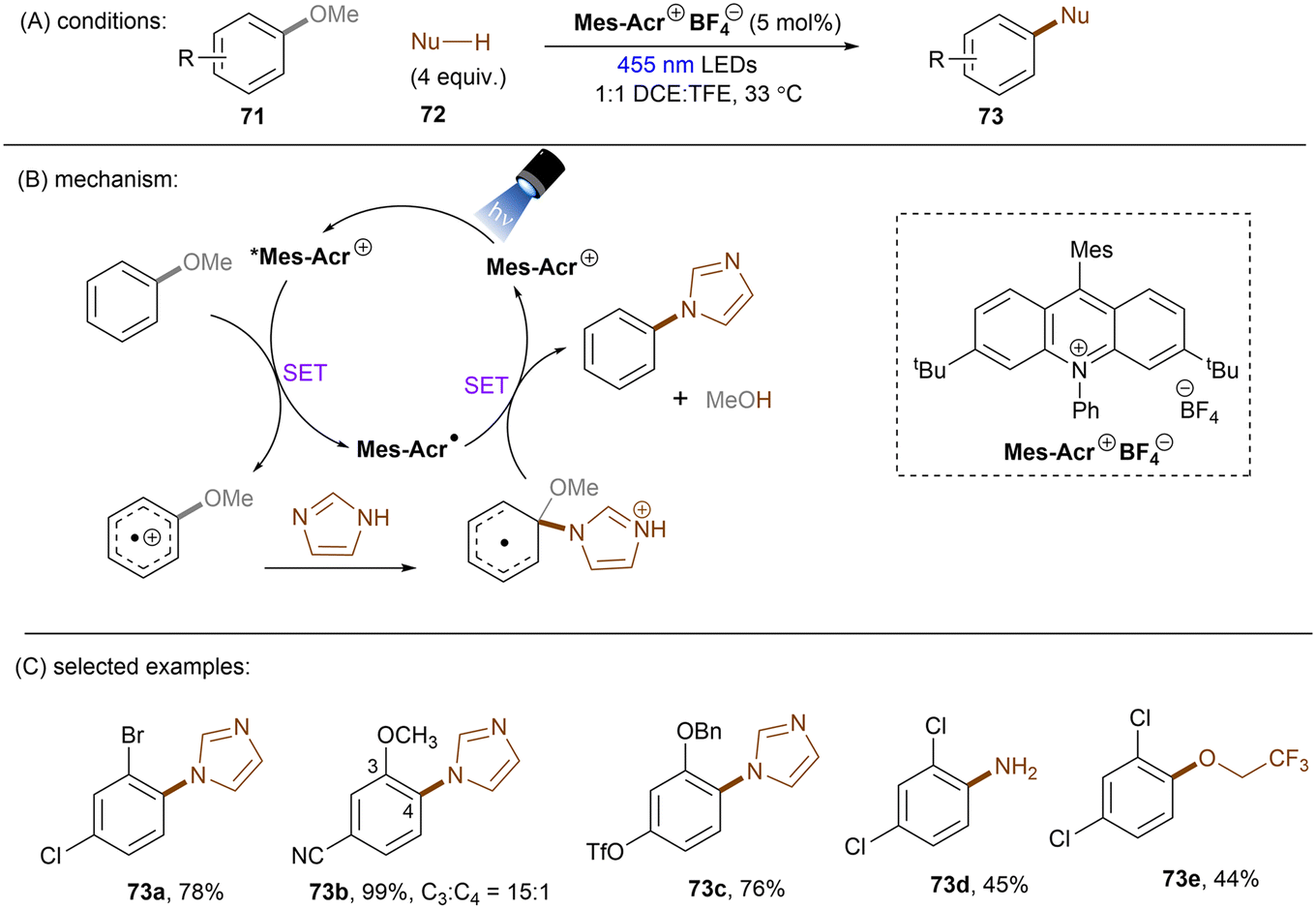
Apart from reductive activations, in a complementary fashion, super photooxidizing PCs have found to be able to oxidatively activate aryl C(sp2)–X (X = F, O) bonds. In 2015, the Nicewicz group established an elegant photoinduced oxidative cross coupling of arenes with nucleophiles.98,99 Key to reactivity was the ability of a newly designed Fukuzumi-type acridinium catalyst (*E1/2 ≈ +2.1 V vs. SCE) to oxidize relatively electron-rich arenes to their radical cations under visible light irradiation. Subsequent nucleophilic attacks by azoles, ammonia or cyanide afforded C(sp2)–H functionalized products. Molecular oxygen was used as an external oxidant to turn over the acridinium photoredox catalyst. This catalyst class was soon employed for redox-neutral nucleophilic aromatic substitution (SNAr) reactions by omitting O2. First substrate used in this reaction is anisole which has a reductively inert C(sp2)–O bond (Scheme 34).100 Under potent oxidizing conditions provided by the photoexcited state of Mes-Acr+BF4− catalyst (*E1/2 = +2.15 V vs. SCE), the C(sp2)–O bonds in anisole substrates 71 were readily converted into C(sp2)–N bonds. Both imidazole (Eox = +1.15 V vs. SCE) and 2,4-dichloroanisole substrate (Eox = +2.04 V vs. SCE) are able to reductively quench *Mes-Acr+BF4−. Therefore, the authors conducted a control reaction by employing a commonly used [Ir]-based photocatalyst whose excited state has a redox half potential of +1.68 V vs. SCE, and no coupling product was yielded. The results indicated that 2,4-dichloroanisole is the species oxidized by the excited *Mes-Acr+ to the arene radical cation, primed to nucleophilic attack by imidazole at the OMe-bearing carbon. Meanwhile Mes-Acr˙ was generated and is capable of reducing the arene-azole adduct to the final product by releasing MeOH. This reaction showed excellent chemo- and regioselectivities. 2-Bromo-4-chloroanisole 71a selectively underwent C(sp2)–O cleavage with C–Cl and C–Br bonds untouched. 3,4-Dimethoxybenzonitrile 71b was functionalized in a high regioselectivity of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The more electrophilic carbon was attacked by the nucleophile. Such selectivity contrasts sharply to the photoredox-mediated C(sp2)–X (X = F, Cl, Br) bond radiofluorination of halogenated anisoles reported by Nicewicz and Li.101 Demethoxyamination still dominated in the presence of a benzyloxy substituent. Direct amination with ammonium carbamate to form aniline is also feasible. Alcoholysis occurred in 2,2,2-trifluoroethanol (TFE) as the solvent in the absence of other nucleophiles. A follow-up study by using alkylamines as nucleophiles for the synthesis of diverse N-alkylated anilines was also reported by the Nicewicz group.102 Under similar PRC conditions, cyanide, generated from acetone cyanohydrin, was also reported as a nucleophile to attack SET oxidized methoxyarenes, effecting a mild demethoxycyanation strategy.103
:1. The more electrophilic carbon was attacked by the nucleophile. Such selectivity contrasts sharply to the photoredox-mediated C(sp2)–X (X = F, Cl, Br) bond radiofluorination of halogenated anisoles reported by Nicewicz and Li.101 Demethoxyamination still dominated in the presence of a benzyloxy substituent. Direct amination with ammonium carbamate to form aniline is also feasible. Alcoholysis occurred in 2,2,2-trifluoroethanol (TFE) as the solvent in the absence of other nucleophiles. A follow-up study by using alkylamines as nucleophiles for the synthesis of diverse N-alkylated anilines was also reported by the Nicewicz group.102 Under similar PRC conditions, cyanide, generated from acetone cyanohydrin, was also reported as a nucleophile to attack SET oxidized methoxyarenes, effecting a mild demethoxycyanation strategy.103
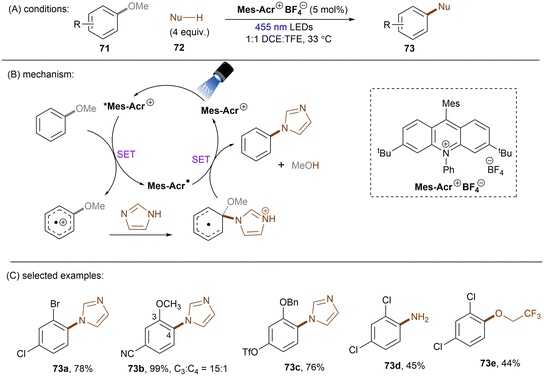 |
| | Scheme 34 Oxidative C(sp2)–O activations using an acridinium catalyst. | |
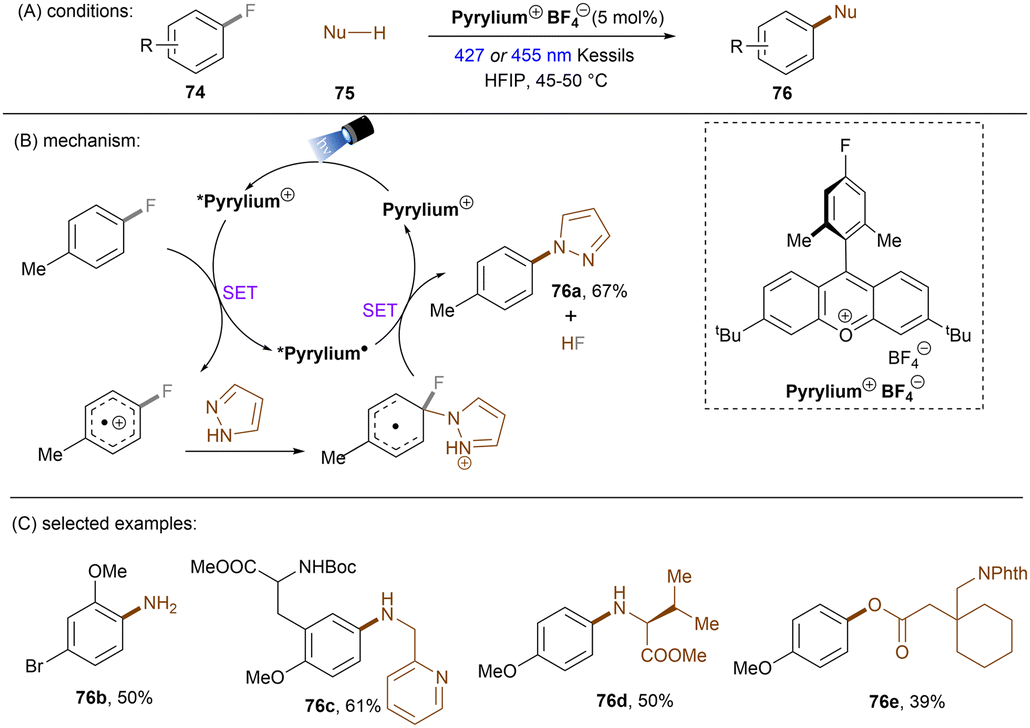
This SNAr strategy was also applicable to C(sp2)–F bond activation. The Nicewicz group developed an operationally simple photocatalytic fluoroarene functionalization method (Scheme 35).104 The reaction proceeded through the oxidation of fluoroarenes by the excited cationic photocatalyst followed by SNAr reactions with diverse nucleophiles such as azoles, alkylamines and carboxylic acids. 4-Fluorotoluene (Eox = +2.24 V) and pyrazole were taken as model substrates. While acridinium catalysts gave poor yields under blue light irradiation, a more oxidizing pyrylium catalyst (*E1/2 > +2.1 V vs. SCE) proved to be effective. HFIP was chosen as the solvent because it can stabilize the radical cations via H-bond interactions. The nucleophilic attack on the radical cation was proposed as the rate-determining step as supported by further kinetic and computational studies.105 This reaction features an impressive functional group tolerance. Fluoroarenes bearing Cl, Br or OMe smoothly and selectively underwent C–F aminations. Alkylamine-based drugs such as rimantadine were successfully installed at the arene ring. Furthermore, carboxylic acids such as gabapentin phthalimide could also be used as nucleophiles. This reaction offers a mild alternative to classic transition metal-catalysed C–N bond cross coupling reactions omitting the use of strong bases and high temperatures. Of note was the need for an electron-withdrawing substituent on the fluoroarenes. Alternative technologies that involve radical cation photocatalysts (conPET, synthetic PEC) are likely needed to engage truly unactivated fluoroarenes in oxidation (vide infra).
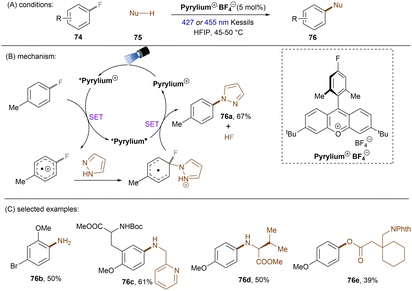 |
| | Scheme 35 Oxidative C(sp2)–F activations using a pyrylium catalyst. | |
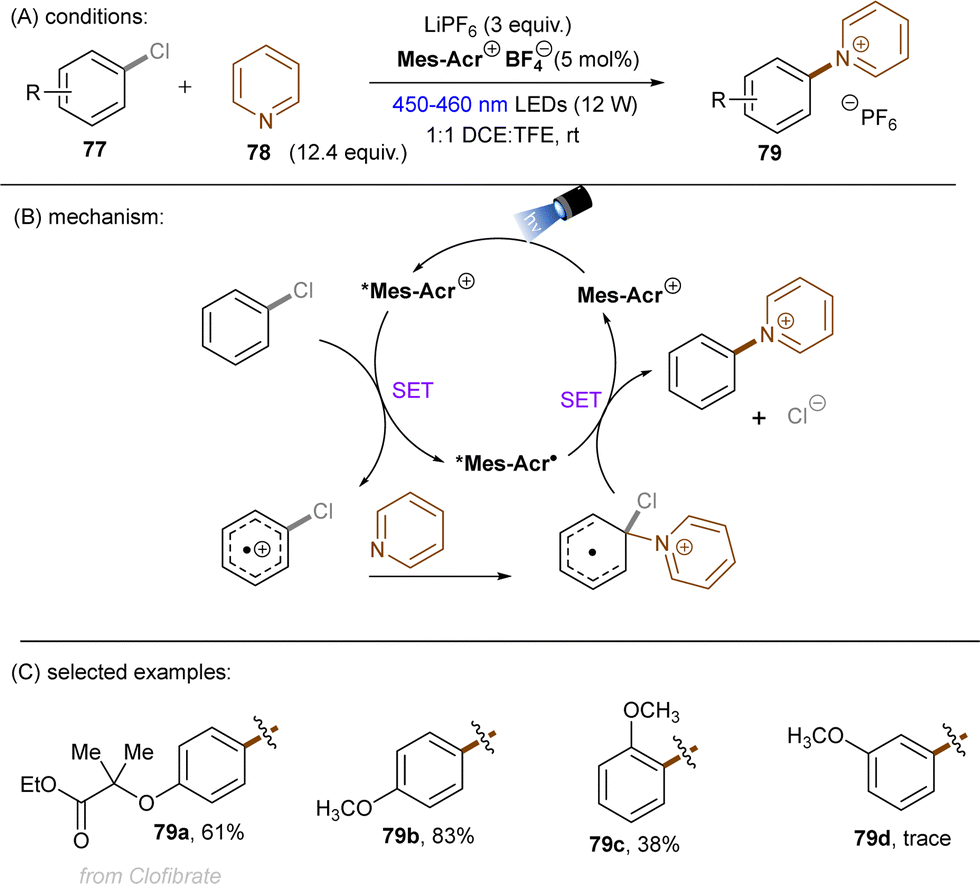
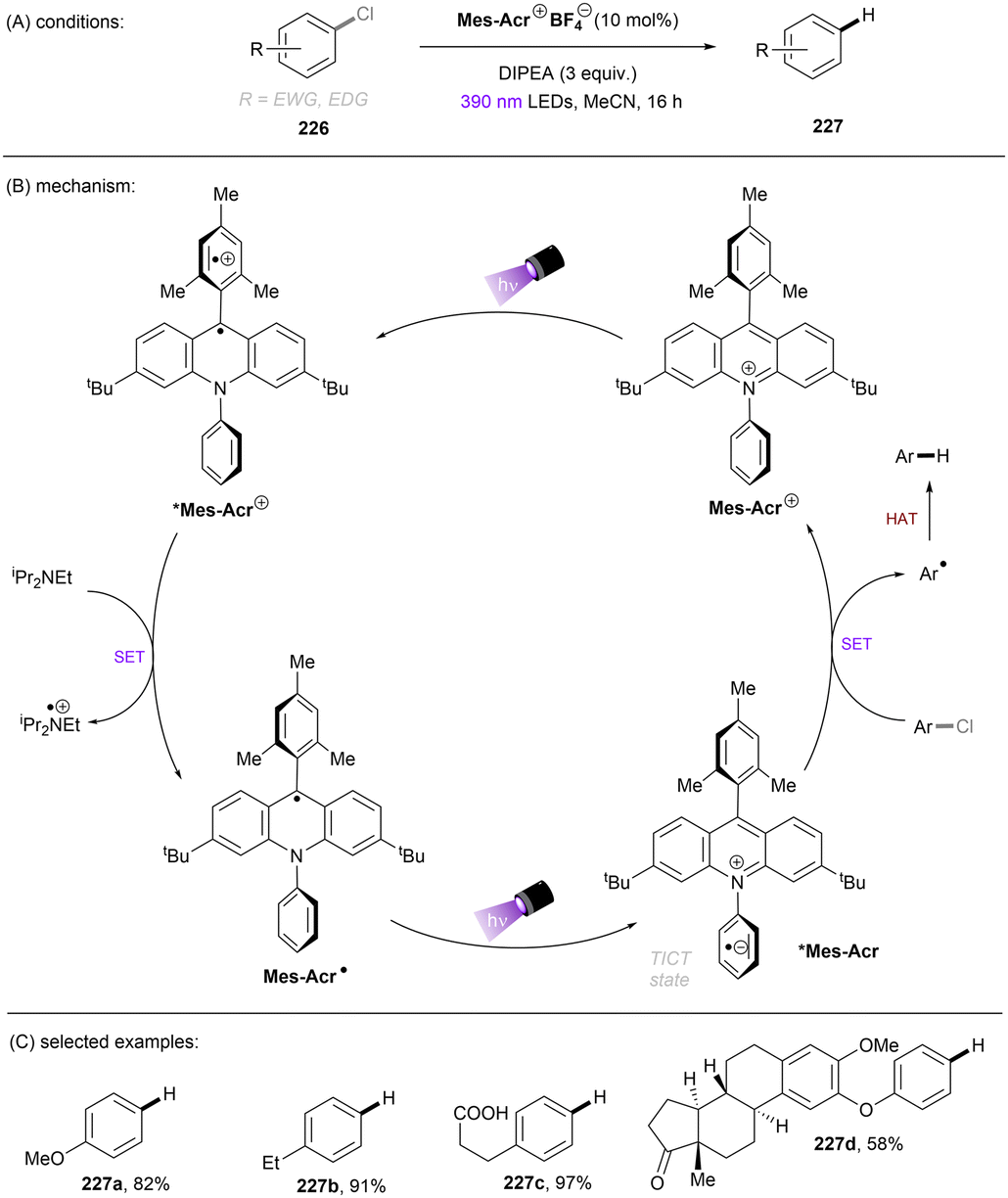
A more recent photocatalytic SNAr reaction using Mes-Acr+BF4− catalyst was reported by Sanford and co-workers (Scheme 36).106 Different to the work from Nicewicz where the C(sp2)–O in chloroanisoles was funtionalized,104 herein the C(sp2)–Cl bond of 4-chloroanisole underwent an SNAr-type pyridination reaction leaving the C(sp2)–O bond untouched. An excess of pyridine (12.4 equiv.) was utilized as a nucleophile to attack the SET-oxidized chloroarene followed by a SET reduction. Loss of chloride anion then affords an arylpyridinium product. This reaction is suitable for aryl chlorides bearing an EDG. While para-alkoxy-substituted chloroarenes, including pharmaceutical clofibrate, deliver generally good yields, ortho-methoxy chlorobenzene gave rise to the desired product 79c in a low yield of 38%. A less electron-rich chloroarene, 3-chloroanisole, led to traces amount of product 79d, revealing an upper limit of the oxidative excited state power of *Mes-Acr+BF4−.
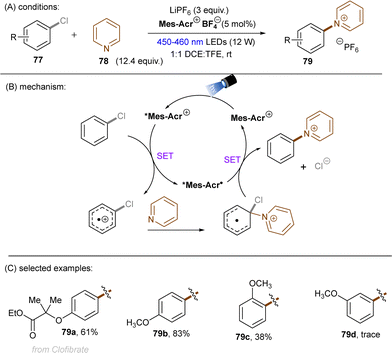 |
| | Scheme 36 Oxidative C(sp2)–Cl activations using an acridinium catalyst. | |
3.1.2. C(sp3)–X (X = F, Cl, O) cleavage.
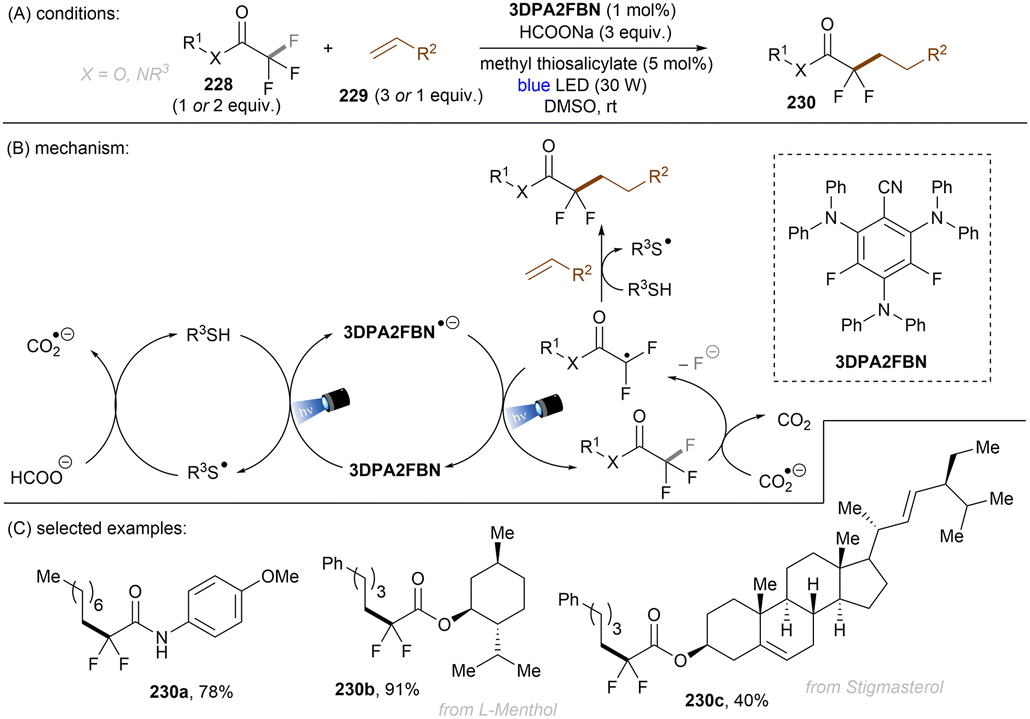
The trifluoromethyl (CF3) group is among the most common functional groups in C(sp3)–F bond activation reactions because the CF3 motif is privileged in medicinal chemistry. The development of new methods towards the late-stage modification of C(sp3)–F bonds in pharmaceuticals is highly beneficial to drug discovery. Along with the well-established benefits imparted by fluorine atoms to pharmaceuticals,107–110 the difluoromethyl group has recently attracted significant attention in medicinal chemistry.111,112 Difluoromethyl groups exhibit H-bond donor behavior. They are bioisosteres of SH and OH groups in cystine and serine moieties respectively, in addition to NH2 groups, while being metabolically inert. They provide advantages to pharmacokinetics and potency by increasing lipophilicity and rigidity.113 Selective reductive functionalization of CF3 groups represents a step-economic strategy for the synthesis of difluoromethyl compounds.114,115 However, mono C–F activation of CF3 compounds is challenging because the BDEs of the C(sp3)–F bonds in them are higher than those in their di- and mono-fluoromethyl derivatives which may lead to double or complete hydrodefluorination (HDF). In the last years, photoredox-catalysed C(sp3)–F bond activations have emerged as mild alternatives to conventional methods suffering harsh conditions.116 As will be seen in this section, PRC reductive C(sp3)–F bond activations often require not only a sufficiently potent photoreductant to enable SET but generally also a promoter to drive the mesolytic C–F cleavage and avoid BET.
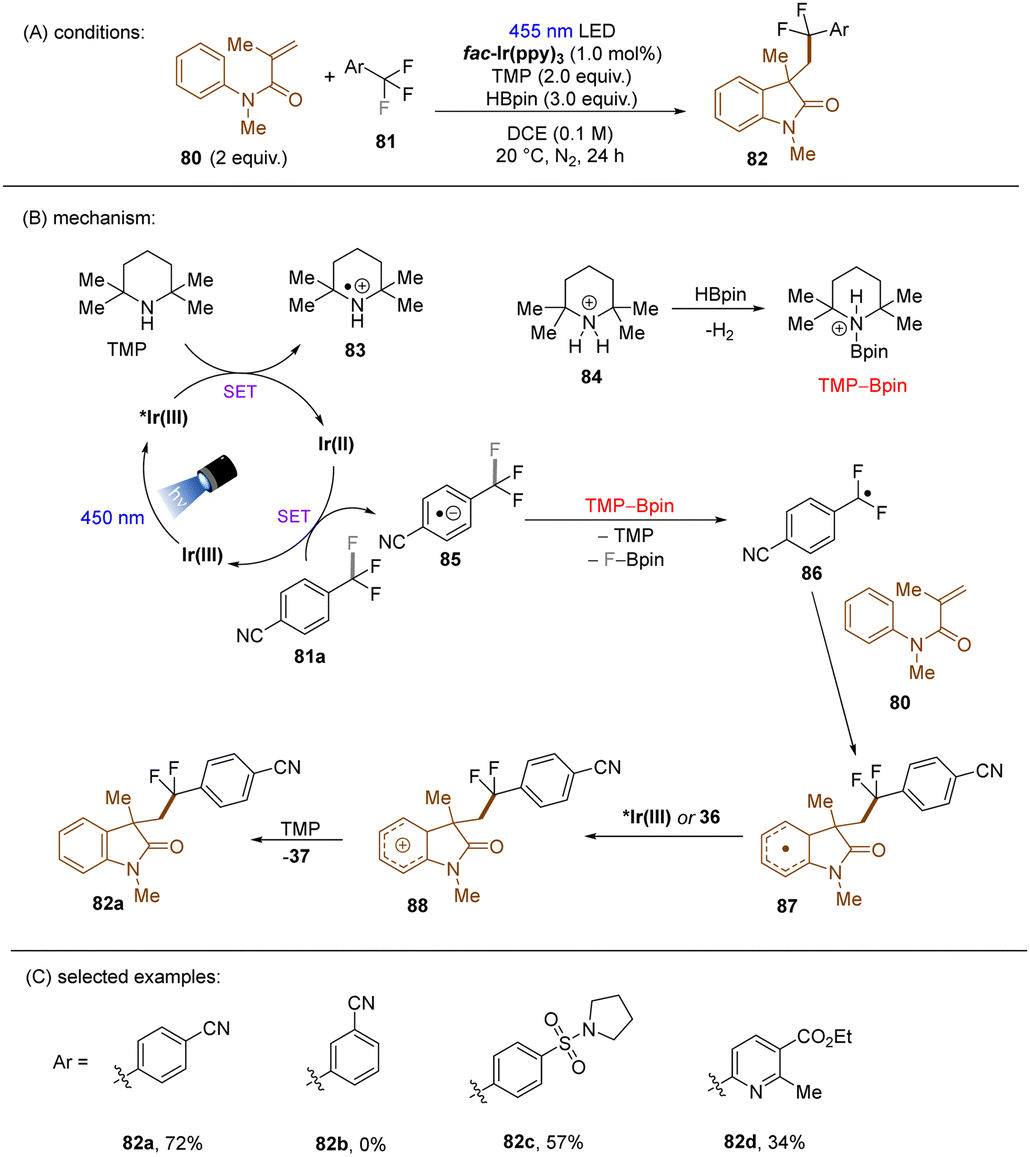
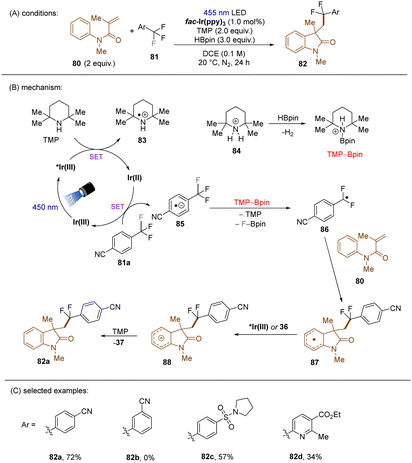
König and co-workers developed the first visible light-induced mono C(sp3)–F bond activation of in trifluoromethylarenes having EWG(s) and coupled the resulting difluorobenzylic radicals with methylacrylamides (Scheme 37).117fac-Ir(ppy)3 was used as the catalyst to reduce trifluoromethyl arenes under visible light irradiation. TMP-Bpin – generated from HBPin and TMPH+ – was a crucial intermediate, proposed to provide the driving force for the C(sp3)–F bond fragmentation of the SET reduced ArCF3. The reaction scope was narrow. Only trifluorotoluenes with a strong electron-withdrawing group at the ortho- or para-positions of the arene ring were suitable substrates, even the reduction of meta-CF3 benzonitrile under the standard conditions was elusive.
 |
| | Scheme 37 C(sp3)–F bond cleavage in trifluoromethylarenes using visible light photocatalysis. | |
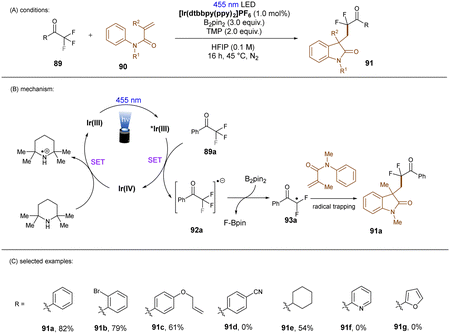
In a related study, Chatterjee and co-workers118 achieved selective mono C(sp3)–F bond activation of substituted trifluoromethyl ketones using Ir(dtbbpy)(ppy)2PF6 as a visible light PC (Scheme 38), offering a complementary method for the preparation of difluoromethylene-containing compounds in the absence of TMSCl and constant current needed under electrolysis conditions (vide infra).119 Instead of HBPin, B2Pin2 was proposed to trap the eliminated fluoride ion. DFT calculations and control experiments indicated that solvent 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was beneficial to the C–F cleavage step and suppressed double and triple hydrodefluorination processes. This reaction tolerates trifluoroacetophenones and aliphatic trifluoromethyl ketones. However, neither heteroaryl trifluoromethyl ketones nor trifluoroacetophenones bearing a strong electron-withdrawing group reacted under the standard conditions.
 |
| | Scheme 38 C(sp3)–F bond cleavage in trifluoromethyl ketones using visible light photocatalysis. | |
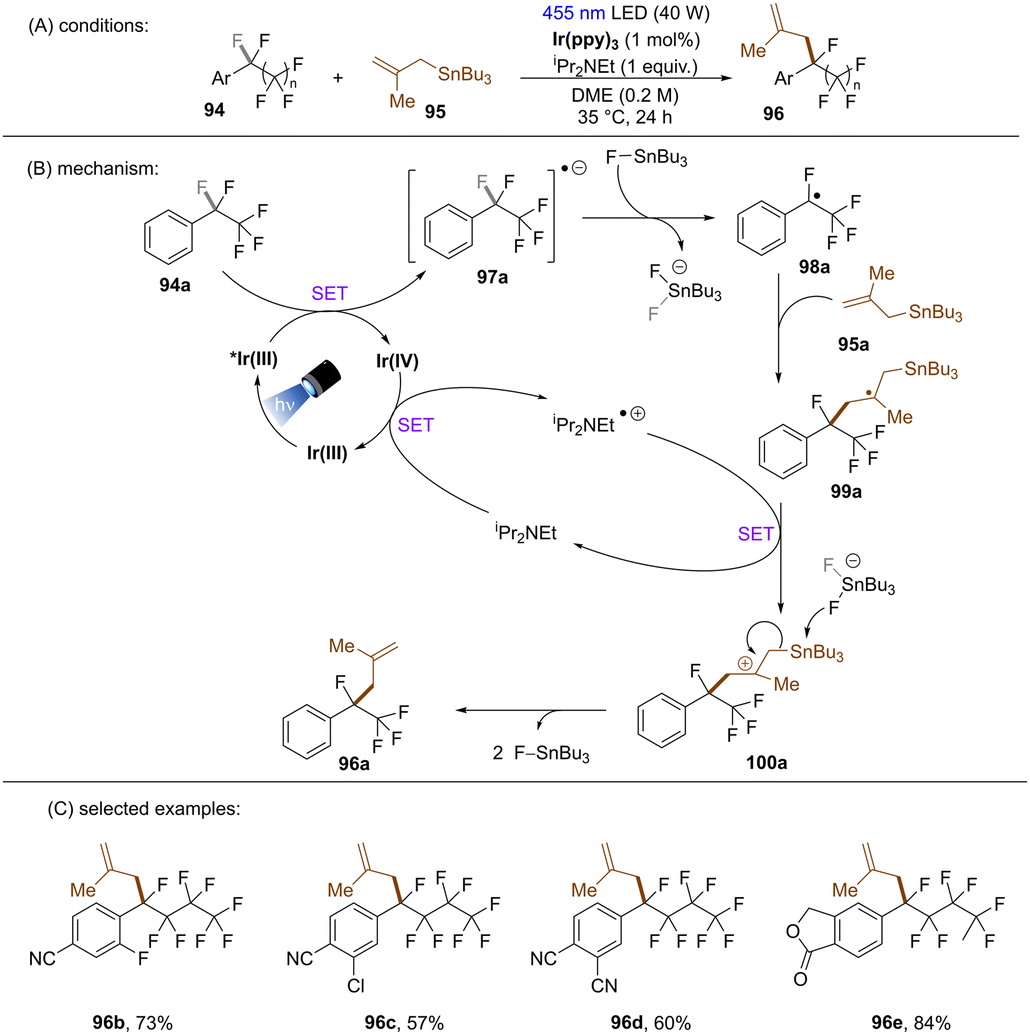
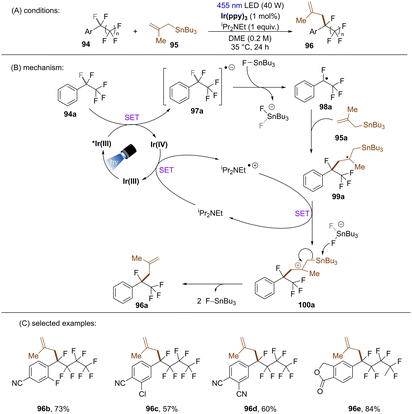
Yasuda and co-workers developed an Ir-catalysed boron-free benzylic C(sp3)–F allylation of perfluoroalkylarenes (Scheme 39).120 The proposed mechanism was supported by Stern–Volmer quenching and DFT calculations. A key feature is the abstraction of fluoride anion from radical anion 97a by FSnBu3 to afford fluorobenzyl radical 98a. Except for the use of comparatively toxic allylic stannanes, this new strategy also requires iPr2NEt (1 equiv.) as an electron shuttle to complete the catalytic cycle. Similarly to König's work,117 this methodology features a limited substrate scope with regard to perfluoroalkylarenes, only more electron-deficient arene substrates were competent substrates.
 |
| | Scheme 39 Defluoroallylation of perfluoroalkylarenes using allylic stannanes. | |
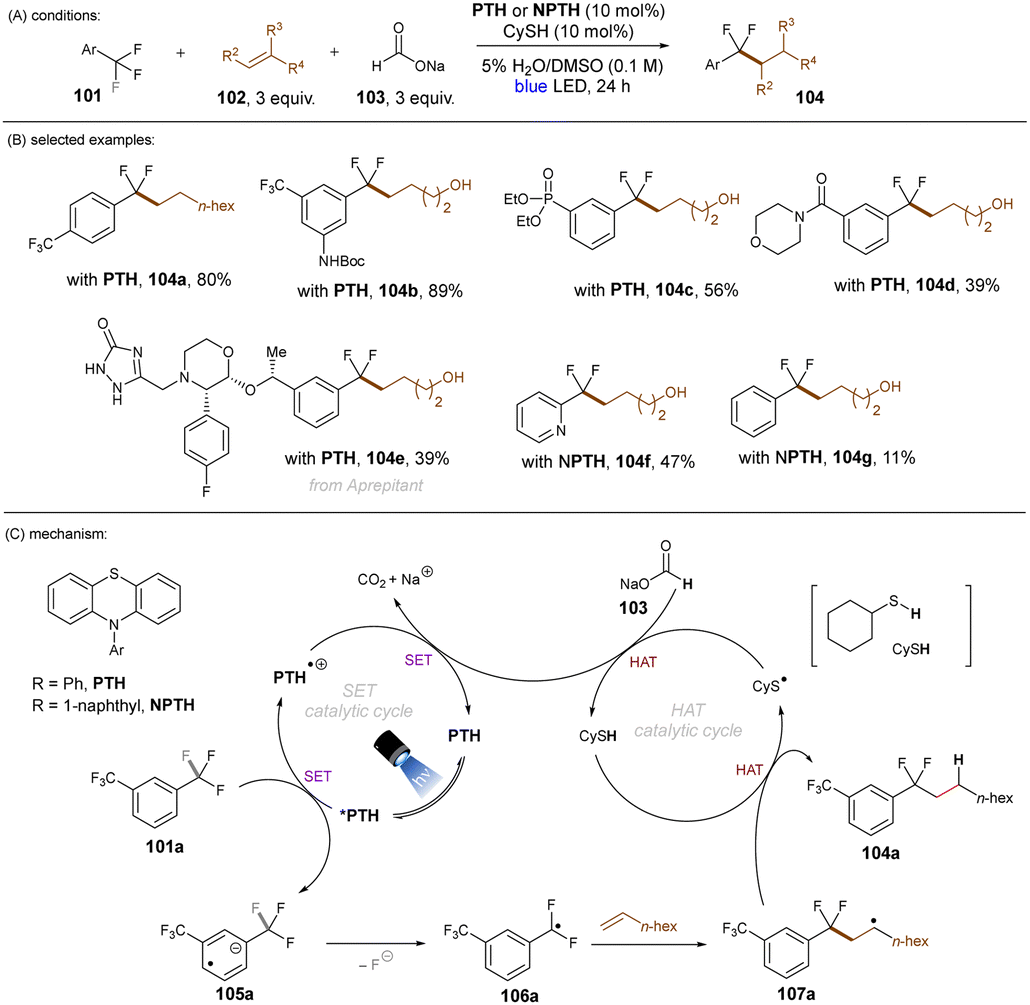
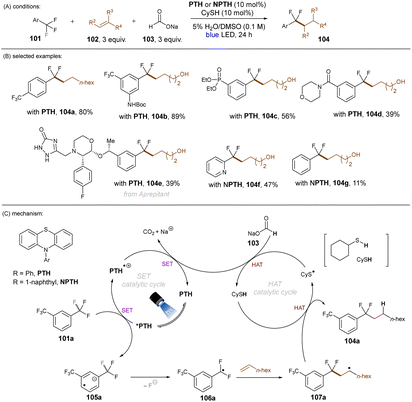
Jui and co-workers demonstrated an elegant approach for the activation of C(sp3)–F bonds in trifluorotoluenes and their coupling with simple alkenes (Scheme 40).121 For the transformation the organic photoredox catalyst N-phenylphenothiazine (PTH) was used in the presence of sodium formate under irradiation with a blue light LED. PTH was chosen for its highly reducing excited state (*E1/2 = −2.10 V vs. SCE) which is sufficient to reduce several trifluoromethylarenes. The authors proposed a dual catalytic mechanism (Scheme 40). Here, 101a is reduced by the *PTH to give radical anion 105a. Upon loss of F−, radical 106a would be formed, which could couple with a simple alkene to give 107a. HAT of 107a with CySH would furnish product 104a. CySH would be regenerated by HAT of CyS radical with sodium formate leading to concomitant reductive regeneration of PTH and release of Na+ and CO2. A large variety of alkenes and trifluoromethylarenes gave the coupling product in moderate to good yields. A wide range of functional groups – either on the alkene or the arene – were tolerated under the mild reaction conditions.
 |
| | Scheme 40 Defluoroalkylation of trifluoromethyl arenes using PTH as an orgPC. | |
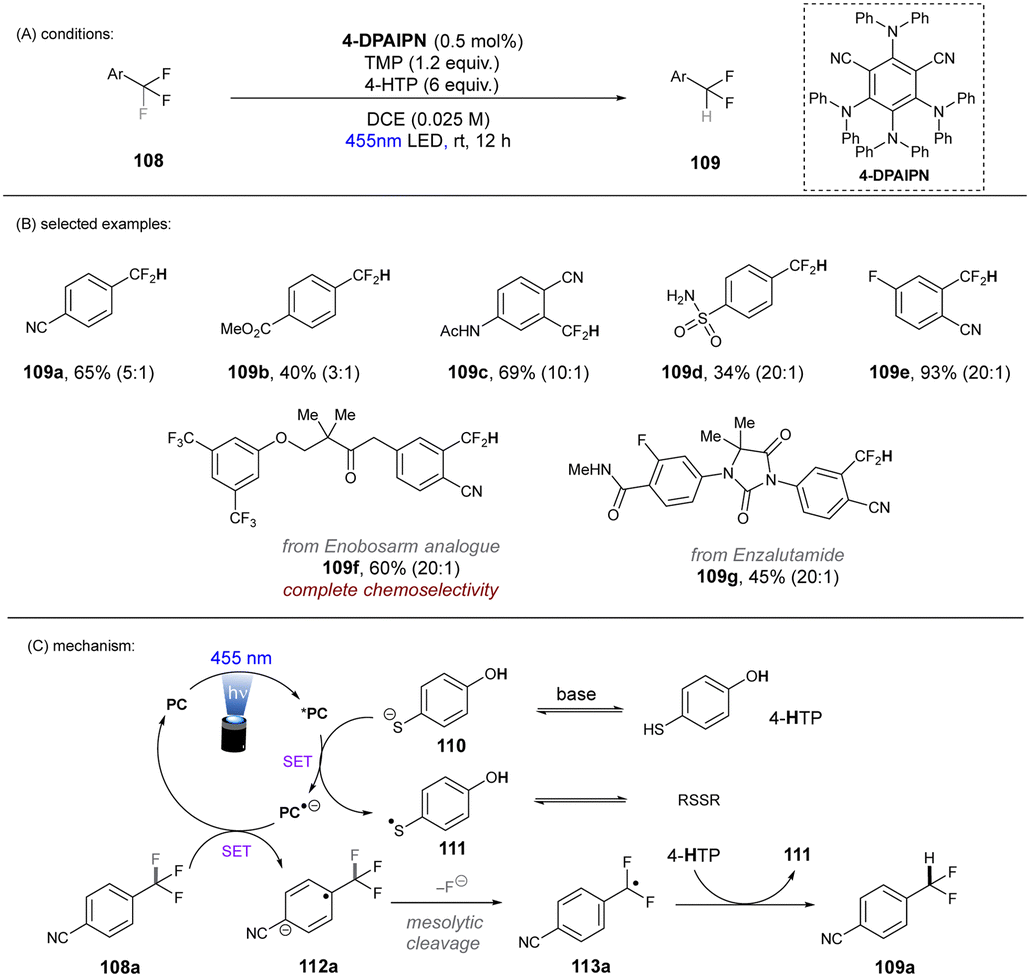
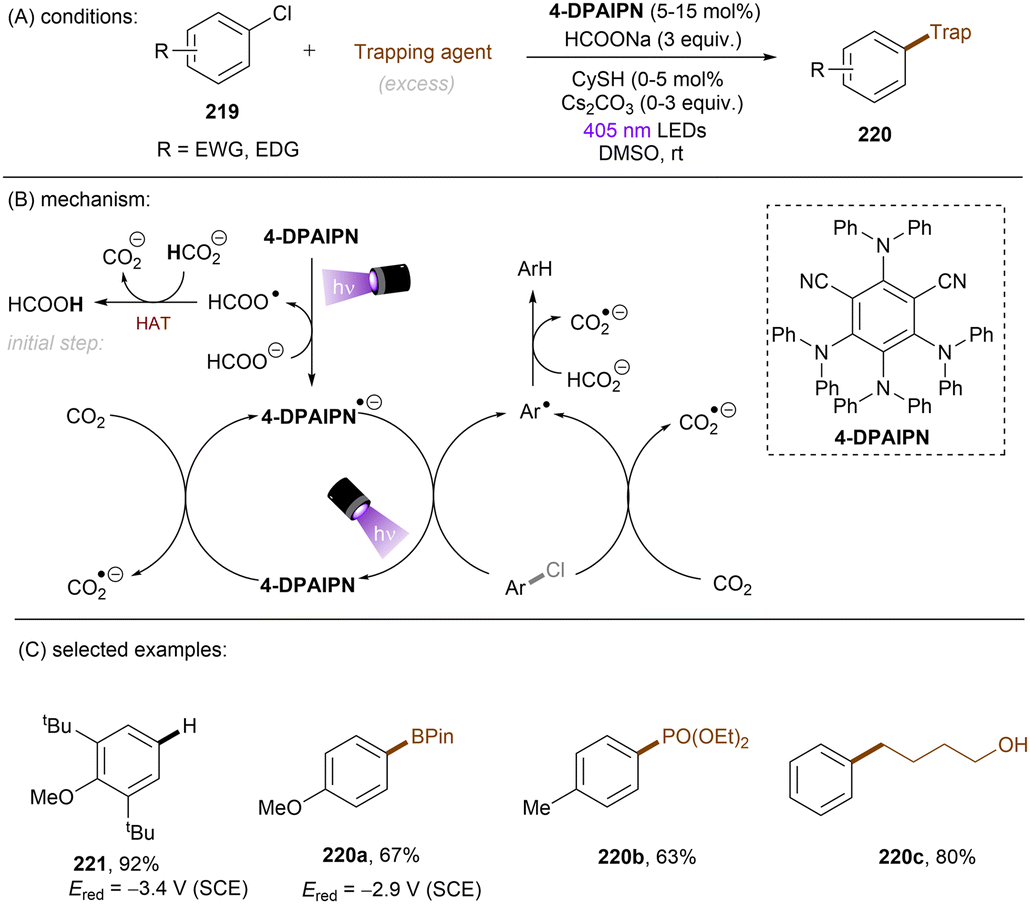
In 2020, Gouverneur and co-workers published HDF of highly activated trifluoromethylarenes using an organophotoredox catalyst under blue light irradiation (Scheme 41).122 One key problem for the desired mono-HDF of trifluoromethylarenes is over-HDF. Even though the reduction potential for formation of radical anion decreases slightly when going from 108 to 109 the rate for mesolytic cleavage of F− increases. Therefore, a suitable system had to be developed to avoid over-HDF. Providing such a compromise as a cheap, metal-free orgPC; 2,4,5,6-tetrakis(diphenylamino)-isophtalonitrile (4-DPAIPN) was found to be the ideal system. Several electron-deficient trifluoromethylarenes could undergo HDF in moderate to good yields. It was also possible to achieve selective HDF for different pharmaceuticals. For the transformation the authors proposed a mechanism as shown in Scheme 41. Excess 4-hydroxythiophenol (4-HTP) functions as both an electron and hydrogen atom donor.
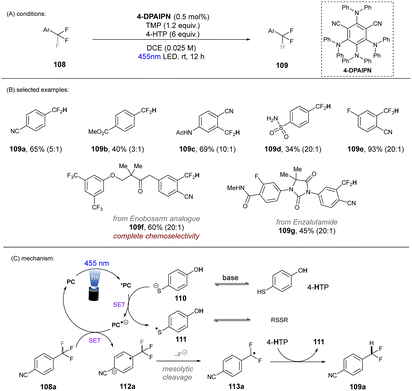 |
| | Scheme 41 Hydrodefluorination of activated trifluoromethylarenes by 4-DPAIPN. Selectivity of –CF2H over –CFH2 is given in parentheses. | |
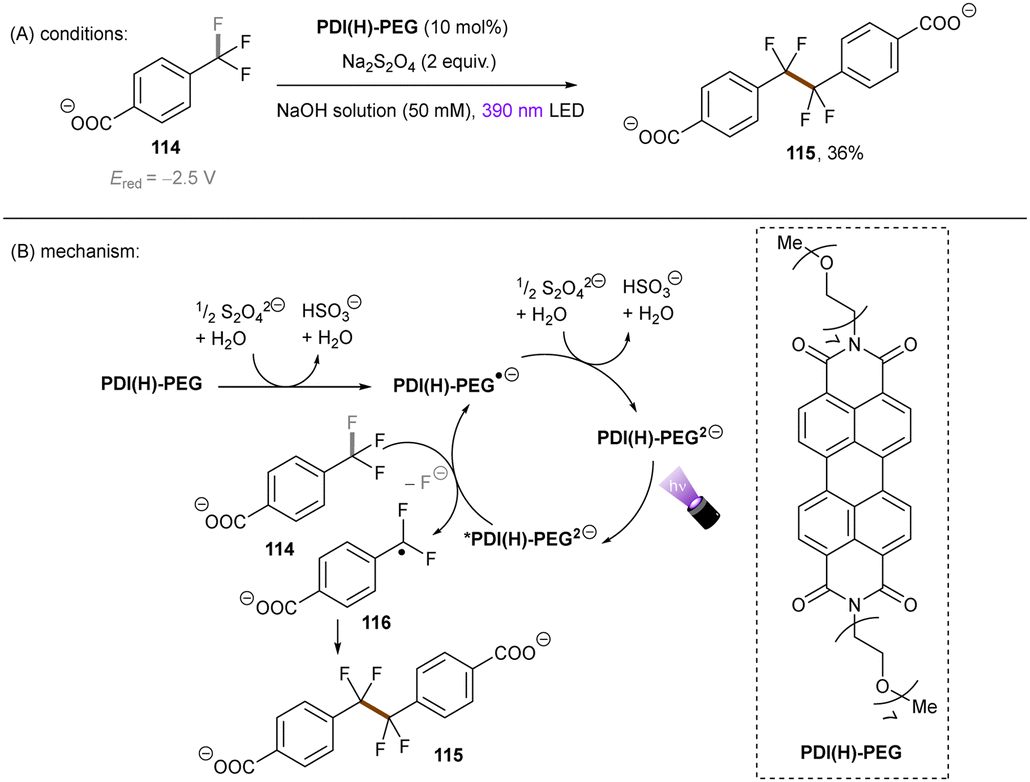
The Wenger group123 investigated the catalytic utility of perylene diimide (PDI) dianions (Scheme 42). Although PDI2− have been explored previously, the investigation was mainly on their photophysics.124,125 These closed-shell species were prepared by treatment of PDI with Na2S2O4 and (i) are more reducing (*Eox1/2 = −2.5 V vs. SCE) and (ii) their longer-lived excited states (presumably singlet states, ca. 6 ns) than those of the open-shell radical (mono) anions which are well-known as photoreductants to reduce activated haloarenes.126,127 The excited state reductive behaviour of the polyethylene glycol chain (PEG)-linked PDI was tested in aqueous solution by employing trifluoromethyl benzoate anion 114 (Ered = −2.5 V vs. SCE). The desired reaction did not occur under irradiation with 525 nm light, however a dimerization product was formed in 36% yield by using 390 nm light, confirming the reducing ability of the photoexcited PDI2−. To explain the different performances of 390 nm and 525 nm light, the authors speculated that 390 nm light excites the PDI2− to a higher-order excited state and leads to the liberation of a solvated electron,128–130 although no direct spectroscopic evidence was yet provided for its detection or to rule out direct PET to the substrate from a higher excited state enabled by a preassembly. Despite the reaction scope being quite narrow, this pioneering work demonstrates the great synthetic potential of dianion catalysts originated from organic dyes. Future applications of these catalysts for synthetic reactions in organic solvents may lead to a pronounced use of this new strategy, as well as opportunities to degrade organic pollutants in water.
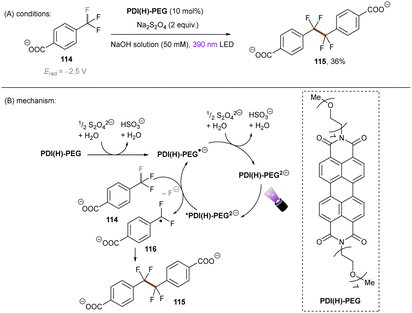 |
| | Scheme 42 Reductive activation of benzylic C(sp3)–F bonds using PDI(H)-PEG dianion. | |
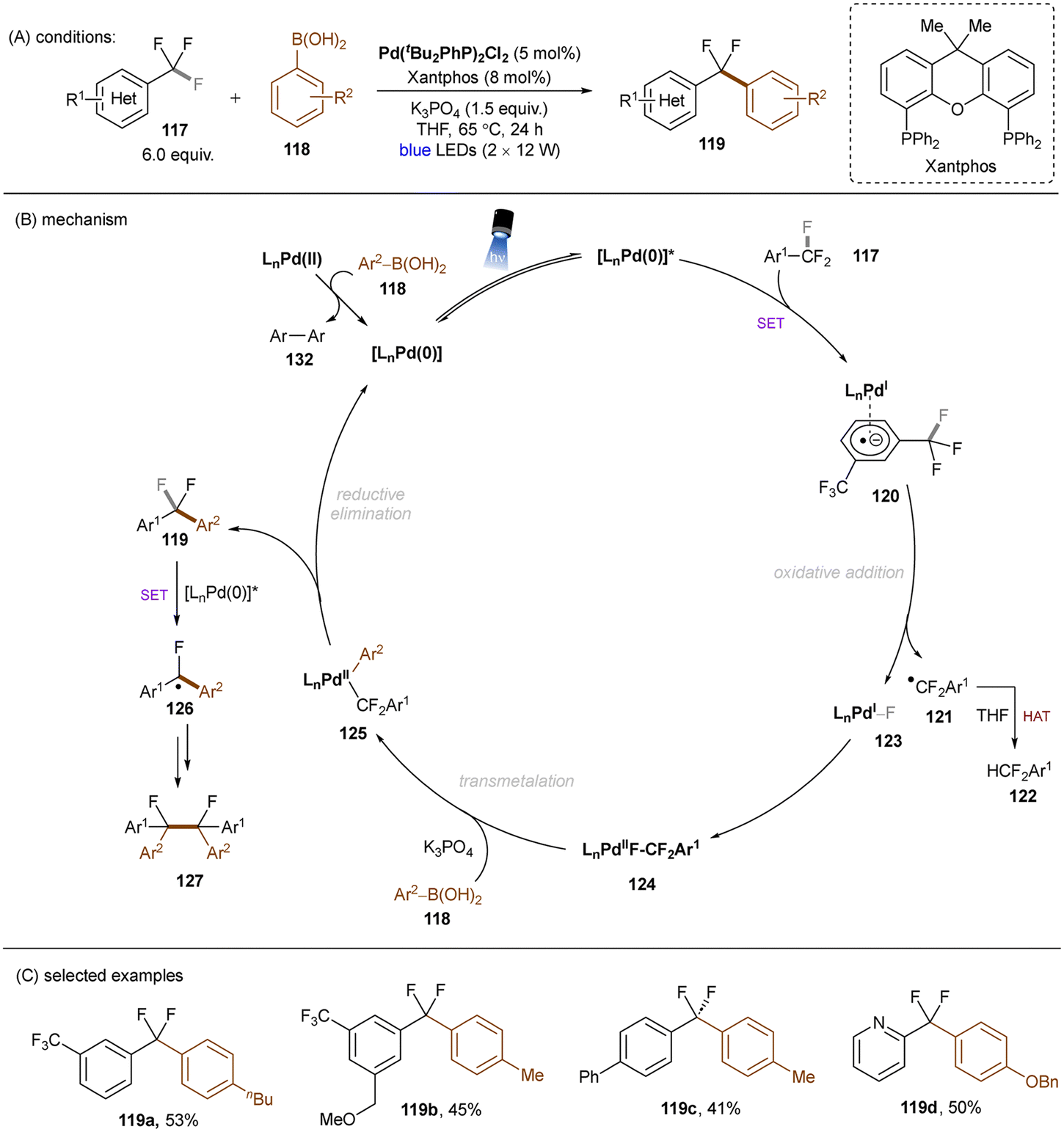
Excited palladium catalysis are also effective for C–F bond activations. The Zhang group developed a photoinduced Pd-catalysed monoarylation of trifluoromethyl C(sp3)–F bonds with arylboronic acids (Scheme 43).131 SET from photoexcited LnPd(0) catalyst to Ar–CF3117, followed by C(sp3)–F bond cleavage, generates a difluorobenzyl radical and LnPd(I)F, which then recombine to afford a Pd(II) complex 124. Analogously to previous work on HDF reactions of trifluorotoluenes,132 herein the electrophilic difluorobenzyl radical is able to abstract a hydrogen atom from THF to yield the hydrodefluorinated product 122. 124 undergoes further transmetalation with arylboronic acid 118 followed by reductive elimination to deliver the desired coupling products 119. Further defluoro dimerization to afford 127 is also feasible. This reaction tolerates electron-deficient trifluorotoluenes and electronically differential arylboronic acids. Substrates bearing other cleavable bonds such as haloarenes were not included. This work elegantly combines PRC with Pd catalysis and clearly demonstrates the utility of oxidative addition to an excited Pd(0) catalyst vs. a ground-state Pd(0) catalyst for achieving strong bond activations.
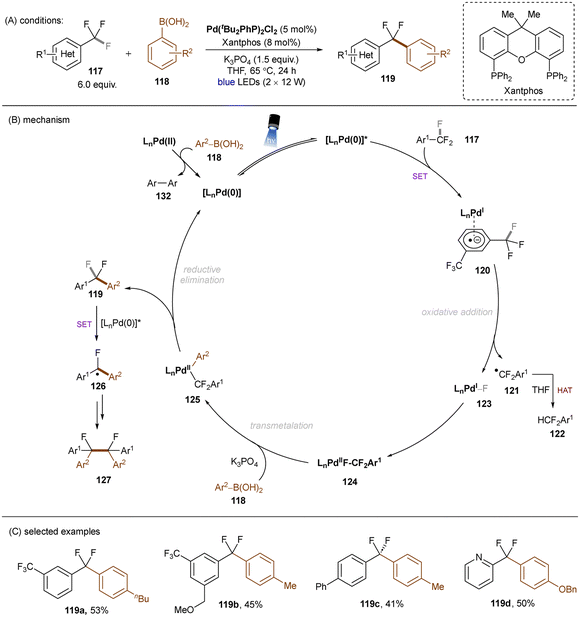 |
| | Scheme 43 Photoinduced Pd-catalyzed reductive C(sp3)–F arylation. | |
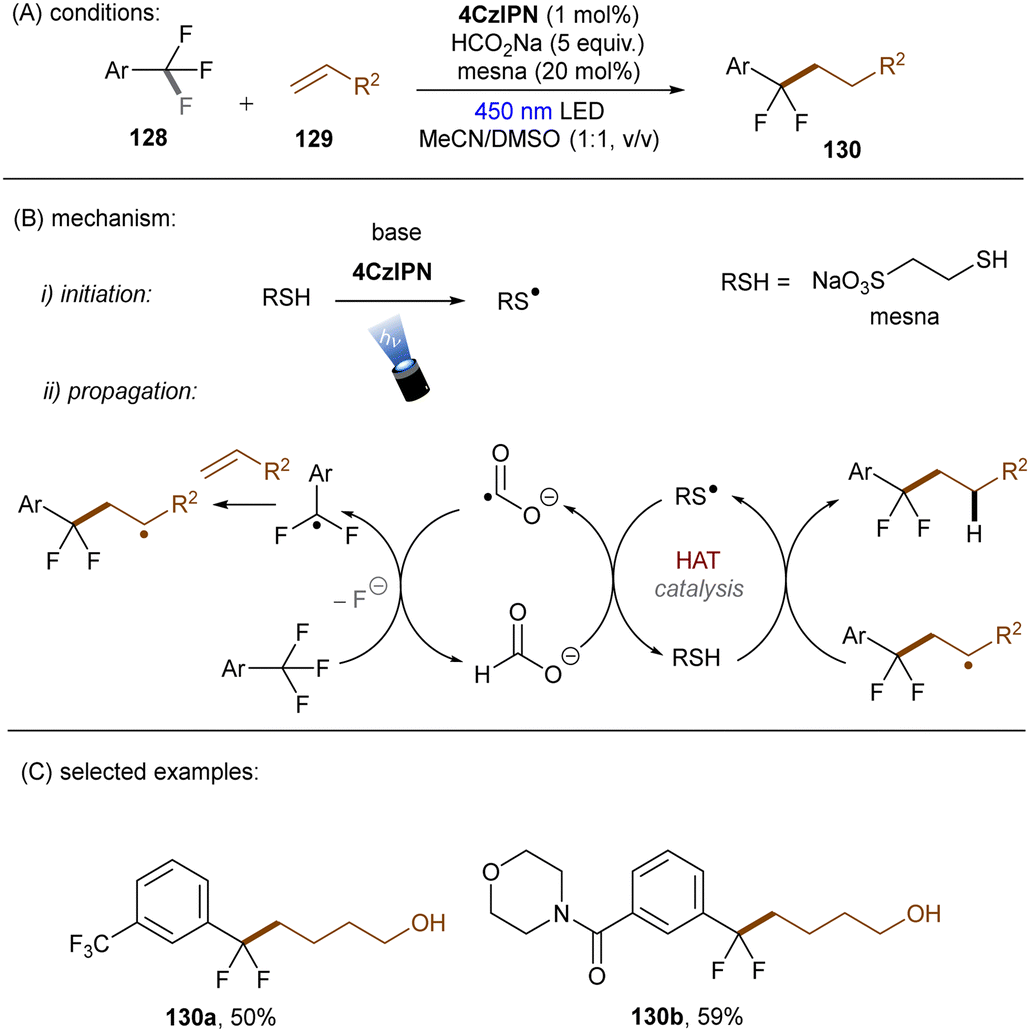
In 2020, the Li group reported that CO2˙− (E0 = −2.21 V vs. SCE), generated by SET and HAT from sodium formate, is able to reduce electron-deficient aryl chlorides.133 In a related study, the Jui group reported a photoinduced defluoroalkylation of trifluoromethyl arenes 128 by using 4-CzIPN as an orgPC in the presence of HCOONa and a thiol co-catalyst, sodium 2-mercaptoethane-1-sulfonate (mesna) (Scheme 44).1344-CzIPN initiates the radical chain by photooxidizing mesna to its thiyl radical. Instead of being proposed as a reductant to return PTH catalyst from its radical cation in their early report,121 CO2˙− (generated as aforementioned) was proposed to reductively cleave activated aryl trifluoromethyls to difluorobenzyl radicals in this work. Subsequent radical addition to an alkyl olefin gives rise to a secondary alkyl radical which then abstracts a hydrogen atom from the thiol to afford the final product and regenerate the thiyl radical. This radical chain has been confirmed by replacing the 4-CzIPN/thiol combination with other initiators such as the persulfate/thiol combination or a disulfide. Shortly afterwards, Molander and co-workers identified a HAT strategy for the generation of CO2˙− from formate salt for mono-defluoroalkylations of trifluoroacetates and trifluoroacetamides with alkyl olefins (Scheme 45).135 A modified diaryl ketone, Benzophenone-1,136 was employed and its triplet excited state was proposed to initiate the radical chain by HAT from formate to generate CO2˙−. Although inexpensive, cyclohexyl thiol as a HAT catalyst is still needed in addition to the Benzophenone-1 orgPC. This reaction proceeded smoothly with a diverse suite of trifluoro- and difluoroacetates including some biologically-relevant ones. Its scalability was confirmed by an efficient 50 mmol batch reaction. In contrast, the activation of trifluoroacetamides further required Zn(OTf)2 as a Lewis acid.
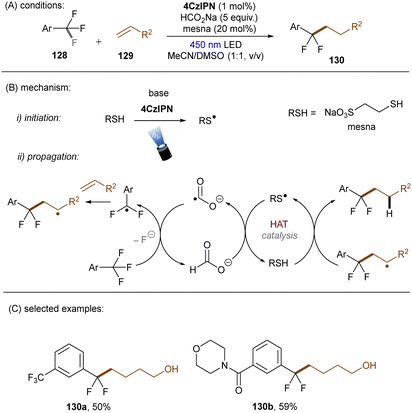 |
| | Scheme 44 Reduction of activated aryl chlorides by CO2˙−. | |
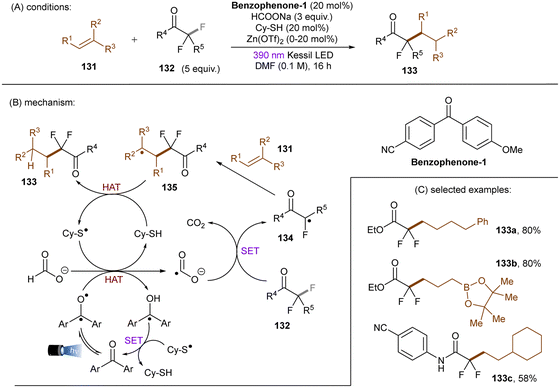 |
| | Scheme 45 Trifluoroacetate and trifluoroacetamide C(sp3)–F bond reductive cleavages by CO2˙−. | |
Very recently, S. Zhu, Guo, X. Zhu and co-workers reported a reductive defluoroalkylation of azolyl trifluoromethanes 135 by using a fac-Ir(ppy)3PC/thiol/formate system (Scheme 46).137 The authors proposed a radical chain mechanism similar to the aforementioned reports. However, direct reduction of substrates by the excited reduced [Ir] complex was not ruled out because the use of (NH4)2S2O8 as initiator led to a markedly lower yield. The reaction was amenable to trifluoromethylated heteroarenes, however, more challenging trifluorotoluene derivatives (137c) remained a limitation. In a distinct fashion, the Yu group utilized CO2˙− for defluorocarboxylations of mono-, di-, and trifluoroalkylarenes, where CO2 gas substituted for super-stoichiometric amounts of external formate salts (Scheme 47).138 CO2 acts as both a CO2˙− source and a carboxylating reactant. Triethylsilane was used as a terminal reductant, which reacts with CO2 in the presence of Cs2CO3 to deliver both formate and silanolate anions. A reductive quenching of the photoexcited PC by the silanolate affords PC˙− and a siloxy radical. This siloxy radical then abstracts a hydrogen atom from the formate anion to give CO2˙− and a silanol. CO2˙− is able to reduce different fluoroalkylarenes and difluoroacetates as well as difluoroacetamides to their corresponding stabilized benzylic radicals 144 (with liberation of fluoride), which are then reduced to carbanions 145 by PC˙−. Nucleophilic addition of the carbanions to CO2 ultimately yielded the desired products. Two different PCs, 4-CzIPN (*E1/2 = −1.21 V vs. SCE) and 3-DPAFIPN (*E1/2 = −1.59 V vs. SCE), were examined, chosen based on the reduction potential required for the generated benzyl radical intermediates. Notably, CO2˙− could also be formed through a three-component reaction of fluoride anion, hydrosilane and CO2. Spectroscopic and computational studies supported the proposed mechanism.
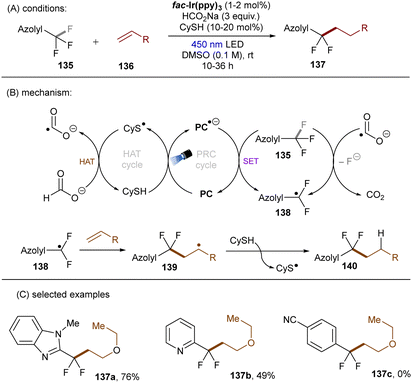 |
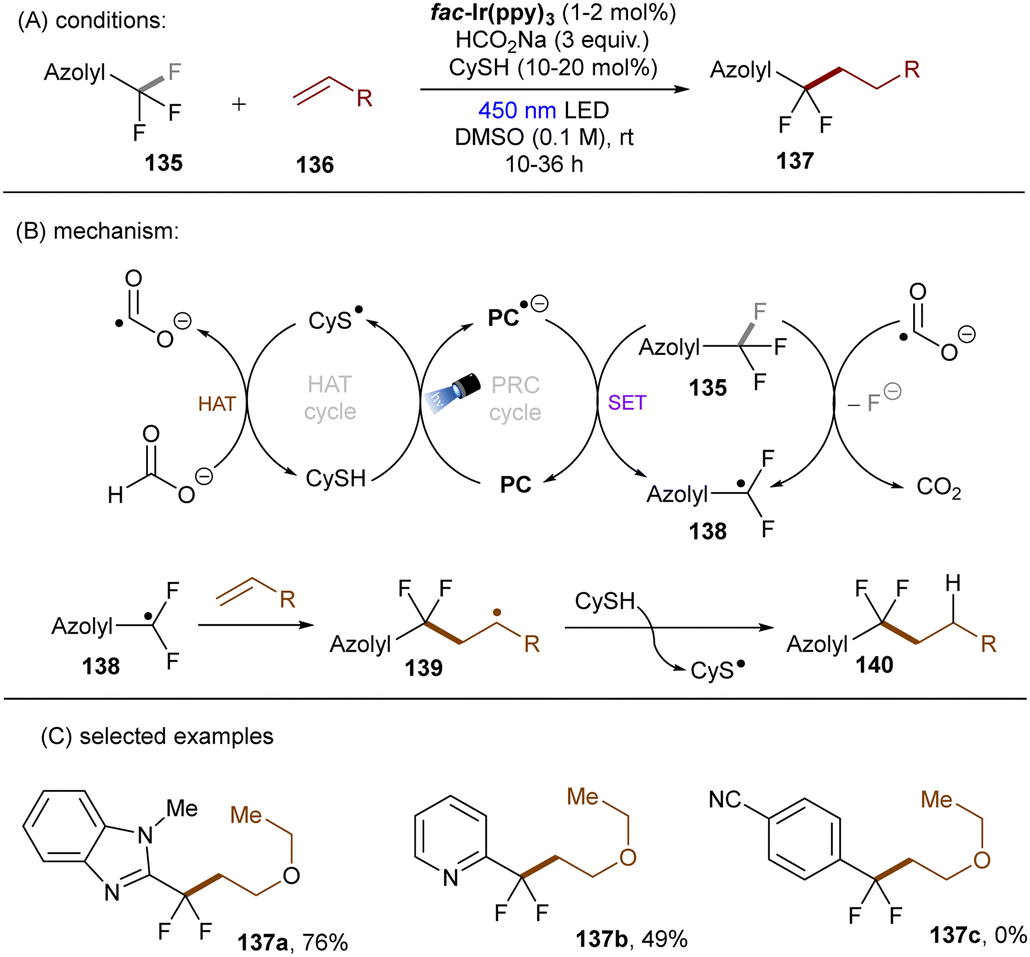
| | Scheme 46 Reductive cleavages of C(sp3)–F bonds in azolyl trifluoromethanes by CO2˙−. | |
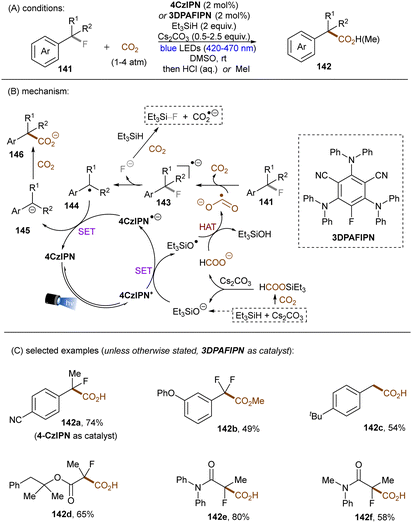 |
| | Scheme 47 Dual roles of CO2 in photochemical C(sp3)–F bond carboxylation. | |
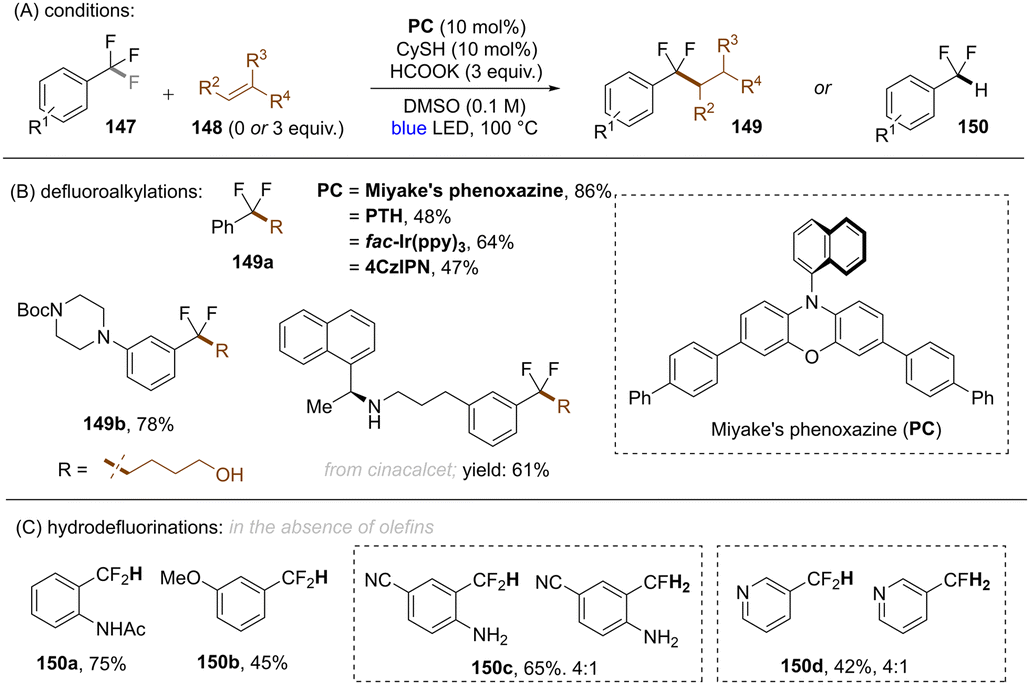
A general limitation in all these works on reductive C(sp3)–F bond cleavage, was that only trifluoromethylarenes bearing electron withdrawing groups could be activated.117,118,120–123,131,134,137 In a later study, Jui and co-workers achieved selective C(sp3)–F bond cleavages of electronically-diverse trifluoromethylarenes by leveraging higher temperature conditions under visible light photoredox catalysis (Scheme 48).139 Among the four different catalysts tested at different temperatures: PTHEred(PC˙+/PC = −2.10 V vs. SCE), fac-IrIII(ppy)3 (Ered(PC˙+/PC = −1.73 V vs. SCE)), Miyake's phenoxazine (Ered(PC˙+/PC = −1.70 V vs. SCE)) and 4CzIPN (Ered(PC˙+/PC = −1.18 V vs. SCE)), the reaction with Miyake's phenoxazine at 100 °C was found to be most effective in their transformation, even though the reducing power is considerably lower than that of PTH. Additionally, 4-CzIPN exhibited the same efficiency as PTH at 100 °C despite having a much less potent excited state reductive power, suggesting that C(sp3)–F mesolytic cleavage of the radical anion may be rate determining rather than the initial PET. Based on computational studies and reactivity patterns, the authors claimed that the excited state lifetimes of the catalysts (presumably, in their triplet states) had a stronger bearing on conversion at higher temperature than their excited state reductive power. The authors were able to couple a large number of electron-rich and electron-poor trifluoromethyl(hetero)arenes with a multitude of alkenes in moderate to excellent yields. Moreover, C(sp2)–F functionalization of different pharmaceutical compounds were achieved in good yields. Furthermore, in the absence of an olefin, the authors were achieved mono-HDF of ArCF3.
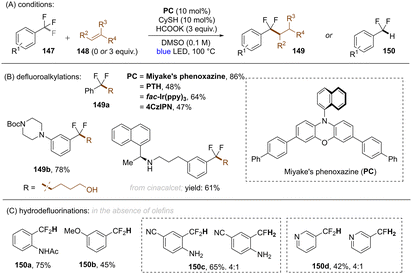 |
| | Scheme 48 Defluoroalkylation or hydrodefluorination of trifluoromethyl arenes using Miyake's phenoxazine at 100 °C. | |
As an advantage over the report of Gouverneur and co-workers,122 the conditions of Jui and co-workers achieved mono-HDF of different electron-rich trifluoromethylarenes. However, in terms of highly activated (featuring electron withdrawing groups) trifluoromethylarenes, over-reduction was unavoidable and a mixture of mono- and double-HDF products were obtained.
Shang and co-workers140 reported a milder reduction of trifluoromethyl compounds, including trifluoroacetamides, trifluoroacetates and trifluoromethyl arenes, to difluoroalkyl compounds by merging previously reported thiol/formate HAT strategy108,126 with visible light phenolate catalysis at room temperature (Scheme 49). The authors claimed a direct reduction of trifluoromethyl compounds by *PO− to afford difluoroalkyl radicals. Such electrophilic radicals either undergo HAT to form hydrodefluorinated products or add to an electron-rich olefin 35 followed by HAT to deliver defluoroalkylated products. CO2˙−, generated in situ by HAT, was proposed to regenerate PO− from PO˙ rather than reduce trifluoromethyl substrates. This method is efficient for the activations of diverse trifluoroacetamides with reduction potentials as deep as −2.5 V. Numerous olefins including terminal olefin-based natural products were successfully employed as radical acceptors. Given the general drawbacks of contemporary photoredox catalysis methods for C(sp3)–F reduction – that trifluoromethyl arenes require either a strong electron withdrawing group on the arene or otherwise require assistance of high reaction temperatures117,118,120–123,131,134,137,139 – a noteworthy advantage of this catalytic system is it offers an unprecedented approach to reductive activations of C(sp3)–F bonds in electron-rich trifluorotoluenes such as trifluoromethyl anisole under benign visible light PRC conditions. Highly related to phenolates are thiolates, which have attracted attention recently in the PRC field.141 Simple thiolates upon visible light excitation have proven to be super reducing species for inert chemical bond activations. Given Miyake and co-workers' report on catalyst-free photochemical synthesis of diaryl sulfides from thiols and aryl halides (mostly bromides and iodides) through the formation of EDA complexes between thiolates and aryl halides,142 the Shang group pursued defluorofunctionalizations of trifluoromethyls using thiophenols as photoredox catalysts, although thiols frequently serve as HAT catalysts in the literature (Scheme 50).143 4-Methoxythiophenol (ArSH) was identified as optimal and its thiolate form absorbs visible light. An advantage of this new catalytic system is that it avoids using an additional exogenous thiol catalyst as ArSH acts as both a HAT and photoredox catalyst. Taking together the small, simple thiol catalyst and formate as both electron and hydrogen donors, respectively, this comprised an elegant approach to facile mono C–F activation of trifluoroacetamides, trifluoroacetates and trifluoromethyl (hetero)arenes. Especially, trifluoromethyl arenes bearing EDGs (Me, OMe) were converted to their difluoroalkyl products in good yields. It is worth noting that substrates such as trifluoromethyl benzadehyde were not included, potentially due to promiscuous nucleophilicity of ArS− in the presence of electrophilic (formyl) groups. This could be a potential drawback of this method.
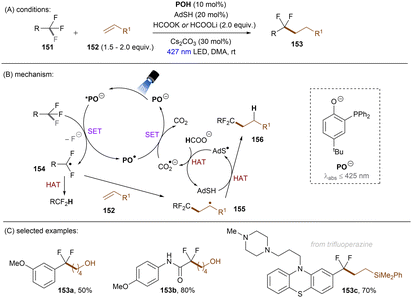 |
| | Scheme 49 Photoexcited deprotonated phosphanyl phenol (POH) for the reduction of less activated trifluoromethyl groups. | |
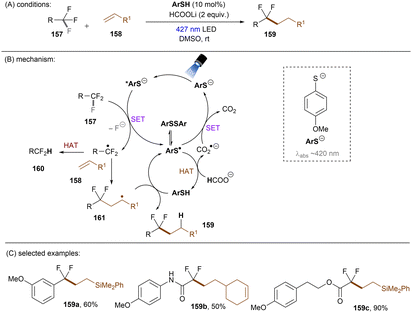 |
| | Scheme 50 A photoexcited thiolate for the reduction of unactivated trifluoromethyl groups. | |
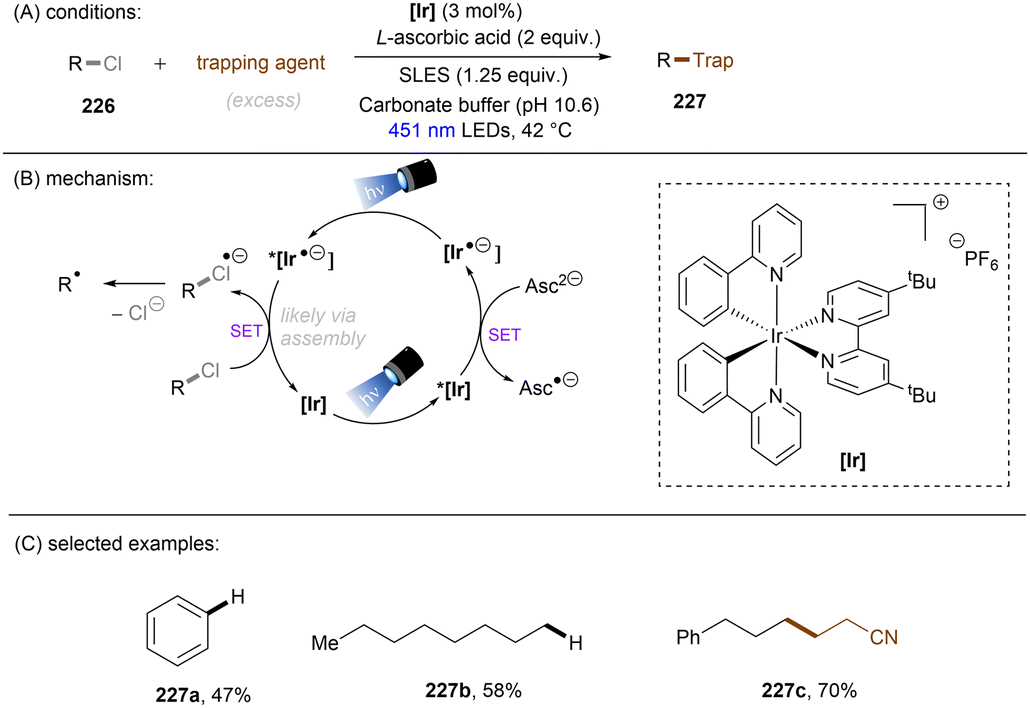
Unlike benzyl chlorides and α-chloro carbonyls, unactivated alkyl chlorides generally have reduction potentials deeper than −2.5 V vs. SCE. Only a limited number of photoreducing systems are able to reductively engage these inert compounds. As well as unactivated aryl chloride/fluoride reductions, the highly photoreducing orgPC, CAR1, developed by the Matsubara group (Scheme 51), was applicable to the activation of unactivated alkyl chlorides.61 Primary, secondary and tertiary alkyl chlorides were found to undergo hydrodechlorination to deliver the corresponding products in 56–87% yields (Scheme 51). The primary one reacted fastest, presumably due to its more easily accessible redox potential. However, alkyl fluorides (166) could not be engaged. Beyond hydrodechlorinations, more synthetically useful transformations were achieved by trapping the generated alkyl radicals with different reaction partners. As a continuation of their ArOH-catalysed photoreduction of electronically different aryl chlorides, the Xia group disclosed the reduction of unactivated alkyl chlorides (Scheme 52).144 The generated alkyl radicals are rapidly intercepted by the in situ formed TEMPO to deliver the desired products. Diverse primary, secondary and tertiary alkyl chlorides were compatible under the standard conditions. Importantly, this reaction proved promising for the late-stage C(sp3)–Cl bond functionalization of natural products and pharmaceuticals. However, TEMPO was required as the radical coupling partner, delivering rather niche products. The use of different radical trapping partners represents room for further development.
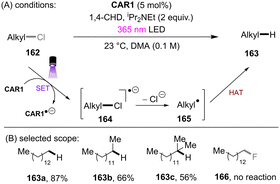 |
| | Scheme 51
CAR1 as a potent photoreductant for reductive activations of unactivated alkyl chlorides. | |
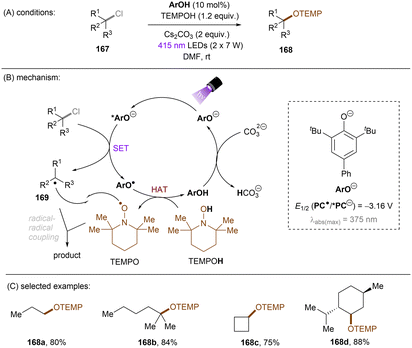 |
| | Scheme 52 A photoexcited phenolate for the reduction of unactivated alkyl chlorides. | |
As eluded to earlier in the work of Zhang (Scheme 43), photoexcited palladium complexes have also found applications in organic synthesis.145,146 The Hong group developed a photoinduced Pd-catalysed Mizoroki–Heck coupling of alkyl chlorides and olefins (Scheme 53).147 Alkyl radicals 173 – generated by SET from the excited state Pd(0) complex to alkyl chlorides 170 – are prone to add to styrenes 171. This forms radicals 174 which further undergo chlorine atom transfer to give alkyl chlorides 175. A base-promoted elimination from 175 yields the final products 172. The elimination step was confirmed by the consistency of KIE values of the standard reaction with values for the K2CO3-assisted elimination of a pre-generated alkyl chloride intermediate. The intermediacy of alkyl radicals was supported by both a radical inhibition experiment using TEMPO and a radical clock experiment using a cyclopropylmethyl chloride. The reaction did not work at 100 °C in the dark, demonstrating the advantage of this method over classic thermal Pd catalysis. A variety of primary, secondary and tertiary alkyl chlorides were tolerated. As coupling partners, not only styrenes, but also enamines and acrylamides were competent. Shortly after this report, the Rueping group described a cascade alkylation/annulation reaction for the synthesis of indolin-2-one derivatives (Scheme 54).148 A comparable mechanism involving a photoexcited-state Pd catalyst was proposed. Similarly to the Hong group, the reaction scope with respect to alkyl chlorides was broad. Ligand tuning led to remarkable changes in product yields; a bulky ligand was found to be essential for reaction efficiency. The replacement of Pd(OAc)2/Ph3P with Pd(PPh3)4 led to no significant loss of efficiency.
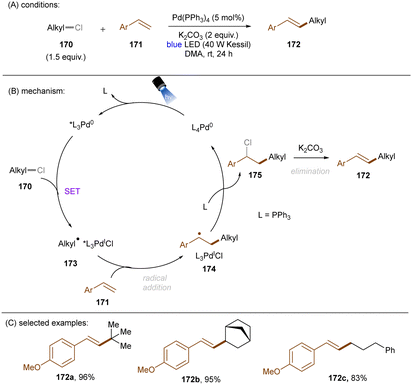 |
| | Scheme 53 A photoexcited [Pd(0)] complex as the active species for the reduction of unactivated alkyl chlorides. | |
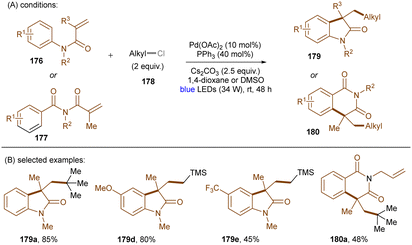 |
| | Scheme 54 Photoinduced Pd-catalyzed cascade alkyl chloride activation/annulation for the synthesis of indolin-2-ones. | |
Reductions of alkyl chlorides by low-valent Ni(I) intermediates were further reported by the Lloret-Fillol group. In 2019, they devised an elegant strategy to activate unactivated alkyl chlorides for intramolecular radical cyclizations.149 It was claimed that the cleavage of C(sp3)–Cl bonds is driven by an Ni(I) complex generated in situ by photoredox catalysis. Recently, a more synthetically useful intermolecular version of this chemistry was reported by the same group (Scheme 55). Again, the Ni catalyst is responsible for alkyl chloride activation, the generated alkyl radical can be intermolecularly trapped by a styrene to form a benzylic radical.150 The intermediacy of the alkyl radical was confirmed by a radical clock experiment using a cyclopropyl-substituted styrene. As indicated by computational modelling, the C(sp3)–Cl activation step proceeded via an oxidative addition, then homolysis of the weak Ni(III)–C bond promoted by photoirradiation directly or by EnT from the excited [Ir] catalyst. This is in stark contrast to the report of Radius and Marder where a direct chlorine atom abstraction by Ni(I) complex was proposed as the key step for aryl chloride activations.151 However, based on computations, a concerted halogen atom abstraction mechanism (the greyed arrows in the middle catalytic cycle) was not completely ruled out as both mechanisms had comparable activation barriers. The mechanism of the C(sp3)–Cl activation step is probably substrate-dependent. The radical anion of the employed [Ir] photocatalyst, generated by reductive quenching with DIPEA, is responsible for returning Ni(I) and reducing the benzyl radical to carbanion. This bimetallic system was found to be very general, with a wide range of styrenes and unactivated alkyl chlorides tolerated. While 1° and 2° alkyl chlorides were suitable substrates, sterically hindered 3° alkyl chlorides were found to be a limitation, as further evidence for the proposed SN2 oxidative addition mechanism. Notably, a tandem intramolecular reduction, cyclization of 6-chloro-1-hexenes and subsequent intermolecular cross-coupling with styrenes for the synthesis of 183e was also feasible.
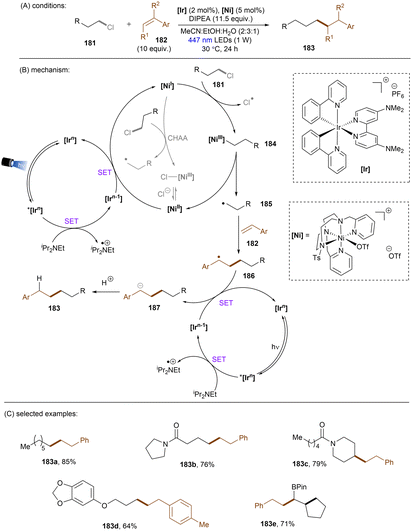 |
| | Scheme 55 Photogenerated [Ni(I)] complex as the active species for the reduction of unactivated alkyl chlorides. | |
Deoxygenation of alcohols through C(sp3)–O bond cleavage is a highly useful strategy for drug discovery as alcohols are ubiquitous in pharmaceuticals and natural products.152–155 However, direct reductive activation of dialkyl ethers is difficult and rare as alkoxides are generally worse leaving groups compared to halides. The stronger C(sp3)–O bond BDE (vide supra, Fig. 1) renders mesolytic cleavage more difficult following SET. This can be compensated by employing a phenolate as the leaving group, that is to say; an alkoxyarene (188) as the substrate. In 2021, the Matsubara group disclosed a PCET-enabled C(sp3)–O bond cleavage of aryl alkyl ethers 188 (Ered < −2.6 V vs. SCE) by employing N-unprotected 3,6-diamino carbazole (DAC) as an orgPC in DMSO in the presence of Cs2CO3 and 1,4-cyclohexadiene (1,4-CHD), culminating in a new method for phenol deprotection (Scheme 56).156 The reaction worked in a comparable efficiency in the absence of 1,4-CHD, suggesting that DMSO could also serve as a hydrogen atom donor. Other control experiments by varying the amount of Cs2CO3 showed that the conversion and product yield correlated with the loading of Cs2CO3. For comparison, the N–Et carbozole derivative – previously used for the activation of aryl chlorides61 – afforded a much lower yield, confirming the critical role of the N–H moiety in the catalyst.
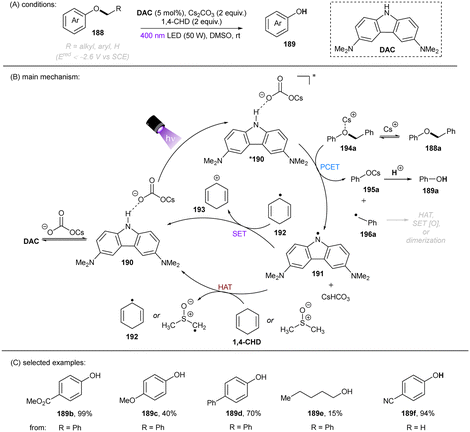 |
| | Scheme 56 Reduction of aryl alkyl ethers enabled by proton-coupled electron transfer. | |
Further spectroscopic studies including 1H NMR, UV-vis absorption and fluorescence quenching experiments indicated an interaction of the ground state DAC with Cs2CO3 through hydrogen bonding forming complex 190, which upon photoexcitation donates an electron to the Cs+-coordinated phenyl benzyl ether 194a. The C(sp3)–O bond then fragments to give cesium phenolate 195a and benzyl radical 196a. The former is protonated to give phenol product, the latter undergoes HAT, dimerization, or SET oxidation to ultimately afford toluene, 1,2-diphenylethane, or benzaldehyde. However direct single-electron reduction of the substrate by the photoexcited DAC in a simple photoredox mechanism could not be ruled out since fluorescence quenching of *DAC by 188a was observed, although less pronounced than that in the presence of Cs2CO3. Despite the moderate product yields for dialkyl ethers and alkoxy benzene substrates bearing strong EDGs, the reaction offers an attractive reductive method for deprotections of phenols compared to catalytic hydrogenations with precious metal catalysts, and a strategy for the deoxygenation of alcohols by using an arene as the auxiliary. However, the difficulties in installing the arene auxiliary directly from the alcohol limits the synthetic applications of this deoxygenation method.
Very recently, the Wickens group157 developed a more general and practical PRC deoxygenation strategy based on previous highly reducing PRC HAT/CO2˙− systems developed by Li,133 Jui134 and Wickens (Scheme 57).158 The benzoyl group is an inexpensive and readily-installable auxiliary for the protection of alcohol substrates. The resulting benzoates (197) generally possess reduction potentials of ca. −2.2 V vs. SCE, which is comparable to the reducing power of CO2˙− (E1/2(CO2/CO2˙−) = −2.2 V vs. SCE). CO2˙− was proposed as the active species that reduces the benzoate substrate 197 to its radical anion 199. This undergoes protonation to radical species 200 instead of a direct C(sp3)–O cleavage to deliver radical 201 with the release of benzoate anion (BzO−). Radical 201 undergoes a thiol-assisted HAT to deliver the deoxygenated product 189 and regenerate CO2˙− to propagate a chain reaction. The mechanism was supported by a parallel computational study of the C(sp3)–O cleavage steps in the presence/absence of a proton. The energy needed for the C(sp3)–O cleavage in 199 is 10 kcal mol−1 higher than that in 200. The authors proposed this was key to engaging aliphatic (not only benzylic) protected alcohols, allowing a broad substrate scope to be achieved. Benzoates originated from activated or unactivated primary, second and tertiary alcohols were successfully defunctionalized. Noteworthy is that the method was applicable to sugars, representing a promising deoxygenating strategy for the synthesis of mutant deoxy-sugars. Questions arise over the innocence of the Zn additive in activation of substrates as a Lewis acid (and promoting reduction steps) or the innocence of the benzoyl group which has been shown to be a photoactive auxiliary under 400 nm irradiation.159 It should be noted that an analogous deoxygenation reaction was achieved in 2014 by Reiser and co-workers using electron-deficient (3,5-trifluoromethyl) benzoate esters and a conventional Iridium PC, including non-benzylic (albeit activated) systems.153 However, truly unactivated alkyl alcohol-derived benzoates were unsuccessful, and here competing C(sp3)–F cleavage of the auxiliary was an issue as fluoride was detected in these reactions.
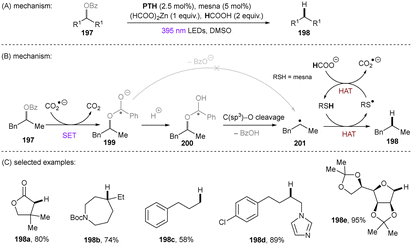 |
| | Scheme 57 PRC reduction of benzoates assisted by in situ generated CO2 radical anion. | |
3.1.3. Reduction of π systems (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C).
In many radical reactions for alkene functionalization, olefins participate as radical acceptors. There have also been reports on alkene functionalization achieved via direct SET reduction of the C
C).
In many radical reactions for alkene functionalization, olefins participate as radical acceptors. There have also been reports on alkene functionalization achieved via direct SET reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. The Wagenknecht group published a series of papers on Markovnikov addition of alcohols to olefins under highly photoreducing conditions.160–162 Aiming for a red-shifted and more photoreducing PTH derivative, the authors designed and tested a new series of N-arylphenothiazines, with N-(para-dialkylaminophenyl)phenothiazines performed well in the reductive activation of olefins (Scheme 58).162 The photoexcited PTH1 and PTH2 derivatives were able to reduce unactivated olefin 202 to its radical anion 204 by SET. Protonation of 204 by MeOH afforded radical 205 which is SET oxidized by the radical cation catalyst to regenerate the PC and afford cation 206. Finally, addition of MeOH and deprotonation furnishes ether 207. A series of alkenes including styrenes and alkyl olefins with reduction potentials as deep as −3.0 V vs. SCE were well tolerated. A limitation of this reaction is that only 1,1-disubstituted olefins were suitable substrates, possibly due to the need to stabilize a 3° carbocation.
C double bond. The Wagenknecht group published a series of papers on Markovnikov addition of alcohols to olefins under highly photoreducing conditions.160–162 Aiming for a red-shifted and more photoreducing PTH derivative, the authors designed and tested a new series of N-arylphenothiazines, with N-(para-dialkylaminophenyl)phenothiazines performed well in the reductive activation of olefins (Scheme 58).162 The photoexcited PTH1 and PTH2 derivatives were able to reduce unactivated olefin 202 to its radical anion 204 by SET. Protonation of 204 by MeOH afforded radical 205 which is SET oxidized by the radical cation catalyst to regenerate the PC and afford cation 206. Finally, addition of MeOH and deprotonation furnishes ether 207. A series of alkenes including styrenes and alkyl olefins with reduction potentials as deep as −3.0 V vs. SCE were well tolerated. A limitation of this reaction is that only 1,1-disubstituted olefins were suitable substrates, possibly due to the need to stabilize a 3° carbocation.
 |
| | Scheme 58 Reductive PRC hydroetherification of olefins. | |
Very recently, the Chiba group revealed that a combination of polysulfide anions and formate is capable of facilitating photoinduced dearomatization of substituted naphthalenes, indoles, and related heteroaromatic compounds, the reduction potentials of which are more positive than −2.5 V (Scheme 59).163 For example, dearomatization of 1-naphtamide 208a (Ered = −2.35 V) to 1,4-dihydronaphthalene 209a proceeded smoothly in the presence of polysulfide anions, formate, and methanol in DMSO under irradiation with 427 nm light. A radical-polar crossover mechanism induced by SET from photoexcited polysulfide anions *Sn2− operates to form dearomatized carbanion intermediates, which are subsequently protonated by MeOH. The resulting polysulfide radical anions Sn˙− undergo HAT from formate to regenerate polysulfide anions Sn2− upon deprotonation. This HAT process concomitantly forms highly reducing CO2˙− (Ered(CO2/CO2˙− = −2.2 V vs. SCE164)), which could serve as another SET reductant to facilitate the process.
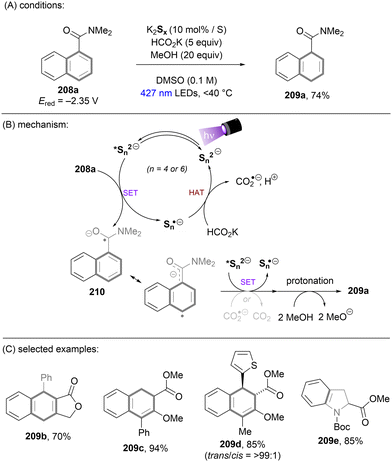 |
| | Scheme 59 Birch-type reduction of arenes by photoexcited polysulfide anions. | |
3.2. Two-photon processes
While visible light photoredox catalysis has enabled a diverse set of organic transformations under mild conditions, the energy limitation of single visible photon limits synthetic applicability to the activations of inert substrates such as aryl fluorides and chlorides; attractive targets as cheaper feedstock chemicals or for the valorization of persistent chemical waste. The deeply negative reduction potentials and high BDEs render the activation of inert substrates highly challenging. Efforts have been made towards the activation of energy demanding organic compounds under mild conditions.165 Except for designing new neutral visible light photocatalysts (PCs),166,167 the consecutive photoinduced electron transfer (conPET) strategy has successfully been employed for super reductions,10,168 in which the energies of two photons are harvested to promote the desired process. In a typical example, a neutral state PC is photoexcited to *PC which can be easily quenched by an electron donor, such as triethylamine, to afford the one-electron-reduced form of PC (PC˙−) – the ‘active’ photocatalyst. This can be excited by a second photon (Scheme 60), which compensates for the energy mismatch and thus enables the activation of challenging substrates.
 |
| | Scheme 60 General mechanistic paradigm for conPET reduction reactions. | |
3.2.1. C(sp2)–X activation.
In 2014, the König group first reported the reductions of electron-deficient aryl chlorides by using perylene diimide (PDI) as the PC in a conPET strategy (Scheme 61).126 Although a follow-up study by Ceroni raised questions over whether the reductant was really *PDI˙− or a catalyst decomposition product that promotes the reduction of aryl chlorides,169 this conPET strategy has nonetheless attracted considerable attention from synthetic community. Other PCs including Rhodamine-6G (Rh-6G+),170,171 and 1,8-dihydroxyanthraquinone (AqOH)172 were reported by the same group to activate aryl bromides (aryl chlorides were not achieved) through a two-photon conPET mechanism.
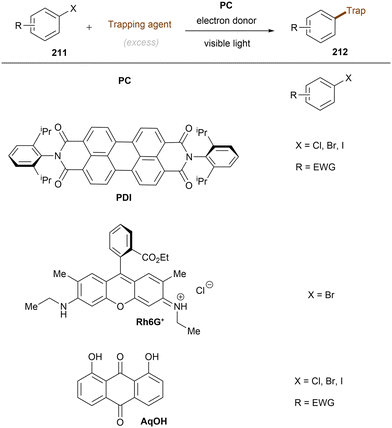 |
| | Scheme 61 Pioneering reports by König on reductive conPET reactions grouped by structure. | |
The conPET strategy for the activation of aryl chlorides had been limited to substrates bearing strong acceptors at their arene ring until a follow-up study by the same group (Scheme 62). Although Rh-6G+ as a conPET catalyst was not able to reduce aryl chlorides,170 the authors found that the issue could be overcome by combining conPET with lanthanide catalysis, termed 'metal ion coupled consecutive photoinduced electron transfer'.173 This affords a more reducing system for difficult reduction events as adding metal ions impacts greatly on the rate of SET by activating either the substrates or the PCs. Key to the enhanced reducing power is the addition of catalytic amount of LnI2 salt or its non-classic forms, DyI2 and NdI2 (Scheme 62).174 The combination of the high reducing power of a lanthanide(II) ion and its regeneration by a Rh-6G+-based conPET strategy offers a new platform for the reduction of unactivated chloroarenes. The authors proposed that a Ln3+ ion, generated in situ by oxidative quenching of Ln2+ by ArCl, forms a complex with ground state Rh-6G+. Photoexcitation of this complex followed by reductive quenching by DIPEA delivers Rh-6G˙–Ln3+. The presence of Ln3+ suppresses BET to DIPEA˙+. Rh-6G˙–Ln3+ absorbs a second photon to enable SET between Rh-6G˙ and Ln3+, regenerating Rh-6G+ and Ln2+ to close the catalytic cycle.
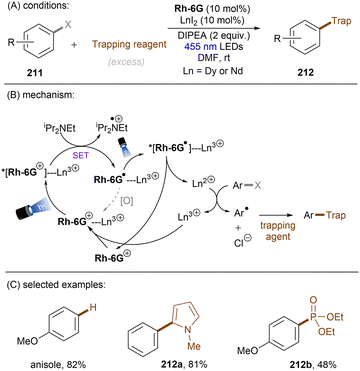 |
| | Scheme 62 Combining Ln(II) ions with conPET for reductive C(sp2)–Cl bond activations. | |
Following these pioneering studies, diverse new conPET catalysts have been designed to engage energy demanding substrates. Jacobi von Wangelin and Pérez-Ruiz explored the candidacy of dicyanoanthracene (DCA) as a conPET catalyst.175 In the presence of DIPEA (1.2 equiv.), DCA is able to catalyse the cross coupling of aryl chlorides 213 with N-methyl pyrrole 214 under irradiation with white LEDs (Scheme 63). Control experiments showed that this reaction requires two different wavelengths, since either 455 nm or 525 nm light only delivered a low product yield. This was explained by the spectroscopic study of DCA and its radical anion where the former absorbs blue light and DCA˙− absorbs green light. The green light excited DCA˙− has a reductive power of −2.5 V (vs. SCE), rendering it a strong reductant that readily donates an electron to electron-deficient aryl chlorides (Ered > −2.5 V). The reaction scope was well investigated and a clear trend could be observed: the reaction efficiency is contrary to the electron richness of the chloroarenes. Compared to PDI, the DCA system was able to engage less electron-deficient aryl chlorides such as 1-chloro-4-(trifluoromethoxy)benzene 212e. However, reductions of more electron-rich substrates including 4-chloroanisole 213d (Ered = −2.9 V) remained elusive, limiting the synthetic utility of the process. Even though the redox potential criteria should render this process feasible (and electrogenerated and authentically isolated DCA˙− were both subsequently shown to engage 4-cloroanisole) (vide infra) this speaks to a crucial threshold concentration of DCA˙− needed for reactivity that is seemingly inaccessible in the conPET strategy due to BET to DIPEA˙+. It could also speak to the in situ decomposition of DCA˙− to another active photoreductant species (for a discussion, see section 5 on PEC, vide infra).
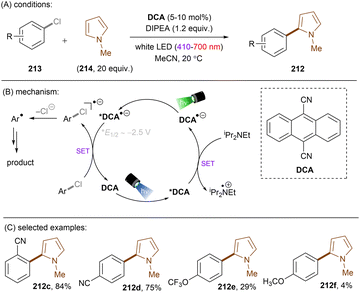 |
| | Scheme 63
DCA as conPET catalyst for the activation of aryl chlorides. | |
Polyzos, Francis and co-workers disclosed the reductions of activated aryl chlorides (215) using Ir(dtbby)(ppy)2+ ([Ir]) as PC with super-stoichiometric (10 equiv.) triethylamine as the electron donor (Scheme 64).176 The authors claimed that the substrates were reduced by the photoexcited [Ir-H], generated in situ by light-promoted SET reduction of [Ir] followed by HAT from Et3N˙+ to its bpy˙− ligand, rather than the unstable photoexcited [Ir˙−]. Interestingly, the replacement of Et3N by DABCO led to no emission colour change although quenching of the photoexcited [Ir] was observed. DABCO suppresses the formation of [Ir-H] as the deprotonation and HAT steps become more difficult. Due to its benign reducing power, *[Ir-H] could only reduce activated aryl chlorides such as methyl 4-chlorobenzoate 215a and 2-chlorobenzonitrile 215b.
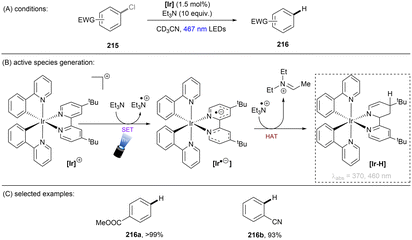 |
| | Scheme 64 A transformed [Ir] complex as a conPET catalyst for the activation of C(sp2)–Cl bonds. | |
Jan Kutta, Cibulka, König and co-workers described the activation of 4-chloroanisole 6 using deazaflavin reductive catalysis (Scheme 65).177 A two-photon reaction pathway was also proposed. Photoexcitation of dFl catalyst generates a singlet 1dFl* and a triplet 3dFl* followed by subsequent intersystem crossing. While radical pair 1[dFl˙−,DIPEA˙+], generated by reductively quenching of 1dFl* by DIPEA through SET, recombines to turn ground state dFl through BET, radical pair 3[dFl˙−,DIPEA˙+] allows the accumulation of stable dFl˙−, i.e. the semiquinone anion, followed by excitation by a second photon. The generated *dFl˙− is highly reducing (*E1/2(dFl/dFl˙−) = −3.3 V) and thus able to reduce 4-chloroanisole. The generated 4-methoxyphenyl radical was converted into anisole through HAT. Intercepting these radicals for more synthetically useful transformations was still a challenge.
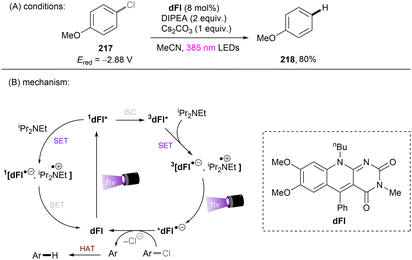 |
| | Scheme 65 Deazaflavin (dFl) as conPET catalyst for dehalogenation of 4-chloroanisole. | |
Isophthalonitrile-based photocatalysts, such as 4-CzIPN and 4-DPAIPN, are generally used as conventional visible-light sensitizers with appreciable oxidizing abilities (E1/2(*PC/PC˙−) > +1.0 V) under single photonic PRC. The Wickens group recently demonstrated that the radical anion of 4-DPAIPN generated is visible light-excitable and could be used as potent reductants for cleaving strong C(sp2)–Cl bonds (Scheme 66).158 Instead of commonly used tertiary amines, sodium formate was identified as an electron donor in this reaction to reduce *4-DPAIPN to 4-DPAIPN˙−, subsequent photoexcitation of which delivers *4-DPAIPN˙−. This highly reducing species is capable of reducing truly unactivated aryl chlorides 8, even those with deep reducing potentials (Ered < −2.9 V), to aryl radicals. UV-Vis absorption experiments offered evidence to support this claim. Once the aryl radical is formed, it can be trapped by phosphites, B2Pin2 or alkenes or abstract a hydrogen atom from formate to afford the dechlorinated product and CO2 radical anion. CO2˙− is also a potent reductant which is able to activate electron-deficient aryl chlorides as reported independently by the Li group133 in 2020 and the Jui group134 in 2021. To figure out whether the aryl chloride substrates were reduced by *4-DPAIPN˙− or CO2˙−, the authors attempted to generate CO2˙− intermediate in situ by UV-promoted homolysis of PhSSPh and subsequent HAT from formate to the thiyl radical in the absence of 4-DPAIPN. As a result, while less reductively challenging substrate 4-chlorobenzonitrile was fully converted, poor conversions of less activated substrates (chlorobenzene and 4-chloroanisole) were observed under these conditions. By contrast, comparable conversions of these three substrates were observed in the presence of 4-DPAIPN catalyst. These results imply that CO2˙− intermediate participates in the reduction of more easily reducible substrates possibly in parallel with the *4-DPAIPN˙−. *4-DPAIPN˙− is the active species for the reduction of those inert substrates and CO2˙−, in this case, is responsible for reducing the photocatalyst to its radical anion form without requiring persistent two-photon excitation. In addition to electron-rich aryl chlorides, a phenyl ammonium salt and a phenyl phosphate could be reductively cleaved under the reaction conditions. The discovery of 4-DPAIPN˙− as a potent photo-reductant herein potentially offers a reasonable explanation for Jui's recent work where 3-DPAFIPN was reported to reduce activated alkyl chlorides beyond its redox potential.178
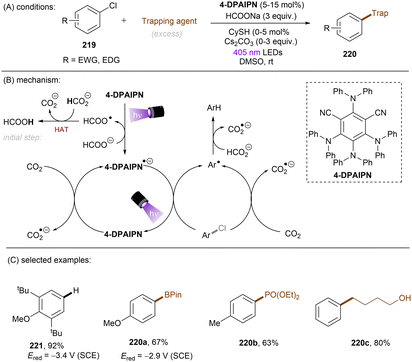 |
| | Scheme 66 Functionalizations of less activated aryl chlorides using 4-DPAIPN. | |
In parallel with the study of the Wickens groups, the Wu group disclosed the use of a new isophthalonitrile catalyst, 3-CzEPAIPN, for the activation of unactivated aryl chlorides 13 through a conPET mechanism (Scheme 67).179 A species was detected with a lifetime of 12.95 ns, that displayed a (weak) linear photoluminescence lifetime quenching by time-resolved emission spectroscopy. While the authors proposed this emitting species could be 3-CzEPAIPN˙−, this would contrast with the observation that most organic radical anion species are ultrashort-(ps)lived in their doublet excited states.22 More likely is the in situ transformation of isophthalonitrile species by CN-substitution occurs in the reaction that gives rise to another isophthalonitrile emitter, as found synthetically and studied in detail spectroscopically.180,181 The generated aryl radical intermediates were intercepted by B2Pin2, triarylphosphine or methyl phosphate to forge C(sp2)–B/–P bonds. The potent reducing ability of *3-CzEPAIPN˙− was proven by the activation of inert C(sp2)–Cl bonds, such as that in 1-chloro-4-ethoxybenzene 13a with a reduction potential as deep as −2.94 V. This catalytic system also effects benzylic C(sp3)–F activation in 4-cyanotrifluorotoluene 224. Under the standard conditions, over-reduction was unavoidable and 4-trifluoromethyl benzonitrile underwent full HDF to afford tolunitrile 225 in 50% yield. Importantly, prospects for scale-up were demonstrated via continuous flow, resulting in a high yield and a decent productivity of 13.1 g day−1.
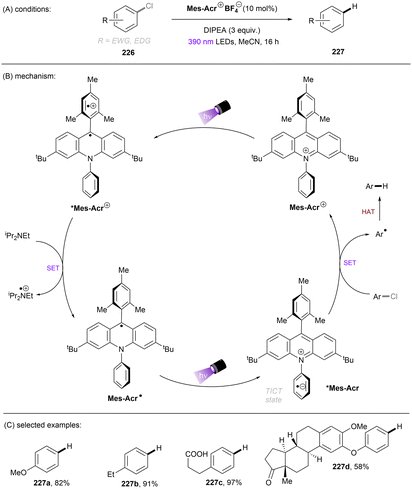 |
| | Scheme 67 Functionalization of C(sp2)–Cl and C(sp3)–F bonds using 3CzEPAIPN. | |
Another type of super photoreductant – that can circumvent the ultrashort lifetime issue of radical ions and prerequisite for assembly – is an in situ formed neutral radical intermediate. On the basis of their discovery with acridinium catalysts in the oxidation direction,98–100 the Nicewicz group recently revealed the applications of these catalysts in the activation of reductively challenging organic compounds through a two-photon pathway (Scheme 68).182 Neutral acridine radical Mes-Acr˙, generated through reductive quenching of excited Mes-Acr+ with DIPEA, is a weak reductant unable to reduce aryl chlorides. However, it becomes deeply reducing (Epox = −3.36 V vs. SCE) after absorbing a second visible photon. The photoexcited state is a ‘twisted intramolecular charge-transfer (TICT) state that features both an N-phenyl radical anion and acridinium, as supported by photophysical, TD-DFT and experimental investigations. This TICT doublet state is potent enough to reductively cleave aryl chlorides to aryl radicals. The generated aryl radicals then underwent HAT to give hydrodechlorinated products. The reaction scope was very well explored. Apart from efficient reduction of electron-deficient aryl halides, more reductively challenging chloroarenes including 1-chloro-4-ethylbenzene 62b (Ered = −2.72 V) and 4-chloroanisole 62a (Ered < −2.9 V) were also competent substrates. This report clearly shows how a commonly used potent photo-oxidant can be converted in situ into an extreme photo-reductant.
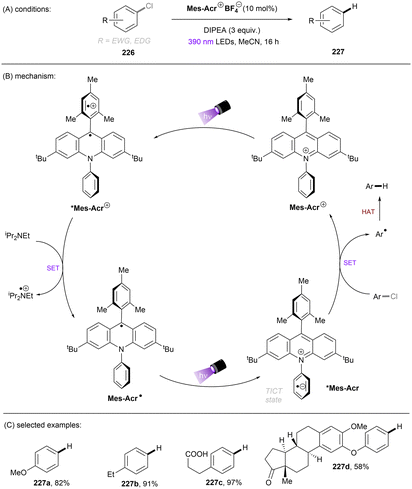 |
| | Scheme 68 Photoexcited acridine radical as the active species for the reduction of inert aryl chlorides. | |
N,N′-Di-n-propyl-1,13-dimethoxyquinacridine radical (nPr-DMQA˙), first synthesized and characterized by spectroscopy in 2014,183 was isolated by the Gianetti group in 2020.184 This radical is visible-light-absorbing (391 nm, 440 nm, 467 nm, and 557 nm) and the authors assigned the excited double state a (surprisingly long) lifetime of 4.6 ± 0.2 ns and an oxidation potential of −3.31 V vs. SCE. Importantly, this persistent radical can be prepared in situ by reductive quenching of photoexcited nPr-DMQA+ with electron donors such as alkylamines,185 rendering its use in conPET reduction of substrates with reduction potentials as deep as −3.0 V vs. SCE. The Gianetti group reported such a reaction by employing nPr-DMQA+ −BF4 as a conPET catalyst (Scheme 69).186 Under blue light irradiation, nPr-DMQA+ −BF4 was able to activate chloroarenes 64 with pyrrolidine as the sacrificial reductant. Both aryl chlorides bearing electron-withdrawing (CN, CF3) and -donating (OMe, Me) groups can be reduced to the corresponding aryl radicals. These radicals can be captured by HAT, B2Pin2, P(OEt)3 or N-methylpyrrole to provide C–Cl functionalized products in good to excellent yields. The reaction mechanism was thoroughly investigated by spectroscopic, computational and experimental studies on the isolated radical catalyst. The authors proposed that solvated electrons might be generated from *nPr-DMQA˙.
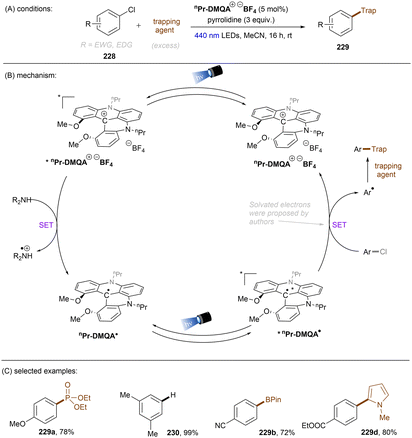 |
| | Scheme 69 Photoexcited nPr-DMQA radical as the active species for the reduction of inert aryl chlorides. | |
A simple CeIII complex, [CeIIICl6]3−, was identified as a potent photoreductant (*E1/2 = −3.45 V vs. Fc0/+) by the Schelter group. It was employed to dehalogenate aryl halides in quartz vessels.187 This catalyst was then applied to effect a Miyaura type borylation of aryl chlorides (Scheme 70).188 The reaction is initiated by the formation of [CeIIICl6]3− from CeCl3 and Cl− primed to photo-excitation. The generated excited state of [CeIIICl6]3− (233) reduces aryl chlorides 230 to form aryl radicals which are trapped by B2Pin2 to form a diverse set of Ar–BPin products 231. Although efficient, a drawback of this highly potent catalytic reducing system is the need for of less user-friendly/energy efficient 350 nm UV light and (thus) the expensive quartz glassware. Longer wavelengths such as 380 nm and 450 nm delivered no product.
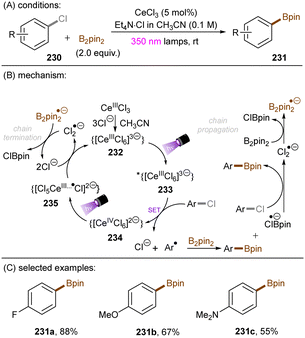 |
| | Scheme 70
In situ generated [CeIIICl6]3− as potent reductant for the borylation of unactivated aryl chlorides. | |
3.2.2. C(sp3)–X activation.
In sharp contrast to the work by Polyzos, Francis and co-workers where Ir(dtbby)(ppy)2+ as the conPET catalyst is not able to activate electron-rich chloroarenes,176 Kunz, König and co-workers showcased the potent reducing ability of the same [Ir] complex in aqueous media for the reduction of not only unactivated chlorobenzene but also inert alkyl chlorides (e.g.226b/c) (Ered < −2.8 V) (Scheme 71).189 The reaction occurred in a microheterogeneous aqueous solution, and water-soluble L-ascorbate (Asc2−) was utilized as the electron donor. The stability of the in situ generated radical anion catalyst [Ir˙−] was examined. It was found that, in a sodium lauryl oligoethylene glycol sulfate (SLES) aqueous solution, [Ir˙−] is more stable than that generated by the typical conPET system using an organic solvent and a trialkylamine electron donor. The low stability of [Ir˙−] under typical conPET reaction conditions is consistent with the report of Polyzos, Francis and co-workers where [Ir˙−] tended to undergo HAT rather than photoexcitation.176 The higher stability in the aqueous solution offers a wider opportunity for [Ir˙−] to absorb a second photon. Control experiments showed that the ground state Ir˙− cannot reduce the substrate, this means that [Ir˙−] can absorb a second photon and the generated *[Ir˙−] is able to reduce even unactivated alkyl chlorides. The authors also claimed that the microstructured solutions not only improve the solubility of alkyl chlorides, but also binds the catalyst and substrate together to facilitate SET. The reaction scope is broad. Diverse unactivated alkyl and aryl chlorides underwent hydrodechlorination to afford corresponding hydrocarbons. Interestingly, the generated alkyl radicals could also be trapped by electron-deficient alkenes such as acrylonitrile to afford 227c, although these radical acceptors are easier to reduce than the organochlorides based on reduction potentials. This excellent chemoselectivity was rationalized by a preassembly of [Ir˙−] with organic chlorides enforced by the microstructured solutions.
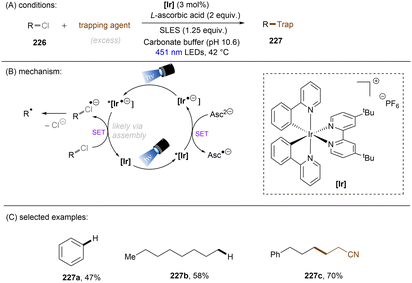 |
| | Scheme 71 An [Ir] complex as conPET catalyst for the activation of inert C–Cl bonds. | |
A method for the activation of C(sp3)–F bonds in trifluoroacetamides and trifluoroacetates 228 was then established by the Glorius group. 3DPA2FBN was used as the PC (Scheme 72).190 The authors proposed a mechanism that the in situ generated, photoexcited 3DPA2FBN˙− and CO2˙− reduce substrates simultaneously. However, different from the report of Wickens,158 a thiol catalyst is essential to the reaction. This reaction features a broad substrate scope, a promising scalability and applicability as demonstrated by over 50 difluorinated products including 10 pharmaceutically-relevant compounds synthesized.
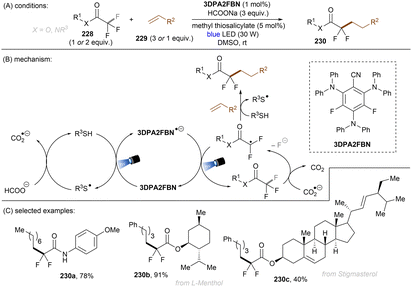 |
| | Scheme 72 Functionalizations of C(sp3)–F bonds in trifluoroacetamides and traifluoroacetates using 3DPA2FBN. | |
3.2.3. Reduction of π systems (CC).
Two-photon PRC reduction of π systems through a π bond breaking is also feasible. Traditionally, the Birch-type reaction in liquid ammonia with solvated alkali metals is utilized for partially reducing arenes.191 These harsh reaction conditions, however, are a challenge for industrial applications. Although recent ammonia-free innovations employing Na dispersion192 or Li/ethylenediamine193 in THF solvent have improved greenness, a solvated alkali- and ammonia-free method is still highly desirable. With the rise of photoredox catalysis, new methodologies for Birch-type reduction emerged. König and co-workers developed an elegant method for Birch-type reduction by combining energy transfer and SET (Scheme 73).194 Anthracene was chosen as the simplest substrate for reduction due to its low aromatic stabilization energy and low triplet energy (ET = 42.6 kcal mol−1) as well as a moderately deep reductive power (*Ered = −1.98 V vs. SCE). Different photocatalysts were screened and Ir[dF(CF3)ppy]2(dtbpy)PF6 (PC1) was found to be the most efficient, even though it has an excited state reductive power (E1/2(*IrIII/IrIV) = −0.89 V vs. SCE) less than fac-[Ir(ppy)3] (PC2) (E1/2(*IrIII/IrIV) = −1.73 V vs. SCE). However, the triplet energy of PC1 is higher (ET = 61.8 kcal mol−1) than PC2 (ET = 58.1 kcal mol−1). As the reaction works considerably better with PC1, the authors took it as support for their hypothesis that both energy transfer and SET are necessary for this catalyst to achieve Birch-type reductions. With the optimized reaction conditions, König and co-workers engaged several large/conjugated electron-rich and electron-deficient aromatic systems in Birch-type reductions to afford products in moderate to excellent yields. Electron-poor nitrogen-containing heterocycles could also be reduced in poor to moderate yields. Nevertheless, the method could not be applied for Birch-type reduction of simple benzene derivatives/benzyl groups probably because of their higher aromatic stabilization energy and higher triplet energy. After performing Stern–Volmer quenching, UV-vis and EPR experiments and further synthetic mechanistic experiments, König and co-workers proposed a two-photon mechanism. First, an EnT from the photoexcited [Ir(III)] catalyst to substrate 231a gives an easily reducible excited intermediate 232. In another cycle, photoexcited *[Ir(III)] is reductively quenched by DIPEA to give an [Ir(II)] complex and DIPEA radical cation. [Ir(II)] reduces triplet excited 232, which is easier to reduce than 231a, to give anthracene radical anion 233. Subsequently, 233 abstracts a hydrogen atom from DIPEA radical cation and the resulting anion 234 is protonated by MeNH3+ to furnish the Birch reduction product 231a. This method boasts an elegant conceptual example of harnessing two blue photons for two distinct processes and offers facile reduction of bi- and tricyclic π systems. However, arenes employed are generally rather activated/conjugated – PRC reduction of more inert simple (mono)arenes like benzyl groups achieved by Murphy and co-workers (vide supra, Scheme 16) was not achieved.
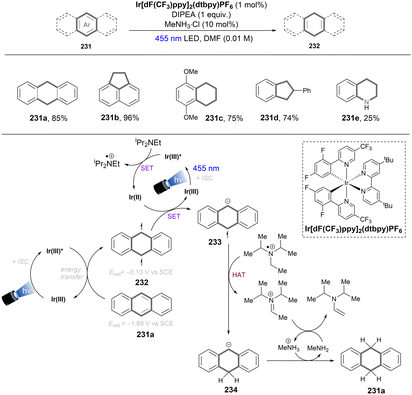 |
| | Scheme 73 Visible-light-induced Birch-type reduction of bicyclic and tricyclic π systems. | |
Although the following reports do not relate directly to reductive activation of π systems, they are conceptually important for discussions of other reports vide infra. In some conPET reactions, the photoexcited radical anion catalyst is proposed to eject a solvated electron (esolv˙−) by a fast photoionization event instead of reducing substrates by SET. Such solvated electrons have served as powerful reductants for the degradation of halogenated pollutants195 and the fixation of N2196 as well as CO2,197 in aqueous solvent. The hydrated electron eaq˙−, when generated in an aqueous solution, has a long lifetime (1–2 μs) and is able to offer a reduction potential of −2.9 V vs. SCE, which is over the energy threshold for the reduction of substrates such as aryl halides and simple ketones, as demonstrated by recent pioneering studies by Goez,198–201 Kerzig and Wenger.129,130 The Goez group disclosed a [Ru(bpy)3]2+/Asc2− system for the generation of hydrated electrons eaq˙− in basic aqueous solutions (pH > 11) with water-soluble Asc2− as the electron donor under irradiation by a green laser.198 Improvements have been made by replacing expensive pulsed lasers with easy-to-handle light from green LEDs (Scheme 74A).199 Decisive to reactivity was the stabilization of the one electron reduced photocatalyst (PC˙−) and suppression of BET by a sodium dodecyl sulphate micelle. Solvated electrons (eaq−) generated under these conditions have found remarkable applications in the reduction of energy-demanding water-soluble organic compounds (Ered < −2.2 V) such as tert-butyl methyl ketone and cinnamate (Scheme 74C). Kerzig and Wenger reported the generation of eaq˙− for the reductions of strong C(sp3)–X bonds in chloroacetate, trifluoromethyl benzoate and benzyltrimethylammonium in neutral aqueous solution by using Irsppy as a water-soluble [Ir] photocatalyst with turnover numbers up to 200 (Scheme 74B).129 Therein, it was proposed that – different from Goez’ report where photoionization of the one electron reduced Ru complex releases a hydrated electron – eaq˙− is formed through photoionization of excited Irsppy (3Irsppy), which has been confirmed by sequential two-pulse laser experiments (Scheme 74B). Interestingly, catalytic activities could be directly controlled by switching the laser light intensity.130
 |
| | Scheme 74 Generation of solvated electrons as the proposed active species for reductions of ketones and olefins by two photon processes. | |
Although solvated electrons are well-known in H2O/NH3 solvents, they have been implicated in reductive transformations in organic solvents as well, for the reduction of more inert compounds.186,202 The Miyake group reported a metal-free photoredox catalysed Birch-type reduction of substituted benzenes ((mono)arenes) to 1,4-cyclohexadienes (Scheme 75).202 Compared to larger/conjugated π systems which were successfully reduced by the EnT/PRC Birch-type strategy developed by the König group,176 this protocol could reduce simple (mono)arenes which represents a substantial advance due to the high triplet energy of the latter compounds.
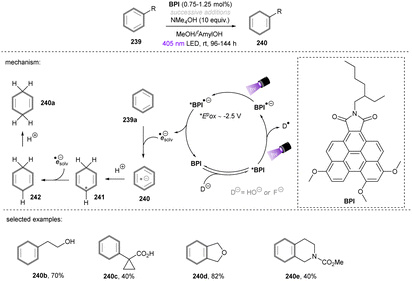 |
| | Scheme 75 conPET Birch reduction of simple arenes by the solvated electron. | |
A visible light absorbing benzo[ghi]perylene monoimide (BPI) was used as PC whose radical anion exhibits strong visible light absorption. Rather than trialkylamines, OH− was proposed to reduce BPI to BPI˙− under photoirradiation. Spectroscopic and computational studies indicated that BPI˙− was generated through an addition of OH− to the carbonyl group of BPI followed by a photoinduced decomposition of the in situ generated adduct. A mixture of alcohols as the solvent could stabilize the radical anion catalyst. Although ground state BPI˙− [E1/2 (BPI/BPI˙−) = −1.24 V vs. SCE] was unable to reduce benzene, it is sufficiently long-lived in the ground state to be excited by a second photon. The excited BPI˙− was found to possess an emissive component with a much longer lifetime than other doublet states of imide and diimide catalysts, this led the authors to propose the generation of *4BPI˙− through intersystem crossing. Further investigation showed that *4BPI˙− is insufficiently reducing to activate benzene directly, nor is the doublet state *2BPI˙−. One proposal for the mechanism of reduction was SET from a higher lying *2BPI˙− to the benzene substrate (in an anti-Kasha fashion). Preassociation of BPI˙− with the benzene substrate was hypothesized, however no direct spectroscopic evidence was yet reported. Another proposal favored by the authors was generation of a solvated electron by photoionization of *BPI˙−. While solvated electrons were proposed, the solvent was not aqueous in this case and catalyst needed successive additions, suggesting that the former proposal cannot be ruled out.
Despite the long reaction time of 96–144 h and successive addition of catalyst that make the reaction untenable on scale, this reaction featured a (overall) low catalyst loading and wide substrate scope, providing an attractive alternative to the classical conditions for the Birch reduction.
4. Electrochemistry
As a result of a transition toward renewable energy-driven and ‘green’ chemistry, over the past two decades, synthetic organic electrochemistry (SOE) has gained considerable attention both as a tool for synthetic chemistry and an interdisciplinary field of research,15,16,18–21,203–212 which is illustrated by whole journal issues being dedicated to it.213,214 One reason behind SOE's rise in popularity is that this methodology can fulfill several important criteria that are needed to develop environmentally compatible processes. It allows to directly utilize electricity from renewable sources, or to store this energy in the form of chemical bonds. By using electrons as reagents and a source of energy, SOE offers very mild, atom-efficient, cost-effective, and safer methods to achieve selective oxidation or reduction processes. Therefore, it allows to circumvent the deployment of toxic or dangerous oxidizing or reducing agents that are frequently required in stoichiometric amounts.21,205,209 In contrast to processes relying on conventional chemical or photochemical redox agents – that operate only at a fixed potential – SOE grants a wider range of operating potentials that is totally user-selectable with high precision. Potentials can even be alternated on demand by alternating current (AC) operation, allows to discriminate between different functional groups and achieve selectivity. These are just a few of the most important attributes that render SOE environmentally friendly and conceptually attractive to the synthetic chemist.
Electrochemical reactions can also be performed at a constant potential, or at constant current, the latter being simpler and faster. However, due to the general inability of electrode surfaces to discriminate between molecules, the electrochemical potential of the target species to be oxidized or reduced should be known and should be more redox accessible than other species present in solution; such as additives, electrolyte or the solvent. Care should be taken to stop reactions when the required Faradays are passed.
Although reactor configurations are entirely different, in some respects, SOE shares similar conceptual principles and intermediates as photochemistry (at least with respect to the target substrate). Akin to photoredox catalysis, electrochemical reactions occur through SET processes, usually through formation of radical ion or ionic species which can then react further to ultimately form a desired product. However, there are key differences in reactivity and the control over it. Firstly, SOE involves a heterogeneous interfacial process, where high local concentrations of intermediates develop in proximity to the electrode surface. This may encourage intermediates to react in different pathways, for example with each other (dimerization) rather than with a partner in bulk solution. Secondly, SOE can involve further SET transformations of the downstream intermediates in the same redox trajectory as the initial redox event (i.e. net double electron oxidation or double electron reduction pathways, or ‘radical polar crossover’). This fundamentally differs from ‘redox neutral’ PRC in which the initial and downstream redox events must occur in different directions. Finally, in net-oxidative or reductive PRC processes with sacrificial redox agents, typically only one redox step is potent and user-selectable by a photoexcited state while the other is limited by the inherent redox properties of a ground-state intermediate. On the other hand, SOE allows redox power to be tuned uniformly for both initial and downstream redox events.
Consequently, SOE can offer new reaction pathways that are complimentary to the traditional synthetic chemistry procedures and can be particularly interesting for late-stage functionalization of complex molecules due to its mild and selective nature of operation.210 However, in contrast to typical photochemical/photocatalytic reactions, the heterogeneicity of SOE means it is often governed by mass transport to/from an electrode surface rather than chemical kinetics, and the interfacial nature of the catalysis makes it harder to mechanistically probe and reproduce reactions.
Despite the many benefits of SOE, it is only in the last few years that the synthetic chemistry community has rediscovered this powerful tool for routine synthetic applications.5,7 One of the issues that presents a high barrier for chemists to adopt this exciting technique was the general perception, that specialized equipment and expert knowledge is required. This has now been eroded by numerous reviews/tutorial articles designed for synthetic chemists without prior experience in physical electrochemistry.215–217 In this review, we seek to highlight the utility of SOE specifically for strong bond activations, allowing comparisons to be made in this context with other contemporary SET platforms like PRC.
4.1. C(sp2)–X bond cleavage
Early examples on electrochemical activation of aryl halide C(sp2)–X bonds through radical chain aromatic substitution (SRN1) date back as early as the 1970s.218 Mechanistically, aryl radicals generated by cathodic reductive cleavages of aryl halides (mainly bromides and iodides) tend to combine with a nucleophile (Nu−) to form ArNu˙−, which propagates the reaction by SET reducing another ArX molecule to deliver ArX− and product ArNu (Scheme 76). Subsequently, Savéant and co-workers developed the electrochemical reduction of electron-deficient aryl chlorides in liquid ammonia with Hg/Au or Au/Al as electrodes.219 In the presence of a mediator such as naphthonitrile – that is electroreduced and then reduces the aryl halides – the generated aryl radicals add to a styrene or an acrylonitrile trapping agent. A second cathodic SET reduction affords the C–Cl alkylation products. However, electroreduction of electron-neutral or electron-rich aryl chlorides remained elusive.
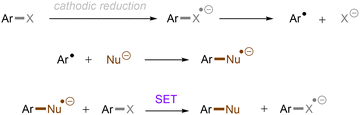 |
| | Scheme 76 Radical SRN1 reaction of aryl halides. | |
This challenge was addressed in 2019, when Pan, Chi and co-workers reported an electrochemical hydrodehalogenation of electronically different aryl chlorides without using any sacrificial metal electrode (Scheme 77).220 Carbon graphite rods were used as both anode and cathode. A trialkylamine (2 equiv.) was used as a sacrificial reductant which undergoes overall a two-electron oxidation process (and loss of a proton) at the anode. The direct cathodic SET reduction of ArX to Ar˙ (via ArX˙−) followed by a further reduction at cathode delivers Ar−. Hydrodechlorinated products ArH are formed by protonation of Ar−. This reaction was compatible with electron-rich aryl chlorides and even 2-naphthyl fluoride was engaged. A related electrochemical deuterodechlorination of electron-deficient aryl chlorides employing D2O was developed by the Lei group.221 Although powerful, direct electrolysis conditions with high constant currents inevitably deliver hydrodechlorinated arenes as over-reduced products. This is due to intermediate aryl radicals having substantially more accessible redox potentials for reduction than their aryl halide precursors,222 leaving no scope for synthetically useful aryl radical trappings. This limitation can be overcome to an extent with indirect electrolysis approaches using mediators, however, such approaches are generally limited by the ground state redox power of the mediators. Photoexcitation of the mediators offers a solution (vide infra, Section 5). An even greater challenge is presented when targeting less activated aryl fluorides. A pioneering report on the indirect electroreduction of fluorobenzene was disclosed by Kariv-Miller and Vajtner in 1984 (Scheme 78).223 Previous studies from the same group demonstrated that electrolytes containing tetrabutylammonium (nBu4N+) and dimethylpyrrolidinium (DMP+) cations were needed to catalyze the electroreductions of organic compounds on mercury.224 The effects of DMP·BF4, nBu4N·BF4, water, and current density were investigated. A catalytic amount of DMP·BF4 (0.01 M) with nBu4N·BF4 (0.2 M) as an electrolyte and a low concentration of water as a proton source (0.5%) in diglyme with mercury pool electrodes (1.25 mA cm−2) were found to be optimum conditions to reduce fluorobenzene. Electrolysis of 1,3-difluorobenzene under constant current at −2.65 V gave 85% fluorobenzene and 9% unreacted starting material, while no formation of benzene was observed. According to their mechanistic proposal, DMP+ forms an “amalgam” by a reversible one electron process on the cathode. In turn, the “amalgam” reduces the fluoroarene substrate to its radical anion 252 that undergoes cleavage to liberate fluorine anion and aryl radical 253. The aryl radical 253 is further reduced at the cathode to an aryl anion 254 that is rapidly protonated. The intermediacy of an aryl radical was further proved by a defluorinated radical clock experiment using o-(3-butenyl)fluorobenzene under similar reaction conditions.225
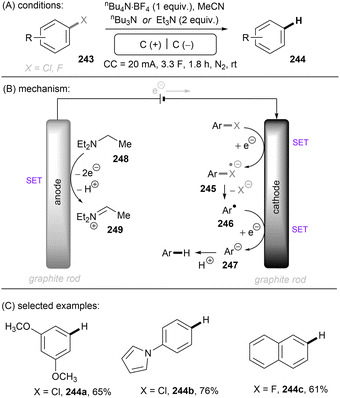 |
| | Scheme 77 Electrochemical hydrodehalogenation of aryl chlorides and fluorides. | |
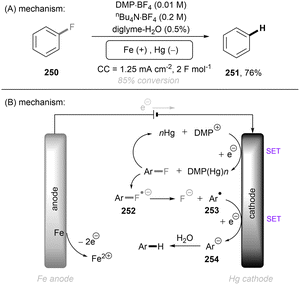 |
| | Scheme 78 Indirect electroreductive hydrodefluorination of fluorobenzene. | |
A contemporary example of electrochemical aromatic hydrodefluorination was reported by Huang and co-workers in 2015 (Scheme 79).226 They reported defluorination of aryl fluorides using platinum electrodes in an undivided cell under air. Diglyme turned out to be the best solvent choice that – together with nBu4N·BF4 as an electrolyte and NaBH4 as an additive – provided 98% of the desired product. Diverse aryl fluoride substrates including difluoro- and trifluoroarenes were hydrodefluorinated to their corresponding aromatics. The mechanism for electrochemical hydrodefluorination was proposed where BH4− acts as a sacrificial material for the anode oxidation. In contrast to the indirect reduction of fluorobenzene with DMP(Hg)n as a mediator as previously reported,189 in this Hg-free protocol, fluorobenzene is proposed to undergo direct reduction to its radical anion. This undergoes C(sp2)–F cleavage to generate phenyl radical in proximity to the cathode surface. The phenyl radical is reduced further, and the resulting phenyl anion deprotonates the solvent or the electrolyte.
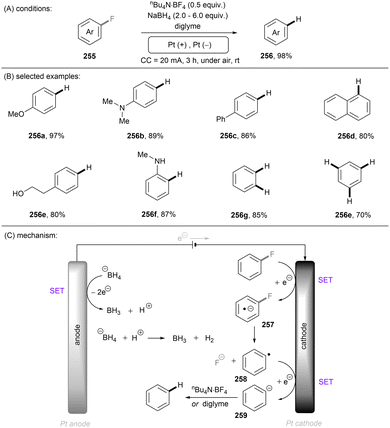 |
| | Scheme 79 Direct electrochemical hydrodefluorination of aryl fluorides. | |
Electrochemical reductive cleavage of C(sp2)–O bonds in diaryl ethers was reported by Bartak and co-workers.227 The products of electroreductive cleavage of diphenyl ether are phenol and benzene (Scheme 80) which is accomplished through formation of radical anion of substrate 1 followed by its rapid cleavage that results in formation of phenyl radical and phenoxide ion. The phenyl radical either abstracts a hydrogen atom from DMF to give benzene, or is reduced again at the cathode to the phenyl anion. Both the phenoxide and phenyl ion are protonated to give the products. Although efficient, poor selectivity would result if an ether with two different aryl groups was employed as the substrate.
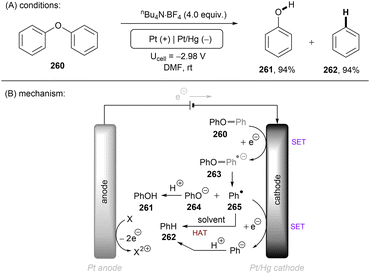 |
| | Scheme 80 Electroreductive cleavage of the C(sp3)–O bond in diphenyl ether. | |
Yanilkin and co-workers reported the electrochemical reduction of triphenyl phosphate as early as 1990 (Scheme 81).228 Using nBu4N·BF4 as electrolyte in DMF as solvent, electroreduction formed butyl diphenyl phosphate. To probe the mechanism of this reduction, they performed polarography, CV, and preparative electrolysis. They proposed the mechanism involves the formation of a radical anion 269, which undergoes spontaneous C(sp2)–O mesolytic cleavage to produce diphenyl phosphate anion 270 and phenyl radical 271. Further reduction of 271 and protonation of 272 afforded benzene. The anion 270 evidently reacts with nBu4N+ to form the observed product. Again, achieving a selective C(sp2)–O bond cleavage would be challenging with respect to phosphates bearing different aryl groups.
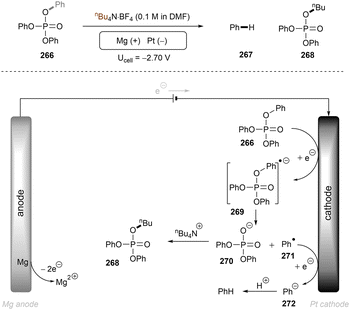 |
| | Scheme 81 Electrochemical reduction of triphenyl phosphate. | |
4.2. C(sp3)–X bond activation
Electrochemical reduction of alkyl halides was classically limited to alkyl bromides/iodides or α-chloro carbonyls as substrates229–232 until a recent contribution by the Lu group.233 They disclosed an electrochemical borylation of unactivated alkyl chlorides using sacrificial Mg anode and carbon cloth cathode (Scheme 82). The indirect cathodic reduction of alkyl chlorides was mediated by a complex 276 of bis(catecholato)diboron (B2cat2) and DMAc as solvent. The proposed active species – radical anion 277 – is a potent reductant that can reduce alkyl chloride 273 to alkyl radical 278 through SET and liberation of chloride. Subsequent trapping of 278 with 274 affords Alkyl–Bcat 280 and anionic species 279. The final product Alkyl–BPin 275 was formed by adding Et3N and pinacol. Primary and secondary alkyl chlorides worked well, however no tertiary alkyl chloride reactions were reported. This reaction reinforces the key advantage of mediated electrolysis: engaging carbon radicals in radical coupling/addition transformations instead of undergoing a further SET reduction.
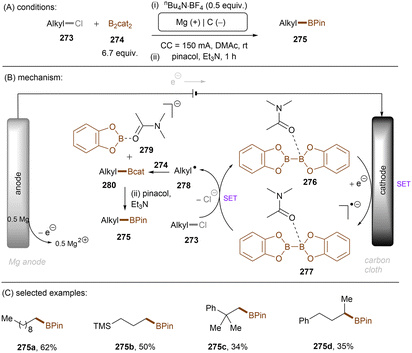 |
| | Scheme 82 Electrochemical borylation of alkyl chlorides. | |
More recently, the Qiu group reported a deutero-dechlorination of unactivated alkyl chlorides (Scheme 83).234 Instead of a sacrificial anode, DIPEA was used as an external reductant which was oxidized at the graphite felt anode. The alkyl chloride 281 underwent overall a two-electron reduction (and loss of chloride) in proximity to the surface of the lead plate cathode to deliver the alkyl carbanion 285, which was deuterated with D2O. The reaction featured an impressive substrate scope; successfully engaging primary, secondary and tertiary chloroalkanes with diverse substituents and electronics. Interestingly, the C(sp2)–Cl bond in 282a was intact with the C(sp3)–Cl being selectively deuterated.
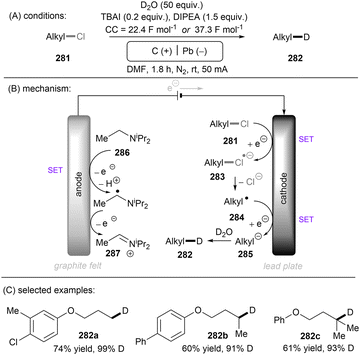 |
| | Scheme 83 Electrochemical deuterodehalogenation of alkyl chlorides. | |
Activation of C(sp3)–F bonds using electroreduction is a highly desirable target,235,236 since difluoroalkyl groups are hydrogen bond donating bioisosteres.237 Troupel and co-workers reported a one-step electrochemical reduction protocol to synthesize difluorobenzyl derivatives from unactivated trifluorotoluene in good yields.238 By using a sacrificial Mg/Al anode, the authors achieved electroreductive couplings of trifluorotoluene with electrophiles such as carbon dioxide, acetone and DMF (Scheme 84). The electrolyses were performed in undivided cells at room temperature in DMF and nBu4N·Br as a supporting electrolyte. For electrocarboxylation, a magnesium anode was used, and CO2 was bubbled through the solution at atmospheric pressure. Methyl carboxylate 289 was obtained by a further esterification using H2SO4 and MeOH. For the acetone coupling experiment, acetone was used as a co-solvent in 10% v/v. Here, aluminium anode provided a better yield than magnesium. For the electroformylation, DMF was proposed to serve both as a reactant and as a solvent. Mono C(sp3)–F activation in PhCF3 was achieved in sharp contrast to another indirect electrochemical C(sp3)–F reduction strategy for the activation of PhCF3 using SmCl3 as a catalyst;239 which delivered a mixture of mono, double- and fully defluorinated products. A follow-up study by Clavel and co-workers revealed that electrochemically generated di- and mono-fluoromethyl anions could also be trapped by TMSCl to give silyl fluoromethyl products 292 and 293.240
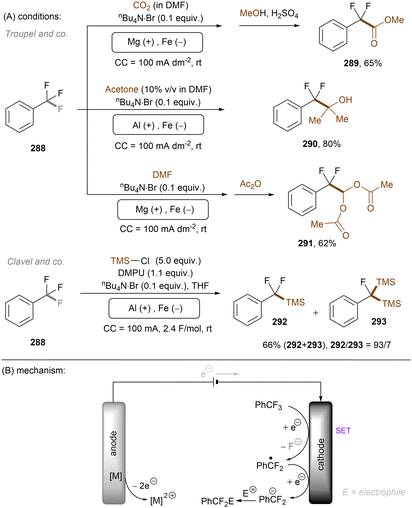 |
| | Scheme 84 Electroreductive coupling of trifluorotoluene with CO2, acetone and DMF. | |
In a more recent study, Xia and co-workers developed an electroreductive hydrodefluorination reaction of trifluoromethylarenes 295 (Scheme 85).241 Similar to Chi's work,220 Et3N was used as an electron donor instead of using a sacrificial metal anode. The reaction only worked for electron-deficient substrates. Fully reduced product toluene derivatives 296 were obtained as a result of the constant high-current electrolysis, which contrasts with the organophotoredox selective mono-HDF of ArCF3 reported by Gouverneur and co-workers (Scheme 41, vide supra).
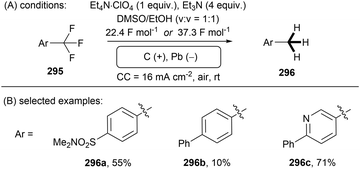 |
| | Scheme 85 Complete electroreduction of trifluorotoluene derivatives. | |
Very recently, the over-defluorination issue in electrochemical reduction of Ar–CF3 compounds was tackled by the Lennox group (Scheme 86).242nBu4N·Br was employed as a milder reductant in lieu of Et3N or to eliminate the need for a sacrificial metal anode. TMSCl as a fluoride trap is beneficial to product formation and selectivity control. Utilizing Ni as the cathode was observed to ensure a high level of selectivity of mono defluorinated product 298 over double defluorinated product 299 after 2 F charge were passed. Double defluorination became dominant after 4.2 F when more electrolyte, TMSCl and nBu4N·Br were added. Trifluoromethyl arenes with various substituents were smoothly reduced under two different reaction conditions (A and B), and the product ratios (298/299, 299/298) varied. A distinct pattern is evident: while the reduction of electron-rich ArCF3 (like 297b), is easier to halt at mono defluorination under conditions A, substrates bearing an electron-withdrawing group or a larger π-system tended to accept a second electron to furnish a double defluorinated product. In contrast to previous photoredox methods117,121,122,134 that can only activate electron-deficient trifluoromethyl arenes, this work offers a switchable defluorinative alternative that can engage electron-sufficient substrates, and again demonstrates that electrochemistry is a user-friendly technique for selective organic synthesis.
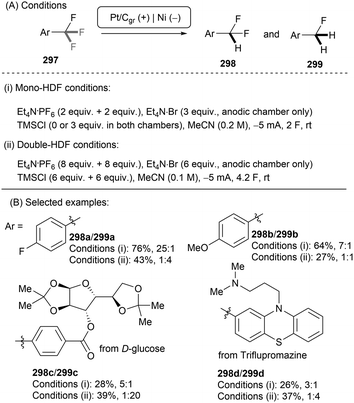 |
| | Scheme 86 Controllable and switchable electroreductive hydrodefluorinations of trifluoromethyl arenes. | |
Electroreductions of α,α,α-trifluoroacetyl groups have also been achieved (Scheme 86). In 1974, Inesi and Rampazzo reported the electrochemical reduction of methyl trifluoroacetate 300 to difluoroacetate 301 and fluoroacetate 302 (Scheme 87A).243 Subsequently, Evans and Kopilov conducted the electroreduction of trifluoroacetanilide 303 in ethanol which provided mixture of 65% α-fluoroacetanilide 301 and 35% acetanilide 305.232 The same reaction in MeCN provided less extensive defluorination products, such as mixture of 46% α,α-difluoroacetanilides 306, 51% 304 and only 3% acetanilide 305 (Scheme 87B). The results confirmed a carbanion was generated by two-electron electrochemical reduction. Such unavoidable over-reduction – caused by the constant current – contrasts to milder photocatalytic strategies for mono C–F activations in trifluoroacetates and trifluoroacetamides devised by Molander,135 Glorius,190 and Shang140,143 independently (vide supra). Of course, the use of Hg electrodes is not ideal from a chemical safety point of view.
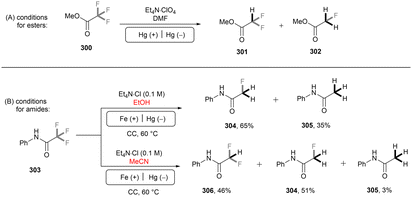 |
| | Scheme 87 Electroreduction of α,α,α-trifluoroacetate and α,α,α-trifluoroacetamide. | |
A highly selective mono hydrodefluorination of trifluoromethyl ketones was achieved via electroreduction by Lennox and co-workers (Scheme 88).244 TMSCl was employed as a radical anion trapping agent and Et4N·PF6 as a proton donor. The HDF protocol was successfully implemented on a series of trifluoromethylketones having electron-rich and electron-poor substituents in conjunction with heterocyclic substrates in moderate to excellent yields (Scheme 87B). Compared to the photoredox C(sp3)–F activation of trifluoromethyl ketones reported by Chatterjee and Qu,118 the electrochemical protocol of Lennox and co-workers features a comparably broad scope. According to their mechanistic proposal, the reaction proceeds through formation of difluoro enol ether 311 (Scheme 88C). Although 311 is a transient intermediate and was never observed, the importance of TMSCl to trap and stabilize radical anion of substrate 307 was confirmed by DFT calculations, which suggested that without TMSCl the reaction is thermodynamically highly endergonic. On the surface of the anode, bromide is oxidized to anionic tribromide (Br3−). Since the latter does not rapidly migrate to the cathodic chamber, side reactions can be mitigated.
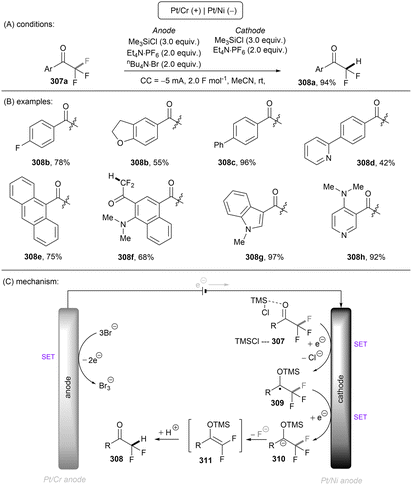 |
| | Scheme 88 Electrochemical hydrodefluorination of trifluoromethylketones. | |
Following the less synthetically useful electrochemical C(sp3)–O cleavage in tributyl phosphate reported by Yanilkin and co-workers in 1990,228 Lam and Markó reported a synthetically useful electrochemical reductive C(sp3)–O cleavage for an overall deoxygenation of alcohols (Scheme 89).245 The authors performed electrochemical reductions of diphenylphosphinate esters with reduction potentials as deep as −3.0 V vs. SCE to their corresponding deoxygenated products. nBu4N·BF4 was chosen as the electrolyte in DMF and reactions were complete in only 10 min. In the optimization, a 100 mA cm−2 current density and a 60 °C temperature were found to provide the best results. They were able to reduce primary, secondary and tertiary phosphinates, including a natural product derivative, in up to 95% yield. However, substrates bearing other reductively labile functional groups, such as halogen atoms, were not evaluated. The observations of mechanistic investigations strongly suggest that the mechanism involves electrochemical and chemical steps. Initially, diphenylphosphinate ester 312 is reduced to the corresponding radical anion 315 (electrochemical step), which undergoes subsequent decomposition to diphenylphosphonate anion 313 and the radical of the alkyl part 316 (chemical step). The authors proposed the reformation of the phosphorus-oxygen double bond as the main driving force of this reaction, when phosphorus(IV) intermediate 315 is decomposed to the phosphorus(V) species 313. The rate-determining step is the decomposition of radical anion 315, which explains the need for assistance of temperature. The more stable alkyl radical 316 is, the faster the mesolytic cleavage of radical anion 315. Finally, radical 316 picks up a hydrogen atom, presumably from solvent, to afford 314.
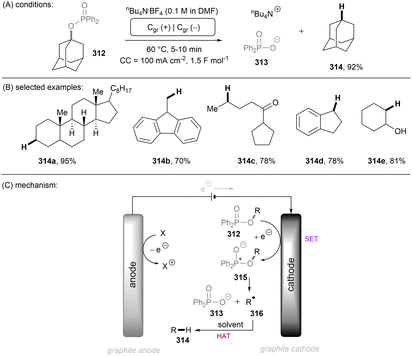 |
| | Scheme 89 Electroreductive cleavage of C(sp3)–O bonds in alkyl phosphinates. | |
One more very interesting example of C(sp3)–O bond cleavage is a Barbier-type reaction that was reported by Tian, Wang and co-workers.246 Through the electroreduction of phosphate protected alcohols with aldehyde or ketone mediators, they achieved effective deoxygenative C(sp3)–C(sp3) bond formations (Scheme 90). With the two aryl C(sp2)–O bonds untouched, the alkyl C(sp3)–O bond was selectively cleaved under constant current at 20 mA in an undivided cell using stainless steel anode and graphite cathode, representing a more useful strategy compared to the work by Yanilkin.228 The substrate scope was explored by using a variety of aldehydes, ketones, and phosphate derivatives. According to their mechanistic proposal, the phosphate protected alcohol is first cathodically reduced to form radical anion intermediate 320, that undergoes spontaneous C–O cleavage to produce alkyl radical 321. From here, 321 may undergo either (i) further reduction to carbanion intermediate 322, which subsequently is trapped by the carbonyl group of 318 to form the anionic intermediate 323, or (ii) it may undergo radical cross-coupling with the reduced aldehyde (the reduction potentials of aldehydes are similar to phosphate protected alcohols) to give the anionic intermediate 323. After protonation, the desired product (319g) is formed.
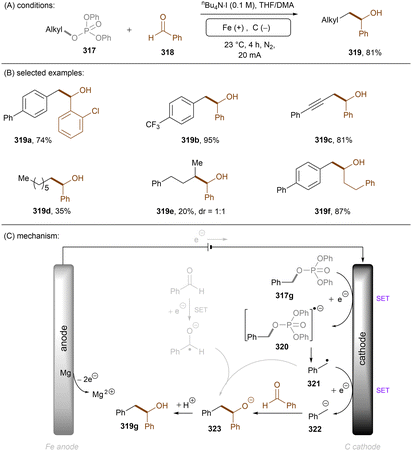 |
| | Scheme 90 Electroreductive cleavage of C(sp3)–O bonds in alkyl phosphates for C(sp3)–C(sp3) formation. | |
Senboku and co-workers made a significant improvement by using a cheaper sulfonyl group.247 They reported electrochemical reduction of alkyl mesylates under mild conditions to give the corresponding alkanes in moderate to good yields (Scheme 91). They were able to selectively reduce mesylates in the presence of other functional groups such as epoxide, olefin, acetal, hydroxy and cyano groups. Electrolysis under constant current at 20 mA in an undivided cell equipped with a Pt cathode and a Mg anode in the presence of biphenyl and tBuOH in DMF provided the best results. Their mechanistic proposal suggests that biphenyl acts as an electron-transfer mediator and tBuOH as a proton donor.
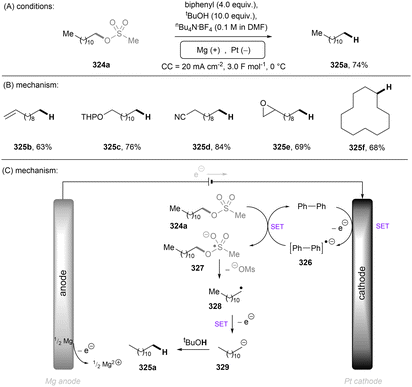 |
| | Scheme 91 Electrochemical reduction of alkyl mesylates. | |
In an attempt to test their hypothesis, they carried out CV of the substrate and biphenyl. The CV of the substrate showed no reduction peak at >−3.2 V vs. Ag/Ag+, however the CV of biphenyl revealed a reduction peak at ca. −3.2 V vs. Ag/Ag+. When the CV of biphenyl was measured in the presence of the increasing amount of the substrate, the reduction current −3.2 V vs. Ag/Ag+ increased, which showed that biphenyl is an electron-transfer mediator.
Thus, the reaction mechanism suggests that biphenyl is first reduced on the surface of the cathode to form a radical anion intermediate 326, that reduces the substrate 324a to form radical anion intermediate 327. The intermediate 327 undergoes spontaneous C(sp3)–O cleavage to produce free alkyl radical 328, that undergoes further reduction at the cathode (or by 327) and protonation to form the product 325a.
4.3. C(sp2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) X cleavage
X cleavage
The hydrogenation of π-systems such as carbonyls, alkenes, alkynes and arenes is a fundamental transformation for the synthesis of fine chemicals, pharmaceuticals, and natural products both in industry and academia.248–251 Reductive electrolysis at a cathode is able to offer transition metal-free hydrogenation methods omitting the use of molecular H2 as an external reductant.
Cheng and co-workers reported an electrochemical hydrogenation of alkenes, alkynes and ketones employing gaseous ammonia as the proton source (Scheme 92).252 Owing to its energy density, ease of transportation and safety in storage, ammonia serves as an ideal hydrogen source yielding dinitrogen as the only benign side product.253 Therefore, direct application of ammonia in organic synthesis is a desirable target for the future of organic synthesis, especially in synergy with the benefits of SOE. Control experiments revealed an atmospheric pressure (balloon) of gaseous ammonia at a cell potential (Ucell =) −5.0 V is crucial for the reaction to achieve selectivity and high yield. Using water as a hydrogen source provided only traces of the desired products. Replacing MeCN with MeOH decreased the yield from 73% to 61%. An extensive variety of alkenes and alkynes with different heterocyclic and functional groups were tolerated. Ketones were also reduced to corresponding alcohols at a working voltage of 4 V in the presence of LiClO4 as an electrolyte in DMF, in poor to excellent yields. Simply by increasing the cell potential to −6.0 V, the ketones were reduced to their corresponding methane derivatives in good yield. The mechanism proposed claims that alkene hydrogenation occurs via a rapid stepwise process, where SET from the cathode and a proton transfer from ammonia results into formation of radical intermediate 335, that undergoes the same sequence to yield the hydrogenation product 332e. The amine anion undergoes oxidation on the anode surface to generate hydrazine 336. This by-product is itself a plausible reducing agent.
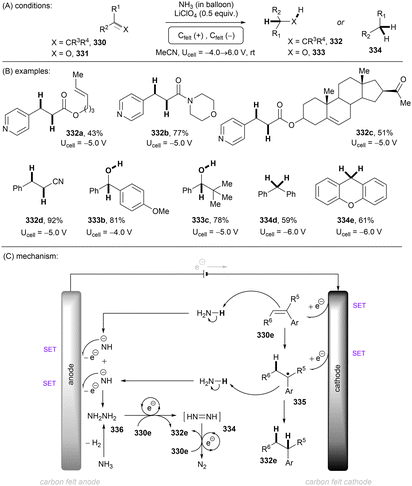 |
| | Scheme 92 (A) Electrochemical hydrogenation of alkenes and ketones using gaseous ammonia. | |
Yang and Wen reported hydrogenation of electron-deficient alkenes and alkynes using methanol as an easy-to-handle hydrogen donor to access saturated ketones (Scheme 93A and B).254 Anodic oxidation of KSCN or PhSPh (2.0 equiv.) was proposed as the counter half-reaction. Diverse ynones and dithioacetals were hydrogenated with high yields. Another electrochemical reduction that proceeds under air was reported by Xia and co-workers,255 where ammonium chloride and methanol both act as hydrogen donors (Scheme 93C). DMSO was used as an electron donor that is oxidized on the surface of the anode to the dimethyl sulfone. A common limitation of these three reactions is that substrates that are non-aromatic/non-conjugated cannot be reduced.
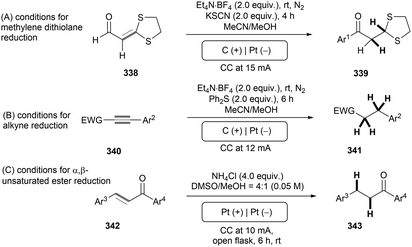 |
| | Scheme 93 Electrochemical hydrogenation of alkenes and alkynes. | |
Considering the importance of deuterated compounds,256–260 Cheng and co-workers developed an electrochemical deuteration of electron-deficient olefins using graphite felt for both electrodes under mild conditions without external reductants (Scheme 94).259 With readily available D2O as a deuterium source, products were achieved in up to 91% yield in as short as 2 h reaction times. A vast library of substrates were well-tolerated by the electrochemical deuteration transformation. Due to the large area and porous structure of the graphite felt, the authors proposed that electron transfer and deuteration occur in a concerted manner that mitigates side reactions such as radical dimerization.
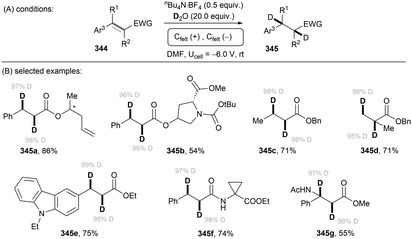 |
| | Scheme 94 Electrochemical deuterogenation of alkenes. | |
4.4. Dearomatizations
Electrochemical Birch-type reduction of simple feedstock arenes to 1,4-cyclohexadiene derivatives is another significant topic. The first example of successful electrochemical reduction of aromatic compounds selectively to either cyclohexene or 1,4-cyclohexadiene derivatives was reported by Benkeser and Kaiser in 1963.260 The cell configuration was crucial to selectivity: when benzene was subjected to electrolysis in methylamine and 4.0 equiv. LiCl as electrolyte in a divided cell, cyclohexene was formed in 49% yield (Scheme 95). When benzene was subjected to the same conditions in an undivided cell, equally efficient formation of 1,4-cyclohexadiene (49%) occurred, revealing the impact of spacial/chemical separation of electrodes. By simply changing the cell configuration (divided or undivided) and concentration of LiCl (4.0 or 8.0 equiv.), the authors were able to reduce 5 different aromatic substrates selectively to either cyclohexene or 1,4-cyclohexadiene derivatives in moderate to excellent yields (44–85%). The authors hypothesized that elemental lithium metal is generated at the cathode and that this is the active reducing agent in this system.
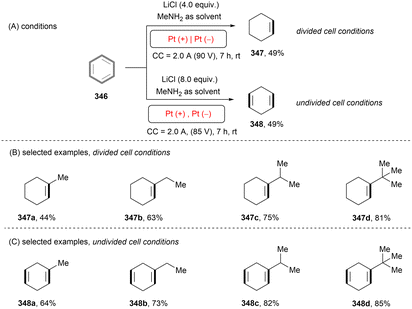 |
| | Scheme 95 Electrochemical Birch reduction of simple arenes in methylamine. | |
Electroreduction of electron-rich alkoxyarenes was first achieved by Kariv-Miller and co-workers in 1983.261 Their conditions passed constant current between a mercury pool cathode and stainless steel anode in an undivided cell, through an aqueous solution of tetrabutylammonium hydroxide (nBu4N·OH) (Scheme 96). Similarly to their aforementioned study on aryl halide reductions (Scheme 78), the authors proposed that the cathodic reduction was mediated by an in situ generated “amalgam” – this time involving tetrabutylammonium counter cations-, that reduces the aromatic substrates. Thiebault and co-workers reported electrochemical reductions in liquid ammonia in an undivided cell.262 As the electroreduction conditions they used Mg bar as the anode, Al sheet wrapped around another Mg bar as the cathode, an alkaline salt as an electrolyte and an aliphatic alcohol as a proton donor in liquid ammonia (Scheme 97). They have demonstrated electro-Birch dearomatizations of arenes such as phenylglycine and 1-naphtol.
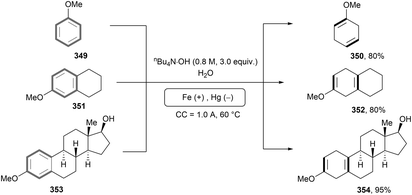 |
| | Scheme 96 Electroreduction of methoxyaromatics in aqueous solution. | |
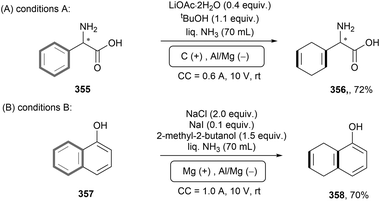 |
| | Scheme 97 Electro-Birch reduction of phenylglycine and 1-naphtol. | |
Kashimura and Ishifune reported milder and safer conditions for regioselective electrochemical Birch reductions of aromatics, including challenging to reduce methoxybenzenes, in up to 91% yields (Scheme 98).263 The conditions consist of Mg as both electrodes, LiClO4 as an electrolyte, tBuOH as a proton donor and THF as a solvent. Constant current at 50 mA and Mg as electrodes provided the best results. The authors proposed that direct electron transfer to aromatics was unlikely, and instead that solvated Li0 – generated by the cathodic reduction of LiClO4 – mediated the SET reduction of aromatics. Among the safest and most scalable conditions reported for Birch reductions so far was the electrochemical protocol reported by Neurock, Minteer, Baran and co-workers in 2019.264 They demonstrated how dimethylurea (DMU) served as a cheap, non-toxic, and water-soluble proton source. Combined with Mg or Al as sacrificial anode material and tris(pyrrolidino)phosphoramide (TPPA) as an overcharge protectant (inspired by battery technology), this enabled multigram-scale reductive electrochemical access to and functionalizations of pharmaceutical building blocks. Impressive functional group tolerance was observed (Scheme 99).
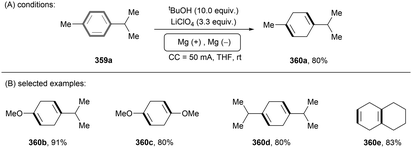 |
| | Scheme 98 Birch-type electroreductions of electron-rich arenes. | |
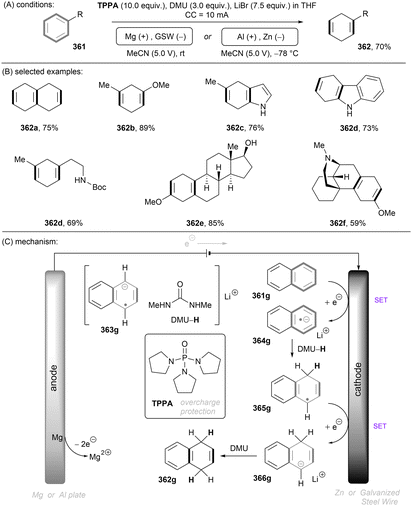 |
| | Scheme 99 Electrochemical Birch reduction of aromatic compounds using conditions inspired by battery technology. | |
The proposed mechanism involved iterative electrochemical SET reductions and protonations of the substrate, akin to that of the classical Birch reaction. The authors hypothesized that strong coordination of Li+ with DMU co-localized DMU with the radical anion of the substrate (361g). The authors conclusively showed that (i) Li0 is not a direct solution-phase arene reductant (in contrast to the proposal or Kashimura and Ishifune) and (ii) solvated electrons are not involved. The authors were able to readily scale up the process from a 0.1 mmol scale to a 100 g scale in comparable yield using a flow reactor, showing industrial amenability (not shown in Scheme, see the publication concerned).
5. By photoelectrochemistry
The 21st century resurgences of photoredox catalysis (PRC)4,6,265–267 and synthetic organic electrochemistry (SOE)11–13,15,16 have provided organic chemists versatile protocols to build molecular complexity through radical pathways avoiding using conventional hazardous radical initiators or hazardous conditions to access radicals. However, both these two techniques feature important drawbacks.268 While PRC has enabled a wide range of redox-neutral transformations under mild conditions, stoichiometric amounts of redox mediators are essential to achieve net-reductive/oxidative reactions. SOE guarantees high redox power, circumventing stoichiometric sacrificial redox mediators in favour of a user-defined applied constant current or cell potential. However, this often leads to side reactions including over-reductions, over-oxidations, product decompositions or solvent redox processes. Organic chemists have merged these two topical techniques to address these aforementioned long-standing issues via synthetic molecular photoelectrochemistry.22–26,269 The combination of the advantages of PRC and SOE has encouraged chemists to explore the emerging concept of electrochemically-mediated PRC (e-PRC) in two main sub-categories (at least in the molecular catalytic context): (i) electro-recycling of PCs by replacing redox mediators with electrical current (Scheme 100A) and (ii) electrochemical generation of colored radical ion PCs – a strategy coined ‘electro-activated’ photoredox catalysis, also termed ‘electron-primed’ or ‘hole-primed’ depending on the nature of catalyst electro-activation (Scheme 100B). The former strategy (Scheme 100A) has grown rapidly in its synthetic practicality, from early simple alcohol oxidations,270–272 oxidative C–X couplings273–279 to recent asymmetric C–H functionalizations.280 The latter strategy (Scheme 100B) offers a substantial conceptual advance on its ability to access ‘super’ redox potentials under mild conditions without requiring either a second photoexcitation event or super stoichiometric amounts of sacrificial redox mediators. The mildness of this strategy avoids typical side reactions in direct electrolysis (such as over-reductions) due to the low concentrations of substrate-derived reactive species, as first demonstrated by Lund and Eriksen in 1985.281 This holds great promise for engaging unactivated substrates never before engaged in redox. However, the growth of the field is hampered by (i) the typical need for divided/semi-divided cells – in order to build up sufficient concentrations of catalyst-derived active species – resulting in high cell resistances that are not conducive to scale up at least as far as organic electrolytes are concerned, and (ii) a lack of understanding around the mechanism and the key requirement for catalyst–substrate assemblies owing to the picosecond excited state lifetimes of radical ion photocatalysts (doublet states). Such understanding will be necessary in order to design more general/effective catalysts and predict reactive transformations in the future.
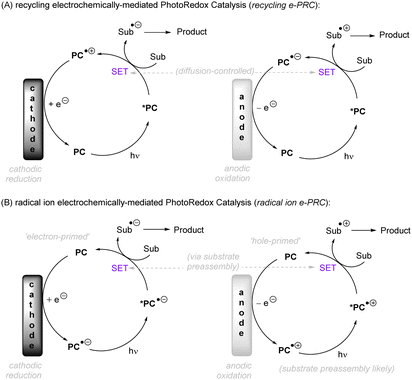 |
| | Scheme 100 Two main sub-categories for synthetic molecular photoelectrochemistry. | |
A seminal contemporary example of electrochemically-mediated photoredox catalysis (e-PRC) was disclosed by the Lambert group. Trisaminocyclopropenium cation (TAC+) was employed as an electro-activated photoredox catalyst.282 The excited state of the radical dication (*TAC˙2+) – generated by anodic oxidation and subsequent visible light excitation – is a super-oxidant (*E1/2 = +3.33 V) sufficient to oxidize simple arenes to arene radical cations susceptible to intermolecular attacks by N-nucleophiles. In the direction of reduction, Lambert, Lin and co-workers reported the e-PRC reduction of aryl chlorides using 9,10-dicyanoanthracene (DCA) as a catalyst (Scheme 101).283 A cell potential of −3.2 V was applied to generate DCA˙− through cathodic reduction of DCA. DCA˙− is a weak reductant (E1/2 = −0.82 V) but it becomes a more reducing species after photoexcitation. The resulting *DCA˙− has a reductive power of −3.2 V, and thus it is able to engage aryl chlorides 367 in SET reduction and subsequent radical coupling reactions with B2Pin2, (hetero)arenes and Sn2Me6 afford the products (369a–e). As sacrificial anodic material, Zn was oxidized to Zn2+. Adding 20 mol% pyridine was found to be beneficial to the product formation. Although a quite high cell potential was applied, it did not encourage serious side reactions as demonstrated by the broad scope and generally good product yields. A distinct advantage of this e-PRC strategy over the previously reported conPET strategy by Jacobi von Wangelin, Pérez-Ruiz and co-workers (Scheme 63)175 is the expansion of substrate scope to electron-rich chloroarenes. This may suggest that the photoactive species (DCA˙−) was not the same in these reports. Indeed, inspection of the UV-vis data in the e-PRC report reveals signals of a cyanoanthrolate species that was earlier reported to form rapidly upon cathodic treatment of DCA in presence of adventitious O2 in solvent/atmosphere.284,285 Alternatively, it may imply that the steady-state concentration of photoactive species is sufficiently different to impact reactivity.265 In the conPET case, attenuation of the steady-state concentration of DCA˙− by BET may prevent productive reactivity with electron-rich chloroarenes. However, for e-PRC, larger steady-state concentrations of DCA˙− may arise and enhance reactivity, potentially via assembly with ArCl.
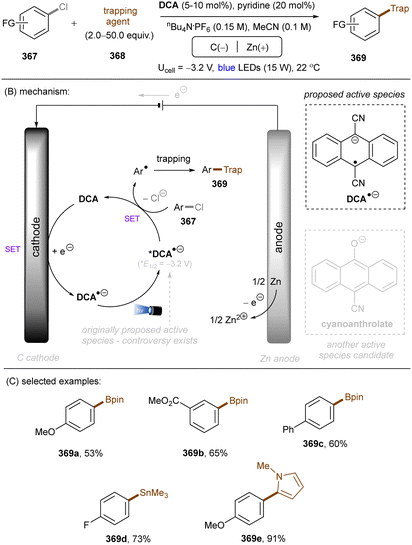 |
| | Scheme 101 e-PRC reduction of aryl chlorides using electro-activated, photoexcited DCA. | |
Concurrently, the Wickens group screened a series of imide-based catalysts, including PDI, naphthalene diimide (NpDI), naphthalene monoimide (NpMI) and phthalimide (PhMI) for e-PRC functionalization of aryl chlorides (Scheme 102).286NpMI was found to be the optimal e-PRCat under blue light irradiation. Differently from Lambert and Lin's work using a sacrificial Zn anode, reticulated vitreous carbon (RVC) was used as both the cathode and the anode material. Ttriethylamine (2 equiv.) was employed in the anodic chamber as a sacrificial reductant. Challenging substrates such as 4-chloroanisole 370b (Ered = −2.9 V vs. SCE) and 2,6-di-tert-butyl-4-chloroanisole 370e (Ered = −3.4 V vs. SCE) with highly negative reduction potentials were readily activated under reaction conditions using a low applied cell potential (−1.3 V) or constant current (0.4 mA). The generated aryl radicals were captured by triethylphosphite (products 370a–e) or N-methyl pyrrole. The authors compared this catalytic system to direct electrolysis and photocatalysis. It was found that while direct electrolysis suffered from unselective reactions yielding both dechlorinated products and radical coupling products, almost no reaction occurred by photocatalysis using a PTH catalyst. These results clearly showcased the great advantages and synthetic potential of the e-PRC strategy with NpMI-type catalysts to achieve deeply reductive SET under mild conditions.
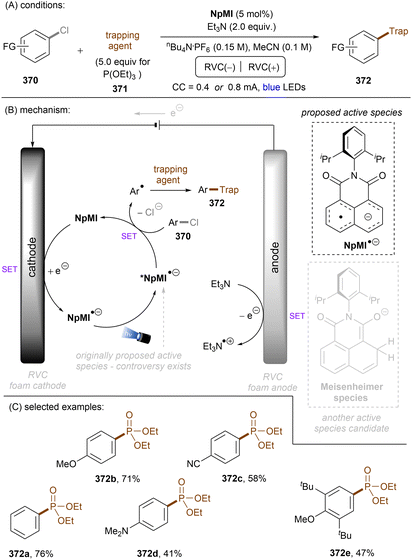 |
| | Scheme 102 e-PRC reduction of aryl chlorides using electro-activated, photoexcited NpMI. | |
Controversy arose over the nature of the active species, since Nocera and co-workers reported preparation of a Meisenheimer species by chemical reduction of NpMI that matched by UV-vis a product resulting from electrochemical decomposition of NpMI˙− at high (Ucell = −3.0 V) applied potentials.287 However, the Meisenheimer species was only demonstrated to engage electron-deficient aryl chlorides. Scott and co-workers later demonstrated how authentically prepared and photoexcited NpMI˙− (and DCA˙−) does lead to SET reduction of more electron-rich 4-chloroanisole/chlorobenzene.288 However, this does not rule out the participation of the NpMI-derived Meisenheimer species when e-PRC is applied to more activated aryl chlorides and it may be possible that more than one active species carries catalytic turnover. After the aforementioned e-PRC reports, the Bardagi group synthesized and screened a series of NpDI catalysts for the reduction of 4-bromobenzonitrile in the presence of excess benzene (Scheme 103).289N,N′-Dihexyl NpDI (NDI2) was found to perform best, delivering the desired product 372 in 27% yield.
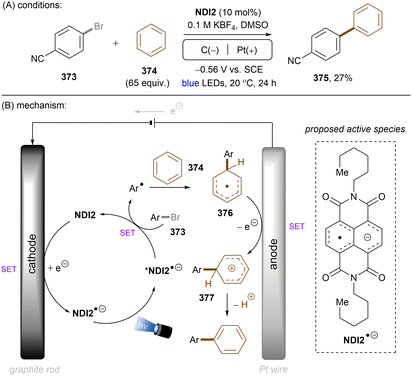 |
| | Scheme 103 e-PRC reduction of bromoarenes with electro-activated, photoexcited NDI2. | |
The e-PRC strategy was also demonstrated for strong aliphatic C(sp3)–X bond activations. König, Barham and co-workers who challenged the activations of strong C(sp3)–O bonds in phosphinates 375 (Ered = −2.2 to −2.6 V vs. SCE) (Scheme 104).290 The e-PRC SET reduction of phosphinates 375 followed by C(sp3)–O bond fragmentation affords alkyl radicals 378, which are further reduced to carbanions 379 at the cathode. Such anionic species undergo either olefinations through elimination of chloride to afford alkenes 376 (X = Cl), or protonations to deliver deoxygenated products 377 (X = H). Electrode materials were found to impact greatly on product formation, and Fe(−)/Zn(+) electrodes were optimal (cheaper than commonly used RVC/Pt).212 Different e-PRCats were examined. With stilbene-forming substrate 376a (R1 = R3 = Ph, R2 = R4 = H), previously reported e-PRC conditions (DCA, Ucell = −3.2 V, 440 nm light)283 led to stilbene 377a and a product of over-reduction, 1,2-diphenyl ethane. A lower applied cell potential of −1.0 V completely circumvented the formation of 1,2-diphenylethane and instead gave rise to E-stilbene in 78% yield. NpMI287 as the e-PRCat provided Z-stilbene as a major product through a tandem e-PRC reduction and energy transfer process. However, neither DCA nor NpMI was able to promote the reduction of more challenging substrates such as 375b, suggesting that redox potential is not the only factor governing reactivity. A newly synthesized NpMI derivative, nBuO-NpMI, was competent in catalyzing these challenging reductions, delivering products in consistently better yields than NpMI. More importantly – and in stark contrast to the report of Wickens and co-workers – this new catalyst allowed selective activations of C(sp3)–O bonds in phosphinates in the presence of other reducible chemical bonds such as the C(sp2)–halogen bonds in aryl halides 375c-d and the C(sp2)–O bond in aryl phosphinate 375f. For comparison, direct electrolysis of 375c-d with a high applied cell potential (−3.2 V) led to dehalogenations rather than olefinations, clearly demonstrating the advantage of this nBuO-NpMI e-PRC system. Synthesis of dienes from allylic phosphinates is also feasible, however an attempt to synthesize alkyl olefin 376h from substrate 375h was unsuccessful. A higher reaction temperature could be beneficial.245
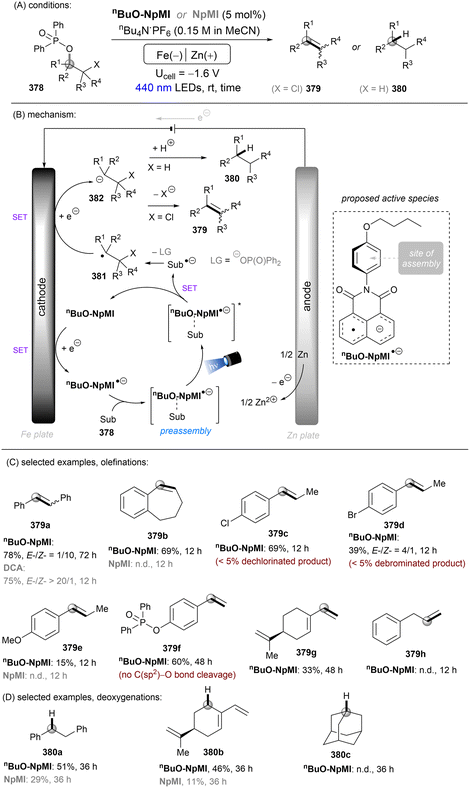 |
| | Scheme 104 e-PRC reductive cleavages of alkyl phosphinates using electro-activated photoexcited nBuO-NpMI. | |
Based on EPR, CV and computational studies, the authors concluded that the C(sp3)–O cleavage was likely the rate-determining step since (i) the SET step was feasible for both catalysts, and (ii) as calculated BDEs of the phosphinate radical anion C(sp3)–O bond increased, synthetic yields decreased. Taken together, this suggests that nBuO-NpMI is able to promote this C(sp3)–O cleavage step in a way that NpMI can not. Given the detailed spectroscopic and computational evidence for preassemblies that have been shown for radical cationic photocatalysts,291,292 the authors proposed a preassembly of nBuO-NpMI˙− with a phosphinate substrate despite a lack of direct spectroscopic evidence.293 Photoexcitation of this complex facilitates SET. Further catalyst structure variation showed that the product yield trended contrary to the steric hindrance at the ortho-position of the aniline moiety. By comparison, varying the electronics/substituents of the naphthalene moiety did not lead to improved catalytic activity compared to NpMI. These results corroborated a preassembly occurring at the aniline moiety. Further computations suggested an interaction of the aniline moiety with two aromatic groups of the phosphinate substrate. This work clearly shows how the tunable cell potential impacts on product types and yields, highlights the wide opportunities for net-reductive transformations provided by e-PRC, and emphasizes the importance of substituent effects in the development of ePRCats, given the cruciality of preassembly on photochemical reactivity and selectivity outcomes.
The Wickens group continued their interest in e-PRC reduction by exploring the scope of possible e-PRCat architectures. They screened 12 commonly used metal-based or organic PCs in order to find a new e-PRCat for reductive activations of strong, halogen-free C(sp2)–X (X = O, N+) bonds in aryl phosphates and trialkylanilinium salts (Scheme 105).294 The minimum one-electron reduction potential – indicated by CV studies of the e-PRCats – was chosen as the cell voltage to reduce these catalysts to their radical anions, followed by irradiation with a proper wavelength, determined by evaluation of wavelength dependent photocurrent, to form corresponding excited state radical anion species. All 12 catalysts were reported to afford the hydrodefunctionalized product in different yields, with an isophthalonitrile-based e-PRCat, 4DPAIPN, performing best at a −1.6 V cell potential and under 405 nm light irradiation. For comparison, direct electrolysis of aryl phosphate substrates under constant current led to uncontrollable reductions, resulting in markedly lower product yields. However, the e-PRC system was able to defunctionalize a diverse suite of trialkylanilinium salts and aryl phosphates (383). In 2012, the Makriyannis group devised a phenol-directed alkylation-defunctionalization synthetic strategy using Li0 to remove a phenol directing group.295 The Wickens group demonstrated their e-PRC defunctionalization strategy as a mild alternative to the Li0-promoted deoxgenation developed by Makriyannis. The generated aryl radicals could also be trapped by P(OEt)3, B2Pin2 and N-methyl pyrrole to form C–P, C–B, and C–C bonds respectively in the products (385a–c). The importance of this study is not limited to discovering a new reductive e-PRC transformation. It also highlights the ability of readily-tunable cathodic potentials to repurpose neutral PCs for mild photooxidations into radical ions as potent photoreductants, a concept that was previously demonstrated in the opposite direction by the Barham group in e-PRC super-oxidation reactions with triarylamine catalysts.291
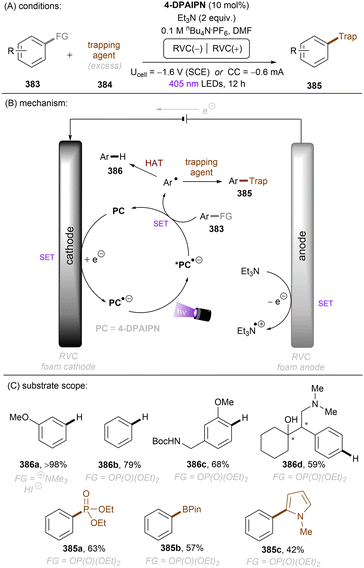 |
| | Scheme 105 e-PRC reductive cleavages of trialkylanilinium salts and aryl phosphates using electro-activated photoexcited 4DPAIPN. | |
In the oxidative direction, the Lambert group reported SNAr substitutions of unactivated aryl fluorides 387 by recycling e-PRC using catalytic amount of 2,3-dichloro-5,6-dicyanoquinone (DDQ) (Scheme 106).296 Given previous reports on SNAr reactions of unactivated aryl fluorides that required strong bases,297–299 this recycling e-PRC strategy offered a base-free alternative. The excited state e-PRCat, *DDQ, is a super-oxidant with a potential of +3.18 V and thus can be reductively quenched even by fluoroarenes. The generated aryl fluoride radical cation 390 undergoes nucleophilic attack by an azole to afford adduct 391. A further cathodic reduction delivers the final product. Meanwhile, DDQ returns upon anodic oxidation of DDQ˙−. This reaction was compatible with both electron-rich and -deficient aryl fluorides. Diverse azoles including pyrazoles (e.g.389a–d), trizaoles and tetrazoles were suitable nucleophiles. A direct amidation with BocNH2 was also successful, although product 389e was afforded in a modest (35%) yield. Defluoroalkoxylations using alcohols were also feasible; the generated alkoxyarene products (e.g.389f) did not undergo further SNAr reactions although such transformations have been reported under acridinium photocatalysis.100 Moreover, H2O was a competent coupling partner, leading to phenol product 389g in a modest (28%) yield. This work demonstrates how recycling e-PRC is able to offer milder alternatives to classical SNAr reactions (that typically required harsher conditions).300,301 With DDQ being promiscuous as a ground-state oxidant for many functional groups, and given its high catalytic loading, modified acridinium-type closed-shell photocatalysts for recycling e-PRC have also been developed that can engage electron-poor arenes in SET oxidations.302
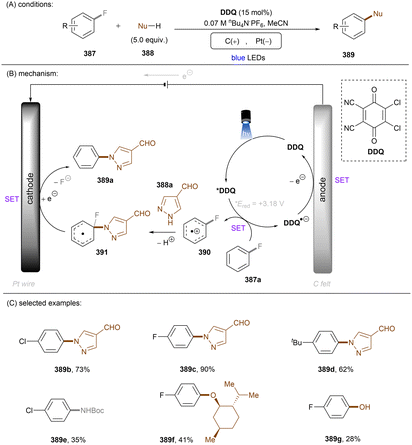 |
| | Scheme 106 SNAr reactions of unactivated aryl fluorides by recycling e-PC. | |
6. Conclusions
Inert C–X bonds are ubiquitous in diverse organic compounds and it is challenge to convert them in a highly selective and efficient manner because of the high energy input required to break these bonds. In the review, we summarized the recent reports on strong bond activations driven by photochemistry and electrochemistry. As the pioneering field, high-energy UVC light (<280 nm) was employed for direct inert bond activations. Later, various stable neutral, cationic or anionic photocatalysts were developed to achieve sufficient redox power via their excited states for inert bond activations under relatively low-energy violet and blue light irradiation. To achieve highly reducing or oxidizing power, unstable or transient photoactive species were utilized and two-photon photocatalysts were designed to obtain extremely redox active photoexcited species for inert bonds activation. On the other hand, electrochemical methods are also promising, if they can be achieved selectively. As the cell potential can be user-adjusted (even rapidly alternated) in electrolysis reactions, we envisage excellent opportunities to realize selective C–X bond activations. When this is not possible, combined photo- and electrochemical strategies provide alternative solutions to realize inert C–X bond activations that avoid the poor selectivity associated with high cell potentials in direct electrolytic processes. Despite the great achievements were made in this recent field, thusfar most of the reactions can only be achieved on a relatively small scale due to limitations of reaction efficiency and reaction set-up. We envisage that inert bond transformations will be crucial for large scale and industrial production of chemicals in the future, especially given the urgent need to use biomass and persistent chemical waste products as feedstocks.
Author contributions
X. T. drafted the pre-proposal for the review and wrote the majority of the manuscript, contributing majorly to the photoredox catalysis section, writing solely the two-photon examples and the photoelectrochemistry section. X. T. guided S. Y. and J. S. with the content of their contributions. Y. L. wrote the section on UV activation and contributed to the introduction. S. Y. wrote the section on electrochemistry. J. S. contributed to the section on photoredox catalysis. J. P. B. supervised X. T., S. Y. and J. S. in their contributions. S. C. guided and supervised Y. L. in her contribution. J. P. B. and S. C. conceived the project, edited the draft of the manuscript and dealt with peer review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
X. T. and J. P. B. thank the Alexander von Humboldt Foundation for funding, provided within the framework of the Sofja Kovalevskaja Award endowed to J. P. B. by the German Federal Ministry of Education and Research. S. Y. is grateful for funding provided by the SynCat programme of the Elite Network of Bavaria. J. P. B. is an associated member of DFG TRR 325 ‘Assembly Controlled Chemical Photocatalysis’ (444632635). J. P. B. is a member of the International Doctoral College (IDK) ‘Photo-Electro Catalysis’ funded by the Elite Network of Bavaria. We thank members of the TRR and IDK for helpful discussions. Y. L. and S. C. thank Nanyang Technological University (NTU Singapore) for the financial support. S. C. is grateful for the funding from the Singapore National Research Foundation (NRF-CRP27-2021-0001) and the Singapore Ministry of Education (Academic Research Fund Tier 2: MOE-T2EP10122-0007).
References
- A. Maercker, Angew. Chem., Int. Ed. Engl., 1987, 26, 972–989 CrossRef.
- W. Liu, J. Li, C.-Y. Huang and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 1786–1796 CrossRef.
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS.
- J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2021, 122, 2292–2352 CrossRef PubMed.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS.
- H.-C. Xu and K. D. Moeller, Journal, 2021, 86, 15845–15846 CAS.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed.
- M. Ghosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710–2746 CrossRef CAS.
- R. D. Little and K. D. Moeller, Journal, 2018, 118, 4483–4484 CAS.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS.
- J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58, 17508–17510 CrossRef CAS PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- M. Freccero, M. Fagnoni and A. Albini, J. Am. Chem. Soc., 2003, 125, 13182–13190 CrossRef CAS PubMed.
- M. Mella, P. Coppo, B. Guizzardi, M. Fagnoni, M. Freccero and A. Albini, J. Org. Chem., 2001, 66, 6344–6352 CrossRef CAS PubMed.
- M. Fagnoni, M. Mella and A. Albini, Org. Lett., 1999, 1, 1299–1301 CrossRef CAS.
- S. Protti, D. Dondi, M. Mella, M. Fagnoni and A. Albini, Eur. J. Org. Chem., 2011, 3229–3237 CrossRef CAS.
- S. Lazzaroni, D. Dondi, M. Fagnoni and A. Albini, Eur. J. Org. Chem., 2007, 4360–4365 CrossRef CAS.
- B. Guizzardi, M. Mella, M. Fagnoni, M. Freccero and A. Albini, J. Org. Chem., 2001, 66, 6353–6363 CrossRef CAS PubMed.
- B. Guizzardi, M. Mella, M. Fagnoni and A. Albini, Tetrahedron, 2000, 56, 9383–9389 CrossRef CAS.
- V. Dichiarante, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 6495–6498 CrossRef CAS PubMed.
- B. Guizzardi, M. Mella, M. Fagnoni and A. Albini, J. Org. Chem., 2003, 68, 1067–1074 CrossRef CAS PubMed.
- S. Protti, M. Fagnoni and A. Albini, Org. Biomol. Chem., 2005, 3, 2868–2871 RSC.
- S. Protti, M. Fagnoni and A. Albini, J. Am. Chem. Soc., 2006, 128, 10670–10671 CrossRef CAS PubMed.
- S. Protti, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2005, 44, 5675–5678 CrossRef CAS PubMed.
- V. Dichiarante, M. Fagnoni and A. Albini, Chem. Commun., 2006, 3001–3003 RSC.
- A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985–2988 CrossRef CAS.
- K. Chen, M. S. Cheung, Z. Lin and P. Li, Org. Chem. Front., 2016, 3, 875–879 RSC.
- T. Ichimura, Y. Mori, H. Shinohara and N. Nishi, Chem. Phys., 1994, 189, 117–125 CrossRef CAS.
- A. M. Mfuh, B. D. Schneider, W. Cruces and O. V. Larionov, Nat. Protoc., 2017, 12, 604–610 CrossRef CAS PubMed.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328–8331 CrossRef CAS PubMed.
- S. C. Lu, X. X. Zhang, Z. J. Shi, Y. W. Ren, B. Li and W. Zhang, Adv. Synth. Catal., 2009, 351, 2839–2844 CrossRef CAS.
- L. Zhang, E. M. Israel, J. Yan and T. Ritter, Nat. Synth., 2022, 1, 376–381 CrossRef.
- W. Liu, X. Yang, Y. Gao and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 8621–8627 CrossRef CAS PubMed.
- Q. Dou, Y. Lang, H. Zeng and C.-J. Li, Fundam. Res., 2021, 1, 742–746 CrossRef CAS.
- W. Liu, J. Li, P. Querard and C.-J. Li, J. Am. Chem. Soc., 2019, 141, 6755–6764 CrossRef CAS PubMed.
- E. Doni, B. Mondal, S. O’Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934–10937 CrossRef CAS PubMed.
- J. Mortensen and J. Heinze, Angew. Chem., Int. Ed. Engl., 1984, 23, 84–85 CrossRef.
- C. Portella, H. Deshayes, J. Pete and D. Scholler, Tetrahedron, 1984, 40, 3635–3644 CrossRef CAS.
- S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed.
- J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 7266–7270 CrossRef CAS PubMed.
- J. Xie, J. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416–9421 CrossRef CAS.
- W. Xu, H. Jiang, J. Leng, H.-W. Ong and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 4009–4016 CrossRef CAS.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- R. M. Pearson, C.-H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef CAS PubMed.
- R. Matsubara, T. Yabuta, U. Md Idros, M. Hayashi, F. Ema, Y. Kobori and K. Sakata, J. Org. Chem., 2018, 83, 9381–9390 CrossRef CAS PubMed.
- D. Meng, Q. Zhu, Y. Wei, S. Zhen, R. Duan, C. Chen, W. Song and J. Zhao, Chin. J. Catal., 2020, 41, 1474–1479 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- S. Yakubov and J. P. Barham, Beilstein J. Org. Chem., 2020, 16, 2151–2192 CrossRef CAS PubMed.
- N. Toriumi, K. Yamashita and N. Iwasawa, Chem. – Eur. J., 2021, 27, 12635–12641 CrossRef CAS PubMed.
- Y. Li, Z. Ye, Y.-M. Lin, Y. Liu, Y. Zhang and L. Gong, Nat. Commun., 2021, 12, 2894 CrossRef CAS PubMed.
- F. Glaser, C. B. Larsen, C. Kerzig and O. S. Wenger, Photochem. Photobiol. Sci., 2020, 19, 1035–1041 CrossRef CAS.
- M. Till, V. Streitferdt, D. J. Scott, M. Mende, R. M. Gschwind and R. Wolf, Chem. Commun., 2022, 58, 1100–1103 RSC.
- M. Schmalzbauer, M. Marcon and B. König, Angew. Chem., Int. Ed., 2021, 60, 6270–6292 CrossRef CAS PubMed.
- M. Schmalzbauer, I. Ghosh and B. König, Faraday Discuss., 2019, 215, 364–378 RSC.
- K. Liang, T. Li, N. Li, Y. Zhang, L. Shen, Z. Ma and C. Xia, Chem. Sci., 2020, 11, 2130–2135 RSC.
- K. Liang, Q. Liu, L. Shen, X. Li, D. Wei, L. Zheng and C. Xia, Chem. Sci., 2020, 11, 6996–7002 RSC.
- H. Hayashi, B. Wang, X. Wu, S. J. Teo, A. Kaga, K. Watanabe, R. Takita, E. K. L. Yeow and S. Chiba, Adv. Synth. Catal., 2020, 362, 2223–2231 CrossRef.
- M. Patil, J. Org. Chem., 2016, 81, 632–639 CrossRef CAS PubMed.
- J. P. Barham, T. N. J. Fouquet and Y. Norikane, Org. Biomol. Chem., 2020, 18, 2063–2075 RSC.
- K. Kolodziejczak, A. J. Stewart, T. Tuttle and J. A. Murphy, Molecules, 2021, 26, 6879 CrossRef CAS PubMed.
- N. Shen, R. Li, C. Liu, X. Shen, W. Guan and R. Shang, ACS Catal., 2022, 12, 2788–2795 CrossRef CAS.
- Q. Wang, M. Poznik, M. Li, P. J. Walsh and J. J. Chruma, Adv. Synth. Catal., 2018, 360, 2854–2868 CrossRef CAS.
- E. Doni, S. O'Sullivan and J. A. Murphy, Angew. Chem., Int. Ed., 2013, 52, 2239–2242 CrossRef CAS PubMed.
- S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476–482 RSC.
- S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818–17826 CrossRef CAS PubMed.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
- M. E. Budén, J. I. Bardagí, M. Puiatti and R. A. Rossi, J. Org. Chem., 2017, 82, 8325–8333 CrossRef PubMed.
- J. T. Moore, M. J. Dorantes, Z. Pengmei, T. M. Schwartz, J. Schaffner, S. L. Apps, C. A. Gaggioli, U. Das, L. Gagliardi, D. A. Blank and C. C. Lu, Angew. Chem., Int. Ed., 2022, 61, e202205575 CrossRef CAS PubMed.
- B. S. Kim and S. M. Park, J. Electrochem. Soc., 1993, 140, 115 CrossRef CAS.
- P. Leghié, J.-P. Lelieur and E. Levillain, Electrochem. Commun., 2002, 4, 406–411 CrossRef.
- Q. Zou and Y.-C. Lu, J. Phys. Chem. Lett., 2016, 7, 1518–1525 CrossRef CAS PubMed.
- H. Li, X. Tang, J. H. Pang, X. Wu, E. K. Yeow, J. Wu and S. Chiba, J. Am. Chem. Soc., 2020, 143, 481–487 CrossRef PubMed.
- H. Li, Y. Liu and S. Chiba, Chem. Commun., 2021, 57, 6264–6267 RSC.
- S. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS PubMed.
- S. Wang, H. Wang and B. König, Chem, 2021, 7, 1653–1665 CAS.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS PubMed.
- L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124–9128 CrossRef CAS PubMed.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef CAS PubMed.
- T.-H. Ding, J.-P. Qu and Y.-B. Kang, Org. Lett., 2020, 22, 3084–3088 CrossRef CAS PubMed.
- M. P. Drapeau, I. Fabre, L. Grimaud, I. Ciofini, T. Ollevier and M. Taillefer, Angew. Chem., Int. Ed., 2015, 54, 10587–10591 CrossRef PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- J. B. McManus and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS PubMed.
- N. E. Tay and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 16100–16104 CrossRef CAS PubMed.
- W. Chen, H. Wang, N. E. S. Tay, V. A. Pistritto, K.-P. Li, T. Zhang, Z. Wu, D. A. Nicewicz and Z. Li, Nat. Chem., 2022, 14, 216–223 CrossRef CAS PubMed.
- N. J. Venditto and D. A. Nicewicz, Org. Lett., 2020, 22, 4817–4822 CrossRef CAS PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Org. Lett., 2019, 21, 7114–7118 CrossRef CAS PubMed.
- V. A. Pistritto, M. E. Schutzbach-Horton and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 17187–17194 CrossRef CAS PubMed.
- V. A. Pistritto, S. Liu and D. A. Nicewicz, J. Am. Chem. Soc., 2022, 144(33), 15118–15131 CrossRef CAS PubMed.
- M. A. Mantell, M. R. Lasky, M. Lee, M. Remy and M. S. Sanford, Org. Lett., 2021, 23, 5213–5217 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed.
- Y. Zafrani, G. Sod-Moriah, D. Yeffet, A. Berliner, D. Amir, D. Marciano, S. Elias, S. Katalan, N. Ashkenazi, M. Madmon, E. Gershonov and S. Saphier, J. Med. Chem., 2019, 62, 5628–5637 CrossRef CAS PubMed.
- D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed.
- G. Yan, X. Gong and Q. Zhou, Org. Biomol. Chem., 2022, 20, 5365–5376 RSC.
- Q. Shen, Y.-G. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, J. Fluor. Chem., 2015, 179, 14–22 CrossRef CAS.
- H.-J. Ai, X. Ma, Q. Song and X.-F. Wu, Sci. China: Chem., 2021, 64, 1630–1659 CrossRef CAS.
- K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed.
- S. Ghosh, Z.-W. Qu, S. Pradhan, A. Ghosh, S. Grimme and I. Chatterjee, Angew. Chem., Int. Ed., 2022, 61, e202115272 CrossRef CAS PubMed.
- J. R. Box, A. P. Atkins and A. J. Lennox, Chem. Sci., 2021, 12, 10252–10258 RSC.
- N. Sugihara, K. Suzuki, Y. Nishimoto and M. Yasuda, J. Am. Chem. Soc., 2021, 143, 9308–9313 CrossRef CAS PubMed.
- H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef CAS PubMed.
- J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, M. Médebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noël, C. W. am Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS PubMed.
- H. Li and O. S. Wenger, Angew. Chem., 2022, 134, e202110491 CrossRef.
- E. Shirman, A. Ustinov, N. Ben-Shitrit, H. Weissman, M. A. Iron, R. Cohen and B. Rybtchinski, J. Phys. Chem. B, 2008, 112, 8855–8858 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt and F. Würthner, Chem. Sci., 2015, 6, 1663–1667 RSC.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- L. Zeng, T. Liu, C. He, D. Shi, F. Zhang and J. Duan, J. Am. Chem. Soc., 2016, 138, 3958–3961 CrossRef CAS PubMed.
- M. Goez, C. Kerzig and R. Naumann, Angew. Chem., Int. Ed., 2014, 53, 9914–9916 CrossRef CAS PubMed.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023–11029 RSC.
- Y.-C. Luo, F.-F. Tong, Y. Zhang, C.-Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971–13979 CrossRef CAS PubMed.
- J. B. Sap, N. J. Straathof, T. Knauber, C. F. Meyer, M. Médebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noël and C. W. Am Ende, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS PubMed.
- H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122–8129 CrossRef CAS PubMed.
- C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS PubMed.
- M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 CrossRef CAS PubMed.
- M. W. Campbell, M. Yuan, V. C. Polites, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 3901–3910 CrossRef CAS PubMed.
- P. Xu, X.-Y. Wang, Z. Wang, J. Zhao, X.-D. Cao, X.-C. Xiong, Y.-C. Yuan, S. Zhu, D. Guo and X. Zhu, Org. Lett., 2022, 24, 4075–4080 CrossRef CAS PubMed.
- S.-S. Yan, S.-H. Liu, L. Chen, Z.-Y. Bo, K. Jing, T.-Y. Gao, B. Yu, Y. Lan, S.-P. Luo and D.-G. Yu, Chem, 2021, 7, 3099–3113 CAS.
- D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed.
- C. Liu, N. Shen and R. Shang, Nat. Commun., 2022, 13, 354 CrossRef CAS PubMed.
- H. Li, Y. Liu and S. Chiba, JACS Au, 2021, 1, 2121–2129 CrossRef CAS PubMed.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed.
- C. Liu, K. Li and R. Shang, ACS Catal., 2022, 12, 4103–4109 CrossRef CAS.
- D. Wei, X. Li, L. Shen, Y. Ding, K. Liang and C. Xia, Org. Chem. Front., 2021, 8, 6364–6370 RSC.
- P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed.
- W.-J. Zhou, G.-M. Cao, Z.-P. Zhang and D.-G. Yu, Chem. Lett., 2019, 48, 181–191 CrossRef CAS.
- G. S. Lee, D. Kim and S. H. Hong, Nat. Commun., 2021, 12, 991 CrossRef CAS PubMed.
- K. Muralirajan, R. Kancherla, A. Gimnkhan and M. Rueping, Org. Lett., 2021, 23, 6905–6910 CrossRef CAS PubMed.
- M. Claros, F. Ungeheuer, F. Franco, V. Martin-Diaconescu, A. Casitas and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2019, 58, 4869–4874 CrossRef CAS PubMed.
- J. Aragón, S. Sun, D. Pascual, S. Jaworski and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2022, 61, e202114365 CrossRef PubMed.
- Y.-M. Tian, X.-N. Guo, I. Krummenacher, Z. Wu, J. Nitsch, H. Braunschweig, U. Radius and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 18231–18242 CrossRef CAS PubMed.
- P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed.
- D. Rackl, V. Kais, P. Kreitmeier and O. Reiser, Beilstein J. Org. Chem., 2014, 10, 2157–2165 CrossRef CAS PubMed.
- E. Speckmeier, C. Padié and K. Zeitler, Org. Lett., 2015, 17, 4818–4821 CrossRef CAS PubMed.
- F. J. A. Troyano, F. Ballaschk, M. Jaschinski, Y. Özkaya and A. Gómez-Suárez, Chem. – Eur. J., 2019, 25, 14054–14058 CrossRef PubMed.
- T. Yabuta, M. Hayashi and R. Matsubara, J. Org. Chem., 2021, 86, 2545–2555 CrossRef CAS PubMed.
- O. P. Williams, A. F. Chmiel, M. Mikhael, D. M. Bates, C. S. Yeung and Z. K. Wickens, Angew. Chem., Int. Ed., 2023, 62, e202300178 CrossRef CAS PubMed.
- A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed.
- S. Yakubov, W. J. Stockerl, X. Tian, A. Shahin, M. J. P. Mandigma, R. M. Gschwind and J. P. Barham, Chem. Sci., 2022, 13, 14041–14051 RSC.
- S. Hermann, D. Sack and H.-A. Wagenknecht, Eur. J. Org. Chem., 2018, 2204–2207 CrossRef CAS.
- F. Speck, D. Rombach and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2019, 15, 52–59 CrossRef CAS PubMed.
- F. Seyfert, M. Mitha and H.-A. Wagenknecht, Eur. J. Org. Chem., 2021, 773–776 CrossRef CAS.
- E. Y. K. Tan, A. S. Mat Lani, W. Sow, Y. Liu, H. Li and S. Chiba, Angew. Chem., Int. Ed., 2023, e202309764 CAS.
- W. H. Koppenol and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 CrossRef CAS.
- L.-L. Liao, L. Song, S.-S. Yan, J.-H. Ye and D.-G. Yu, Trends Chem., 2022, 4, 512 CrossRef CAS.
- Y. Lee and M. S. Kwon, Eur. J. Org. Chem., 2020, 6028–6043 CrossRef CAS.
- F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- M. Lepori, S. Schmid and J. P. Barham, Beilstein J. Org. Chem., 2023, 19, 1055–1145 CrossRef CAS PubMed.
- M. Marchini, A. Gualandi, L. Mengozzi, P. Franchi, M. Lucarini, P. G. Cozzi, V. Balzani and P. Ceroni, Phys. Chem. Chem. Phys., 2018, 20, 8071–8076 RSC.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed.
- A. Das, I. Ghosh and B. König, Chem. Commun., 2016, 52, 8695–8698 RSC.
- J. I. Bardagi, I. Ghosh, M. Schmalzbauer, T. Ghosh and B. König, Eur. J. Org. Chem., 2018, 34–40 CrossRef CAS.
- S. Fukuzumi and K. Ohkubo, Coord. Chem. Rev., 2010, 254, 372–385 CrossRef CAS.
- A. U. Meyer, T. Slanina, A. Heckel and B. König, Chem. – Eur. J., 2017, 23, 7900–7904 CrossRef CAS PubMed.
- M. Neumeier, D. Sampedro, M. Majek, V. A. de la Pena O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem. – Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658 CrossRef CAS PubMed.
- A. Graml, T. Neveselý, R. J. Kutta, R. Cibulka and B. König, Nat. Commun., 2020, 11, 3174 CrossRef CAS PubMed.
- K. A. McDaniel, A. R. Blood, G. C. Smith and N. T. Jui, ACS Catal., 2021, 11, 4968–4972 CrossRef CAS PubMed.
- J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. Yeow and J. Wu, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef CAS PubMed.
- S. Grotjahn and B. König, Org. Lett., 2021, 23, 3146–3150 CrossRef CAS PubMed.
- Y. Kwon, J. Lee, Y. Noh, D. Kim, Y. Lee, C. Yu, J. C. Roldao, S. Feng, J. Gierschner, R. Wannemacher and M. S. Kwon, Nat. Commun., 2023, 14, 92 CrossRef CAS PubMed.
- I. A. MacKenzie, L. Wang, N. P. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- T. J. Sørensen, M. F. Nielsen and B. W. Laursen, ChemPlusChem, 2014, 79, 1030–1035 CrossRef.
- A. C. Shaikh, J. Moutet, J. M. Veleta, M. M. Hossain, J. Bloch, A. V. Astashkin and T. L. Gianetti, Chem. Sci., 2020, 11, 11060–11067 RSC.
- L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS PubMed.
-
A. C. Shaikh, M. M. Hossain, R. Kaur, J. Moutet, A. Kumar, B. Thompson, V. M. Huxter and T. L. Gianetti, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-6qpb8.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef CAS PubMed.
- Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS PubMed.
- M. Giedyk, R. Narobe, S. Weiss, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- J. H. Ye, P. Bellotti, C. Heusel and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202115456 CrossRef CAS PubMed.
-
A. A. Akhrem, Birch Reduction of Aromatic Compounds, 2012 Search PubMed.
- P. Lei, Y. Ding, X. Zhang, A. Adijiang, H. Li, Y. Ling and J. An, Org. Lett., 2018, 20, 3439–3442 CrossRef CAS PubMed.
- J. Burrows, S. Kamo and K. Koide, Science, 2021, 374, 741–746 CrossRef CAS PubMed.
- A. Chatterjee and B. König, Angew. Chem., Int. Ed., 2019, 58, 14289–14294 CrossRef CAS PubMed.
- X. Li, J. Ma, G. Liu, J. Fang, S. Yue, Y. Guan, L. Chen and X. Liu, Environ. Sci. Technol., 2012, 46, 7342–7349 CrossRef CAS PubMed.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed.
- L. Zhang, D. Zhu, G. M. Nathanson and R. J. Hamers, Angew. Chem., Int. Ed., 2014, 53, 9746–9750 CrossRef CAS PubMed.
- R. Naumann, C. Kerzig and M. Goez, Chem. Sci., 2017, 8, 7510–7520 RSC.
- R. Naumann, F. Lehmann and M. Goez, Angew. Chem., Int. Ed., 2018, 57, 1078–1081 CrossRef CAS PubMed.
- R. Naumann, F. Lehmann and M. Goez, Chem. – Eur. J., 2018, 24, 13259–13269 CrossRef CAS PubMed.
- R. Naumann and M. Goez, Chem. – Eur. J., 2018, 24, 17557–17567 CrossRef CAS PubMed.
- J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS PubMed.
- H. Lund, J. Electrochem. Soc., 2002, 149, S21 CrossRef CAS.
- J.-I. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC.
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213 CrossRef CAS.
- J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC.
- S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- S. Mohle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef PubMed.
- Q. Jing and K. D. Moeller, Acc. Chem. Res., 2020, 53, 135 CrossRef CAS PubMed.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- R. D. Little and K. D. Moeller, Chem. Rev., 2018, 118, 4483–4484 CrossRef CAS PubMed.
- S. D. Minteer and P. Baran, Acc. Chem. Res., 2020, 53, 545 CrossRef CAS PubMed.
- L. R. Faulkner, J. Chem. Educ., 1983, 60, 262 CrossRef CAS.
- P. T. Kissinger and A. W. Bott, Curr. Sep. Drug Dev., 2002, 20, 51 CAS.
-
D. T. Sawyer, J. L. Jr. and A. Sobkowiak, Electrochemistry for chemists, Wiley, New York (NY), 1995 Search PubMed.
- J. F. Bunnett, Acc. Chem. Res., 1978, 11, 413–420 CrossRef CAS.
- Z. Chami, M. Gareil, J. Pinson, J. M. Saveant and A. Thiebault, J. Org. Chem., 1991, 56, 586–595 CrossRef CAS.
- J. Ke, H. Wang, L. Zhou, C. Mou, J. Zhang, L. Pan and Y. R. Chi, Chem. – Eur. J., 2019, 25, 6911–6914 CrossRef CAS PubMed.
- L. Lu, H. Li, Y. Zheng, F. Bu and A. Lei, CCS Chem., 2021, 3, 2669–2675 CrossRef CAS.
- Z. Chami, M. Gareil, J. Pinson, J.-M. Savéant and A. Thiebault, J. Org. Chem., 1991, 56, 586–595 CrossRef CAS.
- E. Kariv-Miller and Z. Vajtner, J. Org. Chem., 1985, 50, 1394–1399 CrossRef CAS.
- E. Kariv-Miller, C. Nanjundiah, J. Eaton and K. E. Swenson, J. Electroanal. Chem. Interfacial Electrochem., 1984, 167, 141–155 CrossRef CAS.
- D. M. Loffredo, J. E. Swartz and E. Kariv-Miller, J. Org. Chem., 1989, 54, 5953–5957 CrossRef CAS.
- W.-B. Wu, M.-L. Li and J.-M. Huang, Tetrahedron Lett., 2015, 56, 1520–1523 CrossRef CAS.
- T. A. Thornton, N. F. Woolsey and D. E. Bartak, J. Am. Chem. Soc., 1986, 108, 6497–6502 CrossRef CAS.
- V. Yanilkin, Y. G. Budnikova, Y. M. Kargin, E. Gritsenko and V. Strelets, Bull. Acad. Sci. USSR, Div. Chem. Sci, 1990, 39, 1149–1152 CrossRef.
- J. A. Cleary, M. S. Mubarak, K. L. Vieira, M. R. Anderson and D. G. Peters, J. Electroanal. Chem. Interfacial Electrochem., 1986, 198, 107–124 CrossRef CAS.
- W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS PubMed.
- X. Yan, S. Wang, Z. Liu, Y. Luo, P. Wang, W. Shi, X. Qi, Z. Huang and A. Lei, Sci. China: Chem., 2022, 65, 762–770 CrossRef CAS.
- J. Kopilov and D. H. Evans, J. Electroanal. Chem. Interfacial Electrochem., 1990, 280, 435–438 CrossRef CAS.
- B. Wang, P. Peng, W. Ma, Z. Liu, C. Huang, Y. Cao, P. Hu, X. Qi and Q. Lu, J. Am. Chem. Soc., 2021, 143, 12985–12991 CrossRef CAS PubMed.
- P. Li, C. Guo, S. Wang, D. Ma, T. Feng, Y. Wang and Y. Qiu, Nat. Commun., 2022, 13, 3774 CrossRef CAS PubMed.
- J. L. Röckl, E. L. Robertson and H. Lundberg, Org. Biomol. Chem., 2022, 20, 6707–6720 RSC.
- Z. Wang, Y. Sun, L.-Y. Shen, W.-C. Yang, F. Meng and P. Li, Org. Chem. Front., 2022, 9, 853–873 RSC.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed.
- C. Saboureau, M. Troupel, S. Sibille and J. Périchon, J. Chem. Soc., Chem. Commun., 1989, 1138–1139 RSC.
- H. Hebri, E. Duñach and J. Périchon, Synth. Commun., 1991, 21, 2377–2382 CrossRef CAS.
- P. Clavel, M. P. Léger-Lambert, C. Biran, F. Serein-Spirau, M. Bordeau, N. Roques and H. Marzouk, Synthesis, 1999, 829–834 CrossRef CAS.
- B. Huang, L. Guo and W. Xia, Green Chem., 2021, 23, 2095–2103 RSC.
- J. R. Box, M. E. Avanthay, D. L. Poole and A. J. J. Lennox, Angew. Chem., Int. Ed., 2023, 62, e202218195 CrossRef CAS PubMed.
- A. Inesi and L. Rampazzo, J. Electroanal. Chem. Interfacial Electrochem., 1974, 49, 85–93 CrossRef CAS.
- J. R. Box, A. P. Atkinsa and A. J. J. Lennox, Chem. Sci., 2021, 12, 10252–10258 RSC.
- K. Lam and I. E. Markó, Org. Lett., 2011, 13, 406–409 CrossRef CAS PubMed.
- H. Wang, Z. Wang, G. Zhao, V. Ramadoss, L. Tian and Y. Wang, Org. Lett., 2022, 24, 3668–3673 CrossRef CAS PubMed.
- H. Senboku, M. Takahashi, T. Fukuhara and S. Hara, Chem. Lett., 2007, 36, 228–229 CrossRef CAS.
- A. Stanislaus and B. H. Cooper, Catal. Rev. – Sci. Eng., 1994, 36, 75–123 CrossRef CAS.
- H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- J. Li, L. He, X. Liu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2019, 58, 1759–1763 CrossRef CAS PubMed.
- R. F. Service, Science, 2018, 361, 120 CrossRef CAS PubMed.
- H. Qin, J. Yang, K. Yan, Y. Xue, M. Zhang, X. Sun, J. Wen and H. Wang, Adv. Synth. Catal., 2021, 363, 2104–2109 CrossRef CAS.
- B. Huang, Y. Li, C. Yang and W. Xia, Chem. Commun., 2019, 55, 6731–6734 RSC.
- J. Yokoyama, T. Matsuda, S. Koshiba, N. Tochio and T. Kigawa, Anal. Biochem., 2011, 411, 223–229 CrossRef CAS PubMed.
- P. V. Dayal, H. Singh, L. S. Busenlehner and H. R. Ellis, Biochemistry, 2015, 54, 7531–7538 CrossRef CAS PubMed.
- L. Wei, Y. Shen, F. Xu, F. Hu, J. K. Harrington, K. L. Targoff and W. Min, ACS Chem. Biol., 2015, 10, 901–908 CrossRef CAS PubMed.
- X. Liu, R. Liu, J. Qiu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2020, 59, 13962–13967 CrossRef CAS PubMed.
- R. A. Benkeser and E. M. Kaiser, J. Am. Chem. Soc., 1963, 85, 2858–2859 CrossRef CAS.
- K. E. Swenson, D. Zemach, C. Nanjundiah and E. Kariv-Miller, J. Org. Chem., 1983, 48, 1777–1779 CrossRef CAS.
- J. Chaussard, C. Combellas and A. Thiebault, Tetrahedron Lett., 1987, 28, 1173–1174 CrossRef CAS.
- M. Ishifune, H. Yamashita, Y. Kera, N. Yamashita, K. Hirata, H. Murase and S. Kashimura, Electrochim. Acta, 2003, 48, 2405–2409 CrossRef CAS.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen and S. Udyavara, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed.
- Y. Yu, P. Guo, J.-S. Zhong, Y. Yuan and K.-Y. Ye, Org. Chem. Front., 2020, 7, 131–135 RSC.
- J.-C. Moutet and G. Reverdy, Tetrahedron Lett., 1979, 20, 2389–2392 CrossRef.
- J.-C. Moutet and G. Reverdy, J. Chem. Soc., Chem. Commun., 1982, 654–655 RSC.
- W. Zhang, K. L. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS PubMed.
- H. Yan, Z.-W. Hou and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS PubMed.
- X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed.
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed.
- Y. Qiu, A. Scheremetjew, L. H. Finger and L. Ackermann, Chem. – Eur. J., 2020, 26, 3241–3246 CrossRef CAS PubMed.
- P. Xu, P.-Y. Chen and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed.
- L. Capaldo, L. L. Quadri, D. Merli and D. Ravelli, Chem. Commun., 2021, 57, 4424–4427 RSC.
- H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2021, 60, 11163–11167 CrossRef CAS PubMed.
- C.-Y. Cai, X.-L. Lai, Y. Wang, H.-H. Hu, J. Song, Y. Yang, C. Wang and H.-C. Xu, Nat. Catal., 2022, 5, 943–951 CrossRef CAS.
- P. Nelleborg, H. Lund and J. Eriksen, Tetrahedron Lett., 1985, 26, 1773–1776 CrossRef CAS.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- J. S. Beckwith, A. Aster and E. Vauthey, Phys. Chem. Chem. Phys., 2022, 24, 568–577 RSC.
- D. T. Breslin and M. A. Fox, J. Phys. Chem., 1994, 98, 408–411 CrossRef CAS.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- A. J. Reith, M. I. Gonzalez, B. Kudisch, M. Nava and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 14352–14359 CrossRef PubMed.
- S. J. Horsewill, G. Hierlmeier, Z. Farasat, J. P. Barham and D. J. Scott, ACS Catal., 2023, 13, 9392 CrossRef CAS PubMed.
- S. Caby, L. M. Bouchet, J. E. Argüello, R. A. Rossi and J. I. Bardagi, ChemCatChem, 2021, 13, 3001–3009 CrossRef CAS.
- X. Tian, T. A. Karl, S. Reiter, S. Yakubov, R. de Vivie-Riedle, B. König and J. P. Barham, Angew. Chem., Int. Ed., 2021, 60, 20817–20825 CrossRef CAS PubMed.
- S. Wu, J. Žurauskas, M. Domański, P. S. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug, J. Hauer and J. P. Barham, Org. Chem. Front., 2021, 8, 1132–1142 RSC.
- A. Kumar, P. Malevich, L. Mewes, S. Wu, J. P. Barham and J. Hauer, J. Chem. Phys., 2023, 158, 144201 CrossRef CAS PubMed.
- A lack of change in steady-state UV-vis of the radical ion photocatalyst in the presence of substrate cannot be relied upon to rule out their preassembly. Preassemblies have been confirmed by transient absorption spectroscopy for both radical cations (ref. 289) and radical anions, see: D. Y. Jeong, D. S. Lee, H. L. Lee, S. Nah, J. Y. Lee, E. J. Cho and Y. You, ACS Catal., 2022, 12, 6047–6059 CrossRef CAS.
- C. P. Chernowsky, A. F. Chmiel and Z. K. Wickens, Angew. Chem., Int. Ed., 2021, 60, 21418–21425 CrossRef CAS PubMed.
- D. Lu, S. P. Nikas, X.-W. Han, D. A. Parrish and A. Makriyannis, Tetrahedron Lett., 2012, 53, 4636–4638 CrossRef CAS PubMed.
- H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2020, 59, 658–662 CrossRef CAS PubMed.
- Y. Zheng, A. S. Batsanov, V. Jankus, F. B. Dias, M. R. Bryce and A. P. Monkman, J. Org. Chem., 2011, 76, 8300–8310 CrossRef CAS PubMed.
- C. Borch Jacobsen, M. Meldal and F. Diness, Chem. – Eur. J., 2017, 23, 846–851 CrossRef CAS PubMed.
- F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2012, 51, 8012–8016 CrossRef CAS PubMed.
- R. Jitchati, A. S. Batsanov and M. R. Bryce, Tetrahedron, 2009, 65, 855–861 CrossRef CAS.
- A. Huang, F. Liu, C. Zhan, Y. Liu and C. Ma, Org. Biomol. Chem., 2011, 9, 7351–7357 RSC.
- J. Žurauskas, S. Bohacova, S. Wu, V. Butera, S. Schmid, M. Domański, T. Slanina and J. P. Barham, Angew. Chem., Int. Ed., 2023, 26, e202307550 Search PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b and
Joshua P.
Barham
*b and
Joshua P.
Barham