
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-pH and time-humidity scaling of ionic conductivity spectra of polyelectrolyte multilayers†
Jannis
Schlicke
 ab,
Cornelia
Cramer
*ab and
Monika
Schönhoff
*ab
ab,
Cornelia
Cramer
*ab and
Monika
Schönhoff
*ab
aInstitute of Physical Chemistry, University of Münster, Corrensstraße 28/30, 48149 Münster, Germany. E-mail: schoenho@uni-muenster.de
bCenter for Soft Nanoscience, Busso-Peuss-Str. 10, 48149 Münster, Germany
First published on 9th October 2024
Abstract
In this systematic study, ionic conductivity spectra of poly(diallyl-dimethylammonium)/poly(acrylic acid) (PDADMA/PAA)n polyelectrolyte multilayers (PEMs) are investigated regarding superposition principles. In this context, charge transport as well as charge compensation processes in polyelectrolyte assemblies are discussed. The validity of different scaling concepts is tested to differentiate between changes in the mobility and charge carrier density, caused by the variation of a parameter X, where X is either relative humidity during measurement, or salt concentration or pH during preparation. For the first time, time-X scaling for conductivity spectra of PEMs is reported for all three parameters X, resulting in individual mastercurves. Furthermore, a super-mastercurve can be obtained including variations of all three parameters. Changes in plasticization caused by either varied humidity, pH or ionic strength imply non-constant charge carrier mobilities in accordance with a Summerfield-type of scaling, while the charge carrier density remains constant. Interestingly, for preparation conditions which favor extrinsic charge compensation, significant deviations from such Summerfield-type scaling are observed, indicating a variation of the number density of mobile charge carriers with humidity.
Introduction
Throughout the last decades, materials prepared by the self-assembly of polyelectrolytes have been intensely investigated and discussed for a wide range of applications including drug delivery,1–4 filtration5–9 or sensing.10–13 These materials comprise polyelectrolyte multilayers as well as polyelectrolyte complexes or coacervates. Furthermore, several publications have shed light on the fundamental aspects of the entropically driven assembly.14–17 The formation process, structure, composition and physical properties of these materials as a function of preparation conditions and post-preparational treatments were correlated to variations in polyelectrolyte chain conformation or other fundamental considerations, mostly concerning electrostatic interactions.14,17–23 In certain cases, other interactions like ion–dipole24–26 or hydrophobic interactions,26,27 as well as hydrogen bonding28,29 are discussed. In spite of the wealth of literature, studies on the ionic conductivity of these versatile materials are so far scarce, though potential applications of self-assembled polyelectrolyte materials in the field of solid state ionics are plausible.10,30–36 In this context, especially the charge transport processes in polyelectrolyte multilayers (PEMs) are hard to clarify. This is mostly due to the challenging determination of the composition and conformation of these thin films, which are formed by layer-by-layer processes.37 Considering their beneficial and tunable geometry when discussing admittances of solid ion conductors, PEMs might, however, prove especially suitable candidates for applications where ultrathin films are required. Therefore, the presented publication aims at resolving some critical uncertainties in this field.A fundamental question concerns the nature of the mobile charge carriers in polyelectrolyte materials. Due to the entropy-driven complexation process17,38–40 intrinsic charge compensation of oppositely charged polyelectrolyte chains is highly favored over the extrinsic charge compensation by small counterions. Consequently, the number of mobile extrinsic charge carriers, i.e. counterions, is largely correlated to the stoichiometry of positive and negative polyelectrolyte charges.41–46 For polyelectrolyte complexes including the dialyzed supernatant, this stoichiometry can be easily defined during preparation. Therefore, the ionic conductivity of polyelectrolyte complexes (PECs) with non-equimolar stoichiometry of polyelectrolyte charges is dominated by extrinsic charge carriers.47–51 The charge stoichiometry within PEMs is however expected to be close to equimolar, without extrinsic charge carriers. Indeed, the ionic conductivity of PEMs was found to be small in the dry state.52 In the hydrated state, charge transport is significantly enhanced and was claimed to be dominated by protons.31,53–55 These findings agree with findings on the plasticizing effect of water on glass transition temperatures and chain dynamics.56,57 Recently, when investigating (PDADMA/PAA)n PEMs from certain preparation conditions, we had questioned the latter statement and postulated significant contributions of small counterions to the conductivity.58
Ionic conductivities of polyelectrolyte assemblies have been shown to vary by orders of magnitude depending on both preparation and measurement conditions.52,55,58 On this matter, plasticization by water was proven crucial. For hydrated PEMs, conductivities increased by orders of magnitude due to small changes in hydration caused by systematic variations of the relative humidity (RH).55 For hydrated PECs, the water content was found to be generally proportional to the relative humidity for low water content.50 Deviations for high relative humidity were shown,59–61 and the plasticizing effect of hydration on the chain dynamics has been studied thoroughly in recent years.20,56,57,62–66
Furthermore, in PEMs the nature of the employed polyelectrolytes as well as the preparation conditions influence the conductivity.31,32,34,52,53,55,58,67
In certain cases, changes of the conductivity σ can be attributed to variations in the mobility μ and/or number density N of charge carriers without an alteration of the overall conduction mechanism. This is reflected by a constant general shape of conductivity spectra, which can be scaled onto a mastercurve according to time-parameter superpositions. The utilized scaling concepts grant additional information on the underlying effects concerning a more quantitative separation of variations in μ and N.
For PECs, superposition concepts of the ionic conductivity have been reported for variations concerning preparation and measurement conditions. A time-temperature superposition was found for the ionic conductivity spectra of dry47 and hydrated49 PDADMA/poly(styrene sulfonate) PECs. De et al. furthermore reported a time-humidity superposition for these materials.48 From the latter, a Summerfield-type scaling, see also the following Theory section, was concluded and it was confirmed that the conduction in these PECs is dominated by hydrated counterions. Although for polyelectrolyte multilayers a similarly significant enhancing effect of increased humidity on the conductivity was demonstrated,55 a systematic scaling analysis for conductivity spectra of PEMs has not yet been reported in the literature. To the best of our knowledge, an analysis of superposition concepts concerning conductivity spectra of varied preparation conditions of polyelectrolyte multilayers is also lacking.
The present work provides an analysis on how changes in the preparation and measurement conditions affect the shape of conductivity spectra and thus charge transport processes in PEMs. Employing PAA as a weak polyanion, effects of preparation pH are included in addition to the effect of salt during preparation. Furthermore, the relative humidity during measurement is varied over a wide range. For all three parameter variations, we find a global mastercurve of the conductivity spectrum. Thus, time-pH, time-salt and time-RH superposition of conductivity spectra are shown to be valid. Based on the superposition approach, we are able to separate the effects of varied charge carrier mobility and their number density on the conductivity.
Theory section: scaling analysis of conductivity spectra
A number of studies utilize scaling concepts for conductivity spectra to answer some of the key questions concerning the charge transport in structurally disordered ion conductors.48,68–70 This brief theory section will encompass the method of superpositions as well as the key conclusions, which can be drawn from scaling concepts. While the concepts have been mainly developed using data of inorganic glasses, we apply them to PEMs in our work.The complex frequency-dependent conductivity consists of a real part, denoted as σ′(ν), and an imaginary part denoted as σ′′(ν). The latter is connected to σ′(ν) via the Kramers–Kronig relations and, therefore, contains the same information about ion movements as the real part does. Throughout the manuscript we will therefore represent and discuss σ′(ν) spectra only.
In general, time-parameter (time-X) superposition principles imply that a set of conductivity spectra measured for a varied parameter X can be shifted onto a single mastercurve. The existence of such a mastercurve indicates a constant general conduction mechanism which is independent of X. The variations in conductivity σ′(ν) are attributed to the X-dependences of the mobility μ and number density N of charge carriers, respectively:
| σ′(ν,X) = N(X) · μ(ν,X) · q | (1) |
In this equation ν stands for the experimental frequency in Hz and q for the charge of the mobile ions. Here we assume that only a single mobile species is to be discussed. Increases in mobility μ(ν,X) may, for example, arise from an increase in temperature or plasticization. Furthermore, the number density N(X) might change directly due to a variation in composition or indirectly via a variation of the fraction of free, mobile charge carriers as opposed to immobile ions. In contrast to strong electrolytes, this fraction might depend on X (for example pH) for weak electrolytes.
The method of scaling required to achieve a time-X superposition allows for separation of contributions of varied μ and N. Shifting of the different individual spectra is performed by scaling the frequency and conductivity axes by characteristic values (σ0 and ν0, see Fig. 1). Commonly, the X-dependent dc conductivities σdc(X) are used for σ0. In many cases, ν0 is defined via σ′(ν*(X)) = 2 σdc(X) using the X-dependent transition frequencies ν*(X), which mark the transition from long-time to short-time dynamics. The corresponding time t* = 1/(2πν*) describes the time required for the charge transport to become long-range, with the latter being determined by the ion mobility μ. Consequently, this characteristic frequency is proportional to the mobility of charge carriers.
 | ||
| Fig. 1 Illustration of scaling concepts for conductivity spectra: conductivity spectra for a varied parameter X (but a constant temperature) show a different type of scaling behaviour. The grey spectra in (a) can be superimposed via a Summerfield-type scaling by a shift along the dotted diagonal line, yielding (b), while the red spectra show deviations from this behaviour, which are quantified by the additional scaling parameter F ((b) and (c)). | ||
In a simple case, ν0 = ν* is furthermore proportional to the dc conductivity times temperature, σdc·T, which implies that changes in the dc conductivity with X are only due to changes in the ion mobility, whereas the number density of mobile ions does not change with X. This so-called Summerfield-type scaling68 was found applicable for example for time-temperature superposition or humidity-dependent studies in the case of polyelectrolyte complexes.48 In cases where the temperature is constant, the proportionality of ν0 to σdc implies the variations in conductivity to be solely attributed to changes in μ, see Fig. 1.
In inorganic glasses, for which superpositions concepts of conductivity spectra are frequently applied, some materials show Summerfield-type scaling,70,71 while in other materials deviations from Summerfield-type scaling have been reported.69,71,72 However, in some of the latter cases, a mastercurve can still be constructed, if an additional temperature-dependent shift factor is introduced.69 Here, the authors give two alternative explanations for the deviations from Summerfield scaling: either the number of available pathways for the ion transport decreases or the number density of mobile ions increases with increasing temperature.69 The latter effects are present in addition to the stronger changes in ion mobility μ.
In the present work, scaling according to the equation given in Fig. 1(c) was performed. Shift factors F and dc conductivities are discussed with respect to changes in μ and N.
Experimental
Materials
Polyelectrolyte multilayers were prepared from the weak polyelectrolyte poly(acrylic acid) (PAA, Mw ≈ 100 kDa, 35 wt% aqueous solution) and the strong polyelectrolyte poly(diallydimethylammonium chloride) (PDADMAC, Mw = 100 kDa to 200 kDa, 20 wt% aqueous solution), which were purchased from Sigma-Aldrich and used as received. Further materials for the preparation of polyelectrolyte solutions, surface pretreatment and the adjustment of measurement conditions, were acquired from VWR chemicals (ammonium hydroxide, ammonium nitrate, hydrochloric acid, lithium chloride, magnesium chloride, magnesium nitrate), Sigma-Aldrich (hydrogen peroxide, lithium hydroxide) and Carl Roth GmbH & Co. KG (potassium acetate), respectively. All solutions were prepared by dissolving the respective materials in ultrapure water (MERCK MILLIPORE, Elix®Essential, ρ > 18 MΩ cm−1). The pH was adjusted by potentiometric titration with hydrochloric acid and lithium hydroxide using a pH meter (inoLab Multi720) and a suitable electrode (Sen®Tix Mic). Indium tin oxide (ITO) coated glass substrates were pretreated in a cleaning solution (volumetric ratio: 5 water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ammonium hydroxide (25%):1 hydrogen peroxide (≥35%)) at 70 °C for 20 min.
:1 ammonium hydroxide (25%):1 hydrogen peroxide (≥35%)) at 70 °C for 20 min.
Sample preparation
(PDADMA/PAA)n PEMs were prepared from 0.01 M (referring to the monomer concentration) polyelectrolyte solutions. ITO-coated glass substrates were dip coated using a programmable robot (Riegler and Kirstein; Berlin). The automated dipping procedure consisted of consecutive polyelectrolyte adsorption and rinsing steps lasting for 20 min and 5 min, respectively. Multilayers were obtained by the successive, alternating adsorption of oppositely charged polyelectrolytes. The preparation conditions (pH and salt content) were kept constant and uniform throughout all solutions involved in the coating process. Such conditions were systematically varied to obtain PEMs prepared either from salt-free solution or 0.1 M LiCl solutions, both adjusted to pH values ranging from 3 to 9. PEMs were stored in boxes of controlled relative ambient humidity, which was achieved via small containers of saturated salt solutions (see Table S1 in ESI†). An equilibration period of 7 days at these conditions was awaited before further processing. The humidity after equilibration was determined with a hygrometer and is given in Table S1 (ESI†).Thickness determination
The film thickness was determined by atomic force microscopy (Nanowizard II, JPK, Berlin) or profilometry (DektakXT, Bruker, Germany) at ambient conditions (20% to 49% RH, 22 °C to 32 °C). The AFM was operated in tapping mode using ACTA probes (AppNano, tip < 10 nm, k = 25 N m−1 to 75 N m−1). In both cases, height profiles across scratches inflicted by a brass blade were measured on multiple positions along the dipping direction for each sample. The thickness was derived as the difference between the respective height of the film surface and the substrate. An average thickness![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) was calculated from multiple measurements on each sample. Error propagation included the standard deviation accounting for the roughness and different positions on the sample.
was calculated from multiple measurements on each sample. Error propagation included the standard deviation accounting for the roughness and different positions on the sample.
Conductivity measurement
The conductivity of PEMs was studied via impedance spectroscopy using an Alpha AN High Performance Frequency Analyzer (Novocontrol Technologies GmbH & Co. KG, Montabaur). To perform impedance spectroscopy measurements on PEMs, the films were prepared in a sandwich geometry as described earlier.67 To this end, an array of 21 circular gold dots (diameter 1 mm, thickness approximately 50 nm), acting as upper electrodes, was sputtered (BALTEC SCD 055; BALTIC Präparation e. K., Niesgrau) onto the PEMs, while the electrically conducting ITO-coating of the substrates acts as the lower electrode. For each measurement an electrode dot and an uncoated area of the ITO-coated substrate were contacted via spring-loaded electrodes, which were connected to the impedance setup. The sample stage was situated inside a custom-made airtight box67 accessible by gloves.Utilizing the described setup, humidity-dependent impedance spectroscopy measurements of PEMs, obtained for different preparation conditions, were performed. Different saturated salt solutions were used to vary the relative humidity inside the box (see Table S1, ESI†).
A frequency range of 1 Hz to 1 MHz was scanned at a voltage amplitude of 50 mV. Multiple measurements were performed on different electrode dots of the same sample, as well as on multiple samples with the same preparation and measurement conditions. Complex admittance spectra were averaged neglecting the data from spots close to the edge of the substrates. Complex conductivity spectra were calculated from the arithmetic average of complex admittance spectra by multiplication with the cell constant /A employing the average sample thickness and the area of the dot electrode, A. Changes in the cell constant due to swelling of the PEM at varied hydration was expected to be negligible as compared to the drastic changes of admittance.
Dc conductivities were determined in a Nyquist plot of the complex impedance by fitting an equivalent circuit consisting of an Ohmic resistor in parallel to a constant phase element, see ref. 55, 58, 67.
The dc conductivities were then obtained by multiplication of the inverse bulk resistance with the cell constant. Error propagation from standard deviations of multiple spectra from different electrode dots on multiple samples and the error of the individual cell constants was used to determine the errors of σdc.
Results and discussion
This section systematically investigates the influence of preparation and measurement conditions on the conductivity spectra of PEMs. After a qualitative discussion of the observed trends, data are evaluated regarding time-parameter superpositions to gain insight into the underlying effects driving conductivity variations. Changes in number densities (N) and mobilities (μ) of charge carriers are analyzed based on scaling parameters. The effect of varied relative humidity and preparation conditions (pH, salt) are discussed separately, where for the latter dc conductivities have been analyzed earlier.58Influence of relative humidity
The significant effect of varied relative humidity on the ionic conductivity spectra of (PDADMA/PAA)n PEMs is illustrated in Fig. 2(a) based on PEMs prepared from salt-free solutions adjusted to pH 4.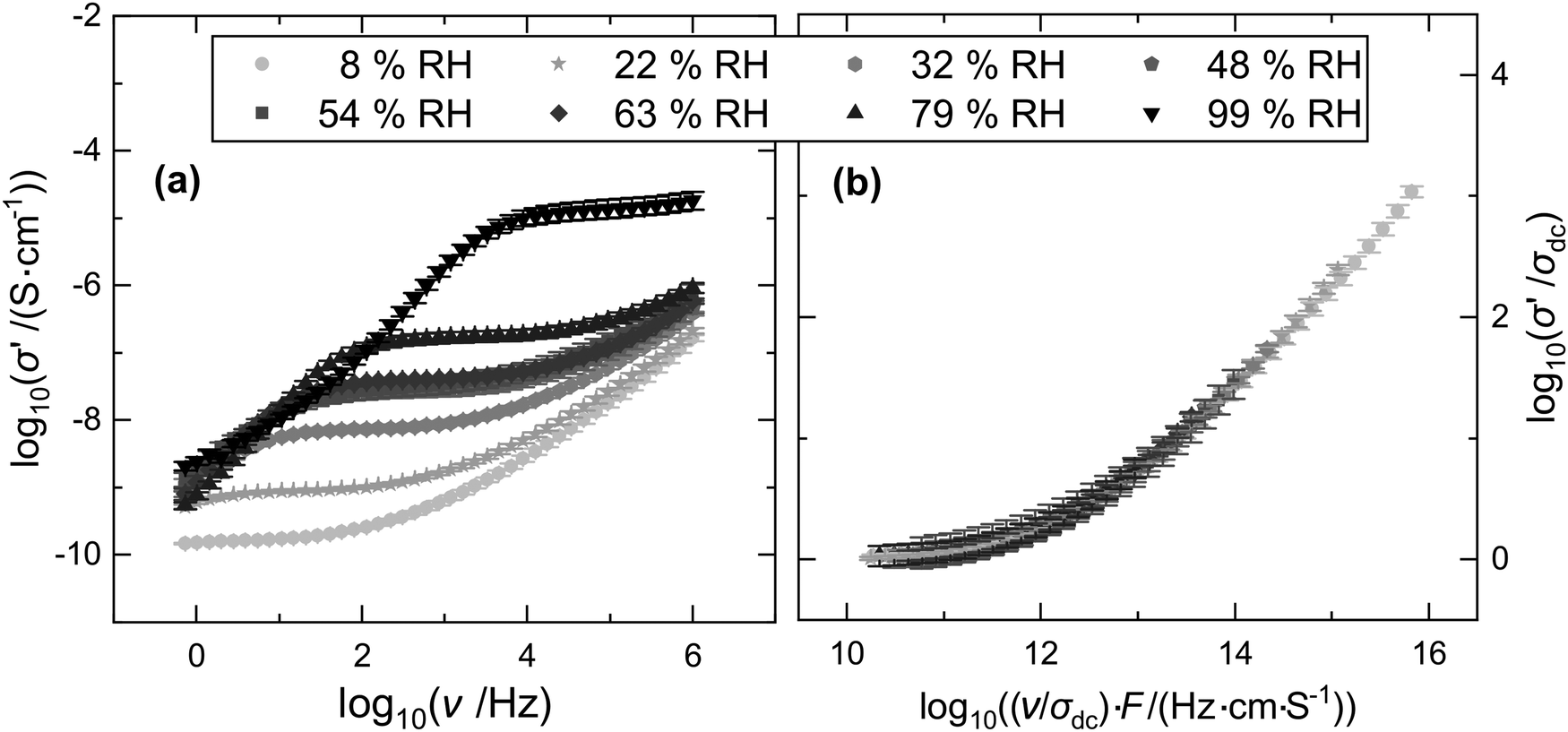 | ||
| Fig. 2 Time-humidity superposition of conductivity spectra of (PDADMA/PAA)n PEMs prepared at pH 4. (a) Conductivity spectra measured at different relative humidity (RH). (b) Spectra scaled according to a time-humidity superposition. | ||
The decrease of the conductivity towards low frequencies, which is significant at high RH values, is due to electrode polarization effects and will not be discussed further. In Fig. 2(b) these regimes are, therefore, omitted from the scaling analysis.
Apart from the frequency regimes dominated by electrode polarization effects, the conductivity spectra of Fig. 2 show a plateau regime which can be identified with the dc conductivity and a dispersive regime at higher frequencies where σ′(ν,RH) increases with ν.
The dc conductivity increases significantly with RH. As σdc is determined by the long-range ion transport, this implies that the humidity of the environment influences the ion movements in the PEMs on long time and length scales. At the same time, the onset of the dispersive regime, which reflects the transition from long-range to short-range transport, is shifted to higher frequencies. This both indicates an acceleration of the charge transport. The fact that the ion dynamics become faster with RH can be attributed to an increasing plasticization of the films due to water uptake when RH increases. The effect of hydration on the glass transition temperatures and dynamic mechanical properties of polyelectrolyte complexes has been discussed in the literature.56,60–62,73–75 Lowered glass transition temperatures at higher degrees of plasticization can be expected to accelerate charge transport. The effect of increasing ionic conductivities with relative humidity has been previously reported by Akgöl et al.,55 who found for several polyelectrolyte systems exponentially increasing dc conductivities with RH according to log(σdc) = aRH + b. The following evaluation will shed further light on the processes driving such a strong conductivity enhancement.
Time-RH superposition
As evident from Fig. 2(b) a mastercurve can be constructed from the individual spectra of Fig. 2(a) which were scaled according to the superposition process described in the theory section above. While the validity of time-RH-scaling of conductivity spectra was shown in PEC-materials,48Fig. 2 provides evidence that such scaling is also valid for polyelectrolyte multilayers. The validity of a time-RH superposition of the conductivity spectra of (PDADMA/PAA)n PEMs prepared from water adjusted to pH 4, implies, that also in PEMs the conduction mechanism is independent of RH while the velocity of the underlying ion migration changes with RH. According to earlier work,55 the conductivity enhancement can be attributed to increasing proton mobilities. On the other hand, the number density of charge carriers, N, might also vary. In the case of PEMs prepared from salt-free solution adjusted to pH 4, the deviations from Summerfield-type scaling, which are quantified by shift factors F, are, however, small. A detailed discussion of the shift factors and their implications is provided in a following paragraph.Since in the present case Summerfield scaling achieves a reasonable superposition of spectra, variations of charge carrier densities due to variations in relative humidity are negligible. This proves the hypothesis of the dominant role of charge carrier mobilities. Here, for X = RH the mobility is the only parameter in eqn (1) which is X-dependent, controlling the X-dependence of the conductivity resulting in σ′ (ν,X) = N · μ(ν,X) · q. In other words, the number density of the mobile protons contributing to the ion transport is not changed by the water uptake with increasing RH, but the mobility increases. These results are in good agreement with published results on polyelectrolyte complexes, where RH-scaling was also achieved using a Summerfield-type of superposition principle.48 Superposition concepts were furthermore found to apply for the humidity-dependent dynamic mechanical behaviour of PECs.60,61 Here, relaxation times were found to be inversely proportional to the volume fraction of water.61 This confirms faster dynamics at higher hydration, which correlates with increased charge carrier mobilities.
Influence of pH and salt during preparation
The structure and growth laws of (PDADMA/PAA)n PEMs, which have been discussed previously, significantly depend on the preparation conditions.24,58 For the most part, the trends in layer thickness can be explained by the hit-and-stick model,19 which assumes the polyelectrolyte conformation in solutions to be largely retained after adsorption. Conclusively, thin, rigid films are obtained from solutions of highly charged chains with high persistence length. This is the case for (PDADMA/PAA)n PEMs prepared from salt-free solutions adjusted to pH values exceeding the pKa of PAA. For low pH values – i.e. low charge degree of PAA – thick and soft films form from PAA chains in a more coiled conformation. Similarly, when PEMs are prepared at increased ionic strengths I, electrostatic screening due to counterions reduces the chain extension and the obtained film thickness increases with the pH of preparation. As discussed previously, such electrostatic screening might imply the incorporation of extrinsic charge carriers into the films.58As Fig. 3 shows, a variation of preparation conditions significantly affects the ionic conductivities. The effect on the dc conductivities has been previously discussed with regard to the PEM structure.58 In general, conductivities increase with the preparation pH value.
 | ||
| Fig. 3 Conductivity spectra of (PDADMA/PAA)n PEMs in dependence on pH and their time-pH superposition. (a) and (b) Data for samples prepared from salt-free solution adjusted to different pH values. (c) and (d) Data for samples prepared from 0.1 M LiCl solutions adjusted to different pH values. In (b) and (d) spectra obtained for different preparation pH values at constant humidity RH = 54% during the measurement are shifted with respect to the reference spectrum. Samples prepared at pH 4 and measured at 8% relative humidity are used as reference. | ||
The pH-dependent increase is enhanced for films prepared from 0.1 M LiCl solutions as compared to the salt-free case (see Fig. 3(a) and (c) as well as Fig. 4(a)). This was attributed to a significant enhancement of charge carrier mobilities caused by enhanced plasticization, while in addition, contributions of extrinsic charge carriers were found likely for the preparations from salt solutions.58 Considering the full spectra, it becomes evident that the onset of dispersion shifts to higher frequencies with increasing pH. This implies that not only the long-range transport, as described by the dc conductivity, but even the short-range charge transport, as probed in the dispersive regime, is accelerated by an increasing pH value. In the following, we analyze the full spectra by scaling approaches.
 | ||
| Fig. 4 (a) pH-dependent dc conductivities of PEMs and (b) pH-dependent shift factors F as derived from the scaling procedure displayed in Fig. 3. Black symbols refer to PEMs prepared in water and red symbols to those prepared in 0.1 M LiCl solution. | ||
Time-pH superposition
Fig. 3(b) and (d) show that the time-pH superposition principle is applicable to both types of PEMs prepared from salt-free and 0.1 M LiCl solutions, respectively. In both cases a mastercurve can be constructed by scaling of both axes from Fig. 3(a) and (c). In conclusion, neither the humidity nor the preparation conditions change the mechanism of charge transport in the investigated PEM samples. Only the velocity of ion displacements is varied, and this variation is described by a global factor, independent on the frequency considered.As evident from Fig. 4(a), the dc conductivities increase with the pH of preparation. While the evaluation of the dc conductivities in dependence on pH and salt concentration only allowed conclusions on trends of the product of N and μ, the scaling approach allows a separation of their respective trends. To achieve the mastercurves in Fig. 3(b) and (d), scaling of the spectra required an additional shift factor, F. Its values are given in Fig. 4(b) and compared to dc conductivities in Fig. 4(a).
Interestingly, the shift factor F increases significantly with the preparation pH for the PEMs prepared from 0.1 M LiCl solutions, where also a strong increase of σdc was observed. This is in direct contrast to the relatively constant values for the samples prepared from salt-free solution. Accordingly, stronger deviations from Summerfield-type scaling occur for the PEMs prepared from salt solutions. These increasing deviations, which are quantified by the F values, indicate enhanced charge carrier densities with increasing pH. The dc conductivity is therefore influenced by both, a changing number density and a changing mobility of the mobile ions, as implied by eqn (1). Such increasing number densities of charge carriers can be attributed to the increasing incorporation of extrinsic charge carriers at higher pH values. This was a hypothesis in our previous work,58 which is now legitimized by the shift factors. The incorporation of extrinsic charges occurs most prominently for films prepared at conditions dominated by the electrostatic screening of counterions, as is the case for high pH and added salt. In addition, the number of mobile protons might further increase due to the increase in hydration. This enhanced hydration could furthermore increase the fraction of mobile charge carriers. The factor of F = 10 implies a 10-fold increase of the charge carrier density.
Time-pH/RH superposition
In a final, combined scaling approach, aiming to distinguish the effects which dominate the observed influences on ionic conductivity spectra, pH-humidity superpositions for differently prepared samples are compared. Here, the most interesting cases according to the time-pH superposition were chosen. Samples prepared at pH 4 in water are compared to samples dominated by electrostatic screening containing strongly charged PAA (prepared at pH 6 and 7.5 in 0.1 M LiCl solution), see Fig. 5. The corresponding spectra are presented in Fig. S1 (ESI†). | ||
| Fig. 5 (a) RH-dependent dc conductivities of PEMs and (b) RH-dependent shift factors F as derived from the scaling procedure displayed in Fig. 1. Black symbols refer to PEMs prepared at pH 4 in water, red symbols stand for PEMs prepared in 0.1 M LiCl solution at pH 6 and blue symbols for PEMs prepared in 0.1 M LiCl solution at pH 7.5. | ||
Fig. 5(a) shows that the dc conductivity increases significantly with the relative humidity for all PEMs which were prepared under varying conditions. The increase is, however, more pronounced when the PEMs were made in the presence of LiCl. On the other hand, the effect of the pH at which the PEMs were formed in LiCl solutions is small. In case of the salt-free preparation at low pH, the increase of the dc conductivity is mainly due to the ion mobility, as evidenced by the shift factor values on the order of 1 (see Fig. 5(b)). In contrast, the effect of increasing plasticization on σdc is more pronounced for samples prepared at higher pH values. Here, shift factors (see Fig. 5(b)) deviate significantly from 1. Again, these are conditions, where samples were prepared at high charge degree and ionic strength. Here, contributions of extrinsic charge carriers were concluded (see above). The more significant effect of RH on the shift factors could be attributed to a more pronounced enhancement of the fraction of free, mobile charge carriers in contrast to immobile, condensed counterions in films containing extrinsic charge carriers. In addition, a more macroscopic explanation could be a more significant effect of the humidity on the hydration of PEM films containing more highly charged PAA. It should be highlighted though, that the increase of the conductivity with RH is mainly due to the increase in ion mobility. As seen in Fig. 5(b) the highest F values are about 10, implying that the number density increases by roughly the same factor. The dc conductivity, however, increases by several orders of magnitude. For PEMs prepared in the presence of LiCl, σdc increases by roughly 4 orders of magnitude, see Fig. 5(a). This implies that the increase in ion mobility with RH is about 1000 times larger than the increase of the number density of mobile ions.
Interestingly, the shape of the conductivity spectra does not vary with the conditions of preparation for all investigated samples (see Fig. S1 in ESI†). Therefore, all mastercurves can be additionally shifted resulting in a “super-mastercurve” comprising pH-, humidity- and ionic strength-dependent data sets (see Fig. 6).
 | ||
| Fig. 6 Super-mastercurve combining a superposition of pH-, humidity-, and ionic strength-dependent conductivity data of (PDADMA/PAA)n PEMs. The super-master curve combines data from Fig. 2(b), 3(b), (d) and Fig. S1(b), (d) (ESI†). The spectrum of samples prepared at pH 4 and measured at 8% relative humidity is used as reference (solid line). | ||
Thus, over a wide range of preparation parameters, the underlying mechanisms of ion transport remain identical. The most influential parameter determining the dynamics at all time scales is the hydration of layers, which dominates the charge carrier mobilities. All parameters, i.e. the preparation parameters pH and salt, as well as RH contribute to the mobility, and can also enhance the charge carrier density, if regimes with highly charged chains and counterion condensation are considered.
Conclusions
This paper shows for the first time the time-X superposition of humidity-, pH- and ionic strength-dependent conductivity spectra of PEMs containing a weak polyacid. This universal superposition indicates that the overall mechanism of conduction is not changing upon significant parameter variations. From the superpositions, variations of charge carrier mobilities and number densities are discussed separately. Increased mobilities are clearly correlated to the hydration of films, which can be varied via the charge degree of PAA, the ionic strength of preparation solutions as well as the humidity during the measurement. Number densities of mobile ions were found to increase for high charge degrees of PAA and increased ionic strength. For these films, a strong dependence of N on the relative humidity was shown, which can be attributed to the mobilization of condensed ions in the film with increasing water content. Nevertheless, the effect of the varying ion mobility on the conductivity is much more pronounced than the influence of the number density of mobile ions.It is unclear whether increases of the number density of mobile ions are due to the increasing fraction of free charge carriers (for example protons) as induced by hydration, or as a result of contributions of extrinsic charge carriers. For a more conclusive interpretation the use of selective electrodes and more information on water content is required.
This paper demonstrates that the decoupling of changes in charge carrier mobilities and number densities, as obtained from scaling approaches, reveals interesting insights on the implications of parameter variations on the charge transport processes in PEMs. Understanding the ion transport mechanism is not only interesting from a fundamental point of view, but also important for the applications of PEMs in the field of solid state ionics and sensing devices. For example, the incorporation of volatile analytes like NH3, after dissociation leading to increased number densities of extrinsic charge carriers, might become distinguishable from effects of increased mobilities due to enhanced plasticization.
Author contributions
J. Schlicke conducted and designed the experiments and wrote the majority of the manuscript. C. Cramer and M. Schönhoff were involved in conceptualizing and planning of the experiments and wrote parts of the manuscript. All authors contributed to the interpretation and discussion of data. M. Schönhoff provided resources for the project.Data availability
The data supporting this article have been included as part of the ESI.† This comprises humidity-dependent raw data (conductivity specta) for samples prepared at different pH values, and their scaling on a mastercurve.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Katja Hoffmann and Martin Lorenz for their assistance in the preparation of PEMs.Notes and references
- K. C. Wood, H. F. Chuang, R. D. Batten, D. M. Lynn and P. T. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10207–10212 CrossRef CAS.
- V. Sharma and A. Sundaramurthy, Beilstein J. Nanotechnol., 2020, 11, 508–532 CrossRef CAS PubMed.
- D. Wu, L. Zhu, Y. Li, X. Zhang, S. Xu, G. Yang and T. Delair, Carbohydr. Polym., 2020, 238, 116126 CrossRef CAS PubMed.
- S. O. Solomevich, E. I. Dmitruk, P. M. Bychkovsky, D. A. Salamevich, S. V. Kuchuk and T. L. Yurkshtovich, Int. J. Biol. Macromol., 2021, 169, 500–512 CrossRef CAS.
- J. de Grooth, R. Oborný, J. Potreck, K. Nijmeijer and W. M. de Vos, J. Membr. Sci., 2015, 475, 311–319 CrossRef CAS.
- S. Ilyas, S. M. Abtahi, N. Akkilic, H. D. W. Roesink and W. M. de Vos, J. Membr. Sci., 2017, 537, 220–228 CrossRef CAS.
- S. Ilyas, N. Joseph, A. Szymczyk, A. Volodin, K. Nijmeijer, W. M. De Vos and I. F. Vankelecom, J. Membr. Sci., 2016, 514, 322–331 CrossRef CAS.
- W. M. de Vos, A. de Keizer, M. A. C. Stuart and J. M. Kleijn, Colloids Surf., A, 2010, 358, 6–12 CrossRef CAS.
- Á. Bóna, Á. Varga, I. Galambos and N. Nemestóthy, Membranes, 2023, 13, 283 CrossRef PubMed.
- J. L. Lutkenhaus and P. T. Hammond, Soft Matter, 2007, 3, 804–816 RSC.
- H. An, T. Habib, S. Shah, H. Gao, A. Patel, I. Echols, X. Zhao, M. Radovic, M. J. Green and J. L. Lutkenhaus, ACS Appl. Nano Mater., 2019, 2, 948–955 CrossRef CAS.
- J. Movilli, S. S. Choudhury, M. Schönhoff and J. Huskens, Chem. Mater., 2020, 32, 9155–9166 CrossRef CAS.
- I. Škugor Rončević, D. Krivić, M. Buljac, N. Vladislavić and M. Buzuk, Sensors, 2020, 20, 3211 CrossRef PubMed.
- A. S. Michaels, Ind. Eng. Chem., 1965, 57, 32–40 CrossRef CAS.
- J. B. Schlenoff, H. Ly and M. Li, J. Am. Chem. Soc., 1998, 120, 7626–7634 CrossRef CAS.
- J. B. Schlenoff and S. T. Dubas, Macromolecules, 2001, 34, 592–598 CrossRef CAS.
- M. Ghasemi, S. Friedowitz and R. G. Larson, Soft Matter, 2020, 16, 10640–10656 RSC.
- P. M. Biesheuvel and M. A. Cohen Stuart, Langmuir, 2004, 20, 2785–2791 CrossRef CAS.
- M. Schönhoff, J. Phys.: Condens. Matter, 2003, 15, R1781 CrossRef.
- H. Li, S. M. Lalwani, C. I. Eneh, T. Braide, P. Batys, M. Sammalkorpi and J. L. Lutkenhaus, Langmuir, 2023, 39, 14823–14839 CrossRef CAS.
- W. Yuan, G.-M. Weng, J. Lipton, C. M. Li, P. R. Van Tassel and A. D. Taylor, Adv. Colloid Interface Sci., 2020, 282, 102200 CrossRef CAS.
- E. Guzman, R. G. Rubio and F. Ortega, Adv. Colloid Interface Sci., 2020, 282, 102197 CrossRef CAS.
- C. Naas, U. Scheler and U. Lappan, Macromolecules, 2021, 54, 1043–1051 CrossRef CAS.
- D. Bütergerds, C. Kateloe, C. Cramer and M. Schönhoff, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 425–434 CrossRef.
- E. A. Litmanovich, E. V. Chernikova, G. V. Stoychev and S. O. Zakharchenko, Macromolecules, 2010, 43, 6871–6876 CrossRef CAS.
- P. K. Jha, P. S. Desai, J. Li and R. G. Larson, Polymers, 2014, 6, 1414–1436 CrossRef.
- M. Gopinadhan, O. Ivanova, H. Ahrens, J.-U. Günther, R. Steitz and C. A. Helm, J. Phys. Chem. B, 2007, 111, 8426–8434 CrossRef CAS PubMed.
- S. T. Dubas and J. B. Schlenoff, Macromolecules, 2001, 34, 3736–3740 CrossRef CAS.
- C. Sung, A. Vidyasagar, K. Hearn and J. L. Lutkenhaus, Langmuir, 2012, 28, 8100–8109 CrossRef CAS PubMed.
- Z. Liu, Z. Yin, J. Wang and Q. Zheng, Adv. Funct. Mater., 2019, 29, 1806092 CrossRef.
- D. M. DeLongchamp and P. T. Hammond, Chem. Mater., 2003, 15, 1165–1173 CrossRef CAS.
- D. M. DeLongchamp and P. T. Hammond, Langmuir, 2004, 20, 5403–5411 CrossRef CAS.
- A. A. Argun, J. N. Ashcraft and P. T. Hammond, Adv. Mater., 2008, 20, 1539–1543 CrossRef CAS.
- J. N. Ashcraft, A. A. Argun and P. T. Hammond, J. Mater. Chem., 2010, 20, 6250–6257 RSC.
- W. Noh, Y. Go and H. An, Sensors, 2023, 23, 1977 CrossRef CAS.
- F. Wu, J. Li, Y. F. Su, J. Wang, W. Yang, N. Li, L. Chen, S. Chen, R. J. Chen and L. Y. Bao, Nano Lett., 2016, 16, 5488–5494 CrossRef CAS PubMed.
- G. Decher, J. D. Hong and J. Schmitt, Thin Solid Films, 1992, 210, 831–835 CrossRef.
- J. Fu and J. B. Schlenoff, J. Am. Chem. Soc., 2016, 138, 980–990 CrossRef CAS.
- Z. Ou and M. Muthukumar, J. Chem. Phys., 2006, 124, 154902 CrossRef PubMed.
- X. Xu, Q. Ran, P. Dey, R. Nikam, R. Haag, M. Ballauff and J. Dzubiella, Biomacromolecules, 2018, 19, 409–416 CrossRef CAS PubMed.
- J. A. Jaber and J. B. Schlenoff, Langmuir, 2007, 23, 896–901 CrossRef CAS.
- T. R. Farhat and J. B. Schlenoff, J. Am. Chem. Soc., 2003, 125, 4627–4636 CrossRef CAS.
- V. A. Kabanov, Russ. Chem. Rev., 2005, 74, 5–23 CrossRef.
- T. Kremer, D. Kovačević, J. Salopek and J. Požar, Macromolecules, 2016, 49, 8672–8685 CrossRef CAS.
- H. M. Fares and J. B. Schlenoff, Macromolecules, 2017, 50, 3968–3978 CrossRef CAS.
- G. Allegri, J. Huskens, R. P. Martinho and S. Lindhoud, J. Colloid Interface Sci., 2024, 672, 654–663 CrossRef CAS.
- A. Imre, M. Schönhoff and C. Cramer, Phys. Rev. Lett., 2009, 102, 255901 CrossRef CAS PubMed.
- S. De, C. Cramer and M. Schönhoff, Macromolecules, 2011, 44, 8936–8943 CrossRef CAS.
- S. De, A. Ostendorf, M. Schönhoff and C. Cramer, Polymers, 2017, 9, 550 CrossRef PubMed.
- A. Ostendorf, M. Schönhoff and C. Cramer, Phys. Chem. Chem. Phys., 2019, 21, 7321–7329 RSC.
- A. Bhide, M. Schönhoff and C. Cramer, Solid State Ionics, 2012, 214, 13–18 CrossRef CAS.
- M. F. Durstock and M. F. Rubner, Langmuir, 2001, 17, 7865–7872 CrossRef CAS.
- S. P. Jiang, Z. Liu and Z. Q. Tian, Adv. Mater., 2006, 18, 1068–1072 CrossRef CAS.
- Y. Daiko, K. Katagiri and A. Matsuda, Chem. Mater., 2008, 20, 6405–6409 CrossRef CAS.
- Y. Akgöl, C. Cramer, C. Hofmann, Y. Karatas, H.-D. Wiemhöfer and M. Schönhoff, Macromolecules, 2010, 43, 7282–7287 CrossRef.
- Y. Chen, M. Yang and J. B. Schlenoff, Macromolecules, 2021, 54, 3822–3831 CrossRef CAS.
- J. Fu, R. L. Abbett, H. M. Fares and J. B. Schlenoff, ACS Macro Lett., 2017, 6, 1114–1118 CrossRef CAS.
- J. Schlicke, K. Hoffmann, M. Lorenz, M. Schönhoff and C. Cramer, J. Phys. Chem. C, 2020, 124, 16773–16783 CrossRef CAS.
- J. Wang, Y. F. Xue, X. C. Chen, M. Hu, K. F. Ren and J. Ji, Adv. Healthcare Mater., 2020, 9, 2000381 CrossRef CAS.
- P. C. Suarez-Martinez, P. Batys, M. Sammalkorpi and J. L. Lutkenhaus, Macromolecules, 2019, 52, 3066–3074 CrossRef CAS.
- S. M. Lalwani, P. Batys, M. Sammalkorpi and J. L. Lutkenhaus, Macromolecules, 2021, 54(17), 7765–7776 CrossRef CAS.
- Y. Zhang, F. Li, L. D. Valenzuela, M. Sammalkorpi and J. L. Lutkenhaus, Macromolecules, 2016, 49, 7563–7570 CrossRef CAS.
- X. Lyu, B. Clark and A. M. Peterson, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 684–691 CrossRef CAS.
- P. Batys, Y. P. Zhang, J. L. Lutkenhaus and M. Sammalkorpi, Macromolecules, 2018, 51, 8268–8277 CrossRef CAS PubMed.
- C. I. Eneh, T. Kastinen, S. Oka, P. Batys, M. Sammalkorpi and J. L. Lutkenhaus, ACS Polym. Au, 2022, 2, 287–298 CrossRef CAS PubMed.
- C. I. Eneh, M. J. Bolen, P. C. Suarez-Martinez, A. L. Bachmann, T. J. Zimudzi, M. A. Hickner, P. Batys, M. Sammalkorpi and J. L. Lutkenhaus, Soft Matter, 2020, 16, 2291–2300 RSC.
- Y. Akgöl, C. Hofmann, Y. Karatas, C. Cramer, H.-D. Wiemhöfer and M. Schönhoff, J. Phys. Chem. B, 2007, 111, 8532–8539 CrossRef.
- S. Summerfield, Philos. Mag. B, 1985, 52, 9–22 Search PubMed.
- S. Murugavel and B. Roling, Phys. Rev. Lett., 2002, 89, 195902 CrossRef CAS PubMed.
- B. Roling, A. Happe, K. Funke and M. Ingram, Phys. Rev. Lett., 1997, 78, 2160 CrossRef CAS.
- C. Cramer, S. Brückner, Y. Gao and K. Funke, Phys. Chem. Chem. Phys., 2002, 4, 3214–3218 RSC.
- A. Šantić, J. Nikolić, L. Pavić, R. D. Banhatti, P. Mošner, L. Koudelka and A. Moguš-Milanković, Acta Mater., 2019, 175, 46–54 CrossRef.
- W. Huang, J. Li, D. Liu, S. Tan, P. Zhang, L. Zhu and S. Yang, ACS Appl. Polym. Mater., 2020, 2, 2119–2125 CrossRef CAS.
- A. Vidyasagar, C. Sung, R. Gamble and J. L. Lutkenhaus, ACS Nano, 2012, 6, 6174–6184 CrossRef CAS PubMed.
- E. Yildirim, Y. Zhang, J. L. Lutkenhaus and M. Sammalkorpi, ACS Macro Lett., 2015, 4, 1017–1021 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Humidity values and humidity-dependent spectra and their superposition. See DOI: https://doi.org/10.1039/d4cp03482a |
| This journal is © the Owner Societies 2024 |