
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysical mechanisms of the Sec machinery operation†
Ekaterina
Sobakinskaya
 and
Frank
Müh
*
and
Frank
Müh
*
Institute for Theoretical Physics, Johannes Kepler University Linz, Altenberger Strasse 69, A-4040 Linz, Austria. E-mail: frank.mueh@jku.at
First published on 9th October 2024
Abstract
The Sec complex, composed of a motor protein SecA and a channel SecYEG, is an ATP-driven molecular machine for the transport of proteins across the plasma membrane in bacteria. Today, there is a consensus about a general “rough” model of the complex activation and operation, which, however, lacks understanding of the physical mechanisms behind it. Molecular dynamics simulations were employed to address a way of allosteric activation, conformational transition of SecYEG from the closed to the open state, and driving forces of protein transport. We found that binding of SecA (in the ATP-bound state) and the protein signal sequence leads to a transmembrane helix rearrangment that weakens contacts inside the hydrophobic core of SecYEG and provides a driving force for plug opening. The conformational transitions are enabled by a delicate interplay between hydrophobic forces on one side and PEES (proton motive force, external – due to binding with the translocation partners – entropic, and solvent-induced) on the other side. In the open state, SecYEG still provides a barrier for bulky residues that contributes to the driving forces of transport. Other important contributions come from SecA and the membrane potential acting in different stages of protein transport to guarantee a nearly constant driving force. Given that the different forces act on different types of residues, the suggested mechanisms taken together provide a directional motion for any substrate, thereby maximizing the efficiency of the Sec machinery.
Introduction
About one third of all proteins are secreted across or integrated into biological membranes.1 Many of them are transported in an unfolded state by the Sec machinery. The complex consists of two main components: protein-conducting channel and translocation partner. The channel, also called translocon, is formed by the hetero-trimeric membrane complex SecYEG in prokaryotic cells2,3 and Sec61 in eukaryotes.4 For co-translational transport, Sec joins the ribosome and forms a ribosome nascent chain complex. Post-translational pathways involve binding of specialized energy-transducing factors, e.g., the SecA protein motor as a translocation partner (in bacteria). While co-translational transport is the main pathway for protein secretion in eukaryotes5 and for membrane insertion in all organisms,6 the post-translational transport is used mainly by prokaryotes for protein secretion.7 In our work, we focus our attention mainly on the latter system, best described for bacteria. Let us consider the architecture of the Sec machinery components.SecA includes two nucleotide-binding domains (NBD1, NBD2), together forming the nucleotide-binding site (NBS), where ATP hydrolysis occurs, stimulated by the channel, acidic lipids and pre-protein substrate8 (Fig. 1a). Other domains important for translocation are the polypeptide cross-linking domain (PPXD) and the two helix finger (2HF). PPXD and the adjacent NBS form a clamp for the transported pre-protein. 2HF is inserted into the entrance of the channel and believed to interact with the translocating substrate, pushing it into the translocon.9 The motor protein cycles between high- and low-affinity states, dissociating from and re-associating with the translocon: ATP-bound SecA has a tight coupling with the channel, while the ADP-bound SecA corresponds to a weakly associated form.8,9
 | ||
| Fig. 1 Architecture of the Sec machinery. (a) Overall structure with a peptide inside, cytosol top (PDB 6ITC). Colors: SecY, ice blue; SecE violet; translocating peptide, magenta. Main domains of SecA: NBD1, blue; NBD2, green; 2HF, red; PPXD, yellow. Other domains not important for the present work are shown in grey. (b) View along the membrane plane on SecYEG, cytosol top (PDB 5AWW). LG helices TM7 and TM2b are displayed in light green and purple, respectively, the plug in pink. SecE, violet; SecG, cyan. (c) View onto the PR from the cytosolic side (PDB 5AWW). Four hydrophobic PR residues (Ile 81, Ile 188, Ile 275, Ile 403) are shown explicitly. | ||
The prokaryotic translocon consists of a large transmembrane (TM) subunit SecY, which spans the membrane ten times, and two small subunits SecE and SecG.4 The subunit SecY has an hourglass shape with a constriction inside the membrane and is divided into N- and C-terminal halves, TM segments 1–5 and 6–10, respectively, which are pseudosymmetrical and linked by the loop on the extracellular side (Fig. 1b). Important structural elements of SecY include (i) the central pore ring (PR) at the constriction composed of hydrophobic amino acids (Fig. 1c), (ii) the lateral gate (LG) formed by transmembrane helices TM2b and TM7 (marked purple and light green in Fig. 1b) and (iii) the re-entrant loop TM2a, called the plug domain (marked pink in Fig. 1b), which closes the channel at the periplasmic side of the membrane. The PR consists of six aliphatic amino acids, usually isoleucines, which project their hydrophobic side chains inside the channel. Four of them are of particular interest, since they determine the opening width of the PR, and, thus, the space available for a translocating substrate. They are located on helices TM5, TM10, TM2 and TM7 (Fig. 1c). Taking into account that two aliphatic amino acids belong to the LG helices, one can readily see that opening or closing of the lateral gate changes the PR opening width. Furthermore, the central part of the translocon includes the hydrophobic core stabilized by hydrophobic interactions between the plug and helices TM7, TM2b as well as the PR residues.10 SecE contains an amphipathic helix and plays the role of a clamp, keeping the two halves of SecY together. The subunit SecG makes only few contacts with both SecY and SecE and seems to have minor importance for the channel's function.
Extensive theoretical and experimental research is performed to reveal how the Sec machinery transports the proteins.2,4,8,11 Today, there is consensus on a general “rough” model of activation, which includes a series of steps: (i) association of the channel with protein and SecA following ATP binding, (ii) opening of the LG by the signal sequence (SS) of the transported protein (resulting from movements of TM7 and TM2b), and (iii) plug relocation, termed “unlocking”.2,12 However, despite significant efforts, no understanding yet emerged of the physical mechanism underlying activation and operation of the SecY machinery. Moreover, several important phenomena are not yet taken into account in the above model.
For example, mutations in the loop between TM8 and TM9 (the TM8/TM9 linker), a binding site of SecA,13,14 were shown to abolish movement of the plug towards SecE (evidenced by diminished crosslinking), indicating on transmission of signals between remote parts of the channel15 (Fig. 1a) that implies allosteric activation. Furthermore, an important factor in the function is the proton motive force.16 (To avoid confusion of the proton-motive force with the potential of mean force (PMF), we use the abbreviation ΔΨH+ for the former.) ΔΨH+ reduces significantly the concentration of ATP required for protein translocation.17 There is evidence that it interacts with the helices of SecYEG,18 although the mechanism by which ΔΨH+ influences translocation is not clear. Integrating the above phenomena into a consistent biophysical model will provide detailed knowledge about activation and operation of the Sec machinery and contribute to the understanding of how the free energy difference between ATP and ADP is transduced into directed mechanical forces driving protein transport.
In the literature, abundant data are accumulated that shed light on the “main players” and reveal important interactions between them. (Available structures of the open SecYEG13,19 are at low resolution and cannot provide much detailed information.) The first periplasmic loop (P1) as well as helices TM7, TM10, TM2, and TM5 are demonstrated to be the structural sites of prlA mutations,20,21 which affect the functionality of the channel.22 Among the most extreme mutations, which cause lethality, combined with L108R (prlG1) in SecE, are the changes I408N and L407R of adjacent residues in TM10 and I278N in TM7.23,24 At the same time, none of the residues of TM7 and TM10 is in direct contact with residue Leu 108 of SecE, but coupled with TM525 (see ESI† A1 for more details). All helices, mentioned above, form the LG and the PR and tightly interact with the plug.
The importance of the coupling between the plug and helices TM5, TM10, TM2b, and TM7 for the Sec machinery function is revealed in electrophysiology experiment. Mutants with the plug fully or partly deleted showed transient channel opening, resulting in ion conductance.26,27 In this case, a new plug is formed, which lost all interactions with TM5 and TM10 and most interactions with TM2b and TM7.28 As a result, SecYEG becomes less “tight” and easier to open. This hypothesis can also explain why plug-deleted mutants allow the translocation of proteins with defective or missing signal sequences, similar to the prlA mutations in and around the PR.21 Furthermore, disulfide bridges between cysteine (Cys) residues mutated into the plug and TM10 lead to inactivation of SecYEG, while the application of long crosslinkers restores normal translocation.29 Mutation of residue Leu 406 to lysine in TM10, located in the hydrophobic core domain of SecYEG (M. janaschii), leads to displacement of the plug toward the extracellular side.30 Interestingly, opening of the plug implies a large-amplitude motion as is evident from other cross-linking experiments: If residues 67 in SecY (plug) and 120 in SecE (E. coli), which are more than 20 Å apart,25 are mutated to Cys and cross-linked by a disulfide bridge through addition of sodium tetrathionate (NaTT), channel opening with massive ion flux is observed.27 Addition of dithiothreitol (DTT) to reduce the disulfide bridge restores the channel in the closed state. Another interesting observation is that NaTT-treatment of a single-Cys mutation in the plug (position 67 in SecY) results in fluctuations of the plug between closed and open states. This result suggests that modification of Cys 67 by NaTT31 destabilizes the plug in the closed position, most likely by perturbing the hydrophobic core.10 The above results raise the following question: What forces trigger the plug movement during Sec machinery activation under native conditions?
In the case of the eukaryotic channel Sec61, relocation of the plug away from the PR was stimulated by movement of TM7 and TM8 in the presence of Sec62 and Sec63, as is evident from cryo-EM structures.32 However, these structures did not include a translocating substrate and did not feature the movement of other helices. In contrast to the bacterial counterpart, helices TM5 and TM7 of Sec61 are more polar than the ones of SecYEG, suggesting that hydrophobic interactions in the center are much weaker. Thus, to enable opening of the bacterial channel, the plug must be released from a quite tight hydrophobic core that requires significant weakening of many contacts.
Here, we present an all-atom molecular dynamics (MD) study, aimed at revealing the pathway of the Sec machinery activation and operation and identifying the physical mechanisms that enable it. The activation process involves the opening of the channel stimulated by the movement of helices upon binding with SecA and the SS. In our work, we determined a key set of helices, whose displacement is crucial for the opening. Special focus was put on the conformation of the plug, which is important for the peptide transport. Starting from a high-resolution crystal structure of the closed channel (PDB ID 5AWW33), we have created two open structures. In one of them – open – the displacement of the key-set helices stimulated the release of the plug and its transition to the open position. (The latter was determined with metadynamics simulations.) The second structure – half-open – represents SecYEG with an open LG and the plug in a closed state. For both conformations, we determined the barrier to peptide translocation, which shows the role of the plug during protein transport. As follows from the review above, a prerequisite for SecYEG operation, especially in vivo, is the proton motive force ΔΨH+. The obtained open structure was analyzed in terms of the influence of the membrane potential ΔΨ, which is an important part of ΔΨH+. Here, we revealed the role of ΔΨ on the Sec machinery in both ATP- and ADP- bound states. Based on the results, we describe the forces stimulating activation of the Sec machinery and suggest ways of its regulation. Furthermore, all findings are integrated into a consistent biophysical model of the Sec machinery and discussed based on the fundamental principles of molecular machine operation.
Results
Main players of the conformational transition: the key set of helices
To understand the allosteric mechanism leading to an opening of the SecYEG channel, first of all, we performed a structural analysis of the closed and open states. Here, the goal was to identify helices, which play an active role in conformational transitions of the channel. As in our earlier work,10 we selected the 5EUL structure19 as a model of the open state. The latter represents a chimera of SecYE from Geobacillus thermodenitrificans and SecA from Bacillus subtilis containing the SS of OmpA (outer membrane protein A) and a short polypeptide following it (for amino acid sequence, see Methods – Systems assembly). It was crystallized in the presence of ADP and beryllium fluoride (BeFx). The latter locks SecA into a conformation close to its ATP-bound state.9,34 Thus, the 5EUL structure corresponds to the active channel with an SS in the open LG and a peptide loop inside the pore.Overlay of the 5AWW (closed) and 5EUL structures reveals a drastic shift of TM7, TM8, and TM2b (Fig. 2a) that is, obviously, induced by SS binding. Other helices feature a less pronounced displacement, which can be a result of both opening of the translocon and inaccuracy in atom coordinates (ESI,† Fig. S1). Moreover, in contrast to the structures of the closed SecYEG, the plug in 5EUL is completely disordered. This can be due to the fact, that the plug is shifted down to the periplasmic side, so that it is easy accessible for the detergents, which solvate non-polar groups. Hence, in our structural analysis, we take into consideration also other data from the literature. Furthermore, a criterion for a helix to play an important role in channel opening is a contribution to loosening of the plug from the hydrophobic core.
 | ||
| Fig. 2 Comparison of the 5AWW and 5EUL structures. Only selected helices are displayed. (a) Side view of the displacement of helices in the open channel (5EUL, red) with respect to the closed state (5AWW, iceblue). (b)–(d) Interaction between the residues of the plug and TM10 (b), TM5 (c), and TM2b (d) in the closed channel (5AWW). Interacting residues are shown explicitly. All residues are colored according to their type: hydrophobic (white), polar (green), positively charged (blue), negatively charged (red). | ||
The results of Lycklama a Nijeholt et al.29 indicate a proximity of TM10 and the plug that is confirmed by the structure 5AWW of closed SecYEG (Fig. 2b). The structure also reveals hydrophobic interaction between the TM9/TM10 linker and the plug (Fig. 2b). On the other hand, the plug interacts only with the loop in the open translocon. Superposition of 5AWW and 5EUL gives a hint that helix TM10 might shift upwards and slightly backwards during opening of SecYEG (Fig. 2a; ESI,† Fig. S1), thus weakening coupling with the plug. The displacement of TM10 can be coupled to the upwards motion of TM9 (ESI,† Fig. S1), driven by the binding between the TM8/TM9 linker and the translocation partner.35 Further examination of the resting state 5AWW indicates that the plug interacts with residues of TM5 (Fig. 2c).
In the active translocon, the plug moves away from the PR to the periplasmic side, losing these contacts. At the same time, overlay of the two structures demonstrates a slight “backside” shift of TM5 (Fig. 2a and ESI,† Fig. S1) that can contribute to weakening the interaction with the plug in the closed state, thus, facilitating its movement to the open configuration. TM5 can be involved in the SecYEG opening through its contacts with TM10 and SecE (see Introduction).
The closed conformation reveals coupling between residues of TM2b and the plug that is a part of the hydrophobic core (Fig. 2d). Obviously, opening of the LG breaks this interaction and contributes to unlocking of the plug. Thus, we can say that a decisive role in the transition of the channel to the active state is played by the key set of helices TM7, TM2b, TM8, TM9, TM10 and TM5 (see also Introduction), which respond to the binding of the translocation partners and release the plug from the hydrophobic core. Moreover, while movement of LG helices and TM8 is stimulated by the incoming SS, other helices react likely to binding of SecA.
To create the channel conformation “open” – resulting from insertion of the SS and binding of SecA – the above key-set helices were pulled to their positions in the 5EUL structure by means of steered molecular dynamics (SMD; see Methods and ESI† M1). The resulting conformation represents a transition state and is called “transit-open” structure (ESI,† Fig. S18 and Table S2). Next, to determine the location of the plug, we computed the free-energy landscape as a function of pairwise distances between the plug and helices TM5, TM2b and the TM9/10 linker (see Methods and ESI† M2, M3) in the transit-open structure. This computation was done with metadynamics, which is an MD technique for reconstructing the free-energy surface as a function of a few selected degrees of freedom, referred to as collective variables (CVs).36 As detailed in the ESI† (M2, M3), we have the three CVs d1, d2, and d3, corresponding, respectively, to the center-of-mass (COM) distances between (1) plug residues 62 to 64 and TM2b residues 76 to 78, (2) plug residues 54 to 61 and TM5 residues 188 to 191, and (3) plug residue 54 to 61 and TM9/10 linker residues 392 to 394. To monitor the convergence of metadynamics trajectories, we ran different simulations with lengths ranging from 2 to 60 ns and used the generalized distance metric as described in ESI,† M3 as convergence criterion. Based on this measure, the 40 ns trajectory was chosen to determine the open conformation of the plug. When the plug position was settled, the obtained structure was complemented with a translocating substrate (see Methods and ESI,† Fig. S18 and Table S2). The resulting open channel was used to create both ATP- and ADP-bound states without explicitly modeling SecA.
To mimic the ATP-bound state in further simulations, we imposed soft constraints on the backbone of helices TM3, TM8 partly, TM9, TM10, as well as the TM6/7 and TM8/TM9 linkers (see ESI,† M6 and Fig. S14). The ADP-bound state of the Sec machinery was modeled by applying soft constraints only on the backbone of residues 344–365 of TM9, thus imitating weak binding with SecA and preventing drifting of the channel.
To better understand the role of the helix movement during the transition from the closed to the open state, we prepared a channel conformation “half-open”, where the LG is open, but the plug is closed, thus corresponding to an intermediate step (ESI,† Fig. S18 and Table S2). This configuration features a widened LG accommodating the SS and a peptide loop inside SecYEG. SMD was used to pull helices TM2b, TM7 and TM8 apart, widening the LG (for details, see Methods and ESI† M1). As a result we obtained an intermediate structure, named “transit-half”, which was used to insert the SS with peptide loop to obtain the structure “half-open”. Note that in our earlier work,10 we investigated a similar structure (called “active state”), which was obtained by putting the SS without the peptide loop into the structure “transit-half”.
Half-open channel: conformation and protein transport
Overlay of selected helices for the closed and half-open conformations demonstrates significant shifts of TM7, TM8, TM2b (Fig. 3a). At the same time, changes in the positions of TM5, the plug, and other helices, originating from adaptation to the presence of the peptide during equilibration, are not so pronounced (Fig. 3a; ESI,† Fig. S2). Hence, accommodation of the translocating substrate inside SecYEG requires only opening of the lateral gate and shift of TM8 to provide room for the movement of TM7.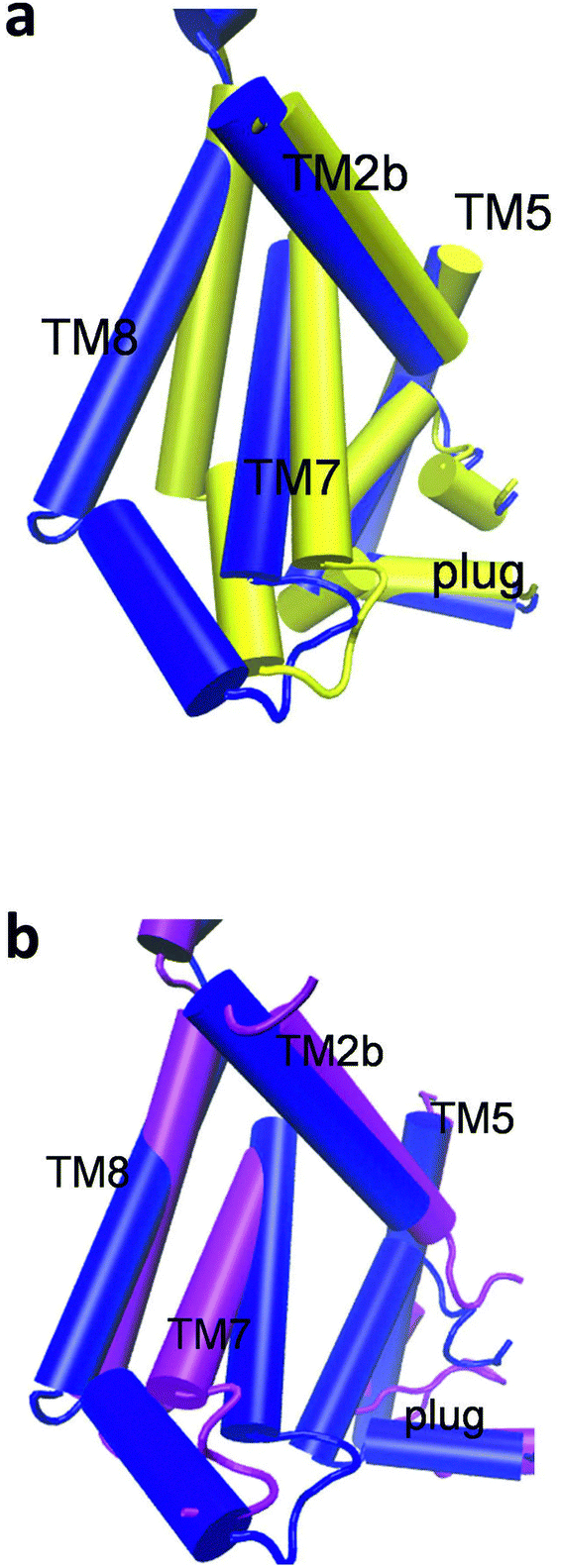 | ||
| Fig. 3 Overlay of SecYEG in the half-open conformation (blue) with (a) the 5AWW structure (yellow) and (b) the 5EUL structure (magenta). | ||
In Fig. 4, we show the free-energy profile for Lys 31 of the peptide being pulled through the translocon along the z-axis (red curve). This profile was obtained from umbrella sampling (US)37 with the reaction coordinate being the distance in the z-direction of the Cα atom of Lys 31 to that of Ile 403, where the z-axis is normal to the membrane. The zero of this reaction coordinate axis is basically the PR position (ESI,† Fig. S12; see ESI† M4 for details; see also our earlier work10 for similar simulations with ions). One can readily see that despite the LG is widened through insertion of the SS, the channel provides a high barrier for peptide transport as represented by the free energy profile of Lys 31. Note, that the PMF is highest, when Lys 31 has passed the PR (z = 0), since the major contribution to the barrier comes from interaction of the peptide with the plug around z = 10 (ESI,† Fig. S3). Interestingly, the plug did not shift significantly, while the peptide was pulled during the SMD (ESI,† Fig. S4). This result underscores that strong forces fix the plug in its closed conformation.
 | ||
| Fig. 4 PMF profile for Lys 31 in the peptide in SecYEG along the z-axis for the half-open channel (red), the open channel (blue) and the open channel with the mutant peptide W26A/Y27G (green). | ||
Open channel: conformation and protein transport
The SecYEG conformation obtained after shifting the key-set helices and metadynamics simulation of the plug’ s position is shown in Fig. 5a–d. In Fig. 5e, we show a contour plot of the PMF landscape as a function of the CVs d2 and d3 at d1 = 17 Å. Plots for other values of d1 can be seen in ESI,† Fig. S11. The PMF profiles for all trajectories reveal a complex structure with several minima reflecting the flexibility of the plug (Fig. 5e; ESI,† Fig. S11). For larger values of d1, a major minimum in the PMF landscape around d2 = 14 Å and d3 = 11 Å becomes evident. As the open conformation of the plug, we finally chose a structure corresponding to the minimum in PMF with collective variables d1 = 17.1 Å, d2 = 14.2 Å, d3 = 11.4 Å (Fig. 5e). This conformation is the closest to the 5EUL as seen from the overlays in Fig. 5c and d.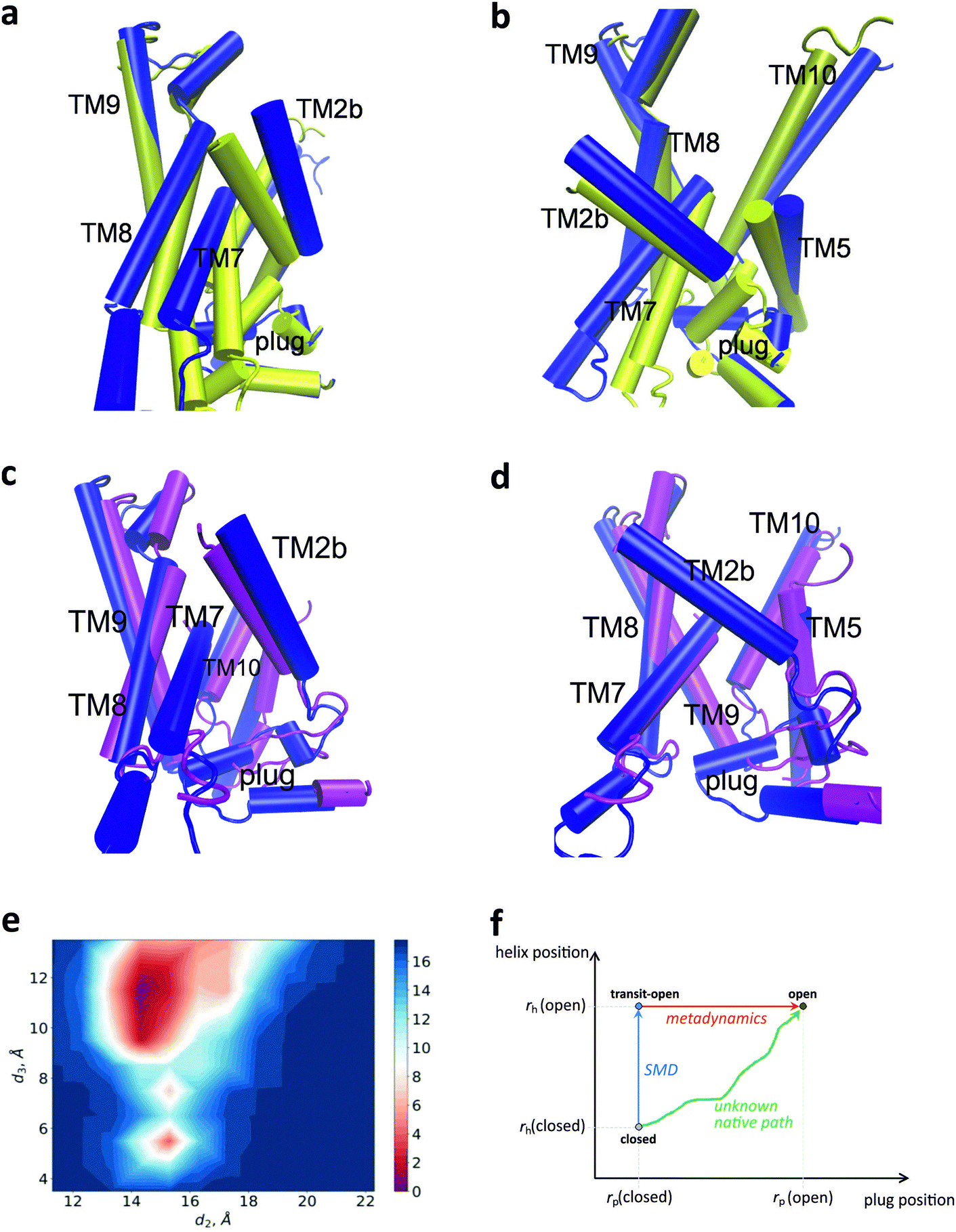 | ||
| Fig. 5 Structure of the open channel. (a) and (b) Overlay of the conformation (blue) obtained after metadynamics simulations (corresponding to d1 = 17.1 Å, d2 = 14.2 Å, d3 = 11.4 Å) with the 5AWW structure (yellow) in two side views. Only key-set helices and the plug are shown. (c) and (d) Same as in (a) and (b), but overlain with the 5EUL structure (magenta). Only key-set helices and the plug are shown. (e) PMF landscape (in kcal mol−1) for the collective coordinate d1 = 17.1 Å. (f) Scheme of the conformational transitions of the translocon in the two dimensional space of coordinates rp representing the plug position and rh representing the positions of the key-set helices. SMD is used to change the positions of the key-set helices from those in the closed structure (5AWW) to those corresponding to the 5EUL structure (transit-open channel), leaving the plug in a closed conformation (blue arrow). Metadynamics is used to find an open conformation of the plug while restraining the helix positions (red arrow). The native path from closed to open, in which key-set helices and the plug possibly move together, remains unknown (green arrow). | ||
Here we underline, that the main goal of the opening procedure was to provide a configuration, which enables releasing the plug. The position of the helices TM7, TM5, and TM8 were mimicked with higher precision, than the ones of TM9 and TM10. The latter were moved slightly to adjust the location of the TM9/10 linker, which is significant for interaction with the plug. TM2b underwent a larger shift, than given by the 5EUL structure. Matching TM2b exactly with the position of the helix in 5EUL still preserves a quite strong coupling with the plug (data not shown).
The procedure outlined above to find a conformation with an open plug involves two steps as sketched schematically in Fig. 5f: first, the key-set helices are moved and then the plug is relaxed. This two-step procedure (blue and red arrows in Fig. 5f) is not meant to represent the native opening mechanism. The latter actually remains unknown (green arrow in Fig. 5f). The simulations merely indicate that moving the key-set helices in response to the interaction with SecA in the ATP-bound state and insertion of the SS contributes to the plug opening. We hypothesize that this interplay between helix and plug movement is the key physical mechanism of “unlocking” the plug. However, the details of conformational changes in this process remain elusive.
An important aspect of the plug transition from the closed to the open state is the interplay of different forces. While the PMF profile, obtained in our work, explicitly displayed the role of the interactions between the plug and selected TM helices, the free energy landscape depends also on the coupling to the solvent (water). The latter can also influence conformational transitions significantly. To analyze the situation, we follow the theoretical philosophy of Ben-Naim38 and partition the protein into groups (numbered by the index i) with location vector ri. The free energy landscape depends on the position vectors of all groups, denoted here collectively by r′. To find the probable direction into which group i will move, we have to determine the gradient of the free energy with respect to ri. For example, the force acting on the plug is given by:
| F(rp) = −∇pU(r′) + FSI(rp) | (1) |
 | (2) |
Hourglass shape and hydrophobic core lead to water depletion in the central regions of SecYEG.10 Thus, a change in the hydration level experienced by the plug can lead to a gradient in the coupling with the solvent along the translocon axis. However, calculation of the force in eqn (2) exerted on the plug along the transition path is a complicated task. Here, we merely estimate the difference in plug-water coupling between the half-open and open conformations (see ESI,† M5 for details). The enthalpic difference amounts to −18.2 ± 3.8 kcal mol−1 and represents a significant gradient in interaction energy that can contribute to the transition to the open state. We can speculate that the contribution of plug-water interaction to the enthalpy difference between closed and open conformations of the translocon is in a similar order of magnitude.
The opening of the plug has an effect on the PMF for the transport of Lys 31 of the peptide: The translocation barrier is significantly reduced (Fig. 4, blue curve), but there remains a still pronounced barrier up to z = 7 Å and a higher barrier for z > 10 Å. Comparison of the PMF profile for the wild type (WT) peptide (blue curve) and that of a mutant, in which the bulky residues Trp 26 and Tyr 27 of the peptide are replaced by the small amino acids Ala and Gly, respectively (green curve), reveals that the major part of the barrier in the WT is caused by the translocation of these bulky residues. The PMF increase for z > 10 Å can be traced back to Trp 34 being present in both the WT and the mutant peptide. Therefore, the barrier for transport is primarily determined by interaction of bulky residues with the channel especially on the periplasmic side. In this context, it is noteworthy that the positively charged Lys 31, being present in the mutant peptide, does not give rise to a significant barrier by itself.
Translocation through the open SecYEG reveals that, in contrast to the half-open channel, the movement of the peptide stimulates an additional shift of the plug (cf. ESI,† Fig. S4 and S5), which is in agreement with ref. 15. This increased mobility of the plug in the open conformation results, obviously, from weakening of interactions with other helices.
Influence of the membrane potential
To test the influence of the electrostatic part ΔΨ of the proton motive force ΔΨH+, we performed simulations with an applied voltage oriented such that the positive potential is at the periplasmic side. These simulations were performed with the open channel in both the ADP- and ATP-bound states. Note that in our simulations, SecA is absent and hence is the NBS. The effects of SecA in the ADP- and ATP-bound states are mimicked by imposing different constraints on backbone atoms of SecY as described above. To observe effects along a relatively short trajectory of 400 ns, we stimulated the system by applying a voltage of 1.1 V during the first 200 ns, which was then reduced to 0.3 V during the second half of the run (for further details of these simulations, see ESI,† M6).In the case of the ADP-bound state, we also performed a reference simulation of 400 ns length with the same starting conformation, but with zero voltage. These two simulations are referred to in the following as “volt-“ and “relax”-simulation/trajectory. For the ADP-bound state, ΔΨ stimulates motion of all channel helices and the SS. The most significant impact is observed for the positions of helices TM2b, TM7 and the plug (Fig. 6a and b; further structures comparing the volt- and relax-trajectories are presented in ESI,† Fig. S6 and S16).
 | ||
| Fig. 6 Overlay of the conformations obtained in the volt- and relax-simulations of the ADP-bound state. The last frames of the volt- and relax-trajectories are red and blue, respectively, while the starting conformation is colored cyan. Presentations in (a) and (b) are side views along the y- and x-axis, respectively. Changes in the dipole orientation of the helices TM2b and signal sequence (SS), when the external electric field is applied (black arrow), are shown in (c). Movements of selected helices, stimulated by the voltage, are shown in (d)–(f) with side views along the y- and x-axis, respectively, in (d) and (e), and a view from the cytoplasm in (f). The small black arrows indicate the motions of the corresponding helices. | ||
Upon application of the external field (volt-simulation), TM2b moves outwards. In contrast, in the relax-simulation, we observe motions driving the helix inwards that result in a pronounced decrease in the PR distance TM2b-TM10 (Fig. 6b; ESI,† Fig. S16). The latter is defined as in our earlier work10 as the COM distance between residues Ile 81 and Ile 403 quantifying the opening of the PR between helices TM2b and TM10 (Fig. 1c; cf. also Fig. 1a in ref. 10). Displacement of the plug in the volt-trajectory brings it closer to TM5, in contrast to the relax-simulation, where it undergoes an upward shift only (Fig. 6b). The dynamics of TM7 reveals rotations in opposite directions for the two simulations (Fig. 6a and ESI,† Fig. S16). This leads to a widening of the LG in the case of the applied voltage, and the opposite effect for the relax-trajectory. Furthermore, in the latter case, motion of both TM7 and the plug reduces the space available for the substrate, which should raise a barrier for translocation. In contrast, the distance TM7-plug increases, when the voltage is applied (ESI,† Fig. S16).
Therefore, according to our results, the major effect of the membrane potential in the ADP-bound state involves keeping the LG open and shifting the plug away from the center of the translocon. Based on the observed dynamics, we can suggest the following simplified model of the above phenomena: The reorientation of the helix dipoles of TM2b and SS in the external field brings these helices closer to each other (Fig. 6c and d). To avoid overlap, the SS helix undergoes a lateral displacement versus TM7. The latter moves aside, thereby widening the LG and PR (Fig. 6d). Ending up in the proximity of the TM2b/3 linker, the N-terminus of the SS enforces a lateral shift that stimulates the consecutive motions of TM2b, TM3 and TM4 (Fig. 6e). As a result, the TM3/4 linker pulls the plug closer to SecE and fixes it in this position (Fig. 6f). This, together with the rearrangement of the TM9/10 linker contributes to the backside shift of the plug.
In the case of the strong coupling between SecA and SecYEG, corresponding to the ATP-bound state, the most significant changes are observed also for TM2b, TM7 and the plug (see ESI,† Fig. S7 and S15), but the movements are less pronounced compared to the ADP-bound state.
The above results and considerations can be used to interpret ion conductivity data from electrophysiological measurements.39 To this end, we calculated the single channel conductivity g. In the experiment, g was estimated to be 700 pS for a salt concentration of 450 mM. At the same time, the registered leak current through a single channel containing a stalled peptide amounted to 0.8 A (for 75 mV and an average salt concentration of 300 mM) that corresponds to g ≈ 11 pS.
To compute the conductivity in the ADP-bound state, we notice that the main passage of ions lies between TM7 and TM10. The distance TM7-TM10 undergoes significant fluctuations in the first half of the trajectory (ESI† M6, Fig. S16f) that induces a large flux of ions between 100 and 170 ns (ESI† M6, Fig. S17). Thus, to separate this effect from the rest of the simulations, where the translocon conformation was getting stabilized, we computed g averaged over the first and the second half of the volt trajectory separately, yielding 59 and 9 pS, respectively. Taking into account, that in our simulations the salt concentration was 100 mM, we can approximately recalculate our values for comparison with experiment, obtaining g ≈ 266 pS and 27 pS, respectively.
In the ATP-bound state, the ion conductivity was observed only for the first half of the trajectory and amounts to g ≈ 4.4 pS that yields 13 pS for experimental conditions of 300 mM salt. Considering the short simulation time and the approximate nature of the computations, the conductivity obtained in our work matches the experimental values well by the order of magnitude. We also note that the translocon in in vitro measurements is likely more flexible than in the MD simulations.
Having in mind the above results and considerations, we can give the following interpretation of the experimental observations. Application of the membrane potential destabilizes the channel's conformation causing fluctuations of helix positions and tilt angles, especially for TM7 and TM2b in the ADP-bound state. This results in an electric current as registered in the experiment. Here, according to our results, the distance between TM7 and TM10 plays an important role. Stabilization of the SecYEG open conformation leads to a decrease of the current. Upon removal of the external voltage, ion flux is still observed experimentally.39 According to our results, we hypothesize that under these conditions, it is mostly the ATP-bound state that conducts the ions. Similar to the ADP-bound state, the ions flow between helices TM7 and TM10. Therefore, one can check our hypothesis experimentally, for example, by cross-linking the peptide with TM7 and TM10.
Physical model of the Sec machinery activation: PEES forces
The PFM profiles from US simulations demonstrate that the half-open channel is very tight engendering barriers for the peptide transport. The plug is held in place by the interactions within the hydrophobic core and provides the major contribution to the barrier for a substrate. Thus, we suggest that in addition to the SS intercalation, external forces exerted by the translocation partner are needed to stimulate complete SecYEG opening and activation of the Sec machinery. It is the addition of SecA and ATP that fully promotes the plug displacement. The latter significantly reduces the barrier for transport as demonstrated by the PMF profile of the open channel. Here, the remaining barrier is due to the interaction of bulky amino acids of the substrate with the PR and the plug in accordance with ref. 40 (see ESI† A2). This barrier for bulky residues contributes to preventing backwards movement of the peptide.Another characteristic of the channel's open conformation is its non-equilibrium nature that implies a need for external forces to maintain it. In the ATP-bound state, it is the coupling with SecA (simulated here with appropriate constraints) that keeps the channel in this conformation. Upon detachment of SecA (simulated here by less stringent constraints to mimic the ADP-bound state), the channel tends to a more closed conformation (see relax-simulations above). However, the membrane potential ΔΨ can counteract this relaxation (see volt-simulations above) and thus plays an important role in keeping the translocon active. This finding can explain the significant drop of the ATP concentration necessary for protein transport in the presence of the proton motive force.17 Similarity in action of ATP-bound SecA and ΔΨH+ is also supported by experiments, where it was shown that the requirement for ΔΨH+ can be partially suppressed by a large amount of SecA or with prlA4 mutations that promote an increased affinity to SecA.41–43 An additional influence of ΔΨH+ on the initial stages of translocation can originate from the stabilization of the SecYEG open conformation, thus facilitating binding with activated SecA.17
Taking into account data from the literature (see Introduction) and the results of the present study, we can suggest the following physical model of the Sec machinery activation. Binding of SecA in ATP-bound state induces conformational changes in SecYEG and helps to overcome barriers originating from interactions between helices, especially, in the hydrophobic core. More precisely, binding of SecA, presumably, shifts TM9 by the TM8/TM9 linker, which as a consequence can drive TM10 by the TM9/TM10 linker (ESI,† Fig. S8). Intercalation of the signal sequence into the lateral gate further contributes to weakening of the hydrophobic core, which leads to the side movement of helices TM2b and TM7 and stimulates the “domino” stacking movement of other helices (Fig. 7). Sequential shifts are transferred to SecE, pushing it backwards, which pulls TM10 and TM5 back as well. “Domino” stacking of helices also leads to a displacement of SecG helices and linkers between them (Fig. 7), which facilitates the release of TM7 and TM5.
 | ||
| Fig. 7 Overlay of the closed (5AWW, green) and open (blue) conformations of SecYEG: (a) and (b) correspond to side views along the y and x axes, respectively; (c) and (d) are perspectives from the cytoplasmic side. Only selected helices are shown. SecG in the closed and open channel is colored yellow and red, respectively. SecE in the closed and open channel is depicted in violet and magenta, respectively. The small black arrows indicate motions of the corresponding helices upon opening of the translocon. | ||
The above rearrangements weaken the contacts of the plug with TM7, TM5, TM2b, and TM10. At the same time, the TM9/TM10 linker, following the movement of the corresponding helices, pulls the plug to the side. The translocating substrate, in turn, promotes downward motion of the plug, blocking upward displacement. The movement of the plug away from the pore ring leads to an increased exposure to water, which penetrates the channel after widening of the pore. Interaction with water molecules can provide favoring enthalpy for plug polar residues, facilitating the movement on the periplasmic side. Remaining interactions of the plug with TM9/10 in the open state help to prevent a further shift on the periplasmic side, which guarantees the plug can return to the closed position, when translocation is over.
The coupling with the translocation partner SecA fixes the channel in the open state. The latter is characterized by a widened interior room that reduces the barrier for translocation and provides sufficient water environment for partitioning of a secreted substrate into the periplasm. Upon hydrolysis of ATP, ΔΨH+, if applied, helps to maintain the open state and, probably, also provides the driving force for a substrate.44,45 In the cells, where in contrast to in vitro systems only a limited amount of ATP is available, the PMF is especially required. So, we suggest, that consecutive cycles of ATP hydrolysis lead to accumulation of ADP in the environment that would considerably weaken the SecA-driving force for translocation. As a support of this hypothesis, we refer to the results of ref. 17, where upon addition of a significant quantity of ADP, the translocation rate in the presence of ΔΨH+ was three times higher than in its absence.
The physical mechanism of the translocon conformational transition from the closed to the open state partially resembles the folding–unfolding process of globular proteins, driven by the interplay of the dominant forces - hydrophobic interactions and entropy.46 (It should be noted, however, that the dominant role of the hydrophobic effect in protein folding has been questioned by Ben-Naim.38) In the case of SecYEG, hydrophobic core interactions are the main forces responsible for folding of the channel and keeping it in the closed state. They are quite tight (in the WT translocon), since leakage of ions would be lethal for a cell. So, external forces due to the binding with translocation partners are needed to break them. (The softening of these interactions can be also reached by mutations in the critical regions.) When weakening is reached, other mechanisms – solvent-induced forces and conformational entropy – come into play. Release of the plug from the hydrophobic core presumably raises the conformational entropy, which can destabilize the channel similar to effects in globular proteins.46 In particular, deletion of long-range contacts (i.e. pairwise interactions of residues with a large separation in the amino acid sequence, not spatial distance), which removes configurational constraints from the protein in its unfolded state, leads to an increase in the entropy of the unfolded conformation and its stabilization (see ref. 47 and references therein). ΔΨH+ works together with the above forces and contributes to shift the free-energy balance between closed and open conformations of SecYEG in favor of the latter. Thus, we have an ensemble of forces, including the proton motive force, external forces (due to binding with the translocation partner), entropic and solvent-induced forces (PEES), which enable the channel to transition from the closed to the open conformation and maintain the latter. Therefore, it is the delicate interplay between the above forces – hydrophobic on the one side and PEES on the other side – that provides flexibility of the translocon and ensures functionality of the Sec machinery.
Discussion
The presented model of the Sec machinery activation, which involves the PEES forces, relies strongly on the interaction between helices, especially in the hydrophobic core. Hence, disruption of the latter, by deletion of the residues or their mutations, strengthens the solvent-induced and entropic forces that shifts the free-energy balance to the open state, and as a consequence, affects the Sec complex function. Examples include channel leakage and growth defect upon replacement of a few isoleucines by glycine in the pore ring26 and lethality of the double mutant I408N(SecY)-L108R(SecE).23,24 The plug-deleted mutants are shown to transport preproteins with defective or missing signal sequences and show transient openings.21,27 In this case, by losing most interactions with the hydrophobic core, the new plug becomes more flexible, which enlarges its conformational space. To further verify our model, we suggest the following mutations. (The analysis below is based on the E. coli structure 5GAE.25)The residues Ile 191, Ile 192, and Ile 195 as well as Ala 197 and Val 196 of TM5 are involved in coupling with TM7, TM10 and SecE. Moreover, Ile 195 – part of the hydrophobic core – contacts also the plug residue Phe 67. Hence, substituting them with polar or charged amino acid can weaken interactions and influence the opening of the translocon. Additional mutations, which destabilize the coupling between TM9 and TM10, include substitution of Ile 381, Ile 384, and Glu 389 of TM9 as well as Leu 407 and Gly 402 of TM10. Breaking of interactions between the TM9/10 linker and the plug should interfere with the movement of the latter. Residues of the plug – important for this coupling – include Met 63 and Met 66. They are located in close proximity to Phe 399 in E. coli. Furthermore, substitution of Phe 67 in the plug with a polar or charged residue should destabilize the hydrophobic core of SecYEG. In particular, introduction of a negatively charged amino acid should result in an effect similar to NaTT-treatment27,31 (cf. Introduction).
Many mutations described in the literature (see above) and suggested in our work break long-range contacts between different domains of SecYEG. Furthermore, the binding sites of SecA and SS are coupled to the important conformational variables – distances between TM9/10 linker, TM2b and the plug (see ESI† M3) – representing long-range contacts as well. The domino staking motion resulting from propagation of the local deformations in the binding sites affects the third conformational variable related to the long-range contact between the plug and TM5. The disruption of the above interactions presumably leads to significant entropy changes – mostly due to release of the plug – that destabilize the closed conformation. Therefore, the channel activation is accomplished through an allosteric mechanism that employs long-range interactions coupled to intrinsic conformational variables. We suggest that such an interplay can be an effective allosteric mechanism also in other complex proteins (see ref. 48 and references therein), especially in the case of remote entropy compensation.49 Here, ligand binding accomplishes rigidification of one part of the protein, while another domain gains flexibility and thus compensates for the entropy loss upon binding.
Finally, we will analyze the operation of the Sec complex to understand how it enables directed motion of a substrate inside the channel. The molecular machines in the cell operate in the regime, where viscous forces dominate inertial forces.50 So, movement is completely overdamped, and any direction of motion is rapidly randomized by collisions with the surroundings. There are two requirements to provide separation between forward and reverse trajectories: spatial asymmetry, non-equilibrium driving or a combination of both (see ref. 50 and references therein). The non-equilibrium driving provides temporal asymmetry to molecular machine dynamics, whereas spatial asymmetry permits a directed response to the energy input.
In the case of the Sec complex, we observe a quite high translocation barrier for bulky residues. (It was demonstrated that the presence of a barrier, where a system can transiently “get stuck”, provides distinct forward and reverse trajectory distributions.51) The probability to overcome the barrier under equilibrium conditions is low, so a non-equilibrium driving force is required. This can be supplied as a pushing force by the 2HF of SecA.9 There are also hints from experimental data, that the membrane potential can exert a pulling force. While several thousand ATP molecules are hydrolyzed for each proOmpA in the absence of ATP, this ratio falls to under 200 in the presence of ΔΨH+.16 In addition, a stepwise mechanism of translocation coupled to ATP hydrolysis would be very slow and inefficient.8 Action of both mechanisms (2HF and ΔΨH+) in different stages of protein transport would guarantee a nearly constant driving force. Thus, the translocation barrier together with external forces would be an efficient combination of spatial asymmetry and non-equilibrium driving guaranteeing separation between forward and reverse trajectories. Furthermore, given their influence on different types of residues (bulky or charged), the suggested mechanisms – barrier, 2HF and ΔΨH+, – taken together would provide a directional motion for any substrate, which significantly increases the efficiency of the machine. From this point of view, the models suggested in the literature that lack either spatial asymmetry or non-equilibrium driving are much less effective.9,52
Our physical model still needs further building blocks. The complete understanding of the physical mechanism underlying protein transport by the Sec complex requires elucidating the partitioning process (membrane insertion vs. secretion) and a more detailed investigation of the role of ΔΨH+, in particular, the H+ part (i.e. ΔpH). As shown above, ΔΨ can significantly improve the efficiency of the machinery. ΔpH might be related to pKa shifts that may lead to (de)protonation of the charged residues (such as Lys 31) with consequences for a uni-directional pulling force that may be exerted by ΔΨ. A cornerstone of the proposed mechanism is related to the properties of water inside the channel, which are proven to be different from those of bulk water,53 and water attached to the plug. This, in turn, can also influence the partitioning of the proteins between insertion into the lipid bilayer and secretion.
Building a complete biophysical model of the Sec machinery will contribute to an understanding of the molecular machine's operation and suggest a method of control of selective protein traffic (e.g., by ΔpH, ΔΨ) in artificial channels. This, in turn, will promote the development of high-demand applications, related to the design of active surfaces and biological nanopore technology.
Methods
Systems assembly
The assembly protocol involved the following procedures: (i) CHARMM-GUI54–56 was used to add missing atoms to the 5AWW structure, which subsequently was embedded in a lipid bilayer composed of 296 lipid molecules: 70% dioleoyl-glycero-phosphatidyl-ethanolamine (DOPE) and 30% dioleoyl-glycero-phosphatidyl-glycerol (DOPG) as well as a water box with dimensions 105 × 105 × 113 Å, containing 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 888 water molecules as well as 100 mM KCl to mimic a bacterial membrane and physiological ion conditions.39,57 The whole system was charge neutral and included 48 Cl− and 123 K+ ions. (ii) To create the half-open and open conformations, the TM helices were pulled by an external constant force, using steered molecular dynamics (SMD),58 to the positions of the corresponding helices in the 5EUL structure. The resulting structures are referred to as transit-half and transit-open, respectively (ESI,† Fig. S18). (iii) In the case of the open channel, the position of the plug was determined with the help of well-tempered metadynamics.59 (iii) CHARMM55 was used to place the peptide inside transit-half as well as inside the structure obtained from metadynamics from transit-open. The peptide was composed of 37 residues, including the SS: Lys Lys Thr Ala Ile Ala Ile Ala Val Ala Leu Ala Gly Phe Ala Thr Val Ser Leu Ser Leu Ser Asp Asn Thr Trp Tyr Thr Gly Ala Lys Leu Gly Trp Ser Gln Tyr. To reveal the role of bulky residues, we have also created a mutant peptide with the two substitutions W26A and Y27G. (iv) The last step of the procedure was minimization and equilibration of the final setup (ESI,† Fig. S18). As an example, we show the half-open structure embedded in the membrane in ESI,† Fig. S19.
888 water molecules as well as 100 mM KCl to mimic a bacterial membrane and physiological ion conditions.39,57 The whole system was charge neutral and included 48 Cl− and 123 K+ ions. (ii) To create the half-open and open conformations, the TM helices were pulled by an external constant force, using steered molecular dynamics (SMD),58 to the positions of the corresponding helices in the 5EUL structure. The resulting structures are referred to as transit-half and transit-open, respectively (ESI,† Fig. S18). (iii) In the case of the open channel, the position of the plug was determined with the help of well-tempered metadynamics.59 (iii) CHARMM55 was used to place the peptide inside transit-half as well as inside the structure obtained from metadynamics from transit-open. The peptide was composed of 37 residues, including the SS: Lys Lys Thr Ala Ile Ala Ile Ala Val Ala Leu Ala Gly Phe Ala Thr Val Ser Leu Ser Leu Ser Asp Asn Thr Trp Tyr Thr Gly Ala Lys Leu Gly Trp Ser Gln Tyr. To reveal the role of bulky residues, we have also created a mutant peptide with the two substitutions W26A and Y27G. (iv) The last step of the procedure was minimization and equilibration of the final setup (ESI,† Fig. S18). As an example, we show the half-open structure embedded in the membrane in ESI,† Fig. S19.
Simulation protocol: general information
NAMD version 2.13 along with the CHARMM36m force field was applied for MD simulations.56,60,61 The TIP3P model was used for water molecules. All simulations were run under NPT conditions: constant temperature control was regulated with Langevin dynamics62 with a 5 ps−1 damping coefficient coupled to all heavy atoms (T = 300 K); constant pressure was maintained at 1 atm by a Nosé–Hoover Langevin piston barostat63,64 with decay period 100 fs and a damping time of 50 fs. To compute electrostatic interactions, the particle-mesh Ewald summation was used with a grid spacing of 1 Å, and the short-range real-space interactions were cut off at 12 Å using a smooth switch function beginning at 10 Å. The equations of motion were integrated in 2 fs or 1 fs steps using the velocity Verlet algorithm. The number of time steps between evaluations of the short-range non-bonded interactions and the full electrostatic interactions were equal to 2 and 4, respectively. The SHAKE algorithm was employed to constrain the length of the bonds involving hydrogen atoms. Coordinates were saved every 2 ps. The images were prepared with VMD.65Minimization and equilibration
All assembled systems were energy minimized with CHARMM using 500 steps of steepest descent followed by 1000 steps of the Newton–Raphson algorithm.After energy minimization, structures were equilibrated. For transit-half with the peptide inside, equilibration was performed with nine consecutive runs. In the first run, harmonic-restraint forces with a force constant of 5 kcal mol−1 Å−2 were applied to the protein backbone and of 0.5 kcal mol−1 Å−2 to atoms of lipids. In the next six runs, the restraints of the protein backbone were released to the following values: 2, 1, 0.5, 0.1, 0.05, 0.01 kcal mol−1 Å−2. The restraints for lipids were 0.2 kcal mol−1 Å−2 in the second run and set to 0 in the subsequent runs. For the eighth and ninth runs, we released all constraints. The total equilibration time was 12 ns.
A similar protocol was applied to the structure obtained from metadynamics from transit-open with the peptide inside. The procedure included eight consecutive runs, resulting in a total simulation time of 17 ns. In the first run, harmonic-restraint forces with a constant of 2.5 kcal mol−1 Å−2 were applied to a backbone of the key-set helices, the plug and the peptide, and of 0.5 kcal mol−1 Å−2 to lipids and ions. In the next runs, the constraints on the peptide were released: 1.5, 0.8, 0.4, 0.2, 0.1, 0.05 kcal mol−1 Å−2. The force constants for lipids decreased in the following two runs: 0.3 and 0.1 kcal mol−1 Å−2. For ions, we applied 0.5 kcal mol−1 Å−2 for the next three runs and then released up to 0.2 and 0.1 in the 5th and 6th run, respectively. Constraints on the backbone of the key-set helices and the plug were kept throughout the whole procedure to mimic strong coupling with SecA.
Details of other protocols are given in ESI† M1-M6.
Author contributions
E. S. developed ideas, performed the simulations and calculations, analyzed the results, and wrote the initial draft. F. M. obtained the financial support for the project leading to this publication, supervised the research, and provided critical review and commentary for the manuscript. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI† and uploaded at https://doi.org/10.5281/zenodo.13936173 entitled "SecYEG channel (with a peptide inside) in open and half-open conformations".Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank T. Renger and H. Krobath for valuable support in the initial phase of the project. This research was supported by hardware grants (PI Kramer, JKU Linz) from NVIDIA and utilized NVIDIA A100 GPUs. The work was supported by the Doctoral Program “Nano-Analytics of Cellular Systems (NanoCell)” of the Austrian Science Fund (FWF) (grant W 1250), project P 33154-B funded by the FWF in conjunction with the federal state of Upper Austria, and the Linz Institute of Technology (grant LIT-2019-8-SEE-120).Notes and references
- C. Song, A. Kumar and M. Saleh, Genomics, Proteomics Bioinf., 2009, 7, 37–46 CrossRef CAS PubMed.
- J. Ostwald, R. Njenga, A. Natriashvili, P. Sarmah and H. G. Koch, Front. Mol. Biosci., 2021, 8, 664241 CrossRef PubMed.
- J. A. Lycklama a Nijeholt and A. J. M. Driessen, Philos. Trans. R. Soc., B, 2012, 367, 1016–1028 CrossRef CAS PubMed.
- T. A. Rapoport, L. Li and E. Park, Annu. Rev. Cell Dev. Biol., 2017, 33, 369–390 CrossRef CAS PubMed.
- D. Görlich and T. A. Rapoport, Cell, 1993, 75, 615–630 CrossRef PubMed.
- M. Müller, H. G. Koch, K. Beck and U. Schäfer, Prog. Nucleic Acid Res. Mol. Biol., 2001, 66, 107–157 Search PubMed.
- F. U. Hartl, S. Lecker, E. Schiebel, J. P. Hendrick and W. Wickner, Cell, 1990, 63, 269–279 CrossRef CAS PubMed.
- I. Collinson, R. A. Corey and W. J. Allen, Philos. Trans. R. Soc., B, 2015, 370, 20150025 CrossRef PubMed.
- B. W. Bauer, T. Shemesh, Y. Chen and T. A. Rapoport, Cell, 2014, 157, 1416–1429 CrossRef CAS PubMed.
- E. Sobakinskaya, H. Krobath, T. Renger and F. Müh, Phys. Chem. Chem. Phys., 2021, 23, 25830–25840 RSC.
- D. G. Knyazev, R. Kuttner, M. Zimmermann, E. Sobakinskaya and P. Pohl, J. Membr. Biol., 2018, 251, 329–343 CrossRef CAS PubMed.
- E. Park and T. A. Rapoport, Annu. Rev. Biophys., 2012, 41, 21–40 CrossRef CAS PubMed.
- C. Y. Ma, X. F. Wu, D. J. Sun, E. Park, M. A. Catipovic, T. A. Rapoport, N. Gao and L. Li, Nat. Commun., 2019, 10, 2872–2880 CrossRef PubMed.
- R. M. Voorhees and R. S. Hegde, Curr. Opin. Cell Biol., 2016, 41, 91–99 CrossRef CAS PubMed.
- P. C. K. Tam, A. P. Maillard, K. K. Y. Chan and F. Duong, EMBO J., 2005, 24, 3380–3388 CrossRef CAS PubMed.
- E. Schiebel, A. J. M. Driessen, F. U. Hartl and W. Wickner, Cell, 1991, 64, 927–939 CrossRef CAS PubMed.
- K. Shiozuka, K. Tani, S. Mizushima and H. Tokuda, J. Biol. Chem., 1990, 265, 18843–18847 CrossRef CAS PubMed.
- D. G. Knyazev, R. Kuttner, A. N. Bondar, M. Zimmerman, C. Siligan and P. Pohl, Biomolecules, 2020, 10, 78 CrossRef CAS PubMed.
- L. Li, E. Park, J. Ling, J. Ingram, H. Ploegh and T. A. Rapoport, Nature, 2016, 531, 395–399 CrossRef CAS PubMed.
- P. J. Schatz and J. Beckwith, Annu. Rev. Genet., 1990, 24, 215–248 CrossRef CAS PubMed.
- M. A. Smith, W. M. Clemons, C. J. DeMars and A. M. Flower, J. Bacteriol., 2005, 187, 6454–6465 CrossRef CAS PubMed.
- F. Duong and W. Wickner, EMBO J., 1999, 18, 3263–3270 CrossRef CAS PubMed.
- R. S. Osborne and T. J. Silhavy, EMBO J., 1993, 12, 3391–3398 CrossRef CAS PubMed.
- A. M. Flower, R. S. Osborne and T. J. Silhavy, EMBO J., 1995, 14, 884–893 CrossRef CAS PubMed.
- A. Jomaa, D. Boehringer, M. Leibundgut and N. Ban, Nat. Commun., 2016, 7, 10471 CrossRef CAS PubMed.
- E. Park and T. A. Rapoport, Nature, 2011, 473, 239–242 CrossRef CAS PubMed.
- S. M. Saparov, K. Erlandson, K. Cannon, J. Schaletzky, S. Schulman, T. A. Rapoport and P. Pohl, Mol. Cell, 2007, 26, 501–509 CrossRef CAS PubMed.
- W. Li, S. Schulman, D. Boyd, K. Erlandson, J. Beckwith and T. A. Rapoport, Mol. Cell, 2007, 26, 511–521 CrossRef CAS PubMed.
- J. A. Lycklama a Nijeholt, M. Bulacu, S. J. Marrink and A. J. M. Driessen, J. Biol. Chem., 2010, 285, 23747–23754 CrossRef CAS PubMed.
- A. N. Bondar, C. del Val, J. A. Freites, D. J. Tobias and S. H. White, Structure, 2010, 18, 847–857 CrossRef CAS PubMed.
- D. J. Parker and W. S. Allison, J. Biol. Chem., 1969, 244, 180–189 CrossRef CAS PubMed.
- S. Itskanov, K. M. Kuo, J. C. Gumbart and E. Park, Nat. Struct. Mol. Biol., 2021, 28, 162–172 CrossRef CAS PubMed.
- Y. Tanaka, Y. Sugano, M. Takemoto, T. Mori, A. Furukawa, T. Kusakizako, K. Kumazaki, A. Kashima, R. Ishitani, Y. Sugita, O. Nureki and T. Tsukazaki, Cell Rep., 2015, 13, 1561–1568 CrossRef CAS PubMed.
- J. Zimmer, Y. S. Nam and T. A. Rapoport, Nature, 2008, 455, 936–943 CrossRef CAS PubMed.
- R. A. Corey, W. J. Allen, J. Komar, S. Masiulis, S. Menzies, A. Robson and I. Collinson, Structure, 2016, 24, 518–527 CrossRef CAS PubMed.
- A. Barducci, M. Bonomi and M. Parrinello, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 826–843 CAS.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 Search PubMed.
- A. Ben-Naim, Molecular Theory of Water and Aqueous Solutions - Part II: The Role of Water in Protein Folding, Self-Assembly and Molecular Recognition, World Scientific Publishing, Singapore, 2011 Search PubMed.
- D. G. Knyazev, L. Winter, B. W. Bauer, C. Siligan and P. Pohl, J. Biol. Chem., 2014, 289, 24611–24616 CrossRef CAS PubMed.
- K. Sato, H. Mori, M. Yoshida, M. Tagaya and S. Mizushima, J. Biol. Chem., 1997, 272, 5880–5886 CrossRef CAS PubMed.
- H. Yamada, S. Matsuyama, H. Tokuda and S. Mizushima, J. Biol. Chem., 1989, 264, 18577–18581 CrossRef CAS PubMed.
- N. Nouwen, B. de Kruijff and J. Tommassen, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5953–5957 CrossRef CAS PubMed.
- J. P. W. van der Wolk, P. Fekkes, A. Boorsma, J. L. Huie, T. J. Silhavy and A. J. M. Driessen, EMBO J., 1998, 17, 3631–3639 CrossRef CAS PubMed.
- N. Ismail, R. Hedman, M. Lindén and G. von Heijne, Nat. Struct. Mol. Biol., 2015, 22, 145–149 CrossRef CAS PubMed.
- M. J. M. Niesen, A. Müller-Lucks, R. Hedman, G. von Heijne and T. F. Miller III, Biophys. J., 2018, 115, 1885–1894 CrossRef CAS PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed.
- L. S. Bigman and Y. Levy, Isr. J. Chem., 2020, 60, 705–712 CrossRef CAS.
- G. H. Lorimer, A. Horovitz and T. McLeish, Philos. Trans. R. Soc., B, 2018, 373, 20170173 CrossRef PubMed.
- D. Kern and E. R. P. Zuiderweg, Curr. Opin. Struct. Biol., 2003, 13, 748–757 CrossRef CAS PubMed.
- A. I. Brown and D. A. Sivak, Chem. Rev., 2020, 120, 434–459 CrossRef CAS PubMed.
- A. Zarrin, D. A. Sivak and A. I. Brown, Phys. Rev. E, 2019, 99, 062127 CrossRef CAS PubMed.
- W. J. Allen, R. A. Corey, P. Oatley, R. B. Sessions, S. A. Baldwin, S. E. Radford, R. Tuma and I. Collinson, eLife, 2016, 5, e15598 CrossRef PubMed.
- S. Capponi, M. Heyden, A. N. Bondar, D. J. Tobias and S. H. White, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9016–9021 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks, 3rd, A. D. Mackerell, Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. Brooks, 3rd, A. D. MacKerell, Jr., J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- J. R. Willdigg and J. D. Helmann, Front. Mol. Biosci., 2021, 8, 634438 CrossRef CAS PubMed.
- H. Lu and K. Schulten, Proteins, 1999, 35, 453–463 CrossRef CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Henin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kale, K. Schulten, C. Chipot and E. Tajkhorshid, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- X. Zhu, P. E. Lopes and A. D. Mackerell, Jr., Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 167–185 CAS.
- T. Schlick, Molecular Modeling and Simulation: An Interdisciplinary Guide, Springer, New York, 2010 Search PubMed.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- S. E. Feller, Y. H. Zhang, R. W. Pastor and B. R. Brooks, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, details of structural analysis, method details. See DOI: https://doi.org/10.1039/d4cp03201b |
| This journal is © the Owner Societies 2024 |