
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular interactions of photosystem I and ZIF-8 in bio-nanohybrid materials†
Sebastian
Reiter
 *a,
Igor
Gordiy‡
a,
Kathrin L.
Kollmannsberger
b,
Feng
Liu
c,
Erling
Thyrhaug
d,
Dario
Leister
c,
Julien
Warnan
b,
Jürgen
Hauer
*d and
Regina
de Vivie-Riedle
a
*a,
Igor
Gordiy‡
a,
Kathrin L.
Kollmannsberger
b,
Feng
Liu
c,
Erling
Thyrhaug
d,
Dario
Leister
c,
Julien
Warnan
b,
Jürgen
Hauer
*d and
Regina
de Vivie-Riedle
a
aDepartment of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstr. 11, 81377 Munich, Germany. E-mail: sebastian.reiter@cup.uni-muenchen.de
bChair of Inorganic and Metal–Organic Chemistry, Department of Chemistry and Catalysis Research Center (CRC), TUM School of Natural Sciences, Technical University of Munich, Lichtenbergstr. 4, 85748 Garching, Germany
cFaculty of Biology, Ludwig-Maximilians-Universität München, Großhaderner Str. 2-4, 82152 Planegg-Martinsried, Germany
dProfessorship of Dynamic Spectroscopy, Department of Chemistry and Catalysis Research Center (CRC), TUM School of Natural Sciences, Technical University of Munich, Lichtenbergstr. 4, 85748 Garching, Germany. E-mail: juergen.hauer@tum.de
First published on 22nd August 2024
Abstract
Bio-nanohybrid devices featuring natural photocatalysts bound to a nanostructure hold great promise in the search for sustainable energy conversion. One of the major challenges of integrating biological systems is protecting them against harsh environmental conditions while retaining, or ideally enhancing their photophysical properties. In this mainly computational work we investigate an assembly of cyanobacterial photosystem I (PS I) embedded in a metal–organic framework (MOF), namely the zeolitic imidazolate framework ZIF-8. This complex has been reported experimentally [Bennett et al., Nanoscale Adv., 2019, 1, 94] but so far the molecular interactions between PS I and the MOF remained elusive. We show via absorption spectroscopy that PS I remains intact throughout the encapsulation-release cycle. Molecular dynamics (MD) simulations further confirm that the encapsulation has no noticeable structural impact on the photosystem. However, the MOF building blocks frequently coordinate to the Mg2+ ions of chlorophylls in the periphery of the antenna complex. High-level quantum mechanical calculations reveal charge-transfer interactions, which affect the excitonic network and thereby may reversibly change the fluorescence properties of PS I. Nevertheless, our results highlight the stability of PS I in the MOF, as the reaction center remains unimpeded by the heterogeneous environment, paving the way for applications in the foreseeable future.
1. Introduction
Harnessing solar energy for catalytic applications is a key aspect in the much-needed transformation to clean energy. Multiple avenues are being explored to create new materials and devices with increased durability and efficiency. These include functionalized photoelectrodes,1,2 carbon-based nanomaterials,3 or metal–organic frameworks (MOFs).4–6 However, the multitude of free parameters in the design of novel photocatalysts poses a significant challenge.7 A promising alternative to developing catalytic materials from scratch may be the incorporation of natural light-harvesting systems into bio-nanohybrid devices.8–17 In such applications, the natural photosystems I and II (PS I and PS II) are prime candidates as catalytic units. Of the two, cyanobacterial PS I is especially attractive due to the strong reductive potential (Em = −1.3 V vs. SHE) of its electron donor.18 Additionally, the spectral and redox properties of PS I are very diverse across different species,19,20 allowing fine-grained adaptations by selecting a different species as donor.18 The cyanobacterial PS I (T. elongatus), which is the focus of this work, occurs naturally as a trimeric membrane protein complex.21 Each monomeric subunit is composed of 11 protein chains and a plethora of cofactors, namely 11 carotenoids, 3 structurally relevant lipid molecules, 2 phylloquinones, 3 iron–sulfur clusters and 96 chlorophylls. The latter can be categorized into an antenna complex of 90 chlorophylls and a reaction center of 6 chlorophylls. In the light-harvesting process, the antenna complex captures light and conducts the energy towards the reaction center via resonance energy transfer. The chlorophylls in the antenna complex are loosely arranged in two layers on the lumenal and stromal side of the membrane, and positioned around the perimeter of the protein complex.21 The function of bio-nanohybrids incorporating PS I has been successfully demonstrated in combination with plasmonic nanoparticles,22–24 carbon nanotubes,8,9 and semiconductor surfaces.10,13,14,17,25 However, any real-world applications of such bio-nanohybrids face the challenge of stabilizing the photosystem against harmful environments.26 Another challenge to overcome is the weak absorption of PS I monolayers,25 which decreases device efficiencies. Here, the encapsulation in a MOF offers the prospect of both, providing a stable microenvironment27,28 and enabling the controlled assembly of multilayered nanostructures.The integration of PS I in a MOF has been demonstrated experimentally in a recent study,29 where the zeolitic imidazolate framework (ZIF-8) was chosen as the encapsulating agent. ZIF-8 is a MOF composed of divalent zinc cations and 2-methylimidazolate (MIm−) and offers a series of attractive physicochemical properties for applications in photocatalytic bio-nanohybrids. In particular, ZIF-8 can be synthesized under mild aqueous conditions and remains stable across a wide range of solvents and temperatures.29 At the same time, it is optically transparent in the visible part of the spectrum, allowing its integration in light-harvesting devices.29 Pump–probe experiments showed that PS I retains its function both upon encapsulation in ZIF-8 and after digestion of the MOF in acidic conditions.29 However, the fluorescence signal of the encapsulated PS I exhibited an additional intense peak at 676 nm, apart from the characteristic broad excitonic band between 660 nm to 800 nm.29 This new signal is attributed to the emission of excitonically uncoupled chlorophyll a and would normally be indicative of a denatured PS I releasing its chlorophylls into the solution. However, after digestion of the MOF, the authors observed the fluorescence returning back to the original, excitonic signal.29 This allows the hypothesis that molecular interactions at the PS I/ZIF-8 interface can reversibly alter the emission properties of the photosystem, while preserving its principal structure and function.
As the mechanism of this process remains elusive to experimental techniques, theoretical investigations can provide new insights into the molecular interactions at an atomistic level. In particular, molecular dynamics (MD) simulations are a valuable tool in the study of photosynthetic processes.30 We recently presented a new computational model of PS I,31 which accounts for the molecular dynamics of the chromophores in their natural environment, as well as the extensive multireference nature of chlorophyll excitations.32 This enabled the calculation of the excitonic energies at an unprecedented high level of theory. In this work, we build on our previous results to investigate the interactions of PS I with ZIF-8, both in the beginning of the ZIF-8 self-assembly around PS I and after formation of the ZIF-8 crystal. We discuss the structural impact of the encapsulation via MD simulations and the coordination of ZIF-8 building blocks to the chlorophyll network. High-level quantum-mechanical calculations give insights into the electronic structure at the PS I/ZIF-8 interface and reveal possibly undesirable electron transfer from the MOF into the photosystem. Our results provide a potential explanation for the previously observed spectral anomalies upon encapsulation29 and give general theoretical insights on potential pitfalls in the future design of bio-nanohybrid devices.
2. Methods
2.1. Preparation and purification of PS I
The PSI purification and monomerization protocol was adapted from Baker et al.33 and Dobson et al.34 Cells from the model cyanobacterium Synechocystis sp. PCC6803 (wild type) were grown in BG11 liquid medium, supplemented with 15 mM glucose under continuous white light (50 μmol photons m−2 s−1) at 30 °C. Cells in the log phase were harvested by centrifugation at 5000g for 10 min at room temperature. The cell pellets were stored at −80 °C for further use. Frozen cell pellets were thawed and resuspended in STN1 buffer (30 mM Tricine–NaOH pH 8, 15 mM NaCl, 0.4 M sucrose) and glass beads (212 μm to 300 μm, 425 μm to 600 μm, Sigma®) were added. Cells were broken using a Tissue Lyser II bead mill (Qiagen®) for 5 cycles, each cycle consisting of 3 min at 30 Hz, followed by 5 min cooling on ice. The lysate was cleared of cell debris and glass beads by centrifuging at 5000g for 5 min at 4 °C. Thylakoid membranes were pelleted by ultracentrifugation using Beckman SW40i at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 h at 4 °C. The membranes were then resuspended and incubated in STN2 buffer (30 mM Tricine–NaOH pH 8, 150 mM NaCl, 0.4 M sucrose) on ice for 30 min. The ultracentrifugation was repeated to pellet thylakoid membranes again. The membranes were resuspended in resuspension buffer (30 mM Tricine–NaOH pH 8, 15 mM NaCl) and n-dodecyl β-maltoside (DDM, ANAGRADE®) was added to achieve a mass ratio of 15:1 DDM-to-chlorophyll. The samples were gently mixed by pipetting and incubated on ice for 30 min. Insoluble materials were removed by ultracentrifugation at 40000 rpm for 1 h at 4 °C, and the supernatant was collected and applied to an ion-exchange column (Toyopearl DEAE-650M, 5 mL, TOSOH BIOSCIENCE®) on Äkta. Proteins were eluted with a linear NaCl gradient from 15 m to 350 m in a buffer of 30 mM Tricine–NaOH (pH 8) and 0.2% DDM. The dark green fractions were collected and loaded on 10% to 30% sucrose gradient, followed by centrifugation at 36000 rpm for 16 h at 4 °C using a Beckman SW40i rotor. The lower green bands, corresponding to the PS I trimer, were collected and 8% PEG3350 was added to precipitate the protein. Precipitated protein was resuspended in resuspension buffer for further use.
000 rpm for 1 h at 4 °C. The membranes were then resuspended and incubated in STN2 buffer (30 mM Tricine–NaOH pH 8, 150 mM NaCl, 0.4 M sucrose) on ice for 30 min. The ultracentrifugation was repeated to pellet thylakoid membranes again. The membranes were resuspended in resuspension buffer (30 mM Tricine–NaOH pH 8, 15 mM NaCl) and n-dodecyl β-maltoside (DDM, ANAGRADE®) was added to achieve a mass ratio of 15:1 DDM-to-chlorophyll. The samples were gently mixed by pipetting and incubated on ice for 30 min. Insoluble materials were removed by ultracentrifugation at 40000 rpm for 1 h at 4 °C, and the supernatant was collected and applied to an ion-exchange column (Toyopearl DEAE-650M, 5 mL, TOSOH BIOSCIENCE®) on Äkta. Proteins were eluted with a linear NaCl gradient from 15 m to 350 m in a buffer of 30 mM Tricine–NaOH (pH 8) and 0.2% DDM. The dark green fractions were collected and loaded on 10% to 30% sucrose gradient, followed by centrifugation at 36000 rpm for 16 h at 4 °C using a Beckman SW40i rotor. The lower green bands, corresponding to the PS I trimer, were collected and 8% PEG3350 was added to precipitate the protein. Precipitated protein was resuspended in resuspension buffer for further use.
For monomerization, the PS I trimer was diluted to 1 mg mL−1 chlorophyll concentration and 0.375% detergent octylthioglucoside (OTG) was added. The mixed samples were incubated at 55 °C for 5 min and cooled down on ice for 2 min. This cycle was repeated 18 times. The treated sample was loaded on 10% to 30% glucose gradient, followed by centrifugation at 36000 rpm for 16 h at 4 °C, and the upper green bands, corresponding to the PS I monomer, were collected and precipitated by 12% PEG3350. Proteins were then resuspended in resuspension buffer for further use.
MOF-encapsulated PS I complexes were formed in one cycle by precipitating the ZIF-8 matrix from PS I-containing solution, in line with earlier published procedures.29 The resulting dispersions were decanted without centrifugation and the powders were washed with water, before redispersion in aqueous phosphate buffer.
2.2. Optical spectroscopy
The UV/Vis spectra were measured on a Cary 60 UV/Vis spectrometer from Agilent Technologies. All spectra were measured at room temperature in the respective solvent. Measurements were taken in the range 200–800 nm. The utilized cuvette was made out of SUPRASIL® quartz glass with a layer thickness of 10 mm.The diffuse-reflectance UV/Vis spectra were measured on a UV-3600 Plus with integrating sphere unit of the company Shimadzu. The reflection of the sample was measured in the range 200–800 nm. For the recording of the UV/Vis spectra, the samples were put between two quartz microscope slides. As a reference barium sulphate was used.
2.3. Computational model of PS I
The molecular model and force-field parameters for PS I were adapted from a previous publication and only a brief overview is given here. For details, please refer to the original publication.31,35The structural model35 is based on the crystal structure of cyanobacterial PSI21 in T. elongatus (PDB: 1JB0). In contrast to our previous study, a PS I monomer was used to reduce computational cost. The protein was described with Amber14sb36,37 and all other parameters were carefully selected to be compatible with the Amber protocol, which has been shown to yield reasonable structures for photosynthetic complexes.38,39 In this context, parameters for chlorophyll a and β-carotene were taken from the literature.40,41 Iron–sulfur clusters and the coordinating cysteine (CYS) residues were described with parameters for oxidized, proximal Fe/S clusters.42 The lipids 2,3 dipalmitoyl-D-glycero-1-phosphatidylglycerol (LHG) and 1,2-distearoyl-monogalactosyl-diglyceride (LMG), which occur naturally as cofactors within PS I, were described with the LIPID17 force field.43–45 Parameters for the head group of LMG were generated with antechamber46 using the GAFF force field47 and RESP charges derived according to the standard Amber protocol for lipids.43
For the MD simulations of pure PS I in water, the model was placed in a triclinic box with dimensions 25 × 25 × 20 nm and solvated by 394405 water molecules. Charge neutralization was achieved by adding 15 Na+ ions. The final system contained 1232600 atoms.
2.4. Molecular models of ZIF-8 and PS I@ZIF-8
ZIF-8 was modeled with the non-bonded force-field nb-ZIF-FF,48 which we adapted for use with Gromacs. In contrast to many other MOF force-fields, nb-ZIF-FF models the interactions between the ionic MOF building blocks by a purely non-bonded potential, allowing the breaking and formation of bonds over the course of an MD simulation. To retain information about the coordination symmetry, each Zn2+ ion and each coordinating N atom is surrounded by a set of charged dummy atoms.48–50 In the original formulation,48 a Morse potential was used to model the interaction between the building blocks. However, as Gromacs does not natively support Morse potentials, a custom Lennard-Jones potential was fitted to the original potential and used instead in this work (see ESI† for details).Two MD simulations including ZIF-8 were conducted in the present work: First, the self-assembly of the MOF around PS I was investigated up until the amorphous stage. Here, the PS I monomer was placed in a triclinic box with an edge length of 23 × 23 × 18 nm and solvated with water. Subsequently, 4272 Zn2+ ions and 8544 MIm− ions were randomly placed in the box by replacing water molecules. The number of molecules was chosen to model the experimentally used MIm− concentration of 1.49 mol L−1. Differing from the experimental conditions, a stoichiometric amount of Zn2+ was used in the calculations to achieve charge neutralization. The remaining negative charge, due to anionic residues in the PS I backbone, was neutralized by adding 15 Na+ ions. The final system contained 890474 atoms.
As the actual crystallization process of ZIF-8 takes place on a timescale of several minutes and is out of reach even for metadynamics simulations,48 a second MD was performed with PS I embedded in a fully-formed ZIF-8 crystal. Here, a unit cell was constructed based on the experimentally determined crystal structure51 (CCDB: 864310). Water and hydrogen atoms were removed and dummy atoms required by the force-field48 were added by superimposing models of Zn2+ and MIm− onto the crystal structure. Next, missing hydrogen atoms were added to the building blocks with the Gromacs function pdb2gmx. Redundant building blocks were removed in order to apply periodic boundary conditions. The resulting unit cell was replicated 12 times in x- and y-directions and 9 times in z-direction. Subsequently, monomeric PS I was embedded in the crystal center by deleting any ZIF-8 atoms in a distance of 5 Å around PS I. The composite was solvated with water and placed in a triclinic box with dimensions 23 × 23 × 18 nm. The charge imbalance caused by the creation of the crystal cavity was neutralized by randomly replacing solvent molecules outside the ZIF-8 crystal with 128 free Zn2+ ions. The remaining negative charge from PS I was neutralized by adding 15 Na+ ions. The final system contained 1014077 atoms.
2.5. Classical molecular dynamics
Molecular dynamics simulations were performed with Gromacs.52 A double-precision installation of Gromacs 2022.2 was used for the simulation of encapsulated PS I, while the simulations of amorphous ZIF-8 and PS I in water were performed with a single-precision version of Gromacs 2023.2 with GPU support.Multiple MD simulations were conducted, which differ in the details but adhere to the same general protocol. The total energy of the system was minimized with the steepest descent algorithm until the maximum force fell below 1000 kJ nm−1.
In all following simulations, the leap-frog integrator was used with a time step of 2 fs and bonds to hydrogen atoms were constrained with the LINCS algorithm.53 Short-range electrostatics were evaluated with Verlet lists54 using a cutoff distance of 1.2 nm. The smooth Particle-mesh Ewald (SPME) scheme55,56 was used to calculate long-range electrostatics, using fourth-order interpolation and a Fouier grid spacing of 0.16 nm.
The system was equilibrated in three phases: first, to relax the system further, it was annealed from 10 K to 100 K over 50 ps in an NVT ensemble, before propagating for another 50 ps at a constant temperature of 100 K. Temperature control was achieved with the V-rescale thermostat,57 using a time constant τT of 0.1 ps. In the second step, the ensemble was switched to NPT, controlled by the V-rescale thermostat57 (τT = 0.1 ps) and the Berendsen barostat58 (τp = 2.5 ps). Here, the system was heated from 100 K to the target temperature of 300 K within 100 ps and propagated at 300 K for another 900 ps at constant temperature and pressure. The pressure was equilibrated to an isotropic reference pressure of 1 bar. An isothermal compressibility of 4.5 × 10−5 bar−1 was used for simulations in water. In the final equilibration step, the system was propagated for 10 ns in an NPT ensemble at 300 K and 1 bar, controlled by the Nosé–Hoover thermostat59,60 (τT = 2.5 ps) and the Parrinello–Rahman barostat61,62 (τp = 10.0 ps).
Production simulations for PS I in water and PS I@ZIF-8 were carried out for 100 ns and 30 ns, respectively, in the fully equilibrated ensemble. The 100 ns production simulation of the ZIF-8 self-assembly around PS I was carried out already after the second equilibration step, to be able to follow the aggregation of the ZIF-8 nanoparticles. The MD trajectories were analyzed with the python library MDAnalysis 2.7.0.63,64 Molecular visualizations were created with VMD 1.9.3.65
2.6. Quantum chemical calculations
Geometry optimizations in the ground state were performed with Orca 5.0.3 using the r2SCAN-3c composite method, which has been shown to provide reasonable structures for a wide variety of organic molecules at much lower cost than hybrid density functionals.66 Optimized structures were verified as minima by the absence of imaginary vibrational frequencies. Excited state geometry optimizations were performed at the TD-CAM-B3LYP level67 with the def2-TZVP basis set.68 The RIJCOSX approximation69–71 was used to speed up the calculations, in conjunction with the def2/J Coulomb fitting basis.72 The range-separated hybrid functional CAM-B3LYP was chosen to deal with possible charge-transfer interactions. It has proven to provide qualitatively correct spectra and structures for chlorophylls,73–75 even though recent benchmarks indicate that final excitation energies should be evaluated at higher levels of theory.76,77Therefore, excited states were calculated in the Tamm–Dancoff approximation78 using the range-separated double-hybrid functional SCS-ωPBEPP8679 in combination with the def2-TZVP basis. Again, the RIJCOSX approximation69–71 was used with the def2/J72 and def2-TZVP/C80 auxiliary basis sets. Eight roots were calculated. The excited state wavefunctions were analyzed with the TheoDORE 2.4.0 program package81–83 to identify charge-transfer (CT) states.
The TD-DFT results were compared against DFT/MRCI calculations,84–86 which include multireference effects. Here, the DFT reference was calculated with Orca 5.0.3 using the BHLYP87,88 functional and the def2-SVP68 basis set. Coulomb and exchange integral evaluation was accelerated with the RI-JK approximation69 using the def2/J72 and def2-SVP/C80 bases. SCF convergence was tightened to 10−7Eh (Orca keyword SCFCONV7). Corresponding to the Gouterman model,89 a CISD expansion of four electrons in the four frontier orbitals was used as an initial guess for the MRCI reference space. This reference space was iteratively optimized until it contained all leading configurations of the first 8 roots, using the R2018 Hamiltonian90 with a selection threshold of 0.8Eh and the tight parameter set.
QM/MM excited state calculations using the DFT/MRCI method in the QM region were performed for eight Chl a chromophores in the PS I@ZIF-8 composite, which were mostly affected by the coordination with MIm−. DFT/MRCI has been used before in the context of QM/MM calculations and has repeatedly performed well in reproducing experimental reference energies.31,91–93 For every chlorophyll molecule, 20 evenly spaced snapshots were sampled from the crystal MD simulation. The QM region contained the chlorophyll and any MIm− units within 4 Å of the central Mg2+ ion. The phytyl chain was capped at the first carbon by a hydrogen link atom.94,95 Electrostatic embedding was used to describe the polarization of the QM wave function by the MM environment. Point charges were taken directly from the force field, including the dummy charges on the ZIF-8 building blocks. To prevent excessive polarization, point charges were shifted away from the link atom, and artificial charges were introduced along the bond axis to maintain the dipole moment.95 In each QM/MM calculation, the QM subsystem was centered in the box by applying periodic boundary conditions.
2.7. Statistical analysis
The sampled data points were divided into three groups, based on the number of MIm− ions within a 4 Å sphere around the chlorophyll's Mg2+ ion in the respective MD snapshot. A one-way ANOVA analysis96 was conducted on the CT numbers and vertical excitation energies into S1, followed by Tukey's HSD test97 to evaluate the statistical significance of the differences between the three groups. Reaction center chlorophylls were excluded from the analysis as they remained unaffected by the ZIF-8 encapsulation.3. Results and discussion
Embedding an intricate biomolecular system like PS I in a MOF can affect its properties in two ways. First, the encapsulation process may alter the structure of the photosystem, either unfolding the protein itself or releasing previously bound chlorophyll molecules into the reaction mixture. Second, electronic interactions between the charged MOF building blocks and the PS I chromophores may alter the spectral properties of the latter. Both types of effects, structural and electronic, will be discussed in detail in the following.3.1. Absorption spectroscopy on PS I in diverse environments
In order to provide benchmarks and guidelines for our calculations, we measure optical spectra under experimental conditions covering the diverse range of PS I local environments. In particular, we rely on UV/Vis absorption and diffuse reflectance spectroscopies to monitor the integrity of the PS I during the stages of MOF encapsulation.In the top panel of Fig. 1 we show the UV-Vis absorption spectra of PS I in buffer solution and in the presence of the MOF matrix building block MImH (1.5 mM, pH 9). The direct comparison with the spectra of an aqueous phosphate buffer solution of PS I reveals that the presence of MImH has no significant effect on the optical properties of the photosynthetic complex. This suggests that electronic interactions between the MOF linker and PS I are negligible in solution.
 | ||
| Fig. 1 Top panel: absorption spectra of PS I in phosphate–(gray, 0.05 M, pH 7.4) and acetate buffer (blue, 0.5 M, pH 5.3). PS I absorption spectrum in phosphate buffer in the presence of 2-methylimidazole (1.5 mM MImH, pH 9) acting as a building block of the ZIF-8 framework shown in red. Bottom panel: comparison of PS I absorption spectra in phosphate buffer (gray), after MOF encapsulation (blue), and after release by digestion of the MOF (red). The scattering spectrum of a pure ZIF-8 MOF suspension shown as dashed gray line. | ||
While simply the presence of the MOF linker MImH clearly has negligible impact on the optical properties of PS I, interaction between the linker and PS I appears more substantial after formation of the MOF framework, as witnessed by the UV/Vis absorption spectra in the bottom panel of Fig. 1. While the overall chlorophyll a-like spectral shape of PS I is similar before and after encapsulation, we observe peak broadening and slight shifts in the transition energies in both Q- and B-band regions. These effects are often observed on embedding chromophores in a more strongly interacting and heterogeneous environment, in agreement the expected behavior on changing the local PS I environment from aqueous buffer to the much more highly structured MOF framework.
Importantly, after digestion of the MOF by acidification of the dispersion, PS I is released back into solution. The corresponding spectrum, shown as a red line in the bottom panel of Fig. 1, shows complete recovery of the initial in-solution PS I absorption spectrum, which provides strong evidence that the complex remains structurally intact and presumably functional throughout the entire encapsulation-release cycle. Thus, our experimental results corroborate previous accounts29 and provide a starting point for theoretical investigations.
3.2. Structural integrity of PS I in ZIF-8
The structural integrity of PS I was investigated via MD calculations both during the crystallization process of ZIF-8 around PS I and after the crystal had fully formed. As the crystalliation of ZIF-8 takes place on a timescale of several minutes,29,48 only the early stages of the self-assembly process, up until an amorphous phase, are accessible with MD simulations.48 Therefore, we simulated the first 100 ns of self-assembly around PS I, starting from a homogeneous, stoichiometric mixture of Zn2+ and MIm− ions, solvated in water. Early on, already after 1 ns, clusters of the MOF building blocks start to form and also attach to PS I. As time progresses, the clusters grow and form an amorphous mass, which is loosely bound to PS I (Fig. 2). Visual analysis of the MD trajectories (Fig. S9, ESI†) confirms that both Zn2+ and MIm− permeate up to 1 nm into the protein matrix, especially in the peripheral antennas. However, these interactions induce no apparent changes in the secondary or tertiary structure of PS I. Most importantly, all 96 chlorophylls remain bound to the photosystem and are not being released into the solvent. | ||
| Fig. 2 Visualization of the structural integrity of PS I in the early stages of the ZIF-8 self-assembly process. (a) Protein and chlorophyll network in 50 equidistant snapshots over 100 ns, colored by the time-averaged RMSD of each residue with respect to the first frame of the trajectory after alignment of the protein backbone. Higher RMSD indicates more mobile residues. (b) Selected snapshots of the MD trajectory, showing the clustering of the ZIF-8 building blocks. Water molecules are omitted for clarity. View from the stromal side, left panel in (a) rotated by 90° around x. | ||
The visual impression from Fig. 2 can be quantified by calculating the RMSD of relevant PS I components with respect to an experimental crystal structure21 over the course of the self-assembly process (Fig. 3a). The time-dependent, mass-weighted RMSD is generally defined as
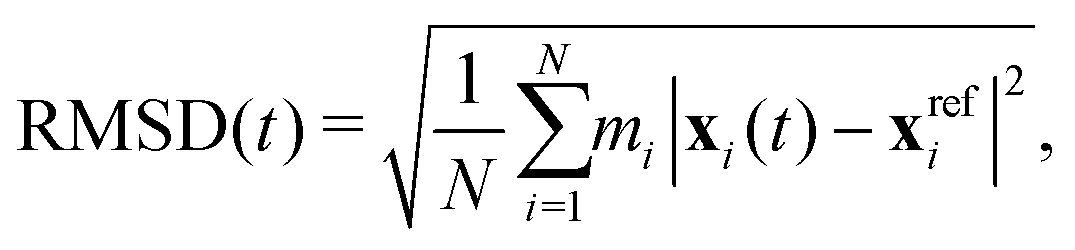 | (1) |
 | ||
| Fig. 3 RMSD of key PS I components (a) in the early stage of cystallization and (b) in the fully formed ZIF-8 crystal. Cα: protein backbone, CLA: chlorophyll a, BCR: β-carotine, PQN: phylloquinone. (c) Protein radius of gyration during and after crystallization. The RMSD in the reference trajectory of PS I in water is depicted in Fig. S5 in the ESI.† | ||
As a measure of the protein stability, the radius of gyration Rg was calculated. Rg measures the compactness of the protein and gives a quantitative estimate on whether the protein unfolds over time. It is calculated in each time step t as the mass-weighted mean distance from the center of mass xCOM:
 | (2) |
Nevertheless, the amorphous phase may interact differently with PS I than a crystal. Therefore, a second MD simulation was performed with PS I encapsulated in a fully formed ZIF-8 crystal (Fig. 4). Due to the molecular setup, where the crystal cavity was perfectly shaped to accommodate the photosystem, PS I is tightly bound and its rotational and translational degrees of freedom are heavily restricted by the ZIF-8 crystal. Apart from the cavity, the periodicity of the crystal is perfectly preserved during the MD, as evident from the Zn2+–Zn2+ radial distribution function (Fig. S4, ESI†). In a real crystal, the packing at the ZIF-8/PS I interface is possibly not as tight and may contain more imperfections. While this would increase the flexibility of the photosystem, it is unlikely to change the fundamental interactions at the interface. As for the amorphous phase, the encapsulation in the crystal induces no structural change observable in the RMSD or in the protein radius of gyration (Fig. 3b and c). Thus, the photosystem is structurally stable both during and after encapsulation in ZIF-8. Although this is an encouraging result in the context of bio-nanohybrid applications, it does not explain the observed spectroscopic changes upon encapsulation.29
 | ||
| Fig. 4 PS I embedded in the ZIF-8 crystal and solvated by water. Chlorophylls are highlighted in red. Top view from the stromal side, box dimensions after equilibration. | ||
Therefore, we now focus more closely on the interactions between the ZIF-8 building blocks and the chlorophylls at the ZIF-8/PS I boundary. In particular, the anionic MIm− building blocks are able to coordinate axially to the chlorophyll's Mg2+ ions if the chlorophylls are exposed to the outside of the photosystem. To quantify the extent of such coordination, we analyzed the coordination of each of the 96 chlorophylls in PS I over the time of the crystal MD trajectory (Fig. S6, ESI†). Most chlorophylls do not interact with the ZIF-8 crystal, because they are deeply embedded in the protein framework. Few chlorophylls show weak interactions, where atoms belonging to MIm− diffuse in and out of the pre-defined coordination sphere with radius 4.0 Å around the Mg2+ ion. In total, there are 32 chlorophylls which experience at least one coordination event over the course of the 30 ns MD trajectory in the ZIF-8 crystal. The eight most affected chlorophylls, coordinated for at least 40% of the total simulation time, are all located on the outskirts of the photosystem and thus not well-shielded by the protein against the environment (Fig. S12, ESI†). Looking at the early stages of the self-assembly, even more chlorophylls interact with MIm−, due to the higher mobility of the ZIF-8 building blocks when they are not bound in a crystal. Here, 40 chlorophylls experience at least one coordination event over 100 ns, with 19 of them remaining coordinated for at least 40% of the trajectory (Fig. S6, ESI†). As the MD trajectory only samples a fraction of the phase space, it is conceivable that in reality, all chlorophylls located on the outer border of PS I experience significant coordination at one time or another in the self-assembly process. Such coordination can alter the spectroscopic properties of the photosystem via electronic interactions between ZIF-8 and the chlorophylls. These interactions at the PS I/ZIF-8 interface will therefore be investigated more closely in the following.
3.3. Electronic interactions at the PS I/ZIF-8 interface
Quantum chemical calculations were performed to investigate the electronic effects of coordination by MIm− on the chlorophylls at the PS I/ZIF-8 interface. In a first step, the structure of an isolated chlorophyll was optimized at the r2SCAN-3c level of theory.66 Additionally, the structure of a chlorophyll axially coordinated by MIm− was optimized at the same level. Excited states were calculated using both the range separated double-hybrid functional SCS-ωPBEPP8679 and the DFT/MRCI method.86 Both methods have shown to yield excellent absorption properties for chlorophylls, which exhibit non-negligible multireference character, even in low-energy excited states.31,32,74 SCS-ωPBEPP86 performs especially well for charge transfer excitations99 and was thus used to double-check the DFT/MRCI results. For simplicity, the rest of the environment was not considered at this stage; its effect will be discussed later.At the DFT/MRCI level, the first excited state of the Chl a⋯MIm− aggregate has non-negligible CT character at the Franck–Condon point, transferring electron density from the anionic ligand to the chlorophyll (Fig. 5a). At the SCS-ωPBEPP86 level, this CT is slightly higher in energy and predicted to be the third excited state. In both theoretical frameworks, the main character of the first excited state remains unchanged with respect to the isolated chlorophyll and the transition dipole moment stays oriented along the molecular y-axis. However, upon excited state relaxation, modeled by optimizing the geometry of the first excited state, the CT character begins to dominate and the transition dipole moment reorients until it is orthogonal to the chlorophyll's molecular plane (Fig. 5b and c). Additionally, the new S1 at the excited state minimum is significantly lower in energy and almost dark, with a vertical emission energy of 0.65 eV and an oscillator strength of 7 × 10−5. It is noteworthy that the CT occurs only if the ligand is in its fully deprotonated, anionic form. In a buffer solution, as used in the spectroscopy experiments in Fig. 1, 2-methylimidazole exists as a mixture of MImH and MImH2+, which do not induce a CT (Tables S10–S14, ESI†), even though they also tend to coordinate (Fig. S7, ESI†).
 | ||
| Fig. 5 Difference density (DFT/MRCI) for Chl. a⋯MIm− at (a) the Franck–Condon point and (b) at the S1 minimum. Electron density is transferred from blue to yellow regions (isovalue 0.002), indicating a CT from MIm− to the chlorophyll. (c) The transition dipole moment (DFT/MRCI) for uncoordinated Chl. a (black), is similar to that of Chl. a⋯MIm− at the Franck–Condon point (orange) but rotates out of the molecular plane upon relaxation to the S1 minimum (short orange arrow, scaled up by factor 15 for visualization). | ||
Starting from the initial CT, two pathways are conceivable: either the chlorophyll relaxes back to the original ground state, regenerating a neutral chlorophyll with an anionic MIm− ligand, or the CT creates a chlorophyll anion. The latter case would require the two molecular units to separate for the CT state to become the new ground state. This is unlikely, given the tight encapsulation by the crystal. Nevertheless, we investigated the spectral impact of chlorophyll anion formation by calculating excited states at the optimized geometry of a Chl a− anion with the same protocol as before. The first excited state of Chl a− is a weakly absorbing state with an oscillator strength of 0.0735 (SCS-ωPBEPP86). Its vertical excitation energy is 1.26 eV, significantly lower than that of a neutral Chl a (1.87 eV). This means that if an anion is formed, it will not be excitonically coupled with adjacent neutral chlorophylls due to the large energy gap. Instead, a chlorophyll anion should give rise to an additional fluorescence band in the NIR spectral range. In practice, the fluorescence signal of PS I@ZIF-8 only shows the characteristic sharp Qy peak of isolated chlorophylls at 661 nm.29 However, the measurement of NIR signals in the presence of ZIF-8 is challenging, due to the strong scattering by the nanoparticles. We therefore can not rule out formation of chlorophyll anions, although it appears unlikely.
To investigate the frequency of the discussed CT events, we performed a series of QM/MM excited state calculations at the DFT/MRCI level on the eight most-coordinated chlorophylls in the PS I@ZIF-8 crystal and on the reaction center chlorophylls labeled eC-A1 and eC-B1. Geometries were sampled from 20 evenly spaced MD snapshots. Out of 200 calculations, 195 terminated normally and were used for further analysis. The vertical excitation energies into the first excited state are summarized in Fig. 6, where each data point represents the respective chlorophyll in one snapshot. Additionally, the CT character of each state was quantified via analysis of the transition density.81–83 The amount of CT is quantified by the CT number, which ranges between 0 (no CT) and 1 (single electron transfer).
 | ||
| Fig. 6 Distribution of vertical excitation energies (DFT/MRCI) into the S1 state of selected, frequently MIm−-coordinated chlorophylls in PS I. Data points are colored by the amount of CT character of the excitation. The shape of the data points signifies the number of coordinating MIm− ions within 4 Å of the respective Mg2+ ion. The horizontal gray line illustrates the experimental excitation energy into Qy in Chl a. | ||
It is immediately apparent from Fig. 6 that the reaction center chlorophylls remain unaffected by encapsulation in ZIF-8, corroborating that PS I retains its function in the bio-nanohybrid.29 In contrast, the chlorophylls interacting directly with the MOF exhibit very different photophysics. The sampled data contains 104 coordination events, corresponding to 67% of the total number of data points outside the reaction center. Out of these, there are 11 cases, where two MIm− ions are closer than 4 Å to the chlorophyll's Mg2+ ion, which will be referred to as double coordination in the following. 92 of the 104 coordination events lead to a CT number >0.5. A one-way ANOVA analysis was carried out to compare the effects of MIm− coordination on the CT numbers and vertical excitation energies into S1 between the uncoordinated, singly coordinated and doubly coordinated chlorophylls. There is a significant effect of the coordination on the CT number for the three groups of samples [F(2192) = 568.617, p<0.001]. Post-hoc analysis using Tukey's HSD test reveals that coordination significantly (p < 0.001) raises the mean CT number of the singly and doubly coordinated samples by 0.800 and 0.893 with respect to the uncoordinated samples, whose CT number is 0 by definition. However, the difference between single and double coordination is not significant. Moreover, coordination by MIm− has a significant effect on the vertical excitation energies into S1 [F(2192) = 71.962, p < 0.001]. Single coordination lowers the mean energy for excitation into S1 by 0.46 eV, double coordination by 0.70 eV. Both energy differences are statistically significant (p < 0.001). We note that the interplay between force field and QM method can affect these results, as both structures and point charges are taken directly from the classical MD simulations.100 The strength of the non-bonded interactions between MIm− and the Mg2+ ions is of particular importance in this regard, as it controls the distance between the two molecules. The average N–Mg distance in the snapshots sampled in Fig. 6 is 4.21 Å, slightly longer than in the optimized geometry of the Chl a⋯MIm− aggregate (3.75 Å). Based on this, the QM/MM sampling likely rather underestimates the amount of CT events. We therefore conclude that coordination by MIm− significantly decreases the vertical excitation energy into S1 and induces a CT towards the chlorophyll.
3.4. Effects on the light-harvesting network
The results presented so far allow to draw conclusions about possible deactivation pathways following photo-excitation of the chlorophylls in PS I@ZIF-8. After population of the Qy state of one of the chlorophylls, the exciton can delocalize via Coulomb interactions with another chlorophyll, lowering the energy of the collective excited state. Without the MOF, this delocalized exciton would eventually fluoresce back to the ground state, resulting in the characteristic exciton emission band in PS I. In the presence of MIm− however, a CT state localized on one of the chlorophylls is energetically below the excitonic state. The large energy difference to the S1 state of uncoordinated chlorophylls as well as the vanishing and reoriented transition dipole moment of this CT state effectively decouples the affected chlorophyll from the rest of the excitonic network. To assess the impact of these changes in the light-harvesting antenna, we simulated the exciton distribution once including all chlorophylls and once without the eight most frequently coordinated chlorophylls (Fig. S12, ESI†).Compared to the unperturbed light-harvesting system in PS I,31 the energy distribution changes only minutely, as the coordinated chlorophylls are only weakly coupled to begin with. No changes are observed in the reaction center and in the red chlorophylls,31 corroborating that the primary function of PS I remains unimpaired. However, a small part of previously delocalized excitons now localize more strongly on single pigments. For example, one exciton that was formerly delocalized across 11 pigments with a larger contribution by chlorophyll A8 (46%) is now localized by 88% on A8. Given that excitons generally quench fluorescence and chlorophyll on its own is a strong fluorophor, this MOF-induced excited state localization could contribute to the strong emission peak observed29 in the PS I@MOF bio-nanohybrid.
4. Conclusions
In this work, we have investigated the structural and electronic impact of encapsulating cyanobacterial PS I in the MOF ZIF-8. Such encapsulation is desirable in the context of bio-nanohybrid applications, where the near-unity efficiency of PS I can be harnessed to catalyze chemical reactions10,13,14,17,25 while the MOF protects the photosystem from harsh environments.MD simulations show how PS I remains structurally intact upon encapsulation in ZIF-8, in both the early and late stages of the crystallization process. Neither does the protein unfold, nor is there any significant structural change in the chlorophyll network. Any observed spectral anomalies29 must therefore stem from electronic interactions at the PS I/ZIF-8 interface. In this context, analysis of the MD trajectories reveals how the ZIF-8 building blocks coordinate to the Mg2+ ion of peripheral chlorophylls in PS I. Such coordination enables CT excitations at the boundary, where the MOF can effectively photoreduce some of the chlorophylls. High-level QM calculations show that such photoinitiated CT lowers the energy of the first excited state, reorients the transition dipole moment and reduces its magnitude. QM/MM sampling along the MD trajectory show that these perturbations occur frequently at the PS I/ZIF-8 interface. As such, they decrease the excitonic coupling between the chlorophylls in PS I, which normally quenches the fluorescence signal. The resulting uncoupled chlorophylls provide a possible explanation for the reversible strong fluorescence signal in the spectrum of PS I@ZIF-8, compared to pure PS I.29 Nonetheless, the reaction center and thus the primary function of the photosystem remains unaffected by the encapsulation in the MOF, encouraging future breakthroughs on this road to artificial photosynthesis.
Author contributions
Sebastian Reiter: conceptualization, methodology, software, validation, formal analysis, investigation, data curation, visualization, writing – original draft, writing – review & editing; Igor Gordiy: methodology, software, validation, formal analysis, investigation, visualization, writing – review & editing; Kathrin L. Kollmannsberger: investigation, formal analysis, writing – review & editing; Feng Liu: investigation, writing – original draft, writing – review & editing; Erling Thyrhaug: investigation, formal analysis, visualization, writing – original draft, writing – review & editing; Dario Leister: resources, supervision, funding aquisition, writing – review & editing; Julien Warnan: resources, supervision, funding aquisition, writing – review & editing; Jürgen Hauer: conceptualization, resources, supervision, funding aquisition, writing – review & editing; Regina de Vivie-Riedle: conceptualization, resources, supervision, funding aquisition, writing – review & editing, project administration.Data availability
The data supporting this article have been included as part of the ESI.† Parameter files for the Gromacs port of nb-ZIF-FF can be found at https://doi.org/10.5281/zenodo.12546403. Structure files and raw data used in this article are available at https://doi.org/10.5281/zenodo.13120983.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the cluster of excellence e-conversion under Germany's Excellence Strategy – EXC 2089/1 – 390776260.Notes and references
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- J. Wang, G. Ni, W. Liao, K. Liu, J. Chen, F. Liu, Z. Zhang, M. Jia, J. Li, J. Fu, E. Pensa, L. Jiang, Z. Bian, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202217026 CrossRef CAS PubMed.
- S. Singla, S. Sharma, S. Basu, N. P. Shetti and T. M. Aminabhavi, Int. J. Hydrogen Energy, 2021, 46, 33696–33717 CrossRef CAS.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS PubMed.
- P. M. Stanley, J. Haimerl, N. B. Shustova, R. A. Fischer and J. Warnan, Nat. Chem., 2022, 14, 1342–1356 CrossRef CAS PubMed.
- P. M. Stanley, V. Ramm, R. A. Fischer and J. Warnan, Nat. Synth., 2024, 3, 307–318 CrossRef.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- S. M. Kaniber, F. C. Simmel, A. W. Holleitner and I. Carmeli, Nanotechnology, 2009, 20, 345701 CrossRef PubMed.
- S. M. Kaniber, M. Brandstetter, F. C. Simmel, I. Carmeli and A. W. Holleitner, J. Am. Chem. Soc., 2010, 132, 2872–2873 CrossRef CAS PubMed.
- G. LeBlanc, G. Chen, E. A. Gizzie, G. K. Jennings and D. E. Cliffel, Adv. Mater., 2012, 24, 5959–5962 CrossRef CAS PubMed.
- A. K. Manocchi, D. R. Baker, S. S. Pendley, K. Nguyen, M. M. Hurley, B. D. Bruce, J. J. Sumner and C. A. Lundgren, Langmuir, 2013, 29, 2412–2419 CrossRef CAS PubMed.
- T. Kothe, S. Pöller, F. Zhao, P. Fortgang, M. Rögner, W. Schuhmann and N. Plumeré, Chem. – Eur. J., 2014, 20, 11029–11034 CrossRef CAS PubMed.
- S. C. Feifel, H. Lokstein, M. Hejazi, A. Zouni and F. Lisdat, Langmuir, 2015, 31, 10590–10598 CrossRef CAS PubMed.
- S. C. Feifel, K. R. Stieger, H. Lokstein, H. Lux and F. Lisdat, J. Mater. Chem. A, 2015, 3, 12188–12196 RSC.
- F. Zhao, A. Ruff, M. Rögner, W. Schuhmann and F. Conzuelo, J. Am. Chem. Soc., 2019, 141, 5102–5106 CrossRef CAS PubMed.
- A. H. Teodor and B. D. Bruce, Trends Biotechnol., 2020, 38, 1329–1342 CrossRef CAS PubMed.
- M. Izzo, M. Jacquet, T. Fujiwara, E. Harputlu, R. A. Mazur, P. Wróbel, T. Góral, C. G. Unlu, K. Ocakoglu, S. Miyagishima and J. Kargul, Int. J. Mol. Sci., 2021, 22, 8396 CrossRef CAS PubMed.
- K. Nguyen and B. D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1553–1566 CrossRef CAS PubMed.
- M. Schenderlein, M. Cetin, J. Barber, A. Telfer and E. Schlodder, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1400–1408 CrossRef CAS PubMed.
- A. Nakamura, T. Suzawa, Y. Kato and T. Watanabe, Plant Cell Physiol., 2011, 52, 815–823 CrossRef CAS PubMed.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauß, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- I. Carmeli, I. Lieberman, L. Kraversky, Z. Fan, A. O. Govorov, G. Markovich and S. Richter, Nano Lett., 2010, 10, 2069–2074 CrossRef CAS PubMed.
- R. Pamu, V. P. Sandireddy, R. Kalyanaraman, B. Khomami and D. Mukherjee, J. Phys. Chem. Lett., 2018, 9, 970–977 CrossRef CAS PubMed.
- M. Szalkowski, D. Kowalska, J. D. J. Olmos, J. Kargul and S. Mackowski, Int. J. Mol. Sci., 2022, 23, 2976 CrossRef CAS PubMed.
- P. I. Gordiichuk, G.-J. A. H. Wetzelaer, D. Rimmerman, A. Gruszka, J. W. de Vries, M. Saller, D. A. Gautier, S. Catarci, D. Pesce, S. Richter, P. W. M. Blom and A. Herrmann, Adv. Mater., 2014, 26, 4863–4869 CrossRef CAS PubMed.
- K. D. Wolfe, D. Dervishogullari, J. M. Passantino, C. D. Stachurski, G. K. Jennings and D. E. Cliffel, Curr. Opin. Electrochem., 2020, 19, 27–34 CrossRef CAS.
- F. C. Herbert, S. S. Abeyrathna, N. S. Abeyrathna, Y. H. Wijesundara, O. R. Brohlin, F. Carraro, H. Amenitsch, P. Falcaro, M. A. Luzuriaga, A. Durand-Silva, S. D. Diwakara, R. A. Smaldone, G. Meloni and J. J. Gassensmith, Nat. Commun., 2021, 12, 2202 CrossRef CAS PubMed.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS PubMed.
- T. H. Bennett, M. D. Vaughn, S. Ali Davari, K. Park, D. Mukherjee and B. Khomami, Nanoscale Adv., 2019, 1, 94–104 RSC.
- N. Liguori, R. Croce, S. J. Marrink and S. Thallmair, Photosynth. Res., 2020, 144, 273–295 CrossRef CAS PubMed.
- S. Reiter, F. L. Kiss, J. Hauer and R. de Vivie-Riedle, Chem. Sci., 2023, 14, 3117–3131 RSC.
- S. Reiter, L. Bäuml, J. Hauer and R. de Vivie-Riedle, Phys. Chem. Chem. Phys., 2022, 24, 27212–27223 RSC.
- D. R. Baker, A. K. Manocchi, M. L. Lamicq, M. Li, K. Nguyen, J. J. Sumner, B. D. Bruce and C. A. Lundgren, J. Phys. Chem. B, 2014, 118, 2703–2711 CrossRef CAS PubMed.
- Z. Dobson, S. Ahad, J. Vanlandingham, H. Toporik, N. Vaughn, M. Vaughn, D. Williams, M. Reppert, P. Fromme and Y. Mazor, eLife, 2021, 10, e67518 CrossRef CAS PubMed.
- S. Reiter, F. L. Kiss, J. Hauer and R. de Vivie-Riedle, Thermal site energy fluctuations in photosystem I, 2023, Dataset (v1.1.0), Zenodo, https://doi.org/10.5281/zenodo.7610935.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- G. E. Milanovsky, V. V. Ptushenko, J. H. Golbeck, A. Y. Semenov and D. A. Cherepanov, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1472–1483 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, J. Am. Chem. Soc., 2020, 142, 18174–18190 CrossRef CAS PubMed.
- L. Zhang, D.-A. Silva, Y. Yan and X. Huang, J. Comput. Chem., 2012, 33, 1969–1980 CrossRef CAS PubMed.
- M. Ceccarelli, P. Procacci and M. Marchi, J. Comput. Chem., 2003, 24, 129–142 CrossRef CAS PubMed.
- D. M. A. Smith, Y. Xiong, T. P. Straatsma, K. M. Rosso and T. C. Squier, J. Chem. Theory Comput., 2012, 8, 2103–2114 CrossRef CAS PubMed.
- A. g A. Skjevik, B. D. Madej, R. C. Walker and K. Teigen, J. Phys. Chem. B, 2012, 116, 11124–11136 CrossRef CAS PubMed.
- C. J. Dickson, B. D. Madej, A. G. A. Skjevik, R. M. Betz, K. Teigen, I. R. Gould and R. C. Walker, J. Chem. Theory Comput., 2014, 10, 865–879 CrossRef CAS PubMed.
- Z. Wu and P. Biggin, Gromacs Port of the Amber LIPID17 Force Field, 2019, Dataset (v0.21), Zenodo, https://doi.org/10.5281/zenodo.3562014 Search PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graph. Model., 2006, 25, 247–260 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- S. R. G. Balestra and R. Semino, J. Chem. Phys., 2022, 157, 184502 CrossRef CAS PubMed.
- Y.-P. Pang, J. Mol. Model., 1999, 5, 196–202 CrossRef CAS.
- S. Jawahery, N. Rampal, S. M. Moosavi, M. Witman and B. Smit, J. Chem. Theory Comput., 2019, 15, 3666–3677 CrossRef CAS PubMed.
- W. Morris, C. J. Stevens, R. E. Taylor, C. Dybowski, O. M. Yaghi and M. A. Garcia-Garibay, J. Phys. Chem. C, 2012, 116, 13307–13312 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Páll and B. Hess, Comput. Phys. Commun., 2013, 184, 2641–2650 CrossRef.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Nosé and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, J. Domanski, D. L. Dotson, S. Buchoux, I. M. Kenney and O. Beckstein, Proc. 15th Python Sci. Conf., 2016, pp. 98–105 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Kossmann and F. Neese, Chem. Phys. Lett., 2009, 481, 240–243 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- J. R. Reimers, Z.-L. Cai, R. Kobayashi, M. Rätsep, A. Freiberg and E. Krausz, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- J. P. Götze, F. Anders, S. Petry, J. F. Witte and H. Lokstein, Chem. Phys., 2022, 559, 111517 CrossRef.
- S. Petry, J. C. Tremblay and J. P. Götze, J. Phys. Chem. B, 2023, 127, 7207–7219 CrossRef CAS PubMed.
- A. Sirohiwal, R. Berraud-Pache, F. Neese, R. Izsák and D. A. Pantazis, J. Phys. Chem. B, 2020, 124, 8761–8771 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, J. Chem. Theory Comput., 2021, 17, 1858–1873 CrossRef CAS PubMed.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- M. Casanova-Páez and L. Goerigk, J. Chem. Theory Comput., 2021, 17, 5165–5186 CrossRef PubMed.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- F. Plasser, M. Wormit and A. Dreuw, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- S. Grimme and M. Waletzke, J. Chem. Phys., 1999, 111, 5645–5655 CrossRef CAS.
- M. Kleinschmidt, C. M. Marian, M. Waletzke and S. Grimme, J. Chem. Phys., 2009, 130, 044708 CrossRef PubMed.
- C. M. Marian, A. Heil and M. Kleinschmidt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 9, e1394 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- M. Gouterman, G. H. Wagnière and L. C. Snyder, J. Mol. Spectrosc., 1963, 11, 108–127 CrossRef CAS.
- A. Heil, M. Kleinschmidt and C. M. Marian, J. Chem. Phys., 2018, 149, 164106 CrossRef PubMed.
- S. Nakagawa, O. Weingart and C. M. Marian, J. Phys. Chem. B, 2017, 121, 9583–9596 CrossRef CAS PubMed.
- S. Salzmann, M. R. Silva-Junior, W. Thiel and C. M. Marian, J. Phys. Chem. B, 2009, 113, 15610–15618 CrossRef CAS PubMed.
- M. R. Silva-Junior, M. Mansurova, W. Gärtner and W. Thiel, ChemBioChem, 2013, 14, 1648–1661 CrossRef CAS PubMed.
- D. Bakowies and W. Thiel, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- R. Fisher, Statistical Methods for Research Workers, Oliver & Boyd, 1925 Search PubMed.
- J. W. Tukey, Biometrics, 1949, 5, 99 CrossRef CAS PubMed.
- D. L. Theobald, Acta Crystallogr., Sect. A: Found. Crystallogr., 2005, 61, 478–480 CrossRef PubMed.
- D. Mester and M. Kállay, J. Chem. Theory Comput., 2022, 18, 1646–1662 CrossRef CAS PubMed.
- S. Chandrasekaran, M. Aghtar, S. Valleau, A. Aspuru-Guzik and U. Kleinekathöfer, J. Phys. Chem. B, 2015, 119, 9995–10004 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on porting the force field; further structural analyis of PS I@ZIF-8; detailed results of excited state calculations and excitons. See DOI: https://doi.org/10.1039/d4cp03021d |
| ‡ Present address: ETH Zürich, Vladimir-Prelog-Weg 1-5/10, 8093 Zürich, Switzerland. |
| This journal is © the Owner Societies 2024 |