
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoelectron spectrum and breakdown diagram of ethanolamine: conformers, excited states, and thermochemistry†
S.
Kechoindi
 ab,
S.
Ben Yaghlane
b,
M.
Mogren Al Mogren
c,
A.
Bodi
*d and
M.
Hochlaf
*a
ab,
S.
Ben Yaghlane
b,
M.
Mogren Al Mogren
c,
A.
Bodi
*d and
M.
Hochlaf
*a
aUniversité Gustave Eiffel, COSYS/IMSE, 5 Bd Descartes 77454, Champs-sur-Marne, France. E-mail: majdi.hochlaf@univ-eiffel.fr
bLaboratoire de Spectroscopie Atomique, Moléculaire et Applications – LSAMA, Université de Tunis El Manar, Tunis, Tunisia
cDepartment of Chemistry, College of Sciences, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
dLaboratory for Synchrotron Radiation and Femtochemistry, Paul Scherrer Institute, PSI, 5232, Villigen, Switzerland. E-mail: andras.boedi@psi.ch
First published on 3rd September 2024
Abstract
Advanced theoretical methodologies and photoelectron photoion coincidence spectroscopy were used to investigate the photoionization of ethanolamine in the 8–18 eV energy range. We identified the low-lying cation conformers and the excited cation electronic states after vertical excitation from the most stable neutral conformer computationally. The TPES is composed of broad, structureless bands because of unfavorable Franck–Condon factors for origin transitions upon ionization, populating the D0–D7 cationic states from the most stable neutral conformer, g′Gg′. The adiabatic ionization energy of ethanolamine is calculated at 8.940 ± 0.010 eV, and the 0 K appearance energies of aminomethylium, NH2CH2+ (+CH2OH), and methyleneammonium, NH3CH2+ (+H2CO), are determined experimentally to be 9.708 ± 0.010 eV and 9.73 ± 0.03 eV, respectively. The former is used to re-evaluate the ethanolamine enthalpy of formation in the gas and liquid phases as ΔfH⊖298K[NH2(CH2)2OH, g] = −208.2 ± 1.2 kJ mol−1 and ΔfH⊖298K[NH2(CH2)2OH, l] = −267.8 ± 1.2 kJ mol−1. This represents a substantial correction of the previous experimental determination and is supported by ab initio calculations.
I. Introduction
Ethanolamine (NH2CH2CH2OH, 2-aminoethanol, or monoethanolamine, Fig. 1) is the smallest stable organic compound combining an amino and an alcohol functional group. It is a pollutant in Earth's atmosphere, present due to its wide use in industry, such as in amine-based aqueous solutions in CO2 capture technology,1–4 as a gas stream scrubbing agent for, e.g., CO2 and H2S,5 in the synthesis of emulsifies, detergents, chemicals, and corrosion inhibitors,6 and in the pharmaceutical and cosmetics industries.7 Several studies have addressed its oxidative degradation and decomposition reactions initiated by atmospheric radicals such as Cl° or OH°.8–10 Besides, a number of theoretical and experimental studies were performed to investigate its structure as well as vibrational and rotational spectra in order to help its identification and characterization in the gas phase and in solution.11–24 These works showed that ethanolamine possesses several low-lying conformers, as shown in Fig. 1. These conformers are named following the nomenclature defined by Pople and co-workers.25 Accordingly, the conformers are named using three letters where the first one is for the NH2 position, the central one is for the rotation around the central CC bond and the third one is for the OH group position. For the rotation angles around the central CC bond, the conformers are labeled by G or G′ or T when this angle is equal to 60° or −60° or 180°, respectively. For the positions of the NH2 and OH groups, the g, g′ and t letters are used where the corresponding dihedral angles τCCNL (where L is the N lone pair) or τCCOH are taken to be 60°, −60° and 180°, respectively.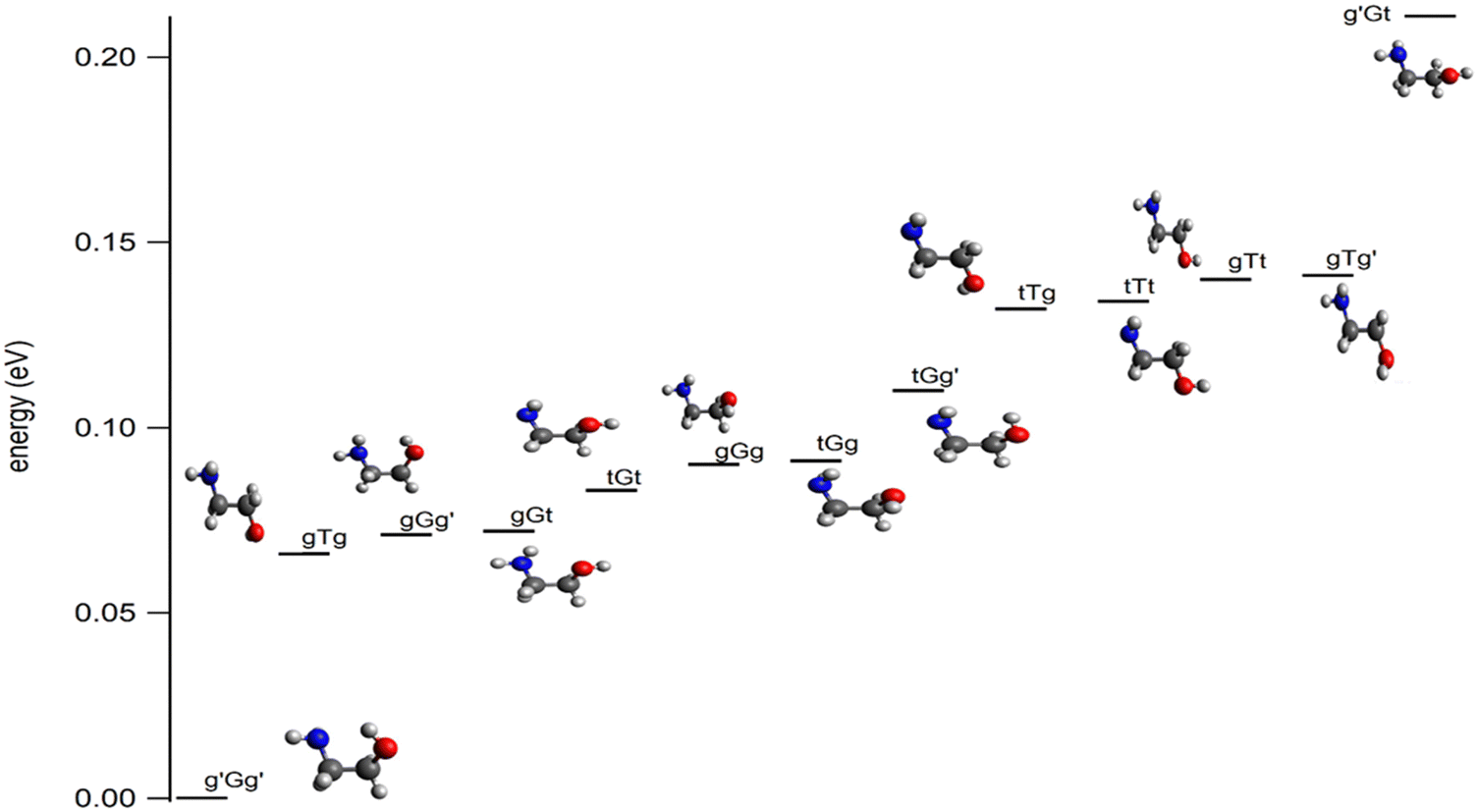 | ||
| Fig. 1 Most stable conformers of ethanolamine as computed at the CCSD(T)-F12/cc-pVDZ-F12(opt) + ΔCV + ΔSR + ΔZPVE level of theory. See Table S1 and Figure S1 and text for more details (ESI†). | ||
In particular, the intramolecular H-bond involving the amino and the alcohol groups was investigated since it is responsible for the stabilization of some conformers20–24 and affects macroscopic properties of ethanolamine, such as state change temperatures and thermodynamic functions.23,24 Various experimental and advanced ab initio methods were used, including rare gas cold matrix isolation probed by IR,14 jet-cooled Fourier transform IR vibrational spectroscopy coupled to synchrotron radiation,21 intracavity laser photoacoustic spectroscopy,20 cavity ringdown spectroscopy,20 microwave spectroscopy,11,12 and explicitly correlated coupled cluster techniques.20 It turns out that, upon molecular beam jet cooling, solely the most stable conformer, g′Gg′ (Fig. 1),21 is present. This conformer exhibits an intramolecular OH⋯NH-bond explaining its stability. Previous works also showed that the conformational isomerism is hindered at low temperatures, preventing the conversion of g′Gg′ into the less stable conformers. Such a large data set of spectroscopic parameters, in particular in the microwave domain, allowed the identification of g′Gg′ in the interstellar medium in 2021.26 This discovery is of primary importance in astrophysics and astrochemistry since ethanolamine is the simplest phospholipid head group and also a precursor of glycine,27 the simplest amino acid.
Compared with the neutral species, much less is known about the ethanolamine cation. To the best of our knowledge, data are limited to early determinations of the ionization energy of ethanolamine using photoelectron spectroscopy28–33 and (2D-)penning ionization,32,33 and to the appearance energy of the formation of CH4N+ + CH2OH fragments upon unimolecular decomposition of the ethanolamine cation by electron ionization.34,35 These early experiments had limited accuracy, and we can only estimate the adiabatic ionization energy (AIE) as ∼8.9 eV. The appearance energy (AE) of the CH4N+ fragment in the dissociative photoionization of ethanolamine has been reported as ∼9.49 eV by Lossing and co-workers,34,35 a value that will be shown to be far too low herein. The dissociative ionization energy can be used to connect the well-known enthalpy of formation of the CH2NH2+ fragment ion to the ethanolamine neutral. However, for this ion thermochemical cycle to be useful, all but one enthalpies of formation and the dissociative ionization energy all have to be known accurately, and the literature experimental values appear to suffer from large discrepancies. The NIST Chemistry Webbook36 lists the condensed phase enthalpy of the formation of ethanolamine as −507.5 kJ mol−1, as reported by Baroody and Carpenter.37 Although Baroody and Carpenter explicitly forwent error bars and intended to report propellant thermochemistry accurate enough for the energetic evaluation of potential fuels, this value is in stark contrast with the group additivity enthalpy of formation,38 ΔfH⊖298K[NH2(CH2)2OH, g] = −203.05 kJ mol−1, which, when combined with the experimentally determined ΔvapH∅[NH2(CH2)2OH] = 59.6 ± 0.3 kJ mol−1,39 gives ΔfH⊖298K[NH2(CH2)2OH, l] = −262.65 kJ mol−1. While the uncertainty in group additivity results is expected to be significant, the close to 250 kJ mol−1 difference between these two values is hard to reconcile.
In recent years, several groups established rigorous procedures for the determination of adiabatic ionization energies and of appearance energies of gas phase small and medium sized compounds.40,41 These techniques are based on synchrotron vacuum ultraviolet single photon ionization followed by the detection of close to zero kinetic energy photoelectrons in coincidence with photoions. The resulting photoion mass-selected slow and threshold photoelectron spectra can be compiled42 and interpreted using, e.g., Franck–Condon simulations in the double-harmonic approximation43 and beyond.44–46 Adiabatic ionization energies (AIE) and dissociative photoionization onsets (appearance energies, AE) can now also be accurately calculated using advanced post-Hartree–Fock methodologies. Overall, AIEs and AEs can be evaluated with an accuracy better than 0.003 eV (0.3 kJ mol−1) both experimentally and theoretically in moderately large systems in favorable cases.41,47,48 Regarding ionization energies, vanishing Franck–Condon factors for the origin transition represent the main hurdle to their experimental determination. Computational results highlighted several cases in which the adiabatic ionization energy was erroneously assumed to lie at the ionization onset.49,50 As far as appearance energies are concerned, kinetic and competitive shifts call for a statistical model with associated uncertainties.51 If the lowest-energy fragmentation channel is fast on the instrumental time scale, all parent ions that have sufficient energy to fragment will do so. Thus, the 0 K threshold to dissociative photoionization is found where all parent ions have sufficient energy to fragment, and the parent ion signal goes to zero in the breakdown diagram. For such channels, the experimental resolution is only limited by the instrumental and the photon energy resolution.52 Furthermore, if the cation fragments along a purely attractive potential, i.e., without a reverse barrier, the appearance energy is equal to the dissociative ionization reaction energy and can be used in the ion cycle to address the enthalpies of formation of the participating species. In the present study, we combine photoelectron photoion coincidence spectroscopy and ab initio computations at the explicitly correlated levels to determine the AIE of ethanolamine and the AE of CH4N+. For these purposes, we map the ground potential energy surface and explore the lowest-lying fragmentation channels of the ethanolamine cation to identify its most stable forms and reveal the dissociative ionization mechanism. Because of large-amplitude motions and the large geometry change upon ionization, harmonic Franck–Condon simulations do not describe the nuclear wave function overlap suitably. In the absence of vibrational fine structure, the ionization onset in the experimental threshold photoelectron spectrum only represents an upper limit to the adiabatic ionization energy and is discussed in the context of the neutral conformational space and the cation isomers. Excited electronic cation states have been computed and compared with the experimental spectrum. The ethanolamine cation is weakly bound and starts to fragment already in the energy range of the ground-state band of the threshold photoelectron spectrum (TPES). It forms fragment ions at m/z 30 and 31, corresponding to CH4N+ and CH5N+, respectively. Based on the breakdown diagram, an accurate appearance energy can be determined for the former, which, when used in a thermochemical cycle, yields an updated and accurate enthalpy of the formation of gaseous ethanolamine. With the help of the well-known vaporization enthalpy,39 this also allows us to update the enthalpy of formation of liquid ethanolamine.
II. Methods
a. Experimental
Photoelectron photoion coincidence experiments were carried out at the VUV beamline of the Swiss Light Source.53,54 The headspace of a vial filled with degassed room-temperature ethanolamine (99%, Chemie Brunschwig) was sampled through a needle valve, so that the pressure in the ionization chamber of the CRF-PEPICO endstation55 was ca. 1.5 × 10−6 mbar. Synchrotron radiation was generated using a bending magnet, collimated, dispersed by a 600 lines per mm laminar grating, and focused into a differentially pumped rare gas filter to suppress higher harmonic radiation of the grating. The 10 cm long absorption chamber of the gas filter was filled with 8.5 mbar of a mixture of Ar:Kr:Ne or pure Ne to provide purely monochromatic radiation in the 7–14 eV or 11–21 eV photon energy range, respectively. The photon energy was scanned between 8.7 and 19.6 eV. The step size ranged from 5 meV in the energy region of the disappearance of the parent ion in the breakdown diagram (see below) through 15 meV for the TPES scans below 14 eV and 100–150 meV for the overview TPES scans. The integration time ranged from 50 to 180 s per point. The light entered the ionization chamber, where it interacted with the sample in the 2 × 2 mm2 ionization volume. Photoelectrons and -ions were extracted by a constant 240 V cm−1 extraction field and velocity map imaged56 on two delay-line Roentdek detectors. The cations were detected in space-focusing conditions,57 as well, so that the time-of-flight mass spectrum could be plotted by evaluating delayed coincidences between electron and ion detection events.58 As electrons are detected according to their lateral momentum, threshold (virtually zero kinetic energy) electrons arrive in the detector center together with kinetic energy electrons with negligible off-axis momentum. The latter leads to the hot-electron contamination of the threshold signal. The hot-electron background is well represented by a ring area around the center spot, and can, thus, be subtracted to obtain the threshold electron signal and threshold ionization mass spectra.59The threshold electron signal was renormalized using a previously recorded flux curve to obtain the TPES. The parent ethanolamine cation at m/z 61 disappears from the threshold ionization mass spectrum in the 9.20–9.80 eV photon energy range, while the m/z 30 and, to a lesser degree, m/z 31 peaks grow in intensity. These correspond to a loss of a neutral fragment at 31 and 30 Da, respectively. The breakdown diagram is plotted in this energy range as the fractional ion abundances of these three peaks as a function of photon energy.
Thanks to the low extraction field and long acceleration region, metastable fragmentations on the experimental time scale can be identified based on asymmetric peak shapes,60 which are a measure of the unimolecular rate constant. In ethanolamine, all peaks were observed to be symmetric, which means that the fragmentation is not affected by a kinetic shift and all the cations with energetically allowed fragmentation channels will dissociate promptly while still in the ionization volume.
b. Computations
We performed the following computations: (i) geometry optimizations in the C1 point group with all coordinates relaxed, followed by Hessian calculations to confirm that all harmonic vibrational frequencies were real and thereby attest that the stationary point is a minimum on the ground-state potential energy surface. These computations were carried out using the explicitly correlated coupled clusters singles and doubles with perturbative treatment of triples ((R)CCSD(T)-F12b61–63) method as implemented in MOLPRO 2015,64 using the cc-pVDZ-F12 basis set.65 (ii) For better accuracy, we carried out single point (SP) computations to evaluate the scalar relativistic (ΔSR) and the core–valence (ΔCV) corrections. These computations were carried out at the (R)CCSD(T)-F12/cc-pVDZ-F12 equilibrium geometry, and involved the evaluation of the energy difference between (R)CCSD(T)/cc-pVTZ-DK66–68 and (R)CCSD(T)/cc-pVTZ total energies to obtain ΔSR and of the energy difference between the (R)CCSD(T)/cc-pwCVTZ69 total energies while treating or omitting the core electron correlation to obtain ΔCV. We computed the zero-point vibrational energy correction (ΔZPVE) at the PBE0/aug-cc-pVDZ level as implemented in GAUSSIAN 16,70 using the anharmonic frequencies obtained after VPT2 treatment. (iii) We treated the electronic excited states using the internally contracted multireference configuration interaction (MRCI)71–73 computation or its explicitly correlated version (MRCI-F12)74–76 on top of a state-averaged complete active space self-consistent field (SA-CASSCF) technique.77,78 In the CASSCF calculation, three active spaces were used. Active Space I, Active Space II, and Active Space III were constructed by including molecular orbitals (MOs) ranging from HOMO−4 to LUMO+3, from HOMO−6 to LUMO+4 and from HOMO−8 to LUMO+3 in the active space, respectively. The lower-lying occupied and higher-lying virtual orbitals were inactive. In these computations, we considered all valence electrons. Afterwards, we constructed the MRCI or MRCI-F12 active spaces by selecting all configurations having a coefficient larger than 0.01 in the CI expansion as a reference. This led to more than 5 × 109 configuration state functions (CSFs) when computing the doublet states of the ethanolamine cation to be considered. We requested 6 or 10 doublet cationic states, which were averaged together in CASSCF and MRCI.For the investigation of the unimolecular decomposition of ethanolamine upon ionisation, the ground-state cation potential energy surface was explored at the B3LYP/6-311++G(d,p) level of theory using Gaussian 16.70 Rotational constants and harmonic vibrational frequencies were obtained at the stationary points and used as input parameters for the statistical model of the breakdown diagram. Afterwards, composite method calculations using W1,79 CBS-APNO,80 and G481 theories and the (R)CCSD(T)-F12 based composite scheme (vide supra) were used to obtain isodesmic reaction energies and derive a computed ethanolamine enthalpy of formation, as well as to refine the energetics of the fragmenting ethanolamine cation.
III. Results and discussion
To complement previous theoretical works that treated the low-lying conformers of ethanolamine, we performed a search for the minimum structures on the ground potential energy surface of this molecular species at the higher, CCSD(T)-F12/cc-pVDZ-F12, level of theory. Fig. 1 shows these structures and their relative energies with respect to g′Gg′. These computations confirm the nature and the energy ordering of these conformers as determined in ref. 14–18 and 21–23. In the 0–0.2 eV energy range, thirteen neutral species are found. In particular, g′Gg′ is confirmed to be the most stable form at the presently used advanced theoretical level. Assuming equilibrium conditions at room temperature, the most stable g′Gg′ conformer dominates the neutral population at 55% abundance (Table S3, ESI†), although the population of the gGt, gGg′, and gTg conformers is also non-negligible at 6%, 17%, and 8%, respectively. As will be shown below, ionization to the cation minima always involves large conformational changes, and the transition moments for the different conformers are unlikely to vary by more than a trivial amount. In light of this, it is expected that the g′Gg′ conformer will dominate the spectrum, which may be perturbed by contributions from the less abundant conformers, notably gGg′.a. Structures and relative energies of the most stable forms of ethanolamine cation
Using these neutral ethanolamine conformers as starting structures, we mapped the ground-state doublet potential of the ethanolamine cation and searched for its conformers at the RCCSD(T)-F12/cc-pVDZ-F12 level. Only four cation minima were found in the 0–0.2 eV range with respect to the most stable one. They are named EtA1+, EtA2+, EtA3+, and EtA4+. EtA2+, EtA3+ and EtA4+ are located close in energy and more than 0.1 eV above EtA1+. They are depicted in Fig. 2 and their structural parameters are given in Table S5 (ESI†).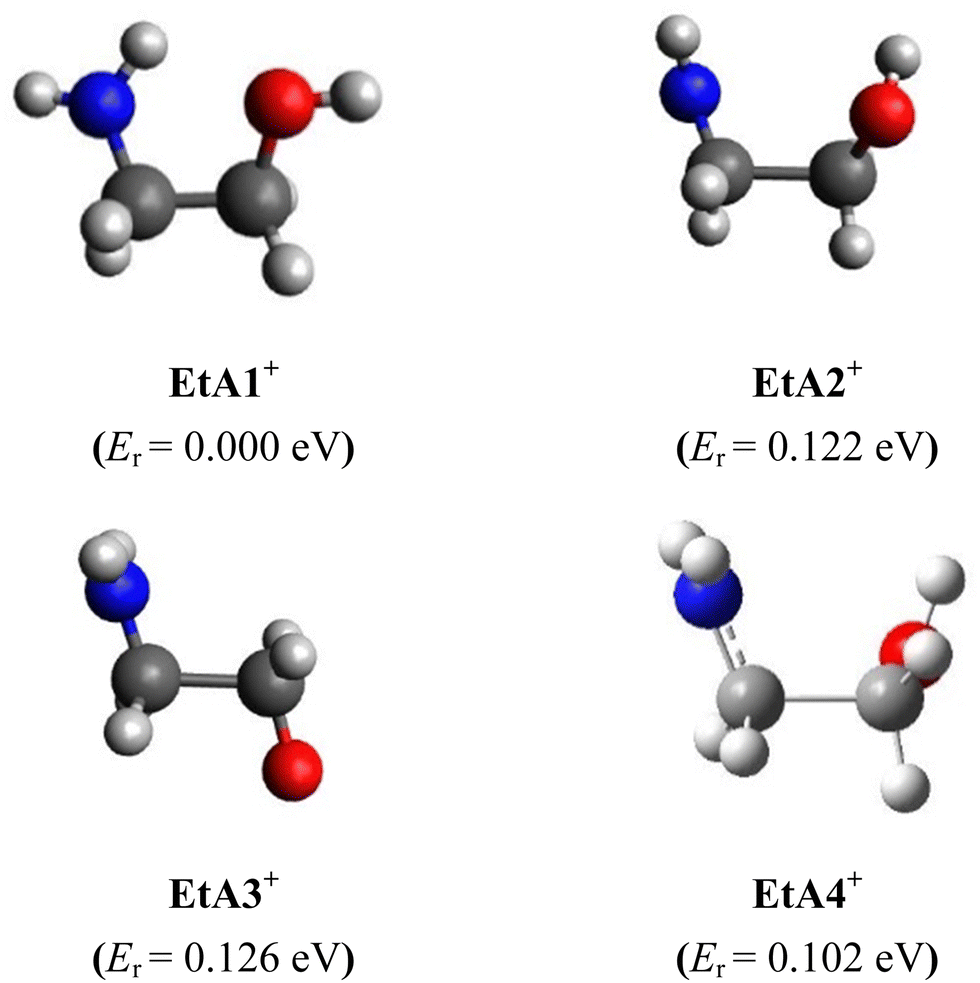 | ||
| Fig. 2 RCCSD(T)-F12/cc-pVDZ-F12 optimized structures of the ethanolamine cation ground electronic state. Conformer energies (Er, eV) with respect to EtA1+ are given in parentheses at the RCCSD(T)-F12/cc-pVDZ-F12 + ΔCV + ΔSR + ΔZPVE level. See Table S9 for more details (ESI†). | ||
The most stable form, EtA1+, has an intramolecular H-bond involving the (N–)H of the amine group and a lone pair of the hydroxyl O. This contrasts with g′Gg′, which exhibits an O–H⋯N H-bond. As can be seen in Table 1, we observe slight changes in the bond lengths upon ejecting an electron from g′Gg′ to form EtA1+. In contrast, the bond and dihedral angles are strongly affected and large deviations of up to 5–6° for bonds and several tens of degrees for dihedral angles can be seen for internal coordinates involving atoms establishing this intramolecular H-bond. Indeed, this constitutes a major structural change associated with ejecting a valence electron from g′Gg′ to form EtA1+. Besides, the NH2 group is tetrahedral in neutral ethanolamine and it is planar in all cation minima. Accordingly, unfavorable Franck–Condon factors (FCFs) are expected upon single photon ionization of g′Gg′ populating the ground electronic state of EtA1+. Computations also suggest unfavorable FCFs for the g′Gg′ → EtA2+ + e−, g′Gg′ → EtA2+ + e−, g′Gg′ → EtA2+ + e− and g′Gg′ → EtA2+ + e− ionization transitions.
| Parameters | g′Gg′ | EtA1+ | Parameters | g′Gg′ | EtA1+ | Parameters | g′Gg′ | EtA1+ |
|---|---|---|---|---|---|---|---|---|
| Distances | In-plane angles | Dihedral angles | ||||||
| C1C2 | 1.516 | 1.531 | C1C2O3 | 110.8 | 104.1 | O3C2C1N4 | 55.0 | 38.6 |
| C1N4 | 1.458 | 1.424 | N4C1C2 | 108.3 | 109.2 | H5O3C2C1 | −39.3 | 173.2 |
| C2O3 | 1.405 | 1.414 | H5O3C2 | 105.0 | 110.8 | H6C2C1N4 | 174.4 | 159.1 |
| O3H5 | 0.964 | 0.961 | H6C2C1 | 110.4 | 108.9 | H7C2C1N4 | −67.3 | −81.1 |
| C2H6 | 1.095 | 1.090 | H7C2C1 | 109.5 | 109.8 | H8C1C2O3 | −179.7 | 163.9 |
| C2H7 | 1.104 | 1.093 | H8C1C2 | 109.8 | 115.4 | H9C1C2O3 | −61.9 | −75.0 |
| C1H8 | 1.101 | 1.090 | H9C1C2 | 108.7 | 109.0 | H11N4C1C2 | −163.2 | −26.7 |
| C1H9 | 1.096 | 1.109 | H10N4C1 | 110.9 | 122.9 | H10N4C1C2 | 77.5 | 158.7 |
| N4H10 | 1.013 | 1.017 | H11N4C1 | 111.6 | 116.9 | |||
| N4H11 | 1.011 | 1.024 | H8C1N4 | 107.9 | 110.5 | |||
| H9C1N4 | 114.2 | 104.5 | ||||||
| H6C2O3 | 107.8 | 112.8 | ||||||
| H7C2O3 | 110.7 | 111.8 | ||||||
b. TPES of ethanolamine and the pattern of the electronic states of the ethanolamine cation
Table 2 lists the vertical ionization energies (VIEs) for the g′Gg′ + hν → g′Gg′+ + e− ionization transitions to the electronic ground and excited states. These energies were computed at the MRCI/aug-cc-pVXZ and at the MRCI-F12/aug-cc-pVXZ (X = D, T) levels, where 6 or 10 electronic states were requested at CASSCF and MRCI/MRCI-F12 levels. In fact, these benchmarks were used to check the effects of the atomic basis set, the number of averaged electronic states, and the consideration of explicit correlation on the ionization energies. Explicit correlation is assumed to provide data using smaller basis set, which are as accurate as those obtained without explicit correlation but with a much larger basis set. As discussed in ref. 74, the MRCI-F12/aug-cc-pVDZ level has indeed a similar accuracy as standard MRCI with the aug-cc-pVQZ basis set, but the computational costs (both CPU time and disk occupancy) are strongly reduced.| States | Electronic configuration | Active space | MRCI/aug-cc-pVDZ | MRCI/aug-cc-pVTZ | MRCI-F12/aug-cc-pVDZ | MRCI-F12/aug-cc-pVTZ | |||
|---|---|---|---|---|---|---|---|---|---|
| a VIE for g′Gg′ → g′Gg′+ + e− computed at the (R)CCSD(T)/aug-cc-pVTZ level. | |||||||||
| D0 | 13a214a215a2 16a217a1 (0.88) | I | 10.30 | 10.26 | 10.24 | 10.21 | 10.55 | 10.45 | 10.28 |
| II | 10.45 | ||||||||
| III | 10.12a | 10.12a | |||||||
| D1 | 13a214a215a2 16a117a2 (0.88) | I | 10.82 | 10.75 | 10.76 | 10.67 | 11.07 | 10.93 | 10.76 |
| II | 10.91 | ||||||||
| III | 10.62 | 10.62 | |||||||
| D2 | 13a214a215a1 16a217a2 (0.68) and 13a114a215a2 16a217a2 (0.66) | I | 12.80 | 12.37 | 12.71 | 12.27 | 13.07 | 12.55 | 12.34 |
| II | 12.41 | ||||||||
| III | 12.20 | 12.20 | |||||||
| D3 | 13a214a115a2 16a217a2 (0.94) | I | 14.32 | 13.89 | 14.26 | 13.81 | 14.56 | 14.05 | 13.87 |
| II | 13.91 | ||||||||
| III | 13.71 | 13.70 | |||||||
| D4 | 13a214a215a1 16a217a2 (0.69) and 13a114a215a2 16a217a2 (0.69). | I | 16.50 | 14.65 | 16.43 | 14.55 | 16.80 | 14.83 | 14.62 |
| II | 14.63 | ||||||||
| III | 14.48 | 14.49 | |||||||
| D5 | 13a214a215a2 16a218a1 (0.68) and 13a214a215a2 17a218a1 (0.61) | I | 17.39 | 17.59 | 17.40 | 17.62 | 17.67 | 17.84 | 17.74 |
| II | 16.66 | ||||||||
| III | 15.87 | 15.86 | |||||||
| D6 | 13a214a215a2 16a117a118a1 (0.89) | I | 17.80 | 17.82 | 18.08 | ||||
| II | |||||||||
| III | 16.60 | 16.61 | |||||||
| D7 | 13a214a215a2 16a117a119a1 (0.85) | I | 18.68 | 18.70 | 18.96 | ||||
| II | |||||||||
| III | 17.18 | 17.19 | |||||||
| D8 | 13a214a215a2 16a117a119a1 (0.92) | I | 19.07 | 19.09 | 19.36 | ||||
| II | |||||||||
| III | 18.00 | 18.04 | |||||||
| D9 | 13a214a215a1 16a217a118a1 (0.91) | I | 19.88 | 19.87 | 20.19 | ||||
| II | |||||||||
| III | 18.62 | 18.66 | |||||||
These benchmarks show that the basis set has almost no effect on the pattern of the electronic states of EtA1+. Also, MRCI and MRCI-F12 computations lead to very close results for a given active space and basis set. Calculating 6 or 10 electronic states does not change the energies of VIEs, which shows that the initial guesses for these states are consistent. In contrast, Table 2 shows that the choice of active space is crucial. In fact, the energies of D0–D4 are almost independent of the active space, the largest nevertheless showing the best agreement with the features of the experimental spectrum (vide infra). However, the VIEs of the D5–D9 electronic states strongly depend on the active space: Active Space I and Active Space II lead to a gap in the 15.0–17.5 eV energy range, which is inconsistent with experiments and not the case when using the larger Active Space III. In this case, the D5–D9 states shift down in energy and populate this energy range. In sum, solely Active Space III allows us to correctly describe the full set of doublet cationic electronic states located in the 8–18 eV energy range. Therefore, the inclusion of the 9a and 10a inner valence MOs is crucial for the description of these states. Fig. 3 shows that these MOs are located on the H2NCC moiety. As suggested earlier by Maruyama and Ohno,32 these doublet cationic states are obtained after an ejection of an electron from these MOs.
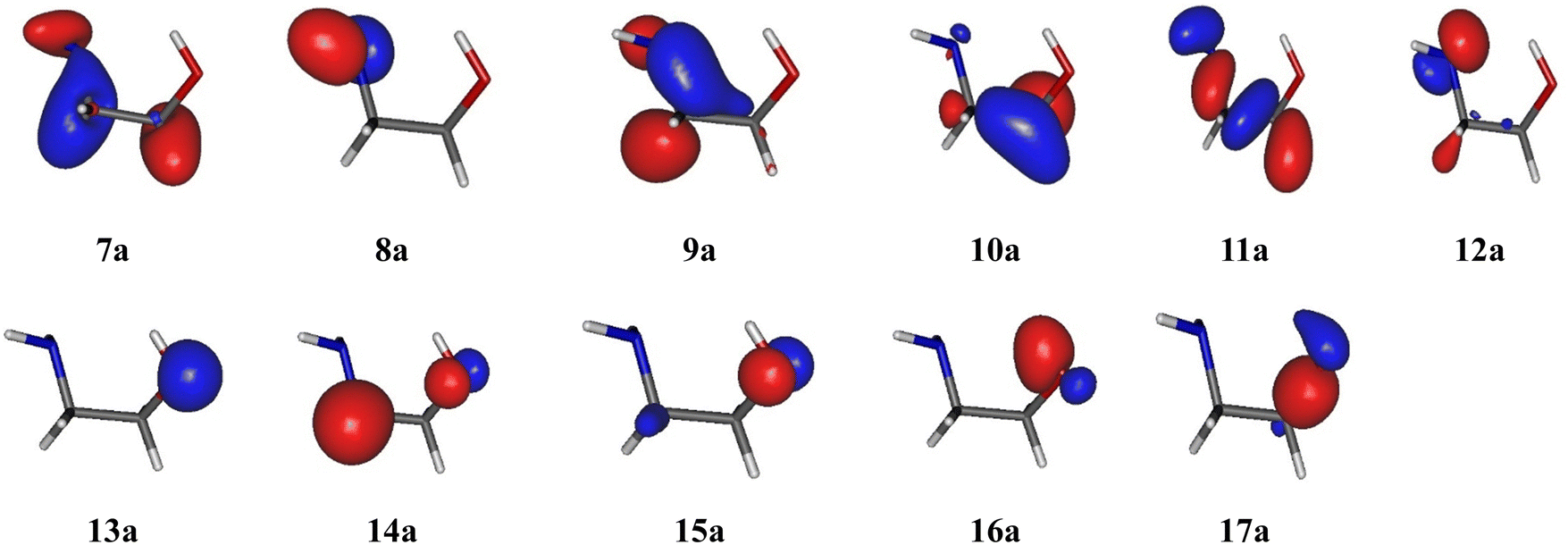 | ||
| Fig. 3 HOMO−10 (7a) to HOMO (17a) molecular orbitals of ethanolamine at the CASSCF/aug-cc-pVTZ level. | ||
Fig. 4 shows the TPES spectrum of ethanolamine in the 8–18 eV energy range. As predicted by the large geometry relaxation upon ionization and the resulting poor Franck–Condon factors for transitions to any of the cation minimum conformers, the origin transition cannot be identified, and the spectrum exhibits a slowly rising onset above 9 eV photon energy. Therefore, neither the photoelectron spectrum nor other direct ionization techniques, such as electron or Penning ionization, are suitable to determine the adiabatic ionization energy of ethanolamine incontrovertibly. Previously published experimental values were based on the ionization onset, which only represents an upper limit to the ionization energy in the absence of hot bands.
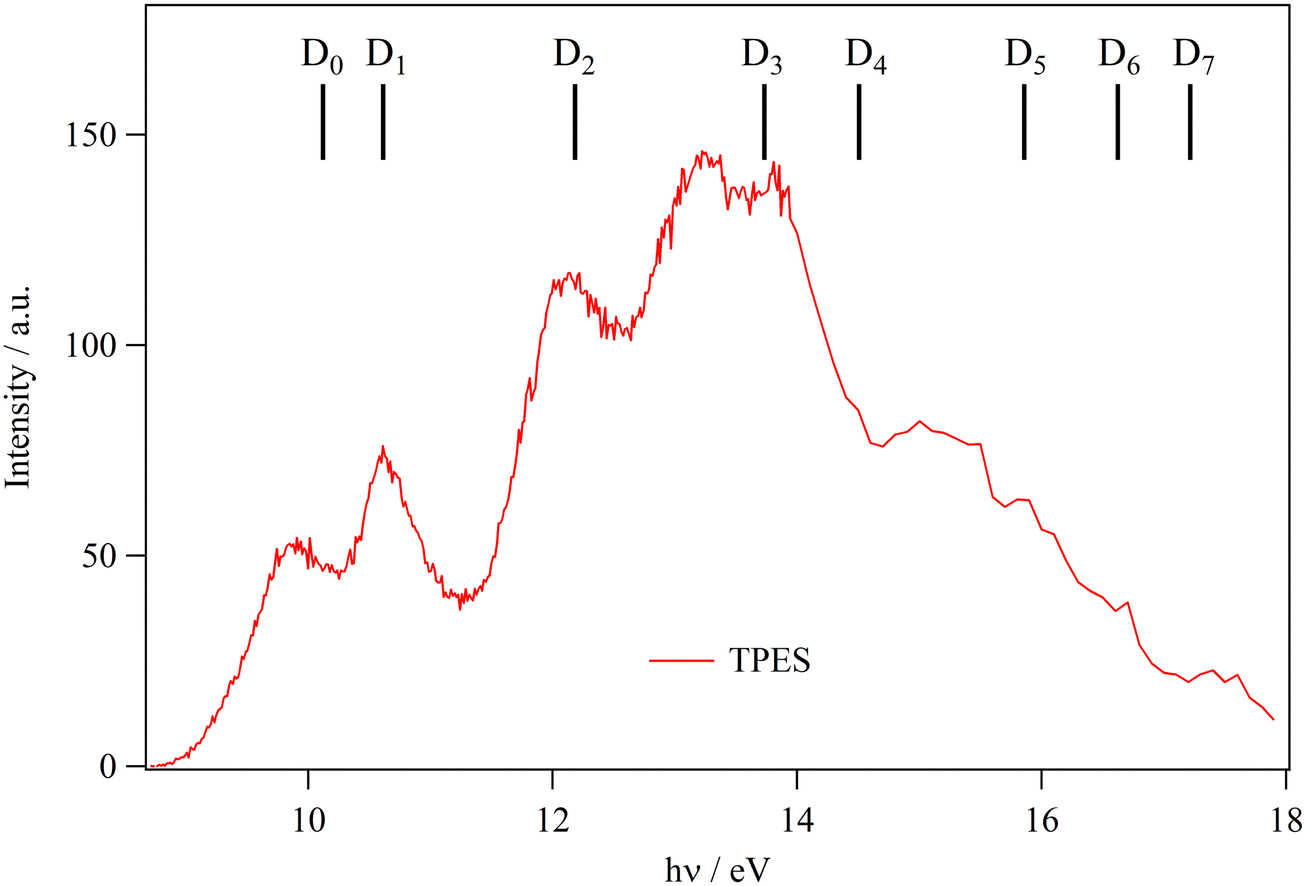 | ||
| Fig. 4 Ethanolamine TPES. Combs correspond to the vertical ionization energies as computed at the MRCI-F12/aug-cc-pVTZ level with Active Space III (cf.Table 2). | ||
It is interesting to compare the general intensity trends in the TPES with the previously published ultraviolet photoelectron spectra (UPS), based on HeI radiation at 21.2 eV. In the spectral compendium of Kimura et al.,30 the peak intensities are comparable between 9 and 15 eV and start rising thereafter in the 15–18 eV range. Ohno et al.32 reported slightly rising intensities in a mostly flat spectrum as far as the peak maxima are concerned with a close pronounced Franck–Condon gap below 12 eV, where the signal returned to the baseline.32 In the UPS by Maruyama and Ohno,33 the 10.5 eV peak is the most intense, and the spectrum trends downward thereafter with the peaks in the 15–18 eV range approx. half as intense as this maximum. In our data, there is a marked intensity maximum in the 12–15 eV range, and both the lower and the higher lying peaks exhibit ca. half the signal observed in this range. A drift in the beamline flux since recording the flux curve may have led to normalization errors and to the dropping signal above 16.5–17 eV, as the 600 lines per mm grating is rarely used in this energy range. The increased intensity of the D2–D4 bands with respect to the HeI photoelectron spectra is probably due to enhanced threshold ionization cross sections to these states.
The peak positions agree with the literature data up to a photon energy of 14 eV. Above this energy, the step size was increased to 100 meV, and the signal-to-noise ratio of the spectrum becomes inferior compared with the literature data, which typically indicate two bands, centered at 16 and slightly below 18 eV together with shoulders on the former. The calculations predict the vertical ionization energy of D0–D2 quite well, and the computed D3 and D4 levels are blue-shifted by ca. 0.5 eV with respect to the experiment. This is also likely true for D5–D7, which populates the 16–18 eV range with D8 calculated at 19.07 eV. According to previous assignments, these four states likely all show up in the 15.5–18.0 eV photon energy range.30,33
c. Adiabatic ionization energy of ethanolamine
The adiabatic ionization energy (AIE) of ethanolamine corresponds to the transition from the most stable g′Gg′ conformer to the cationic minimum, i.e., to the g′Gg′ → EtA1+ + e− transition, with both species in their electronic ground state. The AIE has been computed using the accurate (R)CCSD(T)-F12/cc-pVDZ-F12 (+ΔCV + ΔSR + ΔZPVE) composite scheme, as described in ref. 41. Briefly, the equilibrium geometries of both molecular species are first optimized at the (R)CCSD(T)-F12/cc-pVDZ-F12 level. Then, we evaluate the core-valence (ΔCV), the scalar relativistic (ΔSR) and the anharmonic (ΔZPVE) corrections as described in the previous section. The resulting (R)CCSD(T)-F12/cc-pVDZ-F12 (+ΔCV + ΔSR + ΔZPVE) AIE is 8.940 eV (Table 3). This value lies quite close to the ionization onset, which was relied on in previous experimental determinations. In the absence of a well-resolved origin transition and because of the convoluted Franck–Condon envelope resulting from multiple conformers ionizing into multiple cation conformer minima with multiple anharmonic large-amplitude motions being active upon ionization, the good agreement of the ionization onset with the ionization energy is serendipitous.d. Dissociative ionization
The breakdown diagram in the 9.20–9.80 eV photon energy range is shown in Fig. 5. The symmetric fragment ion peak shapes imply fast dissociation. This means that the disappearance of the parent ion from the breakdown diagram corresponds to the photon energy, at which even the originally zero internal energy species gain enough energy to dissociate, i.e., the 0 K appearance energy of the lowest-energy fragment ion. The dominant m/z 30 signal tells us that this fragment ion is likely the stable NH2CH2+ species, isoelectronic with ethylene, and produced together with the hydroxymethyl (CH2OH) neutral fragment by simple C–C bond breaking. According to the Active Thermochemical Tables,82,83 the alternative 31 amu loss, the methoxy radical (CH3O) can be ruled out because it is 39.3 kJ mol−1 (0.41 eV) less stable, and its formation would involve a tight H-transfer transition state, too. Because of the high ionization energy of formaldehyde (10.88 eV), the m/z 30 signal is exclusively NH2CH2+ in the energy range of its appearance in the mass spectrum. The derived 9.708 ± 0.010 eV AE is distinctly larger than the earlier determinations of 9.49 ± 0.05 eV.34,35 Nevertheless, this AE is in excellent agreement with the G4-computed barrier to dissociative ionization, 9.71 eV (Fig. 6) and the AE computed at the (R)CCSD(T)-F12/cc-pVDZ-F12 (+ΔCV + ΔSR + ΔZPVE) level of 9.724 eV. Note that the AE for the CH2NH2+ + CH3O, evaluated at the accurate (R)CCSD(T)-F12/cc-pVDZ-F12 (+ΔCV + ΔSR + ΔZPVE) level, is 10.029 eV, predicting a slightly lower energy gap of 0.305 eV between CH3O and CH2OH than the 0.41 eV by ATcT. | ||
| Fig. 5 Ethanolamine breakdown diagram in the 9.20–9.80 eV photon energy range. Squares indicate experimental data, to which the statistical model (continuous lines) was fitted. The obtained 0 K appearance energies for hydroxymethyl radical (CH2OH) and formaldehyde loss are also shown. | ||
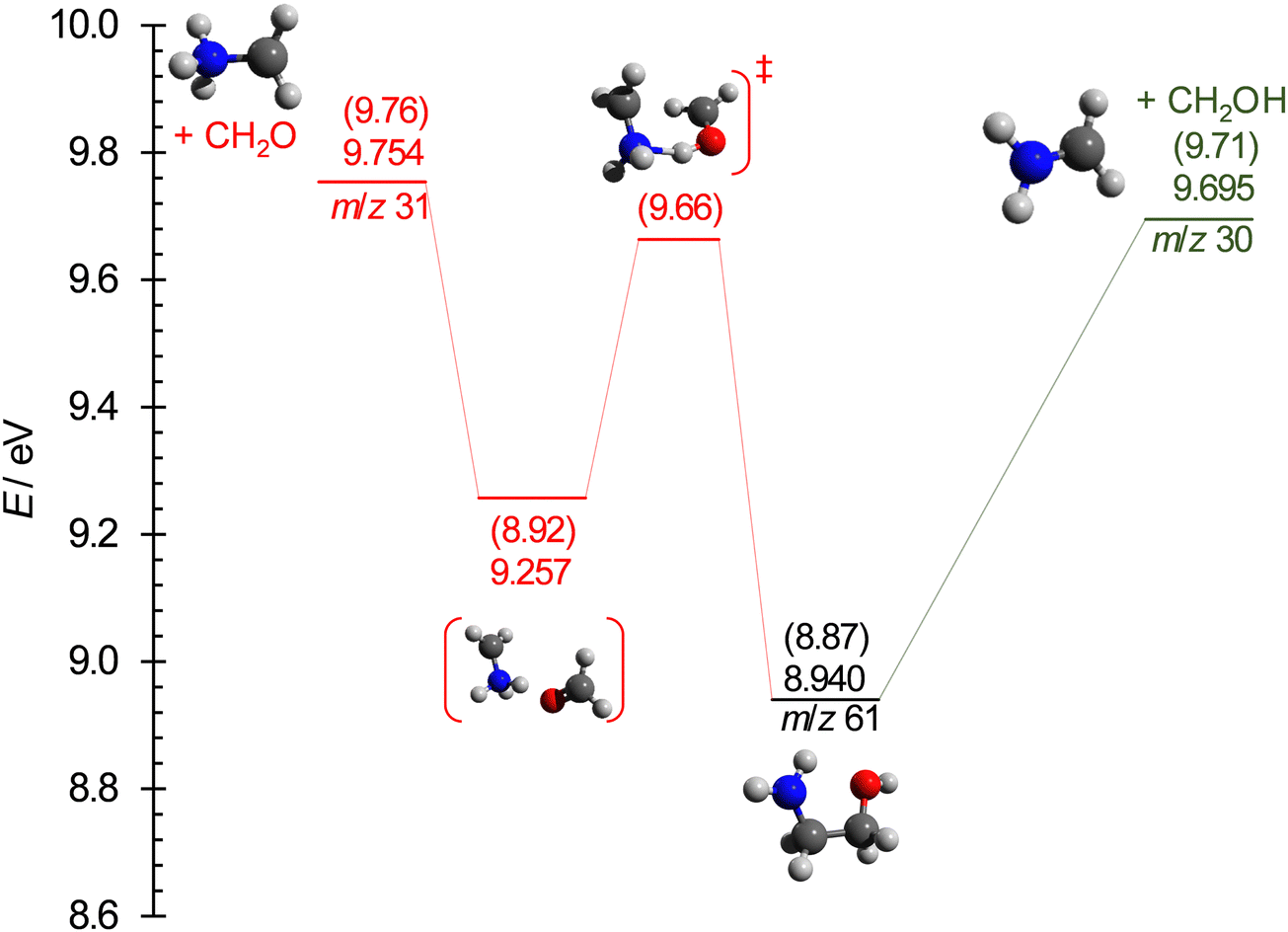 | ||
| Fig. 6 Computed potential energy surface for the lowest-energy dissociative ionization processes of ethanolamine. The energies are listed at the (R)CCSD(T)-F12/cc-pVDZ-F12 + ΔCV + ΔSR + ΔZPVE and at the G4 levels (in parentheses), relative to neutral ethanolamine. | ||
According to our calculations, the m/z 31 peak is best explained by formaldehyde loss, which is readily possible after an intramolecular H-transfer between the hydroxyl and the amine groups over a submerged transition state of 9.66 eV (G4 value). In the statistical model, we assumed this transition state to be the bottleneck in the fragmentation process and relaxed the transitional frequencies of both channels to fit the relative rates and, thus, the branching ratio observed in the breakdown diagram. This fit resulted in an AE of 9.73 ± 0.03 eV for the CH2O-loss channel, slightly below the computed 9.76 eV and 9.754 eV values at the G4 and (R)CCSD(T)-F12/cc-pVDZ-F12 (+ ΔCV + ΔSR+ ΔZPVE) levels, respectively. However, the submerged but tight transition state and the loose but more energetic CH2O-loss step thereafter mean that the phase space bottleneck along the dissociation coordinate is likely energy-dependent, and the extrapolation yields a phenomenological barrier between the H-transfer transition state and the dissociative photoionization energies.
e. Thermochemistry
In the absence of the reverse barrier, the AE for the hydroxymethyl radical loss corresponds to the 0 K reaction enthalpy (i.e., 0 K reaction energy), ΔrH⊖0K, of the following reaction:| NH2(CH2)2OH → NH2CH2+ + CH2OH | (1) |
| ΔrH⊖0K = ΔfH⊖0K(CH2OH) + ΔfH⊖0K(CH2NH2+) − ΔfH⊖0K[NH2(CH2)2OH] |
| ΔfH⊖298K[NH2(CH2)2OH] = ΔfH⊖0K[NH2(CH2)2OH] + (H⊖298K − H⊖0K)[NH2(CH2)2OH] − (H⊖298K − H⊖0K)[½N2 + 3.5H2 + 2C + ½O2] | (2) |
The G4-calculated thermal enthalpy of ethanolamine is (H⊖298K − H⊖0K)[NH2(CH2)2OH] = 15.7 kJ mol−1, while the standard elemental thermal enthalpies are 1.05, 8.67, 8.47, and 8.68 kJ mol−1 for C, N2, H2, and O2, respectively.86 Consequently, the standard enthalpy of formation of gaseous ethanolamine is obtained at room temperature as ΔfH⊖298K[NH2(CH2)2OH, g] = −208.2 ± 1.2 kJ mol−1. This value agrees surprisingly well with the group additivity result of −203.05 kJ mol−1. It can also be converted to ΔfH⊖298K[NH2(CH2)2OH, l] = −267.8 ± 1.2 kJ mol−1 using the 59.6 kJ mol−1 vaporization enthalpy of ethanolamine.39 This value, however, stands in marked contrast with the −507.5 kJ mol−1 result of Baroody and Carpenter.37 This discrepancy is such that we decided to carry out isodesmic reaction energy calculations for reaction (3) to confirm the experimental results. The enthalpies of formation for ethanol, ethylamine, and ethane are taken from the Active Thermochemical Tables (ΔfH⊖0K = −217.31 ± 0.20, −28.06 ± 0.45, and −68.38 ± 0.12 kJ mol−1 for C2H5OH, C2H5NH2, and C2H6, respectively). Based on the G4, CBS-APNO, and W1 computed reaction energies of −8.46, −9.48, and −8.04 kJ mol−1, respectively, the ethanolamine enthalpy of formation can be estimated computationally as ΔfH⊖0K[NH2(CH2)2OH] = −185.7 ± 2.0 kJ mol−1. The corresponding reaction energy at the (R)CCSD(T)-F12/cc-pVDZ-F12 (+ΔCV + ΔSR + ΔZPVE) level is −13.49 kJ mol−1, implying ΔfH⊖0K[NH2(CH2)2OH] = −190.5 kJ mol−1 and further confirming the experimental result.
| C2H5OH + C2H5NH2 → NH2(CH2)2OH + C2H6 | (3) |
IV. Conclusions
We used post-Hartree–Fock methodologies to investigate the conformers of neutral and cationic ethanolamine. The low-lying neutral conformers were confirmed, and the cation isomers newly identified. Computations reveal large geometrical change upon ethanolamine ionization, resulting is unfavorable Franck–Condon factors for the origin transitions between any isomers in the ethanolamine → ethanolamine+ + e− process. The recorded TPES spectrum confirms these findings. Indeed, the spectrum is composed of broad bands corresponding, in the 8–18 eV photon energy range, populating the D0–D7 cationic states from the most stable g′Gg′ neutral conformer. We calculate the adiabatic ionization energy of ethanolamine at 8.940 ± 0.010 eV accordingly. The two lowest-energy dissociative photoionization processes of ethanolamine have been identified as hydroxymethyl radical and formaldehyde losses. The appearance energy of the former could firmly be established by modeling the breakdown diagram as AE = 9.708 ± 0.010 eV. Together with the literature enthalpies of formation of the fragmentation products, this yields ΔfH⊖0K[NH2(CH2)2OH, g] = −183.5 ± 1.2 kJ mol−1 and ΔfH⊖298K[NH2(CH2)2OH, g] = −208.2 ± 1.2 kJ mol−1. Combined with the literature enthalpy of vaporization, the enthalpy of formation in the liquid phase is obtained as ΔfH⊖298K[NH2(CH2)2OH, l] = −267.8 ± 1.2 kJ mol−1. These values supersede and correct the group additivity estimate of ΔfH⊖298K[NH2(CH2)2OH, g] = −203.05 kJ mol−1 and the previously reported calorimetric result of ΔfH⊖298K[NH2(CH2)2OH, l] = −507.5 kJ mol−1, respectively.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project (RSPD2024R808) of King Saud University, Riyadh, Saudi Arabia. PEPICO experiments were carried out at the VUV beamline of the Swiss Light Source of the Paul Scherrer Institute.References
- Y. Liu, L. Zhang and S. Watanasiri, Ind. Eng. Chem. Res., 1999, 38, 2080 CrossRef CAS.
- H. B. Xie, J. K. Johnson, R. J. Perry, S. Genovese and B. R. A. Wood, J. Phys. Chem. A, 2011, 115, 342 CrossRef CAS PubMed.
- G. Puxty, R. Rowland, A. Allport, Q. Yang, M. Bown, R. Burns, M. Maeder and M. Attalla, Environ. Sci. Technol., 2009, 43, 6427 CrossRef CAS.
- Y. Kubota, T. Ohnuma and T. Bučko, J. Chem. Phys., 2017, 146, 094303 CrossRef.
- K. Weissermel, H.-J. Arpe, C. R. Lindley and S. Hawkins, Chapter 7. Oxidation Products of Ethylene, Industrial Organic Chemistry, Wiley-VCH, 2003, pp. 159–161, ISBN 3-527-30578-5 Search PubMed.
- M. Ernst, J.-P. Melder, F. I. Berger and C. Koch, Ethanolamines and Propanolamines, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2022, ISBN 978-3527306732 DOI:10.1002/14356007.a10_001.pub2.
- F. Carrasco. Ingredientes Cosméticos, Diccionario de Ingredientes Cosméticos, 4th edn, 2009, p. 306. ISBN 978-84-613-4979-1, https://www.imagenpersonal.net Search PubMed.
- A. J. Sexton and G. T. Rochelle, Ind. Eng. Chem. Res., 2011, 50, 667 CrossRef CAS.
- H.-B. Xie, F. Ma, Y. Wang, N. He, Q. Yu and J. Chen, Environ. Sci. Technol., 2015, 49, 13246 CrossRef CAS.
- H.-B. Xie, C. Li, N. He, C. Wang, S. Zhang and J. Chen, Environ. Sci. Technol., 2014, 48, 1700 CrossRef CAS PubMed.
- R. E. Penn and R. F. Curl, Jr., J. Chem. Phys., 1971, 55, 651 CrossRef CAS.
- V. K. Kaushik and R. C. Woods, Z. Phys. Chem., 1982, 132, 117 CrossRef CAS.
- M. V. Korolevich, V. V. Sivchik, N. A. Matveeva, R. G. Zhbankov, V. A. Lastochkina, M. L. Frenkel’, A. I. Ladut’ko, A. V. Pavlov and E. P. Petryaev, J. Appl. Spectrosc., 1987, 46, 400 CrossRef.
- M. Rasanen, A. Aspiala, L. Homanen and J. Murto, J. Mol. Struct., 1982, 96, 81 CrossRef CAS.
- K. Wang, X. Shan and X. J. Chen, J. Mol. Struct., 2009, 909, 91 CrossRef CAS.
- L. G. Vanquickenborne, B. Coussens, C. Verlinde and C. De Ranter, J. Mol. Struct., 1989, 201, 1 CrossRef.
- A.-M. Kelterer, M. Ramek, R. F. Frey, M. Cao and L. Schafer, J. Mol. Struct., 1994, 310, 45 CrossRef.
- C. F. P. Silva, M. L. T. S. Duarte and R. Fausto, J. Mol. Struct., 1999, 482–483, 591 CrossRef CAS.
- T. D. Smith, J. B. Gerken, P. V. Jog and J. D. Roberts, Org. Lett., 2007, 9, 4555 CrossRef CAS.
- D. L. Thomsen, J. L. Axson, S. D. Schrøder, J. R. Lane, V. Vaida and H. G. Kjaergaard, J. Phys. Chem. A, 2013, 117, 10260 CrossRef CAS.
- P. Asselin, B. Madebène, P. Soulard, R. Georges, M. Goubet, T. R. Huet, O. Pirali and A. Zehnacker-Rentien, J. Chem. Phys., 2016, 145, 224313 CrossRef.
- I. Vorobyov, M. C. Yappert and D. B. DuPré, J. Phys. Chem. A, 2002, 106, 668 CrossRef CAS.
- Y.-P. Chang, T.-M. Su, T.-W. Li and I. Chao, J. Phys. Chem. A, 1997, 101, 6107 CrossRef CAS.
- Y. V. Novakovskaya and M. N. Rodnikova, Russ. J. Inorg. Chem., 2014, 59, 1290 CrossRef CAS.
- L. Radom, W. A. Lathan, W. J. Hehre and J. A. Pople, J. Am. Chem. Soc., 1973, 95, 693 CrossRef CAS.
- V. M. Rivilla, I. Jiménez-Serra, J. Martín-Pintado, C. Briones, L. F. Rodríguez-Almeida, F. Rico-Villas, B. Tercero, S. Zeng, L. Colzi, P. de Vicente, S. Martín and M. A. Requena-Torres, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2101314118 CrossRef.
- X. Zhang, G. Tian, J. Gao, M. Han, R. Su, Y. Wang and S. Feng, Orig. Life Evol. Biosph., 2017, 47, 413 CrossRef CAS PubMed.
- U. H. Molder, R. J. Pikver and I. A. Koppel, Org. React., 1983, 20, 355 CAS.
- I. A. Koppel, U. H. Molder and R. J. Pikver, Org. React., 1983, 20, 45 CAS.
- K. Kimura, S. Katsumata, Y. Achiba, T. Yamazaki and S. Iwata, Ionization energies, Ab initio assignments, and valence electronic structure for 200 molecules, Handbook of HeI Photoelectron Spectra of Fundamental Organic Compounds, Japan Scientific Soc. Press, Tokyo, 1981 Search PubMed.
- S. Leavell, J. Steichen and J. L. Franklin, J. Chem. Phys., 1973, 59, 4343 CrossRef CAS.
- K. Ohno, K. Imai and Y. Harada, J. Am. Chem. Soc., 1985, 107, 8078 CrossRef CAS.
- R. Maruyama and K. Ohno, J. Phys. Chem. A, 2004, 108, 4211 CrossRef CAS.
- J. L. Holmes and F. P. Lossing, Int. J. Mass Spectrom. Ion Processes, 1984, 58, 113 CrossRef CAS.
- F. P. Lossing, Y. T. Lam and A. Maccoll, Can. J. Chem., 1981, 59, 2228 CrossRef CAS.
- https://webbook.nist.gov .
- E. E. Baroody and G. A. Carpenter, Heats of formation of propellant compounds (U), Rpt. Naval Ordnance Systems Command Task No. 331-003/067-1/UR2402-001 for Naval Ordnance Station, Indian Head, MD, 1972, pp. 1–9 Search PubMed.
- K. G. Joback and R. C. Reid, Chem. Eng. Commun., 2007, 57, 233 CrossRef.
- S. Kapteina, K. Slowik, S. P. Verevkin and A. Heintz, J. Chem. Eng. Data, 2005, 50, 398 CrossRef CAS.
- T. Baer and R. P. Tuckett, Phys. Chem. Chem. Phys., 2017, 19, 9698 RSC.
- M. Hochlaf, Phys. Chem. Chem. Phys., 2017, 19, 21236 RSC.
- P. Hemberger, Z. Pan, X. Wu, Z. Zhang, K. Kanayama and A. Bodi, J. Phys. Chem. C, 2023, 127(34), 16751 CrossRef CAS.
- J. Bouwman, H. R. Hrodmarsson, G. B. Ellison, A. Bodi and P. Hemberger, J. Phys. Chem. A, 2021, 125, 1738 CrossRef CAS PubMed.
- J. Bourgalais, Z. Jiang, J. Bloino, O. Herbinet, H.-H. Carstensen, G. A. Garcia, P. Arnoux, L.-S. Tran, G. Vanhove, L. Nahon, F. Battin-Leclerc and M. Hochlaf, Phys. Chem. Chem. Phys., 2022, 24, 10826 RSC.
- K. Voronova, K. M. Ervin, K. G. Torma, P. Hemberger, A. Bodi, T. Gerber, D. L. Osborn and B. Sztáray., J. Phys. Chem. Lett., 2018, 9(3), 534 CrossRef CAS.
- H. Zhao, K.-C. Lau, G. A. Garcia, L. Nahon, S. Carniato, L. Poisson, M. Schwell, M. Mogren Al-Mogren and M. Hochlaf, Phys. Chem. Chem. Phys., 2018, 20, 20756 RSC.
- Z. Chen, K.-C. Lau, G. A. Garcia, L. Nahon, D. K. Božanić, L. Poisson, M. Mogren Al-Mogren, M. Schwell, J. S. Francisco, A. Bellili and M. Hochlaf, J. Am. Chem. Soc., 2016, 138, 16596 CrossRef CAS PubMed.
- M. Schwell and M. Hochlaf, Photoionization spectroscopy of nucleobases and analogues in the gas phase using synchrotron radiation as excitation light source, in Photoinduced phenomenain nucleic acids I: Nucleobases in the gas phase and in solvants, ed. M. Barbatti, A. C. Borin and S. Ullrich, Topics in Current Chemistry, 2015, vol. 355, pp. 155–208 Search PubMed.
- A. Bodi, M. D. Brannock, B. Sztáray and T. Baer, Phys. Chem. Chem. Phys., 2012, 14, 16047 RSC.
- M. F. Heringa, J. G. Slowik, A. S. H. Prévôt, U. Baltensperger, P. Hemberger and A. Bodi, J. Phys. Chem. A, 2016, 120, 3397 CrossRef CAS PubMed.
- B. Sztáray, A. Bodi and T. Baer, Mol. Phys., 2010, 45, 1233 Search PubMed.
- A. Bodi, J. Csontos, M. Kállay, S. Borkar and B. Sztáray, Chem. Sci., 2014, 5, 3057 RSC.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597 CrossRef CAS.
- A. Bodi, P. Hemberger, T. Gerber and B. Sztaray, Rev. Sci. Instrum., 2012, 83, 083105 CrossRef PubMed.
- B. Sztáray, K. Voronova, K. G. Torma, K. J. Covert, A. Bodi, P. Hemberger, T. Gerber and D. L. Osborn, J. Chem. Phys., 2017, 147, 013944 CrossRef.
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477 CrossRef CAS.
- W. C. Wiley and I. H. McLaren, Rev. Sci. Instrum., 1955, 26, 1150 CrossRef CAS.
- A. Bodi, B. Sztaray, T. Baer, M. Johnson and T. Gerber, Rev. Sci. Instrum., 2007, 78, 084102 CrossRef.
- B. Sztáray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763 CrossRef.
- B. Sztáray, A. Bodi and T. Baer, J. Mass Spectrom., 2010, 45, 1233 CrossRef.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef.
- H.-J. Werner, G. Knizia and F. R. Manby, Mol. Phys., 2011, 109, 407 CrossRef CAS.
- H. J. Werner and P. J. Knowles, et al., MOLPRO version 2015 is a package of ab initio programs, University of Cardiff Chemistry Consultants (UC3), Cardiff, Wales, UK, 2015. See https://www.molpro.net Search PubMed.
- K. A. Peterson, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89 CAS.
- G. Jansen and B. A. Hess, Phys. Rev. A, 1989, 39, 6016 CrossRef PubMed.
- W. A. de Jong, R. J. Harrison and D. A. Dixon, J. Chem. Phys., 2001, 114, 48 CrossRef CAS.
- K. A. Peterson and T. H. Dunning, J. Chem. Phys., 2002, 117, 10548 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, revision A.02, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- H.-J. Werner and P. J. Knowles, J. Chem. Phys., 1998, 89, 5803 CrossRef.
- P. J. Knowles and H.-J. Werner, Chem. Phys. Lett., 1988, 145, 514 CrossRef CAS.
- K. R. Shamasundar, G. Knizia and H.-J. Werner, J. Chem. Phys., 2011, 135, 054101 CrossRef CAS PubMed.
- T. Shiozaki and H.-J. Werner, Mol. Phys., 2013, 111, 607 CrossRef CAS.
- T. Shiozaki, G. Knizia and H.-J. Werner, J. Chem. Phys., 2011, 134, 034113 CrossRef.
- T. Shiozaki and H.-J. Werner, J. Chem. Phys., 2011, 134, 184104 CrossRef.
- H.-J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053 CrossRef CAS.
- P. J. Knowles and H.-J. Werner, Chem. Phys. Lett., 1985, 115, 259 CrossRef CAS.
- J. M. L. Martin and G. de Oliveira, J. Chem. Phys., 1999, 111, 1843 CrossRef CAS.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery, J. Chem. Phys., 1996, 104, 2598 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 84108 CrossRef PubMed.
- https://atct.anl.gov .
- B. Ruscic, R. E. Pinzon, G. V. Laszewski, D. Kodeboyina, A. Burcat, D. Leahy, D. Montoy and A. F. Wagner, J. Phys.: Conf. Ser., 2005, 16, 561 CrossRef CAS.
- B. Ruscic, Int. J. Quantum. Chem., 2014, 114, 1097 CrossRef CAS.
- A. Bodi, J. P. Kercher, C. Bond, P. Meteesatien, B. Sztáray and T. Baer, J. Phys. Chem. A, 2006, 110, 13425 CrossRef CAS PubMed.
- M. W. Chase, NIST-JANAF Thermochemical Tables. https://janaf.nist.gov/ DOI:10.18434/T42S31 (accessed 2021-08-01).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03015j |
| This journal is © the Owner Societies 2024 |