
Role of EDTA protonation in chelation-based removal of mercury ions from water†
Halyna
Butovych
 *ab,
Fatemeh
Keshavarz
b,
Bernardo
Barbiellini
be,
Erkki
Lähderanta
bd,
Jaroslav
Ilnytskyi
ac and
Taras
Patsahan
*ac
*ab,
Fatemeh
Keshavarz
b,
Bernardo
Barbiellini
be,
Erkki
Lähderanta
bd,
Jaroslav
Ilnytskyi
ac and
Taras
Patsahan
*ac
aInstitute for Condensed Matter Physics of the National Academy of Sciences of Ukraine, 1 Svientsitskii str., 79011 Lviv, Ukraine. E-mail: halyna@icmp.lviv.ua; tarpa@icmp.lviv.ua
bDepartment of Physics, School of Engineering Science, LUT University, FI-53850 Lappeenranta, Finland
cInstitute of Applied Mathematics and Fundamental Sciences, Lviv Polytechnic National University, 12 S. Bandera str., 79013 Lviv, Ukraine
dDepartment of Physics, Universitat de les Illes Balears, Cra Valldemossa, km. 7.5, 07122 Palma, Spain
eDepartment of Physics, Northeastern University, Boston, MA 02115, USA
First published on 25th September 2024
Abstract
A robust method of hazardous metal ion removal from an aqueous environment involves the use of chelating agents, such as ethylenediaminetetraacetic acid (EDTA). Here, we focus on mercury (Hg2+) uptake by EDTA using both molecular dynamics and density functional theory simulations. Our results indicate that the deprotonation of the EDTA carboxylate groups improves the localization of negative charge on the deprotonated sites. This mechanism facilitates charge transfer between the metal ions and EDTA, and provides a stronger and more stable EDTA–Hg2+ complex formation improving the efficiency of the chelation process. The best metal removal conditions are achieved using the fully deprotonated form of EDTA, which naturally occurs at pH levels above 3.
1 Introduction
Despite the rules imposed on mercury application, some industrial activities still cause mercury (Hg) pollution. Examples include paint and paper manufacturing, mining, coal burning, and the incineration of waste.1–3 Pollution can endanger the health of both humans and animals by poisoning vital organs.4–6 As a result, the World Health Organization and the environmental protection agency have listed mercury as one of the most toxic heavy metals.7–9The toxicity issue is further exacerbated by the fact that mercury can be found in different forms10 and sources, from the atmosphere to soil and groundwater.11 This has motivated the removal of mercury using different techniques,12 for example coagulation and flocculation,13,14 precipitation,15 ion exchange,16 reverse osmosis,17,18 membrane filtration,19–21 and adsorption. Adsorption is the most attractive approach because of its simplicity and low cost.22
Adsorption of mercury is typically based on chelation by a chelator (or a chelating agent) with two or more groups that are able to donate electron pairs for forming coordination bonds with metal ions and create a ring-like structure known as a chelate ring.23 The most commonly used chelators for Hg removal and preconcentration are ethylenediaminetetraacetic acid (EDTA),24–26 hydroxyethylethylenediaminetriacetic acid (HEDTA),27 2,3-dimercaptopropanesulfonic acid (DMPS),28 and 2-3-dimercaptosuccinic acid (DMSA).29 In this work, we focus on EDTA. EDTA not only adsorbs many metal ions, such as mercury (Hg2+), iron (Fe2+/Fe3+), and calcium ions (Ca2+),30–34 but also outperforms many other commercial chelators in terms of adsorption energy/reactivity.35
The understanding of Hg uptake by EDTA can help to improve the efficiency of chelation mechanisms in wastewater treatment. While some insight about such mechanisms is already available,35–37 the chelation mechanism is not fully understood.
To date, an EDTA–Hg2+ structure containing fully deprotonated EDTA, a Hg2+ ion and a single molecular water in the form of a distorted octahedral has been proposed using an X-ray absorption spectroscopy.38 Furthermore, some density functional theory (DFT)-based studies have addressed the interaction of various metal ions with EDTA. The studies have, for example, focused on Na, K, Rb, Mg, Mn, Ca, and Sr,39 Ba, Y and Zr,40 Al, Sc, and V-Co,41 and Fe, Co, Cu.42 The interactions between Hg2+ and EDTA have also been studied37 by applying the B3LYP/LANL2DZ DFT level to some experimental structural data,43–45 suggesting the positive influence of K+ presence on the complexation process. The impact of water on EDTA complex formation with some metals (other than Hg2+) has also been addressed. For instance, some quantum mechanical/molecular mechanical (QM/MM) simulations46 have revealed that Mg2+ binds to EDTA in a 6-fold octahedral complex, whereas Ca2+ prefers the formation of a 7-fold coordination complex with rapidly exchanging water molecules. In another work,47 a combination of classical molecular dynamics simulations and extended X-ray absorption fine structure (EXAFS) has elucidated the impact of protonation on the binding of Eu3+ to EDTA in aqueous solutions. An additional example is DFT-based ab initio molecular dynamics simulation of EDTA complexation with Ln3+, La3+, Eu3+ and Gd3+ ions in aqueous solution at different pHs and temperatures.48
Considering the possibility of water impact on the Hg2+–EDTA complex formation and the necessity of understanding metal ion chelation mechanisms, in this study, we apply both DFT calculations and atomistic molecular dynamics (MD) simulations to investigate the process in aqueous environment. We consider both realistic and theoretical EDTA protonation scenarios to highlight the impact of both amino and carboxylate protonation on Hg2+–EDTA complex formation. This approach not only guarantees the realistic study of the complex formation process over a wide pH range but also adds to the computational merit of the work by following a wide spectrum of changes in the protonation and charge distribution of EDTA. In the framework of our study, the MD simulations are particularly designed to sample all potential configurations of a complex and extract the most probable structures. The obtained Hg2+–EDTA structures and the corresponding adsorption energies are then validated through DFT simulations to study the impact of charge distribution/protonation state on the efficiency of the chelation process.
2 Computational details
The uptake of Hg2+ by EDTA was studied using MD simulations and DFT calculations. The MD simulations were performed to follow the uptake processes in explicit water solvent. The DFT simulations were used as a higher-level computational technique to re-evaluate the uptake behavior with higher accuracy. Using both approaches, we considered a number of protonation states for EDTA to study the impact of charge distribution on the uptake process. To include a wide range of protonation states in our studies, we considered two series of protonation states – one realistic and one theoretical – with the fully deprotonated EDTA4− state as their starting point. For the theoretical states, we protonated one to four carboxylate groups, and distinguished them from the realistic protonation states by adding a ‘th’ prefix to their names. These structures are schematically shown in Fig. 1, where thEDTA0 is the neutral structure with 4 protonated carboxylate groups, and thEDTA1−, thEDTA2−, thEDTA3− correspond to the different deprotonation levels with 3, 2, and 1 protonated carboxylate groups, respectively. We note that the thEDTA2− can have two forms, which are labeled as thEDTAa2− and thEDTAo2−.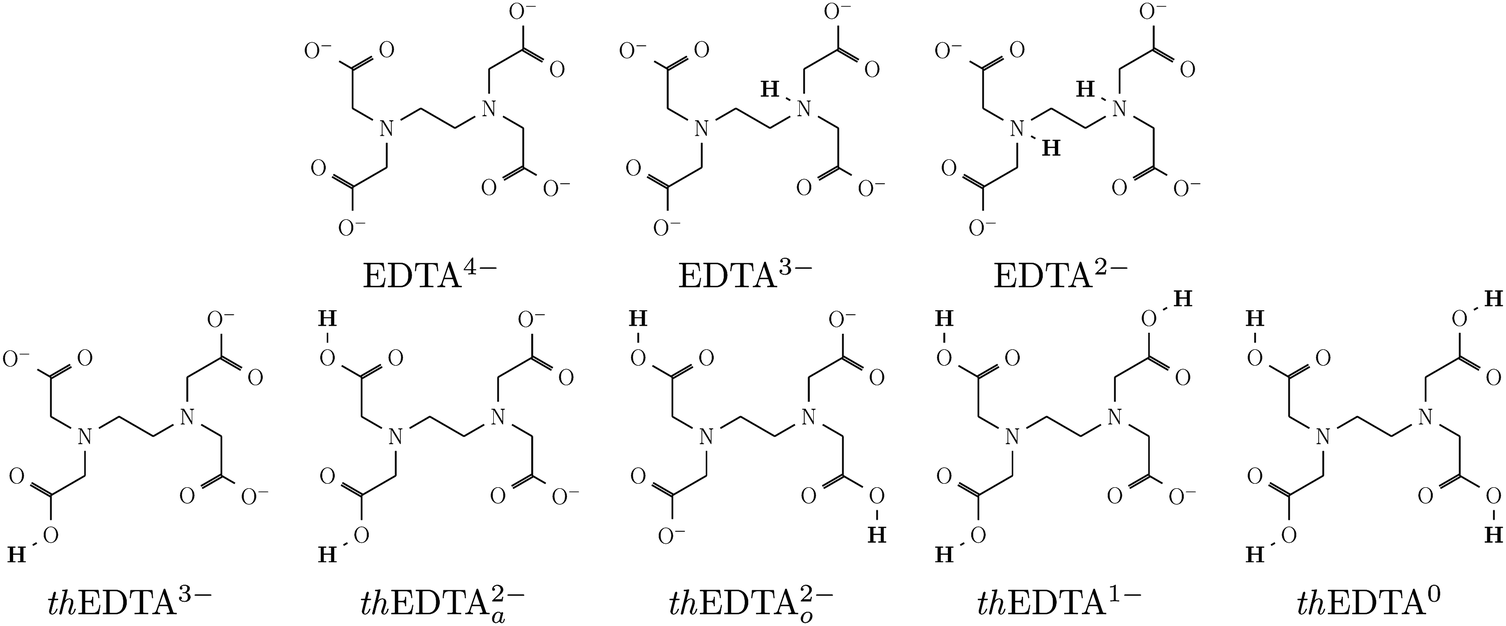 | ||
| Fig. 1 Schematic view of the chemical structure of EDTA at the examined protonation states. | ||
The inclusion of the thEDTA protonation states helps to isolate the impact of carboxylate protonation on the charge distribution and the complex formation process. However, such a protonation trend is only theoretical as EDTA protonation actually begins with the protonation of the amino groups rather than the carboxylates.49 To address realistic conditions, we are concerned with the EDTA protonation states associated with various pH conditions. EDTA molecules in a complex with a heavy metal ion can exist in its fully deprotonated state at above pH 3.38,47,50,51 This is because, over pH 3, the proton of EDTA dissociates into the bulk solution, leaving the molecules in its fully deprotonated state (Hg2+–EDTA4−).38,47,52 Around pH 3–4, the EDTA protonation process starts, and Hg2+–EDTA3− complexes with one protonated amino group purple are also present in the solution, while the concentration of Hg2+–EDTA4− drastically decreases upon lowering pH.38,47 At low pH, the second amino protonation step takes place (Hg2+–EDTA2−),47 resulting in a zwitterionic neutral state when complexed with divalent metal ions. Carboxylate protonation starts at very low pH conditions,49 which is not relevant to industrial metal ion removal. Therefore, we added the EDTA3− and zwitterionic EDTA2− with one and two protonated amino groups, respectively, to our studied protonation states. Their corresponding schematic structures are shown in Fig. 1.
2.1 Molecular dynamics simulations
The formation of the aqueous mercury ion (Hg2+) complexes with the EDTA molecules was investigated through molecular dynamics simulations using the LAMMPS software (version 2Aug2023).53 To describe the interactions between the atoms, the OPLS/AA force field54 and the SPC/E model55 were applied to the EDTA and water molecules, respectively. For the mercury ion, the OPLS/AA and SPC/E-consistent parameters were taken from ref. 56–58. The adopted parameters are summarized in Tables S1–S5 (ESI†). The Moltemplate software59 was used to generate the molecular structure of EDTA and the related input files.The simulations were performed in four stages: (1) simulation of EDTA and Hg2+ chelation at constant volume and temperature T = 298.15 K (the NVT ensemble) to form a stable complex in the absence of water, while maintaining the total electroneutrality of the system by placing two counterions at fixed distances relative to the Hg–EDTA complex; (2) insertion of water molecules in the system obtained at the previous stage and running simulation at the constant pressure 1 atm and temperature T = 298.15 K (the NPT ensemble) until reaching the appropriate system density; (3) equilibration of the system obtained at the previous stage at the same NPT conditions; and (4) the production run at the same conditions and storing the trajectory for further analysis. During all these steps, EDTA and the counterions were kept apart at significant distances to focus exclusively on the EDTA–Hg complex and to avoid any unnecessary interaction between the complex and the counterions.
In stage 1, the system components were introduced in a cubic box of size Lx = 150 Å, Ly = Lz = 75 Å. EDTA was initially positioned at the maximum distance from Hg2+. No water molecules were added to the box. To maintain system electroneutrality, two counterions were placed in the box at a distance larger than 60 Å from EDTA and Hg2+. The distance was sufficient to minimize the effect of any counterion interactions with the complex. Undoubtedly, in the absence of water this effect remained significant. As such, all ions (i.e., Hg2+ and the counterions) were immobilized during the simulation, but EDTA could move. The MD simulation was performed in the NVT ensemble at 298.15 K and resulted in the formation of a stable EDTA–Hg2+ complex within the time scale of 100 ps. The time step was 0.5 fs. The Nosé–Hoover thermostat was used to maintain the temperature using the relaxation parameter of 10 fs.
The system obtained from stage 1 was then immersed in 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 randomly positioned water molecules for stage 2. The minimum overlap distance between the molecules during the insertion was 1.5 Å. The aqueous system was then relaxed at T = 298.15 K and P = 1 atm using a relatively short simulation (100 ps) to achieve the expected density. The Nosé–Hoover thermostat-barostat with the relaxation parameters of 0.1 ps and 1 ps were respectively used to maintain the temperature and pressure. The position of Hg2+ was unfixed to allow the complex to move freely, but the position of the counterions remained fixed. At the end of this stage, the size of the simulation box decreased to Lx = 134 Å, Ly = Lz = 67 Å, and the density of water reached the value of 0.99 g cm−3. Because of the decrease in the simulation box size and the motion of the complex, the distance between the complex and the counterions decreased. However, it remained large enough (over 50 Å) to avoid any significant counterion–EDTA interaction.
000 randomly positioned water molecules for stage 2. The minimum overlap distance between the molecules during the insertion was 1.5 Å. The aqueous system was then relaxed at T = 298.15 K and P = 1 atm using a relatively short simulation (100 ps) to achieve the expected density. The Nosé–Hoover thermostat-barostat with the relaxation parameters of 0.1 ps and 1 ps were respectively used to maintain the temperature and pressure. The position of Hg2+ was unfixed to allow the complex to move freely, but the position of the counterions remained fixed. At the end of this stage, the size of the simulation box decreased to Lx = 134 Å, Ly = Lz = 67 Å, and the density of water reached the value of 0.99 g cm−3. Because of the decrease in the simulation box size and the motion of the complex, the distance between the complex and the counterions decreased. However, it remained large enough (over 50 Å) to avoid any significant counterion–EDTA interaction.
In stage 3, the resultant system was equilibrated for the longer period of 1 ns to ensure stability at the ambient conditions. The other simulation details were identical to stage 2. The distance between the complex and the counterions was still large enough.
Finally, in stage 4, we performed 5 ns of the production run under the same conditions addressed in stages 2 and 3. At this stage, we monitored the positions of the complex and the counterions to ensure sufficient separation between them. According to our observations, the complex needed over 5 ns time to diffuse toward the counterions. Stage 4 generated a series of atomic position trajectories, which were stored every 1 ps for further analysis.
In all MD simulations, the periodic boundary conditions were applied in all directions. The long-range electrostatic interactions were computed using the particle–particle–particle–mesh (PPPM) scheme with 0.0001 accuracy and 12 Å cutoff radius. The cutoff of the Lennard-Jones (LJ) interactions was set to 10 Å. The bond lengths and angles of the rigid SPC/E water molecules were constrained using the SHAKE technique. The LJ cross-interactions were computed through the geometrical mixing rules. The non-bonding intramolecular interactions were considered for the 1–4 adjacent atoms of EDTA and scaled by a factor of 0.5 according to the OPLS/AA force field.
2.2 DFT calculations
The DFT calculations were performed using Gaussian 16 Rev. C1.60 The stability of the structures was validated by the absence of any imaginary frequencies. To calibrate the required parameters, we initially considered several DFT approximations suggested for the simulation of ion uptake by EDTA and similar chelators,61–67 including B3LYP,68,69 M05-2X,70 PBE,71 PBE0,72 and ωB97X-D.73 HSE0674 was also included because of its potential to provide accurate electronic structures.75 For basis sets, we chose the ones that were accessible in the Gaussian package and contained all parameters needed to describe the Hg2+–EDTA complex, i.e., Def2-TZVP,76,77 Def2-TZVPP,76,77 and LanL2DZ.78,79 The SMD,80 CPCM,81 and PCM82 models were used to describe the water environment implicitly.The experimentally resolved Hg2+–EDTA4−–water structure38 was optimized at different computational levels, and the resultant geometry was compared to the experimental counterpart. The comparison was quantified as root mean square displacement (RMSD) of the backbone structure by ChemCraft 1.8.83 The results are summarized in Table S6, ESI† (along with the geometries displayed in Fig. S2, ESI†). The M05-2X/LanL2DZ/SMD scheme was found to be the most accurate computational level with the lowest RMSD value (0.694 Å). Consequently, we adopted this level for our DFT simulations. We noticed some discrepancies between DFT and the experiment regarding the attachment of the water to the complex. Therefore, we did not focus only on the experimental structure but considered the possibility of alternative structures and the possible merits of the other DFT approximations. In the following, we started with the structures of the complex obtained from the MD simulations and refined them using different DFT computational levels.
3 Results and discussion
The chelation process was evaluated from two main perspectives: the structure of the complexes and the impact of the charge distribution/protonation state on their stability. The mechanisms of complex formation and the corresponding structures revealed by the MD simulations are discussed in Section 3.1. The charge distribution variations and the complex formation energies obtained from the DFT calculations are presented in Section 3.2.3.1 Complex formation scenarios
The mechanism of EDTA–Hg2+ complex formation was studied using MD simulations. Different EDTA protonation states were considered by protonating the amino and carboxylate groups of EDTA. The EDTA protonation/charge states included thEDTA0, thEDTA1−, thEDTAa2−, thEDTAo2−, thEDTA3−, EDTA3−, EDTA2− and EDTA4−, where thEDTA0 is the EDTA structure with fully protonated carboxylate groups, EDTA3− and EDTA2− are the structures with protonated amino groups and EDTA4− is the fully deprotonated counterpart. In our notation of the thEDTA2− structures, the ‘a’ and ‘o’ indices indicate the protonation of the carboxylic acid groups positioned adjacent or opposite to the ethylene bridge of the EDTA molecule. The snapshots of the observed complexes are shown in Fig. 2. The corresponding radial distribution functions and running coordination numbers averaged over the collected MD trajectories are presented in Fig. 3–6 and Fig. S3–S7 (ESI†), where N and Np refer to deprotonated and protonated nitrogen atoms, respectively, Oc denotes oxygen atoms of the carboxylate groups, and Ow corresponds to oxygen atoms of water molecules. The radial distribution functions indicate preferable distances at which the atoms are located with respect to each other. The running coordination numbers allow us to estimate the number of atoms in the first coordination shell of the Hg2+ ion. The thEDTA0 case is not reported, because it did not form a complex with Hg2+ in our simulations. | ||
| Fig. 2 The EDTA–Hg2+ complexes and the water molecules in their first coordination shell. The complexes are obtained from the MD simulations at ambient conditions. The green, blue, red, grey and orange colours represent the carbon, nitrogen, oxygen, hydrogen and Hg atoms, respectively. | ||
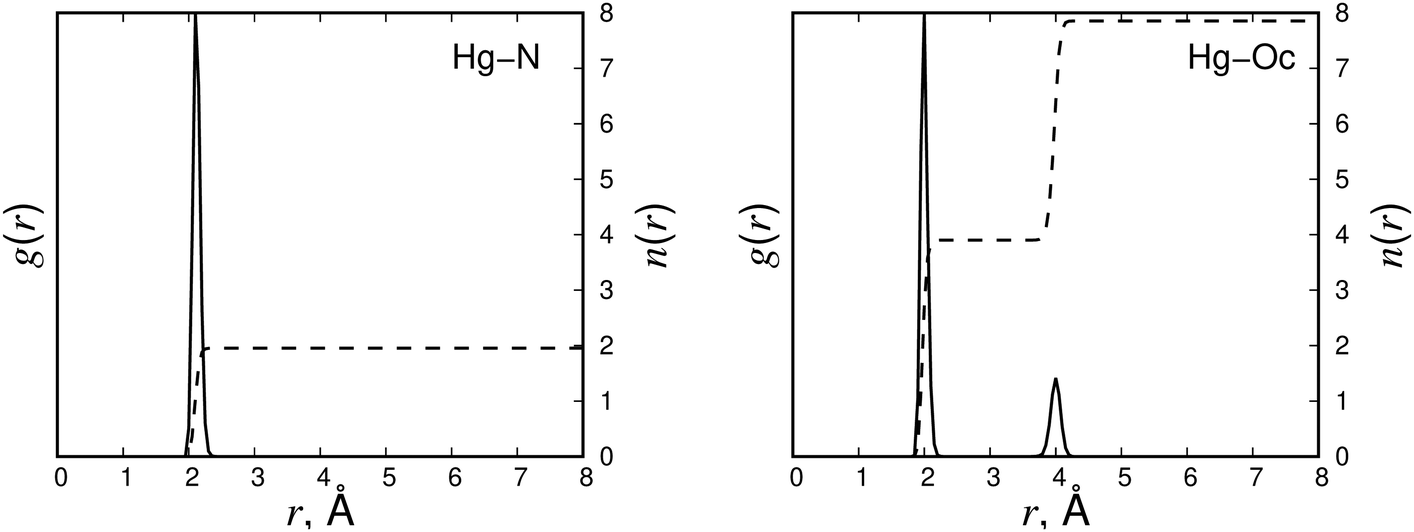 | ||
| Fig. 3 The radial distribution functions g(r) (solid line) and running coordination functions n(r) (dashed line) for EDTA4− + Hg2+. | ||
 | ||
| Fig. 4 The radial distribution functions g(r) (solid line) and running coordination functions n(r) (dashed line) for EDTA3− + Hg2+. | ||
 | ||
| Fig. 5 The radial distribution functions g(r) (solid line) and running coordination functions n(r) (dashed line) for EDTA2− + Hg2+. | ||
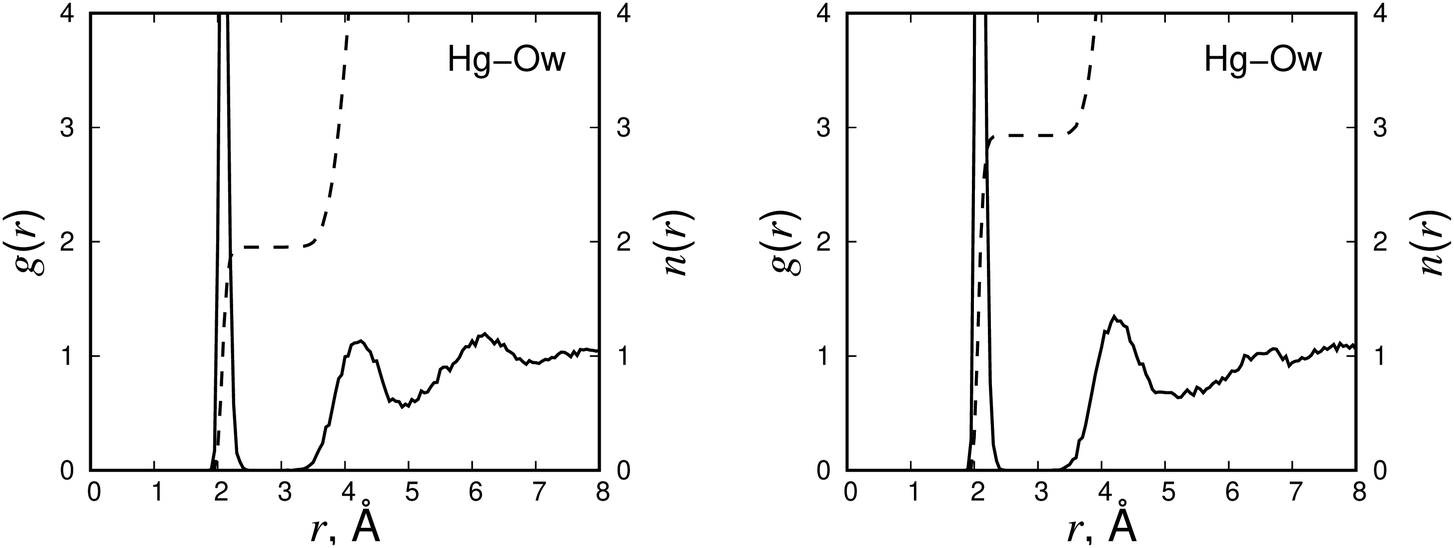 | ||
| Fig. 6 The radial distribution functions g(r) (solid line) and running coordination functions n(r) (dashed line) of Hg2+–Ow in the complexes of EDTA3− + Hg2+ (left panel) and EDTA2− + Hg2+ (right panel). | ||
According to Fig. 3, the formation of a complex from EDTA4− and Hg2+ results in a single sharp peak in the radial distribution function of Hg–N around 2.1 Å, indicating the strong binding of Hg2+ to the two EDTA nitrogen atoms. Also, the radial distribution function of Hg–Oc indicates the coordination of Hg2+ with four Oc atoms at the distance of 2.0 Å. This octahedral structure is the most stable EDTA–Hg2+ complex throughout the simulation runs. The observed relative stability is an important finding because EDTA is naturally observed in its fully deprotonated state at pH levels greater than 3.38,47,50,51
Moving from the fully deprotonated EDTA4− state to the EDTA3− structure with one protonated amino group (Fig. 4) we observe that Hg2+ detaches from one nitrogen atom, while it sustains its strong interaction with the second amino group. At the same time, it binds with three Oc atoms and dissociates from one carboxylate group. These changes in the coordination shell provide an opportunity for two water molecules to enter the first coordination shell of Hg2+ (Fig. 6, left panel). The protonation of the second amino group and the creation of EDTA2− (Fig. 5) causes further disturbance in the adsorption of Hg2+ by hindering Hg2+ binding to the amino groups. The hindrance pushes Hg2+ from the distance of 2.1 Å observed for the fully deprotonated EDTA to the distances above 3.5 Å. This does not affect the coordination of Hg2+ with the carboxylate groups. However, it frees up a coordination site to accommodate an additional water molecule (Fig. 6, right panel). Therefore, we can conclude that the protonation of the amino groups weakens the tight binding of Hg2+ to EDTA and promotes its hydration.
A similar effect can be observed by following the theoretical protonation trend, which refers to the EDTA structures with protonated carboxylate and deprotonated amino groups. According to Fig. S3 (ESI†), the protonation of one carboxylate group (thEDTA3−), leads to the permanent positioning of Hg2+ at 2.0–2.5 Å from the two nitrogen atoms of EDTA, and about 2.0 Å from the three deprotonated oxygens.
When two adjacent carboxylate groups are protonated (thEDTAa2−), Hg2+ binds to the two deprotonated Oc atoms with an average bond length of 2 Å (Fig. S4, ESI†) and to one deprotonated nitrogen atoms at about 2.2 Å. The same is obtained by protonating two opposite carboxylate groups of EDTA (thEDTAo2− + Hg2+), but the different conformation of EDTA molecule is observed in this case (Fig. S5, ESI†). It should be noted that we also found several alternative thEDTAo2−–Hg2+ configurations, where an additional bond appeared between Hg2+ and an oxygen of one carboxylate group, as well as a bond between Hg2+ and the second nitrogen atom. Nevertheless, these configurations of the complex appeared to be short-lived, lasting less than 1.5 ns. The most frequently observed thEDTAo2−–Hg2+ structure is illustrated in Fig. 2.
When three carboxylate groups are protonated (thEDTA1−), the Hg2+ ion binds to the deprotonated carboxylate oxygen at around 2 Å (Fig. S6, ESI†). Hg2+ stays rather far from the nearest nitrogen atom at the distance of about 3.9 Å, as in the case of EDTA2− with protonated amino groups.
As noted for the EDTA structures with protonated amino groups, the unbinding of Hg2+ from the N and Oc atoms in the theoretical protonation series, enhances Hg2+ hydration. Based on Fig. S7 (ESI†) there are 1, 3, 1–3 (with 3 being the most probable), and 5 water molecules in the first coordination shell of Hg2+ in the thEDTA3−, thEDTAa2−, thEDTAo2− and thEDTA1− complexes, respectively. In the EDTA4−–Hg2+ complex, the coordination shell of the ion contains no water molecules, in agreement with the DFT results presented in Fig. S2 (ESI†), displaying water detachment from Hg2+ after structural optimization using several DFT approximations.
The observed ion unbinding from the protonated N and Oc atoms and the increased ion hydration are in perfect agreement with the findings of Licup et al.47 regarding Eu3+ chelation to EDTA. Using extended X-ray absorption fine structure (EXAFS) measurements at pH 3 and 11, Licup et al. observed the increase of Eu–N bond distances resulting from carboxylate protonation and the exposure of Eu3+ to water. They also reported full detachment of Eu3+ from fully protonated EDTA, which is consistent with the fact that a stable Hg2+–thEDTA0 complex was not observed in our MD simulations.
3.2 Complex formation energies
One can expect that the changes in the hydration shells and the structure of the complex implicate varying Hg2+ adsorption energies and, therefore, uptake efficiencies. To confirm this, we first calculated the formation energies of the complexes obtained from the MD simulations. Then, we exported the complexes to DFT simulations and reoptimized the complex structures (and their isolated constituting components) at different computational levels. The complexation Gibbs free energies (Gcmpx) were calculated as the difference between the energy of the complex (GEDTA+Hg[+nH2O]) and its isolated components including the Hg2+ ion (GHg), water (GH2O), and the corresponding the EDTA conformer (GEDTA) through formula Gcmpx = GEDTA+Hg[+nH2O] – GEDTA – nGH2O – GHg, where n is the number of explicit water molecules (see Fig. 2). The results are summarized in Table 1.| Complex | MD | B3LYP/LanL2DZ | M05-2X/LanL2DZ | ωB97X-D/Def2-TZVPP |
|---|---|---|---|---|
| EDTA4− + Hg2+ | −181.7 ± 33.2 | −197.2 | −223.6 | −247.5 |
| EDTA3− + Hg2+ | −112.1 ± 33.7 | −72.8 | −100.4 | −108.9 |
| EDTA2− + Hg2+ | −141.9 ± 35.8 | −45.3 | −92.6 | −70.5 |
| thEDTA3− + Hg2+ | −115.4 ± 32.8 | −143.5 | −185.6 | −191.1 |
| thEDTAa2− + Hg2+ | −82.4 ± 33.1 | −81.0 | −82.5 | −89.6 |
| thEDTAo2− + Hg2+ | −77.6 ± 35.5 | −60.3 | −61.6 | −88.3 |
| thEDTA1− + Hg2+ | −63.4 ± 35.3 | −10.0 | −18.6 | 26.6 |
The MD results in Table 1 show that the higher the degree of EDTA protonation, the higher the Gibbs free energy of Hg2+ complex formation and the lower the stability of the complex. The results strongly suggest that the naturally occurring fully deprotonated EDTA structure provides the highest efficiency of Hg2+ uptake by enabling an octahedral chelation mode (see Fig. 2). Similarly, the DFT results show a significant increase in the free energy of complex formation upon protonation of the amino and carboxylate groups of EDTA. All levels of theory present a similar trend, though they display some numerical differences resulted from various DFT approximations. They all suggest the fully deprotonated EDTA4− state for Hg2+ removal. This suggestion is in line with the findings of earlier studies, which reported the formation of stable metal ion–EDTA4− complexes for Ln,48 and La, Ce, Pr, and Nd.84 Furthermore, all results point out that even partial EDTA deprotonation facilitates Hg2+ uptake, guaranteeing Hg2+ chelation over a wide range of pH.
A question that needs addressing is how the protonation state of EDTA affects the energies. The adsorption energy calculated from MD simulations equals the difference between the potential energy of the Hg2+ ion in the complex and its potential energy in bulk water. In MD, these energies consist of van der Waals interactions (computed using LJ parameters) and electrostatic contributions (estimated using partial atomic charges). The OPLS/AA force field suggests interaction parameters that differ depending on the protonation/deprotonation of the carboxylate or amino groups of EDTA, as detailed in Table S1 (ESI†). Specifically, there are significant changes in the partial charges of the oxygen and nitrogen atoms belonging to these groups (cf. O1/O2 vs. O3, N1 vs. N2), which play a crucial role in Hg–EDTA complexation. However, in classical MD, these changes are not influenced by the conformation of EDTA or the arrangement of the Hg2+ ion and water molecules. Further insights into the charge distribution in the EDTA molecule can be provided by DFT calculations, which we performed for a series of protonation states of EDTA at the M05-2X/LanL2DZ level of theory.
According to the atomic polar tensor (APT) charges reported in Fig. S8 and S9 (ESI†), protonation changes the charge distribution throughout the EDTA molecule. The extent of the changes depends on the proximity of atoms to the protonated centres and the conformation of the EDTA molecule. In the deprotonated carboxylate groups, oxygen atoms have a larger negative charge compared with protonated ones. The absence of positively charged hydrogen atoms and more negatively charged oxygens of deprotonated carboxylate lead to strong binding of EDTA with the positive Hg2+ ion. The deprotonated nitrogen atoms in amino groups also bear significant negative charges, thus they are involved in binding with the ion as well. In a completely deprotonated EDTA molecule (EDTA4−), this results in the formation of 6-fold coordination geometry around Hg2+ ion consisting of four carboxylate oxygen atoms and two amino nitrogen atoms, similar to that observed in MD simulations.
When one of the amino groups is protonated (EDTA3−), the negative charge accumulated on the nitrogen atom decreases, and the attached positively charged hydrogen atom shields the amino group from electrostatic interaction with the Hg2+ ion. This obviously weakens the attractive interaction between the ion and the protonated nitrogen atom, causing dissociation between them. The same is observed in MD simulations, where the reduction in the charge of protonated nitrogen is even more pronounced compared to the corresponding DFT predictions. On the other hand, the DFT calculations show that interaction between EDTA molecule and Hg2+ ion induces the transfer of some additional negative charge to the negatively charged carboxylate and deprotonated amino atoms, which balances the excessive positive charge of the Hg2+ ion acquired through its interaction with certain oxygen atoms of the water molecules. Simultaneously, the carboxylate oxygen atoms of EDTA and the hydrogen atoms of the water molecules can form hydrogen bonds and transfer some charge to stabilize the generated EDTA–Hg–water complex. It is observed, that only three oxygen atoms of carboxylate groups remain in the first coordination shell of the ion, what is in agreement with our MD simulations.
The ion dissociates from both nitrogen atoms when two amino groups are protonated (EDTA2−–Hg2+). Due to this, the structure of the Hg–EDTA complex becomes distorted, and Hg2+ ion binds only with one carboxylate group of EDTA. Two other bonds are created between the ion and the oxygen atoms of water molecules, which, at the same time, are bound to carboxylate groups through hydrogen bonds. Contrary to this, in MD simulations, the Hg2+ ion binds directly to three carboxylate groups.
Similarly, in the theoretical series, we observe significant changes in the partial charge of the protonated oxygen atoms. The changes are more significant for the protonated oxygen atom and lead to the localization of less negative charge on them. This is simultaneous with a slight decrease in the charge of the adjacent carbon atom, as well as a slight increase in the charge of the closest nitrogen atom. These changes weaken the electrostatic interaction of Hg2+ with the negatively charged centres, transferring less charge during the process and reducing the stability of the resultant complex. The charge transfer process manifests itself by increasing the partial charge of Hg2+ and the carbon atom bonded to the deprotonated oxygen atom and reducing the partial charge of the deprotonated oxygen and nitrogen atoms.
Therefore, the charge analysis of both theoretical and realistic EDTA protonation series ascertains that a higher degree of deprotonation can guarantee stronger binding of the mercury ion with the chelating agent. The deprotonated carboxylate groups and the deprotonated amino groups atoms are preferred for binding between the Hg2+ ion and EDTA molecule, offering more negatively charged centres for electrostatic interaction.
4 Conclusions
The combination of the MD and DFT simulation approaches demonstrates the importance of EDTA deprotonation in the uptake of mercury ions from water. According to our results, the availability of more deprotonated amino and carboxylate sites facilitates mercury interaction with EDTA, leading to stronger adsorption and a higher efficiency of mercury removal. As EDTA molecule complexed with mercury ion predominantly exists in the fully deprotonated state over a wide range of pH (greater than 3), its high chelation efficiency is expected at both alkaline and acidic conditions. Our findings about the effect of EDTA protonation on Hg–EDTA complex formation agree with earlier studies on EDTA chelation to other metal ions, which also highlight protonation effects on similar chelating agents such as HEDTA, NTA, DTPA, HEIDA, and GLDA.85 The control of pH conditions, and thus the protonation and charge distribution of chelating agents, is a crucial factor in optimizing chelation processes. The most efficient protonation state can be determined through both simulation and experimental methods.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
CSC (Finnish IT Center for Science) is acknowledged for computing resources. F. K. is grateful for the financial support of the Research Council of Finland (grant no. 346846). H. B. thanks the Finnish National Agency for Education (EDUFI Fellowship for doctoral students from Ukraine, reference number OPH-4602-2022) for financial support. J. I. expresses gratitude to the Research Council of Finland for financial support (reference number 334244). T. P. gratefully acknowledges the financial support from the National Research Foundation of Ukraine (grant no. 2023.05/0019).References
- I. Wagner-Döbler, H. von Canstein, Y. Li, K. N. Timmis and W.-D. Deckwer, Removal of mercury from chemical wastewater by microoganisms in technical scale, Environ. Sci. Technol., 2000, 34, 4628–4634 CrossRef.
- C. T. Driscoll, R. P. Mason, H. M. Chan, D. J. Jacob and N. Pirrone, Mercury as a global pollutant: Sources, pathways, and effects, Environ. Sci. Technol., 2013, 47, 4967–4983 CrossRef CAS PubMed.
- B. Gworek, W. Dmuchowski and A. H. Baczewska-Dabrowska, Mercury in the terrestrial environment: a review, Environ. Sci. Eur., 2020, 32, 128 CrossRef CAS.
- F. Zahir, S. J. Rizwi, S. K. Haq and R. H. Khan, Low dose mercury toxicity and human health, Environ. Toxicol. Pharmacol., 2005, 20, 351–360 CrossRef CAS PubMed.
- R. A. Bernhoft, Mercury toxicity and treatment: A review of the literature, J. Environ. Public Health, 2012, 2012, 1–10 CrossRef.
- E. Paduraru, D. Iacob, V. Rarinca, A. Rusu, R. Jijie, O.-D. Ilie, A. Ciobica, M. Nicoara and B. Doroftei, Comprehensive review regarding mercury poisoning and its complex involvement in Alzheimer's disease, Int. J. Mol. Sci., 2022, 23, 1992 CrossRef CAS.
- S. Venkateswarlu and M. Yoon, Surfactant-free green synthesis of Fe3O4 nanoparticles capped with 3, 4-dihydroxyphenethylcarbamodithioate: stable recyclable magnetic nanoparticles for the rapid and efficient removal of Hg(II) ions from water, Dalton Trans., 2015, 44, 18427–18437 RSC.
- National Primary Drinking Water Regulations, https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations, Accessed: May 2024.
- N. Ballav, R. Das, S. Giri, A. M. Muliwa, K. Pillay and A. Maity, L-cysteine doped polypyrrole (PPy@L-Cyst): A super adsorbent for the rapid removal of Hg2+ and efficient catalytic activity of the spent adsorbent for reuse, Chem. Eng. J., 2018, 345, 621–630 CrossRef CAS.
- Q. Wang, L. Zhang, X. Liang, X. Yin, Y. Zhang, W. Zheng, E. M. Pierce and B. Gu, Rates and Dynamics of Mercury Isotope Exchange between Dissolved Elemental Hg(0) and Hg(II) Bound to Organic and Inorganic Ligands, Environ. Sci. Technol., 2020, 54, 15534–15545 CrossRef CAS.
- I. Robles, J. Lakatos, P. Scharek, Z. Planck, G. Hernández, S. Solís and E. Bustos, in Environmental Risk Assessment of Soil Contamination, ed. M. C. Hernandez-Soriano, IntechOpen, Rijeka, 2014, ch. 29, pp. 827–830 Search PubMed.
- N. A. A. Qasem, R. H. Mohammed and D. U. Lawal, Removal of heavy metal ions from wastewater: a comprehensive and critical review, npj Clean Water, 2021, 4, 36 CrossRef CAS.
- B. Karwowska and E. Sperczyńska, Organic matter and heavy metal ions removal from surface water in processes of oxidation with ozone, UV irradiation, coagulation and adsorption, Water, 2022, 14, 3763 CrossRef.
- W. Ding, Y. Wang, W. Zeng, H. Xu and B. Chen, Preparation of heavy metal trapping flocculant polyacrylamide-glutathione and its application for cadmium removal from water, Polymers, 2023, 15, 500 Search PubMed.
- Y. Zhang and X. Duan, Chemical precipitation of heavy metals from wastewater by using the synthetical magnesium hydroxy carbonate, Water Sci. Technol., 2020, 81, 1130–1136 Search PubMed.
- Z.-J. Fu, S.-K. Jiang, X.-Y. Chao, C.-X. Zhang, Q. Shi, Z.-Y. Wang, M.-L. Liu and S.-P. Sun, Removing miscellaneous heavy metals by all-in-one ion exchange-nanofiltration membrane, Water Res., 2022, 222, 118888 Search PubMed.
- L. Gajdošová, M. Búgel and T. Bakalár, Heavy metal removal using reverse osmosis, Acta Montan. Slovaca, 2009, 14, 250–253 Search PubMed.
- B. Thaçi and S. Gashi, Reverse osmosis removal of heavy metals from wastewater effluents using biowaste materials pretreatment, Pol. J. Environ. Stud., 2018, 28, 337–341 CrossRef PubMed.
- K. C. Khulbe and T. Matsuura, Removal of heavy metals and pollutants by membrane adsorption techniques, Appl. Water Sci., 2018, 8, 19 CrossRef.
- H. Xiang, X. Min, C.-J. Tang, M. Sillanpää and F. Zhao, Recent advances in membrane filtration for heavy metal removal from wastewater: A mini review, J. Water Process Eng., 2022, 49, 103023 CrossRef.
- C. I. Covaliu-Mierlă, O. Păunescu and H. Iovu, Recent advances in membranes used for nanofiltration to remove heavy metals from wastewater: A review, Membranes, 2023, 13, 643 CrossRef PubMed.
- N. Singh, G. Nagpal, S. Agrawal and Rachna, Water purification by using adsorbents: A review, Environ. Technol. Innovation, 2018, 11, 187–240 CrossRef.
- D. Mercier and M.-G. Barthés-Labrousse, The role of chelating agents on the corrosion mechanisms of aluminium in alkaline aqueous solutions, Corros. Sci., 2009, 51, 339–348 CrossRef CAS.
- C. Oviedo and J. Rodríguez, EDTA: The chelating agent under environmental scrutiny, Quim. Nova, 2003, 26, 901–905 CrossRef CAS.
- E. K. B. Busch, EDTA: Ethylene diamine tetra acetic acid – A review, Occup. Med. Health, 2016, 04, 1–5 Search PubMed.
- S. L. Escayo and E. P. Enriquez, EDTA-modified magnetic graphite oxide for preconcentrating Hg2+ from water for XRF analysis, Phil. J. Nat. Sci., 2020, 25, 10–18 Search PubMed.
- S. Elkatatny, M. Mahmoud and B. Bageri, Evaluation of using HEDTA chelating agent to clean up long horizontal heterogeneous sandstone wells without divergent, J. Pet. Explor. Prod. Technol., 2017, 8, 165–173 CrossRef.
- E. Blaurock-Busch, DMPS in metal chelation, Biomed. J. Sci. Tech. Res., 2020, 30, 23206–23210 Search PubMed.
- G. N. George, R. C. Prince, J. Gailer, G. A. Buttigieg, M. B. Denton, H. H. Harris and I. J. Pickering, Mercury binding to the chelation therapy agents DMSA and DMPS and the rational design of custom chelators for mercury, Chem. Res. Toxicol., 2004, 17, 999–1006 Search PubMed.
- E. Repo, T. A. Kurniawan, J. K. Warchol and M. E. Sillanpää, Removal of Co(II) and Ni(II) ions from contaminated water using silica gel functionalized with EDTA and/or DTPA as chelating agents, J. Hazard. Mater., 2009, 171, 1071–1080 CrossRef CAS.
- E. Repo, J. K. Warchoł, A. Bhatnagar, A. Mudhoo and M. Sillanpää, Aminopolycarboxylic acid functionalized adsorbents for heavy metals removal from water, Water Res., 2013, 47, 4812–4832 CrossRef CAS PubMed.
- F. Zhao, E. Repo, D. Yin, Y. Meng, S. Jafari and M. Sillanpää, EDTA-Cross-Linked β-cyclodextrin: An environmentally friendly bifunctional adsorbent for simultaneous adsorption of metals and cationic dyes, Environ. Sci. Technol., 2015, 49, 10570–10580 CrossRef CAS.
- F. Zhao, E. Repo, Y. Meng, X. Wang, D. Yin and M. Sillanpää, An EDTA-β-cyclodextrin material for the adsorption of rare earth elements and its application in preconcentration of rare earth elements in seawater, J. Colloid Interface Sci., 2016, 465, 215–224 CrossRef CAS.
- K. Zhang, Z. Dai, W. Zhang, Q. Gao, Y. Dai, F. Xia and X. Zhang, EDTA-based adsorbents for the removal of metal ions in wastewater, Coord. Chem. Rev., 2021, 434, 213809 CrossRef CAS.
- M. H. Ridzwan, M. K. Yaakob, Z. M. Zabidi, A. S. Hamzah, Z. Shaameri, F. N. A. A. Rashid, K. Kassim, M. F. Mohammat, N. H. Pungot, M. A. M. Hamali, A. S. M. Sauri, F. Jaafar Azuddin, E. S. Majanun, Y. A. Sazali and M. Zuhaili Kashim, Computational insight into the quantum chemistry, interaction and adsorption energy of aminopolycarboxylic acid chelating agents towards metal cations, Comput. Theor. Chem., 2022, 1208, 113579 CrossRef CAS.
- A. Kovács, D. S. Nemcsok and T. Kocsis, Bonding interactions in EDTA complexes, J. Mol. Struct.: THEOCHEM, 2010, 950, 93–97 CrossRef.
- I. Robles, M. García, S. Solís, G. Hernández, Y. Bandala, E. Juaristi and E. Bustos, Electroremediation of mercury polluted soil facilitated by complexing agents, Int. J. Electrochem. Sci., 2012, 7, 2276–2287 CrossRef CAS.
- S. A. Thomas and J.-F. Gaillard, The molecular structure of aqueous Hg(II)-EDTA as determined by X-ray absorption spectroscopy, J. Phys. Chem. A, 2015, 119, 2878–2884 CrossRef CAS.
- M. Gajewski and M. Klobukowski, DFT studies of complexes between ethylenediamine tetraacetate and alkali and alkaline earth cations, Can. J. Chem., 2009, 87, 1492–1498 CrossRef CAS.
- N. A. F. Abdullah and L. S. Ang, Binding sites of deprotonated citric acid and ethylenediaminetetraacetic acid in the chelation with Ba2+, Y3+, and Zr4+ and their electronic properties: A density functional theory study, Acta Chim. Slovaca, 2018, 65, 231–238 CrossRef CAS.
- Q. Yuan, X.-T. Kong, G.-L. Hou, L. Jiang and X.-B. Wang, Electrospray ionization photoelectron spectroscopy of cryogenic [EDTA M(II)]2− complexes (M = Ca, V–Zn): Electronic structures and intrinsic redox properties, Faraday Discuss., 2019, 217, 383–395 RSC.
- H. J. Cho, S. H. Kim, J. Kang, K. Cho, H. Y. Lee, H. Kim, H. Ju, J. W. Choi, S. G. Kim, S.-G. Oh, C.-W. Lee and H. C. Yoon, Experimental and DFT studies on the equilibrium properties, kinetics, and mechanism of nitric oxide removal using metal-EDTA and ferrous thiochelates, Chem. Eng. J., 2022, 431, 134010 CrossRef.
- A. P. Shapiro and R. F. Probstein, Removal of contaminants from saturated clay by electroosmosis, Environ. Sci. Technol., 1993, 27, 283–291 CrossRef.
- J. Fábry and B. A. Maximov, Structure of bis(tetraethylammonium) hexaiododimercurate(II) diiodomercury(II), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 51–53 CrossRef.
- J.-Z. Ramírez, R. Vargas, J. Garza and B. P. Hay, Performance of the effective core potentials of Ca, Hg, and Pb in complexes with ligands containing N and O donor atoms, J. Chem. Theory Comput., 2006, 2, 1510–1519 CrossRef PubMed.
- A. O. Tirler and T. S. Hofer, A Comparative study of [CaEDTA]− and [MgEDTA]−: Structural and dynamical insights from quantum mechanical charge field molecular dynamics, J. Phys. Chem. B, 2015, 119, 8613–8622 CrossRef.
- G. L. Licup, T. J. Summers, J. A. Sobrinho, A. de Bettencourt-Dias and D. C. Cantu, Elucidating the structure of the Eu-EDTA complex in solution at various protonation states, Eur. J. Inorg. Chem., 2024, e202400042 CrossRef.
- R. D. OBrien, T. J. Summers, D. S. Kaliakin and D. C. Cantu, The solution structures and relative stability constants of lanthanide-EDTA complexes predicted from computation, Phys. Chem. Chem. Phys., 2022, 24, 10263–10271 RSC.
- Z. Szakács, S. Béni and B. Noszál, Resolution of carboxylate protonation microequilibria of NTA, EDTA and related complexones, Talanta, 2008, 74, 666–674 CrossRef.
- G. Schwarzenbach, R. Gut and G. Anderegg, Komplexone XXV. Die polarographische Untersuchung von Austauschgleichgewichten. Neue Daten der Bildungskonstanten von Metallkomplexen der Äthylendiamin-tetraessigsäure und der 1, 2-Diaminocyclohexan-tetraessigsäure, Helv. Chim. Acta, 1954, 37, 937–957 CrossRef.
- M. Latva, J. Kankare and K. Haapakka, Solution structures of europium (III) complexes of ethylenediaminetetraacetic acid, J. Coord. Chem., 1996, 38, 85–99 CrossRef.
- R. D. OBrien, T. J. Summers, D. S. Kaliakin and D. C. Cantu, Correction: The solution structures and relative stability constants of lanthanide-EDTA complexes predicted from computation, Phys. Chem. Chem. Phys., 2023, 25, 30735–30736 RSC.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun., 2022, 271, 108171 CrossRef.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The missing term in effective pair potentials, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef.
- M. B. Singh, V. H. Dalvi and V. G. Gaikar, Investigations of clustering of ions and diffusivity in concentrated aqueous solutions of lithium chloride by molecular dynamic simulations, RSC Adv., 2015, 5, 15328–15337 RSC.
- K. Anitha, S. Namsani and J. K. Singh, Removal of heavy metal ions using a functionalized single-walled carbon nanotube: A molecular dynamics study, J. Phys. Chem. A, 2015, 119, 8349–8358 CrossRef PubMed.
- A. K. Giri and M. N. D. Cordeiro, Heavy metal ion separation from industrial wastewater using stacked graphene membranes: A molecular dynamics simulation study, J. Mol. Liq., 2021, 338, 116688 CrossRef.
- A. I. Jewett, D. Stelter, J. Lambert, S. M. Saladi, O. M. Roscioni, M. Ricci, L. Autin, M. Maritan, S. M. Bashusqeh, T. Keyes, R. T. Dame, J.-E. Shea, G. J. Jensen and D. S. Goodsell, Moltemplate: A tool for coarse-grained modeling of complex biological matter and soft condensed matter physics, J. Mol. Biol., 2021, 433, 166841 CrossRef.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson, H. Nakatsuji, et al., Gaussian 16 Revision C.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- L. Chen, T. Liu and C. Ma, Metal complexation and biodegradation of EDTA and S,S-EDDS: A density functional theory study, J. Phys. Chem. A, 2010, 114, 443–454 CrossRef.
- M. Jaworska, G. Stopa and Z. Stasicka, Photochemical NO-removal and NOx-release in the presence of Fe-EDTA complexes. DFT calculations of electronic structure and spectroscopy of the [Fe (edta)(NO)]2− complex, Nitric oxide, 2010, 23, 227–233 CrossRef PubMed.
- S. Mitra, K. Werling, E. J. Berquist, D. S. Lambrecht and S. Garrett-Roe, CH mode mixing determines the band shape of the carboxylate symmetric stretch in Apo-EDTA, Ca2+-EDTA, and Mg2+-EDTA, J. Phys. Chem. A, 2021, 125, 4867–4881 CrossRef PubMed.
- M. M. Foreman, M. Alessio, A. I. Krylov and J. M. Weber, Influence of transition metal electron configuration on the structure of Metal-EDTA complexes, J. Phys. Chem. A, 2023, 127, 2258–2264 CrossRef.
- J. C. Belmont-Sánchez, M. E. García-Rubiño, A. Frontera, A. Matilla-Hernández, A. Castiñeiras and J. Niclós-Gutiérrez, Novel Cd(II) coordination polymers afforded with EDTA or trans-1,2-Cdta chelators and imidazole, adenine, or 9-(2-Hydroxyethyl) adenine coligands, Crystals, 2020, 10, 391 CrossRef.
- C. Liu, Y. Ouyang, B. Jia, Z. Zhu, J. Shi and H. Chen, Lead-enhanced gas-phase stability of multiply charged EDTA anions: a combined experimental and theoretical study, J. Mass Spectrom., 2012, 47, 769–777 CrossRef CAS.
- F. Adeowo, B. Honarparvar and A. Skelton, The interaction of NOTA as a bifunctional chelator with competitive alkali metal ions: a DFT study, RSC Adv., 2016, 6, 79485–79496 RSC.
- A. D. Becke, Density-functional thermochemistry. I. The effect of the exchange-only gradient correction, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- L. Fang, J. Liu, S. Ju, F. Zheng, W. Dong and M. Shen, Experimental and theoretical evidence of enhanced ferromagnetism in sonochemical synthesized BiFeO3 nanoparticles, Appl. Phys. Lett., 2010, 97, 242501 CrossRef.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- H. Zhu, Y. Liu, D. Li and K. Kang, Structural, electronic, and optical properties of Cu2XSnS4 (X = Be, Mg, Ca, Cd, Hg) semiconductors: first-principles calculations, Appl. Phys. A: Mater. Sci. Process., 2023, 129, 539 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- H. Schaefer, Methods of Electronic Structure Theory, Springer, US, 1977 Search PubMed.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- Chemcraft – graphical software for visualization of quantum chemistry computations. Version 1.8, build 654., https://www.chemcraftprog.com.
- A. Smerigan, S. Biswas, F. D. Vila, J. Hong, J. Perez-Aguilar, A. S. Hoffman, L. Greenlee, R. B. Getman and S. R. Bare, Aqueous structure of lanthanide-EDTA coordination complexes determined by a combined DFT/EXAFS approach, Inorg. Chem., 2023, 62, 14523–14532 CrossRef CAS PubMed.
- I. S. Pinto, I. F. Neto and H. M. Soares, Biodegradable chelating agents for industrial, domestic, and agricultural applications- a review, Environ. Sci. Pollut. Res., 2014, 21, 11893–11906 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The force field parameters used in the molecular dynamics simulations are given in Tables S1–S5. The radial distribution functions and running coordination numbers obtained for the theoretical series of EDTA molecules are shown in Fig. S3–S7. The DFT results at different computational levels are presented in Table S6, along with the root-mean-square deviation of the structures from the experimentally resolved Hg2+–EDTA–water complex. The corresponding structures are shown in Fig. S2, while the partial atomic charges of EDTA and Hg2+–EDTA complexes at the different protonation states are shown in Fig. S8 and S9. The atom coordinates for the considered configurations are provided in the last section. See DOI: https://doi.org/10.1039/d4cp02980a |
| This journal is © the Owner Societies 2024 |