
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Observation of metastable structures of the ethylene glycol–water dimer in helium nanodroplets†
Daniel W.
Polak
 ,
Lewis J. P.
Turnbull
,
Owen D.
Bass
,
Shengfu
Yang
and
Andrew M.
Ellis
*
,
Lewis J. P.
Turnbull
,
Owen D.
Bass
,
Shengfu
Yang
and
Andrew M.
Ellis
*
School of Chemistry, George Porter Building, University Road, Leicester, LE1 7RH, UK. E-mail: andrew.ellis@leicester.ac.uk
First published on 8th October 2024
Abstract
Ethylene glycol (EG) is the simplest organic diol. Here we measure infrared spectra of the EG monomer and its dimer with water, the complex, EG(H2O), embedded in superfluid helium nanodroplets. For the monomer, only a single, gauche, conformation is observed. For EG(H2O), no trace of the global energy minimum is seen, a structure that would maximize the hydrogen bonding contacts. Instead, only metastable structures are formed, suggesting that dimerization in a superfluid environment leads to kinetic trapping in local energy minima. In addition, we obtain evidence for a dimer where the conformation of EG switches from gauche to trans on account of dimerization with a water molecule. This observation is assumed to be driven over an energy barrier by utilizing the energy released as hydrogen bonding occurs.
Introduction
Ethylene glycol (EG), is the simplest organic diol and has the chemical formula (CH2OH)2. EG is familiar to many through its use as an antifreeze agent. As a sugar alcohol, EG is the reduced form of glycolaldehyde, the simplest aldehyde sugar species. Both ethylene glycol and glycolaldehyde have been observed in space, including the observation of EG in cometary ices.1–3 EG has also been identified in meteorites.4 The presence of these species in astrochemical environments has been linked to reactions allowing the formation of prebiotic sugars, a precursor to complex life.1–3 Laboratory experiments have also shown that the presence of water has an impact on these reaction pathways.2 Given that amorphous water is expected on the surface of dust grains in the interstellar medium,5 it is therefore important in an astrochemical context to understand how simple sugar/sugar alcohols interact with water.Computational studies, sometimes in combination with experimental work, have explored the potential energy landscape of the EG monomer.6–14 This is quite complex owing to the possibility of internal rotation around the CO and CC bonds. Ten distinct stereoisomeric energy minima were initially identified by theoretical work.9–11 The notation commonly used for these conformers comprises three letters representing a gauche or trans orientation for groups around the C–C and two C–O bonds, where an upper case letter (T/G) is employed for the C–C bond and lower case (t/g) for the C–O bonds.9–11 With this notation the global energy minimum is labelled as tG+g−, where the + and − indicate, in effect, the relative orientations of the two OH groups. In view of its energy relative to other conformers, the global energy minimum is expected to make up 58% of ethylene glycol at room temperature.10 The three lowest energy conformers are all gauche with respect to rotation about the C–C bond, so are designated with a G as the central letter. The second and third lowest energy conformers are thought to be the g+G+g− and g−G+g− conformers, which have been calculated to lie 122 and 297 cm−1 above the global minimum,14 respectively. To simplify the notation for the rest of this paper, we will label the three lowest energy conformers referred to above as G1, G2 and G3, with G1 being the global energy minimum. It is noteworthy that the G1 and G2 conformers gain added stability because they are the only two conformers of EG which are capable of significant intramolecular hydrogen bonding. Three trans conformers lie ca. 900 cm−1 above the global energy minimum, and are expected to make a negligible contribution to EG at room temperature.14
Experimental measurements using electron diffraction,15 infrared (IR) spectroscopy16–20 and microwave spectroscopy1,3,21–25 are all in agreement that the most abundant conformer of the EG monomer is the G1 conformer. While the electron diffraction and microwave studies could find no evidence for any of the other low energy conformers, IR spectroscopy of EG in a cryogenic argon matrix yielded additional peaks of significant intensity in the OH stretching region.17–20 These additional peaks have been suggested to originate from either the coexistence of G1 with a subpopulation of G2 and G317,19,20 or a distortion of the G1 conformer by the argon matrix.18 The lack of low temperature measurements without possible interference from site effects in a rigid argon matrix limits the ability to confirm the assignment.
The few experimental studies conducted on the interaction of EG with water have focused on bulk solutions26 or at air/liquid interfaces.27 In the bulk solution, Guo et al. measured the relative strength of IR absorption features assigned to conformers with a gauche or trans orientation of the central C–C bond.24 By modulating the relative fraction of water and EG, the proportion of trans conformers was shown to become significant, with only 42% of the population being gauche conformers in the EG rich water/EG solution.26 This is a remarkable change given the almost complete absence of trans conformers in pure EG at room temperature. Alongside the experimental work, several computational studies have been conducted for EG/water complexes to search for conformations and hydrogen bonding motifs, starting from the C–C bond in both gauche11,28–30 and trans31,32 orientations. However, the complex conformational landscape, alongside the challenge in interpreting experimental observations from relatively low-resolution measurements on macroscopic systems, suggests the need to explore a simpler system, and indeed there is currently no experimental information on the most elementary dimer comprising a single EG molecule and a water molecule.
Here we address this deficiency by recording IR spectra of the EG(H2O) dimer embedded in helium nanodroplets. Encapsulation in helium droplets offers a number of advantages over similar experiments in the gas phase. First, molecules will naturally cluster together inside a helium droplet, making it easy to form molecular clusters and complexes. Second, this can be achieved at low temperature, which helps minimise spectral congestion through the depopulation of multiple rotational states. Third, mass-selective IR spectra can be readily recorded using a signal depletion technique.33
Remarkably, we see no evidence for any formation of the global energy minimum for the dimer. Instead, only metastable structures are formed under our experimental conditions. In particular, we find that the addition of a single water molecule can promote trans conformations of EG, an observation in line with previous studies of bulk solutions.
Materials and methods
Experimental
The experimental setup used for this study has been described previously.34–36 In brief, helium nanodroplets were prepared via supersonic expansion of pre-cooled helium gas (16 K, 32 bar) into a low-pressure chamber through a 5 μm aperture. Under these conditions a mean droplet size of approximately 5000 helium atoms is expected.33 The droplets were formed into a beam by passage through a skimmer before entering a second vacuum chamber, where dopant molecules could be added. Two dopants were used, ethylene glycol ((CH2OH)2) and water, at room temperature. Each could be independently added by passing the appropriate vapour through a needle valve heated to 40 °C, the heating being necessary to enable stable and consistent flow of dopant vapour. For ethylene glycol monomer, this delivered a partial pressure of 1 × 10−6 mbar and was used throughout these experiments. To make EG(H2O) complexes, a second pickup region was employed, and water was added at a partial pressure of 3 × 10−6 mbar.After doping, the droplets were illuminated by the output of an optical parametric oscillator/amplifier (Laservision; tuneability 2500–4000 cm−1, 4 cm−1 bandwidth, 6–10 mJ pulse energy) pumped by the 1064 nm output of a Nd:YAG laser (Continuum Surelite II; 5 ns pulse duration, 650 mJ per pulse, 10 Hz repetition rate). After excitation by the laser pulse, the droplets travel into the ionization region of a quadrupole mass spectrometer (Extrel MAX-1000), where they can collide with an electron beam (90 eV). The mass spectrometer was tuned to focus on a specific m/z channel and the signal was collected as a function of laser wavelength using a photon counter (Stanford Research Systems, SR400). When the laser is resonant with a vibrational transition in the dopant molecule/cluster, energy is eventually transferred into the droplet, causing evaporative loss of helium atoms and hence a reduction in the droplet size. The resulting smaller electron ionization cross section reduces the ion signal compared to the baseline level obtained without the laser. Recording the reduction in ion signal as a function of laser wavelength then delivers an IR spectrum.
Computational details
Geometry optimizations and subsequent vibrational frequency calculations were conducted via second-order Møller–Plesset perturbation theory (MP2) using Gaussian 16.37 For both EG and EG(H2O), aug-cc-pVTZ basis sets were used. Some calculations were also performed on EG dimers and EG–(H2O)2 to aid the spectral assignment process and in these cases a smaller basis set was selected (aug-cc-pVDZ) to keep the computational cost down. In all cases counterpoise corrections were employed to account for any basis set superposition error.38,39 Anharmonic vibrational calculations were also conducted at the same level of theory for each system using the generalised second-order vibrational perturbation theory (GVPT2) approach.40–42Results and discussion
Mass spectrometry
A mass spectrum obtained from the addition of EG to helium droplets is shown in Fig. 1(a). For reference, the mass/charge (m/z) ratio of the parent ion of ethylene glycol is at 62. The main features in the mass spectrum in Fig. 1(a) are consistent with the electron ionization mass spectrum of EG in the gas phase. Thus, for example, the most abundant ion arises from the C–C fission process in EG, yielding CH2OH+ ions (at m/z 31).43,44 A significant fragment ion is also expected at m/z 33. Another group of peaks, spanning m/z 43–45, are also expected by comparison with the gas phase spectrum, and these presumably arise from C2HnO+ ions, where n = 3–5. The EG parent ion, at m/z 62, is very weak in Fig. 1(a), and this is expected by comparison with the gas phase spectrum. A peak is seen at m/z 61 which is much more intense than anticipated when compared with the gas phase mass spectrum, and which is presumably due to C2H5O2+. In other words, the parent ion, minus one hydrogen atom, is a significant product. We have seen similar behaviour for other alcohols45 and, more broadly, it is worth emphasising that significant differences in ion fragmentation branching ratios can often be seen in the electron ionization mass spectrum of molecules in the gas phase when compared to those in helium nanodroplets.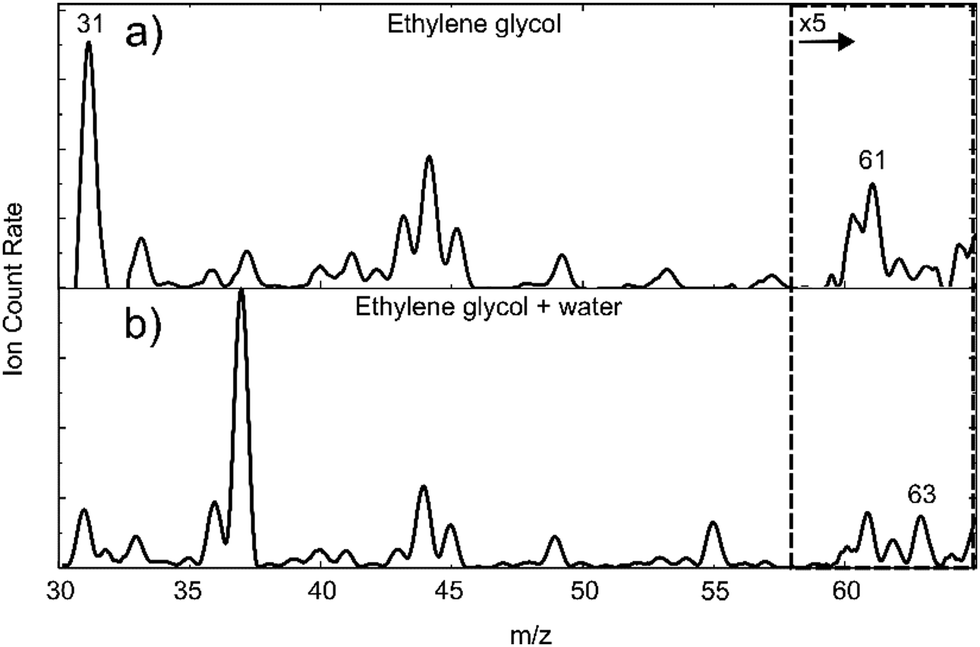 | ||
| Fig. 1 Mass spectrum spanning m/z 30–65 for (a) ethylene glycol and (b) ethylene glycol with water in helium nanodroplets. For both samples a mass spectrum without dopant(s) was recorded and subtracted to remove background signals from the spectra shown above. For the full spectra prior to subtraction, see Fig. S1 and S2 in the ESI.† All mass spectra were taken in the absence of a laser pulse. | ||
In Fig. 1(b) a mass spectrum is shown where water vapour is added downstream of the EG pickup cell. This introduces new peaks into the mass spectrum when compared with Fig. 1(a). The most prominent are at m/z 37 and m/z 55 and are attributed to (H2O)2H+ and (H2O)3H+ ions. Most significant to the present work is the peak at m/z 63, which we assign to the protonated EG monomer. This ion cannot be formed from the EG monomer alone, so it must come from a larger species involving EG. We can rule out the (EG)2 dimer, as this peak would also have to be prominent in Fig. 1(a). It must therefore be a consequence of the ionization of EG(H2O), with possible additional contributions from EG(H2O)2 and larger complexes.
IR spectroscopy
 | ||
| Fig. 2 (a) IR depletion spectrum of ethylene glycol in helium nanodroplets and (b) calculated anharmonic spectra for the four lowest energy conformers, G1, G2, G3 and T1. The structure of these four conformers are shown in images (c)–(f). The energies shown beneath the structures are relative to the lowest energy conformer, G1. | ||
As mentioned earlier, studies of EG in an argon matrix previously identified the same dominant conformer but additional strong peaks were seen which were attributed either to a subpopulation of higher energy conformers17,19,20 or spectral splitting caused by the occupation of distinct sites within the solid argon matrix.18 In the superfluid environment of a helium nanodroplet site-splitting is not possible. Although the temperature within a helium nanodroplet is typically lower than that used for argon matrix studies, the observation of multiple conformers with significant abundances in an argon matrix does seem unlikely and so we suspect that the observation of additional features in the argon matrix work was a consequence of multiple site effects.
In addition to the OH stretching region, we also observe a series of peaks in the CH stretching region (see Fig. S3 in the ESI†). The observed peaks in this region are once again consistent with the broader features measured previously for room temperature EG vapour.16 Unlike the OH region, the CH stretching peaks are expected to be influenced by strong anharmonic effects and are not diagnostic of the structures present, so will not be discussed any further.
 | ||
| Fig. 3 (a) The IR spectrum in the OH stretching region from an EG/H2O mixture in helium nanodroplets. (b) Anharmonic IR spectra calculated for the conformers EW4, 6, 7, and 8 shown in Fig. 4. For the calculated spectra the intensities were weighted at ratios of 1.2/5/1/1.5 for EW4/EW6/EW7/EW8, in order to give the best agreement with experiment. | ||
In effect we have already ruled out a significant contribution from (EG)2 earlier, as there is no clear peak at m/z 63 in the mass spectrum obtained by picking up EG molecules in the absence of water (Fig. 1(a)), and nor are there any additional features in the IR spectrum of Fig. 2(a) beyond those associated with the monomer. Nevertheless, we can categorically eliminate any such contribution through a comparison of the IR spectrum in Fig. 3(a) with those calculated for the various dimer structures, and further details can be found in the ESI.† As expected, given that we operated with deliberately low partial pressures of EG, the abundance of (EG)2 dimers is too low to be observed. The only plausible assignment is therefore that the peaks seen in Fig. 3(a) arise from EG(H2O)n water clusters.
As we also used a relatively low partial pressure of water vapour, we expect clusters containing only one water molecule to dominate the IR spectrum. To confirm this, we have compared the calculated anharmonic IR spectra of the ten EG(H2O)2 structures to the experimentally observed spectrum (see Fig. S7 in the ESI†). For all structures the number of peaks, peak positions and intensities are very different from the relatively simple spectral features seen in Fig. 3(a). Furthermore, no combination of predicted EG(H2O)2 spectra is able to mimic the observed features, allowing us to rule out contributions from complexes containing two water molecules.
We therefore attribute the spectral features seen in Fig. 3(a) to EG(H2O). In order to arrive at a more specific structural assignment, ab initio calculations were carried out in which the position of the water molecule and the starting structure of EG were systematically varied. Nine distinct energy minima were found, including both gauche and trans orientations about the CC bond of the EG monomer. Fig. 4 shows all nine calculated structures, which are labelled as EW1-9, and their energies relative to the lowest energy conformer. All five gauche11,28–30 (EW1, 3–6) and two trans31,32 (EW8, EW9) minima identified for the EG(H2O) system in previous computational work were obtained along with two additional structures (EW2, EW7) involving the water molecule sitting out of plane, both of which were not identified in earlier work.
 | ||
| Fig. 4 All nine minimum energy structures calculated for the EG(H2O) dimer with energies given relative to the minimum energy structure, EW1. | ||
For all structures we observe the formation of intermolecular hydrogen bonds, and these can be categorized into two distinct groups. In one group a single hydrogen bond forms between an OH bond of water and an oxygen atom in EG, such as for EW2, or between the OH of EG and an oxygen atom of water, as for EW5. In the other group are EW1, EW3 and EW6, all of which have two intermolecular hydrogen bonds forming a ring structure. For EW6 this involves the two OH bonds of EG acting as proton donors to the O atom on the water molecule, leaving both OH bonds of the water molecule outside of the hydrogen bonding system. For EW1 and EW3 a ring structure is constructed via formation of two different types of hydrogen bond, one involving OH on EG as the donor and the other involving an OH on water as the donor.
A comparison of the calculated anharmonic IR spectra for EW1, EW2, EW3, EW5, and EW9 with the experimental spectrum is shown in Fig. S8 in the ESI.† For all five of these conformers the calculations predict an intense peak red-shifted by 100 cm−1 beyond anything seen in our experiment, enabling elimination of any contribution from these conformers. It is immediately noteworthy that the global energy minimum, EW1, is omitted as a possible spectral contributor and we will discuss the significance of this shortly.
From the conformers that remain under consideration, no single conformer yields features matching all of those seen in Fig. 3(a). It is therefore clear that at least two, and possibly more, conformers must be contributing. We have considered various possible contributions from different conformers but our best explanation of the experimental findings is that four distinct conformers contribute, namely EW4, EW6, EW7 and EW8. All four have peaks which align with one of the four major features in the experimental spectrum. The largest peak in the spectrum, at ca. 3715 cm−1, is in reasonable agreement with the intense peak predicted for EW6 and is assigned to the symmetric stretch of the OH bonds of the water molecule, neither of which are involved in hydrogen bonding and so are oriented away from EG in this conformer (Fig. 4). This peak is located at a frequency which is characteristic of ‘dangling’ OH bonds, as it does not show the strong red-shift expected for OH bonds involved in hydrogen bonding. Note that a second peak is predicted for this conformer at just below 3600 cm−1. A weak and broad feature is seen at this position in the experimental spectrum but is not as intense as that implied from the calculations.
Three of the conformers, EW4, EW7, and EW8, all contain a peak which aligns with the triple peak structure around 3525 cm−1. In all cases these peaks are strongly red-shifted from the free OH stretch, suggesting they all derive from OH bonds involved in hydrogen bonding. Here, one of the OH bonds of water acts as a proton donor and an O atom on the EG molecule as a proton acceptor, with the other OH bond acting as a free OH group. The OH stretching features from this free OH group in these conformers are expected to lie close to the strong band attributed to EW6, and in all likelihood will be masked by the latter. With these assignments, we can account for all of the main features in the experimental IR spectrum.
Interestingly, all four assigned conformers, EW4, EW6, EW7 and EW8, lie significantly above the global energy minimum. To identify the contribution from each structure, weighting factors were applied to each predicted spectrum to best fit the experimental data, resulting in estimated population ratios. On this basis the majority contributor (57%) is EW6. The reason for the preferred formation of these conformers, and EW6 in particular, is unclear. Clearly, if the system was fully equilibrated the majority species seen would be EW1. However, formation of clusters and complexes in helium droplets frequently favours the formation of higher energy isomers, as is the case for the water hexamer or acetic acid dimers,34,46–48 for example. This has been attributed to the low temperature and the rapid cooling made possible by superfluid helium, which can trap clusters in shallow energy minima as they begin to form. Thus, if there is some dynamical effect which steers molecules initially into a specific conformation as they approach each other, then that conformer may cool so rapidly that it is unable to surmount any energy barrier. We have, in effect, kinetic trapping taking place in the case of EG(H2O) clusters.
Another interesting finding is that one of the assigned structures, EW8, involves EG in a trans conformation, and specifically the g+Tg− conformation. According to our analysis this structure constitutes ca. 14% of the overall EG–H2O mixture in helium nanodroplets. The observation of a trans conformer of EG in EG–H2O clusters is in stark contrast to the IR spectrum of the monomer in Fig. 2(a), which can be assigned exclusively to the lowest energy gauche conformer. The likely explanation for the trans conformation in EG(H2O) is structural rearrangement of the EG as it forms an intermolecular hydrogen bond with the water molecule. According to our calculations, the trans conformer is 0.121 eV above the lowest energy monomer. The intermolecular hydrogen bond energy is roughly twice this energy difference and so, on hydrogen bond formation, there is sufficient energy released to exceed the energy needed to rearrange the EG into the all trans configuration. The likelihood of this annealing process happening will also depend on the subtle dynamics of the collisional encounter between the EG and H2O molecules and the rate of cooling by the surrounding helium, so by no means all of the collisions would be expected to lead to the all-trans structure. Nevertheless, if our assignment is correct, the presence of a single water molecule delivers some significant population of trans EG, which ties in with the apparent observation of trans conformers in bulk EG/water solutions.26
Conclusions
IR spectra of ethylene glycol monomer and ethylene glycol clustered with a single water molecule have been recorded in helium nanodroplets. In the case of EG(H2O), this is the first time an optical spectrum of this dimer has been recorded.For the monomer, the spectroscopy is simple and consistent with the presence of only the lowest energy conformer, which has a gauche arrangement of the substituents about the C–C bond. However, for the EG(H2O) heterodimer several peaks are observed in the IR spectrum. Our experiments show that the lowest energy conformer does not contribute to the IR spectrum; instead, the observed spectra are attributed to four distinct higher energy conformers. Furthermore, one of these involves the all-trans structure of the ethylene glycol monomer, despite this not being observed in the absence of water. In order to form this, the ethylene glycol would need to change conformation as it interacts with the water molecule. Our findings imply that even a single water molecule can stimulate the formation of trans conformers of ethylene glycol.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was made possible by funding from the Leverhulme Trust through the grant RP10G0600.References
- J. M. Hollis, F. J. Lovas, P. R. Jewell and L. H. Coudert, Astrophys. J., 2002, 571, 59 CrossRef.
- R. L. Hudson, M. H. Moore and A. M. Cook, Adv. Space Res., 2005, 36, 184 CrossRef CAS.
- J. Crovisier, D. Bockelee-Morvan, N. Biver, P. Colom, D. Despois and D. C. Lis, Astron. Astrophys., 2004, 418, L35 CrossRef CAS.
- G. Cooper, N. Kimmich, W. Belisle, J. Sarinana, K. Brabham and L. Garrel, Nature, 2001, 414, 879 CrossRef CAS PubMed.
- F. Dulieu, L. Amiaud, E. Congiu, J. H. Fillion, E. Matar, A. Momeni, V. Pirronello and J. L. Lemaire, Astron. Astrophys., 2010, 512, A30 CrossRef.
- F. Podo, G. Némethy, P. L. Indovina, L. Radics and V. Viti, Mol. Phys., 1974, 27, 521 CrossRef CAS.
- P. I. Nagy, W. J. Dunn, G. Alagona and C. Ghio, J. Am. Chem. Soc., 1991, 113, 6719 CrossRef CAS.
- P. I. Nagy, W. J. Dunn, G. Alagona and C. Ghio, J. Am. Chem. Soc., 1992, 114, 4752 CrossRef CAS.
- C. J. Cramer and D. G. Truhlar, J. Am. Chem. Soc., 1994, 116, 3892 CrossRef CAS.
- D. L. Howard, P. Jørgensen and H. G. Kjaergaard, J. Am. Chem. Soc., 2005, 127, 17096 CrossRef CAS PubMed.
- D. L. Crittenden, K. C. Thompson and M. J. T. Jordan, J. Phys. Chem. A, 2005, 109, 2971 CrossRef CAS PubMed.
- O. Guvench and A. D. MacKerell, J. Phys. Chem. A, 2006, 110, 9934 CrossRef CAS PubMed.
- P. I. Nagy, Int. J. Molec. Sci., 2014, 15, 19562 CrossRef CAS PubMed.
- R. Boussessi, M. L. Senent and N. Jaïdane, J. Chem. Phys., 2016, 144, 164110 CrossRef CAS PubMed.
- O. Bastiansen, Acta Chem. Scand., 1949, 3, 415 CrossRef CAS.
- P. Buckley and P. A. Giguére, Can. J. Chem., 1967, 45, 397 CrossRef CAS.
- T. K. Ha, H. Frei, R. Meyer and H. H. Günthard, Theoret. Chim. Acta., 1974, 34, 277 CrossRef CAS.
- H. Frei, T. Ha, R. Meyer and H. H. Günthard, Chem. Phys., 1977, 25, 271 CrossRef CAS.
- H. Takeuchi and M. Tasumi, Chem. Phys., 1983, 77, 21 CrossRef CAS.
- C. G. Park and M. Tasumi, J. Phys. Chem., 1991, 95, 2757 CrossRef CAS.
- D. Christen, L. H. Coudert, R. D. Suenram and F. J. Lovas, J. Mol. Spectrosc., 1995, 172, 57 CrossRef CAS.
- D. Christen, L. H. Coudert, J. A. Larsson and D. Cremer, J. Mol. Spectrosc., 2001, 205, 185 CrossRef CAS PubMed.
- L. P. Kuhn, J. Am. Chem. Soc., 1951, 74, 2492 CrossRef.
- E. Walder, A. Bauder and H. H. Günthard, Chem. Phys., 1980, 51, 223 CrossRef CAS.
- W. Caminati, J. Mol. Spectrosc., 1981, 90, 512 CrossRef.
- Y. C. Guo, C. Cai and Y. H. Zhang, AIP Adv., 2018, 8 Search PubMed.
- E. L. Hommel, J. K. Merle, G. Ma, C. M. Hadad and H. C. Allen, J. Phys. Chem. B, 2005, 109, 811 CrossRef CAS PubMed.
- P. Manivet and M. Masella, Chem. Phys. Lett., 1998, 288, 642 CrossRef CAS.
- R. A. Klein, J. Comput. Chem., 2002, 23, 585 CrossRef CAS PubMed.
- R. M. Kumar, P. Baskar, K. Balamurugan, S. Das and V. Subramanian, J. Phys. Chem. A, 2012, 116, 4239 CrossRef CAS PubMed.
- M. A. Krest’yaninov, A. G. Titova and A. M. Zaichikov, Russ. J. Phys. Chem. A, 2017, 91, 305 CrossRef.
- A. Chaudari and S. A. Lee, J. Chem. Phys., 2004, 120, 7464 CrossRef PubMed.
- J. P. Toennies and A. F. Vilesov, Angew. Chem., Int. Ed., 2004, 43, 2622 CrossRef CAS PubMed.
- J. A. Davies, M. W. D. Hanson-Heine, N. A. Besley, A. Shirley, J. Trowers, S. Yang and A. M. Ellis, Phys. Chem. Chem. Phys., 2019, 21, 13950 RSC.
- J. A. Davies, M. Mugglestone, S. Yang and A. M. Ellis, J. Phys. Chem. A, 2020, 124, 6528 CrossRef CAS PubMed.
- M. I. Sulaiman, S. Yang and A. M. Ellis, J. Phys. Chem. A, 2017, 121, 771 CrossRef CAS PubMed.
- M. J. Frisch et al. Gaussian 16, Revision C.01., Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. F. Boys and F. Bernardi, Molec. Phys., 1970, 19, 553 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024 CrossRef CAS.
- J. A. Bloino, J. Phys. Chem. A, 2015, 119, 5269 CrossRef CAS PubMed.
- J. Bloino and V. A. Barone, J. Chem. Phys., 2012, 136, 124108 CrossRef PubMed.
- P. R. Franke, J. F. Stanton and G. E. Douberly, J. Phys. Chem. A, 2021, 125, 1301 CrossRef CAS PubMed.
- R. N. Katz, T. Chaudhary and F. H. Field, Int. J. Mass Spectrom. Ion Processes, 1987, 78, 85 CrossRef CAS.
- G. Leseigneur, J. H. Bredehöft, T. Gautier, C. Giri, H. Krüger, A. J. MacDermott, U. J. Meierhenrich, G. M. Muñoz Caro, F. Raulin, A. Steele, C. Szopa, W. Thiemann, S. Ulamec and F. Goesmann, ChemPlusChem, 2022, 87, e202200116 CrossRef CAS PubMed.
- S. Yang, S. M. Brereton, M. D. Wheeler and A. M. Ellis, Phys. Chem. Chem. Phys., 2005, 7, 4082 RSC.
- K. Nauta and R. E. Miller, Science, 1999, 283, 1895 CrossRef CAS PubMed.
- K. Nauta and R. E. Miller, Science, 2000, 287, 293 CrossRef CAS PubMed.
- J. D. Pickering, B. Shepperson, L. Christiansen and H. Stapelfeldt, J. Chem. Phys., 2018, 149, 154306 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02899f |
| This journal is © the Owner Societies 2024 |