
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-dimensional antimonene as a potential candidate for dioxin capture†
Moyassar
Meshhal
 *a,
Ashour A.
Ahmed
a,
Mohamed F.
Shibl
b,
Saadullah
Aziz
c,
Oliver
Kühn
a and
Kamal A.
Soliman
*d
*a,
Ashour A.
Ahmed
a,
Mohamed F.
Shibl
b,
Saadullah
Aziz
c,
Oliver
Kühn
a and
Kamal A.
Soliman
*d
aInstitute of Physics, University of Rostock, Albert-Einstein-Str. 23-24, 18059 Rostock, Germany. E-mail: moyassar.meshhal@uni-rostock.de
bDepartment of Chemistry, Faculty of Science, Cairo University, Giza, Egypt
cChemistry Department, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia
dDepartment of Chemistry, Faculty of Science, Benha University, P.O. Box 13518, Benha, Egypt. E-mail: kamal.soliman@fsc.bu.edu.eg
First published on 5th August 2024
Abstract
Among the serious environmental problems that attracted much attention from the broader public is the high toxicity of dioxins. Considerable efforts have been made to develop techniques and materials that could help in their efficient removal from the environment. Due to its high specific surface area, numerous active sites, and outstanding structural and electronic properties, antimonene is considered for a variety of potential applications in different fields such as energy storage, electrocatalysis, and biomedicine. The present study adds to this portfolio by suggesting antimonene as a promising candidate for dioxin capture. Using density functional theory calculations, we studied the adsorption of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on pristine as well as Ca-, Ti-, and Ni-doped antimonene. Three spatial configurations of the adsorption of TCDD on antimonene were analyzed. The results obtained from the calculation of adsorption energies, charge transfer, and densities of states provide evidence that antimonene outperforms other nanomaterials that have been previously suggested for dioxin capture applications. Therefore, we propose these substrates (i.e., pristine and doped antimonene) as potential capture agents for removing such toxic organic pollutants.
1 Introduction
The widespread environmental issue of the toxicity of organic pollutants has garnered significant attention and concern within both the scientific community and the general public. Dioxins, such as polychlorinated dibenzodioxins (PCDD) and polychlorinated dibenzo-furans (PCDF), represent a subset of these harmful organic pollutants. They originate from various sources like waste incineration, cigarette smoke, car exhaust, and natural combustion processes.1–3 These substances, known for their chemical stability, readily accumulate in fatty tissues and persist for approximately 11 years, posing a threat to both humans and animals.In particular, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), illustrated in Fig. 1, is identified as one of the most perilous dioxins. The International Agency for Research on Cancer (IARC) has classified TCDD as a human carcinogen.4 Numerous studies have focused on addressing this problem by exploring diverse chemical and physical processes for dioxin degradation. These approaches encompass photocatalysis,5–7 hydrothermal degradation,8 burning,9 catalytic degradation,10 radiolysis,11,12 electrochemical oxidation,13,14 and biodegradation.15
 | ||
| Fig. 1 Schematic chemical structure of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). | ||
Recent studies have investigated various advanced sorbents, such as zeolites, metal–organic frameworks (MOFs), and activated carbons, for the capture and removal of dioxins, particularly TCDD. Zeolites, especially those doped with metals like silver, copper, and iron, have demonstrated adsorption capabilities due to their high surface area and the ability to form strong interactions with dioxin molecules.16,17 For instance, the desorption activation energy of dioxins on Ag–zeolites is significantly higher than that on other zeolites, indicating stronger binding and more effective capture.16 Activated carbons have also been extensively used for dioxin removal,18 benefiting from their large specific surface area and porosity. Recent comparisons have shown that carbon nanotubes (CNTs) exhibit superior adsorption efficiency and capacity compared to traditional activated carbons, primarily due to their unique structure and higher distribution of mesopores, which facilitate the adsorption of highly chlorinated dioxins.17 Moreover, MOFs, with their tunable porosity and functionalizable surfaces, have also shown dioxin capture capabilities, surpassing conventional materials in efficiency.19
The peculiar properties of two-dimensional (2D) materials, as well as the great potential that they have shown to contribute to the solution of several problems, including environmental ones,20,21 have attracted the attention of researchers from several disciplines (e.g., condensed matter physics, materials science, chemical engineering, and nanotechnology).22,23 The successful preparation of one-atom-thick graphene sheets by Novoselov et al.24 triggered the discovery and investigation of other 2D materials with remarkable properties such as aluminene,25 silicene,26,27 germanene,28 phosphorene,29,30 and antimonene.31
Several studies investigated the ability of various 2D and other materials to capture and remove TCDD. For instance, Ganji et al.32 studied the interaction of TCDD with CNTs and boron nitride nanotubes (BNNTs). They found that the adsorption energy of TCDD on the BNNTs is higher than that on CNTs and the adsorption capability of pristine BNNTs is better than that of defected ones. Moreover, Fagan et al.33 investigated the interaction between TCDD and CNTs and reported that it is enhanced by the defects of CNTs. Zhang et al.34 studied the interaction of metal-doped graphene with TCDD molecules and the results revealed that Ti-doped graphene enhances the capability for TCDD capture. Similarly, Kang35 theoretically studied the binding of TCDD with metal-doped graphene and CNTs, concluding that TCDD exhibits strong binding to Fe-doped CNTs. Additionally, pristine and doped phosphorene have been proposed as promising materials for adsorbing TCDD molecules by Zhang et al.36 They reported that doping phosphorene with Ca improved the adsorption of TCDD on the phosphorene surface.
In recent times, materials based on antimonene37–40 have exhibited distinct physical properties, positioning them as promising candidates for various applications, including energy storage,41–44 gas sensors,45,46 and spintronics.47 Despite these advancements, the application of antimonene in capturing organic pollutants like TCDD has remained unexplored. Consequently, this study delves into the interaction of TCDD with pristine antimonene and 2D antimonene doped with calcium (Ca), titanium (Ti), and nickel (Ni) concerning TCDD, utilizing DFT calculations. The aim is to assess the potential of these materials in binding and ultimately eliminating this highly toxic compound.
2 Molecular models and computational methods
2.1 Model systems
The TCDD molecule has an aromatic planar structure (cf.Fig. 1), while antimonene has a buckled geometry. First, the isolated structure of TCDD has been optimized and analyzed based on selected structural parameters. The values of these selected parameters calculated at the DFT level of theory (see details below) are given in Table 1, together with the corresponding experimental values reported by Boer et al.48| Method | Structural parameters | |||
|---|---|---|---|---|
| C–O (Å) | C–C (Å) | C–Cl (Å) | C–O–C (°) | |
| DFT | 1.38 | 1.40 | 1.73 | 115.98 |
| Experiment | 1.37 | 1.36 | 1.73 | 115.70 |
To construct the antimonene crystal model, we adopted the lattice and geometrical criteria of the antimonene primitive unit cell calculated by Bafekry et al.49 In principle, the antimonene primitive unit cell consists of two Sb atoms in the buckled form with two intersecting atomic planes. The values of the structural parameters that fully describe the monolayer antimonene unit cell, namely, lattice constant, Sb–Sb bond length, Sb–Sb–Sb angle, and the height of the two atomic planes of antimonene, are 4.12 Å, 2.95 Å, 88 Å and 1.75 Å, respectively. Using this primitive unit cell, a 5 × 5 × 1 supercell was built. Following this step, three doped-antimonene (namely, doped with Ni, Ca or Ti) supercells were created by replacing one Sb atom with the dopant atom.
Modifying antimonene by inserting a dopant atom is expected to induce some distortion in the vicinity of this atom. Therefore, prior to studying the adsorption of TCDD on the surfaces of antimonene and doped-antimonene, cell relaxation and geometry optimization calculations for all the constructed supercells were performed to ensure that a minimum energy lattice structure for each case was reached. Fig. 2 displays the optimized molecular structures of isolated TCDD and the antimonene supercells. The values of their lattice and geometrical parameters are given in Table 2. The computational details (e.g., level of theory at which the calculations were performed and the utilized software package) are given in the next section.
 | ||
| Fig. 2 The optimized molecular structures of isolated TCDD and pristine and doped antimonene. | ||
| Systems | a (Å) | b (Å) | Sb–X (Å) | Sb–X–Sb (Å) | Torsion angle (°) |
|---|---|---|---|---|---|
| Sb | 20.784 | 20.784 | 2.905 | 91.4 | 53.6 |
| Ni-doped-Sb | 20.708 | 20.550 | 2.516 | 95.3 | 44.4 |
| 2.520 | 109.0 | ||||
| 2.532 | 112.1 | ||||
| Ca-doped-Sb | 20.825 | 20.669 | 3.167 | 97.7 | 46.8 |
| 3.135 | 103.1 | ||||
| 3.087 | 105.1 | ||||
| Ti-doped-Sb | 20.698 | 20.707 | 2.780 | 104.6 | 40.4 |
| 2.772 | 105.0 | ||||
| 2.777 | 106.8 |
Noteworthily, during cell relaxation the c vector was kept fixed at a value of 50 Å to ensure enough vacuum preventing any interaction with the image of the cell in the z direction. In addition, the lattice angles were also kept fixed at the values of 90°, 90° and 120°. In other words, only a and b vectors, as well as the atomic position, were allowed to change freely during the relaxation.
The orientation of a molecule relative to the surface is crucial for the interaction between the adsorbent and the adsorbate. In this study, three configurations of TCDD/antimonene complexes were considered: (1) TCDD parallel to the antimonene layer through hexagonal rings (denoted as Sb–TCDD), (2) TCDD standing perpendicular to antimonene through Cl atoms (denoted as Sb–TCDD–Cl), and (3) TCDD standing perpendicular to antimonene through two hydrogen atoms with a bridged oxygen atom (denoted as Sb–TCDD–O). These three configurations were also examined for the adsorption of TCDD on doped antimonene. In total, 12 complex structures were initially considered, and the optimized complexes are depicted in Fig. 3.
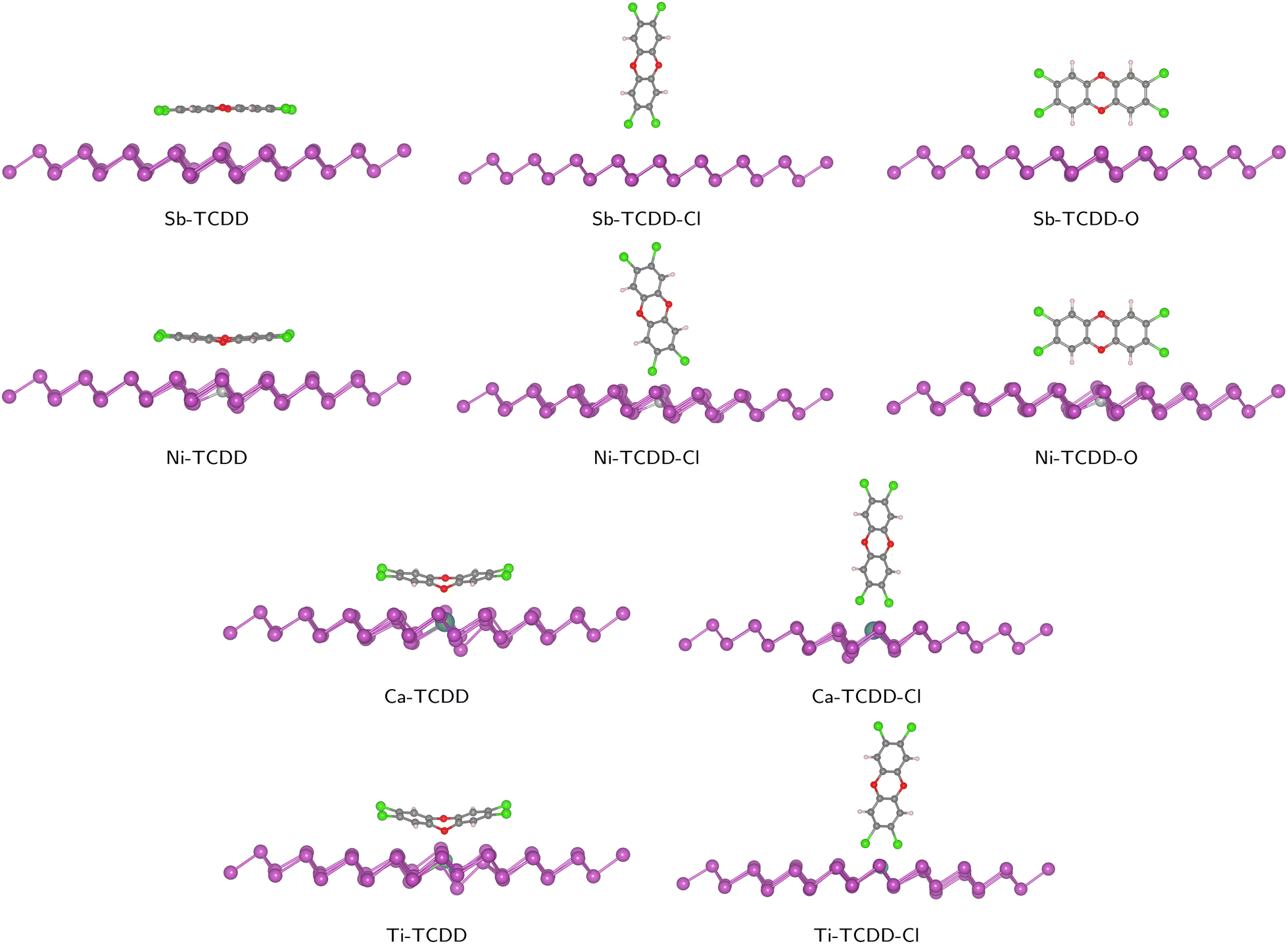 | ||
| Fig. 3 Optimized geometries of all considered configurations of the TCDD/Sb and TCDD/doped-Sb complexes. | ||
2.2 Computational details
Density functional theory (DFT) has been used with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional, a widely adopted generalized gradient approximation (GGA) functional.50 The calculations were performed with periodic boundary conditions (PBC) using the CP2K software package,51,52 employing the hybrid Gaussian and plane wave (GPW) method,53 implemented in the QuickStep module.54 The core electrons of all atoms were treated using the Goedecker–Teter–Hutter (GTH)-PBE pseudopotential,55 while the valence electrons of Sb, Ca, Ni, and Ti are described with the short-range double-ζ valence polarized MOLOPT (DZVP-MOLOPT-SR-GTH) basis set and those for all other atoms are described with the DZVP-MOLOPT-GTH basis set.56 To account for van der Waals (vdW) interactions, crucial in TCDD adsorption on the 2D antimonene surface, the Grimme D3 dispersion correction57 was applied. The adsorption energies of TCDD on antimonene and doped-antimonene were calculated as follows:| ΔE = Eantimonene+TCDD − Eantimonene − ETCDD | (1) |
Another factor that should be taken into account when calculating adsorption energies using methods that include an atomic-centered (i.e., localized) basis set is the basis set superposition error (BSSE). Specifically, as the atoms of interacting molecules/fragments approach each other, their basis functions overlap, which may introduce an error when comparing energies.58 This error becomes more pronounced for smaller basis sets compared to larger basis sets. Therefore, the adsorption energies presented in this work were corrected according to the following equation:
| ΔEcorrected = ΔE − BSSE, | (2) |
To gain insights into the adsorbate/adsorbent interaction, further calculations (namely, the charge density differences (Δρ(r)) and partial density of states (PDOS)) were performed on the optimized structures of selected complexes. The charge density difference is given by the formula:
Δρ(r) = ρTCDD–antimonene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) complex − ρantimonene − ρTCDD complex − ρantimonene − ρTCDD | (3) |
In some cases, the obtained adsorption complex exhibits bonding between one of the O atoms of TCDD and the dopant atom of the doped antimonene surface. This bonding interaction caused a noticeable deformation of the planar structure of TCDD. On the other hand, for non-bonded interactions, TCDD can be mobile on the surface. To quantify this lateral mobility of the TCDD on the antimonene surface, molecular dynamics (MD) simulations were performed for selected cases. Namely, 5 ps of MD simulations for these selected cases were performed at T = 300 K with a time step of 0.5 fs in the NVT ensemble, employing canonical sampling through a velocity rescaling (CSVR) thermostat61 with a time constant of 100 fs. The initial structures for the MD simulations were the optimized structures, and since there is no solvent, the equilibration is reached in the very beginning, taking only a few steps. Energy and force were computed at the same DFT level of theory as employed in the geometry relaxation calculations.
3 Results and discussion
3.1 Adsorption of TCDD on pristine and doped antimonene
Twelve different configurations were considered for the adsorption of TCDD on the pristine and doped antimonene surfaces. Interestingly, the geometry relaxation calculations revealed that the adsorption of TCDD on both Ca- and Ti-doped-antimonene through the orientation in which TCDD is standing perpendicular to the surface via the H and O atoms cannot be observed. In these particular cases, the initial configurations (i.e., X–TCDD–O where X is Ca or Ti) yield the parallel configurations (i.e., X–TCDD). This observation is partially consistent with a recent study by Zhang et al.36 on the adsorption of TCDD on the surface of phosphorene; “partially” because the authors reported that neither Ca-doped-phosphorene nor Ti-doped-phosphorene could adsorb TCDD through standing orientations (regardless of being attached via Cl or O atoms).Except for the two cases mentioned above (i.e., Ca–TCDD–O and Ti–TCDD–O), the remaining 10 complexes were optimized achieving the desired orientations of TCDD relative to antimonene and doped-antimonene surfaces. With respect to the initial configuration, slight adjustments to the TCDD direction towards the antimonene surface took place in a manner that maximizes the binding strength between TCDD and the surface. For instance, when TCDD is positioned on antimonene with two chlorine atoms, the geometry optimization process rotates and tilts the TCDD molecule slightly. This leads to the situation where the two chlorine atoms are positioned as closely as possible to two Sb atoms in the case of pristine antimonene, or to a dopant atom and an Sb atom in the case of Ca- and Ti-doped antimonene. All optimized complexes can be found in Fig. 3.
Interestingly, in the aforementioned study of TCDD adsorption on phosphorene by Zhang et al.,36 the authors reported that the phosphorene layer tends to bend due to its strong interaction with TCDD. In contrast to phosphorene, neither pristine antimonene nor doped antimonene exhibits such a phenomenon. As mentioned earlier, only a slight local distortion around the dopant atom compared with pristine antimonene was observed, while the adsorption of TCDD did not cause any significant change to the adsorbents compared with the isolated surfaces. To assess the extent of structural changes in the surfaces upon adsorption, we calculated the root mean square deviation (RMSD) values by comparing the post-adsorption surface structures to their respective isolated forms. This allows us to quantify the differences between the adsorbed and isolated states. For instance, for the antimonene surfaces in the parallel configuration (i.e., X–TCDD models where X = Sb, Ni, Ca or Ti), we obtained RMSD values of 0.15 Å for bare antimonene, and 0.08 Å, 0.17 Å and 0.3 Å for the Ni-, Ca- and Ti-doped antimonene, respectively. Such small RMSD values confirm that the antimonene layer sustains its planarity in all perpendicular cases and for TCDD being parallel to pristine antimonene. In the latter case, this could be attributed to the equal effect of all the Sb atoms (i.e., the bare antimonene surface is uniform). However, when TCDD is lying flat on Ca- and Ti-doped antimonene, the dopant atom attracts the oxygen atoms resulting in deformation of the planarity of TCDD (i.e., TCDD is bent) showing that the dopant atoms in these cases have superior interactions with TCDD. As a consequence of this bending, the closest TCDD–adsorbate distance decreases from 3.88 Å (i.e., in the case of Sb–TCDD) to 2.35 Å and 2.64 Å (in the cases of Ti–TCDD and Ca–TCDD, respectively). In contrast to Ti–TCDD and Ca–TCDD, one can see that the structure of Ni–TCDD is, to a large extent, similar to that of Sb–TCDD in terms of the planar structure of TCDD and the Ni–O distance ≈ 3.85 Å. This discrepancy in structural deformation could be attributed to differences in electron affinity. Namely, the electron affinities of Ni and Sb are quite similar, both demonstrating larger positive values ranging from 100 to 111.65 kJ mol−1,62,63 whereas Ti and Ca showcase smaller positive values in the range of a few kJ mol−1.64,65
Hence, Ti and Ca, along with TCDD (particularly with its electronegative atoms, i.e., O and Cl), possess a greater affinity to bind more strongly compared to Ni and Sb. Consequently, a higher binding energy and greater deformation would be observed on the surface, as well as in TCDD, for the doped Ti– and Ca–antimonene cases compared to the pristine and Ni-doped counterparts.
The stability of the investigated complexes in Fig. 3 and the trend of the strength of the adsorbate–adsorbent interaction in these complexes have been studied based on their adsorption energies that were calculated according to eqn (2). Fig. 4 summarizes the calculated energies, while the actual values are given in Table 3. Inspecting these data, we first notice that all configurations are stable (i.e., their adsorption energies have negative signs).
 | ||
| Fig. 4 BSSE-corrected adsorption energies for the complexes depicted in Fig. 3, the relaxation energies of their fragments (higher values indicate larger deformation of TCDD upon adsorption), and the corresponding BSSE values. | ||
| System | BSSE-corrected adsorption energy | Fragment relaxation energy | BSSE values |
|---|---|---|---|
| Sb–TCDD | −0.83 | −0.07 | −0.08 |
| Ni–TCDD | −0.81 | −0.11 | −0.06 |
| Ca–TCDD | −1.02 | −0.33 | −0.12 |
| Ti–TCDD | −1.10 | −0.33 | −0.08 |
| Sb–TCDD–Cl | −0.25 | −0.03 | −0.02 |
| Ni–TCDD–Cl | −0.33 | −0.08 | −0.03 |
| Ca–TCDD–Cl | −0.67 | −0.06 | −0.04 |
| Ti–TCDD–Cl | −0.87 | −0.38 | −0.08 |
| Sb–TCDD–O | −0.45 | −0.04 | −0.04 |
| Ni–TCDD–O | −0.43 | −0.05 | −0.04 |
Ping Zhang et al.36 and Zhou et al.66 reported that, regardless of the kind of substrate material, the strongest interaction between TCDD and a 2D surface takes place when TCDD is lying parallel to the surface. Inspection of the data displayed in Fig. 4 reveals that, similar to other 2D materials, TCDD prefers the parallel configuration on both pristine and doped antimonene. The next preference in orientation is the standing-via-O-atoms orientation. Lastly comes the perpendicular one through the Cl atoms. This preference can be rationalized by considering the subjected surface area (SSA) of TCDD to the surface. As the SSA (particularly containing electronegative atoms) increases, the binding of TCDD with bare and doped antimonene increases. The SSA of TCDD to the antimonene surface increases in the order of TCDD–Cl (two electronegative atoms) < TCDD–O (three electronegative atoms) < parallel TCDD (6 electronegative atoms + electron density of the aromatic ring).
It is noteworthy that many similar studies34,36,66 on different materials used almost the same setup of calculation and level of theory as those employed in the current study. Hence, one can directly compare the values of adsorption energies for our systems with their counterparts in these previous studies. Interestingly, it has been found that the adsorption energy in the case of pristine antimonene remarkably exceeds those of previously studied materials, including graphene,34 carbon nanotubes,32 BN nanotubes,32 and phosphorene,36 by roughly 50% to 250%.
Given that pristine antimonene has such a superior ability to bind TCDD, particularly in the parallel configuration, there arises a question of whether doping improves its performance. Comparing the values of adsorption energies of the pristine antimonene complexes with their doped antimonene counterparts, it is observed that, in the case of vertical adsorption, i.e. in the X–TCDD–Cl configurations (where X = Ni, Ca or Ti), one can see how doping significantly improved the performance and raised the adsorption energy from −0.25 eV (in pristine antimonene) to −0.87 (in Ti-doped antimonene). To be specific, doping with Ni, Ca or Ti increased the adsorption energies by 32%, 168%, or 248%, respectively. For the parallel configuration, doping with Ca and Ti enhances the adsorption energies by 23% and 32%, respectively, while for doping with Ni no significant effect is observed. For the standing-via-O (i.e., X–TCDD–O) configuration, doping with Ni showed almost no effect on the adsorption energy.
Finally, by examining the data in Fig. 4 and Table 3, the relaxation energy of the fragments (i.e., the difference between the adsorption energy calculated considering optimized separate fragments and the adsorption energy calculated considering fragments at the adsorption structure) shows the degree of structural deformation that occurs in the TCDD structure upon adsorption. For instance, comparing pristine antimonene with Ni-doped antimonene, one can see that the adsorption does not affect the planar structure of TCDD. In contrast, comparing pristine antimonene with Ca- and Ti-doped antimonene, it is clear that the adsorption significantly affects the geometry of TCDD, particularly the planar structure of TCDD in the parallel configurations. As mentioned above, the significant effect of doping antimonene with Ca or Ti is attributed to their higher electron affinity (i.e., lower positive values) compared to Sb and Ni. One may also notice that in the case of Ti–TCDD–Cl the relaxation energy of the fragments is prominent (−0.38 eV) compared to its counterpart complexes (X–TCDD–Cl models), even though the planar structure of TCDD is preserved. Upon investigating the origin of this high fragment relaxation energy, we found that it arises equally from both the TCDD and the Ti-doped antimonene surface.
As demonstrated earlier, the most stable adsorption case, specifically the Ti–TCDD configuration, involves Ti–O bonding, causing a noticeable deformation in the planar structure of TCDD. While this deformation indicates stronger interactions and thus more overall stability of such systems, it raises a question regarding the lateral mobility and dynamical behavior of TCDD on bare and doped antimonene surfaces. To address this, MD simulations were carried out. In particular, we performed 5 ps of DFT-based MD simulations (refer to Section 2 for details) for TCDD adsorption on pure antimonene (Sb–TCDD) and Ti-doped antimonene (Ti–TCDD).
The analysis of the MD trajectories was conducted with the primary objective of investigating the ability of the surface to trap the TCDD molecule in the examined complexes. Therefore, we computed the time evolution of the RMSD of the TCDD molecule in individual directions throughout the MD trajectories, and the results are depicted in Fig. 5. The RMSD along the x- and y-axes provided insights into the diffusion of the TCDD molecule in the xy plane, whereas the small values of RMSD along the z-axis confirm the strong binding of TCDD to the surface. Fig. 5(a–c) illustrates the results, indicating that the TCDD molecule is effectively immobilized in the three directions by the binding of its O atom with the Ti atom in the Ti-doped antimonene surface. In contrast, on the pure antimonene surface, TCDD exhibits unrestricted movement across the xy plane due to the uniformity of the surface, where it shows no preference for being trapped at a specific location.
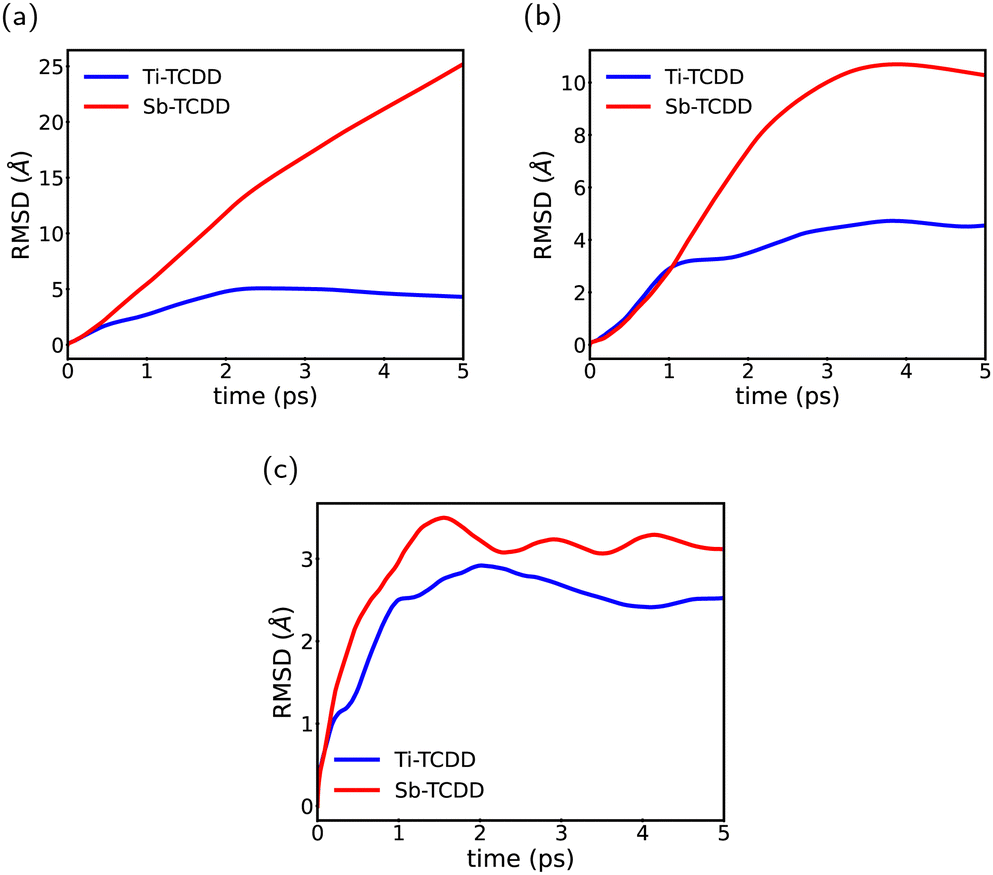 | ||
| Fig. 5 RMSD of TCDD in the individual axis: (a) in the x-axis, (b) in the y-axis and (c) in the z-axis throughout the MD trajectories. | ||
3.2 Electronic properties
Since the adsorption energy calculations indicated that the parallel configuration is the most preferred one for both pristine and doped antimonene, the subsequent analysis will focus solely on this subset of systems. | (4) |
The isosurfaces of Δρ(r), together with the Δρ(z) and ΔQ(z) curves, are displayed in Fig. 6.
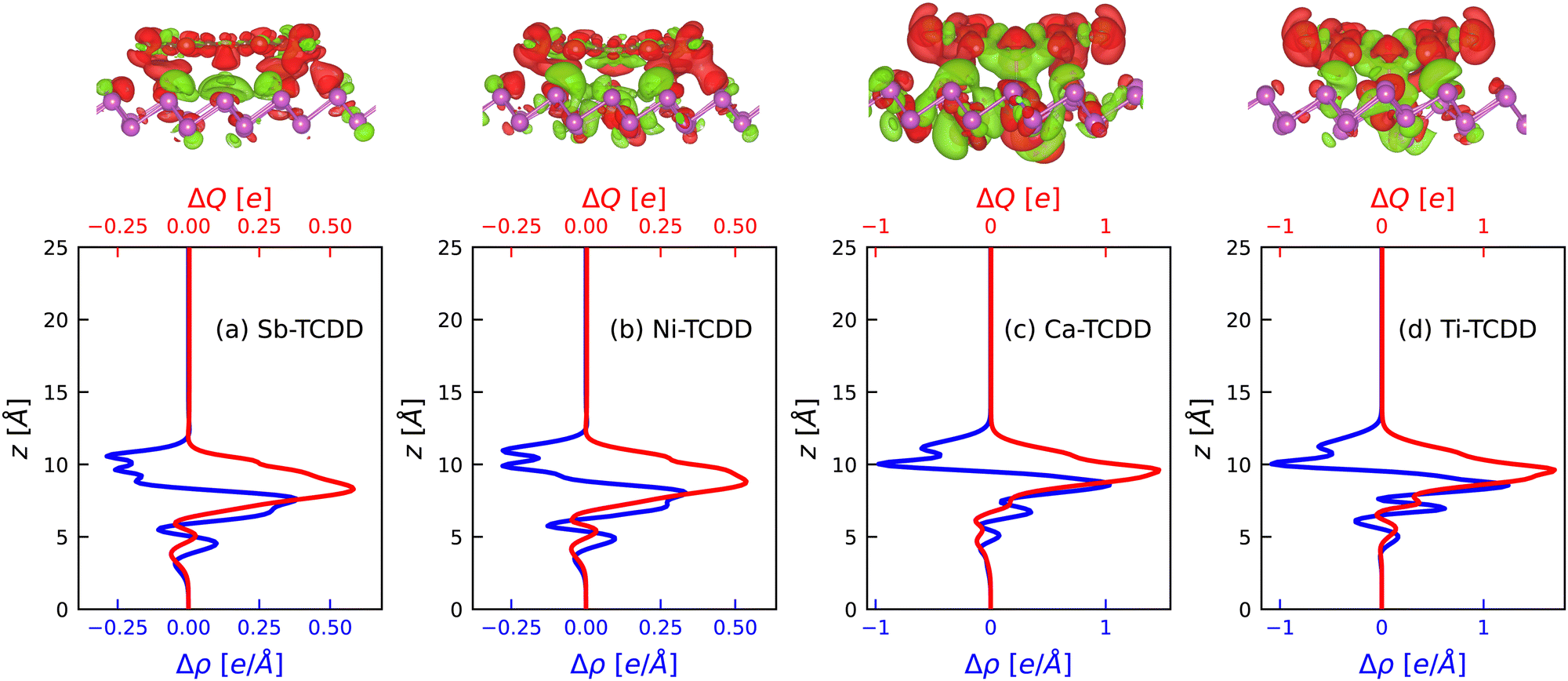 | ||
| Fig. 6 Top row: Charge density difference isosurface plots for parallel TCDD over (a) pristine antimonene, (b) Ni-doped antimonene, (c) Ca-doped antimonene, and (d) Ti-doped antimonene. The color map indicates charge accumulation in green and charge depletion in red. Bottom row: Δρ(z) and ΔQ(z) curves corresponding to the respective isosurfaces (note the different scales). The position z = 0 corresponds to the bottom of the simulation box. | ||
Fig. 6 shows that, in all cases, the charge is transferred from TCDD to the antimonene surface. This can be seen from the isosurface plots in the top row of Fig. 6 as well as the Δρ(z) and ΔQ(z) curves. Moreover, the charge density difference plots reveal a clear modification in the charge distribution (as well as the amount of charge transferred) in Ca- and Ti-doped-antimonene relative to pristine and Ni-doped antimonene. Namely, the maximum ΔQ(z) values in the case of Ca–TCDD and Ti–TCDD are about two times those of their counterparts (i.e., Sb–TCDD and Ni–TCDD). Moreover, the charge distribution in the case of Ca–TCDD and Ti–TCDD aligns with the relatively shorter distance between TCDD and doped antimonene as compared to the distance between TCDD and pristine and Ni-doped-antimonene. In general, these observations are in line with the behaviour expected from the differences in the electron affinities of Ca and Ti compared with Sb and Ni. Based on these results, one may conclude that the interaction between the O atom of TCDD and the dopant atom (Ca or Ti) in the Ca–TCDD and Ti–TCDD complexes is mainly a coordination bond.
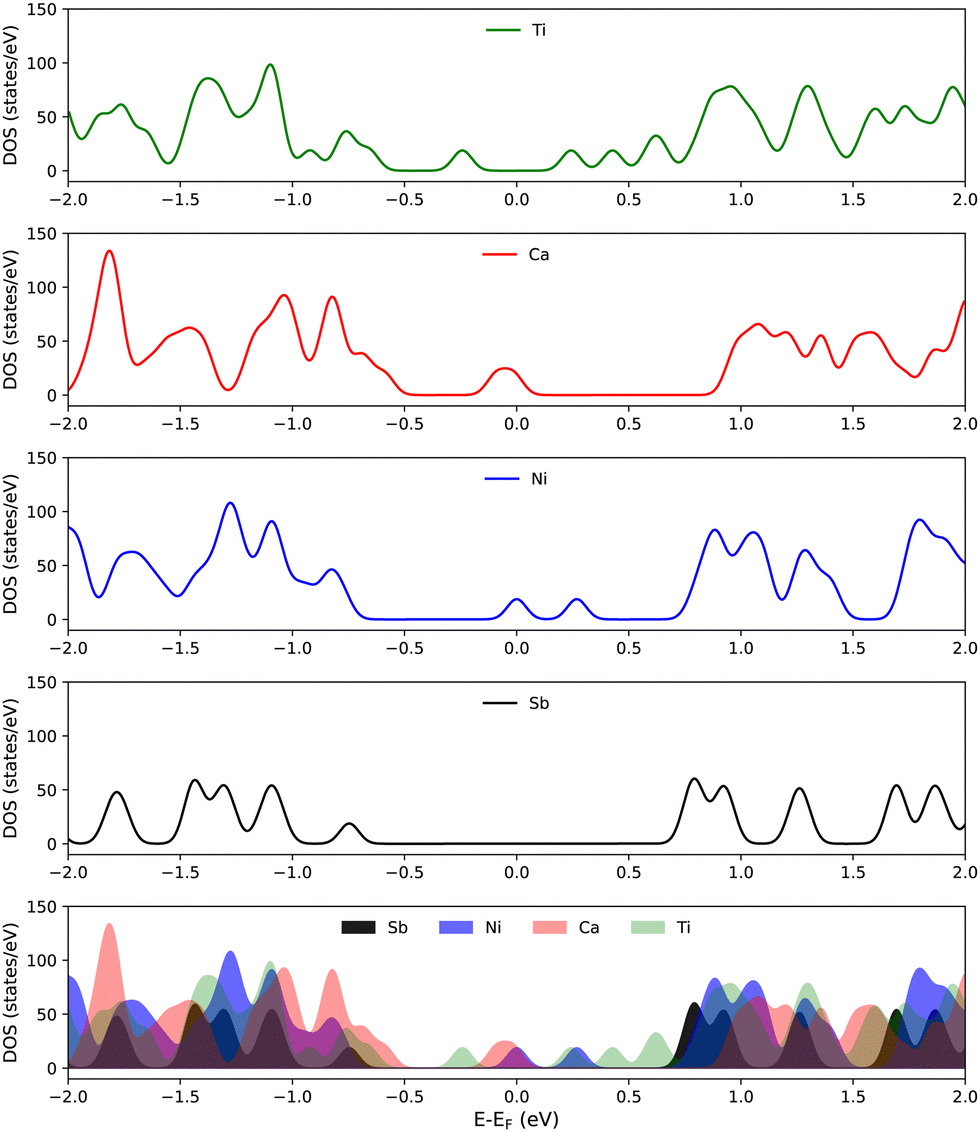 | ||
| Fig. 7 DOS of antimonene and doped antimonene. In the bottom row a direct comparison is given. | ||
It is clear from Fig. 7 that doping has a substantial effect on the electronic properties of 2D antimonene. Namely, some peaks appear around the Fermi level in the DOS of Ni- and Ca-doped antimonene which are missing in the pristine case. Although Ti-doped antimonene has no peaks at the Fermi level, the band gap is significantly reduced relative to that of pristine antimonene. On the other hand, the adsorption of TCDD only marginally affects the electronic properties of antimonene. This can be seen in Fig. S3 (ESI†) for TCDD adsorption on both pristine and doped antimonene substrates.
The origin of the peaks around the Fermi level that appear in the DOS of doped antimonene can be traced by inspecting the PDOS of the dopant atoms (as well as the O atoms of TCDD) given in Fig. 8. Although it is clear that these peaks exist because of the dopant atoms, the PDOS plots show that their magnitudes do not coincide with the corresponding peaks in the total DOS. This implies that the Sb atoms surrounding the dopants may be also involved (see Fig. S4 in the ESI†). Finally, the PDOS of the O atoms of TCDD confirms that TCDD adsorption on these substrates has no direct contribution to the electronic states around the Fermi level.
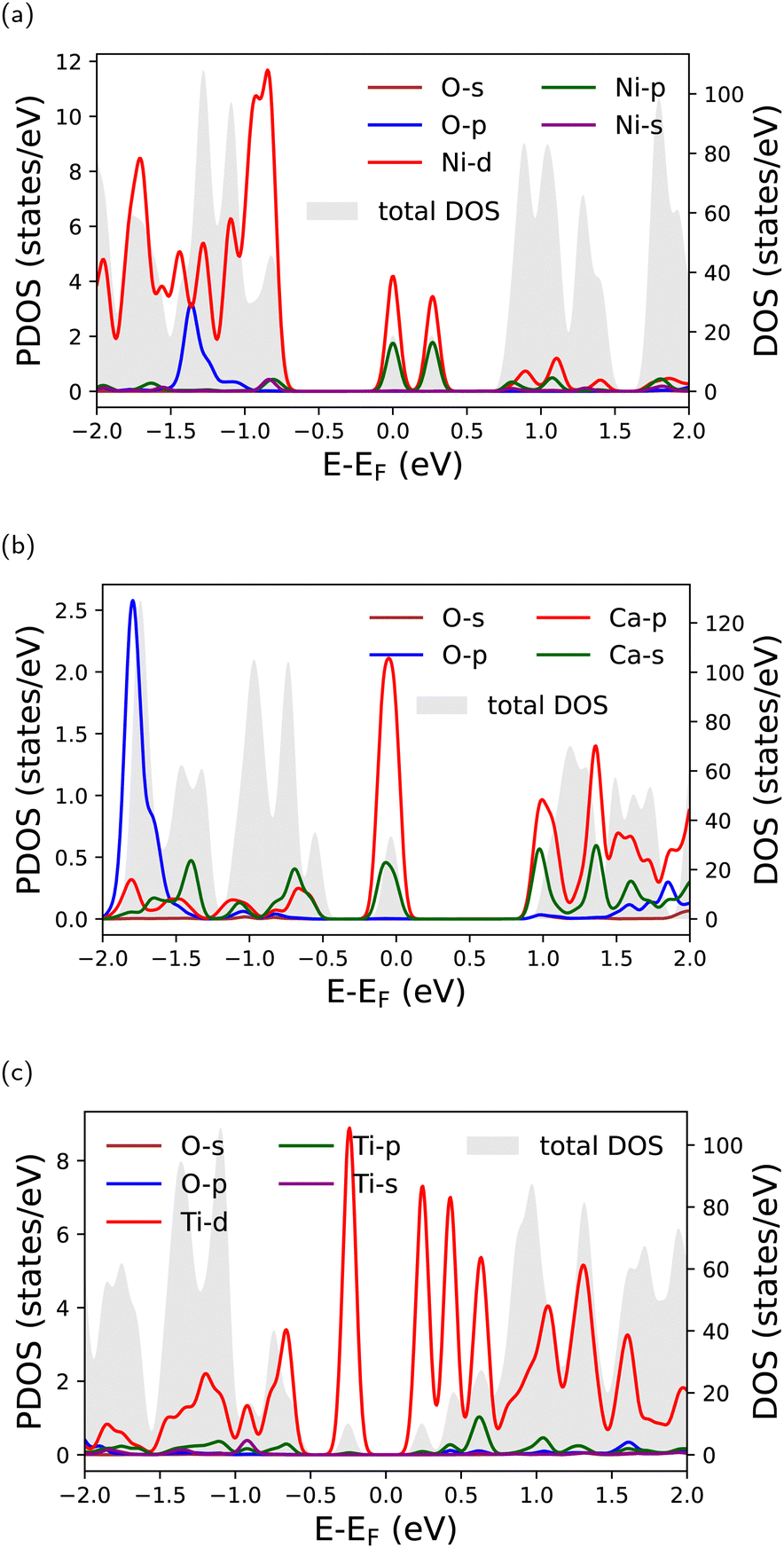 | ||
| Fig. 8 PDOS of oxygen (TCDD) and the dopant atoms in doped antimonene for (a) Ni-TCDD, (b) Ca-TCDD, and (c) Ti-TCDD. The corresponding total DOS for each system is shown in the background. | ||
4 Conclusions
We have explored the capability of antimonene as a potential candidate for capturing the highly toxic 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). DFT has been utilized to calculate the stable structures of several adsorption configurations of TCDD on the surface of pristine, as well as Ni-, Ca- and Ti-doped antimonene. TCDD was found to bind preferentially in the parallel configuration. While all the studied antimonene forms showed a remarkable capability to strongly bind TCDD, it has been found that doping (especially with Ca and Ti) could even improve the performance of pristine antimonene. Specifically, doping antimonene with Ca or Ti enhances its ability to bind TCDD by 23% or 32%, respectively, whereas Ni-doped antimonene showed TCDD capture capability quite similar to pristine antimonene. In addition, doping antimonene with Ca and Ti significantly increased its ability to bind TCDD through the configuration in which TCDD is perpendicular to the antimonene surface through two Cl atoms. Moreover, the stability of the TCDD–complexes that involve bonding between the O atom of TCDD and a dopant atom of the doped antimonene surface has been confirmed by further DFT-based MD simulations.The findings of the current study have been discussed in light of the adsorption energies, the charge transfer between the adsorbate and the adsorbent, and the electronic properties of the complexes under investigation. Interestingly, the comparison between the results reported in the present work and the findings of similar studies revealed that, in terms of binding strength, pristine antimonene outperforms the other pristine nanomaterials (including phosphorene, pristine graphene, and BN nanotubes). Therefore, this study not only proposes antimonene as a promising candidate for dioxin capture but it could also be the basis for further investigations on tuning the properties of antimonene to maximize its capability to be utilized in similar environmental applications.
Author contributions
Moyassar Meshhal: conceptualization, methodology, investigation, formal analysis, writing – original draft, writing – review & editing. Ashour A. Ahmed: conceptualization, methodology, writing – review & editing, supervision. Mohamed F. Shibl: conceptualization, methodology, writing – original draft. Saadullah Aziz: resources. Oliver Kühn: conceptualization, resources, methodology, writing – review & editing, supervision. Kamal A. Soliman: conceptualization, methodology, investigation, formal analysis, writing – original draft.Data availability
The data supporting the findings of this study, including the density functional theory (DFT) calculations, molecular dynamics (MD) simulation data, and (ESI†), have been included as part of the ESI.† Additionally, detailed analysis of the MD simulations and density of states (DOS) plots are available. The datasets generated and analyzed during the current study are available from the corresponding authors on reasonable request. The data have been provided in the ESI† submitted with this manuscript. Any additional data related to this study can be provided by the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB 1477 “Light–Matter Interactions at Interfaces” (project no. 441234705).References
- N. S. C. K. Yive and M. Tiroumalechetty, J. Hazard. Mater., 2008, 155, 179–182 CrossRef CAS PubMed.
- P. S. Kulkarni, J. G. Crespo and C. A. M. Afonso, Environ. Int., 2008, 34, 139–153 CrossRef CAS PubMed.
- Y. Mizukami, J. Mol. Struct. Theochem., 2005, 713, 15–19 CrossRef CAS.
- R. T. Yang, R. Q. Long, J. Padin, A. Takahashi and T. Takahashi, Ind. Eng. Chem. Res., 1999, 38, 2726–2731 CrossRef CAS.
- W. Choi, S. J. Hong, Y.-S. Chang and Y. Cho, Environ. Sci. Technol., 2000, 34, 4810–4815 CrossRef CAS.
- F. Samara, T. Ghalayini, N. A. Farha and S. Kanan, Catalysts, 2020, 10, 957 CrossRef CAS.
- J. Choi, W. Choi and B. J. Mhin, Environ. Sci. Technol., 2004, 38, 2082–2088 CrossRef CAS PubMed.
- Y. Qi Jin, X. Jun Ma, X. Guang Jiang, H. Mei Liu, X. Dong Li and J. Hua Yan, Energy Fuels, 2013, 27, 414–420 CrossRef.
- P. C. Hung, Q. H. Chen and M. B. Chang, Chemosphere, 2013, 92, 857–863 CrossRef CAS PubMed.
- Y. Ukisu and T. Miyadera, Chemosphere, 2002, 46, 507–510 CrossRef CAS PubMed.
- C. Zhao, K. Hirota, M. Taguchi, M. Takigami and T. Kojima, Radiat. Phys. Chem., 2007, 76, 37–45 CrossRef CAS.
- R. J. Hilarides, K. A. Gray, J. Guzzetta, N. Cortellucci and C. Sommer, Water Environ. Res., 1996, 68, 178–187 CrossRef CAS.
- M. Vallejo, M. F. S. Román, A. Irabien and I. Ortiz, Chemosphere, 2013, 90, 132–138 CrossRef CAS PubMed.
- N. Palanisami, S. J. Chung and I. S. Moon, J. Ind. Eng. Chem., 2015, 28, 28–31 CrossRef CAS.
- S. Saibu, S. A. Adebusoye and G. O. Oyetibo, Bioresour. Bioprocess., 2020, 7, 1–21 CrossRef.
- H. Xi, Z. Li, H. Zhang, X. Li and X. Hu, Sep. Purif. Technol., 2003, 31, 41–45 CrossRef CAS.
- X. Zhou, X. Li, S. Xu, X. Zhao, M. Ni and K. Cen, Environ. Sci. Pollut. Res., 2015, 22, 10463–10470 CrossRef CAS PubMed.
- J. Atkinson, P. Hung, Z. Zhang, M. Chang, Z. Yan and M. Rood, Chemosphere, 2015, 118, 136–142 CrossRef CAS PubMed.
- J. Cheng, B.-e Li, J. He and P. Wang, ACS Sens., 2023, 8, 2115–2123 CrossRef CAS PubMed.
- M.-Z. Qin, W.-X. Fu, H. Guo, C.-G. Niu, D.-W. Huang, C. Liang, Y.-Y. Yang, H.-Y. Liu, N. Tang and Q.-Q. Fan, Adv. Colloid Interface Sci., 2021, 297, 102540 CrossRef CAS PubMed.
- J. Di, J. Xiong, H. Li and Z. Liu, Adv. Mater., 2018, 30, 1704548 CrossRef PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han and G.-H. Nam, et al. , Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- X. Yu, H. Cheng, M. Zhang, Y. Zhao, L. Qu and G. Shi, Nat. Rev. Mater., 2017, 2, 1–13 Search PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Eng Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. H. Yeoh, T. L. Yoon, D. S. Ong and T. L. Lim, et al. , Appl. Surf. Sci., 2018, 445, 161–166 CrossRef CAS.
- A. Y. Galashev and A. S. Vorob'ev, Electrochim. Acta, 2021, 378, 138143 CrossRef CAS.
- G. L. Lay, B. Aufray, C. Léandri, H. Oughaddou, J.-P. Biberian, P. D. Padova, M. E. Dávila, B. Ealet and A. Kara, Appl. Surf. Sci., 2009, 256, 524–529 CrossRef.
- E. Bianco, S. Butler, S. Jiang, O. D. Restrepo, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 4414–4421 CrossRef CAS PubMed.
- F. B. Baghsiyahi and M. Yeganeh, Phys. E: Low-dimens. Syst. Nanostruct., 2020, 121, 114088 CrossRef.
- J. H. Wu, F. Zhai, J. Q. Lu, J. Wu and X. Feng, Mater. Today Commun., 2020, 24, 101154 CrossRef CAS.
- X. Wang, X. Yu, J. Song, W. Huang, Y. Xiang, X. Dai and H. Zhang, Chem. Eng. J., 2021, 406, 126876 CrossRef CAS.
- M. D. Ganji, H. Alinezhad, E. Soleymani and M. Tajbakhsh, Phys. E: Low-dimens. Syst. Nanostruct., 2015, 67, 105–111 CrossRef.
- S. B. Fagan, E. J. G. Santos, A. G. S. Filho, J. M. Filho and A. Fazzio, Chem. Phys. Lett., 2007, 437, 79–82 CrossRef CAS.
- H. Zhang, W. He, X. Luo, X. Lin and X. Lu, J. Mol. Model., 2014, 20, 1–7 Search PubMed.
- H. S. Kang, J. Am. Chem. Soc., 2005, 127, 9839–9843 CrossRef CAS PubMed.
- H. Ping Zhang, J. Liang Hou, Y. Wang, P. Ping Tang, Y. Ping Zhang, X. Yan Lin, C. Liu and Y. Tang, Chemosphere, 2017, 185, 509–517 CrossRef PubMed.
- T. Li, C. He and W. Zhang, Appl. Surf. Sci., 2018, 427, 388–395 CrossRef CAS.
- D. R. Kripalani, A. A. Kistanov, Y. Cai, M. Xue and K. Zhou, Phys. Rev. B, 2018, 98, 085410 CrossRef CAS.
- R.-S. Meng, M. Cai, J.-K. Jiang, Q.-H. Liang, X. Sun, Q. Yang, C.-J. Tan and X.-P. Chen, IEEE Electron Device Lett., 2016, 38, 134–137 Search PubMed.
- A. A. Kistanov, Y. Cai, D. R. Kripalani, K. Zhou, S. V. Dmitriev and Y.-W. Zhang, J. Mater. Chem. C, 2018, 6, 4308–4317 RSC.
- W. Tian, S. Zhang, C. Huo, D. Zhu, Q. Li, L. Wang, X. Ren, L. Xie, S. Guo and P. K. Chu, et al. , ACS Nano, 2018, 12, 1887–1893 CrossRef CAS PubMed.
- J. Liu, Z. Yang, J. Wang, L. Gu, J. Maier and Y. Yu, Nano Energy, 2015, 16, 389–398 CrossRef CAS.
- Z. Li, X. Tan, P. Li, P. Kalisvaart, M. T. Janish, W. M. Mook, E. J. Luber, K. L. Jungjohann, C. B. Carter and D. Mitlin, Nano Lett., 2015, 15, 6339–6348 CrossRef CAS PubMed.
- J. Gu, Z. Du, C. Zhang, J. Ma, B. Li and S. Yang, Adv. Energy Mater., 2017, 7, 1700447 CrossRef.
- V. Nagarajan and R. Chandiramouli, J. Mol. Graph. Model., 2019, 88, 32–40 CrossRef CAS PubMed.
- P. Srivastava and N. K. Jaiswal, et al. , Mater. Today: Proc, 2020, 28, 65–69 CAS.
- L. F. Yang, Y. Song, W. B. Mi and X. C. Wang, Appl. Phys. Lett., 2016, 109, 022103 CrossRef.
- F. P. Boer, F. P. V. Remoortere, P. P. North and M. A. Neuman, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1023–1029 CrossRef CAS.
- A. Bafekry, M. Ghergherehchi and S. F. Shayesteh, Phys. Chem. Chem. Phys., 2019, 21, 10552–10566 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- T. D. Kühne, M. Iannuzzi, M. D. Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt and F. Schiffmann, et al. , J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- B. G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- T. V. Mourik, A. K. Wilson, K. A. Peterson, D. E. Woon and T. H. D. Jr, Adv. Quantum Chem., Elsevier, 1998, vol. 31, pp. 105–135 Search PubMed.
- D. V. Oliveira, J. Laun, M. F. Peintinger and T. Bredow, J. Comput. Chem., 2019, 40, 2364–2376 CrossRef PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Scheer, H. K. Haugen and D. R. Beck, Phys. Rev. Lett., 1997, 79, 4104 CrossRef CAS.
- M. Scheer, C. A. Brodie, R. C. Bilodeau and H. K. Haugen, Phys. Rev. A, 1998, 58, 2051 CrossRef CAS.
- R. Tang, X. Fu and C. Ning, J. Chem. Phys., 2018, 149, 134304 CrossRef PubMed.
- V. V. Petrunin, H. H. Andersen, P. Balling and T. Andersen, Phys. Rev. Lett., 1996, 76, 744 CrossRef CAS PubMed.
- Q. Zhou, X. Su, Y. Yong, W. Ju, Z. Fu and X. Li, Vacuum, 2018, 149, 53–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI contains further analysis of the MD simulations and the DOS plots of the isolated antimonene surfaces versus their counterparts after TCDD adsorption. In addition, four movies of the MD simulation that visualize the trajectories of Sb-TCDD and Ti-TCDD (top and side views for each) have also been generated to provide a clearer understanding of the difference in the dynamical behaviour of TCDD for the two adsorption cases. See DOI: https://doi.org/10.1039/d4cp02589j |
| This journal is © the Owner Societies 2024 |