
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas-phase vibrational spectroscopy of the dysprosium monoxide molecule and its cation
Sascha
Schaller
 ,
Sandy
Gewinner
,
Wieland
Schöllkopf
,
Gerard
Meijer
and
André
Fielicke
*
,
Sandy
Gewinner
,
Wieland
Schöllkopf
,
Gerard
Meijer
and
André
Fielicke
*
Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: fielicke@fhi-berlin.mpg.de
First published on 26th July 2024
Abstract
Rotationally resolved vibrational spectra of DyO and DyO+ in a molecular beam are obtained by IR excitation from the X8 ground state and from high-n Rydberg states of DyO using an infrared free electron laser. Vibrational excitation is detected either by resonance enhanced multiphoton ionisation from X8(v = 1) or by autoionisation of Rydberg states converging to DyO+(v = 1). For most heavy molecules, the large spectral width of an infrared free electron laser does not allow for rotational resolution. In DyO and DyO+ the P, Q, and R transitions can be resolved due to the high angular momentum in their ground states. For 164DyO a vibrational constant of ωe = 847.5(2) cm−1 and a vibrational anharmonicity of ωeχe = 2.9(1) cm−1 are deduced. For the 161DyO+ cation a transition frequency of ΔG1/2 = 907(1) cm−1 is found.
Introduction
The elements within the lanthanoid series are characterised by a successive filling of the 4f shell starting with one electron for cerium up to 14 electrons for ytterbium. Their open 4f shell and energetically low-lying excitations generally can lead to extremely rich and complicated electronic spectra. Recently, related to significant developments in experimental and theoretical methods, there has been a renaissance in the studies and the understanding of isolated, lanthanoid atom containing small molecules.1–5 In part, this is also related to the finding of interesting thermochemical properties, e.g., for the lanthanoid monoxides, LnO, see below.6The isolated dysprosium monoxide molecule, DyO, has been intensively studied experimentally, e.g., in the gas phase by emission and absorption spectroscopy,7,8 using spectrally dispersed laser induced fluorescence spectroscopy of an effusive DyO oven beam9,10 or Stark effect measurements.11 The thermodynamics of DyO (and DyO+) has attracted particular interest during the search for molecules with the more unusual property that their ionisation energies are below the ground-state bond dissociation energies.6,12 Furthermore, the vibrational properties of DyO and its ions have been studied using cryogenic matrix isolation IR spectroscopy.13–15 Knowledge on the vibrational properties of lanthanoid monoxides has led to rather fundamental insights into the electronic structure of these molecules and related species.16
The low-lying electronic states of DyO are described in ligand field theory as resulting from the states of the Dy2+ ion, perturbed by the closed-shell O2− ligand.16–18 The ground state of DyO originates from the [Xe](4f9(6H)6s) configuration of Dy2+.17 The lowest state of the 4f9 core is 6H7.5. The angular momentum Jc = 7.5 of the core couples with the 6s electron to give a pair of states, split by the exchange interaction, with total atomic angular momentum Ja = 8 and 7. As the 4f shell is more than half filled, the Ja = 8 level is the lowest in energy. The interaction with the O2− ligand quantizes the projections of each Ja state of Dy2+ on the internuclear axis to give Ω = Ja, Ja − 1, … 0. The state with Ω = Ja is the lowest in energy, and the ground state of DyO thus has Ω = 8.9 It is referred to as the X8 state and the lowest rotational level has a total angular momentum of J = Ω = 8. The X8 state is closest to a 7H8 state,9i.e. with total electron spin S = 3 and total electron angular momentum L = 5, even though only Ω, the sum of the projections of L and S on the internuclear axis, is well-defined. The removal of one electron results in Ω = 7.5, and thus in a lowest rotational level with J = 7.5 for the 6H7.5 ground state of DyO+ which can be described as Dy3+(4f9(6H))O2−.19 Ghiassee et al. use an alternative, nevertheless equivalent, description as Dy+(6I)O(3P), see the ESI of ref. 12.
Recently, we have started gas-phase spectroscopic studies of DyO embedded in a supersonic molecular beam using several variants of laser-based ionisation techniques to obtain mass resolved excitation spectra with rotational resolution. This allowed the characterisation of new electronically excited states, the identification of several Rydberg series as well as accurate measurements of the ionisation energy, bond dissociation energy of DyO and the rotational constant of DyO+.20 We report here on our study of the rotationally resolved vibrational spectra of DyO and DyO+, using excitation with a tunable infrared free electron laser (IR-FEL) in combination with sensitive ionisation detection schemes.
The motivation for the IR spectroscopic study of neutral DyO was in part to characterise the absolute frequency and spectral profile of the light emitted by the IR-FEL operated at the Fritz Haber institute (FHI-FEL)21 using well-known molecular transitions. After having characterised the IR-FEL radiation, we have applied infrared photoinduced Rydberg ionisation (IR-PIRI) using the FHI-FEL to obtain rovibrational spectra of DyO+.
Experimental methods
The experiments on DyO are performed in a molecular beam machine22 that is coupled to the beamline of the FHI-FEL. The experimental setup includes two pulsed dye lasers that allow for the preparation and detection of DyO in different electronic states with rotational resolution, as well as a Nd:YAG laser with 4th harmonics unit that can be used for non-resonant ionisation. The experiment runs at 10 Hz repetition rate, while the FHI-FEL may also be operated at half that rate for subsequent on/off measurements.In a laser ablation source, a rotating and translating dysprosium target (MaTeck, 6.35 mm diameter rod) is vaporized by the second harmonic output of a pulsed Nd:YAG laser (Continuum Minilite II, 2–5 mJ per pulse). In the presence of an Ar gas pulse seeded with ∼0.2% O2 the DyO molecules are formed. Upon supersonically expanding in vacuum through a 1 mm diameter nozzle, the DyO molecules get cooled down to a rotational temperature of about 20 K. The thus formed molecular beam is shaped by a skimmer (3 mm diameter) and charged species are deflected out of the beam by the electric field (800 V cm−1) of a pair of electrodes.
The molecular beam enters the extraction region of a time-of-flight mass spectrometer (Jordan TOF Products, Inc.) that can be run either in linear or reflectron mode. In the linear configuration, the use of a small separation field of typically 1–5 V cm−1 in combination with time-delayed pulsed field ionisation in a field of 100–300 V cm−1, allows to distinguish between prompt and delayed ions in a mass analysed threshold ionization (MATI) configuration.23 Different resonance enhanced multiphoton ionisation (REMPI) schemes have been used to either detect the population of selected rotational levels of X8(v = 1) or to prepare DyO in Rydberg states n(J) of different principal quantum number n, in series that converge to different rotational levels J in the cation. The spectroscopy in the X8 ground state is performed using the most abundant isotopologue, 164Dy16O, while the preparation of Rydberg states makes use of a resonant two-color excitation step that has been identified specifically for 161Dy16O, see ref. 20.
Laser pulses in the visible region of the spectrum (about 5 ns duration) are produced by dye lasers (Sirah PrecisionScan and Radiant Dyes NarrowScan). Their wavelengths are calibrated with a wavemeter (HighFinesse WS6-600) to an accuracy better than the laser linewidth (<0.05 cm−1). The visible laser beams intersect the molecular beam perpendicularly between the extraction plates of the time-of-flight mass spectrometer. The IR beam from the FHI-FEL is loosely focussed counterpropagating the molecular beam. For the spectroscopy of ground-state DyO, the FEL macropulse (about 10 μs duration) ends 5 μs before probing the population in the X8(v = 1) state, while for excitation of the n(J) Rydberg states the FEL pulse starts 0.5 μs after their preparation. In these experiments, typical energies of the IR macropulse are a few tens of mJ in an approximately 5 mm diameter beam. Characterisation of the spectral profile of the FEL macropulse is done using an in-vacuum Czerny–Turner grating spectrometer (Acton VM-504, 75 lines per mm) combined with a pyroelectric linear array detector with 128 elements and a total width of 1/2 inch (DIAS Infrared 128LT).21 By coupling a fraction of the light emitted from the IR-FEL into the spectrometer this allows for a shot-to-shot recording of its spectrum.
When first set up, the combination of grating spectrometer and array detector has been calibrated using a He–Ne laser, operating the grating in 2nd to 20th order. In the present study, the goal was to map the spectrum of the IR-FEL using well-known molecular transitions to obtain an independent and more accurate frequency calibration. For this one has to keep in mind that the pulse structure of the FEL, which contains micropulses of an (adjustable) duration between several 100 fs to a few ps, results in a comparably large Fourier transform-limited spectral bandwidth. The lowest achievable bandwidth in the mid-IR (around 1000 cm−1) is on the order of 2 cm−1 (FWHM). In addition, inherent instabilities of the FEL's electron acceleration system result in a jittering around the central frequency which further broadens the spectral profile. The rovibrational spectrum of a reference molecule must therefore have large spacings between the rotational lines to be able to clearly resolve the spectral profile of the IR-FEL pulses. A large splitting can be realised in molecules with a large rotational constant, B, and/or by selectively studying transitions between levels with a high total angular momentum quantum number J. DyO is well suited for this purpose because the spacing between the two lowest rotational levels in the X8 state is already about 6.5 cm−1.9,10 Moreover, the large value of Ω ensures that the Ω-doubling, i.e. the splitting between the opposite parity levels for each J, will be negligibly small. Furthermore, we can selectively record the IR transitions of the monoxide of the most abundant isotope 164Dy with nuclear spin I = 0. The intrinsic spectral line-shape of IR transitions between rotational levels that can be considered fully degenerate parity doublets and that have no hyperfine structure basically is a delta-function. Given that for the v = 1 ← v = 0 band in the X8 state of 164DyO the absolute frequencies of the ΔJ = 0,±1 transitions are known to better than 0.005 cm−1, these transitions are ideally suited for our purpose.
Results and discussion
IR excitation in ground state neutral DyO
In earlier works, Cheng and Linton9,10 have characterised the optical spectra of 164DyO with high accuracy. Particularly relevant for the study here is that they determined ΔG1/2 as well as the rotational constants of v = 0 and v = 1 in the X8 state, from which the frequencies for the rotational lines of the v = 1 ← v = 0 band can be accurately determined. As all rotational levels are degenerate parity doublets, the requirement for a change of parity in an electric dipole transition is fulfilled for all possible ΔJ = 0,±1 lines. To characterise the FEL spectrum in the mid-IR, the lowest R-branch lines of the v = 1 ← v = 0 band appear to be best suited as these are rather isolated and go from the highest populated J levels in v = 0 to much less populated v = 1, J + 1 levels. The frequency of the R(8) line is precisely known as 848.108 cm−1 while the R(9) line is at 848.793 cm−1.Experimental IR spectra of 164DyO that reflect the change in population of the three lowest rotational levels (J = 8–10) in the X8, v = 1 state induced by IR excitation, are shown in Fig. 1. For this, the population in either one of these three rotational levels is probed via resonant excitation to rotational levels in the electronically excited [17.1]7 state.9 After a short delay of about 20 ns, the electronically excited molecules are ionised using the fourth harmonic of a Nd:YAG laser (266 nm), and the ions are mass-selectively detected. A two-color (1 + 1′)-REMPI spectrum in the relevant spectral range is shown in the lower part of Fig. 2, in which the P(8), P(9), and P(10) lines that have been used for rotationally selective detection are marked with an asterisk. The P-lines are stronger than the Q-lines, which are again stronger than the R-lines in this electronic spectrum, as expected for a ΔΩ = −1 transition. In the corresponding (1 + 1′)-REMPI spectrum from the X8, v = 0 state, shown in the upper part of Fig. 2, the P-lines are saturation broadened, and from their intensities, the Q- and R-lines are seen to be saturated as well. Note that the spectrum from the X8(v = 1) state has been up-shifted by 841.7 cm−1 to allow for a direct comparison between both spectra and for a rapid assignment of the rotational lines.
 | ||
| Fig. 1 IR spectra of 164DyO obtained by monitoring the change of population in three different rotational levels of X8(v = 1) upon IR excitation. The fits (solid lines) take both v = 1 ← v = 0 (growth) and v = 2 ← v = 1 (depletion) transitions into account. A typical time-averaged spectral profile of the IR radiation as recorded by the spectrometer is plotted in gray. The traces are shifted vertically and plotted on a common y-scale. | ||
 | ||
| Fig. 2 Two-color (1 + 1′)-REMPI spectra of the [17.1]7(v = 0) ← X8 transitions for 164DyO. The frequency axis is for the upper spectrum, that starts from X8, v = 0. The lower spectrum starts from X8, v = 1 and is up-shifted by 841.7 cm−1 to show the transitions on the same x-axis. Asterisks mark the three lowest lines of the P-branch that have been used to probe the population in X8, v = 1 for the measurements in Fig. 1. | ||
The middle and upper IR spectrum in Fig. 1, in which the J = 9 and J = 10 levels in X8, v = 1 are probed, show the R(8) and R(9) lines, respectively, as the strongest and rather isolated lines. As there is hardly any overlap on their lower frequency sides with the Q-lines, the shape of both of these lines directly reflects the spectral shape of the IR-FEL. It is seen to compare well to the spectral shape as measured by the grating spectrometer, shown in the inset of Fig. 1. To match the centre frequencies of the R(8) and R(9) lines as determined by the spectrometer with the experimentally known frequencies, the spectrometer reading had to be adjusted by +0.8 cm−1. Even though the required adjustment is only a fraction of the bandwidth of the IR-FEL radiation, this more accurate calibration is significant.
Closer inspection of the spectra in Fig. 1 reveals that in addition to contributions from v = 1 ← v = 0, which increase the population in the probed levels, also transitions to the next higher vibrational level, v = 2 ← v = 1, are seen that decrease the population. These opposing contributions have an overall shift relative to each other given by twice the vibrational anharmonicity, i.e. by 2ωeχe, and will have slightly different spacings due to the slightly different rotational constants in v = 0 and v = 2. Both contributions consist of only two rotational lines when probing J = 8, and it appears that the P(9) and Q(8) lines of the v = 1 ← v = 0 band largely coincide with the Q(8) and R(8) lines of the v = 2 ← v = 1 band, respectively. From this, it is concluded that 2ωeχe is about equal to the spacing between the two lowest rotational levels in X8, i.e. about 6 cm−1. For a more detailed analysis, the energy of the (v, J) levels in the X8 ground-state are calculated from the parameters given in Table 1via:
| E(v,J) = Tv + Bv[J(J + 1) − Ω2] |
| Tv = T0 + ωev − ωeχev(v + 1) and Bv = B0 − αv |
Note that in defining it in this way, T1 − T0 = ωe − 2ωeχe, commonly referred to as ΔG1/2. Table 1 lists the known values for T0 and T1 as well as for B0 and B1, from which B2 has been linearly extrapolated.
The only free parameters for fitting the IR spectra are the value of T2 and the widths and intensities of the lines; the correction to the frequency axis of the grating spectrometer of +0.8 cm−1 has already been applied in Fig. 1. The spectral lines are assumed to be Gaussian and to all have the same width. The best fit is obtained when a width (FWHM) of 3.3 cm−1 is assumed, about 20% larger than the width determined with the spectrometer, indicative of a slight saturation of the transitions.
From these fits a T2 value of 1677.6(2) cm−1 is obtained which results, together with the known value of T1, in a vibrational constant of ωe = 847.5(2) cm−1 and a vibrational anharmonicity parameter of ωeχe = 2.9(1) cm−1 for 164DyO. The uncertainty in the value of ωeχe is mainly related to the ambiguity in locating the weak v = 2 ← v = 1 signals. As an example, the decomposition of the IR spectrum obtained when monitoring the population in J = 10 of X8, v = 1 into the six Gaussian peaks is shown in Fig. 3.
 | ||
| Fig. 3 Fitted composition of the IR spectrum of 164DyO obtained when monitoring the population in J = 10 of X8, v = 1. The experimentally observed IR transitions are indicated in the level scheme (red arrows) together with the P(10) probing transition. The fitted spectrum consists of lines belonging to the v = 1 ← v = 0 band (solid) and to the v = 2 ← v = 1 band (dashed). The sum of both (thick grey) reproduces the experimentally observed change in population upon IR excitation (red crosses) very well. | ||
In earlier gas-phase measurements only the transition frequency for v = 1 ← v = 0 (ΔG1/2) had been determined. From this and the assumed dissociation limit of 6.29 eV24 estimates for ωe and ωeχe of 849 cm−1 and 3.5 cm−1 have been reported.25 These values agree well with the values experimentally determined here. Using density functional theory calculations with the B3LYP functional a vibrational constant of 769 cm−1 was predicted,26 considerably lower than the experimental gas-phase value.
The relative intensities of the lines in the spectra shown in Fig. 1 can be qualitatively understood based on an estimated factor three reduction in the population with each increase of the vibrational quantum number and based on an approximate factor two reduction in the population with each higher rotational quantum number; the latter can be seen from Fig. 2. The Hönl–London factors (SΔJ) for the P, Q, and R-lines are given by:27,28


adding up to the degeneracy (2J′′ + 1) of the starting level J′′. Based on these Hönl–London factors and the abovementioned ratio of the vibrational and rotational populations in the X8 state, one would expect the Q-lines to be by far the strongest in the spectra show in Fig. 1. In these experiments, the transitions are saturated, however, making the R-lines the strongest while the Hönl–London factors are less important. Assuming complete saturation of the transitions and an overlap of the P(9) and Q(8) lines of the v = 1 ← v = 0 band with the Q(8) and R(8) lines of the v = 2 ← v = 1 band, respectively, the observed fractional increase of the population around 841 cm−1 and the observed fractional decrease around 835 cm−1 in the lower spectrum of Fig. 1 can be well understood.
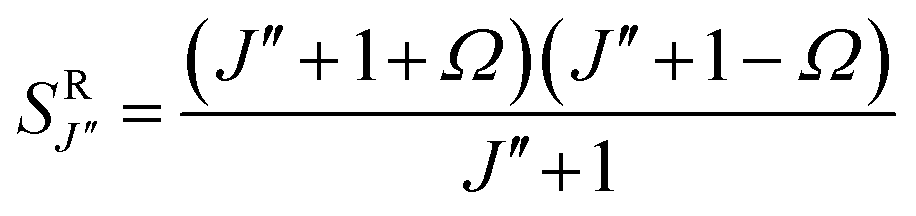
IR-PIRI spectroscopy of DyO+
Using the revised calibration of the FHI-FEL, IR spectra of gas-phase cationic DyO+ have been measured. These experiments rely on the general idea that core-nonpenetrating Rydberg states of the neutral molecule, i.e., Rydberg states with a high value of the principal quantum number n and with an orbital angular momentum l ≥ 2, may be understood as a cationic molecular core with an only weakly interacting electron on a large orbit. This Rydberg electron can be seen as spectator, and the molecule in the Rydberg state is similar to a weakly bound messenger complex.29 Photoexcitation of the cationic core such that the total energy in the molecule is above its ionisation energy will lead to autoionisation.30 An IR excitation spectrum measured via autoionisation of high-n molecular Rydberg states thus provides vibrational data on the cationic core.31 This experimental measurement method has been termed infrared photoinduced Rydberg ionisation (IR-PIRI)32 and its basic concept has been supported by a number of previous studies where, for instance, the influence of the principal quantum number n on the IR-PIRI spectra has been studied. In short, over a wide range of n = 7–93 no detectable influence on band positions in IR-PIRI spectra has been found.33,34 It should be noted, however, that these studies were for rather large molecules like trans-2-butene, cis-dichloroethene, or trichloro-ethene and that the thus obtained IR spectra had a rather limited resolution, possibly related to unresolved rotational band structures. While the band positions have been found to be independent of n, relative intensities of IR bands may be well affected as the spectral intensities in IR-PIRI depend not only on the IR absorption cross-sections but also on the decay dynamics of the different Rydberg states.34,35For the IR-PIRI studies, DyO molecules have been prepared in different Rydberg states. Details of the two-color laser excitation scheme that is used to selectively prepare the 161Dy16O isotopologue in specific n(J) levels are given elsewhere.20 In the excitation region, a constant electric field of 5 V cm−1 is applied. About 1 μs after laser preparation of the Rydberg states an electric field is applied for ion-extraction. Three isolated Rydberg states n(J), namely 34(7.5), 34(8.5), and 44(7.5), have been used for these experiments. In 34(7.5) and 34(8.5), the Rydberg electron is bound by about 95 cm−1, in 44(7.5) by about 57 cm−1. The electric field used for ion-extraction is kept sufficiently low that none of these states will be field-ionised. Ions are only produced when the molecules in the Rydberg states interact with the IR radiation of the FHI-FEL. This interaction starts shortly after laser preparation of the Rydberg states, and lasts until the ions have been extracted, i.e. for less than a μs. The IR radiation produces prompt ions as well as highly excited neutral molecules that are subsequently field-ionised. The constant, weak electric field separates the prompt ions from the neutrals. After field-ionisation and ion-extraction the prompt ions and the delayed ions are separately detected. The IR-PIRI signal will appear in the prompt ion signal, as autoionisation is expected to be fast upon excitation of the ion core. As the highly excited neutral molecules can autoionise before the extraction field is applied, these can also partly contribute to the prompt ion signal.
In Fig. 4 the difference of the signal of the prompt ions and the delayed ions is plotted as a function of the IR excitation frequency in the 880–930 cm−1 range. The signal of the delayed ions is subtracted after being multiplied with a factor that depends linearly on the IR frequency, such as to create a flat baseline in the resulting difference spectrum. This procedure is rationalised by the assumption that the autoionisation rate of the highly excited neutral molecules is a smooth function of the excitation energy. We have studied the effect of IR excitation in a much wider range of 770–1000 cm−1 but only around 910 cm−1 excess prompt ion signal has been reproducibly observed.
 | ||
| Fig. 4 IR-PIRI spectra of 161DyO in Rydberg states n(J), i.e. with principal quantum number n and converging to the rotational level with total angular momentum J in the cation. The solid curves are fits of the rotational structure, taking a rotational constant of 0.377 cm−1 for v = 1 of the cation and relative line intensities governed by the Hönl–London factors. Gaussian lineshapes with a width (FWHM) of 2.8 cm−1 are assumed. The traces are shifted vertically and plotted on a common y-scale. | ||
The IR-PIRI spectra obtained when starting from the Rydberg states 34(7.5) and 44(7.5) show a strong Q(7.5) line and a weaker but clearly discernible R(7.5) line. When starting from the 34(8.5) state, the P(8.5) line can be recognised below the (not so well resolved) Q(8.5) and R(8.5) lines. The observed spectra have been simulated using Gaussian lineshapes with a width (FWHM) of 2.8 cm−1. A rotational constant of 0.377 cm−1 is taken, about 0.5% less than the measured rotational constant for v = 0 of the cation, see ref. 20. The relative intensities of the P, Q and R lines are obtained from the expressions for the Hönl–London factors given earlier, and are seen to agree rather well with the experimental observations. As we are using less than 1 μs of the 10 μs duration macropulse of the FHI-FEL in these experiments, these transitions appear not to be saturated. The values obtained for ΔG1/2 in the cation are not precisely identical and for 34(7.5), 34(8.5), and 44(7.5) values of 907.4(2) cm−1, 907.4(4) cm−1, and 906.6(2) cm−1 have been found, respectively. While the first two values agree very well, the value found when starting from n = 44 is slightly lower, even though the difference is still only a fraction of the bandwidth of the IR-FEL. This difference might be caused by the Stark-shift of the Rydberg levels in the weak electric field, which will be most prominent for the highest n, or due to a perturbation, just as it has been seen for Rydberg states of CO, for instance.36 We therefore take a somewhat more conservative value for the overall uncertainty and determine the transition frequency as 907 ± 1 cm−1.
Comparison of lanthanoid monoxides, LnO and LnO+
The experimental value for ΔG1/2 of the 161DyO+ cation of 907(1) cm−1 that we have found, fits well into the general trend for cationic lanthanoid monoxides. Existing theoretical predictions scatter that much, that a reasonable assignment based on those appears difficult. From DFT calculations vibrational frequencies in the range of 936–983 cm−1 have been reported for the cationic ground state, depending on exchange–correlation functional and basis sets,12,26 all well above the experimental value. On the other hand, potentially more accurate coupled cluster calculations (CCSD(T,25)) using a frozen core predict a significantly lower value of 873 cm−1.12 This value appears to be unrealistically small, because it is even less than the experimental value found in a Ne matrix. Different types of relativistic calculations predict an harmonic ωe in the rather wide range between 843 and 1086 cm−1, while Morse fits to the calculated potential curves give 841 cm−1 for CASCI/CASSCF and 873 cm−1 for X2C-CASSCF with an anharmonicity ωeχe of 7 cm−1.19A comparison of vibrational frequencies for lanthanoid monoxides and their cations is given in Table 2 and Fig. 5 and contains the values measured via matrix isolation IR spectroscopy13,14,37,38 in cryogenic Ar and Ne matrices as well as gas-phase data. Values for gas-phase species have been obtained via a number of different techniques, i.e., emission,39–41 (spectrally dispersed) laser induced fluorescence,42–48 mass analysed threshold ionisation (MATI),4,5 (anion) photoelectron3,49,50 spectroscopies or IR dissociation spectroscopy of He messenger complexes.51 When available, also the vibrational anharmonicity is included in the table.
| ν(LnO) matrix | ΔG1/2,gas | ω e,gas | ω e χ e | ν(LnO+) matrix | ΔG1/2,gas | ω e,gas | ω e χ e | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ar | Ne | Ar | Ne | ||||||||
| a Unpublished, see ref. 15. b MATI, uncertainty ±5 cm−1.5 c IR-PD of SmO+Hen (n = 1–3) complexes, extrapolated from observed shifts of ∼2 cm−1/He. d This work, value for 164DyO. e MATI.4 f This work, for 161DyO, uncertainty ±1 cm−1. | |||||||||||
| La | 796.7 | 811.07 | 817.26 | 3.097 | 838.2 | 864.0a | 877e | 4, 37 and 39 | |||
| Ce | 808.3 | 820 | 824.3 | 827.7 | 1.7 | 849.4 | 874.8a | 887e | 4, 13, 38 and 42 | ||
| Pr | 816.9 | 828.0 | 835b | 857.4 | 882.3 | 909b | 5, 13 and 38 | ||||
| Nd | 814.2 | 825.1 | 829.9 | 834.1 | 2.1 | 848.3 | 878.4 | 889.8 | 892.4 | 1.3 | 3, 13, 38, 43 and 49 |
| Pm | — | — | — | — | — | — | — | — | — | — | |
| Sm | 807.4 | 819 | 822.32 | 840.0 | 878c | 13 | |||||
| Eu | 667.8 | 756.9 | 38 | ||||||||
| Gd | 812.7 | 824 | 824.99 | 829.25 | 2.13 | 844.8 | 13, 38 and 40 | ||||
| Tb | 823.9 | 837.1 | 856.5 | 883.7a | 13, 14 and 45 | ||||||
| Dy | 829.0 | 839.0 | 841.69 | 847.5d | 2.9d | 861.2 | 888.6 | 907f | 13 and 14 | ||
| Ho | 828.1 | 838.1 | 841.4 | 860.5 | 887.9 | 13, 14 and 46 | |||||
| Er | 828.5 | 861.8 | 13 and 14 | ||||||||
| Tm | 832.0 | 864.2 | 13 and 14 | ||||||||
| Yb | 660.0 | 683.1 | 788.7 | 14 and 47 | |||||||
| Lu | 829.3 | 838.3 | 844.5 | 3.1 | 864.9 | 13, 14 and 41 | |||||
 | ||
| Fig. 5 Comparison of experimental values of ν(LnO) for cationic (red) and neutral (blue) lanthanoid monoxides obtained via IR spectroscopy in cryogenic Ar and Ne matrices, as well as in the gas phase (ΔG1/2 values). There are no data for radioactive Pm. | ||
Overall, the stretching frequencies of both, LnO and LnO+, are slightly increasing with atomic number, but show pronounced minima for Eu and Yb, as discussed below. Values for ν(LnO) in a cryogenic Ar matrix have been determined for all lanthanoid monoxides, while in the less interacting Ne matrix only about half of the lanthanoids have been studied.13–15,38 Due to the interaction with the matrices, ν(LnO) values determined in Ar and Ne are significantly lowered compared to those found in the gas phase. These shifts are on average 15 cm−1 (4 cm−1 for Ne) in case of the neutral monoxides and ∼40 cm−1 (10–20 cm−1 for Ne) for the cations.
The monoxides of Eu and Yb show significantly lower ν(LnO) in the neutral and cation, related to their configuration of the metal dication being 4f76s0 and 4f146s0, while the other lanthanoids have a 4fn−16s1 configuration of the dication. The 6s electron forms a non-bonding σ orbital in LnO and upon ionisation it is removed for all Ln except for Eu and Yb leading to an increase of ν(LnO+) compared to ν(LnO). On the other hand, removal of a 4f electron (ionisation of EuO and YbO) leads to a larger increase of ν(LnO+) as it affects the shielding of the metal core stronger and, hence, results in a stronger Coulomb interaction between the metal and O2−. In the same vein, the 4f7 and 4f14 configurations in EuO and YbO effectively screen the metal core leading to a significantly reduced force constant also in the neutral species compared to other LnO.16
Conclusions
Vibrational spectroscopic information has been obtained for gas-phase DyO and DyO+ using an infrared free electron laser. Due to the large value of the total angular momentum in the ground state of both species, rotationally resolved IR spectra could be measured. For neutral DyO the vibrational constant and the vibrational anharmonicity have been determined as ωe = 847.5(2) cm−1 and ωeχe = 2.9(1) cm−1, which agree with previous estimates. For the cation the vibrational transition frequency ΔG1/2 has been obtained as 907(1) cm−1, which is the first gas-phase value for ν(DyO+). The assignment for ν(DyO+) relies on the assumption that the IR-PIRI scheme probes an essentially undisturbed cationic core. The latter is substantiated by the observation of P, Q and R lines in the IR-PIRI spectra with relative intensities just as expected for a v = 1 ← v = 0 transition in the ion.The various excitation and detection schemes applied here are rather unusual in the spectroscopy of diatomic molecules for which vibrational data is frequently extracted from rotationally and vibrationally resolved electronic spectra. Similar experiments to ours, using an IR free electron laser but without resolving any rotational structures in the IR spectra, have been performed before only for CO.52 The combination of IR excitation with rotationally state-selective ionisation detection schemes as shown here provides an alternative way to obtain vibrational data for systems with complex electronic spectra.
Author contributions
Sascha Schaller: investigation (equal); data curation (lead); writing – original draft (supporting); writing – review and editing (equal). Sandy Gewinner: investigation (supporting); writing – review and editing (equal). Wieland Schöllkopf: investigation (supporting); writing – review and editing (equal). Gerard Meijer: supervision (lead); conceptualization (equal); writing – review and editing (lead). André Fielicke: conceptualization (equal); investigation (equal); writing – original draft (lead); writing – review and editing (equal).Data availability
The spectroscopic data supporting this article is included in the figures of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Open Access funding provided by the Max Planck Society.Notes and references
- J. L. Mason, H. Harb, J. E. Topolski, H. P. Hratchian and C. C. Jarrold, Acc. Chem. Res., 2019, 52, 3265–3273 CrossRef CAS PubMed.
- C. Huizenga, H. P. Hratchian and C. C. Jarrold, J. Phys. Chem. A, 2021, 125, 6315–6331 CrossRef CAS PubMed.
- M. C. Babin, M. DeWitt, J. A. DeVine, D. C. McDonald, S. G. Ard, N. S. Shuman, A. A. Viggiano, L. Cheng and D. M. Neumark, J. Chem. Phys., 2021, 155, 114305 CrossRef CAS PubMed.
- W. Cao, Y. Zhang, L. Wu and D.-S. Yang, J. Phys. Chem. A, 2021, 125, 1941–1948 CrossRef CAS PubMed.
- Y. Zhang, T. Nakamura, L. Wu, W. Cao, G. Schoendorff, M. S. Gordon and D.-S. Yang, J. Chem. Phys., 2022, 157, 114304 CrossRef CAS PubMed.
- K. Schofield, J. Phys. Chem. A, 2006, 110, 6938–6947 CrossRef CAS PubMed.
- B. R. Yadav, S. B. Rai and D. K. Rai, Pramana, 1980, 14, 379–388 CrossRef CAS.
- L. A. Kaledin and E. A. Shenyavskaya, J. Mol. Spectrosc., 1981, 90, 590–591 CrossRef CAS.
- C. Linton, D. M. Gaudet and H. Schall, J. Mol. Spectrosc., 1986, 115, 58–73 CrossRef CAS.
- C.-H. Cheng, PhD thesis, University of New Brunswick, Canada, 1992.
- C. Linton and B. Simard, J. Chem. Phys., 1992, 96, 1698–1703 CrossRef CAS.
- M. Ghiassee, E. G. Christensen, T. Fenn and P. B. Armentrout, J. Phys. Chem. A, 2023, 127, 169–180 CrossRef CAS PubMed.
- R. L. DeKock and W. Weltner, Jr., J. Phys. Chem., 1971, 75, 514–525 CrossRef.
- S. P. Willson and L. Andrews, J. Phys. Chem. A, 1999, 103, 6972–6983 CrossRef CAS.
- Y. Gong, M. Zhou and L. Andrews, Chem. Rev., 2009, 109, 6765–6808 CrossRef CAS PubMed.
- R. W. Field, Ber. Bunsenges. Phys. Chem., 1982, 86, 771–779 CrossRef CAS.
- C. Linton, J. Phys., Colloq., 1987, 48, C7–611 Search PubMed.
- P. Carette and A. Hocquet, J. Mol. Spectrosc., 1988, 131, 301–324 CrossRef CAS.
- V. D. Dergachev, D. D. Nakritskaia and S. A. Varganov, J. Phys. Chem. Lett., 2022, 13, 6749–6754 CrossRef CAS PubMed.
- S. Schaller, J. Seifert, G. Valtolina, A. Fielicke, B. G. Sartakov and G. Meijer, arXiv, 2024, preprint, arXiv:2406.03160 DOI:10.48550/arXiv.2406.03160.
- W. Schöllkopf, S. Gewinner, H. Junkes, A. Paarmann, G. von Helden, H. Bluem and A. M. Todd, Proc. SPIE, 2015, 9512, 95121L CrossRef.
- A. Fielicke, G. von Helden and G. Meijer, Eur. Phys. J. D, 2005, 34, 83–88 CrossRef CAS.
- L. Zhu and P. Johnson, J. Chem. Phys., 1991, 94, 5769–5771 CrossRef CAS.
- M. S. Chandrasekharaiah and K. A. Gingerich, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1989, vol. 12, pp. 409–431 Search PubMed.
- R. J. M. Konings, O. Beneš, A. Kovács, D. Manara, D. Sedmidubský, L. Gorokhov, V. S. Iorish, V. Yungman, E. Shenyavskaya and E. Osina, J. Phys. Chem. Ref. Data, 2014, 43, 013101 CrossRef.
- Z. J. Wu, W. Guan, J. Meng and Z. M. Su, J. Cluster Sci., 2007, 18, 444–458 CrossRef CAS.
- I. Kovács, Rotational Structure in the Spectra of Diatomic Molecules, Akadémiai Kiadó, Budapest, 1969 Search PubMed.
- G. H. Herzberg, Molecular Spectra and Molecular Structure. I Spectra of Diatomic Molecules, Van Nostrand Inc., 1950 Search PubMed.
- M. Okumura, L. I. Yeh, J. D. Myers and Y. T. Lee, J. Chem. Phys., 1986, 85, 2328–2329 CrossRef CAS.
- P. M. Johnson, in Photoionization and Photodetachment, ed. C. Y. Ng, World Scientific, 2000, vol. I Search PubMed.
- A. Fujii, A. Iwasaki, T. Ebata and N. Mikami, J. Phys. Chem. A, 1997, 101, 5963–5965 CrossRef CAS.
- M. Gerhards, M. Schiwek, C. Unterberg and K. Kleinermanns, Chem. Phys. Lett., 1998, 297, 515–522 CrossRef CAS.
- H. K. Woo, P. Wang, K. C. Lau, X. Xing and C. Y. Ng, J. Chem. Phys., 2004, 120, 1756–1760 CrossRef CAS PubMed.
- P. Wang, H. K. Woo, K. C. Lau, X. Xing, C. Y. Ng, A. S. Zyubin and A. M. Mebel, J. Chem. Phys., 2006, 124, 064310 CrossRef CAS PubMed.
- H. Nakamura, Int. Rev. Phys. Chem., 1991, 10, 123–188 Search PubMed.
- M. Komatsu, T. Ebata, T. Maeyama and N. Mikami, J. Chem. Phys., 1995, 103, 2420–2435 CrossRef CAS.
- L. Andrews, M. Zhou, G. V. Chertihin and C. W. Bauschlicher, J. Phys. Chem. A, 1999, 103, 6525–6532 CrossRef CAS.
- S. P. Willson and L. Andrews, J. Phys. Chem. A, 1999, 103, 3171–3183 CrossRef CAS.
- P. Carette, J. Mol. Spectrosc., 1990, 140, 269–279 CrossRef CAS.
- B. R. Yadav, S. B. Rai and D. K. Rai, Can. J. Phys., 1976, 54, 2429–2434 CrossRef CAS.
- A. Bernard and C. Effantin, Can. J. Phys., 1986, 64, 246–251 CrossRef CAS.
- C. Linton, M. Dulick and R. W. Field, J. Mol. Spectrosc., 1979, 78, 428–436 CrossRef CAS.
- C. Linton, C. Effantin, P. Crozet, A. J. Ross, E. A. Shenyavskaya and J. d’Incan, J. Mol. Spectrosc., 2004, 225, 132–144 CrossRef CAS.
- G. Bujin and C. Linton, J. Mol. Spectrosc., 1991, 147, 120–141 CrossRef CAS.
- N. Kulikov, L. A. Kaledin, A. I. Kobyliansky and L. V. Gurvich, Can. J. Phys., 1984, 62, 1855–1870 CrossRef.
- Y. C. Liu, C. Linton, H. Schall and R. W. Field, J. Mol. Spectrosc., 1984, 104, 72–88 CrossRef CAS.
- S. A. McDonald, S. F. Rice, R. W. Field and C. Linton, J. Chem. Phys., 1990, 93, 7676–7686 CrossRef CAS.
- J. R. Schmitz, A. T. Le, T. C. Steimle, A. Rodriguez and M. C. Heaven, J. Phys. Chem. A, 2022, 126, 7210–7220 CrossRef CAS PubMed.
- R. A. VanGundy, T. D. Persinger and M. C. Heaven, J. Chem. Phys., 2019, 150, 114302 CrossRef PubMed.
- M. L. Weichman, B. Vlaisavljevich, J. A. DeVine, N. S. Shuman, S. G. Ard, T. Shiozaki, D. M. Neumark and A. A. Viggiano, J. Chem. Phys., 2017, 147, 234311 CrossRef PubMed.
- A. Lachowicz, E. H. Perez, N. S. Shuman, S. G. Ard, A. A. Viggiano, P. B. Armentrout, J. J. Goings, P. Sharma, X. Li and M. A. Johnson, J. Chem. Phys., 2021, 155, 174303 CrossRef CAS PubMed.
- Y. Ogi, T. Endo, K. Tsukiyama, H. Kondoh, K. Tono, Y. Ogawa, Y. Hamada, T. Ohta and H. Kuroda, J. Electron Spectrosc. Relat. Phenom., 2003, 128, 67–73 CrossRef CAS.
| This journal is © the Owner Societies 2024 |