
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, photophysics and two-photon absorption of imidazole-centred tripodal chromophores†
Jiří
Kulhánek
 a,
Zuzana
Burešová
a,
Milan
Klikar
a,
Lampros
Sdralias
b,
Alexandros
Katsidas
b,
Oldřich
Pytela
a,
Patrik
Pařík
a,
Aleš
Růžička
c,
Mihalis
Fakis
*b and
Filip
Bureš
*a
a,
Zuzana
Burešová
a,
Milan
Klikar
a,
Lampros
Sdralias
b,
Alexandros
Katsidas
b,
Oldřich
Pytela
a,
Patrik
Pařík
a,
Aleš
Růžička
c,
Mihalis
Fakis
*b and
Filip
Bureš
*a
aInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic. E-mail: filip.bures@upce.cz; Web: https://bures.upce.cz Tel: +420 46 603 7099
bDepartment of Physics, University of Patras, Patras, 26504, Greece. E-mail: fakis@upatras.gr; Tel: +30 2610 996794
cDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic
First published on 11th July 2024
Abstract
Tripodal push–pull chromophores with D–(π–A)3 arrangement were synthesized using 1-methyl-2,4,5-triphenyl-1H-imidazole as a central electron donor, and their thermal, electrochemical, photophysical and non-linear optical properties were studied and corroborated with quantum-chemical calculations. Their facile synthesis involved Suzuki–Miyaura and Knoevenagel reactions, allowing the installation of various peripheral electron acceptors such as formyl, cyano, ester, trifluoromethyl and more complex moieties such as malonic/acetic acid derivatives, indan-1,3-dione and rhodanine. All phenyl rings appended at the central imidazole core were more or less twisted depending on the peripheral substitution. Although imidazole undergoes reversible one-electron oxidation, peripheral acceptors are reduced irreversibly in a multi-electron process. This behaviour is further seen as a variation of the LUMO, while the HOMO remained almost unaltered across the whole series. TD-DFT calculations revealed centrifugal charge transfer from the central imidazole to all C2, C4 and C5 branches occupied by the LUMO, LUMO+1 and LUMO+2. The HOMO–LUMO gap is tuneable within the range of 3.55–2.31 eV, while the longest-wavelength absorption/emission maxima were found within the broad range of 304–448/393–612 nm. Although the absorption spectra are solvent-independent, the emission depends strongly on the solvent polarity and the electron-withdrawing power of the peripheral acceptors. Extended chromophores with complex electron acceptors were investigated as two-photon absorbers, revealing relatively good cross-section values of up to 521 GM and a figure-of-merit (ΦF × δ2PA) of around 190 GM.
Introduction
Linear (D–π–A), dipodal ((D–π)2–A/(A–π)2–D) and tripodal ((D–π)3–A/(A–π)3–D) push–pull chromophores continue to attract steady attention due to their peculiar (optical) properties dictated mostly by the extent of intramolecular charge transfer (ICT) from the donor (D) to the acceptor (A).1 In principle, di- and tri-podal molecules can adopt either donor- or acceptor-centred arrangements (centrifugal/centripetal; Fig. 1), while these push–pull systems may resemble letters of the alphabet.2 The central acceptor is mostly represented by aromatic six-membered (hetero)cycles such as 1,3,5-trisubstuted benzene,3–6 unsymmetrical pyridine,7,8 diazines9–13 or symmetrical triazines.14–16 Tripodal T-shaped chromophores can also be built on indandione central acceptors.17–19 However, centrifugal chromophores are more frequent, especially those based on triphenylamine cores trisubstituted with peripheral acceptors.20–24N-Phenylcarbazoles25 and N-heterotriangulenes,26 representing planarized triphenylamines, can also be used as useful scaffolds to construct tripodal chromophores. Five-membered 1H-imidazole can be principally employed as an electron-rich heterocycle, while its 2,4,5-trisubstitution affords tripodal systems.27 In our former investigation, we utilized 2-substituted imidazole-4,5-dicarbonitrile28–31 and bis(4-(N,N-dimethylamino)phenyl)imidazole32–34 either as electron-withdrawing or -releasing moieties of various Y-shaped push–pull molecules. Hence, modulation of the appended substituents1 or eventual N-quaternization towards (benz)imidazolium (Fig. 1)35 can significantly affect the imidazole electron properties. | ||
| Fig. 1 The most explored acceptor- and donor-centred tripodal push–pull systems along with the general structure of lophine-derived derivatives 1–6. | ||
Imidazole-centred tripodal molecules have found various applications as pharmacophores,36 charge-transfer (CT) chromophores,37 fluorophores,38–42 molecular sensors capable of detecting various analytes such as F−, BF4−/ClO4−, Hg2+, Zn2+, cysteine or picric acid,43–51 viscosity probes,52 pH probes,53,54 luminescent solar concentrators (LSCs),55 emitters for organic light-emitting diodes (OLEDs)56–59 and robust heterocycles for dye-sensitised solar cells (DSSCs).60–62 Besides interesting linear optical properties, imidazole push–pull molecules have also been investigated for their nonlinear optical (NLO) activity. For instance, a combination of two ferrocene donors and one ester acceptor imparted imidazole second-order nonlinearity with μβ = 780 × 10−48 esu, as measured in the EFISH experiment.63
N,N-Dimethylamino-terminated imidazole-4,5-dicarbonitrile systems showed hyperpolarizability switchable via protonation with the hyperpolarizability ratio between the amine ↔ ammonium forms exceeding 20.31 Imidazole-centred molecules were also utilized as third-order NLOphores, especially for two-photon absorption (2PA). It has been demonstrated that the tripodal Y-shaped arrangement in combination with π-system extension and N-substitution allows tuning of the imidazole two-photon absorptivity with 2PA cross-section coefficients δ2PA up to 1700 GM47,64–73 while molecules with large δ2PA can be applied in applications such as optical limiting,74,75 photodynamic therapy76,77 and 2PA-induced microfabrication.78,79 It is somewhat curious that the 2PA of the parent 2,4,5-triphenylimidazole (lophine) peripherally substituted with electron acceptors has not been addressed so far. Hence, as part of our ongoing efforts towards disclosing the NLO activity of organic fundamental building blocks, such as triphenylamine,80–83 (di/tri)azines11,14,82,83 and indan-1,3-dione,17–19 we report herein the synthesis of two series of lophine-based push–pull chromophores 1–6 (Fig. 1) and the investigation of their thermal, electrochemical, linear and third-order NLO properties corroborated with quantum-chemical calculations.
Results and discussion
Synthesis
The synthetic approach towards trisubstituted N-methyl lophines 1a–4a is outlined in Scheme 1a.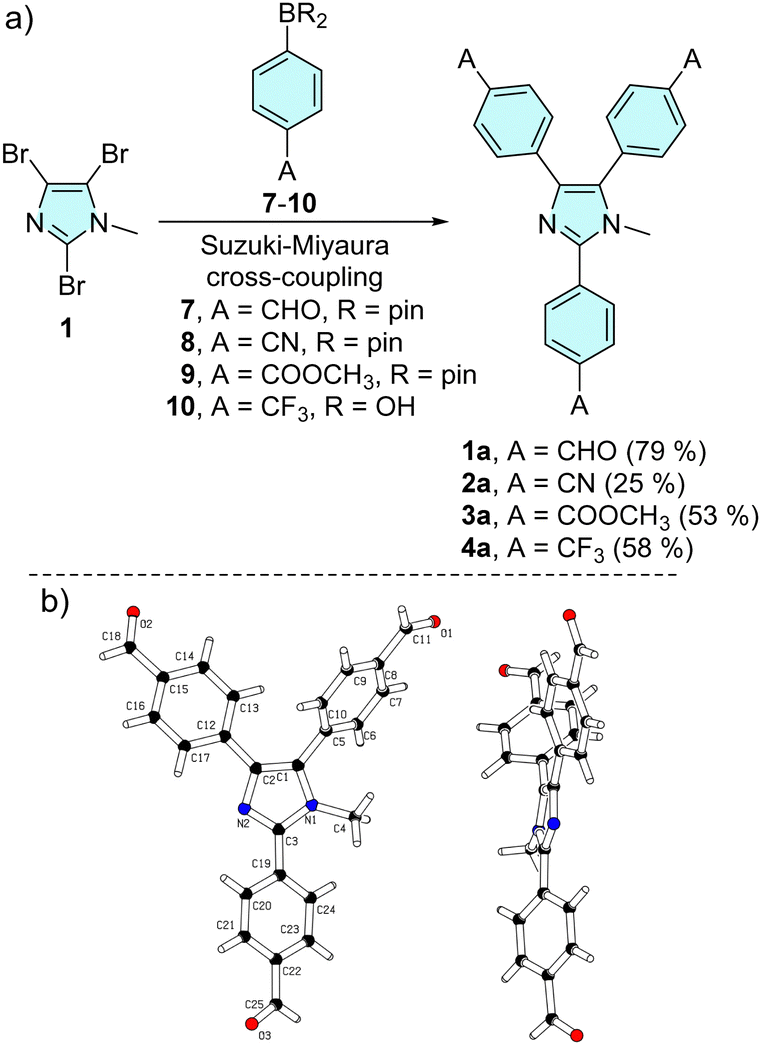 | ||
| Scheme 1 Threefold cross-coupling reaction of tribromoimidazole towards triphenylimidazole series a (a) and X-ray molecular representation (b) of 1a (CCDC 2331438†). | ||
The synthesis involves threefold Suzuki–Miyaura cross-coupling of 2,4,5-tribromo-1-methylimidazole 1 with 4-substituted phenylboronic acids/pinacol esters 7–10 catalysed by [Pd(PPh3)4].84 Chromophores 1a, 3a and 4a were obtained in reasonable 50–80% isolated yields, while the synthesis of tricyano derivative 2a (25%) was accompanied by tedious purification via multiple column chromatography. The starting imidazole 1 was prepared by N-methylation of commercially available 2,4,5-tribromo-1H-imidazole with dimethylsulfate (see the synthesis part in the ESI†).28
The molecular structure of tricarbaldehyde 1a was confirmed by X-ray diffraction (Scheme 1b and Table S1, Fig. S25–S27 in the ESI†). The expected nonplanar arrangement of the central triphenylimidazole corresponds to the related X-ray molecular structures.39,40,48 For instance, N-ethyllophine (CCDC 765921) and 1a showed a similar nonplanar arrangement of the phenyls appended at the imidazole C2/C4/C5 (torsion angles 41/18/71 vs. 27/21/55°). With tricarbaldehyde 1a in hand, we further extended our investigation to series b (Scheme 2). Aldehyde 1a underwent facile threefold Knoevenagel condensation with activated methylene compounds, such as malonic/acetic acid derivatives 11–12/15–16, indan-1,3-dione 13 and 3-ethylrhodanine 14, which allowed the coupling of complex peripheral withdrawing units of various strengths.85,86 Target chromophores 1b–6b were isolated with yields ranging from 50 to 70%. Knoevenagel condensations using unsymmetrical C-acids 12 and 14–16 afforded products 2b and 4b–6b with an exclusive stereochemistry. Three separate signals of the three vinylic protons found in the 1H-NMR spectra point to a single isomer (Fig. S11, S15, S17 and S19, ESI†). Appropriate configuration assignment was accomplished via proton-coupled 13C-NMR spectroscopy allowing the determination of 3J(C–H) interaction constants of the vinylic proton and the neighbouring cyano (2b, 5b, 6b) and carbonyl (4b) carbon atoms (Fig. S21–S24, ESI†). A 3J(C–H) of 13.8 Hz was found for 2b, 5b and 6b, indicating their (E)-configuration and chromophore 4b showed 3J(C–H) = 6.3 Hz pointing to the (Z)-isomer.87
 | ||
| Scheme 2 Knoevenagel condensation of tricarbaldehyde 1a with various electron-withdrawing moieties towards extended chromophores in series b. | ||
Thermal analysis
The thermal properties of imidazoles 1–6 were determined using differential scanning calorimetry. The native DSC thermograms, deduced melting points Tm and temperatures of thermal decomposition Td are provided in the ESI† (Fig. S28–S30 and Table S2). With the same central imidazole, the thermal properties of 1–6 are dictated by the peripheral withdrawing substituents, and their nature clearly affects the thermal robustness of particular imidazole derivatives. Lophines 1a–4a terminated by fundamental acceptors exhibited a distinctive endothermic melting process between 166 and 273 °C (ester 3a → nitrile 2a). Aldehyde 1a showed two melting peaks, probably due to the presence of a second minor crystalline form. Further heating of the melts of 1a–4a resulted in either evaporation or thermal decomposition of the sample. Trifluoromethyl derivative 4a was completely evaporated above 380 °C with no apparent residue in the crucible after measurement. In contrast, a neat exothermic decomposition peak was recorded for aldehyde 1a above 290 °C. Gradual evaporation of the melts of 2a and 3a was not observed, and these samples thermally degraded at temperatures above 400 °C. Although nitrile 2a gradually degraded in a typical exothermic direction, ester 3a decomposed vigorously endothermically with a clear charred residue in the crucible after analysis. The highest thermal robustness with Tm = 273 °C and Td > 400 °C was observed for nitrile derivative 2a.Imidazole derivatives in series b with extended acceptor units afforded more complex thermograms, while a distinct and sharp peak of melting was observed only for cyanoester derivative 2b (Tm = 196 °C). Compound 6b underwent a broad and blurred melting process, which was further confirmed visually by measuring the sample in a capillary.
In contrast, no melting was visually observed for indandione derivative 3b, while the broad endothermic peak above 300 °C was probably related to an enantiotropic solid–solid transition following glass transition at around 270 °C. The other molecules 1b, 4b and 5b decomposed directly without melting. The endothermic peak of 1b between 180 and 210 °C is probably associated with the solid–solid transition. In general, the molecules with olefinic π-linkers in series b showed distinct thermal degradation ranging between 260 and 330 °C. Only rhodanine derivative 4b exhibited a very gradual exothermic decomposition with a hardly distinguishable onset, probably above 220 °C. Nevertheless, the sample was obviously carbonized in a crucible after the DSC analysis. Overall, the thermal stability of the imidazoles in series a is higher as compared to those of the extended derivatives in series b, which is in line with our previous observations.19,88
Electrochemistry
The electrochemical behaviour of the target imidazoles 1–6 was investigated in acetonitrile (ACN) containing 0.1 M Bu4NPF6 in a three-electrode cell by cyclic voltammetry (CV). The recorded peak potentials are plotted vs. the silver/silver chloride electrode (SSCE). The acquired electrochemical data are summarized in Table 1, and further experimental details and CV diagrams are given in the ESI† (Fig. S31–S39). Indan-1,3-dione derivative 3b was poorly soluble in ACN and other solvents and, therefore, it was excluded from the CV analysis. The first oxidation of imidazoles 1–6, except for 4b, was detected as a reversible process localized on the N-methylimidazole unit. The corresponding values of peak-to-peak separation (ΔEp ∼60–70 mV) imply one-electron oxidation. These observations are consistent with the reported electrochemical behaviour of N-methyltriphenylimidazole redox catalysts.89–91 The first oxidation of the rhodanine derivative 4b was recorded as a fully irreversible process due to the preferential oxidation of the sulphur atom instead of the imidazole, which is consistent with our previous studies.85,86 The first reduction involves peripheral acceptors and, therefore, it was observed as a multi-electron irreversible process followed by further reductions. This corresponds to the D–(π–A)3 arrangement of the investigated chromophores bearing three unequivocal branches (acceptors). A (quasi)reversible first reduction was observed only for compounds 2a and 3a bearing CN and COOCH3 acceptors. The current maxima generally imply that the first reduction is a two-electron process, but more than 10 electrons were exchanged during the reduction of 4a terminated with trifluoromethyl groups.| Comp. | E ap(ox1)a [V] | E cp(ox1)a [V] | E cp(red1)b [V] | E ap(red1)b [V] | E HOMO [eV] | E LUMO [eV] | ΔEf [eV] |
|---|---|---|---|---|---|---|---|
| a E ap and Ecp are anodic and cathodic peak potentials of the first reversible oxidation process, respectively. b E ap and Ecp are anodic and cathodic peak potentials of the first reversible reduction process, respectively. All CVs were measured at a scan rate of 100 mV s−1, and all potentials were given vs. SSCE. c Irreversible process. d Deducted as a shoulder potential. e Energies of the HOMO/LUMO levels calculated according to −EHOMO/LUMO = (Eap(ox1) + 0.036) or (Ecp(red1) + 0.036) + 4.429 (vs. SCE).92,93 The increment of +0.036 V corresponds to the difference between SCE (0.241 vs. SHE) and SSCE (0.205 vs. SHE). f HOMO–LUMO gap ΔE = |EHOMO − EHOMO| resp. Eap(ox1) − Ecp(red1). | |||||||
| 1a | 1.60 | 1.54 | −1.47 | —c | −6.07 | −3.00 | 3.07 |
| 2a | 1.79 | 1.72 | −1.76 | −1.63 | −6.26 | −2.71 | 3.55 |
| 3a | 1.57 | 1.50 | −1.90 | −1.80 | −6.04 | −2.57 | 3.47 |
| 4a | 1.63 | 1.57 | −2.04 | —c | −6.10 | −2.43 | 3.67 |
| 1b | 1.73 | 1.66 | −0.72 | —c | −6.20 | −3.75 | 2.45 |
| 2b | 1.59 | 1.52 | −0.97 | —c | −6.06 | −3.50 | 2.56 |
| 4b | 1.45 | —c | −0.86d | —c | −5.92 | −3.61 | 2.31 |
| 5b | 1.65 | 1.58 | −0.81 | —c | −6.12 | −3.66 | 2.46 |
| 6b | 1.66 | 1.59 | −0.79 | —c | −6.13 | −3.68 | 2.45 |
Since half-wave potentials are not available for all molecules, the peak potentials Eap(ox1) and Ecp(red1) were used to consistently calculate the HOMO and LUMO energies (Table 1). The EHOMO/LUMO energies, ranging between −6.26 and −5.92/−3.75 and −2.43 eV, are shown in Fig. 2. Except for rhodanine derivative 4b (EHOMO = −5.92 eV), the EHOMO found at around −6.12 eV confirms that imidazole is a central donor in all target compounds. The HOMO is slightly altered, reflecting the electron-withdrawing power of the appended acceptors. For instance, when comparing compounds in series a, the CN groups in 2a deepened the HOMO down to −6.26 eV, as a result of its strongest electronic effect. The Hammett constants σp of the CHO/CN/COOCH3/CF3 substituents (0.42/0.66/0.45/0.54) roughly correspond to their impact on the HOMO levels of 1a–4a shown in Fig. 2.1 The LUMO levels clearly reflect the variation of the peripheral acceptors attached to the lophine core and their readiness towards electrochemical reduction as well as further extension of the π-system. Hence, tricarbaldehyde 1a proved to be the most easily reducible, followed by cyano, methoxycarbonyl and trifluormethyl derivatives 2a–4a. More complex acceptor moieties in conjunction with the additional olefinic linker induced a significant deepening of the LUMO levels, thus resulting in a diminished HOMO–LUMO gap compared to series a. Although the ΔE values of 1a–4a range between 3.67 and 3.07 eV, the gap of 1b–6b was found at around 2.45 eV (excluding 4b with the overestimated HOMO level). Hence, there is not much difference in attaching various acceptor moieties to the N-methyllophine core via an additional olefinic linker as in 1b–6b.
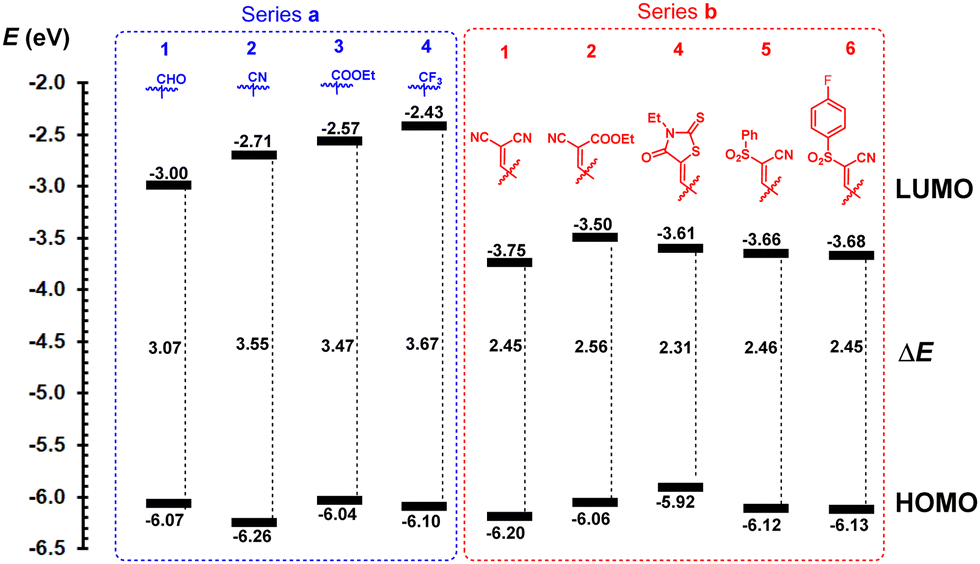 | ||
| Fig. 2 Energy level diagram of investigated imidazoles 1–6. | ||
Some differences can be seen in replacing one CN with COOCH3 (1bvs.2b, ΔE = 2.45 vs. 2.56 eV), but the presence of an additional fluorine atom in 6b is diminished compared to 5b (ΔE = 2.45 vs. 2.46 eV).
Linear optical properties
The fundamental optical properties of chromophores 1–6 were investigated via electronic absorption and emission spectra measured in four solvents of different polarities (toluene, CHCl3, THF, and ACN; c = 1 × 10−5 M). Fig. 3 and Table 2 show representative spectra/data in THF; see the ESI† for a complete list of spectra (Fig. S41–S44) and data (Tables S3–S5). When going from less polar toluene to more polar ACN, the longest-wavelength absorption band slightly shifts hypsochromically (Δλmax = 0–10 nm), a typical feature of CT chromophores.1 The absorption spectra of imidazoles in series a exhibit one single band, whereas the spectra of derivatives in series b feature two bands and a significantly increased molar absorption coefficient. As demonstrated in Fig. 3a, chromophore 2a exhibits one absorption band in the UV area (λmax = 325 nm; ε = 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 M−1 cm−1), whereas its extended analogue 1b features two bands appearing at 303 (ε = 51900 M−1 cm−1) and 411 nm (ε = 70000 M−1 cm−1). Variation of the acceptor moiety in 1a–4a and 1b–6b (Table 2) allowed the tuning of the absorption maxima within the ranges of 304–344 and 399–448 nm, respectively. The measured longest-wavelength absorption maxima vary with different peripheral acceptor substitutions in the same way as observed by electrochemistry. The correlation between the measured optical (1240/λAmax) and electrochemical gaps (ΔE) is almost perfectly linear with R2 = 0.99 (see Fig. S40 in the ESI†).
100 M−1 cm−1), whereas its extended analogue 1b features two bands appearing at 303 (ε = 51900 M−1 cm−1) and 411 nm (ε = 70000 M−1 cm−1). Variation of the acceptor moiety in 1a–4a and 1b–6b (Table 2) allowed the tuning of the absorption maxima within the ranges of 304–344 and 399–448 nm, respectively. The measured longest-wavelength absorption maxima vary with different peripheral acceptor substitutions in the same way as observed by electrochemistry. The correlation between the measured optical (1240/λAmax) and electrochemical gaps (ΔE) is almost perfectly linear with R2 = 0.99 (see Fig. S40 in the ESI†).
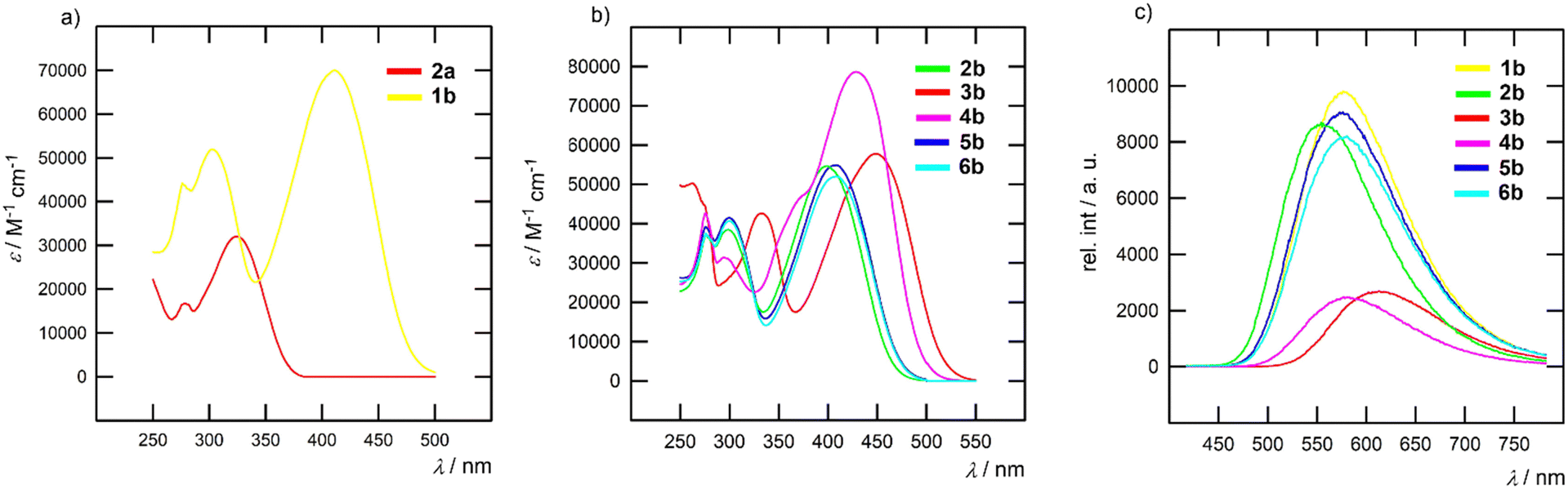 | ||
| Fig. 3 Representative absorption (a) and (b) and emission (c) spectra of the imidazole chromophores measured in THF. | ||
| Comp. | λ Amax [nm eV−1] | ε [M−1 cm−1] |
λ
Emaxa [nm eV−1] |
Φ [%] | Stokes shift [cm−1 eV−1] | 〈τ〉c [ns] | δ 2PA[GM]/λmax2PA [nm] | Φ F × δ2PA [GM] |
|---|---|---|---|---|---|---|---|---|
a Excited at λAmax.
b Relative to anthracene/perylene (Φ = 0.36/0.94 in cyclohexane)94 for series a/b.
c Average lifetime calculated using nanosecond fluorescence decay  . .
|
||||||||
| 1a | 344/3.60 | 35300 |
452/2.74 | 17 | 7000/0.86 | — | — | — |
| 2a | 325/3.82 | 32100 |
421/2.95 | 36 | 7019/0.87 | — | — | — |
| 3a | 327/3.79 | 32300 |
426/2.91 | 34 | 7110/0.88 | — | — | — |
| 4a | 304/4.08 | 23000 |
393/3.16 | 32 | 7440/0.92 | — | — | — |
| 1b | 411/3.02 | 70000 |
576/2.15 | 55 | 7330/0.87 | 2.04 | 286/760 | 157 |
| 2b | 399/3.11 | 54700 |
554/2.24 | 56 | 7010/0.87 | 2.82 | 340/740 | 190 |
| 3b | 448/2.77 | 57800 |
612/2.03 | 21 | 5980/0.74 | 1.36 | 286/810 | 60 |
| 4b | 429/2.89 | 78600 |
580/2.14 | 17 | 6070/0.75 | 1.34 | 521/750 | 88 |
| 5b | 409/3.03 | 54900 |
573/2.16 | 52 | 7000/0.87 | 2.76 | 325/750 | 169 |
| 6b | 408/3.04 | 52000 |
578/2.15 | 50 | 7210/0.89 | 3.23 | 390/740 | 195 |
The longest-wavelength absorption band of rhodanine-terminated imidazole 4b features an additional high-energy shoulder as well as the largest molar absorption coefficient in all used solvents (Fig. 3b). This feature was previously observed for rhodanine molecules95,96 and can be attributed to the formation of H-aggregates, which is further supported by the diminished fluorescence of 4b. Indan-1,3-dione acceptors in 3b induced the most bathochromic shift among all acceptors used, which is in line with our recent study.85 Analogous to the aforementioned electrochemical measurements, the introduction of a fluorine atom (5b → 6b) has no effect.
In contrast to the steady absorption spectra, the fluorescence spectra (Fig. 3c) are solvent-dependent and red-shifted with increasing solvent polarity (see Fig. S41–S44 in the ESI†). For example, the emission maximum (λEmax) of 1b shifts from 517 nm in toluene to 617 nm in ACN. The Stokes shifts range between 5.000–9.000 cm−1 and increase in the series of solvents in the following order: ACN > THF > CHCl3 > toluene. Hence, we can assume a significant structural re-arrangement of 1–6 upon excitation and more polar excited states stabilized by polar solvents. The quantum yields of imidazoles in series a showed minor solvent dependency, except for 1a with increasing ΦF when going from less polar toluene (ΦF = 10%) to more polar ACN (ΦF = 43%). This behaviour is in contrast to that of 1b–6b, whose emission intensity was significantly suppressed with increased solvent polarity. A comparison of the imidazoles in series b clearly indicates that increasing the electron-withdrawing power of the appended acceptor results in diminished fluorescence intensity. Fluorescence quenching is especially obvious for indan-1,3-dione and rhodanine derivatives 3b and 4b. The pronounced ICT and reduced optical gap in polar solvents offer significant competitive decay pathways, e.g. internal conversion and diminishing emissive behaviour of chromophores in series b.
To reveal the effect of the electron-withdrawing power of the complex acceptors in 1b–6b on the excited state properties, we performed nanosecond fluorescence dynamics measurements (Fig. 4 and Fig. S45, Tables S6–S9 in the ESI†).
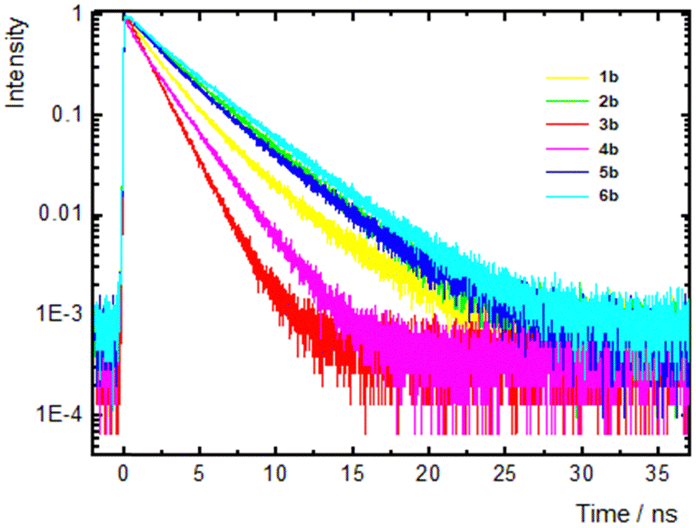 | ||
| Fig. 4 Fluorescence dynamics on the nanosecond timescale of imidazole chromophores 1b–6b measured in THF. Dynamics were detected at the peaks of the fluorescence spectra. | ||
In THF (Fig. 4 and Table S8, ESI†), the decay shows multiexponential dynamics. Specifically, for 1b and 4b, a fast component of 0.2–0.6 ns was found, which is tentatively ascribed to a structural relaxation of the excited state, while two slower components of 1–2 ns and >2 ns were also found in all molecules. For 2b, 5b and 6b, the fluorescence dynamics were detected at various emission wavelengths in order to identify and separate the contribution of the emissive states. For this reason, the decays were fitted using a global method (Fig. S46 and Table S10, ESI†). In these three molecules, the contributions of the fast/slow components (A1 and A2, respectively) were found to decrease/increase as the detection wavelength shifted to the low-energy edge of the spectrum. However, these changes are small, and therefore, the two components cannot originate from emissive states of different natures (e.g. a locally excited and an ICT state), but they can rather be ascribed to different conformers existing in the elongated compounds in series b due to rotation around the quasi-single bonds. The average lifetime ranges from 1.3 to 3.2 ns, which is smaller for 3b and 4b. The latter finding is in accordance with the reduced ΦF in these two chromophores with the strongest indan-1,3-dione and rhodanine electron acceptors, indicating increased non-radiative pathways. The fluorescence decays in all other solvents also present complex multiexponential dynamics, while a common conclusion is that 3b and 4b exhibit the fastest dynamics. Besides, on comparing the dynamics in the two non-chlorinated solvents, toluene and THF, it was observed that the lifetime of the slower component increased by increasing the polarity of the solvent from toluene to THF (e.g. τ = 2.58 and 3.74 ns in toluene and THF were recorded for 6b).
This is correlated to an emission from a more relaxed emitting state.97,98 In the highest polarity solvent, ACN, the lifetime drops significantly due to increased non-radiative transitions followed by a decrease of the energy gap between the final emitting and the ground state.
Two-photon absorption
The 2PA spectra were measured for extended chromophores 1b–6b in toluene and THF (Fig. 5). The maximum δ2PA values in THF are listed in Table 2. Relatively good values were found for all compounds in both solvents, with δ2PA reaching ∼350 to 400 GM for chromophores 2b and 6b in THF, as well as adequate quantum yield ΦF. The maximum values, however, were detected for 4b with rhodanine acceptors, which also possessed a small quantum yield. Consequently, the smallest 2PA action cross-section values, i.e. ΦF × δ2PA were found for 3b and 4b, bearing the strongest indan-1,3-dione and rhodanine peripheral acceptors. Larger δ2PA are expected for systems possessing large transition dipole moments and differences in the permanent dipole moments in the ground and final states, with the latter being related to the Stokes shift. Chromophores 1b–6b exhibit significant transition dipole moments while 2b and 6b experience the most spread LUMO towards the C5-branch (vide infra), which is possibly related to their better 2PA activity. The peaks in the 2PA spectra were observed in the short wavelength region within our experimental range, i.e. 730–760 nm. A comparison of the spectral shape and position of the 2PA spectra vs. the rescaled 1PA spectra is shown in the inset of Fig. 5d for 2b in THF. As expected for tripodal compounds, the 2PA peak is obviously hypsochromically shifted with respect to twice the peak wavelength of the 1PA spectrum, indicating that the lower energy state, produced via excitonic coupling, is only partially 2-photon allowed while the main contribution to the 2PA originates from the higher energy state. Such a behaviour resembles the one previously found for other triphenylamine- or triazine-centred D–(π–A)3 and A–(π–D)3 chromophores.14,99,100 | ||
| Fig. 5 2PA spectra of compounds 1b–6b in THF (a) and (b) and toluene (c); (d) shows the comparison of the 2PA spectrum vs. the rescaled 1PA one for 2b in THF. | ||
Next, we tried to establish a more quantitative aspect of the effect of the different electron-withdrawing groups by calculating the difference between the permanent dipole moments of the ground and excited states, Δμ2PA, using the measured lowest energy peak 2PA cross-sectional values δ2PA(0–0) and the following relationship:14,101–103
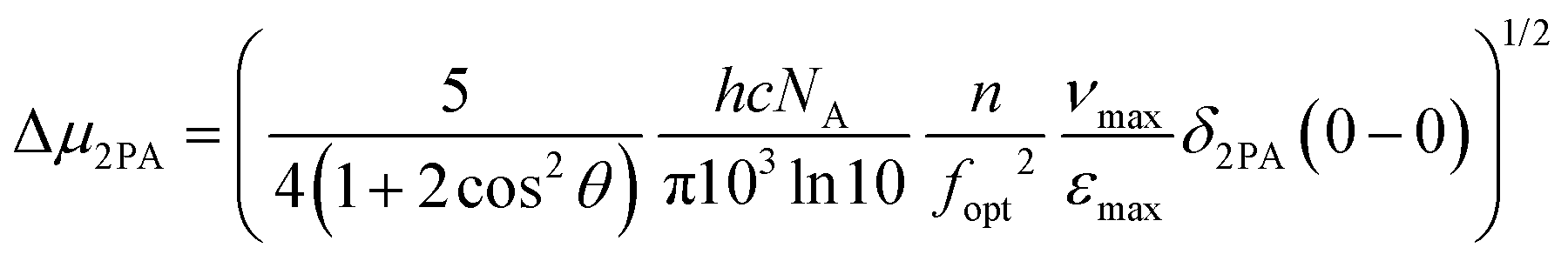
DFT calculations
The spatial and electronic properties of all target derivatives 1–6 were investigated at the DFT level by using the Gaussian® 16 software package.104 Optimized geometries of the chromophores 1a–4a/1b–3b were calculated using the DFT B3LYP/6-311+G(2d,p) method, while the sulphur-containing structures 4b–6b were optimized using DFT B3LYP/6-311+G(2df,p). The same methods were used to calculate the energies of the HOMO (EDFTHOMO) and the LUMO (EDFTLUMO), their differences (ΔEDFT) and ground-state dipole moments μ (Table 3).| Comp | E DFTHOMO [eV] | E DFTLUMO [eV] | ΔEDFT [eV] | μ [D] |
λ
Amaxc [nm eV−1] |
λ
Amaxd [nm eV−1] |
Wavelength [nm]: transitions (f); μtransde [D] |
|---|---|---|---|---|---|---|---|
| a Calculated as the corresponding methyl ester and 3-methylrhodanine derivatives. b Ground state dipole moment. c Calculated using the TD-DFT B3LYP/6-311+g(2df,p) method. d Calculated using the TD-DFT CAM-B3LYP/6-311+g(2df,p) method. e H/L corresponds to the HOMO/LUMO; f is the oscillator strength; and μtrans is the ground to excited state transition dipole moment. | |||||||
| 1a | –6.16 | –2.47 | 3.69 | 8.60 | 380/3.26 | 310/4.00 | 315: H → L/H → L+1 (0.79); 7.25 |
| 305: H → L+1 (0.72), 6.85 | |||||||
| 2a | –6.22 | –2.15 | 4.07 | 7.65 | 342/3.63 | 293/4.23 | 296: H → L/H → L+1 (0.84); 7.28 |
| 290: H → L+1 (0.69); 6.53 | |||||||
| 3a | –6.06 | –2.06 | 4.00 | 7.91 | 349/3.55 | 292/4.25 | 298: H → L (0.89); 7.50 |
| 289: H → L/H → L+1 (0.69); 6.50 | |||||||
| 4a | –6.14 | –1.70 | 4.44 | 6.84 | 310/4.00 | 272/4.56 | 276: H → L+1 (0.59); 5.90 |
| 271: H → L/H → L+1 (0.56); 5.70 | |||||||
| 1b | –6.17 | –3.34 | 2.83 | 5.18 | 489/2.54 | 389/3.19 | 396: H → L/H → L+1 (0.99); 9.14 |
| 384: H → L+1 (1.83); 12.22 | |||||||
| 2b | –6.04 | –3.11 | 2.93 | 7.70 | 482/2.57 | 379/3.27 | 386: H → L/H → L+1 (1.03); 9.17 |
| 373: H → L+1 (1.87); 12.19 | |||||||
| 3b | –5.96 | –3.23 | 2.73 | 6.37 | 522/2.38 | 405/3.06 | 408: H → L (1.15); 10.00 |
| 396: H → L/H → L+1 (2.51); 14.53 | |||||||
| 4b | –5.83 | –3.10 | 2.73 | 7.81 | 517/2.40 | 402/3.08 | 407: H → L+1 (1.18); 10.08 |
| 398: H → L/H → L+1 (2.49); 14.52 | |||||||
| 5b | –6.12 | –3.14 | 2.98 | 13.12 | 477/2.60 | 376/3.30 | 381: H → L/H → L+1 (1.13); 9.56 |
| 371: H → L+1 (1.89); 12.20 | |||||||
| 6b | –6.04 | –3.05 | 2.99 | 9.23 | 469/2.64 | 376/3.30 | 381: H → L/H → L+1 (1.13); 9.55 |
| 371: H → L+1 (1.89); 12.20 | |||||||
The optimized geometries shown in Fig. S47 (ESI†) reveal nonplanar arrangement similar to that observed in the solid state by X-ray analysis (Scheme 1). In general, the phenyl ring appended at C5 proved to be most forced out of the imidazole plane because of its repulsion with the neighbouring N–CH3 group. Sulfones 5b and 6b show a tetrahedral arrangement of the sulphur atoms, resulting in the most nonplanar molecules. The ground state dipole moment of imidazoles in series a is relatively steady (6.74–8.60 D) and extension of the π-system end-capped with peripheral (complex) electron-withdrawing units of various geometries in series b resulted in large fluctuations in the dipole moments (5.18–13.12 D, Table 3). The aforementioned nonplanar arrangements of sulfones 5b and 6b most likely also account for their large ground state dipole moments. The lowest HOMO–LUMO gap was calculated for 1a (3.69 eV) and 4b (2.73 eV), which is in line with the electrochemical measurements (Table 1). In general, the DFT-calculated energies of the HOMO/LUMO are slightly overestimated but the trends seen by electrochemical measurements are clearly observed, as electrochemical and DFT-calculated gaps correlate very tightly (Fig. S51, ESI†).
The electrostatic potentials showed for representative chromophores 1a/2a and 1b/3b/6b in Fig. 6 imply that, except for 1a and 3b, the central imidazole core is neutrally charged and negative and positive charges appear rather on the peripheral withdrawing groups bearing heteroatoms (see Fig. S48 for complete visualization, ESI†). However, the HOMO/LUMO localization (Fig. 6 and Fig. S49, S50 in the ESI†) reveals the imidazole- and 2-phenylimidazole-centred HOMO and that the LUMO spreads on the appended phenyl rings, the C5-branch in particular. Hence, centrifugal charge separation with a central imidazole donor can be deduced. The calculated energies of the HOMO/LUMO are slightly overestimated, but the trends seen from the electrochemical measurements are clearly obeyed as both quantities correlate very tightly (Fig. S51, ESI†). Considering the identical π-systems in both series a (triphenyl-N-methylimidazole) and b (tri(4-vinylphenyl)-N-methylimidazole), the variation of electronic properties must be ascribed to the appended substituents. In series a, the calculated LUMO energies roughly correspond to the Hammett constants of the simple CN, COOCH3 and CF3 substituents (except for 1a). The influence of peripheral CN, COOEt, indan-1,3-dione, 3-ethylrhodanine, PhSO2 and 4-FPhSO2 substituents is more complex. Due to their various spatial arrangements along the peripheral olefinic linker(s), their electronic effects are differently pronounced. However, sulfones 5b and 6b showed a deepened LUMO due to the strong electron-withdrawing power of SO2Ph (0.68) and, expectedly, even stronger SO2PhF.
 | ||
| Fig. 6 DFT-calculated ESP charges, the HOMO/LUMO(+1/2) localization, and CT transitions, including oscillator strengths in representative chromophores in series a and b. | ||
Electronic absorption spectra were predicted using the TD-DFT B3LYP/6-311+g(2df,p) and CAM-B3LYP/6-311+g(2df,p) methods in ACN. Fig. 7 shows the spectra calculated using TD-DFT CAM-B3LYP/6-311+g(2df,p) along with the experimental spectra for the selected chromophores; see Fig. S52 and S53 (ESI†) for complete spectra. Compared with the experimental data in ACN (Fig. S44, ESI†), the spectra calculated using B3LYP/CAM-B3LYP are red/blue-shifted, but the number and shape of the peaks are identical. A correlation of the experimental (Table 2) and calculated (Table 3) optical gaps (calculated as 1240/λAmax) using both functionals is tight (Fig. S54, ESI†), which indicates that both methods are capable of describing trends in the electronic properties of imidazole derivatives 1–6. However, the slope of the regression implies better agreement for the CAM-B3LYP calculated λAmax values (Fig. S54, ESI†).
 | ||
| Fig. 7 UV-Vis absorption spectra in ACN calculated using TD-DFT CAM-B3LYP/6-311+g(2df,p) (blue) along with the experimental spectra (black). The red vertical lines represent oscillator strength (f). | ||
The single absorption band of imidazoles in series a is due to the HOMO → LUMO and HOMO → LUMO+1 transitions (Table 3 and Fig. 6) accompanied by a weaker transition from the HOMO to the LUMO+2. Extension of the π-system and installation of the complex peripheral acceptors as in series b resulted in red-shifted spectra with two particularly evolved absorption bands. The longest-wavelength absorption band is generated by the HOMO → LUMO transition along with the HOMO → LUMO+1 excitation of an even larger oscillator strength. Hence, considering that the LUMO, LUMO+1 and LUMO+2 in 1a–4a spread across all three branches and likewise the LUMO and LUMO+1 in 1b–6b (Fig. 6), we can assume a centrifugal charge transfer from the imidazole-centred HOMO to the peripheral electron acceptors. The transition from the imidazole-centred HOMO to the peripheral branches occupied by the LUMO(+1/2) is accompanied by significant transition dipole moments, which are larger for 1b–6b, as listed in Table 3.
Conclusions
Two series of lophine-based tripodal chromophores were prepared. The synthesis of chromophores 1a–4a involved threefold cross-coupling, while subsequent Knoevenagel condensation allowed the installation of complex peripheral acceptors based on acetic/malonic acids, indan-1,3-dione and rhodanine to afford 1b–6b. Structural analysis revealed a nonplanar arrangement of the central N-methyltriphenylimidazole and also exclusive stereochemistry of olefinic chromophores 1b–6b. DFT calculations further identified sulfones 5b and 6b as the most nonplanar compounds with the largest ground state dipole moments. The thermal stability measured by DSC was high with the temperature of decomposition ranging between 220 and 400 °C. However, the extension of the π-system diminished thermal stability, and the compounds in series b are thus less thermally robust compared to those in series a. Electrochemical measurements revealed a reversible one-electron oxidation centred on imidazole, while the irreversible multi-electron reduction involved peripheral acceptor moieties. The D–(π–A)3 arrangement of 1–6 further resulted in a steady HOMO and the LUMO reflecting the electron-withdrawing power of the acceptors. The centrifugal ICT from the imidazole-centred HOMO to the peripheral LUMO/LUMO+1/LUMO+2 spread among the branches was further corroborated by TD-DFT calculations. In general, lower HOMO–LUMO gaps were recorded/calculated for extended chromophores in series b bearing more powerful electron acceptors and extended π-system. The absorption spectra are solvent-independent while the variation of the acceptor allows tuning of the absorption maxima from 300 to 450 nm. The calculated spectra followed the same trend. In contrast, the fluorescence spectra red-shifted with increasing polarity of the environment and the fluorescence was obviously dependent on the electron-withdrawing power of the peripheral acceptors and solvent polarity. Tripodal imidazoles 1b–6b showed relatively good and tuneable two-photon absorption with δ2PA within the range of 286 to 521 GM. Although the maximum value was measured for rhodanine chromophore 4b, ester 2b and sulfone 6b showed the largest figure of merit (ΦF × δ2PA). The recorded δ2PA values correspond to the reported data for centrifugal imidazole-based chromophores.65 However, generally, a larger cross-section can be achieved when imidazole is applied as a central π-linker of unsymmetrical D–π–(A)2 or A–π–(D)2 tripodal chromophores.47,64 The results presented herein demonstrate the profound effect of the ICT process on the photophysics and NLO properties of the imidazole compounds. The strength and dynamics of this process, depending on the molecular structure, architecture and environment, have been the subject of intense scientific research in recent years.105–107 Due to the positive dipole moment change upon excitation, the excited state is more prone to environmental changes that experience a larger solvent reaction field. Here, molecules with stronger electron-accepting groups exhibit a reduced energy gap, ΦF and excited state lifetime.Author contributions
Investigation: JK, ZB, MK, LS, AK, OP, PP and AR; methodology: JK, MF and FB; writing – original draft: FB; writing – review and editing: MF and FB.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1a has been deposited at the CCDC under 2331438 and can be obtained from https://www.ccdc.cam.ac.uk.Conflicts of interest
There are no conflicts to declare.References
- F. Bureš, RSC Adv., 2014, 14, 58826 RSC.
- M. Klikar, P. Solanke, J. Tydlitát and F. Bureš, Chem. Rec., 2016, 16, 1886 CrossRef CAS.
- F. Terenziani, C. Le Droumaguet, C. Katan, O. Mongin and M. Blanchard-Desce, Chem. Phys. Chem., 2007, 6, 723 CrossRef PubMed.
- B. Mu, X. Quan, Y. Zhao, X. Li and W. Tian, Mater. Chem. Front., 2019, 3, 1671 RSC.
- S. Pelz, J. Zhang, I. Kanelidis, D. Klink, L. Hyzak, V. Wulf, O. J. Schmitz, J. C. Gasse, R. Frahm, A. Pütz, A. Colsmann, U. Lemmer and E. Holder, Eur. J. Org. Chem., 2013, 4761 CrossRef CAS.
- T. Shimasaki, Y. Takiyama, Y. Nishihara, A. Morimoto, N. Teramoto and M. Shibata, Tetrahedron Lett., 2015, 56, 260 CrossRef CAS.
- C. K. R. Namboodiri, P. B. Bisth, R. Mukkamala, R. Chandra and I. S. Aidhen, Chem. Phys., 2013, 415, 190 CrossRef CAS.
- H. F. Huang, S. H. Xu, Y. B. He, C. C. Zhu, H. L. Fan, X. H. Zhou, X. C. Gao and Y. F. Dai, Dyes Pigm., 2013, 96, 705 CrossRef CAS.
- S. Achelle, M. Hodeé, J. Massue, A. Fihey and C. Katan, Dyes Pigm., 2022, 200, 110157 CrossRef CAS.
- S. Achelle and F. Robin-le Guen, J. Photochem. Photobiol., A, 2017, 348, 281 CrossRef CAS.
- M. Fecková, P. le Poul, F. Bureš, F. Robin-le Guen and S. Achelle, Dyes Pigm., 2020, 182, 108659 CrossRef.
- S. Achelle, J. Rodríguez-López and F. Robin-le Guen, Org. Biomol. Chem., 2023, 21, 39 RSC.
- S. Achelle, J. Rodríguez-López, F. Bureš and F. Robin-le Guen, Chem. Rec., 2020, 20, 440 CrossRef CAS PubMed.
- P. Šimon, M. Klikar, Z. Burešová, Ch Vourdaki, A. Katsidas, J. Tydlitát, J. Kulhánek, J. Zelenka, M. Fakis and F. Bureš, J. Mater. Chem. C, 2023, 11, 7252 RSC.
- N. P. Tripathi, S. Jain, R. K. Singh and S. Sengupta, Chem. - Eur. J., 2023, e202303244 Search PubMed.
- A. Maggiore, X. Tan, A. Brosseaus, A. Danos, F. Miomandre, A. P. Monkman, P. Audebert and G. Clavier, Phys. Chem. Chem. Phys., 2022, 24, 17770 RSC.
- P. Solanke, F. Bureš, O. Pytela, M. Klikar, T. Mikysek, L. Mager, A. Barsella and Z. Růžičková, Eur. J. Org. Chem., 2015, 5339 CrossRef CAS.
- T. Adhikari, P. Solanke, D. Pathak, T. Wagner, F. Bureš, T. Reed and J. M. Nunzi, Opt. Mater., 2017, 69, 312 CrossRef CAS.
- P. Solanke, O. Pytela, F. Bureš and M. Klikar, Dyes Pigm., 2019, 162, 755 CrossRef CAS.
- P. Blanchard, C. Malacrida, C. Cabanetos, J. Roncali and S. Ludwigs, Polym. Int., 2019, 68, 589 CrossRef CAS.
- A. Karak, S. K. Manna and A. K. Mahapatra, Anal. Methods, 2022, 14, 972 RSC.
- X. Lian, Z. Zhao and D. Cheng, Mol. Cryst. Liq. Cryst., 2017, 648, 223 CrossRef CAS.
- A. Mahmood, Sol. Energy, 2016, 123, 127144 CrossRef.
- H. J. Yen and G. S. Liou, Polym. Chem., 2018, 9, 3001 RSC.
- L. S. Cui, S. C. Dong, Y. Liu, Q. Li, Z. Q. Jiang and L. S. Liao, J. Mater. Chem. C, 2013, 1, 3967 RSC.
- N. Hammer, T. A. Schaub, U. Meinhardt and M. Kivala, Chem. Rec., 2015, 15, 1119 CrossRef CAS PubMed.
- J. Kulhánek and F. Bureš, Beilstein J. Org. Chem., 2012, 8, 25 CrossRef PubMed.
- J. Kulhánek, F. Bureš, O. Pytela, T. Mikysek, J. Ludvík and A. Růžička, Dyes Pigm., 2010, 85, 57 CrossRef.
- F. Bureš, J. Kulhánek, T. Mikysek, J. Ludvík and J. Lokaj, Tetrahedron Lett., 2010, 51, 2055 CrossRef.
- J. Kulhánek, F. Bureš, W. Kuznik, I. V. Kityk, T. Mikysek and A. Růžička, Chem. – Asian J., 2013, 8, 465 CrossRef PubMed.
- A. Plaquet, B. Champagne, J. Kulhánek, F. Bureš, E. Bogdan, F. Castet, L. Ducasse and V. Rodriguez, ChemPhysChem, 2011, 12, 3245 CrossRef CAS PubMed.
- J. Kulhánek, F. Bureš, T. Mikysek, J. Ludvík and O. Pytela, Dyes Pigm., 2011, 90, 48 CrossRef.
- J. Kulhánek, F. Bureš, O. Pytela, T. Mikysek and J. Ludvík, Chem. – Asian J., 2011, 6, 1604 CrossRef PubMed.
- N. Dey, J. Kulhánek, F. Bureš and S. Bhattacharya, J. Org. Chem., 2021, 86, 14663 CrossRef CAS PubMed.
- T. Inouchi, T. Nakashima, M. Toba and T. Kawai, Chem. – Asian J., 2011, 6, 3020 CrossRef CAS PubMed.
- S. Chettri, S. Tamang, P. Rai, Y. Rai, U. K. Singha, K. Pradhan, D. Brahman and B. Sinha, J. Heterocyclic Chem., 2023, 60, 1394 CrossRef CAS.
- A. Patel, F. Bureš, M. Ludwig, J. Kulhánek, O. Pytela and A. Růžička, Heterocycles, 2009, 78, 999 CrossRef CAS.
- O. L. Al Sharif, L. M. Nhari, R. M. El-Shishtawy and A. M. Asiri, Mater. Today Chem., 2023, 29, 101453 CrossRef CAS.
- P. Ganesan, P. Ganesan, Z. Zhang, J. Xu, R. Rajalingam and P. Gao, J. Org. Chem., 2023, 88, 4077 CrossRef CAS.
- K. Feng, F. L. Hsu, D. Van DerVeer, K. Bota and X. R. Bu, J. Photochem. Photobiol., A, 2004, 165, 223 CrossRef CAS.
- H.-N. Peng, Y. Yu, X.-H. Liu, J. Zheng, H. Cheng and X. Hu, J. Chem. Res., 2018, 1, 20 CrossRef.
- R. Babar, M. Ali Munawar, M. N. Tahir and M. Arif, Spectrochim. Acta, Part A, 2019, 217, 223 CrossRef CAS PubMed.
- S. S. Razi, R. C. Gupta, R. Ali, S. K. Dwivedi, P. Srivastava and A. Misra, Sens. Actuators, B, 2016, 236, 520 CrossRef CAS.
- S. Tsuchiya, K. Sakai, K. Kawano, Y. Nakane, T. Kikuchi and T. Akutagawa, Chem. - Eur. J., 2018, 24, 5868 CrossRef CAS PubMed.
- X. He, S. Zhu, H. Chen, Y. Wang and H. Li, J. Lumin., 2016, 173, 218 CrossRef CAS.
- K. G. Harsha, Ch Madhu, N. Puvvada, T. R. Rao Baggi, V. J. Rao and N. R. Chereddy, Chemistry Select, 2020, 5, 6059 Search PubMed.
- M. Zhang, M. Li, Q. Zhao, F. Li, D. Zhang, J. Zhang, T. Yi and Ch Huang, Tetrahedron Lett., 2007, 48, 2329 CrossRef CAS.
- T. Khamrang, A. Kathiravan, Ch Ponraj and D. Saravanan, J. Mol. Struct., 2021, 1238, 130442 CrossRef CAS.
- C. I. C. Esteves, R. C. M. Ferreira, M. M. M. Raposo and S. P. G. Costa, Dyes Pigm., 2018, 151, 211 CrossRef CAS.
- H. E. Okda, S. El Sayed, R. C. M. Ferreira, S. P. G. Costa, M. M. M. Raposo, R. Martínez-Mánez and F. Sancenón, Dyes Pigm., 2018, 159, 45 CrossRef CAS.
- H. E. Okda, S. El Sayed, I. Otri, R. C. M. Ferreira, S. P. G. Costa, M. M. M. Raposo, R. Martínez-Mánez and F. Sancenón, Dyes Pigm., 2019, 162, 303 CrossRef CAS.
- S. Aryamueang, K. Chansaenpak, A. Watwiangkham, S. Suthirakun, P. Muangsopa, W. Wattanathana, R.-Y. Lai and A. Kamkaew, J. Photochem. Photobiol., A, 2024, 447, 115268 CrossRef CAS.
- R. P. C. L. Sousa, R. B. Figueira, B. R. Gomes, S. P. G. Costa, M. Azenha, R. F. P. Pereira and M. M. M. Raposo, RSC Adv., 2021, 11, 24613 RSC.
- R. P. C. L. Sousa, R. B. Figueira, B. R. Gomes, S. Sousa, R. C. M. Ferreira, S. P. G. Costa and M. M. M. Raposo, Nanomaterials, 2021, 11, 3401 CrossRef CAS PubMed.
- E. Rosadoni, F. Bellina, M. Lessi, C. Micheletti, F. Ventura and A. Pucci, Dyes Pigm., 2022, 201, 110262 CrossRef CAS.
- G. Li, M. Xia, J. Liu, J. Zheng, B. Zhao and H. Peng, Dyes Pigm., 2023, 213, 111183 CrossRef CAS.
- Y.-D. Lin, W.-W. Tsai and C.-W. Lu, Chem. - Eur. J., 2023, 29, e202203040 CrossRef CAS PubMed.
- G. Umasankar, H. Ulla, Ch Madhu, G. R. Reddy, B. Shanigaram, J. B. Nanubolu, B. Kotamarthi, G. V. Karunakar, M. N. Satyanaryan and V. J. Rao, J. Mol. Struct., 2021, 1236, 130306 CrossRef CAS.
- M. Pokladko-Kowar, N. Nosidlak, E. Gondek, I. V. Kityk, F. Bureš F, J. Kulhánek and P. Karasinski, Opt. Quantum Electron., 2016, 48, 82 CrossRef.
- D. Unny, G. R. Kandregula and K. Ramanujan, J. Photochem. Photobiol., A, 2022, 426, 113735 CrossRef CAS.
- J. Sivanadanam, I. Aldhen and K. Ramanujam, New. J. Chem., 2020, 44, 10207 RSC.
- M. Velusamy, Y.-C. Hsu, J. T. Lin, C.-W. Chang and C.-P. Hsu, Chem. – Asian J., 2010, 5, 87 CrossRef CAS.
- S. Prabu, F. Francesco, A. Colombo, C. Dragonetti, P. Biagini, F. Melchiorre and N. Palanisami, New J. Chem., 2024, 48, 394 RSC.
- K. Feng, L. De Boni, L. Misoguti, C. R. Mendonca, M. Meador, F. L. Hsu and X. R. Bu, Chem. Commun., 2004, 1178 RSC.
- G. C. Zheng, Z. B. Cai, Y. L. Pan, L. Bai, Y. T. Zhou, S. L. Li and Y. P. Tian, Tetrahedron, 2016, 72, 2988 CrossRef CAS.
- Z. B. Cai, Q. X. Lou, S. L. Li, L. J. Chen, Q. Ye and Y. P. Tian, Analyst, 2022, 147, 5495 RSC.
- K. Skonieczny, A. I. Ciuciu, E. M. Nichols, V. Hugues, M. Blanchard-Desce, L. Flamigni and D. T. Gryko, J. Mater. Chem., 2012, 22, 20649 RSC.
- M. Zhang, M. Li, F. Li, Y. Cheng, J. Zhang, T. Yi and C. Huang, Dyes Pigm., 2008, 77, 408 CrossRef CAS.
- Y. X. Yan, Y. H. Sun, L. Tian, H. H. Fan, H. Z. Wang, C. K. Wang, Y. P. Tian, X. T. Tao and M. H. Jiang, Opt. Mater., 2007, 30, 423 CrossRef CAS.
- Y. F. Sun, W. Huang, C. G. Lu and Y. P. Cui, Dyes Pigm., 2009, 81, 10 CrossRef CAS.
- Y. X. Yan, H. H. Fan, C. K. Lam, H. Huang, J. Wang, S. Hu, H. Z. Wang and Z. M. Chen, Bull. Chem. Soc. Jpn., 2006, 79, 1614 CrossRef CAS.
- N. Lin, X. Zhao, J. X. Yang, M. H. Jiang, J. C. Liu, C. K. Wang, W. Shi, J. Meng and J. Weng, J. Chem. Phys., 2006, 124, 024704 CrossRef PubMed.
- R. C. M. Ferreira, S. P. G. Costa, H. Goncalves, M. Blesley and M. M. M. Raposo, New. J. Chem., 2017, 41, 12866 RSC.
- D. Zhang, H. Zhu and X. Sheng, Phys. Chem. Chem. Phys., 2023, 25, 7508 RSC.
- H. Zhu, D. Zhang, X. Sun, S. Qian, E. Feng and X. Sheng, Phys. Chem. Chem. Phys., 2024, 26, 12150 RSC.
- W. Feng and Y. Qian, J. Mater. Chem. B, 2024, 12, 2413 RSC.
- L. Shi, Z. Sun, N. Richy, M. Blanchard-Desce, O. Mongin, F. Paul and C. O. Paul-Roth, Chem. Eur. J., 2024, 30, e202303243 CrossRef CAS PubMed.
- M. Durko-Maciag, G. Ulrich, J. Massue, J. Mysliwiec and K. Cyprych, Eur. Pol. J., 2023, 195, 112235 CrossRef CAS.
- T. Wloka, M. Gottschaldt and U. S. Schubert, Chem. - Eur. J., 2022, 28, e202104191 CrossRef CAS PubMed.
- D. Cvejn, E. Michail, I. Polyzos, N. Almonasy, O. Pytela, M. Klikar, T. Mikysek, V. Giannetas, M. Fakis and F. Bureš, J. Mater. Chem. C, 2015, 3, 7345 RSC.
- D. Cvejn, E. Michail, K. Seintis, M. Klikar, O. Pytela, T. Mikysek, N. Almonasy, M. Ludwig, V. Giannetas, M. Fakis and F. Bureš, RSC Adv., 2016, 6, 12819 RSC.
- M. Klikar, D. Georgiou, I. Polyzos, M. Fakis, Z. Růžičková, O. Pytela and F. Bureš, Dyes Pigm., 2022, 201, 110230 CrossRef CAS.
- F. Kournoutas, A. Fihey, J. P. Malval, A. Spangenberg, M. Fecková, P. le Poul, C. Katan, F. Robin-le Guen, F. Bureš, S. Achelle and M. Fakis, Phys. Chem. Chem. Phys., 2020, 22, 4165 RSC.
- S. K. Sachan and G. Anantharaman, Inorg. Chem., 2021, 60, 9238 CrossRef CAS PubMed.
- M. Klikar, V. Jelínková, Z. Růžičková, T. Mikysek, O. Pytela, M. Ludwig and F. Bureš, Eur. J. Org. Chem., 2017, 2764 CrossRef CAS.
- J. Kulhánek, O. Pytela, F. Bureš and M. Klikar, Eur. J. Org. Chem., 2021, 3223 CrossRef.
- T. Szotkowski, F. Bureš, O. Pytela, J. Kulhánek and Z. Trávníček, J. Heterocycl. Chem., 2006, 43, 1583 CrossRef CAS.
- E. Novotná, I. V. Kityk, O. Pytela, F. Bureš, M. Ludwig, M. Klikar, K. Ozga and J. Jedryka, Chem. Plus. Chem., 2020, 85, 1549 CrossRef PubMed.
- N. Zhang, C. Zeng, C. M. Lam, R. K. Gbur and R. D. Little, J. Org. Chem., 2013, 78, 2104 CrossRef CAS PubMed.
- K. Y. Zhang, N. Lu, S. J. Yoo, L. M. Hu, R. D. Little and C. C. Zeng, Electrochim. Acta, 2016, 199, 357 CrossRef CAS.
- T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem. Int. Ed., 2014, 53, 3173 CrossRef CAS PubMed.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894 CrossRef CAS PubMed.
- D. T. Sawyer, A. Sobkowiak and J. L. Roberts, Electrochemistry for Chemists, J. Wiley and Sons Inc, 2nd edn, 1995, ISBN: 978-0-471-59468-0 Search PubMed.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213 CrossRef CAS.
- V. Gopi, S. Subbiahraj, K. Chemmanghattu and P. C. Ramamurthy, Dyes Pigm., 2020, 173, 107887 CrossRef CAS.
- J. E. Barnsley, W. Pelet, J. McAdam, K. Wagner, P. Hayes, D. L. Officer, P. Wagner and K. C. Gordon, J. Phys. Chem. A, 2019, 123, 5957 CrossRef CAS PubMed.
- M. Fakis, V. Petropoulos, P. Hrobárik, J. Nociarová, P. Osuský, M. Maiuri and G. Cerullo, J. Phys. Chem. B, 2022, 126, 8532 CrossRef CAS PubMed.
- A. Cesaretti, T. Bianconi, M. Coccimiglio, N. Montegiove, Y. Rout, P. L. Gentili, R. Misra and B. Carlotti, J. Phys. Chem. C, 2022, 126, 10429 CrossRef CAS.
- J. C. Collings, S. Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L. O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W. Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem. - Eur. J., 2009, 15, 198 CrossRef CAS PubMed.
- N. S. Makarov, S. Mukhopadhyay, K. Yesudas, J. L. Brédas, J. W. Perry, A. Pron, M. Kivala and K. Müllen, J. Phys. Chem. A, 2012, 116, 3781 CrossRef CAS PubMed.
- M. G. Vivas, D. L. Silva, J. Malinge, M. Boujtita, R. Zalesny, W. Bartkowiak, H. Angren, S. Canuto, L. De Boni, E. Ishow and C. R. Mendonca, Sci. Rep., 2014, 4, 4447 CrossRef PubMed.
- A. Rebane, M. Drobizhev, N. S. Makarov, E. Beuerman, S. Tillo and T. Hughes, J. Lumin., 2010, 130, 1619 CrossRef CAS.
- A. Rebane, M. Drobizhev, N. S. Makarov, E. Beuerman, J. E. Haley, D. M. Krein, A. R. Burke, J. L. Flikkema and T. M. Cooper, J. Phys. Chem. A, 2011, 115, 4255 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian 16, Wallingford, CT, 2016 Search PubMed.
- H. J. Womer, C. A. Arrell, N. Banerji, A. Gennizzo, M. Chergui and A. K. Das, et al. , Struct. Dyn., 2017, 4, 061508 CrossRef PubMed.
- R. Misra and S. P. Bhattacharyya, Intramolecular Charge Transfer: Theory and Applications, Wiley-VCH Verlag GmBH & Co, 2018, Germany Search PubMed.
- P. K. Samanta and S. R. Misra, J. Appl. Phys., 2023, 133, 020901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR spectra, X-ray data, thermal analysis, electrochemistry, optical properties, and DFT data. CCDC 2331438. For ESI and crystallographic data in CIF or other electronic format, see DOI: https://doi.org/10.1039/d4cp02227k |
| This journal is © the Owner Societies 2024 |