
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The elusive phenylethynyl radical and its cation: synthesis, electronic structure, and reactivity†
Ginny
Karir‡
 a,
Enrique
Mendez-Vega‡
a,
Adrian
Portela-Gonzalez
a,
Mayank
Saraswat
a,
Wolfram
Sander
*a and
Patrick
Hemberger
*b
a,
Enrique
Mendez-Vega‡
a,
Adrian
Portela-Gonzalez
a,
Mayank
Saraswat
a,
Wolfram
Sander
*a and
Patrick
Hemberger
*b
aLehrstuhl für Organische Chemie II, Ruhr-Universität Bochum, Bochum 44780, Germany. E-mail: wolfram.sander@rub.de
bLaboratory for Synchrotron Radiation and Femtochemistry, Paul Scherrer Institute (PSI), Villigen CH-5232, Switzerland. E-mail: patrick.hemberger@psi.ch
First published on 21st June 2024
Abstract
Alkynyl radicals and cations are crucial reactive intermediates in chemistry, but often evade direct detection. Herein, we report the direct observation of the phenylethynyl radical (C6H5C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C˙) and its cation (C6H5CC+), which are two of the most reactive intermediates in organic chemistry. The radical is generated via pyrolysis of (bromoethynyl)benzene at temperatures above 1500 K and is characterized by photoion mass-selected threshold photoelectron spectroscopy (ms-TPES). Photoionization of the phenylethynyl radical yields the phenylethynyl cation, which has never been synthesized due to its extreme electrophilicity. Vibrationally-resolved ms-TPES assisted by ab initio calculations unveiled the complex electronic structure of the phenylethynyl cation, which appears at an adiabatic ionization energy (AIE) of 8.90 ± 0.05 eV and exhibits an uncommon triplet (3B1) ground state, while the closed-shell singlet (1A1) state lies just 2.8 kcal mol−1 (0.12 eV) higher in energy. The reactive phenylethynyl radical abstracts hydrogen to form ethynylbenzene (C6H5CCH) but also isomerizes via H-shift to the o-, m-, and p-ethynylphenyl isomers (C6H4CCH). These radicals are very reactive and undergo ring-opening followed by H-loss to form a mixture of C8H4 triynes, along with low yields of cyclic 3- and 4-ethynylbenzynes (C6H3CCH). At higher temperatures, dehydrogenation from the unbranched C8H4 triynes forms the linear tetraacetylene (C8H2), an astrochemically relevant polyyne.
C˙) and its cation (C6H5CC+), which are two of the most reactive intermediates in organic chemistry. The radical is generated via pyrolysis of (bromoethynyl)benzene at temperatures above 1500 K and is characterized by photoion mass-selected threshold photoelectron spectroscopy (ms-TPES). Photoionization of the phenylethynyl radical yields the phenylethynyl cation, which has never been synthesized due to its extreme electrophilicity. Vibrationally-resolved ms-TPES assisted by ab initio calculations unveiled the complex electronic structure of the phenylethynyl cation, which appears at an adiabatic ionization energy (AIE) of 8.90 ± 0.05 eV and exhibits an uncommon triplet (3B1) ground state, while the closed-shell singlet (1A1) state lies just 2.8 kcal mol−1 (0.12 eV) higher in energy. The reactive phenylethynyl radical abstracts hydrogen to form ethynylbenzene (C6H5CCH) but also isomerizes via H-shift to the o-, m-, and p-ethynylphenyl isomers (C6H4CCH). These radicals are very reactive and undergo ring-opening followed by H-loss to form a mixture of C8H4 triynes, along with low yields of cyclic 3- and 4-ethynylbenzynes (C6H3CCH). At higher temperatures, dehydrogenation from the unbranched C8H4 triynes forms the linear tetraacetylene (C8H2), an astrochemically relevant polyyne.
Introduction
The carbon–carbon triple bond (CC) is one of the most versatile functional groups of high energy in organic chemistry, playing a key role in biochemistry and materials science. For instance, cis-enediynes based antitumor antibiotics undergo Bergmann cyclization and efficiently cleave DNA.1 Oligoynes (–CC–)n are promising candidates for molecular wires with applications in molecular electronics and optoelectronics.2 Moreover, the CC functional group is ubiquitous in the cold interstellar medium (ISM) and hot circumstellar envelopes of carbon stars, forming closed- and open-shell neutral and charged species.3
Alkynyl radicals (RCC˙) are reactive intermediates underlying alkyne transformations but challenging to detect and characterize since they are among the most reactive carbon-centered radicals.4,5 With a bond dissociation energy (BDE) of about 133 kcal mol−1,6,7 the C(sp)–H bond in ethyne, HCCH, is the strongest C–H bond among all hydrocarbons (Fig. 1). Consequently, the ethynyl radical, HCC˙, is destabilized by 28 kcal mol−1 with respect to methyl radical CH3˙,4 as derived from the difference between the BDEs of the corresponding hydrocarbons RH and CH4 reference.4 Combining the ethynyl moiety with an aromatic core yields the phenylethynyl radical 1,8 which is destabilized by 26 kcal mol−1 compared to CH3˙ (Fig. 1 and Table S1, ESI†).4
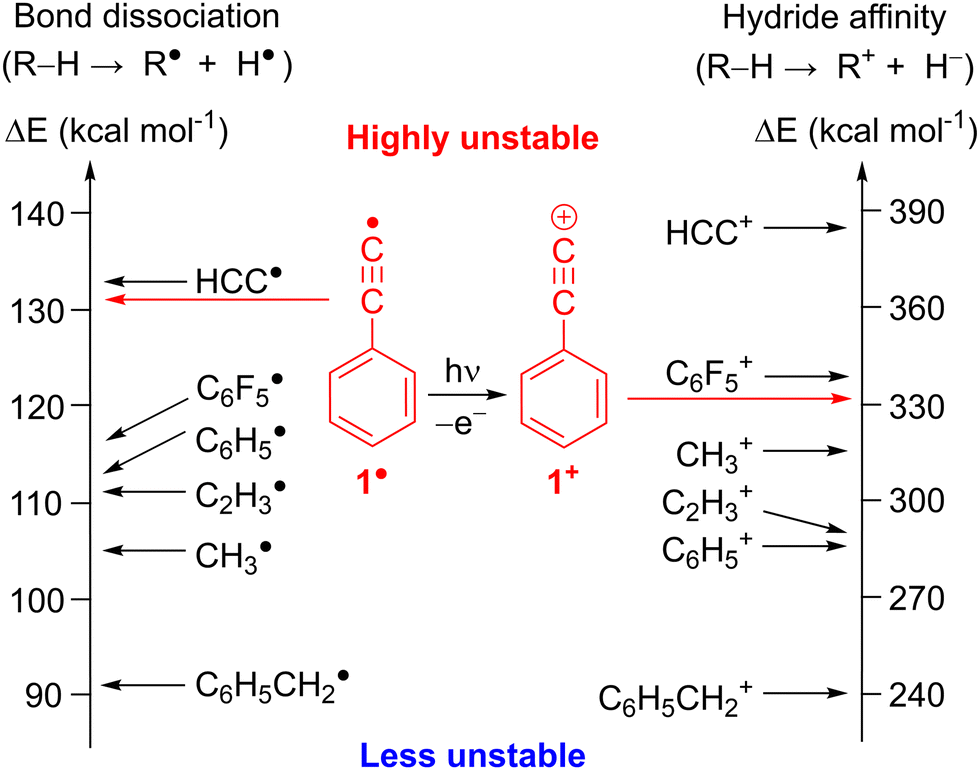 | ||
| Fig. 1 Thermodynamic stability of radicals and cations based on R–H bond dissociation energies and hydride affinities derived from experimental ΔHf (298 K).7 Corresponding values for C6F5 and 1 were calculated at the G4 level of theory. | ||
Radical 1 and the related ethynylbenzene 2 received a lot of attention for the understanding of the chemistry in extreme environments.9,10 Radical 1 and 2 have been proposed as potential precursors towards the gas-phase synthesis of polycyclic aromatic hydrocarbons (PAHs) in combustion flames11,12 as well as in the ISM.13,14 The recent detection of 2 in the Taurus molecular cloud (TMC-1)15 supports the proposed reaction sequence from benzene to 2 and subsequently to naphthalene via hydrogen-abstraction-acetylene-addition (HACA),16,17 as backbone of PAH formation in the ISM.
Radical 1 was synthesized in Ar matrices by Kasai et al. via UV irradiation of (iodoethynyl)benzene 3 and characterized by EPR spectroscopy.18,19 The unpaired electron of radical 1 is located in the π system, while the σ orbital at the terminal C(sp) atom is doubly occupied, resembling a vinylidene, hence 1 is classified as a π (2B1) radical (Scheme 1).20 This radical is so electrophilic that it even forms an adduct with Xe at low temperatures, C6H5CC–Xe–H which could be characterized by IR spectroscopy.21 Pyrolysis of 2 at 1300 K and subsequent analysis of the decomposition products with gas chromatography–mass spectrometry (GC–MS) indicated the presence of radical 1 along with the isomeric o-, m-, and p-ethynylphenyl radicals 5–7 (Scheme 2).22,23 Isomers 5–7 are computed to be 18 kcal mol−1 more stable than 1, which correlates to the difference between the BDEs of 2 and benzene (Fig. 1).20 Radical 1 was also synthesized in the gas-phase by Kaiser et al. using the crossed-beams reaction of dicarbon C2 with benzene under single collision conditions.8 Addition of small alkynes to radical 1 and 2 is found to yield naphthalene derivatives, in line with PAH growth models.14,16,17,24
 | ||
| Scheme 1 Electronic structure of the phenylethynyl radical 1. | ||
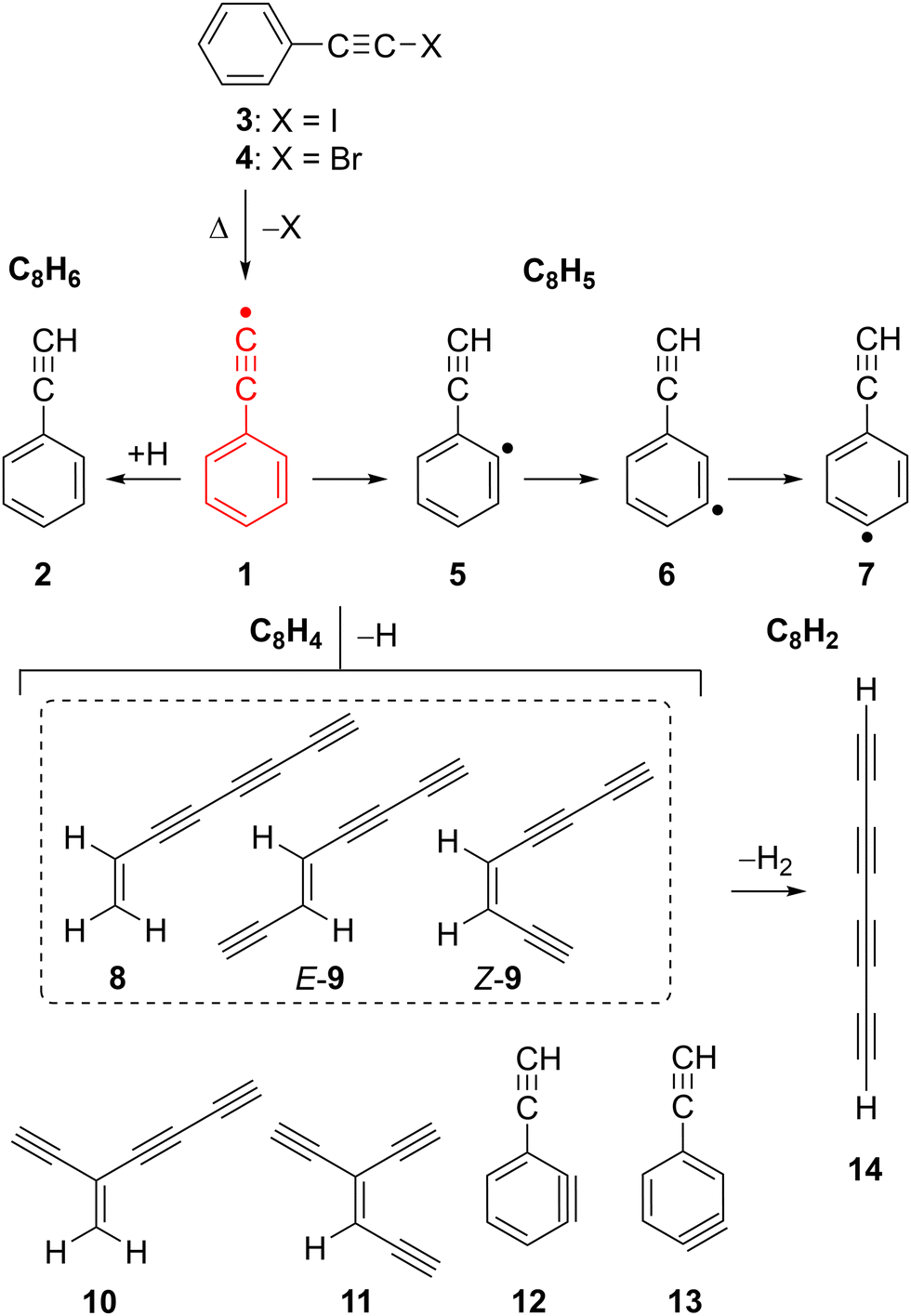 | ||
| Scheme 2 Thermal generation and decomposition of the phenylethynyl radical 1. | ||
Electron removal from radical 1 is a gateway to the most unstable class of carbenium ions, the alkynyl cations.25 The (in)stability of cations is assessed by their hydride affinity (HA) which measures the energy release of the reaction R+ + H− → R − H (Fig. 1, right).26 The HAs of the ethynyl cation, HCC+ and the phenylethynyl cation, C6H5CC+, 1+ are estimated to 384 and 331 kcal mol−1, respectively, which makes them even more unstable than the methyl and the phenyl cation (Fig. 1 and Table S1, ESI†).27 Experimental evidence for alkynyl cations is scarce since these species could not be synthesized in super acidic media. Diazonium precursors, that readily yield aryl cations in solution,25 fail to produce alkynyl cations R–CC+ under similar conditions (Scheme 3).28 In fact, unimolecular loss of N2 in HCC–N2+ to form HCC+, is highly endothermic and has an activation barrier of 150 kcal mol−1. The nuclear decay of tritiated alkynes R–CC–T was an unconventional route for the preparation of derivatives of the phenylethynyl cation 1+ in solution (Scheme 3).29 The formal reaction products of alkynyl cations with xenon, the alkynylxenonium tetrafluoroborates [R–CC–Xe]+[BF4]−, could be isolated and characterized by NMR spectroscopy at low temperatures, stressing the high reactivity of these alkynyl cations.30
 | ||
| Scheme 3 Synthesis of alkynyl cations. | ||
Photoelectron photoion coincidence (PEPICO) spectroscopy coupled to flash-vacuum pyrolysis (FVP) is a broadly applicable tool to identify reactive intermediates in the gas phase,31,32 by measuring vibrationally-resolved photoion mass-selective threshold photoelectron (ms-TPE) spectra.33 This suite of techniques34 allows to unravel reaction mechanisms relevant in combustion,35,36 astrochemistry37–39 and catalysis.40,41 In addition, experimental vibrational information of the ground and excited states, and in some cases the singlet–triplet energy splitting (ΔEST), has been obtained for the ethyl,42 vinyl,43 phenyl,44 and benzyl cations.45 In particular, the ethynyl cation in its triplet ground state was unveiled via direct photoionization of the ethynyl radical.46,47
In this work, we report the pyrolytic generation of radical 1 from (bromoethynyl)benzene 4 at 1500–1700 K, and its characterization using ms-TPE spectroscopy. Furthermore, we investigated the electronic structure of the highly electrophilic phenylethynyl cation 1+, in its ground and excited state. Leveraging the isomer selectivity of ms-TPE spectroscopy, we identified the thermal decomposition products of radical 1 at elevated temperatures leading to triynes and substituted benzynes (C8H4), as well as to tetraacetylene (C8H2), of great relevance to combustion flames as well as astrochemical environments.48–50
Methodologies
Experimental details
The thermal decomposition of (iodoethynyl)benzene 3 and (bromoethynyl)benzene 4 was studied using a pyrolysis microreactor connected to the CRF-PEPICO setup at the vacuum ultraviolet (VUV) beamline of the Swiss Light Source (SLS) at the Paul Scherrer Institute (PSI).51,52 Precursors 3 and 4 were synthesized and purified following a procedure from the literature.53,54 Precursors 3 and 4 were sublimed, mixed with a He flow of 20–40 sccm, and the gas mixture subsequently expanded through a 100 μm nozzle into a pyrolysis microreactor. The reactor consists of a 40 mm long SiC tube with an inner diameter of 1 mm that is electrically heated in the temperature range of 1500–1800 K over a length of 15 mm.55 The pressure and the residence time inside the reactor are estimated to be 10–20 mbar and ∼25–50 μs.56 After skimming (2 mm), an effusive molecular beam reaches the spectrometer chamber and is ionized by tunable VUV synchrotron radiation. During experiments, the pressure in the source and experimental chamber is about 3 × 10−5 and 3 × 10−6 mbar, respectively.The electron–ion pairs formed in an ionization event are vertically accelerated in opposite directions using a 218 V cm−1 field and are recorded in delayed coincidence. Electrons are velocity map imaged on a RoentDek delay-line detector, and their arrival time serves as the start time for the time-of-flight (TOF) measurement of the associated cation. A second RoentDek detector on the opposite end records the position and arrival time of the space focused ions. The photoions are analyzed using TOF mass spectrometry and velocity map imaging (VMI) in Wiley–McLaren space focusing conditions, which enables to distinguish the molecular beam (MB) emanating from the hot FVP reactor from the rethermalized background (BG) signals.57 Threshold electrons with a kinetic energy of <10 meV and photoions are selected in coincidence to record mass-selected threshold photoelectron (ms-TPE) spectra.58 The latter were corrected for false coincidences and the hot electron contribution was subtracted using the approach by Sztáray and Baer.59 Coincidence data were recorded over a photon energy range of 8.0–10.5 eV using a step size of 20–30 meV, and for an integration time of 60 s per energy point resulting in uncertainties of ±0.035 and ±0.05 eV for the reported AIE of 1 and 8–13 respectively. Due to the absence of field dependent TPES scans, we did not correct for Stark shifts.60
Quantum chemical calculations
Geometries and vibrational frequencies of the neutral and cationic species in their ground and excited states were calculated at the ZPE-corrected ωB97XD/6-311++G** level of theory with Gaussian 1661 and Turbomole 7.5.62 Stationary points on the C8H5 and C8H4 potential energy surfaces (PES), and intrinsic reaction coordinate (IRC) calculations were computed with ωB97XD/6-311++G** and energies refined with CCSD(T)/aug-cc-pVTZ. Insufficient HF exchange in hybrid DFT functionals like B3LYP wrongly predicts the out-of-plane bending of the ethynyl moiety in alkynyl radicals,20,63 but that undesired distortion is suppressed with ωB97XD.24 Hence, we did not use G4 and W1BD composite methods64 to calculate the AIE of radical 1 or the PES because these approaches contain geometry optimizations at the B3LYP functional. Nevertheless, AIEs of species 5–13 were refined at the G4 level of theory.For a rigorous treatment of multi-configurational systems and proper recovering of both static and dynamic electron correlation,65 the lowest-energy states of radical 1 and cation 1+ were computed with state-specific CASSCF, NEVPT2, CIPT2, and CASPT2, as well as CCSD(T) (for single-configurational states) using correlation-consistent aug-cc-pV(D/T)Z basis sets. The complete active space (CAS) consisted of the σ, low-lying filled and empty π orbitals as well as the corresponding electrons, denoted CAS(11,11) for 1 and CAS(10,11) for 1+. Coupled cluster and multi-configurational calculations were conducted with Molpro 2012.66
TPE spectra were simulated by calculating Franck–Condon (FC) factors in the double harmonic approximation at 1500 K for radical 1, and 300 K for the decomposition products (8–13). FC factors were calculated from optimized geometries and vibrational normal modes obtained at the ωB97XD/6-311++G** level of theory using Gaussian 16.61 The resulting stick spectra were convoluted with a Gaussian function with a full-width-at-half maximum of 40–48 meV to simulate the rotational envelope and facilitate comparison with the experimental data.
Results and discussion
The phenylethynyl radical 1 was generated by flash vacuum pyrolysis (FVP) of 4 and monitored using mass spectrometry and photoion mass-selected threshold photoelectron (ms-TPE) spectroscopy at various photon energies and temperatures of 1500–1700 K. Isomer-specific identification of the FVP products was performed by comparison of the ms-TPE spectra with experimental data or Franck–Condon spectral modelling.Thermal generation of the phenylethynyl radical 1
The mass spectra obtained upon FVP of the iodo- and the bromo-substituted precursors 3 and 4, respectively, show a similar product distribution (Fig. S1 and S2, ESI†). Therefore, we only discuss the FVP of 4 in detail. The mass spectrum of 4 recorded at room temperature (pyrolysis off) and photon energy of 9.0 eV shows two peaks of similar intensity at m/z 180 and 182 due to the contribution of the 79Br and 81Br isotopologues, respectively (Fig. 2a). The parent ion 4+ is photostable at up to 10.5 eV, within the typical range of TPE spectroscopy (Fig. S2–S4, ESI†).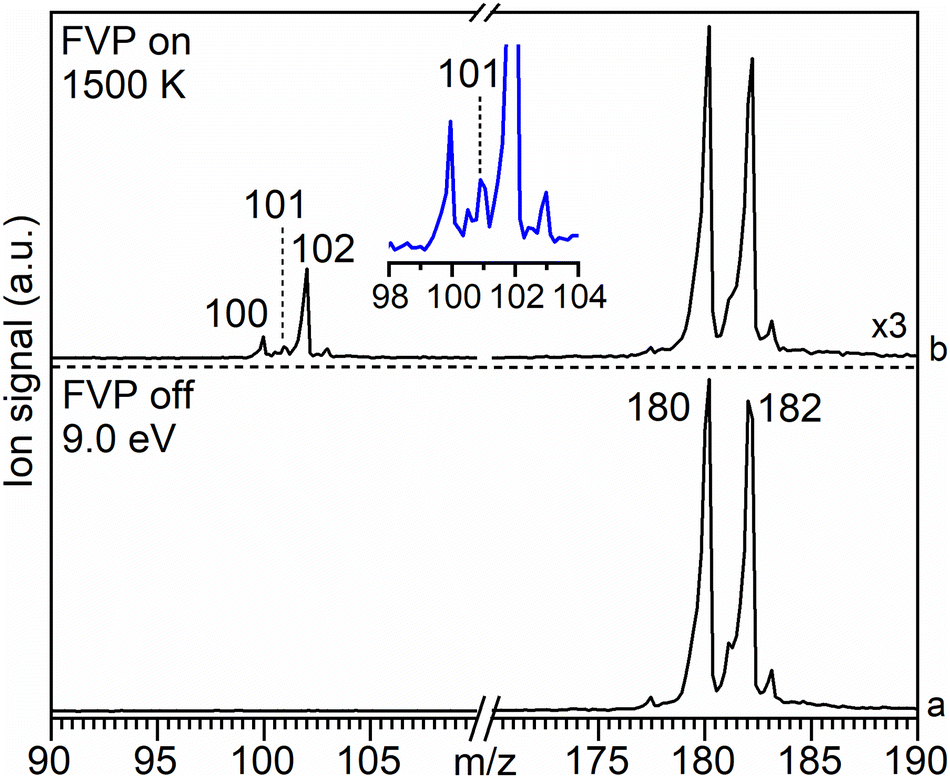 | ||
| Fig. 2 Molecular beam associated mass spectra of 4 (m/z 180) recorded at a photon energy of 9.0 eV at (a) RT (FVP off) and (b) upon pyrolysis (FVP on) at 1500 K (for details see Fig. S5, ESI†). Trace (b) is scaled up by a factor of 3. Inset shows the expansion of the peaks at m/z 100–103. | ||
Pyrolysis of 4 at 1500 K results in the depletion of its signal at m/z 180 and 182 by 60%, concomitantly with the appearance of a strong signal at m/z 102 as well as smaller signals at m/z 100 and 101 (Fig. 2b). Increasing the FVP temperature to 1700 K leads to higher intensities of the peaks at m/z 100 and 102, however, full conversion of precursor 4 is still not achieved (Fig. S3, ESI†).
The peak at m/z 101 starts appearing above 1500 K and is attributed to C8H5 species, particularly to the phenylethynyl radical 1, as discussed in detail in the next section. It originates from direct ionization of the thermally generated radical 1 (Fig. 2b and Fig. S5, ESI†). The signal at m/z 102 is assigned to ethynylbenzene 2 (C8H6) by comparison of its ms-TPE spectrum, containing a fundamental transition at 8.81 eV, with that reported in the literature (Fig. S6, ESI†).38,67,68 The strong peak at m/z 102 is also accompanied by a small satellite peak at m/z 103 corresponding to its 13C isotopologue. The neutral species 2 is formed via H-abstraction from radical 1 through reaction with the precursor in the SiC tube or with contaminants on the chamber walls (Fig. S4, ESI†), vide infra.57
Radical 1 is highly reactive and undergoes extensive thermal unimolecular decomposition through H-loss to yield a peak at m/z 100 or H-abstraction generating m/z 102. The peak at m/z 100 can be assigned to multiple C8H4 isomers (8–13). Smaller decomposition products at m/z 74, 76, 77, and 78 are assigned to triacetylene (C6H2), E- and Z-hexa-1,5-diyne-3-ene (C6H4), phenyl radical (C6H5), and benzene (C6H6), respectively, by comparison of their ms-TPE spectra with those reported in the literature as well as using Frank–Condon simulations (Fig. S7–S9, ESI†).69–71 A small peak observed at m/z 202 is tentatively assigned to the dimer of 1, while other higher mass PAHs were not observed due to the low concentrations and short reaction times in the SiC reactor (Fig. S2, ESI†). In contrast, a much richer chemistry was previously observed in high-pressure shock tube pyrolysis of 2 at 1100–1700 K.17
The pyrolysis of 3 at temperatures up to 1800 K additionally results in the formation of a species with m/z 98 which is assigned to tetraacetylene 14 (C8H2), in excellent agreement with literature data (Fig. S1 and S10, ESI†).72
Characterization of the phenylethynyl cation 1+
The ms-TPE spectrum of the radicals with m/z 101 (C8H5), generated by pyrolysis of 4 at 1500 K is shown in Fig. 3. The signal at m/z 101 partially overlaps with the signals at m/z 100 (C8H4) and 102 (C8H6) (Fig. 2b, inset). To rule out any contributions of m/z 100 and 102 we carefully compared the ms-TPES of all individual species(Fig. S11 and S12, ESI†). The difference spectrum of m/z 101, obtained by subtracting the spectral contributions from m/z 100 and m/z 102, enables a clear determination of the pure ms-TPE signal of m/z 101, free from contamination by overlapping peaks (Fig. S12, ESI†). The strongest vibronic transition of 2 (m/z 102) is reported at 8.81 eV,38,67,68 while that of isomers 8–9 (m/z 100) appears at 8.75 eV (see full assignment in the next section). Since only small peaks are observed between 8.70 and 8.85 eV in the ms-TPE spectrum of m/z 101, the spectrum above 8.90 eV can reliably be assigned to radical 1 (see comparison between the ms-TPE spectra in Fig. S11 and S12, ESI†). Subtracting 8.7% 13C-isotopic contribution of m/z 100 C8H4, from the ms-TPES of m/z 101, only leads to a slight decrease in intensity, without altering the spectral pattern (Fig. S13, ESI†). Moreover, a good signal-to-noise ratio of the ms-TPE spectrum of m/z 101 effectively distinguishes spectral peaks from the background noise (Fig. S13, ESI†). | ||
| Fig. 3 Comparison of the molecular beam associated ms-TPE spectrum of the signal m/z 101, recorded upon FVP of precursor 4 at 1500 K (black trace), with Franck–Condon (FC) simulations at 1500 K of the vibronic transitions of radical 1 to cation 1+ in its 3B1 (blue trace) and 1A1 (green trace) electronic states along with the sum of FC simulations (red trace). The spectrum of m/z 101 has been corrected for the contribution of the isotopic 13C signal of m/z 100. The FC simulations are convoluted using 48 meV fwhm Gaussians over the vibrational frequencies computed with ωB97XD/6-311++G**. The peaks marked with (*) are due to possible contamination of the overlapping signal from m/z 100 and m/z 102. Peaks below 8.80 eV are due to hot band transitions and might have contributions from other isomeric radicals such as 5–7. | ||
The experimentally observed transition at 8.90 eV is compared to the calculated adiabatic ionization energy (AIE) of 1, performed at high-level CCSD(T) and multi-configurational methods (Table 1). The peak at 8.90 ± 0.05 eV is assigned to the AIE of radical 1 in its π (2B1) ground state to cation 1+ in its triplet (3B1) ground state. The peak at 9.02 ± 0.05 eV likely corresponds to the first excited state of cation 1+, the closed-shell singlet (1A1) state. The spectral pattern is reasonably well reproduced by Franck–Condon (FC) simulations of the respective vibronic transitions at 1500 K (Fig. 3) along with a decent fit of sum of FC simulations. This allows us to determine the singlet–triplet gap of 1+ to 0.12 ± 0.05 eV (2.8 ± 1.2 kcal mol−1).
| Method | AIEa (3B1) | AIEb (1A1) | ΔESTc |
|---|---|---|---|
| a Transition 1+ (3B1) ← 1. b Transition 1+ (1A1) ← 1. c Singlet–triplet energy gap (ΔEST) of 1+. d Geometry optimization. e Energy calculation over optimized geometry with CASSCF/aug-cc-pVTZ. | |||
| ms-TPES (exp.) | 8.90 ± 0.05 | 9.02 ± 0.05 | +0.12 |
ωB97XD/6-311++G**![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
8.95 | 9.14 | +0.19 |
| NEVPT2/aug-cc-pVTZd | 8.78 | 8.83 | +0.05 |
| CIPT2/aug-cc-pVTZd | 8.92 | 8.97 | +0.05 |
| CASPT2/aug-cc-pVTZe | 8.80 | 9.00 | +0.20 |
| CCSD(T)/aug-cc-pVTZd | 9.02 | 8.90 | −0.12 |
However, no reasonable fit of the FC simulations by swapping the order of singlet and triplet state or by allowing flexibility of adiabatic ionization energies could be achieved (Fig. S14, ESI†). This confirms our original assignment, to which more conservative experimental error bars of ±50 meV for the adiabatic ionization energies is included, which are higher than the photon (5 meV) and electron energy resolution (10 meV) of the beamline and spectrometer, but in good agreement with a rotational envelope of the TPES at 1500 K. The remaining differences between the experimental and simulated spectra are associated with underestimated FC factors for hot band transitions, a known challenge for simulating high temperature spectra.57
A comparison of the ms-TPE spectrum of m/z 101, obtained by pyrolysis of 4 at a higher temperature (1700 K), consistently shows the bands assigned to the cation 1+ in its singlet and triplet state, lends further credibility to our assignment (Fig. S15, ESI†). However, in case of FVP of precursor 3, the assignment remains tentative as a clean ms-TPES of radical 1 could not be observed due to contamination of precursor 3 with phenylacetylene (Fig. S16, ESI†).
The electronic configuration of 1 is (b1)2 (a1)2 (b1)2 (b2)2 (a2)2 (b1)1, according to CASSCF calculations (Fig. S17, ESI†). Electron removal from doubly occupied (a1, b1, b2, and a2) and singly occupied (b1) orbitals via one-photon absorption (Koopmann transition) leads to several electronic states within 0.7 eV (15 kcal mol−1) for cation 1+ (Table S2, ESI†) with singlet and triplet multiplicities. In addition, other electronic states of cation 1+ are accessible by subsequent electronic excitation into empty π* orbitals, corresponding to multi-photon non-Koopmann transitions. Hence, assignment of the ms-TPE spectrum of the electronically rich cation 1+ is even more challenging than the already complex spectrum of the ethynyl cation, HCC+.46,47
AIEs of the transitions 1+ (3B1) ← 1 and 1+ (1A1) ← 1 are calculated to 8.8–9.0 eV with multi-configurational methods and DFT, in good agreement with the experimental peaks at 8.90 and 9.02 eV. However, the energetic order of the 1A1 and 3B1 states is reversed in the CCSD(T) calculations, which is explained by an overstabilization of the closed-shell singlet (1A1) state. This state resembles a π-cation with a doubly-occupied a1 (σ) and an empty b1 (π) orbital in the ethynyl group. However, the higher-energy configurations also contribute to the wavefunction of the 1A1 state, and configuration mixing is properly taken into account by multi-configurational methods.63 In contrast, the ground state of the triplet (3B1) cation with a dominant (a1)1(b1)1 configuration resembles a σ,π diradical, similar to the triplet phenyl cation.73 The geometries of the singlet and triplet cations 1+ are remarkably different. While a C–CC bonding pattern is found for the triplet (3B1) cation, a fully conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC allene moiety is present in the singlet (1A1) cation (Fig. 4). Thus, ionization into the triplet (3B1) and singlet (1A1) cationic states leads to an activity of the ν28 (CC stretch) vibration. Experimentally observed vibrational spacing of 0.27 ± 0.05 eV (2178 ± 403 cm−1) and 0.21 ± 0.05 eV (1694 ± 403 cm−1), corresponding to the ν28 (CC stretch) vibration of the triplet (3B1) and the singlet (1A1) cation, is in good agreement with the calculated unscaled frequencies of 2104 cm−1 and 2002 cm−1 respectively, at the ωB97XD/6-311++G** level of theory.
CC allene moiety is present in the singlet (1A1) cation (Fig. 4). Thus, ionization into the triplet (3B1) and singlet (1A1) cationic states leads to an activity of the ν28 (CC stretch) vibration. Experimentally observed vibrational spacing of 0.27 ± 0.05 eV (2178 ± 403 cm−1) and 0.21 ± 0.05 eV (1694 ± 403 cm−1), corresponding to the ν28 (CC stretch) vibration of the triplet (3B1) and the singlet (1A1) cation, is in good agreement with the calculated unscaled frequencies of 2104 cm−1 and 2002 cm−1 respectively, at the ωB97XD/6-311++G** level of theory.
 | ||
| Fig. 4 Geometry and occupied σ and π orbitals of the lowest-energy states of radical 1 and singlet and triplet cation 1+, optimized at the NEVPT2/aug-cc-pVTZ level of theory. Selected C–C bond distances in Å are shown. | ||
The TPE signal between 9.07 and 9.4 eV might indicate the presence of the excited open-shell singlet (1B1) cation with a leading (a1)1(b1)1 configuration, due to one-photon electron removal of radical 1 (Table S2, ESI†). The singlet and triplet A2 states of 1+ are calculated to be energetically close and are formally obtained via non-Koopmans transitions. The short lifetime of these excited states may result in broad and unstructured bands, which are difficult to identify in our experimental spectrum. In addition, the bands at 8.48 and 8.57 eV at 1500 K (Fig. 3) and 8.70 eV at 1700 K (Fig. S15, ESI†) are tentatively assigned to ethynylphenyl radicals 5–7 (Fig. 3), the lowest-energy C8H5 isomers. The AIEs of radicals 5–7 (Cs symmetry) are computed to be ∼8.4 eV at ωB97XD as well as CCSD(T) and G4 methods (Table S3, ESI†), in line with earlier experimental reports.74,75 The transitions to excited triplet states of ions 5+–7+ are computed to 8.6–8.9 eV, which can also have a minor contribution to the spectra. However, radicals 5–7 are only formed in low concentrations, due to their efficient decomposition reactions (vide infra). Hot band transitions are also likely responsible for the broad and unstructured signal below 8.80 eV, making a definitive assignment challenging.76
Thermal decomposition of the phenylethynyl radical 1
The decomposition of radical 1 at high temperature was investigated by comparison of the ms-TPE spectrum of m/z 100 recorded at 1700 K reactor temperature with computed AIEs and FC simulations of the lowest-energy C8H4 isomers 8–13 (Fig. 5, Table 2, and Scheme 2). Ethynyl-substituted o-benzynes 12 and 13 are calculated at the ωB97XD/6-311++G** level of theory to be the lowest-energy isomers, while open-ring triynes 8–11 lie 6–16 kcal mol−1 higher in energy (see ΔE0K values in Table 2). This contrasts to the observations in the ms-TPES, which shows a strong transition at 8.76 eV. This transition is matched by the calculated AIEs of isomers 8, E-9 and Z-9. However, if entropic contributions are taken into account at 1700 K, the order is reversed and triyne 8 is the most stable isomer followed by structurally related 9–11 lying 3–9 kcal mol−1 higher in energy (see ΔG1700K values in Table 2). At 1700 K, the Boltzmann population analysis based on Gibbs free energies, suggests that triynes 8–9 are likely to dominate the gas mixture, making up to 87% of the total population. In contrast, isomers 10–13 are predicted to be present in much lower quantities. However, in the absence of tabulated photoionization cross sections of isomers 8–13, the spectral compositions do not necessarily represent the accurate concentration of isomers. Additionally, unimolecular decomposition channels of C8H4 isomers may lower their actual abundances in the reactor too.
 | ||
| Fig. 5 ms-TPE spectrum of the signal at m/z 100, recorded upon FVP of precursor 4 at 1700 K (black trace) reactor temperature. Due to background selection in the ion images (Fig. S5, ESI†) hot and sequence bands are cooled and resemble a 300 K distribution, justifying a fit with RT Franck–Condon (FC) simulations (red trace) of the individual C8H4 isomers 8–13. FC simulations and AIE of individual isomers are also presented. FC line spectra are convoluted using 40 meV fwhm Gaussians and were computed at the ωB97XD/6-311++G** level of theory. | ||
| C8H4 | ΔE0Ka |
ΔG1700Ka |
Pop. | AIE (exp.) | AIE (exp.) | AIE (calc.) | AIE (calc.) |
|---|---|---|---|---|---|---|---|
| Isomers | (ωB97XD) | (ωB97XD) | this work | literature48 | (W1BD) | (G4) | |
| a All energies are ZPE corrected. | |||||||
| 8 | 6.5 | 0 | 49.2 | 8.74 | 8.78 | 8.77 | 8.77 |
| E-9 | 9.7 | 3.2 | 19.3 | 8.75 | 8.77 | 8.77 | 8.80 |
| Z-9 | 10.0 | 3.4 | 18.2 | 8.76 | 8.80 | 8.79 | 8.83 |
| 10 | 13.7 | 7.1 | 6.1 | 9.08 | — | 9.03 | 9.05 |
| 11 | 15.9 | 9.5 | 3.0 | 8.84 | — | 8.85 | 8.84 |
| 12 | 0 | 10.7 | 2.1 | ∼9.16 | — | 9.15 | 9.14 |
| 13 | 0 | 10.7 | 2.1 | ∼9.16 | — | 9.14 | 9.14 |
The ms-TPE spectrum of m/z 100 recorded at 1700 K reactor temperature was plotted by taking into account only the room temperature background cooled ions in the VMI (Fig. S5, ESI†), which justifies the use of 300 K FC simulations for our model.57 The spectrum shows a very strong and broad peak centered at 8.76 eV, followed by additional less intense bands at 8.84, 8.94, 9.02, 9.08, and 9.16 eV (Fig. 5). The broad peak at 8.76 eV is assigned to a mixture of triynes 8–9, in excellent agreement with the computed AIEs obtained by composite method calculations (Table 2). The discrepancy between our experimental values and the literature AIEs of 8.78–8.80 (Table 2) could arise from the well-defined spectral transitions in the ms-TPES in comparison to earlier photoionization mass spectrometry studies, which lack a vibrational structure.48 The less intense peaks at 8.84 and 9.08 eV are tentatively assigned to isomers 11 and 10, respectively. However, the peaks at 8.94 and 9.02 eV correspond to overlapping transitions from several isomers rather than assignment to any particular C8H4 isomer.
The very small band at 9.16 eV is tentatively assigned to the ethynyl-substituted o-benzynes 12 and 13 (Table 2) although the weak, broad and unstructured vibronic bands might be due to very low estimated populations at 1700 K along with the low FC factors associated to a large change in geometry from their neutral to cationic states.77 In addition, since the parent unsubstituted benzyne ionization is dominated by up to three cationic states, a similar spectral behavior is likely to be expected for the ethynyl-substituted benzynes as well, leading to an additional spectral congestion in this energy range.77 Other higher energy isomers (15–21) were ruled out based on lower predicted AIEs as well as higher relative energies (Table S4, ESI†).
Fitting of experimental ms-TPE spectra with FC simulations experimentally determined a vibrational frequency of 0.26 ± 0.035 eV (2097 ± 282 cm−1) corresponding to the ν26 (CC stretching) mode of ions 8+–9+, in fair agreement with unscaled vibrational frequencies of 2275–2319 cm−1 computed at the ωB97XD/6-311++G** level of theory.
Isomerization and decomposition pathways of the phenylethynyl radical 1
To understand the high temperature chemistry of radical 1, which mainly undergoes isomerization and thermal decomposition, computations were performed at 1700 K mimicking the temperature of the pyrolytic reactor (Fig. 6 and Fig. S18–S24, ESI†). The study reveals that radical 1 formed at 1700 K, initially isomerizes to radical 5via intermediate i1. This isomerization step, progressing through consecutive 1,2-H migrations, is exergonic by 15 kcal mol−1 and involves a very high energy barrier of 93 kcal mol−1 for the first 1,2-H migration. An alternative bimolecular pathway from 1 to 5via H-addition and H-elimination must pay the energetic penalty of breaking a C–H bond in the phenyl ring with a BDE of about 113 kcal mol−1 and is thus less probable.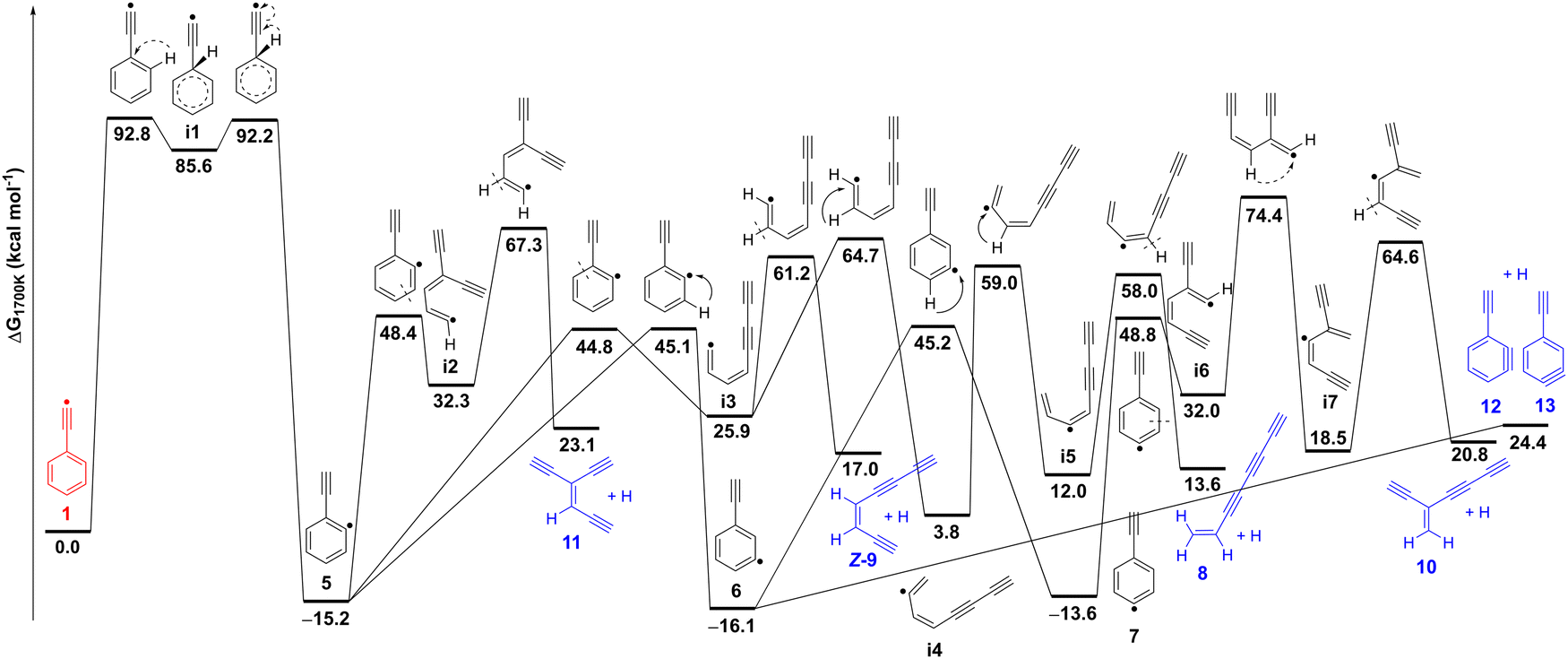 | ||
| Fig. 6 Potential energy surface for the isomerization and thermal decomposition of the phenylethynyl radical 1 calculated at the ZPE-corrected ωB97XD/6-311++G** level of theory. Relative Gibbs free energies at 1700 K are shown for comparison to pyrolysis experiments. Complementary relative electronic energies at 0 K computed with CCSD(T)/aug-cc-pVTZ over the ωB97XD geometries are shown in Table S5 (ESI†). | ||
Radical 5 further decomposes yielding C8H4 isomers (8–13), through various reactions, including ring-opening, H-shifts, and H-loss. These subsequent steps have considerably lower energy barriers as compared to the initial H-shift (1 → i1 → 5). Ring opening at different positions of thermally excited 5 leads to open chain intermediates i2 or i3, which after H-loss, result in the formation of triynes 11 and Z-9, respectively. Isomer E-9 could also be generated via intermediate i3 following a similar pathway. Consecutive 1,2-H shifts further drive the downhill conversion of intermediate i3 to i5, which finally dissociates to afford triyne 8, the most stable C8H4 isomer at 1700 K.
Competing channels on the C8H5 surface proceed through H-shift from radical 5 to isomers 6 and 7, which directly connects to triyne 10via a multistep (7 → i6 → i7 → 10) process. Dissociation of radical 6 also provides an alternative pathway to generate triynes Z-9 and 11 (Fig. S24, ESI†). Closed-shell benzynes 12 and 13 are formed via direct H-loss from radicals 5 and 6. On the other hand, benzynes 12 and 13 can directly ring-open and form triynes 8–11, a process that is entropically favorable. Our proposed reaction mechanism, based on the potential energy surface, is in good agreement with the observed product distribution at m/z 100, however we cannot fully exclude other bimolecular or hydrogen-assisted rearrangement reactions to play a role, although less likely due to the high dilution of the precursor in the reaction mixture.
Conclusions
Alkynyl cations are extremely electrophilic high-energy species that cannot be synthesized in the condensed phase. Even with inert species such as molecular nitrogen or xenon, these cations readily react with high exothermicity. Consequently, experimental evidence for the formation of alkynyl cations is very limited (e.g. mass spectrometric detection in the gas phase), and a spectroscopic characterization of these elusive species was not possible, so far.25The ionization of alkynyl radicals in the gas phase is a viable approach for the synthesis of alkynyl cations. However, alkynyl radicals are also highly unstable and difficult to synthesize. A comparison of the radical stability of the phenylethynyl radical 1 with that of the phenyl radical, which is already a highly unstable radical, reveals that 1 is less stable than the phenyl radical by 18 kcal mol−1 (see definition of radical and cation stability in Fig. 1). Ionization produces the corresponding cations, and now the phenylethynyl cation 1+ is even destabilized by 45 kcal mol−1 compared to the phenyl cation.
Radical 1 was synthesized in the gas phase by FVP of (bromoethynyl)benzene 4 as precursor and characterized via ms-TPE spectroscopy. Vibrationally-resolved ms-TPE spectroscopy in combination with high-level ab initio calculations allowed us to unravel the complex electronic structure of both the radical 1 and the corresponding cation 1+. We conclude that radical 1 exhibits a π (2B1) ground state, in agreement with EPR observations.18,19
Cation 1+ was produced via photoionization of radical 1, and an adiabatic ionization energy (AIE) of 8.90 ± 0.05 eV was measured. The analysis of the ms-TPE spectra reveals that cation 1+ has a triplet 3B1 ground state with the closed-shell singlet 1A1 state lying 2.8 kcal mol−1 higher in energy. This energy difference corresponds to the experimental singlet–triplet gap (ΔEST) of cation 1+, which is a key chemical descriptor in the spin-selective chemistry of carbocations. The ground state of 1+ is thus described as a π1σ1 triplet diradical, in analogy to triplet cyclopentadienyl,78 (dimethylamino)phenyl,79 and naphthyl cations,80 and is expected to exhibit chemoselective reactivity against nucleophiles.79 The excited π0σ2 singlet state of 1+ resembles both a singlet vinylidene and a π-cation and therefore should exhibit ambiphilic character.
Radical 1 efficiently abstracts H-atoms from the environment to form ethynylbenzene 2. Observation of various open chain C8H4 isomers along with potential energy surface calculations supports the isomerization mechanism of radical 1 to the lower-energy ethynylphenyl radical isomers 5–7. This chemistry might be of relevance in the context of high-temperature astrochemical environments like circumstellar envelopes of carbon stars and planetary nebulae as well as cold dense molecular clouds. The rearranged radicals 5–7 were only detected in small quantities since they undergo rapid ring-opening followed by H-loss affording a mixture of C8H4 isomers. The gas mixture mostly contains unbranched triynes 8–9, while branched triynes 10–11 and ethynylbenzynes 12–13 are minor species. At 1800 K reactor temperature, unbranched triynes 8–9 undergo H2-loss to form tetraacetylene 14 (C8H2), which is believed to be an astrochemical precursor of the C8H radical and anion, both detected in the ISM.81,82 The extreme reactivity of the hydrocarbons 1 and 1+ and the rich chemistry observed in our experiments suggests that the thermal isomerization of radical 1 might play a role in the synthesis of polycyclic aromatic hydrocarbons in an astrochemical context.24
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
GK: formal analysis (lead), investigation (supporting), writing – original draft (equal). EMV: conceptualization (lead), investigation (supporting), formal analysis (supporting), writing – original draft (equal). APG: investigation (lead), formal analysis (supporting), writing – review & editing (supporting). MS: investigation (supporting), formal analysis (supporting), writing – review & editing (supporting). WS: supervision (equal), funding acquisition (lead), writing – review & editing (supporting). PH: supervision (equal), resources (lead), writing – review & editing (supporting), formal analysis (supporting).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This project received funding from the European Union's Horizon 2020 research innovation programme under the Marie Skłodowska-Curie grant agreement no. 801459 – FP-RESOMUS and the Deutsche Forschungsgemeinschaft (DFG) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. Open access funds were gratefully received via Lib4RI (Library for the Research Institutes within the ETH Domain). We thank Pritam Kadam (RUB) for conducting preliminary experiments on C6H5CC˙ in cryogenic matrices. Patrick Ascher (PSI) is thankfully acknowledged for technical assistance. The experiments were carried out at the VUV (x04db) beamline of the Swiss Light Source (SLS), located at Paul Scherrer Institute in Villigen, Switzerland.
References
- K. C. Nicolaou and A. L. Smith, in Modern Acetylene Chemistry, eds. P. J. Stang and F. Diederich, VCH, Weinheim, 1995, ch. 7, pp. 203–283 Search PubMed.
- M. R. Bryce, J. Mater. Chem. C, 2021, 9, 10524–10546 RSC.
- B. A. McGuire, Astrophys. J., Suppl. Ser., 2022, 259, 30 CrossRef CAS.
- M. L. Coote, C. Y. Lin and H. Zipse, in Carbon-Centered Free Radicals and Radical Cations, ed. M. D. E. Forbes, John Wiley & Sons, Inc., Hoboken, New Jersey, 2009, ch. 5, pp. 83–104 Search PubMed.
- C. Alcaraz, I. Fischer and D. Schröder, in Encyclopedia of Radicals in Chemistry, Biology and Materials, eds. C. Chatgilialoglu and A. Studer, John Wiley & Sons, Ltd., 2012, pp. 1–27 Search PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.130 of the Thermochemical Network, Argonne National Laboratory, Lemont, Illinois, 2023, available at https://ATcT.anl.gov Search PubMed.
- X. B. Gu, Y. Guo, F. T. Zhang, A. M. Mebel and R. I. Kaiser, Chem. Phys. Lett., 2007, 436, 7–14 CrossRef CAS.
- X. B. Gu and R. I. Kaiser, Acc. Chem. Res., 2009, 42, 290–302 CrossRef CAS PubMed.
- X. B. Gu, F. T. Zhang, Y. Guo and R. I. Kaiser, Angew. Chem., Int. Ed., 2007, 46, 6866–6869 CrossRef CAS PubMed.
- A. Hamadi, W. Y. Sun, S. Abid, N. Chaumeix and A. Comandini, Combust. Flame, 2022, 237, 111858 CrossRef CAS.
- N. Hansen, M. Schenk, K. Moshammer and K. Kohse-Höinghaus, Combust. Flame, 2017, 180, 250–261 CrossRef CAS.
- B. Jones, F. Zhang, P. Maksyutenko, A. M. Mebel and R. I. Kaiser, J. Phys. Chem. A, 2010, 114, 5256–5262 CrossRef CAS PubMed.
- A. M. Mebel, V. V. Kislov and R. I. Kaiser, J. Am. Chem. Soc., 2008, 130, 13618–13629 CrossRef CAS PubMed.
- D. Loru, C. Cabezas, J. Cernicharo, M. Schnell and A. L. Steber, Astron. Astrophys., 2023, 677, A166 CrossRef CAS.
- D. S. N. Parker, R. I. Kaiser, T. P. Troy and M. Ahmed, Angew. Chem., Int. Ed., 2014, 53, 7740–7744 CrossRef CAS PubMed.
- W. Y. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, Combust. Flame, 2020, 220, 257–271 CrossRef CAS.
- J. S. Coleman, A. Hudson, K. D. J. Root and D. R. M. Walton, Chem. Phys. Lett., 1971, 11, 300–301 CrossRef CAS.
- P. H. Kasai and H. C. Mcbay, J. Phys. Chem., 1984, 88, 5932–5934 CrossRef CAS.
- R. K. Sreeruttun, P. Rarnasami, C. S. Wannere, A. C. Simmonett and H. F. Schaefer, J. Phys. Chem. A, 2008, 112, 2838–2845 CrossRef CAS PubMed.
- L. Duarte and L. Khriachtchev, Sci. Rep., 2017, 7, 3130 CrossRef PubMed.
- K. Guthier, P. Hebgen, K. H. Homann, J. Hofmann and G. Zimmermann, Liebigs Ann., 1995, 637–644 CrossRef CAS.
- J. Hofmann, G. Zimmermann, K. Guthier, P. Hebgen and K. H. Homann, Liebigs Ann., 1995, 631–636 CrossRef CAS.
- S. J. Goettl, Z. H. Yang, S. Kollotzek, D. Paul, R. I. Kaiser, A. Somani, A. Portela-Gonzalez, W. Sander, A. A. Nikolayev, V. N. Azyazov and A. M. Mebel, J. Phys. Chem. A, 2023, 127, 5723–5733 CrossRef CAS PubMed.
- P. J. Stang, in Dicoordinated Carbocations, eds. Z. Rappoport and P. J. Stang, Wiley & Sons, Chichester, 1997, ch. 10, pp. 451–460 Search PubMed.
- J. P. Cheng, K. L. Handoo and V. D. Parker, J. Am. Chem. Soc., 1993, 115, 2655–2660 CrossRef CAS.
- D. H. Aue, in Dicoordinated Carbocations, eds. Z. Rappoport and P. J. Stang, Wiley & Sons, Chichester, 1997, ch. 3, pp. 105–156 Search PubMed.
- K. Laali and G. A. Olah, Rev. Chem. Intermed., 1985, 6, 237–253 CrossRef CAS.
- G. Angelini, M. Hanack, J. Vermehren and M. Speranza, J. Am. Chem. Soc., 1988, 110, 1298–1299 CrossRef CAS.
- V. V. Zhdankin, P. J. Stang and N. S. Zefirov, J. Chem. Soc., Chem. Commun., 1992, 578–579 RSC.
- J. M. Dyke, Phys. Chem. Chem. Phys., 2019, 21, 9106–9136 RSC.
- I. Fischer and P. Hemberger, Chem. Phys. Chem., 2023, 24, e202300334 CrossRef CAS PubMed.
- T. Baer and R. P. Tuckett, Phys. Chem. Chem. Phys., 2017, 19, 9698–9723 RSC.
- P. Hemberger, J. A. van Bokhoven, J. Pérez-Ramírez and A. Bodi, Catal. Sci. Technol., 2020, 10, 1975–1990 RSC.
- E. Reusch, F. Holzmeier, M. Gerlach, I. Fischer and P. Hemberger, Chem. – Eur. J., 2019, 25, 16652–16659 CrossRef CAS PubMed.
- X. K. Wu, Z. H. Zhang, Z. Pan, S. Bjelic, A. Bodi and P. Hemberger, Energy Fuels, 2022, 36, 7200–7205 CrossRef CAS.
- J. Bouwman, M. N. McCabe, C. N. Shingledecker, J. Wandishin, V. Jarvis, E. Reusch, P. Hemberger and A. Bodi, Nat. Astron., 2023, 7, 423–430 CrossRef.
- M. N. McCabe, P. Hemberger, D. Campisi, J. C. Broxterman, E. Reusch, A. Bodi and J. Bouwman, Phys. Chem. Chem. Phys., 2022, 24, 1869–1876 RSC.
- M. N. McCabe, P. Hemberger, E. Reusch, A. Bodi and J. Bouwman, J. Phys. Chem. Lett., 2020, 11, 2859–2863 CrossRef CAS PubMed.
- P. Hemberger, V. B. F. Custodis, A. Bodi, T. Gerber and J. A. van Bokhoven, Nat. Commun., 2017, 8, 15946 CrossRef PubMed.
- Z. Y. Pan, A. Puente-Urbina, S. R. Batool, A. Bodi, X. K. Wu, Z. H. Zhang, J. A. van Bokhoven and P. Hemberger, Nat. Commun., 2023, 14, 4512 CrossRef CAS PubMed.
- N. Genossar-Dan, S. Atlas, D. Fux, S. H. Lavan, U. Zamir, I. Rozenberg, T. L. Nguyen, P. Hemberger and J. H. Baraban, Angew. Chem., Int. Ed., 2023, 62, e2023058 CrossRef PubMed.
- X. K. Wu, X. G. Zhou, P. Hemberger and A. Bodi, Phys. Chem. Chem. Phys., 2019, 21, 22238–22247 RSC.
- W. R. Stevens, B. Ruscic and T. Baer, J. Phys. Chem. A, 2010, 114, 13134–13145 CrossRef CAS PubMed.
- J. D. Savee, J. Zador, P. Hemberger, B. Sztaray, A. Bodi and D. L. Osborn, Mol. Phys., 2015, 113, 2217–2227 CrossRef CAS.
- B. Gans, G. A. Garcia, F. Holzmeier, J. Krüger, A. Röder, A. Lopes, C. Fittschen, J. C. Loison and C. Alcaraz, J. Chem. Phys., 2017, 146, 011101 CrossRef CAS PubMed.
- B. Mehnen, R. Linguerri, S. Ben Yaghlane, M. M. Al Mogren and M. Hochlaf, Faraday Discuss., 2018, 212, 51–64 RSC.
- N. Hansen, S. J. Klippenstein, P. R. Westmoreland, T. Kasper, K. Kohse-Höinghaus, J. Wang and T. A. Cool, Phys. Chem. Chem. Phys., 2008, 10, 366–374 RSC.
- J. Cernicharo, A. M. Heras, A. Tielens, J. R. Pardo, F. Herpin, M. Guélin and L. Waters, Astrophys. J., 2001, 546, L123 CrossRef CAS.
- X. Gu, Y. S. Kim, R. I. Kaiser, A. M. Mebel, M. C. Liang and Y. L. Yung, Proc. Natl. Acad. Sci., 2009, 106, 16078–16083 CrossRef CAS PubMed.
- B. Sztaray, K. Voronova, K. G. Torma, K. J. Covert, A. Bodi, P. Hemberger, T. Gerber and D. L. Osborn, J. Chem. Phys., 2017, 147, 013944 CrossRef PubMed.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 CrossRef CAS.
- M. X. W. Jiang, M. Rawat and W. D. Wulff, J. Am. Chem. Soc., 2004, 126, 5970–5971 CrossRef CAS PubMed.
- M. V. Russo, C. Lo Sterzo, P. Franceschini, G. Biagini and A. Furlani, J. Organomet. Chem., 2001, 619, 49–61 CrossRef CAS.
- D. W. Kohn, H. Clauberg and P. Chen, Rev. Sci. Instrum., 1992, 63, 4003–4005 CrossRef CAS.
- Q. Guan, K. N. Urness, T. K. Ormond, D. E. David, G. B. Ellison and J. W. Daily, Int. Rev. Phys. Chem., 2014, 33, 447–487 Search PubMed.
- P. Hemberger, X. K. Wu, Z. Y. Pan and A. Bodi, J. Phys. Chem. A, 2022, 126, 2196–2210 CrossRef CAS PubMed.
- A. Bodi, B. Sztáray, T. Baer, M. Johnson and T. Gerber, Rev. Sci. Instrum., 2007, 78, 084102 CrossRef PubMed.
- B. Sztáray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763–3768 CrossRef.
- P. Hemberger, Z. Pan, X. Wu, Z. Zhang, K. Kanayama and A. Bodi, J. Phys. Chem. C, 2023, 127, 16751–16763 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, M. C. X. Li, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, C. A. M. Klene, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- V. B. Oyeyemi, J. A. Keith, M. Pavone and E. A. Carter, J. Phys. Chem. Lett., 2012, 3, 289–293 CrossRef CAS PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS.
- K. Andersson, P. Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schutz, WIREs Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- J. M. Dyke, H. Ozeki, M. Takahashi, M. C. R. Cockett and K. Kimura, J. Chem. Phys., 1992, 97, 8926–8933 CrossRef CAS.
- C. H. Kwon, H. L. Kim and M. S. Kim, J. Phys. Chem. A, 2003, 107, 10969–10975 CrossRef CAS.
- F. Brogli, E. Heilbronner, V. Hornung and E. Kloster-Jensen, Helv. Chim. Acta, 1973, 56, 2171–2178 CrossRef.
- M. Gerlach, E. Karaev, D. Schaffner, P. Hemberger and I. Fischer, J. Phys. Chem. Lett., 2022, 13, 11295–11299 CrossRef CAS PubMed.
- J. D. Savee, B. Sztáray, P. Hemberger, J. Zádor, A. Bodi and D. L. Osborn, Faraday Discuss., 2022, 238, 645–664 RSC.
- M. Allan, E. Heilbronner, E. Kloster-Jensen and J. P. Maier, Chem. Phys. Lett., 1976, 41, 228–230 CrossRef CAS.
- A. Nicolaides, D. M. Smith, F. Jensen and L. Radom, J. Am. Chem. Soc., 1997, 119, 8083–8088 CrossRef CAS.
- W. R. Stevens, B. Ruscic and T. Baer, J. Phys. Chem. A, 2010, 114, 13134–13145 CrossRef CAS PubMed.
- K. C. Lau and C. Y. Ng, J. Chem. Phys., 2006, 124, 044323 CrossRef PubMed.
- P. Hemberger, X. Wu, Z. Pan and A. Bodi, J. Phys. Chem. A, 2022, 126, 2196–2210 CrossRef CAS PubMed.
- D. Kaiser, E. Reusch, P. Hemberger, A. Bodi, E. Welz, B. Engels and I. Fischer, Phys. Chem. Chem. Phys., 2018, 20, 3988–3996 RSC.
- H. J. Worner and F. Merkt, Angew. Chem., Int. Ed., 2006, 45, 293–296 CrossRef CAS PubMed.
- S. M. Gasper, C. Devadoss and G. B. Schuster, J. Am. Chem. Soc., 1995, 117, 5206–5211 CrossRef CAS.
- S. Rayne and K. Forest, Theor. Chem. Acc., 2016, 135, 69 Search PubMed.
- J. Cernicharo and M. Guelin, Astron. Astrophys., 1996, 309, L27 CAS.
- S. Brunken, H. Gupta, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2007, 664, L43–L46 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass and ms-TPE spectra of FVP products; space focusing mode imaging; lowest-energy electronic states and relevant orbitals of cation 1+; C8H5 and C8H4 PES; intrinsic reaction paths and optimized geometries. Origin files containing spectral plots, excel files containing calculations and description of the data. See DOI: https://doi.org/10.1039/d4cp02129k |
| ‡ G. K. and E. M. V. contributed equally. |
| This journal is © the Owner Societies 2024 |