
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Urocanic acid as a novel scaffold for next-gen nature-inspired sunscreens: II. Time-resolved spectroscopy under solution conditions†
Jiayun
Fan
a,
Jack M.
Wooley
 b,
Hans
Sanders
a,
Vasilios G.
Stavros
*bc and
Wybren Jan
Buma
*ad
b,
Hans
Sanders
a,
Vasilios G.
Stavros
*bc and
Wybren Jan
Buma
*ad
aVan’t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: w.j.buma@uva.nl
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: v.stavros@bham.ac.uk
cSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK
dInstitute for Molecules and Materials, Radboud University, Toernooiveld 7c, 6525 ED Nijmegen, The Netherlands
First published on 2nd September 2024
Abstract
In recent years the use of synthetic UV filters in commercial skincare formulations has come under considerable scrutiny. Urocanic acid is a naturally occurring UV filter that could serve as a scaffold for developing next-generation biomimetic UV filters. We have carried out time-resolved electronic and vibrational absorption studies on urocanic acid and modified variants in various solvents on timescales spanning eighteen orders of magnitude; from femtoseconds to hours. In combination with quantum chemical calculations these provide vital insight into the photochemical and photophysical properties of urocanic acid and how these are tuned by substitutions and solvents. Moreover, they solve the hitherto conundrum of the wavelength dependence of the photochemistry of trans-urocanic acid in an aqueous environment. Crucially, these studies – together with the accompanying article that reports high-resolution laser spectroscopic studies performed under isolated gas-phase conditions (https://doi.org/10.1039/D4CP02087A) open novel avenues for a rational design of urocanic acid-based UV filters.
Introduction
Urocanic acid (UA, Fig. 1) is a well-known natural ultraviolet (UV) radiation absorber present in the upper layers of the skin. Because of its strong absorption in the UV-A and UV-B regions of the electromagnetic spectrum (400–280 nm), it was for a long time thought that one of its primary functions would be to act as a natural sunscreen to protect DNA from damage due to the absorption of harmful UV radiation. UA is produced in the epidermal layer of the skin in its trans isomeric form, which – ironically – with a Sun Protection Factor (SPF) of 1.58 has been proven not to be very effective as a sunscreen as compared to synthetic sunscreens.1 The performance of UA is further compromised by the immunosuppressive properties of the cis isomer, which is produced when UA is exposed to UV radiation. In fact, under solar illumination conditions, a photo stationary state is reached with 70% cis-UA.2,3 Reviews of solution-phase studies focusing on the photochemistry of trans-UA and their implications for the photobiology and photoimmunology of UA are given in ref. 4–6. | ||
Fig. 1 Molecular structure of urocanic acid (UA) and derivatives studied in the present work (methyl urocanate, N3-methyl methyl urocanate, and N1-methyl methyl urocanate). Structures of UA and its methyl ester are drawn for the N3H tautomer which is the dominant tautomer under the employed experimental conditions. In the article conformers of these compounds are labelled according to the trans/cis configuration of the C5–C6, C6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C7, and C7–C8 bonds. The conformer drawn here for urocanic acid is thus labelled as the N3H-TTC conformer. C7, and C7–C8 bonds. The conformer drawn here for urocanic acid is thus labelled as the N3H-TTC conformer. | ||
The potential adverse effects on health and environment of presently employed UV filters has led to a strong demand for better, safer and more efficient sunscreen agents. In recent years there has been therefore an increasing effort to obtain a better understanding of the photophysics and photochemistry of UV filters in order to come to a rational design of novel sunscreens with improved properties.7,8 Although the negative aspects mentioned above would seem to disfavour its use, UA has several characteristics that make its scaffold a promising starting point for the design of biomimetic sunscreens with a more favourable toxicological profile and with improved sunscreen properties.
Given the above, we have embarked on an extensive research program targeting novel UA-based UV filters to be used as sunscreens as well as other photothermal applications that rely on transforming photon energy into harmless, and useful, heat energy. Underpinning this program are spectroscopic studies on internally cooled trans-UA under isolated conditions, and on derivatives designed to mitigate the immunosuppressive effects of the cis isomer. These studies – of which the results have been discussed in detail in the accompanying article9 – established that the spectroscopy and excited-state dynamics of these compounds are critically determined by two close-lying 1nπ* and 1ππ* states. Importantly, they also showed that previously reported excitation spectra of trans-UA10 – used for many years as the gold standard for assessing the results of theoretical studies and studies of UA-based compounds under non-isolated conditions – suffered from thermal degradation reactions.
In the present studies, we elucidate the solar energy dissipating mechanisms of trans-UA and its derivatives in solution. Our studies address two vital aspects of their photochemistry. First, we investigate the photochemical transformations of these compounds under prolonged irradiation conditions to assess their photostability. Second, we employ femtosecond transient electronic absorption spectroscopy (TEAS) to determine the excited-state dynamics and decay pathways occurring upon excitation. We do such experiments for trans-UA and its substituted derivatives in organic solvents with different polarities and hydrogen-bonding propensity to explore how photostabilities and decay pathways can be optimized using different formulations.
Apart from organic solvents, we perform such experiments as well for pH 5.6 and pH 7.2 buffer solutions of trans-UA. The reason for doing so is twofold. Firstly, at pH 5.6, trans-UA is present as the H+UA− zwitterion (the form dominating in the outer epidermis of the skin) and at pH 7.2, trans-UA is present as the UA− anion (dominating in the inner part of the epidermis). To add, the ordering and nature of the lower-lying electronically excited singlet states of the zwitterion/anion differs significantly from that of the neutral form.11,12 It might thus well be that the photochemistry and photophysics of trans-UA in aqueous solutions – and correspondingly under biological conditions in the skin – are quite different from trans-UA in organic solvents. From a sunscreen formulation point of view and to further our understanding of the photoactivity of UA, it is therefore of key interest to study trans-UA in both organic solvents and under aqueous conditions. Secondly, with such studies we target the intriguing wavelength dependence of the photoisomerization efficiency and intersystem crossing yield of trans-UA under aqueous conditions.2,13–17 This is still a subject of extensive discussions in the context of the photobiological and photoimmunological properties of UA, but clearly also has direct implications for sunscreen applications.
Combined with the results of our aforementioned gas-phase studies,9 the present studies provide unique insight into the spectroscopy and decay dynamics of electronically excited states of UA, how these can be tuned by substituents at judiciously chosen positions within the primary chromophore, and how they are affected by formulation conditions. As such, they break new ground for the development of novel nature-inspired UV filters based on UA.
Results and discussion
In the following, we first discuss the results of the photostability experiments. In combination with the results of theoretical studies these UV/vis, FT-IR, and 1H NMR experiments provide us with information on conformational distributions at room temperature in various solvents and allow us to determine the products that are formed upon UV irradiation. We subsequently discuss femtosecond (fs) TEAS experiments performed on UA and its derivatives in organic solvents, linking these to the photostability studies. Finally, we discuss the results of femtosecond TEAS experiments in two buffer solutions, considering what we have learned from the same studies in organic solvents. In the latter discussion we will also include results of nanosecond TEAS experiments that elucidate the contribution of relaxation pathways involving the triplet manifold.Photostability studies
UV/vis absorption spectroscopy
Fig. 2 shows UV/vis spectra of all samples dissolved in ethanol before and during UV irradiation. Two interesting observations can be made: firstly, for UA a blue-shift of the spectrum is observed while for the substituted compounds irradiation leads to a red-shift of the spectrum. Secondly, the spectra show clear isosbestic points indicating the presence of only two distinguishable species. In view of the conclusion of previous studies that irradiation of UA leads to a photochemical trans–cis isomerization, we assert that these two species are the trans and cis isomers of the studied compounds. Further support for this conclusion is found from: (1) UV/vis spectra measured for samples of the pure isomers (Fig. S2, ESI†), which confirm the spectral blue- and red-shifts observed in Fig. 2(a) and (b) (and Fig. S3–S5, ESI†); and (2) 1H NMR spectroscopy, where the significant chemical shifts of CC protons also provide a direct indication of the occurrence of isomerization during UV radiation (Fig. S6–S9, ESI†). We thus conclude that for all studied compounds trans–cis photoisomerization is responsible for the changes observed in the UV/vis spectrum upon irradiation.
 | ||
| Fig. 2 UV/vis absorption spectra of 5 × 10−5 M solutions in ethanol of (a) UA, (b) methyl urocanate, (c) N3-methyl methyl urocanate, and (d) N1-methyl methyl urocanate before and during irradiation at λmax for the indicated irradiation time given in seconds. Inserts display the change of absorption at each λmax (black dot curves) with the red percentage indicating the overall change in absorption. | ||
To determine the influence of polarity and hydrogen bonding, similar experiments have been performed for UA and its derivatives dissolved in dioxane and acetonitrile (see Fig. S3 and S4, ESI† respectively). Furthermore, the stability of UA in buffer solutions at pH 5.6 and pH 7.2 has been considered as well for which results are reported in Fig. S5 (ESI†). In agreement with the observations made for ethanol solutions, we find in the organic solvents consistently a red-shift of the absorption spectrum of the UA derivatives upon irradiation, while the spectrum of UA undergoes a blue-shift. Interestingly, however, the exception to the rule is formed by UA dissolved in dioxane for which a red-shift of ∼10 nm of λmax is observed as well as an absorption profile that is much more extended. Although in the first instance puzzling, this observation is in line with the stabilities of the various conformers in different solvents as reported in Table S1 (ESI†). From Table S1 (ESI†), it can be concluded that in all solvents, the N3H-TTC tautomer of UA is the most stable trans isomer. Upon irradiation, photoisomerization populates the same tautomer albeit in the N3H-TCT form (Fig. S1, ESI†). In dioxane, however, the most stable cis isomer is predicted to be N1H-TCT (Fig. S1, ESI†). Irradiation of UA in dioxane will thus initially lead to the cis isomer of the N3H tautomer but this product will undergo a further tautomerization reaction to the N1H-TCT conformer. The observed red-shift of the absorption spectrum is indeed in line with the difference in excitation energies predicted for the two tautomers.18,19 As can be concluded from Table S2 and the comparison of Fig. 2 and Fig. S3 (ESI†), a similar reasoning can be applied to methyl urocanate.
Inspection of Fig. 2 and Fig. S3, S4 (ESI†) leads to the conclusion that from a sunscreen point of view, N1-methyl methyl urocanate has the most favourable properties. Irradiation reduces the absorption between 17–20% and hardly shifts the absorption spectrum. For the other compounds much larger reductions and shifts are observed.
FT-IR spectroscopy
Similar photostability studies have been performed using FT-IR spectroscopy. As will be shown below, these studies confirm the conclusions drawn so far on the role of photochemical trans–cis isomerization. Their advantage is, however, that they also give access to conformational distributions, which are much harder – if not impossible – to obtain from UV/vis and NMR spectra. Fig. 3 shows FT-IR spectra of the four samples in ethanol before and during irradiation. Focusing on UA (Fig. 3(a)), this figure shows in the CO stretch region a main band at 1691 cm−1 and a shoulder at 1715 cm−1. From this observation it can directly be concluded that in ethanol UA is not predominantly present as a single conformer since in that case only a single band should have been visible. Upon irradiation the intensities of these bands, as well as that of the CC stretch at 1640 cm−1 decrease, while in the 1660–1680 cm−1 region a new, broader band appears. FT-IR spectra of the pure trans and cis isomers of UA (Fig. S10, ESI†) confirm that the changes in the intensities of bands and the observation of new bands are due to the generation of the cis isomer, and provide a consistent explanation for the broad band in the 1660–1680 cm−1 region as arising from the strong N⋯H–O hydrogen bond in the lowest-energy cis-UA conformer (N3H-TCT) (Table S1, ESI†).
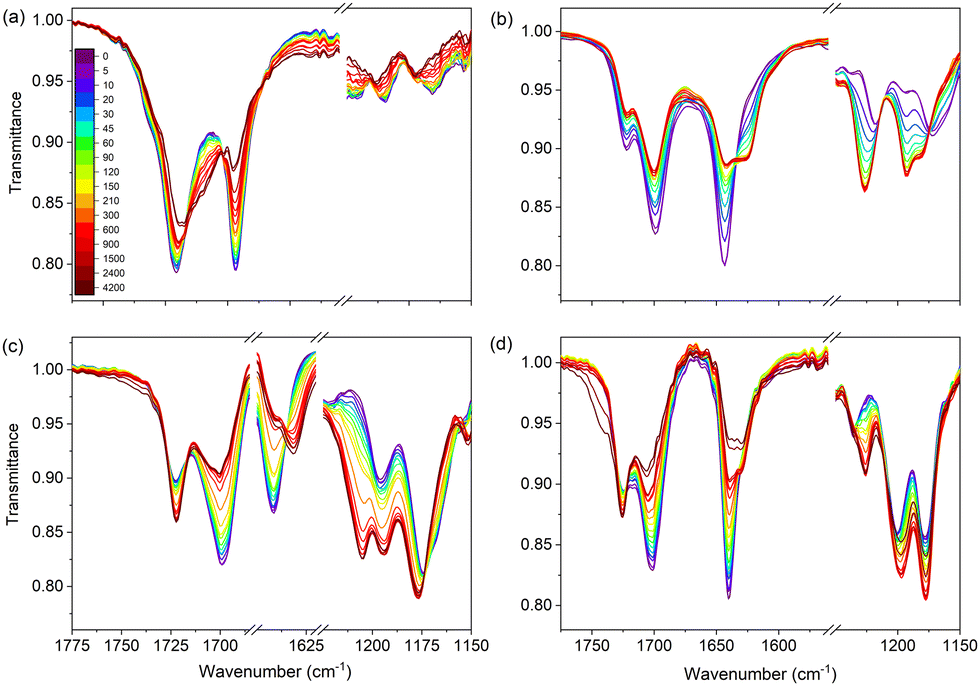 | ||
| Fig. 3 FT-IR spectra of 20 mM solutions in ethanol of (a) UA, (b) methyl urocanate, (c) N3-methyl methyl urocanate, and (d) N1-methyl methyl urocanate before and during irradiation at λmax for the indicated irradiation time given in seconds. | ||
The spectra of methyl urocanate (Fig. 3(b)), N3-methyl methyl urocanate (Fig. 3(c)) and N1-methyl methyl urocanate (Fig. 3(d)) display narrower and well-separated CO stretch bands indicating the presence of multiple conformers for these compounds as well. Upon irradiation these spectra undergo similar changes as observed for UA. Indeed, looking closer, distinct differences can be observed that can be traced back to differences in conformational distributions (vide infra). In the case of methyl urocanate further confirmation that photoisomerization is the underlying reason for the changes in the intensities of bands and the observation of new bands is provided by FT-IR spectra of the pure trans and cis isomers (Fig. S10, ESI†). The importance of taking conformational heterogeneity into account is further emphasized by the FT-IR spectra of methyl urocanate, N3-methyl methyl urocanate and N1-methyl methyl urocanate dissolved in dioxane and acetonitrile (Fig. S11 and S12, ESI† respectively; due to the limited solubility of UA in these solvents, such spectra could not be obtained for UA). In dioxane and acetonitrile all compounds initially show a single CO stretch band. But upon irradiation the spectrum of methyl urocanate evolves to display two CO bands, while the CO band in N3-methyl methyl urocanate and N1-methyl methyl urocanate merely undergoes a slight frequency shift and changes in intensity.
Quantitative information on conformational distributions has been obtained from a comparison of experimentally recorded and theoretically predicted IR absorption spectra with spectra constructed from the sum of the Boltzmann-weighted predicted IR spectra. To this purpose ground-state energies of trans conformers as reported in Tables S1–S4 (ESI†) were employed to make a comparison with the experimentally recorded IR absorption spectra before irradiation. IR spectra after irradiation were constructed using the ground-state energies of trans and cis conformers as reported in Tables S1–S4 (ESI†) in combination with a visual estimate of the overall trans to cis ratio. Comparison of such spectra with the experimental spectra as reported in Fig. S11–S13 (ESI†) shows an impressive agreement and gives credence to the accuracy of the calculations. Overall, it can thus be concluded that several conformers of UA and its derivatives are present in the studied samples, Tables S1–S4 (ESI†) indicating that their specific contribution is solvent dependent. Moreover, the excellent agreement between experiment and theory fully supports the conclusion that upon irradiation trans–cis photoisomerization occurs.
Transient absorption spectra in organic solvents
Fig. 4 displays the transient absorption spectra (TAS) of UA and its derivatives dissolved in ethanol. Inspection of these spectra shows that they are all characterized by three similar features. The first is a relatively strong positive band observed at ∼340 nm for UA, methyl urocanate, and N3-methyl methyl urocanate, while for N1-methyl methyl urocanate it appears at a slightly longer wavelength (∼355 nm). In view of the steady-state UV/vis absorption spectra (Fig. 2), this band is assigned as an excited-state absorption (ESA1) band, which over time undergoes a slight blue-shift. The second feature is a weak negative band appearing around 400 nm attributed to stimulated emission (SE), which appears to decay quite rapidly. The final feature is a weak, rapidly decaying ESA2 band around 500 nm that is slightly blue-shifted for N1-methyl methyl urocanate.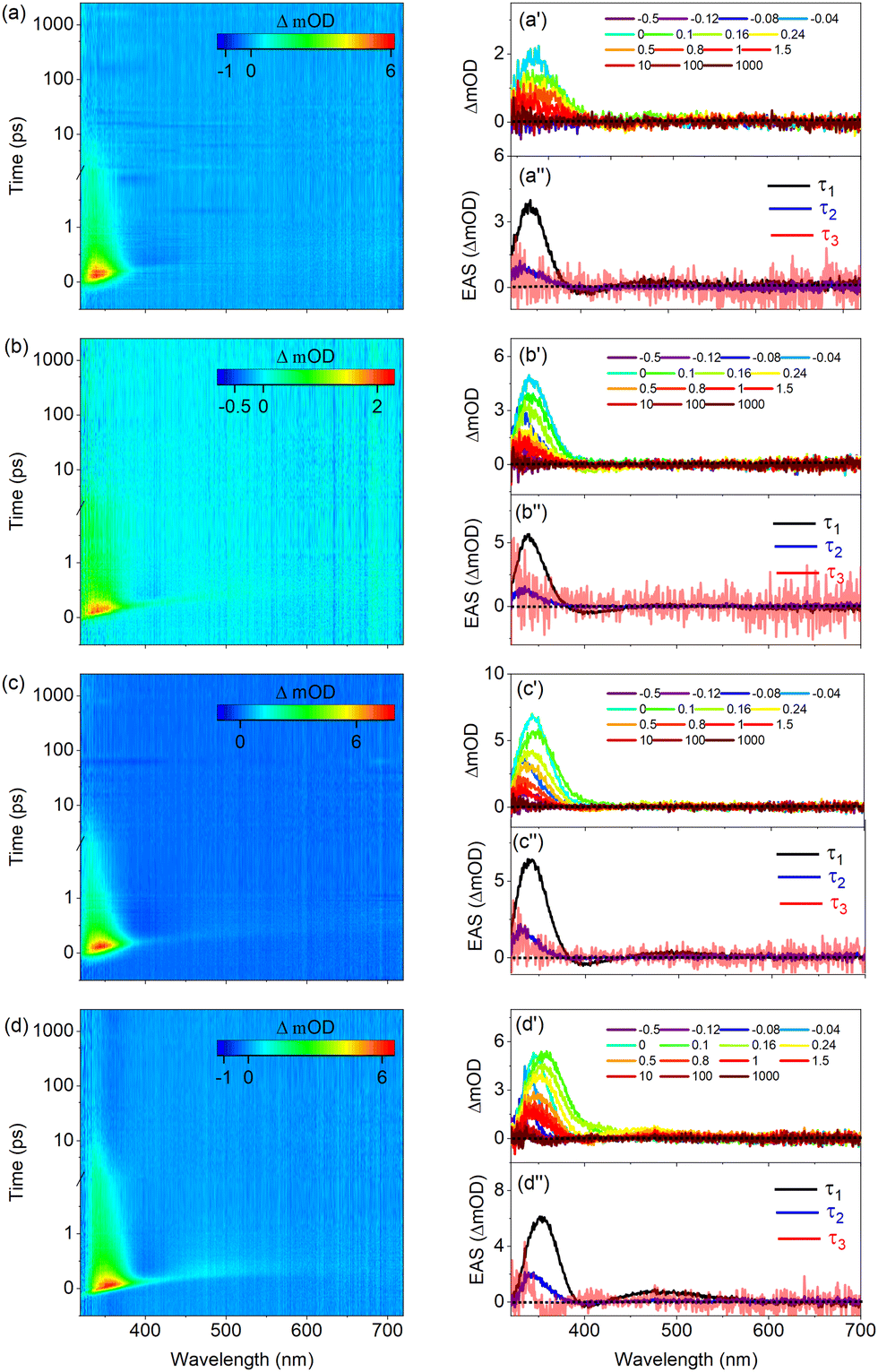 | ||
| Fig. 4 TAS displayed as false colour heat maps of (a) UA, (b) methyl urocanate, (c) N3-methyl methyl urocanate and (d) N1-methyl methyl urocanate in ethanol excited at 285, 287, 287 and 300 nm, respectively, with time delay plotted linearly until 2 ps and then as a logarithmic scale from 2 to 2500 ps. Panels (a′)–(d′) display lineouts at selected pump–probe delays, while panels (a′′)–(d′′) show the evolution associated difference spectra (EADS) resulting from a global analysis of the data with EADS3 multiplied by 5 for visual aid. | ||
To interpret the evolution of these spectra, it is useful to first consider the manifold of the lower-lying electronically excited singlet states. Our previous molecular beam studies9 demonstrated that under isolated gas-phase conditions, the spectroscopy and excited-state dynamics are determined by the nearly degenerate 1nπ* and 1ππ* states. Calculations of the vertical and adiabatic excitation energies of these two states under solvated conditions (Tables S5–S8, ESI†) show that upon solvation the 1ππ* state is lowered in energy while the energy of the 1nπ* state is slightly increased. As a result, for all compounds in polar solvents, the 1ππ* state -which is the state that is excited in our experiments- is both vertically as well as adiabatically the lowest excited singlet state. In such solvents internal conversion processes to a lower-lying electronically excited singlet state will therefore be absent. Tables S5–S8 (ESI†) show that in the more apolar solvent dioxane this excited-state ordering is maintained for the substituted compounds albeit that the energy gap between the 1ππ* and 1nπ* states is smaller. For UA in dioxane, however, the two states are nearly degenerate, and one may expect distinct excited-state dynamics as is indeed confirmed in our experiments (vide infra).
Global fits of the TAS shown in Fig. 4 using the software package Glotaran20 leads to the conclusion that the data can be well described in terms of three exponential decay components with lifetimes that are reported in Table 1. As regards the third component, it should be noted that this component is necessary since for the maximum pump–probe delay there is still a signal in the 300–350 nm region, but the uncertainty in fits in which its decay time is fitted precludes an unambiguous determination. We have therefore chosen to keep it fixed at an -with respect to the experiment- infinitely long value. Application of a sequential kinetic model then leads to evolution associated difference spectra (EADS) that are depicted in Fig. 4(a′′)–(d′′). The first EADS with a decay time τ1 is characterized by the two ESA bands and the SE band discussed previously. The presence of the SE band implies that the species that is excited is associated with the Franck–Condon S1(1ππ*) excited state. This state decays on a sub-ps time scale to a species for which the EADS shows a blue-shifted ESA1 band, and for which the SE and ESA2 bands are absent. We attribute this decay to the evolution out of the Franck–Condon region along an excited-state reaction coordinate that brings the molecule to geometries for which the transition moment to the electronic ground state is significantly reduced.
| Ethanol | Acetonitrile | Dioxane | |||||||
|---|---|---|---|---|---|---|---|---|---|
| τ 1 (fs) | τ 2 (ps) | τ 3 (ns) | τ 1 (fs) | τ 2 (ps) | τ 3 (ns) | τ 1 (fs) | τ 2 (ps) | τ 3 (ns) | |
| a τ 3 with ≫ ns indicates a decay time longer than the maximum pump–probe delay. | |||||||||
| Urocanic acid | 237 ± 40 | 3.23 ± 0.09 | ≫ | 456 ± 40 | 5.43 ± 0.06 | ≫ | 135 ± 40 | 3.84 ± 0.04 | ≫ |
| Methyl urocanate | 371 ± 40 | 5.43 ± 0.12 | ≫ | 467 ± 40 | 9.59 ± 0.04 | ≫ | 373 ± 40 | 5.44 ± 0.12 | ≫ |
| N 3-methyl methyl urocanate | 393 ± 40 | 3.63 ± 0.06 | ≫ | 439 ± 40 | 15.54 ± 0.09 | ≫ | 496 ± 40 | 4.10 ± 0.06 | ≫ |
| N 1-methyl methyl urocanate | 374 ± 40 | 4.94 ± 0.07 | ≫ | 520 ± 40 | 12.54 ± 0.07 | ≫ | 345 ± 40 | 5.42 ± 0.06 | ≫ |
Concurrently with an intramolecular geometry relaxation, solvent relaxation is also expected to occur.21 Both dynamic processes will thus be incorporated into τ1. Interestingly, Table 1 shows that for the methylated compounds, this evolution is about 50% slower than for bare UA irrespective of where methylation has taken place.
The second EADS with a decay time τ2 decays on a time scale of 4–5 ps into a final species whose EADS merely shows a very weak tail of a band that falls outside our probe range (i.e., <330 nm). Our photostability experiments have shown that all compounds are subject to a photochemical trans–cis isomerization process. Previous theoretical studies19,22 indicate that such a process is mediated by a conical intersection (CI) between the 1ππ* and ground state which is characterized by a nearly perpendicular geometry of the central CC double bond. Near, and at such a geometry, the oscillator strength of the S1 → S0 transition is near zero. Taken together leads to the conclusion that the second lifetime τ2 is associated with the traversing of the population through the CI to the ground state with concurrent vibrational cooling. Part of that population ends up in the cis isomer, giving rise to the very weak absorption observed at maximum pump probe delay time. One might be tempted to use Fig. 2 as a guide for assessing whether this band should be positive or negative. However, it should be realized that photoexcitation initially photoisomerizes the predominantly present N3H-TTC conformer into the N3H-TCC conformer. As Table S1 (ESI†) shows, this conformer is energetically much less stable than the N3H-TCT conformer, and will therefore be subject to conformational isomerization on the potential energy surface of the ground state. Such a process will take place on a time scale longer than nanoseconds. Fig. 2 thus reflects the single-bond conformationally equilibrated population distribution, which is different from the population distribution probed at the maximum pump probe delay time in the fs TEAS experiment.
An alternative explanation for the remaining absorption could be that it is associated with ESA from a long-lived electronically excited state, which in this case would be the lowest triplet state. In view of the previously claimed role of this triplet state in the decay dynamics of UA in buffer solutions, it is of interest to further investigate whether this might indeed also be the case for UA dissolved in organic solvents. To this purpose T1 absorption spectra have been calculated and are reported in Fig. 5. This figure shows an absorption band around 350 nm, but also a band with a similar oscillator strength around 575 nm. Although the signal-to-noise ratio of the spectra and the final EADS leave room for an alternative interpretation, considering that for none of the substituted compounds nor in other solvents (vide infra) indications are found for such a band, we conclude that in organic solvents it is unlikely that the triplet manifold is involved in the decay dynamics of UA and its derivatives.
 | ||
| Fig. 5 T1 absorption spectra of UA in various organic solutions as well as in two buffer solutions with pH 5.6 and 7.2 in which UA is present as H+UA− and UA−, respectively. Stick spectra have been calculated using TD-DFT at the wB97XD/cc-pVDZ/PCM level and have been broadened with a 0.222 eV Gaussian profile. | ||
To obtain further insight into the excited-state decay dynamics of UA and its derivatives, TEAS studies have also been performed in a polar, non-hydrogen bonding solvent (acetonitrile) and an apolar solvent (dioxane). Fig. S14 (ESI†) displays TAS as well as the results of a global analysis of these spectra for solutions of these compounds in acetonitrile. These spectra resemble what has been observed for ethanol solutions, that is, they are characterized by two ESA bands and a SE band, the latter being much more prominent than observed for ethanol solutions. In fact, blow-ups of the EADS with a decay time τ2 of the methylated compounds now show a small but distinguishable SE feature indicating that it is associated with an electronically-excited state species, thereby confirming our conclusions from the results on ethanol solutions that population is still in the excited state en route to traversing the CI. Moreover, there remains a long-lived induced absorption in the 350 nm region at long pump–probe delays. From Table 1 in which the results of a global analysis of these data are reported it can be concluded that the initial evolution out of the Franck–Condon region occurs almost twice as slow for UA while for the methylated compounds the process occurs 10–20% slower. Previously we have noticed that in ethanol, UA distinguishes itself from the methylated compounds by a significantly shorter value of τ1. In acetonitrile, on the other hand, the same value is observed throughout. This observation suggests that the hydrogen bonding properties of ethanol are responsible for the enhanced rate at which UA relaxes from the Franck–Condon region, and not an inertial factor resulting from methylation of the carboxylic acid group. As regards to τ2, which is associated with internal conversion to the electronic ground state, slower dynamics (by a factor of 2–3) are observed but with no clear trends between the various compounds.
Results of TEAS studies on UA and its derivatives in dioxane are displayed in Fig. 6. As far as methyl urocanate (Fig. 6(b)) and N3-methyl methyl urocanate (Fig. 6(c)) are concerned only minor differences are observed with respect to the dynamics in ethanol and acetonitrile. This also holds for N1-methyl methyl urocanate (Fig. 6(d)) although compared to methyl urocanate and N3-methyl methyl urocanate the ESA2 and SE bands are more prominent in the first EADS, while the second EADS shows a clearer SE band but also the ESA2 band. TAS spectra of UA in dioxane, on the other hand, show a clearly different behaviour. As visible in Fig. 6(a′), initially an intense ESA band around 350 nm is present. Over time this band undergoes a significant red-shift while rapidly losing intensity and bifurcates to give rise to an absorption spectrum with ESA bands at about 350 and 500 nm. Global fits of these spectra using three decay components show large deviations between observed and fitted spectra for delays up to 0.5 ps. However, attempts to fit the data with more components also did not lead to acceptable fits. We have therefore resorted to fitting the decay profiles up to 1 ps at single wavelengths leading to decay constants for the fast component that are displayed as the dotted trace in Fig. 6(a′′). This figure shows in the 320–420 nm range a constant decay time of about 110 fs, which then steadily increases to about 330 fs at 580 nm.
 | ||
| Fig. 6 TAS displayed as false colour heat maps of (a) UA, (b) methyl urocanate, (c) N3-methyl methyl urocanate and (d) N1-methyl methyl urocanate in dioxane excited at 286, 288, 289 and 307 nm, respectively, with time delay plotted linearly until 2 ps and then as a logarithmic scale from 2 to 2500 ps. Panels (a′)–(d′) display lineouts at selected pump–probe delays, while panels (a′′)–(d′′) show the EADS resulting from a global analysis of the data with EADS3 multiplied by 5 for visual aid. Panel a′′ shows moreover in the dotted trace (right y-axis) the decay constants of the initial fast decay determined at single wavelengths (see main text for details). | ||
Taking stock thus far, we can conclude that UA in dioxane is a special case in the sense that the 1ππ* and 1nπ* states are nearly degenerate while for the substituted compounds in dioxane as well as for both UA and substituted UA compounds dissolved in polar solvents, the 1ππ* state is S1 and well separated in energy from the S2(1nπ*) state. We therefore surmise that the initial spectral evolution that cannot be described by a single exponential is to a large extent associated with the equilibration of the population of the ‘bright’ 1ππ* state excited at time zero and the ‘dark’ 1nπ* state. Further support for such a hypothesis could be obtained from transient vibrational absorption spectroscopy (TVAS) studies but unfortunately the limited solubility of UA in dioxane prevents such studies.
Table 1 shows that for the substituted UAs very similar decay times are found as for ethanol solutions. The same holds for the internal conversion rate of UA back to the ground state (τ2). In view of the single-wavelength fits the initial EADS and the globally fitted decay time τ1 of UA should be considered with some caution. Nevertheless, the evolution from the Franck–Condon state towards the CI appears to be faster in UA than in the substituted compounds.
Transient electronic absorption spectroscopy in buffer solutions
In the following we will discuss the results of TEAS experiments on UA in pH 5.6 and 7.2 buffer solutions in which UA is present as the H+UA− zwitterion and UA− anion, respectively. Key to the interpretation of these experiments is the observation that the ordering of the electronically excited singlet states is entirely different from those in organic solvents. As Table S5 (ESI†) shows and in agreement with previous calculations,11,12 under such conditions the strongly allowed HOMO → LUMO 1ππ* state is neither vertically nor adiabatically S1 but is higher in energy than the two 1nπ* states associated with the lone pair orbitals of the carboxylate anion, and in the zwitterion even higher than the S3(1ππ*) state associated with excitation of antisymmetric combination of the π orbitals on the oxygen atoms to the LUMO. This implies that following excitation of the 1ππ* state in the zwitterion and the anion, decay channels involving internal conversion of the 1ππ* to the 1nπ* states are available which are not present in organic solvents.Fig. 7(a) displays TAS of H+UA− in a pH 5.6 buffer solution when excited at 266 nm, near the maximum of the absorption band. Line traces at specific delay times are depicted in Fig. 7(a′) while the results of a global fit of these spectra are shown in Fig. 7(a′′). Very similar to our observations in organic solvents, the heat maps show at early pump–probe delays an ESA1 band at 335 nm which over time mildly blue-shifts, and a SE feature at 400 nm that initially is overshadowed by ESA but for somewhat longer delays becomes more visible. Finally, a very weak ESA2 band is visible that initially starts at about 400 nm, gradually red-shifts to 450 nm, and decays within about 10 ps. For long delay times, an absorption feature remains at the blue part of the probe spectrum that will be discussed later.
 | ||
| Fig. 7 TAS displayed as false color heat maps of UA in pH 5.6 (H+UA−) ((a) and (b)) and 7.2 (UA−) ((c) and (d)) buffer solutions. In (a) and (c) excitation occurs at 266 nm, while in (b) and (d) an excitation wavelength of 306 nm is employed. Time delay is plotted linearly until 2 ps and then as a logarithmic scale from 2 to 1800 ps. Panels (a′)–(d′) display lineouts at selected pump–probe delays, while panels (a′′)–(d′′) show the EADS resulting from a global analysis of the data with EADS3 multiplied by 5 for visual aid. | ||
Global fits of these data lead to EADS depicted in Fig. 7(a′′) and decay times as given in Table 2. These EADS and decay constants mirror those observed for UA in organic solvents. We therefore conclude that also under the presently used conditions Franck–Condon excitation of the bright state of H+UA− is followed by a geometry relaxation process that brings the compound to a CI with the electronic ground state where the oscillator strength of the S1 ← S0 transition is significantly reduced, and from which the ground state is repopulated. Looking into more detail at the decay times, it appears that the initial evolution out of the Franck–Condon region and the passing through the CI occurs slower than in ethanol and comes close to the rates observed in acetonitrile.
| 266 nm excitation | 306 nm excitation | |||||
|---|---|---|---|---|---|---|
| τ 1 (fs) | τ 2 (ps) | τ 3 (ns) | τ 1 (fs) | τ 2 (ps) | τ 3 (ns) | |
| a τ 3 with ≫ ns indicate a decay time longer than the maximum pump–probe delay. | ||||||
| pH 5.6 (H+UA−) | 451 ± 40 | 7.49 ± 0.24 | ≫ | 797 ± 40 | 56.86 ± 0.10 | ≫ |
| pH 7.2 (UA−) | 531 ± 40 | 10.06 ± 0.08 | ≫ | 720 ± 40 | 38.08 ± 0.33 | ≫ |
We now compare these results with the results shown in Fig. 7(b), (b′), and (b′′) obtained for excitation in the red wing of the absorption spectrum at 306 nm (see Fig. S5(a), ESI†). Inspection of the TAS shows directly after excitation similar spectra as observed when exciting at 266 nm albeit that (i) the ESA1 and SE bands are shifted towards shorter wavelengths (around 330 and 390 nm, respectively), (ii) the SE band is more prominent and therefore (iii) the ESA2 band is difficult to discern. These features are well reflected in the differences between the EADS associated with τ1. What distinguishes the spectra at these two excitation wavelengths is most clearly visible in EADS2. For excitation at 306 nm this EADS no longer shows a prominent ESA band in the 320–350 nm region as observed when exciting at 266 nm and when UA dissolved in organic solvents, but a minor absorption feature at the blue edge of the probe spectrum. Moreover, although τ1 is clearly longer (by almost a factor of two), what is striking is that τ2 is increased by almost an order of magnitude (Table 2). Finally, at long delay times, a TAS remains that shows the onset of absorption around the very blue edge of the spectrum.
Before discussing how these differences should be interpreted, we first compare these results with results obtained for UA in a pH 7.2 buffer solution in which it is present as UA−. Fig. 7(c) displays to the TAS recorded following excitation at 266 nm together with line traces Fig. 7(c′) and results of a global fit of these spectra in Fig. 7(c′′). Although the spectral evolution in the 320–450 nm region resembles to a large extent what is observed for H+UA−, these spectra are notably different from the spectra discussed so far by the presence of the broad induced absorption band starting around 475 nm and covering the entire region to longer wavelengths. This band disappears upon the addition of KNO3, an electron scavenger, (see Fig. S15, ESI†) and we therefore assign it to absorption by solvated electrons. The power dependence of this band indicates that these solvated electrons arise from a multiphoton absorption process, which -together with the solvated electron- also generates the carboxyl radical of UA. It has been suggested that this radical has a main absorption band around 310 nm.23 Since this wavelength region falls outside our white light continuum, it is unfortunately not possible to confirm its presence.
Comparing TAS obtained with and without KNO3 leads to the conclusion that the 320–450 nm region is not affected by this ionization process. Inspection of the heat map and the time traces in this region shows – similar to our observations in organic solvents – at early pump–probe delays an ESA1 band at 335 nm which over time is slightly blue-shifted, and an ESA2 band that initially starts at about 435 nm and then gradually red-shifts, showing an overall decay in about 10 ps. Global fits of the data show a clear SE feature in the first EADS (Fig. 7(c′′)) that is confirmed in additional experiments under tighter focusing conditions which show a clear SE around 400 nm. For long delay times a similar absorption feature remains in the blue part of the probe spectrum as observed in the pH 5.6 buffer solution when excited at 266 nm; we return to discuss this below. Comparison of the EADS and decay times observed under the employed conditions lead to the conclusion that the decay dynamics of UA− and H+UA− upon excitation at 266 nm is quite similar and follow a large extent those observed in organic solvents.
Finally, Fig. 7(d), (d′), and (d′′) depict the TAS and their analysis obtained for excitation of UA− at 306 nm. Like excitation at 266 nm, a broad absorption band covering the wavelength region above 500 nm is visible. Drawing on quenching experiments, this can be attributed to solvated electrons. Inspection of the EADS and decay times that result from a global analysis leads to similar observations made for excitation of H+UA− at 306 nm, that is: (i) a prominent SE directly after excitation, followed by (ii) a dramatic loss of intensity of the ESA1 band, and (iii) a τ2 decay time that is significantly longer than observed for excitation at 266 nm.
Our experiments on H+UA− and UA− show that for both forms, excitation at 266 nm gives rise to excited-state dynamics that are qualitatively similar to the decay dynamics observed in organic solvents. Excitation at 306 nm, in contrast, is distinctly different. Given the sizeable SE feature observed, it can be concluded that the state that is excited is – analogous to all other experiments discussed so far – the strongly allowed 1ππ* electronically excited state (see below, for further discussion). Subsequently, this state evolves to give rise to an absorption spectrum that only shows a hint of an induced absorption band at the blue edge of our probe spectrum. This might indicate that either internal conversion to the ground state or to a lower-lying electronically excited state that does not show absorption bands in the spectral region covered by our white-light continuum. The first scenario would imply that internal conversion to the ground state occurs an order of magnitude faster than observed for excitation at 266 nm and for UA in organic solvents. It would also mean that a further relaxation process with a time constant of 40–50 ps takes place in the electronic ground state, but it is not directly clear which process this might be. Taking these considerations into account and bearing in mind that in buffer solutions the absorbing 1ππ* state is higher in energy than the two 1nπ* states, it seems more plausible that τ1 is associated with internal conversion to the two 1nπ* states from which the ground state is repopulated. This conclusion agrees with the conclusion drawn in ref. 17 that excitation at 308 nm populates a different electronically excited state than excitation at 266 nm. Our studies thus indicate that for excitation at 266 nm either the coupling to the 1nπ* states is reduced or that at those energies a decay path is available to direct the molecule to the (near-) CI discussed previously that is not present at lower excitation energies.
Deprotonation and intersystem crossing in buffer solutions
We finish this discussion by considering the results of the present study in the context of two aspects of previous studies. The first aspect concerns the conclusion that excitation of H+UA− leads to deprotonation on a timescale faster than 200 fs.4,15,16,24 The present experiments agree with such a conclusion and provide further insight. Firstly, Fig. 7 shows that for excitation at 266 and 306 nm, the TAS observed for H+UA− and UA− are very similar. In view of the different energies of the electronically excited states of H+UA− and UA−, this would not have been expected if the spectra would originate from these two different forms of UA. In this respect it is also noteworthy that the same applies for the similarity of the long-term spectra shown in Fig. 8 and Fig. S16–S17 (ESI†) that will be discussed below. Secondly, within the time resolution of our experiments (about 80 fs) we have not observed any indication of an ultrafast process distinguishing the excited-state dynamics of H+UA− from that of UA−. Taking these observations together, we conclude that deprotonation is faster than 80 fs and already occurs during the pump pulse. In the present experiments, we are thus effectively probing the excited-state dynamics of UA−. Deprotonation generates UA− with a different amount of internal energy than when excited directly, which might explain that the fitted lifetimes in the two buffers differ slightly (Table 2). A detailed inspection of the TAS and EADS obtained in a pH 5.6 buffer as well as additional quenching experiments shows that under such conditions solvated electrons are generated as well albeit at a much lower concentration. Moreover, the power dependence of these signals shows a markedly higher intensity dependence. This agrees with a priori expectations since more photons are needed to generate these electrons from H+UA− than from UA−. | ||
| Fig. 8 TAS obtained at −0.5 (black), 100 (blue), and 1000 (red) ps for buffer solutions of UA after adding 0.2 M KNO3 solution. (a) pH 5.6 (H+UA−) excited at 266 nm, and (b) at 306 nm; (c) pH 7.2 (UA−) excited at 266 nm and (d) at 306 nm. | ||
The second aspect to address is the conclusion from photoacoustic calorimetric studies17,25 that for 266 nm excitation of trans-UA in either its zwitterionic or anionic form, efficient intersystem crossing occurs that populates the lowest triplet state with a lifetime >10 ns, while for 306 nm excitation such a pathway is absent. Above we have noticed that the final TAS displays a very small ESA in the blue part of the probe spectrum. To further investigate this, we have performed additional TEAS experiments under carefully optimized conditions and with longer averaging times. The resultant time traces are shown in Fig. 8. These traces show for 266 nm excitation in both buffer solutions a small but distinct ESA band in the 350–450 nm region that does not decrease in intensity between 0.1 and 1.0 ns. Such a band is in good agreement with what is expected based on the theoretically predicted T1 absorption spectrum of UA− shown in Fig. 5 in which an absorption band is predicted at slightly longer wavelengths. Importantly, TAS obtained for excitation at 306 nm do not show such a band. TAS obtained with nanosecond laser excitation (Fig. S16 and S17, ESI†) confirm the above observations. They show TAS that nicely reproduce the 1 ns spectra obtained in the femtosecond TEAS experiments. From global fits of the TAS recorded for the pH 5.6 (pH 7.2) buffer solutions for longer time delays, the triplet lifetime is determined as 29 ± 2 ns (45 ± 2 ns). Such a lifetime is rather short for a spin-forbidden transition and suggests that it is mediated by a minimum energy seam of crossings (MESXs) between the lowest excited triplet state and the ground state similar to what is observed for cinnamates.26
We thus confirm that there is a large difference in the efficiency of intersystem crossing at 266 and 306 nm. However, at the same time it is also noticed that the low intensity of the 350–450 nm band suggests that the triplet quantum yield when exciting at 266 nm is not as high as suggested so far. In this context it is also important to notice that the EADS associated with τ1 and τ2 as well as the τ1 and τ2 values are quite similar under organic solvent conditions and in buffer solutions for excitation at 266 nm, indicating that the major decay pathway – which is associated with internal conversion to the ground state mediated by a (near-)CI – is the same. This points towards the low trans–cis photoisomerization quantum yield when exciting at 266 nm being due to the topology of the CI, which would dominantly repopulate the trans isomer. Quantum chemical calculations might in this respect be quite useful to elucidate these internal conversion dynamics.
Conclusions
The present studies have advanced considerably our understanding of (i) the photophysics and photochemistry of urocanic acid, (ii) how these properties are affected by methyl substitutions at judiciously chosen sites, and (iii) how they depend on a specific solvent environment. Comparison with the gas-phase studies reported in the accompanying article has shown that solute–solvent interactions have far-reaching consequences as they lead to a reordering of electronically excited states such that in organic solvents the strongly allowed HOMO → LUMO 1ππ* state is as a rule the lowest excited singlet state determining the photo dynamics occurring upon excitation. In buffer solutions, on the other hand, the lower-lying electronically excited states are furnished by two 1nπ* states with the states responsible for photon absorption found only at higher excitation energies.These changes manifest themselves prominently in our femtosecond time-resolved studies. From these studies it is concluded that in organic solvents UA and its derivatives all display in essence the same excited-state dynamics, albeit that specific decay rates depend to some extent on the studied compound and solvent. Excitation to the Franck–Condon region of the absorbing state is followed by a structural relaxation that brings the molecule to a region of the potential energy surface where the absence of stimulated emission indicates a significantly reduced dipole transition moment to the ground state. We have argued that this region is associated with a (near-)conical intersection with the ground state thereby explaining the short ps excited-state lifetime, and accounting for the trans–cis photoisomerization observed upon steady-state irradiation. The only exception to the rule is UA dissolved in dioxane. Here, ultrafast changes in the transient absorption spectrum are observed directly after excitation that we have tentatively assigned as due to the near-degeneracy of the 1nπ* and 1ππ* states leading to an equilibration of the population of the initially populated 1ππ* state with that of the 1nπ* state. However, afterwards, the same relaxation pathway is observed as in other solvents.
Femtosecond time-resolved studies in buffer solutions, in which UA is present in its zwitterionic or anionic form, have revealed an intricate set of relaxation pathways that show a distinct excitation energy dependence. Excitation near the absorption maximum leads, at first sight, to decay dynamics that mimic those observed in organic solvents, irrespective of whether UA is excited in its zwitterionic or an ionic form. The latter observation agrees with previous conclusions that excitation of the zwitterionic form leads to very fast deprotonation, the present experiments leading to an upper limit of this process as being faster than 80 fs. A careful inspection of the spectra observed for longer probe delays reveals a small but distinguishable induced absorption band which, based on quantum chemical calculations, can be assigned to absorption from T1. Nanosecond transient absorption experiments confirm these observations and allow us to determine its lifetime as being in the tens of nanoseconds. Although intersystem crossing thus clearly is one of the photon energy dissipation pathways, the overall dynamics appear to be dominated by the same pathway that is at work in organic solvents. Excitation on the red edge of the absorption band, on the other hand, gives rise to clearly different dynamics. In this case, internal conversion to the lower-lying 1nπ* states dominates, with no indications of intersystem crossing playing a role. These states subsequently repopulate the ground state.
Photostability studies using UV/vis and IR absorption spectroscopy to monitor changes under the influence of radiation have shown that all compounds are susceptible to photodegradation. Comparison with spectra obtained for cis isomers as well as with theoretically predicted UV/vis and IR absorptions spectra have shown that in all compounds and irrespective of the solvent or buffer that is used, photoisomerization is predominantly responsible for the observed changes. Apart from affecting the electronically-excited state manifold, solute–solvent interactions also influence the stability of individual conformers, leading to different distributions over low-energy conformers in different solvents. Comparison of measured IR absorption spectra with spectra predicted based on the stabilities and spectra of individual conformers provide excellent agreement and allowed for a detailed determination of the conformational composition of samples before and after irradiation.
The studies performed in buffer solutions are key to furthering our understanding of the processes that occur in the different layers of the epidermis. From a sunscreen point of view, such conditions are not optimal since over a large part of the absorption band the triplet manifold is involved in the decay dynamics. Formulations in which organic solvents are employed appear to be more suited to this purpose as there is no indication of intersystem crossing, and fast internal conversion to the ground state occurs. Overall, it has been found that N1-methylation leads to less photodegradation than observed for N3 compounds. Since such a substitution is also expected to reduce the immunosuppressive effects of bare cis-UA, these compounds would provide a promising scaffold for their further development as safe and efficient sunscreen filters. Such studies are presently being pursued.
Author contributions
H. S. synthesized, purified and characterized substituted UA derivatives. J. F. and W. J. B. performed and analysed the photostability studies and nanosecond transient absorption experiments. J. W., J. G., and V. G. S. acquired the femtosecond time-resolved spectroscopic data and analysed them together with W. J. B., J. F. and W. J. B. performed the quantum chemical calculations. J. F. and W. J. B. conceived the study. W. J. B. and V. G. S. supervised the work. All authors contributed to writing the manuscript.Data availability
Data for this article, including Fig. 2–8 as well as Fig. S2–S17 (ESI†), are available at Zenodo at https://doi.org/10.5281/zenodo.13338188.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the grant agreement no. 828753. Jiayun Fan acknowledges a doctoral fellowship from the China Scholarship Council (no. 201808440365). The authors would like to acknowledge the University of Warwick Research Technology Platform, Warwick Centre for Ultrafast Spectroscopy, for use of the Transient Absorption Spectrometer in the research.Notes and references
- F. F. de Olivarius, H. C. Wulf, J. Crosby and M. Norval, Photodermatol., Photoimmunol. Photomed., 1996, 12, 95–99 CrossRef PubMed.
- H. Morrison, D. Avnir, C. Bernasconi and G. Fagan, Photochem. Photobiol., 1980, 32, 711–714 CrossRef.
- H. Morrison, D. Avnir and T. Zarrella, J. Chromatogr. B: Biomed. Sci. Appl., 1980, 183, 83–86 CrossRef.
- J. D. Simon, Acc. Chem. Res., 2000, 33, 307–313 CrossRef CAS.
- T. Mohammad, H. Morrison and H. HogenEsch, Photochem. Photobiol., 1999, 69, 115–135 CrossRef CAS.
- N. K. Gibbs, J. Tye and M. Norval, Photochem. Photobiol. Sci., 2008, 7, 655–667 CrossRef CAS PubMed.
- B. Rioux, L. M. M. Mouterde, J. Alarcan, T. T. Abiola, M. J. A. Vink, J. M. Woolley, A. A. M. Peru, M. M. Mention, F. Brunissen, G. Berden, J. Oomens, A. Braeuning, V. G. Stavros and F. Allais, Chem. Sci., 2023, 14, 13962–13978 RSC.
- J. Soilán, L. López-Cóndor, B. Peñín, J. Aguilera, M. V. de Gálvez, D. Sampedro and R. Losantos, Photochem, 2024, 4, 128–137 CrossRef.
- J. Fan, A. K. Lemmens, H. Sanders, M. Hilbers, W. Roeterdink and W. J. Buma, Phys. Chem. Chem. Phys., 2024 10.1039/d4cp02087a.
- W. L. Ryan and D. H. Levy, J. Am. Chem. Soc., 2001, 123, 961–966 CrossRef CAS PubMed.
- O. Dmitrenko, W. Reischl, R. D. Bach and J. Spanget-Larsen, J. Phys. Chem. A, 2004, 108, 5662–5669 CrossRef CAS.
- J. Danielsson, J. Uličný and A. Laaksonen, J. Am. Chem. Soc., 2001, 123, 9817–9821 CrossRef PubMed.
- P. McLoone, E. Simics, A. Barton, M. Norval and N. K. Gibbs, J. Invest. Dermatol., 2005, 124, 1071–1074 CrossRef PubMed.
- H. Morrison, C. Bernasconi and G. Pandey, Photochem. Photobiol., 1984, 40, 549–550 CrossRef PubMed.
- B. L. Li, K. M. Hanson and J. D. Simon, J. Phys. Chem. A, 1997, 101, 969–972 CrossRef.
- K. M. Hanson, B. L. Li and J. D. Simon, J. Am. Chem. Soc., 1997, 119, 2715–2721 CrossRef.
- K. M. Hanson and J. D. Simon, Photochem. Photobiol., 1998, 67, 538–540 CrossRef.
- M. Barbatti, Phys. Chem. Chem. Phys., 2011, 13, 4686–4692 RSC.
- D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. B, 2014, 118, 976–985 CrossRef CAS.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef.
- L. Zhao, P. W. Zhou and G. J. Zhao, Chem. Phys., 2016, 145, 044316 Search PubMed.
- J. Brookman, J. N. Chacón and R. S. Sinclair, Photochem. Photobiol. Sci., 2002, 1, 327–332 CrossRef PubMed.
- M. K. Shukla and P. C. Mishra, Spectrochim. Acta, Part A, 1995, 51, 831–838 CrossRef.
- N. Haralampus-Grynaviski, C. Ransom, T. Ye, M. Rôżanowska, M. Wrona, T. Sarna and J. D. Simon, J. Am. Chem. Soc., 2002, 124, 3461–3468 CrossRef PubMed.
- K. Yamazaki, Y. Miyazaki, Y. Harabuchi, T. Taketsugu, S. Maeda, Y. Inokuchi, S. Kinoshita, M. Sumida, Y. Onitsuka, H. Kohguchi, M. Ehara and T. Ebata, J. Phys. Chem. Lett., 2016, 7, 4001–4007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and theoretical procedures, synthesis and characterization, additional figures photostability studies including NMR spectra and analysis population ratios, femtosecond transient absorption spectra in acetonitrile, calculations of vertical and adiabatic excitation energies, electron scavenging studies, nanosecond transient absorption spectra in buffer solutions. See DOI: https://doi.org/10.1039/d4cp02088j |
| This journal is © the Owner Societies 2024 |