
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcentration-dependent aggregation of methylene blue acting as a photoredox catalyst†
Benjamin J.
Thompson
ab,
Anshu
Kumar
ac and
Vanessa M.
Huxter
 *ac
*ac
aDepartment of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, USA. E-mail: vhuxter@arizona.edu
bDepartment of Optical Sciences, University of Arizona, Tucson, Arizona 85721, USA
cDepartment of Physics, University of Arizona, Tucson, Arizona 85721, USA
First published on 8th July 2024
Abstract
Hydroxylation reactions are important in biological processes and synthetic schemes. Many challenging hydroxylation reactions have been achieved using photoredox catalytic methods. For the oxidative hydroxylation of arylboronic acids, methylene blue has been used successfully as a photoredox catalyst to produce phenyl groups. Here we use broadband transient absorption spectroscopy to determine the mechanism of the photoredox catalytic reaction of methylene blue with phenylboronic acid in the presence of N,N-diisopropylethylamine. Our results show that the reaction proceeds through the triplet state of methylene blue in the presence of oxygen, generating superoxide radical anions. In addition, we observe dimerization of the methylene blue at typical catalytic loadings. As these dimers do not participate in the reaction, increasing the concentration of methylene blue is potentially detrimental to the overall yield.
Introduction
Methylene blue, a cationic phenothiazine dye, has been widely used in medial and biological applications, most prominently as a pathology stain and a treatment for methemoglobinemia.1 Its widespread use is due in part to its ability to easily form aggregates with itself,2,3 with proteins,4 or on negatively charged surfaces.5 More recently it has attracted interest as a photoredox catalyst due to its availability, low cost, low toxicity, and extremely high triplet quantum yield.6–9 As a photoredox catalyst, methylene blue generally acts as a triplet sensitizer,10,11 generating singlet oxygen or the superoxide radical anion of oxygen that can initiate oxidative hydroxylation reactions,12,13 however, this pathway is only accessible from the monomer form.Methylene blue is known to readily form H-type dimers in water and acetonitrile solutions which are commonly used for photoredox catalysed oxidative hydroxylation reactions. The formation of co-facial H-dimers results in a hypsochromic or blue shift in the absorption spectrum. In an H-dimer, the higher energy excitonic state at 608 nm (S+) is optically bright while the lower one at 688 nm (S−) is dark, leading to a blue-shifted absorption band relative to the monomer. The lowest energy optical transition of the monomer form of methylene blue appears at 658 nm, while the optically accessible transition of the H-dimer appears at 608 nm.14,15 These features can be seen in steady-state absorption measurements of methylene blue, increasing with higher concentrations. At high concentrations, such as those used in photoredox catalysis, a mixture of dimers and monomers is present in solution.
The previously proposed mechanism16 for the oxidative hydroxylation of phenylboronic acid using methylene blue as a photoredox catalyst involves the photoexcitation of methylene blue to a singlet excited state followed by intersystem crossing to an excited triplet state.8 This mechanism is depicted in Fig. 1. While in the triplet excited state, methylene blue can be quenched by an amine, here N,N-diisopropylethylamine (DIPEA), to generate the semi-reduced form of methylene blue and an amine radical cation.17 Following this reduction, both the alpha-amino radical generated from DIPEA and the semi-reduced methylene blue can transfer an electron to oxygen in solution generating a superoxide radical anion. Generation of the superoxide from either the alpha-amino radical or the semi-reduced methylene blue has been previously demonstrated.12,16,18,19 The superoxide then reacts with the phenylboronic acid to generate phenol. The semi-reduced form of methylene blue is converted to leuco-methylene blue through a second electron transfer event along with a proton transfer. Leuco-methylene blue is easily oxidized by exposure to oxygen in solution, regenerating the cationic form of methylene blue and completing the catalytic cycle. This mechanism relies on the lifetime of the triplet excited state being sufficiently long that methylene blue can encounter DIPEA by diffusion.20 In the dimer form, the excited state lifetime of methylene blue has been reported to be on the order of tens of picoseconds which is significantly shorter than the excited state lifetime of the monomer.21 However, this has not been measured in the solvent mixture used in this paper nor as part of a photoredox catalysis process. The rapid return to the ground state in the dimer, significantly reduces the probability of interaction with the amine while in the excited state. The short lifetime of the excited singlet state also limits the generation of triplet excited states, resulting in a much lower triplet quantum yield in the dimer.
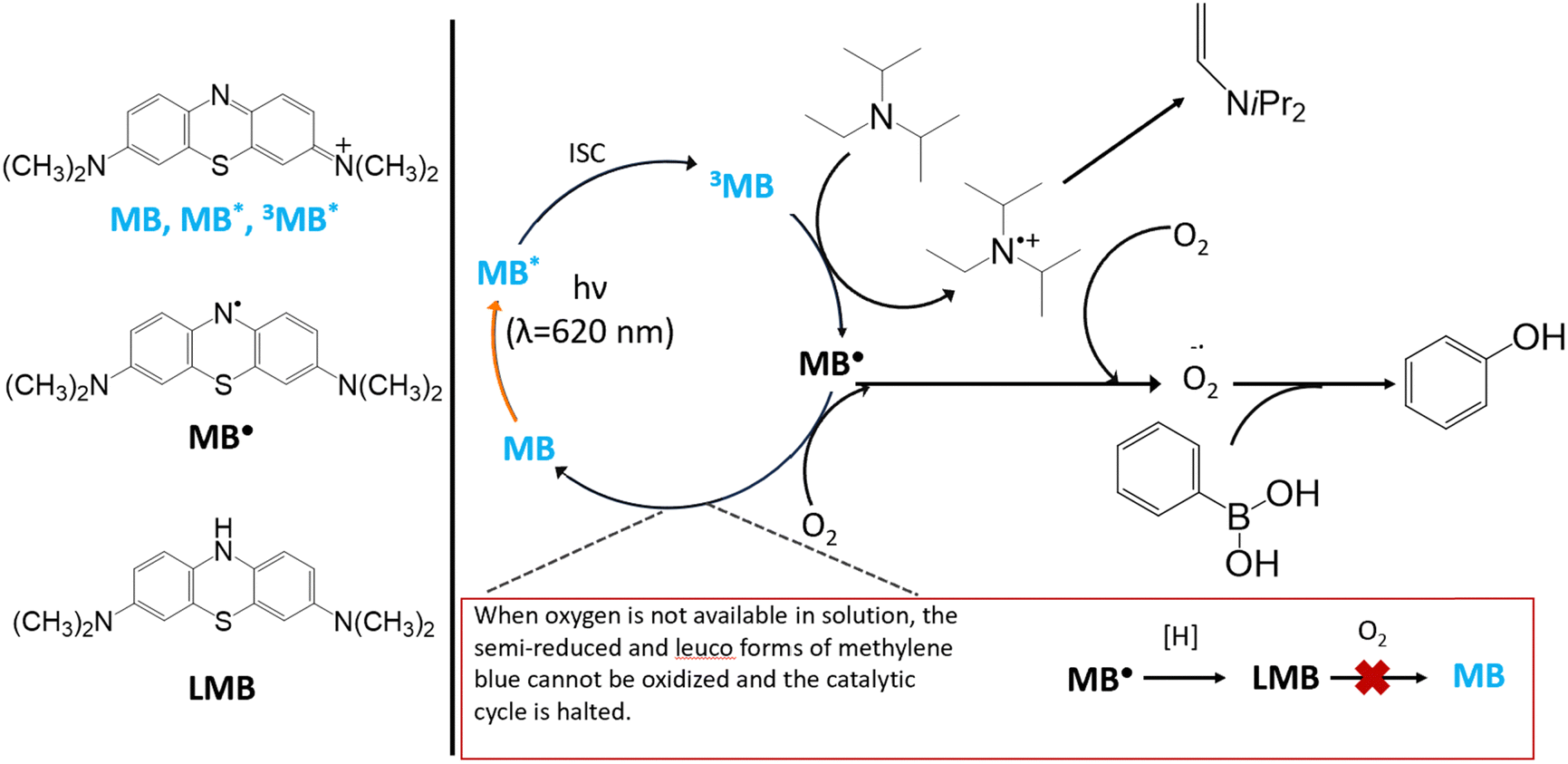 | ||
| Fig. 1 The photoredox catalytic cycle begins with methylene blue as a radical cation with chloride counter-ion (MB) undergoing photoexcitation to the singlet excited state of the monomer. It then undergoes intersystem crossing to the triplet state. This triplet state (3MB) is reduced by DIPEA to form semi-reduced methylene blue (MB˙) and an alpha-aminoalkyl radical. Semi-reduced methylene blue can undergo proton transfer to produce leuco-methylene blue (LMB) or it can reduce oxygen to form a superoxide radical anion. In the presence of oxygen, LMB is quickly oxidized, re-forming MB and closing the catalytic cycle. The alpha-aminoalkyl radical can also react with oxygen to form a superoxide radical anion and an alpha-aminoperoxyl radical. The oxygen superoxide radical anions produced in this process can then react with phenylboronic acid to generate phenol. | ||
The energy of the dimer triplet state is higher than the monomer, raising the triplet reduction potential and inhibiting its ability to act as a photoredox catalyst. Methylene blue catalyst loadings typically used for photoredox reactions are sufficiently high to generate a mixture of dimers and monomers in solution. In water, for a concentration of 0.7 mM, [MB] = 0.248 mM and [MB2] = 0.236 mM.14,21,22 Dimerization also occurs in in solvents with lower dielectric constants.23 As a result, dimerization may limit the overall efficiency of the reaction. While concentration-dependent dimerization is well-known in molecular spectroscopy, it has not been similarly explored in the organic photoredox catalysis literature. A common approach to attempt to increase reaction yield is to raise the concentration of the photocatalyst. In this case, increasing the concentration of the catalyst would have the opposite effect, instead sequestering the catalytically active monomer in a non-interacting dimer species.
In this paper, we investigate the role of the monomer and dimer forms of methylene blue in the photoredox catalytic oxidative hydroxylation of phenylboronic acid. Using steady-state linear absorption and broadband transient absorption (TA) measurements, the excited state dynamics of methylene blue acting as a photoredox catalyst are measured as a function of concentration. Using ultrafast time-resolved spectroscopy, we recover timescales associated with triplet states and excited state lifetimes of both the monomer and the dimer. This information allows for a greater understanding of mechanism of methylene blue as a photoredox catalyst.
Experimental methods
Steady-state spectroscopy
Methylene blue cation (structure shown in Fig. 1) with chloride counter ion, spectrograde (Uvasol) acetonitrile, DIPEA, and phenylboronic acid were used as purchased from Millipore Sigma. For the inert environment light exposure steady-state absorption measurements shown in Fig. 2, methylene blue was dissolved in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v acetonitrile to deionized water with DIPEA and phenylboronic acid. This sample composition corresponds to previously published work on this photoredox reaction system.16 The sample was transferred to a vacuum tube and connected to a nitrogen environment Schlenk line. Four cycles of freeze–pump–thaw were performed to remove oxygen. A syringe was inserted into the degassed Schlenk tube, filled with nitrogen, and used to draw the degassed solution for transfer into a Schlenk line equipped with either a quartz cuvette (Fig. 2) or a 1 cm quartz cuvette (Fig. S3, see ESI†). The changes in the absorption spectrum follow exposure to 4 mW, 100 fs, 1 kHz laser pulses at 620 nm generated by a Libra (Coherent) Ti:Sapphire regenerative amplifier and a TOPAS (Coherent) optical parametric amplifier. Steady-state absorption and fluorescence spectra were measured using a UV/Vis spectrometer (Agilent, Cary 100) and a fluorometer (Agilent, Cary Eclipse). Representative absorption and fluorescence spectra for methylene blue (0.7 mM) in a 4:1 v:v acetonitrile to deionized water solution are shown in Fig. S1 (ESI†). All measurements were made at room temperature.
:1 v:v acetonitrile to deionized water with DIPEA and phenylboronic acid. This sample composition corresponds to previously published work on this photoredox reaction system.16 The sample was transferred to a vacuum tube and connected to a nitrogen environment Schlenk line. Four cycles of freeze–pump–thaw were performed to remove oxygen. A syringe was inserted into the degassed Schlenk tube, filled with nitrogen, and used to draw the degassed solution for transfer into a Schlenk line equipped with either a quartz cuvette (Fig. 2) or a 1 cm quartz cuvette (Fig. S3, see ESI†). The changes in the absorption spectrum follow exposure to 4 mW, 100 fs, 1 kHz laser pulses at 620 nm generated by a Libra (Coherent) Ti:Sapphire regenerative amplifier and a TOPAS (Coherent) optical parametric amplifier. Steady-state absorption and fluorescence spectra were measured using a UV/Vis spectrometer (Agilent, Cary 100) and a fluorometer (Agilent, Cary Eclipse). Representative absorption and fluorescence spectra for methylene blue (0.7 mM) in a 4:1 v:v acetonitrile to deionized water solution are shown in Fig. S1 (ESI†). All measurements were made at room temperature.
 | ||
| Fig. 2 Absorption traces of the high concentration (0.7 mM) methylene blue in in 4:1 v:v acetonitrile to deionized water in the presence of DIPEA (0.3 M) and phenylboronic acid (0.06 M) with increasing light exposure time. The inset shows the ratio of absorption at 658 nm (monomer) to 608 nm (dimer). To the left of the legend, images of the solution show the visibly changing color. | ||
For the TA measurements, samples were prepared at two different concentrations of methylene blue in a 4:1 v:v acetonitrile to deionized water solution. The higher concentration (0.7 mM) was chosen to correspond to that previously used for photoredox catalysis.16 The lower concentration (0.034 mM) is twenty-fold lower than the higher concentration. This concentration was selected to generate a solution with little to no appreciable dimer concentration.21 Phenylboronic acid (0.06 M) and DIPEA (0.3 M) was added to the high and low concentration methylene blue solutions for the TA measurements under photoredox catalytic conditions at the same ratios as had been previously used.16 All samples were filtered using a 0.2 micron Teflon syringe filter. The samples were then transferred to a quartz flow cell and pumped using a peristaltic pump. The sample was measured in a flow cell to minimize heating effects and to use short pathlengths due to the optical density of the high concentration sample. For the high concentration samples, a pathlength of 100 μm was used to generate an OD of 0.5. For the low concentration samples, an OD of 0.3 was produced with a pathlength of 1 mm. All TA measurements were done at room temperature and in air. Steady-state UV/Vis absorption and fluorescence spectra were recorded before and after TA experiments to ensure that no sample breakdown had occurred.
The laser pulses for the TA measurements were generated using a homebuilt white light continuum generated by self-focusing in a argon filled tube.24–26 The resulting white light was compressed using paired chirped mirrors (Laser Quantum, DCM9) and additional chirp compensation was applied to the pump pulse using an acousto-optic programmable dispersive filter (Fastlight, Dazzler). A beam splitter was used to separate the white light into a pump pulse and a probe pulse. The probe pulse spanned 400 nm to 720 nm. The pump pulse was produced by passing the remaining white light through the Dazzler, and its spectrum as a result was narrower, 50 nm full width half maximum with a center wavelength of 620 nm. The pump center wavelength was chosen to excite both the dimer and monomer near the isosbestic point in the absorption spectrum between the two populations.14 Both the pump and the probe spectrum are shown in Fig. S1 (see ESI†). The Dazzler acted as an optical chopper, generating pump on and pump off cycles for the TA measurement. A translation stage (Newport, DL225) was used to produce the population time delays between the pump and the probe. The average power used at the sample position for the TA experiments was 25 μW with a beam spot size of approximately 130 μm. The temporal width of the pulses was measured by frequency-resolved optical gating using carbon disulfide as well as cross-correlation by doubling using mixing in a BBO crystal. Following chirp correction, cross-correlation measurements performed using a BBO crystal showed that the pump–pulse limited time resolution was approximately 35 fs as shown in Fig. S11 (see ESI†).
Results
Oxygen-free steady-state spectra
The proposed mechanism for the oxidative hydroxylation of phenylboronic acid requires oxygen to regenerate the methylene blue catalyst and to form a superoxide radical anion.12,16 Both the semi-reduced form of methylene blue and the superoxide can react with the phenylboronic acid to generate phenol. The semi-reduced and the leuco forms of methylene blue participate in the catalytic cycle where the semi-reduced is a reactive intermediate and the leuco form regenerates the cationic form of methylene blue. However, this has not been directly observed. Both the semi-reduced and the leuco forms of methylene blue absorb in the near UV, making them transparent in the visible.To directly observe the effect of oxygen on methylene blue in the presence of DIPEA and phenylboronic acid, a series of steady-state absorption measurements were performed in an oxygen-free inert environment with varying light exposure times. As shown in Fig. 2 we observed that absorption of the cationic form of methylene blue, whose peak is at 658 nm for the monomer and 608 nm for the dimer, decreased with increasing exposure to light. This agrees with the proposed reaction mechanism. In the absence of oxygen, the cationic form of methylene blue cannot be regenerated via oxidization from the leuco form. In this case, the catalytic cycle stops at the leuco form and the monomer absorption of the cationic form of methylene blue decreases until it is nearly entirely consumed.
The loss of absorption also reveals a relative change in the methylene blue monomer and dimer populations with increasing light exposure times. The initial absorption of methylene blue before the light exposure does not have an obvious feature associated with the dimer. Even though the absorption of the monomer dominates the spectrum, dimers are still present. The linewidth of the absorption of the monomer is broad enough that it is overlapped with the dimer absorption so as the monomer is consumed, the absorption decreases at both wavelengths. However as shown in the inset to Fig. 2, the absorption at 658 nm decreases more rapidly than the absorption at 608 nm. Although, it might be expected that the dimer would dissociate to compensate for the consumption of the monomer in the reaction, these measurements indicate that the dimer is relatively stable. While some dimer likely dissociates as the monomer is consumed, we can still observe the change in the relative ratio of the absorption at 658 nm and 608 nm and so can conclude that the equilibrium constant favors the dimer. This suggests that there is an underlying dimer population that is not consumed in the catalytic cycle. At the start of the experiment, the ratio of absorption at 658 nm to 608 nm is ∼2.3 but this ratio decreases to ∼1.9 after 240 minutes of laser exposure as shown in the inset to Fig. 2. Additionally, loss of absorption associated with the phenylboronic acid being consumed can be observed in Fig. 2 at 280 nm along with changes in absorption between 420 nm and 450 nm, a region associated with absorption of the semi-reduced and leuco methylene blue forms. These have been previously observed as transients in flash photolysis experiments.27–31 At the end of the experiment shown in Fig. 2, when the sample was vented to air, the blue color and absorption spectrum of the methylene blue cation returned as the leuco-methylene blue was oxidized.
One of the methods used to try to improve the yield of a photocatalytic reaction is to increase the amount of catalyst present. This is reasonable in the case where the reaction is limited by the catalyst concentration. For methylene blue, dimers are present at concentrations well below the reported catalytic loading of ∼0.7 mM.16 This occurs even in cases where the dimer absorption is small compared to that of the monomer. If the catalytic reaction proceeds through the monomer, methylene blue present in dimer form will not directly contribute to the reaction. Increasing the concentration of methylene blue will result in a greater percentage of catalysts in dimer aggregates and will likely not produce the expected increase in yield.
Transient absorption studies
To track the dynamics of methylene blue acting as a photocatalyst for the oxidative hydroxylation of phenylboronic acid, transient absorption measurements were performed. These measurements were conducted using two different concentrations of methylene blue to observe the effect of aggregation. One to match the concentration previously used for the reaction (∼0.7 mM)16 and ∼1/20th of this concentration (35 μM). The 20-fold reduction of the methylene blue was used as a comparison since at this concentration, it is expected that the dimer is not prevalent in solution.21TA spectra were obtained for methylene blue alone in solution, methylene blue with DIPEA, methylene blue with phenylboronic acid, and methylene blue in the full reaction mixture containing both DIPEA and phenylboronic acid. TA for methylene blue with only DIPEA or phenylboronic acid was collected as a control and is presented in Fig. S4–S9 (see ESI†). The presence of the phenylboronic acid alone does not change the dynamics. However, DIPEA quenches the excited state, reducing the excited state lifetime from 862 ps to 424 ps.29
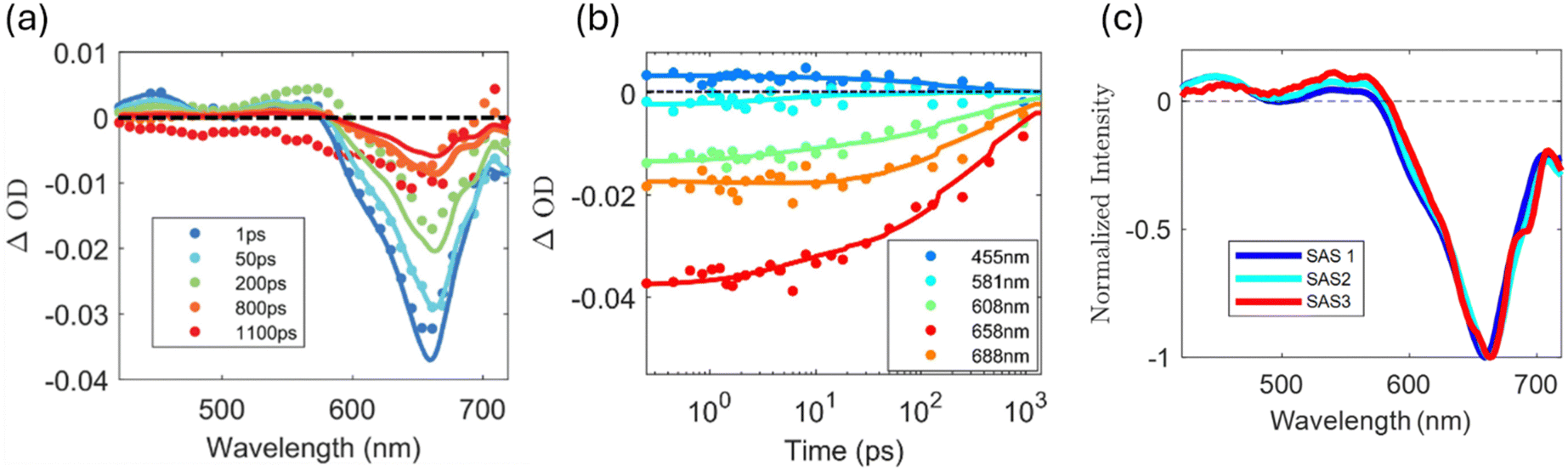
Fig. 3(a) presents TA data for methylene blue in water-acetonitrile solution at the higher concentration. The points are data and the lines are fits generated from a global target analysis32 model developed using the KiMoPack python package.33 We observe a strong, broad ground state bleach (GSB) centred at 665 nm. This bleach is dominated by the S0 to S1 absorption of the methylene blue monomer, but it is also broad enough to include a weak GSB contribution from the dimer at 608 nm. The bleach redshifts to ∼670 nm due to vibrational relaxation. In addition to the GSB, there are two regions of excited state absorption (ESA), one from 420 nm to 480 nm and another from 530 nm to 580 nm. These regions are associated with contributions from both the monomer and the dimer including excited triplet states of the monomer and the transient reduced forms of methylene blue (semi-reduced and leuco forms).29,34Fig. 3(b) shows the time-resolved TA traces at specific wavelengths. The time-dependent traces at 608 nm and at 658 nm, corresponding to the maximum absorbance of the dimer and monomer respectively, show different bleach recovery dynamics.
 | ||
| Fig. 3 (a)–(c) TA data and global fits for methylene blue (0.7mM) in 1:4 water:acetonitrile solution (a) Spectrally-resolved TA traces at different delay times. The points are data and the lines are fits generated from global analysis using a target model (see text for details). (b) Time-resolved TA traces at selected wavelengths presented on a semi-log scale showing the dynamics in different spectral regions. (c) Normalized species associated spectra (SAS) generated from the global analysis for the data presented in (a) and (b). (d)–(f) TA data and global fits for the higher concentration of methylene blue (0.7 mM) in 1:4 water:acetonitrile solution in the presence of DIPEA (0.3 M) and phenylboronic acid (0.06 M). (d) Spectrally-resolved TA traces at different delay times. The points are data and the lines are fits generated from global analysis using a target analysis model. (e) Time-resolved TA traces at selected wavelengths presented on a semi-log scale showing the dynamics in different spectral regions. (f) Normalized SAS spectra generated from the global analysis for the data presented in (d) and (e). | ||
Fig. 3(d) presents transient absorption of the high concentration of methylene blue in with DIPEA and phenylboronic acid present in ratios that have been previously used for photoredox catalytic oxidative hydroxylation reactions (reaction conditions).16 The recovery of the bleach in reaction conditions is almost twice as fast as for methylene blue on its own. This is consistent with the quenching of the excited state reported via tertiary amines.29,35 In addition, the amplitudes of the positive ESA features at 455 nm to 581 nm are different than what is found for methylene blue alone (Fig. 3(a)) as compared to methylene blue in reaction conditions, with the ESA at 455 nm being lower in amplitude than the ESA at 581 nm. Fig. 3(e) presents time-resolved TA traces at specific wavelengths. The trace at 608 nm has a lower absorbance than the trace at the same wavelength in Fig. 3(b), likely associated with quenching of the dimer. The GSB recovers more rapidly as compared to methylene blue alone with the shoulder at 688 nm recovering at the same rate as the main GSB at 658 nm. The rate of decay for both ESAs follows the decay of the main GSB as they do for methylene blue alone.
The fits and species associated spectra (SAS) presented in Fig. 3(c) and (f) were generated using global analysis33 with a custom target analysis model. In our target analysis of the TA data collected for the higher concentration methylene blue samples, we include excitation from both the monomer and dimer populations with decay to their lower lying states and then later access to the triplet state in the case of the monomer. The model includes excitation of the monomer to the S1 state and then vibrational relaxation of this state associated with SAS1. The proposed reaction mechanism proceeds through excitation to the S1 singlet state of monomer followed by intersystem crossing to the T1 triplet state. As the intersystem crossing and singlet decay in parallel, they are both associated with SAS2. From here, DIPEA reduces the triplet methylene blue to form semi-reduced methylene blue (weak absorption around 420 nm) and this subsequently is fully reduced to form leuco-methylene blue, which is only stable in oxygen-free environments. This and the decay of the triplet are parallel and are represented by SAS3. The formation of a reactive intermediate is not included in the modelling of methylene blue alone. The charge transfer from the amine forms an alpha-aminoalkyl radical that later interacts with the phenylboronic acid to produce phenol. Oxygen completes the catalytic cycle by oxidizing the methylene blue.12,16 This reaction is represented in Fig. 1. For high concentrations, the decay of the excited state of the dimer decay pathway proceeds in parallel. It initially is excited to the S+ state of the dimer and then rapidly decays to the lower S− state (SAS4) before subsequently decaying back to the ground state of the dimer (SAS5).
Vibrational relaxation of the excited state of the monomer is fit to a timescale of 9.3 ± 0.8 ps in methylene blue alone and 4.3 ± 0.9 ps under reaction conditions. The vibrationally relaxed excited state singlet of the monomer can decay back to the ground state (fluorescence) or undergo intersystem crossing to the triplet state of the monomer. The spectra decays in parallel, so SAS2 represents the impact of these two rates on the fit.21 Methylene blue alone at high concentration has a singlet decay of 862 ± 52 ps and an intersystem crossing from the singlet to the triplet of 155 ± 17 ps whereas under reaction conditions at high concentration the singlet decay is 424 ± 38 ps and the intersystem crossing is 210 ± 12 ps. Extremely fast intersystem crossing rates have been previously reported for methylene blue21 which has an high triplet yield of greater than 50%. Short intersystem crossing rates have also been reported for other molecules with heteroaromatic rings systems.8,9 The final portion of the monomer decay pathway is associated with the transition from the triplet state back to the ground state. Previous reports have given a timescale on the order of microseconds for this process.14,21,28,36 As this is orders of magnitude beyond the maximum length of the TA measurement, it was set to 1 μs. Under reaction conditions, in parallel to the decay of the triplet state is the formation of the reactive intermediate, semi-reduced methylene blue, which was found to decay at a rate of 1.12 ± 0.14 ns. For the dynamics of the dimer, we observe fast relaxation from the excited S+ dimer state to the S− state with a timescale of 800 ± 123 fs, while for the reaction condition this has a timescale of 865 ± 104 fs. Relaxation from S− to the ground state of the dimer has a timescale of 13.0 ± 1.2 ps and under reaction conditions has a timescale of 6.3 ± 0.9 ps.
TA measurements were also collected for high concentration methylene blue with either only DIPEA or phenylboronic acid. This data was analysed using the same kinetic scheme as for methylene blue at high concentration. In the case of phenylboronic acid and methylene blue, no significant changes to the rate constants were observed. In the case of DIPEA and methylene blue, the timescale of the recovery of the GSB was like that of the methylene blue reaction mixture, however, no component associated with the reactive intermediate was observed. For additional information see the ESI† (Fig. S4–S9).
TA measurements were collected with concentrations of methylene blue 1/20th of that used for high concentration. This lower concentration was chosen as minimal, if any, dimer is expected to be present in solution.21 Target analysis of the low concentration measurements indicated that the monomer was the dominant species in the SAS. No contributions from the dimer were observed and low concentration with methylene blue alone is included for reference in ESI† (Fig. S10a–c).
At low concentration, the transient absorption of methylene blue in the presence of DIPEA and phenylboronic acid (reaction conditions) is presented in Fig. 5(a), where the methylene blue is 1/20th of the high concentration data shown in Fig. 3. The traces in Fig. 5(a) include the same features found in high concentration, only now the GSB is noticeably narrower due to lack of dimer contributions at the shoulders of the bleach. The target analysis model used for global fitting was the same as used for the high concentration reaction mixture except for the contributions from the dimer. The vibrational relaxation of the excited monomer singlet state was found to be 5.14 ± 0.58 ps and its SAS is depicted in Fig. 5(c) as SAS1. The singlet decay and intersystem crossing rates were determined to be 516 ± 76 ps and 189 ± 32 ps respectively and SAS2 is associated with their contributions. The triplet decay and formation of a reaction intermediate were found to be 1 μs (fixed due to being longer than the length of the experiment delays) and 823 ± 112 ps respectively associated with SAS3.
Discussion
Overall, our modelling suggests that the presence of DIPEA reduces the lifetime of the excited state and that phenylboronic acid does not change the transient dynamics. When both DIPEA and phenylboronic acid are present under light excitation, a reactive intermediate is formed. As the relaxation timescale for the dimer is much shorter than that for the generation of this reactive species, we expect that the dimer does not participate in this process. The kinetic scheme used to generate the SAS and fits presented in Fig. 4 was informed by previously published results.21,29,34 Our recovered timescales, specifically the reduction of the excited state lifetimes of the singlet monomer from 862 ps to 424 ps and the singlet dimer from 13 ps to 6.3 ps, are consistent with previous work on methylene blue.34 We observe that the singlet excited state lifetime decreased from 862 ps for methylene blue on its own to 424 ps in the presence of DIPEA and phenylboronic acid. The timescale for relaxation from the singlet dimer state to the ground state decreased from 13 ps to 6.3 ps between methylene blue on its own and under reaction conditions. The timescale associated with intersystem crossing in the monomer increased from 155 ps to 210 ps in the reaction mixture. | ||
| Fig. 4 (a) Kinetic scheme for target analysis of the TA data. The model consists of a parallel excitation of both the monomer (decay rates k0–k3) and the dimer (decay rates k4 and k5), while in the case of reaction, the product forms with decay rate kp (gold dashed arrow). The low concentration fitting did not include dimer contributions (k4 and k5). The monomer consists of a singlet excitation from the lowest transition at 658 nm along with the triplet at a transition of approximately 820 nm. The dimer consists of the optically accessible transition at 608 nm (S+) and the inaccessible state at 690 nm (S−). Chemical models (ChemDraw) appear over the respective models for monomer and dimer. | ||
 | ||
| Fig. 5 (a)–(c) TA data and global fits for the lower concentration of methylene blue (0.035 mM) in 1:4 water:acetonitrile solution in the presence of DIPEA (0.3 M) and phenylboronic acid (0.06 M). (a) Spectrally resolved TA traces at different delay times. The points are data, and the lines are fits generated from global analysis using a target analysis model. (b) Time-resolved TA traces at selected wavelengths presented on a semi-log scale showing the dynamics in different spectral regions. (c) Normalized SAS spectra generated from the global analysis for the data presented in (a) and (b). | ||
The timescales associated with the dimer are less than 20 ps for all high concentration conditions and less than 10 ps in the presence of DIPEA and phenylboronic acid. The decay from the excited state for the dimer is too fast to participate in the triplet-mediated mechanism. As the reaction primarily proceeds through the triplet state of the monomer, aggregation lowers the number of available photocatalysts by sequestering methylene blue in dimers. The relevant timescales for the reaction are then the intersystem crossing through the triplet, 210 ps, the excited state lifetime of the singlet state of the monomer, 424 ps, and the rate of decay of the semi-reduced form of methylene blue, 1.12 ns. All of these timescales are at least 20 times slower than the decay associated with singlet state of the dimer, supporting the proposed mechanism.12 When the concentration is lowered to 1/20 of that of the high concentration, intersystem crossing to the triplet state was faster, going from 210 ps to 189 ps for low concentration in reaction conditions. The semi-reduced methylene blue also decays more quickly with a timescale of 823 ps. While higher concentrations lead to more aggregation, lower concentrations can increase the intersystem crossing rate, implying there is a balance to optimize overall reaction efficiency.
Conclusions
The TA measurements on methylene blue as a photocatalyst presented here support a triplet mechanism for the oxidative hydroxylation of phenylboronic acid. At higher concentrations, methylene blue dimers are formed that do not participate in the reaction. These dimers decay too rapidly to contribute to the triplet reaction mechanism and sequester methylene blue molecules that could otherwise act in the photocatalytic cycle. Many organic photoredox catalysts are based on similar pi-conjugated molecular platforms that can aggregate depending on the concentration and solvent used. For these species, attempting to improve reaction yield by increasing catalyst loading is likely to result in more aggregation and will not lead to the expected increase in product formation. Future work on this system will explore the role of solvent on aggregation and reactivity as well as the photocatalytic efficiency of derivatives of methylene blue.Author contributions
Benjamin J. Thompson: writing – original draft, investigation, conceptualization, validation, data curation, formal analysis, methodology, visualization, software, writing – review & editing. Anshu Kumar: investigation. Vanessa M. Huxter: conceptualization, methodology, formal analysis, investigation, software, resources, data curation, writing – original draft, writing – review & editing, visualization, supervision, project administration, funding acquisition.Data availability
The data supporting this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
V. M. H. gratefully acknowledges support from the donors of the American Chemical Society Petroleum Research Fund through grant no. 65536-ND6 and from the National Science Foundation through CAREER award grant no. 2236610.References
- P. R. Ginimuge and S. D. Jyothi, J. Anaesthesiol. Clin. Pharmacol., 2010, 26, 517–520 CrossRef PubMed.
- L. M. Moreira, J. P. Lyon, A. Lima, L. Codognoto, D. Severino, M. da S. Baptista, A. L. Tessaro, A. P. Gerola, N. Hioka, M. R. Rodrigues, J. A. Bonacin, S. C. dos Santos, A. P. Romani and H. P. M. de Oliveira, Orbital: Electron. J. Chem., 2017, 9, 279–289 CAS.
- G. N. Lewis, O. Goldschmid, T. T. Magel and J. Bigeleisen, J. Am. Chem. Soc., 1943, 65, 1150–1154 CrossRef CAS.
- M.-E. Nedu, M. Tertis, C. Cristea and A. V. Georgescu, Diagnostics, 2020, 10, 223 CrossRef CAS PubMed.
- S. Saita, M. Anzai, N. Mori and H. Kawasaki, Colloids Surf., A, 2021, 617, 126360 CrossRef CAS.
- N. Miyoshi and G. Tomita, Z. Naturforsch. B, 1978, 33, 622–627 CrossRef.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- J. J. Nogueira, M. Oppel and L. González, Angew. Chem., Int. Ed., 2015, 54, 4375–4378 CrossRef CAS PubMed.
- A. Rodriguez-Serrano, V. Rai-Constapel, M. C. Daza, M. Doerr and C. M. Marian, Photochem. Photobiol. Sci., 2012, 11, 1860–1867 CrossRef CAS PubMed.
- T. Ohno and N. N. Lichtin, J. Am. Chem. Soc., 1980, 102, 4636–4643 CrossRef CAS.
- T. Ohno and N. N. Lichtin, J. Phys. Chem., 1982, 86, 354–360 CrossRef CAS.
- S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Acc. Chem. Res., 2016, 49, 1320–1330 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- E. Morgounova, Q. Shao, B. Hackel, D. Thomas and S. Ashkenazi, J. Biomed. Opt., 2013, 18, 056004 CrossRef PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Springer US, Boston, MA, 1983 DOI:10.1007/978-1-4615-7658-7_9, pp. 257–301.
- R. H. Kayser and R. H. Young, Photochem. Photobiol., 1976, 24, 395–401 CrossRef CAS.
- A. D. Klimov, S. F. Lebedkin and V. N. Emokhonov, J. Photochem. Photobiol., A, 1992, 68, 191–203 CrossRef CAS.
- J. C. Scaiano, Chem. Soc. Rev., 2023, 52, 6330–6343 RSC.
- J. C. Dean, D. G. Oblinsky, S. R. Rather and G. D. Scholes, J. Phys. Chem. B, 2016, 120, 440–454 CrossRef CAS PubMed.
- K. Patil, R. Pawar and P. Talap, Phys. Chem. Chem. Phys., 2000, 2, 4313–4317 RSC.
- N. Florence and H. Naorem, J. Mol. Liq., 2014, 198, 255–258 CrossRef CAS.
- A. Kumar, B. Thompson, R. Gautam, E. Tomat and V. Huxter, J. Phys. Chem. Lett., 2023, 14, 11268–11273 CrossRef CAS PubMed.
- K. Kosma, S. A. Trushin, W. Fuß and W. E. Schmid, J. Mod. Opt., 2008, 55, 2141–2177 CrossRef CAS.
- K. S. Wilson, A. N. Mapile and C. Y. Wong, Opt. Express, 2020, 28, 11339–11355 CrossRef CAS PubMed.
- R. M. Danziger, K. H. Bar-Eli and K. Weiss, J. Phys. Chem., 1967, 71, 2633–2640 CrossRef CAS.
- R. Nilsson, P. B. Merkel and D. R. Kearns, Photochem. Photobiol., 1972, 16, 109–116 CrossRef CAS PubMed.
- R. H. Kayser and R. H. Young, Photochem. Photobiol., 1976, 24, 403–411 CrossRef CAS.
- J. M. Kelly, W. J. M. van der Putten and D. J. Mcconnell, Photochem. Photobiol., 1987, 45, 167–175 CrossRef CAS PubMed.
- S. Kato, M. Morita and M. Koizumi, Bull. Chem. Soc. Jpn., 2006, 37, 117–124 CrossRef.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- C. Müller, T. Pascher, A. Eriksson, P. Chabera and J. Uhlig, J. Phys. Chem. A, 2022, 126, 4087–4099 CrossRef PubMed.
- J. Chen, T. C. Cesario and P. M. Rentzepis, Chem. Phys. Lett., 2010, 498, 81–85 CrossRef CAS.
- S. G. Bertolotti and C. M. Previtali, Dyes Pigm., 1999, 41, 55–61 CrossRef CAS.
- J. Chen, T. C. Cesario and P. M. Rentzepis, J. Phys. Chem. A, 2011, 115, 2702–2707 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02026j |
| This journal is © the Owner Societies 2024 |