
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ANI neural network potentials for small molecule pKa prediction†
Ross James
Urquhart
 ,
Alexander
van Teijlingen
and
Tell
Tuttle
*
,
Alexander
van Teijlingen
and
Tell
Tuttle
*
Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: tell.tuttle@strath.ac.uk
First published on 29th August 2024
Abstract
The pKa value of a molecule is of interest to chemists across a broad spectrum of fields including pharmacology, environmental chemistry and theoretical chemistry. Determination of pKa values can be accomplished through several experimental methods such as NMR techniques and titration together with computational techniques such as DFT calculations. However, all of these methods remain time consuming and computationally expensive. In this work we develop a method for the rapid calculation of pKa values of small molecules which utilises a combination of neural network potentials, low energy conformer searches and thermodynamic cycles. We show that neural network potentials trained on different phase and charge states can be employed in tandem to predict the full thermodynamic energy cycle of molecules. Focusing here on imidazolium derived carbene species, the method utilised can easily be extended to other functional groups of interest such as amines with further training.
1 Introduction
Acids ionise in solution by the association or dissociation of a proton. The acid dissociation constant, Ka, describes the strength of an acid in solution and is defined by the equilibrium between the dissociated, A+ and H+, and associated state, HA, of the acid. Consequently, the pKa can be derived as the negative log of the dissociation constant, eqn (1), which can subsequently be related to pH through the Henderson–Hasselbalch equation for dilute acids in aqueous solutions, eqn (2).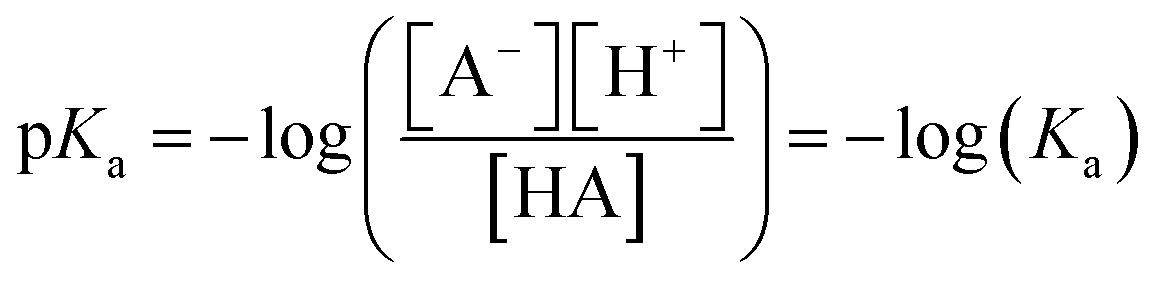 | (1) |
 | (2) |
Many factors are understood to affect the pKa value of a molecule. These factors include solvent present,1 the temperature of the system,2 and the local chemistry present3 within a molecule. Crucially, the understanding of pKa values is important for several branches of chemistry including drug discovery,4 coordination chemistry, and environmental chemistry.
Often determined experimentally through techniques such as titration,5,6 spectrometry6 and NMR,7 several methods have been developed regarding the calculation of pKa theoretically.
Among these include methods such as machine learning (ML), density functional theory (DFT) and constant pH molecular dynamics. In 2004, Yates et al.1 demonstrated the calculation of pKa values for a set of imidazolium derived carbene molecules using a mixture of ab initio and DFT methods in conjunction with the Conductor-like Polarisable Continuum Model (CPCM) for modelling of the aqueous phase. Yates studied a series of imidadzole carbenes in dimethyl sulfoxide and acetonitrile to good accuracy versus available data from experimental pKa values. The study goes on to highlight the importance of conformation within pKa calculations by examining the energies of conformers of the studied carbenes extensively, utilising only the conformers with the lowest energy in each case. The method used by Yates echoes earlier work by Liptak and Shields8 surrounding the use of a thermodynamic cycle for calculation of the relevant thermodynamic properties. This has been visualised in Scheme 1 which shows the deprotonation of a molecule, or in the context of this paper, an imidazolium salt, HA+, resulting in the products, A, a carbene, and H+, a proton.
 | ||
| Scheme 1 Thermodynamic cycle for the deprotonation of an acid (A). These thermodynamic cycles can be used to calculate pKa values more accurately than by direct calculation of ΔG0aq. | ||
Where ΔG0gas and ΔG0aq are the free energies of deprotonation of the gas and aqueous phase, respectively, and the solvation free energy is represented by ΔG0s(X) where X = HA+, A and H. The pKa can then be calculated for a molecule by application of eqn (3)–(5), requiring the free energy of deprotonation for the species in the gaseous and aqueous phase alongside the solvation free energy for each species.
ΔG0aq = −2.303RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logKa logKa | (3) |
| pKa = ΔG0aq/2.303RT | (4) |
| ΔG0aq = G0(Aaq) + G0(H+aq) − G0(HA+aq) = G0(Agas) + ΔG0s(A) + G0(H+gas) + ΔG0s(H+) − G0(HA+gas) − ΔG0s(HA+) | (5) |
Liptak and Shields also drew attention to the demanding nature of theoretical pKa calculations as an energy difference of just 1.36 kcal mol−1 is equal to a change in pKa of 1 unit.8 This naturally requires energies and structures to be calculated with high degree of accuracy. If such an approach as that shown in eqn (5) is used then there are four sources of potential error even when literature values regarding the proton are employed.
Theoretical approaches to calculating pKa have continued to be explored since the work of Yates with studies determining the pKa of molecules such as phenols9 and thiols.10 Studies have also been undertaken to explore the accuracy of methods in the calculation of pKa, with Dutra et al.11 reporting that results via DFT methods are highly dependent on the basis set employed and the number of explicit water molecules in the system. One study by Ho and Coote12 set out to determine if there could be a universal method to be applied to pKa calculations. The study examined four different calculation methods and several different solvent models, concluding that using a proton exchange scheme, wherein a reference acid is employed to conserve charge on both sides of a reaction, gave the most promising results. It was found that for a universal approach, the exchange scheme was much less sensitive to a change in reference than the direct continuum model approaches which are more sensitive to the species upon which the model was parameterised with. Thus methods for calculation of pKa tend to fall within the realms of absolute calculations such as those that employ DFT to calculate thermodynamic cycles like those described above or within the realms of relative methods such as the proton exchange scheme approach.
There have been attempts at pKa prediction using methods other than thermodynamic cycles, one of which includes predicting pKa values by drawing linear correlations between the vibrational frequencies of H–X bonds in hydrogen halides before extrapolating the insights to carboxylic acids.13 The method proved to predict the pKa of a set of aliphatic carboxylic acids to within 0.3 pKa units of their experimental values.
As mentioned above, approaches to pKa calculations like those described above incur many associated errors which need to be minimised in order to achieve any degree of accuracy versus experimental techniques. Amongst these is the value of the solvation free energy of a proton, ΔG0s(H+), a value which is much debated in literature with reported values as high as −254.30 kcal mol−114 and as low as −265.77 kcal mol−1.15 Indeed, the uncertainty surrounding the value of ΔG0s(H+) has led to the popularity of calculating pKa in a relative manner as mentioned above.
Machine learning (ML) has also been employed in the determination of pKa values to good effect. Williams et al. employed ML algorithms including support vector machines, deep neural networks (DNN) and extreme gradient boosting (XGBoost) on a dataset of over 7900 chemicals.16 The best models were capable of producing a coefficient of determination (R2 value) as high as 0.80 and root mean square error (RMSE) values as low as 1.5 pKa units. A 2020 paper by Baltruschat and Czodrowski17 found that a five-fold cross validation random forest model performed best, producing an R2 of 0.82 and RMSE of 1.03 when trained on a compilation of monoprotic compounds assembled from DataWarrior18 and the ChEMBL databases.19 Roszak et al.20 developed a graph neural network for the prediction of pKa values of C–H groups in non-aqueous solvents, with results showing a mean absolute error (MAE) of 2.1 pKa units. The authors reported that the use of descriptors for the chemical properties of the chemical environment alongside topological descriptors led to the low errors in the method when validated on synthetic problems.
ML methods have continued to expand in recent years with efforts at pKa prediction including areas such as proteins21 and drug-like molecules.22 Another area which is developing simultaneously alongside ML models is that of data availability. Datasets of increasing size and quality are becoming available to help drive the training of ML models without which the continued improvements of such models to perform well would suffer. Within the realm of pKa prediction, datasets such as those from DataWarrior18 and ChEMBL,19 allow for access to experimental pKa data, albeit via premium access. Datasets containing theoretical pKa values such as PHMD54923 are also available.
Indeed, there have been papers in recent years which make use of published datasets for pKa prediction. Mayr et al. published a graph NN based method capable of enumerating protonatable sites and predicting their pKa based on training comprised of experimental pKa values and molecules from the ChEMBL dataset. Providing code in both commercial and free-to-use format, the published models show a RMSE of less than 1 pKa unit.24
One of the key requirements to accurate calculation of pKa values is accurate representation of the chemical environment of the molecule/protonatable site of interest. One such way to capture the information computationally is through the use of atomic environment vectors (AEVs), versions of modified symmetry functions that are suited to provide a description of molecular environments on an atomic basis. One of the benefits of describing molecules in such a way is that AEVs are specifically concerned with individual atoms and their local environments rather than molecules as a whole. This allows exact chemical environments to be encoded within them.
Neural network potentials (NNPs), such as ANI,25 SchNet26,27 MACE28,29 and AIMNet30 are able to map chemical information like that within AEVs to the energy of molecules. NNPs use reference data, often from quantum mechanical calculations such as DFT, to build a representation of the potential energy surface to be learning by ML techniques like DNNs. The ANAKIN-ME (ANI) suite of models were first released in 2017. Developed by Smith et al., ANI-125 is an NNP capable of <1.0 kcal mol−1 RMSE values versus DFT calculations at a fraction of the cost. Initially limited to only molecules containing H, C, N and O, the ANI models have since expanded through models ANI-1x,31 ANI-1ccx32 and ANI-2x33 to include H, C, N, O, F, S and Cl alongside also introducing active learning and coupled cluster training data.32 In its latest form, ANI-2x33 is capable of performing 106 times faster than DFT and produces sub-chemical accuracy on test datasets. ANI-2x33 also included improved parameterisation for bulk water, molecular force training (analytical derivatives of the molecular energies) and sampling of torsion angles and chemical space all aimed at improving the accuracy of the model. Much of the success of the ANI suite can be attributed to the development of AEVs. Based on Behler–Parrinello symmetry functions,34 AEVs provide additional terms in order to capture more distinct molecular features allowing the NNP to be capable of better distinguishing between atom types, functional groups, and rings amongst other molecular features. Within the ANI models, the AEVs of a molecule are split up by atom type and fed through separate DNNs to yield an atomic energy which is consequently summed to produce the energy of the conformer (additional information regarding the ANI architecture can be found in the ESI†). ANI-2x33 has also been utilised in the development of pKa-ANI,35 a ML tool that builds upon the existing ANI-2x architecture to predict pKa values for all five titrable amino acid residues within input proteins. pKa-ANI uses the AEVs for each atom type as well as layers from the atomic neural networks as descriptors within the model. The model, based on deep representation learning, performed better than PROPKA,36 a widely used semi-empirical modelling tool, with mean absolute error (MAE) values below 0.5 pKa units for the titrable amino acid residues.
In this study, we evaluate a weighted Boltzmann approach to pKa calculation. Utilising an ensemble of conformers we are better able to consider the molecules under study by including non-equilibrium structures, which is more similar to ensembles of molecules being measured by experimental approaches. A subset of previously investigated carbenes,1Fig. 1, were studied. Most of the carbenes contain an imidazole core. Imidazole-derived carbenes are well known as versatile ligands due to their highly tuneable electronic nature and as such are of great interest in areas such as catalysis.37,38 The previous results by Yates et al.,1 have been studied at multiple levels of theory and provide useful reference points for evaluation of the methods developed within this work. We develop and apply a set of datasets that presents evidence that ANI architecture NNPs can be trained on species in charged and/or aqueous phase so long as the dataset each model is trained on does not overlap in respect to charge/phase and provides a method by which this method of pKa prediction could be extended to any class of small molecules given the appropriate training data.
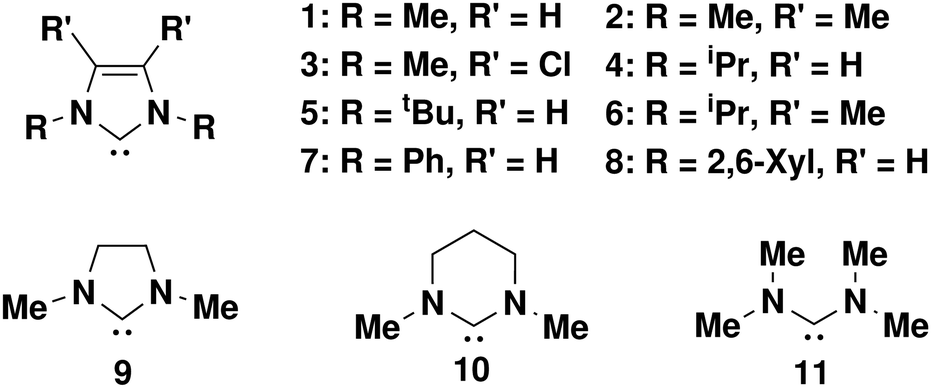 | ||
| Fig. 1 Subset of carbenes studied in this work for pKa prediction. | ||
2 Results and discussion
Calculation of pKa values with DFT at the ωB97X-D4/Def2-SVP level of theory, DFT global minima (GM) (Table 1), gave good comparison to Yates literature values, Yates GM (Table 1), when calculated for carbenes 1–11, Fig. 1. There are of course expected differences between the values owed to a change in level of theory and software package. The Yates values were reported via gas phase calculation optimisation and frequency calculations at B3LYP, CBSB7 level of theory. The solvent phase calculations were completed using CPCM solvent at B3LYP and HF with 6-31+G(d) and 6-31+G(d,p) levels of theory. The carbenes studied here also have the potential to minimise at different conformers. This is noted by Yates et al. in their work and an effort was made in this work to optimise single conformation (GM) reference DFT calculations to the same conformational minima. This is particularly prevalent for structures 7 and 8 where the aromatic R-groups can rotate relative to the imidazole centre of the molecule.| Carbene | Yates GMa | DFT GMb | DFT ensemblec | ML ensembled |
|---|---|---|---|---|
| a pKa values reported by Yates et al.1 b pKa values calculated from the Yates optimised structure, optimised at ωB97X-D4/Def2-SVP level of theory and calculated using the direct aqueous method as detailed in Section 3.7. c pKa values calculated using the DFT optimised (ωB97X-D4/Def2-SVP) geometries of generated crest conformers as detailed in Sections 3.6 and 3.7. d pKa values as predicted by ML models, as detailed in Sections 3.6 and 3.7. | ||||
| 1 | 27.40 ± 0.4 | 28.01 | 28.12 | 29.30 |
| 2 | 29.50 ± 0.3 | 29.94 | 29.98 | 31.16 |
| 3 | 23.40 ± 0.2 | 23.13 | 23.56 | 22.97 |
| 4 | 28.20 ± 0.3 | 28.12 | 28.03 | 27.18 |
| 5 | 28.30 ± 0.1 | 29.33 | 29.36 | 28.60 |
| 6 | 30.40 ± 0.3 | 29.97 | 30.90 | 29.13 |
| 7 | 22.00 ± 0.1 | 23.26 | 23.08 | 28.44 |
| 8 | 22.60 ± 0.1 | 24.32 | 25.65 | 26.48 |
| 9 | 28.50 ± 0.4 | 28.69 | 27.93 | 25.54 |
| 10 | 33.70 ± 0.3 | 32.40 | 32.75 | 31.63 |
| 11 | 34.00 ± 0.3 | 34.68 | 34.29 | 35.11 |
Initial attempts at pKa calculation through direct interface with ANI-2x were unsuccessful. Optimisation from DFT output geometries (Yates et al. ESI1) with ANI as well as optimisation of carbene molecules explicitly solvated with water, Fig. 2, followed by extraction of the relevant carbene AEVs all resulted in large errors. With explicit solvation, the conformational changes to the carbene induced by the water molecules were expected to establish the aqueous structure, facilitating AEV extraction and subsequent summation of atomic energies in order to determine the aqueous energy of the molecule. Despite additional attempts to predict the Gibbs free energy using ANI-2x and ASE's vibrations module, these efforts proved unsuccessful as well. It was thought that the training of ANI on potential energies and not on Gibbs energies could contribute to the large errors when calculating pKa. The Gibbs energy is essential when calculating the pKa through the full thermodynamic cycle. As such it was deemed that ANI-2x was unsuitable for the nature of these calculations.
 | ||
| Fig. 2 Carbene molecule with explicitly solvated water. The AEV's of the molecule being investigated capture the surrounding water molecules without including the atomic energies of the water. | ||
Upon close examination of the ANI-2x dataset, it was found that while it does contain carbene molecules, they comprise a very small amount (247 structures out of 9651712 total, 0.0026%) of the total dataset. Further, the handling of charged states and solvent phase calculations are unavailable within the ANI family of models to date. As such, the development of extensive carbene datasets, both protonated and deprotonated as well as in the gas and aqueous phases have been undertaken in this work, Section 3.4.
There are two routes to calculate the value of ΔG0aq as shown in eqn (5). The first involves the full thermodynamic cycle (the full cycle approach), Fig. 1, which includes calculating gas and aqueous phase energies in order to give the solvation energies. The second involves calculating the aqueous phase directly (the direct aqueous approach), which avoids calculating the gas phase energies. Both routes follow the workflow laid out in Section 3.7, utilising a Boltzmann distribution and weighted average. The direct aqueous approach, while being slightly less accurate, results in a reduction of half the processing time which is the main objective in this study. Indeed, a difference in RMSE of only 0.05 pKa units (1.94 on the full cycle vs. 1.99 direct aqueous) separated using the full cycle versus the direct calculation. Due to the extended time required to compute the full cycle, even if it provides a slight increase in prediction accuracy, results moving forward will be taken from aqueous phase prediction only.
Development of DNN models based on the ANI architecture and calculation of pKa values when carried out on DFT-optimised structures afforded very good results compared to Yates et al.1 Direct calculation via the aqueous phase (GM approach) afforded a RMSE and MAE of 2.03 and 1.19 pKa units respectively. This highlights that our models are capable of calculating pKa values with good accuracy for the studied carbene species. It further indicates that the models have a good grasp of the energetics of carbene species at minima and can reliably predict the pKa of such species. However, using the pre-optimised structures from a DFT calculation doesn't provide any information on whether the models can arrive at the minima when provided with higher energy structures, i.e. those drawn by hand in Avogadro.39
As such, attempts were then made to calculate pKa values from non-ground state structures. The structures were drawn in Avogadro39 and then optimised by the universal force field40 to give a reasonable starting structure for DFT calculations. These structures were then directly used to calculate the pKa of the carbene. As mentioned in Section 1, calculating pKa values is demanding owing to the sensitive nature of the energies involved in their calculation. As such, the structures given as output from Avogadro were not optimised fully to their minima and the trained models struggled to accurately predict pKa values. This led to large errors with an RMSE value of 14.75 and an MAE value of 12.69.
As a result the workflow detailed in Sections 3.6 and 3.7 were developed. First, calculations were completed on the optimised geometries from the ESI of Yates et al.1 at the ωB97X-D4/Def2-SVP level of theory and the pKa calculated via the direct aqueous approach, DFT GM (Table 1). Secondly, conformers for each carbene in both gaseous and aqueous states were generated with Crest (Section 3.6) as detailed below and the Gibbs energy calculated with DFT for each structure. These energies were then used in calculating Boltzmann populations and thus pKa values as detailed in Section 3.7, DFT ensemble (Table 1). Lastly, Crest generated conformers were optimised with the models trained herein and pKa values calculated similarly through Boltzmann populations, ML ensemble (Table 1). More information about the ensemble including the number of conformers generated for each method and analysis of conformer energy and structure can be found in the ESI† (pages S3–S8).
Isayev and Gockan took a different approach to using the ANI DNN potential for pKa predictions by developing the pKa-ANI application.35 The pKa-ANI model provides excellent and efficient computation of pKa values for titrable residues in large proteins due to the calculation of pKa directly from the AEVs without explicit energy calculation. Further, the pKa-ANI model is comprised of five independent models, one for each titrable amino acid residue (His, Asp, Glu, Lys, Tyr). This provides an efficient way to calculate the pKa eliminating compounding errors associated with calculating via thermodynamic cycles. However, this results in the model being limited to protein side chains and their individual pKa ranges. This is in contrast to the model presented here which is more generalisable (though restricted to smaller molecules) due to its use of thermodynamic cycles and global optimisation schemes. As such, in contrast to the pKa-ANI paper, which directly utilises AEVs as features for representation learning, the method employed here only uses AEVs as neural network inputs to predict atomic energies. AEVs are not incorporated directly into the pKa calculation within this workflow. The scope of this model produces higher error measurements by virtue of being able to predict a larger range of pKa values (22–34 pKa units).
pKa-ANI has a range of around ±2 pKa units for each independent residue model. This results in a low RMSE range of between 0.49 and 0.88 units due to the smaller prediction range for each residue. This is reflected in the results of the reported null model which showed that using the mean value as the prediction results in an error of less than 1.5 pKa units. Due to the larger range of pKa values evaluated by our model we report a larger RMSE value of 1.99 pKa units. Although individual predictions generally align well with DFT Ensemble values, Fig. 3, for each carbene, the normalised RMSE (NRMSE) becomes pertinent, calculated by dividing the RMSE by the prediction range. This yields a NRMSE of 0.166 for our model compared to 0.180 for pKa-ANI, indicating comparable performance despite differing prediction ranges.
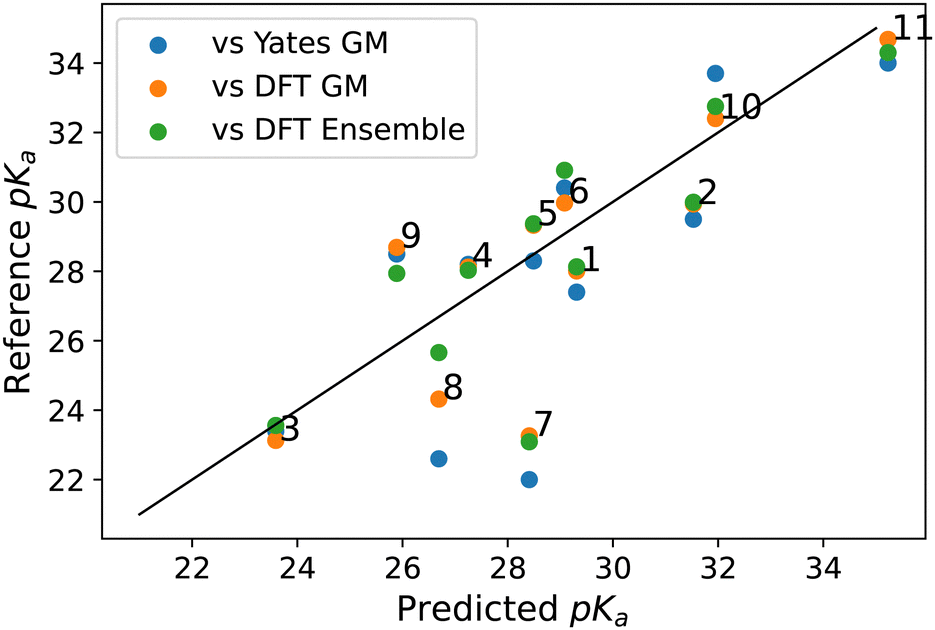 | ||
| Fig. 3 Predicted pKa results of ML Ensemble method vs. reference methods of pKa calculation. We score our model primarily against the DFT ensemble as the DNN was trained on data at the same level of theory as the DFT and used an ensemble of conformers to make its predictions. | ||
When exploring the conformations of carbenes, Yates et al. found that the minima for carbene 8 was found when the xylene R-groups were orthogonal to the core imidazole ring.1 This was due to the methyl groups present at the 2 and 6 positions on the ring of the xylene. Such methyl groups are not present in carbene 7 which gives more rotational freedom to the structure around the C–N bonds connecting the R-groups to the central imidazole. Shown below in Fig. 4(b), the overlap of a conformer of carbenes 1 and 7 highlights the difference between the optimised structures by DFT (blue atoms) and the structure as optimised by the trained models (red atoms). It can be seen that the conformer of carbene 7, as optimised by the models, struggles to match the DFT geometry. This potentially is the cause of the larger error seen in prediction of carbene 7. Conversely, the structure of carbene 8 is modelled well by both methods and this is reflected in the improved prediction of pKa. This could be due to the aforementioned forced conformation of xylene R-groups as orthogonal to the core imidazolium.
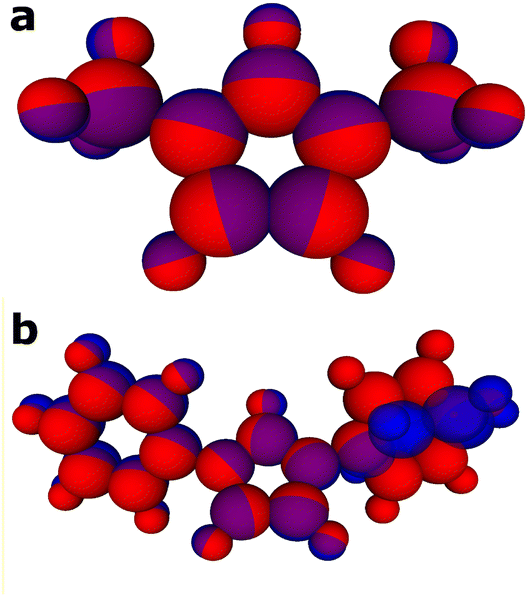 | ||
| Fig. 4 (a) Carbene 1 showing a very close optimisation with an RMSD of 0.08 Å vs. the DFT optimised structure. (b) Carbene 7 showing an overlap of optimised DFT conformer (blue) and conformer optimised with ANI charged aqueous model (red) with an RMSD of 0.81 Å. | ||
Previous studies have reported that the ANI architecture cannot handle charged molecules, however we find that this is only true when trying to mix different charge states into the same training model. This is due to the lack of knowledge of the number of electrons in a system which results in an inability to differentiate between the same conformers with different charge states.33 In this study, we have found that by training separate neural networks based on charge and gas/aqueous phase that the ANI architecture is able to accurately predict energies for charged and/or aqueous phase molecules as long as that was exclusively the type of data it was trained on.
To further examine the internal representation of molecules in the charged vs. neutral models we comparatively analyse the protonated carbenes in both models (neutral and charged). As ANI potentials work by using Atomic Environment Vectors (AEVs) as inputs for neural networks which output the atomic contributions, or absolute energies for each atom, we can thus determine the energy associated to each atom in a molecule by the trained models. This allows us to scrutinise the disparities in energy allocations across atomic components between the neutral and charge models. It should be noted that atomic contributions to the energy are directly outputs of the neural networks within the ML models and do not stem from DFT. It is difficult to infer any understanding of neural network intermediate states between input and final output, however in ANI potentials, the output of a neural network is the atomic energy associated with specific atoms which are in turn summed to give the total molecular energy. We note that the difference between predicted Gibbs energy for a molecule between charged and uncharged models (GZ=+1 − GZ=0) aligns with the expected charge distribution difference. It was found that for protonated carbenes the charged model assigns a greater contribution to the total molecular energy from the ylidine carbon and nitrogen atoms. Moreover, the discrepancy in energetic contributions depends on the N-substituents (Fig. 5). This aligns with the expectation that charge delocalisation increases with the presence of more electronegative N-substituents, as exemplified by the comparison between xylene and methyl substituents (Fig. 5(a)vs.Fig. 5(b)). Within molecules, where greater electron density is present it follows that the electronic energy will be lower due to minimised electrostatic and coulombic repulsion. It is important to note that although there is an approximate correlation between the models energy contribution difference and the difference in charge distribution, it cannot be stated that the ANI potential has explicitly learned how to infer charge distribution.
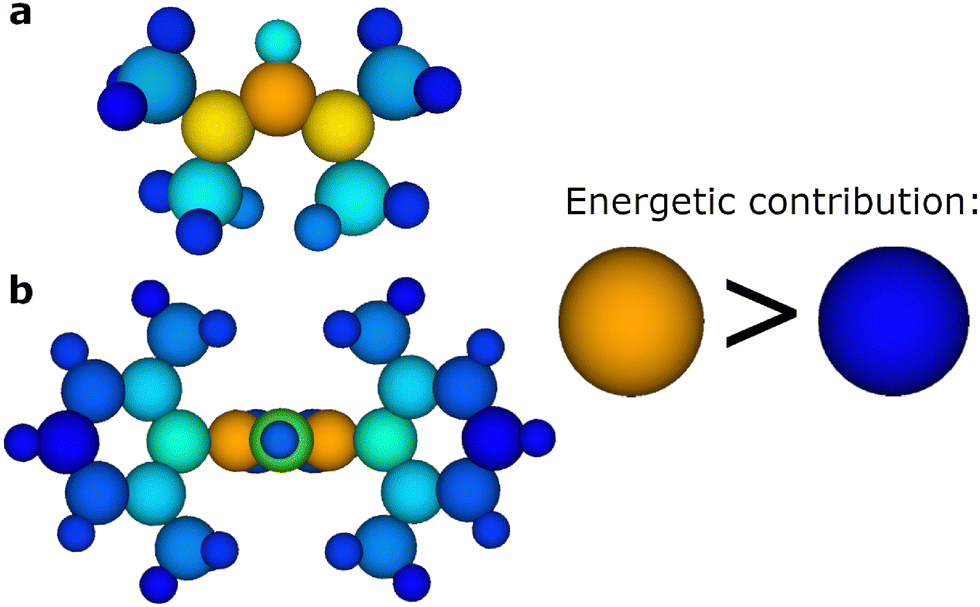 | ||
| Fig. 5 The difference in atomic contributions to the molecular energy derived from the neutral gaseous ANI and charged gaseous ANI models indicates varying degrees of charge delocalisation. These differences are represented as a shift from orange to blue. In the case of carbene 11 (a), the variation is confined to the protonated ylidine carbon and to a lesser extent the bonded nitrogens, representing localised charge distribution on the ylidine carbon. Conversely, in carbene 8 (b), the variation extends further, with the biggest difference being the nitrogen atoms and to a lesser extent encompassing the N-xylene R-groups, indicating an increase of charge delocalisation. | ||
Despite the neural network not being informed of electrons or atomic charge whilst training, the models implicitly capture where the difference in the molecule is located (Fig. 5). This is important as it highlights that, through structure and energies alone, ANI models can infer other chemical properties within its hidden representation. This accounts for the ability of the separate ANI models developed herein to accurately predict pKa values, as charge plays a significant part in the free energy associated with the direct calculation.
3 Methodology
3.1 Level of theory and thermodynamic parameters
DFT calculations were carried out in ORCA V5.0.441 at the ωB97X42/Def2-SVP43 level of theory. Additionally, Grimme's D4 dispersion correction44 was employed alongside RIJCOSX algorithm45 and the Def2/J auxiliary basis set.46 The integration grid was set to DefGrid2 whilst SCF convergence tolerance was set to TightSCF. Orca's DFT module was utilised for geometry optimisations, vibrational analysis, and single point calculations. Implicit water solvation was modelled by the conductor-like polarizable continuum model (CPCM).47In this work, we use benchmark results from Yates et al.1 As such, the value of the proton in the gas phase (Ggas(H+)) has been taken as −4.39 kcal mol−1, a value derived from the Sackur–Tetrode equation but has accounted for a state change into moles per litre from atmospheres. We acknowledge however, that the value of free energy of solvation of the hydrogen atom (ΔG0s(H+)) can contribute as a source of error due to the range in reported values differing significantly.14,15 As the value of the free energy of solvation of the proton in this work we adopt a value of −261.85 kcal mol−1 in accordance with the value used by Yates et al.1 Another source of error in the determination of ΔG0s(H+) is the choice of quantum mechanical method. Although we are not determining the value of the solvation energy of the proton in this work we do utilise a range-separated hybrid functional with additional dispersion corrections. It has been noted that such functionals are suitable for application to such calculations.48
3.2 Calculation with the ANI-2x potential
Calculation of pKa values with the ANI-2x33 potential was carried out in Python via the PyTorch framework.49 The ANI-2x model and parameters were obtained through the torchani module.50 Through interface with the atomic simulation environment (ASE)51 molecules were optimised with the LBFGS52 optimisation algorithm to an maximum force of 0.01. In order to calculate the pKa value, four values were needed. These were the protonated and deprotonated carbene molecules in both the gaseous and aqueous phase.Carbene geometries were generated in Avogadro.39 Gas phase energies were then obtained by optimising with the ANI-2x potential and retrieving the molecular energy. In order to gain access to solvation phase geometries, the isolated carbene molecules were solvated in GROMACS53 with single point charge water within the General Amber Force Field.54 Topology files were generated with the ACPYPE web server.55,56 Boxes of size 3 × 3 × 3 nm with the carbene centred and surrounded with water molecules were then written to XYZ files. The XYZ files were then used as the input for ASE to optimise with the ANI-2x potential. The AEVs corresponding to the water molecules were then extracted and the atomic energies for each of the atoms present in the carbene summed to give the solvation phase energy of the protonated and deprotonated carbene forms. Explicit solvent is utilised only here, in conjunction with the pre-trained ANI-2x model in order to attempt to access the solvation phase structure and energy. Explicit solvent is not used again within this paper.
3.3 Calculation of reference pKa values
ORCA calculations were carried out as described in Section 3.1, upon reference carbene structures. The structures, Fig. 1, were selected to allow comparison between published results by Yates et al.,1 DFT and the developed models. The value of G0aq can be directly calculated from aqueous phase energies, as shown in eqn (5), negating potential error from two additional gas phase calculations (G0(Agas) and G0(HA+gas)).3.4 Development of datasets
Although the original paper by Yates et al.1 calculated the value of 12 carbenes, in this paper, only 11 were studied as we have not extended our models to sulphur due to the large computational effort required to do so. As such our models cover elements H, C, N, O and Cl.As mentioned in Section 3.2, there are four energies required for calculation of pKa, protonated and deprotonated carbene energies in the gaseous and aqueous phase. As such, four different datasets were developed for training of subsequent models. For each distinct molecule there were two optimisation and frequency calculations completed, one in the gas phase and one with implicit CPCM water solvation. Once collated, the coordinates, energies and species of each job were stored in HDF5 format through the H5py python module.57
It is worth noting that as aqueous models were trained on DFT data calculated in the aqueous phase (with implicit CPCM solvation), that when trained, the aqueous models will predict the aqueous structure and energy. The opposite is true for gas state models. This means that there is no solvent present, either implicit or explicit, when predicting energies for pKa calculation with the developed models as it is embedded within the developed models.
Structures under consideration for calculation were limited to contain only H, C, N, O and Cl atoms. This gave a great deal of flexibility in searching for structures to add to datasets whilst also limiting to structures that would have a sizeable impact on what each model could learn. With ANI architecture models scaling in cost in relation to the number of individual atomic networks that have to be trained this also ensured reasonably quick training of each model.
Initial calculations for deprotonated models involved recalculation of the ANI-1 dataset58 at the level of theory indicated in Section 3.1. Further, the calculated geometries were subsequently protonated through RDKit59 and then calculated as before. These calculations, in both gaseous and aqueous phases formed the initial data space. Next, molecules from the QMSpin dataset60 were similarly calculated in the singlet state and in both gas and aqueous phases, both deprotonated and protonated on the -ylidene carbon. Within structures from the QMSpin dataset, any fluorine atoms were replaced by chlorine atoms via RDKit.
Further calculations were completed in order to fine-tune the dataset to be sensitive to energy changes arising from any changes in bond angle and length. To do so, a molecule was scanned to find all connections which returned all bonds, angles and dihedral angles. Each connection was then adjusted over a range and stationary frequency calculations were completed to obtain the Gibbs energy of the structure. For angles and dihedrals, the connection was altered over a range of ±20 degrees. For bonds, alterations were made between a minimum and maximum distance. Maximum distances were calculated using van der Waals radii as reported by Alvarez.61 The radii of both atoms in the bond were summed and then divided by two to obtain the maximum distance the bond should be at. Subsequently the minimum distance was taken as the maximum distance divided by 1.75. For example, for a C–H bond, maximum distance would be the radius of a C atom (1.77 Å) summed with the radius of the H atom (1.2 Å) divided by two, 1.485 Å. The minimum distance would then be 1.485/1.75, or 0.845 Å.
The collected data were also screened to ensure that none of the validation data (Structures 1–11, Fig. 1) appeared within the training and testing datasets. This was achieved by removing all structures that contained the same amount and type of atoms and were within a root mean squared distance (RMSD) of 2.0 Å from any of the validation structures. Unreasonable structures were also removed based on features such as unbound hydrogen atoms present within a structure.
Following data collation, the final dataset sizes can be seen in Fig. 6 and is expanded upon in the ESI† (Table S3). As a result of the fine tuning calculations, the charged datasets have a higher number of data points due to the increased number of calculations associated with fine-tuning the additional proton present in molecules.
 | ||
| Fig. 6 Bar chart depicting the number of distinct data points within each developed dataset. | ||
3.5 Development of auxiliary neural network potential models
The way in which the newly developed models were trained does not differ significantly from the parameters used to train the ANI-2x model. A neural network is defined for each atomic AEV type and default settings were utilised for the AEV parameters. When utilising the ANI-2x model through ASE,51 the generated outputs are the result of a combination of models rather than just one. In line with developers recommendations,50 for each individual model-subset we have trained six models that differ only in batch size with batches of size 256, 512, 1024, 2048, 4096, and 8192. Batch sizes were scaled as powers of 2 in order to generate a wide array of trained models. Training of the models was carried out by applying the PyTorch49 and TorchANI50 Python modules.Models were trained with a decay learning rate on plateau with an MSE loss function until the learning rate reached 1 × 10−5, then training switched to an MAE loss function until the loss plateaued again. This was done to generate a model where the errors were concordant with each other. As mentioned previously, pKa calculations cancel out errors if protonated and deprotonated states have the same sign, i.e. are both positive, but compound the error in pKa value if the errors have different signs. The introduction of the second loss training function works to actively produce errors of the same direction, i.e. both positive.
3.6 Generation, optimisation and filtering of conformers
The workflow for conformer generation and pKa calculation can be seen in Scheme 2, below. Using Avogadro,39 carbene structures were drawn and minimised using the Universal Force Field (UFF).40 The structures were then saved as an XYZ file and used as the input to CREST62 for conformer generation. Through CREST, conformers were generated from the input XYZ using the GFN2-XTB method.63 Generalised Born and solvent accessible surface area implicit solvation (GBSA) available within CREST was used for water solvation for aqueous conformer generation.62 Using the conformers generated by Crest these were then converted into ASE51 and optimised to the default fmax value of 0.05 eV Å−1. Here, the calculators were set to the trained models as described in Section 3.5 and the optimisation algorithm set to FIRE.64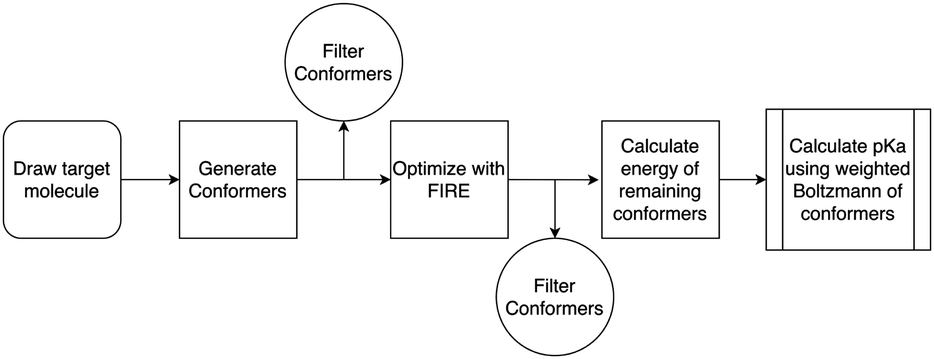 | ||
| Scheme 2 Flowchart detailing the workflow of the pKa prediction. | ||
Conformers were filtered twice during the workflow. On both occasions, the energy of each conformer was obtained and used to calculate a weighted Boltzmann average for each molecule. From the weighted average filtering was completed to retain ∼99% of the Boltzmann population. When the modelled number of conformers is low, this can result in some conformers being filtered out and thus, in this instance, slightly less than 99% of the Boltzmann population was retained. This corresponded to an energy threshold of 3.5 kcal mol−1.65 However, as the filtering was to occur twice, once after CREST conformer generation and once after optimisation, the first threshold was set to a less stringent threshold of 5.0 kcal mol−1 in order to filter any high energy conformers (details of the conformers generated for each structure is provided in the ESI,† pages S3–S6).
3.7 Calculation of pKa values
As shown in eqn (4) and (5), it is possible to calculate the pKa of a compound using Gibbs energy values of different protonation and solvation states as detailed in Fig. 1. In order to calculate ΔG0aq through eqn (5), four values must be calculated. Using each of the newly generated NNP models, for a carbene, A, the values for the deprotonated gas (ΔG(A)) and aqueous phase (ΔGaq(A)) as well as for the protonated gas (ΔG(AH+)) and aqueous phase (ΔGsolv(AH+)) can be obtained. These species are consistent with the thermodynamic cycle shown in Fig. 1. The Gibbs energy of solvation for all species can be calculated as shown in eqn (6). Once the ΔGgas has been calculated as shown in eqn (7), the value of ΔG0aq can be calculated as shown in eqn (8).| ΔG0solv = Gaq − Ggas | (6) |
| ΔGgas = (G(A) + G(H+) − G(AH+)) | (7) |
| ΔG0aq = ΔG0gas + ΔG0solv(A) + ΔG0solv(H+) − ΔG0solv(HA+) | (8) |
As mentioned in Section 3.1, the Gibbs energy values associated with the proton are taken from literature. Once calculated, ΔG0aq can be used in eqn (4) to calculate the pKa value.
Once filtering of optimised conformers was completed as shown in Section 3.6, the pKa was calculated. For every combination of conformers of a molecule, both protonated and deprotonated, the pKa is calculated and then weighted based on the combined Boltzmann probabilities of the protonated and deprotonated conformer used in the calculation. The final pKa for the molecule is then calculated by taking the weighted average of all calculated pKa values, eqn (9).
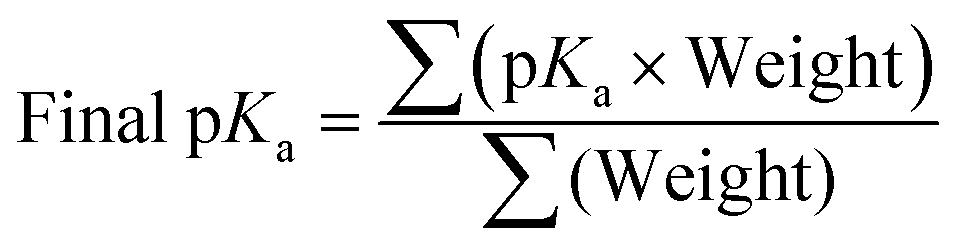 | (9) |
4 Conclusion
We have describe a robust method for the determination of imidazolium-derived carbene pKa values. The method involves a conformer search that ensures the molecule's Boltzmann population is taken into account when calculating pKa. Further, we show that the architecture developed by the ANI family of models can be extended past the neutral singlet gaseous state structures detailed thus far. We show that when trained on aqueous phase energies and/or charged molecules, the models can retain their accuracy, which we demonstrate in this study the prediction of energies and subsequent accurate calculation of pKa values, a problem which is very sensitive to subtle changes in energies and structure.In future, by expanding our models to other functional groups we can overcome some of the limitations with the current developed workflow. Preliminary modelling of butylamine with trained ML models produced a result of 12.79 versus a literature value of 10.70.66 This prediction is almost in line with the results presented in this paper as such, extension to other cationic species is possible with current methods. It should be noted due to the development of charged datasets for this paper, there may be some overlap within the chemical space with protonated amine molecules. However, to better represent this chemical space further calculations may be necessary with emphasis placed on expanding the coverage of amines within the training data.
For expansion to functional groups that include anions within their pKa calculation, e.g. carboxylic acids, there would need to be significant computational effort put towards development of an anionic dataset for accurate modelling of such species. The current level of theory may also need to be reworked as accurate modelling of anionic species often requires diffuse functions, e.g. aug-cc-pVDZ.
Author contributions
Ross Urquhart: methodology, validation, software, investigation, data curation, writing. Alexander van Teijlingen: methodology, validation, software, investigation, writing. Tell Tuttle: conceptualization, methodology, supervision and manuscript editing.Data availability
Relevant files and data underpinning this publication are openly available from the University of Strathclyde KnowledgeBase at: https://doi.org/10.15129/8e3a4b33-c787-43d6-afbb-0c1a9a975af8. All code relevant to this publication can be accessed openly at https://github.com/Tuttlelab/DeepSolv. Other additional data including a description of files and data types have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Results were obtained using the EPSRC-funded ARCHIE-WeSt High Performance Computer based at the University of Strathclyde (https://www.archie-west.ac.uk; EPSRC grant no. EP/K000586/1).Notes and references
- A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717–8724 CrossRef CAS PubMed.
- L. Samuelsen, R. Holm, A. Lathuile and C. Schönbeck, Int. J. Pharm., 2019, 560, 357–364 CrossRef CAS PubMed.
- K. C. Gross and P. G. Seybold, Int. J. Quantum Chem., 2000, 80, 1107–1115 CrossRef CAS.
- G. Cruciani, F. Milletti, L. Storchi, G. Sforna and L. Goracci, Chem. Biodiversity, 2009, 6, 1812–1821 CrossRef CAS PubMed.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC.
- J. H. Berkhout and H. N. Aswatha Ram, Indian J. Pharm. Educ. Res., 2019, 53, S475–S480 CrossRef CAS.
- J. Reijenga, A. Van Hoof, A. Van Loon and B. Teunissen, Anal. Chem. Insights, 2013, 8, 53–71, DOI:10.4137/ACI.S12304.
- M. D. Liptak and G. C. Shields, J. Am. Chem. Soc., 2001, 123, 7314–7319 CrossRef CAS PubMed.
- S. Pezzola, S. Tarallo, A. Iannini, M. Venanzi, P. Galloni, V. Conte and F. Sabuzi, Molecules, 2022, 27, 8590 CrossRef CAS PubMed.
- B. Thapa and H. B. Schlegel, J. Phys. Chem. A, 2016, 120, 5726–5735 CrossRef CAS PubMed.
- F. R. Dutra, C. D. S. Silva and R. Custodio, J. Phys. Chem. A, 2021, 125, 65–73 CrossRef CAS PubMed.
- J. Ho and M. L. Coote, Theor. Chem. Acc., 2010, 125, 3–21 Search PubMed.
- M. Quintano and E. Kraka, Chem. Phys. Lett., 2022, 803, 139746 CrossRef CAS.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, J. Phys. Chem. A, 1998, 102, 7787–7794 CrossRef CAS.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- K. Mansouri, N. F. Cariello, A. Korotcov, V. Tkachenko, C. M. Grulke, C. S. Sprankle, D. Allen, W. M. Casey, N. C. Kleinstreuer and A. J. Williams, J. Cheminf., 2019, 11, 60 Search PubMed.
- M. Baltruschat and P. Czodrowski, F1000Res., 2020, 9, 113 Search PubMed.
- T. Sander, J. Freyss, M. Von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. Magariños, J. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. Radoux, A. Segura-Cabrera, A. Hersey and A. Leach, Nucleic Acids Res., 2018, 47, D930–D940 CrossRef PubMed.
- R. Roszak, W. Beker, K. Molga and B. A. Grzybowski, J. Am. Chem. Soc., 2019, 141, 17142–17149 CrossRef CAS PubMed.
- Z. Cai, F. Luo, Y. Wang, E. Li and Y. Huang, ACS Omega, 2021, 6, 34823–34831 CrossRef CAS PubMed.
- R. C. Johnston, K. Yao, Z. Kaplan, M. Chelliah, K. Leswing, S. Seekins, S. Watts, D. Calkins, J. Chief Elk and S. V. Jerome, et al. , J. Chem. Theory Comput., 2023, 19, 2380–2388 CrossRef CAS PubMed.
- Z. Cai, T. Liu, Q. Lin, J. He, X. Lei, F. Luo and Y. Huang, J. Chem. Inf. Model., 2023, 63, 2936–2947 CrossRef CAS PubMed.
- F. Mayr, M. Wieder, O. Weider and T. Langer, Front. Chem., 2022, 10, 866585 CrossRef CAS PubMed.
- J. S. Smith, O. Isayev and A. E. Roitberg, Chem. Sci., 2017, 8, 3192–3203 RSC.
- K. Schütt, P.-J. Kindermans, H. Sauceda, S. Chmiela, A. Tkatchenko and K.-R. Müller, Advances in Neural Information Processing Systems, 2017, pp. 992–1002 Search PubMed.
- K. T. Schütt, F. Arbabzadah, S. Chmiela, K. R. Müller and A. Tkatchenko, Nat. Commun., 2017, 8, 13890 CrossRef PubMed.
- I. Batatia, D. P. Kovacs, G. N. C. Simm, C. Ortner and G. Csanyi, Advances in Neural Information Processing Systems, 2022 Search PubMed.
- E. Gelžinytė, M. Öeren, M. D. Segall and G. Csányi, J. Chem. Theory Comput., 2024, 20, 164–177 CrossRef PubMed.
- R. Zubatyuk, J. S. Smith, J. Leszczynski and O. Isayev, Sci. Adv., 2019, 5, eaav6490 CrossRef CAS PubMed.
- J. S. Smith, B. Nebgen, N. Lubbers, O. Isayev and A. E. Roitberg, J. Chem. Phys., 2018, 148, 241733 CrossRef PubMed.
- J. S. Smith, B. T. Nebgen, R. Zubatyuk, N. Lubbers, C. Devereux, K. Barros, S. Tretiak, O. Isayev and A. E. Roitberg, Nat. Commun., 2019, 10, 2903 CrossRef PubMed.
- C. Devereux, J. S. Smith, K. K. Huddleston, K. Barros, R. Zubatyuk, O. Isayev and A. E. Roitberg, J. Chem. Theory Comput., 2020, 16, 4192–4202 CrossRef CAS PubMed.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- H. Gokcan and O. Isayev, Chem. Sci., 2022, 13, 2462–2474 RSC.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- F. Nahra, D. J. Nelson and S. P. Nolan, Trends Chem., 2020, 2, 1096–1113 CrossRef CAS.
- D. V. Pasyukov, M. A. Shevchenko, A. V. Astakhov, M. E. Minyaev, Y. Zhang, V. M. Chernyshev and V. P. Ananikov, Dalton Trans., 2023, 52, 12067–12086 RSC.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- B. Helmich-Paris, B. de Souza, F. Neese and R. Izsák, J. Chem. Phys., 2021, 155, 104109 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- T. Matsui, Y. Shigeta and K. Morihashi, J. Chem. Theory Comput., 2017, 13, 4791–4803 CrossRef CAS PubMed.
- A. Paszke, S. Gross, S. Chintala, G. Chanan, E. Yang, Z. DeVito, Z. Lin, A. Desmaison, L. Antiga and A. Lerer, NIPS-W, 2017.
- X. Gao, F. Ramezanghorbani, O. Isayev, J. S. Smith and A. E. Roitberg, J. Chem. Inf. Model., 2020, 60, 3408–3415 CrossRef CAS PubMed.
- H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- D. C. Liu and J. Nocedal, Math. Prog., 1989, 45, 503–528 CrossRef.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- A. W. Sousa Da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 367 CrossRef PubMed.
- L. Kagami, A. Wilter, A. Diaz and W. Vranken, Bioinformatics, 2023, 39, btad350 CrossRef CAS PubMed.
- A. Collette, Python and HDF5, O’Reilly, 2013 Search PubMed.
- J. S. Smith, O. Isayev and A. Roitberg, ANI-1: A data set of 20M off-equilibrium DFT calculations for organic molecules, 2017, https://springernature.figshare.com/collections/ANI-1_A_data_set_of_20M_off-equilibrium_DFT_calculations_for_organic_molecules/3846712/1.
- G. Landrum, RDKit: Open-Source Cheminformatics Software, 2016, https://www.rdkit.org Search PubMed.
- The QMspin data set: Several thousand carbene singlet and triplet state structures and vertical spin gaps computed at MRCISD + Q-F12/cc-pVDZ-F12 level of theory, 2020.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Phys. Rev. Lett., 2006, 97, 170201 CrossRef PubMed.
- I. Iribarren and C. Trujillo, J. Chem. Inf. Model., 2022, 62, 5568–5580 CrossRef CAS PubMed.
- M. Morgenthaler, E. Schweizer, A. Hoffmann-Röder, F. Benini, R. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Müller, ChemMedChem, 2007, 2, 1100–1115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All computational data described in this publication are openly available from the University of Strathclyde knowledge base at https://doi.org/10.15129/8e3a4b33-c787-43d6-afbb-0c1a9a975af8. The code from this paper is available at https://github.com/Tuttlelab/DeepSolv. See DOI: https://doi.org/10.1039/d4cp01982b |
| This journal is © the Owner Societies 2024 |